ВИЗНАЧЕННЯ І КЛАСИФІКАЦІЯ
АГ, за визначенням Комітету експертів ВООЗ — це постійно підвищений САТ та/або ДАТ.
Есенціальна гіпертензія (первинна гіпертензія) — це підвищений АТ за відсутності очевидної причини його підвищення.
Вторинна гіпертензія (симптоматична) — це гіпертензія, причина якої може бути виявлена.
Термін «есенціальна гіпертензія» вперше застосував E. Frank у 1911 р. для визначення підвищення АТ, не зумовленого захворюванням нирок (брайтовою хворобою) або іншою патологією, що спричиняє підвищення АТ. Цей термін не цілком вдалий, тому що англійське слово «essential» означає «істотний, необхідний», у зв’язку з чим поняття «есенціальна гіпертензія» може бути витлумачене як підвищення АТ, необхідне для забезпечення кровопостачання тканин організму. Тому деякі закордонні автори віддають перевагу терміну «первинна гіпертензія». Еквівалентом цих назв є термін «гіпертонічна хвороба» (ГХ), введений Г.Ф. Лангом у 1922 р. і застосовуваний на сьогодні у країнах СНД, зокрема в Росії та Україні. Він більш вдалий, ніж термін «есенціальна гіпертензія», тому що відображає сутність підвищення АТ як хворобливого стану, а не компенсаторного процесу.
ГХ виявляють у 95% осіб з підвищеним АТ. В інших 5% АТ підвищений внаслідок різних захворювань — ураження паренхіми нирок, пухлин надниркової залози, захворювань аорти (коарктація, аортоартеріїт), ниркових артерій і багатьох інших.
Класифікація. Згідно з рекомендаціями ВООЗ, Європейського товариства гіпертензії і Європейського товариства кардіологів (2007) виділяють кілька рівнів АТ (табл. 1.1).
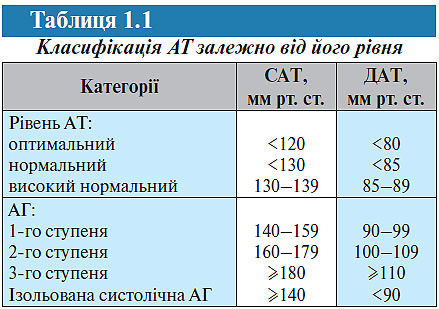
Відповідно до цієї класифікації АГ — підвищення САТ ≥140 мм рт. ст. або ДАТ ≥90 мм рт. ст., якщо таке підвищення стабільне, тобто підтверджується при повторних вимірах АТ (не менше ніж 2–3 рази в різні дні протягом декількох тижнів).
Поділ рівнів АТ на нормальний і високий умовний, тому що розмежувальна риса між ними відсутня. Однак відомо, що рівень АТ і смертність внаслідок серцево-судинних захворювань перебувають у прямому взаємозв’язку: чим вищий АТ, тим вищою є смертність. Навіть АТ 120/80 мм рт. ст. пов’язаний із більш істотним ризиком розвитку серцево-судинних захворювань, ніж, наприклад, АТ 110/75 мм рт. ст. Ризик прогресивно підвищується, коли АТ досягає ≥140/90 мм рт. ст.
Для встановлення стадії АГ використовується класифікація залежно від ураження органів-мішеней (табл. 1.2), рекомендована Українською асоціацією кардіологів (1999; 2004).
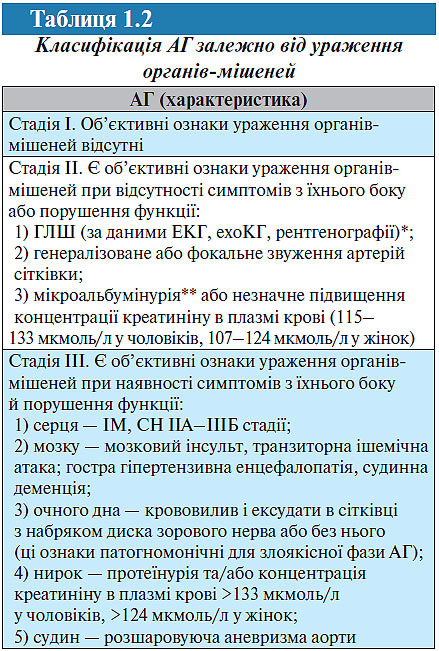
*Критерії ГЛШ: за даними ЕКГ: індекс Соколова — Лайона >38 мм, Корнелльський критерій >2440 мм/мс; за даними ехоКГ: індекс маси міокарда ЛШ ≥125 г/м2 у чоловіків, ≥110 г/м2 у жінок.
**Мікроальбумінурія: екскреція альбуміну 30–300 мг/добу. Протеїнурія: екскреція альбуміну >300 мг/добу.
Вона являє собою незначно модифіковану класифікацію ВООЗ (1996) і відрізняється від останньої тим, що до неї не включені: ультразвукові прояви атеросклерозу судин як критерій II стадії; стенокардія і оклюзивні захворювання артерій як критерій III cтадії. Наявність атеросклерозу судин значно більшою мірою характеризує вираженість атеросклеротичного процесу, ніж АГ. Використання, наприклад, стенокардії або переміжної кульгавості як критерію III стадії ГХ може призводити до невиправданого завищення стадії захворювання.
Цю класифікацію слід використовувати для встановлення стадії як ГХ (есенціальної гіпертензії), так і вторинної АГ.
Діагноз формулюють із указанням стадії захворювання, його ступеня, характеру ураження органів-мішеней, а також ризику ускладнень. Якщо встановлюється діагноз ГХ II стадії, необхідно конкретно вказати, на підставі чого встановлюється ця стадія: наявність ГЛШ або гіпертензивної нефропатії, звуження артерій сітківки. Діагноз ГХ III стадії також необхідно обґрунтувати (наявністю СН, перенесеного мозкового інсульту та ін.).
Відповідно до рекомендацій Української асоціації кардіологів, діагноз ГХ III стадії при наявності ІМ, мозкового інсульту або інших ознак цієї стадії слід встановлювати тільки в тих випадках, коли ці ускладнення з боку серцево-судинної системи розвинулися на фоні тривалого перебігу ГХ, що підтверджується наявними об’єктивними ознаками гіпертензивного ураження органів-мішеней (ГЛШ, генералізоване звуження артерій сітківки та ін.). За відсутності подібних змін слід індивідуально підходити до вирішення питання про наявність ГХ та її стадії. Підвищення АТ на фоні мозкового інсульту або больового синдрому у хворого з ІМ може бути реактивним, минущим. Крім того, ГХ (як і вторинна гіпертензія) може бути в таких хворих супутнім захворюванням на початковій стадії розвитку. У цих випадках встановлюють діагноз ГХ I стадії, незважаючи на гострий або раніше перенесений мозковий інсульт, ІМ або інші захворювання, характерні для III стадії ГХ.
Оцінка ризику. Підвищення АТ — фактор ризику розвитку захворювань серцево-судинної системи. Чим вищий АТ, тим вищий ризик розвитку інсульту, ІХС і передчасної смерті. Тривалий перебіг гіпертензії призводить до ураження органів-мішеней — серця, головного мозку і нирок. Навіть незначне підвищення рівня АТ становить істотну небезпеку для здоров’я. Так, 60% ускладнень з боку серцево-судинної системи виявляють у хворих з помірним підвищенням ДАТ (не вище 95 мм рт. ст.). Нижче наведені дані про вплив АТ на тривалість життя 35-літнього чоловіка, розраховані асоціацією страхових компаній США (1979):
| АТ, мм рт. ст. | Очікувана тривалість життя, років |
| 120/80
130/90 140/95 150/100 |
73,5
67,5 62,5 55 |
Спостерігається позитивна кореляція між рівнем АТ і загальною смертністю: чим нижчий САТ або ДАТ (у будь-якому віці), тим нижча смертність, і навпаки. З підвищенням АТ на кожні 10 мм рт. ст. ризик розвитку захворювань серцево-судинної системи підвищується на 10%.
Максимально корисною для хворого визнана стратегія лікування, яка базується на визначенні загального ризику. Під останнім розуміють той ризик ускладнень, що є в цього хворого внаслідок підвищення АТ, ураження органів-мішеней, наявності супутніх серцево-судинних захворювань і основних факторів ризику (табл. 1.3).

Можна виділити кілька груп ризику (табл. 1.4).

До групи звичайного ризику належать особи з АТ <140/90 мм рт. ст. без додаткових факторів ризику. Група людей, що мають додатковий (до звичайного) ризик ускладнень, але він порівняно невисокий, виділена як група помірного ризику. Її становлять особи з АТ 140–179/90–109 мм рт. ст., які мають не більше 1–2 факторів ризику атеросклерозу, без ураження органів-мішеней, цукрового діабету або інших показників, наведених у табл. 1.3. Іншими словами, це хворі з ГХ I стадії, 1–2-го ступеня, які мають не більше 2 факторів ризику. Підвищення АТ ≥180/110 мм рт. ст. підвищує ймовірність розвитку ускладнень і такі хворі вже становлять групу високого ризику. До групи високого ризику належать також хворі з ГХ II стадії. Пацієнти з ГХ III стадії становлять групу дуже високого ризику.
Відповідно до Фремінгемських критеріїв терміни «низький», «помірний», «високий» і «дуже високий» ризик означають 10-річну ймовірність серцево-судинних ускладнень (фатальних і нефатальних) <15%, 15–20%, 20–30% і >30% відповідно. З 2003 р. у практику європейської кардіології вводиться ще одна модель оцінки ризику — шкала SCORE, яка дозволяє передбачати ймовірність фатальних серцево-судинних подій протягом 10 років. Шкала SCORE відповідає такій імовірності фатальних серцево-судинних ускладнень: <4% — низький, 4–5% — помірний, 5–8% — високий і >8% — дуже високий ризик.
ПАТОГЕНЕЗ
Є підстави вважати, що ГХ — відносно нове в історії цивілізації захворювання. У його розвитку беруть участь як генні механізми, так і зовнішні фактори (табл. 1.5).
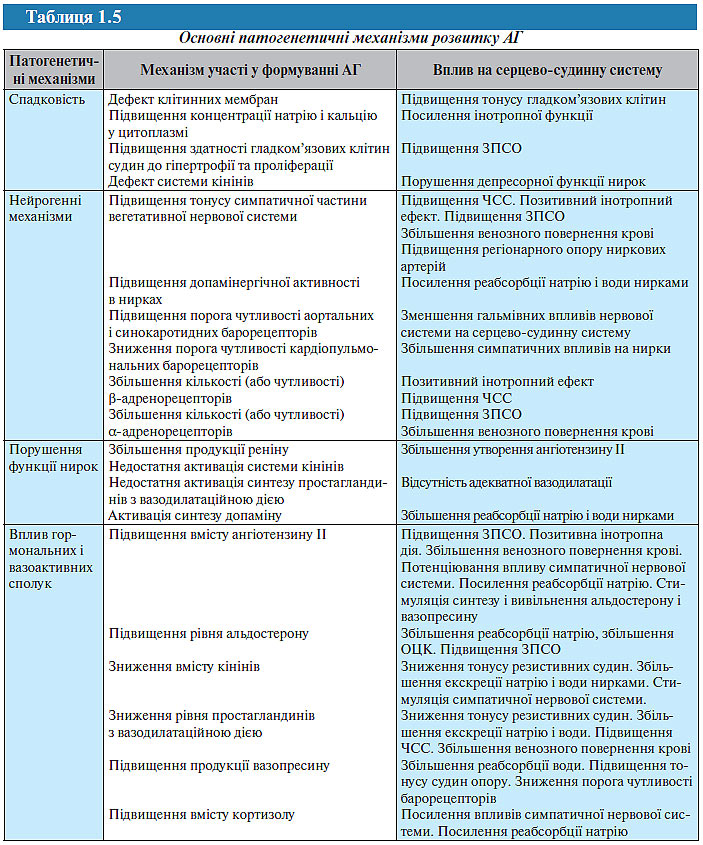
Роль генетичних факторів у розвитку АГ. Спадкова схильність до АГ вважається одним з найбільш достовірних факторів ризику виникнення і прогресування хвороби та часто виявляється у найближчих родичів. У 80% пацієнтів з АГ близькі або далекі родичі також мають підвищений АТ. Відповідно до сучасних уявлень, зазначена схильність реалізується у взаємодії з різними факторами навколишнього середовища, ймовірність успадкування АГ становить близько 30%.
На сьогодні існують кілька теорій успадкування схильності до АГ.
Моногенна теорія заснована на припущенні про єдиний для всіх хворих дефект у серцево-судинній системі або в механізмах регуляції АТ, зумовлений порушеннями на рівні одного гена. Цій теорії, однак, суперечать результати експериментальних досліджень: на сьогодні отримано кілька ліній щурів з генетично зумовленою АГ, що істотно відрізняються за механізмами успадкування АГ.
Полігенна теорія заснована на припущенні про дефект декількох генів (поєднань генів), що контролюють розвиток серцево-судинної системи (метаболізм судинної стінки, який визначає відповідь на регулюючі впливи), або ж групи генів, відповідальних за функціонування систем регуляції кровообігу, у тому числі й АТ. Допускається можливість, що в конкретного хворого який-небудь генний дефект є домінантним і визначає особливості виникнення, розвитку і наслідків АГ.
Теорія граничної моделі генетичної схильності до АГ припускає, що підвищення АТ відображає суму порушень активності різних генів, жоден з яких не є домінантним.
На сьогодні наука ще не має у своєму розпорядженні достатньо фактичних даних, щоб віддати перевагу тій або іншій гіпотезі. Не до кінця зрозумілі також конкретні механізми реалізації спадкової схильності до АГ.
Найбільш важливі докази того, що в підвищенні АТ задіяні полігенні механізми, дає біометричний аналіз, що показує існування кореляції між рівнями АТ у родичів. Іншими словами, в батьків з низьким рівнем АТ вища ймовірність народження дітей з низьким АТ і навпаки. Ця значима залежність може бути пояснена не наявністю одного головного опосередковуючого гена, а лише полігенних послідовностей, у яких кожний ген впливає на АТ. Регіони хромосом або гени, що впливають на АТ, визначаються як такі, у яких молекулярна ідентичність між сибсами асоційована з подібними змінами АТ, що спостерігаються частіше, ніж очікується відповідно до теорії ймовірностей. Подібність може визначатися якісно (наприклад оцінка випадків АГ у сибсів) або кількісно (як похідне чисельних розходжень рівнів АТ між сибсами), причому в сучасних дослідженнях для обох видів оцінки використовують статистичні методи.
Є ряд захворювань, що супроводжуються АГ, для яких визначені генні послідовності і тип успадкування (табл. 1.6).
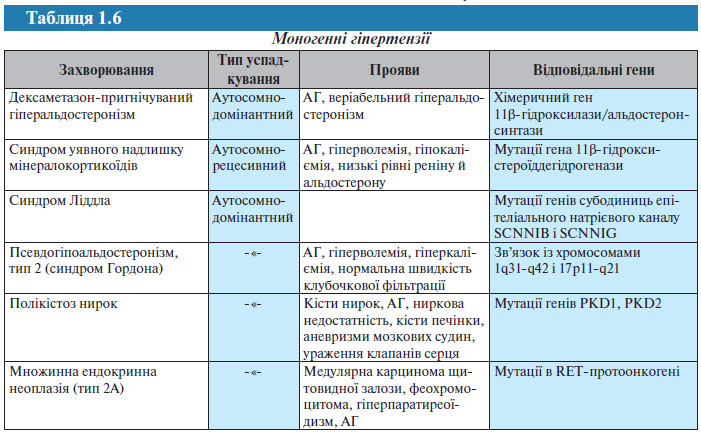
Нижче наведений перелік агентів, що детермінуються генами, які можуть бути відповідальними за розвиток АГ або зумовлюючими підвищення АТ внаслідок мутацій:
- 6-фосфоглюконатдегідрогеназа;
- АПФ;
- ангіотензиноген;
- рецептор глюкокортикоїдів;
- рецептор інсуліну;
- комплемент С3F;
- β2-адренорецептор;
- ліпопротеїнліпаза;
- тип 1А-допаміновий рецептор;
- α1В-адренорецептор;
- ендотеліальна NO-синтаза;
- панкреатична фосфоліпаза;
- α2-адренорецептор;
- рецептор ангіотензину II (АТ1);
- G-протеїн β3-субодиниця;
- простациклінсинтаза;
- гормон росту.
Роль симпатичної нервової системи в розвитку гострого і хронічного підвищення АТ. У класичних роботах Г.Ф. Ланга вказувалося, що початковою патогенетичною ланкою АГ є надмірне тонічне скорочення артеріол у відповідь на появу вогнища застійного порушення вищих центрів, що регулюють АТ. Його послідовник О.Л. М’ясников (1954) підтвердив первинність психогенного порушення функції вазомоторної системи в регуляції АТ. У подальшому був виявлений тісний зв’язок симпатичної нервової системи з іншими пресорними механізмами, що залежить від стадії захворювання і співвідношення пресорних і депресорних механізмів його прогресування.
Результати досліджень D.J. Reis і співавторів (1984; 1989) дозволили встановити роль різних ядер симпатичної нервової системи в коротко- і довгостроковій регуляції АТ. Контроль АТ інтегрований у ростральному вентролатеральному ядрі (РВЯ) довгастого мозку, що іноді називають вазомоторним контролюючим центром. Тіла клітин еферентних стимулюючих серцево-судинну систему симпатичних нейронів перебувають у субрегіоні С1, що взаємодіє з різними центрами ЦНС, одержуючи від них і посилаючи в них нервові імпульси. Найбільш важливі сигнали в РВЯ надходять із суміжного nucleus tractus solitarius (NTS), що одержує аферентні волокна з баромеханорецепторів каротидного синуса і дуги аорти (аортокаротидні барорефлекси). Сигнали з NTS пригнічують симпатичну активність РВЯ, зменшуючи гостре підвищення АТ.
Інгібуючі барорецепторні системи контролюють активність симпатичної ланки нервової системи: одна з них відповідає за регулювання АТ (аортокаротидні барорефлекси), інша — за зміни серцевого об’єму (кардіопульмональні барорефлекси). Ці дві системи працюють узгоджено, зберігаючи сталість ОЦК і АТ.
Артеріальні барорецептори відіграють важливу роль у процесі «хронізації» АГ через властиву їм нездатність реагувати на тривалі зміни АТ (феномен, відомий як барорефлекторне перемикання). В умовах постійно підвищеного АТ барорецептори зберігають здатність реагувати на короткочасні зміни тиску, але не можуть повернути його до нормальних цифр. Отже, симпатична нервова система не пригнічується належною мірою, навіть при високому АТ. Хронічна «нечутливість» барорецепторів пов’язана зі старінням, підвищеною активністю цієї системи і надмірною дією ангіотензину II.
Порушена чутливість кардіопульмональних барорецепторів також може мати велике значення в підтримці тривалого підвищення активності симпатичної нервової системи й АТ. На це вказує, зокрема, такий факт: при зменшенні ХОК в осіб з граничною АГ активація симпатичних нервів більш виражена, ніж в осіб з нормотензією. В експериментах на собаках з нирковою недостатністю і АГ при навантаженні обсягом відсутні як аортокаротидні, так і кардіопульмональні рефлекси. Продемонстровано також, що порушення кардіопульмональних рефлексів впливає на підвищення активності симпатичної нервової системи з віком.
Роль стресу в розвитку АГ. Стимуляція симпатичної нервової системи внаслідок психічних або фізичних навантажень викликає транзиторне збільшення продукції норадреналіну та, відповідно, підвищення АТ. До найбільш важливих стимулів слід віднести фізичні вправи, які короткочасно підвищують АТ, однак при регулярних заняттях сприяють розвитку тренованості й ефективному зниженню базальної та стимульованої активності симпатичної нервової системи і АТ, а отже, знижують ризик серцево-судинних захворювань (рис. 1.1).
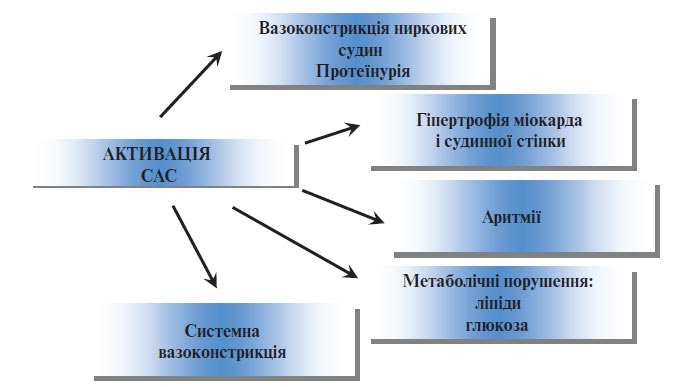
Рис. 1.1. Роль активації CAC
Іншим важливим стимулятором симпатичної нервової системи є тютюнопаління: незважаючи на те, що підвищення АТ після викуреної сигарети короткочасне, тривале тютюнопаління може спричиняти тривале підвищення АТ.
До найсильніших стресових факторів, що викликають різке підвищення АТ, часто з розвитком клініки гіпертензивного кризу, належать опіки, травми головного мозку, хірургічні втручання, загальна анестезія, кожний з яких приводить до вираженої активації симпатичної нервової системи. Холодові навантаження або передозування деяких лікарських препаратів (наприклад опіоїдів) також можуть викликати різку активацію симпатичної нервової системи і підвищення АТ.
З кінця 70-х років ХХ ст. предметом дискусій є гіпотеза, що полягає в тому, що в осіб з гіперреакцією на стрес у вигляді значного підвищення АТ і ЧСС та інших серцево-судинних реакцій високий ризик розвитку хронічної АГ. У дослідження CARDIA (Markovitz J.H. та ін., 1998) були включені >3300 осіб молодого віку, що піддавалися емоційним навантаженням (відеоігри). Період спостереження склав 5 років. Виявлено, що у чоловіків з гіперреакцією на психологічне навантаження у вигляді значного підвищення САТ (на 10–30 мм рт. ст.) був високий ризик розвитку АГ, у той час як у жінок подібної закономірності не було. Такий же зв’язок виявлений у дослідженні A. Steptoe, M. Marmot (2007) — у нормотензивних осіб з уповільненою нормалізацією САТ у постнавантажувальний період (застосовувався ментальний стрес) протягом наступних 3 років гіпертензія розвивалася в 3,5 раза частіше, ніж в осіб з нормальним зниженням АТ у відновлювальний період.
РААС належить до основних регуляторів судинного тонусу, водно-електролітного балансу і рівня АТ. У структурному відношенні вона являє собою каскадну «гормональну вісь», яка включає ланцюг ензиматичних реакцій, внаслідок яких утворюються біологічно активні пептиди — ангіотензини I, II і III. Вивчення вмісту ангіотензину II у крові пацієнтів з ГХ показало відсутність кореляції між рівнем АТ і концентрацією цього пептиду. Разом з тим встановлено, що пригнічення РААС за допомогою препаратів, що блокують утворення або дію ангіотензину II, зумовлює в більшості пацієнтів із ГХ істотне зниження АТ (рис. 1.2).
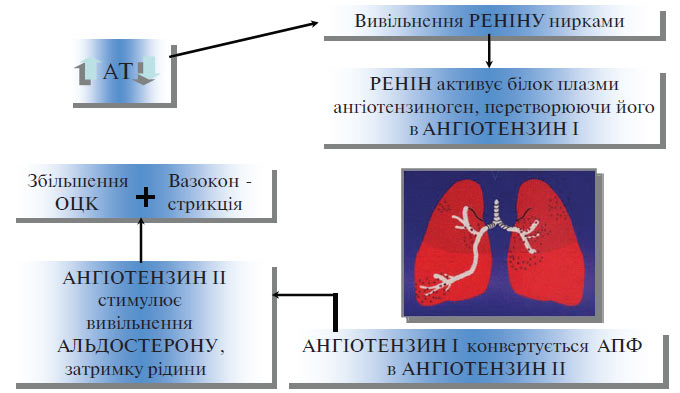
Рис. 1.2. Система ренін — ангіотензин — альдостерон
Ці суперечливі дані знаходять часткове пояснення в гіпотезі, запропонованій J. Laragh і співавторами (1973; 1980). Відповідно до розробленої ними об’ємно-вазоконстрикторної моделі РААС так чи інакше бере участь у всіх видах підвищення АТ. У пацієнтів з ГХ і високою активністю реніну в плазмі крові РААС безпосередньо впливає на вазоконстрикцію і є головним чинником підтримки АГ. У хворих з низькою активністю реніну провідний механізм у підвищенні АТ — це затримка натрію і води; активність реніну знижена внаслідок пригнічення його секреції збільшеним об’ємом крові.
У хворих з нормальною активністю реніну вазоконстрикторний і об’ємний механізми також беруть участь у підтримці АГ. У цих хворих, незважаючи на те що ренін-натрієві профілі перебувають у нормальних межах, рівень реніну неадекватно високий для такого стану натрієвого балансу і такого рівня АТ, тобто має місце непропорційне співвідношення між вазоконстрикторним і об’ємним фактором, що може сприяти підтримці підвищеного рівня АТ.
На сьогодні встановлено, що активація РААС, крім підвищення АТ, є чинником ризику розвитку ускладнень АГ. За даними J. Laragh (1996) у хворих з АГ і однаковим рівнем АТ, але різною активністю реніну в плазмі крові частота розвитку ІХС або інсульту протягом 5 років спостереження становить 11% у групі пацієнтів з помірним підвищенням активності реніну і 14% — з його значною активацією, однак такі ускладнення рідко бувають у хворих з низьким рівнем активного реніну в плазмі крові. Підвищена активність РААС є також незалежним фактором ризику розвитку ІХС та її ускладнень. Очевидно, це пов’язано зі значною роллю РААС у процесах атерогенезу, гіпертрофії і патологічного ремоделювання міокарда. Встановлено, що ангіотензин II спричиняє атерогенну дію, стимулюючи міграцію макрофагів і нейтрофільних гранулоцитів у судинну стінку, підвищуючи окислювання ХС і ЛПНП. У результаті це призводить до ендотеліальної дисфункції з порушенням вивільнення NО і активацією синтезу потужного вазоконстрикторного агента ендотеліну-1, цитокінів і факторів росту, що відіграють важливу роль у структурному ремоделюванні серця і судин.
Ейкозаноїди виконують роль як про-, так і антигіпертензивних субстанцій. Їх внесок у регуляцію АТ не піддається однозначному трактуванню як через чисельність цих речовин, так і внаслідок їх різноспрямованої біологічної дії. До прогіпертензивних ейкозаноїдів належать, зокрема, тромбоксан A2 (TxA2) і простагландин H2 (ПГН2). У багатьох дослідженнях показано, що зміни в системі простагландинів (ПГ) класів Е1 і F2α виявляються ще на етапі граничної АГ і характеризуються підвищенням їхнього сумарного рівня і зсувом співвідношення в бік переваги пресорних фракцій. При прогресуванні захворювання сумарний рівень вищевказаних фракцій знижується, однак зберігається перевага пресорних ПГ, причому спостерігається зниження модулюючого впливу ПГЕ1 на симпатичну нейротрансмісію.
У здорових людей надмірній активації вищезгаданих прогіпертензивних ейкозаноїдів протистоїть система антигіпертензивних ПГ — ПГЕ2 і ПГІ2.
Продукти метаболізму арахідонової кислоти значно впливають на кровоносні судини і транспорт іонів, модуляцію й опосередкування дії вазоактивних гормонів. Таким чином, вони також є частиною системи контролю АТ.
Вторинними посередниками дії ангіотензину II служать і ліпоксигеназні субстанції, зокрема 12-гідроксипероксиейкозатетраєнова кислота і продукт її пероксидації 12-гідроксиейкозатетраєнова кислота, які здатні також пригнічувати синтез ПГІ2.
Медіатори судинної стінки й АГ. Відомо, що ендотелій є високоактивним клітинним шаром, що здійснює багато метаболічних функцій, зокрема регуляцію тонусу судин, тромбоцитарного гемостазу, процесів коагуляції, міграції і проліферації гладком’язових клітин стінки судин.
Ендотеліальні клітини здатні продукувати як медіатори з вазодилатаційною активністю (оксид азоту і простациклін), так і вазоконстриктори (тромбоксан А, ендотелін). Отже, зміни функції клітин ендотелію, вироблення ними специфічних медіаторів можуть бути суттєвою ланкою патогенезу порушень регуляції тонусу судин.
На початку 80-х років ХХ ст. з’явилися повідомлення про те, що ендотеліальні клітини, отримані з аорти бика та вирощені в культурі тканин, продукують вазоконстрикторний пептид, що був виділений із супернатанту культури клітин ендотелію та названий ендотеліном-1 (ЕТ-1). Ендотеліни представляють сімейство регуляторних пептидів, що складаються з 21 амінокислоти, і мають кілька ізоформ: ЕТ-1, ЕТ-2, ЕТ-3 і ЕТ-β.
ЕТ є потужними вазоконстрикторами, що продукуються ендотелієм судин. Роль ЕТ у патогенезі АГ ще недостатньо вивчена: в одних роботах виявлено нормальний вміст цих пептидів у плазмі крові при експериментальній АГ, в інших — парадоксальне зниження відповіді судин на їх введення. Однак більшість дослідників думають, що ефекти ЕТ відіграють важливу роль у патогенезі АГ. Дослідження, проведені з використанням інгібіторів ендотелінперетворювального ферменту (ЕПФ) або блокаторів рецепторів ЕТ свідчать, що ЕТ вносять істотний вклад в підтримку підвищеного АТ (Luscher Th. et al., 1993). Однак рівень циркулюючого ЕТ-1 не завжди визначає регуляцію тонусу судин при АГ, оскільки основним механізмом його дії є локальний вплив на стінку судини.
Роль нирок у розвитку АГ. Рівень АТ регулюється нирками за допомогою механізму тиск — натрійурез: підвищення системного АТ (і відповідно перфузійного тиску в нирках) викликає посилення натрійурезу і діурезу, завдяки чому об’єм позаклітинної рідини, ОЦК і серцевий викид зменшуються до такого рівня, що забезпечує повернення АТ до вихідного. На думку А.С. Guyton та співавторів, у цьому полягає механізм довгострокової регуляції АТ. Він діє за принципом зворотного зв’язку, тобто рівень АТ впливає на натрійурез, що в свою чергу визначає значення системного АТ.
При ГХ функціональні параметри нирки щодо системного АТ істотно зміщені, тому повний обсяг екскреції води і солей можливий лише при підвищеному рівні АТ. Зниження АТ за механізмом зворотного зв’язку активує пресорні механізми, повертаючи його до необхідного для збереження водно-сольового гомеостазу, тобто нирка стає фактором підтримки постійно підвищеного рівня АТ (Постнов Ю.В.) (рис. 1.3).
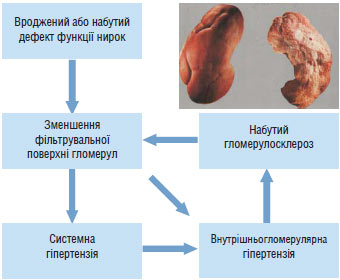
Рис. 1.3. Нирка — і причина, і жертва гіпертензії
Порівняно недавно B.M. Вrenner і S. Anderson (1992) запропонували гіпотезу, яка пояснює вплив нирок на розвиток АГ зменшенням кількості функціонуючих нефронів, що може бути вродженим або набутим внаслідок хронічного захворювання або хірургічного втручання. Зменшення кількості нефронів і пов’язане з цим зниження екскреції натрію і води неминуче призводять до збільшення ОЦК і АТ. Есенціальна гіпертензія зумовлена, принаймні частково, скороченням сумарної фільтрувальної поверхні нирок внаслідок зменшення кількості клубочків або фільтрувальної площі кожного клубочка. Затримка натрію нирками і підвищення АТ у свою чергу дають поштовх до підвищення тиску в капілярах клубочків та їх склерозування. Останнє ще більше зменшує фільтрувальну площу клубочків, замикаючи порочне коло.
Кожна нирка містить близько 1 млн нефронів. Їхня кількість може коливатися від 500 тис. до 1,2 млн. Нові нефрони не утворюються після народження, але їхня кількість починає зменшуватися в процесі нормального старіння після 30-річного віку. B.М. Brenner і S. Anderson вважають, що люди, що народилися з відносно невеликою кількістю нефронів (<700 тис. у кожній нирці), схильні до розвитку АГ, у той час як ті, в кого кількість нефронів перебуває на верхній межі розподілу, мають найнижчі значення АТ у межах фізіологічної норми. Гіпертензія може розвиватися і при нормальній кількості функціонуючих нефронів, якщо відбувається зменшення фільтрувальної площі в кожному нефроні. Зменшення площі базальної мембрани (і відповідно площі фільтрації) призводить до затримки натрію і води та підвищення АТ. Отже, основною патогенетичною детермінантою есенціальної гіпертензії автори гіпотези вважають вроджене зменшення кількості функціонуючих нефронів і/або їх фільтрувальної поверхні, що призводить до зниження здатності нирок екскретувати натрій і воду, особливо в умовах навантаження сіллю. Вторинна гіпертензія, пов’язана із захворюванням нирок, зумовлена набутим зменшенням кількості функціонуючих нефронів.
КЛІНІКА
Клініка АГ зумовлена ураженням органів-мішеней: головного мозку, серця, судин і нирок. Ураження зазначених органів тривалий час протікає безсимптомно і вимагає спеціальних методів для його виявлення: ехоКГ для оцінки ГЛШ, УЗД сонних артерій для оцінки гіпертрофії судин і атеросклерозу, розрахунку кліренсу креатиніну і визначення мікроальбумінурії для виявлення гіпертензивної нефропатії. Хворого необхідно ретельно обстежити для виявлення субклінічних уражень органів-мішеней, тому що вони визначають ризик ускладнень і смерті та впливають на вибір лікування. Тривалий період безсимптомних органних уражень закінчується розвитком ускладнень, які можна розділити на дві великі групи:
- зумовлені ушкодженням судин внаслідок тривалого впливу підвищеного АТ (гіпертензивні ускладнення);
- пов’язані з атеросклеротичним ураженням судин. Ці ускладнення можуть розвиватися і при нормальному рівні АТ, однак наявність АГ зумовлює більш ранню появу і більш тяжкий перебіг.
Судинні (гіпертензивні) ускладнення розвиваються внаслідок прямої механічної дії підвищеного тиску на серце і судини. До них належать гіпертензивна енцефалопатія, крововилив у мозок, субарахноїдальний крововилив, ГЛШ, СН, крововилив у сітківку, набряк соска зорового нерва і втрата зору, первинний нефросклероз і ниркова недостатність, розшаровуюча аневризма аорти, фібриноїдний некроз артеріол і злоякісна АГ (табл. 1.7).
Таблиця 1.7
Класифікація ускладнень АГ
| Орган-мішень | Ускладнення | |
| Гіпертензивні | Атеросклеротичні | |
| Мозок | Гіпертензивна енцефалопатія
Крововилив у мозок Лакунарні інфаркти мозку |
Минуще порушення мозкового кровообігу (транзиторна ішемічна атака)
Інфаркт мозку |
| Серце | ГЛШ
СН |
Стенокардія
ІМ |
| Нирки | Первинний нефросклероз
Фібриноїдний некроз |
Стеноз ниркової артерії |
| Артерії | Аневризми артерій
Розшарування аорти |
Переміжна кульгавість
Гангрена |
Атеросклеротичні ускладнення проявляються ІХС, у тому числі ІМ і раптовою смертю, атеротромботичним інсультом, атеросклеротичним ураженням периферичних артерій, стенозом ниркової артерії та ін. (рис. 1.4).
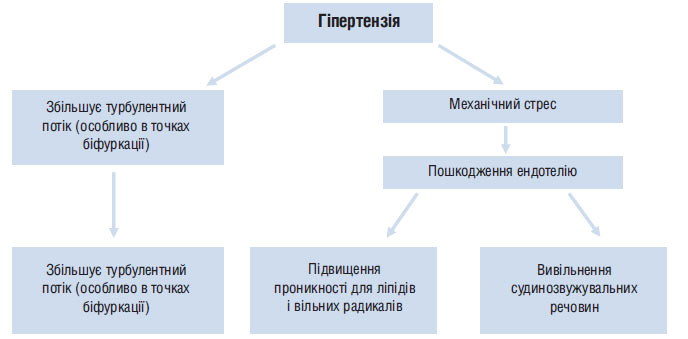
Рис. 1.4. Гіпертензія сприяє розвитку атеросклерозу
Ураження головного мозку внаслідок підвищення АТ зумовлене такими причинами:
- атеросклеротичним ураженням великих артерій з наступним атеротромбозом і розвитком ішемічного інсульту;
- гіпертензивним ураженням дрібних артерій і артеріол, що призводить до внутрішньомозкового крововиливу або формування лакунарних інфарктів мозку, або розвитку судинної деменції;
- гострим порушенням ауторегуляції мозкового кровотоку внаслідок прямої дії високого АТ на судини мозку, що проявляється гострою гіпертензивною енцефалопатією.
Мозковий інсульт і ІХС і на сьогодні лишаються головними причинами смерті хворих з АГ. У той час як у розвинених країнах Європи й Америки смертність від інсульту істотно знизилася, у країнах Східної Європи, Азії, Африки і Південної Америки вона катастрофічно підвищується.
Вважають, що 75% випадків інсульту пов’язані з тромбозами або жировою емболією внаслідок атеросклерозу, 10–15% геморагічних інсультів — з розривами аневризм Шарко — Бушара. Лакунарні інсульти звичайно відбуваються внаслідок оклюзії пенетрувальних судин кільця Віллізієва кола. У значної частини пацієнтів причина розвитку інсульту залишається невідомою (рис. 1.5).
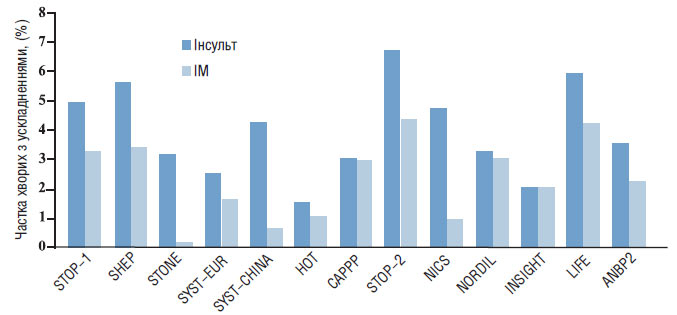
Рис. 1.5. Частота інсульту та ІМ у пацієнтів з ГХ
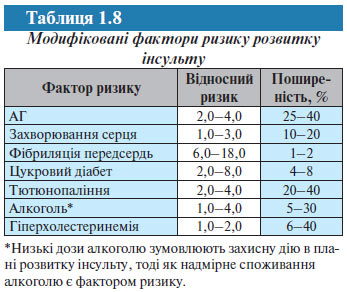
Для зниження частоти розвитку інсульту сьогодні застосовуються різні стратегії, однак безсумнівний пріоритет належить виявленню факторів ризику і розробці методів їх контролю. Основні фактори ризику розвитку інсульту, які можна модифікувати, визначені в епідеміологічних дослідженнях (табл. 1.8).
Лакунарні інфаркти мозку зумовлені оклюзією дрібних пенетрувальних артерій внаслідок фібриноїдного некрозу або (значно частіше) гіалінової дегенерації, яку називають ліпогіалінозом. Лакунарні інфаркти, які реєструють у 2–3 рази частіше, ніж мозкові крововиливи, іноді мають безсимптомний перебіг, їх виявляють тільки при проведенні візуалізуючих процедур (КТ або МРТ). Вони являють собою невеликі глибокі ураження білої речовини мозку, які на томограмах мають вигляд лакун.
Дрібні пенетрувальні артерії мозку особливо чутливі до пошкоджувальної дії високого АТ, тому що вони відходять прямо від головного артеріального стовбура. Це сприяє формуванню аневризм, які вперше були описані Шарко і Бушаром у 1868 р. Аневризми або розриваються незабаром після утворення, викликаючи масивну геморагію, або розтягуються і товщають. Надалі в них може формуватися тромб, що призводить до оклюзії артерії.
Ураження дрібних артерій внаслідок АГ суттєво відрізняється від атеросклеротичного ураження великих артерій — в першу чергу тим, що воно має дифузний характер і охоплює медіальний шар артерії, а не її інтиму, як при атеросклерозі. При цьому порушується нормальна будова судини, гладкі м’язи нерівномірно атрофуються, медіальна оболонка судини некротизується, що призводить до пенетрації компонентів плазми крові (фібрину) і моноцитів всередину судини і закриттю її просвіту.
Лакунарні інфаркти та внутрішньомозковий крововилив часто ускладнюють перебіг АГ в одного хворого. Крім того, невеликий крововилив і лакунарний інфаркт можуть клінічно не відрізнятися. Для діагностики потрібна візуалізація мозку і (зрідка) його артеріографія.
Гіпертензивна енцефалопатія. Зміни системного АТ викликають розширення або звуження судин мозку, що сприяє підтриманню постійного рівня мозкового кровотоку. Цей процес називають ауторегуляцією. Пряме вимірювання мозкового кровотоку в експериментах на тваринах показало, що зниження системного АТ супроводжується розширенням мозкових судин. Ця реакція спрямована на запобігання гіпоперфузії головного мозку. Підвищення АТ, навпаки, зумовлює звуження судин, що запобігає гіперперфузії мозку.
Раптове підвищення АТ, що значно перевищує звичайний для цього хворого рівень, може призводити до порушення ауторегуляції, її «прориву»: звуження судини стає недостатнім для запобігання гіперперфузії мозку. Це супроводжується появою в дрібних артеріях дилатованих ділянок, які чергуються зі звуженими — артерія набуває вигляду чоток або сосисок. з’являються петехіальні крововиливи, вогнищевий, а потім дифузний набряк тканини мозку з розвитком клінічної картини гіпертензивної енцефалопатії, яка являє собою тяжке ускладнення АГ, що призводить, у випадку неефективного лікування, до летального кінця. Навпаки, вчасно розпочате лікування сприяє повній реверсії клінічних симптомів і відновленню порушених функцій.
Судинна деменція — менш часте, ніж інсульт, але настільки ж тяжке ускладнення АГ. У її розвитку, поряд з АГ, важливу роль відіграють вік та гіперліпідемія, що підвищує в’язкість плазми крові і сповільнює мозковий кровотік. Морфологічним субстратом є ураження дрібних артерій (артеріосклероз), що викликає гіпоперфузію субкортикальних відділів мозку. Це сприяє формуванню субкортикальної артеріосклеротичної енцефалопатії, кінцева стадія якої — судинна деменція. Клінічно вона проявляється порушенням пам’яті, особливість якого полягає в раптовому початку і хвилеподібному перебігу в подальшому. У пацієнтів відмічають емоційну лабільність, малорухливість, нестійку ходу, нетримання сечі.
АГ — найбільш значимий фактор ризику розвитку судинної деменції. Зниження АТ у пацієнтів із хронічною АГ поліпшує перфузію мозку, однак надмірне зниження АТ може погіршувати її, тому що ауторегуляція мозкового кровотоку в таких хворих порушена. Швидке зниження САТ <135–150 мм рт. ст. може збільшувати порушення пам’яті і пізнавальних функцій. У доповнення до антигіпертензивної терапії призначають ацетилсаліцилову кислоту: показано, що вона стабілізує перебіг деменції та на 25% знижує ризик розвитку інсульту. Пентоксифілін також може сповільнювати прогресування судинної деменції завдяки зниженню в’язкості крові.
Ураження серця внаслідок АГ — це ГЛШ, СН, ІХС. ГЛШ у пацієнтів з АГ є компенсаторним механізмом подолання підвищеного навантаження, що дозволяє протягом тривалого часу підтримувати задовільний серцевий викид. Її розвиток розглядають як прояв структурної ауторегуляції серцевого м’яза до довгостроково існуючого підвищення АТ. В умовах гострого навантаження, яке раптово виникло, основний механізм підтримки насосної функції полягає в гомеометричній ауторегуляції, тобто посиленні скоротності міокарда. Хронічне навантаження призводить до структурної перебудови міокарда, яка проявляється збільшенням його маси — структурної ауторегуляції (рис. 1.6).
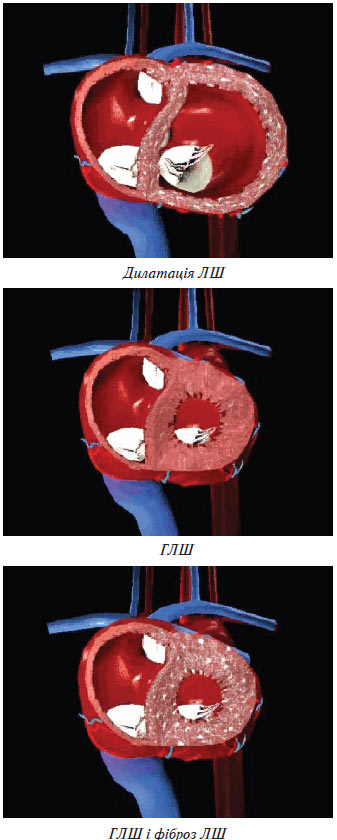
Рис. 1.6. Структурна перебудова міокарда
При прогресуванні ГЛШ втрачає компенсаторне значення і перетворюється на важливий незалежний фактор ризику смерті і серцево-судинних ускладнень.
Компенсаторні зміни геометрії ЛШ називають його ремоделюванням, що включає стовщення стінки ЛШ, спрямоване на нормалізацію його напруження.
Вважають, що розвиток ГЛШ спрямований на підтримку постійного рівня напруження стінки ЛШ. При АГ підвищення постнавантаження збільшує систолічне напруження (стрес) стінки ЛШ і веде до розвитку концентричної ГЛШ, для якої характерне паралельне нагромадження саркомерів у кардіоміоцитах, потовщення стінки ЛШ при збереженні або зменшенні колишнього розміру його порожнини. У випадку збільшення переднавантаження підвищується діастолічне напруження стінки ЛШ. Розвивається ексцентрична ГЛШ, для якої характерне послідовне нагромадження саркомерів і збільшення порожнини ЛШ.
Відповідно до класифікації Ganau (1992) виділяють наступні типи геометричного ремоделювання ЛШ:
- концентричне ремоделювання ЛШ;
- концентрична ГЛШ;
- ексцентрична ГЛШ:
- зі збільшенням порожнини;
- без збільшення порожнини.
Для концентричного ремоделювання ЛШ характерне потовщення його стінки без збільшення загальної маси. Розмір порожнини ЛШ при цьому зменшується. Концентрична ГЛШ полягає в збільшенні м’язової маси ЛШ і підвищенні показника відносної товщини його стінки. Ексцентрична ГЛШ полягає в збільшенні порожнини ЛШ і його маси.
Багато авторів виділяють також асиметричну ГЛШ, що є результатом переважного розвитку ГЛШ в області передньої стінки, верхівки серця й особливо перегородки.
Показники ехоКГ, характерні для ГЛШ, наведені в табл. 1.9.
Таблиця 1.9
Показники ехоКГ, характерні для ГЛШ
| Показник | Значення |
| Діаметр ЛШ, мм
Товщина стінки ЛШ у діастолу, мм Відносна товщина стінки ЛШ Індекс маси ЛШ, г/м2: у чоловіків у жінок |
>57 (ексцентрична ГЛШ)
>11 ≥0,42 (концентрична ГЛШ)
≥125 ≥110 |
Strauer (1984) виділив три типи ГЛШ: нормостресову (адекватну), при якій розвиток ГЛШ приводить до нормалізації напруження стінки ЛШ; високостресову (неадекватну, з недостатнім розвитком ГЛШ і збереженням підвищеного напруження стінки ЛШ); низькостресову, при якій ступінь ГЛШ неадекватний напруженню стінки ЛШ внаслідок надмірної вираженості ГЛШ стосовно такого рівня постнавантаження.
У хворих з нормо- або високостресовою ГЛШ головним механічним стимулом, що індукує розвиток ГЛШ (як концентричної, так і ексцентричної), є напруження стінки ЛШ із підвищенням систолічного стресу в першому випадку і діастолічного — у другому. Однак у хворих з низькостресовою ГЛШ основне значення для розвитку гіпертрофії мають, мабуть, не напруження стінки, а гуморальні фактори (рис. 1.7).
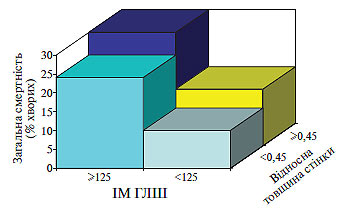
Рис. 1.7. Зв’язок між смертністю і геометрією ЛШ у пацієнтів із ГХ
Важливим функціональним наслідком розвитку ГЛШ є порушення коронарної мікроциркуляції. Розвиток ГЛШ у хворих з АГ супроводжується зниженням коронарного резерву навіть при відсутності стенозу коронарних артерій. Вважають, що це наслідок медіальної гіпертрофії коронарних резистивних судин (коронарної мікроангіопатії), яка спричиняє ішемію міокарда.
Один з найбільш ранніх проявів дисфункції гіпертрофованого ЛШ полягає в порушенні діастолічного наповнення. В останні роки стало очевидним, що застійна СН може бути зумовлена аномальною діастолічною функцією, а не зниженням систолічних властивостей ЛШ. Збільшення маси ЛШ і розвиток інтерстиціального фіброзу призводить до зниження його податливості і порушення наповнення в діастолу, зумовлює посилення систоли передсердя, а надалі, через незначний компенсаторний потенціал лівого передсердя — збільшення його розмірів і підвищення тиску в малому колі кровообігу. Інший фактор, що викликає порушення діастолічного наповнення ЛШ в умовах його гіпертрофії, — це погіршення релаксації, порушення АТФ-залежного процесу розмикання актоміозинових зв’язків внаслідок видалення Са2+ з комплексу тропонін — актоміозин. Уповільнення цього процесу або збільшення кількості нерозімкнутих зв’язків призводить до неповного розслаблення міофібрил, порушення релаксації ЛШ, уповільнення і зменшення обсягу наповнення в ранню фазу діастоли. Для пацієнтів з АГ характерне поєднання сповільненого розслаблення ЛШ із підвищенням його діастолічної пружності і зменшенням розтяжності.
Надзвичайно важливим з клінічної точки зору є питання про те, які з антигіпертензивних препаратів найбільш суттєво сприяють регресії ГЛШ. Встановлено, що майже всі антигіпертензивні засоби, в тому числі блокатори рецепторів ангіотензину II, інгібітори АПФ і антагоністи кальцію, спричиняють регресію ГЛШ. Однак прямі вазодилататори — гідралазин і міноксидил — зменшують масу міокарда ЛШ менше ніж у 50% хворих або навіть сприяють прогресуванню ГЛШ. Це пов’язують із збільшенням ОЦК під їхнім впливом, а також із рефлекторною стимуляцією барорецепторів і вторинним підвищенням рівня катехоламінів і реніну в крові. Діуретики, за винятком індапаміду, незважаючи на їх виражену антигіпертензивну дію, також не завжди сприяють регресії ГЛШ. Очевидно, це пов’язано зі стимуляцією САС, а також із підвищенням рівня реніну й ангіотензину в крові. Блокатори β-адренорецепторів також менш ефективно, ніж препарати, що блокують РААС, або антагоністи кальцію, зменшують масу міокарда ЛШ.
На реверсію ГЛШ впливає не тільки вид лікування, але і його тривалість. Як правило, для істотного зниження маси міокарда потрібно близько 3 міс, хоча є повідомлення і про більш швидке досягненні цієї цілі.
До факторів, що спричиняють вирішальний вплив на регресію ГЛШ, останнім часом стали відносити вихідну вираженість кінцево-систолічного напруження (стресу) стінки ЛШ. Хворі з первинно нормальним напруженням стінки ЛШ відповідають на терапію зменшенням маси міокарда, тоді як хворі з низьким стресом стінки, тобто з непропорційно високою стосовно АТ гіпертрофією міокарда, реагують на антигіпертензивну терапію прогресуванням ГЛШ, незважаючи на аналогічне зниження АТ.
Ураження нирок. Ураження нирок внаслідок АГ або, точніше, в результаті патологічних змін у ниркових артеріях дрібного калібру називають первинним нефросклерозом, на відміну від вторинного нефросклерозу, який розвивається внаслідок захворювань нирок, таких як гломерулонефрит, полікістоз, обструктивні захворювання і т.п. У зарубіжній літературі часто застосовують термін «гіпертензивна нефропатія», що має те ж значення, що й «первинний нефросклероз».
Структурні зміни в нирках, характерні для первинного нефросклерозу, полягають у розвитку фіброзу паренхіми, ураження судин (переважно прегломерулярних дрібних артерій і артеріол) у вигляді їх гіалінозу, фіброплазії інтими, потовщення медії. У пізній стадії клубочки склерозуються, канальці атрофуються. Нирки зменшуються в розмірах, зморщуються, їхня поверхня стає зернистою. Схожі, хоча й менш виражені, зміни в нирках відбуваються в процесі фізіологічного старіння в осіб з нормальним АТ. Тому багато дослідників розцінюють розвиток гіпертензивного нефросклерозу як прискорення природного процесу старіння судинної мережі нирок. Для злоякісної АГ розвиток нефросклерозу є однією з ключових особливостей патогенезу, однак у цьому випадку він має характерну гістологічну картину у вигляді фібриноїдного некрозу в дрібних артеріях і артеріолах.
Індивідуальний ризик розвитку ХНН у хворих з АГ, не зумовленої захворюванням нирок, дуже низький. Однак через надзвичайно високу поширеність АГ серед населення кількість випадків ХНН, зумовлених гіпертензією, досить велика. Це значна проблема для хворого і для системи охорони здоров’я. Хворі з термінальною ХНН підлягають хронічному діалізу, що є дорогою процедурою. Так, у США в 1997 р. на проведення гемодіалізу 300 тис. хворих витрачено 13 млрд дол., у Європі — 10 млрд, у Японії — 9,5 млрд (Remuzzi G., 2000). Наступний після діалізу етап — трансплантація нирки — не менш проблематичний з етичної і матеріальної точки зору. Дешевший шлях — профілактика і лікування АГ. Хоча це не гарантує відсутності ускладнень, але робить їх ймовірність значно нижчою.
Підвищення АТ значно впливає на цей процес. Спостерігається пряма кореляція між рівнем АТ і швидкістю зниження функції нирок. Відповідно до результатів дослідження, проведеного в Балтіморі, швидкість зниження функції нирок і рівень АТ перебувають у прямому взаємозв’язку, однак вона втрачається в осіб із середнім рівнем АТ <107 мм рт. ст. Це значить, що АТ нижче цього рівня, тобто нормальне, перестає впливати на функцію нирок.
У хворих з АГ ризик розвитку ХНН підвищується в міру підвищення АТ: при АТ 160/100–180/110 мм рт. ст. у 11 раз вищий, ніж при оптимальному, а при підвищенні АТ >200/109 мм рт. ст. ризик підвищується ще в 2 рази (дослідження MRFIT).
Клінічні маркери ураження нирок. Специфічних клінічних ознак, які чітко вказували б на наявність гіпертензивної нефропатії (первинного нефросклерозу), немає. У більшості випадків виражений нефросклероз, що виявляється гістологічно, не має клінічних проявів.
Відносно ранніми вказівками на залучення нирок у патологічний процес при есенціальній гіпертензії є мікроальбумінурія, підвищення екскреції із сечею β2-мікроглобуліну, N-ацетилглюкозaмінідази, збільшення вмісту сечової кислоти в плазмі крові (Vermeer S.E. et al., 2002).
Протеїнурією вважають рівень білка в добовій сечі ≥300 мг, якщо він має постійний (персистувальний) характер. Вміст білка в сечі в межах 30–300 мг/добу класифікують як мікроальбумінурію. Останню реєструють у 10–30% пацієнтів з АГ. Вважають, що її наявність вказує на захворювання нирок у початковій стадії. Мікроальбумінурію розцінюють як предиктор явної нефропатії у пацієнтів з цукровим діабетом, а також як провісник серцево-судинної захворюваності і смертності в осіб із цукровим діабетом і без нього. Відповідно до даних дослідження MONICA ймовірність розвитку ІХС за наявності мікроальбумінурії в 2,4 раза вища, ніж при нормоальбумінурії. Значення мікроальбумінурії при есенціальній гіпертензії до кінця не з’ясоване, однак вважають, що її наявність вказує на початкове ураження нирок і/або на ризик прогресуючого порушення функції нирок у майбутньому. Є дані про те, що мікроальбумінурія відображає порушену здатність нирок адекватно реагувати на надмірне надходження білка з їжею. Вважають також, що вона є маркером дисфункції ендотелію ниркових судин (Kannel W.B., 2000; Vasan R.S. et al., 2002).
Екскреція β2-мікроглобуліну підвищується переважно у хворих з важкою гіпертензією. Фермент N-ацетилглюкозaмінідаза виробляється клітинами ниркових канальців. Підвищення його вмісту в сечі у пацієнтів з есенціальною гіпертензією вказує на залучення нирок; антигіпертензивна терапія знижує його рівень. Вміст сечової кислоти підвищений у 25% хворих з нелікованою гіпертензією; він прямо корелює з опором ниркових судин (Vermeer S.E. et al., 2002).
Характерна риса есенціальної гіпертензії полягає в зниженні ниркового кровотоку, яке виявляється за допомогою радіоізотопного дослідження з 123I-ортойодгіпуратом уже на ранніх стадіях хвороби. Швидкість клубочкової фільтрації на початкових стадіях захворювання залишається нормальною, поступово (як правило, дуже повільно) знижуючись у міру збільшення тривалості і тяжкості гіпертензії.
До пізніх проявів патології нирок належать протеїнурія та/або підвищення вмісту креатиніну в плазмі крові. Остання ознака з’являється при зниженні швидкості клубочкової фільтрації приблизно вдвічі порівняно з нормою, тобто коли втрачена половина функціонуючих нефронів.
Для оцінки швидкості клубочкової фільтрації у загальнолікарській практиці використовують розрахунковий кліренс ендогенного креатиніну (рКК), який можна обчислювати за різними формулами. Найбільш широко використовується формула Кокрофта — Голта (1976).
ЛІКУВАННЯ
Чи необхідна антигіпертензивна терапія? Це питання особливо актуальне в тих випадках, коли підвищення АТ протікає безсимптомно. Відповідь на нього вперше отримана в 1967 р., коли були опубліковані результати дослідження Veterans Administration у США (перше велике дослідження, присвячене вторинній профілактиці АГ), які показали, що лікування м’якої, помірної і тяжкої АГ дозволяє знизити частоту розвитку ускладнень протягом 5 років з 55 до 18%. У подальшому було здійснено багато профілактичних програм, що підтвердили ці результати. Результати метааналізу 14 таких досліджень, у яких брали участь 37 тис. хворих, виявили, що зниження ДАТ тільки на 5–6 мм рт. ст. асоціюється зі зниженням смертності внаслідок серцево-судинних захворювань на 21%, частоти фатальних і нефатальних інсультів — 42%, фатальних і нефатальних ІМ — 14% (Mac Mahon, 1990).
Коли починати лікування? Доведено, що зниження навіть незначно підвищеного АТ знижує серцево-судинну захворюваність і смертність. Однак АТ — не єдиний фактор, що визначає прогноз при АГ.
Ураження органів-мішеней, таких як серце або нирки, а також дисліпопротеїнемія, цукровий діабет, метаболічний синдром істотно погіршують прогноз захворювання. Наявність одного або декількох з перерахованих факторів може бути більш важливою детермінантою ризику, ніж невелике підвищення АТ. У зв’язку з цим, ухвалюючи рішення щодо проведення медикаментозної терапії, слід зважити всі обставини — і рівень АТ, і наявність факторів ризику (схема 1.1).
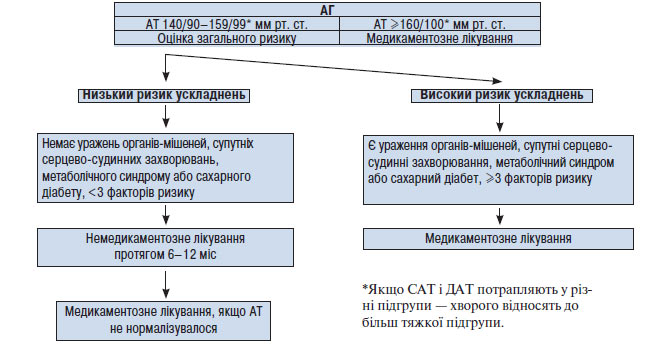
Схема 1.1. Тактика лікаря у разі виявлення у хворого АГ (рекомендації Українського товариства кардіологів, 1999; 2008)
Якщо у хворого діагностовано 2-й або 3-й ступінь АГ, тобто САТ ≥160 мм рт. ст. і/або ДАТ ≥100 мм рт. ст., слід призначити медикаментозне лікування відразу, незалежно від того, чи є в нього додаткові фактори ризику, тому що саме по собі підвищення АТ до такого рівня досить небезпечне з погляду розвитку ускладнень.
Якщо у хворого виявлений 1-й ступінь АГ, тобто САТ <160 мм рт. ст., а ДАТ <100 мм рт. ст., необхідно повторити вимірювання АТ кілька разів протягом 4 тиж, не призначаючи медикаментозного лікування. У 20–30% таких обстежених АТ при повторних вимірюваннях виявляється нормальним. У цьому випадку можна обмежитися немедикаментозною терапією, рекомендацією повторно вимірювати АТ кожні 3 міс протягом 1 року. Якщо АТ при повторних вимірюваннях у цей 4-тижневий період залишається підвищеним (САТ 140–159 мм рт. ст. або ДАТ 90–99 мм рт. ст.), слід розглянути питання про призначення лікарських засобів. При наявності у хворого, крім підвищеного АТ, інших факторів ризику розвитку серцево-судинних захворювань, особливо ураження органів-мішеней, необхідно відразу починати антигіпертензивну терапію.
Ізольовану систолічну гіпертензію часто виявляють у пацієнтів похилого віку. Вона підлягає медикаментозному лікуванню, мета якого — знизити САТ до 140 мм рт. ст.
Ізольовану систолічну гіпертензію (як правило, м’яку) спостерігають також у підлітків і людей молодого віку. Їм показане немедикаментозне лікування. На сьогодні немає доказів на користь медикаментозного лікування таких пацієнтів.
НЕМЕДИКАМЕНТОЗНА ТЕРАПІЯ
Немедикаментозне лікування називають модифікацією способу життя, тому що основні його напрямки — це позбавлення від шкідливих звичок (тютюнопаління, надмірного вживання алкоголю), а також збільшення рухової активності, обмеження вмісту в харчовому раціоні кухонної солі, нормалізація маси тіла.
Збільшення маси тіла тісно корелює з підвищенням АТ, а її зниження у хворих з ожирінням спричиняє досить виражену антигіпертензивну дію. Зменшення маси тіла на 1 кг супроводжується зниженням САТ на 3 мм рт. ст., а ДАТ — на 1–2 мм рт. ст.
Надмірне вживання алкоголю може підвищувати АТ і спричиняти резистентність до проведеної терапії. Відповідно до рекомендацій Національного комітету США по АГ вживання алкоголю чоловіками не має перевищувати 1 унції (30 г) етанолу на день, що еквівалентно 60 мл віски, 300 мл вина або 720 мл пива, жінками — 15 г етанолу на день.
Фізична активність сприяє зниженню АТ. Немає необхідності в інтенсивних вправах і значних витратах часу: досить помірних фізичних навантажень у вигляді швидкої ходьби протягом 30–45 хв щодня або хоча б 3–5 разів на тиждень. Фізичні вправи повинні бути терапією вибору для пацієнтів із ГХ I стадії і використовуватися як доповнення до медикаментозної терапії пацієнтів із захворюванням II–III стадії. У численних епідеміологічних дослідженнях встановлена зворотна кореляція між рівнем фізичної активності й АТ, а в проспективних когортних дослідженнях продемонстровано, що частота нових випадків гіпертензії вища серед тих обстежених, у яких нижча фізична активність.
Дослідження із втручанням також свідчать про сприятливий вплив фізичних вправ на рівень АТ. Проведено близько 45 таких досліджень, у більшості яких використані аеробні навантаження (тобто ритмічні рухи за участю великих м’язів): біг, ходьба, плавання, їзда на велосипеді. Результати метааналізу цих досліджень показали, що динамічні аеробні вправи приводять до зниження АТ на 3/3 мм рт. ст. в нормотензивних осіб, 6/7 мм рт. ст. — в осіб з граничною гіпертензією і на 10/8 мм рт. ст. — у гіпертензивних хворих (Fagard, 1995). У дослідженнях, виконаних найбільш ретельно (з рандомізацією пацієнтів, контролем навантажень у групах втручання і порівняння), ефективність фізичних вправ для пацієнтів з АГ виявилася трохи нижчою: середнє зниження АТ склало 7/5 мм рт. ст. Проте вплив динамічних навантажень на рівень АТ у цих дослідженнях порівняний з дією препаратів, які застосовуються у вигляді монотерапії.
У США запропонована національна настанова, що містить рекомендації щодо бажаної частоти, кількості і якості фізичного навантаження. Ключова ідея цих рекомендацій полягає в тому, що для збереження здоров’я слід присвячувати фізичній активності від 30 до 40 хв (або більше) протягом більшості днів тижня, а бажано всі дні без винятку. Важливо відмітити, що стан серцево-судинної системи поліпшується як у тому випадку, коли фізичне навантаження розподілене на кілька коротких курсів (наприклад тривалістю 10 хв кожний), так і тоді, коли навантаження такої ж інтенсивності виконується за один, але більш тривалий період (наприклад 30 хв).
Більшість пацієнтів, у яких АТ підвищений, але інших факторів ризику немає, можуть без побоювань підвищувати рівень своєї фізичної активності, не проводячи ретельне медичне обстеження. Для пацієнтів віком старше 40 років (чоловіки) або 50 років (жінки) Американська колегія спортивної медицини (1995) рекомендує обстеження в тому випадку, якщо вони збираються зайнятися інтенсивними фізичними вправами або якщо, крім гіпертензії, у них є ще хоча б один фактор ризику ІХС (тютюнопаління, гіперхолестеринемія, абдомінальне ожиріння). Хворі із супутньою ІХС потребують не тільки рутинного обстеження, але й проведення навантажувального тестування, незалежно від того, яка інтенсивність планованих навантажень. Метою тестування є визначення рівня припустимого навантаження та індивідуальної максимальної ЧСС. Відповідно до рекомендацій тієї ж колегії фізичні вправи протипоказані, якщо у хворого є ознаки гострої ішемії, застійної СН, аритмія або гостре інфекційне захворювання. Відносні протипоказання — високий рівень АТ (>200/115 мм рт. ст.), клапанні вади серця, аневризма шлуночка, електролітні порушення, хронічні інфекційні захворювання, період вагітності.
Найефективніші аеробні навантаження. Частота тренувань, що рекомендується — 3–5 днів на тиждень або більше, тривалість — 20–60 хв. Інтенсивність навантаження повинна становити 60–90% максимальної ЧСС, тобто 50–85% максимального споживання кисню. Найбільш точним методом визначення максимальної ЧСС у конкретного хворого є навантажувальне тестування на ергометрі або тредмілі. Однак можливе й непряме визначення максимальної ЧСС: з константи 220 потрібно відняти вік. Для визначення нижньої і верхньої границь ЧСС, що відповідають рівням помірного (60% максимальної) й інтенсивного (90% максимальної) фізичного навантаження, максимальну ЧСС множать на 0,6 або 0,9. Наприклад, для 50-річної людини цільова ЧСС при навантаженні становить: нижня межа = (220–50)‧0,6=102 уд./хв; верхня межа = (220–50)‧0,9=153 уд./хв.
Навантаження, при якому досягається 50–69% максимальної ЧСС, розглядається як помірне, якщо досягається ≥70% максимальної ЧСС — навантаження розцінюють як інтенсивне. У якості альтернативи визначенню ЧСС (для пацієнтів, у яких виникають складності при підрахунку частоти пульсу) можна використовувати такий орієнтир, як відчуття хворим ступеня навантаження і його оцінка навантаження (легке, помірне або важке).
Раціональна дієтотерапія дозволяє знизити рівень АТ у хворих з м’якою гіпертензією тією ж або більшою мірою, що й монотерапія антигіпертензивними препаратами. Зниження вмісту солі в їжі до 6 г асоціюється зі зниженням САТ на 5–10 мм рт. ст. і ДАТ — на 2,2 мм рт. ст. Зниження АТ у відповідь на обмеження солі більш суттєве у людей похилого віку. Пацієнту із ГХ рекомендують зменшити споживання солі до 5–6 г/добу (1 чайна ложка) або готувати їжу без солі, а вміст 1 чайної ложки використовувати для підсолювання страв.
У дослідженні DASH використання хворими низькосольової дієти, що включає фрукти, овочі і продукти з низьким вмістом жирів, дозволило досягти зниження АТ у пацієнтів з АГ на 11,4/5,5 мм рт. ст. Нижче наведені основні компоненти цієї дієти (табл. 1.10).
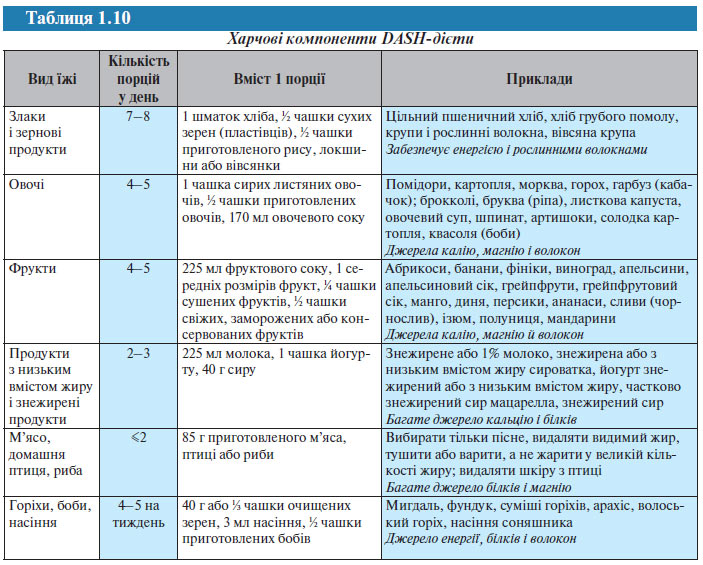
Пацієнтам з АГ і «високим нормальним» АТ, особливо людям похилого віку, рекомендується підтримувати адекватний рівень споживання калію (>100 ммоль/добу). Дієта, багата овочами і фруктами, як джерело калію більш доцільна, ніж вживання таблеток або інших добавок, оскільки така їжа містить і інші необхідні речовини (кальцій, магній, вітаміни), які також можуть позитивно впливати на АТ (табл. 1.11).

У деяких випадках додаткове споживання калію небезпечне. Пацієнтам з нирковою недостатністю, цукровим діабетом, що приймають калійзберігаючі діуретики, інгібітори АПФ, не слід призначати додаткове вживання калію. Необхідно коригувати гіпокаліємію, викликану діуретиками або пов’язану із низьким вмістом калію в дієті, особливо в пацієнтів, що отримують дигоксин, і в осіб з ІХС через ризик розвитку аритмій.
Багате джерело калію — це фрукти, овочі, м’ясо, у тому числі птиця і риба. У денний раціон необхідно включати банани, апельсиновий сік, бобові, картоплю. Додаткове споживання калієвих добавок для зниження АТ можна рекомендувати тільки при призначенні хворому діуретиків, що сприяють виведенню калію.
Зменшення маси тіла у осіб з ожирінням. Встановлено, що у осіб з ожирінням частіше виявляють АГ, а зменшення маси тіла сприяє зниженню АТ. Призначення дієти для зниження АТ у осіб з АГ і ожирінням має цілий ряд переваг порівняно з медикаментозною терапією. Завдяки дієті нормалізується знижена толерантність до глюкози, поліпшуються ліпідний профіль і стан суглобів при їх дегенеративних змінах, підвищується переносимість навантажень.
За даними епідеміологічних досліджень існує пряма залежність між надмірною масою тіла і підвищенням АТ. Так, за даними Фремінгемського дослідження, в осіб з ожирінням АГ розвивається в 2 рази частіше, ніж в осіб з нормальною масою тіла.
Одним з патофізіологічних механізмів, які пояснюють взаємозв’язок між АТ й ожирінням, є гіперінсулінемія. При ожирінні спостерігають резистентність скелетних м’язів до дії інсуліну. Для того щоб підтримувати нормальний гомеостаз глюкози, компенсаторно збільшується вміст циркулюючого інсуліну.
Гіперінсулінемія, що виявляється натще або після прийому їжі, характерна для ожиріння. Збільшення змісту інсуліну в плазмі крові може зумовлювати підвищення АТ через затримку натрію нирками, активацію симпатичної нервової системи і підвищення периферичного опору судин. У перехресних дослідженнях виявлена пряма кореляція між вмістом інсуліну в плазмі крові і АТ. Зниження концентрації інсуліну і підвищення чутливості тканин до нього при зменшенні маси тіла може бути одним із механізмів зниження АТ в осіб з ожирінням.
Зменшення маси тіла за рахунок обмеження енергетичної цінності їжі сприяє зниженню АТ незалежно від рівня екскреції натрію з сечею, тобто для пацієнтів з АГ і ожирінням зменшення маси тіла більш важливе, ніж обмеження споживання натрію.
У численних плацебо-контрольованих дослідженнях показано, що для осіб з надмірною масою тіла (ІМТ >25) зменшення її — найефективніший метод немедикаментозного лікування при АГ і профілактики її розвитку.
МЕДИКАМЕНТОЗНЕ ЛІКУВАННЯ
Результати рандомізованих досліджень, що оцінюють фатальні і нефатальні події, є найбільш значимими доказами з існуючих. Проведено безліч рандомізованих плацебо-контрольованих досліджень, що вивчали переваги зниження АТ у пацієнтів з АГ. Результати можна підсумувати так:
- Антигіпертензивне лікування істотно знижує серцево-судинну захворюваність і смертність, але менш істотно впливає на загальну смертність.
- Поліпшення прогнозу під впливом терапії спостерігають у всіх вікових групах, включаючи хворих з ізольованою систолічною гіпертензією, а також у всіх етнічних групах.
- Антигіпертензивне лікування найбільш істотно знижує ризик фатального і нефатального інсульту (на 30–40%), ризик коронарних подій також знижується, хоча й менш істотно (на 20%). Лікування значно (в 2 рази) знижує ризик розвитку СН.
Дослідження із застосуванням плацебо на сьогодні не проводяться, тому що призначення плацебо вважають неприйнятним через очевидність користі, яку дає антигіпертензивна терапія.
Протягом останніх 10 років всі дослідження, присвячені лікуванню АГ, мають характер порівняння різних препаратів не із плацебо, а між собою. Плацебо може бути використане додатково до базисної терапії, у таких випадках досліджуваний препарат додають до такої ж базисної терапії. Результати цілого ряду подібних досліджень показали, що якщо в одній з таких груп АТ знижується більше хоча б на кілька мм рт. ст., ніж в іншій, то ризик ускладнень у цій групі знижується на 20–30%. Так, у дослідженні HOPE у групі хворих, що отримували додатково з базисною терапією раміприл, зниження САТ було на 3 мм рт. ст. більш істотним, ніж у групі порівняння, що знизило частоту серцево-судинних ускладнень на 22%. У дослідженні EUROPA зниження АТ було на 5/2 мм рт. ст. більше у хворих, що одержували периндоприл, порівняно з тими, кому до базисного лікування додавали плацебо, що забезпечило зниження серцево-судинних подій на 20%. У цілому більш істотне зниження АТ дає більш істотний превентивний ефект у відношенні ускладнень, незалежно від характеру терапії, що застосовується. Порівняння різних класів антигіпертензивних препаратів між собою свідчить, що вони мають певні особливості відносно попередження ускладнень. Антагоністи кальцію, як показує метааналіз Групи дослідників антигіпертензивних препаратів (Blood Pressure Lowering Treatment Trialists, 2003; 2005), дещо більш ефективні, ніж блокатори β-адренорецепторів, діуретики та інгібітори АПФ у попередженні інсульту, але менш ефективні в попередженні СН. Щодо попередження ІХС, зниження серцево-судинної смертності і загальної смертності ці групи препаратів між собою істотно не відрізняються. Ефективність блокаторів β-адренорецепторів у попередженні інсульту нижча, ніж у блокаторів рецепторів ангіотензину II і антагоністів кальцію. Це було продемонстровано у дослідженнях LIFE (2002) і ASCOT (2005), у яких ризик інсульту був вищим у групах, що одержували лікування атенололом поєднано з тіазидним діуретиком, порівняно із групами, яким проводилося лікування лозартаном або амлодипіном поєднано з периндоприлом. Інгібітори АПФ володіють додатковою до зниження АТ здатністю попереджати ускладнення ІХС (BPLTT, 2007).
Відповідно до рекомендацій Європейського товариства гіпертензії і Європейського товариства кардіологів (2007), для лікування АГ слід застосовувати антигіпертензивні препарати п̓яти основних класів:
- тіазидні діуретики;
- антагоністи кальцію;
- інгібітори АПФ;
- блокатори рецепторів ангіотензину II;
- блокатори β-адренорецепторів.
Блокатори β-адренорецепторів не рекомендується застосовувати для лікування хворих з метаболічним синдромом і високим ризиком цукрового діабету, особливо в комбінації з діуретиками.
Діуретики застосовують для лікування АГ близько 50 років і дотепер вони залишаються одними з найбільш широко застосовуваних із цією метою лікарських засобів. За даними численних проспективних досліджень, лікування АГ діуретиками сприяє зниженню частоти розвитку ІМ на 14–16%, інсульту — на 38–42%. Серед хворих похилого віку, що приймають комбінацію тіазидного та калійзберігаючого діуретиків, за даними дослідження EWPHE (1985), виявляють зниження смертності внаслідок ІМ на 60%. У результаті досліджень SHEP (1991), MRC (1992), STOP-Hypertension (1991) встановлено, що діуретики ефективніше запобігають ускладненням з боку серцево-судинної системи у пацієнтів похилого віку, ніж блокатори β-адренорецепторів. У дослідженні SHEР було також показано, що застосування тіазидоподібного діуретика хлорталідону дозволяє знизити частоту розвитку СН на 50%.
З початку 90-х років ХХ ст. у Європі спостерігають тенденцію до зменшенню використання препаратів цієї групи для лікування АГ. Частково це пов’язано з публікацією результатів дослідження з багатофакторної профілактики MRFIT (1985), у якому серед хворих з вихідними змінами ЕКГ у спокої виявляли вищу смертність внаслідок ІХС. Це пов’язували з розвитком фатальних аритмій, зумовлених гіпокаліємією і негативною дією діуретиків на ліпідний профіль. Однак всебічна оцінка результатів інших великих досліджень показала, що роль діуретиків у попередженні смертності внаслідок ІМ недооцінюється. Дослідження HYVET у популяції пацієнтів з АГ похилого і старечого віку продемонструвало достовірне зниження ризику загальної смертності на 21%, фатального інсульту на 39% і СН на 64% при лікуванні індапамідом ретард 1,5 мг. На підставі отриманих у цих дослідженнях даних прийшли до висновку, що діуретики є препаратами вибору при лікуванні хворих похилого віку, пацієнтів із систолічною АГ, а також при супутній СН.
У дослідженні ALLHAT вивчена порівняльна ефективність амлодипіну, лізиноприлу, доксазозину і хлорталідону у попередженні ускладнень ІХС. У січні 2000 р. та частина дослідження, в ході якої вивчалася дія доксазозину, була передчасно зупинена, тому що цей препарат виявився менш ефективним у попередженні ускладнень АГ порівняно з тіазидоподібним діуретиком хлорталідоном. Частота їхнього розвитку в групі хворих, що приймали хлорталідон, була на 25% нижчою, ніж у групі, що приймала доксазозин. Підтвердження про високу ефективність діуретиків були отримані також у дослідженні INSIGHT, у ході якого >6 тис. пацієнтів з АГ одержували лікування або гідрохлоротіазидом у комбінації з амілоридом, або ніфедипіном GITS. Загальна частота розвитку ускладнень з боку серцево-судинної системи, смертність внаслідок їх, частота випадків інсульту і сумарна частота інфаркту були практично однаковими у хворих, що приймали діуретики та ніфедипін, а частота розвитку СН і фатального ІМ — відповідно в 2 і 3 рази нижча у хворих, що приймали діуретики. Дослідження STOP-Hypertension-1 (1991), STOP-Hypertension-2 (1999) і NORDIL (2000) показали, що діуретики не поступаються за ефективністю антигіпертензивним препаратам більш нових класів — антагоністам кальцію та інгібіторам АПФ.
Крім антигіпертензивної дії важливою особливістю діуретиків є їхня здатність попереджати ураження органів-мішеней при АГ. Метааналіз рандомізованих досліджень показав, що діуретики, головним чином індапамід, сприяють регресії ГЛШ. Несподіваними стали результати досліджень VACS (1997) і LIVE (1999), у яких більш виражену дію у відношенні ГЛШ мали діуретики і меншою мірою — інгібітори АПФ. Доведено, що діуретики спричиняють також помірну ренопротекторну дію, яка проявляється в зниженні мікроальбумінурії на 10%.
Таким чином, сечогінні засоби продовжують залишатися ефективними препаратами першого ряду в лікуванні АГ, а використання їх у низьких дозах дозволяє значно знизити ризик появи небажаних метаболічних ефектів.
У лікуванні АГ використовуються переважно тіазидні та тіазидоподібні діуретики (табл. 1.12), а калійзберігаючі та петльові діуретики застосовують тільки в певних ситуаціях. Еталонний представник тіазидних діуретиків — гідрохлоротіазид. До тіазидоподібних діуретиків належать хлорталідон, індапамід, метолазон, клопамід та деякі інші препарати. Діуретичний ефект тіазидних діуретиків реалізується внаслідок гальмування реабсорбції іонів натрію переважно в кортикальному сегменті петлі нефрону.
Петльові діуретики діють впродовж усього висхідного відділу петлі нефрону (петлі Генле), у зв’язку з чим і одержали свою назву. Найбільш широко застосовувані з них — фуросемід і торасемід. У лікуванні АГ фуросемід використовують рідко через нетривалість дії (до 6 год) і незначний антигіпертензивний ефект.
Таблиця 1.12
Діуретики, що застосовуються для лікування АГ
| Препарат | Добова доза, мг | Тривалість дії, год |
| Тіазидні діуретики | ||
| Бендрофлуметіазид
Бентіазид Гідрофлуметіазид Метиклоазид Політіазид Трихлоротіазид Хлоротіазид Циклотіазид |
2,5–5,0
12,5–50 12,5–50 12,5–50 2,5–5,0 1,0–4,0 1,0–4,0 125–500 0,5–2,0 |
18
12–18 12–18 18–24 24 24–48 24 6–12 18–24 |
| Тіазидоподібні діуретики | ||
| Індапамід
Хінетазон Метолазон Хлорталідон |
1,25–2,5
25–100 0,5–1 12,5–50 |
24
6–12 24 24–72 |
| Петльові діуретики | ||
| Буметанід
Етакринова кислота |
0,5–5,0
5–40 25–100 20–480 |
4–6
12 12 4–6 |
| Калійзберігаючі діуретики | ||
| Амілорид
Спіронолактон Тріамтерен |
5–10
25–100 50–150 |
24
8–12 12 |
Петльові діуретики (фуросемід, буметанід, торасемід) застосовують для лікування АГ при наявності ниркової недостатності, коли підвищення рівня креатиніну в крові досягає 177 мкмоль/л і вище, а також у хворих з СН, коли тіазидні діуретики неефективні.
Для калійзберігаючих діуретиків характерна слабка діуретична і антигіпертензивна дія. До них належать амілорид, спіронолактон і тріамтерен. Самостійного значення в лікуванні АГ вони не мають, однак застосовуються в комбінації з іншими діуретиками для зниження рівня гіпокаліємії.
Спіронолактон є препаратом вибору при лікуванні первинного гіперальдостеронізму і застосовується для симптоматичного лікування АГ при синдромі Кона. Калійзберігаючі діуретики реалізують свій ефект переважно в області дистальних канальців, а також в корковому і мозковому відділах збірних трубок. Вони знижують реабсорбцію іонів натрію, пригнічують екскрецію іонів калію і протонів водню.
Механізм антигіпертензивної дії. На початковому етапі лікування (1–2 тиж) антигіпертензивний ефект діуретиків зумовлений зменшенням ОЦК і позаклітинної рідини внаслідок підвищення екскреції натрію. У цей період протягом кількох днів може спостерігатися деяке підвищення ЗПСО і зменшення серцевого викиду. Ці реакції нетривалі і пояснюються зниженням ОЦК. Через 1–2 тиж терапії починає проявлятися вазодилатаційна дія діуретиків. Вазодилатація є основним механізмом зниження АТ при тривалому застосуванні діуретиків. Певний час (до 1 міс) ці два фактори, що викликають антигіпертензивний ефект, діють паралельно. Потім серцевий викид і ОЦК відновлюються до вихідного рівня. Вазодилатаційний ефект діуретиків зберігається протягом усього наступного періоду лікування.
Механізм вазодилатаційної дії діуретиків остаточно не з’ясований. Його пояснюють рефлекторним впливом часткового зменшення ОЦК і тканинного кровотоку на тонус судин. У зниженні судинного тонусу відіграє роль також регресія гіпертрофії м’язового шару судин при тривалому лікуванні.
Діуретики особливо ефективні в таких клінічних ситуаціях:
- похилий вік;
- ізольована систолічна АГ у людей похилого віку;
- затримка рідини і ознаки гіперволемії (периферичні набряки, пастозність);
- супутня СН (петльові діуретики);
- супутня ниркова недостатність (петльові діуретики);
- остеопороз;
- гіперальдостеронізм (спіронолактон).
Діуретики призначають у невеликих дозах (наприклад гідрохлоротіазид — 12,5 мг/добу щодня, індапамід 1,5 або навіть 0,625 мг). Підвищення дози значно збільшує ймовірність побічних явищ. Тривалість дії гідрохлоротіазиду — 12–18 год, тому його можна призначати 1 раз на добу, а хлорталідон, що має пролонговану дію, — 1 раз на 2–3 доби. Для попередження втрати калію тіазидні діуретики рекомендується комбінувати з калійзберігаючими препаратами (амілорид, тріамтерен) або з антагоністами альдостерону (спіронолактон), крім тих випадків, коли діуретики призначаються в низьких дозах або в комбінації з інгібітором АПФ.
Блокатори β-адренорецепторів протягом останніх десятиліть займали лідируючу позицію серед антигіпертензивних засобів. Це зумовлено їх антигіпертензивною ефективністю, що доведена в ході численних досліджень, і здатністю знижувати ризик ускладнень у хворих з АГ.
Однак в останні роки їхня роль у лікуванні АГ переглядається. Це пов’язано з появою результатів порівняльних досліджень, які показали, що блокатори β-адренорецепторів поступаються блокаторам рецепторів ангіотензину II в запобіганні інсульту (дослідження LIFE, 2002) і сприяють появі нових випадків цукрового діабету серед лікованих ними хворих, особливо при застосуванні в комбінації з тіазидними діуретиками. Рекомендації Європейського товариства гіпертензії і Європейського товариства кардіології (2007) залишають за блокаторами β-адренорецепторів місце серед препаратів першого ряду, однак підкреслюють необхідність уникати їхнього призначення хворим, схильним до розвитку цукрового діабету, і особам з метаболічним синдромом. У хворих з уже наявним цукровим діабетом блокатори β-адренорецепторів також не є препаратами вибору. Це не стосується карведилолу і небівололу — адреноблокаторів з вазодилатаційними властивостями. Ці препарати не підвищують резистентність тканин до інсуліну і не мають діабетогенних властивостей.
Незважаючи на критику на їх адресу, блокатори β-адренорецепторів залишаються важливим компонентом антигіпертензивної терапії. Це зумовлено необхідністю їх призначення при багатьох супутніх захворюваннях, таких як стенокардія, перенесений ІМ, СН, порушення ритму серця, а також тим, що вони не поступаються іншим антигіпертензивним засобам у запобіганні ускладнень ІХС і смертності від серцево-судинних захворювань. Вони є необхідним компонентом лікування хворих з явними ознаками активації САС (тахікардія, високий пульсовий тиск у молодих людей), а також при стрес-індукованій гіпертензії. Слід також враховувати, що практично всі дослідження, присвячені оцінці блокаторів β-адренорецепторів при АГ, були проведені з атенололом. Жоден із сучасних блокаторів β-адренорецепторів (бісопролол, бетаксолол, карведилол, небіволол) не вивчався у пацієнтів з АГ у великих багатоцентрових дослідженнях, тому переносити результати оцінки атенололу на ці препарати немає підстав. Низьку активність атенололу в запобіганні інсульту пояснюють недостатньою тривалістю дії (<24 год), а також недостатньою пенетрацією у тканину мозку через гідрофільність препарату.
Виділяють три покоління блокаторів β-адренорецепторів: неселективні (I покоління), β1-селективні (II покоління) і блокатори β-адренорецепторів з вазодилатаційними властивостями (III покоління) (табл. 1.13). Кожне наступне покоління відрізняється від попереднього появою нових, корисних для клініки властивостей.
Таблиця 1.13
Блокатори β-адренорецепторів
| Препарат | Доза, мг/добу | Частота прийому на добу |
| Кардіоселективні | ||
| Без внутрішньої симпатоміметичної активності
Бетаксолол Бізопролол Метопролол Небіволол |
25–100 5–40 2,5–20 50–200 5–10 |
2 1 1 1–3 1 |
| З внутрішньою симпатоміметичною активністю
Ацебутолол Целіпролол |
200–1200 200–400 |
1 1 |
| Некардіоселективні | ||
| Без внутрішньої симпатоміметичної активності
Надолол Пропранолол |
40–120 40–240 20–40 |
1
1–3 |
| З внутрішньою симпатоміметичною активністю
Алпренолол Картеолол Окспренолол Піндолол |
200–800 2,5–10 20–160 10–60 |
4 1 2–3 2 |
| З α-блокуючою здатністю
Карведилол Лабеталол |
12,5–50
200–1200 |
1–2
2 |
Вазодилатаційна дія блокаторів β- адренорецепторів зумовлена або α1-адреноблокуючою активністю (лабеталол, карведилол), або наявністю β2-агоністичної активності (целіпролол), або здатністю індукувати утворення NO клітинами ендотелію (небіволол). Карведилол володіє також додатковою прямою вазодилатаційною дією.
Механізм антигіпертензивної дії. Значну увагу при вивченні антигіпертензивної дії блокаторів β-адренорецепторів приділяли їхньому впливу на активність реніну в плазмі крові. Однак не було виявлено чіткої залежності між зниженням АТ і вихідною активністю реніну плазми крові або ступенем її зниження в процесі лікування блокаторами β-адренорецепторів. Зниження активності реніну плазми крові в результаті зменшення його утворення за допомогою блокади β1-адренорецепторів має більш важливе значення у хворих з переважно гіперреніновою формою АГ. Було показано, що для зниження АТ у хворих з високою активністю реніну плазми крові досить низьких доз пропранололу (160 мг/добу), тоді як при низькореніновій АГ необхідні вищі дози препарату (320–960 мг/добу). Антигіпертензивний ефект в останньому випадку досягається незалежно від впливу на активність реніну плазми крові. Очевидно, зниженням активності РААС повністю пояснити антигіпертензивний ефект блокаторів β-адренорецепторів не можна.
Не одержала повного підтвердження гемодинамічна концепція зниження АТ при блокаді β-адренорецепторів. Відомо, що первинний гемодинамічний ефект блокаторів β-адренорецепторів полягає в зменшенні скоротності міокарда і ЧСС, що в результаті приводить до зменшення ХОК на 15–20% (за винятком піндололу, який має високу внутрішню симпатоміметичну активність). Зниження серцевого викиду супроводжується підвищенням периферичного опору судин у початковий період терапії. Цей ефект пояснюють як підвищенням концентрації катехоламінів у плазмі крові, так і превалюванням α-адренергічного впливу на тонус судин, який супроводжує блокаду β-адренорецепторів. При тривалому застосуванні блокаторів β- адренорецепторів ЗПСО повертається до вихідного або нормального рівня.
Антигіпертензивна дія блокаторів β-адренорецепторів не може бути пояснена лише зменшенням ХОК, оскільки він знижується також у тих випадках, коли блокатори β-адренорецепторів не впливають на рівень АТ. Разом з тим виявлена тісна залежність між зниженням АТ і зменшенням периферичного судинного опору. У тих випадках, коли тонус судин, що підвищився після початку терапії блокаторами β-адренорецепторів, не повертається до вихідного рівня, не спостерігають суттєвого зниження АТ.
Антигіпертензивний ефект блокаторів β-адренорецепторів пов’язували з блокадою пресинаптичних β2-адренорецепторів, стимуляція яких приводить до вивільнення норадреналіну. Встановлено, що рівень норадреналіну в плазмі крові тісно корелює із рівнем ДАТ і вираженістю антигіпертензивного ефекту блокаторів β-адренорецепторів. Проте β1-селективні блокатори мало впливають на пресинаптичні β2-адренорецептори, але мають такий же антигіпертензивний ефект, як і неселективні блокатори β-адренорецепторів. Більше того, отримано дані про те, що β2-селективні блокатори не знижують АТ. Отже, гальмування вивільнення норадреналіну із симпатичних нервових закінчень не є основною ланкою в механізмі дії блокаторів β-адренорецепторів.
Викликає інтерес здатність блокаторів β-адренорецепторів стимулювати синтез простагландинів, що мають судинорозширювальну дію. Блокада β-адренорецепторів підвищує ефективність взаємодії аденілатциклази та цАМФ за допомогою стимуляції утворення простацикліну і підвищення щільності β-адренорецепторів.
Виявлено підвищення рівня передсердного натрійуретичного фактору і зниження вмісту внутрішньоклітинного іонізованого кальцію у гладком’язових клітинах судин при терапії блокаторами β- адренорецепторів, що також має значення в механізмі їх антигіпертензивної дії.
Таким чином, не можна виділити яку-небудь ланку в механізмі зниження АТ під впливом блокаторів β-адренорецепторів. Очевидно, в кожному конкретному випадку превалює той або інший антигіпертензивний механізм. Це залежить від багатьох умов, у тому числі від вихідного стану РААС і САС, гормональних і гемодинамічних факторів.
У пацієнтів з АГ застосування блокаторів β-адренорецепторів має перевагу над іншими антигіпертензивними засобами в наступних випадках:
- молодий і середній вік пацієнта;
- ознаки симпатикотонії (тахікардія, високий пульсовий АТ, гіперкінетичний тип гемодинаміки);
- супутня ІХС (стенокардія та ІМ);
- СН;
- супутня передсердна або шлуночкова екстрасистолія;
- тахікардія;
- гіпертиреоз;
- глаукома;
- мігрень;
- АГ перед хірургічною операцією або після неї.
Антагоністи кальцію. Антагоністи кальцію застосовують для лікування АГ з 1980-х років, хоча й були синтезовані раніше (у 1970-х роках); як і блокатори β-адренорецепторів, вони спочатку розцінювалися як антиішемічні засоби. Зараз антагоністи кальцію належать до найбільш широко застосовуваних антигіпертензивних препаратів в усьому світі. За даними аналізу групи дослідників антигіпертензивних препаратів (BPLTT, 2003) і Staessen (2003) антагоністи кальцію порівняно з плацебо знижують ризик інсульту на 38%, ризик ІХС — 20%, однак підвищують ризик розвитку СН на 21%, хоча це підвищення недостовірне. Порівняно з діуретиками і блокаторами β-адренорецепторів антагоністи кальцію більш ефективно попереджають інсульт (на 8–10%), але менш ефективно — СН (її ризик вищий на 33%, ніж при лікуванні діуретиками і блокаторами β-адренорецепторів). Порівняно з інгібіторами АПФ антагоністи кальцію також більш ефективно попереджають інсульт (на 12%), але менш ефективно — СН.
Антагоністи кальцію представляють неоднорідну за хімічною структурою і фармакокінетичними властивостями групу препаратів із загальним механізмом дії, який полягає в гальмуванні проникнення іонів кальцію в клітину по специфічних повільних кальцієвих каналах (табл. 1.14).
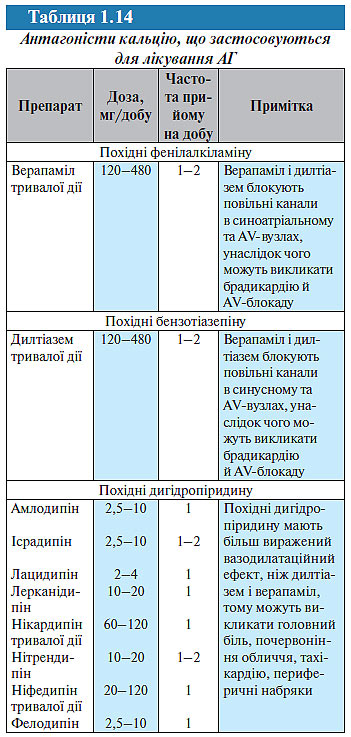
Розрізняють потенціал- і рецепторзалежні кальцієві канали. Головний шлях транспорту Са2+ в клітину лежить через потенціалзалежні кальцієві канали. На їх блокаді заснована терапевтична дія антагоністів кальцію. Ідентифіковано 6 типів кальцієвих каналів: L, T, N, P, Q і R. Клінічне значення встановлене тільки для каналів L-типу (L — від англійського long lasting, що означає «тривалий, довго діючий»). Вони широко представлені в клітинах серцево- судинної системи та інших органів і тканин. Залежно від хімічної структури виділяють чотири основні групи антагоністів кальцію: 1) похідні фенілалкіламіну; 2) похідні бензотіазепіну; 3) похідні дигідропіридину; 4) препарати іншої хімічної структури.
Т. Тоуо-Оkа і W. Nayler у 1996 р. запропонували класифікацію антагоністів кальцію, яка враховує тривалість їхньої дії, їх тканинну селективність, фармакодинамічні і фармакокінетичні особливості. Антагоністи кальцію розділені ними на три групи: препарати першого, другого і третього поколінь (табл. 1.15).
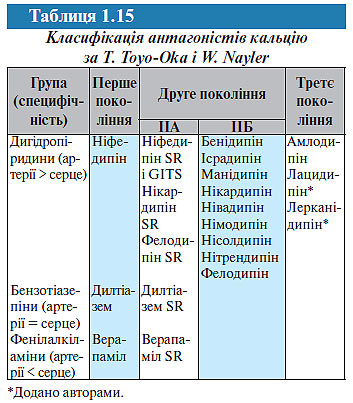
У групу препаратів третього покоління на сьогодні включають також лацидипін, лерканідипін і манідипін, що за тривалістю дії близькі до амлодипіну.
Найвищу вазоселективність мають препарати дигідропіридинової групи. Для верапамілу більш характерна негативна інотропна і хронотропна дія, але у вищих дозах він зумовлює також судинорозширювальний ефект. Дилтіазем, на думку ряду авторів, за вираженістю впливу на судини і міокард займає проміжне положення між ніфедипіном і верапамілом. Інші дослідники вважають, що дилтіазем за своїми фармакологічними характеристиками близький до верапамілу.
Антагоністи кальцію, що спричиняють негативний хронотропний ефект, подібно блокаторам β-адренорецепторів, мають виражену антиішемічну дію. Вона властива також препаратам третього покоління — амлодипіну і лацидипіну, що не впливають на серцевий ритм і знижують споживання міокардом кисню за рахунок зменшення постнавантаження і поліпшення коронарного кровотоку.
Верапаміл і дилтіазем мають здатність блокувати кальцієві канали водіїв ритму й провідної системи серця. Ця властивість не характерна для дигідропіридинових похідних, що пояснюється їхньою дією на різні ділянки L-каналів, а також різною локалізацією місць зв’язування в каналі. Точка прикладання верапамілу, який зумовлює найбільш виражений негативний хронотропний ефект, перебуває всередині каналу, а ніфедипіну — в зовнішній його частині.
Антиаритмічна дія верапамілу і дилтіазему більше виражена, коли ЧСС вища. Це так звана ефективність, що залежить від застосування (usе-depended). Вважають, що верапаміл і дилтіазем діють тільки при відкритому L-каналі. Це зумовлено необхідністю досягнення ними ділянок зв’язування, що знаходяться всередині каналу. Чим довше канали перебувають у відкритому стані (наприклад при суправентрикулярних порушеннях ритму), тим ефективніша дія препаратів.
Дигідропіридинові похідні, що мають високу селективність до судин, діють на N-сегменти кальцієвого каналу, які перебувають у початковій його частині. Їх ефективність не залежить від ступеня відкриття кальцієвого каналу. Очевидно, цим можна пояснити відсутність антиаритмічної дії в препаратів групи дигідропіридину.
Механізм антигіпертензивної дії. Антигіпертензивний ефект антагоністів кальцію зумовлений їх прямою або непрямою дією (рис. 1.8).

Рис. 1.8. Дія антагоністів кальцію на гладкі м’язи і міокард
Пряма дія полягає в блокаді трансмембранного надходження іонів кальцію в клітини через повільні кальцієві канали L-типу, впливі на їх функціональний стан і щільність каналів плазматичних мембран. Зменшуючи надходження Са2+ у гладком’язові клітини судин, антагоністи кальцію сприяють їх релаксації і зниженню судинного тонусу, а також зменшують перевантаження клітини кальцієм за рахунок попередження активації кальцієвих каналів L-типу вазопресорними агентами: ендотеліном, ангіотензином ІІ, катехоламінами та іншими біологічно активними речовинами.
Непряма дія антагоністів кальцію пов’язана з їх впливом на гормони та ендотеліальні фактори, що регулюють тонус судин. Встановлено, що антагоністи кальцію поліпшують функцію ендотелію судин, регулюючи виділення ними вазодилататорних (оксид азоту, брадикінін, простациклін Е) і вазоконстрикторних медіаторів (ЕТ-1, ангіотензин ІІ, простагландин F2α).
Гемодинамічною основою антигіпертензивної дії антагоністів кальцію є зниження ЗПСО. Вони викликають більш виражений вазодилатаційний ефект у хворих з вихідно підвищеним тонусом судин, знижують АТ тим значніше, чим вищим був його рівень до лікування. Препарати не мають ефекту «першої дози» і рідко знижують АТ нижче нормального рівня.
Антагоністи кальцію зумовлюють переважно артеріолодилатаційну дію, не впливаючи на тонус венозних судин. Цим зумовлена відсутність ортостатичної гіпотензії при їх застосуванні. Існує регіональна чутливість судинного русла до дії цих препаратів. Вони істотно знижують тонус коронарних і периферичних артерій. Менш виражений вплив антагоністи кальцію мають на резистивні судини нирок і практично не впливають на тонус судин шкіри і травного тракту.
Антагоністи кальцію ефективно знижують САТ і ДАТ в умовах спокою і при фізичному навантаженні, однак їх дія на САТ в умовах фізичного навантаження проявляється менш значимо, ніж у спокої. Це характерно, зокрема, для ніфедипіну, а також для верапамілу і дилтіазему, якщо вони застосовуються в низьких дозах. Виявлено залежність антигіпертензивного ефекту від віку пацієнта й АРП: ефективність антагоністів кальцію вища у пацієнтів похилого віку з низькореніновою формою АГ.
Вазодилатаційна дія препаратів не супроводжується затримкою рідини в організмі, що пояснюється їхньою здатністю чинити легкий натрійуретичний і діуретичний ефект.
Антагоністи кальцію мають перевагу для лікування АГ у таких випадках:
- середній і похилий вік;
- ізольована систолічна гіпертензія (у пацієнтів похилого віку);
- ГЛШ;
- атеросклероз сонних і коронарних артерій;
- стабільна стенокардія;
- суправентрикулярна тахікардія і екстрасистолія (верапаміл, дилтіазем);
- порушення периферичного кровообігу;
- період вагітності.
Інгібітори АПФ. У клініці використовують чотири способи пригнічення системи ренін — ангіотензин — альдостерон: пригнічення вивільнення реніну за допомогою блокаторів β-адренорецепторів; пряме зниження активності реніну за допомогою аліскірену; зниження активності АПФ за допомогою його інгібіторів; використання конкурентних антагоністів рецепторів ангіотензину II. Інгібітори АПФ — найбільш широко використовувана група препаратів з описаних, а в Україні і деяких інших країнах — і з усіх антигіпертензивних препаратів у цілому (табл. 1.16).

За даними метааналізу BPLTT (2003), інгібітори АПФ порівняно з плацебо знижують ризик розвитку інсульту на 28%, ускладнень ІХС — 22%, СН — 18%. Порівняно з діуретиками або блокаторами β- адренорецепторів інгібітори АПФ не мають істотних відмінностей щодо впливу на ці ускладнення, загальну або серцево-судинну смертність, однак порівняно з антагоністами кальцію вони вірогідно більш ефективно попереджають СН і менш ефективно — інсульт.
Інгібітори АПФ мають добре доведену кардіопротекторну і ренопротекторну дією. Вони рекомендовані для лікування ІХС (хоча не мають антиішемічної дії), тому що в дослідженнях з раміприлом (HOPE) і периндоприлом (EUROPA) установлена їхня здатність знижувати на 20–22% смертність від ІХС і частоту нефатального ІМ. Інгібітори АПФ істотно поліпшують прогноз у пацієнтів із СН. Вони рекомендовані асоціаціями нефрологів, діабетологів і кардіологів до використання у хворих із діабетичною і недіабетичною нефропатією у зв’язку зі здатністю покращувати ниркову гемодинаміку й зменшувати вираженість протеїнурії.
Механізм антигіпертензивної дії. Гальмування перетворення неактивного ангіотензину І в ангіотензин ІІ є основною ланкою в механізмі антигіпертензивної дії інгібіторів АПФ. Вони знижують активність АПФ, зв’язуючись із його активними центрами, що приводить до зменшення перетворення ангіотензину І в ангіотензин ІІ. Порушення утворення останнього супроводжується вазодилатацією і зниженням АТ. На початку лікування інгібіторами АПФ вираженість їх антигіпертензивної дії і зниження концентрації ангіотензину ІІ в плазмі крові залежать від активності РААС: дія препаратів тим сильніша, чим вища її активність. Ця залежність нівелюється через кілька днів або тижнів лікування, тому висока активність РААС не є предиктором ефективності інгібіторів АПФ при тривалій терапії. Разом з тим у хворих з підвищеною активністю РААС і вираженим ефектом «першої дози» лікування інгібіторами АПФ, як правило, дуже ефективне.
Крім основного, існують альтернативні шляхи утворення ангіотензину ІІ за участі хімази, тоніну, катепсину G та інших активних речовин. Інгібітори АПФ не впливають на зазначені механізми. Цей факт, а також те, що в багатьох хворих у процесі терапії інгібіторами АПФ спостерігають подальше зниження АТ, незважаючи на повернення вмісту ангіотензину ІІ до вихідного рівня, свідчить про наявність інших, не пов’язаних із пригніченням АПФ механізмів антигіпертензивної дії цих препаратів. Зменшення розпаду брадикініну, який має вазодилатаційний ефект, — один із них. Існує тісна взаємодія між калікреїн-кініновою системою і РААС. АПФ не тільки сприяє утворенню ангіотензину ІІ, але й, будучи кініназою, викликає руйнування брадикініну.
Характерно, що при цьому концентрація брадикініну в плазмі крові незначно підвищується: основним фактором, що визначає брадикінінзалежні відповіді при лікуванні інгібіторами АПФ, є локальне підвищення продукції кінінів у стінці судини. Брадикінін, стимулюючи брадикінінові рецептори другого типу (БК 2-рецептори), запускає цілий ряд ендотелійзалежних відповідей. Він індукує вивільнення трьох релаксуючих факторів, утворених ендотелієм: оксиду азоту, ендотелійзалежного гіперполяризуючого фактору і простацикліну.
Зменшення секреції альдостерону при лікуванні інгібіторами АПФ пов’язане зі зниженням рівня ангіотензину II. Воно сприяє збільшенню натрійурезу і діурезу, а також зменшенню компенсаторної затримки натрію в нирках у відповідь на вазодилатацію. Тривале застосування інгібіторів АПФ призводить до незначного зниження загального вмісту натрію в організмі.
Зменшення вираженості ендотеліальної дисфункції під впливом інгібіторів АПФ супроводжується поліпшенням релаксації судинної стінки. У дослідженні TREND (1996) встановлено, що інгібітор АПФ квіналаприл у дозі 40 мг 1 раз на добу значно зменшує дисфункцію ендотелію коронарних судин, на 10–20% збільшуючи його відповідь на введення ацетилхоліну в нормотензивних пацієнтів з ІХС.
Гальмування вивільнення ЕТ-1 інгібіторами АПФ за допомогою зниження рівня ангіотензину II викликає безпосереднє поліпшення релаксаційних властивостей судин, а також сприяє зменшенню дисфункції ендотелію.
Основним гемодинамічним механізмом дії інгібіторів АПФ є зниження ЗПСО і системного АТ.
Інгібітори АПФ знижують САТ і ДАТ у стані спокою і при фізичному навантаженні, не впливаючи на ЧСС, оскільки пригнічують контррегуляторні механізми, що активують РААС і САС. У хворих із м’якою й помірною АГ досягти адекватного контролю АТ за допомогою монотерапії інгібіторами АПФ вдається в приблизно 50% випадків. Підвищення ступеня зниження АТ досягається за рахунок обмеження вживання кухонної солі або додаткового призначення діуретиків. Зниження вмісту натрію в організмі стимулює секрецію реніну, підвищуючи тим самим ефективність препаратів.
Зниження інгібіторами АПФ судинного тонусу не супроводжується зміною серцевого викиду. При вихідному порушенні скоротності міокарда інгібітори АПФ покращують його інотропну функцію. Зменшення перед- і післянавантаження, зумовлене зниженням АТ і венодилатаційними властивостями інгібіторів АПФ, сприяє таким позитивним гемодинамічним зрушенням, як зниження тиску наповнення ЛШ та тиску у ЛА і правому передсерді.
Інгібітори АПФ відіграють важливу роль в покращанні коронарного резерву, зменшуючи ендотеліальну дисфункцію судин серця і вазоконстрикцію, зумовлену симпатичними впливами.
При зниженні АТ важливе значення має збереження адекватної церебральної перфузії, особливо у хворих похилого віку. Інгібітори АПФ зумовлюють антигіпертензивну дію без порушення мозкової гемодинаміки. Навіть при їх призначенні хворим з низьким рівнем АТ (лікування СН) погіршення мозкового кровотоку, як правило, не спостерігали. Це пов’язано з тим, що інгібітори АПФ зрушують нижню межу його ауторегуляції до нижчого рівня АТ. У ході експерименту відмічено швидке зниження нижньої границі ауторегуляції церебральної гемодинаміки за допомогою інгібіторів АПФ на 20–30 мм рт. ст. без погіршення перфузії мозку.
Інгібітори АПФ поліпшують ниркову гемодинаміку, що пов’язано з їх здатністю зумовлювати дилатаційний ефект переважно на еферентні (постгломерулярні) ниркові артеріоли. У випадках, коли швидкість клубочкової фільтрації значною мірою залежить від тонусу еферентних артеріол нирок, як це спостерігається при двобічному стенозі ниркових артерій, стенозі артерії єдиної нирки, вираженій дегідратації, тяжкій застійній СН, застосування інгібіторів АПФ протипоказане, оскільки може спровокувати неконтрольоване зниження ниркової перфузії і розвиток ГНН.
Призначення інгібіторів АПФ має перевагу в таких ситуаціях:
- супутня СН;
- безсимптомне порушення функції ЛШ;
- нефропатія (діабетична і недіабетична);
- ГЛШ;
- перенесений ІМ;
- атеросклероз сонних артерій;
- протеїнурія, мікроальбумінурія;
- фібриляція передсердь;
- метаболічний синдром.
Блокатори рецепторів ангіотензину ІІ. Ця група антигіпертензивних препаратів використовується в клініці з 1994 р., однак вона відразу зайняла своє місце в лікуванні АГ серед препаратів першого ряду. Накопичені за цей час дані багатоцентрових рандомізованих досліджень показують, що ефективність блокаторів рецепторів ангіотензину II з точки зору зниження АТ, зниження частоти ІМ, загальної серцево-судинної смертності така ж, як у діуретиків і блокаторів β-адренорецепторів, однак вони ефективніше знижують ризик інсульту (21–24%) і СН (16%). Блокатори рецепторів ангіотензину II так само ефективні в попередженні ускладнень ІХС, як інгібітори АПФ, хоча останні, як показав метарегресійний аналіз BPLTT (2007), мають додатковий сприятливий вплив на ризик розвитку ІМ, який не залежить від зниження АТ. Перевагою блокаторів рецепторів ангіотензину II є їх більш виражена церебропротекторна і добре доведена ренопротекторна дія.
Шляхи утворення ангіотензину ІІ. Відповідно до класичних уявлень, основний ефекторний гормон РААС ангіотензин ІІ утворюється в системному кровотоці в результаті каскаду біохімічних реакцій.
Результати досліджень двох останніх десятиліть свідчать, що ангіотензин ІІ утворюється не тільки в системному кровотоці, але й у різних тканинах, де виявлені всі компоненти РААС (ангіотензиноген, ренін, АПФ, рецептори ангіотензину), а також експресія генів реніну й ангіотензину ІІ. Значення тканинної системи зумовлене її провідною роллю в патогенетичних механізмах розвитку захворювань серцево-судинної системи на органному рівні.
Відповідно до концепції про двокомпонентність РААС циркулюючій ланці відводять головну роль у її короткочасних фізіологічних ефектах. Тканинна ланка РААС забезпечує довгострокову дію на функцію і структуру органів.
Вазоконстрикція і вивільнення альдостерону у відповідь на стимуляцію ангіотензином є негайними реакціями, що виникають протягом секунд, відповідно до їхньої фізіологічної ролі, яка полягає в підтримці кровообігу після крововтрати, дегідратації або ортостатичної гіпотензії. Інші ефекти — гіпертрофія міокарда, СН розвиваються протягом тривалого часу. Для патогенезу хронічних захворювань серцево-судинної системи повільні відповіді, які виконуються на тканинному рівні, важливіші, ніж швидкі, що реалізуються системною ланкою РААС.
Рецептори ангіотензину ІІ. Основні ефекти ангіотензину ІІ здійснюються через його взаємодію зі специфічними клітинними рецепторами. На сьогодні виділено кілька типів і підтипів ангіотензинових рецепторів: АТ1, АТ2, АТ3 і АТ4. У людини виявлені тільки АТ1- і АТ2-рецептори (табл. 1.17).
Таблиця 1.17
Функції рецепторів ангіотензину ІІ
| АТ1-рецептори | АТ2-рецептори |
| Вазоконстрикція
Стимуляція синтезу і вивільнення альдостерону Реабсорбція натрію в проксимальних канальцях нирок Гіпертрофія міокарда Проліферація гладком’язових клітин Підвищення периферичної активності норадреналіну Підвищення центральної симпатоміметичної активності Стимуляція вивільнення вазопресину Зниження ниркового кровотоку Пригнічення синтезу реніну за принципом негативного зворотного зв’язку |
Стимуляція апоптозу
Антипроліферативна дія Ембріональна диференціація і розвиток тканин Ріст ендотеліальних клітин Вазодилатація |
Відомо, що всі серцево-судинні, а також екстракардіальні ефекти ангіотензину ІІ опосередковуються переважно через АТ1-рецептори.
АТ1-рецептори виявлені в тканинах серця, печінки, мозку, нирок, надниркової залози, матки, ендотеліальних і гладком’язових клітинах, фібробластах, макрофагах, периферичних симпатичних нервах, у провідній системі серця.
Про АТ2-рецептори відомо значно менше, ніж про рецептори АТ1-типу. В організмі дорослої людини АТ2-рецептори у високих концентраціях виявлені в мозковому шарі надниркової залози, матці і яєчниках, а також у судинному ендотелії, серці і різних ділянках мозку. У тканинах ембріона АТ2-рецепторів представлені значно ширше, ніж у тканинах дорослого організму, і є в них переважаючими. Незабаром після народження АТ2-рецептор «вимикається» і активується при певних патологічних станах, таких як ішемія міокарда, СН, ушкодження судин. Те, що АТ2-рецептори найбільш широко представлені в тканинах плода і їх концентрація різко знижується в перші ж тижні після народження, свідчить про їх роль у процесах, пов’язаних із клітинним ростом, диференціацією і розвитком.
Вважають, що АТ2-рецептори опосередковують апоптоз — запрограмовану загибель клітини, яка є закономірним наслідком процесів її диференціації і розвитку. Завдяки цьому стимуляція АТ2-рецепторів зумовлює антипроліферативну дію.
АТ2-рецептори вважають фізіологічною противагою АТ1-рецепторам. Очевидно, вони контролюють надмірний ріст, опосередкований через АТ1-рецептори або інші фактори росту, а також урівноважують вазоконстрикторний ефект стимуляції АТ1-рецепторів.
Механізм антигіпертензивної дії. Ефекти антагоністів ангіотензину ІІ зумовлені їх здатністю зв’язуватися зі специфічними рецепторами останнього. Блокада АТ1-рецепторів антагоністами ангіотензину ІІ призводить до пригнічення його основних фізіологічних ефектів — вазоконстрикції, синтезу альдостерону, вивільнення катехоламінів з надниркової залози і пресинаптичних мембран, виділення вазопресину, а також до вповільнення гіпертрофічних і проліферативних процесів в стінці судин і міокарді.
Основним гемодинамічним ефектом блокаторів АТ1-рецепторів є вазодилатація і, отже, зниження рівня АТ. Антигіпертензивна ефективність препаратів залежить від вихідної активності РААС: у хворих з високою активністю реніну вони діють сильніше.
Усі блокатори АТ1-рецепторів зумовлюють тривалу антигіпертензивну дію, що триває протягом 24 год. Вона проявляється через 2–4 тиж терапії і підсилюється до 6–8-го тижня лікування. Більшість препаратів викликають дозозалежне зниження АТ. Вони не порушують його нормальний добовий ритм. Наявні клінічні спостереження свідчать про те, що при тривалому застосуванні блокаторів ангіотензинових рецепторів (протягом ≥2 років) не відбувається звикання до їх дії. Скасування лікування не призводить до «рикошетного» підвищення АТ. Блокатори АТ1-рецепторів не знижують рівень АТ, якщо він перебуває в межах нормальних значень (табл. 1.18).
Таблиця 1.18
Блокатори рецепторів ангіотензину II
| Препарат | Доза, мг/добу | Частота прийому на добу |
| Вальзартан
Епрозартан Ірбесартан Кандесартан Ольмезартан Тельмізартан |
80–320
400–800 150–300 8–32 50–100 20–40 20–80 |
1
1 1 1 1 1 1 |
При порівнянні з антигіпертензивними препаратами інших класів виявлено, що блокатори АТ1-рецепторів, спричиняючи аналогічний антигіпертензивний ефект, викликають менше побічних явищ і краще переносяться хворими.
Відмінна риса нового класу антигіпертензивних препаратів полягає в хорошій, порівняно із плацебо, переносимості. Побічні ефекти при їх прийомі спостерігаються значно рідше, ніж при використанні інгібіторів АПФ. На відміну від останніх, застосування блокаторів ангіотензину ІІ не супроводжується накопиченням брадикініну і появою зумовленого цим кашлю. Значно рідше розвивається також ангіоневротичний набряк.
Подібно інгібіторам АПФ ці засоби можуть викликати досить швидке зниження АТ при ренінзалежних формах АГ. У хворих з двобічним звуженням ниркових артерій можливе погіршення функції нирок. У пацієнтів з ХНН існує ризик розвитку гіперкаліємії у зв’язку з пригніченням вивільнення альдостерону в процесі лікування. Застосування блокаторів АТ1-рецепторів у період вагітності протипоказане через високий ризик порушень розвитку плода і його загибелі.
Незважаючи на вищезгадані небажані ефекти, блокатори АТ1-рецепторів вважаються найбільш добре переносимою хворими групою антигіпертензивних препаратів з найнижчою частотою розвитку побічних реакцій.
Перевагу для лікування АГ їм слід віддавати в наступних клінічних ситуаціях:
- СН;
- перенесений ІМ;
- діабетична нефропатія;
- протеїнурія, мікроальбумінурія;
- ГЛШ;
- фібриляція передсердь;
- метаболічний синдром;
- непереносимість інгібіторів АПФ.
На даний час іде пошук нових напрямків в лікуванні АГ. Прямі інгібітори реніну — перший серед нових класів антигіпертензивних препаратів, який показав свою ефективність у доклінічних та клінічних дослідженнях, аліскірен — перший представник цього класу, який успішно пройшов всі стадії випробувань і отримав реєстрацію для клінічного застосування в США та країнах Європи, зокрема в Україні.
Механізм дії аліскірену полягає в пригніченні активності реніну, а також прореніну, який, згідно із сучасними уявленнями, бере участь у перетворенні ангіотензиногену в ангіотензин I. Крім того, ефект цього препарату виявляється за рахунок зв’язування рецепторів реніну на клітинній мембрані. Відомо, що активність реніну плазми крові вірогідно впливає на ризик розвитку серцево-судинних захворювань, зокрема ІМ. Тому важливо не тільки блокувати його ефекти, але й контролювати активність. Аліскірен знижує активність реніну і, тим самим, може знижувати ризик виникнення тяжких серцево-судинних ускладнень. На вивчення цього факту спрямована великомасштабна програма клінічних досліджень ASPIRE HIGHER з включенням більше ніж 35 000 пацієнтів. Результати вже завершених досліджень цієї програми (AVOID, ALOFT, ALLAY, AGELESS) демонструють високу клінічну ефективність аліскірену щодо лікування АГ, а також його кардіо- та нефропротекторних властивостей.
ГІПЕРТЕНЗИВНІ КРИЗИ
Гіпертензивний криз — це раптове значне підвищення АТ, що майже завжди супроводжується появою або посиленням порушень із боку органів-мішеней або вегетативної нервової системи.
Критеріями гіпертензивного кризу є:
- раптовий початок;
- значне підвищення АТ;
- поява або посилення симптомів з боку органів-мішеней.
Рівень АТ під час кризу набагато вищий, ніж зазвичай у цього хворого, однак у різних пацієнтів він може значно відрізнятися. Так, для дитини з гострим гломерулонефритом кризом є навіть порівняно невелике — до 160/100 мм рт. ст. підвищення АТ. Для пацієнта похилого віку з розшаровуючою аневризмою аорти такий тиск також небезпечний, він вимагає негайного зниження і розцінюється як криз. У той же час вищий рівень АТ, наприклад 220/120 мм рт. ст., у хворого зі стабільною тяжкою гіпертензією може не супроводжуватися істотним погіршенням стану, пацієнт потребує тільки звичайного планового прийому антигіпертензивних препаратів. Отже, рівень АТ не визначає в цілому поняття кризу. Виключення слід зробити тільки при дуже високому АТ (САТ ≥240 мм рт. ст., ДАТ ≥140 мм рт. ст.), навіть якщо симптоми з боку органів-мішеней відсутні. Такий рівень АТ у будь-якого хворого може перевищити можливості ауторегуляції мозкового кровотоку або скорочувальної здатності міокарда і призвести до розвитку гострої гіпертензивної енцефалопатії, СН або інших ускладнень.
Класифікація. Залежно від наявності або відсутності ураження органів-мішеней і необхідності термінового зниження АТ, гіпертензивні кризи можна розділити на такі дві категорії:
- ускладнені — характеризуються гострим або прогресуючим ураженням органів-мішеней, становлять пряму загрозу для життя хворого, вимагають негайного, протягом 1 год, зниження АТ;
- неускладнені — без ознак гострого або прогресуючоого ураження органів-мішеней, становлять потенційну загрозу для життя хворого, вимагають швидкого, протягом декількох годин, зниження АТ (табл. 1.19).
Таблиця 1.19
Класифікація гіпертензивних кризів
| Ускладнені гіпертензивні кризи: характер ускладнення | Неускладнені гіпертензивні кризи |
| ІМ
Інсульт Гостра разшаровуюча аневризма аорти Гостра недостатність ЛШ Нестабільна стенокардія Аритмії (пароксизми тахікардії, миготливої тахіаритмії, шлуночкова екстрасистолія високих градацій) Транзиторна ішемічна атака Еклампсія Гостра гіпертензивна енцефалопатія Кровотеча ГНН |
Церебральний неускладнений криз
Гіпоталамічний пароксизм (діенцефально-вегетативний криз) Кардіальний неускладнений криз Підвищення САТ до 240 мм рт. ст. або ДАТ до 140 мм рт. ст. Значне підвищення АТ у ранній післяопераційний період |
Ускладнені гіпертензивні кризи. Перебіг характеризується клінічними ознаками гострого або прогресуючого ураження органів-мішеней. Такі кризи завжди супроводжуються появою або посиленням симптомів з боку органів-мішеней. Вони загрожують життю хворого і вимагають зниження тиску в проміжок часу від кількох хвилин до 1 год (схема 1.2). Лікування здійснюється в умовах палати інтенсивної терапії із застосуванням парентерального введення антигіпертензивних препаратів. До цієї категорії слід віднести також випадки значного підвищення АТ, коли загроза життю виникає не через ураження органів-мішеней, а через кровотечу, нерідко в післяопераційний період.
Схема 1.2. Тактика лікаря при гіпертензивних кризах
Неускладнені гіпертензивні кризи. Неускладнений криз характеризується відсутністю клінічних ознак гострого або прогресуючого ураження органів, проте при несвоєчасному наданні допомоги хворому може набувати несприятливого перебігу з розвитком ускладнень і навіть з летальним кінцем. Такі кризи супроводжуються, як правило, появою або посиленням симптомів з боку органів-мішеней (інтенсивний головний біль, біль у ділянці серця, екстрасистолія) або вегетативної нервової системи (вегетативно-судинні розлади, тремтіння, часте сечовипускання).
На підставі клінічних особливостей виділяють церебральні і кардіальні неускладнені кризи. Гіпоталамічні пароксизми (за старою термінологією — діенцефально-вегетативні кризи) розглядаються як прояв церебрального кризу. Підвищення САТ до 240 мм рт. ст. або ДАТ до 140 мм рт. ст. слід також розцінювати як гіпертензивний криз незалежно від того, розвинулися симптоми з боку органів-мішеней або ще немає: при середньому рівні АТ, що перевищує 150–180 мм рт. ст., ауторегуляторні механізми, що забезпечують сталість мозкового кровотоку, стають неспроможними, що призводить до розвитку гострої гіпертензивної енцефалопатії або крововиливу. Загрозливим є також значне підвищення АТ у ранній післяопераційний період через ризик кровотечі.
Усі ці клінічні прояви вимагають зниження АТ протягом декількох годин, госпіталізація необов’язкова. Лікування здійснюється шляхом перорального прийому антигіпертензивних препаратів або за допомогою внутрішньом’язових ін’єкцій.
Терміном «гіпертензивна енцефалопатія» позначають клінічний синдром, що характеризується прогресуючими розладами функції мозку внаслідок значного підвищення АТ під впливом будь-якої причини. Він являє собою вкрай тяжкий прояв порушення ауторегуляції мозкового кровотоку. Гіпертензивну енцефалопатію з вираженими клінічними ознаками — сплутаністю свідомості, судомами, вогнищевими неврологічними ознаками виявляють рідко, однак її елементи у вигляді інтенсивного головного болю, нудоти, блювання, порушень зору характерні для будь-якого церебрального кризу.
Лікування. При ускладненому гіпертензивному кризі хворого госпіталізують у палату інтенсивної терапії і негайно приступають до інтенсивної терапії (табл. 1.20).
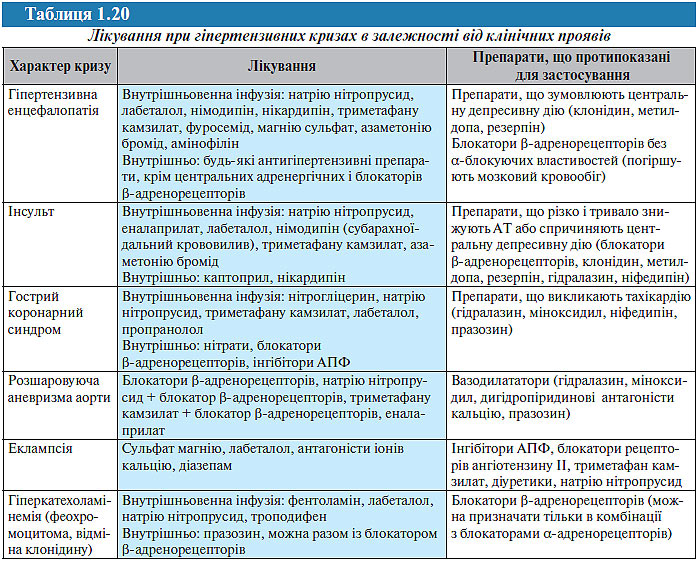
З урахуванням того, що ринок в Україні безупинно поповнюється новими лікарськими засобами (табл. 1.21), наведені практично всі сучасні препарати, навіть ще не зареєстровані в країні, які рекомендують для лікування АГ в екстрених ситуаціях. Однак через дефіцит лікарських засобів згаданої групи в таблицю включені також препарати, які вже виходять із вжитку: триметафану камзилат, клонідин, азаметонію бромід, бендазол.

Ніфедипін у деяких хворих може викликати інтенсивний головний біль, а також неконтрольовану АГ (особливо поєднано з магнію сульфатом), тому його застосування слід обмежити випадками, коли хворі добре реагували на цей препарат раніше (під час планового лікування). Перевагу слід віддавати препаратам короткочасної дії (натрію нітропрусид, нітрогліцерин, триметафану камзилат), оскільки вони дають керований антигіпертензивний ефект. Препарати тривалої дії небезпечні можливим розвитком некерованої гіпотензії. Різке зниження АТ підвищує ризик ускладнень: зменшення мозкового кровообігу (аж до розвитку коми), коронарного кровообігу (напади стенокардії, аритмія, іноді ІМ). Особливо високий ризик ускладнень при раптовому зниженні АТ у хворих похилого віку з вираженим атеросклерозом судин головного мозку.
На першому етапі лікування метою є часткове зниження АТ до безпечного рівня — не обов’язково до нормального. Найчастіше АТ знижують на 20–25%, зазвичай не нижче 160/100 мм рт. ст. У хворих з розшаровуючою аневризмою аорти або гострою лівошлуночковою недостатністю зниження АТ може бути більш агресивним. Хворим із цереброваскулярними порушеннями знижувати АТ слід особливо повільно, при ретельному моніторуванні неврологічного статусу (табл. 1.22).
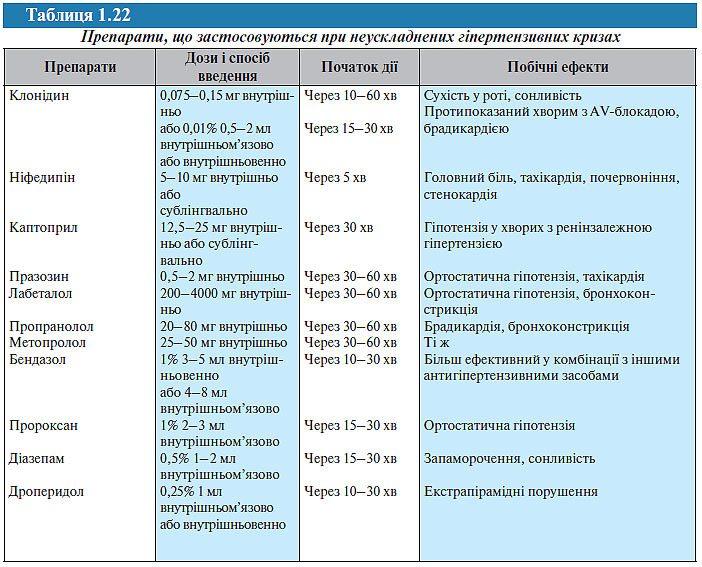
У випадку розвитку неускладненого кризу, як правило, немає необхідності у внутрішньовенному введенні препаратів. Призначають перорально препарати зі швидкою антигіпертензивною дією або внутрішньом’язові ін’єкції. Ефективне застосування клонідину, який не викликає тахікардії, не збільшує серцевий викид, тому його можна рекомендувати при стенокардії, крім того, цей препарат може бути призначений хворим з нирковою недостатністю. Ефект настає через 5–15 хв після парентерального введення й через 30–60 хв після перорального прийому. Препарат протипоказаний хворим із проявами кризу з боку ЦНС: седативний ефект може затруднювати об’єктивну оцінку неврологічного статусу. Клонідин не слід рекомендувати хворим з порушенням серцевої провідності, особливо тим, хто приймає серцеві глікозиди.
Застосовують також ніфедипін, який має властивість знижувати ЗПСО, збільшувати серцевий викид і нирковий кровотік. Зниження АТ спостерігається вже через 15–30 хв після його прийому, антигіпертензивний ефект зберігається протягом 4–6 год. Капсулу ніфедипіну слід розжувати і вміст проковтнути. Зазвичай досить 5–10 мг ніфедипіну. При відсутності ефекту через 30–60 хв прийом повторюють. Слід зазначити, що Об’єднаний національний комітет США по високому АТ вважає недоцільним застосування ніфедипіну під час кризу, щоб уникнути небезпеки розвитку мозкової або коронарної ішемії, оскільки швидкість і ступінь зниження АТ при сублінгвальному прийомі препарату важко контролювати. Інгібітор АПФ каптоприл знижує АТ уже через 30–40 хв після прийому завдяки швидкій абсорбції в шлунку. Перевагою препарату є його здатність зсовувати криву ауторегуляції мозкового кровотоку вліво, в результаті чого мозковий кровотік не погіршується, незважаючи на зниження АТ. Зрідка каптоприл викликає надмірну гіпотензію, особливо у хворих з гіповолемією, нирковою недостатністю або реноваскулярною гіпертензією.
Ефективні комбінації з 2–3 препаратів (наприклад ніфедипін + метопролол або ніфедипін + каптоприл) (див. табл. 1.20).
У 80–90-ті роки ХХ ст. одним з найбільш широко застосовуваних у нашій країні препаратів для купірування кризів був бендазол. Дотепер його використовують лікарі швидкої допомоги і терапевтичних стаціонарів. Ефективність бендазолу не слід переоцінювати. За здатністю знижувати АТ він поступається клонідину, ніфедипіну, каптоприлу, петльовим діуретикам, а також має суттєвий недолік, який полягає в неможливості прийому внутрішньо.
Для профілактики гіпертензивних кризів вирішальне значення має регулярна терапія хронічної АГ. Лікування поліпшує перебіг захворювання і знижує частоту ускладнень. Виявлення вторинних форм АГ на початку захворювання і диференційований підхід до їх лікування також є обов’язковою умовою попередження кризів.
ЛІТЕРАТУРА
- Кобалава Ж.Д., Гудков К.М. (2003) Гипертонические кризы: существуют ли реальные противоречия в классификации и лечении? Сердце, 2(3): 116–127.
- Коваленко В.Н., Свищенко Е.П. и др. (2005) Лечение артериальной гипертензии в особых клинических ситуациях. Каменец-Подольский, 501 с.
- Комитет экспертов Всероссийского научного общества кардиологов (ВНОК) (2004) Профилактика, диагностика и лечение артериальной гипертонии. Российские рекомендации (второй пересмотр). Приложение к журналу «Кардиоваскулярная терапия и профилактика», Москва.
- Кушаковский М.С. (1995) Гипертоническая болезнь. СОТИС, Санкт-Петербург, 311 с.
- Ланг Г.Ф. (1950) Гипертоническая болезнь. Медгиз, Ленинград, 496 с.
- Рогоза А.Н. (2004) Суточное мониторирование артериального давления (по материалам методических рекомендаций ESH 2003). Функциональная диагностика, 4: 29–44.
- Свищенко Е.П., Коваленко В.Н. (2002) Гипертоническая болезнь: вторичные гипертензии. Либiдь, Киев, 504 с.
- Свіщенко Є.П., Багрій А.Є., Коваленко В.М. та ін. (Робоча група з артеріальної гіпертензії Української асоціації кардіологів) (2004) Рекомендації Української асоціації кардіологів з профілактики та лікування артеріальної гіпертензії. Посібник до Національної програми профілактики і лікування артеріальної гіпертензії (3-тє вид., випр. і доп.). Інститут кардіології АМН України, Київ, 86 с.
- Чазова И.Е., Бойцов С.А., Небиеридзе Д.В. (2004) Основные положения проекта второго пересмотра рекомендаций ВНОК по профилактике, диагностике и лечению артериальной гипертензии. Кардиоваскулярная терапия и профилактика, 4: 90–98.
- Чазов Е.И., Чазова И.Е. (ред.) (2005) Руководство по артериальной гипертонии. Media Medica, Москва, 784 с.
- Assmann G., Schulte H. (1988) The Prospective Cardiovascular Münster (PROCAM) study: prevalence of hyperlipidemia in persons with hypertension and/or diabetes mellitus and the relationship to coronary heart disease. Am. Heart J., 116(6 Pt 2): 1713–1724.
- Benetos A., Zureik M., Morcet J. et al. (2000) A decrease in diastolic blood pressure combined with an increase in systolic blood pressure is associated with a higher cardiovascular mortality in men. J. Am. Coll. Cardiol., 35(3): 673–680.
- Chobanian A.V., Bakris G.L., Black H.R. et al.; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee (2003) Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension, 42(6): 1206–1252.
- Devereux R.B., Wachtell K., Gerdts E. et al. (2004) Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA, 292(19): 2350–2356.
- European Society of Hypertension — European Society of Cardiology Guidelines Committee (2003) 2003 European Society of Hypertension — European Society of Cardiology guidelines for the management of arterial hypertension. J. Hypertens., 21(6): 1011–1053.
- Fourth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (Constituted by representatives of nine societies and by invited experts); Graham I., Atar D., Borch-Johnsen K. et al. (2007) European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Eur. Heart J., 28(19): 2375–2414.
- Hansen T.W., Jeppesen J., Rasmussen S. et al. (2006) Ambulatory blood pressure monitoring and risk of cardiovascular disease: a population based study. Am. J. Hypertens., 19(3): 243–250.
- Kannel W.B. (1996) Blood pressure as a cardiovascular risk factor: prevention and treatment. JAMA, 275(20): 1571–1576.
- Kannel W.B. (2000) Risk stratification in hypertension: new insights from the Framingham Study. Am. J. Hypertens., 13(1 Pt 2): 3S–10S.
- Kearney P.M., Whelton M., Reynolds K. et al. (2005) Global burden of hypertension: analysis of worldwide data. Lancet, 365(9455): 217–223.
- Kizer J.R., Bella J.N., Palmieri V. et al. (2006) Left atrial diameter as an independent predictor of first clinical cardiovascular events in middle-aged and elderly adults: the Strong Heart Study (SHS). Am. Heart J., 151(2): 412–418.
- Levey A.S., Eckardt K.U., Tsukamoto Y. et al. (2005) Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int., 67(6): 2089–2100.
- Lewington S., Clarke R., Qizilbash N. et al.; Prospective Studies Collaboration (2002) Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet, 360(9349): 1903–1913.
- Mancia G., Grassi G. (2007) European, American and British Guidelines: similarities and differences. In: Black HR, Elliott W.J., editors. Hypertension. A companion to Braunwald’s Heart diseases. Saunders-Elsevier., 571–574.
- Mancia G., De Backer G., Dominiczak A. et al.; The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) (2007) 2007 Guidelines for the management of arterial hypertension. Eur. Heart J., 28(12): 1462–1536.
- Muiesan M.L., Salvetti M., Monteduro C. et al. (2004) Left ventricular concentric geometry during treatment adversely affects cardiovascular prognosis in hypertensive patients. Hypertension, 43(4): 731–738.
- Neal B., MacMahon S., Chapman N.; Blood Pressure Lowering Treatment Trialists’ Collaboration (2000) Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet, 356(9246): 1955–1964.
- Ohkubo T., Hozawa A., Yamaguchi J. et al. (2002) Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: the Ohasama study. J. Hypertens., 20(11): 2183–2189.
- Thomas F., Rudnichi A., Bacri A.M. et al. (2001) Cardiovascular mortality in hypertensive men according to presence of associated risk factors. Hypertension, 37(5): 1256–1261.
- Turnbull F.; Blood Pressure Lowering Treatment Trialists’ Collaboration (2003) Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. Lancet, 362(9395): 1527–1535.
- Turnbull F., Neal B., Algert C. et al.; Blood Pressure Lowering Treatment Trialists̓ Collaboration (2005) Effects of different blood pressure-lowering regimens on major cardiovascular events in individuals with and without diabetes mellitus: results of prospectively designed overviews of randomized trials. Arch. Intern. Med., 165(12): 1410–1419.
- Vasan R.S., Beiser A., Seshadri S. et al. (2002) Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA, 287(8): 1003–1010.
- Vasan R.S., Larson M.G., Leip E.P. et al. (2001) Assessment of frequency of progression to hypertension in non-hypertensive participants in the Framingham Heart Study: a cohort study. Lancet, 358(9294): 1682–1686.
- Vermeer S.E., Koudstaal P.J., Oudkerk M. et al. (2002) Prevalence and risk factors of silent brain infarcts in the population-based Rotterdam Scan Study. Stroke, 33(1): 21–25.
- Willum-Hansen T., Staessen J.A., Torp-Pedersen C. et al. (2006) Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation, 113(5): 664–670.