
СТАНДАРТ
НАСТАНОВА
СТ-Н МОЗУ 42-8.7:2018
ЛІКАРСЬКІ ЗАСОБИ
НАЛЕЖНІ ПРАКТИКИ ФАРМАКОНАГЛЯДУ
Видання офіційне
Київ
Передмова
1. РОЗРОБЛЕНО: Державне підприємство «ДЕРЖАВНИЙ ЕКСПЕРТНИЙ ЦЕНТР МІНІСТЕРСТВА ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: Т. Талаєва, д-р мед. наук, професор; Т. Думенко, канд. мед. наук; О. Матвєєва, канд. мед. наук; І. Логвіна; В. Яйченя; Л. Пушкар; М. Борецька.
2. Ця настанова відповідає документам:
EMA/541760/2011 «Guideline on good pharmacovigilance practices (GVP). Module I — Pharmacovigilance systems and their quality systems» («Настанова з належних практик фармаконагляду (ННПФ). Модуль І — Фармаконагляд та його система якості»)
EMA/816573/2011 «Guideline on good pharmacovigilance practices (GVP) Module II — Pharmacovigilance system master file» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІ — Майстер-файл системи фармаконагляду»)
EMA/119871/2012 «Guideline on good pharmacovigilance practices (GVP) Module ІІI — Pharmacovigilance inspections» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІІ — Інспекції системи фармаконагляду»)
EMA/228028/2012 «Guideline on good pharmacovigilance practices (GVP) Module IV — Pharmacovigilance audits» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІV — Аудит з фармаконагляду»)
EMA/838713/2011 «Guideline on good pharmacovigilance practices (GVP) Module V — Risk management systems («Настанова з належних практик фармаконагляду (ННПФ) Модуль V — Системи управління ризиками»)
EMA/873138/2011 «Guideline on good pharmacovigilance practices (GVP) Module VI — Management and reporting of adverse reactions to medicinal products» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VI — Управління та звітування про побічні реакції лікарських засобів»)
EMA/816292/2011 «Guideline on good pharmacovigilance practices (GVP) Module VII — Periodic safety update report» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VII — Регулярно оновлюваний звіт з безпеки»)
EMA/813938/2011 «Guideline on good pharmacovigilance practices (GVP) Module VIII — Post-authorisation safety studies» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VIІІ — Післяреєстраційне дослідження з безпеки»)
EMA/395730/2012 «Guideline on good pharmacovigilance practices (GVP) Module VIII Addendum I — Requirements and recommendations for the submission of information on non-interventional post-authorisation safety studies» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VIІІ Додаток І — Вимоги та рекомендації щодо подання інформації про неінтервенційні післяреєстраційні дослідження з безпеки»)
EMA/827661/2011 «Guideline on good pharmacovigilance practices (GVP) Module IX — Signal management» («Настанова з належних практик фармаконагляду (ННПФ) Модуль IX — Управління сигналом»)
EMA/169546/2012 «Guideline on good pharmacovigilance practices (GVP) Module X — Additional monitoring» («Настанова з належних практик фармаконагляду (ННПФ) Модуль X — Додатковий моніторинг»)
EMA/118465/2012 «Guideline on good pharmacovigilance practices (GVP) Module XV — Safety communication» («Настанова з належних практик фармаконагляду (ННПФ) Модуль XV — Процес комунікації з питань безпеки»).
EMA/204715/2012 «Guideline on good pharmacovigilance practices (GVP) Module XVI — Risk minimisation measures: selection of tools and effectiveness indicators» («Настанова з належних практик фармаконагляду (ННПФ) Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності»).
EMA/168402/2014 Corr* «Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations II: Biological medicinal products («Настанова з належних практик фармаконагляду (ННПФ) Частина XIIІ — Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби»).
Ступінь відповідності — модифікований (MOD) Переклад з англійської (en)
3. ВВЕДЕНО ВДРУГЕ
© Міністерство охорони здоров’я України, 2018
© Державний експертний центр МОЗ України
Національний вступ
Здійснення нагляду за безпекою лікарських засобів при їх медичному застосуванні стало одним з основних напрямків у реалізації національної політики обігу лікарських засобів у переважній більшості країн світу. Міжнародний і національний регуляторний механізм нагляду за безпекою лікарських засобів отримав назву — фармаконагляд. Здійснення основної місії фармаконагляду — покращання лікування пацієнтів та їх безпеки при застосуванні лікарських засобів — стає можливим лише за наявності вичерпної, достовірної, якісної та об’єктивної інформації щодо безпеки та ефективності застосування лікарських засобів, джерелами якої є, у першу чергу, власники реєстраційних посвідчень на лікарські засоби, а також — працівники з медичною та/або фармацевтичною освітою, пацієнти та/або їх представники, організації, що представляють чи захищають безпеку та права пацієнтів. У даному документі представлені підходи, які слід використовувати всім учасникам процесу фармаконагляду з метою успішної реалізації визначених законодавством вимог та повноважень у розрізі здійснення фармаконагляду.
Ця настанова є прийнятим зі змінами (версії en) нормативним документом Європейського Союзу (ЄС) «Guideline on good pharmacovigilance practices» (GVP) («Настанова з належних практик фармаконагляду»), де представлено технічні підходи до планування та здійснення належної практики фармаконагляду. Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вносяться до «Guideline on good pharmacovigilance practices» (GVP).
Створення та впровадження в Україні настанови з належних практик фармаконагляду зумовлена потребою усіх зацікавлених сторін (таких як заявники, власники реєстраційних посвідчень, розробники та виробники лікарських засобів, експертні та регуляторні органи) у наявності інструмента, який би дозволив належним чином здійснювати фармаконагляд. Положення вітчизняної настанови з належних практик фармаконагляду повинні бути гармонізовані з положеннями відповідної настанови ЄС, що відповідає абзацу другому пункту 1 розділу II Порядку здійснення фармаконагляду, затвердженого наказом МОЗ України від 27 грудня 2006 р. № 898 (у редакції наказу МОЗ України від 26.09.2016 р. № 996), зареєстрованого в Міністерстві юстиції України 19 грудня 2016 р. за № 1649/29779 [2].
Структурно, настанова складається з окремих модулів. Ця настанова розроблена на підставі Модулів І, ІІ, ІІІ, IV, V, VI, VII, VIII, IX, X, XV, XVI, Частини ХІІІ Настанови з належних практик фармаконагляду (GVP), прийнятої в ЄС:
EMA/541760/2011 «Guideline on good pharmacovigilance practices (GVP). Module I — Pharmacovigilance systems and their quality systems» («Настанова з належних практик фармаконагляду. Модуль І — Фармаконагляд та його система якості») [33];
EMA/816573/2011 «Guideline on good pharmacovigilance practices (GVP) Module II — Pharmacovigilance system master file» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІ — Майстер-файл системи фармаконагляду») [34];
EMA/119871/2012 «Guideline on good pharmacovigilance practices (GVP) Module ІІI — Pharmacovigilance inspections» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІІ — Аудит системи фармаконагляду заявника (власника реєстраційного посвідчення), що проводиться уповноваженою Міністерством охорони здоров’я спеціалізованою експертною установою у сфері здійснення фармаконагляду) [35];
EMA/228028/2012 «Guideline on good pharmacovigilance practices (GVP) Module IV — Pharmacovigilance audits» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІV — Аудит з фармаконагляду») [36];
EMA/838713/2011 «Guideline on good pharmacovigilance practices (GVP) Module V — Risk management systems («Настанова з належних практик фармаконагляду (ННПФ) Модуль V — Системи управління ризиками») [37];
EMA/873138/2011 «Guideline on good pharmacovigilance practices (GVP) Module VI — Management and reporting of adverse reactions to medicinal products» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VI — Управління та звітування про побічні реакції лікарських засобів») [38];
EMA/816292/2011 «Guideline on good pharmacovigilance practices (GVP) Module VII — Periodic safety update report» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VII — Регулярно оновлюваний звіт з безпеки») [39];
EMA/813938/2011 Guideline on good pharmacovigilance practices (GVP) Module VIII — Post-authorisation safety studies (Настанова з належних практик фармаконагляду (ННПФ) Модуль VIІІ — Післяреєстраційне дослідження з безпеки) [41];
EMA/395730/2012 Guideline on good pharmacovigilance practices (GVP) Module VIII Addendum I — Requirements and recommendations for the submission of information on non-interventional post-authorisation safety studies (Настанова з належних практик фармаконагляду (ННПФ) Модуль VIІІ Додаток І — Вимоги та рекомендації щодо подання інформації про неінтервенційні післяреєстраційні дослідження з безпеки) [42];
EMA/827661/2011 Guideline on good pharmacovigilance practices (GVP) Module IX — Signal management (Настанова з належних практик фармаконагляду (ННПФ) Модуль IX — Управління сигналом)[43];
EMA/169546/2012 Guideline on good pharmacovigilance practices (GVP) Module X — Additional monitoring (Настанова з належних практик фармаконагляду (ННПФ) Модуль X — Додатковий моніторинг) [44];
EMA/118465/2012 Guideline on good pharmacovigilance practices (GVP) Module XV — Safety communication (Настанова з належних практик фармаконагляду (ННПФ) Модуль XV — Процес комунікації з питань безпеки) [45];
EMA/204715/2012 Guideline on good pharmacovigilance practices (GVP) Module XVI — Risk minimisation measures: selection of tools and effectiveness indicators (Настанова з належних практик фармаконагляду (ННПФ) Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності) [46];
EMA/168402/2014 Corr* Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations II: Biological medicinal products (Настанова з належних практик фармаконагляду (ННПФ) Частина XIIІ — Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби) [47].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству.
До цієї настанови було внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо в пункти, яких вони стосуються; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Позначення та скорочення», а також національний додаток «Бібліографія». «Зміст» цієї настанови подано в оригінальному вигляді з урахуванням додаткових структурних елементів;
- основні положення викладено в частині І «Модуль І — Фармаконагляд та його система якості», частині ІІ «Модуль ІІ — Майстер-файл системи фармаконагляду», частині ІІІ «Модуль ІІІ — Аудит системи фармаконагляду заявника (власника реєстраційного посвідчення), що проводиться уповноваженою Міністерством охорони здоров’я спеціалізованою експертною установою у сфері здійснення фармаконаглядуN», частині ІV «Модуль IV — «Аудит з фармаконагляду», частині V «Модуль V — Системи управління ризиками», частині VI «Модуль VI — Управління та звітування про побічні реакції лікарських засобів», частині VII «Модуль VII — Регулярно оновлюваний звіт з безпеки», частині VIII «Модуль VIII — Післяреєстраційне дослідження з безпеки», частині IX «Модуль IX — Управління сигналом», частині X «Модуль X — Додатковий моніторинг», частині XI «Модуль XV — Процес комунікації з питань безпеки», частині XII «Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності», частині XIIІ «Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби» при цьому кожний структурний елемент та його номер у цій настанові відповідають таким у настанові «Guideline on good pharmacovigilance practices (GVP)» («Настанова з належних практик фармаконагляду (ННПФ)»);
- розділ «Терміни і визначення понять» складено на основі розділів EMA/541760/2011 «Guideline on good pharmacovigilance practices (GVP). Module I — Pharmacovigilance systems and their quality systems» («Настанова з належних практик фармаконагляду. Модуль І — Фармаконагляд та його система якості») [33];
EMA/816573/2011 «Guideline on good pharmacovigilance practices (GVP) Module II — Pharmacovigilance system master file» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІ — Майстер-файл системи фармаконагляду») [34];
EMA/119871/2012 «Guideline on good pharmacovigilance practices (GVP) Module ІІI — Pharmacovigilance inspections» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІІІ — Аудит системи фармаконагляду заявника (власника реєстраційного посвідчення), що проводиться уповноваженою Міністерством охорони здоров’я спеціалізованою експертною установою у сфері здійснення фармаконаглядуN») [35]. У розділах ІІІ.С.1. — ІІІ.С.4.2. та ІІІ.С.6. представлена інформація про те, які структури та у який спосіб здійснюють процеси моніторингу відповідності здійснення заявниками фармаконагляду вимогам законодавства в Україні. Також у зазначених вище розділах модуля ІІІ описана роль цих структур;
EMA/228028/2012 «Guideline on good pharmacovigilance practices (GVP) Module IV — Pharmacovigilance audits» («Настанова з належних практик фармаконагляду (ННПФ) Модуль ІV — Аудит з фармаконагляду») [36];
EMA/838713/2011 «Guideline on good pharmacovigilance practices (GVP) Module V — Risk management systems («Настанова з належних практик фармаконагляду (ННПФ) Модуль V — Системи управління ризиками») [37];
EMA/873138/2011 «Guideline on good pharmacovigilance practices (GVP) Module VI — Management and reporting of adverse reactions to medicinal products» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VI — Управління та звітування про побічні реакції лікарських засобів») [38];
EMA/816292/2011 «Guideline on good pharmacovigilance practices (GVP) Module VII — Periodic safety update report» («Настанова з належних практик фармаконагляду (ННПФ) Модуль VII — Регулярно оновлюваний звіт з безпеки») [39];
EMA/813938/2011 Guideline on good pharmacovigilance practices (GVP) Module VIII — Post-authorisation safety studies (Настанова з належних практик фармаконагляду (ННПФ) Модуль VIІІ — Післяреєстраційне дослідження з безпеки) [41];
EMA/827661/2011 Guideline on good pharmacovigilance practices (GVP) Module IX — Signal management (Настанова з належних практик фармаконагляду (ННПФ) Модуль IX — Управління сигналом) [43];
EMA/169546/2012 Guideline on good pharmacovigilance practices (GVP) Module X — Additional monitoring (Настанова з належних практик фармаконагляду (ННПФ) Модуль X — Додатковий моніторинг) [44];
EMA/118465/2012 Guideline on good pharmacovigilance practices (GVP) Module XV — Safety communication (Настанова з належних практик фармаконагляду (ННПФ) Модуль XV — Процес комунікації з питань безпеки) [45];
EMA/204715/2012 Guideline on good pharmacovigilance practices (GVP) Module XVI — Risk minimisation measures: selection of tools and effectiveness indicators (Настанова з належних практик фармаконагляду (ННПФ) Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності) [46];
EMA/168402/2014 Corr* Guideline on good pharmacovigilance practices (GVP) (Настанова з належних практик фармаконагляду (ННПФ) Частина XIIІ — Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби [47].
Цей розділ не позначено номером та викладено слідом за розділом «Нормативні посилання». Усі терміни в розділі «Терміни та визначення понять» наведено в алфавітному порядку, вони супроводжуються посиланням на нормативний документ, бібліографічний опис якого наведено в національному додатку «Бібліографія»;
- у розділі «Нормативні посилання» додатково наведено бібліографічний опис нормативних документів, що згадуються у цій настанові;
- у національному додатку «Бібліографія» додатково наведено бібліографічний опис нормативних документів, посилання на які наведено у цій настанові;
- у розділі «Позначення та скорочення» додатково наведено позначення скорочень, що використовуються у цій настанові;
- по всьому тексту внесено редакційні зміни в посилання на структурні елементи цієї настанови, наприклад замість «(see section 3)» вказано «(див. розділ 3 цієї настанови)»;
- додатково до посилань на керівництва ЄС зроблено посилання на відповідні гармонізовані документи, затверджені в Україні.
Ця настанова є рекомендаціями для планування та здійснення нагляду за лікарськими засобами при їх медичному застосуванні.
Правовий статус цієї настанови відповідає правовому статусу відповідного керівництва в ЄС, з яким гармонізовано розроблену настанову. Цю настанову слід розглядати як технічний документ для надання рекомендацій заявникам та власникам реєстраційних посвідчень, компетентним уповноваженим органам та/або іншим зацікавленим особам щодо найкращого та найбільш прийнятного способу виконання положень, визначених фармацевтичним законодавством України. Це наукове керівництво пов’язане зі специфічними питаннями щодо здійснення нагляду за лікарськими засобами при їх медичному застосуванні. Положення цієї настанови відображують гармонізований (у рамках ЄС) підхід; вони базуються на останніх наукових досягненнях у цій галузі знань.
У рамках чинного законодавства ця настанова не має сили нормативно-правового акта, її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану позицію європейського фармацевтичного сектора; дотримання її положень зацікавленими сторонами полегшить оцінку реєстраційних досьє, а також підвищить якість та безпеку лікарських засобів в Україні. Однак можуть бути застосовані альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових керівництв викладений у документі Європейського агентства з лікарських засобів (EMA) Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо керівництв та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005) [48]. Вказаний підхід відповідає позиції ВТО відносно застосування стандартів.
Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вносяться до «Guideline on good pharmacovigilance practices (GVP)» («Настанова з належних практик фармаконагляду (ННПФ)»).
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Належні практики фармаконагляду
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Надлежащие практики фармаконадзора
MEDICINAL PRODUCTS
Guideline on good pharmacovigilance practices
Сфера застосування
Ця настанова визначає положення (рекомендації) щодо належної практики здійснення нагляду за безпекою лікарських засобів при їх медичному застосуванні.
Ця настанова застосовується до лікарських засобів, що реєструються та реалізуються в Україні для медичного застосування, а також вона поширюється на створення та підтримку системи фармаконагляду, проведення аудиту, інспектування у ЄС, аудиту системи фармаконагляду уповноваженою установою в УкраїніN, створення оновленої системи управління ризиками щодо лікарських засобів (якщо застосовано), обмін даних з безпеки, проведення аналізу отриманих у післяреєстраційний період даних з безпеки, менеджмент сигналів, генерацію та подання регулярно оновлюваних звітів з безпеки, комунікації з питань безпеки з медичними працівниками та пацієнтами.
Ця настанова рекомендується для суб’єктів господарювання, які займаються розробкою, поданням заявок на реєстрацію/перереєстрацію лікарських засобів на території України, незалежно від відомчого підпорядкування та форми власності, для відповідних заявників (власників реєстраційних посвідчень) лікарських засобів, які реєструється та реалізуються в Україні, для науково-експертних організацій та регуляторних органів.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128 (Директива 2001/83/ЄC Європейського Парламенту та Ради від 6 листопада 2001 р. про кодекс Співтовариства відносно лікарських препаратів, призначених для застосування людям//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128) [1].
Наказ Міністерства охорони здоров’я України від 27 грудня 2006 р. № 898 «Про затвердження Порядку здійснення фармаконагляду» зареєстровано в Міністерстві юстиції України 29 січня 2007 р. за № 73/13340 (у редакції наказу МОЗ України від 26 вересня 2016 р. № 996) (далі — Порядок здійснення фармаконаглядуN) [2];
EMA/541760/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module I — Pharmacovigilance systems and their quality systems» (Настанова з належної практики фармаконагляду (ННПФ). Модуль І — Фармаконагляд та його система якості. Перегляд 2*) [33].
EMA/816573/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module II — Pharmacovigilance system master file» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІІ — Майстер-файл системи фармаконагляду. Перегляд 2*) [34].
EMA/119871/2012 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module ІІI — Pharmacovigilance inspections» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІІІ — Інспекції системи фармаконагляду. Перегляд 1*) [35].
EMA/228028/2012 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module IV — Pharmacovigilance audits» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІV — Аудит з фармаконагляду. Перегляд 1*) [36].
EMA/838713/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module V — Risk management systems» (Настанова з належних практик фармаконагляду (ННПФ). Модуль V — Системи управління ризиками. Перегляд 2*) [37].
EMA/873138/2011 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module VI — Management and reporting of adverse reactions to medicinal products» (Настанова з належних практик фармаконагляду (ННПФ). Модуль VI — Управління та звітування про побічні реакції лікарських засобів. Перегляд 1*) [38].
EMA/816292/2011 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module VII — Periodic safety update report» (Настанова з належних практик фармаконагляду (ННПФ) Модуль VII — Регулярно оновлюваний звіт з безпеки. Перегляд 1*) [39].
EMA/813938/2011 Rev 2* Corr** «Guideline on good pharmacovigilance practices (GVP). Module VIII — Post-authorisation safety studies» (Настанова з належних практик фармаконагляду (ННПФ). Модуль VIІІ — Післяреєстраційне дослідження з безпеки. Перегляд 2* Corr**) [41].
EMA/827661/2011 «Guideline on good pharmacovigilance practices (GVP). Module IX — Signal management» (Настанова з належних практик фармаконагляду (ННПФ). Модуль IX — Управління сигналом) [43].
EMA/169546/2012 «Guideline on good pharmacovigilance practices (GVP). Module X — Additional monitoring» (Настанова з належних практик фармаконагляду (ННПФ). Модуль X — Додатковий моніторинг) [44].
EMA/118465/2012 «Guideline on good pharmacovigilance practices (GVP). Module XV — Safety communication» (Настанова з належних практик фармаконагляду (ННПФ). Модуль XV — Процес комунікації з питань безпеки) [45].
EMA/204715/2012 Rev 2* «Guideline on good pharmacovigilance practices (GVP). Module XVI — Risk minimisation measures: selection of tools and effectiveness indicators» (Настанова з належних практик фармаконагляду (ННПФ). Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності. Перегляд 2*) [46].
EMA/168402/2014 Corr* «Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations II: Biological medicinal products» ((Настанова з належних практик фармаконагляду (ННПФ). Частина XIIІ — Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби [47].
ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів/І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003 [21].
Настанова СТ-Н МОЗУ 42-1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення/М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005 [22].
Терміни та визначення понять
Нижче наведені терміни, вжиті у цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1, 2, 6, 7, 16] (див. національний додаток «Бібліографія»). Визначення цих термінів можуть відрізнятися в інших нормативних документах або терміни можуть мати інші значенняN.
Аналітичний набір даних (Analytical dataset)
Мінімальний набір даних, що вимагається для виконання статистичних аналізів, що призводить до отримання результатів, необхідних для досягнення первинної(их) цілі(ей) дослідження (ст. 1 Директиви 2001/83/ЄС [1]).
Аудит (Audit)
Систематичний, незалежний і задокументований процес отримання доказів аудиту і об’єктивного їх оцінювання з метою визначення ступеня виконання критеріїв аудиту (див. ISO 19011 (3.1); ДСТУ ISO 19011:2003 (3.1)2).
2International Organisation for Standardisation (ISO); Державний стандарт України (ДСТУ)
Важлива відсутня інформація (Important missing information)
Критичні прогалини в знаннях щодо специфічних питань безпеки лікарського засобу, дозволеного до медичного застосування, або в особливих популяціях, які застосовують лікарський засіб (див. додаток IV, настанова ICHE2С(R2)[28]).
Див. також «відсутня інформація», «питання з безпеки».
Важливий виявлений ризик та важливий потенційний ризик (Important identified risk and Important potential risk)
Виявлений ризик або потенційний ризик, який може вплинути на співвідношення ризик-користь лікарського засобу чи мати наслідки для здоров’я населення (див. настанову ICH-E2F) [22].
Що становить важливий ризик, буде залежати від декількох факторів, включаючи вплив на індивіда, серйозність ризику та вплив на здоров’я населення. Як правило, будь-який ризик, який, імовірно, буде включений до розділів «Протипоказання», «Належні заходи безпеки», «Особливі застереження» інструкції для медичного застосування, повинен вважатися важливим (див. додаток IV, настанова ICH-E2С(R2) [28]).
Див. також «співвідношення ризик-користь», «виявлений ризик», «потенційний ризик», «питання безпеки».
Важливий потенційний ризик (Important potential risk)
Див. «важливий виявлений ризик» та «важливий потенційний ризик».
Валідація сигналу
Процес оцінки даних, що підтримують виявлений сигнал, для підтвердження того, що вони містять достатній доказ існування нового потенційного причинно-наслідкового зв’язку або нового аспекту відомого зв’язку, а тому обґрунтовують подальший аналіз сигналу (ст. 21(1) ІП 520/2012 [6]).
Валідований сигнал (Validated signal)
Сигнал, для якого в процесі валідації даних, що підтримують виявлений сигнал, було підтверджено, що вони містять достатній доказ наявності нового потенційного причинно-наслідкового зв’язку або новий аспект відомого зв’язку, а тому обґрунтовує подальший аналіз сигналу (ст. 21(1) ІП 520/2012 [6]).
Див. також «сигнал».
Високотехнологічний (біотехнологічний) лікарський засіб (Advance therapy medicinal product (АТМР)
Лікарські засоби, що містять діючі речовини, отримані за допомогою методів біотехнології, таких як: генно-інженерна технологія, клітинна інженерія, гібридомні технології, інженерна ензимологія та інженерна імунологія тощо (наказ Міністерства охорони здоров’я України від 26 серпня 2005 р. № 426 «Порядок проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення», зареєстровано в Міністерстві юстиції України 19 вересня 2005 р. за № 1069/11349 (у редакції наказу МОЗ України від 23 липня 2015 р. № 460)(далі — Порядок проведення експертизи реєстраційних матеріалівN [7])).
Виявлений ризик (Identified risk)
Несприятливе явище, між виникненням якого та прийомом лікарського засобу існує причинно-наслідковий зв’язок, що був підтверджений. Наприклад:
побічна реакція, що була належним чином продемонстрована в доклінічних дослідженнях і підтверджена клінічними даними;
побічна реакція, що спостерігається у належним чином спланованих клінічних дослідженнях або епідеміологічних дослідженнях, у яких величина різниці параметрів, що розглядаються, у порівнянні з групою компаратора вказує на причинно-наслідковий зв’язок між виникненням даної побічної реакції та прийомом лікарського засобу;
побічна реакція, по відношенню до якої існують належним чином задокументовані спонтанні повідомлення, де причинно-наслідковий зв’язок між її виникненням та прийомом лікарського засобу підтверджується часовим зв’язком і біологічною імовірністю, наприклад, анафілактичні реакції або реакції у місці введення.
У клінічних дослідженнях компаратором може бути плацебо, діюча речовина або відсутність прийому будь-яких лікарських засобів.
Відсутня інформація (Missing information)
Інформація про безпеку лікарського засобу відсутня на момент подання плану управління ризиками, яка обмежує дані з безпеки для прогнозування безпеки лікарського засобу на ринку.
Приклади відсутньої інформації включають популяції, які не вивчалися (наприклад, вагітні або пацієнти з порушеннями функції нирок), існування високої вірогідності застосування не за показанням.
Див. також «застосування не за показанням».
Вперше виявлений сигнал (Newly identified signal)
У регулярних звітах з оцінки співвідношення користь-ризик сигнал, вперше виявлений у звітний період, який спонукає до подальших дій або подальшої його оцінки (див. додаток IV, настанова ICH-E2С(R2) [28]).
Під це визначення також підпадає раніше закритий сигнал, для якого у звітній період стала відомою нова інформація, що спонукає до подальших дій або подальшої його оцінки (див. додаток IV, настанова ICH-E2С(R2) [28]).
Це визначення також застосовується до регулярно оновлюваних звітів з безпеки.
Див. також «сигнал», «закритий сигнал».
Вплив лікарського засобу, пов’язаний з трудовою діяльністю (Occupational exposure to a medicinal product)
Стосовно повідомлень про випадки підозрюваних побічних реакцій, вплив лікарського засобу в результаті професійної або непрофесійної діяльності.
Генеричний лікарський засіб; синоніми: генерик, взаємозамінний (Generic medicinal product)
Лікарський засіб, який має такий самий кількісний та якісний склад діючих речовин і таку саму лікарську форму, що й референтний препарат, та чия взаємозамінність з референтним препаратом доведена на підставі відповідних досліджень (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Гомеопатичний лікарський засіб (Homeopathic medicinal product)
Будь-який лікарський засіб, виготовлений із продуктів, субстанцій або складових, які називаються гомеопатичною сировиною, відповідно до процедури виготовлення гомеопатичного лікарського засобу, описаної в Державній фармакопеї України (далі — ДФУ), або Європейській фармакопеї, або, у разі відсутності такого опису, у Німецькій гомеопатичній фармакопеї (GHP), Гомеопатичній фармакопеї США (HPUS), Британській гомеопатичній фармакопеї (BHP), Гомеопатичній фармакопеї Шваб (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Дані аудиту (Audit findings)
Результати оцінювання зібраних доказів аудиту за критеріями аудиту (див. ISO 19011 (3.4); ДСТУ ISO 19011:2003 (3.4)3).
Доказ аудиту необхідний для підтвердження результатів оцінювання аудитором, тобто висновку та звіту аудитора. Він кумулятивний по суті та, як правило, отримується з процедур аудиту, що виконуються під час проведення аудиту.
Див. також «аудит».
Дата закриття бази даних для складання наступного звіту (Data lock point)
Для регулярно оновлюваних звітів з безпеки (PSUR) — кінцева дата збору даних, включених у PSUR, визначена на основі міжнародної дати народження.
Для регулярних звітів з оцінки співвідношення користь/ризик (PBRER) — кінцева дата збору даних, включених у PBRER, визначена на основі міжнародної дати народження (див. додаток IV, настанова ICH-E2С(R2) [28]).
Для оновлюваних звітів з безпеки препарату, що знаходяться в стадії розробки (DSUR) — кінцева дата збору даних, включених у DSUR, визначена на основі міжнародної дати народження препарату, що розробляється (див. настанова ICH-E2F[22]).
Дата включає день та місяць (див. настанову ICH-E2F[22]).
Див. також «регулярно оновлюваний звіт з безпеки (PSUR), «оновлюваний звіт з безпеки препарату, що знаходиться в стадії розробки (DSUR)», «міжнародна дата народження», «міжнародна дата народження препарату, що знаходиться в стадії розробки».
Дата, коли розпочато дослідження (Date at which a study commences)
Дата початку збору даних. (ст. 1 Директиви 2001/83/ЄС [1]).
Діяльність з мінімізації ризиків; синонім: заходи з мінімізації ризиків (Risk minimisation activity; synonym: Risk minimisation measure)
Дії з метою запобігання чи зниження ймовірності розвитку побічної реакції, пов’язаної із застосуванням лікарського засобу, або з метою зниження серйозності у разі її виникнення.
Ця діяльність може складатися з рутинних заходів з мінімізації ризиків (наприклад інформація про препарат) або додаткових заходів з мінімізації ризиків (наприклад повідомлення/навчальні матеріали для працівників з медичною та фармацевтичною освітою або пацієнтів).
Довідкова інформація з безпеки (Reference safety information)
У регулярних звітах з оцінки користь-ризик лікарських засобів, вся відповідна інформація з безпеки, що міститься в довідковій інформації про препарат (наприклад в переліку основних даних), підготовленій власником реєстраційного посвідчення, та яку власник реєстраційного посвідчення вимагає реєструвати у всіх країнах, де він продає препарат, за виключенням, коли локальний регуляторний орган вимагає внесення спеціальних змін (див. додаток IV, настанова ICH-E2С(R2) [28]).
Це інформація, що міститься в довідковій інформації про препарат власника реєстраційного посвідчення для регулярного звіту з оцінки користі-ризику. У разі коли довідкова інформація про препарат є переліком основних даних, то довідкова інформація з безпеки є основною інформацією з безпеки (див. додаток IV, настанова ICH-E2С(R2) [28]).
Досліджуваний лікарський засіб (Investigational medicinal product)
Лікарська форма активної субстанції або плацебо, що вивчається або використовується для порівняння у клінічних випробуваннях, включаючи препарати, на які вже видане реєстраційне посвідчення, але вони використовуються або виготовляються (складені або упаковані) в інший спосіб порівняно із зареєстрованою лікарською формою, або використовуються за незареєстрованими показами, або ж використовуються для отримання додаткової інформації про зареєстровану форму лікарського засобу (наказ Міністерства охорони здоров’я України від 23 вересня 2009 р. № 690 «Про затвердження Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань і Типового положення про комісії з питань етики», зареєстровано в Міністерстві юстиції України 29 жовтня 2009 р. за № 1010/17026 (у редакції наказу МОЗ України 12 липня 2012 р. № 523) (далі — Порядок проведення клінічних випробуваньN [16]).
Див. також «клінічне дослідження».
Дотримання вимог якості (Quality adherence)
Виконання завдань та обов’язків відповідно до встановлених вимог якості у фармаконагляді (ст. 8(3) ІП 520/2012 [6]).
Див. також «вимоги щодо якості».
Завершене клінічне випробування (Completed clinical trial)
Дослідження з наявним заключним звітом клінічного випробування (див. настанову ICH-E2F [22]).
Див. також «клінічне дослідження».
Закінчення збору даних (End of data collection)
Дата, починаючи з якої повністю наявний аналітичний набір даних (ст. 37(2) ІП 520/2012 [6]).
Закритий сигнал (Closed signal)
У регулярних звітах з оцінки співвідношення користь-ризик — це сигнал, оцінка якого була завершена під час звітного періоду (див. додаток IV, настанова ICHE2C(R2) [28]).
Це визначення також застосовується у регулярно оновлюваних звітах з безпеки.
Див. також «сигнал».
Запитувані джерела повідомлень про індивідуальні випадки, пов’язані з безпекою (Solicited sources of individual case safety reports)
Системи збору даних, з обов’язковою їх фіксацією, що включають клінічні випробування, реєстри, післяреєстраційні програми застосування препаратів поіменованими пацієнтами, інші програми підтримки пацієнтів та управління захворюваннями, опитування пацієнтів чи медичних працівників, або збір інформації щодо ефективності та прихильності пацієнта до терапії. Запитувані повідомлення не повинні вважатися спонтанними, однак вони класифікуються як повідомлення про індивідуальні випадки, пов’язані з безпекою, з досліджень, а тому повинні містити відповідну оцінку причинно-наслідкового зв’язку працівника з медичною освітою або власника реєстраційного посвідчення (див. додаток IV, настанова ICH-E2D[25]).
Див. також «клінічне дослідження», «післяреєстраційне дослідження з безпеки», «неінтервенційне дослідження».
Застосування не за показанням (Off-label use)
Ситуації, коли лікарський засіб навмисно застосовується в медичній практиці не відповідно до затвердженої інструкції для медичного застосування.
Зловживання (Abuse)
Постійне чи спорадичне навмисне надмірне застосування лікарського засобу, що супроводжується виникненням шкідливих фізичних або психологічних ефектів (ст. 1 Директиви 2001/83/ЄС [1]).
Значна зміна до протоколу дослідження (Substantial amendment to the study protocol)
Зміна до протоколу, що, ймовірно, буде мати вплив на безпеку, фізичне або психічне здоров’я учасників дослідження, або зміна, що може вплинути на результати дослідження та їх інтерпретацію, така як зміни до первинних або вторинних цілей дослідження, досліджуваної популяції, розміру вибірки, визначень основної експозиції, результатів та факторів втручання та аналітичного плану, що описані у протоколі дослідження (ст. 1 Директиви 2001/83/ЄС [1]).
Керівництво (Upper management)
Група осіб, що відповідає за вище виконавче управління організацією. Належність до цієї групи визначається структурою управління організації. Керівництво — це, як правило, група осіб, тому керівник організації несе повну відповідальність за гарантію дотримання організацією відповідного законодавства.
Клінічне випробування (Clinical trial)
Клінічне випробування (дослідження) лікарського засобу — науково-дослідницька робота, метою якої є будь-яке дослідження за участю людини як суб’єкта дослідження, призначене для виявлення або підтвердження клінічних, фармакокінетичних, фармакодинамічних та/або інших ефектів, у тому числі для вивчення всмоктування, розподілу, метаболізму та виведення одного або кількох лікарських засобів та/або виявлення побічних реакцій на один або декілька досліджуваних лікарських засобів з метою оцінки його (їх) безпеки та/або ефективності (Порядок проведення клінічних випробуваньN [16]).
Див. також «поточне клінічне дослідження», «звершене клінічне дослідження», «досліджуваний лікарський засіб».
Лікарський засіб (Medicinal product)
Будь-яка речовина або комбінація речовин (одного або декількох АФІ та допоміжних речовин), що має властивості та призначена для лікування або профілактики захворювань у людей, чи будь-яка речовина або комбінація речовин (одного або декількох АФІ та допоміжних речовин), яка може бути призначена для запобігання вагітності, відновлення, корекції чи зміни фізіологічних функцій у людини шляхом здійснення фармакологічної, імунологічної або метаболічної дії або для встановлення медичного діагнозу (ст. 1 Закону України «Про лікарські засоби»).
Лікарський засіб рослинного походження (Herbal medicinal product)
Будь-який лікарський засіб, що містить виключно діючу(і) речовину(и) з однієї або більше рослинних субстанцій, або один чи більше рослинних препаратів, або одну чи більше рослинних субстанцій у комбінації з одним чи більше рослинним препаратом (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Рослинні субстанції — цілі, подрібнені або порізані рослини, частини рослин, водоростей, грибів, лишайників у необробленій, зазвичай засушеній формі, іноді свіжі. Певні витяжки з рослин (наприклад смоли), не призначені для лікування, також вважаються рослинними субстанціями. Рослинні субстанції чітко визначаються морфологічною частиною рослини, що використовується, та її ботанічною назвою відповідно до біномної системи (рід, вид, різновид та джерело) (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Рослинні препарати — препарати, одержані у результаті обробки рослинних субстанцій шляхом витягування, дистиляції, віджимання, подрібнення, очищення, концентрації та ферментації. Сюди входять потовчені або порошкоподібні рослинні субстанції, настойки, екстракти, ефірні олії, віджаті соки та оброблені витяжки (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Лікарські засоби, отримані з крові або плазми людини (Medicinal product derived from human blood or human plasma)
Лікарські засоби на основі компонентів крові, вироблені промисловим способом на державних або приватних підприємствах; такі лікарські засоби включають, зокрема, альбумін, фактори згортання крові та імуноглобуліни людського походження (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Маркування (Labelling)
Інформація на первинній або вторинній упаковці (ст. 1(25) Директиви 2001/83/ЄС [1]).
Коментар ДЕЦ: маркування лікарського засобу (Labelling) — інформація на первинній (внутрішній) або вторинній (зовнішній) упаковці.
Майстер-файл системи фармаконагляду (Pharmacovigilance system master file — PSMF)
Документ, що містить опис системи фармаконагляду, яка використовується заявником щодо одного або декількох лікарських засобів, вакцин, туберкуліну (Порядок здійснення фармаконаглядуN [2]).
Див. також «система фармаконагляду».
Міжнародна дата народження лікарського засобу, вакцини, туберкулінуN (International birth date — IBD)
Дата видачі заявнику першого дозволу на продаж лікарського засобу, вакцини, туберкуліну в будь-якій країні світу (Порядок здійснення фармаконаглядуN [2]).
Міжнародна дата народження лікарського засобу, що знаходиться в стадії розробки (Development international birth date — DIBD)
Дата першого схвалення (або реєстрації) для проведення інтервенційного клінічного випробування у будь-якій країні (див. настанову ICH-E2F [22]).
Мінімальні критерії для подання повідомлень (Minimum criteria for reporting)
Що стосується повідомлень про випадки підозрюваних побічних реакцій, мінімумом елементів даних для випадку є: ідентифікований повідомник, ідентифікований пацієнт, побічна реакція та підозрюваний лікарський засіб (див. додаток IV, настанова ICH-E2D [25]).
Що стосується перевірки (валідації) повідомлень про індивідуальні випадки, пов’язані з безпекою, див. модуль VI.
Див. також звіт з безпеки для окремого випадку.
Назва лікарського засобу (Name of the medicinal product)
Назва, дана лікарському засобу, яка може бути як вигаданою заявником (виробником), так і загальноприйнятою або науковою, що може супроводжуватися назвою торгової марки або найменуванням заявника (виробника) (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Загальноприйнята назва — це міжнародна непатентована назва (далі — МНН) діючої речовини, рекомендована Всесвітньою організацією охорони здоров’я (далі — ВООЗ), або за відсутності такої звичайна загальноприйнята назва (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Повна назва лікарського засобу — це назва лікарського засобу з силою дії та лікарською формою.
Належна практика фармаконагляду (НПФ) (Good pharmacovigilance practices — GVP)
Настанова для здійснення фармаконагляду в Україні, розроблена на підставі діючих підзаконних та нормативних актів, гармонізована з Європейським законодавством.
Незавершене клінічне випробування (Ongoing clinical trial)
Випробування, яке не має звіту про завершення клінічного випробування (див. настанова ICH-E2F [22], том 10 правил регулювання лікарських засобів в ЄС).
Див. також «клінічне випробування», «завершене клінічне випробування».
Неінтервенційне дослідження (Non-interventional trial; synonym: Noninterventional study)
Дослідження, в якому лікарські засоби призначаються звичайним способом відповідно до затвердженої інструкції з медичного застосування. Залучення пацієнта в групу з визначеним методом лікування в протоколі клінічного дослідження заздалегідь не передбачено, а призначення лікарського засобу диктується сучасною практикою і не залежить від рішення включити пацієнта у випробування. Не застосовують додаткових діагностичних або моніторингових процедур щодо пацієнтів, а для аналізу зібраних даних використовують епідеміологічні методи (Порядок проведення клінічних випробуваньN [16]).
Отже, дослідження буде неінтервенційним за виконання таких вимог:
- лікарський засіб призначається звичайним способом відповідно до затвердженої інструкції для медичного застосування;
- залучення пацієнта в групу з визначеним методом лікування не передбачено заздалегідь у протоколі дослідження, а диктується сучасною практикою і не залежить від рішення включити пацієнта у випробування; та
- пацієнтам не застосовують ніяких додаткових діагностичних або моніторингових процедур, а для аналізу зібраних даних використовуються епідеміологічні методи.
Неінтервенційні дослідження визначаються за методологічним підходом, що використовується, а не за науковими цілями. Неінтервенційні дослідження включають дослідження баз даних або перегляд записів, коли всі явища, що представляють інтерес, вже сталися (включаючи дослідження випадок-контроль, перехресні, когортні та інші дослідження, дані яких використовуються вторинно). Неінтервенційні дослідження також включають дослідження зі збором первинних даних (наприклад, проспективні обсерваційні дослідження та дослідження реєстрів, у яких збираються дані з рутинної клінічної практики), за умови дотримання вищезазначених вимог. У цих дослідженнях інтерв’ю, заповнення анкет та забір зразків крові можуть проводитися в рамках звичайної клінічної практики.
Неінтервенційні дослідження регулюються Порядком проведення клінічних випробуваньN [16].
Непередбачена побічна реакція (Unexpected adverse reaction)
Побічна реакція, характер або тяжкість проявів якої не узгоджується з наявною інформацією про лікарський засіб, вакцину, туберкулін в інструкції для медичного застосування/короткій характеристиці лікарського засобу, вакцини, туберкуліну (Порядок здійснення фармаконаглядуN [2]).
Це поняття також включає реакції, притаманні класу, які зазначені в інструкції для медичного застосування, однак які не описуються як такі, що спостерігалися при застосуванні даного препарату.
Див. також «Інструкція для медичного застосування».
Неправильне застосування (Misuse)
Навмисне та неправильне застосування лікарського засобу не у відповідності до затвердженої інформації про лікарський засіб.
Обґрунтоване повідомлення про індивідуальний випадок, пов’язаний з безпекою (Valid individual case safety report)
Див. «Повідомлення про індивідуальний випадок, пов’язаний з безпекою».
Оновлюваний звіт з безпеки досліджуваного лікарського засобу, що перебуває у стадії розробки (DSUR — Development Safety Update Report)
Формат та зміст оновлюваного звіту з безпеки досліджуваного лікарського засобу, що перебуває у стадії розробки відповідно до вимог Порядку проведення клінічних випробуваньN [16], додаток 15.
Основна інформація з безпеки заявника (Company Core Safety Information — CCSI)
Документ, складений заявником, що містить усю відповідну інформацію з безпеки, яка є складовою переліку основних даних заявника. Основна інформація з безпеки заявника використовується при складанні звітності для визначення, зафіксовано чи ні побічну реакцію в переліку основних даних заявника. Дані основної інформації з безпеки заявника не використовуються для визначення, чи є побічна реакція передбаченою чи непередбаченою, для подання повідомлення про випадок побічної реакції (Порядок здійснення фармаконаглядуN [2]).
Див. також перелік основних даних заявника (CCDS).
Передозування (Overdose)
Введення кількості лікарського засобу, що дається за прийом або кумулятивно, яка є більшою максимальної рекомендованої дози відповідно до інструкції для медичного застосування лікарського засобу. Слід завжди додавати клінічне обґрунтування.
Застосування одноразово чи кумулятивно дози лікарського засобу, що перевищує максимальну рекомендовану дозу, зазначену в діючій інструкції для медичного застосування лікарського засобу. У таких випадках завжди повинна проводитися клінічна оцінка.
Перелік основних даних заявника (Company Core Data Sheet — CCDS)
Документ, складений заявником, що містить інформацію про лікарський засіб, вакцину, туберкулін щодо безпеки пропонованих показань до застосування, сили дії, особливостей застосування, фармакологічних властивостей тощо (Company Core Data Sheet (ССDS)) (див. додаток IV, настанова ICH-E2C(R2) [28]; (Порядок здійснення фармаконаглядуN [2]).
Див. також основну інформацію з безпеки заявника (CCSI).
Першоджерело (Primary source)
Першоджерело інформації про підозрювану(і) побічну(і) реакцію(ї) — це особа, яка повідомила про випадок виникнення побічної реакції. Деякі першоджерела, такі як працівники з медичною та/або фармацевтичною освітою та/або споживачі, можуть надавати інформацію про один і той же випадок. У такій ситуації, згідно з настановою ICH-E2B(R2)[26], слід надати всі відомості про першоджерело, включаючи його кваліфікацію, у розділі «Першоджерело» повідомлення про побічну реакцію, повторивши цей розділ стільки разів, скільки буде необхідно відповідно до вимог ICH-E2B(R2)[26] (див. додаток IV GVP).
Згідно з настановою ICH-E2D[25] (див. додаток IV GVP):
- працівник з медичною та/або фармацевтичною освітою (healthcare professional) — це особа, яка має медичну кваліфікацію, таку як лікар, стоматолог, фармацевт, медсестра, або іншу, передбачену місцевим законодавством;
- споживач — це особа без медичної та/або фармацевтичної освіти, така як пацієнт, юрист, родич пацієнта, знайомий чи опікун.
Медична документація (наприклад дані лабораторних чи інших досліджень), надана споживачем, що підтверджує випадок виникнення підозрюваної побічної реакції, або яка вказує, що ідентифікований працівник з медичною та/або фармацевтичною освітою підозрює обґрунтовану ймовірність існування причинно-наслідкового зв’язку між прийомом лікарського засобу та небажаним явищем, є достатньою умовою для того, щоб вважати спонтанне повідомлення про побічну реакцію підтвердженим працівником з медичною та/або фармацевтичною освітою.
Якщо споживач повідомив про випадок розвитку декількох побічних реакцій і при цьому хоч одна з них медично підтверджена, то таке повідомлення має бути задокументоване як спонтанне повідомлення, підтверджене працівником з медичною та/або фармацевтичною освітою, та надаватися в належні строки. Так само, якщо повідомлення отримане від пацієнта, його знайомого, родича чи опікуна, який має медичну освіту, воно також має розглядатися як спонтанне повідомлення, підтверджене працівником з медичною та/або фармацевтичною освітою (Модуль 6).
Післяреєстраційне дослідження з безпеки та ефективності лікарського засобу, вакцини, туберкуліну (Post-authorisation safety study (PASS), post-authorisation efficacy studies (PAESs))
Будь-яке післяреєстраційне дослідження з безпеки та ефективності дозволеного до медичного застосування лікарського засобу, вакцини, туберкуліну, що проводиться з метою визначення, характеристики чи оцінки загрози безпеці, підтвердження профілю безпеки лікарського засобу, вакцини, туберкуліну та/або оцінки ефективності заходів управління ризиками (Порядок здійснення фармаконаглядуN [2]).
План аудиту (Audit plan)
Опис видів діяльності та заходів з проведення аудиту (див. ISO 19011 (3.12); ДСТУ ISO 19011:2003 (3.12)3).
Див. також «аудит».
3International Organisation for Standardisation (ISO); Державний стандарт України (ДСТУ).
План управління ризиками (Risk management plan — RMP)
Детальний опис системи управління ризиками (Порядок здійснення фармаконаглядуN [2]).
Див. також «система управління ризиками», «діяльність з мінімізації ризиків».
Побічна реакція; синоніми: побічна реакція лікарського засобу, підозрювана побічна реакція (лікарського засобу), побічний ефект, небажаний ефект (Adverse reaction; synonyms: Adverse drug reaction (ADR), Suspected adverse (drug) reaction, Adverse effect, Undesirable effect)
Будь-яка ненавмисна і шкідлива реакція на лікарський засіб; будь-яка ненавмисна і шкідлива реакція на вакцину, туберкулін, якщо вона спричинена чи прискорена активним компонентом (одним з інших компонентів) або пов’язана з порушеннями, що виникають у процесі виробництва вакцини, туберкуліну, включаючи пристрій для введення, що надається виробником (Порядок здійснення фармаконаглядуN [2]).
Реакція у цьому контексті означає, що причинно-наслідковий зв’язок між лікарським засобом та побічним явищем є принаймні обґрунтованою (резонною) можливістю (див. додаток IV, настанова ICH-E2A [29]).
Побічна реакція може виникнути при застосуванні лікарського засобу відповідно чи невідповідно до інструкції для медичного застосування або у разі впливу лікарського засобу, пов’язаного з професійною діяльністю (Директива 2001/83/ЄС ст. 101(1)[1]). Застосування невідповідно до інструкції для медичного застосування включає застосування не за показаннями, передозування, неправильне застосування, зловживання та помилки, пов’язані із застосуванням лікарського засобу.
Дивись також «побічне явище», «серйозна побічна реакція», «непередбачена побічна реакція», «застосування не за показаннями», «передозування», «неправильне застосування лікарського засобу», «зловживання лікарським засобом», «вплив лікарського засобу, пов’язаний з професійною діяльністю».
1У межах клінічних випробувань, побічні реакції — у межах передреєстраційного клінічного випробування нового лікарського засобу або його вивчення за новим використанням, особливо в разі, якщо терапевтичні дози лікарського засобу не встановлені, до побічних реакцій на лікарський засіб треба відносити всі негативні та непередбачені відповіді на введення лікарського засобу будь-якої дози. Термін «відповідь на введення лікарського засобу» означає, що існує принаймні припустима вірогідність причинно-наслідкового зв’язку між застосуванням лікарського засобу та побічною реакцією, тобто взаємозв’язок не можна виключити (Порядок проведення клінічних випробуваньN [16]).
Побічне явище (Adverse event; synonym: Adverse experience)
Будь-який несприятливий медичний прояв у досліджуваного, який не обов’язково має причинний зв’язок із застосуванням лікарського засобу. Побічним явищем може бути будь-який небажаний та непередбачуваний прояв (у тому числі зміни лабораторних даних), симптом або захворювання, які збігаються за часом із застосуванням (досліджуваного) лікарського засобу, незалежно від того, пов’язано це з прийомом (досліджуваного) лікарського засобу чи ні (Порядок проведення клінічних випробуваньN [16]).
Повідомлення про індивідуальний випадок, пов’язаний з безпекою; синонім: повідомлення про побічну реакцію (лікарського засобу) (Individual case safety report (ICSR); synonym: Adverse (drug) reaction report)
Повідомлення, що надається повідомником, про підозрювану(ні) побічну(ні) реакцію(ії), що виникла(и) у окремого пацієнта у певний момент часу.
Див. також «мінімальні критерії для подання повідомлень».
Причинно-наслідковий зв’язок; синонім: причинно-наслідковий зв’язок між клінічними проявами будь-якої побічної реакції/несприятливої події після імунізації/туберкулінодіагностики та застосуванням лікарського засобу, вакцини, туберкуліну (Causality)
Ступінь, який визначається прийнятним методом (якісна методика Всесвітньої організації охорони здоров’я, шкала Наранжо, бінарний метод тощо) за певними критеріями та вказує на взаємопов’язаність/взаємозв’язок побічної реакції/несприятливої події після імунізації/туберкулінодіагностики, що спостерігається, із застосуванням лікарського засобу, вакцини, туберкуліну (Порядок здійснення фармаконаглядуN [2]).
Відповідно до настанови ICH-E2A [29] (див. додаток IV GVP), термін «побічна реакція» передбачає як мінімум обґрунтовану можливість існування причинно-наслідкового зв’язку між застосуванням підозрюваного лікарського засобу та розвитком небажаного явища. Побічна реакція, на відміну від побічного явища, характеризується наявністю підозри існування причинно-наслідкового зв’язку між прийомом лікарського засобу та небажаним ефектом. Як зазначено в настанові ICH-E2D[25] (див. додаток IV GVP), з метою дотримання нормативно-правових вимог щодо звітності про побічні реакції, якщо про побічне явище було поінформовано у вигляді спонтанного повідомлення, навіть якщо зв’язок невідомий або не вказаний, таке явище підпадає під поняття «побічна реакція». Тому всі спонтанні повідомлення, надані працівниками з медичною та/або фармацевтичною освітою1 чи споживачами, вважаються підозрюваними побічними реакціями, оскільки першоджерело повідомлення підозрювало існування причинно-наслідкового зв’язку, за винятком випадків, коли джерело повідомлення однозначно заявляє, що виникнення побічного явища не пов’язане із застосуванням лікарського засобу або вказаний вище зв’язок виключений.
Потенційний ризик (Potential risk)
Небажаний випадок, щодо якого існує певна підозра причинно-наслідкового зв’язку з даним лікарським засобом, але цей зв’язок не був підтверджений (див. настанову ICH-E2F [22], том 10 правил регулювання лікарських засобів в ЄС).
Приклади включають:
- неклінічні токсикологічні дані, які не спостерігалися або не були спростовані в клінічних дослідженнях;
- побічні явища, що спостерігаються у клінічних дослідженнях або епідеміологічних дослідженнях, у яких величина різниці параметрів, які представляють інтерес, порівняно з групою компаратора (плацебо або активна субстанція, або неекспонована група) викликає підозру, але вона недостатньо велика, щоб передбачити причинний зв’язок;
- сигнал, що виникає з системи спонтанних повідомлень про побічні реакції;
- явище, яке, як відомо, пов’язане з іншими активними субстанціями того ж класу, або яке, як передбачається, може виникнути на основі властивостей лікарського засобу (див. настанову ICH-E2F[22], том 10 правил регулювання лікарських засобів в ЄС).
Див. також «побічне явище», «сигнал».
Поточний сигнал (Ongoing signal)
У регулярних звітах з оцінки співвідношення користь-ризик сигнал, який все ще аналізується на момент кінцевого терміну подання даних (див. додаток IV, настанова ICH-E2С(R2) [28]).
Це визначення також застосовується до регулярно оновлюваних звітів з безпеки.
Див. також «сигнал», «кінцевий термін подання даних».
Початок збору даних (Start of data collection)
Дата, з якої інформація про першого суб’єкта дослідження вперше вноситься в набір даних дослідження, або у випадку повторного використання даних, дата, з якої починається витяг даних (ст. 37(1) ІП 520/2012[6]). Простий підрахунок у базі даних для підтримки розробки протоколу дослідження, наприклад для інформування про розмір вибірки та статистичну точність дослідження, не є частиною цього визначення.
Працівники з медичною та/або фармацевтичною освітою (Healthcare professional)
Стосовно повідомлень про підозрювані побічні реакції: працівники з медичною та/або фармацевтичною освітою — це лікарі, провізори, фельдшери, акушери, фармацевти та медичні сестри (Порядок здійснення фармаконаглядуN [2]).
Проблема з безпеки (Safety concern)
Важливий виявлений ризик, важливий потенційний ризик або відсутня інформація.
Програма аудиту (Audit programme)
Один чи декілька аудитів, запланованих на конкретний період часу і спрямованих на досягнення конкретної мети (див. ISO 19011 (3.11); ДСТУ ISO 19011:2003 (3.11)5).
Див. також «аудит».
5International Organisation for Standardisation (ISO); Державний стандарт України (ДСТУ).
Процес управління сигналом (Signal management process)
Діяльність з виявлення сигналу, валідації сигналу, підтвердження сигналу, аналізу та пріоритезації сигналу, оцінки сигналу та рекомендації дій (ст. 21(1) ІП 520/2012 [6]).
Отже, це діяльність, що здійснюється з метою визначення, чи існують нові ризики, пов’язані з активною субстанцією або лікарським засобом, або чи змінилися відомі ризики на основі оцінки повідомлень про індивідуальні випадки, пов’язані з безпекою, сукупних даних з систем активного нагляду або досліджень, інформації з літератури чи інших джерел даних.
Див. також «валідація сигналу».
Регулярно оновлюваний звіт з безпеки лікарського засобу, вакцини, туберкуліну (Periodic safety update report — PSUR)
Письмовий звіт, що містить регулярно оновлювану інформацію з безпеки лікарського засобу (Порядок здійснення фармаконаглядуN [2]).
Рекомендації за результатами аудиту (Audit recommendation)
Описують порядок організації заходів, рекомендованих для усунення виявлених порушень/недоліків та для зниження недоліків у системах контролю управління (див. Sawyer LB et al., 20036). Рекомендації за результатами аудиту повинні бути переконливими та якомога конкретнішими. У них також має вказуватися, хто саме повинен їх виконувати та терміни для їх виконання (див. Sawyer LB et al., 20036)
Див. також аудит.
6Sawyer LB, Dittenhofer MA. Sawyer’s Internal Auditing. 5th ed. Altamonte Springs, FL: The IIA Research Foundation; 2003.
Референтна дата реєстрації в країнах ЄС; синонім: референтна дата ЄС (EU reference date; synonym: Union reference date)
Для лікарських засобів, що містять одну й ту ж саму діючу речовину або одну й ту ж саму комбінацію діючих речовин, дата першого ліцензування в ЄС лікарського засобу, що містить таку діючу речовину або таку комбінацію діючих речовин; або якщо ця дата не може бути встановлена, найраніша з відомих дат торгових ліцензій на лікарські засоби, що містять таку діючу речовину або таку комбінацію діючих речовин (Директива 2001/83/ЄС ст. 107с(5) [1]).
Ризики, пов’язані із застосуванням лікарського засобу (Risks related to use of a medicinal product)
Будь-який ризик, що стосується якості, безпеки або ефективності лікарського засобу для здоров’я пацієнтів або здоров’я населення та будь-який ризик небажаних ефектів для навколишнього середовища (Директива 2001/83/ЄС ст. 1(28) [1]).
Серйозна побічна реакція (Serious adverse reaction)
Будь-яка побічна реакція, що призводить до смерті, становить загрозу для життя, вимагає госпіталізації або збільшення строку госпіталізації, викликає стійку або значну непрацездатність чи інвалідність, або є вродженою аномалією чи вадою розвитку, або має іншу важливу медичну оцінку (Порядок здійснення фармаконаглядуN [2]).
Реакція, що становить загрозу життю, у цьому контексті — це реакція, в момент розвитку якої пацієнт перебував під ризиком смерті; це не стосується реакції, яка гіпотетично могла спричинити смерть, якщо була б більш серйозною (див. додаток IV, настанова ICH-E2D [25]).
При вирішенні питання чи слід вважати серйозними такі реакції, як важливі медичні явища, які можуть не нести негайної загрози для життя та не призводити до смерті або госпіталізації, однак можуть піддавати пацієнта ризику або можуть потребувати втручання для запобігання одного з результатів, перелічених вище, необхідно оцінювати їх з медичної та наукової точки зору. Прикладами таких явищ є інтенсивне лікування у відділенні невідкладної допомоги або вдома алергічного бронхоспазму, дискразії крові або судом, які не призводять до госпіталізації або розвитку залежності чи зловживань (див. додаток IV, настанова ICH-E2D[25]).
Будь-яка підозра щодо передачі збудника інфекції через лікарський засіб також вважається серйозною побічною реакцією.
Див. також «побічна реакція».
Сигнал (Signal)
Інформація, що походить з одного або декількох джерел (у тому числі спостережень і досліджень), яка свідчить про виявлений новий потенційний зв’язок або новий аспект відомого зв’язку між лікарським засобом, вакциною, туберкуліном і явищем або сукупністю взаємопов’язаних явищ, як несприятливих, так і сприятливих, і яка вважається достатньо достовірною, щоб обґрунтувати її перевірку (Порядок здійснення фармаконаглядуN [2]).
З метою моніторингу інформації з баз даних побічних реакцій повинні розглядатися тільки сигнали, що пов’язані з побічними реакціями (ст. 19(1) ІП 520/2012 [6]).
У рамках відповідного розділу регулярного звіту з оцінки користь-ризик сигнали стосуються побічних ефектів (див. додаток IV, настанова ICH-E2С(R2) [28]).
Див. також «валідований сигнал», «нещодавно виявлений сигнал», «закритий сигнал», «поточний сигнал», «процес управління сигналом», «побічна реакція».
Система управління ризиками (Risk management system)
Комплекс процесів та заходів із фармаконагляду, спрямованих на виявлення, характеристику, запобігання або мінімізацію ризиків, пов’язаних із застосуванням лікарського засобу, вакцини, туберкуліну, що включає оцінку їх ефективності (Порядок здійснення фармаконаглядуN [2]).
Система фармаконагляду (Pharmacovigilance system)
Система, що використовується державою та заявником для здійснення фармаконагляду з метою моніторингу безпеки й ефективності лікарських засобів, вакцин, туберкуліну і визначення будь-яких змін співвідношення користь/ризик (Порядок здійснення фармаконаглядуN [2]).
Загалом система фармаконагляду — це система, що використовується організацією для виконання правових завдань та обов’язків з фармаконагляду і призначена для моніторингу безпеки зареєстрованих лікарських засобів та виявлення будь-яких змін у співвідношенні ризик-користь.
Співвідношення ризик-користь лікарського засобу, вакцини, туберкуліну (Risk-benefit balance)
Оцінка позитивних терапевтичних ефектів лікарського засобу, вакцини, туберкуліну щодо будь-яких ризиків, пов’язаних з якістю, безпекою чи ефективністю лікарського засобу, вакцини, туберкуліну, що стосуються здоров’я пацієнтів чи громадського здоров’я (Порядок здійснення фармаконаглядуN [2]).
Оцінка позитивних терапевтичних ефектів лікарського засобу відносно ризиків (Директива 2001/83/ЄС ст. 1(28а) [1]), тобто будь-якого ризику стосовно якості, безпеки або ефективності лікарського засобу для здоров’я пацієнтів або здоров’я населення (Директива 2001/83/ЄС ст. 1(28) [1]).
Див. також «ризики, пов’язані з застосуванням лікарського засобу».
Споживач стосовно повідомлень про випадки підозрюваних побічних реакцій
Особа, яка не є працівником з медичною чи фармацевтичною освітою, така як пацієнт, юрист, друг або родич/батько/дитина пацієнта (див. додаток IV, настанова ICH-E2D [25]).
Спонтанне повідомлення (Spontaneous report, synonym: Spontaneous notification)
Повідомлення, яке працівник з медичною чи фармацевтичною освітою або споживач надсилає заявнику, регуляторному органу або іншій організації (наприклад, Всесвітня організація охорони здоров’я, регіональний центр, токсикологічний центр), що описує одну або більше побічних реакцій у пацієнта, який отримує один або більше лікарських засобів, і яке не походить з дослідження або будь-якої організованої системи збору даних з обов’язковою фіксацією даних (див. додаток IV, настанова ICH-E2D [25]).
У цьому контексті побічна реакція відноситься до підозрюваної побічної реакції.
У певних ситуаціях, наприклад, після оприлюднення прямого повідомлення для медичних та фармацевтичних працівників, публікацій у пресі або після опитування медичних чи фармацевтичних працівників представниками заявника, може мати місце стимульоване надання повідомлень. Такі повідомлення про побічні реакції вважаються спонтанними повідомленнями (див. додаток IV, настанова ICH-E2D[25]), за умови, що вони відповідають вищенаведеному визначенню. Надання повідомлень може також стимулюватися після запрошення організаціями пацієнтів або споживачів до членства у їх організаціях. Надання повідомлень у ранній післяреєстраційній фазі нагляду (early post-marketing phase vigilance — EPPV), наприклад, у Японії, також вважається стимульованим повідомленням.
Див. також «побічна реакція».
Традиційний рослинний лікарський засіб (Traditional herbal medicinal product)
Лікарський засіб, зокрема рослинного походження, який відповідає таким умовам:
- лікарський засіб відповідно до його складу та призначення передбачений для застосування без нагляду лікаря з метою діагностики, без призначення або рецепта або без спостереження за процесом лікування;
- лікарський засіб застосовується в певних концентрації та дозуванні;
- лікарський засіб призначений для орального, зовнішнього або інгаляційного застосування;
- є документальне підтвердження того, що лікарський засіб застосовувався в медичній практиці не менше 30 років у світі та не менше 10 років в Україні;
- є достатньо даних щодо традиційного застосування лікарського засобу (безпека застосування при звичайних умовах, доведена ефективність) (Порядок проведення експертизи реєстраційних матеріалівN [7]).
Фармаконагляд (Pharmacovigilance)
Процес, пов’язаний із виявленням, збором, оцінкою, вивченням та запобіганням виникненню побічних реакцій, несприятливих подій після імунізації/туберкулінодіагностики та будь-яких інших питань, пов’язаних з безпекою та ефективністю застосування лікарських засобів, вакцин, туберкуліну (Порядок здійснення фармаконаглядуN [2]).
Наука і діяльність, спрямовані на виявлення, оцінку, розуміння та запобігання побічним ефектам або будь-яким іншим проблемам, пов’язаним з лікарськими засобами (див. ВООЗ2).
Згідно із цим загальним визначенням основною метою фармаконагляду відповідно до чинного законодавства України є:
- запобігання шкоди від побічних реакцій у людей, що виникають внаслідок застосування зареєстрованих лікарських засобів відповідно або невідповідно до інструкції для медичного застосування або внаслідок трудової діяльності; та
- сприяння безпечному й ефективному застосуванню лікарських засобів, зокрема, шляхом надання своєчасної інформації про безпеку лікарських засобів пацієнтам, працівникам з медичною та фармацевтичною освітою і населенню.
Тому фармаконагляд — це діяльність, що сприяє захисту здоров’я пацієнтів та населення.
Цільова популяція (у контексті медичного застосування лікарського засобу) (Target population (treatment); synonym: Treatment target population)
Пацієнти, які можуть отримувати лікарський засіб відповідно до показання(ь) та протипоказань, що зазначені в інструкції для медичного застосування.
Позначення та скорочення
|
ADI |
Acceptable daily intake (щоденна припустима доза) |
|
ADR |
Adverse reaction; synonyms: Adverse drug reaction, Suspected adverse (drug) reaction, Adverse effect, Undesirable effect (Побічна реакція лікарського засобу, підозрювана побічна реакція (лікарського засобу), побічний ефект, небажаний ефект) |
|
CHMP |
Комітет з лікарських засобів для людини |
|
CMDh |
Coordination Group for Mutual Recognition and Decentralised Procedures — Human (Координаційна група з процедури взаємного визнання та децентралізованої процедури — для людини) |
|
DDD |
Встановлена добова доза |
|
DIBD |
Development international birth date (Міжнародна дата народження лікарського засобу, що знаходиться в стадії розробки) |
|
DSUR |
Development Safety Update Report (Оновлюваний звіт з безпеки досліджуваного лікарського засобу, що перебуває у стадії розробки) |
|
GCP |
Good clinical practice (Належна клінічна практика) |
|
GDP |
Good distribution practice (Належна дистриб’юторська практика) |
|
GLP |
Good Laboratory Practices (Належна лабораторна практика) |
|
GMP |
Good Manufacturing Practice (Належна виробнича практика) |
|
GVP |
Guideline on good pharmacovigilance practices (Належні практики фармаконагляду) |
|
IBD |
International birth date (Міжнародна дата народження лікарського засобу) |
|
ICH |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини) |
|
ICSR |
Individual case safety report (Повідомлення про індивідуальний випадок, пов’язаний з безпекою) |
|
ISO |
International Organization for Standardization (Міжнародна організація зі стандартизації) |
|
PBRER |
Регулярний звіт з оцінки співвідношення користь/ризик PRAC Pharmacovigilance Risk Assessment Committee (Комітет з оцінки ризиків у фармаконагляді) |
|
SmPC |
Summary of Рroduct Сharacteristics (Коротка характеристика лікарського засобу) |
|
SOC |
Класи систем органів |
|
АІСФN |
Автоматизована інформаційна система з фармаконаглядуN |
|
АФІN |
Активний фармацевтичний інгредієнтN |
|
ВМП |
Виріб медичного призначення |
|
ВООЗ |
Всесвітня організація охорони здоров’я |
|
ВТЛЗ |
Високотехнологічні лікарські засоби |
|
ДЗБД |
Встановлена добова доза |
|
ЕМА |
European Medicines Agency (Європейська агенція з лікарських засобів) |
|
ЄЕП |
Європейський економічний простір |
|
ЄС |
Європейський Союз |
|
ЗТД |
Загальний технічний документ |
|
КМУ |
Кабінет Міністрів України |
|
КОВФN |
Контактна особа, відповідальна за фармаконаглядN |
|
Лікарські засоби |
Лікарські засоби, вакцини, туберкулінN |
|
МНН |
Міжнародна непатентована назва |
|
МОЗ |
Міністерство охорони здоров’я |
|
МФСФ |
Майстер-файл системи фармаконагляду |
|
ННПФ |
Настанова з належної практики фармаконагляду |
|
ОІБЗ |
Основна інформація з безпеки заявника |
|
Організації |
Заявники (власники реєстраційних посвідчень) і уповноважений орган щодо здійснення фармаконагляду |
|
ПДБЛЗ |
Післяреєстраційне дослідження з безпеки лікарських засобів |
|
ПДЕЛЗ |
Післяреєстраційні дослідження з ефективності лікарського засобу |
|
ПЗВ |
Програма запобігання вагітності |
|
ПОДЗ |
Перелік основних даних заявника |
|
ПР ЛЗ |
Побічна реакція лікарського засобу |
|
ПУР |
План управління ризиками |
|
РОЗБ |
Регулярно оновлювані звіти з безпеки |
|
СНМР |
Committee for Medicinal Products for Human Use (Комітет з лікарських препаратів для людини) |
|
СОП |
Стандартна операційна процедура |
|
УОВФ |
Уповноважена особа, відповідальна за фармаконагляд |
|
Уповноважений органN |
Міністерство охорони здоров’я України та/або уповноважена ним спеціалізована експертна установа у фармаконаглядіN |
|
ФН |
Фармаконагляд |
Частина І
Модуль I — Фармаконагляд та система якості у фармаконагляді
І.А. Вступ
Цей модуль містить рекомендації щодо створення та підтримки систем фармаконагляду належної якості.
Стаття 1 Директиви 2001/83/ЄС [1] Порядок здійснення фармаконагляду [2]N визначає систему фармаконагляду як систему, що використовується заявником (власником реєстраційного посвідчення), Міністерством охорони здоров’я України та/або уповноваженою ним спеціалізованою експертною установою у фармаконагляді (далі — уповноважений орган)N, для виконання завдань та обов’язків, перелічених у розділі IX Директиви 2001/83/ЄС [1] та у Порядку здійснення фармаконаглядуN [2], і створена з метою моніторування безпеки лікарських засобів, вакцин, туберкулінуN (далі — лікарських засобів) та визначення будь-яких змін співвідношення користь/ризик.
Для здійснення діяльності з фармаконагляду заявники (власники реєстраційних посвідчень) і уповноважений орган щодо здійснення фармаконагляду (далі — Організації) повинні запровадити та використовувати ефективні й відповідні для досягнення цілей системи якості. Законодавчі вимоги щодо систем якості були запроваджені Директивою 2010/84/ЄС [3], що внесла зміни до Директиви 2001/83/ЄС [1], і Постановою (ЄС) № 1235/2010 [4], що внесла зміни у Постанову (ЄС) № 726/2004 [5] для посилення фармаконагляду в ЄС. Мінімальні вимоги, яким повинні відповідати системи якості, викладені в Імплементаційній постанові Комісії (ЄС) № 520/2012 (далі — ІП 520/2012) [6] щодо продуктивності діяльності з фармаконагляду, передбаченої Постановою (ЄС) № 726/2004 [5], Директивою 2001/83/ЄС [1] та положеннями Порядку здійснення фармаконаглядуN (далі — положення Порядку[2]).
Разом з обов’язковим дотриманням вимог законодавства система якості повинна бути адаптована до відповідної організації.
Дотримуючись загальних цілей системи якості, зазначених у підрозділі І.В.4, та керівних принципів, представлених у підрозділі І.В.5, спрямованих на задоволення потреб пацієнтів, спеціалістів системи охорони здоров’я та громадськості стосовно безпеки лікарських засобів, систему якості слід адаптувати до того, наскільки критичним є кожне завдання фармаконагляду для досягнення цілей його якості для кожного лікарського засобу та/або груп лікарських засобів.
Рекомендації щодо систем якості в цьому модулі відповідають основним принципам Стандартів ISO 9000 з належних практик управління якістю, зокрема Стандартів ISO 9001-2015 з систем управління якістю, виданих Міжнародною організацією зі стандартизації (далі — ISO). Загальне застосування принципів управління якістю до систем фармаконагляду викладене в розділі І.В, а специфічні вимоги — у розділі І.С.
І.В. Структури та процеси
І.В.1.Система фармаконагляду
Система фармаконагляду визначається як система, що використовується Організацією для виконання вимог законодавства і обов’язків з фармаконагляду, і призначена для моніторингу безпеки лікарських засобів, дозволених до медичного застосування, та визначення будь-яких змін у співвідношенні користь/ризик (ст. 1(28d) Директиви 2001/83/ЄС [1], положення ПорядкуN [2]).
Система фармаконагляду передбачає наявність власної структури, процесів і результатів. Кожен окремий процес фармаконагляду, включно з необхідними структурами, описується у відповідному модулі Настанови з належної практики фармаконагляду (далі — ННПФ).
І.В.2. Якість, цілі якості, вимоги до якості та система якості
Для цілей ННПФ, що надає рекомендації щодо структур і процесів системи фармаконагляду, система якості може визначатися як сукупність усіх характеристик системи, які вважаються такими, що забезпечують, згідно з оцінкою максимальної правдоподібності, результати, що відповідають цілям фармаконагляду.
Якість — ступенева характеристика, яку можна оцінити. Для встановлення, чи було досягнуто необхідного рівня якості, потрібно мати попередньо визначені вимоги до якості. Вимоги до якості — це ті характеристики системи, які, ймовірно, забезпечать необхідний результат або досягнення цілей якості. Загальні цілі системи якості представлені у підрозділі І.В.4.
Специфічні цілі та вимоги до якості певних структур і процесів системи фармаконагляду належним чином представлені в кожному модулі ННПФ.
Система якості є невід’ємною складовою системи фармаконагляду та має свою власну організаційну структуру, процедури, обов’язки, процеси та ресурси, а також належне управління ресурсами, контроль за дотриманням вимог та ведення документації (ст. 8(2) ІП 520/2012 [6] та положення ПорядкуN [2]).
І.В.3. Цикл якості
Система якості повинна базуватися на всіх перелічених нижче видах діяльності:
- планування якості: створення структур, планування інтегрованих та узгоджених процесів;
- забезпечення якості: виконання завдань і обов’язків відповідно до вимог системи якості;
- контроль і забезпечення якості: моніторинг і оцінка того, наскільки ефективно структури і процеси створені та наскільки ефективно процеси здійснюються, а також
- вдосконалення якості: коригування і вдосконалення структур і процесів, у разі необхідності, ст. 8(3) ІП 520/2012 [6] та положення ПорядкуN [2].
І.В.4. Загальні цілі якості у фармаконагляді
Загальними цілями якості у системі фармаконагляду є:
- дотримання вимог законодавства до завдань і обов’язків з фармаконагляду;
- запобігання шкоді від побічних реакцій у людей, що виникають внаслідок застосування лікарських засобів, дозволених до медичного застосування, відповідно чи невідповідно до інструкції для медичного застосування або у зв’язку з професійною діяльністю;
- сприяння безпечному та ефективному застосуванню лікарських засобів, зокрема, шляхом своєчасного інформування про безпеку лікарських засобів пацієнтів, спеціалістів системи охорони здоров’я і громадськості;
- сприяння захисту здоров’я пацієнтів і суспільства.
І.В.5. Принципи належних практик з фармаконагляду
З метою досягнення загальних цілей якості, описаних у підрозділі І.В.4., при створенні всіх структур і процесів, а також при виконанні усіх завдань та обов’язків слід керуватися такими принципами:
- задоволення потреб пацієнтів, спеціалістів системи охорони здоров’я та громадськості стосовно безпеки лікарських засобів;
- керівництво повинно забезпечувати лідерство у впровадженні системи якості та мотивації для всіх співробітників організації щодо цілей якості;
- залучення всіх співробітників організації до участі у підтримці системи фармаконагляду відповідно до покладених на них завдань та функціональних обов’язків;
- залучення всіх співробітників організації до постійного підвищення якості системи фармаконагляду згідно з циклом якості, описаним у підрозділі І.В.3;
- організація ресурсів і завдання у вигляді таких структур і процесів, які б сприяли активному, ризик-пропорційному, безперервному та всебічному здійсненню фармаконагляду;
- ухвалення рішень повинно відбуватися за умови, коли зібрані всі наявні докази щодо співвідношення користь/ризик лікарських засобів та враховані всі відповідні чинники, що можуть вплинути на співвідношення користь/ризик та застосування лікарського засобу;
- сприяння активній співпраці між заявниками (власниками реєстраційних посвідчень), уповноваженими органами, громадськими організаціями, пацієнтами, спеціалістами системи охорони здоров’я, науковими товариствами та іншими зацікавленими сторонами відповідно до вимог законодавства УкраїниN.
І.В.6. Відповідальність за систему якості в рамках організації
Для здійснення фармаконагляду необхідно мати достатню кількість компетентного, відповідно кваліфікованого і підготованого персоналу (ст.10(1), ст. 14(1) ІП 520/2012 [6] та положень Порядку [2]N). В обов’язки персоналу повинно входити дотримання принципів, описаних у пункті І.В.5.
З метою систематичного підходу до якості у системі фармаконагляду відповідно до циклу якості (див. підрозділ І.В.3.), керівники організації відповідають за:
- забезпечення ведення організацією документації з якості, як описано в підрозділі І.В.11.;
- забезпечення можливості контролю документації, що характеризує систему якості, на етапах її створення, схвалення, впровадження та перегляду;
- забезпечення наявності відповідних ресурсів та навчання персоналу (див. підрозділ І.В.7.);
- забезпечення відповідних та в достатніх обсягах приміщень, умов та обладнання (див. підрозділ І.В.8.);
- забезпечення дотримання законодавства (див. підрозділ І.В.9.1.);
- забезпечення відповідного контролю за веденням документації (див. підрозділ І.В.10.);
- перевірку системи фармаконагляду, включаючи її систему якості, через регулярні проміжки часу способом, що враховує ризики, для підтвердження її ефективності (див. підрозділ І.В.12) та вжиття коригуючих та превентивних заходів, у разі необхідності;
- забезпечення механізмів своєчасної та ефективної передачі інформації, включаючи процедури інформування вищих посадових осіб у рамках організації щодо проблем з безпеки застосування лікарських засобів;
- виявлення і дослідження проблем, що виникають у організації щодо підозрюваного недотримання вимог до систем якості та фармаконагляду та вжиття, у разі необхідності, коригуючих та превентивних заходів;
- забезпечення проведення аудитів (див. підрозділ І.В.12.).
Керівництво організацією слід здійснювати шляхом:
- мотивації усіх співробітників, спираючись на спільні цінності, довіру і свободу говорити й діяти з відповідальністю та з визнанням внеску співробітників у діяльність організації;
- визначення функцій, обов’язків та повноважень співробітників відповідно до їх компетенції, комунікація та імплементація їх у всій організації.
Персонал, який бере участь у процедурах та процесах системи якості, створеної для виконання завдань фармаконагляду, повинен відповідати за належне функціонування цієї системи якості та забезпечувати системний підхід до якості, а також до запровадження та підтримки системи якості (ст. 8(5) ІП 520/2012 [6] та положення Порядку [2]N).
І.В.7. Навчання персоналу з фармаконагляду
Досягнення організацією необхідної якості процесів здійснення фармаконагляду та їх результатів безпосередньо зумовлюється наявністю компетентного, кваліфікованого і навченого персоналу, у кількості, що забезпечую належну якість здійснення фармаконагляду (див. підрозділ І.В.6.).
Увесь персонал, залучений до діяльності з фармаконагляду, повинен проходити первинне та подальше регулярне навчання (ст. 10(3), ст. 14(2) ІП 520/2012 [6], положення Порядку [2]N). Для заявників (власників реєстраційних посвідчень) таке навчання у розрізі здійсненні фармаконагляду має бути спрямоване на процес діяльності відповідно до функціональних обов’язків персоналу (ст. 10(3) ІП 520/2012 [6], положення Порядку [2]N).
Організація повинна зберігати навчальні плани і документовані записи про підтримку та підвищення кваліфікації персоналу. Навчальні плани повинні моніторуватися та базуватися на оцінці потреб у навчанні.
Навчання має забезпечувати безперервне підвищення кваліфікації, запровадження наукових досягнень та професійний розвиток, гарантувати відповідний професійний рівень підготовки співробітників, розуміння ними відповідних вимог фармаконагляду, а також набуття досвіду для виконання доручених завдань і обов’язків. Усі співробітники організації повинні знати, а також мати вільний доступ до інформації про те, як діяти у разі, коли їм стало відомо про проблему з безпеки лікарських засобів.
У організації повинен існувати процес для перевірки того, що навчання призводить до належного рівня розуміння та знання процесів діяльності з фармаконагляду згідно із функціональними обов’язками персоналу. Така перевірка також необхідна для визначення організацією потреб у навчанні, відповідно до планів підвищення кваліфікації, розроблених для організацій та для окремих її співробітників.
Організації слід також навчати власних співробітників, функціональні обов’язки яких безпосередньо не пов’язані зі здійсненням фармаконагляду, але діяльність яких може впливати на систему фармаконагляду або здійснення фармаконагляду. Така діяльність включає, але не обмежується діяльністю, що стосується клінічних випробувань, технічних скарг на лікарські засоби, медичної інформації, термінології, продажу та маркетингу, регуляторних питань, законодавчих питань та аудитів.
Персонал організації повинен мати відповідні інструкції про порядок дій при здійсненні критичних процесів у фармаконагляді, включаючи безперервність діяльності (див. підрозділ І.В.11.3.) (ст. 10(4), ст. 14(3) ІП 520/2012 [6] та положення Порядку [2]N).
І.В.8. Приміщення та обладнання для здійснення фармаконагляду
Для досягнення належної якості процесів з фармаконагляду та їх результатів необхідна наявність відповідних приміщень та обладнання. Приміщення та обладнання включають: офісні площі, системи інформаційних технологій (далі — ІТ), системи зберігання даних, включаючи електронні. Їх слід розташувати, спланувати, сконструювати, адаптувати та підтримувати відповідно до їх призначення та згідно з цілями системи якості (див. підрозділ І.В.4.); вони повинні бути доступними для безперервної діяльності з фармаконагляду (див. підрозділ І.B.11.3.). Приміщення та обладнання повинні бути наявними та доступними для безперервного використання при здійсненні фармаконагляду, що, зокрема, забезпечується наявними ризик-орієнтованими планами підтримки безперервної діяльності з фармаконагляду (див. підрозділ І.В.11.3.). Їх слід належним чином перевіряти на придатність та/або оцінювати для підтвердження відповідності визначеній меті. Необхідно мати в наявності процеси для відслідковування діючих версій термінологій (див. модуль VI ННПФ) та оновлення ІТ-систем.
І.В.9. Спеціальні процедури та процеси системи якості
І.В.9.1. Забезпечення дотримання законодавства заявниками (власниками реєстраційних посвідчень)
Для забезпечення дотримання вимог законодавства заявники (власники реєстраційних посвідчень) повинні виконувати спеціальні процедури і процеси в рамках системи якості з метою:
- безперервного моніторингу даних з фармаконагляду, вивчення можливих варіантів запобігання та мінімізації ризиків та вжиття відповідних заходів (ст. 11(1)(a) ІП 520/2012 [6] та положення Порядку [2]N) (див. модулі IX і XII ННПФ);
- наукової оцінки усієї інформації щодо ризиків, пов’язаних із застосуванням лікарських засобів, для пацієнтів чи суспільного здоров’я, зокрема щодо побічних реакцій, що виникають внаслідок застосування лікарських засобів відповідно або невідповідно до інструкції для медичного застосування, або пов’язаних з професійною діяльністю (ст. 11(1)(b) ІП 520/2012 [6] та положення Порядку [2]N) (див. модулі VI, VII, VIII, IX ННПФ);
- надання точних і підтверджених даних про серйозні та несерйозні побічні реакції уповноваженим органам у терміни, що визначені законодавством (ст. 11(1)(c) ІП 520/2012 [6] та положення Порядку [2]N) (див. модулі VI і IX ННПФ);
- надання якісної, цілісної та повної інформації щодо ризиків, пов’язаних із застосуванням лікарських засобів, включаючи процес запобігання наданню дублюючої інформації та процес підтвердження сигналів (ст. 11(1)(d) ІП 520/2012 [6] та положення Порядку [2]N) (див. модулі V, VI, VII, VIII і IX ННПФ);
- ефективного обміну інформацією між заявником (власником реєстраційного посвідчення) і уповноваженим органом, включаючи обмін інформацією щодо нових або змінених ризиків (див. модуль XII і XV ННПФ), майстер-файлу системи фармаконагляду (далі — МФСФ) (див. модуль II ННПФ), систем управління ризиками (див. модуль V і XVI ННПФ), заходів з мінімізації ризиків (див. модуль V і XVI ННПФ), періодично оновлюваних звітів з безпеки (див. модуль VII ННПФ), коригуючих і запобіжних заходів (див. модулі II, III і IV ННПФ) та післяреєстраційних досліджень з безпеки (див. модуль VIII ННПФ) (ст. 11(1)(e) ІП 520/2012 [6] та положення Порядку [2]N);
- оновлення інформації про лікарський засіб заявником (власником реєстраційного посвідчення) з урахуванням наукових даних (ст. 11(1)(f) ІП 520/2012 [6] та положення Порядку [2]N) (див. модуль XII ННПФ);
- належного інформування з безпеки лікарських засобів спеціалістів системи охорони здоров’я і пацієнтів (див. модулі XII і XV ННПФ) (ст. 11(1)(g) ІП 520/2012 [6] та положення Порядку [2]N).
І.В.9.2. Забезпечення дотримання законодавства уповноваженим органом
Для забезпечення дотримання вимог законодавства уповноважений орган повинен мати спеціальні процедури і процеси в рамках системи якості з метою:
- забезпечення оцінки якості отриманих даних з фармаконагляду, включно з їх повнотою (ст. 15(1) (a) ІП 520/2012 [6] та положення Порядку [2]N);
- забезпечення оцінки даних з фармаконагляду і їх обробки відповідно до термінів, що визначені законодавством (ст. 11(1)(b) ІП 520/2012 [6] та положення Порядку [2]N);
- забезпечення незалежності при здійсненні фармаконагляду (ст. 11(1)(c) ІП 520/2012 [6] та положення Порядку [2]);
- забезпечення ефективного обміну інформацією з пацієнтами, спеціалістами охорони здоров’я, заявниками (власниками реєстраційних посвідчень) та громадськістю (ст. 11(1)(d) ІП 520/2012 [6] та положення Порядку [2]N);
- проведення інспекцій у ЄС, включаючи передреєстраційні інспекції (ст. 11(1)(f) ІП 520/2012 [6] та проведення аудитів системи фармаконагляду заявника (власника реєстраційного посвідчення) в Україні уповноваженою МОЗ спеціалізованою експертною установою у сфері здійснення фармаконагляду (далі — уповноважена установа) відповідно до положень Порядку [2]N).
Під незалежністю при здійсненні фармаконагляду слід розуміти те, що всі рішення, що пов’язані з питаннями безпеки застосування лікарських засобів, повинні прийматися з єдиною метою — захисту здоров’я пацієнтів та суспільства.
І.В.10. Управління документацією
Організація повинна реєструвати всю інформацію з фармаконагляду і гарантувати, що вона обробляється і зберігається у такий спосіб, що забезпечує її точне надання, інтерпретацію та перевірку (ст. 12(1), ст. 16(1) ІП 520/2012 [6] та положення Порядку [2]N).
Система управління документацією повинна охоплювати всі документи, що використовуються організацією при здійсненні фармаконагляду, забезпечувати їх пошук, а також відстеження заходів, вжитих для розслідування питань безпеки, часові рамки таких розслідувань, та рішень з питань безпеки, включаючи їх дати і процес прийняття рішень (ст. 12(1), ст. 16(1) ІП 520/2012 [6] та положення Порядку [2]N).
Система управління документацією повинна забезпечувати:
- управління якістю даних з фармаконагляду, включаючи їх повноту, точність та цілісність;
- встановлений своєчасний доступ до всіх записів;
- ефективний внутрішній і зовнішній обмін інформацією; та
- зберігання документації, що стосується системи фармаконагляду, і здійснення фармаконагляду за окремими лікарськими засобами, відповідно до встановлених організацією періодів зберігання документації.
Заявники (власники реєстраційних посвідчень) повинні запровадити механізми, що забезпечують відслідковування повідомлень про побічні реакції і додаткової (follow-up) інформації (ст. 12(1) ІП 520/2012 [6] та положення Порядку [2]N).
У цьому контексті необхідно гарантувати дотримання основоположного права щодо захисту персональних даних при здійсненні усіх видів діяльності з фармаконагляду відповідно до положень законодавства. Захист суспільного здоров’я представляє значний суспільний інтерес, і тому обробка персональних даних буде виправданою, якщо персональні дані обробляються тільки в разі необхідності і тільки коли всі залучені сторони оцінюють цю необхідність на кожному етапі процесу фармаконагляду (декларативна частина 17 ІП 520/2012 [6] та положення Порядку [2]N). У рамках системи управління документацією на кожному етапі слід вживати відповідних заходів щодо зберігання та обробки даних з фармаконагляду для гарантії безпеки та конфіденційності даних. Такі заходи повинні включати суворе обмеження доступу до документів та баз даних тільки колом офіційно уповноважених осіб, які дотримуються вимог конфіденційності медичних та адміністративних даних.
Слід створити відповідні структури і процеси, що гарантують захист даних та документації з фармаконагляду від знищення протягом необхідного періоду зберігання документації.
Систему управління документацією слід описувати в процесах управління документацією.
І.В.11. Документація системи якості у фармаконагляді
Усі елементи, вимоги та положення, прийняті для системи якості, повинні систематично і регулярно документуватися у формі письмових процесів і процедур, таких як плани з якості, настанови з якості та записи з якості (ст. 8(4) ІП 520/2012 [6] та положення Порядку [2]N).
План з якості — документ, в якому викладені цілі якості та процеси, що будуть запроваджені для їх досягнення. Процедура пояснює, яким чином процес виконується і може мати формат стандартної операційної процедури чи іншої робочої інструкції або настанови з якості. Настанова з якості описує сферу застосування системи якості, процеси системи якості та взаємодію між ними. Запис з якості — це документ, що містить інформацію про досягнуті результати чи надає докази, що підтверджують проведену діяльність.
Для систематичного підходу організації слід заздалегідь визначити:
- цілі якості, специфічні для організації, відповідно до загальних цілей якості, передбачених у підрозділі І.В.4., а також цілі якості, специфічні для структур і процесів, відповідно до кожного модуля ННПФ; та
- методи моніторингу ефективності системи фармаконагляду (див. підрозділ І.В.12.).
Система якості повинна мати документальне відображення у:
- документах з організаційної структури та розподілу функціональних обов’язків для персоналу (див. підрозділи І.В.11.1. та І.В.11.2.);
- навчальних планах та звітах (див. підрозділ І.В.7.) (ст. 10(3), ст. 14(2) ІП 520/2012 [6] та положення Порядку [2]N);
- інструкціях із забезпечення дотримання законодавства (див. підрозділ І.В.9.) (ст. 11(1), ст. 15(1) ІП 520/2012 [6] та положення Порядку [2]N);
- відповідних стандартних операційних процедур та/або інструкціях з процедур, які необхідно використовувати у невідкладних ситуаціях та для забезпечення безперервності діяльності з фармаконагляду (див. підрозділ І.В.11.3.) (ст. 10(4), ст. 14(3) ІП 520/2012 [6] та положення Порядку [2]N);
- індикаторах діяльності, що використовуються для безперервного моніторингу належного здійснення фармаконагляду (ст. 9(1) ІП 520/2012 [6] та положення Порядку [2]N);
- звітах з аудитів якості та наступних (follow-up) аудитів, із зазначенням дат і результатів (ст. 13(2), ст. 17(2) ІП 520/2012 [6] та положення Порядку [2]N).
Навчальні плани та документація повинні зберігатися і бути доступними щодо інспекцій у ЄС (ст. 10(3), ст. 14(2) ІП 520/2012 [6]) та аудиту системи фармаконагляду уповноваженою установою (положення Порядку [2]N).
Рекомендується, щоб документація системи якості також включала:
- методи моніторингу ефективності роботи системи якості;
- правила та принципи управління документацією;
- документи, створені в результаті здійснення фармаконагляду;
- документи і звіти, що стосуються приміщень і обладнання, включаючи перевірки функціонування, атестаційну та валідаційну діяльність, які демонструють, що вжиті всі заходи, які вимагаються згідно з відповідними нормами, протоколами і процедурами;
- документи з інформацією про те, що недоліки і відхилення від встановленої системи якості контролюються, що були вжиті коригуючі і превентивні заходи, що були вирішені питання щодо відхилень чи недоліків, і що ефективність вжитих заходів була підтверджена.
І.В.11.1. Додаткова документація системи якості у фармаконагляді у заявників (власників реєстраційних посвідчень)
Крім документації, що стосується системи якості, зазначеної у підрозділі І.В.11, заявники (власники реєстраційних посвідчень) повинні документувати:
- управління своїм персоналом у МФСФ (див. модуль II) (ст. 2(5)(b) ІП 520/2012 [6] та положення Порядку [2]N);
- посадові інструкції, що визначають функціональні обов’язки керівного та підлеглого персоналу (ст. 10(2) ІП 520/2012 [6] та положення Порядку [2]N);
- організаційну схему, що визначає підпорядкованість керівного та підлеглого персоналу (ст. 10(2) ІП 520/2012 [6] та положення Порядку [2]N);
- інструкції для критичних процесів (див. підрозділ І.В.11.3) у МФСФ;
- і свою систему управління записами у МФСФ (див. модуль II) (ст. 2(5)(с) ІП 520/2012 [6] та положення Порядку [2]N).
Рекомендується, щоб документація з системи якості додатково включала організаційні структури і розподіл функціональних обов’язків та повноважень для всього персоналу, який безпосередньо бере участь у здійсненні фармаконагляду.
Щодо вимог документального оформлення системи якості в МФСФ або його додатках див. модуль II ННПФ.
І.В.11.2. Додаткова документація системи якості у уповноваженого органу
Крім документації системи якості, зазначеної у підрозділі І.В.11, повинні бути доступними в достатній мірі деталізовані організаційна структура та розподіл функціональних обов’язків (ст. 2(5)(с) ІП 520/2012 [6] та положення Порядку [2]N).
Рекомендується, щоб документація системи якості додатково містила організаційні структури та розподіл функціональних обов’язків і повноважень усього персоналу, безпосередньо залученого до виконання завдань фармаконагляду.
Слід призначити контактних осіб (ст. 14(1) ІП 520/2012 [6] та положення Порядку [2]N), зокрема, для сприяння взаємодії між уповноваженим органом, заявниками (власниками реєстраційних посвідчень) та особами, які надають інформацію про ризики для здоров’я пацієнтів або суспільного здоров’я, пов’язані із застосуванням лікарських засобів.
І.В.11.3. Критичні процеси фармаконагляду та безперервність діяльності
Критичними процесами фармаконагляду вважаються:
- постійний моніторинг профілю безпеки та оцінки співвідношення користь/ризик, дозволених до медичного застосування лікарських засобів;
- створення, оцінка та імплементація систем управління ризиками та оцінка ефективності мінімізації ризиків;
- збір, обробка, управління, контроль якості, збір необхідної відсутньої інформації, кодування, класифікація, виявлення дублюючих повідомлень, оцінка та своєчасне подання повідомлень про випадки побічних реакцій (повідомлення про індивідуальний випадок, пов’язаний з безпекою (ICSRs)) від будь-якого джерела;
- управління сигналами;
- планування, підготовка (включаючи оцінку даних та контроль якості), подання та оцінка регулярно оновлюваних звітів з безпеки;
- виконання зобов’язань та відповіді на запити від уповноважених органів з наданням коректної достовірної інформації в повному обсязі;
- забезпечення взаємодії між системою фармаконагляду та системою контролю якості лікарських засобів;
- комунікація з питань безпеки лікарських засобів між заявниками (власниками реєстраційних посвідчень) та уповноваженими органами, зокрема, повідомлення про зміни у співвідношенні користь/ризик лікарських засобів;
- інформування пацієнтів та спеціалістів системи охорони здоров’я щодо змін у співвідношенні користь/ризик лікарського засобу з метою його ефективного та безпечного застосування;
- оновлення інформації про лікарські засоби відповідно до сучасних наукових медичних даних, включаючи висновки з оцінки та рекомендації відповідного уповноваженого органу;
- внесення змін до реєстраційних матеріалів на лікарські засоби протягом терміну дії реєстраційних посвідчень з питань безпеки з урахуванням ступеня терміновості.
Необхідно створити ризик-орієнтовані плани підтримки безперервної діяльності, що повинні включати:
- запобіжні заходи на випадок подій, що можуть значною мірою вплинути на штатний розклад та інфраструктуру організації в цілому, й на структури і процеси фармаконагляду зокрема; та
- дублюючі (резервні) системи для обміну терміновою інформацією в межах однієї організації, між організаціями, що виконують спільні завдання з фармаконагляду, а також між заявниками (власниками реєстраційних посвідчень) та уповноваженими органами.
І.В.12. Моніторинг функціонування та ефективності системи фармаконагляду та її системи якості
Процеси моніторингу функціонування та ефективності системи фармаконагляду та системи якості повинні включати:
- перевірку систем особами, відповідальними за управління нею;
- аудити;
- контроль за дотриманням встановлених законодавством вимог щодо функціонування та ефективності системи фармаконагляду та її системи якості;
- аудити системи фармаконагляду уповноваженою установоюN;
- оцінку ефективності заходів, вжитих з метою мінімізації ризиків лікарських засобів та сприяння їх безпечному та ефективному застосуванню пацієнтами.
Організація може використовувати індикатори ефективності для постійного моніторингу діяльності з фармаконагляду (ст. 9(1) ІП 520/2012 [6] та положення Порядку [2]N в частині вимог до якості. Вимоги до якості кожного з процесів фармаконагляду наведені у кожному відповідному модулі ННПФ.
Ефективність системи якості повинна контролюватися керівниками організації, які повинні регулярно переглядати документацію системи якості (див. підрозділ I.B.11). Періодичність та обсяг таких переглядів визначаються на підставі оцінки ризиків, для чого необхідно мати попередньо сплановані програми перегляду системи якості. Перегляди системи якості повинні включати перегляд стандартних операційних процедур та/або робочих інструкцій, відхилень від встановленої системи якості, звітів про аудити, аудити системи фармаконагляду уповноваженою установоюN за використання індикаторів, про які йшлося вище.
З метою забезпечення дотримання вимог до системи якості, управління кадровими ресурсами, відповідності вимогам законодавства, управління документацією та зберігання даних, а також з метою забезпечення ефективності системи якості необхідно регулярно проводити ризик-орієнтовані аудити системи якості (ст. 13(1), ст. 17(1) ІП 520/2012 [6] та положення Порядку [2]N). Аудити системи якості повинні включати аудит системи фармаконагляду, яка є суб’єктом системи якості. Методи та процеси аудиту викладені в модулі IV ННПФ. Стосовно системи фармаконагляду заявника (власника реєстраційного посвідчення), то за підсумками кожного аудиту якості необхідно складати звіт та надсилати його керівникам, відповідальним за здійснення аудиту (ст. 13(2) ІП 520/2012 [6] та положення Порядку [2]N). Звіт повинен включати результати аудитів організацій чи осіб, яким заявник (власник реєстраційного посвідчення) делегував повноваження, оскільки вони є частиною системи фармаконагляду заявника (власника реєстраційного посвідчення). Звіт про результати аудиту уповноваженого органу повинен бути надісланий керівнику уповноваженого органу, відповідальному за здійснення аудиту (ст. 17(2) ІП 520/2012 [6] та положення Порядку [2]N).
Наслідком моніторингу діяльності та ефективності системи фармаконагляду та її системи якості (включаючи здійснення аудитів) повинна бути імплементація, у разі необхідності, коригуючих та превентивних заходів. Зокрема, результатом здійснення аудитів повинні бути, в разі необхідності, коригуючі дії, включаючи наступний (follow-up) аудит за усуненням недоліків (ст. 13(1), ст. 17(2) ІП 520/2012 [6] та положення Порядку [2]N). Крім того, уповноважений орган повинен мати механізми для моніторингу дотримання заявниками (власниками реєстраційних посвідчень) вимог при виконанні завдань та зобов’язань з фармаконагляду. Він повинен здійснювати подальший моніторинг дотримання вимог законодавства шляхом проведення аудитів системи фармаконагляду уповноваженою установоюN (ст. 111(1) Директиви 2001/83/ЄС [1]) та положення Порядку [2]N (див. модуль III ННПФ). Настанова з моніторингу дотримання вимог законодавства ЄС/УкраїниN для кожного з процесів фармаконагляду міститься у кожному відповідному модулі ННПФ.
Вимоги та методи оцінки ефективності заходів, вжитих до лікарських засобів з метою мінімізації їх ризиків та сприяння їх безпечному та ефективному використанню пацієнтами, викладені в модулі XVI ННПФ.
І.В.13. Планування готовності до фармаконагляду в надзвичайних ситуаціях у галузі охорони здоров’я
Будь-яка система фармаконагляду повинна мати можливість бути адаптованою до надзвичайних ситуацій у галузі охорони здоров’я та, за необхідності, повинні бути розроблені плани готовності до дій.
Планування готовності до таких дій в УкраїніN викладене у підрозділі
І.С. Діяльність з фармаконагляду в Україні
І.С.1. Загальні обов’язки з фармаконагляду заявника (власника реєстраційного посвідчення) в Україні
Заявник (власник реєстраційного посвідчення) несе відповідальність за виконання завдань та зобов’язань з фармаконагляду, визначених законодавством (Директива 2001/83/ЄС [1], Постанова (ЄС) № 726/2004 [5], Імплементаційна постанова Комісії (ЄС) № 520/2012 [6] щодо продуктивності діяльності з фармаконагляду, передбаченої Постановою (ЄС) № 726/2004 [5], Директивою 2001/83/ЄС [1], положення Порядку проведення експертизи реєстраційних матеріалів [7] та положення Порядку [2]N), з метою гарантування відповідальності та зобов’язань щодо його зареєстрованих лікарських засобів та вжиття відповідних заходів у разі необхідності.
З цією метою заявник (власник реєстраційного посвідчення) повинен забезпечити функціонування системи фармаконагляду (ст. 104(1) Директиви 2001/83/ЄС [1], та положення Порядку [2]N) і запровадити й використовувати відповідну та ефективну систему якості для здійснення діяльності з фармаконагляду (ст. 8(1) ІП 520/2012 [5] та положення Порядку [2]N).
За певних обставин заявник (власник реєстраційного посвідчення) може створити більше ніж одну систему фармаконагляду, наприклад, специфічні системи для різних видів лікарських засобів (наприклад, вакцини, безрецептурні лікарські засоби тощо).
Заявник (власник реєстраційного посвідчення) на державну реєстрацію лікарського засобу повинен розробити опис системи фармаконагляду у форматі МФСФ, який буде підтримуватися заявником (власником реєстраційного посвідчення) для всіх лікарських засобів, дозволених до медичного застосування в Україні (див. модуль II ННПФ). Заявник (власник реєстраційного посвідчення) також несе відповідальність за розробку та підтримку систем управління ризиками, специфічних для конкретних лікарських засобів (див. модуль V ННПФ).
Рекомендації щодо структур і процесів, які заявник (власник реєстраційного посвідчення) повинен використовувати при виконанні завдань та зобов’язань з фармаконагляду, представлені у відповідних модулях ННПФ.
І.С.1.1. Зобов’язання заявника (власника реєстраційного посвідчення) стосовно уповноваженої особи, відповідальної за фармаконагляд/контактної особи з фармаконагляду в УкраїніN
Заявник (власник реєстраційного посвідчення) повинен мати у штаті у постійному й безперервному розпорядженні уповноважену особу, відповідальну за фармаконагляд (далі — УОВФ)/призначити єдину контактну особу з фармаконагляду в УкраїніN (далі — КОВФN) відповідної кваліфікації (ст. 104(3)(a) Директиви 2001/83/ЄС [1] та положення Порядку [2]N).
Заявник (власник реєстраційного посвідчення) повинен подати до уповноваженого органу інформацію (прізвище, ім’я, по батькові, інформація про освіту, місцезнаходження, контактні дані)N про УОВФ/КОВФN (останній параграф ст. 104(3) Директиви 2001/83/ЄС) [1] та положення Порядку [2]N). У разі змін до цієї інформації необхідно подати заяву на зміни до реєстраційних матеріалів на лікарські засоби протягом дії їх реєстраційного посвідченняN відповідно до Постанови (ЕС) № 1234/2008 [8] щодо розгляду змін до умов торгової ліцензії (Regulation (EC) No 1234/2008 on variations to the terms of marketing authorisation) та згідно з Повідомленням Комісії — Настанова щодо різних видів змін до умов торгових ліцензій на лікарські засоби для людини та ветеринарні препарати (Communication from the Commission — Guideline on the Details of the Various Categories of Variations to the Terms of Marketing Authorisations for Medicinal Products for Human Use and Veterinary Medicinal Products)3 [9] та положення Порядку проведення експертизи реєстраційних матеріалів [7] та положення Порядку [2]N.
Обов’язки УОВФ/КОВФ повинні бути зазначені в посадовій інструкції (ст. 10(2) ІП 520/2012 [6] та положення Порядку [2]N).
Підпорядкованість УОВФ/КОВФ повинна бути визначена в організаційній структурі організації разом з іншим керівним та контролюючим персоналом (ст. 10(2) ІП 520/2012 [6] та положення Порядку [2]N).
Інформація про УОВФ/КОВФ повинна бути внесена до МФСФ (ст. 2(1) ІП 520/2012 [6] та положення Порядку [2]N) (див. модуль II ННПФ).
Кожна система фармаконагляду може мати лише одну УОВФ. УОВФ може працювати більше ніж на одного заявника (власника реєстраційного посвідчення) для спільних або окремих систем фармаконагляду, або може виконувати роль УОВФ для більш ніж однієї системи фармаконагляду одного заявника (власника реєстраційного посвідчення) за умови, що УОВФ зможе виконувати всі зобов’язання, щоб забезпечити функціонування належної системи фармаконагляду.
Уповноважений орган законодавчо має легітимну можливість вимагати призначення контактної особи з фармаконагляду на національному рівні, підпорядковану УОВФ. Підпорядкованість у даному контексті стосується завдань та зобов’язань з фармаконагляду, а не обов’язково безпосереднього підпорядкування контактної особи з фармаконагляду УОВФ як керівнику. КОВФN на національному рівні може діяти як УОВФ.
Заявник (власник реєстраційного посвідчення) повинен гарантувати, що УОВФ має достатні повноваження для впливу на показники системи якості та діяльність з фармаконагляду заявника (власника реєстраційного посвідчення) (ст. 10(2) ІП 520/2012 [6]). Тому заявник (власник реєстраційного посвідчення) повинен забезпечити УОВФ доступ до МФСФ, повноваження щодо нього та отримання повідомлень про будь-які зміни в МФСФ згідно з модулем II ННПФ (див. підрозділ І.С.1.3). Повноваження УОВФ щодо системи фармаконагляду та МФСФ повинні давати УОВФ можливість внесення змін до системи фармаконагляду, надання пропозицій до планів управління ризиками (див. модуль V ННПФ), а також участі в підготовці регулюючих заходів з питань безпеки у зв’язку з надзвичайними ситуаціями, що змінюють профіль безпеки лікарського засобу.
Загалом заявник (власник реєстраційного посвідчення) повинен гарантувати наявність структур та процесів, які б надавали УОВФ можливість виконувати покладені на неї функціональні обов’язки (див. підрозділ I.C.1.3). Із цією метою заявник (власник реєстраційного посвідчення) повинен забезпечити механізми, за допомогою яких УОВФ матиме змогу отримувати всю релевантну (значущу) інформацію та мати доступ до всієї інформації, яку вважатиме релевантною (значущою), зокрема щодо:
- проблем безпеки та будь-якої інформації, що стосується оцінки співвідношення користь/ризик лікарських засобів, охоплених системою фармаконагляду;
- поточних або завершених клінічних випробувань та інших досліджень, про які стає відомо заявнику (власнику реєстраційного посвідчення) та які можуть мати відношення до безпеки лікарських засобів;
- інформації з інших джерел (не власна інформація заявника (власника реєстраційного посвідчення)), наприклад, від тих, з ким заявник (власник реєстраційного посвідчення) має договірні відносини; та
- процедур, що стосуються фармаконагляду, які заявник (власник реєстраційного посвідчення) має у своєму розпорядженні на кожному рівні з метою забезпечення узгодженості діяльності організації та відповідності вимогам законодавства.
Керівний персонал організації повинен повідомляти УОВФ про результати регулярних переглядів системи якості, про які йшлося в розділах I.B.5., I.B.6. та I.B.12., та вжиті заходи.
УОВФ повинна періодично отримувати інформацію щодо дотримання вимог законодавства. Ця інформація також може використовуватися для надання УОВФ доказів дотримання заявником (власником реєстраційного посвідчення) зобов’язань у частині виконання планів управління ризиками та систем післяреєстраційної безпеки лікарських засобів.
Керівний персонал організації повинен інформувати УОВФ про заплановані аудити з фармаконагляду. УОВФ повинна мати змогу ініціювати аудит у разі його доцільності. Керівникам слід надавати УОВФ копію плану коригуючих та запобіжних заходів після кожного аудиту, що стосується системи факрмаконагляду, за яку УОВФ несе відповідальність з тим, щоб УОВФ могла вжити відповідних коригуючих та запобіжних заходів.
Заявник (власник реєстраційного посвідчення) повинен запровадити процедуру, яка б давала змогу УОВФ отримувати інформацію з бази даних побічних реакцій (або інших систем збору повідомлень про побічні реакції), наприклад, для надання у будь-який час відповіді на термінові запити від уповноважених органів. Якщо ця процедура вимагає залучення додаткового персоналу, наприклад, спеціаліста з баз даних, тоді це потрібно врахувати при створенні заявником (власником реєстраційного посвідчення) механізмів підтримки роботи УОВФ у неробочий час.
У випадках, коли заявник (власник реєстраційного посвідчення) планує розширити номенклатуру своїх лікарських засобів, наприклад, шляхом придбання іншої компанії або окремих лікарських засобів іншого заявника (власника реєстраційного посвідчення), УОВФ повинна бути поінформована про це якомога раніше з метою оцінки потенційного впливу цього розширення на систему фармаконагляду та її відповідної адаптації. УОВФ також може брати участь у визначенні, які дані з фармаконагляду слід вимагати у іншої компанії до або після її придбання. У даній ситуації УОВФ повинна бути ознайомлена з відповідними розділами угоди, що стосуються зобов’язань з фармаконагляду й обміну інформацією з безпеки, та мати повноваження вимагати внесення змін.
Якщо заявник (власник реєстраційного посвідчення) має намір встановити партнерські стосунки з іншим заявником (власником реєстраційного посвідчення), організацією чи особою, яка безпосередньо або опосередковано впливає на систему фармаконагляду, необхідно заздалегідь поінформувати про це УОВФ і залучити її до підготовки відповідних документів (див. підрозділ І.С.1.5.) з тим, щоб до них увійшли всі необхідні положення, які стосуються системи фармаконагляду.
І.С.1.2. Кваліфікації уповноваженої особи, відповідальної за фармаконагляд/контактної особи з фармаконагляду в УкраїніN
Заявник (власник реєстраційного посвідчення) повинен гарантувати, що УОВФ/КОВФ в УкраїніN отримала відповідні практичні та теоретичні знання, необхідні для здійснення діяльності з фармаконагляду (ст. 10(1) ІП 520/2012 [6] та положення Порядку [2]N). УОВФ/КОВФ в Україні повинна мати навички управління системою фармаконагляду та досвід і знання, або мати доступ до знань у таких сферах, як медицина, фармація, а також епідеміологія та біостатистика. У випадках, коли УОВФ/КОВФ в УкраїніN не має вищої медичної освіти відповідно до ст. 24 Директиви 2005/36/ЄС [10] та положень Порядку [2]N, заявник (власник реєстраційного посвідчення) повинен забезпечити доступ УОВФ/КОВФ в УкраїніN до спеціаліста з вищою медичною освітою (тобто відповідно до статті 24 Директиви 2005/36/ЄС [10] та положень Порядку [2]N) з метою отримання від нього допомоги, і це повинно бути належним чином документально оформлено (ст. 10(1) ІП 520/2012 [6] та положення Порядку [2]N).
Заявник (власник реєстраційного посвідчення) повинен оцінити рівень кваліфікації УОВФ/КОВФ в УкраїніN до її призначення, наприклад, шляхом перегляду дипломів про вищу медичну або фармацевтичну освіту (провізор, клінічнийN провізор, визначення рівня знань вимог з фармаконагляду в Україні та досвіду роботи у фармаконагляді. Заявник (власник реєстраційного посвідчення) повинен забезпечити УОВФ/КОВФ в УкраїніN навчання щодо своєї системи фармаконагляду. Таке навчання доцільно провести до того, як УОВФ/КОВФ в УкраїніN обійме посаду та відповідно його задокументувати. У разі потреби можна провести додаткове навчання УОВФ/КОВФ в УкраїніN щодо лікарських засобів, які охоплює система фармаконагляду.
І.С.1.3. Функціональні обов’язки уповноваженої особи, відповідальної за фармаконагляд/контактної особи з фармаконагляду в УкраїніN
УОВФ/КОВФ в УкраїніN є фізичною4 особою.
УОВФ/КОВФ в УкраїніN, призначена заявником (власником реєстраційного посвідчення), повинна мати відповідний рівень кваліфікації (див. підрозділ I.C.1.2.) та постійно і безперервно знаходитися в розпорядженні заявника (власника реєстраційного посвідчення) (див. розділ підрозділ І.С.1.1.) (ст. 104(3)(а) Директиви 2001/83/ЄС [1] та положення Порядку [2]N). УОВФ/КОВФ в УкраїніN повинна проживати та працювати в УкраїніN (останній параграф статті 104(3) Директиви 2001/83/ЄС [1] та положення Порядку [2]). На випадок відсутності УОВФ/КОВФ в УкраїніN необхідно передбачити наявність спеціаліста (ст. 2(1)(d) ІП 520/2012 [6] та положення Порядку [2]N), який виконуватиме обов’язки УОВФ/КОВФ в УкраїніN на період її відсутностіN, і який повинен бути доступним через контакти УОВФ/КОВФ в УкраїніN. УОВФ/КОВФ в УкраїніN повинна гарантувати, що такий резервний спеціаліст володіє усією інформацією, необхідною для виконання функціональних обов’язків УОВФ/КОВФ в УкраїніN.
УОВФ повинна відповідати за створення та підтримку системи фармаконагляду заявника (власника реєстраційного посвідчення) (останній параграф ст. 104(3) Директиви 2001/83/ЄС [1] та положення Порядку [2]N), а тому вона повинна мати достатні повноваження для впливу на результативність системи якості й на діяльність з фармаконагляду (ст. 10(2) ІП 520/2012 [6]) та сприяти, підтримувати і вдосконалювати дотримання вимог законодавства (ст. 2(1)(а) ІП 520/2012 [6]). Таким чином, УОВФ/КОВФ в Україні повинна мати доступ до МФСФ (див. модуль II ННПФ) та мати офіційні повноваження для забезпечення та гарантування того, що інформація в МФСФ є точним та сучасним відображенням системи фармаконагляду, за яку УОВФ несе відповідальність.
Що стосується лікарських засобів, які охоплює система фармаконагляду, УОВФ/КОВФ в УкраїніN може мати такі специфічні обов’язки:
- мати загальну характеристику профілів безпеки лікарських засобів та будь-яких виявлених сигналів з безпеки;
- володіти у повному обсязіN інформацією щодо будь-яких умов чи обов’язків, що були передумовою видачі реєстраційних посвідчень та інших зобов’язань, що стосуються безпеки застосування лікарських засобів;
- володіти у повному обсязіN інформацією щодо заходів з мінімізації ризиків;
- володіти у повному обсязіN інформацією щодо змісту планів управління ризиками та мати достатні повноваження для впливу на них;
- брати участь у рецензуванні та затвердженні протоколів післяреєстраційних досліджень з безпеки лікарських засобів (далі — ПДБЛЗ), що проводяться в УкраїніN, чи згідно з погодженим в УкраїніN планом управління ризиками;
- володіти у повному обсязіN інформацією щодо ПДБЛЗ, що проводяться на вимогу уповноваженого органу, включно з результатами таких досліджень;
- забезпечувати здійснення фармаконагляду та подання всіх пов’язаних із цим документів, згідно із законодавством та належною практикою фармаконагляду;
- забезпечувати необхідну якість, включаючи коректність та повноту даних з фармаконагляду, що подаються до МОЗ України та до уповноваженого органу;
- забезпечувати повну та оперативну відповідь на будь-який запит МОЗ УкраїниN та уповноваженого органу щодо надання додаткової інформації, необхідної для оцінки співвідношення користь/ризик лікарського засобу;
- надавати уповноваженому органу будь-яку іншу інформацію, що стосується оцінки співвідношення користь/ризик;
- надавати пропозиції при підготовці регуляторних заходів у відповідь на проблеми з безпеки, що виникають (наприклад, зміни до матеріалів реєстраційного досьє, термінові обмеження з міркувань безпеки та повідомлення для пацієнтів та спеціалістів системи охорони здоров’я);
- працювати як єдина контактна особа з фармаконагляду для уповноваженого органу з цілодобовим доступом, а також як контактна особа для аудитів системи фармаконагляду уповноваженою установою відповідно до положень Порядку [2]N.
Таке коло відповідальності за систему фармаконагляду означає, що УОВФ/КОВФ в УкраїніN здійснює нагляд за функціонуванням системи фармаконагляду у всіх її аспектах, включно з її системою якості (наприклад, стандартні операційні процедури, контрактні угоди, операції з базами даних, дотримання вимог якості, повноти даних та своєчасності надання термінових повідомлень про побічні реакції та регулярно оновлюваних звітів з безпеки, звіти з аудитів та навчання персоналу з питань фармаконагляду). Зокрема, що стосується бази даних побічних реакцій, УОВФ/КОВФ в УкраїніN повинна знати валідаційний статус бази даних, якщо валідація проводилася, включаючи будь-які помилки, що виникли під час валідації, та коригуючі заходи, вжиті для їх усунення. Слід також інформувати УОВФ/КОВФ в УкраїніN щодо суттєвих змін у базах даних (наприклад, змін, що можуть спричинити вплив на діяльність з фармаконагляду).
УОВФ/КОВФ в УкраїніN може делегувати певні завдання відповідно підготованому та кваліфікованому персоналу з контролем їх виконання. Наприклад, діяти як спеціаліст з безпеки певних лікарських засобів за умови, що УОВФ/КОВФ в УкраїніN здійснює контроль за системою в цілому та нагляд за профілем безпеки всіх лікарських засобів заявника (власника реєстраційного посвідчення). Делегування таких повноважень повинно бути оформлене документально.
І.С.1.4. Специфічні процеси системи якості заявника (власника реєстраційного посвідчення) в УкраїніN
На виконання вимог, зазначених у підрозділі І.В.9.1., заявник (власник реєстраційного посвідчення) повинен запровадити додаткові специфічні процеси системи якості для гарантування:
- подання інформації про побічні реакції до уповноваженого органуN в межах встановлених строків (ст. 11(с) ІП 520/2012 [6] та положення Порядку [2]N);
- моніторингу використання термінології, зазначеної у ст. 25(1) ІП 520/2012 [6], систематичного або шляхом довільної регулярної оцінки (ст. 25(3) ІП 520/2012 [6]);
- зберігання мінімальної кількості елементів МФСФ (модуль II ННПФ) (див. ст. 2 ІП 520/2012 [6] і модуль ІІ ННПФ) до того часу, поки існує система, описана в МФСФ, та щонайменше протягом наступних 5 років після того, як заявник (власник реєстраційного посвідчення) офіційно завершив її використання (ст. 12(2) ІП 520/2012 [6]);
- зберігання даних з фармаконагляду та документів, що стосуються лікарських засобів, дозволених до медичного застосування, протягом дії реєстраційних посвідчень та принаймні 10 років після закінчення їх дії (ст. 12(2) ІП 520/2012 [6]);
- того, що заявник (власник реєстраційного посвідчення) оновлює інформацію про лікарські засоби на сучасному рівні з врахуванням останніх наукових даних включно з оцінкою та рекомендаціями, оприлюдненими через вебсайт уповноваженого органу та на підставі постійного моніторингу інформації, опублікованої на даному вебсайті (ст. 11(1)(g) ІП 520/2012 [6], положення Порядку [2] та положення Порядку проведення експертизи реєстраційних матеріалів [7]N).
Зазначені вище періоди зберігання даних та документів застосовуються у випадках, якщо вимоги законодавства не передбачають більш тривалий термін їх зберігання (відповідно до вимог законодавства в УкраїніN).
Протягом терміну зберігання повинна гарантуватися можливість пошуку (віднаходження) документів. Документи можуть зберігатися в електронному форматі за умови, що електронна система відповідним чином перевірена та існують належні процедури гарантування безпеки системи, доступу до інформації та її резервного копіювання. Якщо документи переводяться з паперового в електронний формат, процес переводу має забезпечувати, щоб вся інформація з оригінального формату трансформувалася в електронний в прийнятному вигляді та щоб носії інформації, що використовуються для її зберігання, з плином часу залишалися зчитуваними.
У випадках, коли бізнес заявника (власника реєстраційного посвідчення) передається іншому суб’єкту господарювання, документи передаються у повному обсязі.
І.С.1.5. Вимоги до системи якості для виконання завдань фармаконагляду, що здійснюються заявником (власником реєстраційного посвідчення) на договірних умовах
Заявник (власник реєстраційного посвідчення) може укладати договори на певні види діяльності у фармаконагляді з третіми сторонами (ст. 6(1) ІП 520/2012 [6] та положення Порядку [2]N), тобто з іншими фізичними та/або юридичними особами (якщо вимоги до фізичних та/або юридичних осіб є однаково прийнятними), включаючи виконання обов’язків УОВФ/КОВФ в УкраїніN. При цьому повна відповідальність за повноту і точність МФСФ залишається за заявником (власником реєстраційного посвідчення) (див. модуль II ННПФ) (ст. 6(1) ІП 520/2012 [6], положення Порядку [2]N). Остаточну відповідальність за виконання усіх завдань та зобов’язань з фармаконагляду, за якість та цілісність системи фармаконагляду завжди несе заявник (власник реєстраційного посвідчення) (положення Порядку [2] та положення Порядку проведення експертизи реєстраційних матеріалів [7]N).
Якщо заявник (власник реєстраційного посвідчення) уклав угоду з третьою стороною на виконання деяких видів діяльності з фармаконагляду, за ним залишається відповідальність за забезпечення ефективної системи якості при виконанні таких завдань (ст. 11(2) ІП 520/2012 [6] та положення Порядку [2]N). Усі настанови з ННПФ є також чинними для тієї фізичної та/або юридичної особи, з якою був укладений договір на виконання зазначених вище завдань.
При передачі частини своїхN завдань з фармаконаглядуN іншій організації (іншій фізичній та/або юридичній особі)N заявник (власник реєстраційного посвідчення) повинен укласти субпідрядні договори (ст. 6(2) ІП 520/2012 [6] та положення Порядку [2]), які повинні бути детальними, відповідати сучасній ситуації та чітко визначати договірні відносини між заявником (власником реєстраційного посвідчення) й іншою організацією, описувати порядок делегування завдань та відповідальності кожної сторони. Опис договірної діяльності та/або послуг повинні бути включені до МФСФ (ст. 6(2) ІП 520/2012 [6] та положення Порядку [2]N), а перелік субпідрядних договорів повинен бути оформленим у вигляді додатку до МФСФ із зазначенням лікарських засобів та відповідних території(й) (ст. 6(2) ІП 520/2012 [6] та положення Порядку [2]N) (див. модуль II ННПФ). Підрядна організація може бути об’єктом аудиту системи фармаконагляду уповноваженою установоюN відповідно до положень Порядку [2]N.
Договори повинні забезпечувати дотримання юридичних вимог кожною зі сторін. Заявник (власник реєстраційного посвідчення) повинен включити в договір детальний опис делегованих завдань, відповідної взаємодії та обміну даними між сторонами, разом з, наприклад, узгодженою термінологією, інструментами, делегованими правами та строками. Договори також повинні містити чітку інформацію щодо практичного управління фармаконаглядом та пов’язаних з ним процесів, включаючи процеси підтримки баз даних з фармаконагляду. Крім того, в договорах повинні бути зазначені процеси, що діють на постійній основі, для перевірки дотримання погоджених положень. Для цього рекомендується використовувати регулярні ризик-орієнтовані аудити або інші методи контролю та оцінки фізичних та/або юридичних осіб заявником (власником реєстраційного посвідчення).
Зобов’язання заявника (власника реєстраційного посвідчення) стосовно УОВФ у даному контексті представлено у підрозділі І.С.1.1.
І.С.2. Загальні зобов’язання з фармаконагляду в рамках регуляторної політики України
МОЗ України як центральний орган виконавчої влади уповноважує спеціалізовану експертну установу у фармаконагляді з метою гарантування вжиття у разі необхідності відповідних заходів (положення Порядку [2]N).
З цією метою уповноважений орган повинен мати систему фармаконагляду (ст. 101(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N), а також запровадити та використовувати адекватну та ефективну систему якості для виконання його діяльності з фармаконагляду (ст. 8(1) ІП 520/2012 [6] та положення Порядку [2]N).
МОЗ УкраїниN та уповноважений орган повинні співпрацювати щодо постійного вдосконалення системи фармаконагляду, здатної досягти високих стандартів захисту здоров’я населення при застосуванні лікарських засобів, включаючи взаємодію для оптимального використання доступних ресурсів (положення Порядку [2]N).
Вимога, представлена у підрозділі І.В.11.2 ННПФ, згідно з якою уповноважений орган повинен забезпечити доступ до чіткого опису організаційних структур, розподілу завдань та обов’язків, а також контактних даних (ст. 14(1) ІП 520/2012 [6], положення Порядку [2]N), повинна стосуватися взаємодії між МОЗ УкраїниN, уповноваженим органом, заявниками (власниками реєстраційних посвідчень) та особами, які надають інформацію про ризики при застосуванні лікарських засобів.
Рекомендації щодо структур та процесів, які дають можливість МОЗ УкраїниN та уповноваженому органу виконувати завдання та зобов’язання з фармаконагляду, наведені у відповідних модулях ННФП.
І.С.2.1. Роль Міністерства охорони здоров’я України
Відповідно до пункту 1 Положення про Міністерство охорони здоров’я України, затвердженого постановою Кабінету Міністрів України від 25 березня 2015 р.N 267, Міністерство охорони здоров’я України (МОЗ) є центральним органом виконавчої влади, діяльність якого спрямовується і координується Кабінетом Міністрів України. МОЗ є головним органом у системі центральних органів виконавчої влади, що забезпечує формування та реалізує державну політику у сфері охорони здоров’я, захисту населення від інфекційних хвороб, протидії ВІЛ-інфекції/СНІДу та іншим соціально небезпечним захворюванням, забезпечує формування та реалізує державну політику у сфері епідеміологічного нагляду (спостереження), забезпечує формування та реалізацію державної політики у сфері створення, виробництва, контролю якості та реалізації лікарських засобів, медичних імунобіологічних препаратів і медичних виробів, у сфері обігу наркотичних засобів, психотропних речовин, їх аналогів і прекурсорів, протидії їх незаконному обігу, а також забезпечує формування державної політики у сфері санітарного та епідемічного благополуччя населенняN.
І.С.2.2. Роль та загальні функції уповноваженого органу
Уповноважений орган здійснює нагляд за безпекою лікарських засобів, дозволених до медичного застосування в Україні, та надає рекомендації щодо заходів, спрямованих на забезпечення їх безпечного та ефективного застосування, шляхом координації процесів оцінки, розробки та впровадження вимог законодавства з фармаконагляду і їх моніторингу.
Уповноважений орган забезпечує координацію діяльності, технічну, наукову та адміністративну підтримку структурного підрозділу з фармаконагляду.
Уповноважений орган здійснює фармаконагляд, залучаючи до цього процесу спеціалістів у сфері охорони здоров’я усіх закладів охорони здоров’я незалежно від форм власності, заявників (власників реєстраційних посвідчень), пацієнтів та організації, що захищають права пацієнтів.
Уповноважений орган уповноважує своїх регіональних представників організовувати та контролювати здійснення фармаконагляду на регіональному рівні за підтримки керівників усіх ланок охорони здоров’я та лікарів закладів охорони здоров’я незалежно від підпорядкування і форм власності, заявників (власників реєстраційних посвідчень), керівників та працівників органу виконавчої влади у сфері якості лікарських засобів.
Уповноважений орган відіграє провідну роль в інформаційному обміні із заявниками (власниками реєстраційних посвідчень). Зобов’язання для кожного процесу фармаконагляду наведені у відповідних модулях ННПФ.
Уповноважений орган з метою виконання завдань та досягнення цілей співпрацює з іншими установчими організаціями та структурами у разі необхідностіN.
Специфічні завдання уповноваженого органу з фармаконагляду охоплюють:
- ведення бази даних побічних реакцій/відсутності ефективності лікарських засобівN (ст. 57(d) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N);
- моніторинг відібраної медичної літератури на предмет повідомлень про побічні реакції на лікарські засоби, що містять певні діючі речовини (ст. 27 Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N) (див. модуль VI ННПФ);
- оцінку регулярно оновлюваних звітів з безпеки (див. модуль VII ННПФ) та нагляд за післяреєстраційними дослідженнями з безпеки лікарських засобів (див. модуль VIII ННПФ) (положення Порядку [2] та положення Порядку проведення експертизи реєстраційних матеріалів [7]N;
- завдання, пов’язані з виявленням сигналів (ст. 28а(1)(с) Постанови (ЄС) № 726/2004 [5], ст.ст. 18–24 ІП 520/2012 [6] та положення Порядку [2]N) (див. модуль IX ННПФ);
- подальше відстеження у часі проблем з безпеки та інших питань з фармаконагляду (див. модуль XII ННПФ);
- сприяння оперативному розповсюдженню інформації з питань безпеки лікарських засобів серед спеціалістів системи охорони здоров’я (ст. 57(е) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N) (див. модуль XV ННПФ);
- розповсюдження інформації щодо проблем з безпеки серед широкого загалу, зокрема, шляхом розміщення інформації на вебсайті уповноваженого органу (ст. 57(f) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N) (див. модуль XV ННПФ);
- оцінка оновлень систем управління ризиками (ст. 28а(1)(b) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N) (див. модуль V ННПФ);
- моніторинг результатів заходів з мінімізації ризиків (ст. 28а(1)(а) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N) (див. модуль XVI ННПФ);
- надання рекомендацій з будь-яких питань, що стосуються фармаконагляду за лікарськими засобами та систем управління ризиками, включаючи моніторинг ефективності таких систем (ст. 56(1)(аа) Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N);
- оцінка заяв та формування висновків, що є підставою для видачі реєстраційних посвідчень, внесення змін до матеріалів реєстраційного досьє, призупинення або заборону дії реєстраційних посвідчень (положення Порядку [2]та положення Порядку проведення експертизи реєстраційних матеріалів [7]N);
- підготовка висновків стосовно проблем з безпеки у післяреєстраційному періоді (положення Порядку [2]та положення Порядку проведення експертизи реєстраційних матеріалів [7]N).
І.C.2.3. Специфічні процеси системи якості уповноваженого органу
Для виконання вимог, викладених у підрозділі І.В.9.2., уповноважений орган повинен запровадити додаткові специфічні процеси системи якості з метою:
- моніторингу та валідації використання термінології, зазначеної у ст. 25(1) ІП 520/2012 [6], систематично або шляхом регулярної довільної оцінки (ст. 25(3) ІП 520/2012 [6], положення ПорядкуN);
- оцінки та обробки даних з фармаконагляду відповідно до строків, передбачених законодавством (ст. 15(1) ІП 520/2012 [6], положення Порядку [2] та положення Порядку проведення експертизи реєстраційних матеріалів [7]N);
- забезпечення ефективної комунікації відповідно до положень ст. 106а Директиви 2001/83/ЄС [1] (ст. 15(1)(d) ІП 520/2012 [6]) та положення Порядку [2]N (див. модуль XV ННПФ);
- організації зберігання основних документів, що описують системи фармаконагляду, протягом часу існування цих систем та протягом щонайменше наступних 5 років після офіційного завершення дії систем фармаконагляду (ст. 16(2) ІП 520/2012 [6] та положення наказу Міністерства юстиції України від 12 квітня 2012 р. № 578/5 «Про затвердження Переліку типових документів, що створюються під час діяльності державних органів та місцевого самоврядування, інших установ, підприємств та організацій, із зазначенням строків зберігання документів», зареєстрованого в Міністерстві юстиції України 17 квітня 2012 р. за № 571/20884N [11])N;
- забезпечення того, щоб дані з фармаконагляду та документи, що стосуються кожного окремо взятого лікарського засобу, дозволеного до медичного застосування в УкраїніN, зберігалися протягом терміну дії реєстраційного посвідчення та щонайменше протягом 10 наступних років після завершення його дії (ст. 16(2) ІП 520/2012 [6] та положення наказу Міністерства юстиції України від 12 квітня 2012 р. № 578/5 «Про затвердження Переліку типових документів, що створюються під час діяльності державних органів та місцевого самоврядування, інших установ, підприємств та організацій, із зазначенням строків зберігання документів», зареєстрованого в Міністерстві юстиції України 17 квітня 2012 р. за № 571/20884 [11])N.
У цьому контексті документи, що стосуються лікарського засобу, включають документи для референтних лікарських засобів у випадках, коли це доцільно.
1. Необхідно дотримуватися зазначених вище термінів зберігання документів, крім випадків, коли законодавство України вимагає, щоб вони були довшими (ст. 16(2) ІП 520/2012 [6] та положення наказу Міністерства юстиції України від 12 квітня 2012 р. № 578/5 «Про затвердження Переліку типових документів, що створюються під час діяльності органів державної влади та місцевого самоврядування, інших установ, підприємств та організацій, із зазначенням строків зберігання документів», зареєстрованого в Міністерстві юстиції України 17 квітня 2012 р. за № 571/20884N [11].
Протягом зазначеного вище терміну зберігання документів повинна гарантуватися можливість доступу до них.
Документи можуть зберігатися в електронному форматі за умови, що електронна система була відповідним чином перевірена/валідована, та існують належні заходи забезпечення її захисту, доступу до даних та їх резервного копіювання. Якщо документи з фармаконагляду переводяться з паперового в електронний формат, процедура переводу має гарантувати, що вся інформація оригіналу зберігається в читабельному вигляді та носії, призначеному для зберігання інформації, з часом залишатимуться придатними для зчитування.
Законодавчі вимоги до ведення документації (див. підрозділ І.В.10. ННПФ) передбачають доступ до записів у межах УкраїниN.
Крім зазначеного вище, уповноважений орган повинен запровадити процедури для збору і реєстрації усіх підозрюваних побічних реакцій, що виникають на території УкраїниN (див. модуль VI ННПФ) (ст. 15(2) ІП 520/2012 [6] та положення Порядку [2]N).
Уповноважений орган повинен запровадити процедури для моніторингу літератури відповідно до ст. 27 Постанови (ЄС) № 726/2004 [5] та положення Порядку [2]N (див. модуль VI ННПФ) (ст. 15(3) ІП 520/2012).
Крім документального оформлення системи якості згідно з І.В.11 та І.В.11.2, уповноважений орган повинен чітко визначити та, до необхідної міри, зробити доступними організаційні структури та розподіл завдань і обов’язків (ст. 14(1) ІП 520/2012 [6] та положення Порядку [2]N), а також контактні дані (ст. 14(1) ІП 520/2012 [6] та положення Порядку [2]N), зокрема, для полегшення взаємодії між уповноваженим органом, заявниками (власниками реєстраційних посвідчень) та особами, які повідомляють про ризики при застосуванні лікарських засобів для здоров’я пацієнтів та громадського здоров’я.
Аудит уповноваженого органу (див. підрозділ І.В.12) повинен здійснюватися за загальноприйнятою методологією (ст. 17(1) ІП 520/2012 [6]) (див. модуль IV ННПФ).
І.С.2.4. Прозорість системи якості уповноваженого органу
За результатами діяльності з фармаконагляду та аудитів системи якості уповноважений орган повинен оприлюднювати звіт.
І.С.3. Захист інформації в Україні
Усі вимоги законодавства, включаючи ті, що стосуються управління записами та викладені в підрозділі І.В.10, повинні застосовуватися без шкоди для зобов’язань уповноваженого органу або для зобов’язань заявників (власників реєстраційних посвідчень) щодо обробки персональних даних відповідно до положень Директиви 95/46/ЄС [12] та законодавства УкраїниN.
В Україні захист інформації гарантований Законом України «Про інформацію»; Закон України «Про захист інформації в інформаційно-телекомунікаційних системах»; Закон України «Про державну таємницю»; Закон України «Про захист персональних даних».
І.С.4. Планування готовності в УкраїніN до фармаконагляду в надзвичайних ситуаціях у галузі охорони здоров’я
Системи фармаконагляду заявників (власників реєстраційних посвідчень) та уповноваженого органу повинні бути здатними до адаптації до надзвичайних ситуацій у сфері громадського здоров’я. При необхідності повинні бути розроблені плани готовності (див. підрозділ І.В.13.).
Надзвичайна ситуація у сфері громадського здоров’я — це загроза громадському здоров’ю, офіційно визнана ВООЗ або ЄС у рамках Рішення № 2119/98/ЄС Європейського парламенту і Ради Європи [13].
Вимоги до фармаконагляду у надзвичайних ситуаціях у сфері громадського здоров’я повинні розглядатися МОЗ УкраїниN та уповноваженим органом окремо для кожного випадку та слід відповідним чином інформувати заявників (власників реєстраційних посвідчень) та громадськість шляхом оприлюднення уповноваженим органом повідомлень на його вебсайті.
Частина ІІ
Модуль II — Майстер-файл системи фармаконагляду
II.A. Вступ
Законодавчу вимогу щодо ведення та надання на запит МФСФ заявниками (власниками реєстраційного посвідчення) було введено Директивою 2010/84/ЄС [3], що внесла зміни відносно фармаконагляду до Директиви 2001/83/ЄС [1] (див. преамбулу (7) і (35), ст. 23(4), ст. 104(3)(b) Директиви 2010/84/ЄС), і Регламентом (ЄС) № 1235/2010 [4], що вніс зміни відносно фармаконагляду лікарських засобів для людини у Регламент (ЄС) № 726/2004 [5] (див. преамбулу (22) і (25), ст. 16(3а) Постанови (ЄС) № 1235/2010) з метою гармонізації та посилення діяльності з фармаконагляду в ЄС та в Україні згідно з положеннями Порядку [2]N.
Визначення МФСФ надано у ст. 1(28е) Директиви 2001/83/ЄC [1] та у Порядку [2]N, а мінімальні вимоги щодо його змісту та підтримки викладені в Імплементаційній постанові Комісії (ЄС) № 520/2012 (далі — ІП 520/2012) [6] щодо ведення діяльності з фармаконагляду, передбаченої Регламентом (ЄС) № 726/2004 [5] та Директивою 2001/83/ЄС [1] та у додатку № 11 до Порядку[2]. Детальні вимоги, викладені в Імплементаційній постанові Комісії та у додатку № 11 до Порядку [2]N, додатково обґрунтовуються настановою у даному модулі ННПФ.
МФСФ повинен бути розміщений у приміщенні за місцем знаходження, де ведеться основна діяльність з фармаконагляду заявника (власника реєстраційного посвідчення), або де працює УОВФ/КОВФN (ст. 7(1) ІП 520/2012 [6], положення Порядку [2]N).
Подання узагальнених даних про систему фармаконагляду до уповноваженого органу є вимогою до заяви на державну реєстрацію (ст. 8(3)(іа) Директиви 2001/83/ЄС та положення Порядку проведення експертизи реєстраційних матеріалів [7]N). Зазначені узагальнені дані про систему фармаконагляду повинні містити інформацію про місцезнаходження МФСФ (див. розділ II.B.2.1).
Цей модуль містить детальний опис щодо вимог до МФСФ, включаючи його ведення, зміст та надання уповноваженому органу.
II.B. Структури та процеси
Наявність МФСФ є законодавчою вимогою ЄС, УкраїниN. У даному модулі представлені вимоги до МФСФ, що застосовуються для будь-якого лікарського засобу, зареєстрованого в ЄС, в УкраїніN, незалежно від процедури реєстрації. Вимоги до змісту та ведення МФСФ діють незалежно від організаційної структури заявника (власника реєстраційного посвідчення), включаючи будь-яку субпідрядну або делеговану діяльність, чи місце знаходження, де вона здійснюється. Незалежно від того, де здійснюються інші види діяльності (окрім фармаконагляду)N, місце постійного проживання УОВФ/КОВФN, місце знаходження, де вона виконує свої функціональні обов’язки, повинні бути в межах ЄС, УкраїниN. Якщо УОВФ не проживає в Україні, на території України призначається єдина КОВФ, яка проживає і працює в Україні та підзвітна УОВФ. КОВФ повинна мати відповідний документ, що підтверджує її повноваження. МФСФ повинен знаходитися або бути доступнимN у межах УкраїниN.
Зміст МФСФ має відображати глобальну доступність інформації з безпеки лікарських засобів, дозволених до медичного застосування в ЄС, УкраїніN, представляючи інформацію щодо системи фармаконагляду, яка застосовується на глобальному, місцевому та регіональному рівні.
II.B.1. Цілі
МФСФ повинен описувати систему фармаконагляду та підтверджувати/документувати її відповідність вимогам законодавства України. Окрім дотримання вимог щодо МФСФ, викладених у положеннях Порядку [2]N та ННПФ, МФСФ повинен також сприяти належному плануванню та проведенню аудитів заявником (власником реєстраційного посвідчення), виконанню контролюючих функцій УОВФ/КОВФN і проведенню аудитів системи фармаконагляду уповноваженою установоюN чи іншого контролю за дотриманням вимог. МФСФ містить огляд системи фармаконагляду, який може вимагатися та оцінюватися уповноваженим органом під час процедури реєстрації або в післяреєстраційний період.
Шляхом створення та ведення МФСФ заявник (власник реєстраційного посвідчення) та УОВФ матимуть змогу:
- гарантувати, що система фармаконагляду була впроваджена відповідно до вимог законодавства;
- підтвердити відповідність системи фармаконагляду вимогам законодавства;
- отримати інформацію про недоліки системи фармаконагляду або недотримання вимог законодавства;
- отримати інформацію про ризики або поточні порушення у реалізації окремих напрямків здійснення фармаконагляду.
Використання цієї інформації має сприяти належному управлінню та вдосконаленню системи фармаконагляду.
Вимоги до подання заявником (власником реєстраційного посвідчення) до уповноваженого органу узагальнених даних про систему фармаконагляду, надання МФСФ та історії внесених змін повинні забезпечувати планування і ефективне проведення уповноваженим органом інспекції в ЄС та аудиту системи фармаконагляду уповноваженою установоюN в Україні на основі ризик-орієнтованого підходу.
Обов’язки щодо МФСФ з боку заявників (власників реєстраційного посвідчення), уповноважених органів та Європейського агентства з лікарських засобів (EMA) докладно описані в розділі С (див. II.C.1.) та в Україні відповідно до положень Порядку [2]N.
II.B.2. Реєстрація та ведення
II.B.2.1. Узагальнені дані про систему фармаконагляду заявника (власника реєстраційного посвідчення)
Згідно зі ст. 8(3) (іа) Директиви 2001/83/ЄС [1] та положення Порядку проведення експертизи реєстраційних матеріалів [7]N у модулі 1.8.1 заяви про державну реєстрацію лікарського засобу повинні бути представлені узагальнені дані системи фармаконагляду заявника (власника реєстраційного посвідчення), що містить наступну інформацію:
- підтвердження того, що заявник (власник реєстраційного посвідчення) має у своєму розпорядженні УОВФ/КОВФN, якщо вона відмінна від УОВФN;
- контактні дані УОВФ/КОВФN, якщо вона відмінна від УОВФN;
- гарантійний лист заявника (власника реєстраційного посвідчення) про забезпечення функціонування належної системи нагляду за безпекою лікарських засобів при їх медичному застосуванні, у тому числі в УкраїніN;
- місце здійснення основної діяльності з фармаконагляду;
- місцезнаходження МФСФ для лікарського засобу.
Від заявників (власників реєстраційного посвідчення) на реєстрацію традиційних рослинних лікарських засобів не вимагається надання узагальнених даних про їх системи фармаконагляду, однак вони зобов’язані мати діючу систему фармаконагляду та створити, вести і надавати на вимогу доступ до МФСФ (ст. 16g (1) Директиви 2001/83/ЄС [1], в Україні відповідно до положення Порядку проведення експертизи[7]N).
Для інших рослинних лікарських засобів, що не підпадають під процедуру реєстрації, як традиційні лікарські засоби, вимагається мати діючу систему фармаконагляду, створити, вести і надавати на запит МФСФ та подавати узагальнені дані про систему фармаконагляду.
Для гомеопатичних лікарських засобів, зареєстрованих за спрощеною процедурою, в Україні згідно з вимогами додатку 7 до Порядку проведення експертизи [7]N не вимагається мати систему фармаконагляду, вести і надавати на вимогу доступ до МФСФ, а також надавати узагальнені дані про систему фармаконагляду (ст. 16(3) Директиви 2001/83/ЄС [1], в Україні відповідно до положення Порядку проведення експертизи [7]N).
Для інших гомеопатичних лікарських засобів, що не підпадають під спрощену процедуру реєстрації, в Україні згідно з вимогами додатку 7 до Порядку проведення експертизи [7]N вимагається мати діючу систему фармаконагляду, створити, вести і надавати на вимогу доступ до МФСФ та надавати узагальнені дані про систему фармаконагляду (ст. 16(3) Директиви 2001/83/ЄС [1] в Україні згідно з вимогами додатку 7 до Порядку проведення експертизи [7]N).
II.B.2.2. Місцезнаходження, реєстрація та зберігання майстер-файлу системи фармаконагляду
МФСФ, незалежно від формату (паперовий або електронний), повинен зберігатися у місці, де ведеться основна діяльність з фармаконагляду заявника (власника реєстраційного посвідчення) або у місці, де працює УОВФ/КОВФN (ст. 7(1) ІП 520/2012 [6]).
У випадку, коли УОВФ територіально знаходиться поза межами України, а на території України її представляє КОВФ, то МФСФ повинен знаходитися у межах доступу, і в ньому повинна бути відображена інформація щодо здійснення нагляду за безпекою лікарських засобів на території УкраїниN.
При поданні заяви на реєстрацію заявник (власник реєстраційного посвідчення) повинен надати інформацію про розміщення МФСФ, використовуючи погоджений формат (ст. 26 1(а) ІП 520/2012 [6]) та послідовно включити в заяву референтний номер МФСФ, який є унікальним кодом, призначеним системою EudraVigilance для майстер-файлу при обробці повідомлення про лікарський засіб EudraVigilance (XEVPRM) (див.5). Після видачі реєстраційного посвідчення МФСФ буде пов’язаний заявником (власником реєстраційного посвідчення) з кодом лікарського засобу зі словника лікарських засобів EudraVigilance (EVMPD). Усі МФСФ мають бути зареєстровані в базі даних за ст. 57 Регламенту (ЄС) № 726/2004 [5]. В Україні при поданні заяви на реєстрацію заявник (власник реєстраційного посвідчення) повинен зазначити дані щодо МФСФ: його наявності, номера, місцезнаходження відповідно до положень Порядку проведення експертизи [7]. Законодавством не передбачено реєстрацію МФСФ в EudraVigilanceN.
Заявники (власники реєстраційного посвідчення) повинні продовжувати гарантувати, що внесені ними дані в базу даних за ст. 57 Регламенту (ЄС) № 726/2004 [5] для лікарських засобів для людини є оновленими, включаючи УОВФ, ім’я та контактні дані (номер телефону та факсу, поштова адреса та електронна пошта) та інформацію про місцезнаходження МФСФ (ст. 4 (4) ІП 520/2012 [6]). Після зміни інформації про УОВФ або місцезнаходження МФСФ база даних згідно зі ст. 57 Регламенту (ЄС) № 726/2004 [5] повинна оновлюватися заявником (власником реєстраційного посвідчення) одразу та не пізніше ніж за 30 днів з метою оновлення інформації в базі даних відповідно до ст. 57 Регламенту (ЄС) № 726/2004 [5] та на Європейському вебпорталі лікарських засобів, що вказано в ст. 26(1) Регламенту (ЄС) № 726/2004 [5] та постійного контролю уповноваженими органами (ст. 4 (4) ІП 520/2012 [6], ст. 57 (2) (с) Регламенту (ЄС) № 726/2004 [5] (див.6). В Україні заявник (власник реєстраційного посвідчення)повинен гарантувати, що інформація, надана ним у короткому описі системи фармаконагляду, є основною, включаючи УОВФ/КОВФ: ім’я, контактні дані (номер телефону, факсу, поштова адреса та електронна пошта) та інформація про місцезнаходження МФСФ. Після зміни інформації про УОВФ/КОВФ або місцезнаходження МФСФ заявник (власник реєстраційного посвідчення) повинен заявити такі зміни у визначеному законодавством порядку (положення Порядку проведення експертизи [7]N).
Необхідна інформація про місцезнаходження МФСФ — це дані про місцезнаходження приміщення заявника (власника реєстраційного посвідчення) або третьої сторони — підрядника. Якщо МФСФ зберігається в електронному форматі, в якості місцезнаходження повинне бути вказане місце, де ці дані безпосередньо доступні, і цього достатньо в розумінні фактичного електронного розміщення (ст. 7(3) ІП 520/2012 [6]).
При визначенні основного місця ведення діяльності з фармаконагляду заявнику (власнику реєстраційного посвідчення) слід розглянути найбільш відповідне для системи фармаконагляду місцезнаходження, оскільки відносна важливість певної діяльності може змінюватися залежно від лікарських засобів та в короткостроковій перспективі. Власник реєстраційного посвідчення повинен відповідно обґрунтувати вибір місцезнаходження МФСФ. У ситуації, коли основна діяльність ведеться за межами ЄС, України, або коли не можна визначити місце, де ведеться основна діяльність з фармаконагляду, місцезнаходженням МФСФ повинне бути приміщення, де працює УОВФ/КОВФN.
II.B.2.3. Передача відповідальності за майстер-файл системи фармаконагляду
Система фармаконагляду з часом може змінюватися. Передача або делегування обов’язків та діяльності, пов’язаних з МФСФ, повинні бути відповідним чином задокументовані (див. підрозділи II.B.4.2. та II.B.4.8.ННПФ) й організовані з тим, щоб гарантувати, що заявник (власник реєстраційного посвідчення) виконує свої зобов’язання. З моменту, коли у функціональних обов’язках УОВФ/КОВФ зазначено про відповідальність за систему фармаконагляду, УОВФ також потрібно інформувати про будь-які зміни до МФСФ з метою підтримки її повноваження щодо удосконалення системи фармаконагляду.
Зміни, про які необхідно регулярно і своєчасно інформувати УОВФ/КОВФN, включають:
- оновлення МФСФ або зміна його місцезнаходження, що доводяться до відома уповноваженого органу;
- внесення коригуючих та/або запобіжних заходів до МФСФ (наприклад, після проведення аудитів, інспекцій та аудитів системи фармаконагляду уповноваженою установою відповідно до положень Порядку [2])N). УОВФ/КОВФN повинна також мати доступ до інформації про відхилення від процесів, визначених у системі управління якістю у фармаконагляді;
- зміни змісту МФСФ, що відповідають критеріям належного нагляду за системою фармаконагляду (з точки зору ресурсів, функціонування та дотримання вимог);
- зміни умов подання МФСФ до уповноважених органів;
- передача значної частини діяльності з фармаконагляду третій стороні (наприклад складання регулярного оновлюваного звіту з безпеки третьою особою);
- включення лікарських засобів до системи фармаконагляду, за яку несе відповідальність УОВФ/КОВФN;
- зміни до лікарських засобів, охоплених системою фармаконагляду, що можуть вимагати змін або збільшення робочого навантаження з фармаконагляду, наприклад, нові показання до застосування, дослідження.
Будь-яка УОВФ/КОВФN повинна офіційно у письмовій формі отримувати інформацію про такі зміни:
- передача УОВФ/КОВФN відповідальності за систему фармаконагляду.
УОВФ/КОВФN повинна мати змогу перевірити і гарантувати, що інформація в МФСФ є точним і актуальним відображенням системи фармаконагляду, за яку вона відповідає (див. модуль I ННПФ).
II.B.3. Репрезентація системи фармаконагляду
Згідно зі ст. 1(28е) Директиви 2001/83/ЄС [1] та Порядком [2]N МФСФ повинен описувати систему фармаконагляду для одного або декількох лікарських засобів заявника (власника реєстраційного посвідчення). Для різних категорій лікарських засобів заявника (власника реєстраційного посвідчення) можуть, якщо доречно, застосовуватися окремі системи фармаконагляду. Кожна така система повинна бути описана в окремому МФСФ. Ці МФСФ повинні в сукупності охоплювати всі лікарські засоби заявника (власника реєстраційного посвідчення), які були зареєстровані відповідно до положень Директиви 2001/83/ЄС чи Постанови (ЄС) № 726/2004 [5] та положень Порядку проведення експертизи реєстраційних матеріалів [7]N:
- Вважається, що в деяких випадках заявник (власник реєстраційного посвідчення) може запровадити більше однієї системи фармаконагляду, наприклад, спеціальні системи для певних груп лікарських засобів (наприклад, вакцини, безрецептурні лікарські засоби тощо), або що системи фармаконагляду можуть охоплювати лікарські засоби більш ніж одного заявника (власника реєстраційного посвідчення). У будь-якому випадку кожну систему повинен описувати окремий МФСФ.
- Згідно зі ст.ст. 8 і 104 Директиви 2001/83/ЄС [1] та положеннями Порядку [2]N повинна бути призначена єдина УОВФ/КОВФN, яка відповідатиме за створення та підтримку системи фармаконагляду, що описана у МФСФ. Кожна система фармаконагляду заявника (власника реєстраційного посвідчення) може мати лише одну УОВФ. Якщо УОВФ не проживає в Україні, на території України призначається єдина КОВФ, яка проживає і працює в Україні та підзвітна УОВФ (положення Порядку [2]N).
- Якщо система фармаконагляду є спільною для декількох заявників (власників реєстраційного посвідчення), кожен заявник (власник реєстраційного посвідчення) несе відповідальність за гарантування наявності МФСФ для опису системи фармаконагляду, що застосовується до його лікарських засобів. Заявник (власник реєстраційного посвідчення) може делегувати (наприклад партнерові або підряднику) шляхом укладання письмової угоди частину або всю діяльність з фармаконагляду для окремого препарату(ів), за яку заявник (власник реєстраційного посвідчення) несе відповідальність. У цьому випадку в МФСФ заявника (власника реєстраційного посвідчення) може даватися посилання на весь чи окремі розділи МФСФ, що регулюється системою сторони, якій була делегована діяльність, відповідно до угоди про доступ до інформації такої системи для заявника (власника реєстраційного посвідчення) та уповноваженого органу. Заявник (власник реєстраційного посвідчення) повинен гарантувати, що зміст документації, що стосується системи фармаконагляду, на яку він посилається, застосовується до його лікарських засобів. Діяльність з підтримки МФСФ в актуальному і доступному стані може бути делегована.
- Коли доцільно, перелік усіх МФСФ, що є власністю заявника (власника реєстраційного посвідчення), повинен надаватися у додатку (див. підрозділ II.B.4.8. ННПФ) (ст. 3(7) ІП 520/2012 [6], положення Порядку [2]N), включно з даними про їх місцезнаходження, інформацією про УОВФ/КОВФN, і відповідний лікарський(і) засіб(засоби).
- Узагальнені дані (короткий опис системи ФНN), що надаються до уповноваженого органу, не можуть містити декілька місцезнаходжень для одного МФСФ. Адреса місцезнаходження МФСФ, що вимагається відповідно до ст. 8(3) Директиви 2001/83/ЄС [1] (та в межах бази даних за ст. 57 Регламенту (ЄС) № 726/2004 [5]) та положень Порядку проведення експертизи реєстраційних матеріалів [7]N, повинна бути адресою місцезнаходження, де ведеться основна діяльність з фармаконагляду заявника (власника реєстраційного посвідчення), або приміщення, де працює УОВФ/КОВФN. Ця адреса може відрізнятися від адреси заявника (власника реєстраційного посвідчення), наприклад, у випадку іншого місцезнаходження заявника (власника реєстраційного посвідчення) або коли третя сторона веде основну діяльність з фармаконагляду.
- Аналогічно, дані УОВФ/КОВФN для конкретного лікарського засобу в базі даних за ст. 57 Регламенту (ЄС) № 726/2004 [5], відповідно до положень Порядку проведення експертизи [7]N можуть бути даними УОВФ/КОВФN підрядника, який відповідає за систему фармаконагляду цього препарату, а не обов’язково УОВФ/КОВФN, безпосередньо найнятої заявником (власником реєстраційного посвідчення).
- При делегуванні будь-якої діяльності, пов’язаної із системою фармаконагляду і МФСФ, заявник (власник реєстраційного посвідчення) несе повну остаточну відповідальність за систему фармаконагляду, надання інформації про місцезнаходження МФСФ, його ведення та надання на вимогу уповноваженому органу (ст. 6 ІП 520/2012 [6] та положення ПорядкуN [2] та Порядку проведення експертизи [7]N). Повинні існувати письмові угоди, які детально описують функції та обов’язки щодо змісту МФСФ, його подання та ведення, а також регулювання здійснення фармаконагляду відповідно до вимог законодавства (на основі ст. 6 ІП 520/2012 [6] та положень Порядку [2])N.
- У випадку спільної системи фармаконагляду рекомендується, щоб партнери узгодили, як спільно вести відповідні розділи своїх МФСФ. Доступ до МФСФ для всіх відповідних заявників (власників реєстраційного посвідчення), а також його подання до уповноваженого органу повинні бути визначені в письмових угодах. Критично важливо, щоб заявник (власник реєстраційного посвідчення) міг отримати підтвердження того, що система фармаконагляду, що використовується для його лікарських засобів, є належною та відповідною.
II.B.4. Інформація, що повинна міститися в майстер-файлі системи фармаконагляду
МФСФ повинен містити принаймні всі документи, перелічені у ст. 2 ІП 520/2012 [6] та у положеннях Порядку [2].
МФСФ повинен містити документи, що описують систему фармаконагляду. Зміст МФСФ повинен відображати глобальну доступність інформації з безпеки лікарських засобів, зареєстрованих в ЄС, Україні. Зміст МФСФ повинен бути пронумерований з тим, щоб забезпечити ефективний пошук у документі, бути організованим за модульною системою, описаною в наступних розділах, і мати заголовки додатків, описані в підрозділі II.B.6.1. ННПФ. Основний принцип структури змісту МФСФ полягає в тому, що основні розділи містять інформацію, необхідну для опису системи фармаконагляду. Детальна інформація потрібна для повного опису системи, і оскільки вона може часто змінюватися, то повинна бути включена у формі посилань та міститися в додатках. Контроль, пов’язаний зі змінами змісту МФСФ, описаний у підрозділі II.B.5.ННПФ.
Допускається, що якщо раніше не видавалося реєстраційне посвідчення (і не існував МФСФ), деяка інформація у МФСФ не може бути надана на початковій стадії, наприклад, інформація про відповідність, однак замість цього повинна надаватися інформація, про те, що буде впроваджено.
II.B.4.1. Розділ майстер-файлу системи фармаконагляду щодо уповноваженої особи, відповідальної за фармаконагляд
Контактна інформація про УОВФ/КОВФ в Україні, якщо вона відмінна від УОВФN, повинна бути зазначена в заяві на реєстрацію (ст. 8(3)(іа) Директиви 2001/83/ЄС [1] та положення Порядку проведення експертизи реєстраційних матеріалів [7])N та/або в базі даних за ст. 57 Регламенту (ЄС) № 726/2004 [5].
Інформація про УОВФ, що міститься в МФСФ (ст. 2(1) ІП 520/2012 [6] та положення Порядку [2])N, повинна включати:
- перелік обов’язків для гарантування того, що УОВФ/КОВФN має достатні повноваження щодо створення системи фармаконагляду, підтримки та покращення її функціонування, у тому числі в УкраїніN;
- резюме з ключовою інформацією про роль УОВФ, включаючи підтвердження реєстрації в базі даних EudraVigilance. В Україні законодавчо не передбачена реєстрація МФСФ в базі даних EudraVigilanceN;
- контактні дані;
- відомості про резервні механізми, що будуть задіяні у випадку відсутності УОВФ;
- інформацію про обов’язки КОВФ, у тому числі в Україні, якщо вона відмінна від УОВФN, включаючи контактну інформацію про неї.
У додатки (див. підрозділ II.B.4.8. ННПФ) необхідно включити перелік завдань, делегованих УОВФ. Необхідно перерахувати діяльність, яку було делеговано, вказати, кому саме, і зазначити доступ до особи з медичною освітою у разі необхідності (див. модуль I ННПФ), ст. 10(1) ІП 520/2012 [6] та положення Порядку [2]N). Цей перелік можна представляти у вигляді копії процедурного документа за умови, що він містить усю необхідну інформацію.
Інформація, що надається стосовно УОВФ, повинна також містити дані про кваліфікацію, досвід роботи та реєстрації відносно фармаконагляду (включаючи реєстрацію в EudraVigilance; в Україні законодавчо не передбачено реєстрацію УОВФ/КОВФ в базі даних EudraVigilanceN). Контактні дані повинні включати прізвище, ім’я, по батькові, поштову адресу, телефон, факс та адресу електронної пошти, які є робочими контактними даними УОВФ/КОВФN і можуть не співпадати з адресою заявника (власника реєстраційного посвідчення). Якщо УОВФ була найнята третьою стороною, навіть якщо її робоча адреса є адресою місцезнаходження заявника (власника реєстраційного посвідчення), це необхідно зазначити і вказати назву компанії, в якій працює УОВФ.
II.B.4.2. Розділ майстер-файлу системи фармаконагляду щодо організаційної структури заявника (власника реєстраційного посвідчення)
У цьому розділі необхідно описати організаційну структуру заявника (власника реєстраційного посвідчення) відносно системи фармаконагляду. Цей розділ повинен містити опис залучених компаній, головних підрозділів з фармаконагляду та взаємозв’язків між організаціями і підрозділами, що мають відношення до виконання зобов’язань з фармаконагляду. В опис необхідно включати третіх осіб, залучених до діяльності, пов’язаної зі здійсненням фармаконагляду. Зокрема, МФСФ повинен описувати:
- організаційну структуру заявника(ів) (власника(ів) реєстраційного посвідчення) із зазначенням посади УОВФ в організації;
- місце(я), де проводиться діяльність з фармаконагляду, що включає збір повідомлень про випадки побічних реакцій, їх оцінку, введення в базу даних з безпеки, генерування регулярно оновлюваних звітів з безпеки, виявлення та аналіз сигналів, управління планом управління ризиками, а також до- та післяреєстраційними дослідженнями, управління змінами з безпеки, короткій характеристиці лікарського(их) засобу(ів)/інструкції для медичного застосування (ст. 2(2) ІП 520/2012 [6], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7])N.
Інформацію у цьому розділі можна наглядно представити у вигляді діаграм із зазначенням назви підрозділу або третьої сторони.
Делегована діяльність
МФСФ повинен містити опис делегованої заявником (власником реєстраційного посвідчення) діяльності та/або послуг з виконання зобов’язань з фармаконагляду, якщо такі мають місце (ст. 2(6) ІП 520/2012 [6], положення Порядку [2])N. Делегована діяльність включає домовленості з іншими сторонами у будь-якій країні світу та на глобальному рівні стосовно системи фармаконагляду, що застосовується до лікарських засобів, зареєстрованих у ЄС, УкраїніN.
У цьому розділі необхідно надати інформацію про зв’язки з іншими організаціями, зокрема угоди про спільну маркетингову діяльність або договори про послуги з фармаконагляду. Потрібно надати інформацію про місцезнаходження і суть договорів та угод, що стосуються виконання зобов’язань з фармаконагляду. Така інформація може бути представлена у вигляді списку/таблиці із зазначенням залучених сторін, їх обов’язків та відповідних лікарських засобів і країн. Список повинен бути сформований відповідно до: постачальників послуг (наприклад, медична інформація, аудитори, організатори програм підтримки пацієнтів, управління даними досліджень тощо), комерційних угод (дистриб’ютори, партнери, спільний маркетинг тощо) та інших технічних послуг (хостинг комп’ютерних систем тощо). У додатках надають список договорів (див. підрозділ II.B.4.8.ННПФ), а самі договори повинні бути доступні на запит уповноваженого органу або під час проведення аудитів, інспекцій, аудиту системи фармаконагляду уповноваженою установоюN.
II.B.4.3. Розділ майстер-файлу системи фармаконагляду щодо джерел даних з безпеки
Опис основних підрозділів зі збору даних з безпеки повинен включати інформацію про всі відповідальні сторони на глобальному рівні для організованого та спонтанного збору даних з безпеки лікарських засобів. Такий опис повинен включати медичні інформаційні сайти, а також філіали організації, і може бути викладений у формі переліку із зазначенням країни, характеру діяльності і лікарського засобу(ів) (якщо діяльність пов’язана із конкретним лікарським засобом) та контактних даних (адреса, телефон і електронна пошта) дільниці. Цей перелік може бути розміщений у додатках МФСФ. Інформація про треті сторони (партнерів або локальні угоди щодо дистрибуції/маркетингових заходів) також повинна бути включена до розділу, що описує договори і угоди (див. II.B.4.2. та II.B.4.8 ННПФ).
Інформацію можна представляти у вигляді блок-схем із зазначенням основних етапів, строків та залучених сторін. Незалежно від представлення опис процесів від збору до подання повідомлень про побічні реакції до уповноважених органів повинен відображати залучені підрозділи та/або треті сторони.
З метою аудиту, інспекції, аудиту системи фармаконагляду уповноваженою установоюN джерела даних з безпеки повинні включати дані, що надходять з досліджень, включаючи будь-які дослідження, реєстри, програми спостережень чи підтримки, що фінансуються заявником (власником реєстраційного посвідчення), за допомогою яких можна збирати повідомлення про побічні реакції. Заявники (власники реєстраційного посвідчення) повинні мати змогу підготувати та надати перелік таких джерел при проведенні аудиту, інспекції, аудиту системи фармаконагляду уповноваженою установоюN та контролю УОВФ/КОВФN. З метою гармонізації рекомендується, щоб перелік був всеохоплюючим для зареєстрованих лікарських засобів, незалежно від їх показань, форми випуску чи способу застосування. У переліку повинні бути зазначені на глобальному рівні статус кожного дослідження/програми, задіяні країни, лікарський(і) засіб(засоби) і головні цілі. Потрібно також зазначити, чи є дослідження інтервенційним, чи неінтервенційним, а сам перелік повинен бути сформований за міжнародною непатентованою назвою діючої речовини лікарського засобу. Перелік повинен бути вичерпним та включати усі поточні дослідження/програми, а також дослідження/програми, завершені протягом останніх двох років, і може міститися в додатку або надаватися окремо.
II.B.4.4. Розділ майстер-файлу системи фармаконагляду щодо комп’ютеризованих систем і баз даних
У МФСФ повинні бути описані розташування, функціональність та практична відповідальність за комп’ютеризовані системи і бази даних, що використовуються для отримання, обробки, запису та надання інформації з безпеки, а також представлена оцінка придатності таких систем і баз даних для виконання їх мети (ст. 2(3) ІП 520/2012 [6], положення Порядку [2])N.
Якщо використовуються декілька комп’ютеризованих систем/баз даних, їх придатність для діяльності з фармаконагляду повинна бути описана таким чином, щоб надати чітке уявлення про ступінь комп’ютеризації в рамках системи фармаконагляду. Також повинен бути описаний валідаційний статус ключових аспектів роботи комп’ютерної системи; інформація про процедури контролю внесення змін, характер тестування, процедури резервування та електронні сховища даних, що важливі для відповідності фармаконагляду, має бути включена в узагальнену інформацію, та повинен бути описаний характер доступної документації. Для систем на основі паперових носіїв (коли електронна система може використовуватися тільки для подачі термінових повідомлень про побічні реакції) слід описати управління даними і механізми забезпечення цілісності та доступності даних про безпеку, зокрема, узагальнення інформації про побічні реакції лікарських засобів.
II.B.4.5. Розділ майстер-файлу системи фармаконагляду щодо процесів фармаконагляду
Важливим елементом будь-якої системи фармаконагляду є наявність чітких письмових процедур. Модуль I ННПФ описує необхідний мінімальний набір письмових процедур для здійснення фармаконагляду. У МФСФ повинні міститися опис доступної процедурної документації (стандартні операційні процедури, інструкції на центральному та/або національному рівнях тощо), тип даних, що зберігаються (наприклад, тип даних випадку, що зберігається для повідомлень про побічні реакції), та спосіб ведення записів (наприклад, база даних з безпеки, паперові документи на дільниці отримання).
У МФСФ має міститися опис процесів, обробки даних і записів для здійснення фармаконагляду, що охоплює наступні аспекти:
- безперервний моніторинг профілю користь/ризик лікарського(их) засобу(ів), результатів оцінки, процесу прийняття рішень для вжиття відповідних заходів; генерація сигналу, його виявлення й оцінка. А також ряд письмових процедур та інструкцій, що стосуються вихідних даних бази даних з безпеки, взаємодії з клінічними департаментами тощо;
- систему(и) управління ризиками і моніторинг результатів заходів з мінімізації ризиків; до даної діяльності можуть бути залучені декілька підрозділів, і їх взаємодія повинна бути регламентована в письмових процедурах або угодах;
- збір повідомлень про індивідуальні випадки, пов’язані з безпекою, узагальнення даних повідомлень, подальше відстеження (follow-up), оцінку та звітування; процедури, що при цьому застосовуються, повинні уточнювати діяльність на глобальному та національному рівнях;
- планування термінів генерації регулярно оновлюваних звітів з безпеки, їх підготовка та подання (див. модуль VII ННПФ);
- інформування з питань безпеки споживачів, спеціалістів системи охорони здоров’я і уповноваженого органу;
- внесення змін з безпеки до короткої характеристики лікарського засобу/інструкції для медичного застосування; процедури повинні охоплювати як внутрішню, так і зовнішню комунікацію (ст. 2(4) ІП 520/2012 [6], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7])N.
Стосовно кожного з цих аспектів заявник (власник реєстраційного посвідчення) повинен бути в змозі надати докази існування системи, що забезпечує належне і своєчасне прийняття рішень і вжиття заходів.
Опис повинен супроводжуватися переліком процесів, зазначених у ст. 11(1) ІП 520/2012 [6], положення Порядку [2]N щодо контролю відповідності, а також взаємодії з іншими функціями. Взаємодія з іншими функціями включає (однак не обмежується) функціональні обов’язки та відповідальність УОВФ/КОВФN, відповіді на запити уповноваженого органу про надання інформації, літературний пошук, медичну наукову літературуN, контроль змін у базі даних з безпеки, договір про обмін даними з безпеки, архівування даних з безпеки, аудит фармаконагляду, контроль якості і навчання персоналу. Цей перелік може бути розміщений у додатках і повинен містити реєстраційний номер процедурного документа, заголовок, дату набуття чинності і тип документа (для усіх стандартних операційних процедур, робочих інструкцій, інструкцій тощо). Повинні бути чітко ідентифіковані процедури, що стосуються постачальників послуг та інших третіх сторін. До переліку не вимагається включати документи, що стосуються відповідних вимог законодавства країни, де зареєстровано лікарський засіб, охоплений цією системою фармаконагляду, але їх перелік може вимагатися на законодавчому рівні країни, де зареєстровані лікарські засоби, що охоплені цією процедурою. Якщо одна або лише деякі країни використовують спеціальні місцеві (національні) процедури, це має бути вказано із зазначенням, де зареєстровані лікарські засоби, що охоплені цією системою.
II.B.4.6. Розділ майстер-файлу системи фармаконагляду щодо ефективності (продуктивностіN) системи фармаконагляду
МФСФ повинен містити дані про проведення постійного моніторингу ефективності (продуктивностіN) системи фармаконагляду, у тому числі дотримання вимог щодо основних результатів фармаконагляду. МФСФ повинен включати опис методів моніторингу, що застосовуються, і містити щонайменше:
- опис процедури оцінки коректності подання повідомлень про індивідуальні випадки, пов’язані з безпекою. У додатку повинні бути надані цифрові дані/графіки, що демонструють своєчасність подання звітів у 15- та 90-денний строк у попередньому році;
- опис цільових показників, що використовуються для контролю якості подання даних та здійснення фармаконагляду. Це включає інформацію, надану уповноваженим органом щодо якості наданих повідомлень про індивідуальні випадки, пов’язані з безпекою, регулярно оновлюваних звітів з безпеки чи іншої інформації, що подається заявником (власником реєстраційного посвідчення);
- загальні відомості про своєчасність подання регулярно оновлюваних звітів з безпеки до уповноважених органів (додаток повинен містити останні дані, що використовувалися заявником (власником реєстраційного посвідчення) для оцінки відповідності);
- короткий опис методів, що використовуються для забезпечення своєчасності подання заяв на зміни з безпеки у порівнянні з внутрішніми граничними строками та строками, передбаченими вимогами законодавства, а також відслідковування необхідних змін з безпеки, що були виявлені, але заяви на які ще не були подані;
- у відповідних випадках надаються узагальнені дані щодо дотримання зобов’язань з виконання плану управління ризиками або інших зобов’язань чи умов, що стосуються фармаконагляду, дотримання яких було умовою видачі реєстраційного посвідчення.
Необхідно описати і пояснити цільові показники ефективності (продуктивностіN) системи фармаконагляду. Перелік показників ефективності (продуктивностіN) потрібно надати в додатку до МФСФ (ст. 3(6), ст. 9 ІП 520/2012 [6], положення Порядку [2]N разом з фактичними результатами оцінки такої ефективності (продуктивностіN).
II.B.4.7. Розділ майстер-файлу системи фармаконагляду про систему якості
Опис системи управління якістю потрібно представити в рамках структури організації і застосування системи якості до фармаконагляду. Такий опис повинен включати:
Контроль документації та записів
Потрібно надати опис механізмів архівування електронних та/або друкованих версій МФСФ, а також короткий опис процедур, що застосовуються до інших систем якості та записів і документів з фармаконагляду (див. також модуль I ННПФ).
Процедурні документи
- Загальний опис документів, що використовуються у фармаконагляді (стандарти, операційні процедури, робочі інструкції тощо), доступність різних документів на глобальному, регіональному або локальному рівні в рамках організації, а також методів контролю їх доступності, впровадження та супроводу.
- Інформація про системи документації, що використовуються для відповідних процесуальних документів під контролем третіх осіб.
Необхідно надати перелік спеціальних процедур і процесів, пов’язаних з діяльністю з фармаконагляду та взаємодією з іншими функціональними підрозділами, з докладною інформацією про те, як ці процедури можуть бути оцінені (ст. 2(5)(а) ІП 520/2012 [6], положення додатку 11 до Порядку [2]N). Детальна інструкція про їх включення описана в підрозділі II.B.4.5 ННПФ.
Навчання
- Опис управління ресурсами для виконання діяльності з фармаконагляду:
- організаційна структура із зазначенням кількості співробітників (повної зайнятості), залучених до діяльності з фармаконагляду, що може бути надана в розділі, що описує організаційну структуру (див. підрозділ II.B.4.3 ННПФ).
- Інформація про місцезнаходження персоналу (ця інформація описана в розділах II.B.4.2 і II.B.4.3 ННПФ), зазначена у МФСФ відповідно до організації кожного виду діяльності з фармаконагляду, і у додатку, в якому зазначена контактна інформація щодо місцезнаходження джерел даних з безпеки. Однак слід надати пояснення про організацію навчання відповідно до інформації про персонал та його місцезнаходження.
- Короткий опис концепції навчання, у тому числі посилання на розміщення навчальних файлів.
Персонал (не тільки співробітники підрозділів з фармаконагляду, а й будь-які працівники, які можуть отримувати повідомлення про індивідуальні випадки, пов’язані з безпекою) повинен бути відповідно навчений для здійснення діяльності з фармаконагляду.
Аудит
Інформація про аудит забезпечення якості у системі фармаконагляду повинна бути включена до МФСФ. У додатку необхідно надати опис підходу, що використовується для планування аудитів системи фармаконагляду, механізму звітності і строків, а також поточний перелік запланованих і проведених аудитів, що стосуються системи фармаконагляду, відповідно до підрозділу II.B.4.8 (ст. 3(5) ІП 520/2012 [6], положення Порядку [2]N. Цей перелік повинен містити дату проведення аудиту, дату подання звіту, мету і стан здійснення аудитів постачальників послуг, специфічних видів діяльності з фармаконагляду або підрозділів, що здійснюють діяльність з фармаконагляду, та їх взаємодію з іншими підрозділами організації, що мають відношення до виконання зобов’язань з фармаконагляду, зазначених у Директиві 2001/83/ЄС [1], положеннях Порядку [2]N, і повинен охоплювати 5-річний період.
МФСФ повинен також містити коротку інформацію про всі аудити, у результаті яких було виявлено значимі результати. Це означає, що необхідно вказати наявні результати, що підпадають під критерії суттєвих або критичних невідповідностей (див. модуль IV ННПФ). Звіт з аудиту повинен бути задокументований у системі якості, а в МФСФ достатньо надати короткий опис коригуючих та/або запобіжних заходів щодо значимих результатів аудиту, дату їх виявлення та очікувану дату(и) їх усунення з перехресним посиланням на звіт аудиту та задокументований план(и) коригуючих та запобіжних заходів. У випадку, якщо плани коригуючих та/або запобіжних заходів або результати не погоджені для певного аудиту, до МФСФ слід включити необхідну примітку, що вказує «плани коригуючих або запобіжних заходів мають бути погоджені». У додатку в переліку проведених аудитів слід вказати аудити, що позначені приміткою «непогоджені» у МФСФ. Примітка та пов’язані коригуючі та/або запобіжні заходи повинні міститися в МФСФ до тих пір, поки коригуючі та/або запобіжні заходи не будуть вжиті в повному обсязі. Тобто примітка видаляється лише після того, як були застосовані відповідні заходи та/або підтверджено незалежною стороною істотне покращення системи (ст. 104(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Доповнення, зміни або видалення інформації про аудити повинні реєструватися в журналі коригувань.
В якості засобу управління системою фармаконагляду та підстави для проведення аудиту, інспекції або аудиту системи фармаконагляду уповноваженою установоюN в МФСФ слід також описувати процес обліку, управління та усунення виявлених відхилень у системі якості. У МФСФ слід також документувати відхилення від процедур фармаконагляду, їх вплив і управління ними до моменту їх вирішення (ст. 4(3) ІП 520/2012 [6], положення Порядку [2]N). Ці відхилення можна документувати у вигляді списку з посиланням на звіт про відхилення, його дату та відповідну процедуру.
II.B.4.8. Додаток до майстер-файлу системи фармаконагляду
Додаток до МФСФ повинен містити такі документи:
- Перелік лікарських засобів, на які розповсюджується МФСФ, включаючи назву лікарського засобу, міжнародну непатентовану назву діючої речовини та назву країни, в якій є діюча реєстрація (ст. 3 ІП 520/2012 [6], положення Порядку [2]N).
Перелік лікарських засобів повинен також включати реєстраційний номер та інформацію про:
- тип процедури реєстрації лікарського засобу та номер процедури (наприклад лікарський засіб зареєстрований за централізованою процедурою, на національному рівні, включаючи ті, які зареєстровані за процедурою взаємного визнання та децентралізованою процедурою);
- наявність на фармацевтичному ринку ЄС, УкраїниN (ст. 23(а) Директиви 2001/83/ЄС [1], ст. 13(4) Регламенту (ЄС) № 726/2004 [5], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7]N);
- інші країни, де лікарський засіб зареєстрований чи наявний на фармацевтичному ринку.
Перелік повинен бути структурований за міжнародною непатентованою назвою діючої речовини, і у відповідних випадках необхідно зазначити специфічні для лікарського засобу вимоги до моніторингу безпеки (наприклад, заходи з мінімізації ризиків, що містяться в плані управління ризиками або були умовою видачі реєстраційного посвідчення, нестандартна періодичність генерації регулярно оновлюваних звітів з безпеки, реферативна процедура за ст. 31 Директиви 2001/83/ЄС [1] або включені в перелік, описаний у ст. 23 Регламенту (ЄС) № 726/2004 [5] та ін.). Більш детальна інформація про моніторинг може бути надана окремим списком.
Для лікарських засобів, що включені у різні системи фармаконагляду, наприклад, коли заявник (власник реєстраційного посвідчення) має більш ніж одну систему фармаконагляду або у випадку делегування системи фармаконагляду за угодою третій стороні, у додатках окремим списком необхідно надати посилання на інші МФСФ таким чином, щоб вся номенклатура лікарських засобів заявника (власника реєстраційного посвідчення) співвідносилася з відповідними МФСФ.
У випадку ведення спільних систем фармаконагляду необхідно включити всі лікарські засоби, для яких використовується така система фармаконагляду, щоб був доступним увесь перелік лікарських засобів, включених до МФСФ. Ці переліки можуть бути представлені у вигляді окремих списків, згрупованих за заявником(ами) (власником(ами) реєстраційного посвідчення) для кожного лікарського засобу, або може включатися окрема примітка для опису лікарського(их) засобу(ів) та заявників (власників реєстраційного посвідчення).
- Перелік письмових норм і процедур з метою відповідності ст. 11(1) ІП 520/2012 (ст. 3 ІП 520/2012) [6], положення Порядку [2]N.
- Перелік угод, що стосуються делегування діяльності, включаючи лікарські засоби та територію(ї), у відповідності зі ст. 6(2) ІП 520/2012 (див. ІІ.В.4.3) (ст. 3(3) ІП 520/2012) [6], положення Порядку [2]N.
- Перелік завдань, делегованих УОВФ/КОВФ (ст. 3(4) ІП 520/2012 [6], положення Порядку [2]N).
- Список усіх завершених аудитів за 5-річний термін та список графіків проведення аудитів (ст. 3(5) ІП 520/2012 [6], положення Порядку [2]N).
- Перелік показників ефективності (продуктивності) діяльності з фармаконагляду організації (якщо доречно) відповідно до ст. 9 ІП 520/2012 [6] (ст. 3(6) ІП 520/2012), положення Порядку [2]N.
- Перелік інших МФСФ, якщо такі ведуться тим же заявником (власником реєстраційного посвідчення) (ст. 3(7) ІП 520/2012 [6], положення Порядку [2]N).
Цей перелік повинен містити номер(и) МФСФ(ів), назву заявника (власника реєстраційного посвідчення) УОВФ/КОВФN, який відповідає за систему фармаконагляду, що застосовується. Якщо до управління системою фармаконагляду залучена третя сторона, що не є заявником (власником реєстраційного посвідчення), необхідно також зазначити назву постачальника послуг.
- Журнал коригувань згідно зі ст. 5(4) ІП 520/2012 (ст. 3(8) ІП 520/2012) [6], положення Порядку [2]N. Іншу документацію щодо контролю змін слід включати у разі необхідності. При документуванні змін у МФСФ необхідно вказати принаймні дату зміни, прізвище, ім’я, по батькові особи, відповідальної за таку зміну, суть зміни (ст. 5(4) ІП 520/2012 [6], положення Порядку [2]N).
II.B.5. Контроль змін, журнал коригувань, версії та архівування
Заявникам (власникам реєстраційного посвідчення) необхідно створити системи контролю змін та мати надійні процеси, що дозволяють постійно бути поінформованими про відповідні зміни з метою належного ведення МФСФ. Уповноважений орган може вимагати інформацію про важливі зміни в системі фармаконагляду. Такі важливі зміни включають, однак не обмежуються, наступними:
- зміни в базі(ах) даних з безпеки або в базах даних, безпосередньо пов’язаних з нею, валідаційного статусу бази даних, а також інформації про передані або перенесені на іншу платформу дані;
- зміни у наданні значних послуг у сфері фармаконагляду, особливо договори, що стосуються подання даних з безпеки;
- організаційні зміни, такі як поглинання, злиття, зміни у приміщеннях, де здійснюється фармаконагляд, або делегування/передача управління МФСФ.
Ці зміни необхідно документувати в МФСФ для контролю змін (у журналі коригувань), і УОВФ/КОВФN повинна завжди бути поінформована про такі зміни.
Зміни до МФСФ повинні реєструватися таким чином, щоб можна було прослідкувати історію змін (із зазначенням дати та суті змін). Опис змін МФСФ повинен міститися в журналі коригувань, описаному у ст. 5(4) ІП 520/2012 [6], положеннях Порядку [2]N.
Історія змін інформації, що міститься в додатках, може бути «на вимогу». У цьому випадку журнал коригувань повинен містити дату перегляду вмісту МФСФ та/або оновлення додатку(ів), необхідно також оновити історію внесення змін у додатки. Інформація, що регулярно оновлюється і міститься в додатках, така як переліки лікарських засобів та стандартних операційних процедур або показники відповідності, може отримуватися з контрольованих систем (наприклад, таких як системи електронного документообігу або бази даних). Версії цих документів можуть підтримуватися окремо від МФСФ за умови, що ведеться історія їх змін і вона може бути надана на вимогу уповноважених органів. Якщо МФСФ не запитувався або залишається незмінним протягом певного часу (наприклад, коли зміни у змісті додатків ведуться поза межами МФСФ), рекомендується проводити його регулярний перегляд. Заявники (власники реєстраційного посвідчення) повинні гарантувати виконання зобов’язання щодо своєчасного надання МФСФ. УОВФ/КОВФN повинна мати доступ до актуальної та точної інформації про систему фармаконагляду, а тому їй повинен бути забезпечений постійний доступ до МФСФ, у тому числі до інформації, що міститься в додатках (безпосередньо через МФСФ або ж через доступ до систем, що використовуються для створення додатків).
Заявники (власники реєстраційного посвідчення) повинні бути в змозі обґрунтувати обраний метод і розробити процедури контролю документації з метою належного управління процесом ведення МФСФ. Як основа для аудиту, інспекції, аудиту системи фармаконагляду уповноваженою установоюN МФСФ повинен містити опис поточного стану системи фармаконагляду, однак може виникати потреба в розумінні того, як функціонувала система фармаконагляду в минулому.
Зміни до МФСФ повинні також враховувати спільні системи фармаконагляду та делеговану діяльність. З метою забезпечення повноти контролю змін необхідно реєструвати та зберігати інформацію про дату та суть повідомлень про зміни, що надані до уповноважених органів, УОВФ/КОВФN та відповідним третім сторонам.
МФСФ повинен зберігатися у спосіб, що забезпечує його точність і доступність (ст. 5 і ст. 7 ІП 520/2012 [6], положення Порядку [2])N.
II.B.6. Представлення майстер-файлу системи фармаконагляду
УОВФ/КОВФN повинна мати постійний доступ до МФСФ (ст. 7(2) ІП 520/2012 [6], положення Порядку [2]N), а уповноважений орган — доступ на вимогу (ст. 16(3а) Постанови (ЄС) № 726/2004 [5], ст. 23(4) Директиви 2001/83/ЄС [1], ст. 7 ІП 520/2012 [6], положення Порядку [2]N). Інформація, що міститься у МФСФ, повинна бути лаконічною, точною і відображати поточний стан системи фармаконагляду. Незалежно від формату МФСФ повинна існувати можливість підтримувати інформацію в актуальному стані і, за необхідності, вносити зміни з урахуванням накопиченого досвіду, технічного і наукового прогресу та змін до вимог законодавства (ст. 4(1) ІП 520/2012 [6], положення Порядку [2]N). МФСФ повинен бути наданий протягом 7 днів на вимогу уповноваженого органу (ст. 23(4) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Проте уповноважений орган може також вимагати негайного доступу до МФСФ або з місцезнаходження МФСФ або з місця роботи УОВФ або КОВФ в Україні (якщо відрізняються)N.
II.B.6.1. Формат та структура
МФСФ може вестися в електронному форматі за умови, що на запит уповноваженого органу може бути видана впорядкована друкована версія (ст. 5(3) ІП 520/2012 [6], положення Порядку [2]N). Незалежно від формату інформація, що міститься у МФСФ, повинна бути розбірливою, повною, викладеною у спосіб, що забезпечує доступність всієї документації і дозволяє повністю відслідковувати зміни. Доцільно обмежити доступ до МФСФ з метою забезпечення належного контролю за його змістом та призначити спеціальні обов’язки з управління МФСФ стосовно управління змінами та архівування.
МФСФ повинен бути викладений українською чиN англійською мовою, проіндексований відповідно до заголовків, описаних у даному модулі (ст. 5(2) ІП 520/2012 [6], положення Порядку [2]N), і забезпечувати простоту навігації за змістом. У разі викладення МФСФ англійською мовою в Україні також повинен бути наявний переклад розділів МФСФ українськоюN. Рекомендується використання електронних закладок і системи пошуку за текстом. Такі документи, як копії підписаних заяв або угод, повинні бути включені у якості додатків і описані в алфавітно-предметному покажчику.
Документи, що входять до МФСФ, та сам МФСФ повинні бути представлені з наступними заголовками, а у друкованому варіанті — у такому порядку:
Титульний аркуш повинен містити:
- унікальний номер; унікальний номер, призначений Системою EV майстер-файлу системи фармаконагляду, коли XEVPRM обробляється у XEVMPD (база даних ст. 57 Регламенту (ЄС) № 726/2004 [5]). Реєстрація в базі даних EudraVigilance на території України не передбачена вимогами законодавства;
- назву заявника (власника реєстраційного посвідчення), назву заявника (власника реєстраційного посвідчення) уповноваженої особи, відповідальної за описану систему фармаконагляду (якщо відрізняється), а також назву третьої сторони уповноваженої особи, відповідальної за описану систему фармаконагляду (якщо застосовується);
- назви інших зацікавлених заявників (власників реєстраційного посвідчення) (у разі спільного використання системи фармаконагляду);
- перелік МФСФ заявника (власника реєстраційного посвідчення) (для лікарських засобів, що мають різні системи фармаконагляду);
- дату складання/останнього оновлення.
Заголовки, описані у розділі II.B.4 ННПФ, повинні використовуватися для упорядкування основного змісту МФСФ. Мінімальний необхідний зміст додатків описано в підрозділі II.B.4.8 ННПФ. У додатки може бути включена додаткова інформація за умови дотримання вимог щодо змісту основних розділів (II.B.1-7). Нижче описується порядок формування додатків; пункти списку є описом можливого змісту (а не обов’язкових заголовків):
УОВФ, додаток А
- Список завдань, які були делеговані уповноваженою особою, відповідальною за фармаконагляд, або відповідний процедурний документ.
- Резюме УОВФ, і пов’язані з ним документи.
- Контактна інформація (у тому числі дані про КОВФ)N. За необхідності, контактна інформація, додаткова до тієї, що міститься у базі даних, що зазначена у ст. 57 Регламенту (ЄС) № 726/2004 [5].
Організаційна структура заявника (власника реєстраційного посвідчення), додаток В
- Списки договорів і угод.
Джерела даних з безпеки, додаток C
- Переліки з описом джерел даних з безпеки, наприклад, партнерів та третіх сторін.
Комп’ютеризовані системи і бази даних, додаток D
Процес фармаконагляду та процедури у письмовому вигляді, додаток Е
- Переліки процедурних документів
Ефективність (продуктивністьN) системи фармаконагляду, додаток F
- Переліки показників ефективності.
- Поточні результати оцінки ефективності по відношенню до показників ефективності.
Система контролю якості у фармаконагляді, додаток G
- Графік аудитів.
- Перелік аудитів, що проводяться, та тих, що завершені.
Лікарські засоби, додаток H
- Перелік(и) лікарських засобів, що охоплені даною системою фармаконагляду.
- Будь-які зауваження щодо заявника (власника реєстраційного посвідчення) щодо кожного лікарського засобу.
Контроль документації та записів, додаток I
- Журнал коригувань.
- Документація щодо історії змін змісту додатків, проіндексована відповідно до порядку додатків, та їх зміст, якщо не надається безпосередньо у відповідному додатку.
Документація щодо повідомлень і підписів, що стосуються МФСФ, якщо передбачено. Якщо вміст додатку відсутній, немає потреби подавати порожні сторінки з заголовками, однак, додатки, які надаються, повинні бути названі відповідно до описаного формату. Наприклад, додаток Е не може бути перейменований у додаток D у випадку, коли додаток про комп’ютеризовані системи і бази даних не використовується, додаток D повинен просто бути позначений як «невикористаний» у покажчику для того, щоб одержувачі МФСФ були впевнені, що такий додаток відсутній не в результаті помилки.
II.C. Функціонування систем фармаконагляду в ЄС, УкраїніN
II.C.1. Обов’язки
II.C.1.1. Заявники (власники реєстраційних посвідчень)
Заявники (власники реєстраційного посвідчення) повинні мати систему фармаконагляду з метою забезпечення контролю та нагляду за безпекою лікарських засобів. Вони також несуть відповідальність за створення та ведення МФСФ, що містить дані про систему фармаконагляду щодо одного або декількох лікарських засобів (ст. 23(4), ст. 104(3)(b) Директиви 2001/83/ЄС [1], ст. 16(3а) Постанови (ЄС) 726/2004 [5]). Згідно зі ст.ст. 8(3)(ia) та 104(3) Директиви 2001/83/ЄС [1], положень Порядку [2]N) повинна бути призначена єдина уповноважена особа, відповідальна за створення та супровід системи фармаконагляду, описаної в МФСФ.
На момент подання заяви на отримання реєстраційного посвідчення заявники (власники реєстраційного посвідчення) повинні мати короткий опис системи фармаконагляду, що буде використовуватися з моменту отримання реєстраційного посвідчення та розміщення лікарського засобу на фармацевтичному ринку країни. Під час проведення експертизи реєстраційних матеріалів від заявника (власника реєстраційного посвідчення) може вимагатися надання копії МФСФ для оцінки.
Заявник (власник реєстраційного посвідчення) несе відповідальність за створення МФСФ у країні ЄС, УкраїніN (на будь-якому місці заявника (власника реєстраційного посвідчення) або партнера, у тому числі — підрядника або маркетингового партнера) та за зазначення місцезнаходження МФСФ у заяві на реєстрацію лікарського засобуN та у базі даних за ст. 57 Регламенту (ЄС) № 726/2004 [5]. МФСФ відповідно до положень Порядку проведення експертизи [7]N повинен описувати існуючу на поточний момент систему фармаконагляду. У МФСФ може бути включена інформація про елементи системи, що будуть реалізовані в майбутньому, але вони повинні бути чітко зазначені як заплановані, а не як впроваджені чи поточні.
Створення та підтримка МФСФ в актуальному і доступному стані (постійно доступному з метою проведення аудитів, інспекції, аудиту системи фармаконагляду уповноваженою установоюN і його подання до уповноваженого органу може бути доручене третій стороні, проте заявник (власник реєстраційного посвідчення) несе повну остаточну відповідальність за дотримання вимог законодавства.
Заявник (власник реєстраційного посвідчення) також несе відповідальність за негайне повідомлення уповноваженого органу про будь-які зміни даних про УОВФ (або КОВФ)N, а також зміни у місцезнаходженні МФСФ. EMA повинна поновити відповідні дані у базі даних EudraVigilance, як зазначено у cт. 24(1) Регламенту (ЄС) № 726/2004 [5] та, за необхідності, на вебпорталі EMA, що зазначено у cт. 26(1) Регламенту (ЄС) № 726/2004 [5] (cтаття 4(4) ІП 520/2012 [6]).
В Україні заявник (власник реєстраційного посвідчення) у разі будь-яких змін даних про УОВФ/КОВФ а також у разі зміни місцезнаходження повинен заявити такі зміни у визначеному законодавством порядку (положення Порядку проведення експертизи [7]N).
II.C.1.2. Уповноважений орган
Уповноважений орган зобов’язаний здійснювати нагляд за системами фармаконагляду заявників (власників реєстраційного посвідчення) (декларативна частина 7 Директиви 2010/84/ЄС [3], положення Порядку [2])N. Відповідно до цієї вимоги уповноважений орган повинен проводити експертизу узагальнених даних про систему фармаконагляду, включених до заяви на реєстрацію лікарського засобу. Повний МФСФ може вимагатися в будь-який момент, наприклад, для перевірки опису системи фармаконагляду заявника (власника реєстраційного посвідчення), який раніше не реєстрував лікарські засоби в УкраїніN або коли існують специфічні питання з приводу системи фармаконагляду та/або профілю безпеки лікарських засобів, а також у рамках підготовки до інспекції, аудиту системи фармаконагляду уповноваженою установоюN (див. модуль III ННПФ). Інформація про зміни в узагальнених даних або змісті МФСФ буде також використовуватися для планування та проведення інспекцій, аудиту системи фармаконагляду уповноваженою установоюN.
Що стосується лікарських засобів, що були зареєстровані за централізованою процедурою, держава ЄС, у якій розміщений майстер-файл, буде контролюючим органом (декларативна частина 22 Регламенту 1235/2010 [4], ст. 18(3) Регламенту (ЄС) № 726/2004 [5]). Відносно систем фармаконагляду, що включають лікарські засоби, зареєстровані за централізованою процедурою, а також національною процедурою, включаючи лікарські засоби, зареєстровані за процедурою взаємного визнання або децентралізованою процедурою, національні уповноважені органи контролюють систему фармаконагляду у співпраці з контролюючим органом та EMA. Що стосується систем фармаконагляду, які не включають лікарські засоби, зареєстровані за централізованою процедурою, національні уповноважені органи країн залишаються відповідальними за контроль системи фармаконагляду та співпрацюють для мінімізації дублювання зусиль.
Національні уповноважені органи спільно застосовують інформацію про системи фармаконагляду та використовують інформацію для інформування національних програм інспекцій, аудитів системи фармаконагляду уповноваженою установоюN, що проводяться на підставі оцінки ризику. Інспектори/аудиториN уповноважених органів/установN повідомлятимуть про недотримання вимог законодавства та ННПФ, включаючи недотримання вимог до МФСФ та системи фармаконагляду (див. модуль III ННПФ).
II.C.1.3. Європейська агенція лікарських засобів/Міністерство охорони здоров’я УкраїниN
Відносно лікарських засобів, зареєстрованих за централізованою процедурою, EMA координує інспекції заявників (власників реєстраційного посвідчення) або їх постачальників послуг. Нагляд за системою фармаконагляду базується на місцезнаходженні МФСФ, при цьому держава ЄС, у якій знаходиться майстер-файл, стає контролюючим органом (ст. 18(3) Регламенту (ЄС) № 726/2004 [5]). EMA може запитувати майстер-файл системи фармаконагляду для виконання своєї координаційної ролі.
Основним обов’язком EMA щодо МФСФ є підтримка всеєвропейської бази даних, розповсюдження інформації та координація діяльності на території ЄС. Для цього EMA, співпрацюючи з державами ЄС та Європейською Комісією, несе відповідальність за створення та підтримку вебпорталу EMA для розповсюдження інформації про лікарські засоби, зареєстровані в ЄС (ст. 26 Регламенту (ЄС) № 726/2004 [5]). EMA координує перелік лікарських засобів, що описаний у ст. 57 Регламенту (ЄС) № 726/2004 [5], який забезпечує практичний механізм поновлення інформації щодо місцезнаходження МФСФ, контактної інформації про УОВФ та лікарські засоби, пов’язані з системою фармаконагляду, яка описана в МФСФ. Перелік місць знаходження в ЄС, де зберігаються майстер-файли системи фармаконагляду, має бути загальнодоступним, завдяки розміщенню на вебпорталі EMA (ст. 26(1)(е) Регламенту (ЄС) № 726/2004 [5]).
В Україні Міністерство охорони здоров’я України встановлює правила, вимоги до заявників (власників реєстраційного посвідчення) лікарських засобів, вакцин, туберкуліну, та їх зобов’язання, що викладені у Порядку [2]. Фармаконагляд здійснює Центр на виконання покладених на нього повноважень щодо фармаконагляду відповідно до вимог законодавства. Центр проводить аудит системи фармаконагляду заявника (власника реєстраційного посвідчення). Центр може вимагати від заявника (власника реєстраційного посвідчення) майстер-файл системи фармаконагляду (положення Порядку [2]N).
II.C.2. Доступ до майстер-файлу системи фармаконагляду
МФСФ повинен підтримуватися в актуальному стані і бути постійно доступним для УОВФ/КОВФN (ст. 4(1), ст. 7(2) ІП 520/2012 [6], положення Порядку [2])N. МФСФ також повинен бути постійно доступним при проведенні інспекції, аудиту системи фармаконагляду уповноваженою установоюN за місцезнаходженням (за вказаною адресою) незалежно від того, чи було заздалегідь попереджено про інспекцію, аудит системи фармаконагляду уповноваженою установоюN чи ні (ст. 7(3) ІП 520/2012 [6], положення Порядку [2])N.
Відповідно до ст. 104 (3)(b) Директиви 2001/83/ЄС [1], положень Порядку [2], положень Порядку проведення експертизи реєстраційних матеріалів [7]N заявник (власник реєстраційного посвідчення) повинен мати та надавати на вимогу уповноваженого органу копію МФСФ. Заявник (власник реєстраційного посвідчення) повинен надати копію не пізніше 7 днів після отримання запиту від уповноваженого органу. МФСФ повинен бути наданий у доступному для читання в електронному форматі або у вигляді друкованої копії.
У разі якщо один МФСФ використовується більш ніж одним заявником (власником реєстраційного посвідчення) (у разі спільного використання системи фармаконагляду), він повинен бути доступним для кожного із заявників (власників реєстраційного посвідчення) з тим, щоб будь-хто з них у разі необхідності був у змозі подати МФСФ до уповноваженого органу протягом 7 днів (ст. 23(4) Директиви 2001/83/ЄС [1], ст. 7(4) ІП 520/2012 [6], положення Порядку [2])N.
МФСФ не повинен вимагатися регулярно при проведенні експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію, однак його надання може вимагатися на ситуативній основі, зокрема якщо впроваджується нова система фармаконагляду або якщо виявлені питання з безпеки, пов’язані з лікарським засобом, чи питання дотримання вимог щодо фармаконагляду.
II.C.3. Прозорість
Для забезпечення прозорості та у комунікативних цілях інформація про місцезнаходження МФСФ має оприлюднюватися на вебпорталі EMA (ст. 26(1)(е) Регламенту (ЄС) № 726/2004 [5]).
В Україні законодавчо не передбачено оприлюднення інформації про місцезнаходження МФСФN.
Частина ІІІ
Модуль III — Аудит системи фармаконагляду заявника (власника реєстраційного посвідчення), що проводиться уповноваженою міністерством охорони здоров’я спеціалізованою експертною установою у сфері здійснення фармаконаглядуN
III.А. Вступ
Цей модуль містить настанову з планування, звітування та проведення аудитів системи фармаконагляду заявників (власників реєстраційного посвідчення) в Україні уповноваженою МОЗ спеціалізованою експертною установою у сфері здійснення фармаконаглядуN (далі — уповноважена установаN) та визначає роль сторін, які беруть у них участь.
З метою встановлення того, чи виконують заявники (власники реєстраційних посвідчень) зобов’язання з фармаконагляду, та з метою сприяння виконанню вимог в Україні уповноважена установаN повинна проводити аудити систем фармаконаглядуN заявників (власників реєстраційних посвідчень) та будь-яких організацій, залучених заявниками (власниками реєстраційних посвідчень) до виконання зобов’язань з фармаконагляду. Такі аудитиN повинні проводитися аудиторамиN, призначеними уповноваженою установоюN, які уповноважені проводити аудитN приміщень, записів, документів та МФСФ заявника (власника реєстраційного посвідчення) або будь-яких організацій, залучених заявником (власником реєстраційного посвідчення) до виконання діяльності, описаної у розділі ІХ директиви 2001/83/ЄС[1] відповідно до ст.ст. 111(1) і 111(1)(d) положень Порядку [2]N, положень Порядку проведення експертизи [7]N. Зокрема, від заявників (власників реєстраційного посвідчення) вимагається надавати на вимогу МФСФ, який буде використовуватися для інформування при проведенні аудиту системи фармаконагляду заявника (власника реєстраційного посвідчення) уповноваженою установоюN, (ст. 23(4) директиви 2001/83/ЄС [1] і ст. 16(4) регламенту (ЄС) № 726/2004 [5], положення Порядку [2])N.
Мета аудиту системи фармаконагляду заявника(власника реєстраційного посвідчення), що проводиться уповноваженою установоюN (далі — аудит системи фармаконагляду уповноваженою установоюN) встановити, що заявник (власник реєстраційного посвідчення) має персонал, системи та приміщення для виконання своїх зобов’язань з фармаконагляду;
- визначити, задокументувати та врегулювати випадки недотримання зобов’язань, що можуть становити загрозу для громадського здоров’я;
- використовувати результати аудиту системи фармаконагляду уповноваженою установоюN як підставу для вжиття відповідних заходів Міністерством охорони здоров’яN у разі необхідності.
В Україні заявник (власник реєстраційного посвідчення) є відповідальним за ефективність, якість та безпеку лікарського засобу згідно з порядком, визначеним чинним законодавством, та мати ресурси для здійснення фармаконагляду в Україні(положення Порядку [2]N та Порядку проведення експертизи [7]N. З метою перевірки надійності та успішності імплементації існуючої чи запропонованої системи фармаконагляду в Україні уповноважена установаN може проводити дореєстраційні аудити систем фармаконагляду заявників (власників реєстраційного посвідчення) (положення Порядку [2])N.
Аудити системи фармаконагляду уповноваженою установоюN повинні здійснюватися за програмами, що включають стандартні планові аудити систем фармаконагляду уповноваженою установоюN, заплановані на основі ризик-орієнтованого підходу, а також аудити систем фармаконагляду уповноваженою установоюN, зумовлені певною причиною, тобто ініційовані для перевірки підозрюваної невідповідності нормативно-правовим вимогам або потенційних ризиків, як правило, стосовно конкретного(их) препарату(ів).
Результати аудиту системи фармаконагляду уповноваженою установоюN повинні надаватися проаудитованому суб’єкту (положення Порядку [2])N з метою забезпечення можливості прокоментувати будь-яку виявлену невідповідність вимогам (положення Порядку [2])N. Заявник (власник реєстраційного посвідчення) повинен своєчасно виправляти будь-яку невідповідність шляхом виконання плану коригуючих і запобіжних заходів.
За результатами аудиту системи фармаконагляду уповноваженою установою Міністерство охорони здоров’яN, якщо необхідно, має вжити необхідних заходів щодо застосування ефективних відповідних заходівN до заявника (власника реєстраційного посвідчення) (положення Порядку [2])N.
Інформація про проведення та результати аудитів системи фармаконагляду уповноваженою установоюN та про контроль і оцінку їх результатів може бути загальнодоступною в рамках загальної прозорості діяльності з фармаконагляду.
III.В. Структури та процеси
III.В.1. Види аудитів системи фармаконагляду уповноваженою установоюN
III.В.1.1. Аудити систем фармаконагляду уповноваженою установоюN на прикладі окремих лікарських засобів
Аудити систем фармаконагляду уповноваженою установоюN призначені для перевірки процедур, систем, персоналу та ресурсів, і встановлення їх відповідності вимогам щодо діяльності з фармаконагляду, встановлених законодавством. При проведенні такої перевірки можуть використовуватися конкретні лікарські засоби у якості прикладів для демонстрації функціонування системи фармаконагляду.
Аудити систем фармаконагляду уповноваженою установоюN щодо конкретного лікарського засобу зосереджуються в основному на питаннях фармаконагляду щодо цього лікарського засобу, а не на перевірці системи фармаконагляду загалом. Проте і в цьому випадку можуть бути оцінені деякі аспекти загальної системи фармаконагляду (наприклад, система фармаконагляду, що використовується для такого лікарського засобу).
III.В.1.2. Планові аудити систем фармаконагляду уповноваженою установоюN, зумовлені певною причиною
Планові аудити систем фармаконагляду уповноваженою установоюN— це аудити систем фармаконагляду, що проводяться уповноваженою установоюN, попередньо заплановані в аудиторськихN програмах/планах (далі — планові аудити системи фармаконагляду уповноваженою установоюN). Для проведення таких планових аудитів системи фармаконагляду уповноваженою установоюN не потрібні специфічні причини, однак з метою оптимізації контролюючої діяльності, слід дотримуватися ризик-орієнтованого підходу. Такі планові аудити систем фармаконагляду уповноваженою установоюN, як правило, стосуються перевірки системи фармаконагляду, однак у якості прикладів, для підтвердження впровадження системи фармаконагляду та забезпечення практичного доказу її функціональності й відповідності вимогам можуть бути вибрані один або декілька конкретних лікарських засобів. У планові аудити систем фармаконаглядуN з метою перевірки можуть також включатися специфічні питання, що виникли у аудиторівN.
Аудити систем фармаконагляду, що проводяться уповноваженою установоюN, викликані певною причиною (далі — позапланові аудити систем фармаконагляду уповноваженою установоюN), проводяться у випадку виявлення причини для ініціювання такого позапланового аудиту системи фармаконагляду уповноваженою установоюN, і при цьому позаплановий аудит системи фармаконагляду уповноваженою установоюN вважається найбільш оптимальним способом для вивчення даної причини. Позапланові аудити систем фармаконагляду уповноваженою установоюN в основному зосереджуються на специфічних процесах фармаконагляду або перевірці дотримання встановлених вимог та їх впливу на конкретний лікарський засіб. Проте позаплановий аудит системи фармаконагляду уповноваженою установоюN може також проводитися з певної причини. Позапланові аудити систем фармаконагляду уповноваженою установоюN можуть проводитися, коли, наприклад, виявлено одну або більше причин, що перераховані нижче:
- співвідношення користь/ризик лікарського засобу:
- зміна співвідношення ризик/користь, коли доцільне подальше її вивчення шляхом позапланового аудиту системи фармаконагляду уповноваженою установоюN;
- зволікання чи неспроможність виявити або повідомити про ризик чи зміну співвідношення користь/ризик;
- оприлюднення інформації з питань фармаконагляду без попереднього чи одночасного повідомлення уповноваженого органу;
- виявлені у процесі моніторингу діяльності фармаконагляду уповноваженим органом невідповідності нормативно-правовим вимогам або проблеми з безпеки;
- призупинення або вилучення лікарського засобу з ринку без попереднього повідомлення про це уповноваженого органу;
- зобов’язання щодо надання звітності (повідомлень про побічні реакції/відсутність ефективності лікарських засобів та регулярно оновлюваних звітів з безпеки (далі — РОЗБ)):
- несвоєчасне надання або ненадання повідомлень;
- надання неповної інформації або повідомлень незадовільної якості;
- невідповідність даних у наданих повідомленнях інформації, отриманої з інших джерел;
- запити від уповноваженого органу:
- ненадання запитуваної інформації чи даних у зазначений уповноваженим органом строк;
- надання даних незадовільної якості чи невідповідних даних;
- виконання зобов’язань:
- занепокоєння щодо стану чи виконання зобов’язань в рамках плану управління ризиками (ПУР);
- затримка з виконанням або невиконання специфічних зобов’язань, пов’язаних з моніторингом безпеки лікарського засобу, виявлених на момент реєстрації;
- незадовільна якість звітів, що вимагаються в рамках виконання специфічних зобов’язань;
- позапланові аудити систем фармаконагляду уповноваженою установоюN:
- зволікання із впровадженням або невідповідне впровадження коригуючих та запобіжних заходів;
- інформація про недотримання нормативно-правових вимог чи проблеми з безпекою лікарських засобів, виявлені при проведенні інших видів аудитів системи фармаконагляду уповноваженою установоюN (GCP, GMP, GLP та GDP);
- інформація про інспекції, отримана від регуляторних органів інших країн, що може свідчити про недотримання нормативно-правових вимог;
- інше:
- проблеми, виявлені при розгляді МФСФ;
- інформація, що стосується невиконання нормативно-правових вимог, виявлена не під час проведення інспекції, отримана від регуляторних органів інших країн;
- інші джерела інформації або скарги.
III.В.1.3. Дореєстраційні аудити систем фармаконагляду уповноваженою установоюN
Дореєстраційні аудити систем фармаконагляду, що проводяться уповноваженою установоюN (далі — дореєстраційні аудити системи фармаконагляду уповноваженою установоюN), проводяться у період до видачі реєстраційного посвідчення. Такі дореєстраційні аудити системи фармаконагляду уповноваженою установоюN проводяться з метою перевірки існуючої або запропонованої системи фармаконагляду, описаної заявником (власником реєстраційного посвідчення) у відповідних матеріалах реєстраційного досьє (ст. 19 регламенту (ЄС) № 726/2004 [5], положення Порядку [2] та положення Порядку проведення експертизи [7]N). Дореєстраційні аудити системи фармаконагляду уповноваженою установоюN не обов’язкові, однак за певних умов все ж може вимагатися їх проведення. Необхідно розробити принципи і процедури щодо вимог до проведення дореєстраційних аудитів системи фармаконагляду уповноваженою установоюN, щоб уникати проведення необґрунтованих аудитів системи фармаконагляду уповноваженою установоюN, що можуть затримати видачу реєстраційного посвідчення. На початковій стадії процесу експертизи реєстраційних матеріалів необхідно розглянути наступні аспекти:
- заявник раніше не мав системи фармаконагляду на території УкраїниN або впроваджує нову систему фармаконагляду;
- наявна інформація (наприклад від регуляторних органів інших країн) про те, що існують нарікання щодо виконання заявником (власником реєстраційного посвідчення) вимог. Якщо заявник (власник реєстраційного посвідчення) має історію серйозної та/або постійної невідповідності вимогам фармаконагляду, то дореєстраційний аудит системи фармаконагляду уповноваженою установоюN може бути єдиним механізмом для підтвердження того, що система фармаконагляду була вдосконалена до видачі нового реєстраційного посвідчення в УкраїніN;
- за наявності проблем з безпеки, специфічних для лікарського засобу, може бути доцільною оцінка можливостей заявника (власника реєстраційного посвідчення) щодо:
- запровадження заходів з мінімізації ризиків, специфічних для конкретного лікарського засобу; або
- належного виконання специфічних вимог з безпеки, що можуть бути висунуті; або
- здійснення рутинного фармаконагляду за конкретним лікарським засобом (наприклад, очікується значне збільшення кількості повідомлень про побічні реакції при його застосуванні у порівнянні з іншими лікарськими засобами).
У більшості випадків до надання запиту на проведення дореєстраційного аудиту системи фармаконагляду уповноваженою установоюN слід здійснити оцінку ризиків на основі комплексної оцінки проблем, що стосуються конкретного лікарського засобу і системи фармаконагляду.
Якщо в результаті проведення дореєстраційного аудиту системи фармаконагляду уповноваженою установоюN виникає занепокоєність щодо здатності заявника (власника реєстраційного посвідчення) дотримуватися вимог, запроваджених регламентом (ЄС) № 726/2004 [5], директивою 2001/83/ЄС [1], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7]N можуть бути надані наступні рекомендації:
- відмова у видачі реєстраційного посвідчення;
- проведення повторного аудиту системи фармаконагляду уповноваженою установоюN до видачі реєстраційного посвідчення для підтвердження того, що були усунені критичні невідповідності та враховані рекомендації;
- видача реєстраційного посвідчення з рекомендацією проведення раннього післяреєстраційного аудиту системи фармаконагляду уповноваженою установоюN. Така рекомендація вплине на графік проведення планових аудитів системи фармаконагляду уповноваженою установоюN (див. розділ III.В.2 ННПФ);
- встановлення вимог з безпеки як умови видачі реєстраційного посвідчення на підставі ст. 21а директиви 2001/83/ЄС [1], ст. 14.8 регламенту (ЄС) № 726/2004, положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7]N.
III.В.1.4. Післяреєстраційні аудити систем фармаконагляду уповноваженою установоюN
Післяреєстраційні аудити систем фармаконагляду, що проводяться в Україні уповноваженою установоюN (далі — післяреєстраційні аудити системи фармаконагляду уповноваженою установоюN), здійснюються після видачі реєстраційного посвідчення та призначені для перевірки виконання заявником (власником реєстраційного посвідчення) зобов’язань фармаконагляду.
III.В.1.5. Оголошені (з попередженнямN) й неоголошені (без попередженняN) аудити систем фармаконагляду уповноваженою установоюN
Більшість аудиторськихN перевірок будуть оголошеними, тобто про проведення такого аудиту системи фармаконагляду уповноваженою установоюN, буде завчасно повідомлено організацію, діяльність з фармаконагляду якої перевірятиметься, з метою забезпечення присутності відповідних осіб для аудиту системи фармаконагляду уповноваженою установоюN. Однак інколи доцільно проводити неоголошені або оголошені напередодні проведення аудити системи фармаконагляду уповноваженою установоюN (наприклад у випадках, коли попередження може скомпрометувати мету аудитуN або коли потрібно провести аудит системи фармаконагляду уповноваженою установоюN у стислий термін через невідкладні проблеми з безпеки).
III.В.1.6. Повторні аудити систем фармаконагляду уповноваженою установою
Повторні аудити систем фармаконагляду уповноваженою установоюN можуть проводитися у плановому порядку в рамках програми планових перевірок. Першочерговість повторних аудитів системи фармаконагляду уповноваженою установоюN буде встановлюватися на основі оцінки факторів ризику. Повторні перевірки на ранніх етапах можуть проводитися у випадках, коли було виявлено значне недотримання зобов’язань, і при цьому необхідно перевірити належність виконання заходів, що вживаються для вирішення виявлених проблем, та оцінити постійне виконання зобов’язань, у тому числі оцінити зміни в системі фармаконагляду. Повторний аудит системи фармаконагляду уповноваженою установоюN на ранньому етапі також може бути доцільним, якщо відомо з досвіду аудиту в минулому, що проаудитованаN сторона не запровадила належні коригуючі та запобіжні заходи у відповідь на рекомендації минулого аудиту системи фармаконагляду уповноваженою установоюN.
III.В.1.7. Дистанційні аудити систем фармаконагляду уповноваженою установоюN
Це аудити систем фармаконагляду уповноваженою установоюN, що проводяться аудиторамиN дистанційно, тобто не у приміщеннях заявника (власника реєстраційного посвідчення) чи компаній, що працюють на заявника (власника реєстраційного посвідчення). При проведенні таких аудитів системи фармаконагляду уповноваженою установоюN можуть застосовуватися такі засоби комунікації, як інтернет або телефон. Наприклад, у випадках, коли основна діяльність з фармаконагляду проводиться за межами УкраїниN або третя сторона, якій делеговані певні види діяльності з фармаконагляду, не доступна на аудитованомуN об’єкті, завдяки віддаленому доступу можна організувати інтерв’ю з відповідним персоналом та перевірку документів, бази даних з безпеки лікарських засобів, первинної документації та МФСФ. Такий підхід може також застосовуватися при наявності логістичних труднощів для проведення аудиту системи фармаконагляду уповноваженою установоюN на місці за виняткових обставин (наприклад, пандемія чи обмеження пересування). Рішення про проведення дистанційних аудитів системи фармаконагляду уповноваженою установоюN приймається на розсуд аудиторівN та має бути узгодженим з уповноваженою установоюN, який здійснює аудит системи фармаконаглядуN. Логістичні аспекти дистанційного аудиту системи фармаконагляду уповноваженою установоюN слід узгоджувати із заявником (власником реєстраційного посвідчення). Якщо в процесі дистанційного аудиту були виявлені проблеми, що вимагають аудиту системи фармаконагляду уповноваженою установоюN на місці або якщо мета аудиту системи фармаконагляду уповноваженою установоюN не може бути досягнута дистанційним шляхом, може бути прийнято рішення про виїзд на об’єкт аудитуN.
III.В.2. Планування аудиту системи фармаконагляду уповноваженою установою
Планування аудиту системи фармаконагляду уповноваженою установоюN має базуватися на системному та ризик-орієнтованому підході з ефективним використанням ресурсів у рамках діяльності з нагляду та контролю і забезпечення високого рівня здоров’я населення. Ризикорієнтований підхід при плануванні аудиту системи фармаконагляду уповноваженою установоюN дозволить визначити періодичність, масштаб та обсяг аудиту системи фармаконагляду уповноваженою установоюN.
З метою забезпечення ефективного використання аудиторськихN ресурсів необхідно створити програми та графік проведення аудитів системи фармаконагляду уповноваженою установоюN. Взаємодія та обмін інформацією між аудиторамиN та експертами є запорукою успішної пріоритезації та визначення цілей аудиту системи фармаконагляду уповноваженою установоюN.
При створенні аудиторськихN програм уповноважена установаN повинна враховувати, однак не обмежуватися, наступні чинники:
- пов’язані з аудитом системи фармаконагляду уповноваженою установоюN:
- інформація щодо дотримання вимог фармаконагляду, виявлена при проведенні аудитів системи фармаконагляду уповноваженою установоюN в минулому або інших видів аудитівN (GCP, GMP, GLP та GDP);
- дата повторного аудиту системи фармаконагляду уповноваженою установоюN, рекомендована аудиторамиN після попереднього аудиту системи фармаконагляду уповноваженою установоюN;
- пов’язані з лікарським засобом:
- лікарський засіб, для якого встановлені додаткова діяльність з фармаконагляду чи заходи з мінімізації ризиків;
- реєстрація лікарського засобу з додатковими умовами, пов’язаними з безпекою його застосування; наприклад, вимога проведення післяреєстраційних досліджень з безпеки або додаткового моніторингу;
- лікарський(і) засіб(оби) з великим обсягом продажів; тобто, препарати, які активно застосовуються в УкраїніN;
- лікарський(і) засіб(и) з обмеженою альтернативою на ринку;
- пов’язані із заявником (власником реєстраційного посвідчення):
- заявник (власник реєстраційного посвідчення), який ніколи не був проаудитованийN з фармаконагляду;
- заявник (власник реєстраційного посвідчення) представляє на фармацевтичному ринку УкраїниN велику кількість лікарських засобів;
- ресурси для проведення діяльності з фармаконагляду, наявні у заявника (власника реєстраційного посвідчення);
- заявник (власник реєстраційного посвідчення) раніше не отримував реєстраційних посвідчень в УкраїніN;
- негативна інформація та/або інформація про проблеми з безпеки, отримана від уповноважених органів інших країн або з інших аудитів системи фармаконагляду уповноваженою установоюN (GCP, GMP, GLP та GDP);
- зміни в організаційній структурі заявника (власника реєстраційного посвідчення), такі як злиття та поглинання;
- пов’язані з системою фармаконагляду:
- заявник (власник реєстраційного посвідчення) має договірні відносини на здійснення діяльності фармаконагляду (наприклад, надання інформації з безпеки лікарських засобів та ін.) та/або залучає до діяльності фармаконагляду декілька компаній;
- зміна УОВФ/КОВФN, з моменту проведення останнього аудиту системи фармаконагляду уповноваженою установоюN;
- зміни у базі(ах) даних з безпеки лікарських засобів, що можуть включати зміну у самій базі даних або у пов’язаних з нею базах, зміну валідаційного статусу бази даних, а також інформації про передані або експортовані дані;
- зміни в договірних угодах з провайдерами послуг з фармаконагляду або зміни місць проведення діяльності з фармаконагляду;
- делегування або передача управління МФСФ.
Уповноважена установаN може вимагати у заявників (власників реєстраційного посвідчення) необхідну для планування ризик-орієнтованих аудитів системи фармаконагляду уповноваженою установоюN інформацію, якщо така недоступна з будь-яких інших джерел.
III.В.3. Об’єкти, що підлягають аудиту системи фармаконагляду
Будь-яка сторона, що здійснює діяльність з фармаконагляду, у повному обсязі або частково, від імені або разом із заявником (власником реєстраційного посвідчення), може бути проаудитованаN з метою підтвердження її здатності до забезпечення належного виконання зобов’язань і нормативно-правових вимог щодо фармаконагляду заявником (власником реєстраційного посвідчення).
Об’єкти, що підлягають проведенню аудиту системи фармаконагляду уповноваженою установоюN, можуть знаходитись як на території УкраїниN, так і за її межами. Аудит системи фармаконагляду уповноваженою установоюN об’єктів за межами УкраїниN може бути доречним у випадках, коли основний центр фармаконагляду, бази даних і/або основна діяльність локалізовані за межами УкраїниN і неможливо у повному обсязі підтвердити їх відповідність вимогам з фармаконагляду з об’єкту в УкраїніN.
Тип і кількість аудитованихN об’єктів повинні вибиратися відповідним чином, щоб забезпечувати досягнення основних цілей аудиту системи фармаконагляду уповноваженою установоюN.
III.В.4. Масштаб аудиту системи фармаконагляду
Масштаб аудиту системи фармаконагляду уповноваженою установоюN буде залежати від їх цілей, масштабу будь-яких попередніх аудитів системи фармаконагляду, що проводилися уповноваженою установоюN, а також від того, чи стосується аудит системи фармаконагляду уповноваженою установоюN системи в цілому чи окремого лікарського засобу (опис видів аудитів системи фармаконагляду уповноваженою установоюN, причин та настанови для різних видів аудитів системи фармаконагляду уповноваженою установоюN надається в розділі III.В.1 ННПФ).
При плануванні масштабу аудиту системи фармаконагляду уповноваженою установоюN слід враховувати:
- інформацію, представлену у МФСФ;
- інформацію, що стосується функціонування системи фармаконагляду, наприклад, дані відповідності вимогам фармаконагляду, наявні у уповноваженого органу, такі як подача повідомлень та аудити якості даних;
- специфічні причини, що ініціювали аудит системи фармаконагляду уповноваженою установоюN (див. розділ III.В.1.2 ННПФ, де наведені приклади причин).
Може бути доцільним запит на надання додаткових даних до проведення аудиту системи фармаконагляду уповноваженою установоюN з метою вибору об’єктів чи з’ясування аспектів системи фармаконагляду.
III.В.4.1. Планові аудити системи фармаконаглядуN
За допомогою планових аудитів системи фармаконагляду уповноваженою установоюN повинна перевірятися відповідність системи фармаконагляду заявника законодавству та настановам УкраїниN. Масштаби таких аудитів системи фармаконагляду уповноваженою установоюN повинні залежати від таких складових (в залежності від конкретного випадку):
- повідомлення про індивідуальні випадки, пов’язані з безпекою лікарських засобів (ICSRs):
- збір, отримання та обмін повідомленнями, отриманими з різних джерел і підрозділів у рамках системи фармаконагляду, включаючи організації, залучені до виконання зобов’язань з фармаконагляду заявника (власника реєстраційного посвідчення), та департаменти, діяльність яких не пов’язана із здійсненням нагляду за безпекою лікарських засобів;
- оцінка повідомлень, включаючи механізми отримання та реєстрації оцінок репортерів, застосування заявником (власником реєстраційного посвідчення) термінології побічних реакцій, визначення їх серйозності, передбаченості та встановлення причинно-наслідкового зв’язку. Крім прикладів повідомлень, отриманих з УкраїниN, слід також розглянути приклади повідомлень, отримані з інших країн (якщо доречно);
- відстеження наступних повідомлень та наслідків побічних реакцій, наприклад, результати застосування лікарського засобу при вагітності чи підтвердження лікарем побічних явищ, повідомлених споживачем;
- звітність відповідно до вимог щодо різних типів повідомлень про побічні реакції та своєчасність такої звітності;
- реєстрація та архівування повідомлень про побічні реакції;
- РОЗБ (якщо застосовано):
- повнота та достовірність включених даних, адекватність рішень стосовно невключення даних;
- вирішення питань безпеки, представлення відповідних результатів аналізу та заходів;
- генерування РОЗБ відповідно до вимог;
- своєчасність подання РОЗБ;
- постійна оцінка безпеки застосування лікарського(их) засобу(ів):
- використання усіх відповідних джерел інформації для виявлення сигналу;
- правильне застосування методології аналізу;
- відповідність процедурN розслідувань та наступних заходів; наприклад, запровадження рекомендацій після оцінки даних;
- реалізація ПУР або інших зобов’язань;
- своєчасне виявлення та надання повних та точних даних уповноваженому органу, зокрема у відповідь на запити;
- імплементація затверджених змін до комунікацій з безпеки та короткої характеристики лікарського засобу/інструкцій для медичного застосуванняN, включаючи внутрішню розсилку та оприлюднення інформації з безпеки лікарських засобів для громадськості та/або медичних працівників;
- інтервенційні (якщо доцільно) та неінтервенційні клінічні дослідження:
- повідомлення про підозрювані непередбачені серйозні побічні реакції (SUSARs) відповідно до директиви 2001/20/ЄС [15], положень Порядку проведення клінічних випробувань [16]N і про випадки з неінтервенційних досліджень відповідно до директиви 2001/83/ЄС [1] та положень Порядку [2]N;
- отримання, реєстрація та оцінка випадків, отриманих при проведенні інтервенційних та неінтервенційних досліджень (див. ICSRs);
- надання результатів дослідження та відповідної інформації з безпеки (наприклад, створення оновлюваних звітів з безпеки ліків, що знаходяться в стадії розробки (DSUR) та представлення інформації в РОЗБ), надання результатів післяреєстраційних досліджень з безпеки чи післяреєстраційних досліджень з ефективності, зокрема тих, що стосуються специфічних зобов’язань чи виконання ПУР;
- відповідний вибір довідкової інформації з безпеки, розробка дослідницьких брошур та інформації з безпеки для пацієнта;
- включення даних дослідження до поточної оцінки безпеки;
- система фармаконагляду:
- роль та обов’язки уповноваженої особи, відповідальної за фармаконагляд (УОВФ)/КОВФN, наприклад, доступ до системи якості, МФСФ, індикаторів ефективності, звітів аудитів системи фармаконагляду уповноваженою установою; здатність УОВФ/КОВФN застосовувати заходи для покращення відповідності вимогам;
- роль та обов’язки заявника (власника реєстраційного посвідчення) щодо системи фармаконагляду;
- точність, повнота та супровід МФСФ;
- якість та відповідність підготовки персоналу, кваліфікація та досвід персоналу;
- повнота охоплення та дотримання системи якості у фармаконагляді, включаючи процеси контролю та гарантування якості;
- відповідність комп’ютеризованих систем поставленим цілям;
- договори та угоди з усіма відповідними сторонами, що відображають їх обов’язки й діяльність з фармаконагляду та забезпечують їх дотримання.
Аудит системи фармаконагляду уповноваженою установоюN може включати систему контролю виконання умов видачі/подовженняN дії реєстраційного посвідчення та впровадження заходів з мінімізації ризиків, якщо вони стосуються безпеки лікарського засобу.
III.В.4.2. Аудити системи фармаконагляду, викликані певною причиною
Масштаб аудиту системи фармаконагляду уповноваженою установоюN буде залежати від конкретної причини(ин). Деякі із цих причин, але не всі, перелічені у розділі III.В.4.1 ННПФ та нижче:
- залучення та поінформованість УОВФ/КОВФN щодо проблем, пов’язаних з конкретним лікарським засобом;
- детальна перевірка процесів, процедурN прийняття рішень, інформаційного обміну та діяльності, що стосуються певної причини, що послужила тригером для проведення аудиту системи фармаконагляду уповноваженою установоюN, та/або лікарського засобу.
III.В.4.3. Повторні аудити системи фармаконагляду
При визначенні масштабу повторного аудиту системи фармаконагляду уповноваженою установоюN слід враховувати такі аспекти:
- оцінка стану системи та/або плану(ів) коригуючих та запобіжних заходів за результатами попереднього(іх) аудиту(ів) системи фармаконагляду уповноваженою установоюN;
- оцінка значних змін, які були зроблені у системі фармаконагляду з моменту останнього аудиту системи фармаконагляду уповноваженою установоюN (наприклад, зміни у базі даних фармаконагляду, злиття або поглинання компаній, значні зміни у контрактній діяльності, зміна УОВФ/КОВФN);
- аналіз процесів та/або специфічних проблем, пов’язаних з конкретним лікарським засобом, що були визначені при оцінці інформації, наданої заявником (власником реєстраційного посвідчення), або які не були охоплені минулим аудитом системи фармаконагляду уповноваженою установоюN.
Масштаб повторного аудиту системи фармаконагляду уповноваженою установоюN буде залежати від досвіду попередніх аудитів системи фармаконагляду уповноваженою установоюN.
Може бути доцільним провести повну оцінку системи фармаконагляду, наприклад, якщо минуло багато часу з моменту останнього аудиту системи фармаконагляду уповноваженою установоюN. У цьому випадку масштаб повторного аудиту системи фармаконагляду уповноваженою установоюN може залежати від елементів, перелічених у розділі III.В.4.1 ННПФ.
III.В.5. Аудиторський процес
Аудити системи фармаконагляду уповноваженою установоюN необхідно планувати, координувати, проводити, звітувати про проведення, контролювати виправлення зауважень та документувати відповідно до аудиторськихN процедур, розроблених та затверджених уповноваженим органом. Процедури аудиту системи фармаконагляду уповноваженою установоюN включають, щонайменше, такі процеси:
- обмін інформацією;
- планування аудиту системи фармаконагляду уповноваженою установоюN;
- дореєстраційні аудити системи фармаконагляду уповноваженою установоюN;
- координація аудитів систем фармаконагляду уповноваженою установоюN заявників (власників реєстраційного посвідчення) в УкраїніN;
- координація аудиту системи фармаконагляду уповноваженою установоюN в третіх країнах (включаючи аудитN підрядників у третіх країнах);
- підготовка аудитів системи фармаконагляду уповноваженою установоюN;
- проведення аудитів системи фармаконагляду уповноваженою установоюN;
- звітування за результатами аудитів системи фармаконагляду уповноваженою установоюN та наступний контроль;
- комунікація та пріоритезація аудиту системи фармаконагляду уповноваженою установоюN та їх результатів;
- ведення обліку та архівування документів, отриманих під час або в результаті проведення аудитів системи фармаконагляду уповноваженою установоюN;
- неоголошені (без попередження) аудити системи фармаконагляду уповноваженою установоюN;
- санкції та заходи у випадку критичної невідповідності вимогам;
- рекомендації з підготовки та досвіду аудиторівN, які проводять аудит системи фармаконагляду уповноваженою установоюN. У разі необхідності ці процеси будуть переглядатися, оновлюватися чи доповнюватися новими процесами.
II.В.6. Післяаудиторський контроль
Якщо в ході аудиту системи фармаконагляду уповноваженою установоюN було виявлено недотримання зобов’язань з фармаконагляду, необхідно здійснювати подальший контроль аж до повного виконання плану коригуючих та запобіжних заходів. У залежності від доцільності слід вжити таких наглядових дій:
- оцінка плану коригуючих та запобіжних заходів заявника (власника реєстраційного посвідчення);
- оцінка періодичних звітів про виконання планів коригуючих та запобіжних заходів (за необхідності);
- повторний аудит системи фармаконагляду уповноваженою установоюN для оцінки відповідності запровадження заходів, передбачених планом коригуючих та запобіжних заходів;
- запити на надання даних, що раніше не надавалися; подання заяв на зміни (наприклад, зміни до короткої характеристики лікарського засобу/інструкцій для медичного застосуванняN); подання аналізу наслідків впливу (наприклад, після аналізу даних, що раніше не розглядалися в процесі рутинної діяльності з виявлення сигналу);
- вимога розповсюдження повідомлення з безпеки, включаючи зміни до реєстраційних матеріалів лікарського засобу та/або рекламної інформації;
- запит на зустрічі із заявником (власником реєстраційного посвідчення) для обговорення невідповідностей, їх впливу та планів заходів;
- комунікація щодо результатів аудиту системи фармаконагляду уповноваженою установоюN з іншим регуляторним органам;
- інші заходи, пов’язані з лікарським засобом, в залежності від впливу виявлених невідповідностей та результатів запроваджених заходів (наприклад, вилучення з ринку, вжиття заходів щодо реєстраційних посвідчень чи щодо дозволів на проведення клінічних досліджень).
Для належного здійснення подальшого післяаудиторськогоN контролю дуже важливим є обмін інформацією та комунікація між аудиторамиN та експертами. Детальний опис процесів взаємодії аудиторівN та експертів, а також здійснення післяаудиторськогоN контролю буде розроблений у подальшому під час розробки процедур, зазначених у розділі III.В.5 ННПФ.
III.В.7. Регуляторні заходи та санкції
Згідно із законодавством УкраїниN з метою захисту громадського здоров’я уповноважена установаN повинна забезпечувати дотримання зобов’язань фармаконагляду. У випадку виявлення невідповідності вимогам фармаконагляду, заходи, яких необхідно вжити, повинні визначатися окремо для кожного конкретного випадку недотримання зобов’язань. Який саме захід буде вжито, залежатиме від потенційного негативного впливу недотримання зобов’язань з фармаконагляду на здоров’я населення, проте для вжиття примусового заходу може розглядатися будь-який випадок недотримання зобов’язань. Як зазначено у ст. 111(8) директиви 2001/83/ЄС [1], положеннях Порядку [2]N, якщо необхідно, уповноважена установаN повинна вжити необхідних заходів для гарантування того, що стосовно заявника (власника реєстраційного посвідчення) вжиті ефективні, відповідні та стримуючі санкції, заходиN. Крім того, регламент (ЄС) № 658/2007 [14] уповноважує Комісію накладати фінансові штрафи на заявників (власників реєстраційного посвідчення) для гарантії примусового виконання певних зобов’язань, пов’язаних з реєстраційними посвідченнями на лікарські засоби, виданими відповідно до регламенту (ЄС) № 726/2004 [5], Порядку проведення експертизи [7]N.
Згідно з настановою і у відповідних випадках з вимогами, встановленими законодавством, у разі недотримання вимог фармаконагляду можливі регуляторні заходи включають наступне:
- навчання та сприяння: уповноважена установаN може вести діалог з представниками заявника (власника реєстраційного посвідчення) (наприклад у вигляді робочих зустрічей) з метою узагальнення виявлених невідповідностей, роз’яснення нормативно-правових вимог і очікувань регуляторного органу та для розгляду пропозицій заявника (власника реєстраційного посвідчення) щодо коригуючих та запобіжних заходів;
- надання інформації уповноваженій установіN чи регуляторним органам третіх країн у рамках домовленостей про конфіденційність;
- аудит системи фармаконагляду уповноваженою установоюN: заявники (власники реєстраційного посвідчення), які не дотримуються вимог фармаконагляду, можуть бути проаудитованіN з метою визначення ступеня недотримання вимог, а потім повторно проаудитованіN для підтвердження того, що відповідність вимогам досягнута;
- лист-попередження, заява про невідповідність або повідомлення про порушення: це інструменти, які може використовувати уповноважена установаN, із зазначенням у них положень законодавства та настанов, що були порушені, для нагадування заявникам (власникам реєстраційного посвідчення) про їх зобов’язання щодо фармаконагляду чи для перерахування заходів, які повинен здійснити заявник (власник реєстраційного посвідчення), та строків їх втілення з метою усунення невідповідності та з метою запобігання подальшим випадкам недотримання вимог фармаконагляду;
- уповноважена установаN може оприлюднювати перелік заявників (власників реєстраційного посвідчення), у яких було виявлено серйозні чи постійні невідповідності вимогам;
- заходи щодо реєстраційного посвідчення, наприклад:
- термінове обмеження, пов’язане з безпекою;
- зміни до реєстраційних матеріалів;
- призупинення дії або анулювання реєстраційного посвідчення;
- затримка видачі нових реєстраційних посвідчень до тих пір, поки не будуть вжиті коригуючі та запобіжні заходи або до нових реєстраційних матеріалів не будуть включені додаткові умови з безпеки;
- вимога проведення дореєстраційного аудиту системи фармаконагляду уповноваженою установоюN;
- відкликання лікарських засобів з ринку, наприклад, у разі, якщо в інформацію про лікарський засіб/коротку характеристику лікарського засобу/інструкцію для медичного застосуванняN не були внесені важливі застереження з безпеки;
- заходи, пов’язані з маркетинговою або рекламною інформацією;
- поправки до протоколу або призупинення клінічних досліджень з міркувань безпеки лікарського засобу;
- адміністративні штрафи, зазвичай фіксовані або нараховуються в залежності від доходів компанії чи стягуються на щоденній основі (в Україні законодавчо не передбаченоN);
- кримінальне переслідування (відповідно до національного законодавства).
III.В.8. Ведення документації та архівування
Принципи та вимоги, яких необхідно дотримуватися, будуть описані у процедурі ведення та архівування документації, отриманої під час або внаслідок аудитів системи фармаконагляду уповноваженою установоюN, як зазначено у розділі III.В.5 ННПФ.
III.В.9. Кваліфікація та підготовка аудиторів
АудиториN, залучені до проведення аудиту системи фармаконагляду уповноваженою установоюN, повинні бути співробітниками уповноваженої установиN і призначатися на посаду на підставі їх досвіду та мінімальних вимог, визначених уповноваженою установоюN. Крім того, необхідно враховувати рекомендації щодо підготовки та досвіду, описані у відповідних процедурах щодо аудиту системи фармаконагляду уповноваженою установоюN, про які йдеться у розділі III.В.5.
АудиториN повинні пройти навчання в обсязі, достатньому для забезпечення компетентності та професійних знань, необхідних для підготовки і проведення аудиту системи фармаконагляду уповноваженою установоюN та звітування про його результати. Вони повинні також пройти навчання щодо процесів та вимог фармаконагляду, щоб у випадку, якщо вони не мають відповідного досвіду, бути спроможними розуміти різні аспекти системи фармаконагляду.
Для забезпечення здійснення аудиту системи фармаконагляду уповноваженою установоюN на належному рівні необхідно створити документально оформлені процеси. Зокрема, аудиториN повинні бути обізнані щодо останніх змін у законодавстві та настанов з фармаконагляду.
Навчання та досвід повинні бути документально підтверджені індивідуально для кожного аудитораN та оцінюватися згідно з вимогами системи якості у фармаконагляді уповноваженої установиN.
III.В.10. Управління якістю процесу аудиту системи фармаконагляду
Якість процесу аудиту системи фармаконагляду уповноваженою установоюN регулюється уповноваженою установоюN і є складовою її системи фармаконагляду та пов’язаної з нею системи якості, що означає, що цей процес також підлягає аудиту. Настанова із впровадження та підтримки системи фармаконагляду належної якості наведена в модулі 1 ННПФ.
Дотриманню належного рівня якості та стабільності роботи аудитів системи фармаконагляду уповноваженою установоюN сприяють процедури, зазначені в розділі III.В.5 ННПФ.
III.С. Діяльність з проведення аудиту системи фармаконаглядуN
III.С.1. Обмін інформацією
В Україні уповноважений орган інформує МОЗ щодо результатів проведеного ним аудиту системи фармаконагляду відповідно до положень Порядку [2], зокрема, коли встановлено, що заявник (власник реєстраційного посвідчення) не відповідає вимогам законодавства та відповідної настановиN.
III.С.2. Роль уповноваженої установи в УкраїніN
III.С.2.1. Загальна роль уповноваженої установи в УкраїніN
В Україні уповноважена установа відповідає за здійснення аудитів/перевірок системи фармаконагляду заявника (власника реєстраційного посвідчення) лікарського(іх) засобу(ів) щодо її відповідності вимогам здійснення фармаконагляду, викладеним у положеннях Порядку [2]. У разі необхідності, вона може проводити аудити систем фармаконагляду заявників (власників реєстраційного посвідчення) з метою перевірки надійності та успішного використання існуючої або запропонованої системи фармаконагляду (положення Порядку [2]).
Уповноважена установа повинна мати достатньо ресурсів та призначати кваліфікованих аудиторів для забезпечення ефективної оцінки дотримання норм належної практики фармаконагляду. За необхідності аудитора(ів) може супроводжувати спеціаліст(и) у відповідних галузях.
Планування, координування, проведення, звітування про результати, здійснення післяаудиторського нагляду та документальне оформлення аудиту системи фармаконагляду повинні здійснюватися уповноваженою установою згідно з розробленими та затвердженими нею процедурами аудиту системи фармаконагляду.
Планування та проведення аудиту системи фармаконагляду уповноваженою установою буде розпочинатися з підготовки аудиторської програми, що ґрунтується на систематичному та ризик-орієнтованому підході, як зазначено в розділі III.В.2 ННПФ.
Уповноважена установа може проводити позапланові аудити системи фармаконагляду, пов’язані із зареєстрованими в Україні лікарськими засобамиN.
III.С.4.3. Програми проведення аудитів системи фармаконагляду уповноваженою установою
Уповноважена установаN повинна розробити програму планових аудитів системи фармаконаглядуN, що проводяться нею. Пріоритетність (першочерговість)N планових аудитів системи фармаконагляду уповноваженою установоюN буде визначатися на підставі потенційного ризику для громадського здоров’я, зважаючи на чинники, перераховані в розділі III.В.5 ННПФ. Загалом, аудит системи фармаконаглядуN заявника (власника реєстраційного посвідчення) повинен здійснюватися уповноваженою установоюN на основі ризик-орієнтованого підходу, однак, принаймні, раз на 4 роки/якщо аудит системи фармаконагляду заявника раніше не проводився уповноваженою установою протягом 5 років після розміщення на фармацевтичному ринку України першого лікарського засобу заявникаN.
Якщо одна й та сама система фармаконагляду застосовується для лікарських засобів, зареєстрованих за різними процедурами, результати проведеного аудиту системи фармаконагляду уповноваженою установоюN можуть застосовуватися до всіх препаратів, охоплених цією системою.
Програма планових аудитів системи фармаконагляду уповноваженою установоюN не буде включати позапланові аудити системи фармаконагляду уповноваженою установоюN, проте якщо проводиться позаплановий аудит системи фармаконагляду уповноваженою установоюN, то вона може замінити необхідність проведення планового аудиту системи фармаконагляду уповноваженою установоюN, в залежності від масштабів останньої.
III.С.5. Роль заявників (власників реєстраційного посвідчення)
Заявники (власники реєстраційного посвідчення), які подали нові заяви на реєстрацію лікарського(их) засобу(ів), підлягають інспектуванню системи фармаконагляду в країнах ЄС чи аудиту системи фармаконагляду, що проводиться в Україні уповноваженою установоюN (див. розділ III.В.1 ННПФ). Їх обов’язки у зв’язку з аудитами системи фармаконагляду уповноваженою установою в УкраїніN включають, але не обмежуються, наступними:
- завжди бути готовими до аудиту системи фармаконагляду в Україні уповноваженою установоюN, оскільки він може проводитися без попередження;
- вести та надавати на вимогу аудиторів системи фармаконагляду в Україні уповноваженою установоюN МФСФ, не пізніше ніж за 7 календарних днів після отримання запиту, відповідно до вимог ст. 23(4) директиви 2001/83/ЄС [1] і ст. 16(4) Регламенту (ЄС) 726/2004 [5], положення Порядку [2]N;
- до початку аудиту системи фармаконагляду уповноваженою установоюN забезпечувати згоду з боку об’єктів, вибраних для перевірки, до яких можуть відноситися організації, залучені заявником (власником реєстраційного посвідчення) для виконання діяльності з фармаконагляду;
- надавати аудиторам в УкраїніN будь-яку інформацію та/або документацію, потрібну для підготовки аудиту системи фармаконагляду уповноваженою установоюN, у зазначені терміни або під час проведення аудиту системи фармаконагляду уповноваженою установоюN;
- під час аудиту системи фармаконагляду уповноваженою установоюN забезпечувати присутність відповідного персоналу, залученого до діяльності з фармаконагляду або пов’язаної з ним діяльності, для інтерв’ю чи роз’яснення питань, що виникають;
- забезпечити доступ до відповідних даних з фармаконагляду, принаймні, з одного місця в межах УкраїниN (ст. 107(1) директиви 2001/83/ЄС [1], положення Порядку [2])N;
- забезпечити належне та своєчасне виконання планів коригуючих та запобіжних заходів для розв’язання проблемних питань, виявлених під час аудиту системи фармаконагляду в Україні уповноваженою установоюN, з встановленням пріоритетності критичних та/або суттєвих невідповідностей.
III.С.6. Фінансування проведення аудиту системи фармаконагляду уповноваженою установою в УкраїніN
В Україні законодавством не передбачено стягування аудиторських внесків та аудиторських витрат за проведення аудиту системи фармаконагляду уповноваженою установоюN.
III.С.7. Прозорість
Інформація про проведення та результати аудиту системи фармаконагляду в Україні уповноваженою установоюN і подальшого нагляду за усуненням недоліків може бути загальнодоступною. Це питання буде конкретизоване при складанні процедур проведення аудиту системи фармаконагляду в Україні уповноваженою установоюN, зазначених у розділі III.В.5 ННПФ.
Частина IV
Модуль IV — Аудит фармаконагляду
IV.A. Вступ
На законодавчому рівні/нормативно-правовими актамиN визначені вимоги до уповноваженого органу та заявників (власників реєстраційного посвідчення) щодо проведення аудиту їх систем фармаконагляду (ст. 101(2), ст. 104(2) Директиви 2004/83/ЄС [1], ст. 28f Регламенту (ЄС) 726/2004 [5], положення Порядку [2])N, включаючи ризик-орієнтований аудит їх систем якості (ст. 13 (1), ст. 17 (1) ІП 520/2012 [6], положення Порядку [2])N.
Терміни «аудит(и) фармаконагляду» та «діяльність з аудиту фармаконагляду» в даному модулі включають аудити систем фармаконагляду та аудит(и) системи якості для діяльності з фармаконагляду.
Мінімальні вимоги до систем фармаконагляду і їх систем якості визначені в Імплементаційній постанові Комісії (ЄС) № 520/2012 (ІП 520/2012) [6] щодо виконання діяльності з фармаконагляду, передбаченої Директивою 2001/83/ЄС [1], Постановою (ЄС) № 726/2004 [5] та положеннями Порядку [2]N. Особливості ризик-орієнтованих аудитів систем якості (для діяльності з фармаконагляду) описані у виконавчих заходах (ст.ст. 8, 10, 11, 12, 13(1) ІП 520/2012 [6] для заявників (власників реєстраційного посвідчення) та ст.ст. 8, 14, 15, 16, 17(1) для національних компетентних органів, ЕМА, уповноваженого органу та положеннях Порядку [2])N.
Загальний опис та завдання системи фармаконагляду та системи якості для діяльності з фармаконагляду наведені в модулі І ННПФ, а специфічні процеси фармаконагляду описані в кожному відповідному модулі ННПФ.
Цей модуль містить рекомендації з планування та проведення аудитів діяльності з фармаконагляду. Метою даного модулю є забезпечення проведення аудитів фармаконагляду, зокрема, шляхом сприяння гармонізації, послідовності та спрощенню процесу аудиту. Принципи, описані в даному модулі, узгоджуються з міжнародно визнаними розробленими відповідними міжнародними організаціями зі стандартизації аудиту7 та оприлюдненими на відповідних вебсайтах та передбачають проведення ризик-орієнтованого підходу до аудиту систем фармаконагляду.
У розділі IV.B ННПФ описані основні структури та процеси, яких необхідно дотримуватися при визначенні видів аудиту фармаконагляду, а також діяльності заявників (власників реєстраційного посвідчення) та уповноваженого органу з планування та проведення аудитів фармаконагляду та звітування за результатами аудиту. У розділі IV.B ННПФ також наведено короткий опис системи якості у фармаконагляді та діловодства, підходу до ведення документації при проведенні аудиту фармаконагляду.
У розділі IV.C ННПФ описуються вимоги та методологія здійснення аудиту фармаконагляду заявниками (власниками реєстраційного посвідчення) та уповноваженим органом.
IV.B. Структури та процеси
IV.B.1. Аудит фармаконагляду, його мета та завдання
Метою аудиту фармаконагляду є перевірка відповідності й ефективності впровадження та функціонування системи фармаконагляду, у тому числі її системи якості, шляхом вивчення та оцінки об’єктивних фактичних даних.
Аудит — це системний, упорядкований, незалежний та документований процес отримання та об’єктивної оцінки фактів з метою визначення ступеня виконання критеріїв аудиту, що сприяє покращенню управління ризиками та здійснення процесів контролю й управління. Аудиторські факти складаються із записів, документальних підтверджень та іншої інформації, що має відношення до критеріїв аудиту та яку можна перевірити. Критеріями аудиту (для кожного завдання аудиту) є стандарти виконання та контролю діяльності, по відношенню до яких буде оцінюватися сторона, яка перевіряється, та її діяльність. Застосовно до фармаконагляду критерії аудиту повинні відображати вимоги до системи фармаконагляду, включаючи систему якості в фармаконагляді, визначені законодавством ЄС/УкраїниN та ННПФ.
IV.B.2. Ризик-орієнтований підхід до аудиту системи фармаконагляду
Ризик-орієнтований — це підхід, що використовує методи для визначення зон ризику. Під ризиком у цьому випадку слід розуміти ймовірність виникнення подій, що можуть негативно вплинути на виконання поставлених завдань, враховуючи тяжкість наслідків та/чи вірогідність того, що такий ризик не буде виявлено за допомогою інших методів. Ризик-орієнтований підхід до аудиту зосереджується на сферах найвищого ризику для системи фармаконагляду організації, включаючи її систему якості діяльності з фармаконагляду. У контексті фармаконагляду найбільш серйозну увагу слід приділити ризику для громадського здоров’я. Ризик можна оцінити на наступних етапах:
- планування аудиту на стратегічному рівні, результатом чого є розробка стратегії аудиту (довгостроковий підхід), яку має схвалити вище керівництво;
- планування аудиту на тактичному рівні, результатом якого є програма аудиту, визначення завдань, обсягів та обмежень аудиту, які часто називають масштабом аудиту; та
- планування аудиту на оперативному рівні, результатом якого є розробка плану аудиту для індивідуальних аудиторських робіт, визначення пріоритетних завдань аудиту на основі оцінки ризику та використання методів ризик-орієнтованого вибіркового дослідження і тестування, звітність за результатами аудиту відповідно до відносного рівня ризику, а також рекомендації аудиту згідно із запропонованою системою оцінювання [див. IV.B.2.3.1 ННПФ].
Проведену оцінку ризику слід належним чином задокументувати для стратегічного, тактичного та оперативного планування аудиту системи фармаконагляду у суб’єкта системи фармаконагляду (див. IV.B.2.1., IV.B.2.2. та IV.B.2.3 ННПФ відповідно).
IV.B.2.1. Планування аудиту на стратегічному рівні
Стратегія аудиту — це документ, у якому визначається, як буде здійснюватися діяльність з аудиту більш ніж впродовж року, зазвичай — протягом 2–5 років. Стратегія аудиту включає перелік аудитів, що обґрунтовано можуть проводитися. Стратегія аудиту використовується для визначення напрямків аудиту, предмету аудиту, а також методів та припущень (включаючи, наприклад, оцінку ризиків), на яких базуватиметься програма аудиту.
Стратегія аудиту повинна охоплювати питання організації управління процесом, управління ризиками та засоби внутрішнього контролю всіх складових системи фармаконагляду, у тому числі:
- усі процеси та завдання фармаконагляду;
- систему якості діяльності з фармаконагляду;
- взаємодію та зв’язки з іншими департаментами організації щодо здійснення діяльності з фармаконагляду, якщо доречно;
- діяльність з фармаконагляду, яку проводять підпорядковані організації, чи діяльність, делеговану іншим організаціям (наприклад, регіональним центрам, які подають інформацію, філіалам заявників (власників реєстраційного посвідчення) і третім сторонам, таким як контрактні організації та інші постачальники послуг).
При проведенні оцінки ризиків слід враховувати, але не обмежуватися наведеними нижче у довільному порядку факторами ризику:
- зміни в законодавстві та у ННПФ;
- істотна реорганізація чи реструктуризація системи фармаконагляду, злиття, поглинання (зокрема, у першу чергу це стосується заявників (власників реєстраційного посвідчення), оскільки це може призвести до значного збільшення кількості лікарських засобів, для яких використовується ця система);
- зміни в ключових управлінських функціях;
- ризик наявності підготованих неналежним чином та недосвідчених співробітників з фармаконагляду, наприклад, внаслідок значного плину кадрів, недоліків навчального процесу, реорганізації, збільшення обсягів роботи, тощо;
- значні зміни в системі фармаконагляду з моменту попереднього аудиту, наприклад, впровадження нової бази (баз) даних для діяльності з фармаконагляду чи значне оновлення існуючої бази (баз) даних, зміни в процесах і діяльності, спрямовані на виконання нових чи змінених нормативно-правових вимог;
- перший лікарський засіб на ринку (для заявника (власника реєстраційного посвідчення);
- лікарський(і) засіб(и) на ринку, щодо якого(их) вимагаються специфічні заходи з мінімізації ризику чи інші специфічні умови безпеки, наприклад, такі, як вимога здійснення додаткового моніторингу;
- ступінь критичності процесу, наприклад:
- для уповноваженого органу: наскільки критичним є вид діяльності/процес для належного функціонування системи фармаконагляду і досягнення загальної мети охорони здоров’я;
- для заявників (власників реєстраційних посвідчень): наскільки критичним є вид діяльності/процес для належного функціонування системи фармаконагляду. При прийнятті рішення стосовно того, коли проводити аудит філіалу чи третьої сторони, заявник (власник реєстраційного посвідчення) повинен врахувати, крім зазначених у цьому переліку факторів, ще й характер та критичність діяльності з фармаконагляду, яку виконує філіал чи третя сторона від імені заявника (власника реєстраційного посвідчення);
- результати попередніх аудитів, наприклад, відомості про те, чи взагалі коли-небудь проводився аудит виду діяльності/процесу (якщо ні, тоді це слід враховувати в залежності від критичності процесу); якщо вид діяльності/процес проходили аудит, то результати аудиту є фактором, який слід враховувати при прийнятті рішення щодо проведення повторного аудиту цього виду діяльності/процесу, включаючи імплементацію погоджених заходів;
- визначення процедурних недоліків, що стосуються специфічних видів діяльності/процесів;
- інша інформація щодо дотримання вимог законодавства та керівних принципів, наприклад:
- для уповноваженого органу: інформація з системи показників дотримання законодавства (як описано у Імплементаційній постанові Комісії щодо здійснення діяльності з фармаконагляду, передбаченої Постановою (ЄС) № 726/2004 [5] Європейського Парламенту і Ради і Директивою 2001/83/ЄС [1], положеннях Порядку [2]N), на основі скарг, із зовнішніх джерел, наприклад, аудитів/оцінок уповноваженого органу зовнішніми організаціями;
- для заявників (власників реєстраційного посвідчення): інформація з системи показників дотримання законодавства (як описано у Імплементаційній постанові Комісії щодо здійснення діяльності з фармаконагляду, передбаченої Постановою (ЄС) № 726/2004 [5] Європейського Парламенту і Ради і Директивою 2001/83/ЄС [1], положеннями Порядку [2]N), за результатами інспекції системи фармаконагляду в країнах ЄС чи аудиту системи фармаконагляду заявника уповноваженою установою в УкраїніN (див. Модуль III), на основі скарг, з інших зовнішніх джерел, наприклад, аудитів;
- інші організаційні зміни, що можуть негативно вплинути на вид діяльності, наприклад, якщо впроваджуються зміни в допоміжні функції (наприклад, підтримка інформаційних технологій) і це може негативно вплинути на діяльність з фармаконагляду.
IV.B.2.2. Планування аудиту на тактичному рівні
Програма аудиту — це опис одного чи декількох аудитів, запланованих на визначений період часу, зазвичай, на рік. Програму аудиту слід розробляти згідно з довгостроковою стратегією аудиту. Програму аудиту має схвалити вище керівництво суб’єкта фармаконагляду, яке несе загальну відповідальність за структуру та організацію поточної діяльності та управління.
Ризик-орієнтована програма аудиту повинна базуватися на належній оцінці ризику та зосереджуватися на наступному:
- система якості діяльності з фармаконагляду;
- критично важливі процеси фармаконагляду (див., наприклад, модуль I ННПФ і ст. 11, ст. 15 ІП);
- ключові системи контролю діяльності з фармаконагляду;
- сфери діяльності високого ризику визначені після того, як були впроваджені елементи контролю чи вжиті заходи для зниження ризику.
Ризик-орієнтована програма аудиту повинна також враховувати види діяльності, недостатньо охоплені аудитом у минулому, а також визначені сфери високого ризику та/чи специфічні вимоги керівництва та/чи осіб, відповідальних за здійснення фармаконагляду.
Документація програми аудиту повинна містити короткий опис плану кожного запланованого аудиту із зазначенням його масштабу та завдань.
Обґрунтування строків, періодичності та масштабів аудитів, включених до програми аудитів, має базуватися на документально оформленій оцінці ризику. Однак ризик-орієнтовані аудити фармаконагляду слід проводити регулярно відповідно до вимог законодавства.
Внесені зміни до програми аудиту повинні бути належним чином задокументовані.
IV.B.2.3. Планування аудиту на оперативному рівні та підготовка звітів
IV.B.2.3.1. Планування аудиту та робота на місцях
Організація повинна забезпечити наявність письмових процедур, що стосуються планування та проведення окремих аудитів. У процедурах з аудиту повинні зазначатися строки всіх етапів, необхідних для проведення аудиту. Організація повинна забезпечити проведення аудитів відповідно до письмових процедур, що узгоджуються із цим модулем ННПФ.
Аудити фармаконагляду повинні проводитися відповідно до затвердженої ризик-орієнтованої програми (див. IV.B.2.2 ННПФ). При плануванні аудиту аудитор визначає та оцінює ризики, що стосуються тієї чи іншої сфери діяльності з фармаконагляду, яка підлягає аудиту, та застосовує найбільш оптимальні ризик-орієнтовані методи вибіркових досліджень і тестування, документуючи метод аудиту в плані аудиту.
IV.B.2.3.2. Звітність
Висновки аудиторів мають бути задокументовані у звіті з аудиту і повинні своєчасно бути доведені до відома керівництва. Процес аудиту повинен передбачати механізм надання інформації про результати аудиту суб’єкту аудиту, отримання зворотного зв’язку і надання результатів аудиту керівництву і зацікавленим сторонам, включаючи осіб, відповідальних за систему фармаконагляду, відповідно до вимог законодавства та рекомендацій з аудиту фармаконагляду. Результати аудиту повинні надаватися відповідно до рівня відносного ризику і класифікуватися з метою визначення їх критичності по відношенню до ризиків, що впливають на процеси чи компоненти процесів. Система класифікації повинна бути визначена в описі системи якості у фармаконагляді та повинна враховувати порогові значення, зазначені нижче, що будуть використовуватися у подальшій звітності відповідно до законодавства, як зазначено в розділі IV.C.2 ННПФ:
- критичні невідповідності — принципові недоліки одного чи декількох процесів або процедур фармаконагляду, що негативно впливають на всю систему фармаконагляду та/чи права, безпеку й благополуччя пацієнтів, чи потенційно становлять ризик для системи охорони здоров’я та/чи є серйозним порушенням законодавства;
- суттєві невідповідності — значні недоліки одного чи декількох процесів або процедур фармаконагляду, чи принциповий недолік частини одного чи декількох процесів або процедур фармаконагляду, що негативно відображається на всьому процесі та/чи може потенційно негативно вплинути на права, безпеку та благополуччя пацієнтів, та/чи потенційно становить ризик для системи охорони здоров’я та/чи є порушенням законодавства, хоча це порушення і не вважається критичним;
- несуттєві невідповідності — недоліки частини одного чи декількох процесів або процедур фармаконагляду, що, як очікується, не спричинять негативного впливу на всю систему фармаконагляду чи її процеси, та/чи права, безпеку та благополуччя пацієнтів.
Питання, що вимагають негайного вирішення, повинні у терміновому порядку доводитися до відома керівництва об’єкта аудиту та вищого керівництва.
IV.B.2.4. Заходи за результатами аудиту та наступний контроль
Заходи, що наведені в даному розділі ННПФ, тобто, негайні дії, оперативні дії, дії в адекватні строки, проблеми, що вимагають негайного вирішення чи термінового інформування, призначені для виконання в належні строки, доцільні та відповідають відносному ризику для системи фармаконагляду. Необхідно встановити першочерговість коригуючих та запобіжних заходів для усунення критичних та суттєвих невідповідностей. Точні строки для проведення заходів, пов’язаних з усуненням конкретної критичної невідповідності, можуть відрізнятися в залежності від особливостей виявлених невідповідностей та запланованих заходів.
Керівництво організації несе відповідальність за забезпечення наявності в організації механізмів для адекватного реагування на проблеми, виявлені під час аудиту фармаконагляду. Заходи повинні включати аналіз першопричин виявлених недоліків, аналіз впливу виявлених невідповідностей, а також підготовку плану коригуючих та запобіжних заходів, якщо необхідно.
Вище керівництво і особи, наділені керівними повноваженнями, повинні забезпечити запровадження ефективних заходів для усунення невідповідностей/недоліків, виявлених у результаті проведення аудиту. Запровадження погоджених заходів має систематично контролюватися. Інформація про стан запровадження повинна періодично доводитися до відома керівництва у відповідності до запланованих дій.
Факти завершення заходів з усунення невідповідностей слід фіксувати для документального підтвердження того, що для усунення проблем, виявлених під час аудиту фармаконагляду, було вжито необхідних заходів.
У програмі аудиту системи фармаконагляду слід передбачити можливість проведення контрольних аудитів. Такі аудити слід проводити за необхідності з метою перевірки завершення погоджених заходів (ст. 13(2), ст. 17(2) ІП 520/2012 [6] в ЄС та положення Порядку в Україні [2])N.
IV.B.3. Система якості та ведення документації
IV.B.3.1. Компетенція аудиторів та управління якістю аудиторської діяльності
IV.B.3.1.1. Незалежність та об’єктивність аудиту та аудиторів
Організація повинна призначити специфічні обов’язки для проведення аудиту фармаконагляду. Аудит фармаконагляду має бути незалежним. Керівництво організації повинно забезпечити незалежність і об’єктивність аудиторівN та документально це зафіксувати.
Аудиторам не повинні перешкоджати у визначенні масштабів аудиту, під час виконання аудиту фармаконагляду, та у інформуванні про його результати. Аудитори повинні співпрацювати з вищим керівництвом, яке несе повну відповідальність за виконавчу і управлінську структуру, що дозволить аудитору(ам) виконувати свої обов’язки та надавати незалежні об’єктивні аудиторські висновки. Аудитори можуть консультуватися з експертами, співробітниками, залученими до процесів фармаконагляду, та з УОВФ (та/або КОВФ); однак аудитори повинні бути неупередженими, переконаними в результатах своєї роботи і не йти на жодні компроміси, які могли б зашкодити якості їх роботи. Принцип об’єктивності вимагає від аудиторів не робити свої висновки з питань аудиту системи фармаконагляду в залежності від думки інших осіб.
IV.B.3.1.2. Кваліфікація, навички й досвід аудиторів та постійне підвищення кваліфікації
Аудитори повинні демонструвати та підтримувати професіоналізм щодо знань, навичок та здібностей, необхідних для ефективного проведення і/або участі у проведенні аудиту фармаконагляду. Професійний рівень аудиторів забезпечується завдяки навчанню, досвіду роботи та підготовки, і колективно вони повинні мати знання, навички та вміння з наступних питань:
- принципи, процедури та методи аудиту;
- чинне законодавство, нормативно-правові акти та інші вимоги, що стосуються фармаконагляду;
- діяльність, процеси та системи фармаконагляду;
- система(и) управління;
- організаційні системи.
IV.B.3.1.3. Оцінка якості аудиторської діяльності
Оцінку якості аудиторської роботи можна проводити за допомогою поточної та періодичної оцінки всієї аудиторської діяльності, на основі коментарів об’єктів аудиту та самостійної оцінки аудиторської діяльності (наприклад, забезпечення якості аудиторської діяльності, дотримання вимог кодексу поведінки, програми аудиту і аудиторських процедур).
IV.B.3.2. Проведення аудиту зовнішніми постачальниками аудиторських послуг
Основна відповідальність за функціонування та ефективність системи фармаконагляду покладена на організацію (наприклад, уповноважений орган чи заявник (власник реєстраційного посвідчення)). Якщо організація вирішить скористатися послугами зовнішнього постачальника аудиторських послуг для запровадження вимог з проведення аудиту фармаконагляду, описаних у даному модулі ННПФ і для проведення аудиту фармаконагляду:
- вимоги і підготовка оцінки аудиторського ризику, стратегії і програми аудиту та окремих аудиторських завдань повинні бути доведені організацією до відома зовнішніх постачальників аудиторських послуг у письмовій формі;
- масштаб, завдання та процедурні вимоги до аудиту повинні бути доведені організацією до відома зовнішніх постачальників аудиторських послуг у письмовій формі;
- організація повинна отримати та документально зафіксувати дані про гарантовану незалежність та об’єктивність зовнішнього постачальника аудиторських послуг;
- зовнішній постачальник аудиторських послуг повинен дотримуватися відповідних положень даного модуля ННПФ.
IV.B.3.3. Зберігання звітів про результати проведеного аудиту
Зберігання звітів про результати проведеного аудиту та матеріали повинні відповідати вимогам, викладеним у розділі I.B.10 модуля І ННПФ.
IV.C. Концепція та організаційна структура
IV.C.1. Заявники (власники реєстраційного посвідчення)
IV.C.1.1. Вимоги до проведення аудиту
Заявник (власник реєстраційного посвідчення) повинен проводити регулярні ризик-орієнтовані аудити своїх систем фармаконагляду (ст. 104(2) Директиви 2001/83/ЄС [1] та положення Порядку [2])N, включаючи аудит(и) своєї системи якості, для забезпечення відповідності системи якості вимогам до системи якості (ст. 8, 10, 11, 12, 13(1) ІП 520/2012 [6] та положення Порядку [2]N). Дати та результати аудитів та контрольних аудитів повинні бути задокументовані (ст. 13(1) ІП 520/2012 [6] та положення Порядку [2])N.
Подальші вимоги до звітів про результати проведеного аудиту заявника (власника реєстраційного посвідчення) див. розділ IV.C.2 ННПФ.
IV.C.1.1.1. Уповноважена особа, відповідальна за фармаконагляд/контактна особа, відповідальна за фармаконаглядN
Обов’язки УОВФ/КОВФN щодо аудиту викладено в модулі І ННПФ. Крім того, УОВФ/КОВФN, повинна отримувати звіти з аудитів та надавати аудиторам інформацію, що стосується оцінки ризиків, включно з даними про стан коригуючих та запобіжних заходів.
УОВФ/КОВФN, повинна бути поінформована про всі результати аудиту, що стосуються системи фармаконагляду, незалежно від того, де він проводився.
IV.C.1.2. Уповноважений орган
IV.C.1.2.1. Вимоги до проведення аудиту
Уповноважений орган повинен регулярно проводити незалежний аудит своїх завдань з фармаконагляду (ст. 28f Постанови 726/2004 [5], положення Порядку [2])N та регулярний аудит своєї системи фармаконагляду (ст. 101(2) Директиви 2001/83/ЄС [1] та положення Порядку [2])N. Крім обов’язкового проведення аудиту системи/завдань з фармаконагляду, уповноважений орган зобов’язаний проводити ризик-орієнтований аудит системи якості, через регулярні інтервали відповідно до загальної методології, з метою гарантування відповідності системи якості вимогам законодавства (ст. 8, 14, 15, 16, 17(1) ІП 520/2012 [6] та положення Порядку [2])N. Дати та результати аудитів та контрольних аудитів повинні бути задокументовані (17(2) ІП 520/2012 [6] та положення Порядку [2])N.
IV.C.1.2.2. Загальна методологія
Для того, щоб система аудиту була ефективною, усі аудиторські перевірки в уповноваженого органу повинні проводитися згідно із загальною методологією Це має забезпечити гармонізовані заходи з планування, реалізації та звітування.
IV.C.2. Вимоги до аудиторських звітів
IV.C.2.1. Звітність заявника (власника реєстраційного посвідчення)
Заявник (власник реєстраційного посвідчення) повинен зазначити критичні та суттєві результати будь-якого аудиту, що стосується системи фармаконагляду, у МФСФ (див. модуль ІІ ННПФ). На основі результатів аудиту заявник (власник реєстраційного посвідчення) повинен забезпечити підготовку та реалізацію відповідного плану коригуючих та запобіжних заходів. Після того як ці заходи буде повністю реалізовано, це повідомлення можна буде видалити з МФСФ (ст. 104(2) Директиви 2001/83/ЄС [1], ст. 13(2) ІП 520/2012 [6], положення Порядку [2])N. Для видалення будь-яких даних/відомостей щодо результатів аудиту з МФСФ необхідні об’єктивні докази (див. модуль ІІ ННПФ).
Заявники (власники реєстраційного посвідчення) повинні забезпечити зберігання списку всіх запланованих та завершених аудитів у додатку МФСФ (ст. 3(5) ІП 520/2012 [6], положення Порядку [2])N, а також дотримання своїх зобов’язань щодо звітування згідно з вимогами законодавства, ННПФ та внутрішніми політиками звітування. Дати та результати аудитів та контрольних аудитів повинні бути задокументовані (ст. 13(2) ІП 520/2012 [6], положення Порядку [2])N.
IV.C.2.2. Звіти про результати проведеного аудиту уповноваженим органом
Уповноважений орган повинен забезпечити надання звітів про результати проведеного аудиту згідно з вимогами законодавства, ННПФ та внутрішніми політиками звітування.
Уповноважений орган повинен звітувати про результати своїх аудитів системи фармаконагляду у терміни відповідно до законодавства.
IV.C.3. Конфіденційність
Документи та інформація, зібрані внутрішнім аудитором, повинні використовуватися з урахуванням рівня конфіденційності та вимог Директиви 95/46/ЄС [12] (Постанова (ЄС) № 45/2001 для установ ЄЕС [18]) та положень Порядку [2]N і національного законодавства із захисту фізичних і юридичних осіб стосовно обробки, використання та надання персональних даних.
IV.C.4. Прозорість
Уповноважений орган оприлюднює звіти з виконання завдань фармаконагляду.
Частина V
Модуль V — Система управління ризиками
V.A. Вступ
Лікарський засіб реєструється за умови, що для певних показань співвідношення користь/ризик є позитивним для цільових популяцій. Як правило, лікарський засіб може спричиняти виникнення побічних реакцій, що будуть варіюватися за ступенем тяжкості, ймовірності виникнення та впливу на окремих пацієнтів і громадське здоров’я. Однак зазвичай не всі побічні реакції та ризики виявляються на момент реєстрації лікарського засобу. Деякі з них виявляються та можуть бути чітко охарактеризовані лише в післяреєстраційний. Метою ПУР є документування системи управління ризиками, що є необхідним для ідентифікації, характеристики та мінімізації важливих ризиків лікарського засобу. ПУР містить інформацію про:
1) ідентифікацію або характеристику профіля безпеки лікарського засобу, при цьому особлива увага звертається на важливі ідентифіковані і потенційні ризики та відсутню інформацію, а також на те, які проблеми безпеки слід вирішувати у проактивний спосіб або додатково досліджувати («специфікація безпеки»);
2) планування заходів з фармаконагляду для описання ризиків та кількісного визначення клінічно релевантних ризиків, а також для виявлення нових побічних реакцій («план з фармаконагляду»);
3) планування і реалізацію заходів з мінімізації ризиків, включаючи оцінку ефективності цих заходів («план мінімізації ризиків»).
Оскільки знання про профіль безпеки лікарського засобу збільшуються з часом, також змінюється ПУР. Регламент (ЄC) № 726/2004 [5], Директива 2001/83/ЄС [1], ІП 520/2012 [6], положення Порядку проведення експертизи [7]N включають положення щодо післяреєстраційних досліджень з безпеки та післяреєстраційних досліджень ефективності як умови видачі реєстраційного посвідчення за певних обставин (ст. 9(4) (cb) та (cc) Регламенту (ЄC) № 726/2004 [5], ст. 10а(1)(а) та (b) Регламенту (ЄC) № 726/2004 [5], ст. 21а(b) та (f) Директиви 2001/83/ЄС [1], ст. 22а(1)(а) та (b) Директиви 2001/83/ЄС [1], Порядок проведення експертизи [7]N) та щодо включення цих досліджень до системи управління ризиками (ст. 14а Регламенту (ЄC) №726/2004 [5], ст. 22с(1) Директиви 2001/83/ЄС [1] та ст. 30(1)(d) ІП 520/2012 [6] та Порядок проведення експертизи [7]N). Законодавство також включає положення щодо додаткових заходів з мінімізації ризиків, що слід включити до системи управління ризиками як умови видачі реєстраційного посвідчення (ст. 9(4) (са) Регламенту (ЄC) № 726/2004 [5], ст. 21а(а) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Заявникам рекомендується планувати на дуже ранній стадії життєвого циклу лікарського засобу, як вони будуть додатково характеризувати та мінімізувати ризики, що пов’язані з лікарським засобом у післяреєстраційний період.
Настанова стосовно форми та подання ПУР оновлюється на сайті EMA8.
В Україні структура та вимоги до подання ПУР зазначені в Порядку [2]N. Цей модуль ННПФ включає принципи мінімізації ризиків, його слід читати разом з модулем XVI ННПФ та Додатком I до модуля XVI ННПФ щодо навчальних матеріалів.
Наступні законодавчі/нормативно-правові актиN містять посилання стосовно правової основи управління ризиком, але додаткові законодавчі/нормативно-правові актиN також можуть бути актуальними:
- Директива 2001/83/ЄС [1]: ст. 8(3)(ia) та (iaa), ст. 21а, ст. 22а(1), ст. 22с(1), ст. 104(3), ст. 106(с), ст. 127а;
- Регламент (ЄC) № 726/2004 [5]: ст. 6(1), ст. 9(4)(с), (са), (cb) (сс), ст. 10а(1), ст. 14а, ст. 26(1)(с);
- ІП 520/2012 [6]: ст. 30, ст. 31, ст. 32, ст. 33, Додаток 1;
- Регламент (ЄC) №1901/2006 [23]: cт. 34(2);
- Регламент (ЄС) № 1394/2007 [21]: cт. 14(2);
- положення Порядку [2]N;
- положення Порядку проведення експертизи реєстраційних матеріалів [7]N.
V.A.1. Термінологія
Визначення, що представлені в розділі «Терміни та визначення понять» ННПФ також застосовуються в контексті цього модулю ННПФ. Однак ПУР повинен приділяти увагу тим ризикам, що мають відношення до заходів з управління ризиками для зареєстрованого лікарського засобу.
З ідентифікованих ризиків лікарського засобу у ПУР слід розглянути тільки ті ризики, що є небажаними несприятливими клінічними наслідками та для яких є достатньо наукових доказів, що вони спричинені лікарським засобом (встановлений причинно-наслідковий зв’язок). Повідомлення про побічні реакції можна отримувати з різних джерел, таких як доклінічні результати, що підтверджені клінічними даними, клінічні випробування, епідеміологічні дослідження та спонтанні джерела даних, включаючи опубліковану літературу. Причинами їх виникнення може бути застосування лікарського засобу за незареєстрованими показаннями, медичні помилки або результат взаємодії з іншими лікарськими засобами. Не всі повідомлені побічні реакції обов’язково вважаються релевантними ризикам лікарського засобу в певному терапевтичному контексті.
З потенційних ризиків лікарського засобу у ПУР слід розглянути тільки ті, що є небажаними клінічними наслідками та для яких є достатньо наукових доказів для припущення щодо можливої наявності причинно-наслідкового зв’язку з лікарським засобом, але на даний момент недостатньо доказів, щоб зробити висновок про цей зв’язок.
У ПУР слід приділяти увагу важливим ідентифікованим ризикам, що, ймовірно, мають вплив на співвідношення користь/ризик лікарського засобу. Важливий ідентифікований ризик, що слід включити до ПУР, як правило, є підставою для:
- додаткової оцінки/вивчення як частини плану з фармаконагляду (наприклад, для вивчення частоти, ступеня тяжкості, серйозності та наслідку ризику за нормальних умов застосування лікарського засобу, або популяцій, які особливо піддаються впливу ризику);
- заходів з мінімізації ризику: інформація про лікарський засіб, що передбачає певні клінічні дії чи рекомендації, що необхідно здійснити для мінімізації ризику (див. V.B.8 ННПФ) або додаткові заходи з мінімізації ризику.
Важливими потенційними ризиками, що слід включити до ПУР, є ті важливі потенційні ризики, що після додаткової характеристики та, у разі підтвердження, матимуть вплив на співвідношення користь/ризик лікарського засобу. Коли існує наукове обґрунтування, що несприятливий клінічний наслідок може бути пов’язаним із застосуванням лікарського засобу не за показаннями, у популяціях, що не вивчені або бути результатом довготривалого застосування лікарського засобу, побічну реакцію слід вважати важливим потенційним ризиком. Якщо це буде визнано важливим, то такі проблеми повинні бути включені до переліку проблем безпеки в якості важливого потенційного ризику. Важливі потенційні ризики, що включені до ПУР зазвичай вимагають подальшої оцінки/вивчення та є частиною плану з фармаконагляду.
Відсутня інформація, що має значення для планування управління ризиками, стосується прогалин у знаннях стосовно безпеки лікарського засобу для певного контексту його застосування (наприклад довготривалого застосування) або для застосування у певних популяціях пацієнтів, відносно яких існує недостатньо даних для визначення, чи відрізняється профіль безпеки лікарського засобу при його застосуванні у цих пацієнтів від раніше відомого. Відсутність самих даних (наприклад, виключення популяції з клінічних випробувань) автоматично не становить проблему безпеки. Між тим, при плануванні управління ризиками увагу слід звернути на ситуації, що можуть відрізнятися від відомого профілю безпеки. Вимагається наукове обґрунтування для включення цієї популяції до ПУР в якості відсутньої інформації.
V.B. Структури та процеси
V.B.1. Принципи управління ризиками
Загальною метою управління ризиками є забезпечення того, щоб користь від застосування окремого лікарського засобу якомога більше переважала ризики. Основною метою і спрямованістю ПУР залишається факт належного планування управління ризиками протягом життєвого циклу лікарського засобу. Система управління ризиками повинна бути пропорційною виявленим ризикам та потенційним ризикам лікарського засобу, а також потребі в даних з безпеки у післяреєстраційний період (ст. 8(3) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
ПУР є динамічним документом, який слід оновлювати протягом життєвого циклу лікарського(их) засобу(ів). Останнє передбачає як додавання проблем безпеки, якщо це необхідно, так і видалення або переоцінку проблем безпеки в міру подальшого вивчення та характеристики профілю безпеки лікарського засобу.
Рекомендації щодо класифікації ризиків у цьому документі можуть сприяти тому, що під час життєвого циклу лікарських засобів перелік проблем безпеки у ПУР буде скорочений (див. також V.A.1. та V.B.5.8 ННПФ):
- можливо, що важливі потенційні ризики будуть видалені із специфікації з безпеки у ПУР (наприклад, коли накопичення наукових та клінічних даних не підтверджує первинне припущення, чи вплив на певну особу, виявляється меншим, ніж очікувалося, що визначає потенційний ризик як неважливий. Потенційні ризики можуть бути видалені також, коли немає жодних обґрунтованих очікувань, що будь-яка діяльність з фармаконагляду може додатково охарактеризувати ризик) або коли їх слід перекласифікувати на «важливі ідентифіковані ризики» (наприклад, якщо наукові та клінічні дані підсилюють зв’язок між ризиком та лікарським засобом);
- у певних обставинах, коли ризик повністю охарактеризований та відповідним чином управляється, важливі ідентифіковані ризики можна видалити зі специфікації з безпеки (наприклад, для лікарських засобів, що знаходяться на ринку протягом тривалого часу, для яких немає жодних невиконаних додаткових дій з фармаконагляду та/або заходів з мінімізації ризику у вигляді певних клінічних заходів для усунення ризику, що стали повністю інтегрованими до стандартної клінічної практики, наприклад, були включені до протоколів лікування або клінічних настанов);
- беручи до уваги загальну мету отримання додаткової інформації про співвідношення користь/ризик у певних популяціях, які не вивчалися у дореєстраційному періоді, очікується, що у міру «дозрівання» лікарського засобу дані, що були розцінені як «відсутня інформація», перестають бути такими після того, як нові дані стають доступними, або коли немає обґрунтованих очікувань, що існуючі або в майбутньому можливі дії дозволять додатково охарактеризувати профіль безпеки лікарського засобу стосовно «відсутньої інформації».
За винятком деяких реєстрів пацієнтів очікується, що з часом додаткові дії з фармаконагляду у ПУР будуть завершені та внаслідок цього видалені з ПУР.
Також може змінюватися і потреба в продовженні додаткових заходів з мінімізації ризиків. Це може бути зумовлене тим, що рекомендації для певних клінічних заходів для мінімізації ризику стають частиною рутинної практики, шляхом включення до протоколів стандартного лікування в ЄС, УкраїніN, або за результатами оцінки ефективності заходів з мінімізації ризиків, можливо, їх потрібно буде замінити на більш ефективні заходи. Може бути необхідним зберігати деякі заходи з мінімізації ризиків протягом життєвого циклу лікарського засобу (наприклад програми профілактики вагітності).
V.B.2. Відповідальність за управління ризиками
У плануванні управління ризиками лікарських засобів ключову роль відіграють заявники (власники реєстраційних посвідчень) та уповноважені органи, що регулюють обіг лікарських засобів.
Заявник (власник реєстраційного посвідчення) несе відповідальність за:
- наявність у нього відповідної системи управління ризиками (8(3)(iaa) Директиви 2001/83/ЄС [1]; ст. 104(3)(с) Директиви 2001/83/ЄС [1], положення Порядку [2] та положення Порядку проведення експертизи [7]N);
- забезпечення критичного огляду знань та розуміння профілю безпеки лікарського засобу, наступне його застосування у клінічній практиці. Заявник (власник реєстраційного посвідчення) повинен проводити моніторинг даних фармаконагляду для визначення, чи існують нові ризики, або чи змінилися ризики, або чи є зміни у співвідношенні користь/ризик лікарських засобів (ст. 104(3)(е) Директиви 2001/83/ЄС [1], положення Порядку [2]N), оцінювати профіль безпеки лікарського засобу та ПУР відповідно, як описано вище. Критичний огляд профілю безпеки лікарського засобу — це постійна діяльність, вона відображається в даних, що надаються у РОЗБ (див. модуль VII ННПФ), коли подання ПУР може або не може бути обґрунтованим. Крім того, є дві особливі стадії, коли заявникам (власникам реєстраційних посвідчень) на лікарські засоби, що зареєстровані за повними досьє, рекомендується розглянути потребу в перегляді переліку проблем безпеки, а також запланованих та поточних дій з фармаконагляду та мінімізації ризику:
- при (першій) перереєстрації через 5 років;
- у період часу, коли перший РОЗБ після (першої) перереєстрації через 5 років підлягає поданню. Очікується, що це подання РОЗБ буде мати місце приблизно через 8–9 років після видачі реєстраційного посвідчення, у той час, коли починається оцінка перших заяв на отримання реєстраційних посвідчень на генеричні лікарські засоби для діючої речовини. Як такий, профіль безпеки лікарського засобу, ймовірно, є достатньо добре охарактеризованим для того, щоб здійснити критичну оцінку та оновити перелік проблем безпеки.
V.B.3. Огляд формату та змісту плану управління ризиками (ПУР)
ПУР складається з семи частин. При поданні ПУР повинен мати відповідну структуру (додаток I ІП 520/2012 [6], додаток 14 до Порядку [2]N). Частина II ПУР — Специфікація з безпеки підрозділяється на модулі (додаток I ІП 520/2012 [6], додаток 14 до Порядку [2]N), тому зміст може бути адаптовано до специфіки даного лікарського засобу. А модулі частини II ПУР, як правило, повторюють назви розділів у специфікації з безпеки настанови ІСН E2E [20] (див. додаток до модуля IV ННПФ). Метою модульної структури є полегшення оновлення ПУР; крім того, за певних обставин до певних модулів ПУР можуть застосовуватися скорочені вимоги до змісту (див. V.C.1.1 ННПФ). Однак очікується, що ПУР буде подаватися як єдиний документ, що включає всі модулі та додатки, якщо необхідно.
Огляд частин та модулів ПУР представлено нижче в таблиці V.1 (додаток I ІП 520/2012 [6], додаток 14 Порядку [2]N).
Таблиця V.1. Огляд частин та модулів ПУР
|
Частина I |
Загальна інформація про лікарський(і) засіб(засоби) |
|
|
Частина II |
Специфікація з безпеки |
|
|
Модуль CI |
Епідеміологія показань до застосування та цільова(і) популяція(ї) |
|
|
Модуль CII |
Доклінічна частина специфікації з безпеки |
|
|
Модуль CIII |
Експозиція пацієнтів, залучених до клінічних випробувань |
|
|
Модуль CIV |
Популяції, які не вивчалися під час клінічних випробувань |
|
|
Модуль CV |
Післяреєстраційний досвід |
|
|
Модуль CVI |
Додаткові вимоги України, ЄС до специфікації з безпеки |
|
|
Модуль CVII |
Ідентифіковані та потенційні ризики |
|
|
Модуль CVIII |
Резюме проблем безпеки |
|
|
Частина III |
План з фармаконагляду (включаючи післяреєстраційні дослідження з безпеки) |
|
|
Частина IV |
Плани щодо післяреєстраційних досліджень ефективності |
|
|
Частина V |
Заходи з мінімізації ризиків (включаючи оцінку ефективності заходів з мінімізації ризиків) |
|
|
Частина VI |
Резюме плану управління ризиками |
|
|
Частина VII |
Додатки |
|
Інформація, представлена в ПУР, зокрема, в його II частині, повинна бути пропорційною ідентифікованим та потенційним ризикам та залежати від типу лікарського засобу, його ризиків і етапу його життєвого циклу (відповідно до ст. 8(3) Директиви 2001/83/ЄС [1], положень Порядку [2], положень Порядку проведення експертизи [7]N).
Ст. 14(2) Регламенту (ЄС) № 1394/2007 [21] передбачає певну структуру для ПУР на високотехнологічні лікарські засоби. Заявники (власники реєстраційних посвідчень) повинні розробляти ПУР високотехнологічних лікарських засобів, враховуючи та обговорюючи очікувані потреби в післяреєстраційному спостереженні та звертаючи увагу на особливості цих лікарських засобів. Особливі вимоги до змісту ПУР для високотехнологічних лікарських засобів слід обговорювати з уповноваженим органом перед поданням. Додаткові рекомендації щодо вимог до подальшого спостереження безпеки та ефективності та управління ризиком для високотехнологічних лікарських засобів надається на сайті EMA.
За необхідності рекомендується, щоб ПУР включав усі відповідні лікарські засоби від одного й того самого заявника (власника реєстраційного посвідчення), що містять однакову(і) діючу(і) речовину(и) (тобто ПУР — це документ, створення якого базується на діючій речовині) (ст. 30(2) ІП 520/2012 [6], положення Порядку [2]N).
Інформація в ПУР повинна бути надана досить докладно, при цьому слід уникати зайвого тексту, що відвертає увагу від основних питань, що слід розглянути для управління ризиками лікарського засобу. Однак у специфікації з безпеки ПУР не повинно бути дублювання даних, що надаються в інших місцях досьє, якщо розділи ПУР не призначені бути спільними модулями з іншими документами, такими як РОЗБ. За необхідності, інформація у ПУР повинна містити консолідований огляд/обговорення з акцентами на найважливіші ризики, що були ідентифіковані або передбачені на підставі доклінічних, клінічних та післяреєстраційних даних, що представлені в інших модулях електронного формату Загального технічного документа (еCTD). Будь-які дані, що включені до ПУР, повинні узгоджуватися з іншими розділами досьє. До ПУР слід включити лінки або посилання на відповідні розділи оглядів та резюме доклінічних та клінічних даних.
Для нової подачі ПУР на лікарські засоби, що реєструються за національною процедурою, з обмеженими даними з безпеки у досьє, цей документ повинен містити відповідні дані з безпеки та обговорення у підтримку обговорення ідентифікації ризику.
Для сприяння відповідності між інформацією, що представлена в досьє та в ПУР, таблиця V.2. вказує на місце розміщення інформації в модулях формату еCTD, що, ймовірно, буде обговорюватися у ПУР. Дані модулів/частин формату еCTD посилаються на подання, що містить ПУР (наприклад заяви на реєстрацію та значні зміни) або на історичні дані, що вже включені до досьє під час попередніх подань.
У контексті централізованої процедури ПУР слід подавати як складову подання формату еCTD; однак для нецентралізованих процедур подання ПУР все ще може бути частиною подання ЗТД. Дані — подання досьє у форматі еCTD у цьому модулі слід читати як формат еСTD або дані/подання CTD, що відповідає типу подання до уповноваженого органу.
Таблиця V.2. Відповідність між модулями ПУР та інформацією в досьє формату еCTD
|
Модуль ПУР |
Досьє у форматі еCTD |
|
Частина I Загальна інформація про лікарський(і) засіб (засоби) |
Модуль 2.3 Загальне резюме з якості Модуль 3 Якість |
|
Модуль CI Епідеміологія показання (ь) та цільової(их) популяції(й) |
Модуль 2.5 Огляд клінічних даних |
|
Модуль CII Доклінічна частина специфікації з безпеки |
Модуль 2.4 Огляд доклінічних даних Модуль 2.6 Резюме доклінічних даних у вигляді опису та таблиць Модуль 4 Звіти про доклінічні дослідження |
|
Модуль CIII Експозиція пацієнтів, включених у клінічні випробування |
Модуль 2.7 Резюме клінічних даних Модуль 5 Звіти про клінічні випробування |
|
Модуль CIV Популяції, які не вивчалися у клінічних випробуваннях |
Модуль 2.5 Огляд клінічних даних |
|
Модуль CV Післяреєстраційний досвід |
Модуль 2.5 Огляд клінічних даних |
|
Модуль CVI Додаткові вимоги УкраїниN, ЄС до специфікації з безпеки |
Дані не представлені в інших розділах eCTD |
|
Модуль CVII Ідентифіковані та потенційні ризики |
Модуль 2.5 Огляд клінічних даних (включаючи висновок щодо оцінки співвідношення користь/ризик) Модуль 2.7 Резюме клінічних даних (SPC) |
|
Модуль CVIII Резюме проблем з безпеки |
Модуль 2.5 Огляд клінічних даних Модуль 2.7 Резюме клінічних даних |
|
Частина III План з фармаконагляду (включаючи післяреєстраційні дослідження з безпеки) |
Модуль 2.5 Огляд клінічних даних Модуль 2.7 Резюме клінічних даних |
|
Частина IV Плани щодо післяреєстраційних досліджень ефективності |
Модуль 2.5 Огляд клінічних даних Модуль 2.7 Резюме клінічних даних |
|
Частина V Заходи з мінімізації ризиків (включаючи оцінку ефективності заходів з мінімізації ризиків) |
Модуль 2.5 Огляд клінічних даних Модуль 2.7 Резюме клінічних даних |
Тільки основні літературні посилання у ПУР повинні бути включені до додатку 7 ПУР. Це слід зробити у форматі електронних лінків або посилань, якщо вони вже включені в інших модулях досьє у форматі еCTD (див. V.B.10 ННПФ).
Опис частин і модулів ПУР у V.B.4 ННПФ містить вказівки про те, які основні теми повинні бути розкриті в кожному з них. Тим не менш, деякі розділи можуть не мати відношення до всіх лікарських засобів, та можуть існувати додаткові теми, що повинні бути включені, але вони не згадуються у цих вказівках. ПУР є частиною наукового досьє лікарського засобу, тому має бути науково обґрунтованим і не повинен включати жодних елементів рекламного характеру.
До вступного розділу ПУР слід включити таку адміністративну інформацію:
- дату закриття бази даних для поточного ПУР;
- дата остаточного підписання та номер версії ПУР;
- перелік усіх частин та модулів. Для оновлень ПУР у цьому розділі слід у табличному форматі зазначати номер версії модулів та дату схвалення. Коментар високого рівня стосовно обґрунтування створення нової версії слід включити для значних змін, що вносяться до кожного модуля;
- доказ контролю з боку УОВФ не вимагається для версій, що подані на оцінку. Реальний підпис УОВФ або доказ, що ПУР переглядався та схвалювався УОВФ, слід включити до заключної схваленої версії документа; для подання досьє у форматі еCTD це буде ПУР з послідовністю процедури останнього досьє у форматі еCTD (наприклад послідовністю закриття). Доказ контролю УОВФ може мати форму заяви, що УОВФ заявника (власника реєстраційного посвідчення) переглянула та схвалила ПУР та електронний підпис знаходиться у файлі.
V.B.4. Частина I ПУР «Загальна інформація про лікарський(і) засіб (засоби)»
Дана частина повинна містити адміністративну інформацію щодо ПУР та огляду лікарського(их) засобу(ів). Представлена інформація повинна бути актуальною та точною в контексті поточної заяви. Інформація повинна включати:
дані щодо діючої речовини:
- назва діючої(их) речовини(н);
- фармакотерапевтична(і) група(и) (Код АТХ);
- найменування:
- заявника — для ПУР, що подані для здійснення післяреєстраційних процедур;
- заявника — для заяв на реєстрацію;
- або
- для заяв за процедурою взаємного визнання/децентралізованою процедурою: найменування очікуваного майбутнього власника реєстраційного посвідчення у референтній державі ЄС, якщо відомо на момент подання заяви;
- лікарський(і) засіб(засоби), на який(і) поширюється ПУР;
- процедура, за якої проходить реєстрація (централізована, взаємне визнання, децентралізована, національна);
- торговельна(і) назва(и) в Європейській економічної зоні (далі — ЄЕЗ), України;
- короткий опис лікарського засобу, включаючи:
- хімічний клас;
- коротке викладення механізму дії;
- важлива інформація про склад (наприклад, походження діючої речовини біологічних лікарських засобів, відповідні ад’юванти або домішки вакцин);
- лінк (посилання) у eCTD на запропоновану інформацію про лікарський засіб, якщо необхідно;
- показання: схвалені та запропоновані (якщо ПУР подається з розширенням/обмеженням показання);
- дозування (стисла інформація — тільки та, що стосується основної популяції; не дублювання розділу короткої характеристики лікарського засобу/інструкції для медичного застосуванняN);
- лікарські форми та сили дії;
- чи є цей лікарський засіб предметом додаткового моніторингу в ЄС/УкраїніN (у висновку до заяви на отримання первинної реєстрації або з оновленнями ПУР).
V.B.5. Частина II ПУР «Специфікація з безпеки»
Метою специфікації з безпеки є забезпечення адекватного огляду профілю безпеки лікарського(х) засобу(ів), із акцентом на ті аспекти, що потребують подальших заходів з управління ризиками. Вона повинна містити резюме важливих ідентифікованих ризиків лікарського засобу, важливих потенційних ризиків та відсутньої інформації. У цій частині слід також розглянути, які популяції перебувають під потенційним ризиком (де лікарський засіб буде застосовуватися за схваленим або за не схваленим показанням), а також будь-які невирішені питання з безпеки, що обґрунтовують подальші дослідження для кращого розуміння співвідношення користь/ризик у післяреєстраційний період. У ПУР специфікація з безпеки є основою для плану з фармаконагляду та плану заходів з мінімізації ризиків.
Специфікація з безпеки містить вісім модулів ПУР, з яких модулі ПУР CI–CV, CVII та CVIII відповідають заголовкам специфікації з безпеки настанови ІСН E2E [20]. Модуль CVI ПУР включає додаткові елементи, що будуть необхідні для подання в ЄС/УкраїніN.
Хоча елементи, що описані у V.B.5.2.–V.B.5.9 ННПФ, є лише керівництвом, рекомендується, щоб заявники (власники реєстраційних посвідчень) додержувалися наданої структури при складанні специфікації з безпеки.
Деталі певних вимог до заяв на отримання першої реєстрації включені до V.C.1.1 ННПФ.
V.B.5.1. Загальні питання стосовно генеричних лікарських засобів та високотехнологічних лікарських засобів
V.B.5.1.1. Генерики
Для генеричних лікарських засобів очікується, що специфікація з безпеки є такою самою, як специфікація з безпеки у референтного лікарського засобу або в інших генеричних лікарських засобів, для яких є ПУР. Якщо існують відмінності між схваленими ПУР на такі лікарські засоби, тоді заявник, як очікується, повинен запропонувати та обґрунтувати найбільш відповідну специфікацію з безпеки для свого лікарського засобу. У виняткових випадках заявник на новий генеричний лікарський засіб може додавати або видаляти проблеми з безпеки порівняно з профілем безпеки референтного лікарського засобу, якщо це відповідно обґрунтовано (наприклад, коли є додаткове нове розуміння поточного профілю безпеки або коли існують відмінності у характеристиках лікарського засобу порівняно з референтним лікарським засобом, наприклад, існує ризик, що пов’язаний з допоміжною речовиною, що наявна лише в певних лікарських засобах, які містять одну й ту саму діючу речовину).
V.B.5.1.2. Високотехнологічні лікарські засоби
Відповідно до Регламенту (ЄC) № 1394/2007 [21] та положень Порядку проведення експертизи [7]N певні лікарські засоби класифікуються в ЄС та УкраїніN як високотехнологічні. Ці лікарські засоби охарактеризовані повною мірою у вказаних вище документах, проте в широкому сенсі включають:
- генно-інженерні лікарські засоби;
- препарати соматоклітинної терапії;
- продукти тканинної інженерії.
Внаслідок властивостей цих лікарських засобів при їх застосуванні можуть виникнути ризики, що не завжди мають місце при прийомі інших лікарських засобів, включаючи ризики для живого донора, ризики трансформації зародкової лінії та передачі векторів. Ці ризики слід враховувати при розробці специфікації з безпеки для високотехнологічних лікарських засобів (див. V.B.5.8 ННПФ).
V.B.5.2. Частина II ПУР, Модуль CI «Епідеміологія показань до застосування та цільова(і) популяція(ї)»
Цей модуль ПУР повинен включати інформацію про такі епідеміологічні показники захворювання, як захворюваність, поширеність, наслідки хвороби-мішені без лікування. Також тут слід зазначити відповідні супутні захворювання і, якщо це важливо для оцінки безпеки та управління ризиками, урахувати вік, стать, расову та/або етнічну належність популяції. Також слід описати фактори ризику виникнення хвороби та основні існуючі варіанти лікування. Акцент повинен бути зроблений на епідеміології зареєстрованого показання для застосування в ЄС та в УкраїніN. Слід обговорити відмінності в епідеміології у різних регіонах (у випадку, коли епідеміологія показань відрізняється залежно від регіону).
Цей розділ також повинен описувати значущі побічні явища, що слід передбачити у цільовій популяції без лікування в ЄС та в УкраїніN (фоновий рівень побічного явища)N, їх частоту та характеристики. Текст повинен допомогти у передбаченні та тлумаченні будь-яких потенційних сигналів та в ідентифікації можливостей для мінімізації ризику. У тексті слід додержуватися чіткості, він не повинен включати жодних елементів рекламного характеру.
V.B.5.3. Частина II, Модуль CII «Доклінічна частина специфікації з безпеки»
У цьому модулі ПУР повинно бути представлене резюме важливих висновків щодо безпеки лікарського засобу за даними доклінічних досліджень, наприклад:
- токсичність (основні проблеми, що виявлені в дослідженнях токсичності при повторному введенні дози, репродуктивна/пренатальна токсичність, генотоксичність, канцерогенність);
- загальна фармакологія (наприклад, вплив лікарського засобу на стан серцево-судинної системи, у тому числі ймовірність пролонгації інтервалу Q–T, на нервову систему тощо);
- інша інформація або дані, що стосуються токсичності.
Що представлятиме собою важливий висновок щодо безпеки лікарського засобу за даними доклінічних досліджень, буде залежати від лікарського засобу, цільової популяції та досвіду застосування інших аналогічних сполук чи лікарських засобів або методів лікування, що належать до одного та того самого класу. Зазвичай також мають бути розглянуті значні особливості токсичної дії (за системою органів-мішеней) і значущість отриманих даних для застосування у людей. Крім того, аспекти якості, якщо вони мають відношення до безпеки (наприклад, генотоксичні домішки), також слід розглянути. Якщо лікарський засіб призначений для застосування у жінок дітородного віку, необхідно докладно представити дані щодо репродуктивної та пренатальної токсичності, а також інформацію щодо наслідків від прийому цього лікарського засобу в цій популяції. Коли за висновками з безпеки на підставі даних доклінічних досліджень може бути виявлений важливий ризик для цільової популяції, він повинен бути включений у якості проблеми з безпеки до модулю CVIII ПУР. Коли висновки з безпеки на підставі даних доклінічних досліджень не вважаються важливими для людини, необхідно надати коротке роз’яснення. Відповідно, їх не потрібно вносити до висновку з безпеки, а також до модулів CVII та CVIII ПУР у якості проблеми з безпеки.
Якщо на підставі оцінки доклінічних або клінічних даних вважаються обґрунтованими додаткові доклінічні дослідження та вони запропоновані в якості складової плану фармаконагляду, це слід стисло обговорити в даному розділі.
Заключні висновки щодо цього розділу, якщо це доцільно, слід відобразити в модулі CVII та в модулі CVIII ПУР.
Зміст цього розділу слід оцінювати з часом на актуальність. Очікується, що в післяреєстраційний період цей розділ буде оновлюватися тільки коли нові доклінічні дані впливатимуть на оцінку проблем безпеки. Проблеми безпеки, що ідентифіковані на підставі доклінічних даних, які згодом втрачатимуть значення та/або не підтвердилися при отриманні достатнього відповідного досвіду та доказів у післяреєстраційний період, можна видалити з переліку проблем безпеки.
V.B.5.4. Частина II ПУР, модуль CIII «Експозиція пацієнтів, залучених до клінічних випробувань»
У цьому модулі ПУР для оцінки обмежень бази даних з безпеки застосування лікарського засобу у людини слід представити стислу інформацію про пацієнтів, які брали участь у клінічних випробуваннях. Необхідно надати у відповідному форматі (наприклад у вигляді таблиць/графіків) на час подання первинного ПУР або у випадку його значного оновлення, у результаті отримання нових даних про експозицію, що вивчалася під час клінічних досліджень (наприклад при новому показанні). Зміст цього розділу слід оцінювати на актуальність з часом та за відсутності нових значущих даних стосовно експозиції пацієнтів, залучених до клінічних випробувань, не потрібно вносити зміни до цього розділу.
Розмір досліджуваної популяції повинен бути вказаний із зазначенням як кількості пацієнтів, так і, за необхідності, тривалості часу, протягом якого пацієнти приймали лікарський засіб. Ці дані слід розподіляти за відповідними категоріями). Розподіл, як правило, відбувається за наступними категоріями:
- вік і стать;
- показання;
- доза;
- інші категорії розподілу слід застосовувати, коли це додає важливої інформації з метою планування управління ризиком (наприклад етнічне походження).
У педіатричній віковій групі дані повинні бути розподілені за категоріями (наприклад згідно з настановою ICH-E119); аналогічно, дані про літніх пацієнтів повинні бути розподілені за категоріями цієї цільової популяції (наприклад 65–74 років, 75–84 і 85 років і старше).
Якщо це не доцільно, дані не потрібно надавати за окремим випробуванням — їх слід об’єднати. Підсумки повинні бути передбачені для кожної таблиці/графіка в залежності від обставин. Якщо пацієнти брали участь більше ніж в одному випробуванні(дослідженні) (наприклад відкрите подовжене дослідження після закінчення основного випробування), вони повинні бути включені до таблиці за віком/статтю/етнічним походженням тільки 1 раз. У разі розходжень між даними таблиць щодо загальної кількості пацієнтів, таблиці повинні супроводжуватися поясненнями щодо причин розбіжностей.
Якщо ПУР подається із заявою на нове показання, нову лікарську форму або шлях введення, дані клінічного дослідження, специфічні для даної заяви, необхідно представити окремо на початку модуля, а також як зведені дані за всіма показаннями.
V.B.5.5. Частина II ПУР, модуль CIV «Популяції, які не вивчалися під час клінічних випробувань»
У цьому модулі ПУР слід надати опис популяцій, які розглядаються у зв’язку з відсутньою інформацією.
У разі наявності та за необхідності слід надати інформацію про експозицію особливих популяцій або її відсутність (наприклад, вагітні, жінки, які годують груддю, пацієнти з нирковою недостатністю, пацієнти з печінковою недостатністю або серцевою недостатністю, популяції із значимим генетичним поліморфізмом, пацієнти з ослабленим імунітетом та популяції різного етнічного походження). За наявності інформації слід зазначати ступінь печінкової або серцевої недостатності, а також тип генетичного поліморфізму.
Якщо очікується, що лікарський засіб застосовуватиметься в популяціях, що не вивчалися у дослідженнях, та якщо існує наукова підстава підозрювати інший профіль безпеки, але наявної інформації недостатньо для визначення, може чи не може застосування лікарського засобу за цих обставин становити проблему безпеки, тоді це слід включити як відсутню інформацію до ПУР. Популяції, які виключалися з програми клінічних випробувань, слід розцінювати як відсутню інформацію, тільки коли вони мають значення для схваленого та запропонованого показання, тобто «за показаннями», та якщо застосування лікарського засобу у таких популяціях може бути пов’язане з ризиками клінічного значення. При обговоренні відмінностей між цільовими популяціями та тими, які вивчалися у клінічних випробуваннях, слід зазначити, що деякі відмінності можуть мати місце скоріше через умови проведення випробування (наприклад лікарня або загальна практика), ніж через визначені критерії включення/виключення. Коли такі популяції розглядаються як «відсутня інформація», тоді модуль CIV ПУР повинен включати обговорення відповідних субпопуляцій.
Якщо існує доказ, що застосування у виключених популяцій пов’язане з небажаним клінічним наслідком, тоді такий наслідок слід включити як важливий (потенційний) ризик.
V.B.5.6. Частина II ПУР, модуль CV «Післяреєстраційний досвід»
Якщо післяреєстраційні дані отримані на підставі післяреєстраційного досвіду в інших регіонах поза ЄС, коли лікарський засіб вже зареєстрований, або на підставі інших зареєстрованих лікарських засобів, що містять таку саму діючу речовину, від того ж самого заявника (власника реєстраційного посвідчення), дані слід обговорити у цьому модулі ПУР.
Слід надавати тільки огляд досвіду в післяреєстраційний період, що корисний для планування управління ризиками. Цей модуль не призначений для дублювання інформації з РОЗБ.
Крім того, обговорення того, як лікарський засіб застосовується на практиці згідно із затвердженими показаннями та за незатвердженими показаннями, включаючи застосування у певних популяціях, що згадані в модулі CIV ПУР можна також включити, якщо це має значення для обговорення ідентифікації ризику в модулі CVII ПУР.
Коли це необхідно та має значення для обговорення у модулі CVII, дані про застосування на ринках поза ЄС, УкраїноюN за показаннями, що не зареєстровані в ЄС, УкраїніN, також слід резюмувати та обговорити наслідки для затвердження таких показань в ЄС, УкраїніN.
V.B.5.7. Частина II ПУР, модуль CVI «Додаткові вимоги України, ЄС до специфікації з безпеки»
Крім питань безпеки, розгляд яких вимагається у настанові ІСН E2E [20] (див. додаток IV ННПФ), у ПУР слід розглянути такі проблеми: потенціал для зловживання та застосування лікарського засобу у протиправних цілях та, якщо необхідно, запропоновані заходи з мінімізації ризиків, наприклад, обмежений розмір пакування, програми контрольованого доступу, особливий медичний рецепт (ст. 71(2) Директиви 2001/83/ЄС [1], додаток 14 до Порядку [2]N) (див. також V.B.8 ННПФ).
V.B.5.8. Частина II ПУР, модуль CVII «Ідентифіковані та потенційні ризики»
У цьому модулі ПУР при обговоренні слід приділити увагу ідентифікації важливих ідентифікованих та важливих потенційних ризиків та відсутній інформації (тобто проблемам безпеки).
Вважається, що такі питання безпеки, що виникли внаслідок певних ситуацій/джерел даних, представляють особливий інтерес при обговоренні ідентифікації ризику у модулі CVII ПУР, їх слід обговорювати, коли вони призводять до ризиків лікарського засобу:
- потенційна шкода від передозування, навмисного чи випадкового, наприклад, у випадках, коли існує вузький терапевтичний інтервал, або потенціал для значної дозозалежної токсичності, та/або коли існує високий ризик навмисного передозування у популяції, що лікується (наприклад при депресії). Коли шкода від передозування виникає під час клінічних випробувань, це слід прямо зазначити та, якщо необхідно, важливі ризики внаслідок передозування слід включити як проблему безпеки до модулю CVIII ПУР, а відповідні заходи з мінімізації ризику запропонувати в частині V ПУР;
- потенціал для ризиків внаслідок медичних помилок, що визначаються як будь-яка ненавмисна помилка у процесі лікування, що призводить або має потенціал призвести до нанесення шкоди пацієнту. Медичні помилки, що приводять до важливих ризиків, виявлених у процесі розробки лікарського засобу, включаючи клінічні випробування, слід обговорити, а також надати інформацію про помилки, їх потенційну(і) причину(и) та можливі заходи для їх усунення. Якщо необхідно, слід вказати, як ці помилки враховані в дизайні кінцевого продукту. Додаткові рекомендації щодо медичних помилок надаються в Посібнику з належної практики мінімізації ризиків та запобігання медичним помилкам, додаток 2 — Характеристики дизайну, що слід враховувати для зниження ризику медичної помилки10, що включає широкий перелік можливих медичних помилок і наслідки для пацієнтів. Важливі ризики, пов’язані з медичними помилками у післяреєстраційний період слід обговорити в оновленому ПУР та запропонувати шляхи зменшення їх виникнення;
- потенціал для передачі збудника інфекції внаслідок характеру процесу виробництва чи використовуваних матеріалів. Для живих атенуйованих вакцин будь-який потенціал передачі мутованого живого вакцинного вірусу, а також потенціал розвитку хвороби в осіб з ослабленим імунітетом, які контактували з вакциною (щепленою особою), слід обговорити, щоб розглядати їх як можливі потенційні ризики;
- потенціал для застосування не за показаннями, коли передбачаються відмінності у проблемах безпеки між цільовою популяцією та популяцією, у якій не схвалювалося застосування лікарського засобу, з метою включення до специфікацій з безпеки потенційних ризиків, що виникають внаслідок застосування за незареєстрованими показаннями;
- якщо важливий ідентифікований або потенційний ризик, що є загальним для інших лікарських засобів, що належать до однієї фармакологічної групи, не вважається важливим ідентифікованим або важливим потенційним ризиком для даного лікарського засобу, докази, що це підтверджують, повинні бути представленні та обговорені;
- важливі ризики, що пов’язані з ідентифікованими та потенційними фармакокінетичними та фармакодинамічними видами взаємодії, слід обговорити у зв’язку з підходами до лікування певної хвороби/стану та застосуванням лікарських засобів, що, як правило, застосовуються у цільовій популяції. Слід резюмувати докази, що підтверджують взаємодію та можливий механізм, обговорити потенційні ризики для здоров’я для різних показань та популяцій, а також описати плани для додаткової характеристики та мінімізації ризиків. Важливі ризики, що виникають внаслідок взаємодій, слід включити як проблему безпеки;
- ризики у вагітних та жінок, які годують груддю, наприклад, тератогенна дія лікарського засобу — пряма або опосередкована дія через дію на сперму. Рекомендації щодо методів контрацепції можна розглядати як заходи з мінімізації ризиків. Додаткову настанову з управління ризиками у випадку розгляду експозиції ембріона/плода, які зазнали впливу лікарських засобів з тератогенною дією можна знайти у модулі III та Модулі XVI ННПФ;
- вплив на репродуктивну функцію — слід розглянути відповідні заходи з мінімізації ризиків, наприклад, рутинні повідомлення про ризики та/або додаткові заходи, що рекомендують збереження репродуктивної функції: кріоконсервацію сперми у чоловіків, а також кріоконсервацію ембріонів та яйцеклітин у жінок;
- ризики, пов’язані з утилізацією лікарських засобів після їх використання (наприклад, трансдермальні пластирі із залишками діючої речовини або залишки засобів для радіоактивних діагностичних заходів);
- ризики, пов’язані процедурою введення (наприклад ризики внаслідок застосування медичного пристрою (несправність, яка впливає на дозу, що вводиться, ризик мінливості при складних техніках введення);
- питання безпеки для педіатричної популяції, що є особливими причинами занепокоєння щодо застосування в педіатричні популяції, як описано в розділі 5 додатку I висновку щодо плану педіатричних досліджень (Потенційні питання безпеки/ефективності при довготривалому застосуванні у зв’язку із застосуванням у педіатричній популяції для врахування у ПУР/діяльності, що пов’язана з фармаконаглядом).
Для ПУР на високотехнологічні лікарські засоби заявники повинні також розглянути можливі особливі ризики при створенні специфікації з безпеки (див. Керівництво з наступного контролю безпеки та ефективності — управління ризиком високотехнологічних лікарських засобів11).
V.B.5.8.1. Частина II ПУР, розділ модулю CVII «Ідентифікація проблем безпеки при первинному поданні ПУР»
Цей розділ ПУР повинен містити первинну ідентифікацію проблем безпеки, також очікується, що дані будуть вноситися до нього при первинному поданні ПУР: на час подання заяви на реєстрацію або в післяреєстраційний період (тобто для зареєстрованих лікарських засобів, у яких раніше не було ПУР).
Очікується, що цей розділ буде «заблокованим» та не буде змінюватися після схвалення первинного ПУР.
V.B.5.8.1.a. Частина II ПУР, розділи модулю CVII «Ризик, що вважається важливим для включення у перелік проблем з безпеки» та «Ризик, що не вважається важливим для включення у перелік проблем з безпеки»
У цьому розділі ПУР слід узагальнити та обговорити таку інформацію:
- серйозність ризику;
- частоту виникнення ризику;
- вплив ризиків на співвідношення користь/ризик.
Для ризиків, що не віднесені до проблем безпеки, інформацію можна групувати за причинами їх невключення як проблем з безпеки.
V.B.5.8.2. Частина II ПУР, розділ модулю CVII «Нові проблеми з безпеки та перекласифікація при поданні оновленого ПУР»
У післяреєстраційний період очікується, що нові ідентифіковані та потенційні ризики лікарського засобу, що представлені в розділі безпеки досьє (наприклад, з оцінкою сигналу, регулярною оцінкою співвідношення користь/ризик або процедурами внесення змін щодо безпеки) разом з оцінкою того, чи слід вважати ризики важливими та додавати їх до специфікації з безпеки в ПУР. Це обговорення не слід дублювати в ПУР, але докладні дані стосовно будь-якого нового важливого ідентифікованого або потенційного ризику слід включати до розділу ПУР, що описаний у V.B.5.8.3 ННПФ.
Коли важливий ідентифікований або потенційний ризик або відсутня інформація перекласифікується або видаляється, у цьому розділі ПУР слід надати обґрунтування з відповідним посиланням на дані з безпеки. Інформація, що включена до цього розділу, може мати форму заяви, що описує попередній регуляторний запит з посиланням на процедуру, у межах якої такий запит був сформульований.
V.B.5.8.3. Частина II ПУР, розділ модулю CVII «Детальна інформація про важливі ідентифіковані ризики, важливі потенційні ризики та відсутню інформацію»
Для ПУР, що містять різні лікарські засоби, якщо існують значні відмінності між лікарськими засобами (наприклад, засоби з фіксованою комбінацією доз), доцільно надати роз’яснення, якого самого лікарського засобу стосуються відповідні проблеми з безпеки.
Цей розділ ПУР є застосовним на всіх стадіях життєвого циклу лікарського засобу.
Представлення даних про важливі ідентифіковані ризики та важливі потенційні ризики:
- найменування ризику (із застосуванням термінів MedDRA, якщо доцільно);
- потенційний механізм;
- джерело(а) і сила доказів (тобто, наукова база для припущення причинно-наслідкового зв’язку);
- характеристика ризику: наприклад, частота, абсолютний ризик, ступінь тяжкості, зворотність, довгострокові наслідки, вплив на якість життя;
- фактори ризику та групи ризику (у тому числі фактори пацієнта, доза, період наявності ризику, адитивні чи синергічні чинники);
- можливість запобігання (тобто передбачуваність ризику; чи були ідентифіковані фактори ризику, що можна зменшити за допомогою рутинних або додаткових заходів з мінімізації ризиків, окрім загальної інформованості завдяки застосування інформації про лікарський засіб; можливість виявлення на ранній стадії, що може знизити серйозність);
- вплив на співвідношення користь/ризик лікарського засобу;
- вплив на громадське здоров’я (наприклад, абсолютний ризик відносно розміру цільової популяції та, отже, реальної кількості уражених, або сукупний наслідок на рівні популяції).
Представлення даних щодо відсутньої інформації:
- найменування відсутньої інформації (із застосуванням термінів MedDRA, якщо доцільно);
- доказ, що, за очікуваннями, профіль безпеки буде відрізнятися від профілю безпеки у загальній цільовій популяції;
- за необхідності, опис популяції, що потребує додаткової характеристики, або опис ризику, що очікується у популяції, яка не вивчалася.
V.B.5.9. Частина II ПУР, модуль CVIII «Резюме проблем з безпеки»
У цьому модулі ПУР слід надати перелік проблем безпеки, розподілений за такими категоріями:
- важливі ідентифіковані ризики;
- важливі потенційні ризики;
- відсутня інформація.
V.B.6. Частина III ПУР «План з фармаконагляду (включаючи післяреєстраційні дослідження з безпеки)»
Мета плану з фармаконагляду в частині III — надати огляд та обговорити, яким чином заявник/власник реєстраційного посвідчення планує в подальшому характеризувати проблеми безпеки, зазначені у специфікації з безпеки. У цій частині ПУР надається структурований план, призначений для:
- дослідження того, чи підтверджується потенційний ризик як ідентифікований або спростований ризик;
- подальшої характеристики проблем з безпеки, у тому числі ступеню тяжкості, частоти виникнення та факторів ризику;
- опису того, яким чином буде відбуватися пошук відсутньої інформації;
- оцінки ефективності заходів з мінімізації ризиків.
План з фармаконагляду не включає заходи щодо зменшення, запобігання чи полегшення ризиків; вони обговорюються в частині V ПУР.
План з фармаконагляду повинен базуватися на проблемах з безпеки, що резюмовані в модулі CVIII II частини ПУР «Специфікації з безпеки», він повинен бути пропорційним користі та ризикам лікарського засобу. На ранніх етапах створення ПУР рекомендується проводити обговорення між уповноваженими органами та заявником (власником реєстраційного посвідчення) з метою з’ясування, чи потрібні додаткові заходи з фармаконагляду, якщо потрібні, то які саме, за узгодження основних етапів. Заходи з фармаконагляду можна розподілити на рутинні та додаткові заходи.
V.B.6.1. Частини III ПУР, розділ «Рутинні заходи з фармаконагляду»
Рутинна діяльність з фармаконагляду — це мінімальний комплекс заходів, що вимагаються для всіх лікарських засобів, що представлені в Директиві 2001/83/ЄС [1], Регламенті (ЄС) № 726/2004 [5] та положеннях Порядку [2]N. Виявлення сигналу, яке є частиною рутинного фармаконагляду, є важливим елементом в ідентифікації нових ризиків для всіх лікарських засобів. У ПУР не потрібно повторювати опис цих заходів, який наданий у МФСФ.
Національні уповноважені органи, в Україні — ЦентрN можуть надавати рекомендації щодо специфічних видів діяльності, пов’язаних зі збором, співставленням, оцінкою та наданням спонтанних повідомлень про побічні реакції, що відрізняються від звичайних вимог до рутинного фармаконагляду (див. модуль I ННПФ). Якщо такі рекомендації включають реєстрацію результатів тестів (у тому числі в структурованому форматі), що є частиною рутинної клінічної практики для пацієнта, у якого виникла побічна реакція, то дану вимогу слід розглядати як рутинну. У таких випадках у розділі щодо рутинного фармаконагляду у плані з фармаконагляду необхідно пояснити, як заявник (власник реєстраційного посвідчення) модифікуватиме свою рутинну діяльність з фармаконагляду на виконання будь-яких спеціальних рекомендацій PRAC, CHMP, CMDh та національного уповноваженого органу, ЦентруN щодо рутинного фармаконагляду.
Однак, якщо рекомендація передбачає надання зразків тканини або крові на аналіз до спеціальної лабораторії (наприклад для тестування на антитіла), що не передбачено звичайною клінічною практикою, то це буде додатковою діяльністю з фармаконагляду.
У цьому розділі ПУР слід описувати тільки ту рутинну діяльність з фармаконагляду, що здійснюється додатково понад надання повідомлень про побічні реакції та виявлення сигналу.
V.B.6.1.1. Спеціальні анкети для подальшого відстеження побічних реакцій
Якщо від заявника (власника реєстраційного посвідчення) вимагається або він планує використовувати спеціальні анкети для отримання структурованої інформації про повідомлені підозрювані побічні реакції, що викликають особливий інтерес, застосування цих матеріалів слід описувати в розділі щодо рутинних заходів з фармаконагляду, а копії форм таких анкет необхідно надавати в додатку 4 ПУР.
Не виключено, що в інтересах громадського здоров’я такі опитувальники будуть використовуватися різними заявниками (власниками реєстраційних посвідчень) з мінімальними змінами формату анкети. У такий спосіб можна буде отримати максимум корисних даних стосовно однакових побічних явищ, що дозволить глибоко їх дослідити, проаналізувати прийняття відповідних регуляторних рішень. Одночасно такий підхід зменшить навантаження на працівників з медичною та/або фармацевтичною освітою. Тому власникам реєстраційних посвідчень суворо рекомендується обмінюватися інформацією про структуру та зміст своїх анкет на вимогу інших власників реєстраційних посвідчень.
V.B.6.1.2. Інші форми рутинних заходів з фармаконагляду
У цьому розділі слід надавати опис інших запланованих форм рутинних заходів з фармаконагляду, наприклад, якісний опис посиленої системи пасивного нагляду, порівняльні аналізи, того, що спостерігається, та що очікується, зведені огляди побічних явищ, що викликають особливий інтерес.
V.B.6.2. Частина III ПУР, розділ «Додаткові заходи з фармаконагляду»
Заявник (власник реєстраційного посвідчення) повинен перерахувати у цьому розділі ПУР свої заплановані додаткові заходи з фармаконагляду, докладно зазначаючи, яку інформацію очікується зібрати, що зможе призвести до більш інформованого розгляду співвідношення користь/ризик.
Додатковими заходами з фармаконагляду є діяльність, пов’язана з фармаконаглядом, що не вважається рутинною. Це можуть бути доклінічні дослідження, клінічні випробування або неінтервенційні дослідження. Наприклад, це може бути довготривале спостереження за пацієнтами з популяції клінічного випробування або когортного дослідження для забезпечення додаткового вивчення характеристики довгострокової безпеки лікарського засобу. Коли існують будь-які сумніви стосовно потреби в додаткових заходах з фармаконагляду, слід розглянути питання проведення консультації з уповноваженим органом.
Метою досліджень у плані з фармаконагляду є ідентифікація та характеристика ризиків, збір додаткових даних, де існують прогалини в необхідній інформації або оцінка ефективності додаткових заходів з мінімізації ризиків. Вони повинні стосуватися проблем безпеки, що ідентифіковані у специфікації з безпеки, бути здійсненними та не повинні включати жодного елементу рекламного характеру.
Дослідження, що включені до плану з фармаконагляду, слід планувати та проводити згідно з існуючим відповідним законодавством, а також рекомендаціями, що дані в модулі VIII ННПФ.
Протоколи дослідження можна включати для оцінки при оновлені ПУР тільки, коли дослідження включені до плану з фармаконагляду, та уповноважений орган вимагав подання протоколів. Проаналізовані та затверджені протоколи для досліджень, що включені до плану з фармаконагляду, слід надавати в додатку 3 — частина С ПУР. Протоколи завершених досліджень слід видалити з додатку 3 ПУР, як тільки заключні звіти про дослідження будуть подані до уповноваженого органу для оцінки, а дослідження видалено з плану з фармаконагляду (див. V.B.10.3).
Етапи подання заключного звіту про дослідження до уповноваженого органу слід включити для всіх досліджень, що внесені до плану з фармаконагляду.
Заявники (власники реєстраційного посвідчення) можуть також надавати до ЕМА або національних уповноважених органів протоколи післяреєстраційних досліджень з безпеки (PASS) для отримання наукової консультації.
V.B.6.3. Частина III ПУР, розділ «Зведена таблиця додаткових заходів з фармаконагляду»
Цей розділ ПУР описує заходи з фармаконагляду, спрямовані на виявлення та характеристику ризиків, пов’язаних із застосуванням лікарського засобу. Деякі з таких заходів можуть бути умовою видачі реєстраційного посвідчення, оскільки є ключовими для співвідношення користь/ризик лікарського засобу (дослідження категорії 1 у плані з фармаконагляду), або тому, що вони є спеціальними зобов’язаннями в контексті реєстраційного посвідчення, що видається за умови виконання певних зобов’язань, або реєстраційного посвідчення, що видається за виняткових обставин (дослідження категорії 2 у плані з фармаконагляду). Якщо таким зобов’язанням є проведення неінтервенційного післяреєстраційного дослідження з безпеки, воно буде контролюватися, як описано у статтях 107(m)–107(q) Директиви 2001/83/ЄС [1] та положеннях Порядку [2]N, та матиме формат і зміст, що визначені в додатку III ІП 520/2012 [6], додатку 15 до Порядку [2]N (див. модуль VIII ННПФ).
Інші дослідження можуть вимагатися у ПУР для дослідження проблем з безпеки або для оцінки ефективності заходів з мінімізації ризиків. Такі дослідження, що включені до плану з фармаконагляду, також мають юридичну силу (дослідження категорії 3 у плані фармаконагляду). Зведена таблиця плану з фармаконагляду повинна бути складена таким чином, щоб для всіх зацікавлених сторін було зрозуміло, до якої категорії відноситься захід у плані з фармаконагляду (див. Таблицю V.3.).
Таблиця V.3. Атрибути додаткових заходів з фармаконагляду
|
Тип заходу |
У додатку II торгової ліцензії (тільки для лікарських засобів, схвалених за централізованою процедурою) |
Категорія дослідження (план з фармаконагляду) |
Статус |
Контролюється
|
|
||
|
|
|
Ст. 107 m | Ст. 107 n-q | ||||
|
Післяреєстраційне дослідження з безпеки, що є умовою |
Інтервенційне* |
Так, у додатку IID |
1 |
Обов’язкове із застосуванням санкцій |
Ні |
Ні |
|
|
Неінтервенційне |
Так, у додатку IID |
Так |
Так |
|
|||
|
Спеціальне зобов’язання |
Інтервенційне* |
Так, у додатку IIЕ |
2 |
Обов’язкове із застосуванням санкцій |
Ні |
Ні |
|
|
Неінтервенційне |
Так, у додатку IIЕ |
Так |
Так |
|
|||
|
Вимагається |
Інтервенційне* |
Ні |
3 |
Законодавчо зумовлене |
Ні |
Ні |
|
|
Неінтервенційне |
Ні |
Так |
Так |
|
|||
*Клінічні інтервенційні дослідження, що проводяться відповідно до нормативно-правових вимог Директиви 2001/20/ЄC [15], положень Порядку проведення клінічних випробуваньN [16]. Доклінічні інтервенційні дослідження є предметом правових та етичних норм, що стосуються захисту лабораторних тварин та належної лабораторної практики.
Дослідження, що вимагаються законодавством країн, що не знаходяться на території ЄС, не слід включати до ПУР, якщо вони не є умовою видачі реєстраційного посвідчення, або як спеціальне зобов’язання, або не вимагаються EMA або національним уповноваженим органом, або МОЗ, або ЦентромN. Дослідження, що не вимагаються EMA або національним уповноваженим органом, не слід включати до плану з фармаконагляду в ПУР. Це не завдає шкоди в аспекті проблем з безпеки, що виявлені під час будь-яких таких досліджень, про які слід звітувати відповідно до застосовного законодавства.
Для генеричних лікарських засобів план з фармаконагляду відображатиме незадоволені потреби в дослідженнях з фармаконагляду на момент їх реєстрації. У деяких випадках проведення поточних або запланованих післяреєстраційних досліджень з безпеки для оригінального лікарського засобу також буде вимагатися для генеричних лікарських засобів (наприклад повинні існувати реєстри для включення більшості/усіх пацієнтів, яких лікують лікарським засобом, чи то генеричним, чи то оригінальним. Якщо необхідно, заявникам (власникам реєстраційних посвідчень) рекомендовано проведення спільного післяреєстраційного дослідження з безпеки, наприклад, у випадку ведення спільних реєстрів, або коли в результаті розгляду проблеми рекомендовано проведення післяреєстраційного дослідження з безпеки для всіх зареєстрованих лікарських засобів, що містять певну діючу речовину, при певному показанні.
V.B.7. Частина IV ПУР «Плани післяреєстраційних досліджень ефективності»
Ця частина ПУР повинна включати перелік післяреєстраційних досліджень ефективності, що є умовою видачі реєстраційного посвідчення або, коли вони є спеціальними зобов’язаннями при видачі реєстраційного посвідчення, що видається за умови виконання певних зобов’язань, або за умови його видачі за виняткових обставин. Якщо жодні такі дослідження не вимагаються, частину IV ПУР можна не заповнювати.
V.B.8. Частина V ПУР «Заходи з мінімізації ризиків (включаючи оцінку ефективності заходів з мінімізації ризиків)»
У частині V ПУР слід надати докладну інформацію про заходи з мінімізації ризику, що вживатимуться для зниження ризиків, що пов’язані з відповідними проблемами з безпеки.
Для окремих лікарських засобів, що містять одну й ту ж саму діючу речовину, що мають значні відмінності у показаннях або цільових популяцій, може бути прийнятним мати план з мінімізації ризиків, специфічний для кожного лікарського засобу, тобто лікарські засоби з різними показаннями для застосування мають різні проблеми з безпеки, що пов’язані з цим; лікарські засоби, ризики яких відрізняються відносно цільової популяції; лікарські засоби з різною категорією відпуску пацієнтам можуть мати окремі плани з мінімізації ризиків.
Потребу у продовженні заходів з мінімізації ризиків слід переглядати на регулярній основі, а також оцінювати ефективність заходів з мінімізації ризиків (див. V.B.8 ННПФ). Настанова із заходів мінімізації ризиків та оцінки ефективності заходів з мінімізації ризиків надається у модулі XVI ННПФ та у Додатку I — Навчальні матеріали модулю XVI ННПФ.
Рутинні заходи з мінімізації ризиків
Рутинними заходами з мінімізації ризиків є заходи, що застосовуються до кожного лікарського засобу. Вони стосуються:
- короткої характеристики лікарського засобу/інструкції для медичного застосування лікарського засобуN;
- маркування (наприклад первинне та вторинне пакування);
- листка-вкладки;
- розміру(ів) пакування;
- умов відпуску лікарського засобу.
Навіть саме формулювання може відігравати важливу роль у мінімізації ризику лікарського засобу.
Коротка характеристика лікарського засобу/інструкція для медичного застосування лікарського засобуN та листок-вкладка
Коротка характеристика лікарського засобу/інструкція для медичного застосування лікарського засобуN та листок-вкладка — важливі інструменти з мінімізації ризику, оскільки вони являють собою контрольований і стандартизований формат для інформування про лікарський засіб працівників з медичною та фармацевтичною освітою і пацієнтів. Настанова щодо короткої характеристики лікарського засобу містить інструкції щодо представлення даної інформації.
Коротка характеристика лікарського засобу/інструкція для медичного застосуванняN та листок-вкладка надають рекомендації щодо рутинних заходів з мінімізації ризиків; однак існують два типи повідомлень, що застосовуються у цих документах:
- рутинні інформаційні повідомлення про ризик: зазвичай, їх можна знайти в розділі 4.8 «Побічні реакції» короткої характеристики лікарського засобу/розділ «Побічні реакції» інструкції для медичного застосуванняN або розділі 4 «Можливі побічні ефекти» листка-вкладки; ці повідомлення інформують працівників з медичною та фармацевтичною освітою і пацієнтів про небажані ефекти лікарського засобу, щоб уможливити прийняття інформованого рішення стосовно лікування;
- рутинні заходи з мінімізації ризику, що рекомендують особливі клінічні заходи для вирішення питання ризику: зазвичай, їх можна знайти в розділах 4.2 «Дозування та спосіб введення» та 4.4 «Спеціальні застереження та запобіжні заходи для використання» короткої характеристики лікарського засобу/«Спосіб застосування та дози» та «Особливості застосування» інструкції для медичного застосуванняN, крім того, їх можна знайти в розділах 4.1 «Терапевтичні показання», 4.3 «Протипоказання», 4.5 «Взаємодія з іншими лікарськими засобами та інші форми взаємодії», 4.6 «Вагітність та лактація», 4.7 «Здатність впливати на керування та використання машини» та 4.9 «Передозування» короткої характеристики лікарського засобу/«Показання», «Протипоказання», «Взаємодія з іншими лікарськими засобами та інші види взаємодій», «Застосування у період вагітності або годування груддю», «Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами» та «Передозування» інструкції для медичного застосуванняN, а також у розділах 2 «Що потрібно знати, перш ніж приймати (назва лікарського засобу)» та 3 «Як приймати (назва лікарського засобу)» листка-вкладки; попереджувальні та застережні повідомлення та рекомендації у короткій характеристиці лікарського засобу/інструкції для медичного застосуванняN включатимуть інформацію стосовно вирішення питання ризику лікарського засобу, наприклад, за допомогою:
- проведення тесту до початку лікування;
- моніторингу лабораторних параметрів під час лікування;
- моніторингу певних ознак та симптомів;
- коригування дози або припинення лікування, якщо спостерігаються побічні явища або зміни лабораторних параметрів;
- проведення процедури «вимивання» після перерви у лікуванні;
- надання рекомендацій з контрацепції;
- заборона застосування інших лікарських засобів під час застосування даного лікарського засобу;
- лікування або запобігання факторам ризику, що можуть призвести до виникнення побічного явища лікарського засобу;
- рекомендації довготривалого клінічного спостереження для виявлення віддалених побічних явищ на ранніх стадіях.
Розмір пакування (Pack size)
Оскільки кожний розмір пакування окремо затверджується для лікарського засобу, планування кількості «одиниць дозування»/готових лікарських засобівN у кожній упаковці і діапазон її наявних розмірів можна вважати однією з форм рутинних заходів з управління ризиками. Теоретично, контроль кількості «одиниць дозування»/готових лікарських засобівN в упаковці означає, що пацієнтам буде необхідно звертатися до працівників з медичною та/або фармацевтичною освітою через певні проміжки часу, і це збільшить імовірність моніторингу і зменшить проміжок часу, коли пацієнт знаходиться не під наглядом. У крайніх випадках, можна обмежити доступність лікарського засобу тільки одним розміром упаковки і таким чином спробувати пов’язати призначення лікарського засобу з необхідністю проведення медичного огляду.
Невеликий розмір упаковки також може бути корисним, особливо, коли передозування або недотримання схеми прийому вважаються значними ризиками.
Умови відпуску
Контроль умов, за яких можна отримати доступ до лікарського засобу, може знизити ризики, пов’язані з його застосуванням або зловживанням.
Реєстраційне посвідчення повинно містити інформацію про будь-які умови чи обмеження на постачання чи застосування лікарського засобу, в тому числі, за яких лікарський засіб може бути доступний пацієнтам. Такі умови, зазвичай, називають умовами відпуску, «правовим статусом» лікарського засобу. Як правило, ці умови включають інформацію про те, чи вимагає відпуск лікарського засобу наявності рецепта, заповненого лікарем (ст. 71 (1) Директиви 2001/83/ЄС) [1] та наказу Міністерства охорони здоров’я України від 19 липня 2005 р. № 360 «Про затвердження Правил виписування рецептів на лікарські засоби і вироби СТ-Н МОЗУ 42-8.7:2018 144 медичного призначення, Порядку відпуску лікарських засобів і виробів медичного призначення з аптек та їх структурних підрозділів, Інструкції про порядок зберігання, обліку та знищення рецептурних бланків», зареєстрованого в Міністерстві юстиції України 20 липня 2005 р. за № 782/11062N [49]).
Вони можуть також обмежувати місця, де лікарський засіб може застосовуватися (наприклад тільки в умовах стаціонару) або коло осіб, які можуть призначати лікарський засіб (наприклад тільки спеціаліст).
До лікарських засобів, що відпускаються лише за рецептом, можуть пред’являтися додаткові умови шляхом їх класифікації на ті, що доступні лише або за обмеженим медичним рецептом, або за спеціальним медичним рецептом.
За обмеженим рецептом (Restricted medical prescription)
Може використовуватися для контролю за тим, хто може ініціювати лікування, призначати лікарський засіб, і заклад, у якому лікарський засіб може призначатися або застосовуватися. Відповідно до законодавства ЄС для вирішення питання, чи вимагає відпуск лікарського засобу наявності обмеженого рецепта, слід враховувати такі фактори (ст. 71(3) Директиви 2001/83/ЄС) [1]):
- лікарський засіб, через його фармацевтичні характеристики або новизну, або в інтересах охорони здоров’я може застосовуватися тільки в умовах стаціонару;
- лікарський засіб застосовується для лікування захворювань, що повинні діагностуватися в умовах стаціонару або в закладах з належним діагностичним обладнанням, хоча його застосування та подальше спостереження можуть здійснюватися в іншому місці;
- лікарський засіб призначений для амбулаторного застосування, однак його прийом може призвести до дуже серйозних побічних реакцій, а тому лікарський засіб вимагає рецепта спеціаліста і особливого нагляду за пацієнтом протягом усього періоду лікування.
За спеціальним рецептом (Special medical prescription)
При віднесенні лікарського засобу до категорії «за спеціальним рецептом», слід враховувати такі фактори (ст. 71(2) Директиви 2001/83/ЄС) [1]:
- лікарський засіб містить у підконтрольній кількості речовину, що визначається міжнародними конвенціями, такими як конвенції ООН 1961 і 1971 р., як наркотичний засіб або психотропна речовина;
- лікарський засіб, який при неправильному застосуванні з великою ймовірністю може представляти значний ризик зловживання, призвести до виникнення залежності або до неналежного використання з протизаконною метою;
- лікарський засіб містить речовину, що, в силу її новизни або властивостей, можна в якості запобіжного заходу віднести до групи, зазначеної в другому абзаці.
Розподіл за категоріями на рівні країн — членів ЄС
Існує можливість реалізації додаткових підкатегорій на рівні держави — члена ЄС, яка дозволить державам — членам ЄС адаптувати загальні класифікації, описані вище, до своєї національної ситуації. Визначення і, отже, реалізація варіюється в тих державах-членах, де існують підкатегорії.
В Україні лікарські засоби поділяються на дві категорії:
- лікарські засоби, які відпускаються за рецептом;
- лікарські засоби, які відпускаються без рецепта.
Лікарські засоби, які відпускаються за рецептом, можуть бути поділені на окремі групи згідно з такою класифікацією:
- лікарські засоби, які відпускаються за разовими чи багаторазовими рецептами;
- лікарські засоби, які відпускаються за спеціальними рецептами;
- лікарські засоби, які відпускаються за рецептами і мають обмежену галузь застосування (Критерії визначення категорій відпуску лікарських засобів, затверджено наказом Міністерства охорони здоров’я України від 17.05.2001 р. № 185, зареєстрованим у Міністерстві юстиції України 31 травня 2001 р. за № 464/5655 [24])N.
Додаткові заходи з мінімізації ризиків
Додаткові заходи з мінімізації ризиків слід пропонувати тільки якщо вони необхідні для безпечного та ефективного застосування лікарського засобу. Якщо пропонується додатковий захід з мінімізації ризиків, необхідно надати його детальний опис та обґрунтування доцільності. Необхідність у продовженні такого заходу слід регулярно переглядати.
За необхідності ключову інформацію про додаткові заходи з мінімізації ризиків слід надавати в додатку 6 ПУР — Докладна інформація про запропоновані додаткові заходи з мінімізації ризиків.
Для лікарських засобів, що зареєстровані не за централізованою процедурою, у ситуаціях, коли потреба в додаткових заходах з мінімізації ризиків може варіюватися між державами ЄС, ПУР може відображати, що необхідність (та зміст) додаткової мінімізації ризику може узгоджуватися на національному рівні. В Україні для лікарських засобів необхідність додаткових заходів з мінімізації ризиків може узгоджуватися з уповноваженим органомN.
Подальша настанова із додаткових заходів з мінімізації ризиків надається в модулі XVI ННПФ.
Оцінка ефективності заходів з мінімізації ризиків
Коли ПУР оновлюється, план з мінімізації ризиків має включати опис впливу додаткових заходів з мінімізації ризиків. Якщо доцільно, таку інформацію можна представляти за регіонами ЄС/УкраїниN.
Слід включити опис результатів будь-якої оцінки заходів з мінімізації ризиків, за наявності. Якщо певна стратегія мінімізації ризику виявилася неефективною або такою, що викликає надмірне або неналежне навантаження на пацієнтів або систему охорони здоров’я, тоді слід приділити увагу альтернативним заходам. Заявник (власник реєстраційного посвідчення) повинен прокоментувати у ПУР, чи потрібні для кожної проблеми безпеки додаткові або інші заходи з мінімізації ризиків; чи, на його думку, можна не вживати додаткових заходів з мінімізації ризиків (наприклад, коли заходи з мінімізації ризиків стали частиною стандартної клінічної практики).
Якщо дослідження для оцінки ефективності заходів з мінімізації ризиків вимагається або призначається уповноваженим органом, дослідження слід включити до плану з фармаконагляду частини III ПУР.
Настанову з моніторингу ефективності заходів з мінімізації ризиків включено до модулю XVI ННПФ.
V.B.8.1. Частина V ПУР, розділ «План з мінімізації ризиків»
У розділі ПУР стосовно плану з мінімізації ризику для кожної проблеми безпеки у специфікації з безпеки слід надати таку інформацію:
- рутинні заходи з мінімізації ризиків, включаючи докладну інформацію про те, чи передбачено тільки внесення інформації до короткої характеристики лікарського засобу/інструкції для медичного застосування лікарських засобівN та листка-вкладки, чи запропоновані інші заходи з мінімізації ризику;
- додаткові заходи з мінімізації ризиків (якщо є), включаючи певні цілі і обґрунтування необхідності у них, а також, як буде оцінюватися їх ефективність.
V.B.8.2. Частина V ПУР, розділ «Резюме заходів з мінімізації ризиків»
У цьому розділі ПУР необхідно надати таблицю, в якій перераховуються рутинні й додаткові заходи з мінімізації ризиків, що розподілені за проблемами з безпеки (наприклад, номер розділу в короткій характеристиці/інструкції для медичного застосуванняN лікарського засобу, де зазначена інформація про ризик, перелік навчальних матеріалів). Слід включити додаткове резюме дій, що пов’язані з фармаконаглядом, що описано в Настанові ЕМА з формату плану управління ризиком у ЄС12.
V.B.9 Частина VI ПУР, розділ «Резюме плану управління ризиками»
Резюме ПУР для кожного зареєстрованого лікарського засобу повинно оприлюднюватися та містити ключові елементи ПУР (ст. 26(1)(с) Регламенту (ЄС) 726/2004 [5], ст. 106(с) Директиви 2001/83/ЄС [1], ст. 31(1) ІП 520/2012 [6], положення Порядку [2]N).
Частину VI ПУР повинні надавати заявники (власники реєстраційних посвідчень) на лікарські засоби, які мають ПУР, незалежно від того, чи вони зареєстровані за централізованою або національною процедурою в ЄС та в УкраїніN. На підставі інформації, що міститься в частині VI ПУР, для лікарських засобів, що зареєстровані за централізованою процедурою, EMA повинно опублікувати резюме ПУР на сайті ЕМА на момент прийняття рішення Європейською Комісією разом з іншими документами Європейського публічного оціночного звіту на цей лікарський засіб. Для лікарських засобів, що зареєстровані за національною процедурою, резюме ПУР слід оприлюднювати на сайті національних уповноважених органів.
Для лікарських засобів, зареєстрованих в Україні, резюме ПУР розміщується на сайті Центру (положення Порядку [2]N).
Резюме ПУР слід оновлювати, коли важливі зміни вносяться до повного ПУР. Зміни слід вважати важливими, якщо вони стосуються наступного:
- нові важливі ідентифіковані або потенційні ризики, або важливі зміни до проблем з безпеки, або видалення проблем з безпеки;
- включення або видалення додаткових заходів з мінімізації ризиків або рутинних заходів з мінімізації ризиків, що рекомендують певні особливі клінічні заходи для вирішення питання ризику;
- значні зміни до плану фармаконагляду (наприклад, додання нових досліджень або завершення досліджень, що тривають).
Аудиторія, якій адресується резюме ПУР, дуже широка. Для забезпечення можливості задоволення за допомогою резюме різних потреб його слід писати та представляти у чіткій формі, застосовуючи підхід «проста мова»13. Однак це не означає, що слід уникати технічних термінів. У документі слід чітко роз’яснювати його мету та зв’язок з іншою інформацією, зокрема, інформацією про лікарський засіб (тобто, короткою характеристикою лікарського засобу/інструкцією для медичного застосуванняN, листком-вкладкою та маркуванням).
Резюме частини VI ПУР має узгоджуватися з інформацією, що представлена в модулях CVII, CVIII частини II та частинах III, IV та V ПУР. Воно має містити таку інформацію:
- лікарський засіб, та для чого він зареєстрований;
- резюме проблем безпеки та відсутньої інформації;
- рутинні та додаткові заходи з мінімізації ризиків;
- додаткові заходи, що стосуються фармаконагляду.
V.B.10. Частина VII ПУР «Додатки до плану управління ризиками»
ПУР повинен включати додатки, що перераховані нижче (за доцільності). Якщо ПУР охоплює більш ніж один лікарський засіб, зазвичай, очікується, що додатки будуть доречні для усіх лікарських засобів. Певні аспекти, що не застосовні до всіх лікарських засобів у ПУР, слід відмітити (наприклад, форма відстеження в додатку 4 може стосуватися тільки лікарських засобів, що містять діючу речовину, що мають причинно-наслідковий зв’язок з явищем).
V.B.10.1. Додаток 1 ПУР
Додаток 1 ПУР є структурованим представленням в електронному вигляді ПУР. Його подання у форматі eCTD не вимагається, файл в електронному вигляді слід надавати відповідно до V.C.2. та застосовної настанови14, положень Порядку [2], додатку 14 до Порядку [2], положень Порядку проведення експертизи реєстраційних матеріалів [7]N. Цей додаток можна залишати пустим у ПУР, якщо застосовно.
V.B.10.2. Додаток 2 ПУР: зведена таблиця про включені до плану з фармаконагляду дослідження, що заплановані, що тривають, що завершені
До цього додатку слід включити таблицю, що містить інформацію про дослідження, які включені до плану з фармаконагляду (поточну або попередні версії ПУР; дослідження категорії 1, 2, 3), а саме:
- дослідження, що заплановані та ті, що тривають, включаючи цілі, проблему безпеки, що розглядається, а також заплановані дати подання проміжних та заключних результатів;
- завершені дослідження, включаючи цілі, проблеми з безпеки, що розглядаються, а також дату подання результатів до уповноважених органів (фактичних результатів, запланованих результатів або зазначити причину неподання результатів).
V.B.10.3. Додаток 3 ПУР: протоколи для включених до плану з фармаконагляду досліджень, що заплановані, що тривають та тих, що завершені
До додатку 3 не слід включати протоколи досліджень, що не призначаються та не вимагаються уповноваженим органом (тобто, ті, що не включені до плану з фармаконагляду). У цей додаток можна включати електронні лінки або посилання на інші модулі досьє у форматі еCTD, до яких включені протоколи, замість повних документів протоколів.
V.B.10.3.1. Частина А — Додаток 3 ПУР: запитувані протоколи для включених до плану з фармаконагляду досліджень, що подаються для регуляторної оцінки з цією оновленою версією ПУР
Якщо уповноважений орган вимагає надати протоколи для оцінки, а заявник (власник реєстраційного посвідчення) вибирає подання протоколу дослідження для оцінки в межах тієї самої процедури, що й для подання ПУР, то цей протокол слід включити до частини А; в іншому випадку протокол можна оцінювати в межах окремої процедури, та після того, як він буде погоджений, включити до частини С (В) додатку 3 ПУР. Регуляторну процедуру для подання протоколу слід узгодити з уповноваженим органом.
V.B.10.3.2. Частина В (Б) — Додаток 3 ПУР: запитувані зміни до раніше схвалених протоколів, включених до плану з фармаконагляду досліджень, що подаються для регуляторної оцінки з цією оновленою версією ПУР
Якщо уповноважений орган вимагає надати зміни до протоколів для оцінки, а заявник (власник реєстраційного посвідчення) вибирає подання зміни до протоколу дослідження для оцінки в межах тієї самої процедури, що й для подання ПУР, до частини В (Б) слід включити оновлений протокол; в іншому випадку зміну до протоколу можна оцінювати в межах окремої процедури, та після того, як вона буде погоджена, включити до частини С (В) додатку 3 ПУР. Регуляторну процедуру для подання протоколу слід узгодити з уповноваженим органом.
Після схвалення протоколи з частин А або В(Б) слід перенести до частини С(В), разом з наступною обґрунтованою оновленою версією ПУР.
V.B.10.3.3. Частина С (В) — Додаток 3 ПУР: раніше погоджені протоколи для досліджень, що тривають, та заключні протоколи, що не оцінювалися уповноваженим органом
Раніше погоджені протоколи для досліджень, що тривають, та заключні протоколи, що не оцінювалися уповноваженим органом, слід включити до цієї частини С (В) додатку 3 ПУР, а саме:
- повні протоколи, що раніше були оцінені уповноваженим органом та погоджені (тобто повторне подання протоколу не вимагалося). У протоколах слід зазначати найменування процедури, за якою протокол був схвалений, та дату схвалення. Сюди можна включати електронний лінк або посилання на інші модулі досьє у форматі еCTD, у яких раніше подавалися протоколи, замість повних документів протоколів;
- заключні протоколи інших досліджень категорії 3: протоколи, які уповноважені органи не вимагали подавати для оцінки, та які подаються власником реєстраційного посвідчення тільки для інформації.
Протоколи завершених досліджень слід видалити з цього додатку, як тільки заключні звіти дослідження будуть подані до уповноваженого органу для оцінки.
V.B.10.4. ПУР, додаток 4: спеціальні форми відстеження побічного явища
Цей додаток повинен включати всі форми відстеження, що використовуються заявниками (власниками реєстраційних посвідчень) для збору додаткових даних щодо спеціальних питань з безпеки. Використання форм відстеження, що включені у цей додаток, слід детально описати в плані з фармаконагляду в ПУР як рутинну діяльність з фармаконагляду.
Форми, що повинні включатися у цей додаток, інколи відомі як «анкета відстеження явища», «засіб збору/запису даних щодо побічного явища» або «форма відстеження побічної реакції».
V.B.10.5. ПУР, додаток 5: протоколи запропонованих та поточних досліджень у частині ІV ПУР
Цей додаток повинен включати посилання на інші частини досьє у форматі еСТD, що вже містять протоколи встановленого дослідження з ефективності, для досліджень, включених у частину ІV ПУР.
V.B.10.6. ПУР, додаток 6: детальні дані запропонованих додаткових дій з мінімізації ризиків
Якщо застосовно, цей додаток повинен включати проєкт (та затверджений варіант) запропонованих ключових повідомлень додаткових заходів з мінімізації ризиків.
V.B.10.7. ПУР, додаток 7: інші підтверджуючі дані (включаючи матеріали, на які посилаються)
Для уникнення дублювання матеріалів, що представлені як посилання, цей додаток повинен включати посилання на досьє у форматі еСТD на інші документи, що включені в інші модулі досьє.
V.B.10.8. ПУР, додаток 8: резюме змін до плану управління ризиками протягом часу
Перелік усіх значних змін до ПУР у хронологічному порядку повинен надаватися у цьому додатку. Він повинен включати короткий опис змін та дату і номер версії ПУР, коли:
- додаються, видаляються або повторно класифікуються питання з безпеки;
- дослідження додаються або видаляються з плану з фармаконагляду;
- у заходах з мінімізації ризиків рекомендують спеціальні клінічні заходи для розгляду ризиків, або додаткові заходи з мінімізації ризиків були змінені у плані з мінімізації ризиків.
V.B.11. Зв’язок між ПУР та регулярно оновлюваним звітом з безпеки
У післяреєстраційний період основними документами з фармаконагляду для контролю безпеки є ПУР та РОЗБ. Хоча зазначені документи певною мірою перекривають один одного, їх основні цілі різні, а ситуації, коли вимагається їх надання, не завжди однакові. Так, основною метою створення РОЗБ є ретроспективна, комплексна оцінка співвідношення користь/ризик у післяреєстраційний період, а основною метою ПУР є проспективне дореєстраційне та післяреєстраційне управління ризиками та планування активностей з фармаконагляду та мінімізації ризиків. Це два взаємодоповнюючі документи.
У разі якщо РОЗБ і ПУР повинні надаватися одночасно, ПУР повинен відображати висновки РОЗБ. Наприклад, якщо новий сигнал оцінюється в РОЗБ як важливий ідентифікований або важливий потенційний ризик, то він має включатися до ПУР, а оновлена версія ПУР повинна подаватися разом з РОЗБ. План з фармаконагляду і план з мінімізації ризиків повинні оновлюватися для того, щоб відображати точку зору заявника (власника реєстраційного посвідчення) щодо подальшого вивчення проблем з безпеки і мінімізації ризиків.
Таблиця V.4. РОЗБ та ПУР містять подібну інформацію (проте вона може бути не в однаковому форматі та може бути невзаємозамінною)
|
Розділ ПУР |
Розділ РОЗБ |
|
Частина II, модуль СІІІ — «Експозиція пацієнтів, залучених до клінічних випробувань» |
Підрозділ 5.1 — «Кумулятивна експозиція учасників клінічних випробувань» |
|
Частина II, модуль СV — «Післяреєстраційний досвід» |
Підрозділ 5.2 — «Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційний період» |
|
Частина II, модуль СVII — «Ідентифіковані та потенційні ризики» та частина ІІ, модуль СVІІІ — «Резюме проблем з безпеки» |
Підрозділ 16.1 — «Резюме проблем з безпеки», та 16.4 «Характеристика ризиків» |
|
Частина V — «Заходи з мінімізації ризиків», розділ «Ефективність заходів з мінімізації ризиків» |
Підрозділ 16.5 — «Ефективність заходів з мінімізації ризиків (якщо застосовувалися)» |
V.B.12. Системи якості та управління записами
Хоча у підготовці ПУР можуть брати участь багато експертів, остаточна відповідальність за його якість, точність та наукову цілісність лежить на заявникові (власникові реєстраційного посвідчення). Таким чином, УОВФ в ЄС, УкраїніN, КОВФ в УкраїніN повинна усвідомлювати і мати достатні повноваження для контролю його змісту. УОВФ/КОВФ в Україні мають обов’язки (але не виключно) щодо складання (для КОВФ — у разі необхідності) та/або надання планів управління ризиками (положення Порядку [2])N. Заявник (власник реєстраційного посвідчення) несе відповідальність за оновлення ПУР у випадку появи нової інформації, та повинен застосовувати принципи якості, викладені в модулі I ННПФ. Заявник (власник реєстраційного посвідчення) повинен зберігати записи, коли ПУР надавався уповноваженим органам, а також коли вносилися значні зміни у версіях ПУР. Ці записи, ПУР, а також будь-які документи, що стосуються інформації, представленої у ПУР, можуть бути предметом аудиту, інспекцій, аудитів системи фармаконагляду уповноваженою установоюN з боку інспекторів/аудиторівN з фармаконагляду.
V.C. Функціонування системи управління ризиками в ЄС, УкраїніN
V.C.1. Вимоги до заявника (власника реєстраційного посвідчення) в ЄС, УкраїніN
При реєстрації лікарських засобів заявник (власник реєстраційного посвідчення) повинен надати ПУР, що описує систему управління ризиками разом з резюме такої (ст. 8(3) (іаа) Директиви 2001/83/ЄС [1], положення ПорядкуN [2]).
У післяреєстраційний період може виникнути необхідність подати оновлений ПУР та новий ПУР у будь-який час:
- на запит EMA або уповноваженого органу в країні ЄС, в УкраїніN, коли існує питання щодо ризику, що впливає на співвідношення користь/ризик;
- із заявою на зміну до існуючого реєстраційного посвідчення, якщо включені дані призводять до зміни в переліку питань з безпеки, або якщо необхідні нові додаткові заходи з фармаконагляду, або нові заходи з мінімізації ризиків, або їх пропонується видалити. Оновлення ПУР може гарантуватися в результаті поданих даних із заявами, таких як нова або значна зміна в показанні, нова лікарська форма, новий шлях введення, новий виробничий процес препаратів, отриманих методом біотехнологій.
Необхідність у ПУР або в оновленні ПУР слід обговорити з EMA або уповноваженим органом в країні ЄС, в УкраїніN завчасно до подання заяви на значну зміну до існуючого реєстраційного посвідчення.
V.C.1.1. Плани управління ризиками, які подаються із реєстрацією
При реєстрації лікарських засобів за повним досьє ПУР повинен мати всі частини (див. V.В.4.ННПФ). При реєстрації лікарських засобів за іншими типами заяв до ПУР висуваються певні вимоги, що відповідають концепції пропорційності до важливих ідентифікованих та потенційних ризиків лікарського засобу і необхідності в післяреєстраційних даних з безпеки (ст. 8(3) Директиви 2001/83/ЄС [1], положення ПорядкуN [2]); тому певні частини або модулі можуть бути пропущені, представлені у певному форматі або бути незаповненими, якщо це застосовано.
Таблиця V.5. Резюме вимог до ПУР для заяв на реєстрацію (для повного опису див. текст нижче)
|
Препарат |
Частина І |
Частина ІІ |
Частина ІІІ |
Частина IV |
Частина V |
Частина VI |
|||||||
|
|
|
СI |
СII |
СIII |
СIV |
СV |
СVI |
СVII | СVIII | ||||
|
0. Заява на РП за повним досьє |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
|
1. Генеричний лікарський засіб |
√ |
╪ |
√ |
√ |
* |
∫ |
√ |
||||||
|
2. Інформована згода1 |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
|
3. Гібридний лікарський засіб |
√ |
+ |
+ |
+ |
√ |
√ |
√ |
∫ |
√ |
||||
|
4.а. Фіксована комбінація — нова діюча речовина |
√ |
⊥ |
⊥ |
⊥ |
⊥ |
⊥ |
⊥ |
√ |
√ |
√ |
√ |
√ |
√ |
|
4.б. Фіксована комбінація — не нова діюча речовина |
√ |
+ |
+ |
╪ |
√ |
√ |
* |
∫ |
√ |
||||
|
5. «Добре вивчене медичне застосування» |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
||||||
|
6. Біоподібний лікарський засіб |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
√ |
|
√ = вимагається;
╪ = вимагається лише якщо «оригінальний» лікарський засіб не має ПУР та профіль безпеки його діючої речовини не публікувався на сайті CMDh;
* = вимагається лише якщо післяреєстраційні дослідження з ефективності (PAES) були обов’язковими для «оригінального» препарату;
∫ = приведення у відповідність інформації з безпеки в інформації щодо лікарського засобу є достатнім;
+ = вимагається згідно з принципом пропорційності ризику, що передбачає наявність нових даних або відмінності у порівнянні з «оригінальним» лікарським засобом;
⊥ = увага зосереджена на новій діючій речовині/АФІN.
V.C.1.1.1. При реєстрації генеричного лікарського засобу
Для лікарських засобів, що реєструються відповідно до ст. 10(1) Директиви 2001/83/ЄС [1], до Порядку проведення експертизи [7]N, вимоги до ПУР є такими:
- ПУР частина І. Елементи є такими ж, як для заяв на реєстрацію за повним досьє;
- ПУР частина ІІ. Існує 3 можливі варіанти:
1. Оригінальний лікарський засіб має ПУР, тоді модулі ПУР СI-СVII можуть не надаватися. Модуль СVIIІ повинен включати резюме питань з безпеки, як у оригінального лікарського засобу. Якщо заявник вважає, що наявний доказ обґрунтовує видалення або зміну питання з безпеки, тоді ці дані слід представити в модулі СVII, де розглянути питання з безпеки з детальною аргументацією. Аналогічно, якщо заявник визначив/виявив нове питання з безпеки, що є специфічним для генерика (наприклад, ризики пов’язані з новою допоміжною речовиною або нове питання з безпеки виникає з будь-яких отриманих клінічних даних), це слід детально обговорити як нове питання з безпеки в модулі СVII.
2. Оригінальний лікарський засіб не має ПУР, але питання з безпеки його діючої речовини опубліковані на вебсайті CMDh. У цьому разі ПУР повинен містити модулі CVII та CVIII. Якщо більш ніж одне питання з безпеки, опубліковане на вебсайті CMDh, стосовно однієї й тієї ж діючої речовини, заявник повинен обґрунтувати вибір запропонованих питань з безпеки в модулі СVIII.
3. Оригінальний лікарський засіб не має ПУР та питання безпеки його діючої речовини не опубліковані на вебсайті CMDh. У цьому разі модулі СVII та СVIII повинні включатися в ПУР. У модулі СVII слід критично проаналізувати наявну відповідну інформацію (наприклад, власні доклінічні та клінічні дані, наукову літературу, інформацію про лікарський засіб оригінального лікарського засобу) та запропонувати перелік важливих ідентифікованих та потенційних ризиків, а також відсутньої інформації, що повинні бути відображені в модулі СVIII.
- ПУР частина ІІІ повинна включати опис стандартної діяльності з фармаконагляду, як детально описано в V.В.6.1ННПФ.
Заявникам генеричних лікарських засобів доцільно брати участь у запланованих або поточних дослідженнях, що проводяться заявником (власником реєстраційного посвідчення) оригінального лікарського засобу, якщо важливо, щоб усі наявні (майбутні) дані були отримані в результаті одного дослідження. Наприклад, дані пацієнтів, які застосовують новий лікарський засіб, є важливими для подальшої характеристики профілю безпеки його речовини. Однак залучення пацієнтів в окремі дослідження з тими самими або подібними цілями створює непотрібне навантаження на пацієнтів, лікарів або дослідників (наприклад, реєстри вагітності, реєстри хвороб, будь-які післяреєстраційні дослідження з безпеки, що оцінюють довготривале застосування).
Уповноважений орган може також розглянути доцільність проведення досліджень, що повинні проводитися для генериків (наприклад, у контексті процедури, коли включаються генерики, або в результаті процедури, що визначає дослідження для оригінального лікарського засобу).
- Частина IV ПУР. Ця частина ПУР може залишатися незаповненою, якщо післяреєстраційні дослідження з ефективності не рекомендувалися для проведення для генериків.
- Частина V ПУР. Якщо для оригінального лікарського засобу не вимагається додаткових дій з мінімізації ризиків, формулювання, що інформація з безпеки в інформації про лікарський засіб генерика відповідає оригінальному лікарському засобу є достатньою для частини V ПУР. Якщо для генерика були виявлені нові ризики, заходи з мінімізації ризиків для таких питань з безпеки слід представити в частині V з тими ж елементами, як і для заяви на реєстрацію за повним досьє.
Якщо для оригінального лікарського засобу не вимагається додаткових заходів з мінімізації ризиків, для генерика вимагається повна частина V.
- Частина VI ПУР. Елементи цієї частини є такими ж, як для заяв на реєстрацію за повним досьє в межах даних, що вимагаються або надаються в інших частинах ПУР, як вказано вище.
- Частина VIІ ПУР. Елементи є такими ж як для заяв на реєстрацію за повним досьє. Для додатків 4 та 5 ПУР слід використовувати дані якомога більше подібні до тих, що є в ПУР оригінального лікарського засобу.
V.С.1.1.2. При реєстрації лікарського засобу за «інформованою згодою»
Для лікарських засобів, що реєструються за ст. 10с Директиви 2001/83/ЄС [1], відповідно до положень Порядку проведення експертизи [7] за інформованою згодою ПУР повинен бути таким, як ПУР референтного лікарського засобу. ПУР буде вимагатися, навіть якщо референтний лікарський засіб не має ПУР. Заявнику (власнику реєстраційного посвідчення) зареєстрованого лікарського засобу слід мати лише один ПУР для своїх лікарських засобів з однаковою діючою речовиною.
V.С.1.1.3. При реєстрації лікарського засобу як «гібридного лікарського засобу»
Для лікарських засобів, що реєструються за ст. 10(3) Директиви 2001/83/ЄС [1], відповідно до положень Порядку проведення експертизи [7]N як «гібридний лікарський засіб», елементи ПУР є такими, як для генеричного лікарського засобу. Проте у разі змін діючої(их) речовини (речовин), терапевтичних показань, сили дії, лікарської форми або шляху введення, заявник повинен розглянути в модулі СVІІ ПУР, чи ці зміни додадуть чи виключать питання з безпеки. Дані дореєстраційних клінічних досліджень повинні розглядатися в ПУР (наприклад, ПУР, частина ІІ, модулі СІ, СІІІ). Частини ПУР повинні узгоджуватися, наприклад, частини V та VІ.
V.С.1.1.4. При реєстрації лікарських засобів з «фіксованою комбінацією»
Для лікарських засобів, що реєструються за заявою на фіксовану комбінацію, можливі два варіанти:
4. Якщо комбінація містить нову діючу речовину, то повинен подаватися повний ПУР у форматі, як для заяви на реєстрацію за повним досьє. У модулях СІ–СVІ ПУР повинна бути представлена інформація про нову діючу речовину.
5. Якщо комбінація не містить нової діючої речовини, то ПУР повинен мати формат, як для генерика. Для встановлення елементів частини ІІ ПУР «оригінальний препарат» слід розглядати як «будь-який/всі зареєстровані лікарські засоби, що містять такі самі діючі речовини, які включені в новий лікарський засіб».
Додатково отримані нові дані для фіксованої комбінації повинні бути представлені в модулях СІІ та СІІІ.
V.С.1.1.5. При реєстрації лікарського засобу з добре вивченим медичним застосуванням
Для лікарських засобів, що реєструються за ст. 10а Директиви 2001/83/ЄС [1], відповідно до положень Порядку проведення експертизи [7]N як «добре вивчене медичне застосування», ПУР матиме такі особливості:
- частина І ПУР: елементи є такими ж, як для заяви на реєстрацію за повним досьє;
- частина ІІ ПУР: можуть бути представлені лише модулі ПУР СVІІ та СVІІІ. Заявник повинен обґрунтувати запропоновані питання з безпеки або відсутність таких, використовуючи наявний доказ з опублікованої наукової літератури (загальнодоступну інформацію);
- частини ІІІ–VІІ ПУР: елементи є такими ж, як для заяви на реєстрацію за повним досьє.
V.С.1.1.6. При реєстрації «біоподібного лікарського засобу»
Вимоги до ПУР біоподібних лікарських засобів описані в ННПФ р.ІІ.
V.С.1.1.7. При реєстрації гомеопатичних та рослинних препаратів, що не підпадають під спрощену реєстрацію
При реєстрації гомеопатичних та рослинних препаратів, що не підпадають під спрощену реєстрацію, відбувається стандартна процедура видачі реєстраційного посвідчення, тому елементи ПУР є такими ж, як визначено типом заяви на реєстраційне посвідчення.
V.C.1.2. Плани управління ризиками, які вперше подаються після реєстрації
V.C.1.2.1. Нові плани управління ризиками на запит уповноваженого органу для розгляду одного або більше питань з безпеки
Елементи є такими ж, що і у ПУР для генеричного лікарського засобу, коли оригінальний лікарський засіб не має ПУР (див. V.C.1.1.1.).
Можливі два варіанти:
1. Заявник (власник реєстраційного посвідчення) може подавати ПУР з модулем СVІІ ПУР, що зосереджений на питанні(ях) безпеки, яке (які) розглядається в процедурі. Інші питання з безпеки повинні включатися при необхідності.
2. Заявник (власник реєстраційного посвідчення) може подавати ПУР, де відображено загальновизначені питання з безпеки.
Уповноважений орган вирішує, який з них є найбільш відповідним за даних обставин.
V.C.1.2.2. Подання плану управління ризиками в післяреєстраційний період
У цьому разі ПУР має елементи, що визначені типом заяви при реєстрації лікарського засобу (тобто, заява на реєстрацію за повним досьє, генеричні лікарські засоби, заяви «інформованої згоди» тощо, див V.С.1.1. ННПФ).
V.C.2. Подання плану управління ризиками уповноваженим органам у ЄС, УкраїніN
Для реєстрації лікарського засобу за централізованою процедурою ПУР повинен подаватися у вигляді PDF-файлів, у форматі eCTD в електронному вигляді,в Україні у складі I модуля реєстраційного досьє (положення Порядку проведення експертизи реєстраційних матеріалів [7] у форматі відповідно до вимог Порядку [2])N. Після рішення Комісії, якщо процедура включає подання ПУР, заявник повинен у вказаний строк подати додаток I до ПУР у форматі XML. Додаток I до ПУР містить у структурованому електронному форматі ключову інформацію стосовно ПУР, що, після валідації в EMA завантажується в базу даних EMA, яка є доступною та дозволяє проводити пошук EMA та уповноваженим органам в країнах ЄС. Підходи до реєстрації лікарських засобів у країнах ЄС за національною процедурою відрізняються, тому їх слід дотримуватися.
Детальна інформація щодо нових вимог до подання та електронного формату ПУР буде надаватися на вебсайтах EMA та країн ЄС, та в майбутньому можуть замінитися вимоги, що представлені в параграфі вище.
Початковий ПУР повинен подаватися у складі реєстраційних матеріалів на реєстрацію, а для тих лікарських засобів, що не мають ПУР, — шляхом відповідної післяреєстраційної процедури.
V.C.2.1. Оновлення плану управління ризиками
Оновлений ПУР, як очікується, повинен подаватися в будь-який час, коли внесена зміна у перелік питань з безпеки, або коли виникає потреба у новій або значній зміні в існуючі додаткові заходи з фармаконагляду, або додаткові заходи з мінімізації ризиків. Значні зміни в існуючих додаткових заходах з фармаконагляду та заходах з мінімізації ризиків можуть включати видалення такої діяльності з ПУР. Наприклад, зміна в цілях дослідження, популяції або даті кінцевих результатів, або додавання нових питань з безпеки в ключові повідомлення навчальних матеріалів, як очікується, будуть відображатися в оновленому ПУР під час процедури, що ініціює ці зміни.
Оновлення ПУР буває потрібним, якщо дані, що подані під час процедури, призводять або, як очікується, призведуть до змін у стандартній діяльності з фармаконагляду, що виходить за межі повідомлень про побічні реакції та діяльності з виявлення сигналу, або у стандартній діяльності з мінімізації ризиків, та потребуватимуть спеціальних клінічних заходів для реагування на ризик. Прикладом, коли зміни рутинних заходів з фармаконагляду, є підставою зміни ПУР, можуть бути значні зміни планів щорічного покращення нагляду за безпекою. Прикладом, коли зміни рутинних заходів з мінімізації ризиків, є підставою для зміни ПУР, може бути ситуація, коли контроль функції нирок додається як рекомендація в «Спеціальних попередженнях та застереженнях до застосування» розділу 4.4 короткої характеристики/розділу «Особливості застосування» інструкції для медичного застосування лікарського засобуN. При такому оновленні ПУР слід розглянути необхідність оновлення планів оцінки ефективності заходів з мінімізації ризиків.
Якщо питання з безпеки, що виникає, все ще розглядається (як вказано в модулі VІ ННПФ), зокрема, в контексті сигналу або потенційного ризику, який може бути важливим ідентифікованим ризиком, може вимагатися оновлення ПУР. У разі, коли питання з безпеки, що виникає, підтверджене, важливий ідентифікований або потенційний ризик необхідно внести до переліку питань з безпеки в ПУР.
Якщо не вимагається інакше, режим відслідковування змін ПУР слід включити при кожному оновленні ПУР для демонстрації внесених змін в останню оновлену версію у порівнянні з «поточною» схваленою версією ПУР.
Лікарський засіб може мати лише одну «поточну» схвалену версію ПУР. Якщо під час процедури подається декілька оновлень до ПУР, версія, що вважається «поточною» затвердженоюN для майбутніх оновлень та режиму відслідковування змін ПУР, повинна бути та, що подається з послідовністю закриття (closing sequence) процедури.
Якщо подається оновлений ПУР під час процедури, ПУР вважається схваленим/затвердженимN наприкінці процедури, коли всі зміни вважаються прийнятними.
У післяреєстраційний період подання нового або оновленого ПУР за межами іншої регуляторної процедури є зміною відповідно до «Настанов про зміни15», Порядку проведення експертизи [7]N. Для отримання детальних настанов щодо відповідних категорій змін та їх класифікації слід дивитися Практичні питання та відповіді ЕМА у підтримку імплементації Настанов щодо змін у централізованій процедурі16, в Україні — Порядок проведення експертизи [7]N.
Управління ПУР з паралельними процедурами
Якщо для лікарського засобу проводиться більш ніж одна супутня поточна процедура, що вимагає подання ПУР, в ідеалі повинен подаватися комбінований ПУР з відповідним відокремленням даних у модулі СІІІ ПУР. Найкращий регуляторний шлях для оновлення ПУР у випадку множинних процедур, що потенційно впливають на зміст ПУР, полягає в обговоренні підходів до оновлення ПУР з уповноваженим органом до його подання.
Оновлення ПУР з регулярно оновлюваним звітом з безпеки
Якщо при підготовці РОЗБ виникає необхідність внесення змін до ПУР (внаслідок визначення нових проблем з безпеки чи нових даних), тоді разом з РОЗБ повинна бути надана оновлена версія ПУР. У цьому випадку не потрібно подавати окрему заяву на зміни до ПУР. Якщо строки подання обох цих документів співпадають, а зміни не пов’язані одна з одною, ПУР повинен подаватися у супроводі окремої заяви на зміни.
Проте в контексті єдиної оцінки РОЗБ у ЄС (PSUSA) оновлення до ПУР не можуть бути подані та прийняті разом з РОЗБ лікарських засобів (зареєстрованих за централізованою процедурою та/або на національному рівні). Заявники (власники реєстраційних посвідчень), у разі необхідності, повинні оновити свої ПУР після процедури єдиної оцінки РОЗБ у ЄС. У якості альтернативи власник реєстраційного посвідчення може надати окрему заяву на зміну у зв’язку з оновленням ПУР.
Для лікарських засобів, зареєстрованих за національною процедурою, ПУР необхідно надавати до уповноважених органів в країнах ЄС, в УкраїніN для його оцінки.
V.C.3. Оцінка плану управління ризиками в рамках регуляторної мережі ЄС, в УкраїніN
У рамках ЄС регуляторний контроль ПУР для лікарських засобів, зареєстрованих за централізованою процедурою, здійснює PRAC. Для оцінки ПУР PRAC призначає доповідача PRAC, який тісно співпрацює зі (спів-)доповідачем(ами), призначеним СНМР та Комітетом з високотехнологічних лікарських засобів (САТ) (для високотехнологічних лікарських засобів) або з референтною країною ЄС. ЕМА може в кожному конкретному випадку проконсультуватися з працівниками з медичною та/або фармацевтичною освітою та пацієнтами під час оцінки ПУР для отримання їх думки щодо запропонованих заходів з мінімізації ризиків.
Для лікарських засобів, які зареєстровані за національною процедурою, за оцінку ПУР відповідають національні уповноважені органи (в Україні — ЦентрN. Національний уповноважений орган може зобов’язати заявника (власника реєстраційного посвідчення) використовувати систему управління ризиками для кожного лікарського засобу, як вказано в ст. 104(3)(с) Директиви 2001/83/ЄС [1], положеннях ПорядкуN[2], положеннях Порядку проведення експертизи [7]N, якщо існують питання щодо ризиків, які впливають на співвідношення користь/ризик зареєстрованого лікарського засобу. У цьому контексті національний уповноважений орган повинен також зобов’язати власника реєстраційного посвідчення подавати детальний опис системи управління ризиками, яку він планує ввести для даного лікарського засобу (ст. 104а(2) Директиви 2001/83/ЄС [1], положення ПорядкуN [2]).
Для лікарських засобів, зареєстрованих за централізованою процедурою, лише заходи з мінімізації ризиків, що рекомендовані PRAC та послідовно погоджені СНМР, повинні включатися до плану з мінімізації ризиків, як додаткові заходи з мінімізації ризиків. Ключові елементи додаткових заходів з мінімізації ризиків, що є умовами для отримання реєстраційного посвідчення, детально описані в додатку ІІ рішення Комісії. У виняткових випадках рішенням Комісії, відповідно до ст. 127а Директиви 2001/83/ЄС [1], країни ЄС можуть бути зобов’язані імплементувати на національному рівні певні умови або обмеження відносно безпечного та ефективного застосування лікарського засобу.
Якщо необхідно, уповноважені органи повинні гарантувати, що всі заявники (власники реєстраційних посвідчень) на лікарські засоби, що містять однакові діючі речовини, вносять подібні зміни до заходів з мінімізації ризиків, коли таких змін зазнали заходи щодо референтного лікарського засобу.
V.C.4. Прозорість
ЕМА та країни ЄС повинні публікувати на Європейському вебпорталі лікарських засобів та національних вебпорталах лікарських засобів звіт з оцінки для громадськості (public assessment reports) та резюме планів управління ризиками (ст. 26(1)(с) Регламенту (ЄС) 726/2004 [5], ст. 106 (с) Директиви 2001/83/ЄC [1]). В Україні резюме плану управління ризиками розміщується на сайті ЦентруN (положення ПорядкуN [2]).
Для лікарських засобів, зареєстрованих за централізованою процедурою, EMA буде:
- публікувати резюме ПУР;
- включати таблиці, що мають відношення до ПУР, у Європейський звіт з оцінки для громадськості (EPAR), включаючи інформацію про лікарський засіб та умови реєстрації.
Національні уповноважені органи будуть надавати детальні дані про те, як вони планують імплементувати заходи з прозорості на національному рівні (з посиланням на ст. 106 Директиви 2001/83/ЄC [1]).
Частина VІ
Модуль VI — Управління та звітування про побічні реакції лікарських засобів
VІ.А. Вступ
Даний модуль стосується законодавчих вимог, викладених у розділі IX Директиви 2001/83/ЄC[1], розділі 3 Регламенту (ЄС) №726/2004 [5] та положеннях ПорядкуN [2], що застосовуються до уповноваженого органу та заявників (власників реєстраційних посвідчень) у процедурах збору, управління даними і звітами про підозрювані побічні реакції (серйозні та несерйозні), пов’язані з прийомом лікарських засобів, зареєстрованих для медичного застосування в ЄС та в УкраїніN. В даному модулі також представлені рекомендації щодо звітування про нові проблеми з безпеки або про підозрювані побічні реакції в особливих ситуаціях. У даному модулі застосовуються і вимоги, передбачені у розділах IV, V і IX Імплементаційної постанови Комісії (ЄС) № 520/2012 (далі — ІП 520/2012 [6], положеннях ПорядкуN [2]).
Настанова, описана в даному модулі, не стосується збору, управління даними та повідомлень про події чи схеми застосування лікарських засобів, внаслідок яких не виникали підозрювані побічні реакції (наприклад, безсимптомне передозування, зловживання, застосування не за показаннями, неправильне застосування чи медична помилка) або що не вимагають надання у вигляді повідомлення про індивідуальний випадок, пов’язаний з безпекою (individual case safety report) чи про нові проблеми з безпеки (emerging safety issues). Однак може бути необхідним надання такої інформації в РОЗБ для інтерпретації даних з безпеки або для оцінки співвідношення користь/ризик лікарських засобів. У такому випадку слід користуватися настановою, представленою в модулі VII ННПФ.
VI.B. Структури та процеси
У розділі В цього модуля надається інформація для уповноважених органів та заявників (власників реєстраційних посвідчень) та інших зацікавлених державних та недержавних структур та осібN про загальні принципи збору, реєстрації та надання повідомлень про підозрювані побічні реакції, пов’язані з лікарським засобом для застосування у людини, що застосовуються. Вимоги ЄС та УкраїниN представлені у VI.C ННПФ.
VI.B.1. Збір повідомлень
Уповноважений орган та заявники (власники реєстраційних посвідчень) повинні вживати належних заходів для збору та обробки всіх повідомлень про виникнення підозрюваних побічних реакцій, що надходять з різних джерел.
З цією метою повинна бути створена система фармаконагляду, що дозволить отримувати відповідну інформацію для наукової оцінки таких повідомлень.
Система повинна бути створена таким чином, щоб гарантувати, що інформація у зібраних повідомленнях є достовірною, розбірливо написаною, точною, послідовною, її можливо перевірити і вона є максимально повною для її клінічної оцінки.
Усі повідомлення, що містять дані з фармаконагляду, повинні реєструватися і архівуватися відповідно до діючих вимог щодо захисту інформації (див. VI.C.6.2.2.8 щодо рекомендацій ЄС та УкраїниN).
Система повинна бути структурована таким чином, щоб існувала можливість проведення процесу перевірки повідомлень про підозрювані побічні реакції (див. VI.B.2) у належний термін, а також обмінюватися ними між уповноваженим органом та заявниками (власниками реєстраційних посвідчень) у рамках передбаченого законодавством терміну звітування (див. VI.B.7.1).
Відповідно до Настанови ICH-E2D [25] у післяреєстраційний період розрізняють два види повідомлень з безпеки: повідомлення, що надходять без запиту (unsolicited reports), а також повідомлення, що походять із джерел з організованою системою збору даних (solicited reports).
VI.B.1.1. Повідомлення, що надходять без запиту (unsolicited reports)
VI.B.1.1.1. Спонтанні повідомлення
Спонтанне повідомлення — це повідомлення, надане за власною ініціативою працівником з медичною та/або фармацевтичною освітою або споживачем до уповноваженого органу, заявника (власника реєстраційних посвідчень) власника реєстраційних посвідчень чи іншої організації (наприклад, регіонального відділення з фармаконагляду, токсикологічного центру управління, представників Центру з питань фармаконагляду в адміністративно-територіальних одиницяхN), у якому описується одна або декілька підозрюваних побічних реакцій у пацієнта, який застосовував один чи більше лікарських засобів, і це повідомлення не походить з дослідження або будь-якої іншої організованої системи збору даних, що вимагає надання даних про небажані явища, як це визначено у розділі VI.B.1.2.
Повідомлення, отримані в результаті стимульованого репортування, що відбувається внаслідок «Прямої комунікації з працівниками з медичною та/або фармацевтичною освітою», публікацій у пресі, опитування працівників з медичною та/або фармацевтичною освітою представниками компанії, комунікації організацій пацієнтів або судових процесів, повинні розглядатися як спонтанні.
Повідомлення про побічні реакції, що надходять від споживачів, слід обробляти як спонтанні повідомлення незалежно від будь-якого подальшого «медичного підтвердження».
Процедури звітування та чинні вимоги щодо строків надання спонтанних повідомлень описані в розділах VI.B.7 і VI.B.8.
VI.B.1.1.2. Повідомлення про побічні реакції, опубліковані в медичній літературі
Наукова та медична література — важливе джерело інформації для моніторингу профілю безпеки і співвідношення користь/ризик лікарського засобу, особливо у зв’язку з виявленням нових сигналів з безпеки або актуальних питань з безпеки. Тому заявники (власники реєстраційних посвідчень) повинні бути поінформовані про можливі публікації шляхом систематичного перегляду літературних джерел або широко використовуваних довідникових баз даних (наприклад, Medline, Excerpta Medica або Embase) не рідше одного разу на тиждень. Заявник (власник реєстраційного посвідчення) повинен забезпечити, щоб перегляд літературних джерел включав використання довідкових баз даних, що містять найбільший перелік посилань на статті, що стосуються властивостей його лікарського засобу17. Крім того, заявники (власники реєстраційних посвідчень) повинні проводити моніторинг наукових і медичних публікацій у місцевих медичних журналах, та доводити отриману з них інформацію до відома департаменту з безпеки компанії.
Заявники (власники реєстраційних посвідчень) повинні переглядати і оцінювати дані про підозрювані побічні реакції, отримані з наукової та медичної літератури, включаючи відповідні опубліковані тези наукових зустрічей та проєкти рукописів, для виявлення та реєстрації повідомлень про індивідуальні випадки, пов’язані з безпекою, що походять зі спонтанних повідомлень або неінтервенційних післяреєстраційних досліджень. Якщо у публікації згадуються декілька лікарських засобів, відповідні заявники (власники реєстраційних посвідчень) повинні розглядати лише ті, що визначені автором публікації як такі, що мають хоча б мінімальний можливий причинно-наслідковий зв’язок з підозрюваною побічною реакцією.
Валідні повідомлення про індивідуальні випадки, пов’язані з безпекою, повинні надаватися відповідно до вимог, зазначених у розділах VI.B.7 та VI.B.8.
Базуючись на характеристиках, наданих у розділі VI.B.2, необхідно створити одне повідомлення для кожного окремого ідентифікованого пацієнта. Потрібно надати всю відповідну медичну інформацію, а першоджерелом таких повідомлень повинен(ні) вважатися автор(и) публікації.
Спеціальні вимоги ЄС та УкраїниN щодо лікарських засобів та наукових і медичних публікацій, що не моніторуються ЕМА, і для яких заявники (власники реєстраційних посвідчень) повинні надавати валідні повідомлення про індивідуальні випадки, пов’язані з безпекою, див. детальніше в VI.C.2.2.3.
VI.B.1.1.3. Повідомлення з інших джерел
Якщо заявнику (власнику реєстраційних посвідчень) стає відомо про повідомлення про підозрювані побічні реакції з немедичних джерел, наприклад, з неспеціалізованої преси або інших засобів масової інформації, його слід розглядати як спонтанне повідомлення. Необхідно докласти всіх зусиль для подальшого спостереження даного випадку з метою збору мінімуму інформації, що уможливить валідність повідомлення про індивідуальний випадок, пов’язаний з безпекою. Для таких повідомлень застосовуються такі ж терміни звітування, як і для інших спонтанних повідомлень.
VI.B.1.1.4. Інформація щодо підозрюваних побічних реакцій, що була знайдена в інтернеті та за допомогою цифрових засобів комунікації
Заявники (власники реєстраційних посвідчень) повинні регулярно проводити моніторинг інтернету чи цифрових засобів комунікації18, що перебувають під їх управлінням або відповідальністю, на предмет існування потенційних повідомлень про випадки виникнення підозрюваних побічних реакцій. У цьому контексті цифровий засіб комунікації вважається спонсорованим компанією, якщо він є власністю, оплачується, та/або контролюється заявником (власником реєстраційного посвідчення).19 Частота проведення такого моніторингу повинна дозволяти вчасне надання потенційного повідомлення про індивідуальний випадок, пов’язаний з безпекою до уповноваженого органу відповідно до строків надання повідомлень про побічні реакції на основі дати, коли така інформація була розміщена на інтернет-сайті/у цифровому середовищі. Заявники (власники реєстраційних посвідчень) можуть також використовувати свої вебсайти для збору повідомлень про випадки виникнення підозрюваних побічних реакцій (див. VI.C.2.2.1).
Якщо заявнику (власнику реєстраційного посвідчення) стає відомо про випадок виникнення підозрюваної побічної реакції, описаний у будь-яких цифрових медіа, що не спонсоруються компанією, його необхідно оцінити для визначення того, чи підпадає він під умови репортування.
Інформація про випадки виникнення підозрюваних побічних реакцій, що отримана з мережі інтернет або цифрових засобів комунікації, що надаються на добровільній основі, повинна оброблятися як спонтанні повідомлення. Відповідно, строки звітування про такі повідомлення такі ж, як і для спонтанних повідомлень (див. VI.B.7).
У ситуації, коли випадок розвитку підозрюваної побічної реакції був знайдений в інтернеті або за допомогою цифрових медіа, ідентифікувати особу, яка надала цю інформацію, можна тільки тоді, коли можна перевірити її контактні дані (наприклад надана у правильному форматі адреса електронної пошти). Якщо не вказана країна першоджерела, то в її якості слід зазначати країну, де була отримана інформація або де був проведений моніторинг.
VI.B.1.2. Повідомлення, що походять із джерел з організованою системою збору даних (solicited reports)
Як визначено в Настанові ICH-E2D [25], повідомлення, що походять із джерел з організованою системою збору даних — це повідомлення, отримані з систем організованого збору даних, що включають клінічні випробування, неінтервенційні дослідження, реєстри, післяреєстраційні програми персоніфікованого застосування, інші програми підтримки пацієнтів і моніторингу захворювань, опитування пацієнтів або медичних працівників чи збір даних з ефективності або комплаєнсу пацієнтів. Повідомлення про випадки виникнення побічних реакцій, отримані з будь-якої з цих систем організованого збору даних, не слід розглядати як спонтанні. Винятком є:
- підозрювані побічні реакції, що відносяться до тих побічних явищ, для яких протоколами неінтервенційних післяреєстраційних досліджень передбачено інакше і не вимагається їх систематичний збір;
- підозрювані побічні реакції, що походять з програм застосування лікарських засобів зі співчуття чи персоніфікованого застосування, якщо у таких програмах не вимагається активний збір даних щодо побічних явищ (див. VI.C.1.2.2).
Повідомлення, що походять із джерел з організованою системою збору даних, повинні класифікуватися як повідомлення з досліджень. Їх необхідно оцінювати на наявність причинно-наслідкового зв’язку з тим, щоб розглянути питання, чи належать вони до підозрюваних побічних реакцій і, отже, відповідають критеріям звітності.
Загальні правила надання повідомлень про випадки виникнення підозрюваних побічних реакцій, що походять з організованих систем збору даних, які здійснюються в ЄС відповідно до положень Директиви 2001/83/ЄС [1], Регламенту (ЄC) №726/2004 [5] чи Директиви 2001/20/ЄС [15]; в Україні відповідно до положень Порядку [2] та положень Порядку проведення клінічних випробувань [16]N, представлені у розділі VI.C.1.
VI.B.2. Валідація повідомлень
Тільки валідні повідомлення про побічні реакції підлягають рапортуванню. Тому всі повідомлення про випадки виникнення підозрюваних побічних реакцій, перед наданням їх до уповноваженого органу, необхідно перевіряти на їх відповідність мінімальним критеріям, що дозволяють вважати це повідомлення про побічну реакцію валідним (див. Настанова ICH-E2D [25]). Мінімальними критеріями для надання повідомлень про побічні реакції є:
- одна чи більше ідентифікованих осіб, які надали інформацію (першоджерело), інформацію про кваліфікацію/освітуN (наприклад, лікар, фармацевт, інший працівник у галузі охорони здоров’я, юрист, споживач або інший працівник, який не має медичної/фармацевтичної освіти), прізвище, ініціали або адресу20. Якщо можливо, повинні бути надані контактні дані особи, яка надала інформацію, що дозволить у подальшому проводити спостереження за випадком. Однак, якщо особа, яка надала інформацію, не бажає надавати контактні дані, повідомлення про індивідуальний випадок, пов’язаний з безпекою, буде валідним за умови, що організація, до якої надійшло повідомлення, змогла отримати підтвердження випадку безпосередньо від першоджерела інформації. Повинні бути ідентифіковані всі сторони, які надають інформацію про випадок або прагнуть отримати таку інформацію, а не тільки першоджерело; в Україні заявник (власник реєстраційного посвідчення) повинен подавати інформацію про випадки побічних реакцій, що мають медичне підтвердженняN;
- один ідентифікований пацієнт, із зазначенням ініціалів, ідентифікаційного номера, дати народження, віку, вікової групи, статі, номера історії хвороби або амбулаторної картиN. Ця інформація повинна бути якомога більш повною;
- одна або декілька підозрюваних діючих речовин/лікарських засобів;
- одна або декілька підозрюваних побічних реакцій. Якщо першоджерело повідомлення чітко заявляє, що причинно-наслідковий зв’язок між прийомом лікарського засобу і небажаним явищем виключається, і отримувач інформації (уповноважений орган або заявник (власник реєстраційного посвідчення)) погоджується з ним, повідомлення не може вважатися валідним, оскільки відсутній необхідний мінімум інформації21. Повідомлення також не буде валідним, якщо повідомляється, що у пацієнта виникла побічна реакція, однак не повідомляється, яка саме. Невалідним буде і повідомлення, в якому зазначено лише наслідок побічної реакції і (І) немає жодної інформації щодо клінічних обставин, які дозволили б вважати це підозрюваною побічною реакцією, або (ІІ) першоджерело не зазначило можливий причинно-наслідковий зв’язок із застосуванням підозрюваного лікарського засобу. Наприклад, заявнику (власнику реєстраційного посвідчення) повідомили, що пацієнт був госпіталізований або помер, без надання будь-якої додаткової інформації. У цьому конкретному випадку, при прийнятті рішення про те, чи розцінювати повідомлену інформацію як побічну реакцію чи як небажане явище, завжди необхідно проводити медичну оцінку. Наприклад, повідомлення про раптову смерть, як правило, необхідно розглядати як випадок виникнення підозрюваної побічної реакції і поінформувати про неї.
Відсутність будь-якого з цих чотирьох елементів означає, що даний випадок вважається неповним і не відповідає вимогам звітності. Уповноважений орган і заявники (власники реєстраційних посвідчень) повинні активно відстежувати випадки з метою збору відсутніх даних. Повідомлення, в яких відсутній мінімум інформації, все ж повинні реєструватися в системі фармаконагляду для використання їх у поточній діяльності з оцінки безпеки. Рекомендації щодо електронного надання валідних повідомлень про індивідуальні випадки, пов’язані з безпекою, після отримання відсутньої інформації, надані у VI.C.6.2.3.8.
При зборі повідомлень про підозрювані побічні реакції через інтернет або цифрові медіа термін «ідентифікований» означає можливість перевірки існування особи, яка надала інформацію і пацієнта (див. VI.B.1.1.4).
Коли одній стороні (уповноваженому органу чи заявнику (власнику реєстраційного посвідчення)) стає відомо, що першоджерело могло також надати інформацію про підозрювану побічну реакцію іншій зацікавленій стороні, повідомлення все одно має розглядатися як валідне. У повідомленні про індивідуальний випадок, пов’язаний з безпекою, повинна бути включена вся відповідна інформація, необхідна для виявлення випадків-дублікатів22.
Валідний випадок виникнення підозрюваної побічної реакції, повідомлений споживачем, не може бути розцінений як повідомлення про побічне явище, в якому виключений причинно-наслідковий зв’язок, якщо працівник з медичною та/або фармацевтичною освітою, контактні дані якого споживач надав для отримання додаткової інформації, не згоден з думкою споживача. У цьому випадку у повідомленні про індивідуальний випадок, пов’язаний з безпекою, потрібно зазначити думку як споживача, так і працівника з медичною та/або фармацевтичною освітою. Необхідно дотримуватися інструкцій щодо надання медичного підтвердження випадку, зазначених у настанові ICH-E2B(R2) [26] розділ A.1.14 («Чи був випадок медично підтверджений, якщо першоджерело — не працівник з медичною та/або фармацевтичною освітою?»).
У випадку повідомлень, що походять із джерел з організованою системою збору, (див. розділ VI.B.1.2), якщо отримувач не згоден з наявністю обґрунтованого причинно-наслідкового зв’язку між застосуванням підозрюваного лікарського засобу і виникненням побічної реакції, описаної першоджерелом, випадок не повинен бути понижений у статусі до повідомлення про непов’язане побічне явище. У такому повідомленні про індивідуальний випадок, пов’язаний з безпекою, повинна бути зазначена думка як першоджерела, так і отримувача повідомлення.
Такий же підхід застосовується і до критерію серйозності повідомлення про індивідуальний випадок, пов’язаний з безпекою: він не може бути понижений до несерйозного, якщо отримувач повідомлення не згоден з критерієм серйозності, повідомленим першоджерелом.
VI.B.3. Подальше відстеження повідомлень
Надана в повідомленнях первинна інформація про випадки виникнення підозрюваних побічних реакцій може бути неповною. Такі випадки, за необхідності, повинні відстежуватися для отримання додаткової детальної інформації, важливої для наукової оцінки. Це особливо стосується моніторингу явищ, що становлять особливий інтерес, проспективних повідомлень про вагітність, повідомлень про смерть пацієнта, про нові ризики або зміни відомих ризиків. Перелічене вище є додатковим до всіх можливих спроб збору відсутнього мінімуму інформації (див. VI.B.2 ННПФ). Усі спроби отримання подальшої інформації повинні бути задокументовані.
Методи подальшого відстеження повинні бути спрямовані на оптимізацію збору відсутньої інформації. Це має бути зроблено таким чином, щоб заохочувати першоджерело надавати нову інформацію, необхідну для наукової оцінки специфічних питань з безпеки. Використання цільових спеціальних форм для заповнення не повинно вимагати від першоджерела повторювати інформацію, вже надану в первинному повідомленні, та/або заповнювати великі анкети, оскільки це може відбити бажання у першоджерела в майбутньому надавати спонтанні повідомлення. Тому слід попередньо заповнити деякі поля даних у таких повідомленнях для подальшого відстеження, щоб зробити їх заповнення простішим для першоджерела.
У випадку отримання інформації про побічну реакцію безпосередньо від споживача, якщо така інформація неповна, необхідно намагатися отримати його згоду зв’язатися із зазначеним ним працівником з медичною та/або фармацевтичною освітою для отримання подальшої інформації про випадок. Якщо випадок, про який спочатку повідомив споживач, був підтверджений (повністю або частково) працівником з медичною та/або фармацевтичною освітою, цей факт необхідно зазначити в повідомленні про індивідуальний випадок, пов’язаний з безпекою23.
Що стосується підозрюваних побічних реакцій, пов’язаних із застосуванням біологічних лікарських засобів, то особливо важливе значення має ідентифікація способу виробництва лікарського засобу. Тому необхідно вжити всіх належних заходів для визначення торговельної назви та номера серії такого лікарського засобу. Блок-схема процесу обов’язкового подальшого відстеження (follow-up) інформації для ідентифікації підозрюваних біологічних лікарських засобів представлена в розділі VI. Додаток 1.
Для повідомлень, пов’язаних із застосуванням вакцин, слід також дотримуватися рекомендацій, наданих у ННПФ та нормативно-правових вимог, викладених у положеннях Порядку [2]N.
VI.B.4. Управління даними
Електронні дані та паперові повідомлення про підозрювані побічні реакції повинні зберігатися і оброблятися так само, як і інша медична документація з належним дотриманням законодавчих вимог щодо конфіденційності даних пацієнтів та осіб, які надають інформацію. Завжди необхідно зберігати конфіденційність історій хвороб/амбулаторних картN пацієнтів, у тому числі ідентифікаторів персональних даних, якщо такі надаються. Необхідно також дотримуватися конфіденційності стосовно персональних даних працівників з медичною та/або фармацевтичною освітою, які надають повідомлення. Що стосується персональних даних пацієнта і репортера, то інформація про випадок повинна передаватися між зацікавленими сторонами (заявники (власники реєстраційних посвідчень) чи уповноважені органи) відповідно до законодавства щодо конфіденційності даних (див. VI.C.6.2.2.8 щодо обробки персональних даних у повідомленнях про індивідуальний випадок, пов’язаний із безпекою).
З метою гарантування безпеки та конфіденційності даних з фармаконагляду необхідно забезпечити суворий контроль за доступом до документів і баз даних і обмежити його тільки компетентним персоналом. Ці умови безпеки розповсюджуються на всі канали передачі даних. У цьому аспекті необхідно дотримуватися всіх вимог для гарантування безпеки й цілісності даних під час їх передачі.
При передачі даних з фармаконагляду в межах організації або між організаціями, які мають укладені договірні умови, цей механізм повинен забезпечувати отримання всіх нотифікацій; для цього повинні бути розроблені процеси підтвердження та/або звірки.
Правильність введення даних, у тому числі належне використання термінології, повинні перевірятися у ході аудиту з якості — систематичного чи регулярного, а також вибіркового. Персонал, який відповідає за введення даних, необхідно навчити користуванню термінологією, і їх знання повинні бути підтверджені.
До даних, отриманих від першоджерела, потрібно ставитися неупереджено, не змінювати інформацію, уникати висновків і приписувань під час введення даних чи електронної передачі інформації. Повідомлення повинні містити дослівний текст, зазначений першоджерелом, або його точний переклад. Оригінальний дослівний текст повинен бути закодований з використанням відповідної термінології, як описано в VI.B.8 ННПФ. Для того, щоб забезпечити узгодженість практик кодування, рекомендується застосувати для кодування дослівного тексту, коли можливо, переклад термінології місцевою/локальною мовою.
Електронні бази даних повинні дозволяти відстеження (аудит) усіх введених або змінених даних, у тому числі дат та джерел отриманої інформації, а також дат і джерела переданої інформації.
Повинна бути створена процедура для виявлення та обробки дублікатів повідомлень під час введення даних та формування зведених звітів (див. VI.C.6.2.4 ННПФ).
VI.B.5. Управління якістю
Уповноважений орган і заявники (власники реєстраційних посвідчень) повинні мати у своєму розпорядженні систему управління якістю з метою забезпечення дотримання необхідних стандартів на кожному етапі документування випадків виникнення побічних реакцій, таких як збір даних, передача даних, управління даними, кодування даних, валідація даних, оцінка випадку, подальше відстеження випадку, надання повідомлення про індивідуальний випадок, пов’язаний з безпекою та архівування випадку (див. VI.C.6.2.4 і модуль I НПФ). Узгодженість збережених даних з первинними та наступними (follow-up) повідомленнями повинна перевірятися за допомогою процедур контролю якості, що дозволяють проводити перевірку збережених даних у порівнянні з оригінальними даними чи їх зображеннями. У зв’язку з цим дані першоджерела (наприклад, листи, електронні листи, записи телефонних розмов, що включають відомості про явище) або зображення вихідних даних повинні бути легко доступними.
Чіткі письмові стандартні операційні процедури повинні гарантувати, що ролі, обов’язки та завдання зрозумілі для всіх залучених осіб, і що забезпечено належний контроль і, якщо необхідно, зміна системи. Це однаковою мірою стосується видів діяльності, що виконуються за контрактом третіми сторонами, чиї процедури повинні бути адекватними та відповідати чинним вимогам.
Необхідно проводити навчання персоналу, який безпосередньо виконує діяльність з фармаконагляду, не тільки щодо процесів, пов’язаних із обробкою повідомлень, за який він відповідає та/або здійснює, а й чинного законодавства та ННПФ. Інший персонал, який може отримувати чи обробляти звіти з безпеки (наприклад, відділ клінічної розробки, продажу, медичної інформації, юридичний, контролю якості), повинен проходити підготовку щодо процесів збору даних про побічні явища та передачу такої інформації відповідно до внутрішньої політики та процедур.
VI.B.6. Особливі ситуації
VI.B.6.1. Застосування лікарських засобів під час вагітності або годування груддю
Вагітність
Повідомлення про можливий вплив лікарських засобів на ембріон або плід (після застосування матір’ю чи шляхом трансмісії лікарського засобу через сперму батька) слід продовжувати відстежувати з метою отримання інформації про результат вагітності та стан розвитку дитини після народження. При проведенні наукової оцінки та моніторингу, збору і надання інформації у таких специфічних ситуаціях слід керуватися рекомендаціями, наданими в Настанові з впливу лікарських засобів під час вагітності: необхідність збору післяреєстраційних даних[24]. Якщо діюча речовина (або один з її метаболітів) має тривалий період напіввиведення, цей факт необхідно враховувати при оцінці можливості впливу на ембріон, якщо такий лікарський засіб застосовувався до зачаття.
Нерідко вагітні жінки або працівники з медичною та/або фармацевтичною освітою будуть звертатися до уповноважених органів з проханням надати інформацію щодо тератогенності лікарського засобу або досвіду його застосування під час вагітності. Необхідно знайти якомога більше даних щодо потенційного впливу лікарського засобу на ембріон або плід.
Повідомлення про застосування лікарських засобів під час вагітності повинні містити якомога більш детальний опис випадків з тим, щоб надати змогу оцінити причинно-наслідковий зв’язок між будь-якими повідомленими побічними явищами і прийомом підозрюваного лікарського засобу. У цьому випадку рекомендується використовувати стандартні структуровані опитувальники.
Повідомлення, що містять інформацію про аномальні результати вагітності, пов’язані з лікарським засобом, що застосовувався під час вагітності, класифікуються як серйозні і повинні надаватися відповідно до вимог, викладених у VI.B.725 та положеннях ПорядкуN [2].
Це, зокрема, стосується повідомлень про:
- вроджені вади чи затримку розвитку у плода або дитини;
- смерть плода або спонтанний аборт;
- підозрювані побічні реакції у новонароджених, що класифікуються як серйозні.
Повідомлення про інші випадки, такі як переривання вагітності без інформації щодо вроджених вад розвитку, повідомлення про застосування лікарських засобів у вагітних без зазначення наслідків або повідомлення з нормальним результатом вагітності, не повинні надаватися, оскільки у них відсутні підозрювані побічні реакції. Такі повідомлення слід збирати й аналізувати в регулярних звітах з безпеки лікарських засобів (див. модуль VII ННПФ).
Проте у деяких випадках може вимагатися надання повідомлень про застосування лікарських засобів під час вагітності без зазначених у них підозрюваних побічних реакцій. Існують ситуації, коли така вимога може бути умовою видачі реєстраційного посвідчення або передбачена в ПУР; наприклад, щодо застосування лікарських засобів, протипоказаних у період вагітності або щодо лікарських засобів, для яких необхідно проводити моніторинг через їх високий тератогенний потенціал (наприклад талідомід, ізотретиноїн).
Сигнал про можливий тератогенний вплив (наприклад, через кластерні або аналогічні патологічні результати вагітності) повинен негайно передаватися до уповноваженого органу відповідно до рекомендацій, наданих у VI.C.2.2.6.
Годування груддю
Про підозрювані побічні реакції, що виникають у немовлят внаслідок застосування лікарського засобу пацієнтками та проникнення його у грудне молоко, потрібно повідомляти відповідно до критеріїв, викладених у VI.B.725.
VI.B.6.2. Застосування лікарських засобів у педіатричній популяції чи літніми пацієнтами
Збір даних з безпеки у педіатричній популяції та осіб літнього віку має важливе значення. Необхідно звернути особливу увагу на збір та надання інформації про вік пацієнта чи вікову групу, до якої він належить, коли інформація про випадок надається працівником з медичною або фармацевтичною освітою або споживачем з тим, щоб мати можливість ідентифікувати потенційні сигнали з безпеки для специфічної популяції.
Що стосується педіатричної популяції, то необхідно дотримуватися опублікованої ЕМА настанови26 при здійсненні фармаконагляду.
VI.B.6.3. Повідомлення про передозування, зловживання, застосування не за показаннями, медичні помилки та вплив, пов’язаний з характером професійної діяльності
У даному модулі термін «медична помилка» означає будь-яку ненавмисну помилку при призначенні, дозуванні або застосуванні лікарського засобу працівником з медичною або фармацевтичною освітою, пацієнтом або споживачем.
Повідомлення про передозування, зловживання, застосування не за показаннями, медичні помилки та вплив, пов’язаний з характером професійної діяльності, внаслідок яких не відбувалося виникнення побічних реакцій, не повинні надаватися у вигляді повідомлень про індивідуальний випадок, пов’язаний з безпекою. Якщо необхідно, такі повідомлення необхідно проаналізувати у РОЗБ лікарського засобу. Якщо такі повідомлення становлять проблему з безпеки, що впливає на співвідношення користь/ризик лікарського засобу, то про них слід негайно повідомити уповноважений орган відповідно до рекомендацій, представлених у розділі VI.C.2.2.6.
Повідомлення, в яких описані випадки виникнення підозрюваних побічних реакцій, необхідно надавати відповідно до критеріїв, зазначених у розділі VI.B.7 і вимог щодо електронної передачі інформації, описаних у розділі VI.C.6.2.3.3. Такі випадки повинні рутинно відслідковуватися для гарантування того, що інформація про симптоми, лікування, наслідки, обставини виникнення (наприклад, помилки при призначенні, застосуванні, видачі, дозуванні, застосуванню за не затвердженими показаннями чи у популяції тощо) є якомога більш повною.
VI.B.6.4. Відсутність терапевтичного ефекту
Повідомлення про відсутність терапевтичного ефекту повинні реєструватися і, у випадку, коли у них була надана неповна інформація, відстежуватися. Зазвичай такі повідомлення не повинні надаватися до уповноваженого органу, а їх слід аналізувати у РОЗБ лікарських засобів. Однак за певних обставин може вимагатися надання вказаних вище повідомлень у 15-денний термін (див. VI.C.6.2.3.4 щодо надання повідомлень в електронній формі). Такі випадки стосуються, наприклад, лікарських засобів, що застосовуються для лікування критичних станів або життєво небезпечних захворювань, вакцин, контрацептивів. Повідомлення про відсутність терапевтичного ефекту таких лікарських засобів необхідно надавати до уповноваженого органу, крім випадків, коли особа, яка надає інформацію, наголошує, що випадок стався внаслідок прогресування захворювання і не був пов’язаний із застосуванням лікарського засобу.
Для вирішення питання щодо необхідності надання інформації про інші випадки відсутності терапевтичного ефекту, слід провести їх клінічну оцінку. Наприклад, не слід повідомляти про відсутність ефективності антибіотика, що застосовували для лікування життєво загрозливого стану, коли цей збудник інфекції насправді не чутливий до даного лікарського засобу. Проте повідомлення про лікування життєво загрозливої інфекції, коли відсутність терапевтичного ефекту може бути пов’язана з появою нових стійких штамів бактерій, що раніше вважалися чутливими, необхідно надавати у 15-денний термін.
Повідомлення про випадки неефективності вакцинації необхідно надавати, зокрема, з метою виявлення потенційних сигналів щодо зниження імуногенності у підгрупі щеплених, послаблення імунітету, зміни штаму. Що стосується останнього, то вважається, що спонтанні повідомлення про випадки неефективності вакцинації, надані працівниками з медичною та/або фармацевтичною освітою, можуть бути сигналом про зміну штаму. Такий сигнал може вимагати оперативних дій та, за необхідності, подальшої оцінки шляхом проведення післяреєстраційних досліджень з безпеки. При цьому можна дотримуватися рекомендацій, наданих у настанові щодо здійснення моніторингу неефективності вакцин, зазначених у звіті Робочої групи CIOMS/ВООЗ з фармаконагляду за вакцинами27.
VI.B.7. Надання повідомлень про індивідуальний випадок, пов’язаний з безпекою
Потрібно надавати тільки валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою (див. VI.B.2). Відлік часу для надання таких повідомлень про побічну реакцію починається з моменту, коли інформація, що відповідає мінімальним критеріям інформування, стала відомою регіональному відділенню з фармаконагляду, представникам Центру з фармаконагляду в адміністративно-територіальних одиницяхN, уповноваженому органу або будь-якому співробітнику заявника (власника реєстраційного посвідчення), включаючи медичних представників і підрядників. Ця дата вважається днем «0». Це перший день, коли отримувачу стає відомо про валідне повідомлення про індивідуальний випадок, пов’язаний із безпекою, незалежно від того, чи була інформація отримана у вихідний або святковий день. Строки репортування базуються на календарних днях.
Якщо заявник (власник реєстраційного посвідчення) має договірні відносини з особою чи організацією, між ними повинні існувати чіткі процедури і детальні домовленості для гарантування того, що заявник (власник реєстраційного посвідчення) може виконувати зобов’язання щодо надання повідомлень. У цих процедурах необхідно, зокрема, описати процеси обміну інформацією з безпеки, у тому числі часові рамки і хто відповідальний за звітність. При цьому необхідно виключати дублювання надання повідомлень до уповноваженого органу.
Щодо повідомлень про індивідуальний випадок, пов’язаний з безпекою, описаних у науковій та медичній літературі (див. VI.B.1.1.2), відлік часу (день «0») починається з моменту ознайомлення з публікацією, що містить необхідний мінімум інформації. Якщо існують договірні відносини з особою чи організацією, що передбачають здійснення моніторингу літератури та/або надання валідних повідомлень про індивідуальний випадок, пов’язаний з безпекою, повинні бути укладені детальні домовленості для того, щоб гарантувати, що заявник (власник реєстраційного посвідчення) може виконувати зобов’язання щодо надання повідомлень.
Відлік часу для надання наступного повідомлення починається з дня отримання важливої додаткової інформації щодо випадку, про який повідомлялося раніше. При цьому «важлива додаткова (follow-up) інформація» означає нову інформацію, що може вплинути на оцінку випадку або може змінити його критерії серйозності; неважлива інформація включає оновлені коментарі щодо оцінки випадку або виправлення друкарських помилок у попередній версії. Щодо різниці між важливими та неважливими даними подальшого відстеження див. також VI.C.6.2.2.7.
VI.B.7.1. Строки надання повідомлень
Надавати валідні повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою, до уповноваженого органу необхідно якомога раніше, але не пізніше 15 календарних днів після отримання інформації регіональним відділенням з фармаконагляду, представниками Центру з фармаконагляду в адміністративно-територіальних одиницях або будь-ким із персоналу заявника (власника реєстраційного посвідчення), включаючи медичних представників і підрядників. Такі строки надання стосуються як для первинних повідомлень, так і для додаткової інформації. Якщо випадок спочатку вважався серйозним, а потім, коли була отримана додаткова інформація, він перейшов у категорію несерйозних, про це необхідно повідомити протягом 15 днів; строки надання повідомлень про несерйозні реакції повинні потім бути застосовані для подальших/наступних/додаткових (follow-up) повідомлень.
Інформація щодо строків звітності про валідні повідомлення про несерйозний індивідуальний випадок, пов’язаний із безпекою, надається в VI.C.3.
VI.B.8. Механізми надання повідомлень про побічні реакції
Враховуючи міжнародні аспекти надання повідомлень про побічні реакції та необхідність досягнення згоди між усіма зацікавленими сторонами, повідомлення про індивідуальний випадок, пов’язаний з безпекою, мають надаватися у вигляді структурованих даних в електронному форматі з використанням контрольованих словників для відповідних елементів даних. Що стосується змісту та формату електронних повідомлень про індивідуальний випадок, пов’язаний з безпекою, заявники (власники реєстраційних посвідчень) повинні дотримуватися наступних узгоджених на міжнародному рівні настанов та стандартів ICH28:
- Термінологія ICH M1 — медичний словник для регуляторної діяльності (ICH M1 terminology — Medical Dictionary for Regulatory Activities (далі — MedDRA);
- Вибір терміна MedDRA: Настанова — остання версія затверджених ICH настанов для користувачів MedDRA (MedDRA Term Selection: Points to Consider Document — The latest version of the ICH-endorsed Guide for MedDRA Users);
- ICH M2 EWG — Електронне надання повідомлення про індивідуальний випадок, пов’язаний з безпекою (ICH M2 EWG — Electronic Transmission of Individual Case Safety reports Message Specification);
- ICH E2B(R2) [26] — настанова ICH з управління даними з клінічної безпеки: Елементи даних для надання повідомлення про індивідуальний випадок, пов’язаний з безпекою (Maintenance of the ICH Guideline on Clinical Safety Data Management: Data Elements for Transmission of Individual Case Reports);
- Робоча група з впровадження ICH E2B — Питання і відповіді (R5) (3 березня 2005 р.) (ICH E2B Implementation Working Group — Questions & Answers (R5) (March 3, 2005).
З розвитком технічних стандартів, зазначені вище документи можуть переглядатися. У цьому контексті, завжди повинна братися до уваги остання версія цих документів.
Інформація, що стосується специфічних механізмів надання повідомлень представлена в VI.C.4.
VI.C. Діяльність зі збору інформації з безпеки
У розділі С цього модуля описано вимоги (згідно з Директивою 2001/83/ЄС [1], Регламенту (ЄС) № 726/2004) [5] та положень ПорядкуN [2]) до збору, обробки та надання повідомлень про випадки виникнення підозрюваних побічних реакцій (серйозних та несерйозних), пов’язаних із прийомом лікарських засобів, незалежно від умов їх застосування. Ці вимоги застосовуються до уповноваженого органу та/або до заявників (власників реєстраційних посвідчень). Розділ C необхідно розглядати в сукупності з визначеннями і загальними принципами, детально описаними в розділах VI.A і VI.B даного модуля і з вимогами, передбаченими в розділах IV, V і IX ІП 520/2012 [6].
VI.C.1. Правила надання звітів з клінічних випробувань і післяреєстраційних досліджень
Нормативно-правові вимоги щодо здійснення фармаконагляду, викладені в Директиві 2001/83/ЄС [1], Регламенті (ЄC) №726/2004 [5] та положеннях ПорядкуN [2], не поширюються на досліджувані і не досліджувані лікарські засоби29, що використовуються у клінічних випробуваннях, що проводяться відповідно до Директиви 2001/20/ЄС [15]30 та положень Порядку проведення клінічних випробуваньN [16].
Післяреєстраційні дослідження з безпеки чи ефективності, що проводяться заявником (власником реєстраційного посвідчення) за вимогою уповноважених органів (відповідно до Директиви 2001/83/ЄС [1], або Регламенту (ЄС) № 726/2004) [5], або положень ПорядкуN [2]) чи добровільно, можуть бути або клінічними випробуваннями або неінтервенційними післяреєстраційними дослідженнями (рис. VI.1). Тому надання звітів з моніторингу безпеки підпадає або під дію Директиви 2001/20/ЄС [15], положень Порядку проведення клінічних випробуваньN [16] у випадку клінічних випробувань, або під дію положень, визначених Директивою 2001/83/ЄС [1], Регламентом (ЄС) № 726/2004 [5] та положеннями ПорядкуN [2] у випадку неінтервенційних післяреєстраційних досліджень. Про підозрювані побічні реакції не слід повідомляти в рамках обох порядків, згідно з Директивою 2001/20/ЄC [15], положеннями Порядку проведення клінічних випробуваньN [16] та згідно з Регламентом (ЄC) № 726/2004 [5], Директивою 2001/83/ЄC [1] та положеннями ПорядкуN [2], так як це створює звіти в двох примірниках.
Детальна настанова з проведення післяреєстраційних досліджень з безпеки надана у модулі VIII ННПФ.
На рис. 1 проілюстровані різні види досліджень і клінічних випробувань, що можуть проводитися. Надання звітів з моніторингу безпеки з клінічних випробувань, позначених на рис. 1 сегментами A, B, C і D відбувається відповідно до вимог Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16]. Ця процедура у випадку неінтервенційних післяреєстраційних досліджень (сегменти E та F на рис VI.1.) відбувається відповідно до вимог Директиви 2001/83/ЄС [1], Регламенту (ЄС) № 726/2004 [5] та положень ПорядкуN [2].
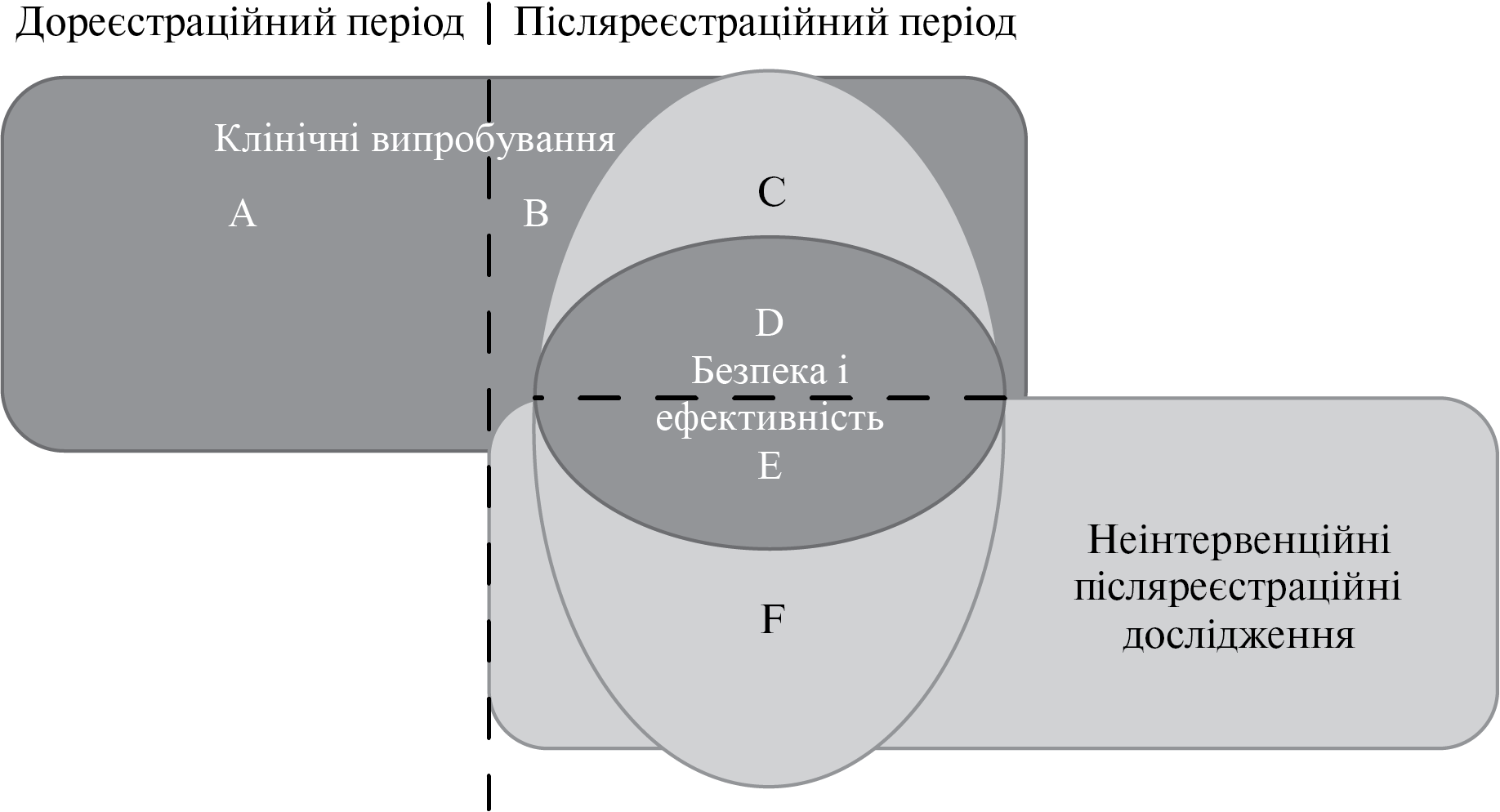
Рис. 1 — Ілюстративна схема різних видів досліджень та клінічних випробувань
|
Сегмент А: |
Клінічні випробування, що підпадають під дію Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16] і проводяться до реєстрації лікарського засобу. |
|
Сегмент В: |
Клінічні випробування, що підпадають під дію Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16] та проводяться у післяреєстраційний період, наприклад, для нових показань. |
|
Сегмент С: |
Післяреєстраційні клінічні випробування, що проводяться згідно із затвердженими показаннями відповідно до короткої характеристики лікарського засобу/інструкції для медичного застосування лікарського засобуN, однак підпадають під дію Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16], оскільки є інтервенційними. |
|
Сегмент D: |
Післяреєстраційні клінічні випробування з безпеки чи ефективності, що проводяться заявником (власником реєстраційного посвідчення) добровільно чи за вимогою уповноваженого органу згідно з Директивою 2001/83/ЄС [1] або Регламентом (ЄС) № 726/2004 [5], положеннями ПорядкуN [2], та які підпадають під дію Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16], оскільки є інтервенційними. |
|
Сегмент Е: |
Неінтервенційні післяреєстраційні дослідження з безпеки чи ефективності, що проводяться заявником (власником реєстраційного посвідчення) добровільно чи за вимогою уповноваженого органу згідно з Директивою 2001/83/ЄС [1] або Регламентом (ЄС) № 726/2004 [5] та положеннями ПорядкуN [2], що підпадають під дію цих же регуляторних актів. |
|
Сегмент F: |
Неінтервенційні післяреєстраційні дослідження, що проводяться відповідно до показань і умов застосування, зазначених у короткій характеристиці лікарського засобу/інструкції для медичного застосування лікарського засобуN, та підпадають під дію Директиви 2001/83/ЄС [1] або Регламенту (ЄС) № 726/2004 [5] та положень ПорядкуN [2]. |
VI.C.1.1. Правила надання звітів з клінічних випробувань
Підозрювана побічна реакція на досліджуваний лікарський засіб, що виникла під час клінічних випробувань та підпадає під дію Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16], розглядається спонсором тільки згідно з вимогами цієї Директиви та положень Порядку проведення клінічних випробуваньN [16]. Тому вона не є предметом даного модуля, навіть якщо такі клінічні випробування є післяреєстраційними дослідженнями з безпеки чи ефективності, що вимагаються відповідно до Директиви 2001/83/ЄС [1] або Регламенту (ЄС) № 726/2004 [5] та положень ПорядкуN [2] чи проводяться заявником (власником реєстраційного посвідчення) добровільно.
Якщо під час клінічних випробувань, що проводяться згідно з Директивою 2001/20/ЄС [15] та положеннями Порядку проведення клінічних випробуваньN [16], було виявлено проблеми з безпеки, що впливають на співвідношення користь/ризик зареєстрованого лікарського засобу, необхідно негайно повідомити про це уповноважений орган відповідно до процедур, детально викладених у VI.C.2.2.6. Це розповсюджується також і на випадки, коли проблеми з безпеки були виявлені у ході клінічних випробувань, що проводилися за межами ЄС та УкраїниN.
Дані з безпеки, отримані з клінічних досліджень, що повинні бути представлені у відповідних розділах РОЗБ зареєстрованого лікарського засобу, детально описані в модулі VII.
Якщо під час проведення клінічного випробування, що проводиться відповідно до Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16], виявляються випадки виникнення побічних реакцій, пов’язані тільки з прийомом недосліджуваного лікарського засобу (або іншого лікарського засобу, що не є частиною протоколу клінічного дослідження) і такі реакції не є результатом можливої взаємодії з досліджуваним лікарським засобом, вони не підпадають під вимоги термінового звітування Директиви 2001/20/ЄС [15] та положень Порядку проведення клінічних випробуваньN [16], що стосуються лише досліджуваних лікарських засобів. Досліднику чи спонсору рекомендується надсилати такі повідомлення до уповноваженого органу або до заявника (власника реєстраційного посвідчення) на підозрюваний лікарський засіб, але не до обох, щоб уникнути дублювання31. Якщо заявнику (власнику реєстраційного посвідчення) стає відомо про такий випадок, він повинен надати інформацію щодо нього уповноваженому органу відповідно до вимог, описаних у VI.C.3, VI.C.4 і VI.C.6. Щодо надання звітів в електронній формі необхідно дотримуватися рекомендацій, зазначених у VI.C.6.2.3.7.
VI.C.1.2. Правила звітування з безпеки при проведенні неінтервенційних післяреєстраційних досліджень, застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами (compassionate use), застосування незареєстрованого лікарського засобу поіменованими пацієнтами (named patient use)
Цей розділ стосується неінтервенційних післяреєстраційних досліджень, застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами (compassionate use) та їх застосування поіменованими пацієнтами (named patient use). Для цих організованих схем збору даних необхідно створити систему для обліку і документування повної і всебічної інформації про випадки побічних явищ, що необхідно збирати, як зазначено у VI.C.1.2.1 та VI.C.1.2.2. Ці побічні явища необхідно систематично оцінювати з метою визначення, чи вони ймовірно пов’язані з прийомом досліджуваного лікарського засобу (або лікарського засобу, що постачається) (див. ICH-E2D [25]). Для оцінки зв’язку між застосуванням досліджуваного лікарського засобу (або лікарського засобу, що постачається) та розвитком побічного явища слід використовувати метод оцінки причинно-наслідкового зв’язку (наприклад, система WHO-UMC для стандартизованої оцінки причинно-наслідкового зв’язку). Побічне явище буде класифікуватися як побічна реакція, якщо існує обґрунтована можливість існування вказаного вище причинно-наслідкового зв’язку. Тільки валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою (див. VI.B.2), щодо побічних реакцій, що першоджерело або отримувач інформації про випадок підозрює, що вони пов’язані з досліджуваним лікарським засобом (або лікарським засобом, що постачається), повинні надаватися відповідно до вимог, представлених у VI.C.3, VI.C.4 та VI.C.6.2.3.7. Інші повідомлення про побічні явища повинні узагальнюватися у складі будь-якого проміжного аналізу даних з безпеки і у заключному звіті з дослідження. У випадках, коли у повідомленнях підозрюється, що виникнення побічної реакції пов’язане із застосуванням недосліджуваного лікарського засобу (чи не з лікарськими засобами, що постачаються), такі повідомлення повинні класифікуватися і надаватися як спонтанні повідомлення про індивідуальний випадок, пов’язаний з безпекою. Першоджерело має відправляти ці повідомлення уповноваженому органу або заявнику (власнику реєстраційного посвідчення) на підозрюваний лікарський засіб, але не обом (з метою уникнення дублювання).
Якщо в рамках таких організованих схем збору даних заявнику (власнику реєстраційного посвідчення) стає відомо про явища, що впливають на співвідношення користь/ризик досліджуваного лікарського засобу (або лікарського засобу, що постачається) і/або на здоров’я населення, він повинен повідомити про це уповноважений орган відповідно до умов, зазначених у VI.C.2.2.6.
Подальші інструкції щодо післяреєстраційних досліджень, що проводяться заявниками (власниками реєстраційних посвідчень), надаються у VI.C.2.2.2.
Вимоги, зазначені у цьому модулі, не застосовуються до неінтервенційних післяреєстраційних досліджень, що проводяться такими організаціями, як наукові співтовариства, доброчинні медичні дослідницькі організації, державні дослідницькі організації. Ці організації повинні повідомляти про випадки виникнення підозрюваних побічних реакцій відповідно до вимог уповноваженого органу. Однак, якщо дослідження, що проводяться цими організаціями, ініціюється, управляється, фінансується заявником (власником реєстраційного посвідчення), або якщо заявник (власник реєстраційного посвідчення) впливає на дизайн таких досліджень (добровільно або внаслідок зобов’язань відповідно зі ст. 21а і 22а Директиви 2001/83/ЄС [1] і ст. 10 чи 10(а) Регламенту (ЄС) № 726/2004 [5] та положеннями ПорядкуN [2]), то до них застосовуються вимоги цього модуля32. У цьому випадку повинні бути укладені договірні угоди з метою чіткого визначення ролі і обов’язків для імплементації цих вимог (див. модуль I НПФ).
VI.C.1.2.1. Неінтервенційні дослідження
Неінтервенційні післяреєстраційні дослідження за дизайном розрізняються на ті, в яких збір первинних даних проводиться безпосередньо у споживачів та працівників з медичною або фармацевтичною освітою, та дослідження, у яких використовуються вторинні дані. Тому вимоги, що вказані нижче, залежать від дизайну дослідження33. За наявності сумнівів щодо того, які саме вимоги до звітування щодо побічних реакцій застосовуються, слід проконсультуватися з уповноваженим органом відповідної країни ЄС, УкраїніN. Слід також дотримуватися національного законодавства, що стосується комітетів з етики.
Неінтервенційні післяреєстраційні дослідження з первинним збором даних (primary data collection)
У ході дослідження слід збирати інформацію від працівників з медичною та/або фармацевтичною освітою та споживачів про всі побічні явища, крім тих, про які інформація не потрібна відповідно до протоколу. Щодо усіх побічних явищ, дані про які необхідні, слід збирати вичерпну та якісну інформацію, що дозволяє у встановлені строки звітувати про валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою (див. VI.C.3).
Із усіх зібраних побічних явищ випадки побічних реакцій, щодо яких першоджерело або отримувач інформації про випадок підозрює зв’язок з прийомом досліджуваного лікарського засобу, слід повідомляти відповідно до вимог, зазначених у VI.C.3 та VI.C.4. Валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, повинні розглядатися як повідомлення із джерел з організованим збором даних (solicited reports) (див. VI.C.2.2.2 та VI.C.6.2.3.7). Див. підсумкову табл. 1.
Усі летальні випадки слід розглядати як побічні явища, що необхідно збирати. У окремих випадках підозрювані побічні реакції з летальним наслідком можуть не підпадати під вимоги термінового надання у вигляді повідомлень про індивідуальний випадок, пов’язаний з безпекою, наприклад, тому що вони відносяться до результатів дослідження (кінцеві точки ефективності), або тому, що у включених у дослідження пацієнтів відмічають захворювання з високим ризиком смерті, або тому що летальні наслідки не є метою дослідження. У цих конкретних ситуаціях у протоколі дослідження повинно бути чітко описано пояснення, чому не буде повідомлятися про окремі побічні реакції з летальними наслідками.
Уся зібрана інформація про побічні явища повинна узагальнюватися при проведенні проміжних оцінок безпеки і у заключному звіті з дослідження.
Таблиця 1 — Неінтервенційні післяреєстраційні дослідження з первинним збором даних: вимоги, що стосуються збору даних про побічні явища і інформування про підозрювані побічні реакції
|
Побічні явища, для яких у протоколі не зазначено інакше та побічні явища, що мали летальний наслідок |
|
|
Вимоги щодо збору |
|
|
Вимоги щодо інформування про підозрювані побічні реакції |
|
|
Вимоги щодо інформування про побічні явища |
Узагальнення усіх зібраних побічних явищ під час проведення проміжного аналізу з безпеки і у заключному звіті з дослідження |
Стосовно побічних явищ, щодо яких у протоколі дослідження зазначено інше і не вимагається їх систематичний збір, у протоколі дослідження (або інших документах дослідження) слід поінформувати працівників з медичною та/або фармацевтичною освітою та споживачів про можливість надання повідомлень про побічні реакції на підозрюваний лікарський засіб (досліджуваний чи ні) (виникнення яких вони пов’язують із застосуванням лікарського засобу) заявнику (власнику реєстраційного посвідчення) або уповноваженому органу через національну систему надання спонтанних повідомлень. Отримувач повідомлень повинен класифікувати валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, та повідомляти про них як про спонтанні. Усі такі випадки повинні узагальнюватися у відповідних звітах з дослідження.
Неінтервенційні післяреєстраційні дослідження на основі вторинного використання даних
Дизайн таких досліджень характеризується вторинним/повторним використанням раніше зібраних даних, отриманих з інших причин від споживачів або працівників з медичною або фармацевтичною освітою. Приклади включають перегляд історій хвороб (включно з додатковими (follow up) даними від працівників з медичною або фармацевтичною освітою), аналіз електронних медичних записів, систематичні огляди або метааналіз.
Надання повідомлень щодо побічних реакцій у вигляді повідомлень про індивідуальний випадок, пов’язаний з безпекою, з неінтервенційних досліджень, що базуються на вторинному/повторному використанні даних, не вимагається. Повідомлення про побічні явища/реакції повинні враховуватися при проведенні будь-якого проміжного аналізу з безпеки та узагальнюватися у заключному звіті з дослідження, крім випадків, коли у протоколі дослідження зазначено інше.
VI.C.1.2.2. Застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами та застосування незареєстрованого лікарського засобу поіменованими пацієнтами
Настанова, представлена у даному модулі, використовується для лікарських засобів, що застосовуються у зв’язку з виключними обставинами, як зазначено у ст. 83 (2) Регламенту (ЄС) № 726/2004 [5] та положеннях Порядку проведення експертизиN [7]. Ця настанова може також використовуватися для лікарських засобів, що застосовуються персонально окремим пацієнтом, як зазначено у ст. 5(1) Директиви 2001/83/ЄС [1]. Слід дотримуватися також локальних вимог у разі необхідності.
Якщо організація34 або працівник з медичною або фармацевтичною освітою, що постачає або призначає лікарський засіб для застосування за вказаними вище програмами (див. VI.A.2.2), отримує інформацію про виникнення побічного явища, така інформація повинна оброблятися відповідно до вимог національного уповноваженого органу наступним чином:
- якщо при проведенні таких програм вимагається активний збір інформації щодо небажаних явищ, пов’язаних з прийомом лікарського засобу, що застосовувався у даних програмах, то необхідно надавати всі повідомлення про виникнення відповідних підозрюваних побічних реакцій, що були отримані від першоджерела та відправника. Такі повідомлення повинні розглядатися як повідомлення з джерел з організованою системою збору даних (див. VI.C.2.2.2 і VI.C.6.2.3.7);
- якщо при проведенні таких програм не вимагається активний збір даних про небажані явища, будь-яка інформація про шкідливі чи небажані реакції на лікарський засіб, що постачається, повинна розглядатися як спонтанне повідомлення про підозрювану побічну реакцію.
VI.C.2. Збір повідомлень
VI.C.2.1. Обов’язки країн ЄС та УкраїниN
Кожна країна ЄС та УкраїнаN повинні мати систему для збору та зберігання повідомлень про підозрювані побічні реакції, що виникають на її території, та про які повідомляють працівники з медичною або фармацевтичною освітою, споживачі та заявники (власники реєстраційних посвідчень)35 (ст. 101(1) і 107а (1) Директиви 2001/83/ЄС [1] та положення Порядку [2]N). З цією метою уповноважений орган повинен запровадити процедури для збору та зберігання всіх повідомлень про підозрювані побічні реакції (ст.15 (2) ІП 520/2012 [6] положення Порядку [2]N). Загальні принципи детально описані в VI.B, а у VI.C.3, VI.C.4 і VI.C.6 представлені процедури надання даних, що повинні застосовуватися до цих повідомлень. Дані з фармаконагляду та документи, що стосуються зареєстрованих лікарських засобів, повинні зберігатися до тих пір, доки лікарський засіб знаходиться на ринку країни і принаймні 10 років після завершення строку дії реєстраційного посвідчення. Ці документи повинні зберігатися протягом більш тривалого період, якщо цього вимагає національне законодавство (ст. 16 (2) ІП 520/2012 [6].
Кожна країна ЄС та УкраїнаN повинні вживати всіх належних заходів для заохочення працівників з медичною та фармацевтичною освітою і споживачів повідомляти про випадки виникнення підозрюваних побічних реакцій до уповноваженого органу. Крім того, уповноважений орган може накладати певні зобов’язання на працівників з медичною або фармацевтичною освітою. З цією метою уповноважений орган повинен створити крім веб-формату альтернативні прості системи звітності про підозрювані побічні реакції, доступні для працівників з медичною та фармацевтичною освітою та споживачів (ст. 102 Директиви 2001/83/ЄС [1] та положення Порядку [2]N). Інформація про різні способи повідомлення про виникнення вказаних вище побічних реакцій, повинна бути доступною для громадськості в тому числі і на офіційному вебсайті уповноваженого органу (ст.106 (e) Директиви 2001/83/ЄС [1] та положення Порядку [2]N). Для підвищення обізнаності про системи інформування, у разі необхідності, можна залучити організації, що представляють інтереси споживачів та працівників з медичною або фармацевтичною освітою (ст.102 Директиви 2001/83/ЄС [1], положення Порядку [2]N).
З метою збору інформації, необхідної для оцінки підозрюваних побічних реакцій включно з медичними помилками, ЕМА у співпраці з уповноваженими органами країн ЄС повинне розробити стандартні структуровані електронні форми для повідомлень про підозрювані побічні реакції для працівників з медичною та фармацевтичною освітою та споживачів (ст. 25 Регламенту (ЄC) №726/2004 [5]). У цьому контексті основні поля даних для звітності будуть надані EMA уповноваженим органам в країни ЄС для використання в своїх національних системах звітності залежно від обставин.
Якщо необхідно, то слід надсилати працівникам з медичною та фармацевтичною освітою і споживачам підтвердження отримання їх повідомлень, і надавати їм запитувану додаткову інформацію, якщо вона доступна.
В Україні, відповідно до положень Порядку, пацієнти та/або їх законні представники надають до Центру інформацію про побічну реакцію лікарського засобу та/або відсутність ефективності лікарського засобу за формою карти-повідомлення для надання пацієнтом, наведеною в додатку 2 до Порядку, або до Автоматизованої інформаційної системи з фармаконагляду (АІСФ) за посиланням aisf.dec.gov.ua.
Особи з медичною або фармацевтичною освітою відповідно до положень Порядку подають до Центру карти-повідомлення про будь-які побічні реакції лікарських засобів, відсутність ефективності лікарського засобу у визначені Порядком строки у паперовому та/або електронному вигляді за формою, наведеною у додатку 6 до Порядку, або до АІСФ за посиланням aisf.dec.gov.ua.
Заявник (власник реєстраційного посвідчення) відповідно до положень Порядку подають до Центру повідомлення з достовірною інформацією про побічні реакції лікарських засобів, відсутність ефективності лікарських засобів у визначені Порядком строки у будь-який спосіб у паперовому та/або в електронному вигляді до АІСФ за посиланням aisf.dec.gov.ua.
АІСФ передбачає можливість зворотного зв’язку з повідомником у разі виникнення необхідності в отриманні додаткової інформації стосовно надісланої до Центру інформації про випадок побічної реакції, що передбачає можливість формування електронного листа до повідомника та отримання від нього необхідної відповіді.
Для повідомлень, представлених заявником (власником реєстраційного посвідчення), уповноважений орган тієї країни ЄС та УкраїниN, на території якої виникла побічна реакція, може залучати заявників (власників реєстраційних посвідчень) до подальшого відстеження повідомлень про підозрювані побічні реакції (ст. 107а (2) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Кожна країна ЄС та УкраїнаN повинні забезпечити, щоб уповноважений орган, був поінформований про виникнення будь-яких підозрюваних побічних реакцій, щодо яких повідомлялося до будь-якого іншого органу, установи чи організації, відповідальної за безпеку пацієнтів у країні, і щоб валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, були надіслані до Європейської бази даних з фармаконагляду (далі — EudraVigilance), в Україні — до автоматизованої інформаційної системи з фармаконагляду (далі — АІСФ))N. У випадку якщо такі повідомлення направляються до інших органів влади, організацій та/або установ в країні, уповноважений органи повинен мати угоди про обмін даними, щоб усі повідомлення були своєчасно внесені до EudraVigilance, в Україні — АІСФ (ст. 107а(5) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Ця вимога стосується і повідомлень про підозрювані побічні реакції, що виникають у результаті помилок, пов’язаних із застосуванням лікарського засобу. Інформація щодо повідомлень про підозрювані побічні реакції, що виникають у результаті помилок, отримана уповноваженим органом, у тому числі з бази EudraVigilance відповідно до ст. 24(4) Регламенту ЄС № 726/2004 [5], в Україні — АІСФ, відповідно до положень Порядку [2]N повинна також бути доведена до відома інших органів, організацій та/або установ, відповідальних за безпеку пацієнтів в країні (cт. 107а(5) Директиви 2001/83/ЄС [1]).
У разі відсутності вагомих підстав, що зазвичай виявляються за допомогою заходів з фармаконагляду, національний уповноважений орган країни ЄС та УкраїниN не покладатимуть будь-яких додаткових зобов’язань на заявника (власника реєстраційного посвідчення) щодо надання інформації про підозрювані побічні реакції (cт. 107а(6) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
VI.C.2.2. Зобов’язання заявників (власників реєстраційних посвідчень)
Кожний заявник (власник реєстраційного посвідчення) повинен мати систему збору та зберігання усіх повідомлень про виникнення підозрюваних побічних реакцій, незалежно від того, чи були вони надані спонтанно працівниками з медичною або фармацевтичною освітою або споживачами, чи виявлені в ході післяреєстраційних досліджень (cт. 104(1) і 107(1) Директиви 2001/83/ЄС [1] та положення Порядку [2]N). Заявники (власники реєстраційних посвідчень) не мають права не приймати до уваги повідомлення про підозрювані побічні реакції, отримані від пацієнтів та працівників з медичною та фармацевтичною освітою в електронному форматі чи надані ними будь-яким іншим належним способом (cт. 107(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Усі такі повідомлення повинні бути доступними в одному місці (cт. 107(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Заявники (власники реєстраційних посвідчень) повинні впровадити механізми відслідковування та подальшого спостереження (follow up) повідомлень про виникнення побічних реакцій з дотриманням законодавства про захист інформації (cт. 12(1) ІП 520/2012 [6], положення Порядку [2]N). Дані з фармаконагляду та документи, що стосуються зареєстрованих лікарських засобів, слід зберігати до тих пір, доки лікарський засіб знаходиться на фармацевтичному ринку країни і принаймні 10 років після завершення строку дії реєстраційного посвідчення. Ці документи повинні зберігатися протягом більш тривалого періоду, якщо цього вимагає законодавство (ст. 12(2) ІП 520/2012 [6]).
Обов’язки щодо збору та зберігання повідомлень про підозрювані побічні реакції заявниками (власниками реєстраційних посвідчень) поширюються і на повідомлення, пов’язані із застосуванням лікарських засобів (див. VI.A.2.2), щодо яких не можна виключити їх належність до заявників (власників реєстраційних посвідчень), виходячи з одного з наступних критеріїв: назва лікарського засобу, назва діючої речовини, склад, номер серії чи спосіб застосування. Винятком із цього правила буде ситуація, якщо заявник (власник реєстраційного посвідчення) на підставі країни першоджерела або країни виникнення побічної реакції може продемонструвати, що підозрюваний лікарський засіб ніколи не постачався на ринок цієї країни, або що лікарський засіб не є препаратом для подорожуючих (наприклад протималярійні лікарські засоби).
Заявник (власник реєстраційного посвідчення) повинен забезпечити, щоб будь-яка інформація про виникнення підозрюваних побічних реакцій, пов’язаних з прийомом принаймні однієї з діючих речовин його лікарських засобів, зареєстрованих в ЄС, в УкраїніN, доводилася до його відома компаніями за межами ЄС, УкраїниN, що належать тій же материнській компанії (або групі компаній)36. Це розповсюджується і на заявника (власника реєстраційного посвідчення), що уклав комерційну угоду з компанією, розташованою за межами ЄС щодо одного із зареєстрованих в ЄС, УкраїніN лікарських засобів. Відлік часу для надання повідомлень (див. VI.B.7) , коли валідне повідомлення отримує одна з цих компаній, розташованих за межами ЄС та УкраїниN.
Крім вимог, описаних у цьому розділі, заявники (власники реєстраційних посвідчень) повинні дотримуватися загальних принципів, детально викладених у розділі VI.B, та процедур надання повідомлень, описаних у VI.C.3, VI.C.4 і VI.C.6, щодо усіх повідомлень про виникнення підозрюваних побічних реакцій.
VI.C.2.2.1. Спонтанні повідомлення
Заявники (власники реєстраційних посвідчень) повинні реєструвати всі повідомлення про випадки підозрюваних побічних реакцій, що виникли як на території ЄС та УкраїниN, так і за межами ЄС та УкраїниN, що спонтанно надають їм працівники з медичною або фармацевтичною освітою або споживачі, незалежно від того, чи отримані вони в електронному форматі чи за допомогою будь-яких інших належних засобів (ст. 107(1), 107(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Заявники (власники реєстраційних посвідчень) можуть використовувати свої вебсайти для розміщення на них форм для збору повідомлень про підозрювані побічні реакції або контактної інформації для прямої комунікації (див. VI.B.1.1.4).
VI.C.2.2.2. Повідомлення, що походять із джерел з організованою системою збору даних
Відповідно до ст. 107(1) Директиви 2001/83/ЄС [1], положень Порядку [2]N заявники (власники реєстраційних посвідчень) повинні реєструвати всі повідомлення про підозрювані побічні реакції, що спостерігалися у післяреєстраційних дослідженнях, що ініційовані, управляються чи фінансуються ними, в ЄС, УкраїніN та за їх межами37. Для неінтервенційних досліджень ця вимога стосується дизайну дослідження, заснованого на зборі первинних даних та настанові, наданій у VI.C.1.2.1. Загальні рекомендації щодо післяреєстраційних досліджень зазначені в VI.C.1.2. Для всіх повідомлень, що походять із джерел з організованою системою збору даних (див. VI.B.1.2) заявники (власники реєстраційних посвідчень) повинні мати механізми для збору повної всебічної інформації про випадки побічних явищ, виявлених при проведенні післяреєстраційних досліджень, та аналізу цієї інформації з тим, щоб забезпечити обґрунтовану оцінку випадків та надання валідних повідомлень про випадки виникнення побічних реакцій (див. VI.B.2), пов’язаних з прийомом досліджуваного лікарського засобу, що постачається. Заявники (власники реєстраційних посвідчень) повинні дуже старанно підійти до створення такої системи, до подальшого відстеження випадків (див. VI.B.3), а також до отримання точки зору першоджерела щодо причинно-наслідкового зв’язку між повідомленим побічним явищем та застосуванням досліджуваного лікарського засобу. Якщо думка першоджерела відсутня, заявник (власник реєстраційного посвідчення) повинен провести власну оцінку випадку на основі наявної інформації для того, щоб вирішити, чи є даний випадок валідним повідомленням про побічну реакцію, що підлягає поданню до уповноваженого органу. Під ці вимоги не підпадають дослідження, що базуються на вторинному/повторному використанні даних, для яких не вимагається надання повідомлень про побічні реакції (див. VI.C.1.2.1).
Дані з безпеки, що повинні бути надані у відповідних розділах РОЗБ лікарського засобу, детально описані в модулі VII.
VI.C.2.2.3. Повідомлення про випадки побічних реакцій, опубліковані в науковій та медичній літературі
Загальні принципи моніторингу випадків виникнення підозрюваних побічних реакцій, описаних у науковій та медичній літературі, надано у VI.B.1.1.2. Що стосується моніторингу наукової та медичної літератури, то вимоги, описані у цьому модулі, є частиною більш широкого процесу моніторингу літератури, який повинен проводитися для формування РОЗБ (див. модуль VII ННПФ).
Відповідно до ст. 107(3) Директиви 2001/83/ЄС [1] з метою уникнення надання дублюючих повідомлень про виникнення побічних реакцій, заявники (власники реєстраційних посвідчень) повинні повідомляти тільки про випадки, описані в науковій та медичній літературі, моніторинг якої не ведеться уповноваженим органом, щодо всіх лікарських засобів, які містять діючі речовини, не включені до переліку, який моніторить ЕМА відповідно до ст. 27 Регламенту (ЄС) №726/2004 [5]. До тих пір, поки переліки наукової і медичної літератури та діючих речовин не будуть опубліковані ЕМА, заявники (власники реєстраційних посвідчень) повинні проводити моніторинг усіх діючих речовин, на які вони мають реєстраційні посвідчення, шляхом перегляду широко використовуваних систематичних літературних оглядів та довідкових баз даних відповідно до принципів, викладених у VI.B.1.1.2 і в VI.Додаток 2.
В Україні заявник (власник реєстраційного посвідчення) повинен здійснювати моніторинг безпеки лікарських засобів, включаючи моніторинг літератури. Вимоги до надання повідомлень за даними літератури визначені Порядком [2]N.
Заявник (власник реєстраційного посвідчення) не повинен надавати повідомлення, якщо у розвитку випадків побічних реакцій, описаних у статті, підозрюється лікарський засіб з торговельною назвою іншої компанії. Якщо у статті не зазначено виробника/заявника лікарського засобу та/або його торговельної назви, приналежність лікарського засобу визначається за діючою речовиною, якщо не існують перелічені нижче причини, коли заявник (власник реєстраційного посвідчення) не повинен вважати, що публікація стосується його лікарського засобу, а саме:
- приналежність лікарського засобу заявникові (власникові реєстраційного посвідчення) може бути виключена на основі критеріїв, детально викладених у VI.C.2.2;
- повідомлення про випадки виникнення побічних реакцій, що містяться у науковій та медичній літературі та що сталися в країні, у якій компанія зареєструвала лікарський засіб, однак ніколи не продавала;
- випадки побічних реакцій, описані в науковій та медичній літературі, що засновані на аналізі інформації з бази даних уповноваженого органу. Однак у випадку повідомлень про побічні реакції, оцінка яких проводилася на основі бази даних уповноваженого органу іншої країни, вимоги щодо надання повідомлень залишаються;
- опубліковані статті, що містять аналіз даних із загальнодоступних баз даних, або узагальнені результати післяреєстраційних досліджень (див. VI.C.1.2). Цей вид публікацій описує побічні реакції, що виникли в групи пацієнтів при прийомі певного лікарського засобу з метою виявлення чи кількісної оцінки ризику, пов’язаного з цим лікарським засобом, а узагальнені дані пацієнтів часто представляються в таблицях або переліках. Основною метою цих досліджень є виявлення/оцінка специфічних ризиків, що можуть вплинути на співвідношення користь/ризик лікарського засобу.
Однак нові і значущі дані з безпеки, представлені в статтях, надання повідомлень з яких не вимагається, повинні обговорюватися у відповідних розділах РОЗБ (див. модуль VII ННПФ) і аналізуватися щодо їх загального впливу на співвідношення користь/ризик лікарського засобу. Крім того, заявник (власник реєстраційного посвідчення) повинен негайно доводити до відома уповноваженого органу будь-яку нову інформацію з безпеки, що може вплинути на співвідношення користь/ризик, відповідно до рекомендацій, що містяться в VI.C.2.2.6.
Детальна інструкція з моніторингу наукової та медичної літератури була розроблена відповідно до ст. 27(3) Регламенту (ЄС) №726/2004 [5] і включена у VI. Додаток 2.
Рекомендації щодо надання повідомлень про підозрювані побічні реакції, опублікованих в науковій і медичній літературі, у електронному форматі містяться у VI.C.6.2.3.2.
VI.C.2.2.4. Підозрювані побічні реакції, пов’язані із застосуванням забракованих або фальсифікованих лікарських засобів
Валідні повідомлення про випадки виникнення підозрюваних побічних реакцій, пов’язані з прийомом підозрюваного щодо фальсифікацію чи підтверджено фальсифікованого лікарського засобу або лікарського засобу з дефектом якості, повинні надаватися до уповноваженого органу. Серйозність такого повідомлення пов’язана із серйозністю підозрюваних побічних реакцій, щодо яких інформується, відповідно до визначень, що містяться в VI.A.2.4. Щодо надання електронної форми повідомлення слід виконувати рекомендації, зазначені у VI.C.6.2.3.5.
У таких випадках з метою охорони здоров’я населення може виникнути необхідність вжити невідкладних заходів, таких як відкликання з ринку однієї чи більше серій лікарських засобів, у яких виявлено дефект(и). Тому заявники (власники реєстраційних посвідчень) повинні мати систему, що забезпечує негайну оцінку і розслідування отриманих повідомлень про виникнення підозрюваних побічних реакцій, спричинені застосуванням фальсифікованого лікарського засобу або лікарського засобу з дефектом якості, і у разі підтвердження наявності дефекту, — негайно повідомити про це безпосереднього виробника лікарського засобу та уповноважений орган відповідно до положень, зазначених у ст. 13 Директиви 2003/94/ЄС [17] та положень Порядку [2]N.
VI.C.2.2.5. Підозрювана передача збудника інфекції з лікарським засобом
Будь-яка підозрювана передача збудника інфекції з лікарським засобом повинна розглядатися як серйозна побічна реакція і інформація про такі випадки повинна надаватися протягом 15 днів відповідно до вимог, викладених у VI.C.438. Якщо до повідомлень про індивідуальний випадок, пов’язаний з безпекою, не може бути застосований жоден з критеріїв серйозності, то його слід розглядати як важливу медичну подію (див. VI.A.2.4). Дані вимоги також поширюються і на вакцини. Щодо надання електронної форми повідомлення слід виконувати рекомендації, зазначені у VI.C.6.2.3.6.
Щодо лікарських засобів, отриманих з людської крові або плазми крові людини, нагляд за безпекою переливання крові (haemovigilance) може здійснюватися відповідно до Директиви 2002/98/ЄC [27]. Тому заявник (власник реєстраційного посвідчення) повинен мати систему інформування виробника, станцій переливання крові і уповноваженого органу про повідомленння про передачу збудника інфекції лікарським засобом, отриманим з людської крові або плазми крові людини.
Збудником інфекції вважається будь-який організм, вірус або інфекційна частка (наприклад, білок пріону, що передає трансмісивну губчасту енцефалопатію), патогенні або непатогенні.
Про передачу збудника інфекції можуть свідчити клінічні ознаки або симптоми, результати лабораторних даних, що вказують на наявність інфекції у пацієнта, який застосовував лікарський засіб.
Слід приділяти особливу увагу виявленню інфекцій/збудників інфекцій, що потенційно можуть передаватися через лікарський засіб, але при цьому слід враховувати також і ризик появи невідомих збудників.
У контексті оцінки підозрюваної передачі збудника інфекції лікарським засобом слід ретельно і обґрунтовано проводити диференціацію, наскільки це можливо, між причиною (наприклад ін’єкція/застосування), джерелом інфекції (наприклад контамінація) і клінічним станом пацієнта на момент інфікування (імуносупресивний стан/вакцинація, що передувала застосуванню лікарського засобу).
Підтвердження контамінації (включаючи невідповідну інактивацію/ослаблення вірулентності (аттенуацію) збудників інфекції як активних речовин) підозрюваного лікарського засобу посилює доказ передачі збудника інфекції та підозру на наявність дефекту якості, і в цьому випадку застосовуються процедури, описані у VI.C.2.2.4.
Лікарські засоби повинні відповідати рекомендаціям, викладеним у Примітці до Настанови з мінімізації ризику передачі збудників трансмісивної губчастої/спонгіформної енцефалопатії тварин через лікарські засоби для людини та тварин39. Для високотехнологічних (біотехнологічних) лікарських засобів залежно від обставин слід дотримуватися ст. 14 (5) Регламенту (ЄС) № 1394/2007 [21] та Настанови з безпеки та ефективності подальшого відстеження (Follow up) — управління ризиками високотехнологічних (біотехнологічних) лікарських засобів40.
VI.C.2.2.6. Нові дані щодо безпеки застосування лікарського засобу
На практиці можуть виникати ситуації, пов’язані з питаннями безпеки лікарського засобу, що безпосередньо не є повідомленнями про побічну реакцію, а, отже, не підпадають під критерії щодо надання повідомлень. Однак така інформація важлива, оскільки може призводити до зміни співвідношення користь/ризик лікарського засобу, наприклад:
- значущі результати з безпеки, отримані за результатами нещодавно завершених доклінічних досліджень;
- значущі питання з безпеки, за результатами неінтервенційного післяреєстраційного або в ході клінічного досліджень;
- сигнали про можливу тератогенну дію або значну загрозу для громадського здоров’я;
- дані про проблеми з безпеки, опубліковані в науковій і медичній літературі;
- проблеми з безпеки, що виникають у результаті виявлення сигналу чи оцінки нового повідомлення про індивідуальний випадок, пов’язаний з безпекою, про непередбачену побічну реакцію та можуть вплинути на співвідношення користь/ризик лікарського засобу;
- проблеми з безпеки, пов’язані із застосуванням лікарського засобу не у відповідності з короткою характеристикою/інструкцією для медичного застосування лікарського засобуN;
- проблеми з безпеки, пов’язані з помилковою/неправильною інформацією, зазначеною в короткій характеристиці/інструкції для медичного застосування лікарського засобуN;
- відкликання, неподовження, анулювання або призупинення дії реєстраційного посвідчення/дозволу на маркетинг на території інших країн з причин, пов’язаних з безпекою лікарського засобу;
- введення термінових обмежень на застосування на території інших країн з причин, пов’язаних з безпекою лікарського засобу;
- проблеми з безпеки, пов’язані з поставками сировини;
- порушення в поставках лікарського засобу.
Зазначені вище приклади ситуацій, що можуть вплинути на співвідношення користь/ризик лікарського засобу, не потрібно надавати у вигляді повідомлень про побічні реакції. Про ці проблеми з безпеки необхідно повідомляти уповноважений орган у письмовому вигляді негайно, як тільки про них стає відомо. У письмовому документі, що надається до цього органу, необхідно описати проблему з безпеки і запропоновані заходи щодо реєстраційного статусу підозрюваного лікарського засобу. Зазначені питання з безпеки підлягають відображенню та аналізу у відповідних розділах РОЗБ.
VI.C.2.2.7. Період між поданням заяви на реєстрацію та отриманням реєстраційного посвідчення
У період між подачею заяви на реєстрацію та отриманням реєстраційного посвідчення у заявника (власника реєстраційного посвідчення) може з’явитися інформація (щодо якості, доклінічних, клінічних даних), що може вплинути на співвідношення користь/ризик лікарського засобу41. Заявник (власник реєстраційного посвідчення) зобов’язаний негайно поінформувати про це уповноважений орган тієї країни, де він подав заяву на реєстрацію, та ЕМА VI.C.2.2.6, уповноважений орган в УкраїніN.
У ситуації, коли заява на реєстрацію лікарського засобу перебуває в процесі розгляду в ЄС, в УкраїніN, і даний лікарський засіб уже зареєстрований у третій країні, повідомлення про індивідуальний випадок, пов’язаний з безпекою, щодо препарату, що пройшов перевірку за межами ЄС, УкраїниN, повинні надаватися відповідно до вимог, передбачених у VI.C.3, VI.C.4 та VI.C.6.
VI.C.2.2.8. Період після призупинення, анулювання або відкликання реєстраційного посвідчення
Заявник (власник реєстраційного посвідчення) повинен продовжувати збирати будь-які дані про побічні реакції лікарського засобу під час тимчасового припинення дії реєстраційного посвідчення та надавати звіти відповідно до вимог, зазначених у VI.C.4.
У випадку відкликання чи анулювання реєстраційного посвідчення, колишньому заявнику (власнику реєстраційного посвідчення) рекомендується продовжувати збирати спонтанні повідомлення про підозрювані побічні реакції, наприклад, з метою проведення оцінки відтермінованих побічних реакцій, або отримання повідомлень про побічні реакції, наданих ретроспективно.
VI.C.2.2.9. Період під час надзвичайної ситуації у галузі охорони здоров’я
Надзвичайна ситуація в галузі охороні здоров’я — це загроза громадському здоров’ю, що належним чином визнана Всесвітньою організацією охорони здоров’я (ВООЗ) або Спільнотою у рамках Рішення № 2119/98/ЄС [13]. У випадку надзвичайної ситуації можуть бути змінені вимоги до надання РОЗБ. Такі зміни будуть розглядатися в кожному конкретному випадку, а інформація про це повинна бути оприлюднена на вебсайті уповноваженого органу.
VI.C.2.2.10. Повідомлення, що є результатом колективних судових позовів
Стимульовані повідомлення, отримані в результаті колективних судових позовів, слід вважати спонтанними. Валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, повинні описувати побічні реакції, виникнення яких пов’язане із застосуванням відповідного лікарського засобу. Їх слід надавати відповідно до термінів і вимог, описаних в VI.C.3, VI.C.4 і VI.C.6.
Якщо заявник (власник реєстраційного посвідчення) отримує велику кількість таких повідомлень, — він може попросити у виняткових випадках відтермінування періоду інформування про серйозні побічні реакції протягом 30 днів з дати їх отримання замість 15 днів. 90-денний термін надання повідомлень про випадки виникнення несерйозних побічних реакцій залишається незмінним. Можна буде подати заявку на таке звільнення тільки після того, як буде встановлена функціональність бази даних EudraVigilance, що зазначено у ст. 24(2) Регламенту (ЄС) № 726/2004 [5]. Запит повинен бути зроблений до департаменту фармаконагляду уповноваженого органа.
VI.C.2.2.11. Повідомлення з програм підтримки пацієнтів і програм дослідження ринку
Програма підтримки пацієнтів — це організована система збору даних, за допомогою якої заявник (власник реєстраційного посвідчення) отримує і збирає інформацію про застосування його лікарського засобу. Прикладами можуть служити післяреєстраційні програми підтримки пацієнтів і лікування захворювань, опитування пацієнтів і медичних працівників, збір інформації про комплаєнс пацієнтів або схеми компенсації/реімбурсації.
Програма досліджень ринку заявником (власником реєстраційного посвідчення) — це систематичний збір та аналіз даних про лікарський засіб, що стосуються маркетингу та розвитку бізнесу.
Повідомлення про виникнення побічних реакцій, зібрані з таких програм, інтерпретуються як повідомлення з джерел з організованою системою збору даних, для яких застосовуються вимоги, описані у VI.C.2.2.2.
VI.C.3. Строки надання повідомлень про побічні реакції
Загальні правила, що стосуються надання первинних та наступних (follow up) повідомлень щодо виникнення побічних реакцій, у тому числі й початку відліку часу, докладно описані в VI.B.7.
Відповідно до ст.ст. 107(3) та 107а(4) Директиви 2001/83/ЄС [1] та положень Порядку [2]N:
- повідомлення про індивідуальний випадок, пов’язаний з безпекою, щодо виникнення серйозних побічних реакцій необхідно надати до національного уповноваженого органу протягом 15 днів з дати отримання інформації;
- повідомлення про індивідуальний випадок, пов’язаний з безпекою щодо несерйозних побічних реакцій — протягом 90 днів з дати отримання інформації.
VI.C.4. Умови надання повідомлень про побічні реакції
Крім рекомендацій, викладених у VI.B.8, національні уповноважені органи та заявники (власники реєстраційних посвідчень) повинні використовувати формати, стандарти і термінологію для передачі в електронній формі даних про підозрювані побічні реакції, викладені в розділі IV ІП 520/2012 [6] та положень Порядку [2]N. Повідомлення про індивідуальні випадки, пов’язані з безпекою, повинні використовуватися для звітності до бази даних EudraVigilance, АІСФN, про підозрювану побічну реакцію на лікарський засіб, що сталася у одного пацієнта в конкретний момент часу (ст. 27 ІП 520/2012 [6], положення Порядку [2]N). Національні уповноважені органи та заявники (власники реєстраційних посвідчень) повинні також забезпечити, щоб усі надані в електронному вигляді повідомлення про індивідуальні випадки, пов’язані з безпекою, були належним чином були задокументовані й містили якомога більш повну інформацію (ст. 28 ІП 520/2012 [6], положення Порядку [2]N).
Строки надання повідомлень про випадки виникнення серйозних і несерйозних побічних реакцій вказані в VI.C.3. При електронному обміні повідомленнями між заявниками (власниками реєстраційних посвідчень), уповноваженим органом та ЕМА, в Україні — ЦентромN слід дотримуватися рекомендацій, описаних у VI.C.6.
Повідомлення про індивідуальний випадок, пов’язаний з безпекою, надані в електронному вигляді до бази даних уповноваженого органу, будуть відкриті для доступу зацікавлених сторін, таких як уповноважені органи, працівники з медичною та/або фармацевтичною освітою, споживачі, а також заявники (власники реєстраційних посвідчень) та науково-дослідні організації у відповідності до ст. 24 (2) Регламенту (ЄС) № 726/2004 [5] та політикою доступу до Європейської бази даних з фармаконагляду42. Дана політика визначає загальні принципи надання доступу до бази даних з безпеки уповноваженого органу відповідно до чинного законодавства та гарантує захист персональних даних. Як зазначено в політиці доступу вказаної вище бази даних, заявники (власники реєстраційних посвідчень) можуть завантажувати відібрані повідомлення про індивідуальний випадок, пов’язаний з безпекою у форматі ICH E2B [26] і відповідно до специфікації ICH M2 з метою сприяння їх діяльності з фармаконагляду.
VI.C.4.1. Проміжні положення
Відповідно до ст. 2 (4), ст. 2 (5) і ст. 2 (6) Директиви 2010/84/ЄС [3], положень Порядку [2]N до тих пір, поки ЕМА/уповноважений органN не зможе забезпечити всі функціональні можливості бази даних EudraVigilance/АІСФN, як зазначено у ст. 24 (2) Регламенту (ЄС) № 726/2004 [5], описані нижче вимоги до надання застосовуватимуться до валідних повідомлень про індивідуальний випадок, пов’язаний з безпекою, що надходять без запиту та тих, що походять із джерел з організованою системою збору даних, що надаються працівниками з медичною та фармацевтичною освітою та іншими особами. Це не залежить від умов використання підозрюваного лікарського засобу і очікуваності побічної реакції.
а. Повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою
- Заявники (власники реєстраційних посвідчень) реєстраційних посвідчень повинні надавати всі повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою, щодо всіх побічних реакцій, які виникають в ЄС та УкраїніN, до уповноваженого органу тієї країни, на території якої вони відбулися.
- Заявники (власники реєстраційних посвідчень) повинні надавати до бази даних EudraVigilance всі повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою, щодо побічних реакцій, які виникли поза межами ЄС, в тому числі повідомлення про індивідуальний випадок, пов’язаний з безпекою, отримані від уповноважених органів. У разі відповідної вимоги країни ЄС ці звіти надаються також до уповноважених органів у країні ЄС, в якій зареєстровано лікарський засіб. В Україні заявник (власник реєстраційного посвідчення) повинен надавати у будь-який спосіб достовірну інформацію про всі випадки серйозних непередбачених побічних реакцій на лікарські засоби, вакцини, туберкулін, що призвели до смерті або становили загрозу для життя пацієнта, про всі підозрювані випадки передавання інфекції лікарським засобом, що були зафіксовані на території іншої країни та мають медичне підтвердження і про які йому стало відомо, за наявності причинно-наслідкового зв’язку між ними та застосуванням лікарського засобу, вакцини, туберкуліну — не пізніше 15 календарних днів з дня отримання такої інформаціїN.
- Уповноважені органи країни ЄС повинні забезпечити, щоб усі повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою, надані до них, щодо побічних реакцій, які виникли на їх території, включаючи повідомлення про індивідуальний випадок, пов’язаний з безпекою, що були отримані від заявників (власників реєстраційних посвідчень), надавалися до бази даних EudraVigilance. Національні уповноважені органи повинні також надавати заявникам (власникам реєстраційних посвідчень) підозрюваних лікарських засобів усі повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою щодо цих лікарських засобів, надані безпосередньо їм.
б. Повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою
У разі відповідної вимоги заявники (власники реєстраційних посвідчень) повинні надавати всі повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою, до уповноваженого органу тієї країни, на території якої виникли підозрювані побічні реакції.
Огляд вимог щодо надання повідомлень про серйозний і несерйозний індивідуальний випадок, пов’язаний з безпекою, протягом проміжного періоду, що застосовуються до заявників (власників реєстраційних посвідчень) або уповноважених органів, представлено в VI.Додаток 3.1, разом з детальною схемою робочого процесу.
Вимоги країн ЄС щодо надання повідомлень про серйозний індивідуальний випадок, пов’язаний з безпекою, з побічними реакціями, які виникли за межами ЄС, а також повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою, щодо побічних реакцій, які мали місце в ЄС, також включені в цей додаток.
VI.C.4.2. Заключні положення
Після того, як будуть забезпечені всі функціональні можливості EudraVigilance, як зазначено у ст. 24 (2) Регламенту (ЄС) № 726/2004 [5], перелічені нижче вимоги, детально викладені в ст. ст. 107(3) і 107А(4) Директиви 2001/83/ЄС [1], будуть діяти протягом 6 місяців після оголошення ЕМА, для валідних повідомлень про індивідуальні випадки, пов’язані з безпекою, що надходять без запиту, та тих, що походять із джерел з організованою системою збору даних, що надаються працівниками з медичною та/або фармацевтичною освітою та іншими особами. Це не залежить від умов застосування підозрюваного лікарського засобу і очікуваності побічної реакції.
а. Повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою
Заявники (власники реєстраційних посвідчень) повинні надавати всі повідомлення про індивідуальний випадок, пов’язаний з безпекою, щодо побічних реакцій, що виникли в межах чи поза межами ЄС, у тому числі повідомлення про індивідуальний випадок, пов’язаний з безпекою, отримані від уповноважених органів поза межами ЄС, до бази даних EudraVigilance.
Уповноважені органи у країні ЄС повинні надавати до бази даних EudraVigilance всі повідомлення про серйозний індивідуальний випадок, пов’язаний з безпекою, про які безпосередньо було повідомлено, щодо побічних реакцій, які виникли на їх території.
б. Повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою
Заявники (власники реєстраційних посвідчень) повинні надавати всі повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою щодо побічних реакцій, які виникли на території ЄС, до бази даних EudraVigilance.
Уповноважені органи країни ЄС повинні надавати всі повідомлення про несерйозний індивідуальний випадок, пов’язаний з безпекою щодо побічних реакцій, які виникли на її території, до бази даних EudraVigilance.
Огляд вимог щодо надання повідомлень про серйозний і несерйозний індивідуальний випадок, пов’язаний з безпекою, що застосовуються до заявників (власників реєстраційних посвідчень) або уповноважених органів країн ЄС після впровадження фінальних заходів представлено в VI. Додаток 3.2 разом з детальною схемою робочого процесу.
Відповідно до вимог, які детально викладено у ст. 24(4) Регламенту (ЄС) № 726/2004 [5] для фінальних заходів, повідомлення про індивідуальний випадок, пов’язаний з безпекою, надані до бази даних EudraVigilance заявниками (власниками реєстраційних посвідчень), повинні автоматично передаватися після отримання до уповноваженого органу країни ЄС, в якій виникла побічна реакція. Детальна схема робочого процесу включена до VI. Додаток 3.3.
VI.C.5. Співпраця із Всесвітньою організацією охорони здоров’я та Європейським центром моніторингу наркотиків і наркозалежності
EMA має надати в розпорядження ВООЗ (на практиці — Центру співпраці ВООЗ з міжнародного моніторингу лікарських засобів) усі повідомлення про підозрювані побічні реакції, що виникли на території ЄС (ст. 28с (1) Регламенту (ЄС) № 726/2004 [5]). Це відбуватиметься на щотижневій основі, після їх передачі в базу даних EudraVigilance уповноваженими органами держав-членів або заявниками (власниками реєстраційних посвідчень). Така процедура прийде на зміну потребі країнам — учасницям програми ВООЗ з міжнародного моніторингу лікарських засобів безпосередньо повідомляти ВООЗ про підозрювані побічні реакції, що відбуваються на їх території. Це буде реалізовано після того, як функції бази даних EudraVigilance, зазначені у ст. 24(2) Регламенту (ЄС) № 726/2004 [5] встановляться.
Детальна Схема робочого процесу для передачі повідомлень з бази даних EudraVigilance до Центру співпраці ВООЗ з міжнародного моніторингу лікарських засобів представлена у VI. Додаток 4.
ЕМА та Європейський центр моніторингу наркотиків і наркозалежності також повинні обмінюватися інформацією щодо зловживання лікарськими засобами, у тому числі інформацією, пов’язаною з розповсюдженням незаконних наркотичних речовин (ст. 28с(2) Регламенту (ЄC) №726/2004 [5]).
В Україні уповноважений орган повинен надавати в розпорядження ВООЗ (Центр співпраці ВООЗ з міжнародного моніторингу лікарських засобів) повідомлення про підозрювані побічні реакції, що виникли на території України (положення Порядку [2]) за використання АІСФ. АІСФ передбачає можливість надання інформації про побічні реакції до бази даних VigiFlow шляхом вибору повідомлень у базі АІСФ та переводу його у формат XML-файлу. Після зберігання повідомлення у XML файлі інформація надходить до ВООЗ. На даний час система передачі інформації таким шляхом модернізується та вдосконалюється з урахуванням зауважень, отриманих від ВООЗN.
VI.C.6. Електронний обмін інформацією з безпеки
Цей розділ висуває на перший план вимоги, зазначені у ст. 24 (1) і 24 (3) Регламенту (ЄС) № 726/2004 [5], положень Порядку [2]N, для встановлення та підтримки бази даних EudraVigilance/АІСФN, за допомогою якої відбувається збір та обмін даними з фармаконагляду в електронному вигляді між національними уповноваженими органами, заявниками (власниками реєстраційних посвідчень) та ЕМА.
Інформація, надана у даному розділі, стосується електронного обміну повідомленнями про індивідуальний випадок, пов’язаний з безпекою в ЄС/в УкраїніN між усіма зацікавленими сторонами та надання до ЕМА/уповноваженого органуN інформації про лікарські засоби в електронному вигляді.
VI.C.6.1. Діючі настанови, визначення, міжнародні формати, стандарти і термінологія
Для класифікації, пошуку, надання, оцінки та аналізу співвідношення користь/ризик, електронного обміну та передачі даних з фармаконагляду і даних щодо лікарських засобів уповноважений орган та заявники (власники реєстраційних посвідчень) повинні дотримуватися нормативно-правових вимог, передбачених у розділі IV ІП 520/2012 [6] та Порядку [2]N.
Крім того, повинні застосовуватися наступні настанови/правилаN:
- Примітки до настанови — EudraVigilance Human — Обробка повідомлень з безпеки та повідомлень про індивідуальний випадок, пов’язаний з безпекою (EMA/H/20665/04/Final Rev. 2) (Бізнес-правила EudraVigilance);
- Примітка до настанови щодо обміну електронними даними (EDI) повідомлень про індивідуальний випадок, пов’язаний з безпекою (ICSR), і звітів щодо лікарських засобів (MPRS) у фармаконагляді у дореєстраційний та післяреєстраційний періоди в Європейському економічному просторі (ЄЕП) (EMEA/115735/2004);
- Настанови ICH, описані в VI.B.8;
- Настанова ICH-M5 «Контрольований словник способів застосування»;
- Правила користування АІСФN.
Завжди необхідно керуватися останньою версією цих документів.
В Україні при наданні повідомлень про індивідуальний випадок, пов’язаний з безпекою, за використання АІСФ, слід користуватися Інструкцією для повідомлень заявниками (власниками реєстраційних посвідчень)N.
VI.C.6.2. Надання повідомлень про індивідуальний випадок, пов’язаний з безпекою, в електронному форматі
Подання валідних повідомлень про індивідуальний випадок, пов’язаний з безпекою, в електронному вигляді національними уповноваженими органами та заявниками (власниками реєстраційних посвідчень) є обов’язковим для всіх лікарських засобів (ст.ст. 107 (3) та 107a (4) Директиви 2001/83/ЕС [1], та положень Порядку [2]N. Невиконання цієї вимоги є недотриманням законодавства ЄС, УкраїниN. Відповідальність у випадку помилки зв’язку (у тому числі дотримання вимог до форми повідомлень) детально описано в розділі IV Примітки до настанови щодо обміну електронними даними (EDI), повідомленнями про індивідуальний випадок, пов’язаний з безпекою, і звітів щодо лікарських засобів (MPRS) у фармаконагляді у дореєстраційний та післяреєстраційний періоди в ЄЕП (EMEA/115735/2004), в Інструкції для повідомлень заявниками (власниками реєстраційних посвідчень)N.
З метою сприяння дотриманню вимог щодо надання звітів про випадки виникнення побічних реакцій в електронному вигляді уповноважений орган надасть технічні засоби (EVWEB) зацікавленим у електронному обміні даними партнерам, у тому числі малим і середнім підприємствам. Більш детальна інформація буде доступна на вебсайті бази даних EudraVigilance43. В Україні інформація для повідомників про індивідуальний випадок, пов’язаний з безпекою, за використання АІСФ, надана в Інструкції для повідомлення пацієнтом, Інструкції для повідомлення медичними працівниками та заявникамиN.
VI.C.6.2.1. Модулі Європейської бази даних з фармаконагляду (EudraVigilance)/Бази даних з фармаконагляду в Україні — автоматизованої інформаційної системи з фармаконагляду
Два модулі бази даних EudraVigilance призначені для збору повідомлень про виникнення підозрюваних побічних реакцій, що пов’язані із застосуванням лікарського засобу людиною, відповідно до законодавства ЄС:
- Модуль післяреєстраційних даних EudraVigilance (EVPM), впроваджений на основі вимог, описаних у Регламенті (ЄС) № 726/2004 [5] та Директиві 2001/83/ЄС [1];
- Модуль клінічних випробувань EudraVigilance (EVCTM), впроваджений на основі вимог, описаних у Директиві 2001/20/ЄС [15].
База даних з фармаконагляду в Україні — АІСФ — призначена для збору повідомлень про виникнення підозрюваних побічних реакцій, що пов’язані із застосуванням лікарського засобу людиною, відповідно до положень Порядку [2]N.
VI.C.6.2.1.1. Збір даних щодо побічних реакцій у Модулі післяреєстраційних даних Європейської бази даних з фармаконагляду)/Бази даних з фармаконагляду в Україні — автоматизованої інформаційної системи з фармаконаглядуN
Повідомлення про виникнення побічних реакцій, що накопичуються у Модулі післяреєстраційних даних EudraVigilance (EVPM) відносяться до звітів, що надходять без запиту та тих, що походять із джерел з організованою системою збору даних, що не підпадають під дію Директиви клінічних випробувань 2001/20/ЄC [15] (див. VI.C.1). В Україні повідомлення про виникнення побічних реакцій, що накопичуються у АІСФ, відносяться до звітів, що надходять без запиту та тих, що походять із джерел з організованою системою збору даних, що не підпадають під дію Порядку проведення клінічних випробувань [16]N.
Повідомлення про індивідуальний випадок, пов’язаний з безпекою, необхідно надавати із поміткою «EVHUMAN» в полі даних «Message receiver identifier» (Ідентифікатор отримувача повідомлення) (ICH M2 M.1.6).
Залежно від типу повідомлення про індивідуальний випадок, пов’язаний з безпекою, класифікуються наступним чином, відповідно до правил бази даних44:
- Поле даних «Type of report» (Тип звіту) (ICH-E2B(R2) [26] A.1.4):
- спонтанне повідомлення;
- інше;
- невідомо;
- звіт з дослідження.
- Крім того, у разі якщо поле даних ICH-Е2В(R2) [26] А.1.4 містить значення «повідомлення з дослідження», поле даних «Тип дослідження, у якому спостерігалася реакція/явище» (ICH- E2B(R2) [26] A.2.3.3) повинен бути заповнений такими даними:
- індивідуальне застосування пацієнтом, наприклад, застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами (compassionate use), застосування лікарського засобу пацієнтами на поіменній основі (named patient use);
- інші дослідження, наприклад фармакоепідеміологічне, фармакоекономічне дослідження, інтенсивний моніторинг, післяреєстраційне спостереження.
В Україні в залежності від типу, повідомлення про індивідуальний випадок, пов’язаний з безпекою, класифікуються наступним чином, відповідно до Інструкції для повідомлення медичними працівниками та заявниками. АІСФN:
- Поле даних «Вид повідомлення»:
- спонтанне;
- з дослідження;
- інше;
- невідомо.
VI.C.6.2.1.2. Збір даних про побічні реакції у Модулі клінічних випробувань Європейської бази даних з фармаконагляду/Порядку проведення клінічних випробуваньN.
Тільки випадки виникнення підозрюваних непередбачених серйозних побічних реакцій (SUSARs), пов’язані з прийом досліджуваних лікарських засобів, що вивчаються в ході клінічних випробувань, які підпадають під дію Директиви 2001/20/ЄC [15] (див. VI.C.1) слід надавати до Модуля клінічних випробувань EudraVigilance (EVCTM). Вимоги, встановлені в розділі II EudraLex, том 10 Правил регулювання лікарських засобів в Європейському Союзі, повинні бути застосовані. Повідомлення про індивідуальний випадок, пов’язаний з безпекою повинні надаватися зі значенням «EVCTMPROD» в полі даних «Message receiver identifier» (Ідентифікатор одержувача повідомлення) (ICH M2 M.1.6) і повинні бути класифіковані наступним чином, відповідно до правил EudraVigilance45:
- Поле даних «Тип звіту» (ICH-E2B(R2) [26]A.1.4):
- звіт з дослідження;
- поле даних «Тип дослідження, у якому спостерігалася реакція/явище» (ICHE2B(R2) [26] A.2.3.3):
- клінічні випробування.
В Україні інформація про випадки виникнення підозрюваних непередбачених серйозних побічних реакцій (SUSARs), пов’язані з прийомом досліджуваних лікарських засобів, що вивчаються в ході клінічних випробувань, надається відповідно до положень Порядку проведення клінічних випробувань [16]N.
VI.C.6.2.2. Підготовка повідомлень про індивідуальний випадок, пов’язаний з безпекою
VI.C.6.2.2.1. Загальні принципи
Усі валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, якими обмінюються в електронному вигляді всі зацікавлені сторони, повинні відповідати вимогам законодавства та настановам, викладеним в ІП 520/2012 [6] і в VI.C.6.1, зазначеним в VI.C.6.1, і, зокрема:
- вимогам, передбаченим у розділах IV та V ІП 520/2012 [6];
- останній версії настанови ICH для користувачів MedDRA;
- бізнес-правилам для електронної передачі повідомлень про побічні реакції, детально описаним у Примітці до настанови — EudraVigilance Human — обробка повідомлень з безпеки та повідомлень про індивідуальні випадки, пов’язані з безпекою (EMA/H/20665/04/Final Rev. 2).
В Україні всі валідні повідомлення про індивідуальний випадок, пов’язаний з безпекою, якими обмінюються в електронному вигляді всі зацікавлені сторони, повинні відповідати вимогам законодавства та настановам, викладеним відповідно до положень Порядку [2]N.
Слід визнати, що часто буває важко отримати всі подробиці про конкретний випадок. Уся доступна відправнику інформація (медичні та адміністративні дані), що стосується валідних повідомлень про індивідуальні випадки, пов’язані з безпекою, повинна бути представлена у структурованому вигляді у відповідних елементах даних ICH-E2B(R2) [26] (що повинні бути повторені в міру необхідності, коли множинна інформація доступна) і в описовій частині (див. VI.C.6.2.2.4). Це стосується всіх типів повідомлень про індивідуальні випадки, пов’язані з безпекою, як первинних та з подальшого відстеження (follow up), так і повідомлень, позначених як анульовані46.
У ситуації, коли очевидно, що відправник повідомлень про індивідуальні випадки, пов’язані з безпекою, передав неповну інформацію про випадок, отримувач повідомлення може звернутися до відправника із запитом на повторну передачу повідомлення протягом 24 годин уже з усією інформацією про випадок в електронному вигляді відповідно до вимог. Необхідність такого запиту слід розглядати у світлі якісного виявлення сигналів та оцінки діяльності, якщо отримувачу повідомлення для проведення медичної оцінки важливо мати всю доступну інформацію про випадок (див. VI.C.6.2.4).
Якщо підозрювані побічні реакції, щодо яких повідомляється в одному повідомленні про індивідуальний випадок, пов’язаний з безпекою, впливають на співвідношення користь/ризик лікарського засобу, то його слід розглядати як нову проблему з безпеки лікарського засобу (див. VI.C.2.2.6), про яку необхідно негайно повідомити уповноважений орган тієї країни, де зареєстрований лікарський засіб, та ЕМА у письмовій формі. Це додаткова вимога до детально викладених у VI.C.4. вимог щодо надання звітів. У полі даних «Коментар відправника» (ICH-E2B (R2) [26] В.5.4) слід надати стислий опис проблеми з безпеки та пропонованих заходів.
В Україні вся доступна відправнику інформація (медичні та адміністративні дані), що стосується валідних повідомлень про індивідуальні випадки, пов’язані з безпекою, повинна бути представлена у структурованому вигляді відповідно до Інструкції для повідомлення медичними працівниками та заявниками. АІСФN. Це стосується вкладок ІІІ «Дані досліджень» й ІV «Інша медично важлива інформація» та поле «Коментар» на будь-якій з вкладок.
VI.C.6.2.2.2. Інформація щодо підозрюваних, взаємодіючих та супутніх лікарських засобів
Підозрювані, взаємодіючі та/або супутні діючі речовини/торговельні назви лікарських засобів, щодо яких надається інформація, повинні бути описані відповідно до ст.28 (3) (g) до (i) ІП 520/2012 [6], настанови ICH-E2B (R2) [26] і Бізнес-правил бази даних EudraVigilance.
Віднесення лікарських засобів до підозрюваних, взаємодіючих або супутніх базується на інформації, наданій першоджерелом повідомлення.
Для комбінованих лікарських засобів, що містять більше ніж одну діючу речовину, кожна з них повинна бути відображена окремо в полі даних «Діюча(і) речовина(и)» (ICH-Е2В(R2) [26] B.4.k.2.2), і це поле необхідно повторити стільки разів, скільки діючих речовин міститься в комбінованому лікарському засобі.
Якщо першоджерело вказує лише торговельну/запатентовану назву підозрюваного або взаємодіючого лікарського засобу без зазначення діючої речовини (речовин) і якщо такий лікарський засіб може бути одним з генериків, що мають різний склад залежно від країни, де цей лікарський засіб зареєстрований, то поля повідомлення про індивідуальний випадок, пов’язаний з безпекою, повинні заповнюватися таким чином:
- поле даних «Торговельна назва лікарського засобу» (ICH-E2B(R2) [26] B.4.k.2.1) має містити торговельну/запатентовану назву лікарського засобу, вказану першоджерелом;
- поле даних «Діюча(і) речовина(и)» (ICH-E2B(R2) [26] B.4.k.2.2) повинно містити назви діючих речовин, що відповідають складу зареєстрованого лікарського засобу тієї країні, де виникла/ло побічна дія/явище.
Однак, якщо відома наступна інформація:
- «Країна, в якій застосовувався лікарський засіб» (поле даних ICH E2B(R2) [26] B.4.k.2.3);
- «Номер реєстраційного посвідчення» (поле даних ICH-E2B(R2) [26] B.4.k.4.1);
- «Країна заявника (власника реєстраційного посвідчення) (поле даних ICHE2B(R2) [26] B.4.k.4.2), та/або
- «Номер серії» (поле даних ICH-E2B(R2) [26] B.4.k.3),
то у полі «Діюча(і) речовина(и)» (ICH-E2B(R2) [26] B.4.k.2.2) необхідно вказати відповідний склад діючої речовини (речовин) зареєстрованого лікарського засобу.
У випадку, коли першоджерело повідомляє тільки про торговельну назву підозрюваного або взаємодіючого лікарського засобу, не вказуючи форму випуску лікарського засобу, а він представлений на ринку у більше ніж одній формі випуску та має різний склад у країні, повідомлення про індивідуальний випадок, пов’язаний з безпекою, необхідно заповнювати таким чином:
- поле даних «Торговельна назва лікарського засобу» (ICH-E2B(R2) [26] B.4.k.2.1) повинно містити назву лікарського засобу, повідомлену першоджерелом;
- поле даних «Діюча(і) речовина(и)» (ICH-E2B(R2) [26] B.4.k.2.2) повинно містити назви діючих речовин, що є спільними для всіх зареєстрованих в країні форм випуску.
Якщо лікарський засіб не можна визначити за діючою речовиною або торговельною назвою, наприклад, коли першоджерело зазначило тільки терапевтичний клас або інші відомості про терапію, для яких немає окремого поля, то цю інформацію потрібно відобразити тільки в полі «Опис випадку» (ICH-E2B(R2) [26] B.5.1). Поля даних «Торговельна назва лікарського засобу» (ICH-E2B(R2) [26] B.4.k.2.1) і «Діюча(і) речовина(и)» (ICH-E2B(R2) [26] B.4.k.2.2) не повинні заповнюватися. Зазначена умова застосовується також у випадку, якщо повідомляється про підозрювану взаємодію з їжею (наприклад грейпфрутовий сік).
Якщо повідомляється про випадок виникнення побічної реакції та вказується тільки фармакотерапевтична група, таке повідомлення вважається неповним і не відповідає вимогам надання звітів щодо побічних реакцій (див. VI.B.2). Слід відстежувати такий випадок для отримання відсутньої інформації про підозрюваний лікарський засіб (див. VI.B.3).
Віднесення взаємодії, про яку повідомляється, до взаємодій між лікарськими засобами (взаємодія лікарський засіб/лікарський засіб (у тому числі між біологічними лікарськими засобами), лікарський засіб/їжа, лікарський засіб/медичний виріб і лікарський засіб/алкоголь) необхідно проводити в розділі «Реакції/явища) (ICH-E2B(R2) [26] B.2) згідно з останньою версією затвердженої ICH Інструкції для користувачів MedDRA — MedDRA вибір терміну: основні пункти розгляду документа (ICH-Endorsed Guide for MedDRA Users — MedDRA Term Selection: Points to Consider Documen). Відомості про діючі речовини/торговельні назви лікарських засобів у повідомленнях про взаємодію лікарський засіб/лікарський засіб, повинні бути надані в розділі «Інформація про лікарський засіб» (ICH-E2B(R2) [26] В.4), зокрема, у полі даних «Характеристика ролі лікарського засобу» (ICH-E2B(R2) [26] B.4.k.1) повинна бути зазначена взаємодія.
Якщо першоджерело підозрює у виникненні побічної реакції один із компонентів (наприклад наповнювач чи допоміжну речовину) лікарського засобу, то у розділі «Інформація про лікарський засіб» (ICH-E2B(R2) [26] В.4), крім інформації про підозрюваний лікарський засіб, необхідно окремо зазначити й підозрюваний компонент. Цей факт також слід зазначити в полі «Опис випадку» (елемент даних ICH-E2B(R2) [26] B.5.1). За наявності результатів досліджень (позитивних або негативних) щодо впливу підозрюваного компоненту, їх потрібно надати в розділі «Результати випробувань та процедур, пов’язані з дослідженням пацієнта» (ICH E2B(R2) [26] В.3).
В Україні інформація щодо підозрюваних, взаємодіючих та/або супутніх діючих речовин/торговельних назв лікарських засобів надається відповідно до положень Порядку [2] через систему АІСФ відповідно до Інструкції для повідомлення медичними працівниками та заявниками. А саме: торговельну(і) назву(и) лікарського(их) засобу(ів), що підозрюється(ються) у спричиненні побічної реакції та/або відсутності ефективності (у базі АІСФ — VІ вкладка Лікарські засоби); підозрювані лікарські засоби, у тому числі супутні лікарські засоби; супутні лікарські засоби, вакцини, туберкуліни або, якщо назва невідома, активний(і) фармацевтичний(і) інгредієнт(и) і будь-які інші дані, що дають змогу ідентифікувати лікарський(і) засіб(оби), вакцину(и), туберкуліни; підозрюваний лікарський засіб*, ручне введення або вибір лікарського засобу із реєстру препаратів, у тому числі найменування заявника (власника реєстраційного посвідчення); виробник, назва, заявник (власник реєстраційного посвідчення), назва, номер реєстраційного посвідчення; номер реєстраційного посвідчення, країну, де зареєстрований лікарський засіб; країна виробника, країна заявника (власника реєстраційного посвідчення), вакцина, туберкулін, форму випуску; форма випуску та спосіб застосування; спосіб введення*, показання для застосування у цьому випадку; показання для призначення, показання для призначення (за можливості — за МКХ-10)*, застосовані дози; разова доза*, кратність приймання*, дату початку застосування; початок терапії* і дату закінчення застосування; закінчення терапії або терапія триває, вжиті заходи щодо усунення проявів побічної реакції, включаючи фармакотерапію (у базі АІСФ –V вкладка Побічні реакції/ВЕ/НППІ); побічні реакції/відсутність ефективності — лікування побічної реакції, лікування побічних реакцій (довільний текст), результат відміни та повторного призначення підозрюваного(их) лікарського(их) засобу(ів); засоби корекції, вакцини. Супутні лікарські засоби (у базі АІСФ – VІ вкладка Лікарські засоби), супутні лікарські засоби, вакцини, туберкулін, що не підозрюються у спричиненні побічної реакції/відсутності ефективності, фармакотерапія пацієнта (батьків) у минулому у разі доцільності.
VI.C.6.2.2.3. Підозрювані побічні реакції
Необхідно надавати всю наявну інформацію для кожного випадку виникнення побічної реакції (ст. 28 (3) (j) ІП 520/2012 [6]). Кодувати діагнози та попередні діагнози з проявами та симптомами в полі даних «Реакція/явище» (ICHE2B(R2) [26] B.2.i.1) слід з використанням найнижчого ієрархічного рівня (Lowest Level Term) термінології MedDRA та відповідно до останньої версієї затвердженої ICH Інструкції для користувачів MedDRA — MedDRA вибір терміну: основні пункти розгляду документа.
У випадку якщо про побічну реакцію повідомляється у вигляді опису діагнозу із зазначенням проявів та симптомів, то в розділі В.2 «Реакції/явища» ICHE2B(R2) [26] слід зазначати MedDRA термін і код лише для діагнозу. Якщо діагноз не вказаний, то в розділі В.2 «Реакції/явища» ICH-E2B(R2) [26] необхідно перелічити всі прояви та симптоми, про які повідомлялося, закодовані за допомогою MedDRA. Якщо ці прояви і симптоми є складовою діагнозу, уповноважений орган чи заявник (власник реєстраційного посвідчення) може закодувати його за допомогою терміну MedDRA у полі даних В.5.3 «Діагноз/синдром та/або перекласифікація реакції/явища, встановлені відправником» ICH-E2B(R2) [26].
Якщо в описі випадка були зазначені інші явища, що не є типовими проявами чи симптомами діагнозу або попереднього діагнозу, встановленого першоджерелом, і такі явища є підозрюваними побічними реакціями, їх необхідно перелічити й закодувати за допомогою MedDRA у розділі В.2 «Реакції/явища» ICH-E2B(R2) [26].
Якщо уповноважений орган або заявник (власник реєстраційного посвідчення) не згоден з діагнозом, вказаним першоджерелом, він може крім цього діагнозу, який необхідно зазначити в розділі В.2 «Реакції/явища» ICH-E2B(R2) [26], вказати альтернативний діагноз у полі даних «Діагноз/синдром та/або перекласифікація реакції/явища, встановлені відправником» ICH-E2B(R2) [26]. У цьому випадку слід надати обґрунтування у полі даних «Коментар відправника» (ICH-Е2В (R2) [26] В.5.4) (див. VI.C.6.2.2.4).
У випадку смерті пацієнта необхідно зазначити дату смерті, причину смерті, включно з паталогоанатомічним діагнозом, якщо доступно (ст. 28 (3) (l) ІП 520/2012 [6]). Якщо смерть не пов’язана з вказаними у повідомленні підозрюваними побічними реакціями, а була наслідком, наприклад, прогресування захворювання, у цьому випадку, критерій серйозності повідомлення про індивідуальний випадок, пов’язаний з безпекою, не вважається летальним.
VI.C.6.2.2.4. Опис випадку, оцінка причинно-наслідкового зв’язку та коментарі
Відповідно до ст. 28 (3) (m) ІП 520/2012 [6] опис випадку (поле даних ICHE2B(R2) [26] В.5.1 «Case narrative») необхідно надавати, якщо це можливо47, у всіх випадках, за винятком несерйозних. Інформація повинна бути представлена в логічній послідовності, у хронології зміни стану пацієнта, включаючи клінічний перебіг, терапевтичні заходи, наслідок і отриману додаткову інформацію. Необхідно також надати стислий опис важливих результатів розтину чи результатів посмертного дослідження.
Опис випадку необхідно зробити відповідно до рекомендацій, наданих у розділі 5.2 настанови ICH-E2D [25]. Він повинен бути всебічним, самостійним «медичним звітом», що містить усю відому важливу клінічну та пов’язану з нею інформацію, включаючи характеристику пацієнтів, дані про лікування, історію хвороби, клінічний перебіг явищ(а), діагноз, побічні реакції і їх наслідки, важливі лабораторні дані (у тому числі показники в межах норми) і будь-яку іншу інформацію, що підтверджує або спростовує виникнення підозрюваних побічних реакцій. Приклад стандартного опису випадку надається у Звіті V Робочої групи CIOMS (Report of the CIOMS Working Group V)48.
Інформація, надана в описі випадку, повинна відповідати даним у всіх інших відповідних полях даних ICH-E2B (R2) [26] повідомлення про індивідуальний випадок, пов’язаний з безпекою.
Опис випадку, наданий заявником (власником реєстраційного посвідчення), включений у повідомлення про індивідуальний випадок, пов’язаний з безпекою, до національного уповноваженого органу, не може бути змінений або видалений при передачі цього повідомлення вказаним вище органом до бази даних EudraVigilance.
За наявності коментарів першоджерела стосовно діагнозу, оцінки причинно-наслідкового зв’язку чи іншої відповідної інформації, їх необхідно надати в полі «Коментар повідомника» (ICH-E2B(R2) [26] B.5.2). Уповноважений орган та заявники (власники реєстраційних посвідчень) можуть провести оцінку випадку і описати незгоду із зазначеним у повідомленні діагнозом та/або вказати інший (див. VI.C.6.2.2.3). Ця оцінка повинна бути представлена в полі «Коментар відправника» (ICH-E2B(R2) [26] В.5.4), у якому також можна описати неточності чи незрозуміле в інформації, наданій першоджерелом. У випадку, якщо повідомлення про індивідуальний випадок, пов’язаний з безпекою, призводить до виникнення нової проблеми з безпеки (див. VI.C.2.2.6), у полі «Коментар відправника» (ICH-E2B(R2) [26] В.5.4) потрібно надати резюме проблем з безпеки та пропонованих заходів. Ступінь підозрюваного зв’язку між прийомом кожного лікарського засобу та виникненням побічної(их) реакції(й) може бути зазначено в полі «Зв’язок лікарського засобу з реакцією(ями)/явищем(ами)» (ICH-Е2В(R2) [26] B.4.k.0.18), що повинен повторюватися, якщо необхідно. У цьому повторюваному полі слід вказати ступінь зв’язку, присвоєного різними джерелами, з можливістю вибору різних методів оцінки.
В Україні інформація про побічну реакцію надається відповідно до положень ПорядкуN [2] через систему АІСФN відповідно до Інструкції для повідомлення медичними працівниками та заявниками. А саме: інформацію про підозрювану(і) побічну(і) реакцію(ї)/відсутність ефективності (у базі АІСФ – V вкладка Побічні реакції/ВЕ/НППІ); опис випадку побічної реакції/зазначення ВЕ/НППІ лікарського засобу* у довільній формі, кожний прояв ПР та засоби корекції потрібно описувати окремо. Натисніть кнопку «Додати». Опис побічної реакції/зазначення ВЕ/НППІ лікарського засобу*; дата початку; початок побічної реакції/ВЕ/НППІ і дата завершення закінчення побічної реакції або тривалість, серйозність (у базі АІСФ — І вкладка Інформація про повідомлення; повідомлення про випадок серйозної побічної реакції, наслідок підозрюваної(их) побічної(их) реакції(й)/відсутності ефективності на момент останнього спостереження пацієнта; наслідок побічної реакції/ВЕ/НППІ*, проміжок часу, що минув від початку застосування підозрюваного лікарського засобу, вакцини, туберкуліну і початку побічної(их) реакції(й)/відсутності ефективності, терміни та дані, зазначені в документації про побічну(і) реакцію(ї)/відсутність ефективності, які використовувало першоджерело для опису побічної(их) реакції(й)/відсутності ефективності, та країна, на території якої виникла(и) побічна(і) реакція(ї) та/або відсутність ефективності).
VI.C.6.2.2.5. Результати аналізів та інструментальних досліджень
Результати аналізів та інструментальних досліджень пацієнта повинні бути надані (ст. 28 (3) (k) ІП 520/2012 [6]).
Як описано в настанові ICH-E2B(R2) [26], у розділ B.3 «Результати аналізів та досліджень пацієнта» потрібно ввести дані результатів аналізів і процедур, що проводилися з метою діагностики чи підтвердження реакції/явища, включаючи аналізи, зроблені для дослідження (виключення) побічних явищ/реакцій, не пов’язаних з прийомом лікарського засобу (наприклад, серологічні тести на інфекційний гепатит при підозрі на гепатит, викликаний лікарським засобом). Слід надавати як позитивні, так і негативні результати досліджень.
Кодування досліджень необхідно проводити у відповідності з останньою версією затвердженої ICH Інструкції для користувачів MedDRA — MedDRA вибір терміну: основні пункти розгляду документа. Якщо неможливо представити інформацію про дослідження і результати досліджень в окремих полях, то її можна надати в текстовому полі «Результати аналізів та процедур під час досліджень» (ICH-Е2В(R2) [26] B.3.2.).
В Україні інформація про результати аналізів та інструментальних досліджень пацієнта надається відповідно до положень ПорядкуN [2] через систему АІСФN відповідно Інструкції для повідомлення медичними працівниками та заявниками. А саме: результати тестів і процедур, що стосуються дослідження пацієнта (у базі АІСФ — ІІІ вкладка Дані досліджень); результати лабораторно-інструментальних досліджень.
VI.C.6.2.2.6. Додаткова інформація
Ключова інформація з медичної документації повинна бути представлена у відповідних полях даних повідомлення про індивідуальний випадок, пов’язаний з безпекою, а наявність доступу до неї зазначена в полі «Перелік документів, наявних у відправника» (ICH-E2B(R2) [26] A.1.8.2).
Інші відомі ідентифікатори випадку, що необхідні для виявлення дублікатів, слід систематично надавати в полі «Інші ідентифікатори випадку в попередніх передачах повідомлень» (ICH-Е2В(R2) [26] A.1.11).
В Україні додаткова інформація про пацієнта надається відповідно до положень ПорядкуN [2] через систему АІСФN відповідно до Інструкції для повідомлення медичними працівниками та заявниками. А саме: додаткова важлива медична інформація (у базі АІСФ — ІV вкладка Інша медично важлива інформація); важлива медична інформація (діагноз, супутні захворювання, алергія, вагітність).
VI.C.6.2.2.7. Інформація з подальшого відстеження
Повідомлення про індивідуальні випадки, пов’язані з безпекою, направляються до багатьох організацій в різні проміжки часу. А тому статус повідомлення (первинне/подальше відстеження) буде залежати від отримувача повідомлення. Саме тому поле, у якому зазначається цей статус, не включене до переліку полів даних ICH E2B(R2) [26]. Проте поля «Дата отримання найостаннішої інформації до цього повідомлення» (ICH-E2B(R2) [26] A.1), «Ідентифікатор відправника» (ICH E2B(R2) [26] А.3.1.2) та «Унікальний номер повідомлення, присвоєний відправником» (ICH-E2B(R2) [26] A.1.0.1) забезпечують механізм, за допомогою якого кожний отримувач зможе визначити статус повідомлення, що передається (первинне чи подальшого відстеження). З цієї причини ці поля дуже важливі, і при передачі в них необхідно завжди вказувати точні дати (день, місяць, рік). Поле «Дата отримання найостаннішої інформації до цього повідомлення» (ICH-E2B(R2) [26] A.1.7) повинно оновлюватися кожного разу при передачі уповноваженим органом чи заявником (власником реєстраційного посвідчення) наступних повідомлень, якщо отримано додаткову важливу інформацію про випадок. При цьому поле даних «Дата отримання початкового повідомлення від першоджерела» (ICH-E2B(R2) [26] A.1.6) повинно залишатися незмінним та містити дату отримання уповноваженим органом чи заявником (власником реєстраційного посвідчення) первинного повідомлення про виникнення побічної реакції.
Нова інформація повинна бути чітко виділена в описі випадку (елемент даних ICH-E2B(R2) [26] B.5.1) та відображена у відповідних полях даних ICHE2B(R2) [26].
Національні уповноважені органи або заявники (власники реєстраційних посвідчень) повинні надавати повідомлення з подальшого відстеження випадку, якщо вони отримали нову клінічно значущу інформацію. Такою інформацією буде, наприклад, отримана інформація про нові підозрювані побічні реакції, зміни ступеня причинно-наслідкового зв’язку, а також будь-які нові або оновлені дані про випадок, що впливають на його медичну інтерпретацію. Тому важлива нова інформація, про яку необхідно повідомляти, завжди вимагає медичної оцінки.
Якщо змінюється критерій серйозності повідомлення та/або ступінь причинно-наслідкового зв’язку (наприклад, отримана нова інформація призводить до зміни статусу повідомлення з серйозного на несерйозний; причинно-наслідковий зв’язок змінюється з пов’язаного на непов’язаний), такі зміни також вважаються важливими і про них необхідно повідомляти (див. VI.B.7.1 щодо терміну надання повідомлень).
Національні уповноважені органи та заявники (власники реєстраційних посвідчень) повинні також повідомляти про доступну нову адміністративну інформацію, яка може вплинути на обробку даних про випадки, наприклад, якщо відправникові стали відомі нові ідентифікатори випадку, що могли використовуватися при передачі попередніх повідомлень (поле даних «Інші ідентифікатори випадку, присвоєні в раніше переданих повідомленнях» (ICHЕ2В(R2) [26] A.1.11). Ця інформація особливо важлива для виявлення потенційних дублікатів. Іншим прикладом може бути інформація, що зазначається в полі даних «Додаткові документи, доступні для відправника» (ICH-E2B(R2) [26] A.1.8), якщо додаткова медична документація, важлива для медичної оцінки випадку, стає доступною для відправника.
У той же час не потрібно надавати повідомлення з подальшого відстеження, що містить незначну інформацію. Таку як, наприклад, незначні зміни в деяких датах, що не впливають на оцінку випадку чи передачу повідомлення, виправлення типографічних помилок в попередній версії повідомлення. У будь-якому випадку при появі нової інформації необхідно проводити її медичну оцінку, оскільки, наприклад, зміна дати народження може бути новою важливою зміною (наприклад з наслідками для даних про вік пацієнта). Так само, зміна коду/терміну MedDRA з актуального на неактуальний у зв’язку зі зміною версії MedDRA буде розглядатися як незначна зміна до тих пір, поки вона не впливає на медичні дані випадку. Однак зміна терміну MedDRA у зв’язку зі зміною в інтерпретації раніше повідомленої підозрюваної побічної реакції може являти собою значну зміну і, отже, про неї слід повідомити.
Якщо змінена в повідомленні інформація про випадок не впливає на його медичну оцінку і при цьому не отримано нової інформації про випадок (наприклад виправлення помилки чи орфографічної помилки), дата отримання останньої інформації, вказана в полі даних «Дата отримання найостаннішої інформації до цього повідомлення» (ICH-E2B (R2) [26] A.1.7), не повинна змінюватися. Однак у будь-якій іншій ситуації це поле повинно оновлюватися на дату, коли отримано нові дані з подальшого відстеження (незалежно від того, істотні, чи ні), або на дату, коли були внесені зміни, що впливають на інтерпретацію даного випадку.
Якщо дані з подальшого відстеження випадку, про який спочатку було повідомлено заявником (власником реєстраційного посвідчення), були отримані напряму від першоджерела уповноваженим органом, то поле «Міжнародний унікальний номер випадку» (ICH-E2B(R2) [26] A.1.10) первинного повідомлення не повинно змінюватися, відповідно до ICH-E2B(R2) [26]. Такого ж принципу необхідно дотримуватися, якщо заявника (власника реєстраційного посвідчення) повідомили щодо даних з подальшого відстеження випадку, про які першопочатково було поінформовано уповноважений орган.
В Україні адміністративна інформація про пацієнта надається відповідно до положень ПорядкуN [2] через систему АІСФN відповідно до Інструкції для повідомлення медичними працівниками та заявниками. А саме: адміністративну інформацію (у базі АІСФ — І вкладка Інформація про повідомлення) (вид повідомлення — Вид випадку*, дата — дата заповнення повідомлення*, дата вперше отриманої інформації про випадок*, унікальний ідентифікаційний номер випадку — міжнародний № повідомлення, а також унікальний ідентифікатор відправника — Дані відправника; дата, коли було вперше отримано інформацію від джерела — дата вперше отриманої інформації про випадок*, та дата отримання найновішої інформації — дата отримання найостаннішої інформації (точні); інші ідентифікатори та їх джерела — джерело повідомлення, кваліфікація джерела повідомлення*, Літературне джерело, Тип відправника *, Відправник *, інформація про джерело повідомлення, а також посилання на додаткові доступні документи — чи наявні додаткові документи?, що належать відправнику повідомлення, про випадок побічної реакції лікарського засобу та/або відсутності ефективності лікарського засобу, коли застосовано). Надається інформацію про першоджерело(а) (інформацію, що ідентифікує джерело повідомлення, включаючи назву країни, де проживає повідомник, та його професійну кваліфікацію — кваліфікація джерела повідомлення*, інформація про джерело повідомлення).
VI.C.6.2.2.8. Що слід враховувати щодо законодавства про захист персональних даних
У процесі діяльності з виявлення, оцінки, розуміння та запобігання розвитку побічних реакцій, ідентифікації ризиків і вжиття заходів для їх мінімізації, збільшення користі від прийому лікарського засобу, а також обробки персональних даних у базі даних EudraVigilance необхідно дотримуватися законодавства ЄС щодо захисту персональних даних (Директива 95/46/ЄС [12], Регламент (ЄС) № 45/2001 [18]).
Якщо згідно з чинним національним законодавством інформація, пов’язана з особистими даними, не може бути передана до бази даних EudraVigilance [10], заявники (власники реєстраційних посвідчень) можуть використовувати псевдонімізацію, тобто заміну персональних даних, таких як ім’я і адреса, псевдонімами або кодами, наприклад, відповідно до Технічної специфікації ISO DD ISO/TS 25237:2008, Медична інформатика — Псевдонімізація (Recital 17 ІП 520/2012 [6]). Застосування псевдонімізації дозволить базі даних EudraVigilance адекватно підтримувати обробку випадків виникнення побічних реакцій та виявляти дублікати. Однак псевдонімізацію слід робити без порушення інформаційного потоку у вказаній вище базі даних, що погіршило б інтерпретацію та оцінку даних з безпеки; такі поля даних, як вік пацієнта, вікова група та стать, необхідно залишити відкритими/видимими.
В Україні інформація, що ідентифікує пацієнта, надається відповідно до положень ПорядкуN [2] через систему АІСФN відповідно до Інструкції для повідомлення медичними працівниками та заявниками. А саме: інформація, що ідентифікує пацієнта (у базі АІСФ — ІІ вкладка Інформація про пацієнта); ініціали пацієнта*; стать; й одного з батьків у випадку повідомлення батьки — дитина, включаючи вік; дата народження*, на момент виникнення побічної реакції/відсутності ефективності, вікову групу; вікова група*, гестаційний вік; вік*, коли реакція/явище спостерігалася(ося) у плода, масу тіла, зріст, стать, дату останнього менструального та/або гестаційного періоду на момент виникнення побічної реакції/відсутності ефективності. Також зазначається анамнез і супутні захворювання пацієнта (у базі АІСФ — ІІІ вкладка Дані досліджень); Результати лабораторно-інструментальних досліджень; (у базі АІСФ — ІV вкладка Дані досліджень); важлива медична інформація (діагноз, супутні захворювання, алергія, вагітність).
VI.C.6.2.2.9. Використання різних мов
Концепція ICH-Е2В(R2) [26] електронного обміну повідомленнями про індивідуальні випадки, пов’язані з безпекою ґрунтується на тому факті, що структурована і закодована інформація використовується для надання даних з фармаконагляду (наприклад переліки) і для виявлення сигналу. Однак для наукової оцінки випадків та оцінки сигналу зазвичай необхідні узагальнені медичні дані, наведені в полі даних «Опис випадку, включаючи клінічний перебіг, терапевтичні заходи, наслідок та іншу відповідну інформацію» (ICHE2B(R2) [26] B.5.1) (див. VI.6.2.2.4).
Якщо про підозрювані побічні реакції повідомляється в описовій частині і текстові описання викладено однією з офіційних мов ЄС, окрім англійської, заявнику (власнику реєстраційного посвідчення) необхідно надати оригінальний дослівний текст і стисле резюме англійською мовою49. Країни ЄС можуть надавати текстові описання випадку своєю офіційною мовою (мовами). Для таких повідомлень, у разі запиту ЕМА або інших країн ЄС, для оцінки потенційних сигналів повинні бути забезпечені переклади англійською мовою (ст. 28 (4) ІП 520/2012 [6]).
Додаткові документи, наявні у відправника повідомлення, доступні лише місцевою мовою, повинні перекладатися тільки на вимогу отримувача.
VI.C.6.2.2.10. Анулювання випадків
Відповідно до настанови ICH-E2B(R2) [26], анулювання випадків побічних реакцій слід використовувати для зазначення того, що раніше надане повідомлення слід розцінювати як недійсне, наприклад, коли виявилося, що випадок помилковий, або якщо це дублювання. Дуже важливо зазначати ті ж самі номери в полях даних «Унікальний ідентифікатор повідомлення, присвоєний відправником) (ICH-E2B(R2) [26] A.1.0.1) і «Міжнародний унікальний номер випадку» (ICH-E2B(R2) [26] A.1.10), що і в раніше відправлених повідомленнях стосовно цього випадку.
Анульований випадок — це випадок, що більше не повинен враховуватися в процедурах оцінки. Процес анулювання випадку супроводжується інформуванням відправником повідомлення отримувача про те, що випадок більше не дійсний. Однак інформація про випадок має зберігатися у базі даних з фармаконагляду відправника для цілей аудиту.
Принципи, що повинні враховуватися при анулюванні випадку, детально викладені в VI. Додаток 5.
В Україні анулювання випадків побічних реакцій відбувається через систему АІСФN.
VI.C.6.2.3. Особливі ситуації
VI.C.6.2.3.1. Застосування лікарських засобів під час вагітності або годування груддю
Загальні рекомендації наведено в VI.B.6.1.
Що стосується надання повідомлень в електронній формі про випадки виникнення побічних реакцій типу «батьки — дитина/плід», необхідно дотримуватися наступних вимог:
- у ситуації, коли плід або немовля піддається впливу одного або декількох лікарських засобів через одного з батьків і у нього виникає одна або більше підозрюваних побічних реакцій (за винятком мимовільного аборту/загибелі плоду), у одному й тому ж повідомленні потрібно надавати інформацію про матір (батька) та дитину/плід. Ці випадки розглядаються як повідомлення «батьки — дитина/плід». Інформація, представлена у розділі B.1 «Характеристики пацієнта» ICH-E2B(R2) [26], стосується тільки дитини/плода. Характеристики, що стосуються батьків (матері або батька), які застосовували підозрюваний лікарський засіб, повинні бути представлені в полі «Інформація про матір(батька) для повідомлення «батьки — дитина/плід» (ICH-E2B(R2) [26] B.1.10). Якщо обоє батьків приймали підозрюваний лікарський засіб, тоді в полі B.1.10 «Інформація про матір (батька) для повідомлення «батьки — дитина/плід» ICH-E2B(R2) [26] повинні бути зазначені дані матері. У полі даних B.5.1 «Опис випадку, включаючи клінічний перебіг, терапевтичні заходи, наслідок та іншу відповідну інформацію» ICH-E2B (R2) [26] необхідно надати всю інформацію про випадок, у тому числі дані батька;
- якщо побічні реакції виникли як у матері (батька), так і в дитини/плода, то потрібно створити два різні повідомлення, тобто одне щодо матері (батька) і одне щодо дитини/плода, але вони повинні бути поєднані за допомогою поля даних A.1.12 «Номер повідомлення, що пов’язане з даним повідомленням» ICH-E2B(R2) [26] у кожному із цих повідомлень;
- якщо у дитини не виникало побічної реакції, то не потрібно використовувати повідомлення «батьки — дитина/плід», тобто розділ B.1 «Характеристики пацієнта» ICH-E2B(R2) [26] включає тільки дані матері або батька, у яких спостерігалися підозрювані побічні реакції.
- у випадках, що стосуються викидня чи раннього мимовільного аборту, повідомлення заповнюється тільки для матері, тобто у розділі B.1 «Характеристики пацієнта» ICH-E2B (R2) [26] зазначаються дані матері. Проте якщо підозрюваний лікарський засіб застосовував батько, у полі даних B.4.k.19 «Додаткова інформація про лікарський засіб» (ICH-E2B(R2) [26]) необхідно це вказати.
В Україні інформація про застосування лікарських засобів під час вагітності або годування груддю надається через систему АІСФN.
VI.C.6.2.3.2. Випадки виникнення підозрюваних побічних реакцій, що були опубліковані в науковій літературі
Вимоги щодо моніторингу випадків виникнення підозрюваних побічних реакцій внаслідок прийому лікарського засобу, описані у науковій та медичній літературі, викладені у VI.C.2.2.3. Що стосується надання повідомлень про вказані вище випадки, що опубліковані в науковій та медичній літературі, в електронному вигляді, то слід дотримуватися наступних вимог:
- літературні джерела повинні бути зазначені в полі даних A.2.2 «Літературне джерело(а)» (ICH-E2B(R2) [26]) відповідно до Ванкуверської системи (відомою як «Ванкуверський стиль»), розробленої Міжнародним комітетом редакторів медичних журналів (ст. 28 (3) (b) ІП 520/2012 [6]). Стандартний формат, а також формати для особливих ситуацій можна знайти за наступним посиланням: International Committee of Medical Journal Editors. Uniform requirements for manuscripts submitted to biomedical journals. N Engl J Med. 1997; 336: 309-1550;
- у полі даних B.5.1 «Опис випадку, включаючи клінічний перебіг, терапевтичні заходи, наслідок та іншу відповідну інформацію» (ICHE2B(R2) [26]) необхідно надати вичерпне резюме статті англійською мовою (ст. 28 (3) (b) ІП 520/2012 [6]);
- на запит ЕМА для специфічного аналізу з безпеки заявник (власник реєстраційного посвідчення), який надав первинне повідомлення, повинен надати копію та переклад відповідної статті англійською мовою з урахуванням обмежень щодо авторських прав (ст. 28 (3) ІП 520/2012 [6]). Слід дотримуватися рекомендацій, викладених у VI. Додаток 2.10 щодо відправки статті поштою.
- Слід дотримуватися рекомендацій, наданих у VI. Додаток 2.10 щодо повідомлення про декілька випадків, опублікованих в одній статті.
В Україні інформація про випадки виникнення підозрюваних побічних реакцій, що були опубліковані в науковій літературі, надається через систему АІСФN.
VI.C.6.2.3.3. Підозрювані побічні реакції, що виникли внаслідок передозування, зловживання, застосування не за показаннями, неправильного застосування, медичної помилки, впливу, пов’язаного з професійною діяльністю
Загальні принципи описані у VI.B.6.3.
Якщо про випадок передозування, зловживання, застосування не за показаннями, медичної помилки або впливу, пов’язаного з професійною діяльністю, повідомляється з клінічними наслідками, у поле даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICH-E2B(R2) [26]) необхідно, крім спостережуваних підозрюваних побічних реакцій, надати код терміна найнижчого рівня термінології MedDRA, що найкраще описує повідомлене передозування, зловживання, застосування не за показаннями, медичної помилки або впливу, пов’язаного з професійною діяльністю, відповідно до рекомендацій, включених в останню версію ICH-Endorsed Guide for MedDRA Users «MedDRA Term Selection: Points to Consider» (MTS:PTC).
В Україні інформація про підозрювані побічні реакції, що виникли внаслідок передозування, зловживання, застосування не за показаннями, неправильного застосування, медичної помилки, впливу, пов’язаного з професійною діяльністю надається через систему АІСФN.
VI.C.6.2.3.4. Відсутність терапевтичного ефекту
Загальні принципи описані у VI.B.6.4.
Якщо першоджерело підозрює відсутність терапевтичного ефекту, у полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICHE2B(R2) [26]) необхідно надати код терміна найнижчого рівня термінології MedDRA, що найкраще описує повідомлену відсутність терапевтичного ефекту, відповідно до рекомендацій, включених в останню версію інструкції ICH для користувачів MedDRA.
За винятком випадків, коли погіршується перебіг захворювання, у полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICH-E2B(R2) [26]) не потрібно включати показання, за яким призначався підозрюваний лікарський засіб.
Ті ж вимоги щодо формування та надання звітів, як для серйозних повідомлень про індивідуальні випадки, пов’язані з безпекою (див. VI.C.4), повинні застосовуватися у тих випадках, пов’язаних з фармакологічною групою, для яких, як описано в VI.B.6.4, повідомлення про відсутність терапевтичного ефекту повинні бути надані впродовж 15 днів. За відсутності критерію серйозності допускається надання повідомлення протягом 15 днів як несерйозного.
В Україні інформація про відсутність терапевтичного ефекту надається відповідно до положень ПорядкуN через систему АІСФN.
VI.C.6.2.3.5. Підозрювані побічні реакції, пов’язані з дефектом якості або фальсифікованими лікарськими засобами
Вимоги щодо повідомлень представлені в VI.C.2.2.4. Нижче надаються рекомендації щодо того, як чітко ідентифікувати випадки, пов’язані із застосування лікарських засобів з дефектами якості чи фальсифікованими лікарськими засобами, при обміні даними між зацікавленими сторонами:
а. Дефекти якості
У полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICH-E2B(R2) [26]) повідомлення про підозрювану побічну реакцію, пов’язану з підозрюваним або підтвердженим дефектом якості лікарського засобу, слід вказати код терміна найнижчого рівня термінології MedDRA, що найкраще описує даний дефект якості.
б. Фальсифіковані лікарські засоби
У полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICH-E2B(R2) [26]) повідомлення про виникнення підозрюваної побічної реакції, пов’язаної із застосуванням підозрюваного або підтверджено фальсифікованого51 інгредієнта, діючої речовини чи лікарського засобу, слід вказати код терміна найнижчого рівня термінології MedDRA, що найкраще описує повідомлену інформацію. Інформація про підозрюваний лікарський засіб, діючу(і) речовину(и) або допоміжну(і) речовину(и), повідомлена першоджерелом, повинна бути зазначена у полі даних B.4.k.2.2 «Назва лікарського засобу» (ICH-E2B(R2) [26] та/або B.4.k.2.2 «Діюча речовина(и)» ICH-E2B(R2) [26].
В Україні інформація про підозрювані побічні реакції, пов’язані з дефектом якості або фальсифікованими лікарськими засобами, надається через систему АІСФN.
VI.C.6.2.3.6. Підозрювана передача збудника інфекції з лікарським засобом
Вимоги щодо надання повідомлень представлені у VI.C.2.2.5.
Кодування підозрюваної передачі збудника інфекції через лікарський засіб у полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» (ICH-E2B(R2) [26]) має проводитися відповідно до останньої версії інструкції ICH для користувачів MedDRA.
Крім того, якщо було зазначено збудника інфекції, то у полі даних B.2.i.1 «Реакція/явище згідно з термінологією MedDRA (Lowest Level Term)» ICHE2B(R2) [26] потрібно вказати код терміна MedDRA найнижчого рівня, що відповідає збуднику інфекції.
В Україні інформація про підозрювану передачу збудника інфекції з лікарським засобом надається через систему АІСФN.
VI.C.6.2.3.7. Повідомлення, що надходять з організованих систем збору даних та інших систем
Загальні вимоги щодо надання повідомлень про випадки виникнення побічних реакцій у післяреєстраційних дослідженнях вказані у VI.C.1 і VI.C.2.2.2. Повідомлення про побічну реакцію з таких досліджень повинні містити інформацію про вид дослідження, його назву та номер, присвоєний спонсором, або реєстраційний номер дослідження (ст. 28 (3) (с) ІП 520/2012 [6]). Ці дані повинні бути вказані в розділі A.2.3 «Ідентифікація дослідження» ICH E2B(R2) [26].
Вимоги щодо надання повідомлень про випадки виникнення побічних реакцій під час програм підтримки пацієнтів або програм маркетингових досліджень надані у VI.C.2.2.11.
Залежно від:
- виду системи збору даних, і
- того, чи належить підозрюваний лікарський засіб до системи збору даних, застосовуються наступні правила надання повідомлень:
1. Для випадків виникнення підозрюваних побічних реакцій, (а) пов’язаних з виникненням тих побічних явищ, для яких у протоколах неінтервенційних післяреєстраційних досліджень не зазначено інше і вимагається їх систематичний збір (див. VI.C.1.2.1), (б) тих, що походять з програм застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами (compassionate use), застосування лікарського засобу пацієнтами на поіменній основі (named patient use), що проводяться в ЄС, у яких вимагається активний збір випадків побічних явищ (див. VI.C.1.2.2), чи (в) тих, що походять з програм підтримки пацієнтів або ринкових досліджень (див. VI.C.2.2.11):
- коли підозрюється, що виникнення побічної реакції пов’язане із застосуванням хоча б одного з досліджуваних лікарських засобів (лікарських засобів, що постачаються):
- повідомлення про виникнення побічної реакції розцінюються як повідомлення, що походять із джерел з організованою системою збору даних (solicited);
- поле даних A.1.4 «Тип повідомлення» ICH E2B(R2) [26] повинно містити значення «звіт з дослідження»;
- поле даних A.2.3.3 «Вид дослідження, у ході якого спостерігалась реакція(ї)/явище(а)» ICH E2B(R2) [26] повинно містити значення «інші дослідження» або «застосування конкретними пацієнтами»;
- коли підозрюється, що виникнення побічної реакції пов’язане тільки із застосуванням лікарського засобу, що не входить до організованої системи збору даних, і немає взаємодії з досліджуваним лікарським засобом, що постачається:
- повідомлення вважається спонтанним тому, що передає підозру первинного джерела;
- поле даних A.1.4 «Тип повідомлення» ICH E2B(R2) [26] повинно містити позначення «спонтанне».
2. Для випадків виникнення підозрюваних побічних реакцій, (і) пов’язаних з тими побічними явищами, для яких у протоколах неінтервенційних післяреєстраційних досліджень зазначено інше і не вимагається їх систематичний збір (див. VI.C.1.2.1), або (іі) тих, що походять з програм застосування незареєстрованих лікарських засобів у зв’язку з виключними обставинами (compassionate use), застосування лікарського засобу пацієнтами на поіменній основі (named patient use), що проводяться в ЄС, у яких не вимагається активний збір випадків побічних явищ (див. VI.C.1.2.2):
- повідомлення вважається спонтанним тому, що передає підозру первинного джерела;
- поле даних A.1.4 «Тип повідомлення» ICH E2B(R2) [26] має містити позначення «спонтанне».
3. Щодо випадків виникнення підозрюваних побічних реакцій під час клінічних випробувань, що підпадають під дію Директиви 2001/20/ЄC [15], у ситуаціях, коли підозрюється, що побічна реакція пов’язана з прийомом не досліджуваного лікарського засобу (або іншого лікарського засобу, що не є предметом клінічного дослідження), і відсутня взаємодія з ним:
- повідомлення вважається спонтанним тому, що передає підозру первинного джерела;
- поле даних A.1.4 «Тип повідомлення» ICH E2B(R2) [26] повинно містити позначення «спонтанне».
Усі повідомлення про побічні реакції, що надаються до бази даних EudraVigilance, та що походять з післяреєстраційних досліджень, та не підпадають під дію Директиви 2001/20/ЄC [15] щодо клінічних випробувань, повинні бути представлені в EVPM (див. VI.C.6.2.1). Зазначена умова діє також для випадків виникнення побічних реакцій, що спостерігалися під час клінічних випробувань, не пов’язаних з досліджуваним лікарським засобом.
В Україні інформація стосовно повідомлень про випадки виникнення побічних реакцій у післяреєстраційних дослідженнях надається відповідно до положень Порядку проведення клінічних випробуваньN.
VI.C.6.2.3.8. Отримання мінімуму інформації, що дозволяє ідентифікувати випадок
Після отримання мінімуму інформації, якої бракувало для того, щоб повідомлення про побічну реакцію вважалося валідним (див. VI.B.2), діють наступні правила:
- у полі даних A.1.6 «Дата отримання початкового повідомлення від першоджерела» (ICH-E2B(R2) [26] потрібно зазначити дату отримання первинного невалідного повідомлення про побічні реакції;
- у полі даних A.1.7 «Дата отримання найостаннішої інформації до цього повідомлення» (ICH-E2B(R2) [26] потрібно зазначити дату, коли стали відомими всі чотири елементи необхідного мінімуму інформації;
- у описі випадку (поле даних B.5.1 ICH-E2B(R2) [26] необхідно пояснити, що деякі з чотирьох елементів були відсутні в первинному повідомленні;
- як і для всіх інших повідомлених випадків, контроль за дотриманням строків надання повідомлень здійснюється за допомогою поля даних «Дата отримання найостаннішої інформації до цього повідомлення» (ICH-E2B(R2) [26] A.1.7).
В Україні дані про мінімум інформації, що дозволяє ідентифікувати випадок надаються відповідно до положень ПорядкуN, а саме: повідомлення про випадок побічної реакції лікарського засобу, вакцини, туберкуліну та/або відсутності ефективності лікарського засобу має містити щонайменше інформацію, за допомогою якої можна ідентифікувати повідомника, пацієнта, одну побічну реакцію/відсутність ефективності і підозрюваний(і) лікарський(і) засіб(оби), вакцину(и), туберкулін.
VI.C.6.2.4. Якість повідомлень про побічні реакції, що надаються в електронній формі, і вирішення проблеми дублювання
Для здійснення діяльності з фармаконагляду база даних EudraVigilance повинна містити дані про всі випадки виникнення підозрюваних побічних реакцій, що підлягають повідомленню відповідно до Директиви 2001/83/ЄС [1] та Регламенту (ЄC) №726/2004 [5]. Це стосується всіх лікарських засобів, дозволених до медичного застосування в ЄС.
Вказана вище база даних повинна відповідати найвищим міжнародно визнаним стандартам якості даних.
Для досягнення цих цілей уповноважені органи країн ЄС та заявники (власники реєстраційних посвідчень) повинні дотримуватися:
- вимог законодавства ЄС до надання повідомлень в електронній формі;
- концепції структурування даних, кодування і надання повідомлень у відповідності до законодавства ЄС, настанов, стандартів і принципів, зазначених у VI.C.6.2.2.1.
Зазначені вище вимоги є необхідною умовою для належного функціонування Європейської бази даних з фармаконагляду.
ЕМА у співпраці із зацікавленими сторонами, які надають повідомлення про побічні реакції до Європейської бази даних з фармаконагляду (EudraVigilance), несе відповідальність за операційні процедури, що гарантують високу якість і цілісність інформації, зібраної в зазначеній базі (ст. 25 (3) Регламенту (ЄС) № 726/2004) [5]. Це включає також моніторинг використання термінології, зазначеної у розділі IV ІП 520/2012 (cт. 25(3) ІП 520/2012 [6]).
Необхідно створити спеціальні процедури і процеси системи якості для забезпечення:
- надання точних і достовірних (що можливо перевірити) даних про випадки виникнення серйозних і несерйозних підозрюваних побічних реакцій до Європейської бази даних з фармаконагляду в 15- або 90-денний термін (cт. 11 (1) (с) ІП 520/2012 [6]);
- якості, цілісності і повноти наданої інформації про ризики лікарських засобів, у тому числі процесів уникнення надсилання повідомлень-дублікатів (cт. 11 (1) (d) ІП 520/2012 [6]).
У зв’язку з цим уповноважені органи країн ЄС та заявники (власники реєстраційних посвідчень) повинні мати систему аудиту, що забезпечує найвищу якість повідомлень про побічні реакції, що надаються в електронному вигляді до EudraVigilance в належний строк та що дозволяє виявляти дублікати повідомлень в їх системах. Надані повідомлення про побічні реакції повинні містити всю відому інформацію про випадок, бути нескороченими за форматом та змістом.
Високорівневі блок-схеми робочих процесів і опис процесів контролю якості повідомлень про побічні реакції, виявлення і управління дублікатами надано в VI. Додаток 6 і VI. Додаток 7. Подальші інструкції щодо виявлення дублікатів повідомлень містяться в Настанові з виявлення та видалення дублікатів повідомлень про побічні реакції (ICSRs) EMA/13432/2009.
ЕМА буде проводити на регулярній основі контроль якості, цілісності та відповідності строків надання повідомлень усіма організаціями, що відправляють їх до Європейської бази даних з фармаконагляду. Зазначеним організаціям будуть надаватися результати цього моніторингу.
В Україні питання дублювання інформації вирішується за допомогою системи АІСФN.
VI.C.6.2.5. Повторне надання в електронному вигляді повідомлень про індивідуальні випадки, пов’язані з безпекою при їх обміні між кількома відправниками та отримувачами
Цей розділ стосується вимог, що повинні виконуватися при електронному обміні повідомленнями між кількома відправниками та отримувачами (наприклад, за наявності договірної угоди, повідомлення про побічні реакції з третіх країн спочатку надаються заявником (власником реєстраційного посвідчення) за межами ЄС іншому заявнику (власнику реєстраційного посвідчення) в ЄС, а звідти — в ЕМА). Дана умова також діє під час періоду тимчасових заходів, коли на основі вимог щодо надання повідомлень, які детально викладені у VI.C.4.1, повідомлення про побічні реакції, що походять з ЄС, надаються власниками реєстраційних посвідчень до уповноваженого органу в країні ЄС, де виникла побічна реакція, а потім повторно надаються до бази даних EudraVigilance.
Під час процесу повторної передачі інформація про випадок побічної реакції не повинна видалятися або змінюватися, крім ситуацій, якщо відправнику, який здійснює передачу даних, доступна нова інформація про випадок.
Винятки поширюються на наступні поля даних та розділи:
- «Унікальний номер повідомлення, присвоєний відправником» (ICH-E2B(R2) [26] A.1.0.1);
- «Дата цієї передачі повідомлення» (ICH-E2B(R2) [26] A.1.3);
- «Дата отримання початкового повідомлення від першоджерела» (ICHE2B(R2) [26] А.1.6) для первинних повідомлень;
- «Дата отримання найостаннішої інформації до цього повідомлення» (ICHE2B(R2) [26] A.1.7);
- «Інформація про відправника та отримувача повідомлення» (ICH-E2B(R2) [26] A.3);
- «Зв’язок лікарського засобу з реакцією(ями)/явищем(ами)» (ICH-E2B(R2) [26] B.4.k.18);
- «Діагноз/синдром відправника та/або декласифікація реакції/явища» (ICHE2B(R2) [26] В.5.3);
- «Коментар відправника» (ICH-E2B (R2) [26] В.5.4).
З метою поліпшення якості даних у випадку виявлення помилок чи неузгодженості інформації у повідомленні, особа, яка здійснює передачу повідомлення, повинна звернутися до його автора, який повинен внести відповідні виправлення. Однак якщо це не може бути зроблено в межах відповідних строків надання повідомлень, особа, яка здійснює передачу повідомлення, може виправити інформацію, що була неправильно структурована.
Крім того, будь-який партнер з обміну електронними даними повинен дотримуватися правил ICH-E2B(R2) [26] щодо надання даних з подальшого відстеження, згідно з якими «Міжнародний унікальний номер випадку» (ICHE2B (R2) [26] A.1.10) повинен підтримуватися відповідно до настанови ICHE2B(R2) [26]. Недотримання цих вимог ставить під загрозу управління електронними даними про випадки і потенційно призводить до непотрібного дублювання повідомлень у базі даних отримувача.
В Україні надання в електронному вигляді повідомлень про індивідуальні випадки, пов’язані з безпекою, при їх обміні між кількома відправниками та отримувачами відбувається через систему АІСФN.
VI.C.6.2.6. Надання повідомлень в електронній формі через центральний офіс компанії
Якщо фармацевтична компанія вирішила централізувати надання повідомлень про побічні реакції в електронній формі (наприклад шляхом їх надання через центральний офіс), необхідно враховувати наступне:
- порядок централізованого надання повідомлень про випадки виникнення побічних реакцій повинен бути чітко описаний у майстер-файлі системи фармаконагляду заявника (власника реєстраційного посвідчення) і у внутрішніх стандартних операційних процедурах;
- центральний офіс компанії, який буде здійснювати надання повідомлень, повинен бути зареєстрований у базі даних EudraVigilance;
- ті ж принципи можуть застосовуватися до надання повідомлень про побічні реакції уповноваженими органами у країнах ЄС заявникам (власникам реєстраційних посвідчень) протягом періоду тимчасових заходів, тобто уповноважені органи у країнах ЄС надають повідомлення в електронному вигляді до центрального офісу компанії, а не в місцеві філії.
В Україні надання повідомлень в електронній формі через центральний офіс компанії відбувається через систему АІСФN.
VI.C.6.3. Надання інформації про лікарські засоби в електронному вигляді
Для підтримки цілей Директиви 2001/83/ЕС [1] та Регламенту (ЄС) № 726/2004 [5], положення, передбачені в другому абзаці ст. 57(2) Постанови (ЄС) № 726/2004 [5], що стосуються електронного надання і оновлення інформації про лікарські засоби для людини, що зареєстровані для продажу або зареєстровані в ЄС, повинні виконуватися заявниками (власниками реєстраційних посвідчень). У цьому аспекті вони застосовують міжнародно узгоджені формати і терміни, описані в розділі IV ІП 520/2012 [6]. Рекомендації, що стосуються електронного надання інформації про лікарські засоби, розміщені на сайті ЕМА52 (в Україні — на сайті ЦентруN).
VI. Додаток 1. Ідентифікація біологічних лікарських засобів

Рисунок 2. Схема робочого процесу — ідентифікація біологічних лікарських засобів
Таблиця 2. Опис процесу — ідентифікація біологічних лікарських засобів
|
№ |
Крок |
Опис |
Відповідальна організація |
|
1 |
Початок. Отримано повідомлення |
День 0. Отримана інформація про випадок побічної реакції, у розвитку якої підозрюється лікарський засіб біологічного походження. |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
2 |
Чи стосується повідомлення біологічного лікарського засобу? |
Якщо так, перейдіть до кроку 3. Якщо ні, перейдіть до кроку 4 |
|
|
3 |
Чи відома інформація про номер серії, торговельну назву і діючу речовину і чи можна їх ідентифікувати? |
Якщо так, створіть повідомлення про випадок, надайте його належному отримувачу (крок 3). Якщо наявний більше ніж один номер серії, введіть номер серії, що відповідає побічній реакції в розділі «Лікарський засіб (ICHE2B(R2) [26] В.4), а інші номери серій зазначте в описі випадку. Якщо ні, створіть повідомлення про випадок і надайте його належному отримувачу (крок 3) та зверніться до джерела за відсутньою інформацією (крок 3.1) |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
3.1 |
Зворотній зв’язок (follow up) з джерелом повідомлення |
Зв’яжіться з репортером з метою отримання відсутньої інформації |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
3.2 |
Чи змогло джерело повідомлення надати необхідну відсутню інформацію? |
Якщо так, то поверніться до кроку 1 — створіть нову версію (follow up) повідомлення і надайте її належному отримувачу. Якщо ні, задокументуйте це (крок 3.3) |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
3.3 |
Задокументувати необхідну відсутню інформацію про випадок |
Задокументуйте в інформації про випадок, що ви запитували необхідну відсутню інформацію, однак джерело повідомлення не змогло або не забажало її надати |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
4 |
Надіслати отримувачу, якщо необхідно |
Якщо вимагається передати інформацію про випадок отримувачу, передайте дані про випадок в електронному вигляді, у форматі ICH-Е2В(R2) [26] у відповідний термін (15 або 90 днів) відповідному отримувачу |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
5 |
Отримати повідомлення в базі даних з фармаконагляду |
Отримати дані про випадок в електронному вигляді і завантажити їх до бази даних з фармаконагляду |
Отримувач |
|
б |
Перевірити лікарські засоби і діючі речовини |
Перевірте наявність даних у полях даних, що містять інформацію про назви лікарських засобів і діючих речовин, щоб переконатися, що торговельна назва, діюча речовина і номер серії наявні і можуть бути ідентифіковані. Це додаткова перевірка, крім звичайної перевірки |
Отримувач |
|
7 |
Перевірка пройшла успішно? |
Якщо так, збережіть повідомлення про випадок побічних реакцій у базі даних з фармаконагляду (крок 8). Якщо ні, зв’яжіться з відправником повідомлення (крок 7.1 |
Отримувач |
|
7.1 |
Зворотній зв’язок з відправником |
Зв’яжіться з відправником щодо відсутніх або неідентифікованих даних |
Отримувач |
|
7.2 |
Чи наявні всі необхідні дані про випадок? |
Після отримання запиту від отримувача перевірте дані про випадок для виявлення того, чи запитувана відсутня або неідентифікована інформація вже міститься у файлі даних про випадок. Якщо вона міститься у файлі, відкоригуйте дані про випадок (крок 7.3). Якщо інформація не міститься у файлі, зв’яжіться з джерелом повідомлення для одержання необхідної інформації (крок 3.1). |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
7.3 |
Відкоригувати дані про випадок |
Внесіть виправлення до даних про випадок, включивши відсутню інформацію, і відправте оновлену версію повідомлення отримувачу (етап 4) |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
8 |
Зберегти дані про випадок у базі даних з фармаконагляду |
Збережіть повідомлення про випадок побічної реакції у базі даних з фармаконагляду. |
Отримувач |
|
9 |
Кінець |
Дані про випадок побічної реакції доступні для виявлення сигналів і якісного аналізу даних |
VI. Додаток 2. Детальна інструкція з моніторингу медичної та наукової літератури
VI. Додаток 2.1. Коли слід починати і завершувати пошук у науковій і медичній літературі
Специфічні вимоги щодо моніторингу наукової та медичної літератури надані у VI.C.2.2.3.
Крім надання повідомлень про серйозні та несерйозні побічні реакції та представлення їх у регулярно оновлюваних звітах з безпеки лікарських засобів, заявник (власник реєстраційного посвідчення) зобов’язаний слідкувати за міжнародним досвідом застосування лікарського засобу у період після подання заяви на державну реєстрацію та отриманням реєстраційного посвідчення. Міжнародний досвід включає опубліковану наукову та медичну літературу. Моніторинг літератури у цей період часу повинен проводитися з метою виявлення публікацій, що містять інформацію, яка може вплинути на співвідношення ризик/користь лікарського засобу. З метою підготовки регулярно оновлюваних звітів з безпеки лікарських засобів (див. Модуль VII) та повідомлень про нові питання з безпеки (див. VI.C.2.2.6) вимога щодо моніторингу літератури відноситься до будь-якого лікарського засобу, що знаходиться на ринку. Моніторинг літератури повинен здійснюватися для всіх зареєстрованих лікарських засобів, незалежно від їх комерційного статусу. Цей моніторинг також повинен розпочинатися з моменту подання заяви на державну реєстрацію та здійснюватися протягом періоду дії реєстраційного посвідчення.
VI. Додаток 2.2. Де вести моніторинг
Статті, що стосуються безпеки застосування лікарських засобів, як правило, публікуються у загальновизнаних наукових і медичних журналах, однак нова та важлива інформація може вперше озвучуватися на міжнародних симпозіумах або в локальних журналах. Хоча найбільш відомі бази даних (наприклад Medline) й охоплюють більшість наукових та медичних журналів, однак найбільш важливі статті можуть публікуватися в дуже спеціалізованих медичних виданнях, зокрема, що стосуються окремих видів лікарських засобів (наприклад лікарських засобів рослинного походження) або коли проблема з безпеки є об’єктом доклінічного дослідження. Заявник (власник реєстраційного посвідчення) повинен визначити найбільш відповідне літературне джерело для кожного лікарського засобу.
Для виявлення повідомлень про побічні реакції найчастіше використовуються такі бази даних літературних джерел, як Medline, EMBASE і Excerpta Medica. Ці бази даних охоплюють широку медичну тематику. Можуть використовуватися й інші загальновизнані відповідні системи. Провайдери баз даних можуть надати поради щодо джерел записів, актуальності даних та особливостей включення джерел до бази даних. Найкращою практикою є обрання однієї або декількох баз даних, що найбільш підходять для конкретного лікарського засобу. Наприклад, оцінка співвідношення ризик/користь, проблеми з безпеки, що виявляються в ході доклінічних досліджень, можуть вимагати регулярного перегляду бази даних, що має менш медичну спрямованість і включає більше публікацій, які базуються на лабораторних дослідженнях.
Для виявлення повідомлень про випадки виникнення побічних реакцій та включення у регулярно оновлювані звіти з безпеки слід переглядати відповідні опубліковані тези конференцій, зустрічей і чорнові рукописи. Заявникам (власникам реєстраційних посвідчень) необов’язково брати участь у всіх таких зустрічах, однак, якщо персонал компанії присутній на них або якщо вони проводяться заявником (власником реєстраційного посвідчення), то вважається, що відповідні статті будуть наявні в його системі фармаконагляду. Крім того, необхідно переглядати літературні джерела, що публікуються чи спонсоруються заявником (власником реєстраційного посвідчення), з тим, щоб усі повідомлення про побічні реакції були надані в належні строки перед публікацією.
Повідомлення про побічні реакції, що заявник (власник реєстраційного посвідчення) отримав з таких джерел, повинні оброблятися так само, як і повідомлення про побічні реакції, отримані в результаті перегляду баз даних літературних джерел або медичних журналів.
У деяких базах даних індексуються та розміщуються тези важливих наукових конференцій, однак отримати з таких джерел стендові доповіді та контакти доповідачів можна дуже рідко.
VI. Додаток 2.3. Пошук у базах даних
Пошук — це більше, ніж набір ключових слів, що використовуються у запиті бази даних. Рішення про їх вибір, підхід до пошуку інформації, підбір ключових слів і застосування обмежень повинні відповідати меті пошуку. Нижче описані деякі рекомендації для організації пошуку в базах даних.
VI. Додаток 2.3.1. Точність і повнота пошуку
Медичні та наукові бази даних — це набір відомостей щодо публікацій. Усі бази даних структуровані таким чином, що полегшує організацію та пошук записів за допомогою різних засобів, від простого тексту до складного індексування термінів з пов’язаними підзаголовками. Для об’єднання поняття, розширення або зниження специфічності запиту ключові слова пошуку (текстові або індексовані) можуть бути пов’язані за допомогою логічних операторів і кодів приблизності. Крім того, можна використовувати фільтри результатів пошуку. Застосування ключових слів при здійсненні пошуку означає, що результат пошуку за обсягом буде меншим від всієї бази наявних публікацій. Успішність пошуку можна визначити на основі точності і вибірки (чутливості) пошуку. Вибірка (чутливість) — це кількісне співвідношення отриманих у результаті пошуку записів («попадань») до загальної кількості релевантних записів, наявних у базі даних. Точність — це співвідношення доречних «попадань» до загальної кількості отриманих записів. Загальновідомо, що чим ширша повнота пошуку, тим нижча його точність.
VI. Додаток 2.3.2. Побудова пошуку
Бази даних відрізняються за структурою, часом затримки при індексації та принципом індексації нових термінів (ключових слів). У деяких базах даних зберігається інформація щодо історії індексації термінів (ключових слів) чи застосування синонімів, в інших, більш примітивних, — ні.
При побудові пошукового запиту для фармаконагляду найбільша повнота результатів пошуку досягається при введенні лише назви лікарського засобу і діючої речовини (у всіх варіантах). Однак на практиці, для збільшення точності і зменшення обсягу результатів пошуку з метою отримання записів, що стосуються саме фармаконагляду, вводяться додаткові терміни (ключові слова) та текстові формулювання. Тобто при побудові пошукового запиту необхідно досягати певного балансу. Тому складні пошукові запити повинні супроводжуватися їх початковим тестуванням з тим, щоб перевірити, чи не відсіюються релевантні джерела. Однак якогось визначеного допустимого показника втрат при пошуку не існує. Набір термінів (ключових слів) повинен відповідати використовуваній базі даних і предметові пошуку.
VI. Додаток 2.3.3. Вибір ключових слів для лікарських засобів
Пошукові запити слід будувати таким чином, щоб відбирати джерела не тільки за торговельними назвами лікарських засобів, а й за діючими речовинами. У пошуковий запит можна також включати назви допоміжних речовин, що можуть мати фармакологічний ефект. Під час вибору пошукових термінів (ключових слів) для лікарських засобів, слід враховувати наступні фактори:
- назва діючої речовини в базі даних — проіндексований термін чи ні?
- які варіанти написання могли використовуватися авторами (зокрема, якщо діюча речовина не проіндексована)?
- які альтернативні назви можуть використовуватися (цифри або коди, використовувані для нещодавно розроблених лікарських засобів, хімічні назви, комерційні назви, активні метаболіти)?
- чи з медичної точки зору виправдано проводити пошук тільки для окремої солі або компонента діючої речовини?
При пошуку повідомлень про побічні реакції може бути можливо побудувати пошуковий запит, який виключає записи про лікарські форми чи способи введення, що відрізняються від предмету пошуку, однак обмеження повинні дозволяти включення статей, у яких ці дані не зазначені. Побудова пошукового запиту повинна також передбачати можливість отримання даних про передозування лікарськими засобами, медичні помилки, зловживання, неправильне застосування, застосування не за показаннями або вплив, пов’язаний з характером професійної діяльності, які можуть бути погано проіндексовані. Пошукові запити також не повинні регулярно виключати записи про немарочні лікарські засоби або записи про інші бренди компанії.
VI. Додаток 2.3.4. Вибір ключових слів для пошуку
- Як описано вище, не існує прийнятного рівня втрат при пошуку літератури з фармаконагляду. Використання пошукових термінів (простий текст або використання індексації) для побудови більш точних пошукових запитів може допомогти в організації результатів. Недоліки, що часто були виявлені в ході перевірок, які проводилися уповноваженим органом, включають: пропуск термінів, що описують результати, наприклад, «смерть» як результат може виявитися єдиним проіндексованим терміном у разі раптової смерті;
- пропуск термінів щодо вагітності при пошуку побічних реакцій, що виникли під час вагітності, для надання повідомлень про побічні реакції;
- пропуск термінів при включенні у пошук спеціальних типів повідомлень, що необхідно також враховувати в регулярно оновлюваних звітах з безпеки лікарських засобів, наприклад:
- повідомлення про безсимптомне передозування, застосування не за показаннями, медичну помилку або вплив, пов’язаний з характером професійної діяльності;
- повідомлення про вагітність, що пройшла без ускладнень.
VI. Додаток 2.3.5. Фільтрація результатів пошуку
Деякі бази даних мають індексацію, що дозволяє застосовувати фільтрацію результатів пошуку, наприклад за віком, статтю, типом публікації. Фільтри результатів пошуку не завжди відображаються в «стратегії пошуку» або пошуковому рядку.
Якщо застосовуються фільтри, вони повинні відповідати меті пошуку. При пошуку у світовій базі даних наукової та медичної літератури назви і тези, як правило, викладено англійською мовою. Використання фільтрів, що обмежують результати пошуку тільки тими, що опубліковані англійською мовою, як правило, не допускається. Фільтри щодо типів пацієнтів або інших аспектів статті, наприклад «люди», повинні бути виправданими в контексті мети пошуку.
Фільтри можуть бути застосовані для отримання результатів за певний часовий проміжок, наприклад, щотижневі моніторинги можуть проводитися шляхом вказання дати початку та закінчення періоду записів. Слід проявляти обережність, щоб гарантувати, що пошук проводиться протягом усього періоду часу, наприклад, записи, які, можливо, були додані пізніше в той же день протягом дня пошуку повинні входити в наступний період пошуку. Пошук повинен також показувати всі записи, внесені протягом окремого періоду, а не тільки первинні, що увійшли або були опубліковані протягом такого періоду (таким чином, що записи, які були оновлені або додані, отримуються ретроспективно). Якщо існують сумніви, це питання має бути узгоджене з провайдером бази даних.
Хоча однією з цілей пошуку є виявлення повідомлень про побічні реакції для їх надання, використання фільтрів за типом публікації не є надійним. Повідомлення про побічні реакції можуть бути представлені у складі публікацій про огляди або дослідження, і, відповідно, такі записи можуть бути не проіндексовані як «звіти про випадки побічних реакцій», у результаті чого вони будуть недоступні для підготовки регулярно оновлюваних звітів з безпеки лікарських засобів на основі результатів пошуку, відфільтрованих за типом публікації.
VI. Додаток 2.4. Ведення записів
Записи про моніторинги літератури повинні вестися відповідно до вимог, описаних в Імплементаційній постанові, ст. 12. Заявники (власники реєстраційних посвідчень) повинні проявляти належну обачність при пошуку опублікованої наукової та медичної літератури. Доброю практикою є збереження пошукових запитів, використаних баз даних і дати проведення пошуку. Крім того, може бути корисним зберігати результати пошуку протягом відповідного періоду часу, зокрема, у випадку нульового результату. Якщо прийняття рішень задокументовано в результатах, особливо важливо зберігати цю інформацію.
VI. Додаток 2.5. Результати
Бази даних можуть показувати результати пошуку по-різному, наприклад, тільки заголовки або заголовки статей та анотації з або без термінів індексування. Деякі публікації можуть бути очевидно актуальними на перший же погляд, у той час як інші необхідно детальніше вивчати. Відповідно до вимоги, при наданні повного тексту статті та повного посилання, завжди необхідно вказувати назву, посилання та анотацію (якщо є).
VI. Додаток 2.6. Перегляд та відбір статей
Визнається, що результати пошуку є замінником актуальної статті. Таким чином, очікується, що людина, яка переглядає результати пошуку, навчена визначати актуальні статті. Це може бути професіонал з обробки даних, який має підготовку з фармаконагляду або спеціаліст з фармаконагляду зі знанням бази даних, що використовується. Документальне підтвердження того, що результати пошуку були переглянуті, допоможе продемонструвати існування системного підходу до збору інформації з літературних джерел про підозрювані побічні реакції. Рекомендується, щоб контроль якості проводився на вибірці оглядів літератури/підбірці статей для перевірки того, як первинний рецензент виявляє актуальні статті.
Поширеною проблемою у виборі відповідних статей з результатів пошуку є той факт, що часто цей процес проводиться з метою ідентифікації виключно повідомлень про індивідуальні випадки, пов’язані з безпекою. Хоча перегляд повинен також використовуватися в якості основи для збору статей для підготовки регулярно оновлюваного звіту з безпеки лікарського засобу. Тому відповідні дослідження, що не містять повідомлень про індивідуальний випадок, пов’язаний з безпекою, також повинні бути ідентифіковані, так само як і повідомлення про явища, які не відповідають вимогам щодо формування та надання повідомлень.
Результати пошуку можуть містити достатньо інформації для того, щоб бути валідним повідомленням про індивідуальний випадок, пов’язаний з безпекою, у такому випадку стаття повинна бути замовлена. Терміновість, з якою це відбувається, повинна відповідати змісту переглянутого матеріалу та відповідним необхідним діям з боку заявника (власника реєстраційного посвідчення). Статті можуть бути виключені заявником (власником реєстраційного посвідчення) зі звітів, якщо підозрюваним лікарським засобом є брендований лікарський засіб іншої компанії. За відсутності даних щодо джерела лікарського засобу та/або його торговельної назви, приналежність лікарського засобу визначається відповідно до статей про діючу речовину. Альтернативні причини для виключення публікацій щодо повідомлень про індивідуальний випадок, пов’язаний з безпекою, докладно описані в VI.C.2.2.3.
VI. Додаток 2.7. Нульовий день
Як описано в VI.B.7, нульовий день є датою ознайомлення організації з публікацією, що містить необхідний мінімум інформації для того, щоб сформувати повідомлення про побічну реакцію, що відповідає вимогам репортування. Ознайомлення з публікацією також може здійснювати будь-який персонал цієї організації або треті сторони, які мають договірні відносини з організацією. Іноді можна визначити дату, на яку запис був доступний у базі даних, хоча при щотижневому пошуку літератури нульовий день для надання інформації про побічну реакцію, описану в анотації, визначається як дата, коли було проведено моніторинг. Для статей, що були замовлені на основі результатів пошуку літератури, нульовий день є датою, коли було отримано інформаційний мінімум для формування валідного повідомлення про побічну реакцію. Організації повинні вживати належних заходів для швидкого отримання статей для того, щоб підтвердити випадок.
VI. Додаток 2.8. Дублікати
Відповідно до вимог до формування та надання повідомлень про побічні реакції випадки, описані в літературі, повинні бути перевірені з метою запобігання наданню дублікатів, і раніше повідомлені випадки повинні бути визначені як такі, коли їх було отримано. Таким чином, очікується, що повідомлення про побічну реакцію перевіряються в базі даних організації для визначення статей, про які вже було повідомлено.
VI. Додаток 2.9. Моніторинг літературних джерел як послуга
Можна використовувати послуги третьої сторони для проведення моніторингу опублікованої наукової та медичної літератури. У цьому випадку відповідальність за виконання моніторингу та подальше надання повідомлень залишається. Передача завдань або функцій з фармаконагляду повинна бути деталізована в договорі між організацією та постачальником послуг. Характер домовленостей з третьою стороною щодо моніторингу літературних джерел може варіювати від доступу тільки до інтерфейсу (доступ до технології) конкретної бази даних до повного моніторингу літературних джерел, аналізу і надання звітів (з використанням професійних послуг фармаконагляду, що надаються іншою організацією). Визнано, що більше однієї організації може використовувати послуги третьої сторони для проведення моніторингу щодо генериків. У цьому випадку кожна організація повинна переконатися в тому, що моніторинг і послуги відповідають її потребам і зобов’язанням.
Якщо організація залежить від конкретного постачальника послуг з моніторингу літературних джерел, оцінка послуги (послуг) повинна проводитися з метою визначення того, чи відповідають вони потребам і зобов’язанням організації. У будь-якому випадку умови повинні бути чітко задокументовані.
Відлік часу для надання повідомлень про побічні реакції починається з отримання інформаційного мінімуму організацією або партнером за відповідним договором (що відбудеться раніше). Дана умова також діє, коли третя сторона проводить огляд або співставлення звіту опублікованої наукової та медичної літератури з метою переконатися, що опубліковані в літературі випадки повідомляються належним чином в належний час. Таким чином, нульовим днем є день початку моніторингу, якщо мінімальні критерії наявні в анотації, а не день, коли інформація була надана організації.
VI. Додаток 2.10. Надання копій статей, опублікованих у науковій та медичній літературі, в електронній формі
До розроблення стандартів для електронної передачі файлів (наприклад, копії статей) не в рамках ICH, відправник повинен дотримуватися правил, зазначених нижче, для надання копій статей, як зазначено в VI.C.6.2.3.2:
Адреса відправлення та формат статей:
1. Статті, що надаються ЕМА, повинні бути представлені у форматі PDF і надіслані електронною поштою на наступну адресу: [email protected].
Щодо копій статей з опублікованої наукової та медичної літератури, заявникам (власникам реєстраційних посвідчень) рекомендується зважати на питання про потенційні проблеми авторського права, особливо щодо питань електронних засобів передачі та обробки електронних копій у рамках нормативної діяльності.
2. Ім’я файлу для статей, що надсилаються в електронному форматі до ЕМА:
Ім’я файлу для статті, що надсилається в форматі PDF, має точно співпадати з Унікальним глобальним ідентифікаційним номером випадку (Worldwide Unique Case Identification Number) (ICH-E2B(R2) [26] A.1.10.1 або A.1.10.2 за наявності), призначеним у кожному окремому випадку, який описаний у статті, і який повідомляється в форматі повідомлення про індивідуальний випадок, пов’язаний з безпекою ICH-Е2В(R2) [26].
Якщо стаття з подальшим відстеженням випадку опублікована в літературних джерелах, ім’я файлу, що містить унікальний глобальний ідентифікаційний номер випадку, повинне бути збережене, але має включати порядковий номер, відокремлений дефісом.
Приклади:
- Первинне повідомлення про побічну реакцію, опубліковане в літературних джерелах: FR-ORGABC-23232321 (унікальний глобальний ідентифікаційний номер випадку (ICH-E2B(R2) [26] A.1.10.1)):
- ім’я файлу статті: FR-ORGABC-23232321.pdf;
- Дані подальшого відстеження, опубліковані в літературних джерелах в окремій статті:
- повідомлення про індивідуальний випадок, пов’язаний з безпекою під номером: FR-ORGABC-23232321 (унікальний глобальний ідентифікаційний номер випадку залишається незмінним (ICH-E2B(R2) [26] A.1.10.1));
- ім’я файлу: FR-ORGABC-23232321-1.pdf.
3. Повідомлення про випадки, описані в науковій та медичній літературі, з посиланням на більше ніж одного пацієнта.
Коли в статі описано інформацію про більш ніж одного пацієнта, копія статті повинна надсилатися тільки один раз.
Ім’я файлу статті, що надсилається в форматі PDF, повинне точно співпадати з Унікальним глобальним ідентифікаційним номером випадку) (поле даних ICH-E2B(R2) [26] A.1.10.1 або A.1.10.2 за наявності), призначеним першому окремому випадку, що описаний у статті.
Крім того, всі повідомлення про побічні реакції, що відносяться до однієї статті, повинні містити перехресні посилання у полі даних Identification Number of The Report Which is Linked to This Report (ідентифікаційний номер повідомлення, що пов’язаний з даним повідомленням) (ICH-E2B(R2) [26] A.1.12). Зазначене поле даних слід повторювати за необхідності, щоб вказати на всі пов’язані випадки (див. таблицю VI.2).
Таблиця 3. Приклади для надання повідомлень про індивідуальні випадки, пов’язані з безпекою, що описані в науковій та медичній літературі, з посиланням на більше ніж одного пацієнта:
|
Сценарій |
Дії |
|
В статті описано підозрювані побічні реакції, що виникли менш ніж у 3 окремих пацієнтів |
Для випадку 1, описаного в статті:
UK-ORGABC-0001 |
|
Повинні бути складені і надані 3 повідомлення про індивідуальний випадок, пов’язаний з безпекою для кожного окремого ідентифікованого пацієнта, описаного в статті Кожне повідомлення про індивідуальний випадок, пов’язаний з безпекою повинне містити всю наявну інформацію про випадок |
UK-ORGABC-0003
Літературні джерела, що відповідають єдиним вимогам до рукописів, які надаються в біомедичні журнали. N Engl J Med. 1997; 336: 309–15.
UK-ORGABC-0001.pdf Для випадку 2, описаного в статті:
UK-ORGABC-0002
UK -ORGABC-0001
UK-ORGABC-0003
N Engl J Med. 1997; 336:309–15.
|
|
Для випадку 3, описаного в статті:
UK-ORGABC-003
UK-ORGABC-0001 |
|
UK-ORGABC-0002
N Engl J Med. 1997; 336: 309–15. Копія статті не вимагається, оскільки вона вже була надана для випадку 1. |
|
|
В статті описано підозрювані побічні реакції, що виникли більш ніж у 3 окремих пацієнтів. Повинні бути складені і надані повідомлення про індивідуальний випадок, пов’язаний з безпекою, по одному для кожного окремого ідентифікованого пацієнта, описаного в статті. Кожне повідомлення про індивідуальний випадок, пов’язаний з безпекою, повинне містити всю наявну інформацію про випадок. Перехресні посилання з усіма пов’язаними повідомленнями про індивідуальний випадок, пов’язаний з безпекою, із цієї статті повинні бути надані тільки в описі першого випадку, у полі даних ICHE2B (R2) [26] A.1.12 Identification Number of The Report Which is Linked to This Report (ідентифікаційний номер повідомлення, що пов’язаний з даним повідомленням). |
Для повідомлень про побічні реакції, що стосуються однієї статті, перехресні посилання в полі даних ICHE2B(R2) [26] A.1.12 Identification Number of The Report Which is Linked to This Report (ідентифікаційний номер звіту, що пов’язаний з даним звітом) слід надавати наступним чином:
Приклад звіту про випадки, вперше описані в науковій та медичній літературі з посиланням на велику кількість пацієнтів: Для випадку 1, описаного в статті:
UK-0RGABC-0001
UK-0RGABC-0002
UK-0RGABC-0003 |
|
Немає необхідності повторювати всі перехресні посилання в інших повідомленнях про індивідуальний випадок, пов’язаний з безпекою. |
UK-0RGABC-0004 |
UK-0RGABC-000N
N Engl J Med. 1997; 336:309-15.
UK-0RGABC-0001.pdf. Для випадку 2, описаного в статті:
UK-0RGABC-0002
UK-0RGABC-0001
N Engl J Med. 1997; 336: 309-15.
Для випадку N, описаного в статті:
UK-ORGABC-000N
UK -ORGABC-0001
Engl J Med. 1997; 336: 309–15.
|
VI. Додаток 3. Процедури подання звітності
VI. Додаток 3.1. Тимчасові заходи
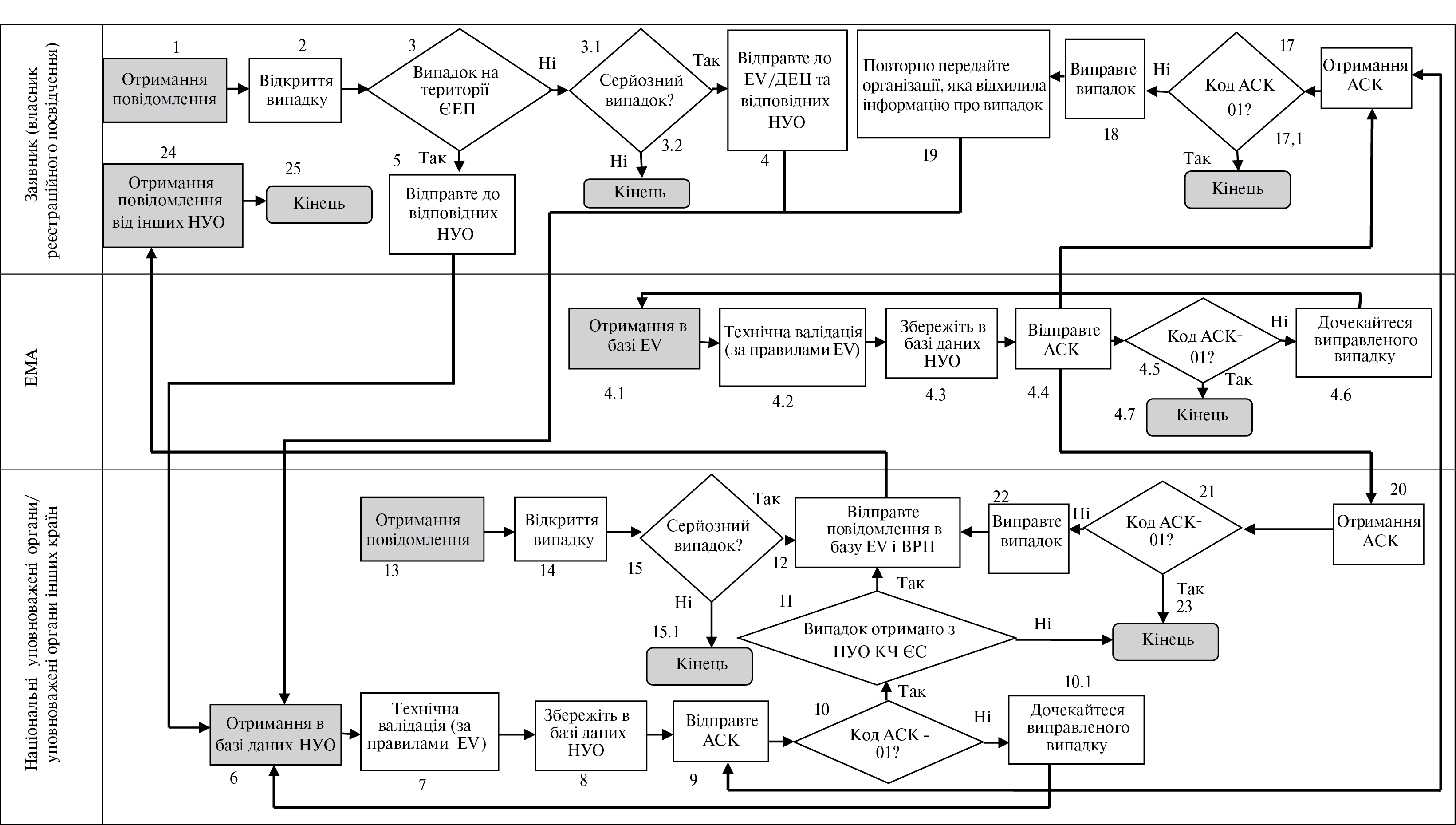
Рисунок 3. Схема бізнес-процесів — повідомлення про підозрювані побічні реакції в ЄС. Тимчасові заходи
Таблиця 4. Описання процесів — повідомлення про підозрювані побічні реакції в ЄС. Тимчасові заходи
|
№ |
Крок |
Описання |
Відповідальна організація |
|
1 |
Початок Отримання повідомлення |
Заявник (власник реєстраційного посвідчення) отримує інформацію про випадки виникнення підозрюваних побічних реакцій від пацієнта, працівника з медичною та/або фармацевтичною освітою або іншої належної особи, що надає інформацію. Якщо дані про випадок було отримано від національного уповноваженого органу ЄС, не треба надавати їх до EudraVigilance (EV) |
Заявник (власник реєстраційного посвідчення) |
|
2 |
Відкрийте випадок |
Відкрийте і створіть повідомлення про побічну реакцію |
Заявник (власник реєстраційного посвідчення) |
|
3 |
Дані про випадок надійшли з ЄЕП? |
Побічна реакція мала місце в ЄС? Якщо ні, перейдіть до кроку 3.1. Якщо так, перейдіть до кроку 5. |
Заявник (власник реєстраційного посвідчення) |
|
3.1 |
Чи є випадок серйозним? |
Якщо ні, перейдіть до кроку 3.2. Якщо так, перейдіть до кроку 4. |
Заявник (власник реєстраційного посвідчення) |
|
3.2 |
Кінець |
Тепер дані про випадок збережено в базі даних з фармаконагляду. Слід продовжувати вести нормальне подальше відстеження. У разі отримання будь-яких даних з подальшого відстеження, поверніться до кроку 1 |
Заявник (власник реєстраційного посвідчення) |
|
4 |
Направте дані до EudraVigilance і до відповідних національних уповноважених органів |
Передайте дані про серйозний випадок в електронному вигляді, у форматі повідомлення ICH-E2B(R2) [26] XML протягом 15 днів до EudraVigilance і відповідного національного уповноваженого органу, у разі необхідності. Обробка даних про випадок, переходять до кроку 4.1 і кроку 6 |
Заявник (власник реєстраційного посвідчення) |
|
4.1 |
Отримайте в EudraVigilance |
Отримайте повідомлення в базі даних EudraVigilance від заявника (власника реєстраційного посвідчення) або національного уповноваженого органу (НУО) |
EMA |
|
4.2 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення, що отримується EudraVigilance, звіряється з правилами EudraVigilance, після чого створюється повідомлення про підтвердження (Acknowledgement message (ACK)), в якому зазначається, чи є валідним повідомлення і випадок (випадки), описані у ньому |
EMA |
|
Валідне повідомлення буде мати код ACK 01. Невалідне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є валідним) або 03 (якщо саме повідомлення має неправильний формат) |
|||
|
4.3 |
Збережіть в EudraVigilance |
Як тільки дані про випадок перевірено, вони зберігаються в EudraVigilance |
EMA |
|
4.4 |
Відправте повідомлення про підтвердження (ACK) |
Повідомлення про підтвердження (ACK), створене у кроці 4.2, передається відправникові даних про випадок не пізніше 2 робочих днів після отримання таких даних. Перейдіть до кроку 16 для заявників (власників реєстраційних посвідчень), що отримують повідомлення про підтвердження (ACK). Перейдіть до кроку 20 для національних уповноважених органів, що отримують повідомлення про підтвердження (ACK). Перейдіть до кроку 4.5 для наступного кроку EMA |
EMA |
|
4.5 |
Чи код повідомлення про підтвердження (ACK) дорівнює 01? |
Якщо ні, перейдіть до кроку 4.6. Якщо так, перейдіть до кроку 4.7 |
EMA |
|
4.6 |
Дочекайтеся надання виправлених даних про випадок |
Відправник повинен виправити дані про кожен випадок повідомлення про підтвердження (ACK), що має код помилки, і повторно передати їх, дотримуючись встановленого терміну звітування. EMA повинне періодично перевіряти всі випадки повідомлення про підтвердження (ACK), що мають код помилки і для яких виправлення не були передані, та зв’язуватися з уповноваженою особою, відповідальною за фармаконагляд (УОВФ)(QPPV) щоб поінформувати її про такі випадки. Якщо відправник не вносить виправлення до даних про випадок, ця інформація повинна бути включена до оцінки якості даних і повинні бути поінформовані відповідні комітети. Поверніться до кроку 4.1 після отримання виправлених даних про випадок |
EMA |
|
4.7 |
Кінець |
Тепер дані про випадок збережено в системі EudraVigilance, подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних |
EMA |
|
5 |
Направте дані до відповідних національних уповноважених органів |
Передайте дані про випадок (серйозний і, якщо потрібно, несерйозний) в електронному вигляді, у форматі повідомлення XML ICH-E2B(R2) [26] у відповідний строк (15 або 90 днів, залежно від обставин) відповідному національному уповноваженому органу тієї країни ЄС, де мала місце побічна реакція. Якщо дана країна ЄС не вказана, тоді країна першоджерела, як правило, вважається країною, де мала місце відповідна побічна реакція |
Заявник (власник реєстраційного посвідчення) |
|
6 |
Отримайте в базі даних з фармаконагляду (PhV DB) |
Отримання повідомлення від заявника (власника реєстраційного посвідчення) в базі даних з фармаконагляду національного уповноваженого органу |
Національний уповноважений орган |
|
7 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення, що отримується в базі даних з фармаконагляду національного уповноваженого органу, перевіряється на відповідність вимогам EudraVigilance, після чого створюється повідомлення про підтвердження (ACK), в якому зазначається, чи валідним є повідомлення і випадок, описаний у ньому. Валідне повідомлення буде мати код ACK 01. Невалідне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є валідним) або 03 (якщо саме повідомлення має неправильний формат) |
Національний уповноважений орган |
|
8 |
Збережіть в EudraVigilance |
Як тільки дані про випадок перевірено, вони зберігаються в базі даних фармаконагляду національного уповноваженого органу |
Національний уповноважений орган |
|
9 |
Відправте повідомлення про підтвердження (ACK) |
Повідомлення про підтвердження (ACK), створене в кроці 7, передається відправникові даних про випадок не пізніше 2 робочих днів після отримання таких даних |
Національний уповноважений орган |
|
Перейдіть до кроку 16 для заявників (власників реєстраційних посвідчень), що отримують повідомлення про підтвердження (ACK). Перейдіть до кроку 10 для наступного кроку національного уповноваженого органу |
|||
|
10 |
Чи код повідомлення про підтвердження (ACK) дорівнює 01? |
Якщо ні, перейдіть до кроку 10.1. Якщо так, перейдіть до кроку 11 |
Національний уповноважений орган |
|
10.1 |
Дочекайтеся надання виправлених даних про випадок |
Заявник (власник реєстраційного посвідчення) повинен виправити дані про кожен випадок, повідомлення про підтвердження (ACK), що містить помилку, і повторно передати їх, дотримуючись встановленого терміну звітування. Національний уповноважений орган повинен періодично перевіряти всі випадки, повідомлення про підтвердження (ACK), що мають код помилки і для яких виправлення не були передані, і зв’язуватися з уповноваженою особою, відповідальною за фармаконагляд (УОВФ)(QPPV) для того, щоб поінформувати її про такі випадки відсутності виправлень. Якщо відправник не вносить виправлення до даних про випадок, ця інформація повинна бути включена до всіх оцінок якості даних і повинні бути вжиті належні заходи. Поверніться до кроку 6 після отримання виправлених даних про випадок |
Національний уповноважений орган |
|
11 |
Дані про випадок надійшли з національного уповноваженого органу країни ЄС? |
Випадок мав місце на території, за яку відповідає національний уповноважений орган, що отримав інформацію? Якщо ні, перейдіть до кроку 11.1. Якщо так, перейдіть до кроку 12. |
Національний уповноважений орган |
|
11.1 |
Кінець |
Тепер дані про випадок збережено в базі даних з фармаконагляду національного уповноваженого органу, подальше виявлення дублікатів та внесення записів може проводитися для виявлення сигналів і якісного аналізу даних |
Національний уповноважений орган |
|
12 |
Направте дані до EudraVigilance і до заявників (власників реєстраційних посвідчень) |
Передайте дані про серйозний випадок в електронному вигляді, у форматі повідомлення ICH-E2B(R2) [26] XML протягом 15 днів до EudraVigilance і заявників (власників реєстраційних посвідчень). Перейдіть до кроку 4.1 для отримання даних про випадок від EudraVigilance. Перейдіть до кроку 24 для отримання даних про випадок відповідним заявником (власником реєстраційного посвідчення) |
Національний уповноважений орган |
|
13 |
Початок. Отримання повідомлення |
Національний уповноважений орган отримує інформацію про випадок виникнення підозрюваної побічної реакції від пацієнта, працівника з медичною та/або фармацевтичною освітою або іншої належної особи, яка надає дану інформацію про випадок, що мав місце на території, за яку відповідає уповноважений орган, що отримав такі дані |
Національний уповноважений орган |
|
14 |
Відкрийте випадок |
Відкрийте і створіть повідомлення про індивідуальний випадок, пов’язаний з безпекою |
Національний уповноважений орган |
|
15 |
Чи є випадок серйозним? |
Якщо ні, перейдіть до кроку 15.1 Якщо так, перейдіть до кроку 12 |
Національний уповноважений орган |
|
15.1 |
Кінець |
Тепер дані про випадок збережено в базі даних з фармаконагляду національного уповноваженого органу, подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних |
Національний уповноважений орган |
|
16 |
Отримайте повідомлення про підтвердження (ACK) |
Отримайте повідомлення про підтвердження (ACK), зв’яжіть його з відповідним випадком і переконайтеся, що інформація про випадок була помічена як валідна |
Заявник (власник реєстраційного посвідчення) |
|
17 |
Чи код повідомлення про підтвердження (ACK) дорівнює 01? |
Якщо так, перейдіть до кроку 17,1. Якщо ні, то відлік часу продовжується і інформація про випадок повинна бути виправлена і повторно передана до EudraVigilance у відповідні нормативні строки. Днем 0 залишається день, коли інформація була отримана вперше. Повідомлення про підтвердження (ACK) з кодом 02 або 03 не є новою інформацією. Перейдіть до кроку 18 (Виправте дані про випадок) |
Заявник (власник реєстраційного посвідчення) |
|
17.1 |
Кінець |
Кінець процесу передачі цієї версії даних про випадок до EudraVigilance або національного уповноваженого органу. Слід продовжувати вести нормальне подальше відстеження. У разі отримання будь-яких даних з подальшого відстеження, поверніться до кроку 1. |
Власник реєстраційних посвідчень |
|
18 |
Внесіть виправлення до даних про випадок |
Виправте дані про випадок для видалення помилок, вказаних у повідомленні про підтвердження (ACK) |
Заявник (власник реєстраційного посвідчення) |
|
19 |
Повторно передайте організації, яка відхилила інформацію про випадок |
Повторно передайте виправлену інформацію про випадок організації, котра відхилила інформацію про випадок, що має код повідомлення про підтвердження (ACK) 02 або 03. Перейдіть до кроку 4.1 та/або кроку 6 залежно від необхідності |
Заявник (власник реєстраційного посвідчення) |
|
20 |
Отримайте повідомлення про підтвердження (ACK) |
Отримайте повідомлення про підтвердження (ACK), зв’яжіть його з відповідним випадком і переконайтеся, що інформація про випадок була помічена як валідна |
Національний уповноважений орган |
|
21 |
Чи код повідомлення про підтвердження (ACK) дорівнює 01? |
Якщо так, перейдіть до кроку 23. Якщо ні, то відлік часу продовжується і інформація про випадок повинна бути виправлена і повторно передана до EudraVigilance у відповідні нормативні строки. Днем 0 залишається день, коли інформація була отримана вперше. Повідомлення про підтвердження (ACK) з кодом 02 або 03 не є новою інформацією. Перейдіть до кроку 22 (Виправте дані про випадок) |
Національний уповноважений орган |
|
22 |
Внесіть виправлення до даних про випадок |
Виправте дані про випадок для видалення помилок, вказаних у повідомленні про підтвердження (ACK), і повторно передайте дані про випадок до EudraVigilance і до відповідного заявника (власника реєстраційного посвідчення) (поверніться до кроку 12) |
Національний уповноважений орган |
|
23 |
Кінець |
Кінець процесу передачі цієї версії даних про випадок до EudraVigilance та відповідного заявника (власника реєстраційного посвідчення). Слід продовжувати вести нормальне подальше відстеження. У разі отримання будь-яких даних подальшого відстеження поверніться до кроку 6 або 13 |
Національний уповноважений орган |
|
24 |
Отримайте звіт від національного уповноваженого органу |
Заявник (власник реєстраційного посвідчення) отримує інформацію про випадок виникнення підозрюваної побічної реакції від національного уповноваженого органу. Дані про цей випадок не повинні повторно передаватися до EudraVigilance і національного уповноваженого органу, який передав їх власнику реєстраційних посвідчень |
Заявник (власник реєстраційного посвідчення) |
|
25 |
Кінець |
Тепер дані про випадок збережено в базі даних фармаконагляду заявника (власника реєстраційного посвідчення) Подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних |
Заявник (власник реєстраційного посвідчення) |
VI. Додаток 3.1.1. Тимчасові заходи, що застосовуються до заявників (власників реєстраційних посвідчень)
Вимоги щодо надання повідомлень про індивідуальні випадки, пов’язані з безпекою для заявників (власників реєстраційних посвідчень), що мають дію протягом перехідного періоду, детально описані в останній версії документа EMA/321386/2012 на вебсайті ЕМА.
VI. Додаток 3.1.2. Тимчасові заходи, що застосовуються до уповноважених органів країн ЄС
Таблиця 5. Вимоги щодо надання повідомлень, що застосовуються до уповноважених органів країн ЄС. Тимчасові заходи VI. Додаток 3.2. Фінальні заходи
|
Процедура видачі реєстраційних посвідчень |
Походження |
Тип побічної реакції |
Призначення |
Термін надання |
|
Централізована Взаємне визнання, децентралізована або підлягають направленню Виключно на рівні країни |
Євросоюз |
Усі серйозні |
|
15 днів |
VI. Додаток 3.2. Фінальні заходи
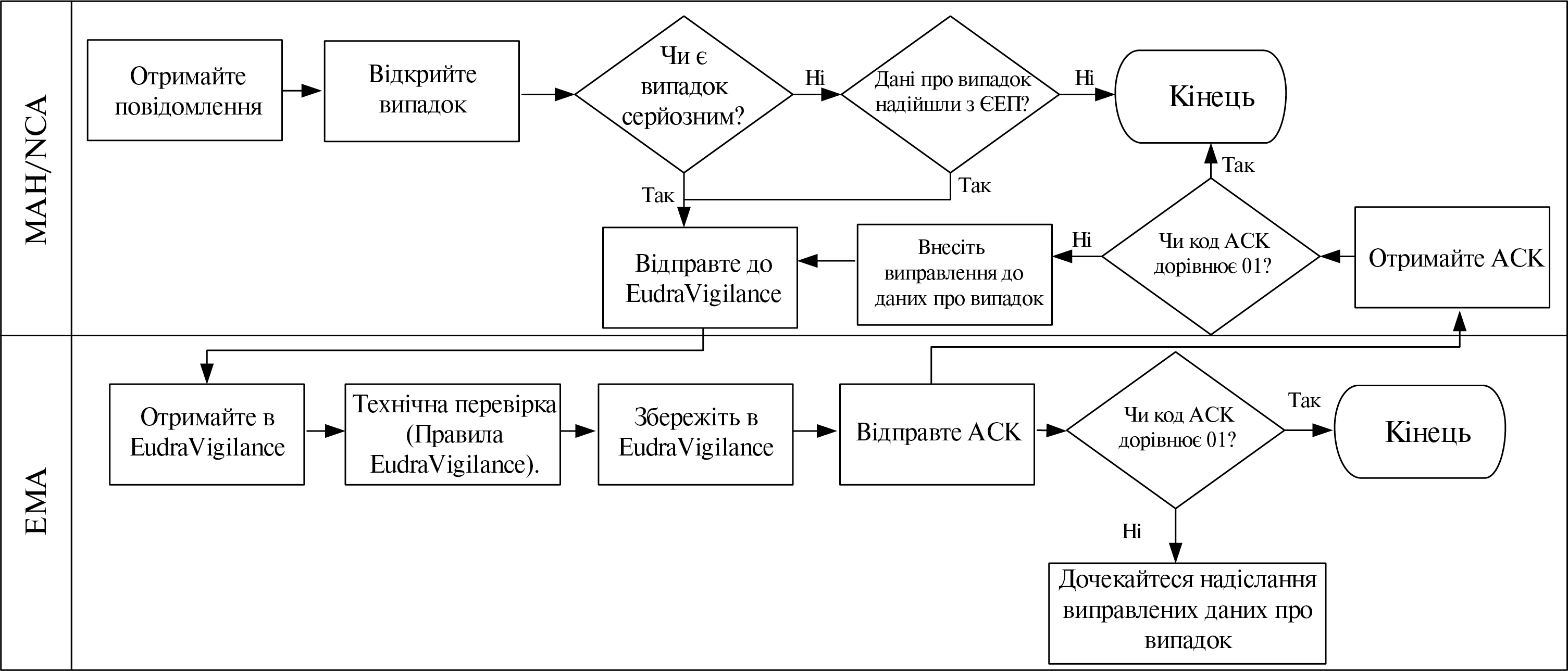
Рисунок 4. Схема робочого процесу – повідомлення про підозрювані побічні реакції в ЄС. Фінальні заходи
Таблиця 6. Описання процесів — Повідомлення про підозрювані побічні реакції в ЄС — Фінальні заходи
|
№ |
Крок |
Описання |
Відповідальна організація |
|
1 |
Початок Отримання повідомлення |
Національний уповноважений орган або заявник (власник реєстраційного посвідчення) отримує інформацію про випадок виникнення підозрюваних побічних реакцій від пацієнта, працівника з медичною та/або фармацевтичною освітою або іншої належної особи, що надає інформацію. Якщо дані про випадок було отримано від національного уповноваженого органу ЄС, не надавайте їх до EudraVigilance (EV). |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
2 |
Відкрийте випадок |
Відкрийте і створіть повідомлення про індивідуальний випадок, пов’язаний з безпекою |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
3 |
Чи є випадок серйозним? |
Якщо ні, перейдіть до кроку 3.1. Якщо так, перейдіть до кроку 4 |
|
|
3.1 |
Дані про випадок надійшли з ЄЕП? |
Якщо ні, перейдіть до кроку 11.1. Якщо так, перейдіть до кроку 4 |
|
|
4 |
Направте дані до EudraVigilance |
Передайте дані про випадок (несерйозні, що сталися в ЄС, та всі серйозні) в електронному вигляді, у форматі повідомлення XML ICH-E2B(R2) [26] у відповідний строк (15 або 90 днів, залежно від обставин) до EudraVigilance |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
5 |
Отримайте в EudraVigilance |
Отримайте повідомлення з EudraVigilance |
EMA |
|
6 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення, що отримується EudraVigilance [10], перевіряється на відповідність правилам EudraVigilance, після чого створюється ACK, у якому зазначається, чи належними є повідомлення і випадок, описаний у ньому. Належне повідомлення буде мати код ACK 01. Неналежне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є належним) або 03 (якщо саме повідомлення має неправильний формат) |
EMA |
|
7 |
Збережіть в EudraVigilance [10] |
Як тільки дані про випадок перевірено, вони зберігаються в EudraVigilance [10] |
EMA |
|
8 |
Відправте повідомлення про підтвердження (ACK) |
Повідомлення про підтвердження (ACK), створене у кроці 6, передається відправникові даних про випадок не пізніше 2 робочих днів після отримання таких даних. Перейдіть до кроку 9 для наступного кроку Європейського агентства з лікарських засобів (EMA). Перейдіть до кроку 10 для наступного кроку заявника (власника реєстраційного посвідчення)/національного уповноваженого органу |
EMA |
|
9 |
Чи код ACK дорівнює 01? |
Якщо ні, перейдіть до кроку 9.1. Якщо так, перейдіть до кроку 9.2 |
EMA |
|
9.1 |
Дочекайтеся надсилання виправлених даних про випадок |
Відправник повинен виправити дані про кожен випадок, ACK якого має код помилки, і повторно передати їх, дотримуючись встановленого терміну. EMA повинне періодично перевіряти всі випадки, ACK яких мають код помилки і для яких виправлення не були передані, та зв’язуватися з кваліфікованою особою, яка відповідає за фармаконагляд (QPPV), щоб поінформувати її про це. Якщо відправник не вносить виправлення до даних про випадок, ця інформація повинна бути включена до оцінки якості даних і повинні бути проінформовані відповідні комітети. Поверніться до кроку 5 після отримання виправлених даних про випадок. |
EMA |
|
9.2 |
Кінець |
Тепер дані про випадок збережено в системі EudraVigilance, подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних. Якщо випадок стався в ЄС і був переданий до EudraVigilance заявником (власником реєстраційного посвідчення), дані про нього буде перенаправлено до відповідного національного уповноваженого органу (див. VI. Додаток 3.3.) |
EMA |
|
10 |
Отримайте ACK |
Отримайте ACK, зв’яжіть його з відповідним випадком і переконайтеся, що інформація про випадок була помічена як належна |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
11 |
Чи код ACK дорівнює 01? |
Якщо так, перейдіть до кроку 11,1. Якщо ні, то відлік часу продовжується і інформація про випадок повинна бути виправлена і повторно передана до EudraVigilance відповідно до нормативних строків. Днем 0 залишається день, коли інформація була отримана вперше. ACK з кодом 02 або 03 не є новою інформацією. Перейдіть до кроку 12 (Виправте дані про випадок) |
Заявник (власник реєстраційного посвідчення) національний уповноважений орган |
|
11.1 |
Кінець |
Кінець процесу для цієї версії даних про випадок. Слід продовжувати вести подальше відстеження. У разі отримання будь-яких даних з подальшого відстеження, поверніться до кроку 1 |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
12 |
Внесіть виправлення до даних про випадок |
Виправте дані про випадок для видалення помилок, вказаних в ACK, і повторно передайте дані про випадок до EudraVigilance (поверніться до кроку 4) |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
VI. Додаток 3.2.1. Фінальні заходи, що застосовуються до заявників (власників реєстраційних посвідчень)
Таблиця 7. Вимоги щодо надання повідомлень, що діють для заявників (власників реєстраційних посвідчень). Фінальні заходи
|
Процедура видачі реєстраційних посвідчень |
Походження |
Тип побічної реакції |
Призначення |
Термін надання |
|
Централізована Взаємне визнання, децентралізована або підлягають направленню Виключно на рівні країни |
ЄС |
Усі серйозні |
|
15 днів |
|
Усі несерйозні |
|
90 днів |
||
|
Не входять до ЄС |
Усі серйозні |
|
15 днів |
VI. Додаток 3.2.2. Фінальні заходи, що застосовуються до уповноважених органів країн ЄС
Таблиця 8. Вимоги щодо надання повідомлень, що застосовуються до уповноважених органів країн ЄС. Фінальні заходи
|
Процедура видачі реєстраційних посвідчень |
Походження |
Тип побічної реакції |
Призначення |
Термін надання |
|
Централізована Взаємне визнання, децентралізована або підлягають направленню Виключно на рівні країни |
ЄС |
Усі серйозні |
|
15 днів |
|
Усі несерйозні |
|
90 днів |
VI. Додаток 3.3 Передача та перенаправлення повідомлень про індивідуальні випадки, пов’язані з безпекою, до уповноважених органів країн ЄС53
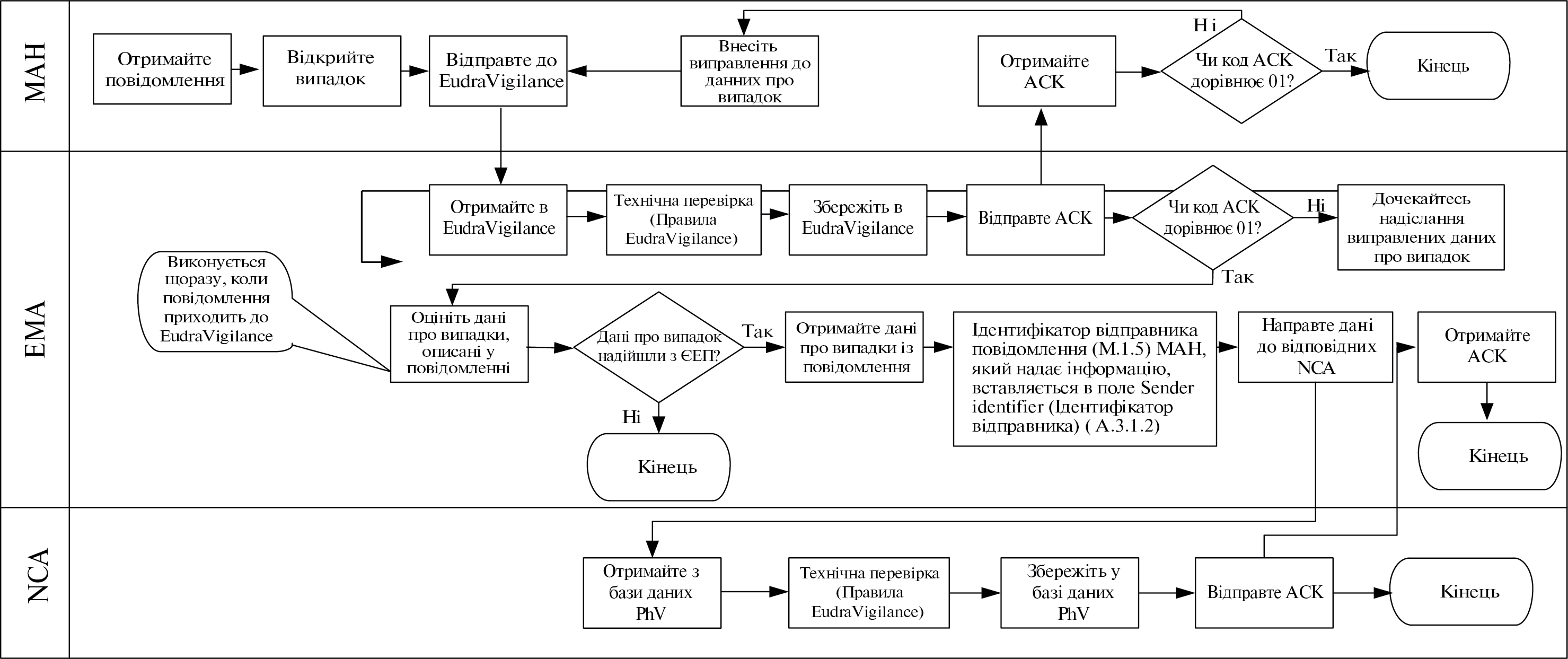
Рисунок 5. Схема робочого процесу — передача та перенаправлення ICSR до уповноважених органів країн ЄС
Таблиця 9. Описання процесів — передача та перенаправлення повідомлень про індивідуальні випадки, пов’язані з безпекою до уповноважених органів країн ЄС54
|
№ |
Найменування |
Описання |
Відповідальна організація |
|
1 |
Початок Отримання повідомлення |
Заявник (власник реєстраційного посвідчення) отримує інформацію про випадки виникнення підозрюваних побічних реакцій від пацієнта, працівника з медичною та/або фармацевтичною освітою або іншої належної особи, що надає інформацію |
Заявник (власник реєстраційного посвідчення) |
|
2 |
Відкрийте випадок |
Відкрийте і створіть повідомлення про індивідуальний випадок, пов’язаний з безпекою |
Заявник (власник реєстраційного посвідчення) |
|
3 |
Направте дані до EudraVigilance |
Передайте інформацію про випадок в електронному вигляді у форматі ICHE2B(R2) [26] як повідомлення XML у відповідний строк (15 або 90 днів, залежно від обставин) до EudraVigilance |
Заявник (власник реєстраційного посвідчення) |
|
4 |
Отримайте в EudraVigilance |
Отримайте повідомлення з EudraVigilance |
EMA |
|
5 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення, що отримується EudraVigilance, перевіряється на відповідність правилам EudraVigilance, після чого створюється ACK, у якому зазначається, чи належними є повідомлення і випадок, описаний у ньому. Належне повідомлення буде мати код ACK 01. Неналежне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є належним) або 03 (якщо саме повідомлення має неправильний формат) |
EMA |
|
6 |
Збережіть в EudraVigilance |
Як тільки дані про випадок перевірено, вони зберігаються в EudraVigilance |
EMA |
|
7 |
Відправте ACK |
ACK, створене на кроці 5, передається відправникові даних про випадок не пізніше 2 робочих днів після отримання таких даних |
EMA |
|
7.1 |
Отримайте ACK |
Отримайте ACK, зв’яжіть його з відповідним випадком і переконайтеся, що інформація про випадок була помічена як належна |
Заявник (власник реєстраційного посвідчення) |
|
7.2 |
Чи код ACK дорівнює 01? |
Якщо так, перейдіть до кроку 7.2.1. Якщо ні, то відлік часу продовжується і інформація про випадок повинна бути виправлена і повторно передана до EudraVigilance у відповідні нормативні строки. Днем 0 залишається день, коли інформація була отримана вперше. ACK з кодом 02 або 03 не є новою інформацією. Перейдіть до кроку 7.2.2 (Виправте дані про випадок) |
Заявник (власник реєстраційного посвідчення) |
|
7.2.1 |
Кінець |
Кінець процесу передачі цієї версії даних про випадок до EudraVigilance. Слід продовжувати вести нормальне подальше відстеження. У разі отримання будь-яких даних з подальшого відстеження, поверніться до кроку 1 |
Заявник (власник реєстраційного посвідчення) |
|
7.2.2 |
Внесіть виправлення до даних про випадок |
ACK, і повторно передайте дані про випадок до EudraVigilance (поверніться до кроку 3) |
Заявник (власник реєстраційного посвідчення) |
|
8 |
Чи код ACK дорівнює 01? |
Якщо так, перейдіть до кроку 9. Якщо ні, не проводьте ніякої подальшої обробки цієї версії даних про випадок і перейдіть до кроку 8.1 |
EMA |
|
8.1 |
Дочекайтеся надіслання виправлених даних про випадок |
Відправник повинен виправити дані про кожен випадок, ACK якого має код помилки, і повторно передати їх, дотримуючись встановленого терміну. EMA повинне періодично перевіряти всі випадки, ACK яких мають код помилки і для яких виправлення не були передані, і зв’язуватися з кваліфікованою особою, яка відповідає за фармаконагляд (QPPV), щоб поінформувати її про такі випадки. Якщо відправник не вносить виправлення до даних про випадок, ця інформація повинна бути включена до оцінки якості даних і повинні бути проінформовані відповідні комітети. |
EMA |
|
9 |
Оцініть дані про випадки, описані у повідомленні |
Коли повідомлення пройшло технічну перевірку, дані про випадки, що містяться в ньому, повинні бути негайно вивчатися для визначення країни, де мала місце реакція, для цілей регуляторної звітності |
EMA |
|
10 |
Дані про випадок надійшли з ЄЕП? |
Для даних про кожний випадок оцінити, чи є країна, де мала місце відповідна реакція, країною ЄС. Якщо так, перейдіть до кроку 11. Якщо ні, перейдіть до кроку 10.1 |
EMA |
|
10.1 |
Кінець |
Тепер дані про випадок збережено в системі EudraVigilance, подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних |
EMA |
|
11 |
Отримайте дані про випадки із повідомлення |
Випадки, що сталися в ЄС, отримуються з повідомлення для їх обробки до повторної передачі |
EMA |
|
12 |
Технічна перевірка. |
Ідентифікатор відправника повідомлення (ICH M2 M.1.5) заявника (власника реєстраційного посвідчення), що надає інформацію, вноситься в поле Sender identifier (Ідентифікатор відправника) (ICH-E2B(R2) [26] А.3.1.2) до повторної передачі. Це надає можливість національному уповноваженому органу, що отримує інформацію, можливість здійснювати однозначну ідентифікацію заявника (власника реєстраційного посвідчення), відповідального за передачу даних про випадок до EudraVigilance |
EMA |
|
13 |
Направте дані до відповідних національних уповноважених органів |
Дані про випадок передаються до відповідного національного уповноваженого органу країни ЄС, де мала місце відповідна побічна реакція, без інших змін. Якщо країна ЄС має більше одного національного уповноваженого органу, відповідального за післяреєстраційні звіти, дані про випадки, що відбуваються в цій країні ЄС, направляються до всіх її національних уповноважених органів |
EMA |
|
14 |
Отримайте з бази даних з фармаконагляду (PhV) |
Відповідний національний уповноважений орган отримує повідомлення в свою базу даних з фармаконагляду |
Національний уповноважений орган |
|
15 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення повинне бути перевірене на предмет відповідності правилам EudraVigilance (ті ж правила, що й в кроці 5). ACK створюється із зазначенням того, чи є належними повідомлення і випадок, описаний у ньому. |
Національний уповноважений орган |
|
Належне повідомлення буде мати код ACK 01. Неналежне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є належним) або 03 (якщо саме повідомлення має неправильний формат) |
|||
|
16 |
Збережіть в базі даних з фармаконагляду (PhV) |
Як тільки дані про випадок перевірено, вони зберігаються в базі даних з фармаконагляду |
Національний уповноважений орган |
|
17 |
Відправте ACK. |
ACK, створене у кроці 15, передається до EudraVigilance не пізніше 2 робочих днів після отримання таких даних |
Національний уповноважений орган |
|
17.1 |
Кінець |
Тепер дані про випадок збережено в базі даних з фармаконагляду національного уповноваженого органу, подальше виявлення дублікатів та внесення записів можуть проводитись для виявлення сигналів і якісного аналізу даних |
Національний уповноважений орган |
|
18 |
Отримайте ACK. |
ACK, відправлене на кроці 17, приймається і зберігається в EudraVigilance |
EMA |
|
19 |
Кінець |
Тепер матеріали про випадок успішно повторно передані до відповідного національного уповноваженого органу |
EMA |
VI. Додаток 4. Передача повідомлень про індивідуальні випадки, пов’язані з безпекою, до Всесвітньої організації охорони здоров’я (ВООЗ)
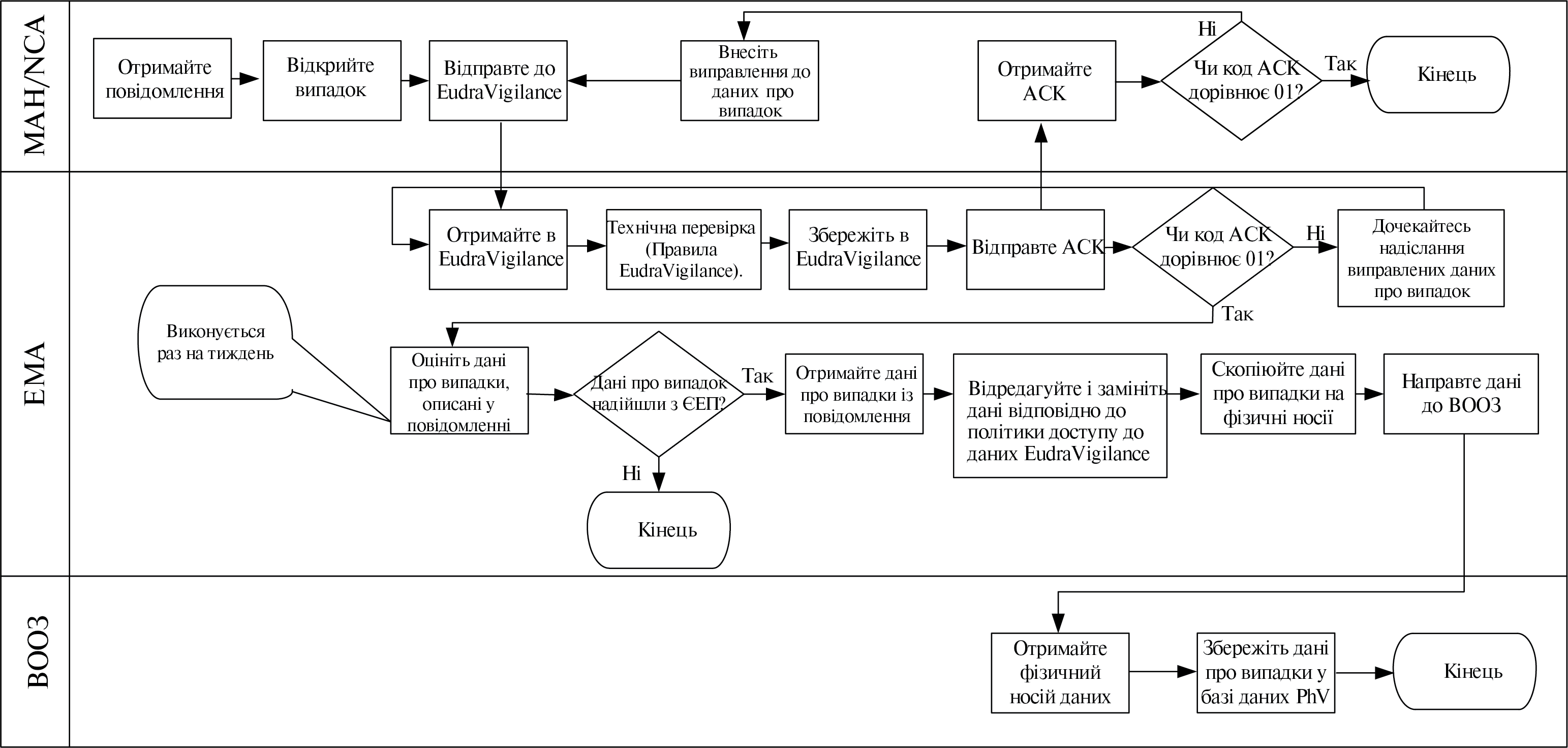
Рисунок 6. Схема робочого процесу — передача повідомлень про індивідуальні випадки, пов’язані з безпекою, до центру співпраці Всесвітньої організації охорони здоров’я (ВООЗ)
Таблиця 10. Описання процесів — передача повідомлень про індивідуальні випадки, пов’язані з безпекою, до Всесвітньої організації охорони здоров’я (ВООЗ)55
|
№ |
Крок |
Описання |
Відповідальна організація |
|
1 |
Початок Отримання повідомлення |
Національний уповноважений орган або заявник (власник реєстраційного посвідчення) отримує інформацію про випадки виникнення підозрюваних побічних реакцій від пацієнта, працівника з медичною та/або фармацевтичною освітою або іншої належної особи, що надає інформацію |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
2 |
Відкрийте випадок |
Відкрийте і створіть повідомлення про індивідуальний випадок, пов’язаний з безпекою |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
3 |
Направте дані до EudraVigilance |
Передайте дані про випадок в електронному вигляді, у форматі повідомлення XML ICH-E2B(R2) [26] у відповідний строк (15 або 90 днів, залежно від обставин) до EudraVigilance |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
4 |
Отримайте в EudraVigilance |
Отримайте повідомлення з EudraVigilance |
EMA |
|
5 |
Технічна перевірка (Правила EudraVigilance) |
Кожне повідомлення, що отримується EudraVigilance, звіряється з правилами EudraVigilance, після чого створюється ACK, в якому зазначається, чи належними є повідомлення і випадок, описаний у ньому. Належне повідомлення буде мати код ACK 01. Неналежне повідомлення буде мати код ACK 02 (якщо випадок, описаний у ньому, не є належним) або 03 (якщо саме повідомлення має неправильний формат) |
EMA |
|
6 |
Збережіть в EudraVigilance |
Як тільки дані про випадок перевірено, вони зберігаються в EudraVigilance |
EMA |
|
7 |
Відправте ACK |
ACK, створене у кроці 5, передається відправнику даних про випадок не пізніше 2 робочих днів після отримання таких даних |
EMA |
|
7.1 |
Отримайте ACK |
Отримайте ACK, зв’яжіть його з відповідним випадком і переконайтеся, що інформація про випадок була помічена як належна |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
7.2 |
Чи код ACK дорівнює 01? |
Якщо так, перейдіть до кроку 7.2.1. Якщо ні, то відлік часу продовжується і інформація про випадок повинна бути виправлена і повторно передана до EudraVigilance у відповідні нормативні строки. Днем 0 залишається день, коли інформація була отримана вперше. ACK з кодом 02 або 03 не є новою інформацією. Перейдіть до кроку 7.2.2 (Виправте дані про випадок). |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
7.2.1 |
Кінець |
Кінець процесу передачі цієї версії даних про випадок до EudraVigilance. Слід продовжувати вести нормальне подальше відстеження. У разі отримання будь-яких даних з подальшого відстеження, поверніться до кроку 1 |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
7.2.2 |
Внесіть виправлення до даних про випадок |
Виправте дані про випадок для видалення помилок, вказаних у ACK, і повторно передайте дані про випадок до EudraVigilance (поверніться до кроку 3) |
Заявник (власник реєстраційного посвідчення)/національний уповноважений орган |
|
8 |
Чи код ACK дорівнює 01? |
Якщо так, перейдіть до кроку 9. Якщо ні, не проводьте ніякої подальшої обробки цієї версії даних про випадок і перейдіть до кроку 8.1 |
EMA |
|
8.1 |
Дочекайтеся надіслання виправлених даних про випадок |
Відправник повинен виправити дані про кожен випадок, ACK якого має код помилки, і повторно передати їх, дотримуючись встановленого терміну. EMA повинне періодично перевіряти всі випадки, ACK яких мають код помилки і для яких виправлення не були передані, і зв’язуватися з кваліфікованою особою, яка відповідає за фармаконагляд (QPPV), щоб поінформувати її про це. Якщо відправник не вносить виправлення до даних про випадок, ця інформація повинна бути включена до оцінки якості даних і повинні бути поінформовані відповідні комітети |
EMA |
|
9 |
Оцініть дані про випадки, описані у повідомленні |
Один раз на тиждень повідомлення, що пройшло технічну перевірку, дані про випадки, що містяться в ньому, повинні вивчатися для визначення країни, де мала місце побічна реакція, з метою надання регулярних звітів |
EMA |
|
10 |
Дані про випадок надійшли з країни ЄС? |
Для даних про кожний випадок оцінити, чи є країна, де мала місце відповідна побічна реакція, країною ЄС Якщо так, перейдіть до кроку 11. Якщо ні, перейдіть до кроку 10.1 |
EMA |
|
10.1 |
Кінець |
Тепер дані про випадок збережено в системі EudraVigilance, подальше виявлення дублікатів та внесення записів можуть проводитись для виявлення сигналів і якісного аналізу даних |
EMA |
|
11 |
Отримайте дані про випадки із повідомлення |
Дані про випадки, що сталися в ЄС, отримуються з повідомлення для їх обробки до повторної передачі |
EMA |
|
12 |
Відредагуйте і замініть дані відповідно до політики доступу до даних EudraVigilance |
До передачі даних про випадки до центру співпраці Всесвітньої організації охорони здоров’я (ВООЗ) отримані копії даних про випадки мають деякі поля, відредаговані і замінені відповідно до політики доступу до даних EudraVigilance з метою забезпечення захисту персональних даних |
EMA |
|
13 |
Скопіюйте дані про випадки на фізичні носії |
Дані про випадки скопіюйте на фізичні носії |
EMA |
|
14 |
Направте дані до Всесвітньої організації охорони здоров’я (ВООЗ) |
Фізичні носії надсилаються до ВООЗ |
EMA |
|
15 |
Отримайте фізичний носій даних |
Центр співпраці ВООЗ отримує фізичний носій |
ВООЗ |
|
16 |
Збережіть дані про випадки в базі даних з фармаконагляду (PhV) |
Як тільки дані про випадки буде перевірено, вони зберігаються в базі даних з фармаконагляду |
ВООЗ |
|
17 |
Кінець |
Тепер дані про випадок збережено в базі даних центру співпраці ВООЗ, подальше виявлення дублікатів та внесення записів можуть проводитися для виявлення сигналів і якісного аналізу даних |
ВООЗ |
VI. Додаток 5. Анулювання випадків
Загальні принципи щодо анулювання випадків описано в VI.C.6.2.2.10.
Наступні рекомендації повинні також виконуватися:
- Значення в полі даних Report nullification (анулювання звіту) (ICH-E2B(R2) [26] A.1.13) повинне бути встановлене на «Так», а причина анулюванням повинна бути вказана у полі даних Reason for nullification (Причина анулювання) (ICH-E2B(R2) [26] A.1.13.1). Тут необхідно чітко і стисло пояснити, чому дана інформація про випадок більше не вважається належним повідомленням. Наприклад, причина анулювання «повідомлення більше не відповідає вимогам щодо формування та надання повідомлень» або «повідомлення направлено з помилкою» є недостатньо докладним поясненням.
- Дані про окремий випадок можуть бути анульовані лише організацією, яка відправила інформацію.
- Після анулювання даних про окремий випадок вони не можуть бути відновлені.
- Якщо виникає необхідність повторно надати дані про випадок, що раніше були анульовані, повинні бути призначені новий Sender’s (case) safety report unique identifier (унікальний ідентифікатор звіту з безпеки, призначений відправником) (ICH-E2B(R2) [26] A.1.0.1) і унікальний глобальний ідентифікаційний номер випадку (ICH- Е2В(R2) [26] A.1.10).
- Індивідуальні версії (тобто, звіт з подальшого відстеження) даних про випадок не можуть бути анульовані, це можливо лише для всього окремого випадку, до якого вони належать.
Таблиця 11. Приклади сценаріїв, за яких повідомлення про індивідуальні випадки, пов’язані з безпекою, повинні бути анульовані
|
№ |
Сценарій |
Дії |
|
1 |
Окремий випадок був визначений як дублікат іншого окремого випадку, дані про який раніше були представлені |
Один з окремих випадків повинен бути анульований. Належний випадок необхідно оновити з включенням додаткових даних з анульованого випадку |
|
2 |
Був випадково використаний неправильний унікальний глобальний ідентифікаційний номер випадку (ICHE2B(R2) [26] A.1.10), що не відноситься до існуючого випадку |
Випадок з неправильним унікальним глобальним ідентифікаційним номером випадку) (ICH-E2B(R2) [26] A.1.10) повинен бути анульований. Повинен бути створений новий випадок з правильним унікальним глобальним ідентифікаційним номером випадку |
|
3 |
Після отримання додаткової інформації було підтверджено, що побічна реакція мала місце до застосування підозрюваного лікарського засобу |
Випадок повинен бути анульований |
|
4 |
Після отримання додаткової інформації було підтверджено, що пацієнт не застосовував підозрюваний лікарський засіб. Мінімальні критерії для повідомлення про індивідуальний випадок, пов’язаний з безпекою, передбачені в VI.B.2, більше не виконуються |
Випадок повинен бути анульований |
|
5 |
Після отримання додаткової інформації від тієї ж особи, яка надає інформацію, підтверджується, що побічна реакція у пацієнта, про яку повідомлялося, не мала місця. Мінімальні критерії для повідомлень про індивідуальні випадки, пов’язані з безпекою, передбачені в VI.B.2, більше не виконуються |
Випадок повинен бути анульований |
|
6 |
Після отримання додаткової інформації було підтверджено, що для даного окремого випадку не існувало належного пацієнта. Мінімальні критерії для ICSR, передбачені в VI.B.2, більше не виконуються |
Якщо неможливо отримати підтвердження існування пацієнта, то випадок повинен бути анульований |
- Окремі випадки, що були анульовані, не повинні використовуватися для наукової оцінки, однак вони повинні залишатися в базі даних для цілей аудиту.
- Крім того, у ситуації, коли одне з повідомлень-дублікатів має бути анульовано, оновлення того, що залишилося, повинне проводитися у вигляді звіту з подальшого відстеження56. Інформація про ідентифікацію анульованого випадку(-ів) повинна бути представлена в полі даних Source(s) of the case identifier (e.g. name of the company, name of regulatory agency) (ідентифікатор джерела даних (наприклад, назва компанії, назва регулюючого органу)) (ICH-E2B(R2) [26] A.1.11.1), і в елементі даних Case identifier (Ідентифікатор випадку) (ICH-Е2В (R2) [26] A.1.11.2).
Таблиця 12. Приклади сценаріїв, за яких повідомлення про індивідуальні випадки, пов’язані з безпекою, НЕ ПОВИННІ бути анульовані
|
№ |
Сценарій |
Дії |
|
7 |
Був випадково використаний неправильний унікальний глобальний ідентифікаційний номер випадку (ICH E2B(R2) [26] A.1.10). Такий неправильний унікальний глобальний ідентифікаційний номер випадку (ICH E2B(R2) [26] A.1.10) належить існуючому випадку |
Повідомлення з неправильним унікальним глобальним ідентифікаційним номером випадку) (ICHE2B(R2) [26] A.1.10) не повинно бути анульоване. Для виправлення раніше наданої інформації повинен бути представлений звіт з подальшого відстеження. Повинно бути створено та надано нове повідомлення про індивідуальний випадок, пов’язаний з безпекою з правильним унікальним глобальним ідентифікаційним номером випадку) |
|
8 |
Після отримання додаткової інформації було підтверджено, що пацієнт не отримував підозрюваний лікарський засіб заявника (власника реєстраційного посвідчення). Однак пацієнт отримав інший підозрюваний лікарський засіб, і мінімальні критерії для ICSR все ще виконуються |
Випадок не повинен бути анульований |
|
9 |
Після отримання додаткової інформації особа, яка надає інформацію, підтвердила, що повідомлення про побічні реакції більше не вважаються пов’язаними з підозрюваним лікарським засобом |
Випадок не повинен бути анульований. Звіт з подальшого відстеження з оновленою інформацією про випадок повинен бути представлений протягом належного строку |
|
10 |
Зміна статусу окремого випадку від серйозного до несерйозного (зниження) |
Випадок не повинен бути анульований. Повинен бути наданий звіт з подальшого відстеження з полем даних Seriousness (Серйозність) (IICHE2B(R2) [26] A.1.5.1), що містить значення No (Ні) без вказання значення поля даних Seriousness criteria (критерії серйозності) (ICHE2B(R2) [26] A.1.5.2). Поле даних Does this case fulfil the local criteria for an expedited report? (чи відповідає випадок місцевим критеріям для експрес-звітності?) (ICHE2B(R2) [26] A.1.9) повинен так само містити значення Yes (Так). |
|
11 |
Змінилася країна першоджерела, що має вплив на умови ICH-E2B(R2) [26] щодо призначення унікального глобального ідентифікаційного номера випадку) (ICH-E2B(R2) [26] A.1.10) |
Випадок не повинен бути анульований. Sender’s (case) safety report unique identifier (Унікальний ідентифікатор звіту з безпеки, призначений відправником) (ICHE2B(R2) [26] A.1.0.1) може бути замінений на код нової країни першоджерела. Однак унікальний глобальний ідентифікаційний номер випадку (ICH-E2B(R2) [26] A.1.10) повинен залишатися незмінним. Якщо з якоїсь технічної причини локальна система відправника не повністю сумісна з ICHE2B(R2) [26] і не може відповідати цій політиці, відправник повинен анулювати випадок-оригінал. Необхідно створити новий випадок з новим унікальним глобальним ідентифікаційним номером випадку) (ICHE2B(R2) [26] A.1.10), що відображає змінений код країни першоджерела. Унікальний глобальний ідентифікаційний номер випадку (ICH-E2B (R2) [26] A.1.10), що був анульований, повинен міститися в полі даних Other case identifiers in previous transmissions (Інші ідентифікатори випадку в попередніх повідомленнях) (ICHE2B(R2) [26] A.1.11) |
|
12 |
Підозрюваний лікарський засіб належить іншому заявнику (власнику реєстраційного посвідчення) (наприклад, лікарський засіб з такою ж діючою речовиною, але має іншу торговельну назву) |
Випадок не повинен бути анульований. Рекомендується, щоб перший відправник повідомив інших заявників (власників реєстраційних посвідчень) про цей випадок (включаючи використаний унікальний глобальний ідентифікаційний номер випадку) (ICH-E2B(R2) [26] A.1.10). Перша організація також повинна надати звіт з подальшого відстеження щоб передати цю нову інформацію. Інший зацікавлений заявник (власник реєстраційного посвідчення) повинен створити новий випадок і вказати індивідуальний номер випадку та ім’я першого заявника (власника реєстраційного посвідчення) — відправника полі даних Source(s) of the case identifier (e.g. name of the company, name of regulatory agency) (ідентифікатор джерела даних (наприклад, назва компанії, назва регулюючого органу)) (ICH-E2B(R2) [26] A.1.11.1) і Case identifier (Ідентифікатор випадку) (ICH-E2B(R2) [26] A.1.11.2). Це дозволить здійснювати групування випадків у базі даних EudraVigilance |
|
13 |
Підозрюваний лікарський засіб не належить заявнику (власнику реєстраційного посвідчення) (та ж діюча речовина, торгова назва невідома, а повідомлення отримано з країни, де заявник (власник реєстраційного посвідчення) не має реєстраційних посвідчень на описуваний лікарський засіб) |
Випадок не повинен бути анульований. Заявник (власник реєстраційного посвідчення) повинен надати звіт з подальшого відстеження з цією інформацією в межах відповідного терміну |
|
14 |
Випадок помилково повідомлений заявником (власником реєстраційного посвідчення) А, хоча заявник (власник реєстраційного посвідчення) B в якості комерційного партнера відповідає за повідомлення щодо даного випадку |
Випадок не повинен бути анульований. Заявником (власником реєстраційного посвідчення) А повинне бути направлене пояснення комерційному партерну — заявнику (власнику реєстраційного посвідчення) B, про те, що про випадок вже повідомлялося. Заявник (власник реєстраційного посвідчення) B повинен надати всю додаткову інформацію у цій справі у формі звіту з подальшого відстеження з тим же унікальним глобальним ідентифікаційним номер випадку) (ICH-E2B(R2) [26] A.1.10 |
VI. Додаток 6. Моніторинг якості даних повідомлень про індивідуальні випадки, пов’язані з безпекою, що передаються в електронному вигляді
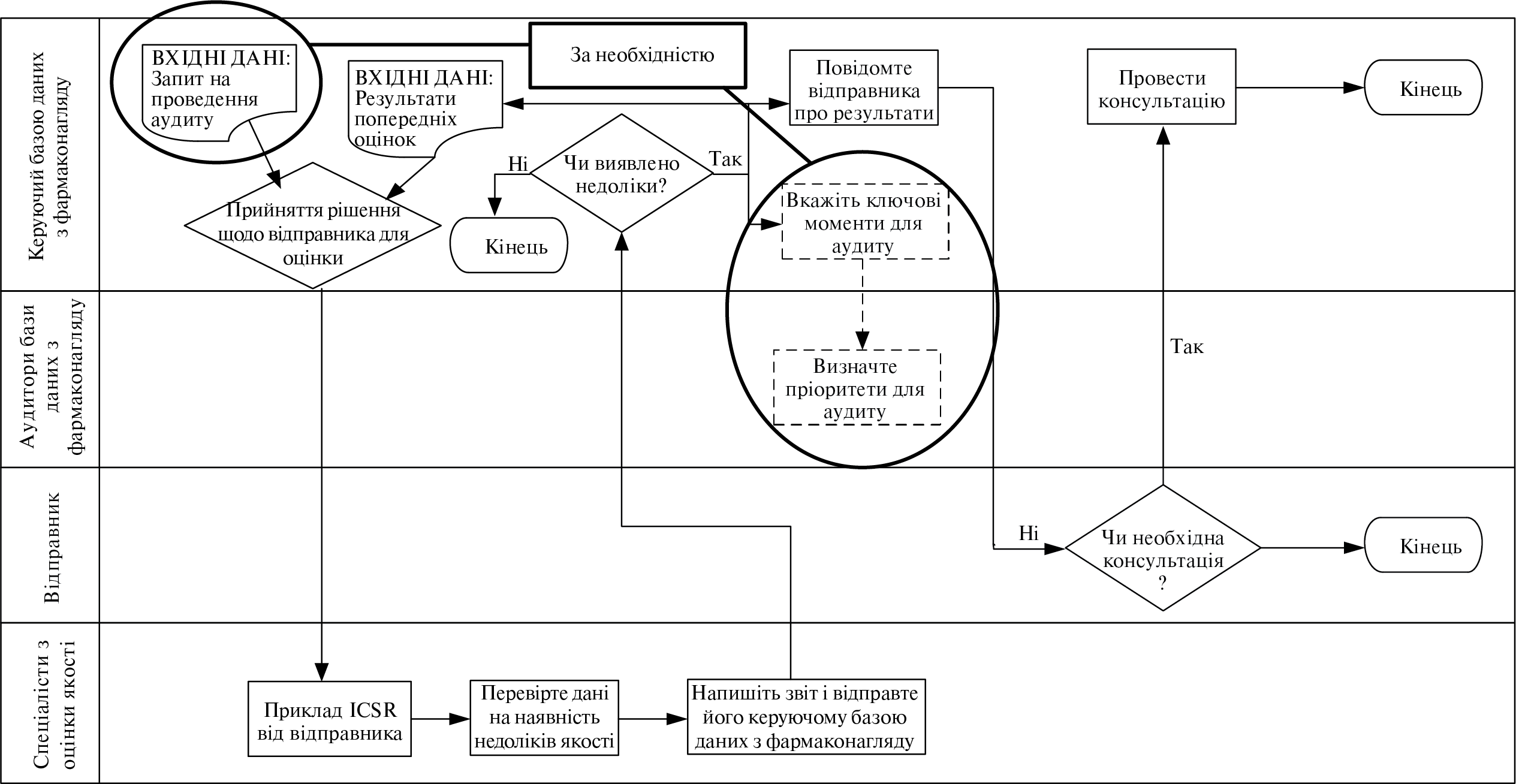
Рисунок 7. Схема робочого процесу — моніторинг якості даних повідомлень про індивідуальні випадки, пов’язані з безпекою, що надаються в електронному вигляді
Таблиця 13. Описання процесу — моніторинг якості даних повідомлень про індивідуальні випадки, пов’язані з безпекою, що надаються в електронному вигляді
Схема та описання процесів демонструють систему, де існує поділ між керуючим базою даних з фармаконагляду, спеціалістами з оцінки якості (QA) інформації бази даних з фармаконагляду і аудиторами бази даних з фармаконагляду; проте це не є обов’язковим, і ці функції можуть виконуватися одними й тими ж особами або групами.
|
№ |
Крок |
Описання |
Відповідальна організація |
|
1 |
Початок. Прийняття рішення щодо відправника для оцінки |
Оберіть одну з організацій, яка надавала повідомлення про індивідуальний випадок, пов’язаний з безпекою до вашої бази даних. Підстави для цього рішення можуть включати результати попередніх оцінок і запити на проведення аудитів, але не обмежуватися ними |
Керуючий базою даних з фармаконагляду |
|
2 |
Приклад повідомлення про індивідуальний випадок, пов’язаний з безпекою від відправника |
Відберіть зразок ICSR, що були передані обраним відправником |
Спеціалісти з оцінки якості |
|
3 |
Перевірте дані на наявність недоліків якості |
Перевірте випадки на наявність недоліків якості даних. Випадки повинні оцінюватися за допомогою відповідних опублікованих стандартів і аналогічних документів, наприклад документа MedDRA Term Selection: питання для розгляду |
Спеціалісти з оцінки якості |
|
4 |
Напишіть звіт і відправте його керуючому базою даних з фармаконагляду |
Результати оцінки якості даних повинні бути зведені в один звіт. Вони можуть включати пов’язані перевірки, такі, наприклад, як дотримання 15-денного звітного періоду, виправлені звіти, що містили помилки, і аналогічну статистичну інформацію |
Спеціалісти з оцінки якості |
|
5 |
Чи виявлено недоліки? |
Чи було при аналізі випадків виявлено будь-які недоліки? Якщо ні, перейдіть до кроку 5.1. Якщо так, перейдіть до кроку 5.2, 5.3 та 6 |
Керуючий базою даних з фармаконагляду |
|
5.1 |
Кінець |
Якщо не було виявлено жодних недоліків, подальші дії не потрібні. Процес може бути завершений до наступної оцінки даних відправника |
Керуючий базою даних з фармаконагляду |
|
Керуючий базою даних з фармаконагляду може прийняти рішення передати дану інформацію відправнику, перевірка якого проводилася, та аудиторам бази даних, які, можливо, виявлять бажання врахувати її при обранні відправника для перевірки |
|||
|
5.2 |
Вкажіть ключові моменти для аудиту |
Якщо організація бази даних з фармаконагляду має відділ аудиту, будь-які значущі результати завжди повинні надаватися до нього |
Керуючий базою даних з фармаконагляду |
|
5.2.1. |
Визначте пріоритети для аудиту |
Відділі аудиту або перевірки повинен використовувати надану йому інформацію для ухвалення рішення про пріоритетність аудиту або перевірки для організації |
Аудитори бази даних з фармаконагляду |
|
5.3 |
ВХІДНІ ДАНІ: результати попередніх оцінок |
Будь-які знайдені недоліки (або навіть їх відсутність) повинні бути враховані при прийнятті рішення про те, перевірку якого з відправників проводити, також необхідно поінформувати про проведення перевірки (наприклад, орієнтованість на конкретні типи випадків) і звіти (зазначення того, чи були вирішені раніше виявлені проблеми) |
Керуючий базою даних з фармаконагляду |
|
6 |
Повідомте відправника про результати |
Повідомте відправника про результати, у тому числі щодо необхідних заходів (наприклад повторна передача даних про окремі випадки) та терміну виконання цих заходів |
Керуючий базою даних з фармаконагляду |
|
7 |
Чи необхідна консультація? |
Відправник повинен мати можливість попросити про проведення консультації для обговорення результатів та належних заходів і часових рамок. Якщо консультація не потрібна, перейдіть до кроку 7.1. Якщо консультація потрібна, перейдіть до кроку 8 |
Відправник |
|
7.1 |
Розгляньте висновки і повторно передайте всі необхідні дані про випадки |
Розгляньте всі висновки, вживайте необхідних заходів для запобігання повторення таких висновків і повторно надайте всі необхідні дані про випадки |
Відправник |
|
7.2 |
Кінець |
Після того, як усі висновки розглянуто, необхідні заходи вжито з метою запобігти повторенню подібних висновків, а всі необхідні дані про випадки повторно надано, процес може закінчитися до наступної оцінки відправника |
Відправник |
|
8 |
Проведіть збори |
На прохання однієї зі сторін проводяться консультація для обговорення результатів оцінки якості і відповідних коригувальних і профілактичних заходів, які гарантують, що дані про випадки в базі даних є правильними і залишатимуться такими в майбутньому. |
Керуючий базою даних з фармаконагляду |
|
9 |
Кінець |
Якщо не визначено подальші дії (наприклад, майбутні консультації або оцінка), процес може закінчитися до наступної оцінки відправника |
Керуючий базою даних з фармаконагляду |
VI. Додаток 7. Виявлення дублікатів та обробка повідомлень про індивідуальні випадки, пов’язані з безпекою
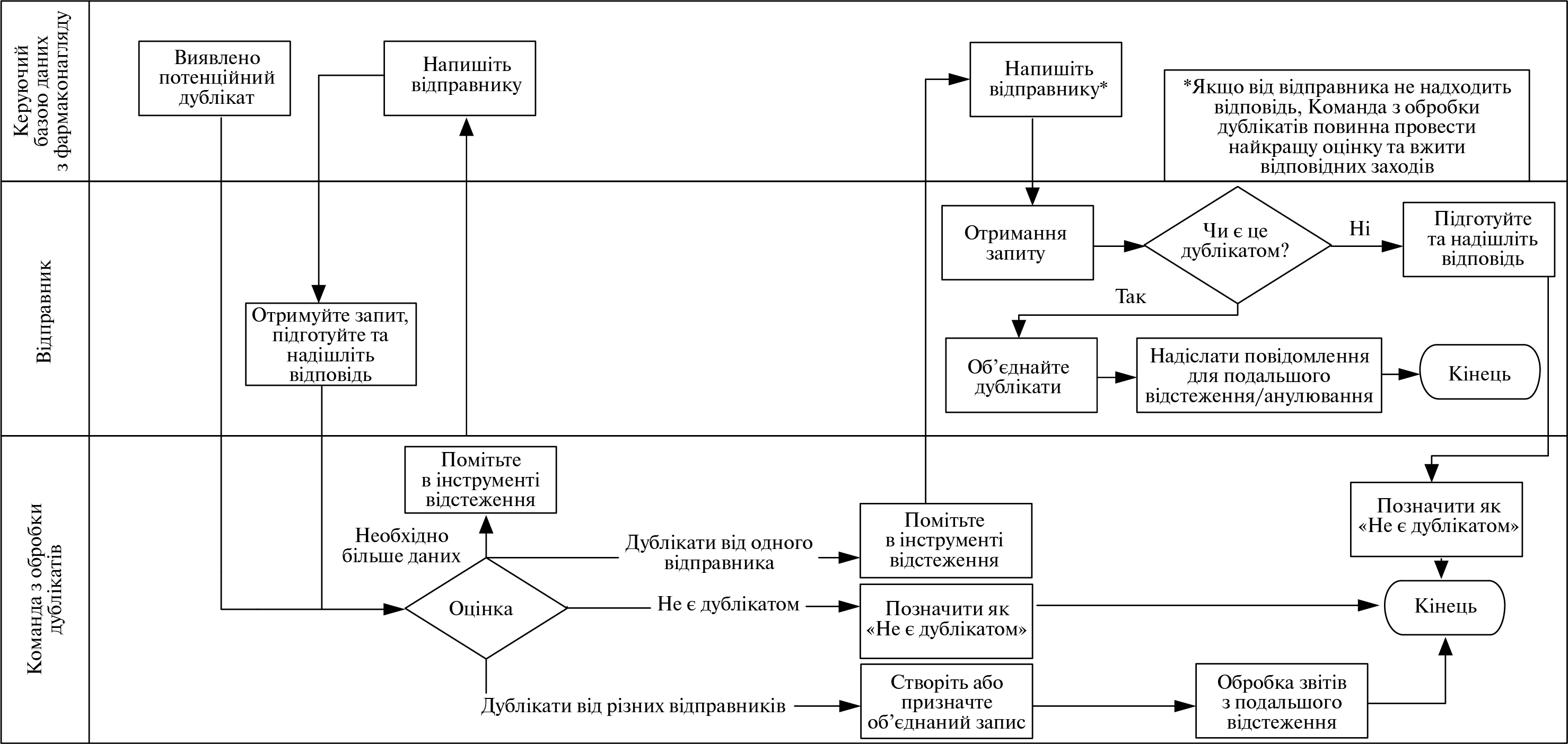
Рисунок 8. Схема робочого процесу — виявлення дублікатів та обробка повідомлень про індивідуальні випадки, пов’язані з безпекою
Таблиця 14. Описання процесів — виявлення дублікатів та обробка повідомлень про індивідуальні випадки, пов’язані з безпекою
|
№ |
Крок |
Описання |
Відповідальна організація |
|
1 |
Початок Виявлено потенційний дублікат |
Потенційні дублікати виявлено організацією — керуючим базою даних з фармаконагляду або організація — керуючий базою даних з фармаконагляду була повідомлена про потенційні дублікати отримувачем інформації про випадок |
Керуючий базою даних з фармаконагляду |
|
2 |
Оцінка |
Усі потенційні дублікати потребують оцінки командою з обробки дублікатів (DMT) організації, щоб підтвердити або спростувати їх статус як дублікатів. Після проведення оцінки існує 4 можливих результати: 1) Не дублікат, перейдіть до кроку 2.1. 2) Необхідно більше даних, перейдіть до кроку 2.2. 3) Дублікати від різних відправників, перейдіть до кроку 2.3. 4) Дублікати від одного відправника, перейдіть до кроку 2.4. Результати проведених оцінок повинні бути збережені, щоб уникнути повторного перегляду тих самих випадків після отримання нових версій. Такі збережені результати також можуть бути використані для оптимізації методів виявлення дублікатів у ході майбутнього розвитку |
Команда з обробки дублікатів |
|
2.1 |
Не є дублікатом: позначити як «Не є дублікатом» |
Якщо випадки, що були оцінені, не дублюють один одного, то помітьте обидва як такі, що не є дублікатами. Перейдіть до кроку 3 (Кінець) |
Команда з обробки дублікатів |
|
2.2 |
Необхідно більше даних: помітьте в інструменті відстеження |
Коли необхідно більше інформації, повинен існувати інструмент для відстеження, у якому помічається, коли кореспонденція була відправлена, чи була отримана відповідь, і, якщо так, то коли |
Команда з обробки дублікатів |
|
2.2.1 |
Напишіть відправнику |
Необхідно більше інформації, щоб мати можливість зробити точну оцінку. Необхідно звернутися до відправника (який надав інформацію про випадок) із запитом на конкретну інформацію, необхідну, щоб підтвердити або спростувати факт дублювання. Захист персональних даних має залишатися першочерговим пріоритетом, таким чином, незахищені способи зв’язку не повинні включати дані, достатні для ідентифікації особи |
Керуючий базою даних з фармаконагляду |
|
2.2.2 |
Отримайте запит, підготуйте та надішліть відповідь |
Після того, як запит на додаткову інформацію отримано, відправник даних про випадок повинен оперативно відреагувати, сформувати звіт з подальшого відстеження випадку або відповідь на запит. Команда з обробки дублікатів повинна знову оцінити випадок на основі нової інформації (поверніться до кроку 2) |
Відправник |
|
2.3 |
Дублікати від різних відправників: створіть або призначте об’єднаний запис |
Після того, як випадки визначено як дублікати, і, коли вони були передані керуючому базою даних з фармаконагляду різними відправниками, їх необхідно об’єднати в одному записі, відповідно до процедур, описаних у розділі 2.3 «Управління дублікатами випадків» Настанови з виявлення та видалення дублікатів окремих випадків і повідомлень про індивідуальні випадки, пов’язані з безпекою (EMA/13432/2009) |
Команда з обробки дублікатів |
|
2.3.1. |
Обробка звітів з подальшого відстеження |
Якщо надаються будь-які звіти з подальшого відстеження для будь-якого випадку, ця інформація може потребувати повторної оцінки об’єднаного запису. Проведіть повторну оцінку і, за необхідності, внесіть зміни до об’єднаного запису відповідно до отриманої інформації. Перейдіть до кроку 3 (Кінець) |
Команда з обробки дублікатів |
|
2.4 |
Дублікати від одного відправника: помітьте в інструменті відстеження |
Після того, як випадки визначено як дублікати, і у разі, якщо вони передані керуючому базою даних з фармаконагляду одним і тим самим відправником, це рішення і листування, зазначене у пункті 2.4.1, повинне бути занесене до інструменту відстеження, вказаного в кроці 2.2 |
Команда з обробки дублікатів |
|
2.4.1 |
Напишіть відправникові |
Організація-відправник як джерело дублікатів повинна бути повідомлена відповідно до розділу 2.3.3 і Настанови з виявлення і видалення дублікатів повідомлень про індивідуальні випадки, пов’язані з безпекою (EMA/13432/2009) Запитати відправника про підтвердження або спростування дублювання і вимагати вжиття належних заходів відповідно до розділу 2.3.1 зазначеної вище настанови |
Керуючий базою даних з фармаконагляду |
|
2.4.2 |
Отримайте запит |
Отримайте і збережіть повідомлення, що містять інформацію про підозрювані дублікати в базі даних відправника. |
Відправник |
|
2.4.3 |
Чи є це дублікатом? |
Оціните потенційні дублікати. Чи є випадки дублікатами один одного? Якщо так, перейдіть до кроку 2.4.3.1. Якщо ні, перейдіть до кроку 2.4.3.2 |
Відправник |
|
2.4.3.1 |
Об’єднайте дублікати |
Об’єднайте дублікати з урахуванням схеми 1 розділу 2.3.1.3 Настанови з виявлення та видалення дублікатів повідомлень про індивідуальні випадки, пов’язані з безпекою (EMA/13432/2009) |
Відправник |
|
2.4.3.1.1 |
Надіслати повідомлення для подальшого відстеження/анулювання |
Для випадків, дані про які були об’єднані в один запис, відправте повідомлення про анулювання керуючому базою даних з фармаконагляду |
Відправник |
|
Відправте об’єднаний запис керуючому базою даних з фармаконагляду як дані з подальшого відстеження. Злиття та передача повинні бути завершені вчасно і в будь-якому випадку протягом 15 днів з дати отримання інформації від утримувача бази даних фармаконагляду про те, що випадки є можливими дублікатами. Цією датою слід вважати дату отримання останньої інформації для цілей регулярного надання звітів |
|||
|
2.4.3.1.2 |
Кінець |
Дублікати видалено як із системи відправника, так і з системи керуючого базою даних з фармаконагляду, і лише об’єднаний запис є доступним для виявлення сигналу і аналізу якості даних. Якщо отримано дані з подальшого відстеження, жодні подальші кроки не вимагаються |
Відправник |
|
2.4.3.2 |
Підготувати та надіслати відповідь |
Надішліть відповідь керуючому базою даних з фармаконагляду, повідомивши його, що випадки не є дублікатами |
Відправник |
|
2.4.3.2.1 |
Позначте як такий, що не є дублікатом |
Після отримання підтвердження від організації-відправника про те, що випадки не є дублікатами, позначте випадки як Not a duplicate (не є дублікатом) і перейдіть до кроку 3 (Кінець) |
Команда з обробки дублікатів |
|
3 |
Кінець |
Для даної пари подальші дії не вимагаються |
Команда з обробки дублікатів |
Ключові слова: безпека застосування лікарських засобів, належна практика фармаконагляду, настанова, побічна реакція лікарського засобу, управління та звітування про побічні реакції лікарських засобів, фармаконагляд.
Частина VII. Модуль VII — Регулярно оновлюваний звіт з безпеки
VII.A. Вступ
РОЗБ — це документ з фармаконагляду, що призначений для надання оцінки співвідношення користь/ризик лікарського засобу та подання заявниками (власниками реєстраційних посвідчень) до уповноваженого органу у визначені строки у післяреєстраційний період.
Нормативно-правові вимоги для подання РОЗБ визначені в Регламенті (ЄC) № 726/2004 [5], Директиві 2001/83/ЄС [1], ІП № 520/2012 [6], положеннях Порядку[2]N, з виконання діяльності з фармаконагляду, що передбачена Регламентом (ЄC) №726/2004 [5], Директивою 2001/83/ЄС [1] (ІП 520/2012 [6]), положеннями Порядку [2]N.
Формат РОЗБ повинен відповідати наведеній структурі, що описана у ст. 35 ІП 520/2012 [6] та у додатку 12 до Порядку [2]N. Цей модуль надає рекомендації з підготовки, надання та оцінки РОЗБ.
Обсяг, цілі, формат та зміст РОЗБ описані в розділі VII.В ННПФ. Формат та зміст РОЗБ ґрунтуються на форматі та змісті, що передбачені для регулярного звіту з оцінки користі та ризику (Регулярний звіт з оцінки співвідношення користь/ризик — PBRER) та описані в настанові ICH-E2C(R2) [28] (див. додаток IV ICH-E2C(R2) [28]. Формат PBRER замінює попередній формат РОЗБ, що був описаний у настанові ICH-E2C(R1). Відповідно до законодавства ЄС у модулях ННПФ звіт описується як РОЗБ.
Більш детальна інформація та рекомендації з подання РОЗБ, включаючи перелік референтних дат ЄС (в Україні — Періодичність подання регулярно оновлюваних звітів з безпеки лікарських засобів, вакцин, туберкуліну за міжнародною непатентованою назвою активного фармацевтичного інгредієнта або комбінацій активних фармацевтичних інгредієнтів (далі — періодичність подання регулярних звітів з безпеки), що наведена у додатку 10 до Порядку [2]N) та частоту подання РОЗБ, надаються в розділі VII.С. ННПФ, який також містить дані про єдиний підхід до оцінки регулярних звітів з безпеки у розділі VII.С.4. Детальна інформація стосовно системи якості та публікації документів, що стосуються РОЗБ у рамках прозорості надаються в розділі VII.С.6. та у розділі VII.С.7 відповідно.
Заявники (власники реєстраційних посвідчень) несуть відповідальність за подання РОЗБ на свої власні лікарські засоби (ст. 107b Директиви 2001/83/ЄС [1], ст. 28(2) Регламенту (ЄC) № 726/2004 [5], положення Порядку [2]N, положення Порядку проведення експертизи реєстраційних матеріалів [7]N) та дотримання строків подання таких звітів у ЄС до Європейського агенства з лікарських засобів (EMA) (див. VII.C.9. щодо перехідного періоду), в Україні до ЦентруN відповідно до наступних строків:
а) протягом 70 календарних днів з дати закриття бази даних (день 0) — для РОЗБ, що охоплюють звітний період 12 міс включно;
б) протягом 90 календарних днів з дати закриття бази даних (день 0) — для РОЗБ, що охоплюють інтервали понад 12 міс;
в) строки для подання спеціальних РОЗБ на запит від уповноваженого органу, як правило, будуть вказані в запитах; в інших випадках спеціальні РОЗБ слід подавати протягом 90 календарних днів з дати закриття бази даних.
Слід зазначити, що детальні переліки індивідуальних випадків, пов’язаних з безпекою, не повинні включатися до РОЗБ систематично (ст. 34(4) ІП 520/2012 [6] та положення до Порядку [2]N). У РОЗБ слід акцентувати увагу на узагальненій інформації, науковій оцінці безпеки та інтегрованій оцінці співвідношення користь/ризик.
Пункт 23 декларативної частини Директиви 2010/84/ЄС [3], положення до Порядку [2]N визначає, що зобов’язання, висунуті до РОЗБ, повинні бути СТ-Н МОЗУ 42-8.7:2018 300 пропорційними ризикам, пов’язаним із застосуванням лікарських засобів. Тому необхідно, щоб подання РОЗБ було пов’язане з системами управління ризиками лікарського засобу (див. модуль V ННПФ). Модульний підхід до формування РОЗБ, що описаний у розділі VII.В.5. ННПФ, має на меті мінімізацію дублювання та підвищення ефективності при підготовці та перегляді РОЗБ та інших регуляторних документів, таких як оновлюваний звіт з безпеки препарату, що знаходиться в стадії розробки (далі — DSUR)58, або специфікація з безпеки у плані управління ризиками, з тим, щоб за необхідності використовувати спільну інформацію певних розділів як взаємозамінну у DSUR та ПУР.
Директива 2001/83/ЄС [1] із змінами та положення Порядку [2]N також відміняють регулярну подачу РОЗБ для генеричних лікарських засобів (зареєстрованих відповідно до ст. 10(1) Директиви 2001/83/ЄС [1], положень Порядку проведення експертизи [7]N), лікарських засобів, що мають добре вивчене медичне застосування (зареєстрованих відповідно до ст. 10а Директиви 2001/83/ЄС [1], положень Порядку [2]N), гомеопатичних лікарських засобів (зареєстрованих відповідно до ст. 14 Директиви 2001/83/ЄС [1], положень Порядку проведення експертизи [7]N) та традиційних рослинних лікарських засобів (зареєстрованих відповідно до ст. 16а Директиви 2001/83/ЄС [1]), (ст. 107b(3) Директиви 2001/83/ЄС [1], положень Порядку проведення експертизи [7]N). Для цих лікарських засобів РОЗБ повинен надаватися, якщо це є умовою реєстраційного посвідчення або якщо цього вимагає уповноважений орган на підставі проблем, пов’язаних з даними з фармаконагляду, або внаслідок відсутності РОЗБ для діючої речовини після її реєстрації (ст. 107b(3)(а) та (3)(b) Директиви 2001/83/ЄС [1] та положення Порядку проведення експертизи [2]N).
Уповноважені органи у державах ЄС та УкраїніN повинні проводити експертну оцінку регулярного звіту з безпеки з метою встановлення, чи з’явилися нові ризики, чи змінилися ризики, чи є зміни у співвідношенні користь/ризик лікарських засобів (ст. 107d Директиви 2001/83/ЄС [1] та положення Порядку [2]N).
З метою ефективного спільного застосування ресурсів уповноваженими органами у країнах ЄС, УкраїниN, в ЄС, УкраїніN слід проводити єдину експертну оцінку РОЗБ для різних лікарських засобів, що містять однакову діючу речовину або однакову комбінацію діючих речовин, дозволених у більш ніж одній країні ЄС, УкраїніN, для яких була встановлена референтна дата ЄС (в Україні — згідно зі встановленою періодичністю подання регулярних звітів з безпекиN) та частота подання РОЗБ. Єдина оцінка у ЄС, в УкраїніN може включати спільну оцінку лікарських засобів, що отримали дозвіл за національною або централізованою процедурами видачі реєстраційного посвідчення. EMA повинно створити перелік референтних дат ЄС та встановити періодичність подання РОЗБ (ст. 26(g) Регламенту (ЄC) №726/2004 [5]), що будуть мати обов’язкову юридичну силу. В Україні періодичність подання регулярних звітів з безпеки визначена положенням Порядку (додаток 10 до Порядку [2]N).
У рамках експертної оцінки слід розглянути доцільність проведення подальших досліджень та необхідність вживання будь-яких заходів до реєстраційних посвідчень на лікарські засоби, що містять однакову діючу речовину (активний фармацевтичний інгредієнт (далі — АФІ)N) або однакову комбінацію діючих речовин, та до інформації щодо цих лікарських засобів.
EMA повинно зробити РОЗБ доступними для уповноважених органів у державах ЄС, PRAC, СНМР та CMDh, а також Європейської Комісії за допомогою архіву РОЗБ (ст. 107d Директиви 2001/83/ЄС [1]).
VII.B. Структури та процеси
VII.B.1. Цілі регулярно оновлюваного звіту з безпеки
Головною метою РОЗБ є надання стислого всебічного та критичного аналізу співвідношення користь/ризик лікарського засобу з урахуванням нової інформації в контексті сукупної інформації з ризиків та користі. Отже, РОЗБ є інструментом післяреєстраційної оцінки у визначені моменти часу впродовж життєвого циклу лікарського засобу.
З метою управління співвідношенням користь/ризик протягом усього життєвого циклу необхідно продовжувати оцінку ризиків та користі лікарського засобу в повсякденній медичній практиці і тривалому застосуванні у післяреєстраційний період. Дана діяльність може поширюватися на оцінку популяцій та кінцевих точок, що не могли досліджуватися в дореєстраційних клінічних випробуваннях.
Співвідношення користь/ризик може змінюватися по мірі виявлення у межах фармаконагляду нової інформації з безпеки. Тому заявник (власник реєстраційного посвідчення) повинен повторно оцінювати співвідношення користь/ризик своїх лікарських засобів в експонованих популяціях. Така структурована оцінка повинна проводитися в контексті безперервного здійснення фармаконагляду та управління ризиками (див. модуль V) з метою сприяння оптимізації співвідношення користь/ризик шляхом ефективної мінімізації ризиків.
Термінову інформацію, що стосується безпеки лікарського засобу, слід повідомляти за допомогою відповідного механізму. Не слід використовувати РОЗБ у якості першочергового засобу для повідомлення нової важливої інформації з безпеки та ефективності або джерела виявлення нових проблем безпеки (див. модуль IХ). Відомо, що аналіз даних РОЗБ може призвести до виявлення нових проблем безпеки.
VII.B.2. Принципи оцінки співвідношення ризик/користь у регулярно оновлюваному звіті з безпеки та обсяг інформації, що повинна бути включена до звіту
Для забезпечення захисту громадського здоров’я та підвищення безпеки пацієнтів шляхом ефективної мінімізації ризиків оцінку співвідношення користь/ризик слід проводити протягом усього життєвого циклу лікарського засобу.
Після видачі реєстраційного посвідчення необхідно продовжувати оцінку користі та ризиків лікарських засобів при їх фактичному застосуванні та/або тривалому застосуванні для підтвердження того, що співвідношення користь/ризик залишається позитивним.
Аналіз співвідношення користь/ризик повинен містити обґрунтовану та відповідну оцінку інформації з безпеки, ефектів та ефективності лікарського засобу, що стає доступною59 у звітний період, у контексті того, що було відомо раніше.
Оцінка ризику повинна базуватися на всіх аспектах застосування лікарського засобу. Вона охоплює оцінку безпеки у повсякденній медичній практиці, включаючи застосування не за показаннями та не відповідно до короткої характеристики/інструкції для медичного застосуванняN лікарського засобу. Якщо існують дані про застосування лікарського засобу у пацієнтів чи в популяціях, щодо яких існують значні прогалини в знаннях з певних питань безпеки, про це слід надати інформацію у РОЗБ (наприклад, застосування в педіатричній популяції або вагітними). Джерелами інформації про випадки застосування не відповідно до короткої характеристики/інструкції для медичного застосуванняN лікарського засобу можуть бути дані щодо споживання лікарського засобу, спонтанні повідомлення та публікації в літературі.
Інформація про користь лікарського засобу повинна включати дані як клінічних випробувань, так і реального споживання лікарського засобу згідно із затвердженими показаннями для застосування.
Інтегрована оцінка співвідношення користь/ризик повинна проводитися для всіх затверджених показань, однак вона повинна також включати й оцінку ризиків при будь-якому застосуванні лікарського засобу (включаючи застосування за незатвердженими показаннями).
Оцінка повинна включати:
1. Критичну оцінку зібраної за звітний період інформації для визначення, чи вона вказує на генерацію нових сигналів, призводить до виявлення нових потенційних чи ідентифікованих ризиків або доповнює дані щодо раніше виявлених ризиків.
2. Критичне узагальнення нової інформації з безпеки, ефектів та ефективності, що може мати вплив на співвідношення користь/ризик лікарського засобу.
3. Проведення інтегрованого аналізу співвідношення користь/ризик для всіх затверджених показань на підставі сукупної інформації, починаючи від міжнародної дати народження лікарського засобу, що знаходиться в стадії розробки (DIBD), дати першого дозволу на проведення інтервенційних клінічних випробувань у будь-якій країні. У випадках, коли міжнародна дата народження лікарського засобу, що знаходиться на стадії розробки, невідома, або заявник (власник реєстраційного посвідчення) не має доступу до даних періоду клінічних випробувань, слід використовувати найранішу дату в якості початкового відліку для включення та оцінки зведеної інформації.
4. Резюме будь-яких заходів з мінімізації ризиків, що застосовувалися чи запроваджувалися у звітний період, а також заходи з мінімізації ризиків, що плануються до запровадження.
5. Складання планів щодо оцінки сигналів чи ризиків, включаючи строки та/або пропозиції щодо додаткових заходів з фармаконагляду.
VII.B.3. Принципи підготовки регулярно оновлюваного звіту з безпеки
Якщо уповноважений орган не вимагає іншого, заявник (власник реєстраційного посвідчення) повинен підготувати єдиний РОЗБ для всіх своїх лікарських засобів, що містять однакову діючу речовину, на основі інформації, що охоплює всі затверджені показання, способи застосування, лікарські форми та схеми застосування, незалежно від того, чи зареєстровані ці лікарські засоби під різними торговельними назвами або за різними процедурами. Якщо доцільно, дані стосовно певного показання, лікарської форми, способу застосування або дозування повинні бути представлені в окремому розділі РОЗБ, та будь-які питання безпеки, що їх стосуються, повинні бути відповідно розглянуті (ст. 34(6) ІП 520/2012 [6], положення Порядку [2]N). У деяких виключних випадках може бути доцільним підготувати окремий РОЗБ; наприклад, для лікарських засобів з різними лікарськими формами для цілком різних показань. У цьому випадку необхідно отримати згоду відповідних уповноважених органів (бажано під час реєстрації).
У розділі з оцінки ризиків РОЗБ описи випадків побічних реакцій повинні надаватися в якості невід’ємної частини наукового аналізу сигналу чи питання безпеки (ст. 34(4) ІП 520/2012 [6],додаток 12 до Порядку [2]N). У цьому контексті термін «опис випадку» стосується скоріше клінічної оцінки окремих випадків побічних реакцій, а не опису випадку відповідно до CIOMS (карти-повідомлення про випадок побічної реакціїN). Тобто, не потрібно надавати дослівний опис випадків відповідно до CIOMS (карти-повідомлення про випадок побічної реакціїN), що включений до повідомлення про індивідуальний випадок, пов’язаний з безпекою (ICSR), а представити клінічну оцінку важливих чи наглядних випадків у контексті оцінки питань безпеки/сигналу.
У випадках коли дані, отримані заявником (власником реєстраційного посвідчення) від партнерів можуть значно вплинути на аналіз безпеки, користі чи співвідношення користь/ризик, а також можуть вплинути на інформацію про лікарський засіб заявника (власника реєстраційного посвідчення), який звітує, такі дані також повинні бути включені та прокоментовані у РОЗБ.
Формат та зміст регулярного звіту з безпеки повинен відповідати структурі, що описана у ст. 35 ІП 520/2012 [6], додатку 12 до Порядку [2]N. Кожний регулярний звіт з безпеки повинен включати дані як за звітний період, так і кумулятивні дані. Оскільки РОЗБ повинен бути окремим документом за звітний період, що ґрунтується на кумулятивних даних, то узагальнені зведені звіти та звіти у вигляді доповнень, запроваджені у настанові ICH-E2C(R1) [28], не будуть прийматися до розгляду.
За необхідності при підготовці РОЗБ слід користуватися даними модулів ННПФ стосовно особливих питань, пов’язаних із лікарськими засобами або популяцією60.
VII.B.4. Довідкова інформація
Заходи з мінімізації ризиків, що оцінюються у РОЗБ, включають оновлення інформації про лікарський засіб.
Довідкова інформація про лікарський засіб при складанні РОЗБ повинна включати компоненти «основної інформації з безпеки» та «затверджені показання до застосування». Для сприяння оцінці користі і співвідношення користь/ризик за кожним показанням у відповідних розділах РОЗБ, у яких проводиться оцінка, довідкова інформація про лікарський засіб повинна містити перелік затверджених показань у ICH країнах61 чи регіонах ICH. Якщо РОЗБ також надається до інших країн, у яких локально затверджені додаткові показання до застосування, ці показання, на розсуд заявника (власника реєстраційного посвідчення), можуть бути додані до довідкової інформації про лікарський засіб або представлятися у вигляді окремого локального додатку. Основою для оцінки користі від застосування лікарського засобу повинна бути базова важлива інформація з його ефекту та ефективності, узагальнена у розділі 17.1 «Важлива базова інформація з ефективності» РОЗБ.
Інформація, пов’язана зі специфічними показаннями, формою випуску чи способом застосування, повинна бути чітко визначеною в довідковій інформації про лікарський засіб.
Заявник (власник реєстраційного посвідчення) може розглядати наступні опції при виборі найбільш відповідної довідкової інформації про лікарський засіб при складанні РОЗБ:
- перелік основних даних заявника (власника реєстраційного посвідчення) (далі — ПОДЗ):
- загальною практикою для заявників (власників реєстраційних посвідчень) є підготовка їх власних ПОДЗ, що включають інформацію з безпеки, показань, дозування та іншу інформацію про лікарський засіб. Основна інформація з безпеки, яка є складовою ПОДЗ, називається основною інформацією з безпеки заявника (власника реєстраційного посвідчення) (далі — ОІБЗ). Для складання РОЗБ практичним варіантом вибору для кожного заявника (власника реєстраційного посвідчення) є використання ПОДЗ, дійсного на момент завершення звітного періоду, у якості довідкової інформації як при генерації розділу ризиків РОЗБ, так і для основних затверджених показань, для яких оцінюється користь;
- якщо ПОДЗ не містить інформації про затверджені показання, заявник (власник реєстраційного посвідчення) повинен чітко зазначити, який саме документ використовувався у РОЗБ у якості довідкової інформації для затверджених показань;
- інші опції для вибору довідкової інформації про лікарський засіб:
- якщо для лікарського засобу не існує ні ПОДЗ, ні ОІБЗ (наприклад, якщо лікарський засіб зареєстрований тільки в одній країні чи регіоні, або для лікарських засобів, що мають добре вивчене медичне застосування/генеричних лікарських засобів, що перебувають на ринку вже багато років), заявник (власник реєстраційного посвідчення) повинен чітко зазначити, яку довідкову інформацію при генерації РОЗБ він використовував. Така інформація може включати національну чи регіональну інформацію про лікарський засіб, наприклад, коротка характеристика/інструкція для медичного застосуванняN лікарського засобу у ЄС/в УкраїніN;
- якщо довідкова інформація для затверджених показань є окремим документом у довідковій інформації з безпеки (ОІБЗ є складовою довідкової інформації про лікарський засіб), її версія, дійсна на завершення звітного періоду, повинна бути включена в якості додатка до РОЗБ (див. розділ VII.В.5.20 ННПФ).
Заявник (власник реєстраційного посвідчення) повинен постійно оцінювати, чи існує необхідність у будь-якому перегляді довідкової інформації про лікарський засіб/довідкової інформації з безпеки лікарського засобу, якщо під час звітного періоду отримується нова інформація, та забезпечувати, щоб усі значні зміни, внесені у звітний період, були описані в розділі 4 «Зміни у довідковій інформації з безпеки» РОЗБ і, якщо доцільно, розглянуті в розділі 16 «Оцінка сигналів та ризиків» РОЗБ. Такі зміни можуть включати:
- зміни до розділів «Протипоказання», «Особливі заходи з безпеки»/«Особливості застосування»;
- доповнення в розділи «Побічні реакції» та «Взаємодія з іншими лікарськими засобами та інші види взаємодій»;
- доповнення значущої нової інформації про передозування лікарським засобом;
- видалення показань чи інші обмеження з причини безпеки чи відсутності ефективності.
Заявник (власник реєстраційного посвідчення) має надати примірники всіх версій довідкової інформації, дійсних на завершення звітного періоду (наприклад, стосовно різних лікарських форм лікарського засобу, що увійшли до одного й того ж РОЗБ), у вигляді додатку до РОЗБ (див. розділ VII.В.5.20 ННПФ). Довідкова інформація повинна бути датованою, а її версії повинні контролюватися.
Якщо протягом періоду від дати закриття бази даних до подання РОЗБ до довідкової інформації про безпеку лікарського засобу була внесена нова інформація з безпеки, що може зумовити зміни до затвердженої інформації про лікарський засіб (наприклад, нові побічні реакції, застереження чи протипоказання), якщо доречно, її потрібно включити до розділу 14 «Інформація, отримана в останній момент» РОЗБ.
Якщо регіональне законодавство вимагає, заявник (власник реєстраційного посвідчення) повинен надати в локальному додатку інформацію про будь-які внесені зміни, зміни, що вносяться чи пропонуються до внесення у національну або локальну коротку характеристику/інструкцію для медичного застосуванняN лікарського засобу (див. розділ VII.С.5 ННПФ).
VII.B.5. Формат та зміст регулярно оновлюваного звіту з безпеки
РОЗБ повинен базуватися на всіх доступних даних та акцентувати увагу на новій інформації, що стала відомою з моменту закриття бази даних останнього РОЗБ (ст. 34(1) ІП 520/2012 [6], положення Порядку [2]N). При проведенні загальної оцінки з безпеки та інтегрованої оцінки співвідношення користь/ризик необхідно враховувати кумулятивну інформацію.
Оскільки клінічна розробка лікарського засобу часто продовжується в післяреєстраційний період, до РОЗБ необхідно також включати відповідну інформацію з післяреєстраційних досліджень чи клінічних випробувань при застосуванні лікарського засобу за незатвердженими показаннями чи у несхвалених популяціях. Подібним чином, оскільки інформацію про безпеку лікарського засобу можна отримувати при оцінці інших даних, пов’язаних із застосуванням лікарського засобу не за затвердженими показаннями, її також слід використовувати при оцінці ризиків, якщо це доцільно та можливо.
РОЗБ повинен надавати узагальнені дані щодо користі та ризиків лікарського засобу, включаючи результати всіх досліджень з урахуванням їх потенційного впливу на реєстраційне посвідчення (ст. 107b(1)(a) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Нижче наведені приклади джерел інформації про ефекти, ефективність та безпеку лікарських засобів, що можуть використовуватися при підготовці РОЗБ:
- доклінічні дослідження;
- спонтанні повідомлення (наприклад база даних з безпеки заявника (власника реєстраційного посвідчення));
- системи активного фармаконагляду (наприклад «контрольні» ділянки);
- дослідження якості лікарського засобу;
- дані про застосування лікарського засобу (usage data) та інформація про його споживання (utilisation information);
- клінічні випробування, включаючи дослідження за незареєстрованими показаннями або у несхвалених популяціях;
- обсерваційні дослідження, включаючи реєстри;
- програми підтримки пацієнтів;
- систематичні огляди та метааналіз;
- вебсайти, що спонсоруються заявниками (власниками реєстраційних посвідчень)62;
- опублікована наукова література чи повідомлення з оглядів, включаючи інформацію, що представлена на наукових заходах;
- неопубліковані рукописи;
- договірні партнери, інші спонсори або наукові установи та дослідницькі центри;
- уповноважені органи (на глобальному рівні).
Наведений вище перелік не є вичерпним. Якщо доцільно, заявник (власник реєстраційного посвідчення) може використовувати додаткові джерела інформації для відображення інформації з безпеки, ефектів та ефективності у РОЗБ та для оцінки співвідношення користь/ризик з урахуванням відомої та нової інформації про важливі переваги та ризики. Якщо заявник (власник реєстраційного посвідчення) вважає за потрібне, він може надати перелік джерел інформації, що використовувалися при складанні РОЗБ, у додатку до РОЗБ.
При оформленні РОЗБ необхідно дотримуватися повної модульної структури, що представлена в додатку II ІП 520/2012 (ст. 35 ІП 520/2012 [6]), додатку 12 до Порядку [2]N.
У рамках цього модуля джерела інформації включають дані стосовно діючої речовини (АФІN), що міститься в складі підзвітного лікарського засобу, або будь-якого іншого лікарського засобу, до якого заявник (власник реєстраційного посвідчення) може мати доступ, та який є важливим для оцінки безпеки та/або співвідношення користь/ризик. Тому що хоч і необхідно дотримуватися однакового формату РОЗБ для всіх лікарських засобів, однак обсяг наданої інформації може відрізнятися, якщо це виправдано залежно від того, що є доступним заявнику (власнику реєстраційного посвідчення). Наприклад, при спонсорованому заявником (власником реєстраційного посвідчення) клінічному випробуванні він матиме доступ до даних на рівні пацієнта, а у випадку неспонсорованого ним клінічного випробування — лише до опублікованого звіту про проведене клінічне випробування.
Рівень деталізації інформації, представленої у певних розділах РОЗБ, залежить від відомої або нової значущої інформації про користь та ризики лікарського засобу. Такий підхід застосовується до тих розділів РОЗБ, у яких оцінюється інформація з безпеки та ефективності, сигналів безпеки та співвідношення користь/ризик.
При підготовці РОЗБ слід також використовувати настанову ICH-E2C(R2) [28] (див. додаток IV настанови ICH-E2C(R2) [28]) з підготовки PBRER. Рекомендації щодо назв, послідовності та змісту розділів РОЗБ надаються у розділах VII.В.5.1.–VII.В.5.21. Якщо для будь-якого з розділів відсутня відповідна інформація, про це слід зазначити.
Частина I: Титульна сторінка, включаючи підпис63.
Частина II: Загальні положення.
Частина III: Зміст.
1. Вступ.
2. Міжнародний реєстраційний статус.
3. Заходи з безпеки, вжиті протягом звітного періоду.
4. Зміни у довідковій інформації з безпеки.
5. Оцінка експозиції та схем застосування.
5.1. Кумулятивна експозиція учасників клінічних випробувань.
5.2. Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційний період.
6. Дані зведених таблиць.
6.1. Довідкова інформація.
6.2. Кумулятивні зведені таблиці про серйозні побічні явища, що виявлені під час клінічних випробувань.
6.3. Кумулятивні та інтервальні зведені таблиці згідно з даними щодо безпеки, отриманими у післяреєстраційний період.
7. Резюме значущих результатів клінічних випробувань протягом звітного періоду.
7.1. Завершені клінічні випробування.
7.2. Поточні клінічні випробування.
7.3. Тривале спостереження.
7.4. Інше терапевтичне застосування лікарського засобу.
7.5. Нові дані з безпеки, пов’язані з фіксованим комбінованим лікуванням.
8. Результати неінтервенційних досліджень.
9. Інформація з інших клінічних випробувань та джерел.
9.1. Інші клінічні випробування.
9.2. Помилки, пов’язані лікарським засобом.
10. Неклінічні дані.
11. Дані з літератури.
12. Інші регулярно оновлювані звіти з безпеки
13. Відсутність ефективності в контрольованих клінічних випробуваннях.
14. Інформація, отримана в останній момент.
15. Огляд сигналів (нові, поточні та закриті).
16. Оцінка сигналів та ризиків.
16.1. Резюме проблем безпеки.
16.2. Оцінка сигналів.
16.3. Оцінка ризиків та нової інформації.
16.4. Характеристика ризиків.
16.5. Ефективність заходів з мінімізації ризиків (якщо застосовано).
17. Оцінка користі.
17.1. Важлива базова інформація з ефектів та ефективності.
17.2. Нова виявлена інформація з ефектів та ефективності.
17.3. Характеристика користі.
18. Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань.
18.1. Контекст співвідношення користь/ризик (медична необхідність і важливі альтернативи).
18.2. Оцінка аналізу співвідношення користь/ризик.
19. Висновки та заходи.
20. Додатки до регулярно оновлюваного звіту з безпеки.
Титульна сторінка регулярно оновлюваного звіту з безпеки
Титульна сторінка має містити назву(и) лікарського(их) засобу(ів)64 та діючої речовини (АФІN), міжнародну дату народження лікарського засобу (дата першої реєстрації будь-якого лікарського засобу, що містить таку саму діючу речовину (АФІN), що видана будь-якій компанії у будь-якій країні світу (дата видачі заявнику першого дозволу на продаж лікарського засобу в будь-якій країні світуN)), звітний період, дату складання звіту, детальні дані про заявника (власника реєстраційного посвідчення) та твердження про конфіденційність інформації, що надана в РОЗБ.
На титульній сторінці повинен стояти підпис.
Основні положення регулярно оновлюваного звіту з безпеки
Основні положення РОЗБ повинні бути розміщені відразу за титульною сторінкою перед змістом. Мета основних положень полягає в наданні стислого викладу змісту та найбільш важливої інформації, що надана у РОЗБ. Вони повинні містити таку інформацію:
- вступ і звітний період;
- лікарський(і) засіб (засоби), хіміко-терапевтична або фармакологічна група(и), механізм(и) дії, показання, форми випуску, доза(и) та спосіб (способи) застосування;
- узагальнена оцінка кумулятивного впливу лікарського засобу на учасників клінічних випробувань;
- узагальнена оцінка кумулятивного та інтервального впливу лікарського засобу на пацієнтів у післяреєстраційний період;
- кількість країн, у яких лікарський засіб зареєстровано;
- резюме загальної оцінки аналізу співвідношення користь/ризик (на основі даних, представлених у підрозділі 18.2 «Оцінка аналізу співвідношення користь/ризик» регулярного звіту з безпеки);
- вжиті та пропоновані заходи з питань безпеки (наприклад, важливі зміни до довідкової інформації про лікарський засіб чи інші заходи з мінімізації ризиків);
- висновки.
Зміст регулярно оновлюваного звіту з безпеки
Після основних положень повинен надаватися зміст РОЗБ.
VII.B.5.1. Розділ регулярно оновлюваного звіту з безпеки «Вступ»
Заявник (власник реєстраційного посвідчення) повинен коротко описати лікарський(і) засіб (засоби) з тим, щоб РОЗБ був «автономним» документом, однак також відображав зв’язок з попередніми регулярними звітами з безпеки та обставинами. Вступ повинен містити таку інформацію:
- міжнародна дата народження лікарського засобу та звітний період;
- лікарський(і) засіб (засоби), хіміко-терапевтична або фармакологічна(і) група(и), механізм(и) дії, показання, форма(и) випуску, доза(и) та спосіб (способи) застосування;
- стислий опис пролікованих та досліджуваних популяцій.
VII.B.5.2. Розділ регулярно оновлюваного звіту з безпеки «Міжнародний реєстраційний статус»
Цей розділ РОЗБ повинен містити стислий опис із зазначенням наступної інформації: дати першої реєстрації у світі, показання, зареєстрованої(их) дози(доз), країни реєстрації лікарського засобу.
VII.B.5.3. Розділ регулярно оновлюваного звіту з безпеки «Заходи з безпеки, вжиті протягом звітного періоду»
Цей розділ РОЗБ повинен містити опис важливих заходів з безпеки, що були вжиті у світі протягом звітного періоду власником реєстраційного посвідчення, спонсорами клінічних випробувань, комітетами з моніторингу даних, етичними комітетами чи уповноваженими органами (незалежно від того, стосувалися вони застосування лікарського засобу чи ні) при проведенні випробувань чи в післяреєстраційний період, та мали:
- значний вплив на співвідношення користь/ризик зареєстрованого лікарського засобу; та/або
- вплив на проведення окремого(их) клінічного(их) випробування(ь) або на загальну програму клінічних випробувань.
Якщо відомо, слід надати обґрунтування кожного заходу та за необхідності включити до РОЗБ будь-яку додаткову важливу інформацію. У цьому розділі слід також надати в узагальненому вигляді відповідні оновлення до попередніх заходів з питань безпеки.
Нижче наведені приклади важливих заходів, вжитих з причин безпеки:
1) заходи, пов’язані із застосуванням лікарського засобу з дослідницькою метою:
- відмова в реєстрації клінічного випробування з етичних причин або з міркувань безпеки;
- часткове (може включати декілька заходів (наприклад, припинення досліджень повторних доз, але продовження досліджень однієї дози; припинення досліджень за одним показанням, але продовження за іншим та/або припинення застосування певної схеми прийому у дослідженні) (Настанова ICH-E2C(R2) [28] (див. додаток IV)) або повне припинення клінічного випробування або дострокове припинення поточного клінічного випробування з причин безпеки або відсутності ефективності;
- відкликання досліджуваного лікарського засобу або препарату порівняння;
- відмова в реєстрації для досліджуваних показань, включаючи добровільне відкликання заяви на реєстрацію;
- заходи з управління ризиками, включаючи:
- зміни до протоколу, що пов’язані з проблемами безпеки та ефективності (наприклад, зміни у дозуванні, зміни критеріїв включення/виключення з дослідження, посилення моніторингу суб’єктів дослідження, обмеження тривалості дослідження);
- обмеження досліджуваних популяцій або показань;
- зміни з проблем безпеки, внесені до інформованої згоди пацієнта;
- зміни у складі лікарського засобу;
- введення регуляторними органами особливих додаткових вимог щодо надання повідомлень, пов’язаних з безпекою;
- надання повідомлень дослідникам або працівникам з медичною чи фармацевтичною освітою;
- плани нових досліджень для вивчення проблем з безпеки;
2) заходи, пов’язані з досвідом перебування на ринку:
- відмова в перереєстрації або неподання заяви на перереєстрацію;
- відкликання або призупинення дії реєстраційного посвідчення;
- заходи, вжиті через проблеми, пов’язані з якістю;
- призупинення постачання лікарського засобу заявником (власником реєстраційного посвідчення);
- заходи з управління ризиками, включаючи:
- значні обмеження дистрибуції лікарського засобу чи вжиття інших заходів з мінімізації ризиків;
- значні зміни з безпеки, що вносяться до інструкції для медичного застосування, включаючи обмеження застосування чи обмеження популяцій, які отримають лікування;
- надання повідомлень працівникам з медичною та фармацевтичною освітою;
- нова(і) вимога(и) регуляторних/уповноважених органів щодо проведення післяреєстраційного дослідження.
VII.B.5.4. Розділ регулярно оновлюваного звіту з безпеки «Зміни в довідковій інформації з безпеки»
У даному розділі РОЗБ мають бути перераховані будь-які значні зміни до довідкової інформації з безпеки у звітний період. Такі зміни можуть стосуватися протипоказань, особливостей застосування, застережень, серйозних побічних реакцій, взаємодій, важливих результатів поточних чи завершених клінічних випробувань або важливих результатів неклінічних досліджень (наприклад, дослідження канцерогенності). Специфічна інформація стосовно таких змін повинна надаватися у відповідних розділах РОЗБ.
VII.B.5.5. Розділ регулярно оновлюваного звіту з безпеки «Оцінка експозиції та схем застосування»
РОЗБ повинен надати точну оцінку популяцій, які піддавалися впливу лікарського засобу, включаючи обсяги продажів та кількість призначень. Така оцінка має супроводжуватися якісним та кількісним аналізом фактичного застосування лікарського засобу. Такий аналіз повинен, якщо доцільно, показувати, наскільки фактичне застосування відрізняється від показаного застосування на підставі всіх даних, доступних власнику реєстраційного посвідчення, включаючи результати обсерваційних досліджень або досліджень застосування (ст. 34 ІП 520/2012 [6], положення Порядку [2]N).
У даному розділі РОЗБ потрібно надати оцінку численності та типу популяції, яка піддавалася впливу лікарського засобу, включно з коротким описом методу(ів), що використовувалися для оцінки впливу на суб’єктів/пацієнтів та із зазначенням обмежень такого методу.
Необхідно використовувати порівняльні методи для оцінки впливу на суб’єктів/пацієнтів у всіх РОЗБ для одного й того самого лікарського засобу. Якщо з об’єктивних причин метод був змінений, обидва методи та розрахунки слід надавати у РОЗБ, із зазначенням зміни та будь-якої важливої відмінності між результатами при використанні обох методів.
VII.B.5.5.1. Підрозділ регулярно оновлюваного звіту з безпеки «Кумулятивна експозиція учасників клінічних випробувань»
Цей підрозділ РОЗБ повинен містити наступну інформацію про учасників клінічних випробувань, спонсорованих заявником (власником реєстраційного посвідчення), подану (за можливості) у вигляді таблиці:
- кумулятивна кількість учасників поточних та завершених клінічних випробувань, які піддавалися впливу досліджуваного лікарського засобу, плацебо та/або препарату(ів) порівняння, починаючи з міжнародної дати народження лікарського засобу, що знаходиться в стадії розробки. Загальновизнано, що для «старих лікарських засобів» детальні дані можуть бути недоступними;
- за можливості, слід надати більш детальні дані про вплив на учасників клінічних випробувань (наприклад, дані, згруповані за віком, статтю та расовою приналежністю, для всієї програми випробування);
- якщо можливо, в таблицях має бути зазначено суттєві розбіжності у випробуваннях у дозах, способах застосування чи популяціях пацієнтів; можна також використовувати окремі таблиці;
- якщо клінічні випробування проводилися або проводяться у спеціальних популяціях (наприклад, вагітні, пацієнти з нирковою, печінковою або серцевою недостатністю; або пацієнти з відповідним генетичним поліморфізмом), необхідно надати дані про їх експозицію;
- якщо існують значні розбіжності у тривалості впливу між суб’єктами, рандомізованими до досліджуваного лікарського засобу чи компаратора, або у випадку невідповідності щодо тривалості експозиції у різних клінічних випробуваннях, потрібно надати інформацію про експозицію лікарського засобу у суб’єкто-часі (суб’єкто-днях, суб’єкто-місяцях або суб’єкто-роках);
- експозиція досліджуваного лікарського засобу у здорових добровольців може бути менш важливою для оцінки загального профілю безпеки (залежно від типу побічної реакції, зокрема, у разі застосування суб’єктами однієї дози). Такі дані можуть бути надані окремо з відповідним поясненням;
- якщо серйозні побічні явища, виявлені під час клінічних випробувань, надані у зведених таблицях за показаннями, експозиція пацієнтів має бути також наведена за показаннями (за можливості);
- для окремих особливо важливих досліджень демографічні характеристики повинні надаватися окремо.
Приклади табличного формату для оцінки експозиції у клінічних випробуваннях представлені в таблицях VII.2, VII.3 та VII.4 додатку 1 розділу VII.
VII.B.5.5.2. Підрозділ регулярно оновлюваного звіту з безпеки «Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційний період»
Якщо можливо, потрібно надати кумулятивну експозицію (починаючи з міжнародної дати народження) та інтервальну експозицію (з моменту закриття бази даних для складання попереднього РОЗБ). Відомо, що часто дуже складно отримати та оцінити експозицію пацієнтів, проте, якщо можливо, слід зазначити кількість пацієнтів, які піддавалися впливу лікарського засобу, а також метод(и), що використовується для оцінки. У випадку неможливості оцінити експозицію пацієнтів, необхідно надати пояснення причин цього. У такому випадку, якщо можливо, слід надати альтернативні розрахунки експозиції, а також методику(и) їх отримання. Прикладами альтернативних розрахунків експозицій є пацієнто-дні та кількість призначень. Лише у випадках, коли такі показники є недоступними, можуть використовуватися розрахунки за обсягами продажу лікарського засобу, такі як тоннаж або одиниці дозування. Для обчислення впливу лікарського засобу можна також використовувати встановлену добову дозу (далі — DDD).
Дані мають бути подані відповідно до таких категорій:
- післяреєстраційна (клінічні дослідження) експозиція:
слід надати загальну експозицію пацієнтів. За можливості дані повинні бути розподілені за статтю, віком, показаннями, дозами, формою випуску та регіонами. Залежно від лікарського засобу може бути важливим розподіл за іншими параметрами, такими як кількість курсів вакцинації, спосіб (способи) застосування та тривалість лікування.
Якщо існують повідомлення про побічні реакції, що вказують на сигнал безпеки, за можливості потрібно надати дані експозиції лікарського засобу в межах відповідних підгруп;
- застосування у спеціальних популяціях у післяреєстраційний період:
якщо у післяреєстраційний період лікарський засіб застосовувався у спеціальних популяціях, необхідно надати інформацію про загальну кількість пацієнтів, які його застосовували, та метод підрахунку. Джерела таких даних можуть, наприклад, включати неінтервенційні дослідження, що проводилися з метою отримання такої інформації, у тому числі реєстри. Такими джерелами можуть бути також дані, зібрані не в умовах дослідження, включаючи інформацію, що зібрана методом спонтанних повідомлень (наприклад, у цьому пункті може бути узагальнена інформація з повідомлень про вплив лікарського засобу в період вагітності без зв’язку з побічними реакціями). Популяції, які необхідно розглянути, включають, але не обмежуються наступними:
- педіатрична популяція;
- пацієнти похилого віку;
- вагітні або жінки, які годують груддю;
- пацієнти з печінковою та/або нирковою недостатністю;
- пацієнти з іншими відповідними супутніми захворюваннями;
- пацієнти з тяжкістю захворювання, що відрізняється від тієї, що досліджувалася у клінічних випробуваннях;
- субпопуляції з відповідним генетичним поліморфізмом(ами);
- пацієнти певного расового та/або етнічного походження;
- інше застосування лікарського засобу у післяреєстраційний період:
якщо заявник (власник реєстраційного посвідчення) має інформацію про схеми застосування лікарського засобу, що можуть бути регіональними та вважаються важливими для інтерпретації даних з безпеки, необхідно надати їх стислий опис. Прикладами таких схем застосування можуть бути випадки передозування, зловживання, неправильного застосування, а також застосування не за показаннями згідно з довідковою інформацією про лікарський засіб (наприклад, протиепілептичний лікарський засіб використовувався для зменшення вираженості невропатичного болю і/або для профілактики мігреневого болю). Якщо ці дані впливають на оцінку безпеки та/або співвідношення користь/ризик, отримана інформація про схеми застосування без зареєстрованих побічних реакцій повинна бути узагальнено подана у цьому підрозділі. Така інформація може бути отримана за допомогою системи спонтанних повідомлень, запитів на надання медичної інформації, скарг пацієнтів чи за допомогою аналізу електронних видань інших інформаційних джерел, що доступні власнику реєстраційного посвідчення. Якщо відомі кількісні показники таких даних, необхідно їх викласти в цьому підрозділі.
Якщо відомо, заявник (власник реєстраційного посвідчення) може стисло прокоментувати, чи може бути пов’язане застосування лікарського засобу не у відповідності з довідковою інформацією про лікарський засіб з клінічними настановами, результатами клінічних випробувань, або ж воно зумовлене відсутністю зареєстрованого альтернативного лікування. З метою виявлення схем застосування, що не відповідають довідковій інформації про лікарський засіб, заявник (власник реєстраційного посвідчення) має використовувати відповідні розділи довідкової інформації про лікарський засіб, що була дійсною на момент завершення звітного періоду РОЗБ (наприклад, зареєстровані показання, спосіб застосування, протипоказання).
Виявлені з таких даних чи джерел інформації сигнали і ризики повинні розглядатися у відповідних розділах РОЗБ.
Приклади формату таблиць для представлення даних експозиції надані в таблиці VII.5 та VII.6 Додатку VII.1.
VII.B.5.6. Розділ регулярно оновлюваного звіту з безпеки «Дані зведених таблиць»
Метою цього розділу РОЗБ є надання даних з безпеки у вигляді зведених таблиць щодо серйозних побічних явищ з клінічних випробувань, спонтанних повідомлень про серйозні та несерйозні реакції на підставі післяреєстраційного досвіду застосування лікарського засобу (включаючи повідомлення працівників з медичною та/або фармацевтичною освітою, споживачів, дані з медичної наукової літератури, повідомлення від уповноважених органів (на міжнародному рівні) та дані про серйозні реакції з неінтервенційних досліджень та інших неінтервенційних джерел інформації про побічні реакції). На розсуд заявника (власника реєстраційного посвідчення) та для більш наочного представлення інформації, для ілюстрування специфічних аспектів даних може використовуватися їх графічне відображення.
При кодуванні термінів побічних явищ/реакцій з використанням термінології медичного словника для регуляторної діяльності (Medical Dictionary for Regulatory Activities — MedDRA)) у зведених таблицях необхідно надавати дані на рівні термінів переважного застосування (preferred term — PT)) та класів систем і органів (system organ class — SOC)).
Серйозність побічних явищ/реакцій у зведених таблицях повинна відповідати серйозності явищ/реакцій у повідомленнях про індивідуальні випадки побічних реакцій, що визначається із застосуванням критеріїв, наведених в настанові ICH-E2А [29]/відповідно до положень Порядку [2]N. Якщо серйозні та несерйозні явища/реакції належать до одного й того ж випадку, у зведених таблицях необхідно надати інформацію про серйозність кожної реакції. Не можна змінювати серйозність побічного явища/реакції спеціально для підготовки РОЗБ.
VII.B.5.6.1. Підрозділ регулярно оновлюваного звіту з безпеки «Довідкова інформація»
У цьому підрозділі РОЗБ необхідно вказати версію(ї) довідкової інформації (словника), що використовувалася(ися) для кодування побічних явищ/реакцій.
VII.B.5.6.2. Підрозділ регулярно оновлюваного звіту з безпеки «Кумулятивні зведені таблиці про серйозні побічні явища, що виявлені під час клінічних випробувань»
У цьому підрозділі РОЗБ потрібно надати основні дані для додатка, в якому надаються зведені таблиці серйозних побічних реакцій з клінічних випробувань заявника (власника реєстраційного посвідчення), починаючи з міжнародної дати народження лікарського засобу, що знаходиться в стадії розробки до дати закриття бази даних для складання наступного РОЗБ. Заявник (власник реєстраційного посвідчення) повинен надавати пояснення з приводу будь-якого невключення даних до таблиць (наприклад, дані клінічного випробування можуть бути недоступними для лікарських засобів, що наявні на ринку впродовж багатьох років). Таблиці мають бути структуровані за MedDRA на рівні класів — систем органів (далі — SOC) (впорядкованими згідно з міжнародними правилами) як для досліджуваного лікарського засобу, так і для груп(и) компаратора (препарати порівняння, плацебо), що використовувалися у програмі клінічних випробувань. Дані можуть бути зібрані з усієї програми. Якщо доцільно та можливо, дані можуть бути розподілені за дослідженнями, показаннями, способами застосування або іншими параметрами.
Цей підрозділ не повинен використовуватися для надання аналізу чи висновків на підставі серйозних побічних явищ.
При підготовці цього підрозділу потрібно враховувати наступне:
- оцінка причинно-наслідкового зв’язку зазвичай корисна для оцінки індивідуальних випадків рідкісних побічних реакцій. Ця оцінка має меншу цінність для аналізу узагальнених даних, коли стає можливим групове порівняння частоти випадків. Тому до зведених таблиць потрібно включати всі серйозні побічні явища, а не лише серйозні побічні реакції на досліджуваний препарат, компаратори та плацебо. Може бути доцільним надавати частоту відповідно до доз;
- загалом таблиці серйозних побічних явищ з клінічних випробувань повинні містити лише терміни, що використовувалися для визначення випадку як серйозного, а несерйозні побічні явища повинні бути включені у звіти про випробування;
- таблиці повинні містити дані сліпих та відкритих клінічних випробувань. Відкриті дані про серйозні побічні явища можуть походити із завершених випрбувань та бути отриманими на підставі індивідуальних випадків, що були розкриті з міркувань безпеки (наприклад термінове повідомлення), якщо необхідно. Спонсори клінічних випробувань та заявники (власники реєстраційних посвідчень) не повинні розкривати дані сліпих досліджень з метою підготовки РОЗБ;
- деякі побічні явища можуть бути виключені зі зведених таблиць клінічних випробувань, однак у звіті такі виключення необхідно обґрунтувати. Наприклад, можна виключити побічні явища, які були визначені у протоколі як такі, що не підлягають спеціальному збору та внесенню до бази даних з безпеки, оскільки їх розвиток передбачався у популяції пацієнтів, а також ті, що представляють кінцеві точки клінічного випробування (наприклад, повідомлення про смерть при дослідженні лікарського засобу для лікування застійної серцевої недостатності, коли смерть незалежно від її причини є первинною кінцевою точкою, чи прогресування захворювання у дослідженнях лікування онкології).
Приклад зведеної таблиці серйозних побічних явищ з клінічних випробувань можна знайти в таблиці VII.7 Додатку 1 розділу VII.
VII.B.5.6.3. Підрозділ регулярно оновлюваного звіту з безпеки «Кумулятивні та інтервальні зведені таблиці згідно з даними щодо безпеки, отриманими у післяреєстраційний період»
У цьому підрозділі РОЗБ потрібно надати основні дані для додатка, в якому надаються кумулятивні та інтервальні зведені таблиці побічних реакцій, зібраних за період від міжнародної дати народження лікарського засобу до дати закриття бази даних для складання наступного РОЗБ. Ці побічні реакції, отримані зі спонтанних повідомлень, що включають повідомлення працівників з медичною та фармацевтичною освітою, споживачів, дані з наукової літератури, повідомлення від уповноважених органів (на глобальному рівні), а також обов’язкові повідомлення про індивідуальний випадок, пов’язаний з безпекою, в межах неінтервенційного організованого збору даних, включаючи неінтервенційні дослідження (настанова ICH-E2D [25]). Управління даними з безпеки у післяреєстраційний період: визначення та стандарти для термінового повідомлення). Дані щодо серйозних та несерйозних реакцій зі спонтанних джерел, а також серйозних побічних реакцій з неінтервенційних досліджень та інших неінтервенційних джерел слід надавати в одній таблиці, представляючи поряд кумулятивні дані та інтервальні. Таблицю слід структурувати згідно з MedDRA на рівні SOC (впорядкувати згідно з міжнародними правилами). Дані зі специфічних проблем безпеки можуть бути представлені в додаткових таблицях з розподілом за показаннями, способом застосування лікарського засобу чи іншими параметрами.
Як описано в настанові ICH-E2D [25], для зареєстрованих лікарських засобів побічні явища, про які спонтанно повідомлено, як правило, означають, що джерело повідомлення принаймні підозрює причинно-наслідковий зв’язок, тому вони повинні вважатися підозрюваними побічними реакціями у контексті надання повідомлень до уповноважених органів.
У цьому підрозділі РОЗБ не потрібно надавати аналіз чи висновки на підставі даних зведених таблиць.
Приклад зведених таблиць побічних реакцій на лікарські засоби з післяреєстраційних джерел даних можна знайти в таблиці VII.8 Додатку VII.1.
VII.B.5.7. Розділ регулярно оновлюваного звіту з безпеки «Резюме значущих результатів клінічних випробувань протягом звітного періоду»
У цьому розділі РОЗБ потрібно надати стислий опис клінічно важливих результатів з ефективності та безпеки лікарського засобу, отриманих за звітний період зі спонсорованих власником реєстраційного посвідчення клінічних випробувань, із джерел, що зазначені у підрозділах, що перераховані нижче. Якщо можливо і доцільно, дані слід розподілити за статтю та віком (особливо діти в порівнянні з дорослими), показанням, дозою та регіоном.
Сигнали, що походять з джерел клінічних випробувань, слід звести в таблиці в розділі 15 РОЗБ «Огляд сигналів (нові, поточні та закриті)». Оцінка сигналів незалежно від того, чи це спростований сигнал, чи потенційний, чи виявлений ризик, що були закриті за звітний період, слід надати в підрозділі 16.2 «Оцінка сигналів» РОЗБ. Нова інформація стосовно вже відомих потенційних чи виявлених ризиків, що не підпадає під поняття нового виявленого сигналу, повинна оцінюватися і характеризуватися у підрозділах 16.3 «Оцінка ризиків та нової інформації» та 16.4 «Характеристика ризиків» відповідно.
Результати клінічних випробувань, що не спонсорувалися заявником (власником реєстраційного посвідчення), мають описуватися у відповідних розділах РОЗБ.
Якщо інформація про відсутність ефективності з клінічних випробувань лікарських засобів для лікування захворювань, що не загрожують життю, відповідно до зареєстрованих показань має значення для оцінки співвідношення користь/ризик, її необхідно стисло описати в цьому розділі. Інформація про відсутність ефективності лікарського засобу, що отримана з клінічних випробувань, у яких лікарський засіб використовувався для лікування та профілактики серйозних або загрозливих для життя захворювань, повинна узагальнюватися в розділі 13 «Відсутність ефективності у контрольованих клінічних випробуваннях» РОЗБ (VII. B.5.13).
Інформація з інших клінічних випробувань/досліджуваних джерел повинна бути включена до підрозділу 9.1 «Інші клінічні випробування» РОЗБ (VII.B.5.9.1).
Крім того, заявник (власник реєстраційного посвідчення) повинен надати у вигляді додатку перелік спонсорованих ним за звітний період завершених та поточних післяреєстраційних інтервенційних досліджень, основною метою яких було виявити, характеризувати чи кількісно визначити загрозу безпеці або підтвердити профіль безпеки лікарського засобу. Перелік повинен містити наступну інформацію щодо кожного дослідження:
- ідентифікатор випробування (наприклад, номер протоколу чи інший ідентифікатор);
- назва випробування (абревіатурна назва дослідження, якщо застосовується);
- вид випробування (наприклад, рандомізоване клінічне випробування, когортне випробування, дослідження випадок-контроль);
- досліджувана популяція, включаючи країну та інші відповідні характеристики популяції (наприклад, педіатрична популяція або дослідження пацієнтів з нирковою недостатністю);
- дата початку (визначена заявником (власником реєстраційного посвідчення)) і планована дата завершення випробування;
- етап клінічного випробування (триває (клінічне випробування почалося) чи завершене (завершено написання звіту про клінічне випробування)).
VII.B.5.7.1. Підрозділ регулярно оновлюваного звіту з безпеки «Завершені клінічні випробування»
У цьому підрозділі РОЗБ потрібно надати стислий опис клінічно важливих результатів з ефективності та безпеки, що отримані з клінічних випробувань, завершених у звітний період. Ця інформація може бути подана у вигляді опису чи синопсису65. Підрозділ може включати інформацію, що підтверджує або спростовує попередні виявлені проблеми безпеки, а також докази нових сигналів з безпеки.
VII.B.5.7.2. Підрозділ регулярно оновлюваного звіту з безпеки «Поточні клінічні випробування»
Якщо заявнику (власнику реєстраційного посвідчення) відомі клінічно важливі результати щодо ефективності та безпеки, виявлені в поточних клінічних випробуваннях (наприклад, при проміжному аналізі даних з безпеки або внаслідок декодування даних щодо пацієнтів з побічними явищами), їх необхідно стисло узагальнити в цьому підрозділі. Це може бути інформація, що підтверджує чи спростовує попередні ідентифіковані проблеми безпеки, а також підтверджує нові сигнали з безпеки.
VII.B.5.7.3. Підрозділ регулярно оновлюваного звіту з безпеки «Тривале спостереження»
Якщо заявник (власник реєстраційного посвідчення) володіє інформацією про тривале спостереження за суб’єктами клінічних випробувань, зокрема, високотехнологічних лікарських засобів (наприклад, продуктів генної інженерії, клітинної терапії та тканинного інжинірингу), її слід надати в цьому підрозділі.
VII.B.5.7.4. Підрозділ регулярно оновлюваного звіту з безпеки «Інше терапевтичне застосування лікарського засобу»
У даному підрозділі РОЗБ має міститися клінічно важлива інформація з безпеки, виявлена в інших програмах, що проводилися власником реєстраційного посвідчення за спеціальним протоколом з обов’язковим поданням повідомлень про побічні реакції згідно з настановою ICH-E2D [25]. Управління післяреєстраційними даними з безпеки: визначення та стандарти для термінового повідомлення) (наприклад, програми розширеного доступу, програми застосування незареєстрованого лікарського засобу при тяжких патологіях, програми застосування певними пацієнтами та інший організований збір даних).
VII.B.5.7.5. Підрозділ регулярно оновлюваного звіту з безпеки «Нові дані з безпеки, пов’язані з фіксованим комбінованим лікуванням»
Якщо інше не передбачено нормативно-правовими актами на національному або регіональному рівні, при поданні даних щодо комбінованого лікування у цьому підрозділі слід враховувати таке:
- якщо діюча речовина, що є предметом РОЗБ, також зареєстрована чи розробляється як компонент фіксованого комбінованого лікарського засобу чи схеми комбінованої терапії, у цьому підрозділі необхідно резюмувати важливі результати з безпеки комбінованої терапії;
- якщо сам лікарський засіб, що є предметом РОЗБ, є фіксованою комбінацією, у цьому підрозділі РОЗБ необхідно узагальнити важливу інформацію з безпеки окремих його компонентів, як вже зареєстрованих, так і тих, що знаходяться в розробці.
Дані, специфічні для комбінації, можуть надаватися в окремому(их) розділі(ах) регулярного звіту з безпеки щодо одного чи всіх його компонентів.
VII.B.5.8. Розділ регулярно оновлюваного звіту з безпеки «Результати неінтервенційних досліджень»
У цьому розділі слід також узагальнити відповідну інформацію з безпеки чи інформацію, що має потенційний вплив на оцінку співвідношення користь/ризик, отриману за звітний період зі спонсорованих власником реєстраційного посвідчення неінтервенційних досліджень (наприклад, обсерваційні дослідження, епідеміологічні дослідження, реєстри та програми активного фармаконагляду). До даного розділу також слід включати відповідну інформацію з досліджень щодо використання лікарських засобів, якщо вона стосується декількох регіонів.
Заявник (власник реєстраційного посвідчення) має надати додаток з переліком спонсорованих ним неінтервенційних досліджень, головною метою яких було ідентифікувати, характеризувати або кількісно визначити загрозу безпеці, підтвердити профіль безпеки лікарського засобу або оцінити ефективність заходів з управління ризиками, що були завершені або продовжувалися протягом звітного періоду. (Див. розділ VII. B.5.7. щодо інформації, яку слід включити до переліку).
Заключні звіти про дослідження, що були завершені у звітний період, слід також включити до локального додатку РОЗБ (див. розділ VII.B.5.20. та розділ VII.С.5.4.).
У цьому розділі можна також надавати підсумкову інформацію, що ґрунтується на комплексній оцінці даних, що отримані з програм підтримки пацієнтів, якщо вони не надавалися в інших розділах РОЗБ. Що стосується інших інформаційних джерел, то власник реєстраційного посвідчення повинен надавати інформацію про сигнали і ризики, виявлені з таких джерел, у відповідних розділах РОЗБ.
VII.B.5.9. Розділ регулярно оновлюваного звіту з безпеки «Інформація з інших клінічних випробувань та джерел»
VII.B.5.9.1. Підрозділ регулярно оновлюваного звіту з безпеки «Інші клінічні випробування»
У цьому пункті РОЗБ слід узагальнити інформацію про оцінку співвідношення користь/ризик лікарського засобу з інших клінічних випробувань/джерел дослідження, які доступні заявнику (власнику реєстраційного посвідчення) під час звітного періоду (наприклад, результати узагальненого або метааналізу рандомізованих клінічних випробувань, інформація з безпеки, надана партнерами з сумісної розробки або отримана з досліджень, що ініційовані дослідником).
VII.B.5.9.2. Підрозділ регулярно оновлюваного звіту з безпеки «Лікопов’язані помилки»
У цьому підрозділі РОЗБ необхідно узагальнити інформацію про лікопов’язані помилки і потенційні помилки, навіть якщо вони не пов’язані з розвитком небажаних ефектів. Потенційні лікопов’язані помилки, — розпізнавання обставин, що можуть призводити до помилок, що пов’язані із застосуванням лікарського засобу, і можуть як включати пацієнта, так і не включати. Ця інформація може стосуватися інтерпретації даних з безпеки чи загальної оцінки співвідношення користь/ризик лікарського засобу. Лікопов’язані помилки можуть виникати на будь-якому етапі процесу медичного застосування і можуть включати пацієнтів, споживачів, працівників з медичною чи фармацевтичною освітою.
VII.B.5.10. Розділ регулярно оновлюваного звіту з безпеки «Неклінічні дані»
У цьому розділі РОЗБ слід узагальнити основні результати з безпеки з неклінічних in vivo та in vitro досліджень (наприклад, дослідження канцерогенності, репродуктивності та імунотоксичності), поточних чи завершених у звітний період досліджень. Результати досліджень, що проводилися з метою вивчення специфічних проблем безпеки, повинні включатися до РОЗБ незалежно від наслідків. Наслідки цих результатів повинні обумовлюватися у відповідних розділах РОЗБ.
VII.B.5.11. Розділ регулярно оновлюваного звіту з безпеки «Дані з літератури»
Цей розділ РОЗБ повинен містити резюме нових та важливих даних з безпеки стосовно лікарського засобу, що опубліковані в спеціальній науковій медичній літературі або доступні у вигляді неопублікованих рукописів, про які стало відомо заявнику (власнику реєстраційного посвідчення) у звітний період.
Літературний пошук для РОЗБ має бути більш розширеним, ніж при пошуку окремих випадків побічних реакцій, оскільки він має також включати дані з безпеки з досліджень для груп пацієнтів та для інших препаратів, що містять однакову діючу речовину.
Нижче наведені особливі види інформації з безпеки, що повинні бути включені до РОЗБ, але яку можна не знайти шляхом літературного пошуку випадків побічних реакцій:
- результати вагітності (включаючи її завершення) без несприятливих наслідків;
- застосування у педіатричних популяціях;
- застосування під контролем незареєстрованого лікарського засобу тяжко хворими пацієнтами чи поіменованими пацієнтами;
- відсутність ефективності;
- безсимптомне передозування, зловживання або неправильне застосування;
- лікопов’язана помилка без виникнення побічних явищ;
- важливі неклінічні результати з безпеки.
Якщо доцільно, слід розглянути інформацію про інші діючі речовини того самого класу. Посилання на публікації слід надавати у стилі Ванкуверської конвенції66, 67.
VII.B.5.12. Розділ регулярно оновлюваного звіту з безпеки «Інші регулярно оновлювані звіти з безпеки»
Цей розділ РОЗБ застосовується тільки за певних обставин відносно фіксованих комбінованих лікарських засобів чи лікарських засобів з багатьма показаннями та/або формами випуску у випадках, коли за погодженням з уповноваженим органом готується декілька РОЗБ. Зазвичай, заявник (власник реєстраційного посвідчення) повинен написати один регулярний звіт з безпеки на одну діючу речовину (якщо уповноважений орган не вимагає інакше). Однак якщо для лікарського засобу з однією діючою речовиною готуються різні РОЗБ, тоді у цьому розділі також необхідно зазначити важливі результати з інших РОЗБ, якщо вони не представлені в інших розділах РОЗБ.
Керуючись контрактними домовленостями, заявник (власник реєстраційного посвідчення) повинен узагальнити важливі дані з РОЗБ, що надані у звітний період іншими сторонами (наприклад, спонсорами, іншими заявниками (власниками реєстраційних посвідчень) або іншими контрактними партнерами), якщо такі є доступними.
VII.B.5.13. Розділ регулярно оновлюваного звіту з безпеки «Відсутність ефективності у контрольованих клінічних випробуваннях»
У цьому розділі слід резюмувати дані з клінічних випробувань, що вказують на відсутність ефективності, або відсутність ефективності лікарських засобів із встановленим/вивченим застосуванням, для лікарських засобів, призначених для лікування і профілактики серйозних чи таких, що загрожують життю, захворювань (наприклад, зростання рівня серцево-судинних побічних явищ у дослідженні нового антиагрегантного лікарського засобу для лікування гострих коронарних синдромів), що можуть відображати значний ризик для популяції, яка отримає лікування.
VII.B.5.14. Розділ регулярно оновлюваного звіту з безпеки «Інформація, отримана в останній момент»
У даному розділі РОЗБ заявник (власник реєстраційного посвідчення) повинен узагальнити потенційно важливі дані з безпеки, ефекту та ефективності лікарського засобу, про які йому стало відомо після дати закриття бази даних, але під час підготовки РОЗБ. Прикладами можуть бути нові клінічно значущі публікації, важливі дані спостережень, клінічні токсикологічні дані та будь-які заходи, вжиті власником реєстраційного посвідчення, комітетом з моніторингу даних або уповноваженим органом (на міжнародному рівні) з міркувань безпеки. Нові повідомлення про випадки побічних реакцій не слід рутинно включати до РОЗБ, за винятком ситуацій, коли вони вважаються важливим репрезентативним випадком (тобто перший приклад важливого явища) або важливим сигналом з безпеки, або коли вони можуть надати додаткову інформацію до оцінки проблем безпеки, що вже представлені у РОЗБ (наприклад, документально підтверджений випадок апластичної анемії, пов’язаний із застосуванням лікарського засобу з відомими побічними ефектами на кістковий мозок, за відсутності можливих альтернативних причин).
До цього розділу РОЗБ потрібно також включати всі значні зміни, що запропоновані до внесення до довідкової інформації лікарського засобу (наприклад, нова побічна реакція, застереження або протипоказання), внесені у цей період (див. розділ VII.B.4).
Наведені у цьому розділі дані потрібно також враховувати при оцінці ризиків та нової інформації (див. розділ VII.B.5.16.3).
VII.B.5.15. Розділ регулярно оновлюваного звіту з безпеки «Огляд сигналів (нові, поточні та закриті)
Загальне розміщення інформації про сигнали і ризики у РОЗБ представлено на рисунку VII.1 (див. VII.B.5.21.). Метою цього розділу є надання високорівневого огляду сигналів68, що були закриті (тобто їх оцінка завершена) протягом звітного періоду, а також поточних сигналів, оцінка яких ще продовжувалася на момент завершення звітного періоду. До РОЗБ сигнал має включатися одразу, як тільки він пройшов початковий скринінг чи був класифікований, і було вирішено, що власник реєстраційного посвідчення69 проводитиме його подальшу оцінку. Необхідно зауважити, що сигнал з безпеки не є синонімом статистичної характеристики диспропорційного подання повідомлень про побічні реакції для комбінації лікарський засіб/явище, оскільки вимагає валідації. Сигнали можуть бути якісними (наприклад, важливий випадок побічної реакції, серія випадків) чи кількісними (наприклад, показник диспропорційності, результати клінічних випробувань чи епідеміологічних досліджень). Сигнали можуть сформуватися як результат інформаційних запитів від уповноваженого органу щодо питання безпеки (на глобальному рівні) (див. модуль IX).
Рішення стосовно класифікації цих сигналів і висновки з їх оцінки вимагають медичного аналізу і наукової інтерпретації доступних даних, що надається в розділі 16 РОЗБ «Оцінка сигналів та ризиків».
Новим сигналом вважається сигнал, виявлений у звітний період. Якщо у звітний період РОЗБ стає відомою нова клінічно важлива інформація стосовно раніше закритого сигналу, вона буде новим сигналом на підставі того, що новий аспект попередньо спростованого сигналу або визнаного ризику потребує подальших кроків для його верифікації. Залежно від стадії оцінки сигналу на момент завершення звітного періоду та подання РОЗБ новий сигнал може бути класифікований як закритий чи поточний.
Приклади нових сигналів будуть включати нову інформацію щодо раніше:
- закритого і спростованого сигналу, наслідком якої є повторне відкриття сигналу;
- виявленого ризику, коли нова інформація передбачає клінічно важливу різницю в серйозності чи частоті ризику (наприклад, отримана нова інформація щодо раніше ідентифікованого ризику «транзиторне підвищення рівня печінкових ензимів», що вказує на більш серйозні наслідки, такі як печінкова недостатність, або отримано документально підтверджений випадок агранулоцитозу без будь-яких альтернативних причин розвитку при відомому ризику «нейтропенія»);
- виявленого ризику, для якого виявлена вища частота чи серйозність (наприклад у вказаній субпопуляції);
- потенційного ризику, який у разі його підтвердження, вимагатиме нового застереження, нових особливостей застосування, нового протипоказання або обмеження показань чи популяції або інших заходів з мінімізації ризиків.
Заявник (власник реєстраційного посвідчення) повинен у цьому розділі або в додатку надати у вигляді таблиці всі сигнали, поточні чи закриті на момент завершення звітного періоду. Ця таблиця повинна включати таку інформацію:
- стислий опис сигналу;
- дата, коли заявнику (власнику реєстраційного посвідчення) стало відомо про сигнал;
- статус сигналу на момент завершення звітного періоду (закритий чи поточний);
- дата закриття сигналу, якщо сигнал закритий;
- джерело сигналу;
- стислий опис ключових даних;
- плани щодо подальшої оцінки;
- вжиті чи заплановані заходи.
Приклад таблиці сигналів наведено в додатку VII.2.
У цьому розділі не потрібно надавати детальну оцінку закритих сигналів, а слід включити її в підрозділ 16.2 «Оцінка сигналу» РОЗБ.
Оцінку нової інформації, пов’язаної з будь-якими раніше відомими виявленими і потенційними ризиками, яка не вважається новим сигналом, слід надавати у підрозділі 16.3 «Оцінка ризиків та нової інформації» РОЗБ.
Якщо уповноважений орган (на міжнародному рівні) вимагає проведення моніторингу особливого питання, що не вважається сигналом, та звітування про нього у РОЗБ, заявник (власник реєстраційного посвідчення) повинен узагальнити результати аналізу у цьому розділі, якщо вони негативні. У разі якщо особливе питання стає сигналом, воно повинно бути включене у таблицю сигналів і бути проаналізованим у підрозділі 16.2 «Оцінка сигналів» РОЗБ.
VII.B.5.16. Розділ регулярно оновлюваного звіту з безпеки «Оцінка сигналів та ризиків»
Мета цього розділу РОЗБ надати:
- короткий опис того, що було відомо про важливі виявлені і потенційні ризики і відсутню інформацію на початок звітного інтервалу, що охоплює РОЗБ (VII. B.5.16.1);
- оцінку усіх сигналів закритих протягом звітного періоду (VII. B.5.16.2);
- оцінку нової інформації стосовно раніше ідентифікованих і потенційних ризиків (VII. B.5.16.3);
- оновлену характеристику важливих потенційних і ідентифікованих ризиків, якщо застосовується (VII. B.5.16.4);
- рефективності заходів з мінімізації ризику в будь-якій країні чи регіоні, що можуть бути корисними в інших країнах чи регіонах (VII. B.5.16.5).
Блок-схема процесу визначення, в яких розділах/підрозділах РОЗБ повинна міститися відповідна інформація про сигнали та ризики, представлена в розділі VII.B.5.21.
У наведених нижче підрозділах не слід дублювати інформацію, надану в попередніх розділах РОЗБ, а надавати інтерпретацію та критичну оцінку інформації з метою характеристики профілю ризиків, що були оцінені як важливі. Крім того, як правило, немає необхідності включати описи випадків у розділи РОЗБ для оцінки ризиків. Проте, якщо вони є частиною наукового аналізу сигналу або ризику, необхідно надати клінічну оцінку важливих або ілюстративних випадків (наприклад, перший випадок підозрюваного агранулоцитозу, пов’язаний із застосуванням діючої речовини, що належить до класу з відомим зв’язком з цією побічною реакцією) (див. VII.B.3).
VII.B.5.16.1. Підрозділ регулярно оновлюваного звіту з безпеки «Резюме проблем безпеки»
Метою цього підрозділу є надання резюме важливих питань з безпеки, відомих на початку звітного періоду, відносно яких можна зробити оцінку нової інформації. Для лікарських засобів з існуючою специфікацією з безпеки цей підрозділ буде або таким як резюме специфікації з безпеки, що дійсна на початок звітного періоду РОЗБ, або походитиме з нього. Тут слід надати таку інформацію з безпеки:
- важливі виявлені ризики;
- важливі потенційні ризики;
- важлива відсутня інформація.
При визначенні важливості кожного ризику слід враховувати наступні чинники:
- медична серйозність ризику, включаючи вплив на окремих пацієнтів;
- його частота, передбачуваність, можливість запобігання та зворотність;
- потенційний вплив на здоров’я населення (частота; чисельність популяції, яка отримає лікування);
- потенціал для уникнення застосування лікарського засобу, призначеного для профілактики захворювання, у зв’язку з непропорційним усвідомленням населенням ризику (наприклад, вакцини).
Для лікарських засобів, що не мають специфікації з безпеки, у цьому підрозділі необхідно надати інформацію про важливі ідентифіковані та потенційні ризики і важливу відсутню інформацію, що пов’язані з їх застосуванням, на підставі дореєстраційного та післяреєстраційного досвіду. Важливі ідентифіковані та потенційні ризики можуть включати, наприклад:
- важливі побічні реакції;
- взаємодії з іншими лікарськими засобами;
- взаємодії з продуктами харчуванням та іншими речовинами;
- помилки, пов’язані із застосуванням лікарського засобу;
- наслідки впливу, пов’язаного з професійною діяльністю;
- ефекти, що властиві фармакотерапевтичному класу.
Щодо відсутньої інформації в резюме слід врахувати, чи критичні прогалини в знаннях стосуються специфічних питань з безпеки або популяцій, які використовують лікарський засіб.
VII.B.5.16.2. Підрозділ регулярно оновлюваного звіту з безпеки «Оцінка сигналів»
У цьому підрозділі РОЗБ необхідно узагальнити результати оцінки всіх сигналів з безпеки, закритих у звітний період, незалежно від того, чи були вони класифіковані як важливі чи ні. Сигнал з безпеки після його оцінки може бути закритий або тому, що він спростований, або тому, що визначений як потенційний чи ідентифікований ризик. До цього підрозділу необхідно включити наступні дві основні категорії сигналів:
- сигнали, які після їх оцінки були спростовані як помилкові на підставі наукової оцінки наявної на даний момент інформації;
- сигнали, які після оцінки були класифіковані як потенційні або ідентифіковані ризики, включаючи відсутність ефективності. З метою чіткого опису підстав для спростування чи визначення ризику як потенційного або ідентифікованого заявник (власник реєстраційного посвідчення) у даному підрозділі повинен надати достатньо детальний опис оцінки кожного сигналу з обох категорій закритих сигналів.
Рекомендується, щоб рівень деталізації при описі оцінки сигналу відображав його медичну значущість (наприклад, тяжкий, незворотний, призводить до підвищення захворюваності або смертності) і потенційну важливість для охорони здоров’я (наприклад, широке застосування, частота, значне застосування не у відповідності до рекомендацій інформації про лікарський засіб) та обсяг доступної доказової бази. Якщо для закритих сигналів обох категорій використовуються різні оцінки, вони можуть бути представлені в такому порядку:
- закриті та спростовані сигнали;
- закриті сигнали, класифіковані як важливі потенційні ризики;
- закриті сигнали, класифіковані як важливі ідентифіковані ризики;
- закриті сигнали, що є потенційними ризиками, що не класифіковані як важливі;
- закриті сигнали, що є ідентифіковані ризиками, що не класифіковані як важливі.
Коли доцільно, оцінку закритих сигналів можна надавати за показанням або популяцією.
Описи оцінки сигналів можуть бути включені до цього підрозділу РОЗБ або до додатка. Кожна оцінка має містити залежно від ситуації таку інформацію:
- джерело або інформацію, що стала підставою для формування сигналу;
- основні дані, важливі для оцінки;
- метод(и) оцінки, включаючи джерела даних, критерії пошуку (якщо можливо, із застосуванням спеціальних MedDRA-термінів (наприклад, на рівні термінів переважного застосування (PT) терміну групи високого рівня (HLT), та класів систем і органів (SOC) тощо), або перегляд стандартизованих запитів до MedDRA (SMQs) та аналітичні підходи;
- результати — резюме та критичний аналіз даних, що враховувалися при оцінці сигналу; можуть містити опис серії випадків або окремого випадку (наприклад, перший випадок детально документованого агранулоцитозу або синдрому Стівенса — Джонсона), якщо вони є частиною оцінки;
- обговорення;
- висновок.
Оцінка та висновки заявника (власника реєстраційного посвідчення) щодо спростованих сигналів повинні бути підкріплені даними та представлені у зрозумілому вигляді.
VII.B.5.16.3. Підрозділ регулярно оновлюваного звіту з безпеки «Оцінка ризиків та нової інформації»
У цьому підрозділі слід надати критичну оцінку новій інформації, пов’язаній з раніше визнаними ризиками, що не була включена у підрозділ 16.2 «Оцінка сигналів» РОЗБ.
Нову інформацію, що є сигналом для раніше визнаного ризику або для раніше спростованого сигналу, слід надати в таблиці сигналів (див. VII.B.5.15.) і оцінити у підрозділі 16.2 «Оцінка сигналів» РОЗБ, якщо сигнал був також закритий протягом звітного інтервалу РОЗБ.
У цей підрозділ слід включити оновлену інформацію, що стосується раніше визнаного ризику, який не становить сигнал. Приклади включають інформацію, що підтверджує потенційний ризик як ідентифікований, або інформацію, що уможливлює подальшу характеристику раніше визнаного ризику.
Нову інформацію можна представити в такому вигляді:
- нова інформація щодо важливих потенційних ризиків;
- нова інформація щодо важливих ідентифікованих ризиків;
- нова інформація щодо інших потенційних ризиків, що не класифіковані як важливі;
- нова інформація щодо інших ідентифікованих ризиків, що не класифіковані як важливі;
- оновлені дані щодо відсутньої інформації.
При оцінці нової інформації основна увага повинна приділятися інформації, що стала відомою у звітний період, що охоплює РОЗБ. Така оцінка повинна була чіткою та пояснювати вплив, якщо він є, на розуміння та характеристику ризику. Відповідно, така оцінка становитиме основу для оновленої характеристики важливих потенційних та ідентифікованих ризиків у підрозділі 16.4 «Характеристика ризиків» РОЗБ. Рекомендується, щоб рівень деталізації оцінки, включеної у цей підрозділ, був пропорційним наявним доказам ризику і його медичної важливості та впливу на громадське здоров’я.
Оцінка нової інформації та оновленої відсутньої інформації може входити до цього підрозділу РОЗБ або надаватися в додатку. Кожна оцінка повинна включати таку відповідну інформацію:
- джерело нової інформації;
- основні дані, важливі для оцінки;
- метод(и) оцінки, включаючи джерела даних, критерії пошуку та аналітичні підходи;
- результати — резюме та критичний аналіз даних, що розглядаються при оцінці ризику;
- обговорення;
- висновок, у тому числі й про те, чи підтверджує дана оцінка необхідність оновлення характеристик будь-яких важливих та ідентифікованих ризиків, зазначених у підрозділі 16.4 «Характеристика ризиків» РОЗБ.
У даному підрозділі слід критично оцінити будь-яку нову інформацію щодо популяцій, які застосовують лікарський засіб, або даних, отриманих для аналізу раніше відсутньої інформації. Слід також описати наявні невирішені проблеми та сумнівні питання.
VII.B.5.16.4. Підрозділ регулярно оновлюваного звіту з безпеки «Характеристика ризиків»
У цьому підрозділі слід охарактеризувати на основі узагальнених даних (тобто, не обмежених звітним періодом) важливі ідентифіковані та потенційні ризики та описати важливу відсутню інформацію.
У залежності від типу джерела даних, характеристика ризику може включати, якщо можливо:
- частоту;
- кількість випадків (чисельник) та точність оцінки, враховуючи джерело даних;
- обсяг застосування лікарських засобів (знаменник), виражений як кількість пацієнтів, пацієнто-час та інше, а також точність оцінки;
- оцінку відносного ризику та точність оцінки;
- оцінку абсолютного ризику та точність оцінки;
- вплив на окремого пацієнта (вплив на симптоми, якість та тривалість життя);
- вплив на громадське здоров’я;
- характеристики пацієнтів, пов’язані з ризиком (наприклад, вік, вагітність/лактація, порушення функції печінки/нирок, відповідні супутні захворювання, серйозність захворювання, генетичний поліморфізм);
- дозу, спосіб застосування;
- тривалість лікування, період ризику;
- можливість запобігання (тобто, передбачуваність, здатність контролювати «сигнальну» побічну реакцію або лабораторний маркер);
- зворотність;
- потенційний механізм
- сила доказу та ступінь його достовірності, включаючи аналіз суперечливого доказу, якщо це є доцільним.
Якщо відсутня інформація становить важливий ризик, то вона повинна розглядатися як проблема безпеки. Необхідно також проаналізувати обмеження баз даних з безпеки (стосовно кількості досліджуваних пацієнтів, кумулятивного впливу, тривалого застосування тощо).
У РОЗБ, що складаються для лікарських засобів з кількома показаннями, формами випуску, способами застосування, коли можуть мати місце значні розбіжності у виявлених та потенційних ризиках, може бути доцільним навести ризики за показаннями, формами випуску або способами застосування. Заголовки можуть бути такими:
- ризики, пов’язані з діючою речовиною;
- ризики, пов’язані з особливою формою випуску або способом застосування (включаючи ризики, пов’язані з впливом, пов’язаним з професійною діяльністю);
- ризики, пов’язані з особливими популяціями;
- ризики, пов’язані із застосуванням безрецептурних лікарських засобів (для діючих речовин/АФІN, що входять до складу як рецептурних, та і безрецептурних лікарських засобів).
VII.B.5.16.5. Підрозділ регулярно оновлюваного звіту з безпеки «Ефективність заходів з мінімізації ризиків (якщо необхідно)»
Заходи з мінімізації ризиків — це заходи, що вживаються в системі охорони здоров’я з метою запобігання виникненню побічних реакцій, пов’язаних із застосуванням лікарського засобу або для зниження їх тяжкості у разі виникнення. Метою заходів з мінімізації ризиків є зниження ймовірності виникнення або тяжкості перебігу побічної реакції. Заходи з мінімізації ризиків можуть складатися з рутинних заходів (наприклад коротка характеристика/інструкція для медичного застосуванняN лікарського засобу) або додаткових заходів з мінімізації ризиків (наприклад лист-звернення до працівників з медичною та фармацевтичною освітою/навчальні матеріали).
РОЗБ повинен містити результати оцінки ефективності заходів з мінімізації ризиків, що стосуються оцінки співвідношення користь/ризик (ст. 34(3) ІП 520/2012 [6], положення Порядку [2]N).
У даному підрозділі РОЗБ слід узагальнити відповідну інформацію щодо ефективності та/або обмежень специфічних заходів з мінімізації важливих ідентифікованих ризиків, що стала доступною у звітний період.
Особливу увагу слід приділити ефективності тих заходів з мінімізації ризиків у будь-якій країні чи регіоні, що можуть застосовуватися в інших регіонах чи країнах. Якщо доречно і можливо, інформацію можна узагальнити за регіонами.
Якщо вимагається надання результатів оцінок ефективності заходів з мінімізації ризиків у звітний період, що стосуються окремого регіону, вони повинні надатися в локальному додатку до РОЗБ (див. розділ VII.B.5.20. та розділ VII.С.5.5.).
VII.B.5.17. Розділ регулярно оновлюваного звіту з безпеки «Оцінка користі»
У підрозділах РОЗБ 17.1 («Важлива базова інформація з ефективності лікарського засобу») і 17.2 («Нова інформація з ефективності лікарського засобу») надається базова та нова виявлена інформація щодо користі, що підтверджує характеристику користі, що описується в підрозділі 17.3 («Характеристика користі»), яка, у свою чергу, враховується при оцінці співвідношення користь/ризик у розділі 18 («Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань»).
VII.B.5.17.1. Підрозділ регулярно оновлюваного звіту з безпеки «Важлива базова інформація з ефекту та ефективності»
У цьому підрозділі РОЗБ узагальнюється відома на початку звітного періоду інформація щодо ефекту та ефективності лікарського засобу, що є основою для оцінки користі. Ця інформація має відповідати зареєстрованим показанням лікарського засобу, що містяться у довідковій інформації про лікарський засіб (див. VII.В.4.).
Для лікарських засобів з різними показаннями, популяціями та/або способами застосування, якщо доцільно, користь лікарського засобу слід характеризувати окремо за цими факторами.
Рівень деталізації інформації, що надається в цьому підрозділі РОЗБ, повинен бути достатнім для підтвердження характеристики користі в підрозділі РОЗБ 17.3 «Характеристика користі» і оцінки співвідношення користь/ризик у розділі 18 «Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань».
VII.B.5.17.2 Підрозділ регулярно оновлюваного звіту з безпеки «Нова виявлена інформація з ефекту та ефективності»
Для деяких лікарських засобів протягом звітного періоду може з’явитися додаткова інформація щодо ефекту та ефективності для зареєстрованих показань. Таку інформацію слід надати у цьому підрозділі РОЗБ. У цьому підрозділі слід також надати нову інформацію щодо ефекту та ефективності для зареєстрованих показань в умовах фактичного застосування, якщо вона доступна. У цьому підрозділі не слід надавати нову інформацію щодо ефекту та ефективності при застосуванні за незареєстрованими показаннями, крім випадків, коли вона є важливою для оцінки співвідношення користь/ризик лікарського засобу за зареєстрованими показаннями.
У цьому підрозділі слід також представити інформацію про показання, що були зареєстровані у звітний період. Рівень деталізації інформації, що надається у цьому підрозділі РОЗБ, повинен бути достатнім для підтвердження характеристики користі у підрозділі 17.3 «Характеристика користі» РОЗБ і оцінки співвідношення користь/ризик у розділі 18 «Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань» РОЗБ.
У цьому підрозділі особливу увагу слід приділити вакцинам, антиінфекційним та іншим лікарським засобам, для яких зміна терапевтичних умов може з часом вплинути на їх ефект/ефективність.
VII.B.5.17.3. Підрозділ регулярно оновлюваного звіту з безпеки «Характеристика користі»
У цьому підрозділі надається сукупна базова та нова інформація щодо користі для зареєстрованих показань, що стала доступною у звітний період.
Рівень деталізації інформації, що надається у цьому підрозділі РОЗБ, повинен бути достатнім для забезпечення оцінки співвідношення користь/ризик у розділі 18 «Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань» РОЗБ.
Якщо немає нових важливих даних щодо користі, у цьому підрозділі слід надати характеристику інформації, наданої в підрозділі 17.1 «Важлива базова інформація щодо ефекту та ефективності» РОЗБ.
Якщо є нова позитивна інформація щодо користі, але немає значних змін у профілі ризику лікарського засобу у цей звітний період, сукупна базова та нова інформація повинна надаватися у цьому підрозділі у стислому вигляді.
У цьому підрозділі слід надати стислу, але критичну оцінку переваг та недостатності доказів щодо ефекту та ефективності лікарського засобу, враховуючи за можливості наступне:
- стислий опис сили доказів користі, зважаючи на компаратора(ів), обсяг впливу, статистичну достовірність, сильні та слабкі сторони методології, а також узгодженість результатів низки випробувань/досліджень;
- нова інформація, що ставить під сумнів обґрунтованість сурогатного маркера, якщо такий використовується;
- клінічна значущість обсягу впливу;
- узагальнення відповіді на лікування для популяції пацієнтів, яким показаний лікарський засіб (наприклад, інформація, що демонструє відсутність лікувального ефекту у субпопуляції);
- відповідність характеристики доза-реакція;
- тривалість ефекту;
- порівняльна ефективність;
- визначення ступеню, до якого дані з ефективності з клінічних випробувань можуть розповсюджуватися на популяції пацієнтів у медичній практиці.
VII.B.5.18. Розділ регулярно оновлюваного звіту з безпеки «Комплексний/інтегрований аналіз співвідношення користь/ризик для зареєстрованих показань»
У цьому розділі РОЗБ заявник (власник реєстраційного посвідчення) повинен надати загальну оцінку користі та ризику лікарського засобу при його застосуванні у клінічній практиці. Тоді як у підрозділах 16.4 «Характеристика ризиків» та 17.3 «Характеристика користі» РОЗБ представлені дані про ризик та користь, у даному розділі слід критично проаналізувати та узагальнити ключову інформацію з попередніх розділів, а не дублювати характеристику користі та ризику, надану у вищезгаданих підрозділах.
VII.B.5.18.1. Підрозділ регулярно оновлюваного звіту з безпеки «Контекст співвідношення користь/ризик (медична необхідність та важливі альтернативи)»
У цьому підрозділі РОЗБ слід надати стислий опис медичної потреби в лікарському засобі за зареєстрованими показаннями та резюме альтернатив (медичних, хірургічних або інших, включаючи відсутність лікування).
VII.B.5.18.2. Підрозділ регулярно оновлюваного звіту з безпеки «Оцінка аналізу співвідношення користь/ризик»
Співвідношення користь/ризик, як правило, специфічне для показань та популяції. Тому для лікарських засобів, зареєстрованих більш ніж за одним показанням, профіль користь/ризик слід оцінювати та представляти за кожним показанням окремо. У випадку наявності важливих розбіжностей у співвідношенні користь/ризик серед різних популяцій у межах одного показання оцінку співвідношення користь/ризик слід надавати для кожної популяції, якщо це можливо.
Оцінку співвідношення користь/ризик слід представити і проаналізувати таким чином, щоб забезпечити порівняння користі і ризиків, при цьому слід враховувати таке:
- хоча у попередніх розділах/підрозділах повинна міститися інформація про всі важливі переваги та ризики, однак не всі переваги та ризики значно впливають на загальну оцінку користі/ризику, а тому при оцінці необхідно зазначити ключові переваги та ризики. Для оцінки співвідношення користі/ризику необхідно узагальнити ключову інформацію щодо користі та ризиків, представлену в попередніх розділах/підрозділах;
- обставини застосування лікарського засобу: захворювання, для лікування, профілактики чи діагностування якого призначений лікарський засіб, його тяжкість та серйозність; та популяція, для якої призначений лікарський засіб (відносно здорові пацієнти; хронічні захворювання; рідкісні захворювання);
- що стосується ключової користі, то слід враховувати її характер, клінічну значимість, тривалість та узагальненість, а також доказ ефективності у пацієнтів з негативною відповіддю на інші види терапії та альтернативне лікування. Розглянути величину ефекту. Слід розглянути всі елементи користі (наприклад, при лікуванні ревматоїдного артриту: зменшення вираженості симптомів та пригнічення прогресування ушкодження суглоба, що було підтверджено рентгенографічно);
- стосовно ризику, то слід розглянути його клінічну значимість (наприклад, характер токсичності, серйозність, частоту, передбачуваність, превентивність, зворотність, вплив на пацієнтів) та чи він був виявлений при застосуванні лікарського засобу за незареєстрованими показаннями чи у нецільових популяцій у клінічних випробуваннях, при застосуванні не за показаннями або при неправильному застосуванні;
- при формулюванні оцінки співвідношення користі/ризику слід розглядати сильні, слабкі сторони та неоднозначність доказів. Слід описати, яким чином невизначеності в користі та ризику впливають на оцінку. Слід також розглянути недоліки оцінки.
Необхідно надати чітке пояснення методології* (методи визначення співвідношення користі/ризику представлені в додатку до цього модуля) та обґрунтуванню, що використовувалися для оцінки співвідношення користь/ризик:
- припущення, міркування, твердження чи оцінка, що обґрунтовують висновки щодо оцінки співвідношення користі/ризику, повинні бути чіткими;
- якщо надається формальна кількісна чи напівкількісна оцінка співвідношення користь/ризику, необхідно надати короткий опис методів;
- при проведенні оцінки співвідношення користі/ризику не повинні братися до уваги економічні міркування (наприклад економічна ефективність).
У випадку якщо існує важлива нова інформація або вимагається надання спеціального (ad hoc) регулярного звіту з безпеки, детальний аналіз співвідношення користь/ризик має ґрунтуватися на кумулятивних даних. І навпаки, якщо протягом звітного періоду отримано небагато нової інформації, першочергова увага при оцінці співвідношення користь/ризик може приділятися оцінці оновлених даних з безпеки за звітний період.
VII.B.5.19. Розділ регулярно оновлюваного звіту з безпеки «Висновки та заходи»
У РОЗБ слід зробити висновки щодо будь-якої нової інформації, отриманої протягом звітного періоду, відносно загальної оцінки співвідношення користі/ризику лікарського засобу для кожного зареєстрованого показання, а також відповідних підгруп пацієнтів.
На основі оцінки сукупних даних з безпеки та аналізу співвідношення користі/ризику заявник (власник реєстраційного посвідчення) повинен оцінити необхідність внесення змін до довідкової інформації і запропонувати зміни, якщо вони необхідні.
Крім того, у разі доцільності у висновки слід включити попередні рекомендації щодо оптимізації чи подальшої оцінки співвідношення користь/ризик для подальшого їх обговорення з відповідним(и) уповноваженим(и) органом(и). У висновки також можуть бути включені пропозиції щодо додаткових заходів з мінімізації ризиків.
Для лікарських засобів, для яких розроблені плани з фармаконагляду чи ПУР, необхідно також обміркувати пропозиції, що повинні бути враховані у плані з фармаконагляду чи у плані мінімізації ризиків (див. модуль V ННПФ).
Заявник (власник реєстраційного посвідчення), ґрунтуючись на оцінці сукупних даних з безпеки та аналізі співвідношення користі/ризику повинен надати в регулярному звіті з безпеки висновки щодо необхідності змін та/або заходів, включаючи зміни до затвердженої короткої характеристики/інструкції для медичного застосуванняN лікарського засобу, на який подається РОЗБ (ст. 34(5) ІП 520/2012 [6], положення Порядку [2]N).
У цьому розділі регулярного звіту з безпеки необхідно описати запропоновані зміни до довідкової інформації лікарського засобу. У локальному додатку слід надати пропозиції щодо інформації про лікарський засіб (короткої характеристики/інструкції для медичного застосуванняN лікарського засобу та листка-вкладки), також інформації про поточні зміни, якщо такі є.
VII.B.5.20. Додатки до регулярно оновлюваного звіту з безпеки
РОЗБ повинен містити наступні додатки з такою нумерацією:
- довідкова інформація (див. VII.B.4.);
- зведені таблиці серйозних побічних явищ з клінічних випробувань; зведені та інтервальні таблиці серйозних і несерйозних побічних реакцій за даними післяреєстраційного нагляду;
- зведена таблиця сигналів з безпеки (якщо зведену таблицю сигналів з безпеки не включено до змісту відповідного розділу РОЗБ, рекомендується включати її до змісту відповідного розділу РОЗБ);
- перелік всіх інтервенційних і неінтервенційних досліджень, спонсорованих заявником (власником реєстраційного посвідчення), головною метою яких є ідентифікація, характеристика чи кількісне визначення загрози безпеці або підтвердження профілю безпеки лікарського засобу, або у випадку неінтервенційних досліджень оцінка ефективності заходів управління ризиком;
- перелік джерел інформації, що використовувалися для підготовки РОЗБ (за бажанням заявника (власника реєстраційного посвідчення));
- локальний додаток: вимоги до складання локального додатку описані у розділі VII.C.5.
VII.B.5.21. Схема відображення у регулярно оновлюваному звіті з безпеки інформації про сигнали і ризики
Представлена нижче блок-схема (див. рис. VII.1) відображає розміщення у РОЗБ інформації про сигнали та ризики.

Рисунок VII.1. Розділи і підрозділи регулярного звіту з безпеки — сигнали та ризики
VII.B.6. Системи якості для регулярно оновлюваних звітів з безпеки на рівні заявників (власників реєстраційних посвідчень)
Заявники (власники реєстраційних посвідчень) повинні мати структури та процеси для підготовки, контролю якості, перегляду та подання регулярного звіту з безпеки, включаючи подальше спостереження під час та після їх оцінки. Ці структури та процеси повинні бути задокументовані у вигляді службових інструкцій та процедур у системі якості заявника (власника реєстраційного посвідчення) (див. модуль I ННПФ).
У процесі здійснення фармаконагляду існують сфери діяльності, що можуть безпосередньо вплинути на якість РОЗБ. Прикладами можуть бути управління випадками спонтанних повідомлень та звітами з досліджень, скринінг літератури, управління сигналами, додаткові заходи з фармаконагляду та післяреєстраційна дослідницька діяльність, процедури об’єднання інформації щодо користі та ризиків з усіх наявних джерел даних та супровід інформації про лікарський засіб. Система якості повинна описувати зв’язки між процесами, канали передачі інформації та обов’язки з метою збору всієї відповідної інформації для підготовки РОЗБ. Повинні бути документовані процедури, включаючи перевірки з контролю якості на місцях для контролю достовірності та повноти даних, представлених у РОЗБ. Для забезпечення повноти даних може бути розроблений шаблон документа або план для отримання даних з різних джерел даних. Важливість інтегрованого підходу до оцінки співвідношення користі/ризику повинна лягти в основу процесів та внеску різних департаментів у підготовку РОЗБ.
РОЗБ повинен також містити оцінку особливих питань безпеки згідно із запитом уповноважених органів у відповідності з узгодженими строками та процедурами. Заявник (власник реєстраційного посвідчення) повинен мати у своєму розпорядженні механізми для забезпечення належного реагування на запити уповноваженого органу під час оцінки РОЗБ.
З метою забезпечення достовірності даних про кількість побічних явищ/реакцій, необхідно перевірити джерела надходження даних, що включені у зведені таблиці (див. розділ VII.B.5.6.), та співставити ці дані з базою даних з безпеки заявника (власника реєстраційного посвідчення). Процес запитів інформації з бази даних з безпеки, параметри, що використовувалися для пошуку даних, та контроль якості повинні бути належним чином задокументовані.
Заявник (власник реєстраційного посвідчення) повинен мати у своєму розпорядженні відповідну систему якості з метою уникнення таких невідповідностей вимогам РОЗБ, як:
- неподання РОЗБ, подання не у відповідності до затвердженого поточного графіка подачі або не у затверджені строки (без попереднього погодження з уповноваженим органом);
- необґрунтоване упущення інформації, що вимагається в розділі VII.B.5.;
- неякісні РОЗБ: незадовільна документація чи недостатня інформація або оцінка, що надані для ретельного аналізу нової інформації з безпеки, сигналів, оцінки ризику, оцінки користі та кумулятивного/інтегрованого аналізу співвідношення користі/ризику, незазначення неправильного застосування лікарського засобу, невикористання стандартизованої медичної термінології (наприклад MedDRA) та необґрунтоване неврахування випадків з неповідомленими факторами ризику у загальних оглядах;
- подання РОЗБ, у яких не надано відповідей на попередні запити уповноважених органів;
- неспроможність надати чітку оцінку співвідношення користі/ризику лікарського засобу;
- неспроможність надати відповідні пропозиції щодо локальної інформації про лікарський засіб.
Будь-яке значне відхилення від процедур підготовки чи подання РОЗБ слід документувати та вживати відповідних коригувальних та профілактичних заходів. Ця документація повинна бути доступною в будь-який час.
Якщо заявники (власники реєстраційних посвідчень) мають договірні угоди (наприклад ліцензіар — ліцензіат), у письмовій угоді необхідно чітко прописати відповідні обов’язки щодо підготовки та подання РОЗБ до уповноважених органів.
Якщо підготовка РОЗБ делегується третім особам, заявник (власник реєстраційного посвідчення) повинен гарантувати, що вони використовують систему якості, що відповідає поточним вимогам законодавства. Повинні бути прописані чіткі процедури взаємодії та детальні домовленості між заявником (власником реєстраційного посвідчення) та третіми сторонами. Домовленості можуть детально описувати предмети аудиту процесу підготовки РОЗБ.
VII.B.7. Навчання персоналу, пов’язане з процесом регулярно оновлюваного звіту з безпеки
Обов’язок осіб, відповідальних за систему фармаконагляду, в усіх організаціях — забезпечення того, щоб персонал, включаючи персонал з фармаконагляду, медичний персонал і персонал з якості, залучений до підготовки, перегляду, контролю якості, подання та оцінки РОЗБ, мав відповідну кваліфікацію, досвід та підготовку згідно з чинними настановами (наприклад настанова ICH E2C(R2) [28]) та цим модулем ННПФ (VII). За необхідності потрібно проводити спеціальні тренінги з різних процесів, завдань та обов’язків, що пов’язані з РОЗБ.
Слід також проводити тренінги для оновлення знань та навичок, якщо необхідно.
Навчання повинно охоплювати пов’язане з РОЗБ законодавство, настанови, наукову оцінку та письмові процедури. Документація щодо проведення тренінгів повинна демонструвати, що відповідне навчання було проведене до виконання діяльності, пов’язаної з РОЗБ.
VII.С. Процеси, пов’язані з регулярним звітом з безпеки
VII.С.1. Загальний процес, пов’язаний з регулярним звітом з безпеки
Представлена блок-схема (див. рис. VII.2) описує загальний цикл процесу для процедури РОЗБ на рівні ЄС, коли були видані рекомендації PRAC. Вона представляє цикл, починаючи з опису загального процесу від підготовки регулярного звіту з безпеки до впровадження рішення регуляторного органу, якщо необхідно. Відповідні окремі етапи цієї процедури складаються з проміжних етапів, які далі пояснюються та розглядаються в розділах цього модуля.
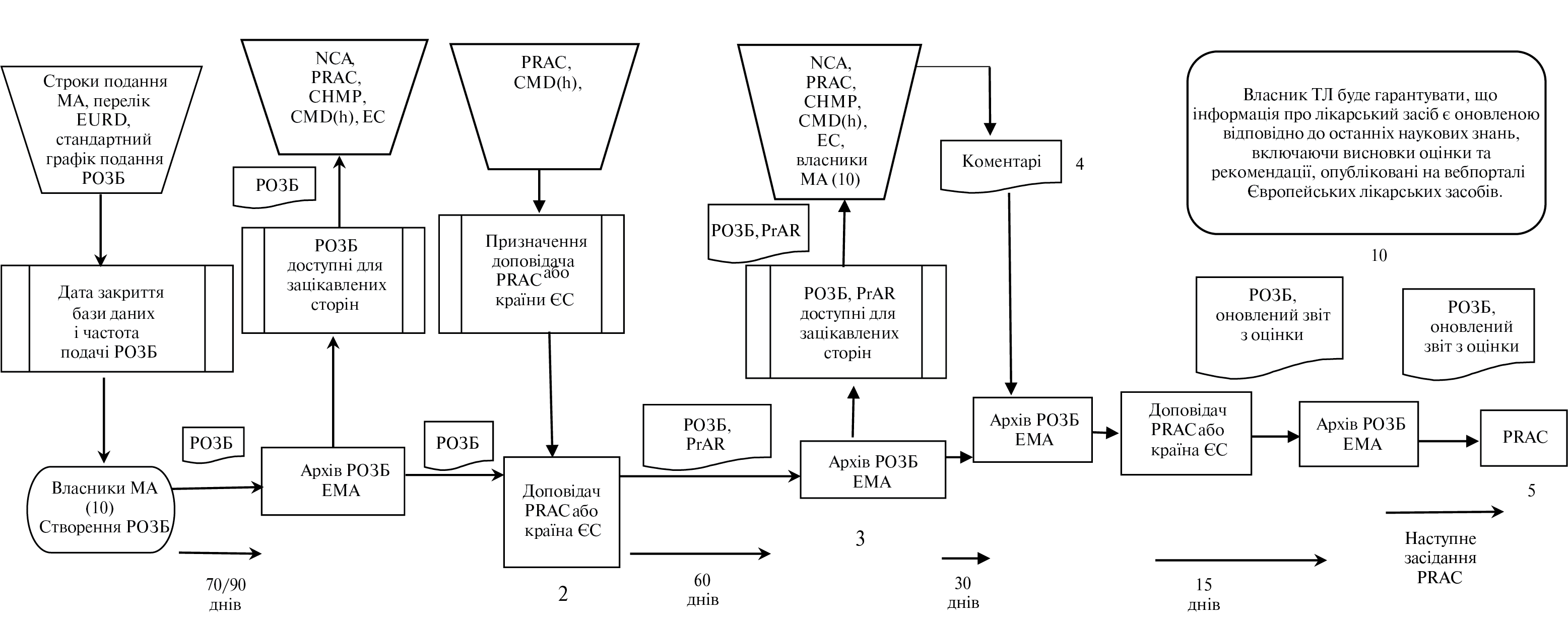
Познаки та скорочення:
MA — торгова ліцензія;
EURD — референтні дати Європейського Союзу;
NCA — національні уповноважені органи;
EC — Європейська Комісія;
PrAR: попередній звіт з оцінки;
CAP — централізовано зареєстрований лікарський засіб, зареєстрований за централізованою процедурою;
NAP — національно зареєстрований лікарський засіб (включаючи взаємне визнання та децентралізовану процедуру), зареєстрований за національною процедурою.
*Стандартний графік подання регулярного звіту з безпеки стосується 6 міс, 1 року або 3 років, як встановлено у 2-му параграфі ст. 107с(2) Директиви 2001/83/ЄС [1].
Рис. VII.2. Загальний цикл процесу для процедури регулярного звіту з безпеки — аркуш 1
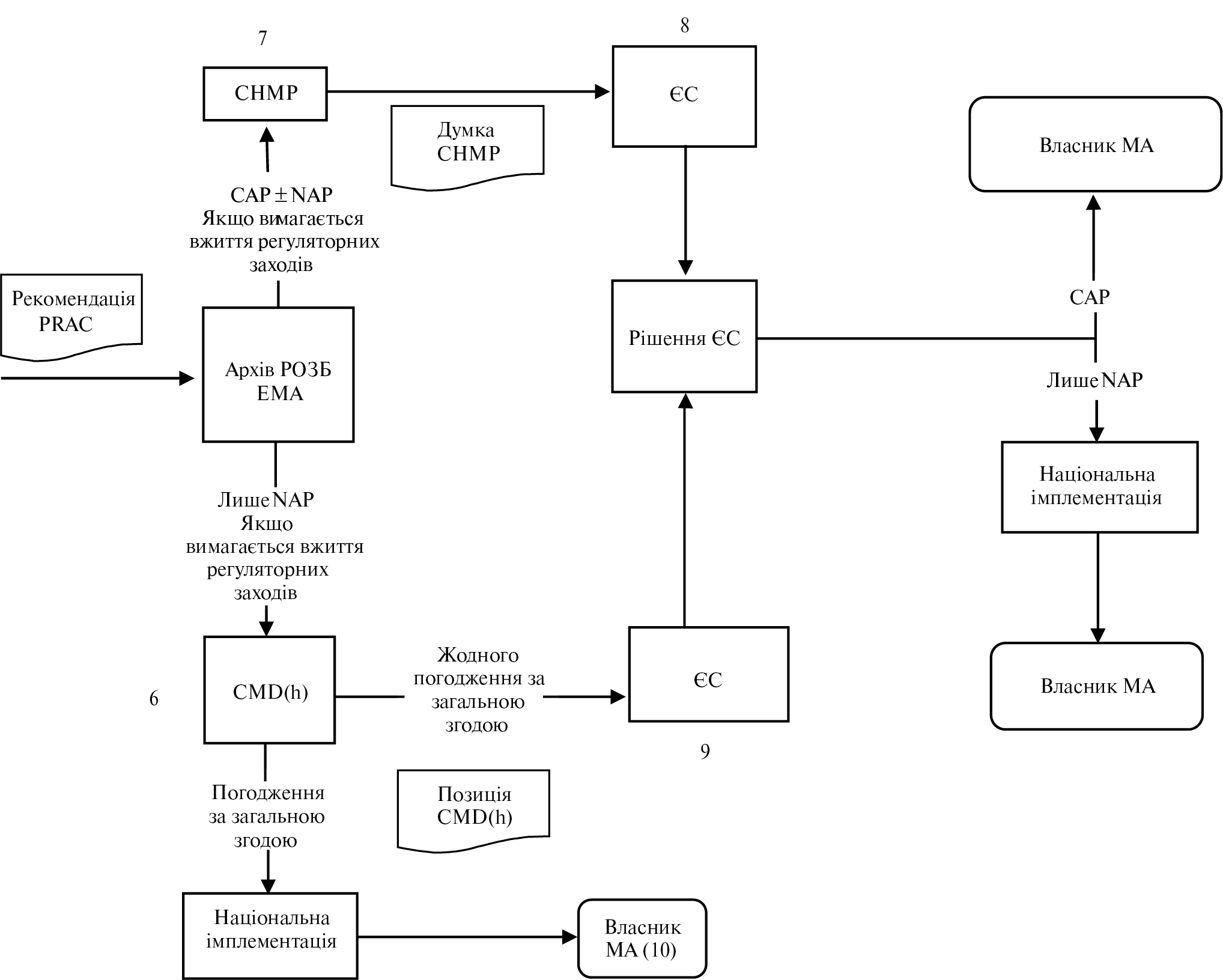
Умовні познаки (правові посилання):
1 — ст. 28(2) Регламенту (ЄC) № 726/2004 [5];
2 — ст. 107e(1) Директиви 2001/83/ЄС [1], 1-й параграф ст. 28(3) Регламенту (ЄC) № 726/2004 [5];
3 — 1-й параграф ст. 107e(2) Директиви 2001/83/ЄС [1], 2-й параграф статті 28(3) Регламенту (ЄC) № 726/2004 [5];
4 — 2-й параграф ст. 107e(2) Директиви 2001/83/ЄС [1], 3-й параграф ст. 28(3) Регламенту (ЄC) № 726/2004 [5];
5 — ст. 107e(3) Директиви 2001/83/ЄС [1], 4-й параграф ст. 28(3) Регламенту (ЄC) № 726/2004 [5];
6 — ст. 107g (1) та (2), 1-й параграф Директиви 2001/83/ЄС [1];
7 — ст. 107g (3) Директиви 2001/83/ЄС [1], ст. 28(4) Регламенту (ЄC)/2004 [5];
8 — ст. 107g (4) Директиви 2001/83/ЄС [1], 2-й параграф ст. 28(4) та (5) Регламенту (ЄC) № 726/2004 [5];
9 — 3-й параграф ст. 107g (2) Директиви 2001/83/ЄС [1];
10 — ст. 23(3) Директиви 2001/83/ЄС [1], ст. 16(3) Регламенту (ЄC) № 726/2004 [5].
Архів
1– ст. 25а Регламенту (ЄC) № 726/2004 [5];
5 –ст. 107e(3) Директиви 2001/83/ЄС [1], 4-й параграф ст. 28(3) Регламенту (ЄC) № 726/2004 [5].
Рисунок VII.2 — аркуш 2
VII.С.2. Стандартний графік подання регулярно оновлюваного звіту з безпеки
Заявники (власники реєстраційних посвідчень) на лікарські засоби, що зареєстровані до 2 липня 2012 р. (лікарські засоби зареєстровані за централізованою процедурою) та 21 липня 2012 р. (лікарські засоби зареєстровані за національною процедурою), для яких частота та дати подання РОЗБ не зазначені в умовах видачі реєстраційного посвідчення чи визначені інакше у переліку референтних дат ЄС, повинні подавати РОЗБ з такою періодичністю (ст. 28(2) Регламенту (ЄC) № 726/2004 [5], ст. 107с(2) Директиви 2001/83/ЄС [1]:
- кожні 6 міс, якщо лікарський засіб зареєстровано, навіть, якщо він не продається;
- якщо лікарський засіб продається, РОЗБ слід подавати кожні 6 міс після початкового розміщення на ринку протягом перших 2 років, потім 1 раз на рік після 2 років, а потім кожні 3 роки.
В Україні для лікарського засобу, що зареєстрований в Україні як у першій країні світу або вперше у будь-якій іншій країні світу, — РОЗБ подається кожні 6 міс протягом перших 2 років (незалежно від наявності лікарського засобу в обігу), один раз на рік протягом наступних 2 років та надалі — 1 раз кожні 3 роки, починаючи з дати реєстрації лікарського засобу. Якщо зазначені вище строки подання РОЗБ збігатимуться зі строками відповідно до періодичності подання РОЗБ, — РОЗБ подається відповідно до строків, зазначених у періодичності подання РОЗБ згідно з додатком 10 до Порядку [2]N.
VII.С.3. Перелік референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN та частота подання регулярно оновлюваного звіту з безпеки70
VII.С.3.1. Мета переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN
EMA публікує перелік референтних дат Європейського Союзу (далі — перелік референтних дат ЄС) та частоту подання РОЗБ за допомогою вебпорталу Європейських лікарських засобів (ст. 107с (7) Директиви 2001/83/ЄС [1], ст. 26(1) (g) Регламенту (ЄC) № 726/2004 [5]). В Україні періодичність подання РОЗБ визначена в положеннях Порядку [2]N, додаток 10 до Порядку [2]N).
Метою переліку референтних дат ЄС (в Україні — періодичність подання РОЗБ) та частоти подання РОЗБ є:
- гармонізація дати закриття бази даних для складання наступного звіту (data lock point) та частоти подання РОЗБ для однієї й тієї ж діючої речовини (АФІN) та комбінації діючих речовин (АФІN).
Для лікарських засобів, що містять одну й ту ж саму діючу речовину (АФІN) або комбінацію діючих речовин (АФІN), що підлягають реєстрації за різними процедурами, слід встановити референтну дату ЄС та гармонізовану частоту та дату подання РОЗБ з метою підготовки єдиної оцінки, що встановлена відповідно до ст. 107e(1) Директиви 2001/83/ЄС [1], положень Порядку [2]N. Така інформація повинна включатися до переліку, що опублікований EMA/наведений у додатку 10 до Порядку [2]N;
- оптимізація роботи з РОЗБ та оцінки РОЗБ у рамках ЄС та УкраїниN.
Перелік відміняє графік подання РОЗБ описаний у ст. 107c(2)(b) Директиви 2001/83/ЄС [1].
«В Україні, якщо строки подання РОЗБ згідно зі стандартним графіком» збігатимуться зі строками відповідно до додатку 10 до Порядку [2]N — РОЗБ повинен подаватися відповідно до строків додатку 10 до Порядку [2]N.
Для діючих речовин (АФІN) або комбінацій діючих речовин (АФІN), що входять до переліку референтних дат ЄС (в Україні — періодичність подання регулярних звітів з безпекиN), заявники (власники реєстраційних посвідчень) повинні змінювати, якщо необхідно, умову, яка викладена в їх реєстраційних посвідченнях з метою подання РОЗБ відповідно до частоти та дати подання, що зазначена в переліку референтних дат ЄС (ст. 107c(4)-(7) Директиви 2001/83/ЄС [1]) (в Україні — періодичність подання РОЗБN (додаток 10 до Порядку [2]N)).
Періодичність подання РОЗБ визначається за допомогою ризик-орієнтованого підходу з метою визначення пріоритетності періодичної переоцінки співвідношення користі/ризику діючих речовин (АФІN) шляхом, який найкраще захищає здоров’я населення (пункт 23 у преамбулі Директиви 2010/84/ЄС [3], додаток 10 до Порядку [2]N);
- єдина оцінка ЄС, УкраїниN та переоцінка співвідношення користі/ризику діючої речовини (АФІN), що базується на всіх доступних даних з безпеки.
Перелік дозволяє гармонізувати подання РОЗБ на лікарські засоби, що містять однакову діючу речовину (АФІN) або однакову комбінацію діючих речовин (АФІN).
Єдина оцінка РОЗБ забезпечує механізм для оцінки всіх наявних даних з користі та ризиків діючої речовини (АФІN) чи комбінації діючих речовин (АФІN). Ефективне застосування принципів розподілу праці важливе для уникнення дублювання зусиль та визначення пріоритету для використання обмежених трудових ресурсів з метою якнайкращого захисту інтересів громадян Європи, УкраїниN.
VII.С.3.2. Опис переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN
Референтною датою ЄС для лікарських засобів, що містять однакову діючу речовину (АФІN) або однакову комбінацію діючих речовин (АФІN) повинна бути (ст. 107с(5) Директиви 2001/83/ЄС [1]):
- дата першої реєстрації на лікарський засіб у ЄС, що містить таку діючу речовину (АФІN) або таку комбінацію діючих речовин (АФІN); або
- якщо дату першої реєстрації неможливо встановити, — то найраніша з відомих дат реєстрації на лікарський засіб, що містить таку діючу речовину (АФІN) чи таку комбінацію діючих речовин (АФІ).
Перелік референтних дат ЄС та частота подання РОЗБ (в Україні — періодичність подання РОЗБN) складається з повного переліку діючих речовин (АФІN) та комбінацій діючих речовин (АФІN) у алфавітному порядку, для яких вимагається подання РОЗБ у відповідності з референтною датою та з частотою, визначеною СНМР та CMDh після консультації з PRAC (ст. 107с(4) та (6) Директиви 2001/83/ЄС [1]) в ЄС, в Україні згідно з положеннями, викладеними в додатку 10 до Порядку [2]N). Цей перелік повинен оновлюватися відповідно до «переліку всіх лікарських засобів для людини, що зареєстровані у ЄС», як вказано у ст. 57(1)(b) Регламенту (ЄC) №726/2004 [5], у разі реєстрації нової АФІ/комбінації АФІ згідно з положеннями, викладеними в додатку 10 до Порядку [2]N) Перелік референтних дат ЄС повинен містити наступну інформацію:
- референтні дати ЄС;
- частоту подання регулярних звітів з безпеки;
- дату закриття бази даних для наступного регулярного звіту з безпеки;
- дату публікації (на Європейському вебпорталі лікарських засобів) частоти подання регулярних звітів з безпеки та дати закриття бази даних для кожної діючої речовини і комбінації діючих речовин. Будь-які зміни дат подання та частоти подання регулярних звітів з безпеки, які зазначені в умовах реєстраційного посвідчення, повинні набути чинності через 6 міс після дати такої публікації (ст. 107с(7) Директиви 2001/83/ЄС [1]).
В Україні періодичність подання регулярних звітів з безпеки містить інформацію про:
- частоту подання регулярних звітів з безпеки;
- кінцеву дату для включення даних до звіту за певний період;
- дату поданняN.
У разі необхідності до переліку слід включити сферу дії РОЗБ та відповідну процедуру єдиної оцінки (див. розділ VII.С.3.3.), зокрема:
- чи повинен РОЗБ охоплювати всі показання діючої речовини (АФІN) або комбінації діючих речовин (АФІN);
- чи повинен РОЗБ включати всі форми випуску/шляхи введення лікарських засобів, що містять діючу речовину (АФІN) або комбінацію діючих речовин (АФІN);
- чи повинні у післяреєстраційний період подаватися РОЗБ для генеричних лікарських засобів, лікарських засобів з добре вивченим застосуванням, традиційних рослинних та гомеопатичних лікарських засобів на запит уповноваженого органу чи у зв’язку з проблемами, пов’язаними з даними з фармаконагляду, або через відсутність РОЗБ відносно діючої речовини (АФІN) (другий підпараграф ст. 107с(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N) (див. розділ VII.С.3.3.2.).
VII.С.3.3. Застосування переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN для подання регулярно оновлюваних звітів з безпеки
VII.С.3.3.1. Подання регулярних звітів з безпеки для лікарських засобів: загальні вимоги
На рисунку VII.3 представлені загальні вимоги при різних потенційних сценаріях подання РОЗБ.
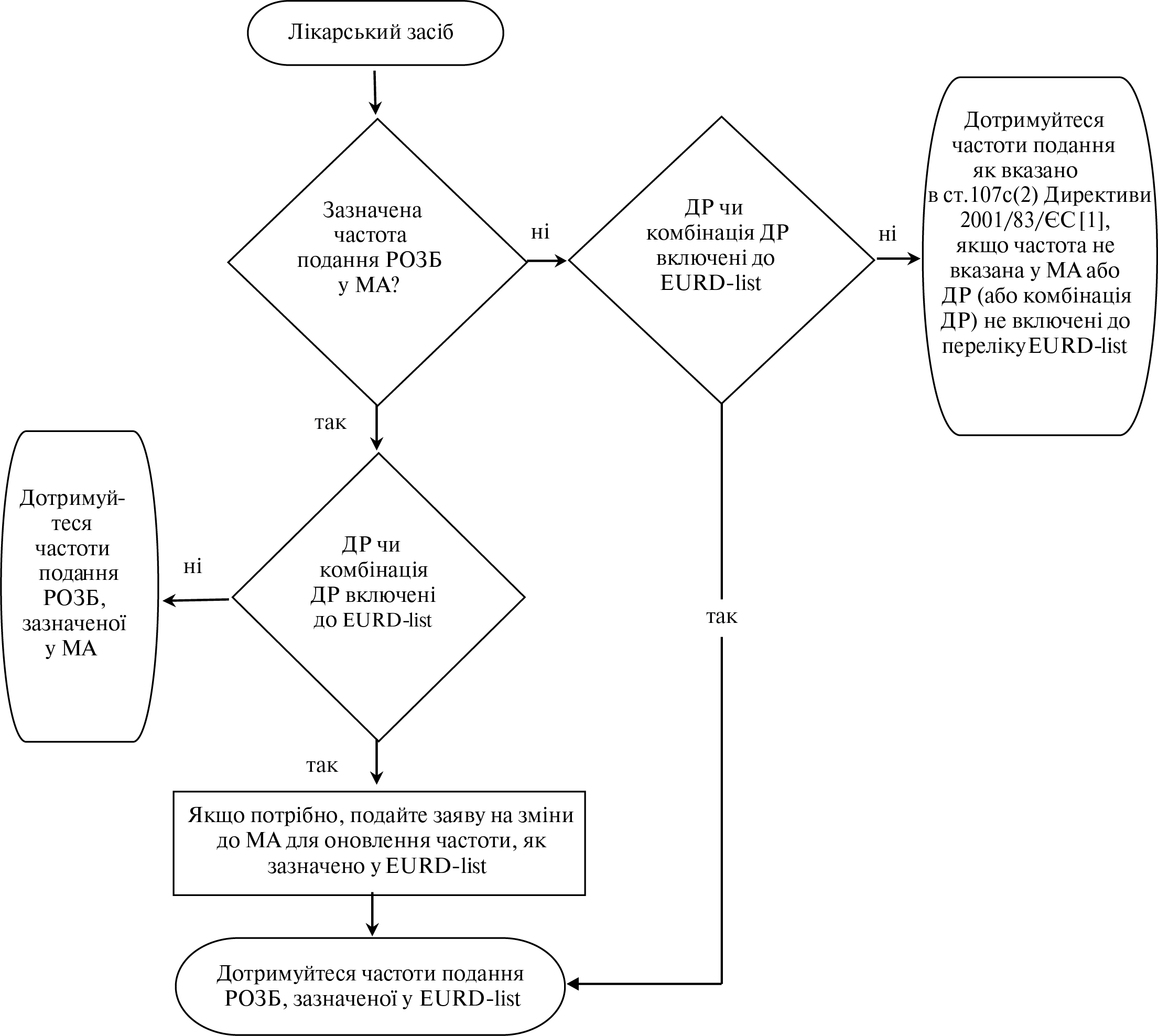
Рисунок VII.3. Загальні вимоги: умови подання РОЗБ
MA — дозвіл на маркетинг;
МДН — міжнародна дата народження;
АФІ — активний фармацевтичний інгредієнт;
РОЗБ — регулярно оновлюваний звіт з безпеки;
ДР — діюча речовина;
EURD-list — перелік строків подання регулярно оновлюваних звітів з безпеки лікарських засобів за міжнародною непатентованою назвою у ЄС.

Примітка: *Дотримуйтеся частоти подання з урахуванням МДН лікарського засобу/генерації на центральному рівні до моменту, коли такі строки подання РОЗБ співпадатимуть з Переліком — надалі РОЗБ подається відповідно до Переліку
Рисунок VII.3а. Загальні вимоги: умови подання РОЗБ в Україні
РП — реєстраційне посвідчення;
МДН — міжнародна дата народження;
АФІ — активний фармацевтичний інгредієнт;
РОЗБ — регулярно оновлюваний звіт з безпеки;
Перелік — Перелік строків подання регулярно оновлюваних звітів з безпеки лікарських засобів за міжнародною непатентованою назвою АФІ або комбінації АФІ, відповідно до додатку 10 до Порядку здійснення фармаконагляду, затвердженого наказом Міністерства охорони здоров’я України від 27 грудня 2006 р. № 898, зареєстрованого в Міністерстві юстиції України 19 грудня 2016 р. за № 1649/29779 (у редакції наказу Міністерства охорони здоров’я України від 26 вересня 2016 р. № 996).
Зазначена в переліку референтних дат дата закриття бази даних дає можливість синхронізувати подання РОЗБ лікарських засобів за різними реєстраційними посвідченнями та здійснювати єдину оцінку в ЄС, УкраїніN. Дата закриття бази даних — це фіксована конкретна дата місяця, що використовується для визначення дати надання регулярного звіту з безпеки (яка має правовий статус). Заявники (власники реєстраційних посвідчень) можуть пропонувати внести зміни до цих дат у відповідності з розділом VII.C.3.5.2.
Крім випадків, коли у переліку референтних дат ЄС та частоті подання, періодичності подання РОЗБ в УкраїніN зазначено інакше, або є погодження з уповноваженими органами в країнах ЄС, або ЕМА, або УкраїніN, повинен готуватися єдиний РОЗБ для усіх лікарських засобів, що містять однакову діючу речовину (АФІN), та зареєстровані одним заявником (власником реєстраційних посвідчень). РОЗБ повинен охоплювати всі показання, способи введення, лікарські форми та схеми прийому незалежно від того, чи зареєстровані лікарські засоби під різними торговельними назвами або за різними процедурами. Якщо доцільно, дані стосовно певних показань, лікарських форм, шляхів введення чи схем застосування слід надавати в окремих розділах РОЗБ, і будь-які питання безпеки повинні розглядатися відповідно (ст. 34(6) ІП 520/2012 [6], положення Порядку [2]N).
Для лікарських засобів, що містять діючу речовину (АФІN) або комбінацію діючих речовин (АФІN), які не увійшли до переліку референтних дат ЄС, періодичності подання РОЗБ згідно з переліком строків подання РОЗБ в УкраїніN РОЗБ повинен подаватися відповідно до частоти, зазначеної в умовах видачі реєстраційного посвідчення, якщо ж така відсутня, то відповідно до графіка подання регулярних звітів з безпеки вказаного в ст. 107с(2) Директиви 2001/83/ЄС [1], ст. 28(2) Регламенту (ЄC) № 726/2004 [5], згідно з частотою подання з урахуванням міжнародної дати народження лікарського засобу (положення Порядку [2]N).
VII.С.3.3.2. Подання регулярно оновлюваного звіту з безпеки для генеричних лікарських засобів, лікарських засобів з добре вивченим медичним застосуванням, традиційних рослинних та гомеопатичних лікарських засобів
Шляхом часткової відміни подання РОЗБ для генериків (зареєстрованих за ст. 10(1) Директиви 2001/83/ЄС [1], згідно з положеннями Порядку проведення експертизи реєстраційних матеріалів [7]N), лікарських засобів з добре вивченим медичним застосуванням (зареєстрованих за ст. 10а Директиви 2001/83/ЄС [1], згідно з положеннями Порядку проведення експертизи реєстраційних матеріалів [7]N), гомеопатичних лікарських засобів (зареєстрованих за ст. 14 Директиви 2001/83/ЄС [1], згідно з положеннями Порядку проведення експертизи реєстраційних матеріалів [7]N) та традиційних рослинних лікарських засобів (зареєстрованих за ст. 16а Директиви 2001/83/ЄС [1], згідно з положеннями Порядку проведення експертизи реєстраційних матеріалів [7]N) не вимагається, крім випадків, коли надання РОЗБ (ст. 107b(3) Директиви 2001/83/ЄС [1], положення Порядку [2]N):
- є умовою видачі реєстраційного посвідчення;
- вимагається уповноваженими органом у зв’язку з питаннями, пов’язаними з даними фармаконагляду, або через відсутність РОЗБ для діючої речовини (АФІN) після видачі реєстраційного посвідчення (наприклад коли «референтний» лікарський засіб більше не продається). Звіти з оцінки РОЗБ, що запитуються, повинні надаватися в PRAC, який має розглянути, чи існує необхідність у єдиному звіті з оцінки для всіх реєстраційних посвідчень на лікарські засоби, що містять одну й ту ж саму діючу речовину (АФІ), та інформувати CMDh та СНМР відповідно з метою застосування процедур, що викладені у ст. 107с(4) та ст. 107е Директиви 2001/83/ЄС [1].
З метою сприяння та оптимізації процесу єдиної оцінки РОЗБ, для уникнення дублювань запитів на надання РОЗБ та забезпечення прозорості й прогнозованості для заявників (власників реєстраційних посвідчень), застосовуються законодавчі положення, що викладені в ст. 107b(3)(b) Директиви 2001/83/ЄС [1], положеннях Порядку [2]N, вказуючи в переліку референтних дат ЄС, у періодичності подання РОЗБ в УкраїніN діючі речовини (АФІN), для яких вимагається надання РОЗБ для генериків, лікарських засобів з добре вивченим медичним застосуванням, традиційних рослинних та гомеопатичних лікарських засобів. Ця специфікація ґрунтується на запиті, який зроблений уповноваженим органом у країні ЄС, УкраїніN під час створення або підтримки переліку референтних дат ЄС, періодичності подання РОЗБ в УкраїніN та на основі питань, що пов’язані з даними фармаконагляду, або внаслідок відсутності РОЗБ стосовно діючої речовини (АФІN).
Гармонізована частота подання РОЗБ та референтні дати ЄС визначаються СНМР та/або CMDh після консультації з PRAC. В Україні періодичність подання регулярних звітів з безпеки складається і переглядається відповідно до положень, викладених у додатку 10 до Порядку [2]N.
Застосування переліку референтних дат ЄС періодичності подання регулярних звітів з безпеки в УкраїніN для подання РОЗБ для генеричних лікарських засобів, лікарських засобів з добре вивченим медичним застосуванням, традиційних рослинних та гомеопатичних лікарських засобів не позбавляє уповноважений орган права вимагати надання РОЗБ у будь-який момент згідно з положеннями, викладеними в другому підпараграфі ст. 107с(2) Директиви 2001/83/ЄС [1] та положеннях Порядку [2]N.
Що стосується лікарських засобів, для яких більше не вимагається регулярне надання РОЗБ, передбачається, що заявники (власники реєстраційних посвідчень) будуть продовжувати регулярну оцінку безпеки своїх лікарських засобів та повідомляти будь-яку нову інформацію з безпеки, що впливає на співвідношення користі/ризику чи інформацію про лікарський засіб (див. модуль VI та IX НПФ).
Рис. VII.4 представляє різні потенційні сценарії відносно подання РОЗБ для генеричного лікарського засобу, лікарського засобу з добре вивченим застосуванням, традиційного рослинного лікарського засобу та гомеопатичного лікарського засобу:

*Чи повинні заявники (власники реєстраційних посвідчень) на генеричний лікарський засіб, лікарський засіб з добре вивченим застосуванням, традиційний рослинний лікарський засіб та гомеопатичний лікарський засіб подавати РОЗБ після запиту уповноваженого органу, внаслідок виникнення питань, що стосуються даних з фармаконагляду або неподання РОЗБ.
Рисунок VII.4. Умови подання РОЗБ для генеричного лікарського засобу, лікарського засобу з добре вивченим застосуванням, традиційного рослинного лікарського засобу та гомеопатичного лікарського засобу в ЄС

*Чи повинні заявники (власники реєстраційних посвідчень) на генеричний лікарський засіб, лікарський засіб з добре вивченим застосуванням, традиційний рослинний лікарський засіб та гомеопатичний лікарський засіб подавати РОЗБ після запиту уповноваженого органу внаслідок виникнення питань, що стосуються даних з фармаконагляду або неподання РОЗБ.
Рисунок VII.4а Умови подання РОЗБ для генеричного лікарського засобу, лікарського засобу з добре вивченим застосуванням, традиційного рослинного лікарського засобу та гомеопатичного лікарського засобу в УкраїніN
VII.С.3.3.3. Подання регулярно оновлюваного звіту з безпеки на лікарські засоби з фіксованою комбінацією
Якщо не зазначено інше у переліку референтних дат ЄС та частоти подання (в Україні — періодичність подання регулярних звітів з безпекиN), якщо діюча речовина (АФІN), що є предметом РОЗБ, також зареєстрована як компонент лікарського засобу з фіксованою комбінацією, заявник (власник реєстраційного посвідчення) повинен подавати або окремий РОЗБ для комбінації діючих речовин (АФІN), що зареєстровані тим самим заявником (власником реєстраційного посвідчення), з перехресним посиланням на РОЗБ для окремих діючих речовин (АФІN), або надати дані щодо комбінації в одному з РОЗБ для однокомпонентного лікарського засобу (ст. 34(7) ІП 520/2012 [6], положення Порядку [2]N).
VII.С.3.3.4. Подання регулярно оновлюваного звіту з безпеки на вимогу уповноваженого органу в країні ЄС та УкраїніN
Заявники (власники реєстраційних посвідчень) повинні надавати РОЗБ негайно на вимогу уповноваженого органу в країні ЄС (ст. 107с(2) Директиви 2001/83/ЄС [1]), та УкраїниN (положення Порядку [2]N). Для сприяння оцінці та уникнення дублювання запитів уповноважені органи в країнах ЄС та в УкраїніN, як правило, будуть використовувати перелік референтних дат ЄС, (періодичність подання регулярних звітів з безпеки в УкраїніN) для запиту на надання РОЗБ, однак в особливих умовах уповноважені органи у країнах ЄС та в УкраїніN можуть безпосередньо вимагати подання РОЗБ. Якщо у запиті уповноваженого органу не обумовлений строк подання, заявники (власники реєстраційних посвідчень) повинні подати РОЗБ протягом 90 календарних днів з моменту закриття бази даних.
VII.С.3.4. Критерії, що використовуються для визначення частоти подання регулярно оновлюваного звіту з безпеки
При відхиленні від графіку подання РОЗБ, що визначений у ст. 107с(2)(b) Директиви 2001/83/ЄС [1] та положеннях Порядку [2]N, слід, щоб частоту подання РОЗБ та відповідні дати закриття бази даних визначив СНМР, в Україні — ЦентрN за допомогою підходу з урахуванням ризиків, якщо принаймні одне відповідне реєстраційне посвідчення було видано за централізованою процедурою, або CMDh після консультації з PRAC, в Україні — МОЗN. Наступні критерії за пріоритетністю слід врахувати при визначенні частоти подання РОЗБ для даної діючої речовини (АФІN) або комбінації діючих речовин (АФІN):
- інформація щодо ризиків чи користі, що може вплинути на громадське здоров’я;
- новий лікарський засіб, для якого на даний момент існує обмежена інформація з безпеки (включаючи дореєстраційний та післяреєстраційний досвід);
- значні зміни лікарського засобу (наприклад, зареєстровано нове показання, нова лікарська форма чи спосіб введення, що розширюють популяцію пацієнтів, які застосовують лікарський засіб);
- вразливі/недостатньо вивчені популяції пацієнтів, відсутня інформація (наприклад, діти, вагітні), коли такі популяції пацієнтів, , можуть застосовувати лікарський засіб у післяреєстраційний період;
- сигнал про/потенційна загроза неправильного застосування, медичної помилки, ризику передозування або залежності;
- обсяг бази даних з безпеки та експозиції лікарського засобу;
- лікарські засоби, що підлягають додатковому моніторингу.
Будь-яка зміна у критеріях, що перераховані вище, для даної діючої речовини (АФІN) або комбінації діючих речовин (АФІN) може призвести до зміни в переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN (наприклад підвищення частоти подання РОЗБ).
VII.С.3.5. Підтримка переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN)
VII.С.3.5.1. Загальні принципи
Підтримка переліку референтних дат ЄС, періодичність подання регулярних звітів з безпеки в УкраїніN повинна сприяти регуляторному реагуванню на питання охорони здоров’я, що виявлені в рамках ЄС та УкраїниN, а отже, перелік буде змінюватися для відображення прийнятих рішень (наприклад комітетами EMA, ЦентромN після виявлення сигналу).
Інформація, що входить до переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN, така як діючі речовини (АФІN) та комбінації діючих речовин (АФІN), частота подання РОЗБ та дата закриття бази даних можуть змінюватися, якщо це вважає необхідним СНМР або CMDh після консультації з PRAC, в Україні — ЦентрN. Зміни до переліку можуть вноситися за однієї з наступних причин:
- поява нової інформації, що може вплинути на співвідношення користі/ризику діючих речовин (АФІN) чи комбінації діючих речовин (АФІN) та потенційно на громадське здоров’я;
- будь-яка зміна критеріїв, що використовуються для визначення частоти подання РОЗБ, та описаних у розділі VII.С.3.4., ННПФ;
- на вимогу заявників (власників реєстраційних посвідчень) за визначенням ст. 107с(6) Директиви 2001/83/ЄС [1], положень додатку 10 до Порядку [2]N;
- нова зареєстрована діюча речовина (АФІN).
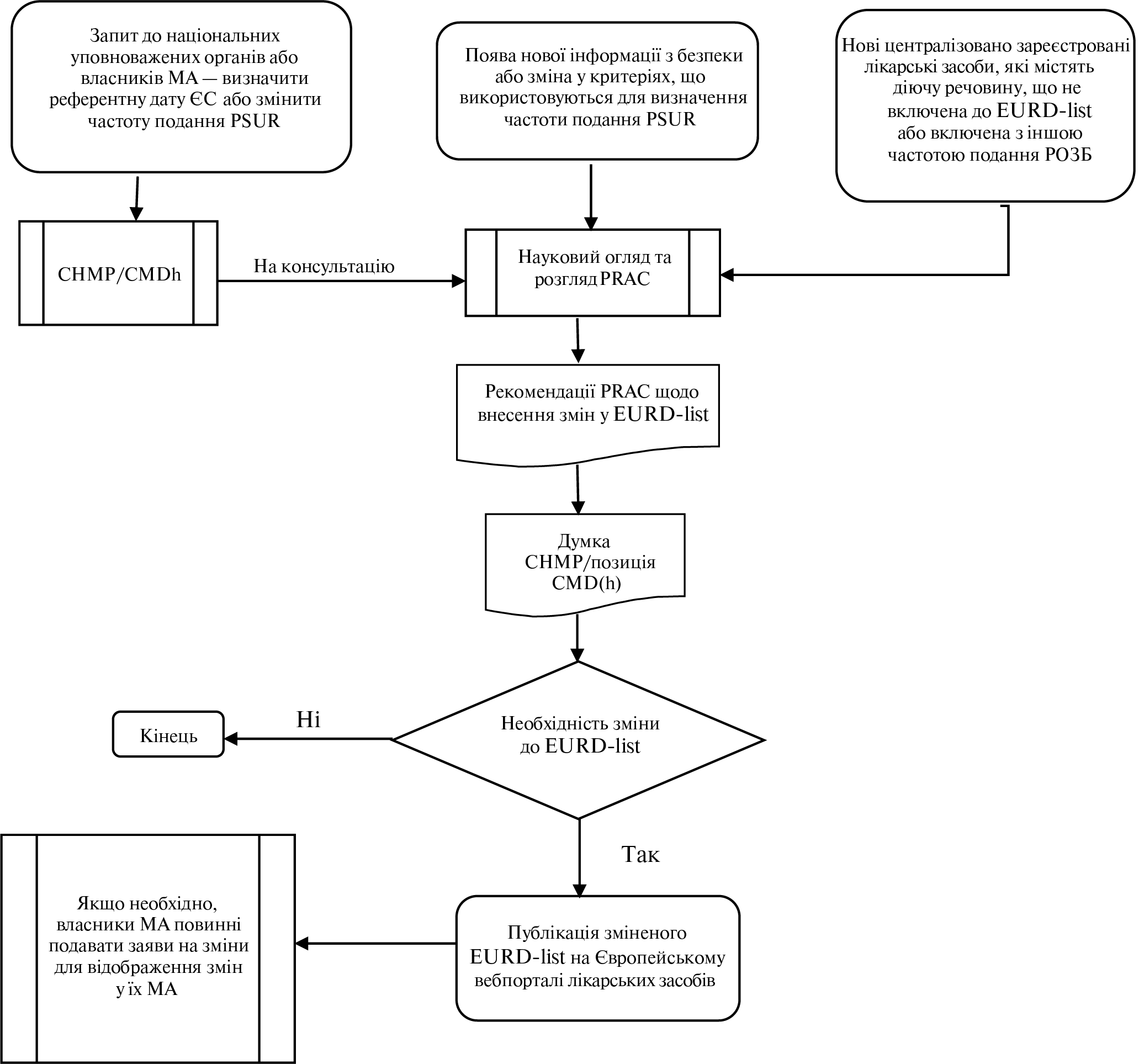
Рисунок VII.5. Підтримка переліку референтних дат ЄС та частоти подання регулярних звітів з безпеки
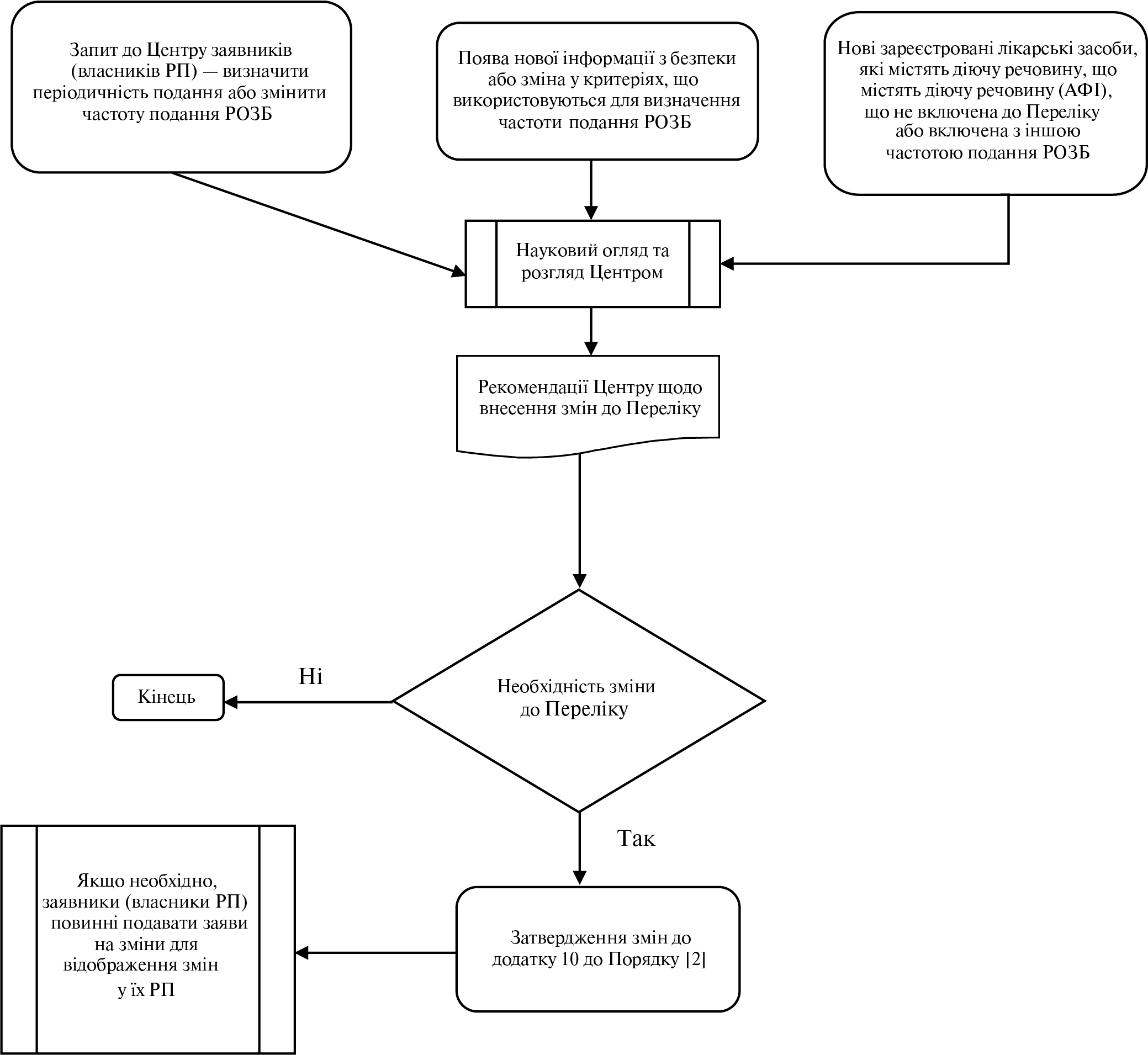
Рисунок VII.5а. Підтримка переліку періодичності подання регулярних звітів з безпеки в УкраїніN
VII.С.3.5.2. Запити заявників (власників реєстраційних посвідчень) на внесення змін до переліку референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN)
Заявники (власники реєстраційних посвідчень) можуть подати запит до CHMP або CMDh, в Україні — до ЦентруN, для визначення референтних дат ЄС, періодичність подання регулярних звітів з безпеки в УкраїніN, чи зміни частоти подання РОЗБ на підставі однієї з наступних причин (ст. 107в(6) Директиви 2001/83/ЄС [1], положення додатку 10 до Порядку [2]N):
- з причин, що стосуються громадського здоров’я;
- з метою уникнення дублювання оцінки;
- з метою досягнення міжнародної гармонізації.
Запит та його причини слід розглянути PRAC та СНМР, Центру — в УкраїніN, якщо він стосується принаймні одного реєстраційного посвідчення, що видане відповідно до централізованої процедури, або CMDh, в Україні — МОЗN в іншому випадку, які (якийN) або схвалять, або відмовлять у запиті.
Після внесення до переліку відповідних змін він повинен бути опублікованим на Європейському вебпорталі лікарських засобів (див. розділ VII.С.3.6.). В Україні зміни до Періодичності подання регулярних звітів з безпеки затверджуються наказом МОЗN.
Щодо детальних даних про те, як подавати запити на внесення змін до переліку, треба звернутися до супровідного коментаря до референтних дат ЄС та відповідної форми, опублікованої на Європейському вебпорталі лікарських засобів71.
VII.С.3.6. Публікація переліку
Перелік референтних дат ЄС та частота подання регулярних звітів з безпеки після його створення та затвердження CHMP та CMDh після консультації з PRAC оприлюднюється на Європейському вебпорталі лікарських засобів. Перелік періодичності подання регулярних звітів з безпеки оприлюднюється на вебсайті Центру (положення додатку 10 до Порядку [2]N).
У випадку внесення змін, оновлений перелік слід публікувати після його затвердження СНМР або CMDh. Передбачається, що оновлення переліку відбуватиметься щомісяця.
В Україні у випадку внесення змін до Переліку подання регулярних звітів з безпеки, оновлений перелік буде опублікований на вебсайті Центру після його затвердження наказом МОЗN.
VII.С.3.7. Внесення змін до умов реєстраційних посвідчень згідно з переліком референтних дат ЄС, періодичності подання регулярних звітів з безпеки в УкраїніN
Будь-які зміни до дат та частоти подання РОЗБ у переліку набувають чинності через 6 місяців з дати його публікації на Європейському вебпорталі лікарських засобів. В Україні дата набуття чинності змін до Переліку буде зазначена у наказі МОЗN. Якщо необхідно, заявники (власники реєстраційних посвідчень) повинні подавати заяви на відповідні зміни з метою відображення змін у своїх реєстраційних посвідченнях (ст. 107с(6) Директиви 2001/83/ЄС [1], положення додатку 10 до Порядку [2], положення Порядку проведення експертизи [7]N), крім випадків, коли реєстраційне посвідчення містить пряме перехресне посилання на перелік референтних дат ЄС (в Україні — періодичність подання регулярних звітів з безпекиN).
VII.С.4. Процеси оцінки регулярно оновлюваного звіту з безпеки в системі ЄС та в Україні
Уповноважені органи в країнах ЄС та в УкраїніN повинні здійснювати оцінку РОЗБ для того, щоб встановити чи існують нові ризики, чи змінилися ризики, чи змінилося співвідношення користь/ризик лікарського засобу (стаття 107d Директиви 2001/83/ЄС [1], положення ПорядкуN [2]).
Для лікарських засобів, що зареєстровані виключно за національною процедурою у одній країні ЄС, в УкраїніN, оцінка РОЗБ проводиться уповноваженим органом у країні ЄС, в УкраїніN, де лікарський засіб зареєстровано (див. розділ VII.С.4.1.).
Для лікарських засобів, які зареєстровані більше, ніж в одній країні ЄС, що містять однакову діючу речовину (АФІN) або однакову комбінацію діючих речовин (АФІN), незалежно від того чи володіють ними однакові заявники (власники реєстраційних посвідчень), та для яких частота та дати подання РОЗБ були гармонізовані в переліку референтних дат ЄС, єдина оцінка ЄС всіх РОЗБ проводиться за рекомендацією PRAC відповідно до процедури, що описана у розділах VII.С.4.2.1. та VII.С.4.2.2., ННПФ.
Додатково до оцінки РОЗБ та думки СНМР або позиції CMDh, якщо необхідно, після рекомендації PRAC уповноважені органи у країнах ЄС або Європейська Комісія для лікарських засобів, що зареєстровані за централізованою процедурою повинні вжити необхідних заходів для внесення змін, призупинення або відкликання реєстраційного(их) посвідчення(ь) відповідно до результату оцінки (стаття 107g(2) Директиви 2001/83/ЄС [1]) (статті 28(4) та (5) Регламенту (ЄC) №726/2004 [5]) (див. розділи VII.С.4.2.3. та VII.С.4.2.4.).
Результат оцінки РОЗБ призводить до юридично обов’язкового рішення або позиції у випадку будь-якої дії щодо внесення зміни, призупинення або відкликання реєстраційного посвідчення на лікарські засоби, що містять дану діючу речовину (АФІN) або комбінацію діючих речовин (АФІN) на підставі позиції CMDh або думки СНМР після отримання рекомендації від PRAC (в Україні — ЦентруN). Більш того заявникам (власникам реєстраційних посвідчень) нагадують про їх зобов’язання оновлювати свої реєстраційні посвідчення відповідно до статті 16(3) Регламенту (ЄC) №726/2004 [5], статті 23(3) Директиви 2001/83/ЄС [1], положень ПорядкуN [2], положень Порядку проведення експертизи реєстраційних матеріалівN [7]. Тому рекомендації застосовуються гармонізовано та вчасно для всіх лікарських засобів в рамках процедури на території ЄС та УкраїниN.
Зміни до короткої характеристики/інструкції для медичного застосуванняN, лікарського засобу, листка-вкладки та етикетки в результаті оцінки РОЗБ слід застосовувати без послідовного подання заяви на внесення змін для централізовано зареєстрованих лікарських засобів та шляхом внесення відповідної зміни до національно зареєстрованих лікарських засобів за національною процедурою, включаючи такі, що зареєстровані шляхом процедури взаємного визнання та децентралізованої процедури.
Якщо пропонується внести до інформації про лікарський засіб нові побічні реакції у розділ 4.8 («Небажані ефекти») короткої характеристики/розділ «Побічні реакції» інструкції для медичного застосуванняN, лікарського засобу чи змінити опис, частоту і серйозність уже існуючих побічних реакцій, заявники (власники реєстраційних посвідчень) повинні у відповідних розділах РОЗБ надати необхідну інформацію для належного опису і класифікації частоти побічних реакцій. Якщо необхідно оновити інші розділи короткої характеристики/інструкції для медичного застосуванняN, лікарського засобу (наприклад, розділ короткої характеристики лікарського засобу 4.4 «Спеціальні застереження і попередження щодо застосування»), уповноваженим органам в країнах ЄС, Центру — в УкраїніN потрібно надати чіткі пропозиції для розгляду під час оцінки РОЗБ72. Пропозиції слід включити в локальний додаток РОЗБ.
Гармонізація повної інформації про лікарський засіб у всіх країнах ЄС, у яких лікарський засіб зареєстровано, та УкраїніN є однією з цілей процедури оцінки РОЗБ. Результатом оцінки повинно бути включення нових застережень з безпеки і ключових рекомендацій для мінімізації ризику, що були виявлені у процесі оцінки даних у РОЗБ, у відповідні розділи інформації про лікарські засоби.
VII.С.4.1. Регулярно оновлюваний звіт з безпеки тільки для національно зареєстрованих лікарських засобів
Обов’язком уповноваженого органу у країні, у якій лікарський засіб зареєстровано, є проведення оцінки РОЗБ для таких ліків, що проводиться відповідно до національного законодавства.
Переліки окремих випадків можуть вимагатися в контексті процедури оцінки РОЗБ для побічних реакцій, що представляють особливий інтерес, та повинні надаватися заявником (власником реєстраційного посвідчення) у межах визначеного періоду часу, що включений у запит. Якщо необхідно для наукової оцінки, запит може супроводжуватися вимогою аналізу окремих випадків побічних реакцій (включаючи інформацію про кількість випадків, детальну інформацію про летальні випадки і, якщо необхідно, аналіз несерйозних випадків). Зазвичай слід надати інформацію щодо контексту або обґрунтування запиту.
Після оцінки РОЗБ уповноважений орган країни ЄС та УкраїниN має розглянути необхідність будь-яких дій стосовно реєстраційного посвідчення на лікарський засіб, що розглядався. Це може бути внесення змін, призупинення або відкликання реєстраційного посвідчення, якщо необхідно, згідно з відповідною процедурою на національному рівні.
Звіт з оцінки РОЗБ та висновки уповноваженого органу країни ЄС, УкраїниN, мають надаватися заявнику (власнику реєстраційного посвідчення).
VII.С.4.2. Лікарські засоби, зареєстровані більше ніж в одній країні ЄС
VII.С.4.2.1. Оцінка регулярно оновлюваного звіту з безпеки для одного лікарського засобу зареєстрованого централізовано
У цьому розділі описується процес оцінки РОЗБ лише для одного централізовано зареєстрованого лікарського засобу згідно з процедурою, зазначеною у ст. 28 Регламенту (ЄC) №726/2004 [5] (див. рис. VII.6).
Оцінка РОЗБ для одного централізовано зареєстрованого лікарського засобу координується EMA та має проводитися призначеним PRAC доповідачем (ст. 28(3) Регламенту (ЄC) №726/2004 [5]) (далі вказується як «доповідач PRAC»).
EMA після отримання повинно провести технічну перевірку РОЗБ для того, щоб впевнитися, що заява на РОЗБ (PSUR application) у відповідному форматі.
При оцінці РОЗБ можуть використовуватися дані про окремі випадки побічних реакцій бази даних EudraVigilance. Після вищезгаданих перевірок процедура оцінки відбувається відповідно до офіційних дат початку, опублікованих на вебсайті EMA. Детальний графік процедури опублікований у вигляді узагальненого календаря на вебсайті EMA.
Опублікований графік визначає дати подання РОЗБ, початку та закінчення процедур, а також проміжні дати/основні етапи процедури.
Під час оцінки додаткові переліки окремих випадків можуть вимагатися доповідачем PRAC через EMA щодо побічних реакцій, які представлять особливий інтерес, та слід, щоб вони подавалися заявником(ами) (власником(ами) реєстраційного(их) посвідчення(ь)) протягом встановленого строку для їх включення у запит.
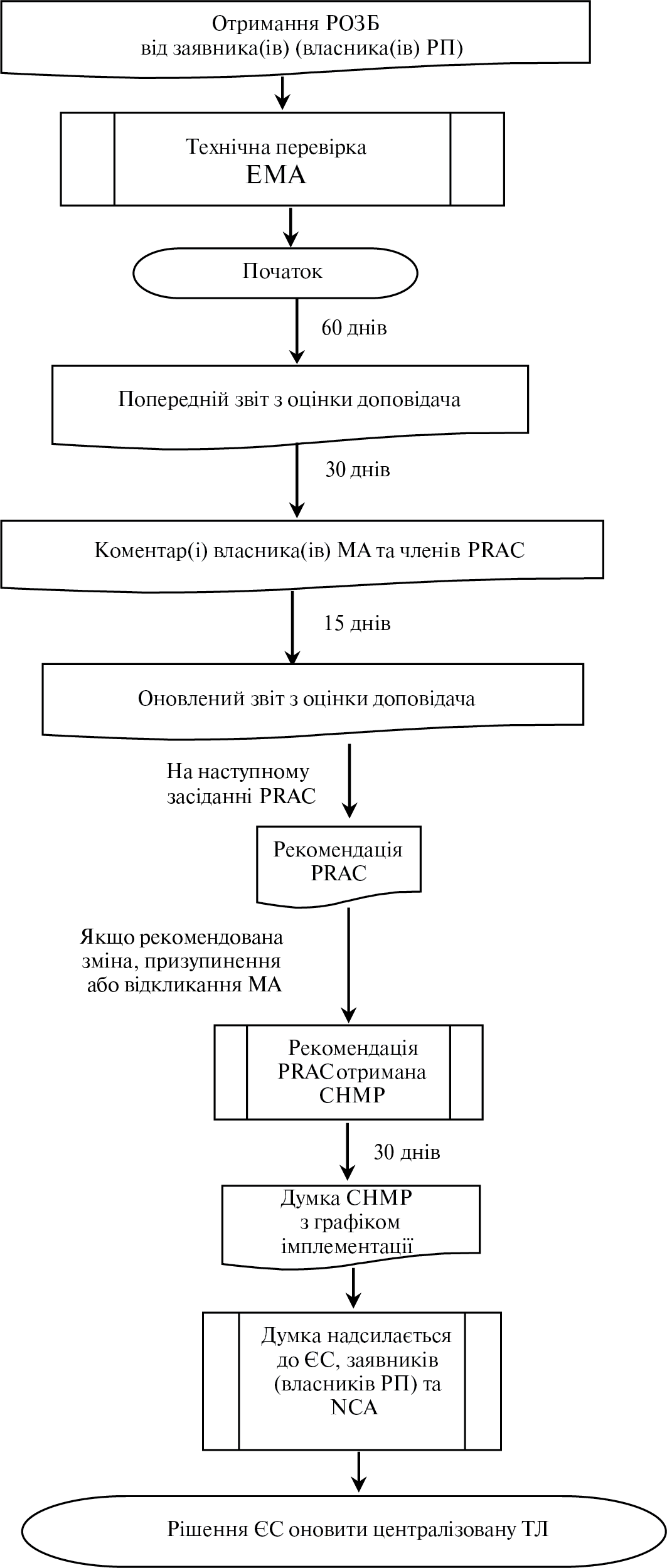
Рисунок VII.6. Процедура оцінки регулярного звіту з безпеки для одного централізовано зареєстрованого лікарського засобу
Заявники (власники реєстраційних посвідчень) повинні надати такі переліки протягом визначеного в запиті строку. Якщо необхідно для наукової оцінки, запит може також містити вимогу аналізу окремих випадків побічних реакцій (включаючи інформацію про кількість випадків, інформацію про летальні випадки, та, якщо необхідно, аналіз випадків несерйозних побічних реакцій). Зазвичай слід надати інформацію щодо контексту та обґрунтування запиту.
Під час складання звіту з оцінки доповідач PRAC повинен тісно співпрацювати з доповідачем СНМР (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]).
Доповідач PRAC повинен підготувати звіт з оцінки та надіслати його до EMA та членам PRAC (ст. 28(3) Регламенту (ЄC) №726/2004 [5]) протягом 60 днів з моменту початку процедури.
EMA повинно надіслати попередній звіт з оцінки доповідача PRAC заявнику (власнику реєстраційного посвідчення) (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]).
На 90-й день заявник (власник реєстраційного посвідчення) та члени PRAC можуть надіслати коментарі щодо попереднього звіту з оцінки доповідача PRAC EMA та доповідачу PRAC. Ці коментарі повинні також включати відповіді на невирішені проблеми або питання, які підняті доповідачем PRAC у попередньому звіті з оцінки, та які можуть розглядатися в межах часових рамок фази коментарів.
Після отримання коментарів доповідач PRAC повинен підготувати оновлений звіт з оцінки (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]) протягом 15 днів (тобто до 105-го дня). Оновлений звіт з оцінки надається членам PRAC та повинен направлятися EMA заявнику (власнику реєстраційного посвідчення).
Усне пояснення для PRAC може проводитися на вимогу PRAC або заявника (власника реєстраційного посвідчення) у випадку рекомендації з відкликання або призупинення реєстраційного посвідчення, нового протипоказання, обмеження показання або зниження рекомендованої дози.
PRAC повинен затвердити оновлений звіт з оцінки з або без подальших змін на своєму наступному засіданні (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]) разом з рекомендацією щодо збереження реєстраційного посвідчення або необхідності внести зміни, призупинити або відкликати реєстраційне посвідчення. Рекомендація PRAC може також вимагати проведення ПДБЛЗ, оновлення ПУР, перегляду питань з безпеки та/або детального моніторингу випадків, що представляють інтерес.
Суперечливі позиції членів PRAC та причини, на яких вони ґрунтуються, повинні відображатися в рекомендаціях, що видаються PRAC (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]).
EMA повинно включати рекомендацію PRAC та затверджений звіт з оцінки в архів та направляти обидва документа заявнику (власнику реєстраційного посвідчення) (ст. 28(3) Регламенту (ЄC) № 726/2004 [5]).
Після затвердження на засіданні PRAC та у випадку, якщо рекомендована будь-яка регуляторна дія, звіт з оцінки та рекомендація PRAC надсилаються до СНМР для схвалення думки щодо даного централізовано зареєстрованого лікарського засобу, як описано у розділі VII.С.4.2.3., ННПФ.
VII.С.4.2.2. Оцінка регулярно оновлюваного звіту з безпеки для лікарських засобів, на які видані різні реєстраційні посвідчення, що місять однакову діючу речовину (АФІN) (єдина оцінка ЄС)
Цей розділ описує оцінку РОЗБ для лікарських засобів, на які видані різні реєстраційні посвідчення, які зареєстровані більш ніж в одній країні ЄС, та які містять однакову діючу речовину (АФІN), або однакову комбінацію діючих речовин (АФІN), незалежно від того, чи володіє ними один і той самий заявник (власник реєстраційного посвідчення), та для яких частота та дати подання РОЗБ були гармонізовані в переліку референтних дат ЄС. Це може бути комбінація з лікарських засобів ,централізовано зареєстрованих, лікарських засобів, зареєстрованих за процедурою взаємного визнання, за децентралізованою процедурою та лікарських засобів, зареєстрованих на національному рівні (ст. 107е — 107g Директиви 2001/83/ЄС [1]) (так звана процедура «єдиної оцінки ЄС» для РОЗБ).

Рисунок VII.7. Процедура оцінки регулярного звіту з безпеки за єдиною оцінкою ЄС — аркуш 1
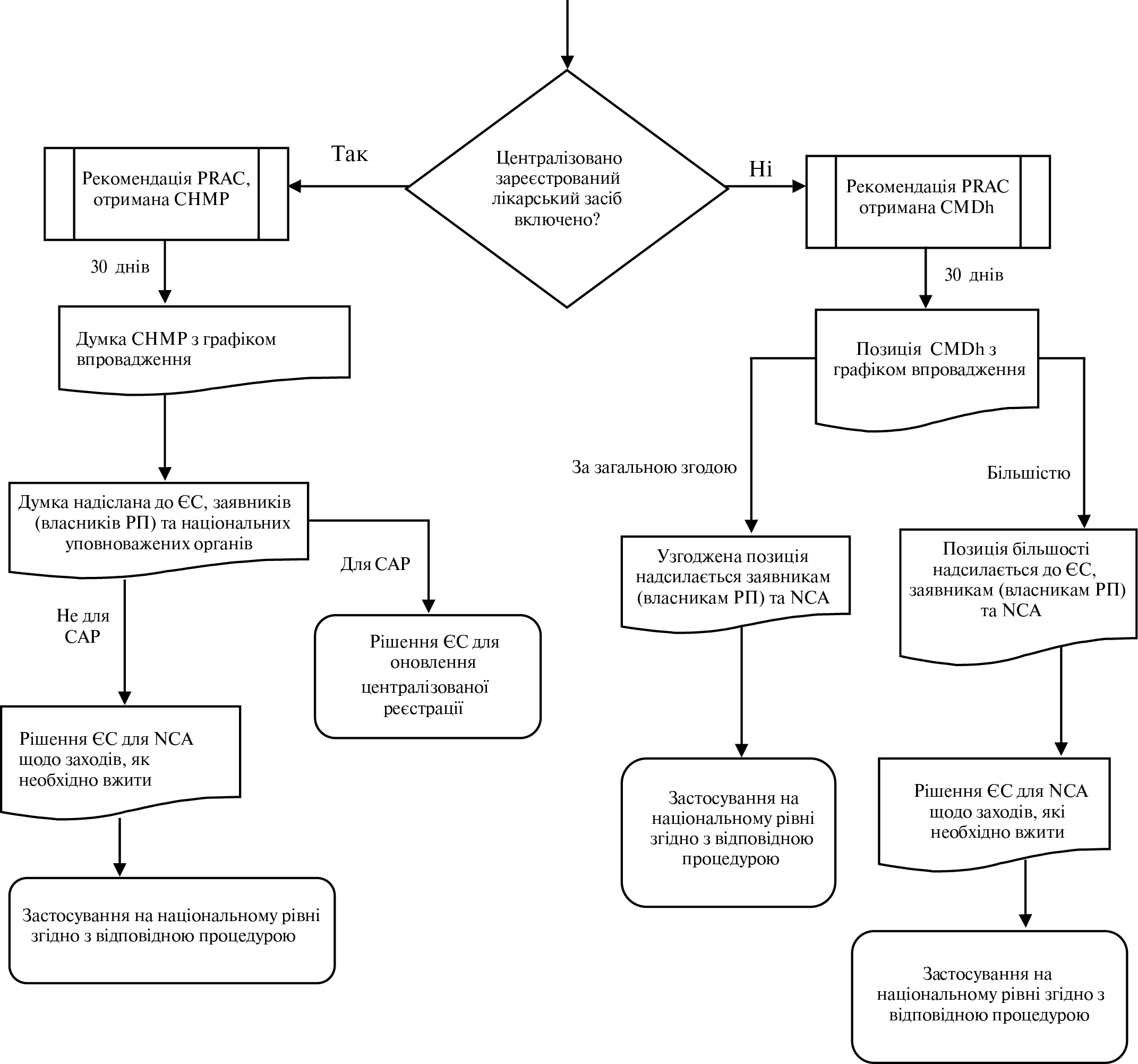
Рисунок VII.7 — аркуш 2
Оцінка РОЗБ для лікарських засобів також називається «єдина оцінка ЄС» та має проводитися (ст. 107е (1) Директиви 2001/83/ЄС [1]):
- країною ЄС, призначеною CMDh, якщо жодне з реєстраційних посвідчень не було видане відповідно до централізованої процедури;
- доповідачем, призначеним PRAC, якщо принаймні одне з даних реєстраційних посвідчень було видане відповідно до централізованої процедури (далі вказується як «доповідач PRAC»).
Процедура єдиної оцінки ЄС РОЗБ координується EMA. Після отримання EMA має провести технічну валідацію звітів для забезпечення застосування РОЗБ у відповідному форматі.
Після створення переліку всіх лікарських засобів для людини, що зареєстровані у ЄС, який вказано у ст. 57 Регламенту (ЄC) № 726/2004 [5], EMA слід гарантувати, що всі заявники (власники реєстраційних посвідчень) на дану речовину надали регулярні звіти з безпеки, згідно з вимогою. У випадку якщо РОЗБ не був наданий, EMA слід зв’язатися з відповідним заявником (власником(ами) реєстраційного(их) посвідчення(ь)). Однак це не перешкоджає початку процедури з єдиної оцінки іншого(их) РОЗБ для такої ж діючої речовини (АФІN).
Для підтримки оцінки РОЗБ можна робити витяг переліків окремих випадків з бази даних EudraVigilance.
Після вищезгаданих перевірок процедури починаються відповідно до офіційних дат початку, опублікованих на вебсайті EMA. Детальні графіки процедур опубліковані у вигляді узагальненого календаря на вебсайті EMA.
Опубліковані графіки визначають дати подання, початку та закінчення процедур, а також інші проміжні дати/основні етапи, що відбуваються під час процедури.
Після початку процедури доповідач від PRAC або країна ЄС проводить єдину оцінку всіх РОЗБ, що подаються на дану діючу речовину (АФІN).
Під час оцінки доповідач від PRAC або країна ЄС може надати запит через EMA на додаткові переліки окремих випадків побічних реакцій на лікарські засоби, які представляють особливий інтерес; відповідь на запит має надаватися заявником(ами) (власником(ами) реєстраційного(их) посвідчення(ь)) в межах зазначеного у запиті часу. Останній може супроводжуватися запитом на аналіз окремих випадків (включаючи інформацію щодо кількості випадків, детальних даних щодо летальних випадків та, якщо необхідно, аналіз несерйозних випадків), якщо необхідно, для наукової оцінки. Як правило, повинна надаватися інформація щодо контексту або обґрунтування запиту.
Доповідач від PRAC або країна ЄС має підготувати звіт з оцінки та надіслати його EMA та відповідним країнам ЄС (ст. 107е(2) Директиви 2001/83/ЄС [1]) протягом 60 днів з моменту початку процедури. Цей попередній звіт з оцінки слід надсилати членам PRAC.
EMA має надіслати попередній звіт доповідача від PRAC/країни ЄС відповідному заявнику (власнику (ам) реєстраційного (их) посвідчення (ь)) (ст. 107е (2) Директиви 2001/83/ЄС [1]). Цей звіт з оцінки повинен розповсюджуватися серед усіх заявників (власників реєстраційних посвідчень), чиї лікарські засоби є частиною єдиної оцінки ЄС.
На 90-й день заявник (власник(и) реєстраційного(их) посвідчення(ь)), країни ЄС та члени PRAC, якщо є доцільним, можуть надсилати коментарі щодо попереднього звіту з оцінки доповідача PRAC/країни ЄС до EMA та доповідачу від PRAC/країни ЄС, якщо необхідно. Ці коментарі повинні також включати відповіді на невирішені проблеми або питання, які підняті доповідачем PRAC/країною ЄС у попередньому звіті з оцінки, та які можуть бути розглянуті в рамках фази коментарів.
Після отримання коментарів доповідач від PRAC/країна ЄС має підготувати оновлений звіт з оцінки (ст. 107е(3) Директиви 2001/83/ЄС [1]) протягом 15 днів (тобто на 105-й день). Оновлений звіт з оцінки направляється членам PRAC та повинен розповсюджуватися EMA серед усіх заявників (власників реєстраційних посвідчень), чиї лікарські засоби є частиною єдиної оцінки ЄС.
Усне пояснення для PRAC може проводитися на вимогу PRAC або заявника (власника реєстраційних посвідчень) у випадку рекомендації відкликати або припинити дію реєстраційного посвідчення, нового протипоказання, обмеження показання або зниження рекомендованої дози.
PRAC має схвалити оновлений звіт з оцінки з або без подальших змін на своєму наступному засіданні (ст. 107e(3) Директиви 2001/83/ЄС [1]) разом з рекомендацією про підтримку реєстраційного посвідчення або необхідність внесення до нього змін, призупинення або відкликання реєстраційного посвідчення. Рекомендація PRAC може також вимагати необхідності проведення ПДБЛЗ (див. модуль VIII), оновлення ПУР (див. модуль V), огляду питання з безпеки та/або детального моніторингу випадків, що представляють інтерес.
Суперечливі позиції членів PRAC та причини, на яких вони ґрунтуються, повинні відображатися в рекомендаціях, що видаються PRAC (ст. 107e(3) Директиви 2001/83/ЄС [1]).
EMA має включити рекомендацію PRAC та затверджений звіт з оцінки до архіву та направити обидва документи заявнику (ам) (власнику (ам) реєстраційного (их) посвідчення (ь)) (ст. 107e (3) Директиви 2001/83/ЄС [1]).
Після затвердження на засіданні PRAC у випадку, якщо рекомендується будь-яка регуляторна дія, звіт з оцінки та рекомендація PRAC надсилаються до:
- СНМР, якщо до єдиної оцінки увійшов принаймні один централізовано зареєстрований лікарський засіб, для схвалення думки, викладеної в розділі VII.С.4.2.3.;
- CMDh, якщо до єдиної оцінки не увійшов жоден централізовано зареєстрований лікарський засіб, для схвалення позиції, викладеної в розділі VII.С.4.2.4.
VII.С.4.2.3. Єдина оцінка, яка включає принаймні один централізовано зареєстрований лікарський засіб, що призводить до винесення думки СНМР
СНМР підтверджує отримання рекомендації та звіту з оцінки PRAC, у випадку будь-якої регуляторної дії, на своєму наступному засіданні після затвердження PRAC. Протягом 30 днів після отримання СНМР має розглянути звіт з оцінки та рекомендації PRAC та ухвалити думку щодо підтримки, внесення змін, призупинення або відкликання даного(их) реєстраційного(их) посвідчення(ь) (ст. 107g(3) Директиви 2001/83/ЄС [1]).
Усне пояснення для СНМР може проводитися на вимогу СНМР або заявника (власника(ів) реєстраційного(их) посвідчення(ь)) лише у випадку розбіжностей з рекомендацією PRAC, якщо СНМР розглядає можливість ухвалити думку про припинення дії або відкликання реєстраційного(их) посвідчення(ь), нове протипоказання, обмеження показань або зниження рекомендованої дози.
Думка повинна містити наступне:
- заключний звіт з оцінки та рекомендацію, схвалену PRAC;
- детальне пояснення наукових причин розбіжностей з рекомендацією PRAC, якщо можливо (ст. 107g(3) Директиви 2001/83/ЄС [1]);
- у випадку думки СНМР щодо внесення змін до реєстраційного посвідчення:
- наукові висновки і обґрунтування рекомендації внесення змін до умов видачі реєстраційного посвідчення;
- для централізовано зареєстрованих лікарських засобів, переглянуту інформацію про лікарський засіб та, якщо можливо, умови, що встановлюються для заявника (власника реєстраційного посвідчення) та, якщо можливо, умови або обмеження, що встановлюються для країн ЄС для безпечного та ефективного використання лікарського засобу відповідно до положення, що надане у ст. 127а Директиви 2001/83/ЄС [1];
- для національно зареєстрованих лікарських засобів, включаючи ті, що зареєстровані за процедурою взаємного визнання та децентралізованою процедурою, додаток, у якому вказані нові попередження з безпеки та основні рекомендації з мінімізації ризиків, що мають бути включені у відповідні розділи інформації про лікарський засіб, якщо необхідно;
- у випадку думки СНМР призупинити дію реєстраційного(их) посвідчення(ь): наукові висновки та причини для призупинення дії реєстраційного(их) посвідчення(ь) та умови поновлення їх дії;
- у випадку думки СНМР відкликати реєстраційне(і) посвідчення, науково обґрунтовані висновки та причини відкликання;
- суперечливі позиції, якщо такі є.
Після затвердження EMA повинно надіслати думку СНМР разом з її додатками та доповненнями Європейській Комісії, власнику(ам) реєстраційного(их) посвідчення(ь) та уповноваженим органам у країнах ЄС.
Висновки та рекомендації кінцевої оцінки публікуються на Європейському вебпорталі лікарських засобів (розділ VII.С.7.).
Заходи після думки СНМР — централізовано зареєстровані лікарські засоби
Якщо думка СНМР передбачає необхідність змін до реєстраційного(их) посвідчення(ь), заявник (и) (власник(и) реєстраційного(их) посвідчення(ь)) на лікарські засоби, що зареєстровані за централізованою процедурою, мають надати переклади інформації про лікарський засіб та наукові висновки і обґрунтування щодо рекомендування зміни до умов видачі реєстраційного посвідчення усіма офіційними мовами ЄС відповідно до графіку підготовки перекладів, ухваленого СНМР.
Після отримання думки СНМР, у якій вказано, що необхідна регуляторна дія щодо відповідного реєстраційного посвідчення, Європейська Комісія повинна прийняти рішення, яке адресоване заявникам (власникам реєстраційних посвідчень), внести зміни, призупинити або відкликати реєстраційне(і) посвідчення на лікарський(і) засіб (засоби), що зареєстрований(і) за централізованою процедурою (ст. 107g(4b) Директиви 2001/83/ЄС [1]).
Після прийняття слід, щоб Європейська Комісія повідомила заявнику (власнику(ам) реєстраційного(их) посвідчення(ь)) про рішення щодо внесення змін до умов реєстраційного посвідчення на лікарські засоби, що зареєстровані за централізованою процедурою.
Заходи після думки СНМР — національно зареєстровані лікарські засоби, включаючи зареєстровані за процедурою взаємного визнання та децентралізованою процедурою
Після отримання думки СНМР, у якій зазначена необхідність регуляторної дії щодо відповідних реєстраційних посвідчень, Європейська Комісія має ухвалити рішення, адресоване уповноваженим органам в країнах ЄС щодо заходів, яких необхідно вжити (ст. 107g(а) Директиви 2001/83/ЄС [1]) стосовно національно зареєстрованих лікарських засобів, включаючи зареєстровані за процедурою взаємного визнання та децентралізованою процедурою.
Після отримання рішення від Європейської Комісії уповноважені органи у країнах ЄС мають вжити необхідних заходів для внесення зміни, призупинення або відкликання реєстраційного(их) посвідчення(ь) протягом 30 днів (ст. 107g(4) Директиви 2001/83/ЄС [1]).
VII.С.4.2.4. Єдина оцінка, що не включає централізовано зареєстрований лікарський засіб, що призводить до позиції СМDh
СМDh підтверджує отримання рекомендації та звіту з оцінки PRAC, у випадку необхідності будь-якої регуляторної дії, на своєму наступному засіданні після затвердження PRAC.
Протягом 30 днів після отримання СМDh має розглянути звіт з оцінки та рекомендацію PRAC та досягти позиції щодо збереження, внесення змін, призупинення, відкликання відповідного(их) реєстраційного(их) посвідчення(ь) (ст. 107g(1) Директиви 2001/83/ЄС [1]).
Усне пояснення для СМDh може проводитися на вимогу СМDh або заявника (ів) (власника(ів) реєстраційного(их) посвідчення(ь)) лише у випадку розбіжностей з рекомендацією PRAC, якщо СМDh розглядає можливість досягти позиції щодо призупинення або відкликання реєстраційного(их) посвідчення(ь), нового протипоказання, обмеження показання або зниження рекомендованої дози.
Позиція має містити наступне:
- кінцевий звіт з оцінки та рекомендацію, що затверджені PRAC;
- детальне пояснення наукових причин розбіжностей з рекомендацією PRAC, якщо можливо (ст. 107g(2) Директиви 2001/83/ЄС [1]);
- у випадку позиції СМDh внести зміни до реєстраційного(их) посвідчення(ь) наукові висновки та причини рекомендування зміни до умов видачі реєстраційного посвідчення та додаток, уякому вказані нові попередження з безпеки та основні рекомендації з мінімізації ризиків, що мають бути включені у відповідні розділи інформації про лікарський засіб, якщо необхідно;
- у випадку позиції СМDh призупинити реєстраційне(і) посвідчення мають бути надані наукові висновки разом з причинами призупинення та умовами відміни призупинення;
- у випадку позиції СМDh відкликати реєстраційне(і) посвідчення мають бути надані наукові висновки разом з причинами відкликання;
- суперечливі позиції членів СМDh, якщо такі є.
Остаточні висновки та рекомендації щодо оцінки мають бути опубліковані EMA на Європейському вебпорталі лікарських засобів (ст. 107l Директиви 2001/83/ЄС [1]) (розділ VII.С.7.).
Якщо позиція СМDh досягнута за взаємною згодою:
Узгоджена позиція, включаючи заходи, що необхідно вжити, записується головуючим у протоколі засідання СМDh після її узгодження.
Головуючий має надіслати узгоджену позицію СМDh (ст. 107g(2) Директиви 2001/83/ЄС [1]) та додатки до неї заявнику (власнику(ам) реєстраційного(их) посвідчення(ь)) та уповноваженим органам у країнах ЄС.
Після отримання позиції СМDh, в якій зазначається необхідність регуляторної дії щодо конкретного реєстраційного посвідчення, уповноважені органи країни ЄС мають ухвалити необхідні заходи для внесення зміни, призупинення або відкликання реєстраційного(их) посвідчення(ь) відповідно до графіку впровадження, визначеного в узгодженій позиції (ст. 107g(2) Директиви 2001/83/ЄС [1]).
У випадку ухвалення позиції СМDh, що вимагає зміни до умов реєстраційного(их) посвідчення(ь), заявник(и) (власник(и) реєстраційного(их) посвідчення(ь)) мають надати відповідну зміну згідно з графіком впровадження (ст. 107g(2) Директиви 2001/83/ЄС [1]), що додається до ухваленої позиції.
Якщо позиція СМDh досягнута більшістю голосів:
Позиція більшості щодо дії, яку слід виконати, записується головуючим у протоколі засідання СМDh, на якому вона ухвалена.
Позиція більшості СМDh, разом з додатками та доповненнями до неї, включаючи переклади всіма офіційними мовами ЄС, якщо це є необхідним, мають направлятися до Європейської Комісії (ст. 107g(2) Директиви 2001/83/ЄС [1]). Позиція СМDh також має направлятися до уповноважених органів країн ЄС.
Після отримання позиції СМDh, у якій зазначена необхідність у регуляторній дії стосовно відповідного реєстраційного посвідчення, Європейська Комісія має ухвалити рішення (ст. 107g(2) Директиви 2001/83/ЄС [1]), адресоване уповноваженим органам країн ЄС з метою внесення ними змін, призупинення або відкликання реєстраційного(их) посвідчення(ь) на лікарський(і) засіб (засоби), що зареєстрований(і) за національною процедурою, що належать заявникам (власникам реєстраційних посвідчень).
Після отримання рішення від Європейської Комісії уповноважені органи країн ЄС мають вжити необхідних заходів для підтримки, внесення зміни, призупинення або відкликання реєстраційного(их) посвідчення(ь) протягом 30 днів (ст. 107g(2) Директиви 2001/83/ЄС [1]).
VII.С.4.3. Взаємозв’язок між регулярно оновлюваним звітом з безпеки та планом управління ризиками
Взаємозв’язок між ПУР та РОЗБ описується в модулі V, а у розділі VII.С.4.3.1 надається опис спільних модулів для ПУР та РОЗБ.
Під час підготовки РОЗБ заявник (власник реєстраційного посвідчення) повинен оцінювати, чи важливі виявлені або потенційні ризики, що розглядаються в РОЗБ, та чи необхідно оновлювати ПУР. У таких випадках в оновлений ПУР включається нова важлива інформація з безпеки, і він подається одночасно з РОЗБ та оцінюється разом з ним відповідно до графіку оцінки РОЗБ, як описано вище.
Якщо під час оцінки РОЗБ національними уповноваженими органами в країнах ЄС виявлені важливі питання безпеки і при цьому не подавався ПУР чи його оновлена версія, необхідно рекомендувати протягом вказаного періоду часу подати оновлений або новий ПУР.
VII.С.4.3.1. Регулярно оновлюваний звіт з безпеки та план управління ризиками — спільні модулі
Запропоновані модульні формати для РОЗБ та ПУР мають на меті запобігти дублюванню та сприяти гнучкості шляхом використання загальних розділів РОЗБ/ПУР взаємозамінно у обох документах.
Загальні розділи вищезазначених документів визначені в таблиці VII.1.
Таблиця VII.1. Спільні розділи для регулярного звіту з безпеки та плану управління ризиками
|
Розділ РОЗБ |
Розділ ПУР |
|
Розділ 3 — «Заходи з безпеки, вжиті протягом звітного періоду» |
Частина II, модуль SV CVN — «Післяреєстраційний досвід», розділ «Заходи з безпеки, вжиті регуляторними органами та/або заявником (власниками реєстраційних посвідчень) з міркувань безпеки» |
|
Підрозділ 5.2 — «Кумулятивна та інтервальна експозиція пацієнтів у післяреєстраційний період» |
Частина II, модуль SV CVN — «Післяреєстраційний досвід», розділ «Експозиція у післяреєстраційний період (за винятком досліджень)» |
|
Підрозділ 16.1 — «Резюме проблем безпеки» |
Частина II, модуль SVIII CVIIIN — «Резюме проблем безпеки» (включене у версію ПУР, що була чинною на початок звітного періоду РОЗБ) |
|
Підрозділ 16.4 — «Характеристика ризиків» |
Частина II, модуль SVII CVIIN — «Ідентифіковані та потенційні ризики» |
|
Підрозділ 16.5 — «Ефективність заходів з мінімізації ризиків (якщо застосовано)» |
Частина V — «Заходи з мінімізації ризиків», розділ «Ефективності заходів з мінімізації ризиків» |
VII.С.5. Особливі вимоги ЄС та УкраїниN до регулярно оновлюваного звіту з безпеки
Наукова оцінка співвідношення користі/ризику лікарського засобу, включена до РОЗБ та детально описана в розділі VII.В.5., повинна базуватися на всіх доступних даних, включаючи дані клінічних випробувань для незареєстрованих показань та популяцій відповідно до положень у ст. 107b Директиви 2001/83/ЄС [1] та ст. 34(1) ІП 520/2012 [6], ПорядкуN [2].
Особливі вимоги ЄС, УкраїниN слід включити в локальний додаток до РОЗБ.
VII.С.5.1. Локальний додаток регулярно оновлюваного звіту з безпеки, підрозділ «Запропонована інформація про лікарський засіб»
Оцінка необхідності внесення змін в інформацію про лікарський засіб включена у процедуру оцінки РОЗБ в ЄС та УкраїніN. Слід, щоб регуляторна думка/позиція включала рекомендації щодо оновлення інформації про лікарський засіб, якщо необхідно. Заявникам (власникам реєстраційних посвідчень) слід надати необхідну підтримуючу документацію та посилання в рамках РОЗБ або в цьому додатку для сприяння цьому.
Заявник (власник реєстраційного посвідчення) повинен розглянути у РОЗБ вплив на реєстраційне посвідчення представлених у РОЗБ даних та оцінок. На підставі оцінки узагальнених даних з безпеки та аналізу співвідношення користі/ризику, заявник (власник реєстраційного посвідчення) повинен у РОЗБ зробити висновки щодо необхідності змін та/або дій, включаючи наслідки для затвердженої короткої характеристики/інструкції для медичного застосуванняN лікарського засобу, на який подається РОЗБ (ст. 34(5) ІП 520/2012 [6], положення Порядку [2]N).
У цьому підрозділі заявник (власник реєстраційного посвідчення) повинен на основі вищезазначеної оцінки надати пропозиції до інформації про лікарський засіб (коротка характеристика/інструкції для медичного застосуванняN лікарського засобу та листок-вкладка). Пропозиції повинні базуватися на всіх зареєстрованих в ЄС, Україні показаннях.
Необхідно надати версію запропонованої короткої характеристики/інструкції для медичного застосуванняN лікарського засобу та листка-вкладки з відслідковуванням змін, що ґрунтуються на оцінці і висновках РОЗБ. Для лікарських засобів, зареєстрованих за централізованою процедурою, запропоновану інформацію про лікарський засіб слід представити в модулі 1.3.1 досьє у форматі CTD.
Усі діючі на момент закриття бази даних короткі характеристики/інструкції для медичного застосуванняN лікарських засобів та листки-вкладки, що включені до РОЗБ, необхідно переглянути з тим, щоб гарантувати, що в них відображена відповідна інформація, що ґрунтується на кумулятивних даних, включених та проаналізованих у РОЗБ.
Зміни до інформації про лікарські засоби не повинні відкладатися або затримуватися до подання РОЗБ, а зміни, не пов’язані з представленою у РОЗБ інформацією, не повинні пропонуватися в рамках процедури РОЗБ. Заявники (власники реєстраційних посвідчень) зобов’язані подавати заяви на зміни відповідно до Регламенту (ЄС) №1234/2008 [8], положень Порядку проведення експертизи реєстраційних матеріалів [7], положень Порядку [2]N щодо змін до реєстраційного посвідчення.
У цьому розділі потрібно надати короткий опис поточних процедур (наприклад зміни) щодо оновлення інформації про лікарські засоби.
VII.С.5.2. Локальний додаток до регулярно оновлюваного звіту з безпеки, підрозділ «Запропоновані додаткові заходи з фармаконагляду та мінімізації ризиків»
Зважаючи на положення, встановлені в ст. 34(5) ІП 520/2012 [6] у цьому підрозділі слід надати пропозиції щодо додаткових дій з фармаконагляду та мінімізації ризиків на підставі висновків та дій, описаних у РОЗБ, включно із зазначенням наміру надати ПУР чи оновлений план управління ризиками, якщо необхідно.
VII.С.5.3. Локальний додаток до регулярно оновлюваного звіту з безпеки, підрозділ «Резюме поточних проблем з безпеки»
З метою підтвердження інформації, наданої у розділі 16.1 «Резюме проблем безпеки» РОЗБ (див. розділ VII.В.5.16.1), у цей підрозділ РОЗБ потрібно включити таблицю 1.10 (відповідно до поточної форми ПУР) «Резюме — Поточні проблеми з безпеки» із версії ПУР, дійсної на початок звітного періоду РОЗБ (див. модуль V).
VII.С.5.4. Локальний додаток до регулярно оновлюваного звіту з безпеки, підрозділ «Повідомлення про результати післяреєстраційних досліджень з безпеки»
У РОЗБ необхідно надати результати як інтервенційних, так і неінтервенційних ПДБЛЗ (PASS) (для подальших інструкцій див. модуль VIII ННПФ). Оскільки заявнику (власнику реєстраційного посвідчення) необхідно негайно повідомляти уповноважені органи в країнах ЄС та в УкраїніN про будь-яку нову інформацію, що може вплинути на співвідношення ризику/користі, у розділах 7 та 8 РОЗБ він повинен надати повну інформацію про результати всіх ПДБЛЗ, як інтервенційних, так і неінтервенційних.
Заключні звіти досліджень, завершених у звітний період, основною метою яких було виявлення, характеристика чи кількісне визначення зміни профілю безпеки, підтвердження профілю безпеки лікарського засобу чи визначення ефективності заходів управління ризиками, необхідно надати у вигляді додатку до РОЗБ. Якщо впродовж звітного періоду проведення таких досліджень призупинялося, то слід пояснити причини їх припинення.
Якщо у дослідженні було виявлено важливу проблему з безпеки, незалежно від того, чи вона була виявлена шляхом попередньо вказаних методів, і чи це ПДБЛЗ, заявник (власник реєстраційного посвідчення) і, зокрема, УОВФ/КОВФN, повинні негайно повідомити про це відповідний уповноважений орган в країнах ЄС та в УкраїніN.
РОЗБ не повинні використовуватися як засіб первинної комунікації для подання заключних звітів дослідження уповноваженим органам в країнах ЄС та в УкраїніN або для повідомлення будь-якої нової інформації, що може вплинути на оцінку співвідношення користі/ризику.
VII.С.5.5. Локальний додаток до регулярно оновлюваного звіту з безпеки, підрозділ «Ефективність мінімізації ризиків»
Заходи з мінімізації ризиків — це втручання в охорону здоров’я з метою запобігання виникненню побічних реакцій, пов’язаних із застосуванням лікарського засобу, або для зниження тяжкості їх прояву у випадку виникнення. Результативність дій з мінімізації ризиків у досягненні цих цілей необхідно оцінювати протягом усього життєвого циклу лікарського засобу з метою забезпечення зведення до мінімуму тяжкості побічних реакцій, а, отже, — оптимізації загального профілю користі/ризику. Відповідно до розділу VII.В.5.16.5., у РОЗБ необхідно відобразити оцінку досвіду застосування лікарського засобу у світі. Що ж стосується ефективності заходів з мінімізації ризиків у будь-якій країні чи регіоні, то особливий інтерес представляють ті з них, що можуть бути застосовані в інших країнах.
У цьому підрозділі РОЗБ необхідно надати оцінку ефективності рутинних та/або додаткових заходів з мінімізації ризиків. При цьому потрібно врахувати й зобов’язання щодо імплементації заходів з мінімізації ризиків, що запроваджені на вимогу регуляторних/уповноваженихN органів, додатково до загальних вимог моніторингу безпеки та співвідношення користь/ризик. Слід надати результати всіх досліджень, що проводилися з метою оцінки впливу чи інших формальних оцінок заходів з мінімізації ризиків в ЄС та в УкраїніN. У рамках цієї важливої оцінки заявник (власник реєстраційного посвідчення) повинен відмітити фактори, що впливають на результативність чи недостатність заходів з мінімізації ризиків. Якщо якась стратегія мінімізації ризиків виявляється неефективною, то необхідно вживати альтернативних заходів. У деяких випадках можна дійти висновку, що за допомогою заходів з мінімізації не вдасться контролювати ризики до ступеня, коли можливо забезпечити позитивне співвідношення користі/ризику, і лікарський засіб або необхідно відкликати з ринку, або обмежити його застосування тільки тими пацієнтами, у яких користь переважає над ризиками. Більш детальна настанова з моніторингу ефективності заходів з мінімізації ризиків міститься в модулі ХVI.ННПФ. У принципі заявник (власник реєстраційного посвідчення) у своїй оцінці повинен розрізняти успішність запровадження заходів та досягнення запланованого результату.
VII.С.6. Системи якості та системи управління записами для регулярно оновлюваного звіту з безпеки в мережі ЄС, УкраїніN
VII.С.6.1. Системи якості та системи управління записами на рівні заявника (власника реєстраційного посвідчення)
Заявник (власник реєстраційного посвідчення) повинен мати в системі якості певні процедури та процеси для забезпечення оновлення інформації про лікарський засіб відповідно до наукових знань, враховуючи оцінки та рекомендації, опубліковані на Європейському вебпорталі лікарських засобів, вебсайті МОЗ, ЦентруN та на основі власного безперервного моніторингу інформації, опублікованої на Європейському вебпорталі лікарських засобів, вебсайті МОЗ, ЦентруN (ст. 11(1)(f) ІП 520/2012 [6]).
З метою забезпечення відповідності вимогам подання РОЗБ щодо своїх лікарських засобів заявники (власники реєстраційних посвідчень) зобов’язані регулярно перевіряти перелік референтних дат ЄС та частоту подання, які опубліковані на Європейському вебпорталі лікарських засобів, перелік строків подання РОЗБ відповідно до додатку 10 Порядку [2]N (див. розділ VII.С.3.).
Слід створити системи планування підготовки РОЗБ відповідно до:
- переліку референтних дат ЄС та частоти подання РОЗБ, переліку строків подання РОЗБ відповідно до додатку 10 Порядку [2]N; або
- умов, викладених у реєстраційному посвідченні; або
- стандартного графіку подання РОЗБ, який встановлено відповідно до ст. 107с(2) Директиви 2001/83/ЄС [1] для лікарських засобів, що зареєстровані до 2 липня 2012р. (для централізовано зареєстрованих лікарських засобів) та 21 липня 2012р. (для національно зареєстрованих лікарських засобів), якщо необхідно (без будь-яких умов у їх реєстраційному посвідченні або які не включені в перелік референтних дат ЄС та частоту подання регулярного звіту з безпеки, та на які не впливає часткова відміна, яка встановлена в ст. 107b(3) Директиви 2001/83/ЄС [1]), стандартного графіку подання РОЗБ, який встановлено Порядком [2] для лікарських засобів (без будь-яких умов у їх реєстраційному посвідченні або які не включені до переліку строків подання РОЗБ відповідно до додатку 10 Порядку [2]N; або
- спеціальних запитів щодо РОЗБ уповноваженого органу в країні ЄС/УкраїніN або EMA.
Для лікарських засобів, для яких не вимагається подання ПУР, з метою підготовки РОЗБ заявник (власник реєстраційних посвідчень) повинен вести файл із специфікацією важливих виявлених ризиків, важливих потенційних ризиків та відсутньою інформацію.
Заявник (власник реєстраційного посвідчення) повинен мати процедури для дотримання вимог, встановлених EMA для РОЗБ.
УОВФ/КОВФN відповідає за встановлення і підтримку системи фармаконагляду (ст. 104(е) Директиви 2001/83/ЄС [1], положення Порядку [2]N), а тому повинна гарантувати, що наявна система фармаконагляду відповідає вимогам, встановленим для підготовки та подання РОЗБ. Стосовно лікарських засобів, охоплених системою фармаконагляду, специфічні додаткові обов’язки УОВФ/КОВФN у зв’язку з РОЗБ включають:
- забезпечення необхідної якості, включаючи правильність та повноту даних, що подаються в РОЗБ;
- забезпечення повної відповіді у відповідні строки та в рамках узгодженої процедури (наприклад наступний РОЗБ) на будь-який запит уповноваженого органу в країні ЄС/УкраїніN та EMA стосовно РОЗБ;
- інформованість щодо РОЗБ і висновків звіту з оцінки, рекомендацій PRAC, думки CHМР та позиції CMDh та рішень Європейської Комісії, МОЗ, ЦентруN з метою гарантування вжиття відповідних заходів.
Зазначені в модулі I строки зберігання документації, пов’язаної з лікарськими засобами, застосовуються і до РОЗБ та документів, що використовуються при його створенні, включаючи документи, пов’язані з вжитими заходами з міркувань безпеки, клінічними випробуваннями та післяреєстраційними дослідженнями, відповідною інформацією щодо користі лікарського засобу, та документи, що використовуються для розрахунку експозиції пацієнтів.
VII.С.6.2. Системи якості та системи управління записами на рівні EMA
Слід, щоб застосування системи якості EMA/ЦентруN (див. модуль I ННПФ) підтримувало дотримання вимог EMA при виконанні своїх завдань та обов’язків для управління процедурами РОЗБ та єдиною оцінкою ЄС/УкраїниN.
EMA/ЦентруN слід мати розроблений процес для технічної валідації повноти подання РОЗБ.
Переліки та зведені таблиці з бази даних EudraVigilance/АІСФN, що використовуються на підтримку оцінки РОЗБ, будуть створені із застосуванням звітів, отриманих за допомогою системи аналізу даних EudraVigilance/АІСФN.
Ефективна комунікація і документообіг РОЗБ та пов’язані з ними документи є критичними для успішного завершення процедури; отже, повинні існувати процеси, що забезпечують документообіг між EMA/ЦентромN, заявники (власниками реєстраційних посвідчень), Комісією та уповноваженими органами в країнах ЄС. Де доцільно, потрібно запровадити процедури необхідні для перевірки якості з метою видалення будь-якої персональної чи комерційної інформації конфіденційного характеру.
Створені письмові процедури повинні відображати послідовність дій, що потрібно виконувати для підтримки переліку референтних дат ЄС та частоти подання регулярних звітів з безпеки, які опубліковані EMA на Європейському вебпорталі лікарських засобів/перелік строків подання РОЗБ відповідно до додатку 10 до Порядку [2] в УкраїніN (див. розділ VII.С.3.).
До публікації резюме звітів з оцінки РОЗБ на Європейському вебпорталі лікарських засобів (див. розділ VII.С.7.) відповідний персонал EMA повинен дотримуватися процедур, що встановлені для вебпублікації документів, що створені EMA або уповноваженими органами в країнах ЄС.
Усі записи, пов’язані з РОЗБ, що створені персоналом, експертами або консультантами EMA, є власністю EMA, а всі отримані РОЗБ та відповідні документи зберігаються в EMA. Обидва типи записів, що стосуються РОЗБ (створені або отримані EMA), підлягають загальному контролю EMA шляхом їх зберігання в архіві РОЗБ, створеному відповідно до положень, що викладені у ст. 25а Регламенту (ЄC) №726/2004 [5].
Політика EMA щодо управління записами (ЕМЕА/590678/2007)73 надає основу для узгодженої, сталої та ефективної програми управління записами; вона була розроблена відповідно до загальновизнаних міжнародних стандартів ведення записів, «ISO 15489-1:2001 Інформація та документація — управління записами74». Відповідно до класифікації записів, встановленої політикою EMA, РОЗБ буде вважатися записами, що мають ділове, правове, доказове, дослідницьке/історичне значення.
Тривалість зберігання документації щодо лікарських засобів, що вказана у модулі I ННПФ стосується документів, пов’язаних з системою РОЗБ (наприклад стандартні операційні процедури), та документів, що стосуються РОЗБ (наприклад, РОЗБ, звіти з оцінки, дані, отримані з бази даних EudraVigilance або інші дані, що використовуються при проведенні оцінки РОЗБ).
VII.С.6.3. Системи якості та системи управління записами на рівні уповноважених органів в країнах ЄС
Кожний уповноважений орган у країнах ЄС повинен мати систему фармаконагляду (ст. 101 Директиви 2001/83/ЄС [1]) для контролю лікарських засобів та для отримання та оцінки всіх даних з фармаконагляду, включаючи РОЗБ. У рамках виконання цих завдань стосовно РОЗБ додатково до системи фармаконагляду національним уповноваженим органам у країнах ЄС слід застосовувати систему якості (див. модуль I НППФ).
Уповноваженим органам у країнах ЄС слід проводити моніторинг заявників (власників реєстраційних посвідчень) на дотримання регуляторних зобов’язань щодо РОЗБ. Додатково уповноваженим органам слід обмінюватися інформацією у випадках недотримання вимог та вживати відповідних регуляторних дій, якщо необхідно.
Оцінка РОЗБ на рівні ЄС не передбачена для виключно національно зареєстрованих лікарських засобів, що зареєстровані лише в одній країні ЄС; тому національному уповноваженому органу у країні ЄС, де зареєстровано лікарський засіб, слід мати процедури для оцінки РОЗБ стосовно таких лікарських засобів.
Слід, щоб процедури, які встановлені національними уповноваженими органами у країнах ЄС для виконання єдиної оцінки ЄС РОЗБ, відповідали процедурам, що встановлені EMA для координації оцінки РОЗБ у регуляторній системі ЄС (див. розділ VII.С.4.). Цими процедурами слід встановити ефективний обмін інформацією в регуляторній системі ЄС та дії, які повинні вживатися відносно внесення зміни, припинення або відкликання реєстраційного посвідчення після рекомендації PRAC, думки СНМР, позиції СМDh та рішення Європейської Комісії, якщо необхідно.
Національні уповноважені органи у країнах ЄС повинні дотримуватися процедур, що встановлені EMA для використання архіву РОЗБ для підтримки єдиної оцінки.
Якщо завдання стосовно процедур РОЗБ делеговані третій стороні, національним уповноваженими органам у країнах ЄС слід гарантувати, що вони підлягають системі якості відповідно до обов’язків, що передбачені Європейським законодавством.
Час зберігання документації, що стосується лікарського засобу, у модулі I ННПФ, також застосовується до відповідних документів системи РОЗБ (наприклад, стандартні операційні процедури) та документів, що стосуються РОЗБ (наприклад, регулярні звіти з безпеки, звіти з оцінки, дані, які отримані з бази даних EudraVigilance, або інші дані, що використовуються для підтримки оцінки РОЗБ).
VII.С.7. Прозорість
VII.С.7.1. Публікація документів, що стосуються регулярних звітів з безпеки, на Європейському та національному вебпорталі лікарських засобів
На Європейському вебпорталі лікарських засобів повинні публікуватися наступні документи (ст. 107l Директиви 2001/83/ЄС [1], ст. 26(g) Регламенту (ЄC) № 726/2004 [5]):
- перелік референтних дат ЄС та частота подання РОЗБ (див. розділ VII.С.3);
- заключні висновки оцінки схвалених звітів з оцінки;
- рекомендації PRAC, включаючи відповідні додатки;
- позиція СМDh, включаючи відповідні додатки та, якщо необхідно, детальне пояснення наукових причин будь-яких розбіжностей з рекомендаціями PRAC;
- думка СНМР, включаючи відповідні додатки та, якщо необхідно, детальне пояснення наукових причин будь-яких розбіжностей з рекомендаціями PRAC;
- рішення Європейської Комісії.
Версія та дата публікації відображаються в кожному документі оскільки вони визначають випуск рекомендації PRAC, думки СНМР, позиції СМDh та рішення Європейської Комісії в певний момент часу.
Зв’язок між Європейським вебпорталом лікарських засобів та національними вебпорталами лікарських засобів слід зробити можливим та відповідним.
Будь-які персональні або конфіденційні дані, що зроблені загальнодоступними EMA або уповноваженими органами у країнах ЄС та УкраїніN, як вказано у параграфах 2 та 3 ст. 106а Директиви 2001/83/ЄС [1], повинні бути видалені, якщо не вважаються необхідними для захисту здоров’я населення (ст. 106а(4) Директиви 2001/83/ЄС [1]).
VII.С.8. Перереєстрація лікарських засобів
Перереєстрація лікарських засобів повинна відбуватися через 5 років на основі повторної оцінки співвідношення користі/ризику з метою подовження строку розміщення лікарського засобу на ринку. Перереєстрація не залежить від того, чи призупинено реєстраційне посвідчення. Подальші детальні дані щодо процедури та вимог до документації можна знайти у поточних версіях «Настанови з обробки заяв на поновлення ліцензії за централізованою процедурою» (ЕМЕА/СНМР/2990/00) для централізованих лікарських засобів та «Настанови щодо найкращих практик СМDh щодо обробки заяв на поновлення ліцензії за процедурою взаємного визнання (MRP)/децентралізованою процедурою (DCP)» (СМDh/004/2005) для інших лікарських засобів, Порядку проведення експертизи [7]N.
У рамках заяви на перереєстрацію не надаються РОЗБ, додаткові звіти та короткі зведені звіти. У клінічний огляд необхідно включити додаток, що містить відповідні розділи повторної оцінки співвідношення користі/ризику лікарського засобу. Ці розділи визначені у вищезгаданих настановах щодо перереєстрації. Заявникам (власникам реєстраційних посвідчень) рекомендується використовувати цей модуль VII ННПФ у якості посібника при підготовці Доповнення до огляду клінічних даних.
Після подання заяви на перереєстрацію можна проконсультуватися з PRAC стосовно лікарських засобів, що зареєстровані шляхом централізованої процедури, відносно питань безпеки. Для національно зареєстрованих лікарських засобів, включаючи ті, що зареєстровані шляхом процедури взаємного визнання або децентралізованої процедури, можна також проконсультуватися з PRAC після запиту уповноваженого органу в країні ЄС з приводу питань безпеки.
Умовні реєстраційні посвідчення слід поновлювати щорічно (ст. 14(7) Регламенту (ЄC) №726/2004) [5]. Подальші дані щодо процедур та документації, яка повинна подаватися, можна знайти в «Настанові з наукового застосування та практичних заходів, які необхідні для виконання Постанови Комісії (ЄС) № 507/2006 щодо умовного реєстраційного посвідчення для лікарських засобів для людини, що є в рамках Регламенту (ЄС) № 726/2004 [5]» (ЕМЕА/509951/2006).
VII.С.9. Перехідні та тимчасові положення
VII.С.9.1. Подання та доступність документів до створення архіву EMA
EMA повинно разом з уповноваженими органами у країнах ЄС та Європейською Комісією створити та підтримувати архів для РОЗБ та відповідних звітів з оцінки так, щоб вони були повністю та постійно доступними Європейській Комісії, уповноваженим органам у країнах ЄС, PRAC, СНМР та СМDh (ст. 25а Регламенту (ЄC) №726/2004 [5]).
Архів повинен підлягати незалежному аудиту до оголошення функціональних можливостей радою директорів EMA (ст. 25а Регламенту (ЄC) № 726/2004 [5]).
Як зазначено в перехідних положеннях, представлених у ст. 2(7) Директиви 2010/84/ЄС [3], до того, як EMA зможе гарантувати функціональні можливості архіву, заявники (власники реєстраційних посвідчень), до кола обов’язків яких входить подання РОЗБ (незалежно від того, чи лікарський засіб зареєстровано в одній або більше країнах ЄС та незалежно від того, чи є діюча речовина (АФІN) або комбінація діючих речовин (АФІN) у переліку референтних дат ЄС), мають подавати РОЗБ до всіх уповноважених органів країн ЄС, у яких дані лікарські засоби є зареєстрованими. Для діючих речовин (АФІN) або комбінацій діючих речовин (АФІN), що підлягають єдиній оцінці в ЄС, або для яких була встановлена референтна дата ЄС, РОЗБ слід також надсилати до EMA.
Вимоги уповноважених органів країн ЄС до подання РОЗБ під час даного перехідного періоду опубліковані на вебсайті EMA75.
Через 12 місяців після встановлення функціональних можливостей архіву та оголошення про це EMA, заявники (власники реєстраційних посвідчень) мають подавати РОЗБ до EMA в електронному вигляді, незалежно від процедури реєстрації лікарського засобу (ст. 107b(1) Директиви 2001/83/ЄС [1]). Уповноважені органи країн ЄС повинні гарантувати, що це зобов’язання виконується відповідно до вимоги (ст. 2(7) Директиви 2001/83/ЄС [1]).
Коли структурований електронний формат «еРОЗБ» на основі узгодженого в настанові ICH-E2C(R2) [28] змісту стає доступним, заявники (власники реєстраційних посвідчень) матимуть можливість подавати РОЗБ та відповідні документи автоматично електронним шляхом.
- До тих пір, поки архів не створено, наступні документи слід розповсюджувати наступним чином: попередній звіт з оцінки, підготовлений доповідачем PRAC/країною ЄС протягом 60 днів з моменту початку процедури буде направлятися EMA та членам PRAC за допомогою відповідної поштової скриньки. EMA буде надсилати звіт відповідному(им) заявнику(ам) (власнику(ам) реєстраційного(их) посвідчення(ь));
- члени PRAC повинні розповсюджувати свої коментарі щодо попереднього звіту з оцінки від доповідача PRAC/країни ЄС за допомогою відповідної поштової скриньки до 90 дня;
- коментарі, що подані заявником(ами) (власником(ами) реєстраційного(их) посвідчення(ь)) на 90-й день після попереднього звіту з оцінки доповідача PRAC/країни ЄС, повинні подаватися до EMA, доповідачу PRAC та всім членами PRAC відповідно до інструкцій щодо подання, що опубліковані EMA;
- оновлений звіт з оцінки від доповідача PRAC/країни ЄС, створений протягом 15 днів (тобто на 105-й день), буде направлений EMA та членам PRAC за допомогою відповідної поштової скриньки. EMA повинно направляти оновлені звіти з оцінки доповідача PRAC/країни ЄС відповідним заявникам (власникам реєстраційних посвідчень).
Після схвалення звіту EMA має надсилати думку СНМР разом з додатками та доповненнями до Європейської Комісії, заявнику(ам) (власнику(ам) реєстраційного(их) посвідчення(ь)) та уповноваженим органам в країнах ЄС через захищені канали електронної пошти, поки не буде запроваджений архів.
VII.С.9.2. Системи якості та системи управління записами на рівні уповноважених органів країн ЄС
Особливу увагу слід приділити управлінню РОЗБ, що подаються відповідним уповноваженим органам країн ЄС, поки EMA не зможе забезпечити функціональні можливості архіву РОЗБ та протягом 12 місяців після запровадження архіву відповідно до перехідних положень.
VII.С.9.3. Публікація переліку референтних дат ЄС/переліку строків подання РОЗБ в УкраїніN та початок єдиної процедури оцінки регулярно оновлюваних звітів з безпеки ЄС/УкраїниN
Як зазначено в розділі VII.С.3.6, перелік референтних дат ЄС/перелік строків подання РОЗБ в УкраїніN та частота подання РОЗБ будуть публікуватися на Європейському вебпорталі лікарських засобів/затверджуватися наказом МОЗN; проте процедура єдиної оцінки ЄС/для діючих речовин (АФІN), що включені лише до національно зареєстрованих лікарських засобів, яка детально представлена в розділах VII.С.4.2.2, та VII.С.4.2.4, буде відкладена до появи фінансування. В Україні процедура єдиної оцінки РОЗБ для АФІ/комбінацій АФІ лікарських засобів, що зареєстровані в Україні буде здійснюватися відповідно до переліку строків подання РОЗБ згідно з додатком 10 Порядку [2]N.
VII. Додатки
VII. Додаток 1. Приклади таблиць для оцінки впливу та даних про побічні явища/реакції
Заявники (власники реєстраційних посвідчень) за необхідності можуть змінювати ці приклади таблиць для відповідності певним ситуаціям.
Таблиця VII.2. Оцінка кумулятивного впливу у учасників клінічних випробувань
|
Лікування |
Кількість суб’єктів |
|
Лікарський засіб |
|
|
Компаратор |
|
|
Плацебо |
Оцінка кумулятивного впливу у учасників ґрунтується на даних експозиції завершених клінічних випробувань та схемах залучення/рандомізації поточних досліджень.
Таблиця VII.3. Оцінка кумулятивного впливу досліджуваного лікарського засобу у суб’єктів завершених клінічних випробувань, розподілених за віком та статтю
|
Кількість суб’єктів |
|||
|
Віковий діапазон |
Чоловіки |
Жінки |
Усього |
Дані завершених досліджень станом на (дата).
Таблиця VII.4. Оцінка кумулятивного впливу досліджуваного лікарського засобу у учасників завершених клінічних випробувань, розподілених за расовою/етнічною групою
|
Расова/етнічна група |
Кількість учасників |
|
Азійська |
|
|
Негроїдна |
|
|
Європейська |
|
|
Інші |
|
|
Невідомо |
|
|
Усього |
Дані завершених досліджень станом на (дата).
Таблиця VII.5. Кумулятивна експозиція у післяреєстраційний період
|
Показання |
Стать |
Вік (Роки) |
Доза |
Форма випуску |
Регіон |
|||||||||||
|
Чоловіки |
Жінки |
2–≤16 |
>16–65 |
>65 |
Невідомо |
<40 |
≤40 |
Невідомо |
Внутрішньовенна |
Пероральна |
ЄС |
Японія |
Колумбія |
УкраїнаN США/Канада |
Інший |
|
|
Усього |
||||||||||||||||
|
Депресія |
||||||||||||||||
|
Мігрень |
||||||||||||||||
Таблиця VII.5. містить кумулятивні дані, отримані за день/місяць/рік впродовж дня/місяця/року, якщо такі дані є доступними.
Таблиця VII.6. Інтервальна експозиція (за звітний період)
|
Показання |
Стать |
Вік (Років) |
Доза |
Форма випуску |
Регіон |
|||||||||||
|
Чоловіки |
Жінки |
2–≤16 |
>16–65 |
>65 |
Невідомо |
<40 |
≤40 |
Невідомо |
Внутрішньовенна |
Пероральна |
ЄС |
Японія |
Колумбія |
УкраїнаN США/Канада |
Інший |
|
|
Депресія |
||||||||||||||||
|
Мігрень |
||||||||||||||||
Таблиця VII.6. Включає дані за певний період, отримані за день/місяць/рік впродовж дня/місяця/року.
Таблиця VII.7. Зведена таблиця серйозних побічних явищ з клінічних випробувань
|
Клас систем органів Вибраний термін |
Досліджуваний лікарський засіб |
Сліпе |
Активний компаратор |
Плацебо |
|
Порушення функції кровоносної та лімфатичної системи |
||||
|
Анемія |
||||
|
Некроз кісткового мозку |
||||
|
Кардіальні розлади |
||||
|
Тахікардія |
||||
|
Ішемічна кардіоміопатія |
Таблиця VII.8. Кількість побічних реакцій з післяреєстраційних джерел*
|
Клас систем органів (SOC) MedDRA Вибраний термін (РТ) |
Спонтанні повідомлення, у тому числі від уповноважених органів (різних країн) та з літератури |
Неінтервенційне післяреєстраційне дослідження та повідомлення з інших необхідних джерел** |
|||||
|
Серйозні |
Несерйозні |
Загалом спонтанних |
Серйозні |
||||
|
|
Інтервальні |
Кумулятивні |
Інтервальні |
Кумулятивні |
Кумулятивні |
Інтервальні |
Кумулятивні |
|
(SOC 1) |
|||||||
|
(РТ) |
|||||||
|
(РТ) |
|||||||
|
(РТ) |
|||||||
|
(SOC 2) |
|||||||
|
(РТ) |
|||||||
|
(РТ) |
|||||||
|
(РТ) |
|||||||
|
(РТ) |
|||||||
*Неінтервенційні післяреєстраційні дослідження, повідомлення з інших необхідних джерел та спонтанні повідомлення про окремі випадки (тобто повідомлення від працівників з медичною та фармацевтичною освітою, споживачів, компетентних органів (різних країн) та наукової літератури).
**Не включають даних інтервенційних клінічних випробувань.
VII. Додаток 2. Приклад зведеної таблиці поточних та закритих у звітний період сигналів з безпеки
Таблиця VII.9. Зведена таблиця, що представлена нижче, є уявним прикладом зведеної таблиці сигналів безпеки — поточних та закритих у звітний період
Звітний період: дд/ммм/рррр — дд/ммм/рррр
|
Назва сигналу |
Дата виявлення |
Статус (поточний чи закритий) |
Дата закриття (для закритих) |
Джерело сигналу |
Мета оцінки та резюме ключових даних |
Метод оцінки сигналу |
Вжиті чи заплановані заходи |
|
Інсульт |
ммм/рррр |
Поточний |
ммм/рррр |
Метааналіз (опубліковані дослідження) |
Статистично значиме підвищення частоти |
Огляд даних метааналізу і доступних даних |
Розглядаються |
|
Синдром Стівенса –Джонсона (ССД) |
ммм/рррр |
Закритий |
ммм/рррр |
Спонтанні повідомлення |
Висип уже ідентифікований ризик; ССД не виявлявся у дореєстраційних КД; 4 повідомлення впродовж 6 місяців після реєстрації; правдоподібний проміжок часу до початку розвитку і відсутні можливі альтернативні причини |
Цільове відстеження повідомлень з відвіданням однієї лікарні. Повний огляд випадків дерматологом, власником реєстраційного посвідчення та літературний пошук |
У довідкову інформацію з безпеки внесено попередження і застереження. Надіслано пряме повідомлення для спеціаліста-медика (DHPC). Аналіз ефективності запланований через 6 місяців після DHPC. Оновлення ПУР. |
Пояснювальні примітки:
Назва сигналу:
- коротка описова назва медичної концепції сигналу. Вона може видозмінюватися та уточнюватися в ході оцінки сигналу. Концепція та контекст можуть обмежуватися або не обмежуватися спеціальним(ними) терміном(ами) MedDRA, в залежності від джерела сигналу.
Дата виявлення:
- місяць та рік, коли заявнику (власнику реєстраційного посвідчення) стало відомо про сигнал.
Статус:
- поточний: сигнал, оцінка якого не завершилася на момент закриття бази даних РОЗБ. Слід зазначити очікувану дату завершення, якщо вона відома;
- закритий: сигнал, оцінка якого була завершена до закриття бази даних РОЗБ.
Примітка: новий сигнал, виявлений заявником (власником реєстраційного посвідчення) впродовж звітного періоду, може бути класифікований як закритий чи поточний в залежності від статусу оцінки сигналу на завершення звітного періоду РОЗБ.
Дата закриття:
– місяць та рік, коли оцінка сигналу була завершена.
Джерело сигналу:
- джерело даних або інформації, з яких походить сигнал. Приклади включають, але не обмежуються, спонтанними повідомленнями про побічні реакції, даними клінічних випробувань, науковою літературою, результатами неклінічних досліджень та інформаційними запитами чи розслідуваннями уповноважених органів (у всьому світі).
Причина оцінки та резюме основних даних:
- коротке резюме основних даних та обґрунтування подальшої оцінки.
Заходи, що вжиті або плануються:
- вкажіть, чи були вжиті або заплановані спеціальні заходи для всіх закритих сигналів, класифікованих як потенційні або виявлені ризики. Якщо стосовно нових або раніше виявлених сигналів, які оцінюються на момент закриття бази даних, були заплановані будь-які подальші заходи, слід надати їх перелік або залишити незаповнене місце для поточних сигналів.
VII. Додаток 3. Підходи до оцінки співвідношення користі/ризику лікарського засобу та добре відомі моделі оцінки співвідношення користі/ризику лікарського засобуN
Концепція оцінки співвідношення користі/ризику щодо лікарського засобу не є унікальною. Подібна практика вже існує в галузі екології, виробництва харчових добавок, хімічних речовин і навіть у хірургії.
Станом на сьогодні на підставі вже наявного досвіду експертами декількох робочих груп були зроблені рекомендації стосовно моделей, якими можуть користуватися виробники/заявники лікарських засобів, експерти регуляторних/експертних органів, лікарі та пацієнти з метою проведення аналізу співвідношення користі/ризику лікарського засобу.
Найбільш відомими та дієвими були ініціативи IV СIOMS групи «Користь/ризик для лікарських засобів, що маркетуються» (Benefit-Risk balance for marketed drugs: evaluating safety signals), Женева, 1998 р.; СНМР (ЕМА) «Звіт робочої групи щодо моделей та методів оцінки користі/ризику», 19 січня 2007 р. (ЕМЕА/СНМР/15404/2007); Переглянута версія 1, 31 серпня 2010 р. (ЕМА/549682/2010); спільного європейського проєкту PROTECT (Європейський консорціум з фармакоепідеміологічних досліджень застосування лікарських засобів (The Pharmacoepidemiological Research on Outcomes of Therapeutics by a European Consortium), координатором якого є EMA, спрямованого на усунення недоліків існуючих методик, що застосовуються в фармакоепідеміологіі і фармаконагляді, а також ініціативної групи Асоціації дослідницьких організацій і фармацевтичних виробників США PhRMA (Pharmaceutical Research and Manufacturers of America).
У цьому додатку представлені узагальнені результати роботи зазначених вище ініціатив та ті добре відомі моделі, що можуть бути використані для оцінки співвідношення користі/ризику лікарського засобу. Слід зауважити, що жоден з представлених методів оцінки не може вважатися повністю об’єктивним, проте Європейський Союз (ЄС) вимагає від виробників здійснювати «адекватний і постійний моніторинг і оцінку співвідношення користі/ризику у післяреєстраційний період з метою забезпечення мінімізації ризиків і максимізації користі лікарського засобу». Україна ж тільки починає переймати подібну практику.
Відповідно до наявних рекомендацій необхідно зібрати, оцінити і зіставити всі доступні наукові дані, включаючи результати експериментальних, клінічних і фармакоепідеміологічних досліджень, врахувати альтернативні методи терапії, а також думку фахівців практичної охорони здоров’я і пацієнтів. При цьому важливо оцінити повноту і якість наданої для аналізу інформації, оскільки неповна, недостовірна або суперечлива інформація може вводити в оману і приводити до неправильних висновків.
Проведення оцінки співвідношення користі/ризику лікарського засобу за допомогою комплексного/інтегрованого аналізу описано у розділах VІІ.В.5.13—VІІ.В.5.18 даного модулю.
Незалежно від того, яким методом буде здійснений аналіз співвідношення користі/ризику, експерти групи IV СIOMS рекомендують використовувати представлену нижче обґрунтовану стандартну схему оцінки, що включає такі розділи:
1) вступ:
- коротка специфікація/опис препарату, де потрібно описати:
- показання до застосування. У разі, якщо існують відмінності щодо показань до застосування у різних країнах, де маркетується лікарський засіб, це потрібно описати;
- ідентифікація одного або кількох альтернативних видів або методів лікування, включаючи хірургічні;
- дуже стислий опис підозрюваної або встановленої важливої/значної проблеми з безпеки.
2) оцінка користі:
- епідеміологія та опис перебігу цільової хвороби (хвороб);
- мета застосування лікарського засобу (лікування, профілактика тощо);
- резюме ефективності та загальні дані про переносимість у порівнянні з:
- іншими методами лікування;
- хірургічним лікуванням або іншими видами втручань;
- відсутністю лікування.
3) оцінка ризику:
- фоновий рівень ризику;
- вагомість доказів щодо підозрюваного ризику (сфера тощо);
- докладні дані про новий підозрюваний ризик та результати їх аналізу;
- ймовірні (правдоподібні) та можливі роз’яснення;
- запобіжність, передбачуваність і зворотність нового ризику;
- надання пояснень, наскільки ризик пов’язаний з альтернативними методами лікування чи відсутністю терапії;
- повний огляд профілю безпеки препарату, за застосування, якщо можливо, візуалізації ризиків у вигляді схем, діаграм тощо. При необхідності сфокусуватися, наприклад, на трьох найбільш поширених або на трьох найбільш серйозних з медичної точки зору побічних реакціях;
- описати профілі безпеки для альтернативних лікарських засобів або видів/методів лікування;
- коли це можливо, оцінити надмірну/незвичайну частоту відомих побічних реакцій, які є спільними для альтернативних видів лікування;
- у разі наявності значущих побічних реакцій, що невластиві препаратам порівняння, слід описати важливі відмінності між препаратами.
4) оцінка користі/ризику:
- представити підсумок переваг від застосування оцінюваного лікарського засобу у порівнянні з серйозністю цільової хвороби, метою і ефективністю лікування;
- представити підсумок домінуючих ризиків за використання таких оціночних критеріїв, як: серйозність/тяжкість, тривалість, частота ризику тощо;
- представити підсумок співвідношення користі/ризику лікарського засобу, кількісно або схематично, якщо це можливо, з урахуванням альтернативних методів лікування або відсутності лікування;
- представити резюме оцінки і висновок.
5) аналіз варіантів вирішення/мінімізації/усунення проблеми з безпеки:
- представити всі відповідні варіанти заходів щодо вирішення/мінімізації/усунення проблеми з безпеки;
- описати докази «за» та «проти», можливі наслідки (аналіз впливу) кожного із запропонованих заходів, у порівнянні з альтернативними методами лікування;
- якщо необхідно, описати плани стосовно вивчення проблеми з безпеки, включаючи проведення досліджень, за допомогою яких можливо було б отримати своєчасну і важливу додаткову інформацію;
- якщо доцільно, слід зазначити кількісні або якісні критерії будь-якого майбутнього доказу, що може сигналізувати про необхідність переоцінки співвідношення користі/ризику;
- представити пропозиції, в який спосіб можна проводити моніторинг та оцінку наслідків запропонованих заходів з вирішення/мінімізації/усунення проблеми з безпеки.
«Принцип трьох» в оцінці співвідношення користі/ризику
При застосуванні цієї моделі користь і ризики можна описувати в контексті інтенсивності (серйозності або тяжкості) хвороби, яка лікується оцінюваним лікарським засобом, або побічної реакції з урахуванням її тривалості, хронічності перебігу, частоти виникнення у популяції, яка лікується.
Модель «Принцип трьох» використовується для оцінки співвідношення користі/ризику лікарського засобу, навіть якщо показань більше одного. Цією методикою передбачена оцінка трьох елементів:
- захворювання;
- впливу лікарського засобу на захворювання;
- побічна реакція (слід вибрати найсерйознішу побічну реакцію (домінуючу) для кожного показання).
Серйозність, тривалість і частота — ті характеристики, за допомогою яких у даній методиці оцінюється захворювання, побічні реакції і вплив лікарського засобу на захворювання.
У свою чергу, перераховані вище характеристики можуть бути високого, середнього і низького ступеня вираженості, що є оціночними критеріями характеристик захворювання, побічної реакції і впливу лікарського засобу на захворювання.
Користь лікарського засобу можна оцінювати та описувати для хвороби-мішені, враховуючи її серйозність, хронічність перебігу (наприклад гостра форма, хронічна форма або тривалість хвороби), ступінь контролю або лікування.
При оцінці характеристики «частота» ризиків слід орієнтуватися на рекомендації, надані в доповіді CIOMS III про ризики, де зазначена така градація: ≥1% (поширені або часті); ≥1 на 1000 але ˂1%(непоширені або нечасті); ≥1на 10 000 але менше 1 на 1000 (поодинокі); ˂1 на 10 000 (рідкісні).
Згідно з рекомендаціями ВООЗ у разі, якщо побічні реакції відмічаються у понад 10% випадків, то побічні реакції вважаються дуже частими, від 1-10% — частими, від 0,1—1% — нечастими, від 0,01—0,1% — поодинокими/рідкісними, менше 0,01% — рідкісними.
З метою вербального опису властивостей користі та ризику представлене вище можна узагальнити у вигляді таблиці.
Таблиця VII.10. Оцінка характеристик користі та ризиків
|
Висока |
Середня |
Низька |
|
|
Серйозність |
Летальний наслідок |
Призводить до непрацездатності/інвалідизації |
Незручності |
|
Тривалість |
Постійно |
Персистуючий характер/тривало |
Тимчасово/короткотривало |
|
Частота |
Часто/поширена |
Нечасто/випадково |
Рідкісна |
Для оцінки серйозності, тривалості та частоти захворювання, впливу лікарського засобу на захворювання і побічну реакцію використовується бальна шкала:
- низька = 1;
- середня = 2;
- висока = 3;
- не впливає = 0.
За кожним з оцінюваних елементів слід спочатку надати описову частину, з посиланнями на джерела інформації, з якої логічно повинна випливати оцінка у балах у вигляді таблиці.
Таким чином, спочатку слід описати і заповнити три таблиці з оцінки захворювання/стану, впливу лікарського засобу на захворювання/стан та побічних реакцій лікарських засобів. Після цього надаються результати оцінки у зведеній таблиці, а також — її описова частина.
Описуючи та оцінюючи елементи даної моделі потрібно вказувати джерела інформації на які здійснюються посилання.
«Метод трьох» передбачає, що може бути оцінена не одна домінуюча побічна реакція, а 3 найбільш серйозні та/або 3 найбільш часті побічні реакції, характерні для оцінюваного лікарського засобу. При такому підході до оцінки побічної реакції кожну побічну реакцію слід оцінити за допомогою характеристик і оціночних критеріїв цієї методики і визначити середнє арифметичне для всіх цих побічних реакцій.
Наприклад, фуросемід при лікуванні гіпертонічної хвороби (ГХ) може спричинити виникнення туговухості (т), аритмії (а) і рабдоміолізу (р) як наслідку гіпокаліємії. Відповідно:
- серйозність туговухості може буде оцінена в 2 бали, тривалість — 3 бали і частота — 1 бал;
- серйозність аритмії може буде оцінена в 3 бали, тривалість — 2 бали і частота — 1 бал;
- серйозність рабдоміолізу може буде оцінена в 2 бали, тривалість — 3 бали і частота — 1 бал.
Склавши бали за оцінкою кожної побічної реакції під час визначення їх серйозності, тривалості та частоти, і розділивши отриманий результат на 3, оскільки мова йде про 3 побічні реакції, в результаті отримуємо 6 балів:
|
(2(т)+3(а)+2(р) + |
3(т)+2(а)+3(р) + |
1(т)+1(а)+1(р)) = |
18:3 = 6 |
|
серйозність |
тривалість |
частота |
При цьому підсумкова таблиця може виглядати таким чином:
Таблиця VII.11. Зведені дані з оцінки характеристик фуросеміду при лікуванні гіпертонічної хвороби
|
Захворювання — ГХ |
Вплив лікарського засобу (фуросемід) на захворювання |
Побічна реакція (т), (а), (р) |
|
|
Серйозність |
2 |
2 |
2 (т), 3(а), 2(р) |
|
Тривалість |
3 |
3 |
3 (т), 2(а), 3(р) |
|
Частота |
2 |
1 |
1 (т), 1(а), 1(р) |
|
Усього |
8 |
6 |
6 |
«Принцип трьох» — це модель, яка ґрунтується на концепції серйозності, тривалості та частотності стосовно показання до застосування при захворюванні, поліпшення перебігу захворювання за допомогою лікарського засобу та побічних реакцій, властивих лікарському засобу. Кожний параметр оцінюється як високий, середній або низький. Методологія ґрунтується на очевидному оцінюванні показників «рівня покращення, який досягається лікарським засобом» у порівнянні з показниками критеріїв «побічні реакції». Третій вимір, тобто серйозність, тривалість та частота захворювання, використовується для визначення, наскільки користь оцінюваного лікарського засобу повинна переважати ризики.
Кількісний підхід в оцінці співвідношення користі/ризику
Коли доступно достатньо даних про користь та ризики, кориснішою буде більш кількісно виражена версія описового методу, що був представлений попередньо («Принцип трьох»). Приклад, представлений нижче, ілюструє запропонований метод.
|
Лікарський засіб: |
А |
|
Ефективність |
40% — показник одужання (ефективності лікування) (хвороба, що призводить до інвалідності) |
|
Побічні реакції |
Шкірний висип (виникав у 20% пацієнтів, яких лікували; тривав близько 3 днів після припинення застосування лікарського засобу); розлад шлунку (виникав у 10% пацієнтів, яких лікували; тривав до 1 дня після припинення застосування лікарського засобу); діарея (виникала у 5% пацієнтів, яких лікували; у деяких пацієнтів тривала до 3 тижнів після припинення застосування лікарського засобу, у 05% випадків спричинювала виснаження пацієнтів); агранулоцитоз (виникав у 0,005% лікованих пацієнтів, з показником смертності 10%). |
Для ранжування властивостей користі та ризику можна вибрати зручну, але довільну градацію, подібну до градації, що застосовується у «Методі трьох», а саме: низький = 10, середній = 20, високий = 30.
Оцінка користі здійснюється за такою формулою:
користь = показник/коефіцієнт одужання · серйозність хвороби · хронічність/тривалість хвороби. Отже,
користь = 0,4 · 30 (хвороба, що призводить до інвалідності) · 20 (перехід з гострої форми у хронічну) = 240.
Загальні побічні реакції (середній баз 13,3).
|
Бал шкірного висипу |
= 0,2 · 10 («низька» серйозність) · 10 («низька» тривалість) = 20 |
|
Бал розладу шлунку |
= 0,1 · 10 · 10 = 10 |
|
Бал діареї |
= 0,05 · 10 · 20 = 10 |
Поодинокі побічні реакції (середній бал 0,078)
|
Бал виснажуючої діареї |
= 0,0005 · 20 · 20 = 0,2 |
|
Бал агранулоцитозу |
= 0,00005 · 30 · 20 = 0,03 |
|
Бал смертельного |
= 0,000005 · 30 · 30 = 0,0045 |
Сукупне «співвідношення користі/ризику» можна обчислити із застосуванням середньої величини всіх балів побічних реакцій [(13,3 + 0,078)/2 = 6,69]: К/Р = 240/6,69 = 35,9.
Цей метод дозволяє порівнювати побічні реакції між різними лікарськими засобами. Він є особливо ілюстративним для порівняння цього співвідношення для двох або більше лікарських засобів. За допомогою цієї моделі можна обчислити та порівняти окремі величини співвідношення користі/ризику для частих та рідкісних побічних реакцій (відповідно 240/13,3 або 18 та 240/0,078 або 3077).
Модель TURBO
Модель TURBO («Прозорий уніфікований огляд ризику/користі») є кількісним та графічним підходом до аналізу користі/ризику. За допомогою цього методу підраховується користь та ризики лікарського засобу за певним показанням і обидва фактори потім відображаються на діаграмі TURBO.
Коефіцієнт ризику «R» визначається як сума двох ризиків: основного та додаткового.
Коефіцієнт користі «B» визначається подібним чином, як сума основної користі та додаткової користі.
Коефіцієнт R та коефіцієнт B розміщуються на діаграмі TURBO та визначається показник Т, який виражає внутрішній аналіз користі/ризику.
R-фактор — визначено як сума Rо + Rс, де Rо — це найсерйозніша побічна реакція при застосуванні лікарського засобу (наприклад гепатит, а не підвищення трансіміназ), а Rс — це наступна найбільш серйозна або найчастіша побічна реакція. Rо розраховується шляхом кількісного визначення частоти побічних реакцій і характеристики тяжкості прояву найбільш серйозної побічної реакції з подальшим вибором відповідного числа.
Значення Rо — береться з більш точних і доступних джерел, наприклад, з клінічних досліджень, епідеміологічні дані (CIOMS IV). У разі, коли йдеться про генеричний лікарський засіб, джерелом таких даних може бути, наприклад, SmPC оригінального препарату, оскільки частота виникнення побічної реакції береться з клінічних досліджень.
Rо може варіюватися від 1 до 5 і залежить від:
- тяжкості проявів побічної реакції;
- частоти виникнення найбільш серйозної побічної реакції.
Тяжкість проявів Rо може бути:
- незначною = 1 бал — коли в результаті побічної реакції виникає деяка незручність, що не впливає на працездатність (вона зберігається);
- легкою = 2 бали — тимчасова/періодична непрацездатність;
- середньою = 3 бали — непрацездатність, що не загрожує життю та не впливає на його тривалість;
- важкою = 4 бали — зменшення тривалості життя, але без загрози для життя;
- дуже важкою = 5 балів — загрожує життю.
Визначаючи Rо, необхідно враховувати можливість управління ризиком:
1) значення Rо можна зменшити, наприклад, якщо:
- виникненню побічної реакції можна запобігти шляхом моніторування (кровотеча при застосуванні антикоагулянтів);
- відновити порушену функцію, якщо правильно управляти побічною реакцією (гепатотоксичність);
- існує можливість вчасно діагностувати побічну реакцію (за умови наявності очевидної симптоматики);
2) значення Rо можна збільшити або зменшити, якщо врахувати частоту, з якою відмічається побічна реакція.
Таблиця VII.12 — Розрахунок Rо
|
Частота*, з якою виникає |
Дуже часто |
3 |
3,5 |
4 |
4,5 |
5 |
|
Часто |
2,5 |
3 |
3,5 |
4 |
4,5 |
|
|
Найбільш серйозна побічна реакція |
Не часто |
2 |
2,5 |
3 |
3,5 |
4 |
|
Рідко |
1,5 |
2 |
2,5 |
3 |
3,5 |
|
|
Дуже рідко |
1 |
1,5 |
2 |
2,5 |
3 |
|
|
Тяжкість прояву |
Незначна |
Легкої тяжкості |
Середньої тяжкості |
Тяжка |
Дуже тяжка |
|
*Критерії оцінки частоти розвитку побічної реакції лікарського засобу відповідно до рекомендацій ВООЗ:
- понад 10% — дуже часті;
- 1–10% — часті;
- 0,1–1% — нечасті;
- 0,01–0,1% — поодинокі/рідкісні;
- менше 0,01% — рідкісні.
Наприклад, побічна реакція має дуже тяжкий прояв і тому має бути оцінена в 5 балів. Однак ця побічна реакція виникає дуже рідко, тому в кінцевому результаті вона може бути оцінена в 3 бали.
І навпаки, побічна реакція має незначну тяжкість прояву, тому спочатку оцінюється в 1 бал. Однак вона виникає дуже часто, тому, у кінцевому результаті, вона може бути оцінена в 3 бали.
Розрахунок Rс здійснюється після оцінки Rо. При цьому Rс може бути оцінений в балах від 0 до 2 залежно від оцінки Rо:
- якщо Rо = 5, то Rс = 2;
- якщо Rо = 4, то Rс = 1;
- якщо Rо ≤ 3, то Rс = 0.
Розрахунок R-фактора здійснюється за формулою: R-фактор = Rо + Rс.
Тому значення R-фактор може коливатися в межах від 1 (Rо = 1 і
Rс = 0) і до 7 (Rо = 5 і Rс = 2) балів.
В-фактор розраховується аналогічно.
В-фактор = Во + Вс, де Во — основна користь, і Вс — додаткова користь.
Во — варіює від 1 до 5. Розраховується шляхом кількісного визначення ймовірності отримання користі і характеристики ступеня користі з подальшим вибором відповідного числа.
Користь лікарського засобу залежить від інтенсивності його впливу на захворювання.
Користь лікарського засобу може бути:
- незначною = 1 бал — захворювання/стан під впливом лікарського засобу менше турбує, але непрацездатність залишається;
- легкою = 2 бали — захворювання/стан під впливом лікарського засобу стає менш вираженим, непрацездатність триває коротше або потреба в ній виникає рідше, менш часта тимчасова непрацездатність;
- середнього ступеню = 3 — непрацездатність виникає набагато рідше, але немає жодних змін щодо очікуваної тривалості життя;
- вираженою = 4 — лікарський засіб позитивно впливає на тривалість життя (виживаність) у пацієнтів з певним захворюванням/станом;
- значною = 5 — під впливом лікарських засобів захворювання/стан миттєво стає небезпечним для життя.
При оцінці Во необхідно враховувати можливість управління користю. Значення Во можна збільшити або зменшити, якщо врахувати ймовірність виникнення користі, яка може зустрічатися:
- рідко;
- нечасто;
- часто;
- дуже часто;
- майже завжди.
Таблиця V.13. Розрахунок Во
|
Ймовірність користі |
Майже завжди |
3 |
3,5 |
4 |
4,5 |
5 |
|
Дуже часто |
2,5 |
3 |
3,5 |
4 |
4,5 |
|
|
Часто |
2 |
2,5 |
3 |
3,5 |
4 |
|
|
Нечасто |
1,5 |
2 |
2,5 |
3 |
3,5 |
|
|
Рідко |
1 |
1,5 |
2 |
2,5 |
3 |
|
|
Ступінь користі |
Незначна |
Легкого ступеню |
Середнього ступеню |
Виражена |
Значна |
|
Вс може бути оцінена в 1 або 2 бали.
Вс = 2, якщо існує додаткова терапевтична дія, яка могла б бути використана при певному показанні, наприклад, ефект зниження рівня холестерину для протидіабетичних препаратів.
Вс = 1, якщо існує додаткова корисна (або практична) властивість, наприклад, однократний прийом лікарського засобу на добу або швидкий початок дії препарату.
Після того, як розраховані показники R-фактора і В-фактора, вони розміщуються в діаграмі, де вісь Х — це користь, а вісь Y — це ризик. Далі розраховується Т-показник — характерне співвідношення користі і ризику, що варіюється від 1 до 7. Для його визначення потрібно співставити результати чисел B-фактора і R-фактора.
Від того, де буде розташований Т-фактор, залежатиме оцінка профілю безпеки лікарського засобу.
Таблиця VII.14. Розрахунок Т-показника
|
R-фактор |
Т = |
|||||||
|
7 |
1 |
1 |
2 |
2 |
3 |
3 |
4 |
|
|
6 |
1 |
2 |
2 |
3 |
3 |
4 |
5 |
|
|
5 |
2 |
2 |
3 |
3 |
4 |
5 |
5 |
|
|
4 |
2 |
3 |
3 |
4 |
5 |
5 |
6 |
|
|
3 |
3 |
3 |
4 |
5 |
5 |
6 |
6 |
|
|
2 |
3 |
4 |
5 |
5 |
6 |
6 |
7 |
|
|
1 |
4 |
5 |
5 |
6 |
6 |
7 |
7 |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
В-фактор |
|
Виходячи з особливостей моделі, якщо при розрахунку Во та/або Rо отримуємо не ціле число, то його необхідно округлити до цілого, для можливості визначення Т-показника, який вкаже на співвідношення користь/ризик.

Рисунок VII.9. Оцінка співвідношення користі/ризику за результатами місцезнаходження Т-показника
Мультикритеріальний аналіз рішень (Multiple criteria decision analysis, MCDA): підхід оцінки співвідношення користі/ризику
Методологія MCDA ґрунтується на теорії прийняття рішення за результатами аналізу з використанням декількох критеріїв. ЕМА започаткувало використання методології MCDA при оцінці співвідношення користі/ризику. Основною метою MCDA є математичне зведення оцінок варіантів за різними критеріями в одну загальну оцінку. Це можливо здійснити з проведенням двох основних етапів: оцінювання та зважування. Оцінювання — це процес встановлення числових значень критеріїв. Зважування забезпечує відмінність між критеріями із призначенням вагових коефіцієнтів і надання їм певної значимості.
Дерево рішення для оцінки співвідношення користі/ризику
Дерево рішення для оцінки користі/ризику у вигляді таблиці наведено нижче (змінено за Musse F.: Оцінка методів для оцінки користі/ризику лікарських засобів та розробка нової моделі із застосуванням багатокритеріального аналізу прийняття рішень, докторська дисертація, Уельський університет).
|
Співвідношення користі/ризику лікарського засобу |
Критерій |
Субкритерії |
Субкритерії |
|
Первинна кінцева точка 1 |
|||
|
Первинна кінцева точка n |
|||
|
Користь |
Опорне випробування від 1 до n |
Ефективність порівняно з препаратом порівняння |
|
|
Доказ ефективності у відповідних підгрупах |
|||
|
Ефективність за результатами значимих непервинних кінцевих точок |
|||
|
Інші критерії користі |
Ефективність за результатами відповідних неопорних випробувань та подовжених фаз випробувань |
||
|
Передбачуване дотримання схеми лікування пацієнтом у клінічній практиці |
|||
|
Ризик |
Побічні реакції |
Загальна частота (показник захворюваності) побічних реакцій |
|
|
Загальна частота (показник захворюваності) серйозних побічних реакцій |
|||
|
Коефіцієнт припинення лікування через побічні реакції |
|||
|
Частота, тяжкість, тривалість та зворотний характер певних побічних реакцій |
|||
|
Інші критерії ризику |
Безпека у підгрупах |
||
|
Взаємодії з іншими лікарськими засобами та харчовими продуктами |
|||
|
Можливість застосування поза затвердженими показаннями, що призводить до загрози безпеці |
|||
|
Ймовірність непродемонстрованого додаткового ризику |
У результаті обмежень клінічних випробувань та/або експозиції пацієнтів: обмежені дані щодо безпеки лікарського засобу при його тривалому застосуванні, у пацієнтів з супутнім захворюванням, що виключені з випробування, додаткові види взаємодії |
||
|
Питання безпеки (включаючи якість та неклінічні сигнали) |
|||
|
Питання безпеки, що спостерігаються відносно інших лікарських засобів тієї самої фармакологічної групи |
Або дивись рисунок, що наведено нижче.
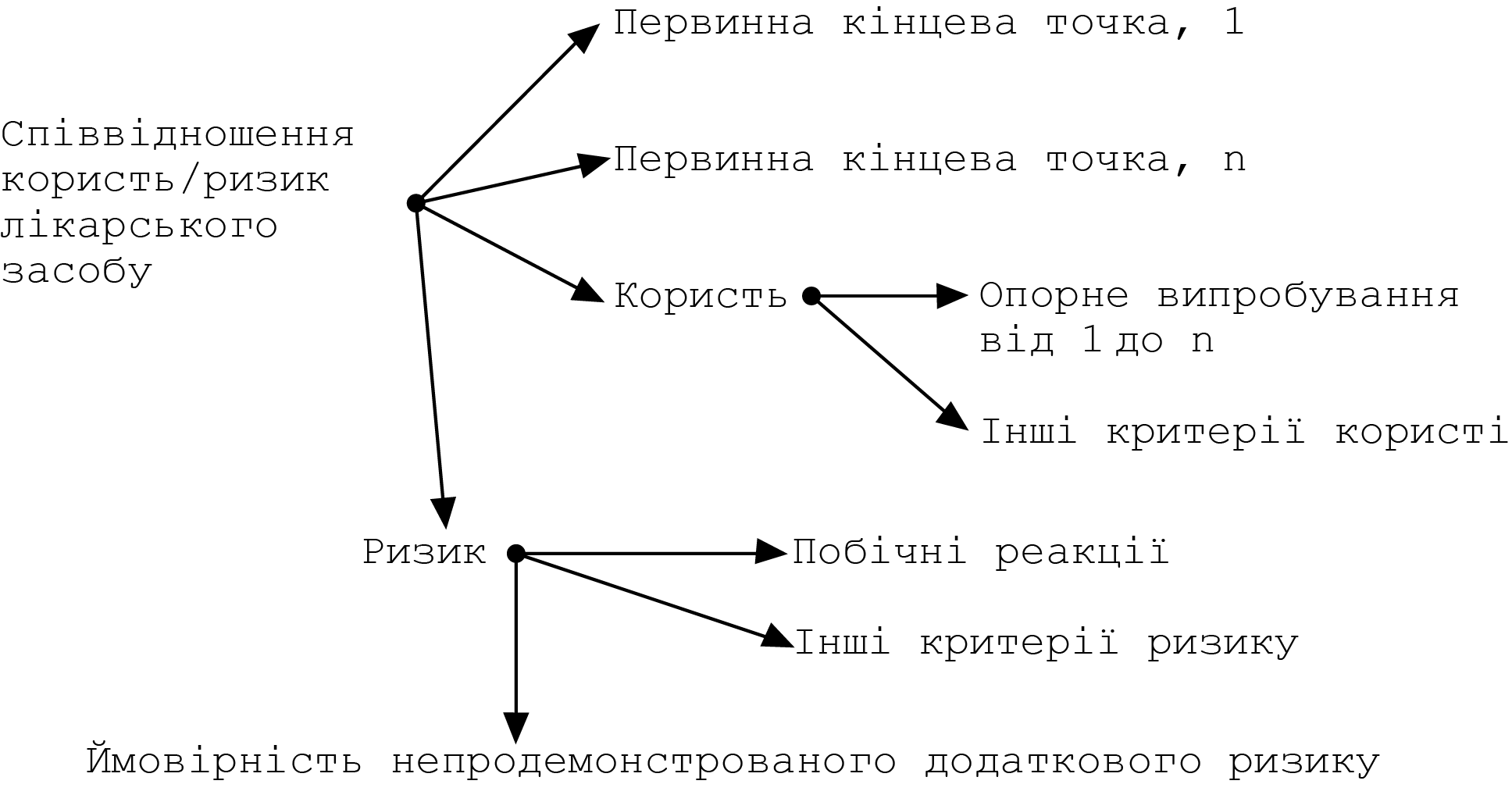
Рисунок. Блок-схема «дерево рішень» для оцінки співвідношення користі/ризику
Алгоритм дій при проведенні оцінки співвідношення користі/ризику лікарського засобу за використання методу мультикритеріального аналізу
1.Вибір критеріїв/субкритеріїв, що будуть використовуватися для проведення оцінки (власні дослідження, літературні дані тощо).
На цьому етапі слід визначити критерії (субкритерії), що будуть використовуватися при проведенні оцінки.
Наприклад, були обрані 4 критерії:
— нормалізація показників дихання за результатами спірометрії;
— нормалізація частоти дихання;
— зникнення вогнищ запалення за результатами КТ;
— зникнення хрипів за результатами аускультації.
2. Ранжування критеріїв, згідно з їх значущістю/важливістю (зважування і визначення значущості/важливості критеріїв).
На цьому етапі слід обговорити важливість критеріїв, визначити важливість кожного критерію і розташувати критерії один за одним у порядку зростання значущості (або навпаки). На цьому етапі критеріями не присвоюються цифрові значення.
Наприклад:
— нормалізація частоти дихання;
— зникнення хрипів за результатами аускультації;
— нормалізація показників дихання за результатами спірометрії;
— зникнення вогнищ запалення за результатами КТ.
3. Розрахунок значущості/вагомості критеріїв шляхом:
3.1. Порівняння критеріїв (попарне).
Слід порівняти критерії між собою попарно, починаючи з порівняння другого по значущості критерію з першим, третього з другим, четвертого з третім і т.д. і виразити відмінності в разах.
Наприклад:
Другий критерій важливіший за перший в 2 рази.
Третій критерій важливіший за другий в 1,5 раза.
Четвертий критерій важливіший за третій в 3 рази іт.д.
3.2. Присвоєння цифрових значень критеріям відповідно до наявних даних (власні дослідження, літературні дані тощо).
Оцінка критеріїв повинна проводитися в числовому еквіваленті, шляхом присвоєння їм балів, з урахуванням раніше проведеного порівняння пар критеріїв і визначення, у скільки разів наступний критерій важливіший за попередній.
Наприклад:
Якщо перший критерій прийняти за 1, а другий критерій важливіший за перший у 2 рази, то другий критерій отримує оцінку 2 бали (1 · 2 = 2).
Якщо третій критерій важливіший за другого в 1,5 раза і другий критерій оцінений у 2 бали, то третій критерій отримує оцінку 3 бали (2 · 1,5 = 3).
Якщо четвертий критерій важливіший за третій в 3 рази і третій критерій оцінений у 3 бали, то четвертий критерій отримує оцінку 9 балів (3 · 3 = 9) і т.д.
3.3. Вираження у відсотках значущості кожного критерію.
На цьому етапі слід скласти всі критерії, які оцінені в цифровому еквіваленті, прийняти суму за 100% і провести розрахунок значущості/вагомості кожного критерію у процентах.
Наприклад:
Складаємо числові значення всіх критеріїв.
У даному випадку 1 + 2 + 3 + 9 = 15
Приймаємо 15 за 100%, отже:
— перший критерій, що отримав оцінку 1 бал, має значущість/вагомість 6,7%;
— другий критерій, що отримав оцінку 2 бали, має значущість/вагомість 13,3%;
— третій критерій, що отримав оцінку 3 бали, має значущість/вагомість 20%;
— четвертий критерій, що отримав оцінку 9 балів, має значущість/вагомість 60%.
Розрахунок значущості/вагомості критеріїв закінчено.
4. Уніфікація значень критерієв (оскільки вони найімовірніше будуть виражені в різних одиницях виміру) шляхом їх вираження у відносних одиницях виміру.
Оскільки критерії можуть бути виражені в різних одиницях виміру (%, мм3, мл, разах та ін.), а значимість/вагомість критеріїв виражається у відсотках і між значеннями критеріїв існує лінійна залежність*, їх відносне значення теж можна виразити у відсотках.
*Якщо лінійна залежність відсутня, то слід розрахувати відносне значення критерію за використання математичних моделей.
5. Проведення розрахунку критерію шляхом перемноження відносних одиниць вимірювання критерію на його значимість/вагомість.
6. Порівняння отриманих результатів.
7. Висновки [51–55].
MCDA моделювання для визначення співвідношення користі/ризику з метою забезпечення проведення аналізу чутливості високого рівня і якісного графічного відображення результатів оцінки, може здійснюватися за використання спеціалізованого програмного забезпечення, а саме:
V∙I∙S∙A (http://www.visadecisions.com/),
Logical Decisions (http://www.logicaldecisions.com/home.htm)
або
Hiview3 (http://www.catalyze.co.uk/).
Якісна методика PROACT-URL для визначення співвідношення користі/ризику лікарського засобу
Якісна методика PrOACT-URL передбачає восьмиетапну модель оцінки співвідношення користі/ризику, що представлена нижче у вигляді таблиці.
|
Етап |
|
Етап І — ПРОБЛЕМА (Problem) 1. Визначити характер проблеми і її контекст, а саме: 1а) лікарський засіб: наприклад, новий, біологічного походження, генерик тощо); 1б) показання до застосування; 1с) терапевтична сфера і епідеміологія захворювання; 1d) незадоволена медична потреба, тяжкість захворювання, популяція ризику, побоювання лікарів і пацієнтів, тимчасовий клінічний результат; 1е) вирішення проблеми (що має бути зроблено і ким, наприклад, фармкомпанією, регуляторним органом, лікарем, пацієнтом). 2. Окреслити проблему, а саме: 2а) проблема в основному стосується того, що невідомо, чи декількох суперечливих проблем, або комбінації перерахованих, або щось інше (наприклад швидкість прогресування захворювання); 2b) фактори, які необхідно враховувати при вирішенні проблеми (наприклад, дизайн дослідження, джерела та адекватність даних, епідеміологія захворювання, наявність альтернативних методів лікування). |
|
Етап ІІ — ЦІЛІ (Objective) 3. Визначити цілі, досягнення яких буде свідчити, що основна мета досягнута, а саме: мета (наприклад, визначити співвідношення користі/ризику, визначити, яка додаткова інформація необхідна, оцінити зміну співвідношення користі/ризику, рекомендувати внести обмеження). 4. Визначити критерії для: a) сприятливих ефектів b) несприятливих ефектів, а саме: усі критерії, що охоплюють сприятливі і несприятливі ефекти (наприклад, кінцеві точки, відповідні стадії захворювання, клінічні результати). Робочі дефініції для кожного критерію поряд з вимірювальною шкалою з двома точками, визначеними таким чином, щоб охопити діапазон характеристик альтернатив (включаючи довірчі інтервали). Міркування щодо клінічної значущості критеріїв: одні мають більшу значущість для вирішення, ніж інші. |
|
Етап ІІІ — АЛЬТЕРНАТИВИ (Alternatives) 5. Визначити опції, які будуть оцінені відповідно до критеріїв, а саме: 5a) передреєстраційний період: дозування, строки лікування, лікарський засіб у порівнянні з плацебо і/або препаратом порівняння; необхідне рішення і рекомендації (наприклад, реєстрація/відмова в реєстрації, обмеження). |
|
5b) післяреєстраційний період: нічого не робити, обмежити тривалість лікування, обмежити показання, призупинити дію реєстраційного посвідчення. |
|
Етап ІV — НАСЛІДКИ (Consequences) 6. На підставі наявних даних порівняти значення критеріїв зі значеннями альтернатив (тобто значення всіх ефектів, і їх бажаність або тяжкість, і їх частота), а саме: результати окремо для кожної альтернативи за кожним критерієм (наприклад клінічно значущі ефекти ефективності і безпеки, позитивні і негативні результатів лікування) узагальнити в «таблицю ефектів» з альтернативами. У кожному осередку уявити якісні і кількісні значення ефектів, включаючи статистичні дані та довірчі інтервали, посилання на вихідні дані і графіки. При цьому слід врахувати, як і для кроків 3 і 4,що дуже рідко всі дані наявні в одному джерелі. Як правило, необхідний пошук інформації. Якщо є дані з більш ніж одного дослідження, для прийняття рішення краще ґрунтуватися на даних одного «якісного» дослідження або на об’єднаних даних. Чи є метааналіз? Чи можна наповнити «таблицю ефектів» результатами декількох досліджень? Оцінити відсутні дані. Кількісна модель потребує значень функції, які виражають клінічну значущість даних. |
|
Етап V — ОЦІНКА СПІВВІДНОШЕННЯ СПРИЯТЛИВИХ І НЕСПРИЯТЛИВИХ ЕФЕКТІВ (Trade-off) 7. Оцінка співвідношення сприятливі/несприятливі ефекти. Висновок про співвідношення користі/ризику і його обґрунтування (PrOACT — на цьому етапі оцінки лікарського засобу, проаналізовані тільки сприятливі і несприятливі ефекти, і їх співвідношення. На наступних трьох етапах (URL) оцінюється, як впливають на співвідношення користі/ризику невраховані фактори) |
|
Етап VІ — НЕВИЗНАЧЕНОСТІ (Uncertainty) 8. Відомості про невизначеності, пов’язані зі сприятливими і несприятливими ефектами, а саме: ступінь впливу невизначеностей на вірогідність статистичних даних (наприклад, можливі відхилення в даних, надійність і нерепрезентативність клінічних випробувань, потенціал для невиявлення побічних ефектів). 9. Оцінити як співвідношення сприятливі/несприятливі ефекти залежить від неврахованих факторів (невизначеностей), а саме: оцінити ступінь зменшення визначеного співвідношення користь/ризик, з урахуванням усіх невизначеностей, надати співвідношення користі/ризику і причини його зменшення. |
|
Етап VІІ — ТОЛЕРАНТНІСТЬ ДО РИЗИКУ (Risk tolerance) 10. Оцінити відносну важливість ставлення до ризику особи, яка приймає рішення щодо даного лікарського засобу, а саме: Прийняти до уваги будь-які міркування, які можуть вплинути, або вплинуть на ставлення особи, яка приймає рішення щодо ризику даного лікарського засобу (наприклад, препарат-сирота, лікарський засіб застосовується у спеціальній популяції, незадоволена медична потреба тощо). |
|
11. Оцінити, як це вплине на співвідношення користі/ризику. Оцінка може включати зміни значень нелінійної функції або зважування критеріїв. Ці зміни можуть імітувати різні рівні толерантності до ризику осіб, які приймають рішення щодо лікарського засобу. |
|
Етап VІІІ — ПОВ’ЯЗАНІ РІШЕННЯ (Linked decisions) 12. Розглянути послідовність цього рішення з аналогічними рішеннями в минулому, і оцінити, наскільки прийняття цього рішення може вплинути на майбутні рішення. Мається на увазі, як це рішення, оціночні судження і дані, на яких воно ґрунтується, можуть створити прецедент або зробити подібні рішення в майбутньому легшими чи складнішими. |
Таблиця VII.15. Зведена таблиця оцінки співвідношення користі/ризику лікарського засобу
|
Ефекти |
Назва критерію |
Опис критерію |
Добре |
Погано |
Одиниці виміру (місяці, роки, оціночні параметри,%) |
Опція (альтернатива) |
Опція (оцінюваний лікарський засіб) |
|
|
Сприятливі ефекти |
Вторинні кінцеві та/або первинні точки* |
Критерій |
||||||
|
Критерій |
||||||||
|
Критерій |
||||||||
|
Несприятливі ефекти |
Критерій |
|||||||
|
Критерій |
||||||||
|
Критерій |
||||||||
*Первинні — тверді точки (смерть, ампутація, видалення, видужання, стійка ремісія тощо).
Вторинні — проміжні точки (покращення лабораторних показників, зниження температури тіла, покращання ЕКГ тощо).
Модель BRAT (Benefit Risk Action Team)
Ініціативна група Асоціації дослідницьких організацій і фармацевтичних виробників США (Pharmaceutical Research and Manufacturers of America — PhRMA) розробила модель BRAT (Benefit Risk Action Team), що являє собою шестиетапний процес оцінки співвідношення користі/ризику лікарського засобу, що представлений нижче у таблиці.
|
Етап |
Опис |
|
1) Визначити контекст рішення, що прийматиметься |
— препарат, доза, хімічний склад, показання, популяція, препарати порівняння, результати лікування, перспектива зацікавлених сторін (регулятора, фармкомпанії, пацієнта або лікаря) |
|
2) Визначити ефекти, пов’язані з лікарським засобом |
— відібрати всі важливі ефекти (позитивні та негативні) і створити початкове дерево рішень; — визначити попередній набір критеріїв оцінки/кінцевих точок; — описати причини для їх включення/виключення для подальшого аналізу; |
|
3) виявлення і включення даних |
— визначити і задокументувати всі джерела даних (наприклад клінічні випробування, обсерваційні дослідження) — внести всі відповідні дані в таблицю даних, включаючи докладні посилання і будь-які інструкції з тим, щоб допомогти в подальшій інтерпретації даних |
|
4) удосконалення моделі |
— змінити дерево рішень, ґрунтуючись на подальшому розгляді даних і клінічної експертизи — уточнити критерії оцінки/кінцеві точки |
|
5) оцінка важливості критеріїв (ефектів) |
— застосувати або оцінити будь-який рейтинг і зважування ефектів, важливих для осіб, котрі приймають рішення і інших зацікавлених сторін |
|
6) відображення та інтерпретація ключових показників користь/ризик |
— узагальнити вихідні дані в табличному і графічному вигляді для інтерпретації — врахувати вплив систематичних помилок, наприклад, при об’єднанні даних тощо — інтерпретувати узагальнену інформацію |
2.1.1. Джерела даних, що можуть бути використані для проведення оцінки співвідношення користі/ризику лікарського засобу.
Проведення інформаційного пошуку доказових даних з метою проведення оцінки співвідношення користі/ризику слід проводити відповідно до поставленого питання. Вважається, що оптимальним є формулювання питання за використання схеми PICO. Рекомендації відносно побудови стратегії пошуку представлені в настановах Cochrane [56], де також описана концепція схеми PICO, яка відображає ключові елементи пошуку, а саме:
Р — population — досліджувана популяція населення;
І — intervention — технологія, яка вивчається;
С — comparator — альтернативна технологія;
О — outcomes — результати лікування.
На практиці рекомендовано використовувати 2-3 найбільш важливі елементи схеми з метою уникнення пропуску даних під час пошуку з використанням операторів «Boolean» (AND, OR, NOT).
Важливими під час проведення пошуку є чітка документальна фіксація баз даних, часу і періоду проведення, використовуваних стратегій та джерел, загальне число результатів, отриманих після кожного пошуку в кожній базі даних і кількість результатів після дедуплікації, експорт, завантаження відповідних даних та їх збереження.
Пошук джерел інформації рекомендується проводити у таких інформаційних ресурсах з доказової медицини:
- Кокрейнівська бібліотека: http://www.cochranelibrary.com;
- база даних PubMed: https://www.ncbi.nlm.nih.gov/pubmed;
- база даних EMBASE: https://www.elsevier.com/solutions/embase-biomedicalresearch;
- база даних TRIP https://www.tripdatabase.com;
- база даних NHS EED https://www.crd.york.ac.uk/CRDWeb;
- INAHTA http://www.inahta.org.
На етапі аналізу знайдених джерел інформації слід представити висновки з виявлених вторинних джерел доказової медицини та обговорити їх обмеження, зокрема в контексті мети та виконання аналізу клінічної ефективності чи безпеки лікарського(их) засобу(ів).
Визначені дослідження також можуть бути використані як джерело аналітичної інформації у вирішенні заданої проблеми. У виправданих та обґрунтованих випадках допускається проводити аналіз клінічної ефективності чи безпеки лікарських засобів, виходячи виключно з результатів знайдених систематичних оглядів (СО) (необхідність проведення оцінки клінічної ефективності за короткий час, виявлені СО є систематичними, оновленими, відповідають на питання дослідження та їх методологія відповідає вимогам щодо якості).
Якість СО слід оцінювати, використовуючи поточну версію шкали AMSTAR [57].
Метою проведення СО первинних досліджень є пошук усіх наукових результатів, які відповідають критеріям включення аналізу. Основними базами даних для пошуку первинних досліджень є:
- PubMed;
- EMBASE;
- Кокрейнівська бібліотека;
- NICE Evidence search;
У виправданих випадках рекомендується також шукати медичну інформацію в інших базах даних.
Цілком імовірно доповнити пошук з баз даних медичної інформації за допомогою інших джерел, у тому числі:
- посилання на літературу, що містяться в публікаціях про клінічну ефективність/практичну ефективність чи безпеку лікарських засобів;
- реєстри клінічних випробувань (clinicaltrials.gov і clinicaltrialsregister.eu), щоб знайти дослідження, які завершені, але не опубліковані.
Також для пошуку додаткової інформації для проведення оцінки користі/ризику можна використовувати зазначені нижче методи:
- консультації з клінічними експертами;
- несистематичний пошук даних, опублікованих у спеціалізованих виданнях у галузі, що не індексовані в медичних інформаційних базах даних;
- звернення до авторів клінічних досліджень, наприклад, для отримання спеціальних неопублікованих матеріалів/даних;
- використання пошукових систем в інтернеті;
- використання даних, що містіться у періодичних звітах з безпеки (PSUR);
- використання даних реєстраційного досьє препарату, доступного на вебсайтах регуляторних органів (наприклад EMA, FDA).
Отже, дані про клінічну ефективність та безпеку лікарських засобів в основному отримуються методом проведення СО рандомізованих клінічних досліджень (РКД).
У випадку рідкісних захворювань та/або етичних проблем, пов’язаних з проведенням клінічних досліджень, виправдано оцінити ефективність та безпеку лікарського засобу, виходячи з непорівняльних досліджень (single arm study), особливо коли такого типу дослідження були рекомендовані регуляторними органами з суворою регуляторною політикою.
Дані щодо практичної ефективності (effectiveness) повинні надходити з надійних досліджень реальних даних (РД), проведених в умовах реальної клінічної практики (реальні дані, RWD/ RWE).
Оцінка ефективності повинна базуватися на даних з найвищим рівнем достовірності. За визначенням робочої групи експертів Міжнародного товариства фармакоекономічних досліджень (ISPOR) термін «реальні дані» (англ. real world evidence) охоплює різноманітні типи інформації, які використовуються для прийняття рішень та отримані не із РКД, а за даними реальної практики. РД відрізняються за типом результатів лікування (клінічні, економічні, отримані від пацієнтів), надійністю і валідністю доказових даних для прийняття рішень. За методологією та підходами до одержання РД класифікуються на:
1) додаткові до традиційних рандомізованих клінічних досліджень — передреєстраційні, з метою реєстрації нового лікарського засобу або нових показань до застосування;
2) прагматичні або натуральні клінічні дослідження, які проводяться в умовах реального надання медичної допомоги, але мають ширші критерії включення та рандомізацію пацієнтів у групи лікування. Неінтервенційне дослідження (non-interventional trial, noninterventional study) — це дослідження, в якому лікарський засіб призначаються звичайним способом відповідно до затвердженої інструкції з медичного застосування. Залучення пацієнта в групу з визначеним методом лікування в протоколі клінічного дослідження заздалегідь не передбачено, а призначення лікарського засобу диктується сучасною практикою і не залежить від рішення включити пацієнта у випробування. Не застосовують додаткових діагностичних або моніторингових процедур щодо пацієнтів, а для аналізу зібраних даних використовують епідеміологічні методи (п. 2.1 Порядку проведення клінічних випробувань[16] та глава 5 розділу V Порядок здійснення фармаконагляду [2]);
3) реєстри — проспективні спостережні когортні дослідження пацієнтів з певним захворюванням, які отримують визначене лікування;
4) адміністративні дані на основі діючих баз даних;
5) опитування та інтерв’ювання фахівців охорони здоров’я, фармацевтичного сектору, пацієнтів;
6) електронні медичні карти.
РД доповнюють і уточнюють інформацію з клінічної ефективності, отриману як «золотий стандарт» із РКД, даними із практичної реальної ефективності та безпеки лікарських засобів, одержаними від ширшої популяції пацієнтів.
2.1.2. Стратегія пошуку
Стратегія пошуку повинна бути розроблена на основі визначеної проблеми рішення і відповідати рекомендаціям Кокрейнівського посібника, зазначених EUnetHTA щодо належного проведення СО [58].
Якщо аналіз клінічної ефективності та безпеки ґрунтується на нерандомізованих дослідженнях, необхідно надати стратегію пошуку як для рандомізованих досліджень (щоб підтвердити їх відсутність), так і стратегію пошуку нерандомізованих досліджень.
Критерії пошуку повинні містити елементи схеми PICO, що представлена вище.
Рекомендовано, щоб кінцевим результатом пошуку був перелік усіх доступних досліджень та даних, необхідних для надійної оцінки ефективності та безпеки, включаючи практичну ефективність та безпеку лікарського засобу.
Процес пошуку даних та інформації слід детально описати в такій формі, щоб його можна було відтворити для верифікації.
При проведенні пошуку слід враховувати та використовувати:
- ключові пошукові слова та дескриптори, використовувані для пошуку;
- використані оператори «Boolean» (AND, OR, NOT);
- використані фільтри;
- проміжок часу пошуку/дата останнього пошуку;
- кількість ідентифікованих записів окремо для кожного запиту, використаного в стратегії пошуку.
2.1.3. Відбір інформації
Процес відбору даних для систематичного огляду повинен бути виконаний принаймні двома незалежними експертами.
Перевірку відповідності ідентифікованих доказових даних у аналізі до критеріїв включення слід виконувати поетапно. Початковий етап передбачає вибір на основі назв і тез, а згодом — на основі повних текстів публікацій.
Вибір дослідження повинен здійснюватися на основі критеріїв включення та виключення, обраних перед початком пошуку, відповідно до визначеної PICO схеми (протокол СО).
VII. Додаток 4. Пояснювальна записка до модуля VII «Настанови з належної практики фармаконагляду»
З початку єдиної оцінки РОЗБ (PSUSA) процедура ставить ряд завдань, що є специфічними для оцінки лікарських засобів, зареєстрованих за національною процедурою. Наступна пояснювальна записка до модуля VII ННПФ має на меті розгляд проблем, з якими стикаються протягом двох років ведення процесу єдиної оцінки РОЗБ. Зрештою, пояснювальна записка слугуватиме як основа для оновлення модуля VII ННПФ, яка з часом його замінить. Слід зазначити, що пункти, які висвітлені у цьому документі, можуть також застосовуватися для оцінки лікарських засобів, зареєстрованих за централізованою процедурою. Цю записку слід розглядати разом з модулем VII ННПФ; якщо необхідно, посилаючись на настанову ІСН Е2С (R2) [28]; її слід використовувати для підготовки РОЗБ, що підлягають єдиній оцінці.
З метою збереження змісту, який погоджений міжнародним консенсусом, уся спеціальна інформація ЄС повинна надаватися в локальному додатку ЄС до РОЗБ (розділ VII С.5. модуля VII ННПФ).
Документ розподілений на розділи, що визначають ключові питання для розробки наступної настанови.
У пояснювальній записці інформація про лікарський засіб вказується як інформація про лікарський засіб ЄС.
Рекомендації, що надані в цьому документі, мають на меті обмежити кількість питань та запитів для роз’яснення, що піднімаються під час періоду оцінки, враховуючи терміни процедури.
Заявники (власники реєстраційних посвідчень) можуть розглянути необхідність навчання за настановою ІСН Е2С (R2) [28] та ICH настановою E2C (R2) — Питання та відповіді (ICH guideline E2C (R2) — questions and answers) [28], що розроблені імплементаційною робочою групою ІСН Е2С (R2).
1. Принципи оцінки співвідношення користі/ризику в рамках регулярного звіту з безпеки та масштаби інформації, яку необхідно включити
1.1. Загальні принципи
Як детально описано в модулі VII ННПФ, мета РОЗБ — представити всебічний, стислий та критичний аналіз співвідношення користі/ризику лікарського засобу, враховуючи нову інформацію або ту, що з’являється, в контексті кумулятивної інформації щодо ризиків та користі.
УОВФ, повинна гарантувати, що РОЗБ готуються та пишуться таким чином, що полегшить розуміння даних, висновків та послідовних дій, тобто, логічне викладення даних має бути чітким та зрозумілим.
Надання інформації відповідного рівня та якості в рамках РОЗБ та протягом процедури оцінки є ключовим для обґрунтування висновків заявників (власників реєстраційних посвідчень) та будь-яких дій, що вживаються або пропонуються. Рівень деталізації інформації, представленої заявником (власником реєстраційного посвідчення) повинен бути достатнім, як для представлення результатів (розділ 6–14), так і для послідовної оцінки (розділ 15 та 16) на основі клінічної значимості представлених даних. Важливо розуміти, що ця оцінка включає медичні та наукові судження, що повинні бути чітко представлені власником реєстраційного посвідчення, що дозволить провести повну повторну оцінку співвідношення користі/ризику лікарського засобу та відповідну характеристику й управління профілем безпеки, зосереджуючись на реальних покращеннях для пацієнта на основі мінімізації ризику. Див. також рекомендації, надані в ICH настанові E2C (R2) — «Питання та відповіді».
РОЗБ загалом не призначений для повідомлення термінової нової інформації з безпеки та ефективності, яка може мати важливий вплив на громадське здоров’я. Натомість слід розглядати інші процеси, які входять до сфери дії модуля ІХ ННПФ (управління сигналом). Також термінову інформацію з безпеки слід повідомляти шляхом відповідного механізму.
1.2. Зміни до терапевтичного показання
Основним є те, що на початку звітного періоду РОЗБ співвідношення користі/ризику лікарського засобу є позитивним на основі даних на момент отримання реєстраційного посвідчення та подальшої оцінки співвідношення користі/ризику, в рамках процедури оновлення, подання РОЗБ та ін.
Оскільки РОЗБ зосереджені на безпеці та можливому впливі нових питань з безпеки на загальне співвідношення користі/ризику лікарського засобу, тільки зміни до показання, що обґрунтовані на основі питань безпеки та ефективності (тобто видалення або обмеження існуючого показання), можуть імплементуватися як результат процедури оцінки РОЗБ.
Якщо існує нова позитивна інформація щодо користі та відсутні значні зміни в профілі ризику у звітний період, викладення основної інформації та нової повинно бути стислим (модуль VII ННПФ, розділ VII.В.5.17.3).
Важливо зазначити, що рекомендація PRAC, у якій вказано, що співвідношення користі/ризику для певних показань, що зареєстровані лише в деяких країнах ЄС, є незмінним, не може використовуватися як основа для розширення показань в інших країнах ЄС/Україні, де ці показання не зареєстровані. Заявки на нові показання необхідно подавати шляхом відповідної регуляторної процедури до відповідного національного уповноваженого органу, включаючи повний пакет даних.
1.3. Регулярний звіт з безпеки для генеричних лікарських засобів
РОЗБ для генеричного лікарського засобу повинен мати той самий формат та зміст, як вказано у модулі VII ННПФ. Інформація повинна мати відповідний рівень якості, що дозволить провести повну повторну оцінку співвідношення користі/ризику лікарського засобу та надати відповідну характеристику профілю безпеки та управління ним з метою його реального покращання для пацієнта на основі мінімізації ризика.
Джерела наявної інформації для діючої речовини є такими, до яких заявник (власник реєстраційного посвідчення) може мати доступ в достатній мірі, та які є відповідними для оцінки профілю безпеки або співвідношення користі/ризику (див. також додаток Е, «Приклади можливих джерел інформації, які можуть використовуватися при підготовці PBRER».
2. Заходи, що вживалися у звітний період з причин безпеки
Якщо значні заходи вживалися в будь-якій країні світу з причин безпеки у міжзвітний період, що охоплюється певним РОЗБ, їх слід описати достатньо детально, щоб дозволити експерту зрозуміти, чи ці заходи безпеки мають будь-який вплив на профіль користі/ризику лікарського засобу.
Наприклад, просте формулювання «припинення дії реєстраційного посвідчення» не слід розглядати як достатньо інформативне.
Приклади значних заходів, що вживалися в міжзвітний період з причин безпеки, наведені у настанові ІСН Е2С (R2) [28] та модулі VII ННПФ (розділ VII.В.5.2) та включають: значні зміни з безпеки до короткої характеристики інструкції для медичного застосуванняN, включаючи обмеження щодо використання та популяції, яка лікується, відкликання або припинення реєстраційного посвідчення, заходи, що вживалися з причин виявлення проблем лікарського засобу та питань якості, заходи з управління ризиками, такі як листи-звернення до спеціалістів системи охорони здоров’я, інспекції або аудити системи фармаконагляду уповноваженою установоюN, ініційовані з питань безпеки, та інші значні заходи, що вживаються органами охорони здоров’я країн ЄС та не ЄС з причин, що наведені в модулі VII (розділ VII. В.5.3.).
3. Референтна інформація
Референтна інформація про лікарський засіб повинна надаватися англійською/українськоюN мовою. Якщо необхідно, короткий опис поточних процедур (наприклад змін) для оновлення інформації про лікарський засіб вимагається згідно з локальним додатком ЄС/УкраїниN та повинен включатися до розділу «Запропонована інформація про лікарський засіб» модуля VII ННПФ розділу VII. С.5.1. Зміни для оновлення інформації про лікарський засіб, що, як очікується, будуть включені, як правило, будуть такими, при яких зміни вносяться до відповідних даних з безпеки в інформації про лікарський засіб.
У той час як референтна інформація про лікарський засіб може надаватися в будь-якому форматі відповідно до настанови ІСН-Е2С(R2) [28], УОВФ/КОВФN повинна мати повний контроль за затвердженою інформацією про лікарський засіб, оскільки цей документ є ключовим стандартним механізмом з мінімізації ризиків у фармаконагляді.
Важливо, що будь-які коментарі та питання щодо запропонованих змін до референтної інформації з безпеки у РОЗБ також завжди були відображені у інформації про лікарський засіб, що зареєстрований в ЄС/УкраїніN. Наприклад, якщо добре відома побічна реакція лікарського засобу ефективно управляється, оскільки зазначена в протипоказаннях та вже включена або пропонується до референтної інформації з безпеки, очікуваним є те, що протипоказання та побічна реакція лікарського засобу не тільки включаються до референтної інформації з безпеки, а і також до відповідної інформації про лікарський засіб. Оскільки це очікування є специфічним для ЄС/УкраїниN, будь-яке обговорення щодо впливу змін референтної інформації з безпеки на інформацію про лікарський засіб слід надавати в локальному додатку ЄС/УкраїниN (розділ VII.С.5.1) разом з формулюванням, у якому заявник (власник реєстраційного посвідчення) підтверджує, що він розглянув вплив даних РОЗБ на інформацію про лікарський засіб. Якщо експерт виявить питання, яке невідповідно відображене в заяві про вплив від заявника (власника реєстраційного посвідчення) (наприклад, заявник (власник реєстраційного посвідчення) дійде висновку, що жодні подальші зміни до інформації про лікарський засіб не гарантуються на основі даних РОЗБ, у той час як дані демонструють відповідний рівень повідомлення про побічні реакції лікарських засобів, що включені до референтної інформації з безпеки, але не до інформації про лікарський засіб), подальші дані можуть запитуватися у заявника (власника реєстраційного посвідчення) під час оцінки.
Відповідно до законодавства на основі оцінки кумулятивних даних з безпеки та аналізу співвідношення користі/ризику заявник (власник реєстраційного посвідчення) повинен прийняти рішення відносно необхідності змін та/або дій, включаючи будь-які наслідки для затвердженої інформації про лікарський засіб (на лікарські засоби), для яких був розроблений РОЗБ (ст. 34 (5) ІП 520/2012 [6]), положення Порядку [2], положення порядку проведення експертизи [7]N.
Зміни в інформації на лікарський засіб, що не стосуються інформації, що подана в регулярному звіті з безпеки, не повинні пропонуватися в цьому контексті.
Якщо зміни необхідні лише в деяких документах, наприклад, якщо важлива інформація з безпеки відсутня в основній інформації з безпеки лікарського засобу деяких препаратів, але не всіх, заявник (власник реєстраційного посвідчення) тим не менш повинен внести відповідне формулювання на основі даних, поданих та оцінених у рамках РОЗБ. PRAC пропонує єдине формулювання для опису побічної реакції/застереження/протипоказання тощо, яке буде застосовне до всіх лікарських засобів, які підлягають процедурі єдиної оцінки без різниці між окремими заявниками (власниками реєстраційних посвідчень). Заявники (власники реєстраційних посвідчень), що видані згідно зі ст. 10 директиви 2001/83/ЄС [1], відповідно до Положень проведення експертизи [7]N повинні гарантувати, що вони повністю привели у відповідність свою основну інформацію з безпеки лікарського засобу до своїх референтних лікарських засобів до дати подання РОЗБ, оскільки РОЗБ не буде використовуватися як механізм для внесення змін до інформації про лікарський засіб, що обумовлені виключно невідповідністю такій інформації.
4. Експозиція пацієнта
Оскільки часто складно отримати та підтвердити дані експозиції, кількість пацієнтів слід надавати відповідно до тривалості експозиції (бажано кількість пацієнтів або пацієнтів/років). Метод слід пояснити. Якщо існує розбіжність (наприклад, інформація, що надається щодо дослідження, при додаванні разом не відповідає загальній сумі) це необхідно пояснити. Відмінності в експозиції пацієнтів, що надаються, від одного РОЗБ до іншого (наприклад, відмінності між попередньою та поточною кумулятивною експозицією, якщо використовуються різні одиниці або дані відсутні) слід обґрунтувати та представити з відповідним рівнем деталізації.
5. Дані в зведених таблицях
Кількість проявів для певних побічних реакцій, що вказані за текстом у РОЗБ, наприклад, для сумарної оцінки закритого сигналу, який представлений у розділі 15, може відрізнятися від кількості проявів відповідного терміна MedDRA в зведених таблицях. Проте, якщо кількість проявів реакцій за текстом РОЗБ відрізняється від кількості повідомлень у відповідних зведених таблицях настільки, що може потенційно змінити висновки щодо оцінки, такі відмінності слід пояснити та обґрунтувати.
6. Надання звітів дослідження
Слід зазначити, що РОЗБ не є механізмом оцінки кінцевих звітів клінічних досліджень. Слід дотримуватися відповідної процедури та повний звіт клінічного дослідження не буде оцінюватися під час PSUSA, за відповідною регуляторною процедурою, як належить. Тип змін для подання звітів клінічних досліджень буде залежати від типу змін інформації про лікарський засіб, на яких зосереджена увага відповідно до настанов Комісії щодо детальних даних різних категорій змін, щодо управляння процедурами.76
Проте, якщо звіт дослідження містить дані з безпеки, всі або відповідні розділи звіту повинні також подаватися як додаток до РОЗБ. На відповідну інформацію слід надавати перехресні посилання та/або включати у розділ 7.1 «Завершені клінічні дослідження» та розділ 8 «Результати неінтервенційних досліджень» РОЗБ та вона повинна оцінюватися власниками реєстраційних посвідчень в регулярному звіті з безпеки відповідно.
Відомо для ПДБЛЗ77 (PASS), що були завершені у звітний період, локальний додаток ЄС до РОЗБ модуля ННПФ VІІ.С.5.4., підрозділ «Повідомлення результатів післяреєстраційних досліджень з безпеки» вимагає, щоб кінцеві звіти дослідження були включені як додаток до РОЗБ. Ця вимога, що може переглядатися під час оновлення модуля VІІ ННПФ, має на меті зібрати всю відповідну інформацію в рамках одного документа, але, як вказано вище, звіт клінічного дослідження не буде оцінюватися не під час єдиної оцінки РОЗБ, а за окремою регіональною процедурою відповідно. У Модулі VІІ ННПФ в розділі VІІ.С.5.4. вказано, що РОЗБ не повинні використовуватися як метод початкового повідомлення або для подання кінцевих звітів дослідження до національних уповноважених органів у країнах ЄС/УкраїниN, або для повідомлення будь-якої нової інформації, що може вплинути на оцінку співвідношення користі/ризику.
7. Інформація, що подається в останній момент
Як вказано у розділі VІІ.В.5.14. «Інформація, що подається в останній момент» модуля ННПФ VІІ, заявники (власники реєстраційних посвідчень) розглядають будь-яку інформацію, що стосується безпеки (наприклад, референтну СТ-Н МОЗУ 42-8.5:2018 428 інформацію, що стала відомою у цей період) або інші важливі питання щодо безпеки, що виникали після закриття бази даних.
8. Огляд сигналів: нових, поточних або закритих
Заявник (власник реєстраційного посвідчення) повинен надати огляд важливих сигналів, оцінка яких була завершена у звітний період, а також поточних сигналів, що проходять оцінку у звітний період. При огляді РОЗБ до цього часу було помічено ряд недоліків, включаючи відсутність кумулятивних оглядів, які розглядалися в попередньому РОЗБ, або виключення сигналу без відповідного пояснення.
Огляд слід представити у відповідному табличному форматі відповідно до модуля ННПФ VІІ, додаток 2:
|
Термін сигналу |
Дата виявлення |
Статус (поточний або закритий) |
Дата закриття (для закритих сигналів) |
Джерело сигналу |
Мета оцінки та резюме основних даних |
Метод оцінки сигналу |
Дії, що вживаються або заплановані |
|
Інсульт |
мм/рррр |
Поточний |
мм/рррр |
Метааналіз (опубліковані дослідження) |
Статистично значиме підвищення в частотності |
Перегляд метааналізу та наявних даних |
На розгляді |
|
Синдром Стівенса —Джонсона (SJS) |
мм/рррр |
Закритий |
мм/рррр |
Спонтанні повідомлення |
Висип — уже виявлений ризик. Про синдром Стівенса — Джонсона не повідомлялося у дореєстраційних клінічних випробуваннях. Було отримано повідомлення протягом 6 місяців після реєстрації. Ймовірний час до початку та жодних можливих альтернативних причин |
Цільове відслідковування повідомлень з візитом до однієї лікарні. Повний перегляд випадків дерматологом заявника (власника реєстраційного посвідчення) та пошуки в літературі |
ОІБЗ оновлюється застереженням та попереджанням. Надсилається DHPC. Аналіз ефективності вжитих заходів запланований через 6 місяців після розповсюдження DHPC. Оновлено ПУР |
Національний уповноважений орган може попросити провести моніторинг та повідомити в РОЗБ певне питання (що не вважається сигналом). Такі запити ретельного моніторингу PRAC будуть належним чином обґрунтовані. Як вказано в розділі VІІ.В.5.15 модуля VІІ, заявник (власник реєстраційного посвідчення) повинен підсумувати результат аналізу в розділі 15 РОЗБ, якщо він негативний. Усі принципи оцінки сигналу, що висвітлені в розділі 9 цієї пояснювальної записки, також застосовуються до будь-яких певних питань (що не вважаються сигналом), що включені до розділу 15 РОЗБ. Якщо результат аналізу негативний, заявник (власник реєстраційного посвідчення) може запропонувати припинити спеціальний моніторинг у майбутніх РОЗБ. Якщо експерт не підтримує позицію заявника (власника реєстраційного посвідчення), наукові причини непогодження повинні бути чітко обґрунтовані у висновку разом з будь-якими рекомендаціями щодо необхідної оцінки в майбутньому РОЗБ. Якщо певне питання стає сигналом, його необхідно включити в таблицю сигналів (якщо сигнал був закритим або поточним під час закриття бази даних) та проаналізувати в підрозділі 16.2 («Оцінка сигналу») РОЗБ, якщо сигнал оцінювався та був закритий у звітний період. Погоджено, що рутинних заходів з фармаконагляду буде достатньо, якщо проводився достатній моніторинг проблеми та вона не була виявлена як сигнал безпеки.
9. Оцінка сигналу
Заявникам (власникам реєстраційних посвідчень) необхідно проводити фармаконагляд на регулярній основі відповідно до законодавства. Регулярний фармаконагляд описується у МФСФ. Тому немає необхідності для заявників (власників реєстраційних посвідчень) детально описувати стандартний фармаконагляд та дії з виявлення сигналу в РОЗБ.
Резюме оцінки сигналу, представлене в розділі 16.2 РОЗБ, є основою для будь-якого рішення та призведе до виключення сигналу або продовження класифікації сигналу як поточного, якщо оцінка є неостаточною. Якщо вагомість доказу не дозволяє спростувати або підтвердити сигнал як проблему безпеки лікарського засобу, його слід продовжувати спостерігати в наступному РОЗБ або за іншою процедурою на основі серйозності або терміновості сигналу. Якщо оцінка в наступному РОЗБ виявиться неостанньою, рішення про необхідність продовжувати цю оцінку повинно прийматися в кожному конкретному випадку, враховуючи медичну важливість сигналу, ступінь експозиції, ймовірний вплив на співвідношення користь/ризик та загальну вагомість доказу.
Якщо питання безпеки (що не вважаються сигналом або важливим ризиком) розглядаються у наступних РОЗБ, проміжні дані слід представити в контексті кумулятивних даних (це один з основних принципів РОЗБ). При виключені випадку з кумулятивного огляду заявники (власники реєстраційних посвідчень) таке виключення повинні обґрунтувати, оскільки воно не може лише ґрунтуватися на факті, що початковий репортер вважав побічну реакцію такою, що не пов’язана з лікарським засобом.
Оцінка сигналу повинна проводитися на основі узагальненого огляду кумулятивних даних, наявних у заявника (власника реєстраційного посвідчення), та слід оцінити ступінь доказу за і проти причинно-наслідкового зв’язку з лікарським засобом. Ця експертиза буде, як правило, проводитися, оцінюючи дані з багатьох джерел інформації та заявник (власник реєстраційного посвідчення) повинен використовувати методику, що найбільше підходить до наявних даних, враховуючи джерело сигналу. Наприклад, при оцінці спонтанних повідомлень важливі фактори, на які слід звернути увагу, це: зв’язок у часі, позитивна динаміка при відміні лікарського засобу, поява при повторному призначенні.
Визнаний метод, такий як, критерій Бредфорда Хілла, може також бути корисним. Який би аналіз не використовувався, оцінка заявника (власника реєстраційного посвідчення) повинна бути чітко представлена та має включати достатньо інформації та пояснення, що дозволить експерту зрозуміти причини висновків та дій заявника (власника реєстраційного посвідчення) (якщо такі вживаються або пропонуються).
Заявник (власник реєстраційного посвідчення) повинен надати чіткий доказ у підтримку або для спростування можливого причинно-наслідкового зв’язку в розділі 16.2 РОЗБ. Представлений аналіз слід зосередити на підтвердженні того, як заявник (власник реєстраційного посвідчення) дійшов висновку, що:
- сигнал був спростований на підставі наявного доказу, що свідчив проти причинно-наслідкового зв’язку;
- сигнал став ідентифікованим ризиком (тобто проблемою безпеки, для якої існує достатньо наукових підстав вважати, що він спричинений лікарським засобом);
- сигнал став потенційним ризиком (тобто проблемою безпеки, для якої існує наукова підстава підозрювати можливість причинно-наслідкового зв’язку з лікарським засобом, але існує недостатньо доказів, щоб зробити висновок, що причинно-наслідковий зв’язок існує).
Коли оцінка сигналу завершена, та заявник (власник реєстраційного посвідчення) визначив сигнал як ідентифікований або потенційний ризик, слід представити відповідне обґрунтування того, чому сигнал/проблема буде/не буде впливати на співвідношення користі/ризику. Оцінки, в яких надано недостатньо роз’яснень, можуть викликати питання. Наприклад, бувають ситуації, коли в оцінці сигналу згадуються летальні випадки, але жодних додаткових роз’яснень не надається, що викликає низку питань у провідної держави ЄС (LMS).
Крім важливої інформації, що вже надана в таблиці сигналів, у розділі з оцінки (розділ 16.2) за необхідності слід надавати мінімум інформації, що перерахована нижче. У цілому інформація, що надана у цьому розділі РОЗБ, повинна забезпечувати чітке обґрунтування висновків заявників (власників реєстраційних посвідчень) щодо:
- опису сигналу;
- джерела виникнення сигналу;
- вихідних даних, що мають значення для оцінки;
- методів оцінки, включаючи джерела даних та критерії пошуку;
- результатів:
- резюме та критичний аналіз даних, що вважаються основними в оцінці сигналу, включаючи огляд випадків (коли це доречно та є невід’ємною частиною оцінки);
- на підставі обґрунтованого кумулятивного огляду та аналізу;
- висновку за результатами оцінки, включаючи необхідність подальшої оцінки та заходів, що вжиті або заплановані.
Оцінка даних є основою для будь-якого рішення та може призвести до закриття сигналу або продовження спостереження. В принципі закритий сигнал не повинен приводити до додаткового моніторингу через превентивні причини, за умови, що PRAC погоджується з його оцінкою. З цього моменту застосовуватиметься рутинний фармаконагляд. Чітке обґрунтування закриття сигналу слід надати разом з даними, що підтверджують висновок (наприклад загальний (кумулятивний) огляд та аналіз) у розділі «Оцінка сигналу». Модуль II ННПФ у розділі В.5.16.2 повинен містити оцінки та висновки заявника (власника реєстраційного посвідчення) відносно закритих сигналів з чітким обґрунтуванням.
У РОЗБ увагу слід зосередити на узагальненій інформації, науковій оцінці безпеки та інтегрованій оцінці співвідношення користі/ризику. Отже, перерахування або описи індивідуальних випадків, у принципі, заявник (власник реєстраційного посвідчення) не повинен систематично включати78, так само LMS не повинна запитувати їх, якщо тільки вони не є невід’ємною частиною наукового аналізу або проблеми, що пов’язана з безпекою. У цьому контексті термін «опис випадку» стосується скоріше клінічних оцінок індивідуальних випадків, ніж описів CIOMS79. У такому випадку клінічна оцінка важливих або ілюстративних випадків у контексті оцінки проблеми, що пов’язана з безпекою/сигналу, може бути важливішою.
Однак у певних ситуаціях для заявників (власників реєстраційних посвідчень) може бути важливим надати, а для експерта — запитати детальний опис основних або ілюстративних випадків, включаючи резюме описів випадків. У таких ситуаціях пошуки в базах даних про безпеку та в літературі повинні включати всі релевантні терміни стосовно сигналу. Стратегія пошуку та терміни, що включені, слід чітко зазначити. Особливу увагу слід приділити визначенню того, чи може сигнал стосуватися медичних концепцій з різною, дуже специфічною термінологією. Прикладами є гематофагоцитарний гістіоцитоз та синдром активації макрофагів, ювенільний ідіопатичний артрит та хвороба Стіла. Визначення найбільш доречних критеріїв пошуку для оцінки сигналу вимагає медичного та наукового обґрунтування.
9.1. Резюме проблем безпеки
Слід надати резюме важливих проблем безпеки, для діючої речовини (АФІ)/речовин для РОЗБ відомих на початку звітного періоду, враховуючи, що РОЗБ є документом, який надається на глобальному рівні, та тому може вимагатися включення додаткових проблем безпеки, що запитують неєвропейські регуляторні органи. Документ міжнародної конференції з гармонізації ICH E2C (R2) «Питання та відповіді» (13.4) пропонує один підхід до вирішення питання, коли проблеми безпеки відрізняються між країнами або регіонами.
Для лікарського засобу, для якого існує ПУР у ЄС, очікується як мінімум включення резюме проблем безпеки, що описані у ПУР ЄС на початку звітного періоду. У локальному додатку ЄС у розділі VII.C.5.3 модуля ННПФ слід зазначити номер версії ПУР, а також надати роз’яснення будь-яких відмінностей або додаткових проблем безпеки в розділі 16.1 РОЗБ порівняно з ПУР ЄС.
Для лікарського засобу, для якого немає ПУР у ЄС, слід враховувати аспекти, що перераховані нижче, з метою визначення, що означає важливий ідентифікований або потенційний ризик або відсутня інформація (тобто проблеми безпеки). Увагу слід приділити тим важливим ідентифікованим/потенційним ризикам, що є критичними та впливають на співвідношення користі/ризику.
- Цей розділ не повинен бути вичерпним переліком усіх побічних реакцій, що внесені до ОІБЗ на лікарський засіб, та тому може бути дуже стислим (наприклад, для добре вивчених лікарських засобів, для яких важливі ризики відсутні завдяки повній характеристиці профілю безпеки). У ньому слід зосередитися на важливих проблемах безпеки/відсутній інформації на початку звітного періоду.
- Для лікарського засобу, для якого немає ПУР у ЄС, слід надати обґрунтування кожного включення. Для лікарських засобів, що не мають ПУР у ЄС, модуль VII ННПФ встановлює, що у розділі 16.1 заявники (власники реєстраційних посвідчень) повинні надати інформацію про важливі ідентифіковані та потенційні ризики, а також відсутню інформацію. Досвід застосування єдиної оцінки РОЗБ демонструє, що дуже «старі» лікарські засоби, що зареєстровані за старими регуляторними процедурами, можуть включати профілі безпеки, які не повністю визначені. Отже, наданої інформації має бути достатньо для обґрунтування проблем безпеки, вона має базуватися на наукових та клінічних підставах.
У розділі 16.1 РОЗБ надається резюме проблем безпеки, що існують на початку звітного періоду та відносно яких можна порівнювати нову інформацію та здійснювати оцінку. Якщо нові проблеми безпеки були додані до специфікації безпеки ПУР у ЄС під час звітного періоду, що охоплюється РОЗБ, їх слід охарактеризувати в розділі 16.4 та можна прокоментувати як «нові» у відповідному підрозділі. Якщо нова проблема безпеки ідентифікована, наприклад як результат оцінки сигналу, що включений до РОЗБ, але все ще проходить процедуру внесення зміни для ПУР у ЄС, пропозицію щодо додання слід включити до висновків регулярного звіту з безпеки. Цей підхід відповідатиме як настанові ICH E2C (R2) [28], так і модулю VII ННПФ. Якщо перекласифікація або видалення підтверджено під час звітного періоду РОЗБ, це слід зазначити у розділі 16.4 або у висновках, якщо необхідно.
Як правило, РОЗБ по суті не є інструментом гармонізації переліку проблем безпеки для всіх лікарських засобів, що містять однакову (і) діючу (і) речовину (и), незалежно від того, має чи не має лікарський засіб пов’язаного ПУР у ЄС. Однак, якщо оцінка РОЗБ визначає новий важливий потенційний або новий важливий ідентифікований ризик або відсутню інформацію, можна рекомендувати, щоб усі заявники (власники реєстраційних посвідчень) включали цей певний ризик до специфікації з безпеки ПУР, що вже існує, для забезпечення того, щоб вперше виявлена проблема з безпеки відповідно управлялася та вирішувалася в майбутніх регулярних звітах з безпеки на даний (і) лікарський засіб (засоби). Будь-які з таких вимог повинні виконуватися всіма власниками реєстраційних посвідчень для забезпечення відповідної оцінки нових проблем безпеки для всіх лікарських засобів при наступній єдиній оцінці. Коли немає ПУР у ЄС, але новий ПУР слід розробити, запит робитиметься в індивідуальному порядку. Коли ПУР для таких лікарських засобів існують, належну увагу слід приділити також з метою внесення змін до специфікації з безпеки у ПУР за допомогою відповідної процедури внесення змін.
Якщо важливі проблеми з безпеки необхідно включити або поновити у специфікації з безпеки ПУР для лікарських засобів, зареєстрованих на національному рівні, їх слід включити як зміну, що вноситься на підставі проведення процедури єдиної оцінки РОЗБ згідно з Постановою Комісії 1234/2008 [8] щодо настанови з класифікації змін. Слід розглянути тільки специфічну проблему безпеки із застосуванням загальної термінології для всіх лікарських засобів, яких стосується процедура єдиної оцінки РОЗБ.
9.2. Оцінка ризиків та нової інформації
Метою цього розділу є надання нової інформації, що має значення для раніше відомих ризиків та стала відомою під час звітного періоду (наприклад, інформація, що отримана у дослідженнях для додаткової характеристики важливого потенційного ризику); у розділі не надається уся наявна інформація стосовно переліку проблем безпеки. Немає необхідності включати докладне обговорення інформації, що стала відомою за звітний період, що лише підтверджує встановлений профіль безпеки або характеристику вже відомих ризиків лікарського засобу.
Заявник (власник реєстраційного посвідчення) повинен не тільки представити нові дані, що стали доступними за період, коли був встановлений сигнал. Також очікується, що заявник (власник реєстраційного посвідчення) представить нові дані, що стали доступними за звітний період та акцентує увагу на змінах у профілі безпеки, без дублювання розділу щодо сигналу та розділу щодо ризику.
Заявники (власники реєстраційних посвідчень) повинні враховувати вплив нової інформації на співвідношення користі/ризику свого(їх) лікарського (их) засобу (ів), зокрема на перелік проблем безпеки та/або діяльності з фармаконагляду/мінімізації ризику, та забезпечити рівень деталізації пропорційний рівню ризику. По суті особливу увагу слід приділяти важливим потенційним ризикам та тому, чи можуть нові дані підтвердити ці ризики. Увага, що приділяється аналізу нових даних, має приділятися в контексті відомої та сукупної інформації (тобто інформацію про випадки, яка надійшла під час звітного періоду, слід аналізувати в контексті узагальненої та вже відомої інформації). Якщо немає жодних нових важливих даних, необхідно це зазначити.
9.3. Характеристика ризиків
У цьому розділі слід відображати характеристику важливих ідентифікованих та/або потенційних ризиків для лікарського засобу, що базується на кумулятивних даних (тобто не тільки на інформації, що отримана під час звітного періоду) та також описувати важливу відсутню інформацію, що пов’язана із застосуванням лікарського засобу. Як і інші розділи РОЗБ, цей розділ слід готувати, застосовуючи дані, до яких заявник (власник реєстраційного посвідчення) обґрунтовано міг мати доступ, та які мають значення для характеристики ризиків. Для лікарських засобів, що не мають ПУР ЄС, заявник (власник реєстраційного посвідчення) може запропонувати зміни до переліку проблем безпеки на підставі інформації, що виникає під час періоду оцінки (наприклад запроваджені успішні заходи мінімізації ризиків). Коли важливий ризик або відсутня інформація перекласифікуються або видаляються, обґрунтування слід надати у цьому розділі.
9.4. Ефективність заходів з мінімізації ризику (якщо застосовуються)
Результати оцінки ефективності вжитих заходів з мінімізації ризиків слід представити в локальному додатку ЄС/УкраїниN (розділ VII.C.5.5).
На підставі цієї оцінки заявник (власник реєстраційного посвідчення) повинен запропонувати запровадження додаткових заходів/змін до існуючих заходів та/або врахувати значимість збереження або видалення відповідної проблеми безпеки.
10. Оцінка аналізу співвідношення користі/ризику
Як правило, той принцип, що на початку періоду РОЗБ співвідношення користі/ризику лікарського засобу є позитивним, базується на даних, що були оцінені під час первинного реєстраційного посвідчення та наступних оцінок його співвідношення користі/ризику, таких як перереєстрація та РОЗБ. У РОЗБ звертають увагу на оцінку нових даних про безпеку та ефективність, що були отримані під час періоду, що розглядається, у контексті сукупного досвіду застосування лікарського засобу, його місця у терапії та того, чи ця інформація впливає на загальне співвідношення користі/ризику лікарського засобу.
Визнано, що увагу при оцінці РОЗБ слід приділяти тому, чи існують нові важливі ризики, або чи змінилися вже визнані важливі ризики, або чи є зміни у співвідношенні користі/ризику лікарських засобів.
Коли існує нова позитивна інформація про користь та не існує жодних змін у профілі ризику у звітний період, опис об’єднаної вихідної та нової інформації має бути стислим. Заявник (власник реєстраційного посвідчення) не повинен включати нову інформацію щодо ефекту/ефективності, що тільки підтверджує те, що вже було відомо щодо лікарського засобу. У цій ситуації повна переоцінка вихідних даних про ефективність не виправдана. Слід враховувати тільки зміни за звітний період.
Відсутність ефективності або дослідження, що піддають сумніву встановлений профіль ефективності, слід обговорювати у розділі 7 або 13 РОЗБ відповідно до модуля VII ННПФ. Коли існує нова важлива інформація, що вказує на відсутність ефективності, виправданий детальний аналіз співвідношення користі/ризику.
Висновки за оцінкою РОЗБ не слід робити на підставі доказів ефективності застосування за новими показаннями, для яких заявник (власник реєстраційного посвідчення) повинен надавати заяву за відповідною процедурою.
Хоча РОЗБ мають відповідні регуляторні можливості для обмеження/призупинення/анулювання реєстраційного посвідчення на підставі причин, пов’язаних з безпекою, припускається, що необхідність у ширшому залученні до ретельного наукового аналізу краще реалізовувати за посередництвом альтернативної процедури (наприклад процедури передачі на розгляд до іншої інстанції).
11. Оцінка та результат
Оцінка РОЗБ є критичним аналізом, що зосереджений на оцінці нових даних про безпеку та ефективність, що були отримані під час періоду, що розглядається. Рівень деталізації, що забезпечується у певних розділах РОЗБ, повинен залежати від відомих або нових даних щодо важливої користі та ризиків лікарського засобу. Тому масштаб інформації, що надається, варіюватиме між окремими РОЗБ. Наприклад, коли існує нова важлива інформація про безпеку, слід включити докладне представлення цієї інформації плюс релевантну інформацію про користь з метою полегшення робастного аналізу співвідношення користі/ризику. Навпаки, коли стало відомо небагато нової інформації про безпеку за звітний період, буде достатньо стислого резюме вихідної інформації про користь, а оцінка співвідношення користі/ризику включатиме в основному оцінку оновлених даних про безпеку за звітний період. Запити, що адресовані власнику реєстраційного посвідчення у попередньому звіті про оцінку або для наступного регулярного звіту з безпеки, будуть ґрунтуватися на врахуванні ризику, та того, як запит, як і графік відповіді на нього, буде обґрунтовуватися у звіті про оцінку та, у міру можливості, буде частиною діючого або наступного РОЗБ. Спостерігається негативна кореляція між рівнем та якістю інформації, що надається у РОЗБ, та кількістю й характером запитів, що зроблені в контексті оцінки.
Зазвичай запити додаткової інформації здійснюються на 60-й день процедури, при цьому очікується, що вони будуть задовільнені протягом 30 днів, а висновки будуть зроблені під час процедури. У виключних випадках, коли це неможливо, наприклад, коли запитувані дані не можна зібрати протягом 30-денного періоду, передбаченого для подання коментарів, можна застосовувати інші процедури. Процес обробки при поданні додаткових даних чітко визначатиметься у звіті з оцінки.
Запитів табличних переліків та звітів CIOMS, як правило, слід уникати, якщо тільки вони відповідним чином не обґрунтовані. Перед тим, як запитувати сукупний огляд або табличні переліки та описи CIOMS, оцінювач повинен уважно обміркувати, чи запропонований запит надаватиме або скоріше надаватиме значиму інформацію (наприклад чи запит про необхідність надання сукупного огляду для лікарського засобу із встановленим профілем безпеки, коли доступні тільки спонтанні дані, дозволить виявити будь-яку нову важливу інформацію, що може призвести до змін в інформації про лікарський засіб).
Гармонізація інформації про лікарський засіб не є метою, оскільки, за необхідності, можна використовувати доречніші процедури, такі як внесення змін, що пов’язані з розподілом навантаження, або процедура передачі на розгляд до іншої інстанції відповідно до ст. 30 Директиви 2001/83/ЄС [1]. Заявники (власники реєстраційних посвідчень) повинні забезпечувати постійне оновлення своєї інформації про лікарський засіб та надавати заяви на внесення змін за відповідною регуляторною процедурою для підтримки координованості своєї інформації про лікарський засіб. Однак у контексті процедури єдиної оцінки РОЗБ можна ввести загальне формулювання, що придатне для всіх лікарських засобів, що охоплюються РОЗБ, коли науковий розгляд певної проблеми безпеки призводить до того, що PRAC/ЦентрN рекомендує привести інформацію про лікарський засіб у відповідність. PRAC/ЦентрN рекомендуватиме формулювання, що придатне для усіх лікарських засобів, та не може бути жодної диференціації за лікарським засобом/компанією, якщо тільки існують специфічні та обґрунтовані причини (наприклад лікарська форма). Однак, за необхідності, слід враховувати відмінності у показанні та/або складі лікарських засобів. Формулювання, що встановлене в рекомендації PRAC/УкраїниN, слід включати цілком. Якщо певне формулювання вже існує в інформації про лікарський засіб для певних лікарських засобів, його слід замінити на нове формулювання, за винятком випадків, коли існує дуже подібне формулювання та його розміщення в інформації про лікарський засіб є таким самим, як у рекомендації PRAC/УкраїниN.
12. Системи якості для регулярних звітів з безпеки на рівні власників торгової ліцензії
Законним обов’язком заявників (власників реєстраційних посвідчень) є подання РОЗБ, що містять резюме даних, що стосуються користі та ризиків лікарського засобу, та наукову оцінку співвідношення користі/ризику лікарського засобу, враховуючи всі наявні дані.
Для уможливлення адекватної оцінки РОЗБ дуже важливо, щоб інформація, яку надає заявник (власник реєстраційного посвідчення), була достатньо належної якості.
Від заявників (власників реєстраційних посвідчень) також вимагається забезпечення адекватного рівня та якості інформації та аналізів під час процедури у відповідь на запит додаткової інформації у попередньому звіті про оцінку або при наданні додаткових даних для попереднього РОЗБ.
У розпорядженні необхідно мати відповідну систему якості для уникнення невідповідності вимогам до РОЗБ, таким як неможливість надати адекватні відповіді на запити уповноважених органів.
Значні проблеми, що пов’язані з якістю даних РОЗБ, можна також виділити для подальшого контролю питання якості та відповідності, що слід покращити при поданні наступного РОЗБ, та може бути причиною для подальшої інспекції з фармаконагляду у ЄС та аудиту системи фармаконагляду уповноваженою установоюN.
Заявник (власник реєстраційного посвідчення) повинен усвідомлювати, що будь-яка виявлена невідповідність буде висвітлена при оцінці РОЗБ, а подальші дії обговорюватимуться на рівні ЄС/УкраїниN.
Ключові слова: безпека застосування лікарських засобів, належна практика фармаконагляду, настанова, РОЗБ, фармаконагляд.
Частина VІІІ
Модуль VIІІ — Післяреєстраційні дослідження з безпеки
VIII.A. Вступ
Регламент (ЄC) №726/2004 [5], Директива 2001/83/ЕС [1], ІП 520/2012 [6] та положення Порядку [2]N включають положення про ПДБЛЗ, що застосовуються в ЄС та УкраїніN.
У ст. 1(15) Директиви 2001/83/ЄС [1], положеннях Порядку [2]N ПДБЛЗ визначається як будь-яке дослідження зареєстрованого лікарського засобу, що проводиться з метою визначення, характеристики або кількісного визначення ступеня загрози безпеці, підтвердження профілю безпеки лікарського засобу або оцінки ефективності заходів з управління ризиками.
ПДБЛЗ може бути інтервенційним або неінтервенційним. Цей модуль стосується інтервенційних та неінтервенційних ПДБЛЗ, однак головна увага приділяється неінтервенційним дослідженням. Він не стосується доклінічних випробувань з безпеки.
Неінтервенційними ПДБЛЗ, що розглядаються у цій настанові, є дослідження, що ініційовані, контролюються або фінансуються заявником (власником реєстраційного посвідчення) добровільно або згідно із зобов’язанням, що встановлюється уповноваженим органом ЄС та УкраїниN (ст. 107m(1) Директиви 2001/83/ЄС [1], ст. 28b Регламенту (ЄC) №726/2004 [5], положення Порядку [2]N).
Неінтервенційні ПДБЛЗ, що розглядаються, можуть:
- встановлюватися як зобов’язання відповідно до ст. 9(4)(cb) та ст. 10а(1)(а) Регламенту (ЄC) № 726/2004 [5], ст. 21a(b) та 22а(1)(a) Директиви 2001/83/ЄС [1], положень Порядку [2], положень Порядку проведення експертизи реєстраційних матеріалів [7]N (дослідження категорії 1 у модулі V ННПФ);
- встановлюватися як спеціальні зобов’язання та є умовою реєстрації за виняткових обставин (дослідження категорії 2 у модулі V ННПФ);
- вимагатися у ПУР для вивчення питань безпеки або оцінки ефективності дій з мінімізації ризиків (категорія 3 досліджень у модулі V ННПФ);
- проводитися з власної ініціативи заявника (власника реєстраційного посвідчення).
Неінтервенційні ПДБЛЗ повинні проводитися відповідно до таких положень:
- ст. 107m Директиви 2001/83/ЄС [1], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7]N для ПДБЛЗ, що ініційовані, контролюються або фінансуються заявником (власником реєстраційного посвідчення) добровільно або відповідно до встановлених зобов’язань;
- ст. 107n-q Директиви 2001/83/ЄС [1], ст. 28b Регламенту (ЄC) №726/2004 [5], ст. 36-38 ІП 520/2012 [6], положення Порядку [2], положення Порядку проведення експертизи реєстраційних матеріалів [7]N для неінтервенційних ПДБЛЗ, що проводяться відповідно до зобов’язання, що встановлюється уповноваженим органом ЄС та/або УкраїниN (категорії 1 та 2 досліджень у модулі V НПФ).
ПДБЛЗ є неінтервенційним, якщо виконуються в сукупності такі вимоги (том 10 Правил регулювання обігу лікарських засобів в ЄС, питання та відповіді, версія 11.0, 15 травня 2013 р., питання 1.1080, положення Порядку [2]N):
- лікарський засіб призначається звичайним способом відповідно до умов реєстраційного посвідчення;
- залучення пацієнта до групи з визначеним методом лікування в протоколі дослідження заздалегідь не передбачено, а призначення лікарського засобу здійснюється відповідно до вимог сучасної медичної практики та не залежить від рішення про залучення пацієнта до дослідження; та
- пацієнтам не призначаються жодні додаткові діагностичні або моніторингові процедури, і для аналізу зібраних даних використовуються епідеміологічні методи.
Неінтервенційні дослідження визначаються методологічним підходом, що використовується, а не їх науковими цілями. Неінтервенційні дослідження включають дослідження баз даних або аналіз облікової документації, коли явища, що розглядаються, вже сталися (це можуть бути дослідження випадок-контроль, перехресні дослідження, когортні або інші види досліджень, що повторно використовують дані). Неінтервенційні дослідження також охоплюють дослідження, пов’язані з первинним збором даних (наприклад, проспективні обсерваційні дослідження та реєстри, в яких дані збирають у ході рутинної клінічної практики) за умови виконання вищезазначених положень. У цих дослідженнях опитування, анкетування, забір зразків крові та спостереження пацієнтів можуть проводитися як частина рутинної клінічної практики.
Якщо ПДБЛЗ є інтервенційним випробуванням, тоді слід дотримуватися положень Директиви 2001/20/ЄС [15], положень Порядку проведення клінічних випробувань [16]N і Тому 10 Правил регулювання обігу лікарських засобів в Європейському Союзі81.
Мета даного модуля полягає у такому:
- надання загальних методичних рекомендацій щодо прозорості, наукових стандартів та стандартів якості неінтервенційних ПДБЛЗ, що проводяться заявниками (власниками реєстраційних посвідчень) за власною ініціативою або згідно із зобов’язаннями, встановленими уповноваженим органом ЄС та/або УкраїниN (див. VIII.В);
- опис процедур, згідно з якими уповноважений орган ЄС та/або УкраїниN може зобов’язати заявника (власника реєстраційного посвідчення) провести ПДБЛЗ (див. VIII.C.1.);
- опис процедур неінтервенційних ПДБЛЗ, що проводяться згідно із зобов’язаннями, встановленими уповноваженим органом ЄС та/або УкраїниN для контролю дотримання протоколу та повідомлення результатів (див. VIII.С.2), а також для внесення змін до реєстраційних матеріалів на лікарські засоби протягом строку дії реєстраційних посвідчень внаслідок отриманих результатів (див. VIII.С.3).
Також застосовуються національні вимоги та вимоги Європейського Союзу для забезпечення благополуччя та прав учасників неінтервенційних ПДБЛЗ (ст. 107m(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
У розділі VIII.B. деякі нормативно-правові вимоги, що є обов’язковими для неінтервенційних ПДБЛЗ, проведених відповідно до зобов’язання, що встановлює уповноважений орган ЄС та/або УкраїниN, рекомендовані для неінтервенційних ПДБЛЗ, що проводяться заявником (власником реєстраційного посвідчення) за власною ініціативою з метою додержання однакового рівня прозорості, наукових стандартів та стандартів якості. Це стосується, наприклад, формату та змісту протоколу дослідження, заключного звіту дослідження та його резюме.
Для неінтервенційних ПДБЛЗ ця настанова застосовується для досліджень, у яких здійснюється первинний збір даних з безпеки безпосередньо від пацієнтів, працівників з медичною та/або фармацевтичною освітою, а також тих, хто повторно застосовує дані, що раніше були зібрані у пацієнтів, працівників з медичною та/або фармацевтичною освітою з іншою метою.
VIII.B. Структури та процеси
VIII.B.1. Принципи
Згідно зі ст. 1(15) Директиви 2001/83/ЄС [1] післяреєстраційне дослідження слід класифікувати як ПДБЛЗ, якщо основна мета ініціалізації дослідження включає будь-яку із зазначених нижче:
- кількісне визначення потенційних або виявлених ризиків (наприклад, характеристика рівня захворюваності, оцінка показників співвідношення ризиків або відмінності ризиків, порівняно з групою досліджуваних, які не застосовували лікарський засіб, або застосовували інший лікарський засіб або лікарський засіб, що відноситься до іншої групи за АТС-класифікацією, в залежності від обставин) та дослідження факторів ризику, у тому числі модифікаторів ефекту;
- оцінка ризиків лікарського засобу, що застосовується у групі пацієнтів, для яких інформація з безпеки обмежена або відсутня (наприклад вагітні, особливі вікові групи, пацієнти з порушенням функції нирок або печінки або інші значущі супутні захворювання або одночасне застосування інших лікарських засобів);
- оцінка ризиків лікарського засобу після тривалого застосування;
- отримання доказів відсутності ризиків;
- оцінка схем застосування лікарського засобу, що доповнюють знання про безпеку лікарського засобу або ефективність заходів з мінімізації ризиків (наприклад, збір інформації про показання, застосування не за показанням, дозування, одночасне застосування інших лікарських засобів або помилки, що пов’язані із застосуванням лікарського засобу у клінічній практиці, що можуть впливати на безпеку, а також дослідження, що забезпечують оцінку впливу будь-якої проблеми з безпеки на громадське здоров’я);
- оцінка ефективності заходів управління ризиками.
Зважаючи на те, що дизайн ПДБЛЗ має відповідати меті та завданням дослідження, класифікація післяреєстраційного дослідження як ПДБЛЗ у випадку, коли воно відповідає критеріям, встановленим у ст. 1(15) Директиви 2001/83/ЄС [1], положеннях Порядку [2]N, не обмежується тільки видом обраного дизайну. Наприклад, в залежності від мети проведення систематичний огляд літератури чи метааналіз можуть вважатися ПДБЛЗ.
Заявникам (власникам реєстраційних посвідчень) та дослідникам при розробці протоколів дослідження, проведенні дослідження та написанні звітів про дослідження, а також PRAC та національним уповноваженим органам при оцінці протоколів та звітів про дослідження, слід орієнтуватися на відповідні наукові настанови. Такі настанови включають Настанову з методологічних стандартів у фармакоепідеміології ENCePP (Європейська мережа центрів фармакоепідеміології та фармаконагляду)82, Контрольний перелік протоколів дослідження ENCePP83, Настанову з проведення фармаконагляду лікарських засобів, що застосовуються педіатричною популяцією84 та Настанову з належної фармакоепідеміологічної практики Міжнародної спільноти з фармакоепідеміології (ISPE GPP)85.
Стосовно досліджень, що фінансуються заявником (власником реєстраційного посвідчення), включно з дослідженнями, що розробляються, проводяться чи аналізуються повністю або частково дослідниками, які не є співробітниками заявника (власника реєстраційного посвідчення), заявник (власник реєстраційного посвідчення) повинен гарантувати, що дослідники мають відповідний рівень освіти, підготовки та досвід для виконання своїх завдань. Договір на проведення дослідження між заявником (власником реєстраційного посвідчення) та дослідниками повинен гарантувати відповідність дослідження до регуляторних зобов’язань, та дозволити застосовувати наукові знання та досвід дослідників протягом усього процесу дослідження. У договорі про дослідження заявнику (власнику реєстраційного посвідчення) слід врахувати положення Кодексу ділової етики ENCePP86 та прописати наступні аспекти:
- обґрунтування, основні цілі та короткий опис запланованих методів дослідження, що повинні здійснюватися дослідником(ами);
- права та обов’язки дослідника(ів) та заявника (власника реєстраційного посвідчення);
- чіткий розподіл завдань та обов’язків;
- процедуру погодження протоколу дослідження;
- положення щодо виконання зобов’язань з фармаконагляду заявником (власником реєстраційного посвідчення), включаючи повідомлення дослідниками про побічні реакції та інші дані з безпеки, коли необхідно;
- права інтелектуальної власності, що виникають внаслідок дослідження та доступ до даних дослідження;
- зберігання та доступ до масиву аналітичних даних та статистичних програм для аудиту, інспекцій для країн ЄС та аудитів системи фармаконагляду уповноваженою установоюN;
- стратегію комунікації, що пов’язана з виконанням запланованого графіку проведення дослідження та заключними звітами;
- стратегію оприлюднення проміжних та кінцевих результатів.
Неінтервенційні ПДБЛЗ не повинні проводитися, якщо акт проведення дослідження заохочує до застосування лікарського засобу (ст. 107m(3) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Ця вимога стосується усіх досліджень та всієї діяльності, що здійснюється в рамках дослідження, включаючи дослідження, що проводяться персоналом заявника (власника реєстраційного посвідчення) і третіми сторонами від імені заявника (власника реєстраційного посвідчення).
Винагорода медичному персоналу за участь у дослідженні повинна обмежуватися компенсацією за час та понесені витрати (ст. 107m(4) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
VIII.B.2. Реєстрація дослідження
Для неінтервенційних ПДБЛЗ, що проводяться на виконання зобов’язань, встановлених уповноваженим органом ЄС, до заключного звіту про дослідження слід включати дату реєстрації дослідження в електронному реєстрі досліджень в якості важливого етапу (додаток III ІП 520/2012 [6]). Для цього Реєстр післяреєстраційних досліджень ЄС (EU PAS Register), що адмініструє EMA, та доступ до якого надається за посередництвом європейського вебпорталу лікарських засобів, слугує електронним реєстром досліджень87.
З метою підтримки прозорості усіх неінтервенційних ПДБЛЗ та для сприяння обміну інформацією з фармаконагляду між EMA, національними уповноваженими органами та заявниками (власниками реєстраційних посвідчень), заявники (власники реєстраційних посвідчень) повинні також вносити до Реєстру післяреєстраційних досліджень ЄС інформацію про усі неінтервенційні ПДБЛЗ, що вимагаються ПУР, що узгоджений в ЄС, або проводяться за власною ініціативою в ЄС.
Неінтервенційне ПДБЛЗ слід реєструвати у Реєстрі післяреєстраційних досліджень ЄС перед початком дослідження або якомога раніше, наприклад, якщо вже розпочато збір даних для дослідження, що включено до плану управління ризиками. Протокол дослідження слід вносити до реєстру якомога скоріше після їх завершення та до початку збору даних. До реєстру слід також вносити оновлення протоколу дослідження у випадку значних змін до нього, коли доцільно — звіти про хід дослідження, а також заключний звіт про результати дослідження (якомога швидше та бажано протягом двох тижнів після їх оформлення). Інформацію про дослідження слід, як правило, надавати англійською мовою. Якщо протокол дослідження або звіт про дослідження написані іншою мовою, заявник (власник реєстраційного посвідчення) повинен додати переклад англійською мовою назви, резюме протоколу дослідження та резюме заключного звіту дослідження.
В Україні вимоги до подання перекладу на українську мову протоколу, звіту про дослідження визначені положеннями Порядку [2]N. Якщо попередня публікація протоколу може загрожувати валідності дослідження (наприклад у дослідженнях із первинним збором даних, коли знання про мету дослідження може призвести до помилок в оцінці інформації) або захисту прав інтелектуальної власності, до реєстру до початку збору даних може вноситися протокол дослідження з редагуванням (вилученням інформації, що вимагає захисту від розголошення), зробленим заявником (власником реєстраційного посвідчення). Таке редагування повинно бути обґрунтованим та зведеним до необхідного мінімуму для досягнення мети редагування. У випадку, коли до початку збору даних реєструється відредагований протокол дослідження, на титульній сторінці протоколу потрібно зазначити «відредагований протокол», а на вимогу EMA або національних уповноважених органів повинна надаватися повна версія протоколу дослідження. Повну версію протоколу дослідження слід вносити до реєстру (найкраще впродовж двох тижнів після завершення збору даних).
VIII.B.3. Протокол дослідження
Неінтервенційні ПДБЛЗ, що проводяться за зобов’язанням, встановленим уповноваженим органом ЄС та/або УкраїниN, або проводяться за власною ініціативою, повинні мати у письмовому вигляді протокол дослідження. Протокол дослідження повинен розроблятися особами з відповідним науковим досвідом та освітою. Огляд дизайнів дослідження та баз даних, що часто застосовуються у ПДБЛЗ, надається у модулі VIII. Додаток 1.
Стосовно неінтервенційних ПДБЛЗ, що встановлюються як зобов’язання, проєкт протоколу дослідження повинен надаватися заявником (власником реєстраційного посвідчення) до PRAC або національного уповноваженого органу країни ЄС/УкраїниN, що запитувала дослідження, якщо дослідження проводиться тільки в одній країні ЄС/УкраїниN (ст. 107n(1) Директиви 2001/83/ЄС [1]) (див. розділ VIII.С.2). В Україні заявник (власник реєстраційного посвідчення) подає до Центру проєкт протоколу до проведення дослідження з безпеки (положення Порядку [2])N.
Національний уповноважений орган може вимагати від заявника (власника реєстраційного посвідчення) надання протоколу дослідження, що надавався до уповноважених органів країн ЄС, у яких проводиться дослідження (ст. 107m(5) Директиви 2001/83/ЄС [1]). Вимоги та рекомендації щодо подання протоколу дослідження до EMA, національних уповноважених органів та уповноваженого органу в УкраїніN зазначені у додатку І до модуля VIII ННПФ, положеннях Порядку [2] та додатку 15 до Порядку [2]N.
З метою забезпечення дотримання заявником (власником реєстраційного посвідчення) зобов’язань з фармаконагляду слід, щоб УОВФ/КОВФN, або особа, яка її заміщує (див. модуль І ННПФ), брала участь в експертизі та підписанні протоколів досліджень, що вимагаються у ПУР, що узгоджений у ЄС/УкраїніN, або проводяться за власною ініціативою в ЄС/УкраїніN (див. модуль I НПФ).
За необхідності КОВФ заявника (власника реєстраційного посвідчення) на національному рівні слід інформувати про будь-яке дослідження, що спонсорується або проводиться заявником (власником реєстраційного посвідчення) у цій країні ЄС, та надати доступ до протоколу.
VIII.B.3.1. Формат та зміст протоколу дослідження
Стосовно неінтервенційних ПДБЛЗ, що проводяться за зобов’язанням, встановленим уповноваженим органом ЄС/УкраїниN, протокол дослідження має бути викладений у форматі, що описаний у цьому розділі (додаток III ІП 520/2012 [6], додаток 15 до Порядку [2]N). Цього формату також повинні дотримуватися у неінтервенційних ПДБЛЗ, що вимагаються у ПУР в ЄС/УкраїніN або проводяться за власною ініціативою в ЄС/Україні.
1. Назва: інформативна назва, що містить загальновживаний термін, що вказує на дизайн дослідження та лікарський засіб, діючу речовину або відповідну фармакологічну групу; у підзаголовку слід зазначити ідентифікатор версії та дату останньої версії. Якщо протокол дослідження був зареєстрований у Реєстрі післяреєстраційних досліджень ЄС, на титульній сторінці наступних версій протоколу слід зазначити «Номер у реєстрі EU PAS» разом з реєстраційним номером.
2. Заявник (власник реєстраційного посвідчення): назва та адреса заявника (власника реєстраційного посвідчення).
3. Відповідальні сторони: прізвища, ім’я, по батькові, посади, кваліфікація, адреси та підпорядкованість (назва організації) всіх основних відповідальних сторін, включаючи основного(их) автора(ів) протоколу, головного дослідника, дослідника-координатора у кожній країні, в якій буде проводитися дослідження, та для інших відповідних баз дослідження. Перелік усіх партнерських установ та дослідницьких структур повинен надаватися EMA та національним уповноваженим органам на вимогу.
4. Резюме: резюме протоколу дослідження у вигляді окремого документа, що містить такі підрозділи:
- заголовок з підзаголовками, включаючи версію та дату протоколу, а також прізвище, ім’я, по батькові та підпорядкованість (назва організації) головного автора;
- обґрунтування та історія питання;
- предмет дослідження та цілі;
- дизайн дослідження;
- досліджувана популяція;
- змінні;
- джерела даних;
- масштаб дослідження;
- аналіз даних;
- основні етапи.
5. Зміни та оновлення: будь-яка значна зміна та оновлення до протоколу дослідження після початку збору даних, включаючи обґрунтування кожної зміни або оновлення, дату кожної зміни та посилання на розділ протоколу, до якого було внесено зміну.
6. Основні етапи: таблиця із запланованими датами для таких основних етапів:
- початок збору даних;
- завершення збору даних;
- звіт(и) про хід дослідження, як вказано у статті 107m(5) Директиви, положеннях Порядку [2]N (див. VIII.B.4.3.1.);
- проміжний(і) звіт(и) про результати дослідження, коли доцільно, відповідно до етапів аналізу даних (див. VIII.B.4.3.1.);
- заключний звіт про результати дослідження (див. VIII.B.4.3.2.).
Потрібно зазначити усі інші важливі етапи дослідження із зазначенням терміну здійснення.
7. Обґрунтування та історія питання: короткий опис загроз(и) безпеці, профілю безпеки або заходів з управління ризиками, які зумовили дослідження за власною ініціативою або за зобов’язанням, та короткий критичний аналіз наявних опублікованих та неопублікованих даних для пояснення прогалин в знаннях, які дане дослідження повинно заповнити. Аналіз може охоплювати відповідні експерименти на тваринах та людині, клінічні дослідження, демографічну статистику та попередні епідеміологічні дослідження. В аналізі слід надати результати схожих досліджень та очікуваний внесок даного дослідження.
8. Предмет та цілі дослідження: предмет дослідження, що пояснює, як у дослідженні буде розглянута проблема, у зв’язку з якою його ініційовано, та цілі дослідження, включаючи будь-які попередньо висунуті гіпотези та основні підсумкові показники.
9. Методи дослідження: опис методів дослідження, включаючи:
9.1. Дизайн дослідження: загальний формат дослідження та обґрунтування його вибору.
9.2. Параметри дослідження: досліджувана популяція, яка представлена в особах, місце, періоди та критерії відбору, включаючи обґрунтування будь-яких критеріїв включення та виключення. Якщо проводиться будь-яка вибірка з вихідної популяції, має бути наданий опис вихідної популяції та детальний опис методики формування вибірки. Якщо дизайн дослідження — систематичний огляд або метааналіз, повинні бути надані пояснення критеріїв вибору та правомірності досліджень.
9.3. Змінні: результати, експозиції та інші змінні, включаючи визначені фактори ризику, слід розглядати окремо, з застосуванням їх діючих визначень; необхідно зазначити також потенційні систематичні помилки та модифікатори ефекту.
9.4. Джерела даних: стратегії та джерела даних для визначення експозиції, результатів та всіх інших змінних, що мають значення для цілей дослідження, таких як потенційні систематичні помилки та модифікатори ефекту. Якщо дослідження буде використовувати існуюче джерело даних, таке як електронна медична документація, слід зазначити будь-яку інформацію щодо валідності записів та кодування даних. У випадку систематичного огляду або метааналізу слід описати стратегію та процедури пошуку, а також будь-які методи для підтвердження даних, отриманих від дослідників. Якщо методи або інструменти збору даних тестуються у пілотному дослідженні, слід представити плани пілотного дослідження. Якщо пілотне дослідження вже було проведене, слід надати стислий виклад результатів. Слід зазначити факт залучення будь-яких експертних комітетів для оцінки коректності діагнозів.
9.5. Масштаб дослідження: будь-який запланований масштаб дослідження, точність оцінок дослідження та будь-який підрахунок розміру вибірки, що принаймні здатні виявити попередньо визначений ризик з попередньо заданою статистичною точністю.
9.6. Управління даними: управління даними та статистичні програми, що будуть використовуватися у дослідженні, включаючи процедури збору, пошуку та підготовки даних.
9.7. Аналіз даних: основні етапи від первинних даних до отримання кінцевого результату, включаючи методи, що використовуватимуться для виправлення невідповідностей або помилок, внесення показників, модифікацію первинних даних, класифікацію, аналіз та представлення результатів, а також процедури контролю джерел систематичних помилок та їх впливу на результати; статистичні процедури, що будуть застосовуватися до даних для отримання точкових оцінок та довірчих інтервалів показників випадків чи зв’язку та аналіз чутливості. Первинні аналізи слід чітко відрізняти від субгрупових аналізів та вторинних аналізів.
9.8. Контроль якості: опис будь-яких механізмів та процедур для забезпечення якості та цілісності даних, включаючи точність та зрозумілість зібраних даних та оригінальних документів, ступінь перевірки джерела даних та валідації кінцевих точок, зберігання записів та архівування статистичних програм. Якщо доцільно, слід включити сертифікацію та/або кваліфікацію будь-якої залученої допоміжної лабораторії або дослідницьких груп.
9.9. Обмеження дослідницьких методів: будь-які потенційні обмеження дизайну дослідження, джерел даних та аналітичних методів, включаючи фактори втручання, систематичні помилки, узагальнення та випадкову похибку. Слід описати ймовірну ефективність зусиль, що докладаються для зменшення кількості помилок.
10. Захист досліджуваних: заходи з безпеки з метою дотримання національних вимог та вимог ЄС щодо гарантування захисту прав учасників неінтервенційних ПДБЛЗ.
11. Управління та повідомлення про побічні явища/побічні реакції: процедури збору, управління та повідомлення про випадки підозрюваних побічних реакцій та будь-які інші медично важливі явища під час проведення дослідження, що можуть вплинути на оцінку співвідношення користі/ризику лікарського засобу під час проведення дослідження.
Для досліджень з первинним збором даних, у яких не збиратиметься інформація про певні побічні явища (див. модуль VI), заявник (власник реєстраційного посвідчення) повинен надати у протоколі обґрунтування загального підходу до збору даних з безпеки. При будь-якому посиланні на побічне явище, інформацію про яке не збиратимуть, слід застосовувати відповідний рівень класифікації MedDRA. Якщо інформація про певні побічні явища не збиратиметься, шляхи та документи, що будуть застосовуватися для інформування працівників з медичною та/або фармацевтичною освітою та споживачів про можливість надання повідомлення про побічні реакції власнику реєстраційного посвідчення або до національної системи спонтанних повідомлень, слід зазначити у цьому розділі (див. модуль VI ННПФ). За певних обставин, коли підозрювані побічні реакції з летальним наслідком не підлягатимуть терміновому повідомленню у вигляді повідомлення про індивідуальний випадок, пов’язаний з безпекою (див. модуль VI ННПФ), кожну з цих побічних реакцій слід перерахувати у таблиці, застосовуючи відповідний рівень класифікації MedDRA з обґрунтуванням неподання повідомлень про них.
Слід надати роз’яснення, якщо дослідження ґрунтується тільки на повторному застосуванні даних, для якого необхідно збирати усі побічні явища, але надання повідомлення про підозрювані побічні реакції у формі повідомлень про індивідуальний випадок, пов’язаний з безпекою, не вимагається (див. модуль VI НПФ).
Стосовно комбінованих дизайнів досліджень із первинним та повторним збором даних слід дотримуватися таких самих вимог, що і стосовно досліджень з первинним збором даних (див. модуль VI НПФ).
12. Плани щодо розповсюдження та комунікації стосовно результатів дослідження, включаючи будь-які плани подання звітів про хід дослідження та заключних звітів.
13. Посилання.
Що стосується формату протоколу дослідження, слід дотримуватися Настанови щодо формату та змісту протоколу неінтервенційних ПДБЛЗ88.
У відповідному розділі протоколу дослідження потрібно надати аналіз доцільності дослідження або пілотні дослідження, що проводилися на підтримку розробки протоколу, наприклад, тестування анкети або простий підрахунок медичних явищ або призначень у базі даних для визначення статистичної точності дослідження, разом з коротким описом їх методів та результатів. Повна версія звіту повинна надаватися EMA та національним уповноваженим органам на вимогу. У протоколі слід описати аналіз доцільності дослідження або пілотні дослідження, які є частиною процесу дослідження, наприклад, пілотна оцінка анкет(и) дослідження, що використовувалася для першого набору пацієнтів, залучених у дослідження.
У додатку повинен міститися перелік усіх окремих документів, а також перелік з чітким посиланням на документи або самі документи, що містять будь-яку додаткову чи допоміжну інформацію щодо особливих аспектів, які раніше не розглядалися (наприклад анкети, індивідуальні реєстраційні форми).
VIII.B.3.2. Значні зміни до протоколу дослідження
До протоколу дослідження слід вносити зміни та оновлення, якщо у них виникає потреба в ході дослідження. Будь-які значні зміни до протоколу після початку дослідження, включно з їх датами, слід документувати в протоколі у спосіб, у який можна їх відслідкувати та перевірити. Якщо зміни до протоколу призводять до того, що дослідження стає інтервенційним клінічним дослідженням, слід негайно інформувати про це національні уповноважені органи та EMA. У подальшому воно повинно проводитися відповідно до положень Директиви 2001/20/ЄС [15], положень Порядку проведення клінічних випробувань [16]N і Тому 10 Правил, що регулюють лікарські засоби у ЄС.
Для неінтервенційних ПДБЛЗ, що проводяться заявником (власником реєстраційного посвідчення) на виконання зобов’язання, встановленого уповноваженим органом ЄС/УкраїніN, див. розділ VIII.С.2. щодо подання значних змін до протоколу дослідження.
Вимоги та рекомендації щодо подання заяви на внесення значних змін до протоколу дослідження представлені у Додатку I модулю III ННПФ.
VIII.B.4. Надання даних з фармаконагляду уповноваженим органам
VIII.B.4.1. Дані, що стосуються співвідношення користі/ризику лікарського засобу
У ході дослідження заявник (власник реєстраційного посвідчення) повинен здійснювати моніторинг отриманих даних та оцінювати їх вплив на співвідношення користі/ризику досліджуваного лікарського засобу (ст. 107m(7) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Про будь-яку нову інформацію, що може вплинути на співвідношення користі/ризику лікарського засобу, слід негайно повідомляти у письмовому вигляді в якості нової проблеми з безпеки до уповноважених органів країн ЄС/УкраїниN, у яких лікарський засіб зареєстрований, та EMA електронною поштою за адресою ([email protected]). Інформація, яка впливає на співвідношення користі/ризику лікарського засобу, може бути результатом аналізу побічних реакцій або зведених даних.
Така комунікація не повинна впливати на інформування про результати досліджень, які слід надавати у РОЗБ (див. модуль VII ННПФ) та в оновленому ПУР (див. модуль V НПФ).
VIII.B.4.2. Повідомлення про побічні реакції/побічні явища
Про побічні реакції/побічні явища необхідно повідомляти уповноважені органи відповідно до положень модуля VI ННПФ. Необхідно запропонувати і стисло описати у протоколі дослідження процедури збору, управління (включаючи оцінку власника реєстраційного посвідчення, коли доцільно) та подання повідомлень про підозрювані побічні реакції/побічні явища. За доцільності слід зробити посилання на МФСФ (див. модуль II ННПФ), але деталі, характерні для конкретного дослідження, слід описати у протоколі дослідження.
VIII.B.4.3. Звіти про дослідження
VIII.B.4.3.1. Звіт про хід дослідження та проміжний звіт про результати дослідження
Звіт про хід дослідження призначений для включення відповідної інформації для документування ходу дослідження, наприклад, кількості пацієнтів, які були включені у дослідження, кількості пацієнтів, які отримують лікування, або кількість пацієнтів, щодо яких є інформація про результати лікування, проблеми, що виникали, а також відхилення від встановленого плану. Звіт про хід дослідження може включати проміжний звіт про результати дослідження.
Проміжний звіт про результати дослідження призначений для включення результатів будь-якого запланованого проміжного аналізу даних дослідження до та після завершення збору даних.
На запит національного уповноваженого органу звіти про хід дослідження для ПДБЛЗ, що проводяться за зобов’язанням або за власною ініціативою, необхідно надавати до уповноважених органів УкраїниN, країн ЄС, де проводиться дослідження (ст. 107m(5) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Ці звіти також може запитувати EMA для ПДБЛЗ стосовно лікарських засобів, зареєстрованих за централізованою процедурою. Запит на подання звіту про хід дослідження може надходити як до початку дослідження, так і в будь-який момент під час його проведення. Такі запити можуть бути зумовлені надходженням інформації про співвідношення користі/ризику лікарського засобу, яка стає відомою в процесі дослідження, або потребою в інформації про хід дослідження в контексті регуляторних процедур або важливим повідомленням про безпеку лікарського засобу. Вимоги та рекомендації для представлення доповідей про хід роботи зазначені в Модулі VIII Додатку I ННФП.
До початку проведення дослідження необхідно узгодити з відповідними уповноваженими органами строки подання звітів про хід дослідження та після узгодження зазначити їх у протоколі дослідження.
VIII.B.4.3.2. Заключний звіт про дослідження
Стосовно неінтервенційних ПДБЛЗ, що проводяться за зобов’язанням, встановленим уповноваженим органом ЄС/УкраїниN, заключний звіт про дослідження повинен мати формат, що описаний у цьому розділі (додаток III ІП 520/2012 [6], додаток 15 до Порядку [2]N), та повинен бути наданий протягом 12 міс після закінчення збору даних (ст. 107p(1) Директиви 2001/83/ЄС[1], положення Порядку [2]N) (див. VIII.C.2.). Цього формату та графіку також повинні дотримуватися ПДБЛЗ, що зазначені у ПУР в ЄС та УкраїніN, або проводяться за власною ініціативою в ЄС та УкраїніN.
Вимоги та рекомендації стосовно подання заключного звіту визначені у Додатку I Модуля VIII.
У випадку припинення дослідження необхідно надати заключний звіт та вказати причини його припинення.
Заключний звіт дослідження повинен містити таку інформацію:
1. Назва: назва, що містить загальновживаний термін дизайну дослідження; підзаголовки з датою заключного звіту, прізвищем та місцем роботи особи, яка підготувала звіт. Якщо дослідження було зареєстровано в реєстрі EU PAS, на титульній сторінці заключного звіту дослідження слід зазначити «Номер в реєстрі EU PAS» та реєстраційний номер, а також посилання на сторінку в інтернеті, де знаходяться записи про дослідження.
2. Резюме: окремий стислий опис у форматі, структура якого наведена нижче (додаток III ІП 520/2012 [6], додаток 15 до Порядку [2]N).
3. Заявник (власник реєстраційного посвідчення): найменування та адреса заявника (власника реєстраційного посвідчення).
4. Дослідники: прізвища, посади, вчені ступені, адреси та місця роботи (назви організацій) головного дослідника та усіх дослідників і співробітників, а також перелік усіх партнерських установ та відповідних дослідницьких баз, де проводиться дослідження. Таку інформацію слід надавати для кожної країни, де планується проводити дослідження, та інших відповідних дослідницьких баз. Перелік усіх партнерських установ та дослідників повинен надаватися EMA та національним уповноваженим органам на вимогу.
5. Основні етапи: дати для таких основних етапів:
- початок збору даних (запланована та фактична дати);
- завершення збору даних (запланована та фактична дати) або дата передчасного завершення, якщо воно мало місце, із зазначенням причин передчасного завершення;
- звіт(и) про хід дослідження (див. VIII.B.4.3.1.);
- проміжний(і) звіт(и) про результати дослідження, якщо є доцільним (див. VIII.B.4.3.1);
- заключний звіт про результати дослідження (запланована та фактична дати);
- будь-які інші важливі етапи, що стосуються дослідження, включаючи дату реєстрації дослідження в реєстрі EU PAS (в Україні — протоколу в переліку дослідженьN) та дату затвердження протоколу експертною радою установи/незалежним етичним комітетом, якщо доцільно.
6. Обґрунтування та історія питання: опис питань безпеки, що призвели до ініціювання дослідження за власною ініціативою або за встановленим зобов’язанням, та короткий критичний аналіз важливих опублікованих та неопублікованих відповідних даних і прогалин у знаннях, які дослідження призначене заповнити.
7. Предмет дослідження та цілі: предмет дослідження та цілі дослідження, включаючи будь-які попередньо висунуті гіпотези, як зазначено у протоколі дослідження.
8. Зміни та оновлення в протоколі: перелік будь-яких значних змін та оновлень до початкового протоколу дослідження після початку збору даних, включаючи обґрунтування кожної зміни або оновлення.
9. Методи дослідження:
9.1. Дизайн дослідження: основні елементи дизайну дослідження та обґрунтування його вибору.
9.2. Параметри дослідження: середовище (тип дослідних центрів), місце проведення та відповідні дати дослідження, включаючи періоди набору пацієнтів, спостереження та збору даних. У разі систематичного огляду або метааналізу вивчити характеристики, що використовуються в якості критеріїв для відбору, з відповідним обґрунтуванням.
9.3. Суб’єкти дослідження: будь-яка вихідна популяція та критерії відбору суб’єктів для дослідження. Слід зазначити джерела та методи відбору учасників, включаючи, якщо доцільно, методи підтвердження випадку, а також кількість учасників дослідження, які вибули, та причини.
9.4. Змінні дані: усі результати, експозиція, прогностичні параметри, потенційні систематичні помилки та модифікатори ефекту, включаючи діючі визначення та діагностичні критерії, якщо доцільно.
9.5. Джерела даних та вимірювання: для кожної суттєвої змінної джерела даних та детальний опис методів оцінки та вимірювання. Якщо в дослідженні використовують існуюче джерело даних, таке як електронна медична документація, слід повідомляти будь-яку інформацію щодо достовірності записів та кодування даних. У випадку систематичного огляду або метааналізу потрібно надати опис усіх інформаційних джерел, стратегії пошуку, методів відбору досліджень, методів збору даних та будь-яких процесів для отримання або підтвердження даних від дослідників.
9.6. Систематичні помилки: будь-які спроби оцінки та розгляду потенційних джерел систематичних помилок на етапі визначення дизайну.
9.7. Масштаб дослідження: масштаб дослідження, обґрунтування будь-якого розрахунку масштабу дослідження та будь-якого методу досягнення запланованого масштабу дослідження.
9.8. Перетворення даних: перетворення, підрахунок або оперування даними, включаючи те, яким чином оброблялися кількісні дані при аналізі, які групи були вибрані та чому.
9.9. Статистичні методи: опис таких елементів:
- основні підсумкові показники;
- усі статистичні методи, що застосовувалися у дослідженні, включаючи ті, що використовувалися для контролю факторів втручання (факторів викривлення даних), а у випадку метааналізу — методи комбінування результатів досліджень;
- будь-які методи, що використовувалися для вивчення підгруп та взаємодій;
- яким чином вирішувалася проблема відсутніх даних;
- будь-які оцінки чутливості;
- будь-які зміни до плану аналізу даних, включеного до протоколу дослідження з обґрунтуванням змін.
9.10. Контроль якості: механізми гарантії якості та цілісності даних.
10. Результати: таблиці, графіки та ілюстрації, що демонструють відповідні дані та відображають проведені аналізи. Слід представляти скориговані та нескореговані результати. Точність оцінки слід визначити кількісно, використовуючи довірчі інтервали. Даний розділ повинен містити такі підрозділи:
10.1. Учасники: кількість учасників на кожному етапі дослідження; наприклад, кількість учасників, які потенційно придатні, які оцінені на придатність, придатність яких була підтверджена, які, включені у дослідження, за якими завершено спостереження, та які проаналізовані, і причини неучасті на будь-якому етапі. У випадку систематичного огляду або метааналізу визначаються кількість відібраних досліджень, оцінених на придатність та включених до огляду, із зазначенням причин виключення на кожному етапі.
10.2. Описові дані: характеристика учасників дослідження, інформація щодо експозиції, потенційних систематичних помилок та кількості учасників з відсутніми даними для кожної змінної, що представляє інтерес. У випадку систематичного огляду або метааналізу — характеристика кожного дослідження, з якого було взято дані (наприклад розмір дослідження, спостереження).
10.3. Дані про результати: кількість учасників за категоріями основних результатів (наслідків).
10.4. Основні результати: нескореговані і, якщо необхідно, скориговані показники щодо факторів втручання, та їх точність (наприклад 95% довірчий інтервал). Якщо доцільно, розрахункові показники відносного ризику потрібно трансформувати в абсолютний ризик за значимий період часу.
10.5. Інші види аналізу: інші проведені аналізи, наприклад, аналіз підгруп та взаємодій, а також аналізи чутливості.
10.6. Побічні явища та побічні реакції: резюме всіх побічних явищ/побічних реакцій, повідомлення про які находять під час дослідженні відповідно до вимог модуля VI ННПФ.
11. Висновки:
11.1. Основні результати: основні результати з посиланням на цілі дослідження, попереднє дослідження на підтримку та на противагу результатам завершеного ПДБЛЗ та, у відповідних випадках, вплив результатів на співвідношення ризику/користі лікарського засобу.
11.2. Обмеження: обмеження дослідження, враховуючи обставини, які вплинули на якість або цілісність даних, обмеження підходу до дослідження і методів, що використовуються для вирішення проблеми цих обмежень (наприклад частина отриманих відповідей відсутня або неповні дані), джерела потенційних систематичних помилок та неточностей, а також оцінка побічних явищ. Описуються тенденція та масштаб потенційних систематичних помилок.
11.3. Інтерпретація: інтерпретація результатів з урахуванням цілей, обмежень, складності аналізу, результатів подібних досліджень та інших відповідних доказів.
11.4. Узагальнення: узагальнення (зовнішня валідність) результатів дослідження.
12. Інша інформація: будь-яка додаткова або уточнююча інформація щодо специфічних аспектів, які раніше не розглядалися.
13. Висновки: основні висновки стосовно дослідження, що зроблені при аналізі даних.
14. Посилання.
Формат заключного звіту про дослідження повинен відповідати Настанові щодо формату та змісту заключного звіту дослідження для неінтервенційних ПДБЛЗ89, додатку 15 до Порядку [2]N.
Резюме заключного звіту про дослідження повинно містити стислий опис методів та результатів дослідження, представлених у такому форматі:
- заголовок та підзаголовки, включаючи дату резюме та прізвище, ім’я, по батькові і місце роботи особи, яка підготувала звіт;
- ключові слова (не більше п’яти ключових слів, які дають основну характеристику дослідження);
- обґрунтування та історія питання;
- предмет дослідження та цілі;
- дизайн дослідження;
- параметри дослідження;
- суб’єкти та масштаб дослідження, включаючи вибулих учасників.
Змінні дані та джерела даних:
- результати;
- обговорення (включаючи, якщо необхідно, оцінку впливу результатів дослідження на співвідношення користі/ризику лікарського засобу);
- висновок;
- заявник (власник реєстраційного посвідчення);
- прізвища, ім’я, по батькові та місця роботи (назви організацій) основних дослідників.
VIII.B.5. Публікація результатів дослідження
Для досліджень, що повністю або частково проводилися дослідниками, які не є співробітниками заявника (власника реєстраційного посвідчення), заявник (власник реєстраційного посвідчення) та дослідник повинні заздалегідь домовитися про принципи публікації, які б дозволяли головному досліднику самостійно готувати публікації на основі результатів дослідження незалежно від прав власності на інформацію. Заявник (власник реєстраційного посвідчення) повинен мати право переглядати результати та пояснення, включені до рукопису, та надавати коментарі до подання рукопису для публікації.
VIII.B.5.1. Подання рукописів, прийнятих до публікації
Заявник (власник реєстраційного посвідчення), який ініціює, здійснює управління або фінансування неінтервенційного ПДБЛЗ, повинен надавати EMA та уповноваженим органам країн ЄС та УкраїниN, у яких зареєстрований лікарський засіб, фінальний рукопис статті протягом двох тижнів після першого прийняття її до публікації з тим, щоб уповноважений орган мав змогу завчасно переглянути результати дослідження та їх інтерпретацію, які публікуватимуться.
VIII.B.6. Захист даних
Заявники (власники реєстраційних посвідчень) та дослідники повинні дотримуватися відповідного національного законодавства та настанов тих країн, де проводиться дослідження (ст. 107m(2) Директиви 2001/83/ЄС [1]). Відповідно до Директиви 95/46/ЄС [12] Європейського Парламенту та Ради про захист осіб з приводу обробки персональних даних та вільного переміщення таких даних слід дотримуватися положень нормативно-правових актів щодо захисту даних.
Для неінтервенційних ПДБЛЗ, що встановлюються як зобов’язання, заявник (власник реєстраційного посвідчення) забезпечує обробку та збереження усієї інформації з дослідження з безпеки, що дає змогу коректного звітування, інтерпретації та перевірки цієї інформації і забезпечує захист конфіденційної інформації про учасників дослідження з безпеки (ст. 36 ІП 520/2012 [6], положення Порядку [2]N). Це положення слід також застосовувати до ПДБЛЗ, яких вимагає погоджений в ЄС та УкраїніN ПУР або які проводяться за власною ініціативою в ЄС/УкраїніN.
VIII.B.7. Системи якості, аудити, інспекції у країнах ЄС та аудити системи фармаконагляду уповноваженою установоюN
Заявник (власник реєстраційного посвідчення) повинен забезпечити виконання своїх зобов’язань з фармаконагляду в рамках дослідження та можливість проведення аудиту, інспекції у країнах ЄС, аудиту системи фармаконагляду уповноваженою установою в УкраїніN і підтвердження цього. Для ПДБЛЗ, встановлених як зобов’язання, заявник (власник реєстраційного посвідчення) забезпечує зберігання в електронному вигляді та доступність для аудитів аналітичного набору даних і статистичних програм, що використовувалися для генерації даних, включених у заключний звіт про дослідження з безпеки (ст. 12 ІП 520/2012, ст. 36 ІП 520/2012 [6], положення Порядку [2]N).
Стосовно ПДБЛЗ, яких вимагає погоджений в ЄС та УкраїніN ПУР, або які проводяться за власною ініціативою в ЄС/УкраїніN, для управління записами та зберігання даних слід дотримуватися положень ст. 12 ІП 520/2012 [6], Порядку [2]N.
VIII.B.8. Вплив на систему управління ризиками
Інформацію про неінтервенційні ПДБЛЗ, що проводяться за зобов’язанням, встановленим уповноваженим органом ЄС/УкраїниN, або вимагаються ПУР, слід включити до ПУР, як описано в модулі V ННПФ.
VIII.С. Функціонування системи ЄС, УкраїниN
VIII.С.1. Процедура призначення післяреєстраційних досліджень з безпеки
В ЄС та УкраїніN вимога проведення ПДБЛЗ може встановлюватися EMA або національним уповноваженим органом, або уповноваженим органом в УкраїніN під час оцінки заяви на реєстрацію (ст. 9(4)(cb) Регламенту (ЄC) № 726/2004 [5], ст. 21а(b) Директиви 2001/83/ЄС [1], положення Порядку проведення експертизи реєстраційних матеріалів [7]N) або в післяреєстраційний період (ст. 10а(1)(а) Регламенту (ЄC) № 726/2004 [5], ст. 22а(1)(а) Директиви 2001/83/ЄС [1], положення Порядку [2]N), якщо існують питання щодо ризику зареєстрованого лікарського засобу, стосовно якого результати ПДБЛЗ значно вплинули б на співвідношення користі/ризику лікарського засобу. Ця вимога повинна належним чином обґрунтовуватися, повинна повідомлятися у письмовому вигляді та включати цілі та строки подання документації і проведення дослідження. Вимога може також включати рекомендації щодо ключових елементів дослідження (наприклад дизайн дослідження, параметри дослідження, експозиція(ї), результат(и), досліджувана популяція).
VIII.С.1.1. Вимога післяреєстраційних досліджень з безпеки в рамках заяви на реєстрацію
Реєстраційне посвідчення може видаватися за умови проведення ПДБЛЗ. Якщо під час оцінки заяви на отримання реєстраційного посвідчення виявлена необхідність в ПДБЛЗ, PRAC буде схвалювати рекомендацію зі звітом з оцінки для СНМР або країни ЄС, яка вимагала такої рекомендації.
VIII.С.1.2. Вимога післяреєстраційних досліджень з безпеки під час післяреєстраційної регуляторної процедури
Необхідність ПДБЛЗ може визначатися EMA або уповноваженим органом під час післяреєстраційної регуляторної процедури, наприклад, розширення або зміни до реєстраційного посвідчення, процедури перереєстрації або процедури подання РОЗБ. Якщо під час оцінки післяреєстраційної процедури визначена необхідність в ПДБЛЗ, PRAC може схвалити консультацію або рекомендацію зі звітом з оцінки для СНМР або країни ЄС, якщо необхідно.
VIII.С.1.3. Вимога післяреєстраційних досліджень з безпеки у зв’язку з виявленим питанням з безпеки
Після видачі реєстраційного посвідчення EMA або уповноважений орган, якщо необхідно, може встановлювати для заявника (власника реєстраційного посвідчення) вимогу провести ПДБЛЗ у разі існування питань, пов’язаних із ризиком зареєстрованого лікарського засобу. Якщо визначена необхідність в ПДБЛЗ, PRAC може схвалити консультацію або рекомендацію зі звітом з оцінки для СНМР або країни ЄС, якщо необхідно.
VIII.С.1.4. Спільні післяреєстраційні дослідження з безпеки
Якщо проблема безпеки стосується більше ніж одного лікарського засобу, EMA або національний уповноважений орган повинен після консультації з PRAC заохочувати відповідних заявників (власників реєстраційних посвідчень) проводити спільні ПДБЛЗ (ст. 10а(1)(а) Регламенту (ЄC) № 726/2004 [5], ст. 22а(1)(а) Директиви 2001/83/ЄС [1]). Вимога до заявників (власників реєстраційних посвідчень) повинна містити обґрунтування вимоги проведення спільного дослідження, а також може включати основні елементи протоколу дослідження. Національний уповноважений орган або EMA повинні підтримувати зв’язок між відповідними заявниками (власниками реєстраційних посвідчень) з наданням міркування щодо пропозиції спільного дослідження.
VIII.С.1.5. Письмові зауваження у відповідь на встановлення вимоги
Протягом 30 днів з моменту отримання письмового повідомлення про встановлену вимогу, заявник (власник реєстраційного посвідчення) може надати письмові зауваження у відповідь на встановлену вимогу (ст. 10а(2) Регламенту (ЄC) № 726/2004 [5], ст. 22а(2) Директиви 2001/83/ЄС) [1]. Національний уповноважений орган або EMA визначать граничний строк для надання цих письмових зауважень. На підставі письмових зауважень, наданих заявником (власником реєстраційного посвідчення), національний уповноважений орган або Європейська Комісія повинні відкликати або підтвердити вимогу проведення дослідження. Якщо вимога підтверджена, до реєстраційного посвідчення слід внести зміну для включення вимоги як умови видачі реєстраційного посвідчення та, якщо необхідно, слід відповідно оновити ПУР (ст. 10а(3) Регламенту (ЄC) № 726/2004 [5], ст. 22а(3) Директиви 2001/83/ЄС [1], відповідно до положення Порядку проведення експертизи реєстраційних матеріалів [7]N) (див. модуль V ННПФ).
VIII.С.2. Контроль за неінтервенційними післяреєстраційними дослідженнями з безпеки, що проводяться відповідно до вимоги
Неінтервенційні ПДБЛЗ, що проводяться на вимогу уповноваженого органу в ЄС (категорія 1 та 2 досліджень в модулі V ННПФ), контролюються та оцінюються PRAC, якщо ПДБЛЗ не вимагалося національним уповноваженим органом однієї з країн ЄС відповідно до ст. 22а Директиви 2001/83/ЄС [1] та не проводилося лише в тій країні ЄС, в якій застосовуються національні процедури контролю (ст. 107n(1) Директиви 2001/83/ЄС [1]).
VIII.С.2.1. Роль та обов’язки заявника (власника реєстраційного посвідчення)
Якщо дослідження неінтервенційне (див. розділ VIII.А.), власник реєстраційного посвідчення повинен гарантувати, що дослідження відповідає вимогам, які застосовуються до неінтервенційних ПДБЛЗ, що викладені у ст. 107m-q Директиви 2001/83/ЄС [1], ст. 28b Регламенту (ЄC) № 726/2004 [5], ст.ст. 36–38 ІП 520/2012 [6], положення Порядку [2]N і у цьому модулі. Заявник (власник реєстраційного посвідчення) повинен забезпечити виконання своїх зобов’язань з фармаконагляду стосовно дослідження та можливість проведення його аудиту, інспекції у ЄС, аудиту уповноваженою установоюN та підтвердження (див. розділ VIII.В.6 та VIII.В.7).
Після встановлення проведення неінтервенційного ПДБЛЗ як умови видачі реєстраційного посвідчення заявник (власник реєстраційного посвідчення) повинен розробити протокол дослідження та подати його до уповноваженого органу або PRAC для огляду (ст. 107n(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N), якщо необхідно. Обов’язком заявника (власника реєстраційного посвідчення) є гарантування того, що дослідження не є клінічним дослідженням, до якого повинна застосовуватися Директива 2001/20/ЄС [15], положення Порядку проведення клінічних випробувань [16]N та том 10 Правил регулювання лікарських засобів в Європейському Союзі90.
Дослідження може розпочинатися лише після отримання письмового схвалення від національного уповноваженого органу або PRAC (в Україні — Центру (положення Порядку [2])N). Якщо PRAC (в Україні — ЦентрN) видав лист-схвалення, заявник (власник реєстраційного посвідчення) повинен надіслати протокол до національного уповноваженого органу країн(и) ЄС/УкраїниN, у якій має проводитися дослідження, та може після цього починати дослідження відповідно до схваленого протоколу (ст. 107n(3) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Повинні виконуватися вимоги ЄС та національні вимоги для гарантії здоров’я та прав учасників дослідження (ст. 107m(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Заявник (власник реєстраційного посвідчення) до подання протоколу може надати запит до національного уповноваженого органу або EMA стосовно засідання до подання документів (з EMA та доповідачем PRAC, якщо запит подається до EMA) з метою уточнення конкретних аспектів дослідження, що будуть вимагатися (таких як цілі дослідження, досліджувана популяція, визначення експозиції та результатів) та сприяння розробці протоколу відповідно до цілей, визначених національним уповноваженим органом або PRAC.
Після початку неінтервенційного ПДБЛЗ, що вимагається, заявник (власник реєстраційного посвідчення) повинен подавати будь-які значні зміни до протоколу перед їх запровадженням до національного уповноваженого органу або PRAC (ст. 107о Директиви 2001/83/ЄС [1]), (в Україні — Центру (положення Порядку [2])N) (див. розділ VIII.А.1. щодо визначення значних змін).
Після завершення дослідження заявник (власник реєстраційного посвідчення) повинен якомога раніше, але не пізніше 12 місяців після закінчення збору даних надати до національного уповноваженого органу або PRAC (в Україні — Центру (положення Порядку [2])N) заключний звіт дослідження, включаючи публічне резюме дослідження, крім випадків, коли національний уповноважений орган або PRAC надав письмову відмову від нього (ст. 107р(1) Директиви 2001/83/ЄС [1]).
Якщо PRAC залучений до контролю дослідження, заявник (власник реєстраційного посвідчення) повинен письмово звернутися із запитом про відмову до EMA принаймні за три місяці до закінчення терміну подання звіту. У цьому запиті слід обґрунтувати відмову. Доповідач PRAC повинен розглянути даний запит та, зважаючи на обґрунтування та терміни його подання, схвалити або відхилити його.
Заявник (власник реєстраційного посвідчення) повинен надати протокол дослідження, резюме заключного звіту про дослідження та заключний звіт про дослідження англійською мовою, за винятком ситуацій, коли дослідження мають проводитись тільки на території однієї країни ЄС, що вимагає проведення дослідження відповідно до ст. 22а Директиви 2001/83/ЄС [1]. Для завершених досліджень власник реєстраційного посвідчення повинен надати англійський переклад назви та резюме протоколу дослідження, а також англійський переклад резюме заключного звіту дослідження (ст. 36 ІП 520/2012 [6]). В Україні заявник подає до Центру заключний звіт про дослідження з безпеки українською мовою, якщо дослідження проводилося тільки на території України, або англійською мовою, якщо дослідження проводилося і в інших країнах. У цьому випадку заявник подає переклад українською мовою назви, резюме (abstract) протоколу дослідження з безпеки (положення Порядку [2])N.
VIII.С.2.2. Роль та обов’язки PRAC та національного уповноваженого органу
Національний уповноважений орган або PRAC, в Україні — Центр (положення Порядку [2]N) повинен протягом 60 днів з моменту отримання проєкту протоколу надати заявнику (власнику реєстраційного посвідчення) лист-повідомлення, що ухвалює проєкт протоколу, або лист-відмову, або лист-повідомлення, що дослідження є клінічним та підпадає під дію Директиви 2001/20/ЄС [15], положень Порядку проведення клінічних випробувань [16]N. Лист-відмова повинен містити детальне обґрунтування відмови в будь-якому з наступних випадків:
- проведення дослідження сприяє просуванню лікарського засобу на фармацевтичний ринок;
- дизайн дослідження не відповідає його цілям (ст. 107n(2) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Якщо дослідження підтверджує статус інтервенційного, PRAC або національний уповноважений орган (в Україні — Центр (положення Порядку [2])N) надає лист-повідомлення заявнику (власнику реєстраційного посвідчення), що дослідження є клінічним дослідженням, яке підпадає під дію Директиви 2001/20/ЄС [15], положення Порядку проведення клінічних випробувань [16]N.
Якщо PRAC залучено до контролю дослідження, PRAC буде призначати доповідача PRAC, що відповідає за контроль ПДБЛЗ. Доповідачу PRAC слід написати звіт з оцінки протоколу та подавати його для огляду та ухвалення PRAC.
У випадку подання змін до протоколу дослідження національний уповноважений орган або PRAC (в Україні — Центр (положення Порядку [2])N) повинен оцінити зміни та поінформувати заявника (власника реєстраційного посвідчення) про згоду або незгоду щодо їх впровадження (ст. 107o Директиви 2001/83/ЄС [1], положення Порядку [2]N). Національний уповноважений орган або PRAC (в Україні — Центр (положення Порядку [2])N) повинен надати заявнику (власнику реєстраційного посвідчення) лист-схвалення або відмову у внесенні змін до протоколу протягом 60 днів з моменту їх подання. У листі-відмові повинен бути зазначений термін, до якого заявнику (власнику реєстраційного посвідчення) слід повторно надати змінену версію протоколу.
Якщо протокол дослідження оцінює національний уповноважений орган, цей національний уповноважений орган повинен надати свою оцінку іншим зацікавленим країнам ЄС, в яких лікарський засіб зареєстрований.
Відносно оцінки результатів дослідження, якщо PRAC залучено до контролю дослідження, PRAC буде готувати звіт з оцінки та надавати рекомендацію для СНМР або CMDh, якщо необхідно.
VIII.С.2.3. Роль та обов’язки EMA
EMA має забезпечити науковий секретаріат для PRAC.
EMA буде інформувати заявника (власника реєстраційного посвідчення) у письмовому вигляді та протягом відповідного терміну про рішення PRAC щодо оцінки наступного:
- протоколу дослідження;
- змін до протоколу дослідження;
- заключного звіту дослідження;
- запиту на відмову від подання заключного звіту дослідження.
Якщо заявник (власник реєстраційного посвідчення) подає запит до EMA щодо засідання до подання документів, EMA буде відповідати за своєчасну підготовку засідання з EMA та доповідачем PRAC.
EMA має оприлюднити на Європейському вебпорталі лікарських засобів протоколи та загальні резюме результатів ПДБЛЗ, на які посилаються у ст. 107n та 107p Директиви 2001/83/ЄС [1].
VIII.С.3. Зміни до реєстраційного посвідчення за результатами неінтервенційних післяреєстраційних досліджень з безпеки
Заявник (власник реєстраційного посвідчення) повинен подати до національного уповноваженого органу або PRAC (в Україні — Центру (положення Порядку [2])N) заключний звіт про дослідження протягом 12 місяців з дати закінчення збору даних, якщо не було видано письмової відмови (ст. 107р(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Заявник (власник реєстраційного посвідчення) повинен оцінювати, чи результати дослідження впливають на реєстраційне посвідчення та повинен, якщо необхідно, подавати до національних уповноважених органів або EMA (в Україні — Центру (положення Порядку [2])N) заяву щодо внесення змін до реєстраційного посвідчення (ст. 107р(2) Директиви 2001/83/ЄС [1], положення Порядку [2], положення Порядку проведення експертизи [7]N). У такому випадку зміну слід подати до національного уповноваженого органу або EMA (в Україні — ЦентруN).
За результатами перегляду заключного звіту про дослідження PRAC або національний уповноважений орган (в Україні — Центр (положення Порядку [2])N) може рекомендувати внести зміни, призупинення або відкликання реєстраційного посвідчення (ст. 28b(2) Регламенту (ЄC) № 726/2004 [5], ст. 107q(2) Директиви 2001/83/ЄС) [1], положення Порядку [2]N. У цій рекомендації PRAC (в Україні — ЦентруN) повинні бути зазначені будь-які розбіжні позиції та причини, на яких вони ґрунтуються (ст. 107q(1) Директиви 2001/83/ЄС [1]), положення Порядку [2]N.
Якщо заключні результати дослідження стосуються принаймні одного лікарського засобу, зареєстрованого за централізованою процедурою, рекомендації PRAC повинні передаватися до СНМР, який має схвалити думку з врахуванням рекомендації. Якщо думка СНМР відрізняється від рекомендації PRAC, СНМР повинен додати до своєї думки детальне пояснення наукових причин відмінностей (ст. 28b(2) Регламенту (ЄC) № 726/2004 [5]).
Якщо заключні результати дослідження стосуються лікарських засобів, зареєстрованих за національною процедурою, країни ЄС, які представлені в CMDh, мають узгодити позицію, враховуючи рекомендацію PRAC, та скласти графік впровадження цієї узгодженої позиції. Якщо домовленість досягнута, CMDh повинен надіслати узгоджену позицію заявнику (власнику реєстраційного посвідчення) та країнам ЄС, яким слід вжити необхідних заходів для зміни, призупинення або відкликання реєстраційного посвідчення згідно з графіком впровадження, який розроблений CMDh. Якщо зміна узгоджена, заявник (власник реєстраційного посвідчення) повинен надати національним уповноваженим органам відповідну заяву щодо внесення зміни, включаючи оновлену коротку характеристику препарату (SmPC) та листок-вкладку, впродовж визначеного графіку впровадження (ст. 107q(2) Директиви 2001/83/ЄС [1]). Якщо домовленість за загальною згодою не може бути досягнута, позицію більшості країн ЄС, що представлені в рамках CMDh, слід направляти до Комісії, яка має застосувати процедуру, що викладена в ст. ст. 33 та 34 Директиви 2001/83/ЄС [1] (ст. 107q(2) Директиви 2001/83/ЄС [1]).
Якщо згода або позиція CMDh відрізняється від рекомендації PRAC, CMDh повинен додати до згоди або позиції більшості детальне пояснення наукових причин розбіжностей разом з рекомендацією (ст. 107q(2) Директиви 2001/83/ЄС [1]).
У певних випадках можуть вимагатися більш термінові заходи, наприклад, на підставі проміжних результатів, що включені до звітів про хід дослідження (див. також розділ VIII.В.4.3.1). У такому випадку буде ініційована відповідна процедура (див. модуль VI ННПФ).
VIII. Додаток 1. Методи післяреєстраційних досліджень з безпеки VIII.
Додаток 1.1. Дизайни дослідження
Залежно від цілей ПДБЛЗ можуть мати різні дизайни. Нижче наведений короткий опис основних видів досліджень, а також типів існуючих джерел даних. Цей додаток неповний і повинен бути доповнений іншими джерелами інформації, такими як Настанова ENCePP (Європейська мережа центрів з фармакоепідеміології та фармаконагляду) з методологічних стандартів в фармакоепідеміології.
VIII. Додаток 1.1.1. Активний фармаконагляд (Active surveillance)
Активний фармаконагляд, на відміну від пасивного, включає намагання встановити більш повний перелік побічних явищ у даній популяції шляхом безперервно організованого процесу. Прикладом активного фармаконагляду може бути спостереження за пацієнтами, які лікуються певним лікарським засобом, із застосуванням системи управління ризиками. Пацієнтів при відпуску їм цього лікарського засобу за рецептом можуть попросити заповнити коротку анкету та надати дозвіл на можливість зв’язатися з ними пізніше. Загалом за допомогою системи активного фармаконагляду ймовірніше отримати більш повні дані щодо окремих випадків побічних явищ, ніж за допомогою пасивної системи повідомлень. Проте деякі обмеження систем спонтанних повідомлень все ще застосовуються, особливо, коли оцінюються відтерміновані у часі ефекти. Наприклад, побічні явища, що виникають через великий проміжок часу після експозиції (наприклад онкологічне захворювання, вроджений дефект) не можуть одразу виявлятися через систему спонтанних повідомлень. Ефективну систему активного нагляду може також забезпечити автоматичне виявлення «нетипових» лабораторних показників в комп’ютеризованих лабораторних звітах у деяких клінічних закладах.
VIII. Додаток 1.1.1.1. Схеми інтенсивного моніторингу (Intensive monitoring schemes)
Інтенсивний моніторинг — це система збору записів у визначених сферах; наприклад, відділення лікарень або лікарі загальної практики. У таких випадках збір даних може проводитися моніторами, які присутні на обходах палат, де вони збирають інформацію щодо небажаних та непередбачених явищ, що вважаються лікарем як такі, що (потенційно) мають причинно-наслідковий зв’язок із застосуванням лікарського засобу. Моніторинг може також зосереджуватися на окремих значних явищах, що можуть бути пов’язані з лікарським засобом, таких як, порушення функції печінки, ниркова недостатність, гематологічні порушення, кровотеча. Перевага таких систем у тому, що спеціалісти-монітори мають змогу документувати важливу інформацію щодо явищ та впливу лікарських засобів. Основним недоліком є необхідність утримувати підготовану групу спеціалістів-моніторів протягом тривалого часу.
Інтенсивний моніторинг можна також здійснювати шляхом перегляду медичних записів чи опитування пацієнтів та/або лікарів/фармацевтів на обраних сигнальних дільницях для забезпечення повноти та точності даних щодо повідомлених побічних явищ. Від обраних дільниць може надходити інформація, така як: дані спеціальних підгруп пацієнтів, які б не були доступними в пасивних системах спонтанних повідомлень. Більш того, збір інформації щодо застосування лікарського засобу, наприклад, ймовірність зловживання, може проводитися у обраних сигнальних дільницях. Основними недоліками сигнальних дільниць є проблеми селективності відбору, невеликої кількості пацієнтів та підвищених витрат. Інтенсивний моніторинг сигнальних дільниць найбільш ефективний для лікарських засобів, що застосовуються в основному в лікувальних закладах, таких як лікарні, будинки престарілих та центри гемодіалізу. У лікувальних закладах можуть частіше використовуватися певні лікарські засоби та може надаватися інфраструктура для спеціальних повідомлень. Додатково автоматизоване відстеження «нетипових» лабораторних показників з комп’ютеризованих лабораторних звітів в певних клінічних закладах може забезпечити ефективну систему інтенсивного моніторингу.
VIII. Додаток 1.1.1.2. Моніторинг реакцій на рецептурні лікарські засоби (Prescription event monitoring)
При моніторингу реакцій на рецептурні лікарські засоби пацієнти можуть бути визначені за допомогою електронних даних рецептурних лікарських засобів або автоматизованих заяв на медичне страхування. Кожному лікарю, який виписує рецепт, або пацієнту через попередньо визначені інтервали часу може надсилатися анкета для отримання інформації щодо результатів застосування лікарського засобу. В анкету можна включити інформацію щодо демографічних даних пацієнта, показань до застосування, тривалості терапії (включаючи дати початку), дозування, клінічних явищ та причин припинення лікування. Моніторинг реакцій на рецептурні лікарські засоби використовується як метод дослідження безпеки одразу після випуску лікарського засобу на фармацевтичний ринок. Основними недоліками моніторингу реакцій на рецептурні лікарські засоби є значна втрата при спостереженні, відносно коротка тривалість спостереження, селективний відбір, селективне повідомлення та обмеження сфери дії досліджуваними лікарськими засобами, що застосовуються виключно в лікарнях. Проте при моніторингу реакцій на рецептурні препарати може бути зібрана більш детальна інформація щодо побічних явищ від більшої кількості лікарів та/або пацієнтів.
VIII. Додаток 1.1.1.3. Реєстри (Registries)
Реєстр — це організована система, що використовує методи спостереження для збору однорідних даних щодо специфічних наслідків лікування у популяції, яка визначена за певним захворюванням, станом організму або експозицією. Реєстр може використовуватися як джерело даних, у рамках якого можна проводити дослідження.
Включення в реєстр, як правило, визначається діагнозом захворювання, призначеним лікарським засобом або обома (пацієнти з певним захворюванням, яких лікують певним лікарським засобом, визначеною діючою речовиною або будь-яким лікарським засобом визначеної фармако-терапевтичної групи). Вибір популяції реєстру та дизайну реєстру повинен визначатися цілями, виходячи з результатів, які будуть оцінюватися, та аналізів і порівнянь, що будуть проводитися.
Реєстри особливо корисні, коли стосуються рідкісних захворювань, рідкісної експозиції або спеціальної популяції. У багатьох випадках реєстри можуть наповнюватися даними про результати, змінні, що заплутують, модифікатори ефектів, що отримані через поєднання з іншими існуючими базами даних, такими як національні реєстри онкологічних захворювань, базами даних виписаних рецептів або записів про смертність.
Залежно від цілі за допомогою реєстрів можуть бути отримані дані про пацієнта, захворювання та результат лікування та про їх детермінанти. Дані щодо результатів можуть включати дані щодо повідомлених результатів лікування у пацієнтів, клінічних станів, моделей застосування лікарських засобів, безпеки та ефективності. Підтверджено, що інколи реєстри можуть бути єдиною можливістю надати уявлення про аспекти ефективності лікарського засобу. Проте реєстри спостережень не повинні використовуватися для демонстрації ефективності. Якщо ефективність лікарського засобу була продемонстрована в рандомізованих клінічних випробуваннях, реєстри пацієнтів можуть бути корисними для вивчення ефективності в гетерогенних популяціях, модифікаторів ефекту, таких як дози, що були призначені лікарями та які можуть відрізнятися від тих, що застосовувалися в рандомізованих клінічних випробуваннях, підгруп пацієнтів, які визначені за змінними, такими як вік, супутні захворювання, застосування супутнього лікування або генетичними факторами, або факторами, пов’язаними з певною країною або системою охорони здоров’я.
Якщо дані наявні або можуть збиратися, реєстри пацієнтів можуть використовуватися для порівняння ризиків між різними групами. Наприклад, дослідження випадок-контроль може проводитися для порівняння експозиції лікарським засобам випадків серйозних побічних реакцій, вибраних з реєстру, з контрольними випадками, відібраними серед пацієнтів у межах або поза межами реєстру. Подібним чином когортне дослідження може включатися в реєстр. Дизайн випадок-контроль може також застосовуватися (див. VIII. Додаток 1.1.2.4.).
Реєстри пацієнтів можуть містити інформацію про вплив лікарського засобу в певних популяціях, наприклад, у вагітних. За пацієнтами можуть спостерігати протягом тривалого часу та включати їх до когортного дослідження для збору даних про побічні явища, використовуючи стандартизовані анкети. За допомогою простих когортних досліджень можна підрахувати частотність, проте без групи порівняння неможливо оцінити будь-який зв’язок між експозицією та результатами. Однак, їх можна застосовувати для посилення сигналу, зокрема, для рідкісних явищ. Цей тип реєстру може бути дуже корисним для вивчення безпеки препаратів-сиріт, зареєстрованих для лікування специфічних захворювань.
VIII. Додаток 1.1.2. Дослідження за даними спостережень (Observational studies)
Традиційні епідеміологічні методи є ключовим компонентом для оцінки побічних явищ. Існує низка дизайнів досліджень за даними спостережень, які застосовні для оцінки сигналів, що виявлені за допомогою спонтанних повідомлень, програм активного фармаконагляду або серії випадків. Основні з них — це дослідження методом поперечного зрізу, дослідження випадок-контроль та когортні дослідження, що ґрунтуються на зборі первинних даних або повторному використанні існуючих даних.
VIII. Додаток 1.1.2.1. Дослідження методом поперечного зрізу (Crosssectional study)
Дослідження методом поперечного зрізу передбачає збір даних щодо групи пацієнтів у певний час (або інтервал часу) незалежно від експозиції або стану захворювання. Ці типи досліджень використовуються переважно для збору даних для аналізу чи для екологічного аналізу. Недоліком досліджень методом поперечного зрізу є неможливість відстеження часового зв’язку між експозицією та результатом, що обмежує їх використання для етіологічного дослідження, крім випадків, якщо експозиція не змінюється з часом. Ці дослідження найкраще використовувати для вивчення розповсюдженості захворювання в конкретний момент часу або для вивчення тенденцій з часом, якщо можна проводити збір даних через певні проміжки часу. Ці дослідження можна також застосовувати для вивчення зв’язку на початковій стадії між експозицією та результатом при проведенні аналізу.
VIII. Додаток 1.1.2.2. Когортне дослідження (Cohort study)
У когортному дослідженні популяція з ризиком розвитку явища, що представляє інтерес, спостерігається протягом деякого часу на предмет виникнення такого явища. Інформація щодо статусу експозиції відома для кожного учасника дослідження впродовж усього періоду спостереження. У деякий момент часу протягом спостереження учасник дослідження може зазнати впливу лікарського засобу, але не зазнати впливу в інший момент часу. Оскільки відома експозиція популяції під час спостереження, можна підрахувати показник частоти. У багатьох когортних дослідженнях, що вивчають експозицію лікарським(и) засобом(ами), групи порівняння, що представляють інтерес, обираються на основі застосування лікарського засобу та спостерігаються протягом деякого періоду часу. Когортні дослідження корисні, коли крім відносних ризиків побічних явищ існує необхідність знати частоту розвитку побічних явищ. Використовуючи одне й те ж когортне дослідження, можна оцінити багато побічних явищ. Складнощі може викликати залучення достатньої кількості пацієнтів, які зазнали впливу лікарського засобу, що представляє інтерес (наприклад у випадку препарату-сироти), або дослідження дуже рідкісних результатів. Відбір пацієнтів для когортних досліджень може здійснюватися з великих автоматизованих баз даних або з наявних даних, зібраних спеціально для даного дослідження. Крім того, когортні дослідження можуть використовуватися для вивчення питань безпеки у особливих популяціях (люди похилого віку, діти, пацієнти з супутніми захворюваннями, вагітні) шляхом додаткового набору цих пацієнтів або розподілу когорти на групи у випадку достатньої кількості пацієнтів.
VIII. Додаток 1.1.2.3. Дослідження випадок-контроль (Case-control study)
У дослідженні випадок-контроль визначаються випадки захворювання (або явища) та включаються у контрольну групу пацієнти з вихідної популяції, у яких спостерігаються випадки, але які на момент відбору не мали захворювання чи явища, що представляє інтерес. Різниця експозиції потім порівнюється між двома групами. Набір пацієнтів може проводитися з існуючої бази даних або може використовуватися підхід польового дослідження, при якому дані збираються спеціально для даного дослідження випадок-контроль. Якщо проводиться пошук інформації з безпеки для особливих популяцій, випадки та контролі можуть класифікуватися за популяцією, яка представляє інтерес (наприклад люди похилого віку, діти, вагітні). Наявні великі бази даних окремих популяцій є практичним та ефективним засобом забезпечення необхідними даними щодо експозиції та медичних результатів у відносно короткий період часу. Дослідження випадок-контроль особливо корисне, якщо метою є дослідити, чи існує зв’язок між лікарським засобом (або декількома лікарськими засобами) та одним певним рідкісним побічним явищем, а також визначити різні фактори ризику побічних явищ. До факторів, що представляють інтерес, належать такі захворювання, як, наприклад, порушення функції нирок та печінки, що можуть змінювати зв’язок між експозицією лікарського засобу та побічним явищем. Якщо всі випадки, що представляють інтерес (або добре визначена частина випадків), у зоні охоплення зареєстровані, а частина контролів з вихідної популяції відома, у дослідженні випадок-контроль можна визначити абсолютну частоту явищ.
У випадку, коли вихідна популяція дослідження випадок-контроль є добре визначеною групою або зоною охоплення, існує можливість провести в ній випадкову вибірку для формування контрольної групи. У цих ситуаціях через те, що вибіркова частина випадків та контролів відома, дослідження випадок-контроль може також надати дані абсолютної частоти явищ. Назва «вкладене дослідження випадок-контроль» була придумана для позначення тих досліджень, у яких контрольна вибірка ґрунтується на густоті (наприклад контрольна група представляє розподіл експозиції в людино-часі у вихідній популяції). Ще одним різновидом є дослідження випадок-когорта, у якому контрольна вибірка виконується серед тих осіб, які складають вихідну популяцію, незалежно від тривалості часу, протягом якого вони входили до її складу. Підхід випадок-контроль може також встановлюватися як постійна схема для виявлення та кількісного визначення ризиків (спостереження випадок-контроль). Цієї стратегії дотримувалися при рідкісних захворюваннях з урахуванням відповідної етіології, що характерна для лікарських засобів, включаючи патологічні зміни крові або серйозні шкірні порушення.
VIII. Додаток 1.1.2.4. Дизайни дослідження з вивчення лише випадків
Дизайни дослідження з вивчення лише випадків були запропоновані для оцінки зв’язку між періодичними експозиціями та короткостроковими явищами, включаючи самоконтрольовані дослідження серії випадків, перехресне порівняння випадків та контрольовані дослідження випадок-час. У цих дизайнах використовують лише випадки, а контрольну інформацію самостійно отримують з «особа-час» досвіду випадків. Однією з важливих переваг цих дизайнів є те, що сплутуючі змінні, які не змінюються з часом у осіб, автоматично приводяться у відповідність. Проте дизайни дослідження з вивчення лише випадків не можуть використовуватися за всіх умов, наприклад, коли точну дату початку захворювання важко встановити або при оцінці хронічних експозицій.
VIII. Додаток 1.1.3. Клінічні випробування (Clinical trials)
Якщо в дореєстраційних клінічних випробуваннях були виявлені важливі ризики, для оцінки механізму дії побічної реакції можуть вимагатися подальші клінічні випробування. Якщо дослідження є клінічним випробовуванням, до нього мають застосовуватися положення Директиви 2001/20/ЄС [15], положення Порядку проведення клінічних випробувань [16]N. У деяких випадках можуть проводитися фармакодинамічні та фармакокінетичні дослідження для визначення, чи може певна схема прийому піддавати пацієнтів підвищеному ризику розвитку побічних явищ. Генетичне дослідження може також надавати інформацію про те, яка група пацієнтів може піддаватися підвищеному ризику виникнення побічних реакцій. Крім того, на підставі фармакологічних властивостей та очікуваного застосування лікарського засобу в клінічній практиці може вимагатися проведення спеціальних досліджень для вивчення потенційних взаємодій лікарських засобів та взаємодій лікарських засобів з продуктами харчування. Такі дослідження можуть включати фармакокінетичні дослідження популяції та клінічну апробацію лікарського засобу у пацієнтів та у здорових добровольців.
Інколи потенційні ризики чи непередбачена користь в особливих популяціях можуть визначатися в дореєстраційних клінічних досліджень, однак їх неможливо повністю оцінити внаслідок невеликого розміру вибірки або через виключення субпопуляцій пацієнтів з цих клінічних досліджень. Ці популяції можуть включати людей похилого віку, вагітних, дітей або пацієнтів з порушеннями функції нирок чи печінки. Метаболізм лікарського засобу у дітей, людей похилого віку та осіб з супутніми захворюваннями може відрізнятися від такого у пацієнтів, які зазвичай залучаються до клінічних досліджень. Тому можуть проводитися подальші клінічні дослідження для визначення та оцінки величини ризику (або користі) у таких популяціях.
VIII. Додаток 1.1.3.1. Масштабні дослідження за спрощеною процедурою (Large simple trials)
Масштабне просте дослідження є особливою формою клінічного дослідження, у якому велика кількість пацієнтів рандомізована для лікування, але збір даних та моніторинг зводиться до мінімуму, що відповідає меті дослідження — бути відносно невеликим навантаженням. Подібним чином у популяції пацієнтів може проводитися стандартизований нагляд, який, як правило, відповідає стандартній клінічній практиці. Такий дизайн може використовуватися у фармаконагляді для з’ясування профілю співвідношення користі/ризику лікарського засобу за межами формальних/традиційних параметрів клінічного дослідження та/або для повної оцінки ризику критичного, але відносно рідкісного побічного явища. Термін «спрощений» стосується структури, а не збору даних. Він використовується відносно ситуацій, в яких збирається обмежена інформація щодо експозиції, результатів та потенційних заплутуючих факторів для гарантування можливості залучення великої кількості пацієнтів в експериментальний дизайн, а термін не може адекватно відображати складність проведених досліджень. Ці дослідження відносяться до клінічних. У цьому контексті визначення «прагматичне дослідження» та «масштабне просте дослідження» є синонімічними.
VIII. Додаток 1.1.4. Дослідження застосування лікарського засобу (Drug utilization studies)
Дослідження застосування лікарського засобу (DUS) описують призначення та застосування лікарського засобу в стандартній клінічній практиці у великих популяціях, у тому числі у осіб похилого віку, дітей, вагітних чи пацієнтів з порушенням функції печінки або нирок. Ці популяції часто неприйнятні для включення в рандомізовані клінічні дослідження. Розподіл за віком, статтю, супутнім лікуванням та іншими характеристиками дозволяє всебічно охарактеризувати пацієнтів, які проходять лікування, включаючи розподіл тих чинників, що можуть вплинути на клінічні, соціальні та економічні результати. Такі дослідження дають змогу отримати дані знаменника для визначення частоти побічних явищ. Дослідження застосування лікарських засобів (DUS) використовувалися для вивчення впливу регуляторних заходів та уваги засобів масової інформації до застосування лікарських засобів у щоденній медичній практиці, для вивчення зв’язку між рекомендованою та дійсною клінічною практикою, для моніторингу медичних помилок та для визначення загрози зловживання лікарським засобом шляхом з’ясування, чи застосовують пацієнти підвищені дози, або існування доказів невідповідного повторного призначення. Дослідження застосування лікарських засобів (DUS) особливо практичні, як перший етап у дизайні ПДБЛЗ для отримання достатнього розуміння характеристик популяції споживачів досліджуваних лікарських засобів та визначення найбільш відповідного компаратора, а також важливих потенційних заплутуючих факторів, які слід розглянути. Вони також корисні, тому що вони першими вказують на рівень впливу на здоров’я населення, який передбачається, якщо існує дійсний причинно-наслідковий зв’язок між експозицією, що представляє інтерес, та побічним явищем, наприклад, встановлений розмір популяції, що експонується, масштаб застосування не за показанням та ін. З регуляторною метою дослідження застосування лікарських засобів (DUS), для яких головною метою є поглиблення знань про безпеку лікарських засобів або ефективність заходів з мінімізації ризиків можуть класифікуватися як ПДБЛЗ (див. модуль VIII.B.1.)
VIII. Додаток 1.2. Джерела даних
Фармакоепідеміологічні дослідження можуть проводитися з використанням різних джерел даних. Традиційно польові дослідження вимагалися для отримання необхідних даних щодо експозиції, результатів лікування, потенційних заплутуючих факторів та інших змінних шляхом опитування відповідних суб’єктів (наприклад пацієнтів, родичів) або перегляду медичної документації на паперових носіях. Поява автоматизованих медичних баз даних значно підвищила ефективність фармакоепідеміологічного дослідження. Існує два основних види автоматизованих баз даних: такі, що містять загальну медичну інформацію, включаючи, призначення, діагностику, направлення, виписку з історії хвороби, та такі, що створюються з адміністративною метою, що вимагає зв’язку між аптечними та медичними базами даних. Такі бази даних можуть включати мільйони пацієнтів та дозволяють проводити великі дослідження. Значним обмеженням, проте, часто є відсутність довготривалого спостереження та послідовного лівого та правого цензурування даних. Також ці бази даних можуть не містити детальної та точної інформації, необхідної для деяких досліджень, такої як валідована діагностична інформація або лабораторні дані, тому необхідно переглядати медичну документацію на паперових носіях для встановлення та валідації результатів тесту та медичних діагнозів. Залежно від результату, що представляє інтерес, валідація може вимагати підходу в залежності від кожного конкретного випадку або лише огляду рандомізованої вибірки випадків. Інші ключові аспекти можуть вимагати валідації, якщо необхідно. Існує багато баз даних для потенційного використання в фармакоепідеміологічних дослідженнях або в їх валідаційній фазі.
Заявники (власники реєстраційних посвідчень) повинні обирати найкраще джерело даних з урахуванням його валідності (наприклад повнота відповідної інформації, можливість перевірки результатів) та критеріїв ефективності (наприклад, часовий діапазон для надання результатів). Слід також враховувати і зовнішню валідність. Обране для дослідження джерело даних, за можливості, повинно охоплювало популяцію, у якій виявлена проблема з безпеки. У випадку залучення іншої популяції заявнику (власнику реєстраційного посвідчення) слід оцінити розбіжності, що можуть існувати у відповідних змінних (наприклад вік, стать, спосіб застосування лікарського засобу), та їх потенційний вплив на результати дослідження. При статистичному аналізі слід дослідити потенційний вплив відхилень таких змінних.
При використанні будь-якого джерела даних слід дотримуватися правил забезпечення конфіденційності персональних даних.
VIII. Додаток I. Вимоги та рекомендації щодо подання інформації про неінтервенційні післяреєстраційні дослідження з безпеки
VIII. Додаток I.1. Вступ
У цьому Додатку надається додаткова інформація про нормативно-правові вимоги (що позначаються модальним дієсловом «повинен») та рекомендації (що позначаються модальним дієсловом «слід») щодо подання протоколів дослідження, звітів про дослідження та заключних звітів про дослідження для ПДБЛЗ до національних уповноважених органів та EMA. У ньому також надається додаткова інформація про реєстрацію неінтервенційних ПДБЛЗ у Реєстрі післяреєстраційних досліджень ЄС (EU PAS Register). У ньому не надаються рекомендації щодо передання інформації до комітетів з етики, національних наглядових рад або інших органів, що існують на місцях відповідно до національного законодавства.
VIII. Додаток I.2. Реєстрація дослідження
Відповідно до додатку III.3 ІП 520/2012 [6] (Формат заключного звіту про дослідження) для неінтервенційних ПДБЛЗ, що проводяться на виконання зобов’язань, заключний звіт дослідження повинен включати дату реєстрації дослідження в електронному реєстрі досліджень у якості важливого етапу. У розділі VIII.B.2. також стверджується, що власникам реєстраційних посвідчень слід також реєструвати всі неінтервенційні ПДБЛЗ, що проводяться за власною ініціативою в ЄС або вимагаються планом управління ризиками, що узгоджений у ЄС. Неінтервенційне ПДБЛЗ слід реєструвати перед початком дослідження або якомога раніше, а також до реєстру слід завантажувати протокол дослідження (та зміни до нього), звіти про дослідження, а також заключний звіт про результати дослідження.
EU PAS Register — це реєстр післяреєстраційних досліджень, вільний доступ до якого надається на вебсторінці EU PAS Register91, що слугує в якості електронного реєстру досліджень, що згадується у Додатку III ІП 520/2012 [6]. Інформація, що вимагається на момент реєстрації дослідження у EU PAS Register, включає адміністративні деталі, цілі дослідження та методологічні аспекти. Можна завантажувати протокол дослідження, звіт про дослідження та інші документи. Адміністративна інформація включає інформацію про те, чи дослідження проводиться на вимогу регуляторного органу, категорію ПУР, якщо застосовується, інформацію про долю фінансування з різних джерел та країн, де буде проводитися дослідження. Якщо у записах стосовно нового зареєстрованого дослідження зазначено, що дослідження проводиться на вимогу регуляторного органу, фінансується навіть частково фармацевтичною компанією та проводиться принаймні в одній країні ЄС, EMA надсилає повідомлення з повною назвою дослідження, назвою організації(й), що фінансує(ють), назвою країни, де проводитиметься дослідження, та посилання на вебсторінку із записами про поточне дослідження усім національним уповноваженим органами держав ЄС. Це повідомлення призначене для систематичного інформування держав ЄС про публічну реєстрацію ПДБЛЗ, що проводиться на вимогу регуляторного органу, фінансується заявником (власником реєстраційного посвідчення) та проводиться на його території.
Завантаження протоколу дослідження, звіту(ів) про хід дослідження та заключного звіту про дослідження у EU PAS Register не є правовим зобов’язанням. Отже, реєстрація неінтервенційних ПДБЛЗ в EU PAS Register не може бути єдиними каналом подання цих документів до національних уповноважених органів та EMA.
VIII. Додаток I.3. Вимоги та рекомендації для неінтервенційних післяреєстраційних досліджень з безпеки, що проводяться на виконання зобов’язань, встановлених уповноваженим органом ЄС
Ці дослідження включають неінтервенційні ПДБЛЗ категорій 1 та 2 досліджень, що описані у Модулі V ННПФ.
Проєкт протоколу, оновлений протокол дослідження після внесення значущої зміни та заключний звіт про дослідження повинні надаватися відповідно до встановленої процедури до PRAC, EMA або національного уповноваженого органу держави ЄС/УкраїниN, що зобов’язала проводити дослідження, якщо дослідження проводиться тільки в одній державі ЄС або в УкраїніN (ст. 107n-107p Директиви 2001/83/ЄС [1]), положення Порядку [2]N. Заключний звіт про дослідження слід надати протягом 12 місяців після закінчення збору даних (ст. 107p(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Відповідно до статті 107m(5) Директиви 2001/83/ЄС [1]) національний уповноважений орган може вимагати від власника реєстраційного посвідчення подання звітів про хід дослідження до уповноважених органів держав ЄС, в яких проводиться дослідження. Національні уповноважені органи усіх держав ЄС, в яких проводиться дослідження, за винятком Данії, стверджували, що вони вимагають подання звітів про хід дослідження. Звіти про дослідження слід також надавати до EMA стосовно лікарських засобів, що зареєстровані за централізованою процедурою.
VIII. Додаток I.4. Вимоги та рекомендації для неінтервенційних післяреєстраційних досліджень з безпеки, що проводяться за власною ініціативою
Ці дослідження включають неінтервенційні ПДБЛЗ категорії 3, що описана в Модулі V ННПФ, та інші неінтервенційні ПДБЛЗ, що проводяться за власною ініціативою заявниками (власниками реєстраційних посвідчень).
Відповідно до ст. 107m(6) Директиви 2001/83/ЄС [1], положень Порядку [2]N протягом 12 місяців з дати закінчення збору даних заключний звіт про дослідження повинен надаватися за національними процедурами до уповноважених органів держав ЄС/УкраїниN, в яких проводилося дослідження.
Відповідно до ст. 107m(5) Директиви 2001/83/ЄС [1] національний уповноважений орган може вимагати від заявника (власника реєстраційного посвідчення) подання протоколу дослідження та звітів про хід дослідження до уповноважених органів держав ЄС, у яких проводиться дослідження. Національні уповноважені органи таких держав ЄС, у яких проводиться дослідження, встановили, що вони вимагають подання протоколу дослідження та звітів про дослідження за національними процедурами: Австрія, Болгарія, Хорватія, Чеська Республіка, Франція, Німеччина, Італія, Литва, Нідерланди, Португалія, Румунія, Словаччина, Словенія, Іспанія.
Для досліджень категорії 3 звіти про хід дослідження слід також надавати до EMA (стосовно лікарських засобів, що зареєстровані за централізованою процедурою).
Відповідно до рекомендацій Модуля V ННПФ для досліджень категорії 3 протокол дослідження слід також надавати разом з ПУР.
Частина ІХ
Модуль ІХ — Управління сигналом
IX.A. Вступ
Звіт VIII Робочої групи Ради міжнародних організацій з медичних наук «Практичні аспекти виявлення сигналу у фармаконагляді» (CIOMS, Женева, 2010) визначає сигнал як інформацію, що надходить з одного або декількох джерел (включаючи спостереження та експерименти), яка свідчить про новий потенційно причинно-наслідковий зв’язок або новий аспект відомого зв’язку між втручанням та явищем або низкою пов’язаних явищ, будь-яких несприятливих чи сприятливих подій, що вважається достатньо вірогідним для обґрунтування.
У контексті цього модуля буде розглядатися тільки нова інформація, що має відношення до побічних ефектів.
Для припущення нового потенційного причинно-наслідкового зв’язку або нового аспекту відомого зв’язку будь-який сигнал слід підтвердити з урахуванням інших значущих джерел інформації.
Процес управління сигналом можна визначити як послідовність дій, що здійснюються для визначення, на підставі вивчення повідомлень про індивідуальний випадок, пов’язаний з безпекою (ICSR), згрупованих даних активних систем нагляду або досліджень, інформації з літератури або інших джерел даних, чи існують нові ризики, що пов’язані з діючою речовиною або лікарським засобом, або відомі ризики змінилися. Процес управління сигналом має включати всі етапи: початкове виявлення сигналу; його валідації та підтвердження; аналіз та визначення пріоритетів; оцінку сигналу для рекомендації заходів, а також відстеження заходів, що вжиті, та будь-яких рекомендацій, що надаються (ст. 21(1) ІП 520/2012 [6], положення Порядку [2]N).
У ЄС та Україні процес управління сигналом здійснюється усіма зацікавленими сторонами, що залучені до моніторингу безпеки лікарських засобів, включаючи пацієнтів, працівників з медичною та/або фармацевтичною освітою, власників реєстраційних посвідчень, регуляторні органи, наукові комітети та органи, що приймають рішення (такі як уповноважені органи країн ЄС та Європейська Комісія (ЄК)).
У той час як база даних EudraVigilance (в Україні — АІСФN) є основним джерелом інформації щодо фармаконагляду, процес управління сигналом охоплює сигнали, що надходять не лише з бази даних EudraVigilance (в Україні — АІСФN), або прямо не підтримуються базою даних EudraVigilance (в Україні — АІСФN). Для моніторингу даних в базі даних EudraVigilance (в Україні — АІСФN) розглядаються лише сигнали, що пов’язані з побічними реакціями (ст. 19(1) ІП 520/2012 [6]).
Регламент (ЄС) № 1235/2010 [4], що вносить зміни до Регламенту (ЄС) № 726/2004 [5], Директива 2010/84/ЄС [3], що вносить зміни до Директиви 2001/83/ЄС[1] та ІП 520/2012 [6] щодо впровадження заходів з фармаконагляду, що передбачені у Регламенті (ЄС) № 726/2004 [5] та Директиві 2001/83/ЄС [1], включають положення щодо управління сигналом в Європейському Союзі.
Метою цього модуля є:
- надання загальних рекомендацій та вимог до структур та процесів, що включені до управління сигналом (розділ IX.B.);
- опис ролі цих структур та процесів при здійсненні фармаконагляду та функціонуванні регуляторної системи (розділ IX.C.).
IX.B. Структури та процеси
IX.B.1. Джерела даних та інформація
Джерела ідентифікації нових сигналів різноманітні. Вони потенційно включають всю наукову інформацію про застосування лікарських засобів, у тому числі про якість, доклінічні дані, клінічні дані, дані з фармаконагляду та фармакоепідеміологічні дані. Певні джерела для сигналів включають системи повідомлень про спонтанні побічні реакції, системи активного нагляду, неінтервенційні дослідження, клінічні дослідження, наукову літературу та інші джерела інформації.
Сигнали зі спонтанних повідомлень можна виявити під час моніторингу повідомлень про індивідуальний випадок, пов’язаний з безпекою, у базах даних побічних реакцій, у статтях наукових публікацій або при розгляді інформації, що надана заявниками (власниками реєстраційних посвідчень) в контексті регуляторних процедур (наприклад, внесення змін, перереєстрація, післяреєстраційні зобов’язання, РОЗБ, нових редакцій ПУР або в контексті іншої діяльності, що має відношення до моніторингу співвідношення користі/ризику лікарських засобів).
Спонтанні повідомлення про побічні реакції можна також надавати до токсикологічних центрів, інформаційних тераторологічних служб, програм нагляду за вакцинами, систем повідомлень, що встановлені заявниками (власниками реєстраційних посвідчень), та будь-яких інших структурованих та організованих схем збору даних, що дозволяють пацієнтам та працівникам з медичною та/або фармацевтичною освітою повідомляти про підозрювані побічні реакції на лікарські засоби. Уповноваженим органам слід зв’язуватися з іншими установами або організаціями, що управляють такою системою повідомлень, щоб бути інформованими щодо цих підозрюваних побічних реакцій.
Завдяки збільшенню кількості спонтанних повідомлень, введенню електронного подання повідомлень про безпеку пацієнтами або заявниками (власниками реєстраційних посвідчень) до уповноважених органів, виявлення сигналу наразі все більше ґрунтується на регулярному моніторингу великих баз даних, таких як база даних EudraVigilance (в Україні — АІСФN).
Сигнали можуть виникати з широкого спектру різних типів досліджень, включаючи дослідження якості, доклінічні, інтервенційні та неінтервенційні дослідження, системні огляди та метааналізи. До інтервенційних та обсерваційних досліджень можна залучати та спостерігати певну популяцію досліджуваних, у яких можуть розвинутися побічні реакції. Огляд згрупованих даних та статистичні аналізи можуть також вказувати на підвищений ризик побічного явища, який буде в подальшому досліджуватися як сигнал.
Заявникам (власникам реєстраційних посвідчень) слід ідентифікувати опубліковані результати релевантних досліджень шляхом скринінгу наукової літератури. Для отримання загальних рекомендацій з проведення пошуку даних у літературі див. модуль VI ННПФ.
Заявникам (власникам реєстраційних посвідчень) слід регулярно відстежувати за допомогою всіх наявних та доступних ресурсів, включаючи інтернет, у межах відповідальності, що зазначені у модулі VI, потенційні повідомлення про підозрювані побічні реакції, які можуть характеризувати новий сигнал. Заявникам (власникам реєстраційних посвідчень) або уповноваженим органам слід у подальшому шукати додаткову інформацію щодо підозрюваних побічних реакцій, про які їм стало відомо з будь-якого джерела. Підозрювані побічні реакції слід підтверджувати, якщо можливо, за допомогою інших джерел даних, таких як EudraVigilance (в Україні — АІСФN).
IX.B.2. Методологія виявлення сигналу
У якості загального принципу для виявлення сигналу слід додержуватися визнаної методології, що може варіювати залежно від типу лікарського засобу, який вона має охоплювати. Наприклад, вакцини можуть мати інші методологічні підходи, ніж лікарські засоби у традиційному їх розумінні. Виявлення сигналів має ґрунтуватися на багатосторонньому підході.
Виявлення сигналу у базі даних EudraVigilance (в Україні — АІСФN) необхідно доповнювати статистичним аналізом (ст. 19(2) ІП 520/2012 [6]).
Для визначення доказової цінності (тобто підтвердження доказу) сигналу необхідно застосовувати визнану методологію із врахуванням клінічного значення, кількісної сили зв’язку, послідовності даних, співвідношення експозиції/реакції, біологічної вірогідності, експериментальних даних, можливих аналогій, а також характеру та якості даних (ст. 20(1) ІП 520/2012 [6]).
Різні фактори можна враховувати для визначення пріоритетності сигналів, а саме: чи зв’язок або діюча речовина/лікарський засіб є новими, сила зв’язку, серйозність даної реакції та документування повідомлень у базі даних EudraVigilance (в Україні — АІСФN) (ст. 20(2) ІП 520/2012 [6], положення Порядку [2]N).
IX.B.3 Процес управління сигналом
IX.B.3.1 Введення
Процес управління сигналом включає такі стадії:
- виявлення сигналу;
- валідація сигналу;
- аналіз ти визначення пріоритетності сигналу;
- оцінка сигналу;
- рекомендація щодо дій;
- обмін інформацією.
Управління сигналом, як правило, передбачає здійснення зазначених вище етапів у логічній послідовності. Однак широкий діапазон джерел інформації, наявних для виявлення сигналу, може вимагати певної гнучкості в здійсненні управління сигналом, наприклад:
1) коли виявлення сигналу головним чином ґрунтується на розгляді повідомлень про індивідуальні випадки, пов’язані з безпекою, ця діяльність може включати підтвердження та попереднє визначення пріоритетності будь-якого виявленого сигналу;
2) коли сигнал виявляється на підставі результатів дослідження, як правило, неможливо або недоцільно оцінювати кожний індивідуальний випадок, та для підтвердження може знадобитися збір додаткових даних;
3) рекомендація заходів (що супроводжуються рішенням згідно із законодавством) та обмін інформацією, яку слід розглядати на кожному етапі процесу.
У контексті цієї настанови сигнали, що надходять з моніторингу даних систем спонтанних повідомлень, розглядаються як початкова точка для процесу управління сигналом. Ті самі принципи слід застосовувати до даних, що надходять з інших джерел.
IX.B.3.2. Виявлення сигналу
Детальну настанову щодо методів виявлення сигналу можна знайти у Звіті VIII Робочої групи Ради міжнародних організацій з медичних наук «Практичні аспекти виявлення сигналу у фармаконагляді» (CIOMS, Женева, 2010) та в Настанові із застосування статистичних методів виявлення сигналу в Системі аналізу даних EudraVigilance (Doc. Ref.EMEA/106464/2006 rev. 1).
Будь-який метод, що використовується для виявлення сигналів, повинен враховувати зазначені нижче принципи:
- метод, що застосовується, повинен відповідати набору даних (наприклад складні статистичні інструменти не застосовуються для аналізу незначних даних);
- слід розглядати дані з усіх відповідних джерел;
- на місцях слід встановити системи для забезпечення якості діяльності з виявлення сигналу;
- будь-які результати огляду сукупних даних повинна своєчасно оцінювати особа з відповідною кваліфікацією;
- процес слід адекватно задокументувати, включаючи обґрунтування методу та зазначення періодичності діяльності з виявлення сигналу.
Виявлення сигналів може проводитися на підставі огляду повідомлень про індивідуальні випадки, пов’язані з безпекою, на підставі статистичного аналізу у великих базах даних або на підставі їх обох.
IX.B.3.2.1. Розгляд (аналіз) індивідуальних випадків, пов’язаних з безпекою
Як зазначено в модулі VI, повідомлення про індивідуальний випадок, пов’язаний з безпекою, можуть надходити із системи спонтанного рапортування, ПДБЛЗ та моніторингу літератури. Навіть одного повідомлення про серйозну або тяжку побічну реакцію (наприклад один випадок токсичного епідермального некролізу, апластичної анемії або трансплантації печінки) може буде достатньо для формування сигналу та впровадження подальших заходів. У цьому разі потрібно здійснювати огляд спонтанних повідомлень про побічні реакції та враховувати кількість випадків (після виключення дублікатів), демографічні дані пацієнтів (включаючи вік та стать), підозрюваний лікарський засіб (включаючи дозу, що вводиться, склад) та підозрювану побічну реакцію (включаючи ознаки та симптоми), зв’язок у часі, клінічний результат відносно продовження або припинення застосування лікарського засобу (тобто інформацію про відміну/повторне призначення). Під час оцінки причинно-наслідкового зв’язку слід враховувати наявність потенційних альтернативних причин, включаючи інші лікарські засоби, що одночасно приймаються, основне захворювання, оцінку інформатором причинно-наслідкового зв’язку та ймовірності біологічного та фармакологічного зв’язку.
IX.B.3.2.2. Статистичні аналізи
Виявлення сигналу все частіше базується на регулярному періодичному моніторингу великих баз даних спонтанних повідомлень про побічні реакції. Такі бази даних дозволяють створювати статистичні звіти, що надають інформацію про побічні реакції, що одержані за певний період часу відносно певних діючих речовин або лікарських засобів. Розроблені різні методи для статистичного визначення непропорціонального подання повідомлень, наприклад, більш високий показник повідомлень, ніж очікувалося для підозрюваної побічної реакції на діючу речовину/лікарський засіб, що розглядається, порівняно з усіма іншими діючими речовинами/лікарськими засобами у базі даних, (виражені, наприклад, як нижня межа пропорційного показника повідомлень ≥1). Беручи до уваги обмеження цих методів, сама статистика непропорційної подачі повідомлень не обов’язково вказує на те, що існує сигнал для подальшого дослідження, або на те, що існує причинно-наслідковий зв’язок.
Використання статистичних інструментів може не підходити до всіх ситуацій. Розмір набору даних, повноту наявної інформації та тяжкість побічної(их) реакції(й) слід враховувати при застосуванні статистичних методів та виборі критеріїв для виявлення сигналів.
Періодичність, з якою слід створювати та розглядати статистичні звіти, може варіюватися відповідно до діючої речовини (АФІN)/лікарського засобу, показання до застосування та будь-яких відомих потенційних або ідентифікованих ризиків. Деякі діючі речовини (АФІN)/лікарські засоби можуть також вимагати підвищеної частоти моніторингу даних (див. IX.C.2). Тривалість цієї підвищеної частоти моніторингу також може варіювати та бути гнучкою відносно накопичення знань про профіль ризику, що пов’язаний із застосуванням діючої речовини (АФІN)/лікарський засіб, що оцінюється.
IX.B.3.2.3. Комбінація статистичних методів та огляд повідомлень про індивідуальні випадки, пов’язані з безпекою
Статистичні звіти можуть розроблятися для функціонування інструментів для ідентифікації підозрюваних побічних реакцій, що відповідають заздалегідь визначеним критеріям частоти, тяжкості, клінічного значення, нових даних або статистичного зв’язку. Такі інструменти фільтрування можуть полегшити вибір повідомлень про побічні реакції, що повинні розглядатися на першому етапі. Пороги, що застосовуються в цьому процесі фільтрації (наприклад повідомлено щонайменше про 3 випадки) можуть варіювати відповідно до ступеня застосування лікарських засобів і, таким чином, потенційного впливу на громадське здоров’я.
Незалежно від статистичного методу, що застосується, якщо статистичні звіти використовуються для автоматизації скринінгу бази даних, до виявлення сигналу слід завжди включати клінічну оцінку, а відповідні повідомлення про побічні реакції слід розглядати в індивідуальному порядку з урахуванням їх клінічного значення (IX.B.3.2.1.).
Тому статистичний метод слід вважати підтримуючим інструментом у загальному процесі виявлення сигналу та його наступної валідації.
IX.B.3.3. Валідація сигналу
Валідація сигналу — це процес оцінки даних, що свідчить про виявлений сигнал та підтверджує, що наявна документація містить достатньо доказів, що демонструють наявність нового потенційного причинно-наслідкового зв’язку або новий аспект відомого зв’язку, та тому обґрунтовує подальший аналіз (ст. 21(1) ІП 520/2012 [6]).
Для валідації сигналу до уваги слід брати наступне:
а) клінічне значення, включаючи, наприклад:
- силу доказу причинного впливу (наприклад кількість повідомлень, експозицію, зв’язок у часі, ймовірний механізм, інформацію про відміну/повторне призначення, альтернативне пояснення/фактори, що спотворюють результат);
- серйозність та тяжкість реакції та її наслідки;
- нові дані про реакцію (наприклад нові та серйозні побічні реакції);
- взаємодію з іншими лікарськими засобами;
- реакції, що виникають у певних популяціях.
б) попередня інформованість:
- обсяг інформації, що вже була включена до короткої характеристики лікарського засобу або листа-вкладки;
- чи вже оцінювався зв’язок у РОЗБ або ПУР або обговорювався на рівні наукового комітету, або підлягав регуляторній процедурі.
У принципі слід валідувати тільки новий сигнал, відносно якого відсутня попередня інформація. Однак уже відомий зв’язок може стати причиною нового сигналу, якщо очевидна частота повідомлень, його тривалість та тяжкість або зміна у попередньо повідомлених наслідках побічних реакцій (таких як нова фатальність) припускають появу нової інформації, порівняно з інформацією, що включена до короткої характеристики лікарського засобу або раніше оцінювалася уповноваженим органом.
в) наявність інших релевантних джерел інформації, що забезпечує більший набір даних щодо того самого зв’язку:
- літературні дані щодо подібних випадків;
- експериментальні результати або біологічні механізми;
- скринінг баз даних з більшими наборами даних (наприклад EudraVigilance, коли сигнал сформувався спочатку з національної бази даних або бази даних компаній).
Величину та клінічне значення сигналу можна також вивчати за допомогою описового (дескриптивного) аналізу в інших наявних джерелах даних або за допомогою аналізу характеристик пацієнтів, які зазнали впливу лікарського засобу, та моделей застосування у них лікарського засобу.
Інколи до сигналів, валідність яких не підтверджена, повертаються при проведенні наступного аналізу, оскільки може бути доцільним подовження моніторингу потенційного сигналу, поки не буде достатньо доказів для його підтвердження. Наприклад, у наявності може бути невідповідна документація щодо випадку або доказ причинно-наслідкового зв’язку тільки у деяких повідомленнях про побічну реакцію. У такому разі повідомлення про такі ж прояви побічних реакцій або наступні повідомлення про раніше одержані випадки, слід розглядати через відповідні інтервали часу з метою врахування всіх значущих випадків.
Заявникам (власникам реєстраційних посвідчень) та уповноваженим органам слід встановити системи відстеження для фіксації результату валідації сигналів, включаючи причини, з яких не здійснювалася валідація сигналів, а також інформацію, що може полегшити подальший пошук повідомлень про побічні реакції та валідацію сигналів.
IX.B.3.4. Аналіз та визначення пріоритетності сигналу
Основний елемент процесу управління сигналом — швидка ідентифікація валідованих сигналів, що мають важливий вплив на громадське здоров’я або які можуть значно вплинути на співвідношення користі/ризику лікарського засобу у пацієнтів, яким проводиться лікування. Ці сигнали вимагають негайної уваги та визначення їх пріоритетності для подальших дій без затримки. При визначенні пріоритетності сигналу слід враховувати:
- вплив на пацієнтів залежно від тяжкості, оборотності, потенціалу для запобігання та клінічних наслідків;
- наслідки припинення лікування для хвороби та наявність терапевтичних альтернатив;
- сила та послідовність доказів у підтримку зв’язку, наприклад, біологічна імовірність, велика кількість випадків, про які повідомлено за короткий період часу, показник непропорційності повідомлень і швидке збільшення показника з часом та ідентифікація сигналу у різних закладах (наприклад загальна практика або стаціонари), джерела даних або країни;
- клінічний контекст (наприклад, чи зв’язок припускає клінічний синдром, який може включати інші реакції);
- вплив на громадське здоров’я, включаючи ступінь споживання лікарського засобу у загальній популяції та у спеціальних популяціях (наприклад вагітні, діти або особи похилого віку) та моделі споживання лікарського засобу (наприклад застосування не за затвердженими показаннями або помилкове застосування). Вплив на громадське здоров’я може включати оцінку кількості пацієнтів, на яких вплинула побічна реакція, цю кількість слід розглядати відносно розміру загальної популяції, популяції з хворобою-мішенню та популяцією, якій проводилося лікування;
- підвищення частоти або тяжкості відомої побічної реакції;
- чи є підозрювана побічна реакція новою, наприклад, коли невідома підозрювана побічна реакція виникає незабаром після реєстрації нового лікарського засобу;
- чи заява на одержання реєстраційного посвідчення на нову діючу речовину (АФІ) ще знаходиться на розгляді.
За деяких обставин пріоритет слід віддати сигналам, що ідентифіковані відносно лікарських засобів або явищ, та які представляють потенційно високий інтерес для медіа або зацікавлених сторін (у фармаконагляді), щоб якнайшвидше повідомити результат населенню та працівникам з медичною та/або фармацевтичною освітою.
Результат визначення пріоритетності сигналу має включати рекомендацію щодо термінів для управління сигналом.
Результат процесу визначення пріоритетності сигналу слід ввести до системи відстеження з обґрунтуванням визначеної пріоритетності.
IX.B.3.5. Оцінка сигналу
Метою оцінки сигналу є подальша оцінка валідованого сигналу з метою визначення необхідності у зборі додаткових даних або здійснення будь-яких регуляторний дій. Вона включає оцінку наявних фармакологічних, доклінічних та клінічних даних та інформації з інших джерел. Цей огляд має бути, за можливості, повним відносно джерел інформації, включаючи досьє до заяви, літературних публікацій, спонтанних повідомлень, консультацій експертів та інформації, що відома заявникам (власникам реєстраційних посвідчень) та уповноваженим органам. Коли інформацію отримають із низки джерел, слід розглянути переваги та обмеження кожного джерела з метою оцінки їх внеску, щодо загальної оцінки сигналу у вигляді рекомендації для здійснення відповідних заходів. Підсумовування інформації, що надійшла з різних джерел, також вимагає вибору міжнародно узгодженого визначення випадку (наприклад, визначення випадку реакції на вакцини в межах Брайтонського співробітництва). Якщо не існує такого визначення, слід розробити оперативне визначення.
Може існувати потреба в оцінці сигналу на ширшому рівні, наприклад, на рівні терапевтичного класу або класу систем органів або на рівні стандартизованого запиту MedDRA (тобто SMQ). Пошук інформації для оцінки важливості сигналу може потребувати його розширення на інші лікарські засоби відповідної фармако-терапевтичної групи та на інші побічні реакції, так само на інші терміни, що пов’язані із складним захворюванням (наприклад ретробульбарний неврит як можлива рання ознака розсіяного склерозу), з попередньою стадією реакції (наприклад пролонгацією інтервалу Q–T та тріпотінням/фібриляцією шлуночків) або з клінічними ускладненнями відповідної побічної реакції (наприклад зневоднення та гостра ниркова недостатність).
Збір інформації з різних джерел може займати час. Для нового сигналу про серйозну або тяжку побічну реакцію слід вживати заходів на будь-якій стадії управління сигналом, включаючи виявлення, якщо вже наявна інформація підтверджує висновок про наявність потенційного ризику, якому слід запобігти або який слід вчасно мінімізувати.
IX.B.3.6. Рекомендація щодо дій
У результаті оцінки сигналу на конкретний момент часу може бути прийняте рішення про необхідність подальших дій, або про відсутність необхідності подальших дій. Хоча необхідність дій, як правило, є наступним етапом після оцінки сигналу, необхідність у діях слід розглядати протягом процесу управління сигналом, що залежить від обсягу інформації. Наприклад, перший випадок побічної реакції, що вказує на виробничий дефект, може вимагати негайного відкликання серії лікарського засобу. Наявна інформація на стадіях валідації сигналу або визначення пріоритету сигналу може стати підґрунтям для прийняття висновку, що доказ є достатньо вагомий для введення тимчасових заходів. У таких ситуаціях все ще необхідно продовжувати формальну оцінку сигналу з метою підтвердження чи непідтвердження безпеки для подовження або відміни тимчасових заходів.
Рекомендації щодо дій можуть включати:
- впровадження негайних заходів, включаючи можливість призупинення дії реєстраційного посвідчення на лікарський засіб;
- запит на надання додаткової інформації, що має надати заявник (власник реєстраційного посвідчення), наприклад, для підтвердження, що висновок має силу для всіх показань та груп пацієнтів;
- регулярний огляд сигналу, наприклад, за допомогою РОЗБ (див. модуль VII); · додаткових досліджень або дій для мінімізації ризику;
- перегляду інформації про лікарський засіб за допомогою регуляторної процедури;
- проведення ПДБЛЗ (див. модуль VIII).
Щоразу, коли від заявника (власника реєстраційного посвідчення) вимагаються дії, у вимозі слід зазначати строки, до яких дії слід закінчити, включаючи положення про звіт про виконання та проміжні результати пропорційно до тяжкості та впливу сигналу на громадське здоров’я.
IX.B.3.7. Обмін інформацією
Уповноваженим органам та заявникам (власникам реєстраційних посвідчень) слід обмінюватися інформацією про валідовані сигнали, нові питання щодо безпеки, які виникають, та результати оцінок сигналу.
Заявникам (власникам реєстраційних посвідчень) слід негайно повідомляти уповноваженим органам про сигнали, що можуть мати наслідки для громадського здоров’я та впливати на співвідношення користі/ризику лікарського засобу в якості питання, що виникає (нового) (див. модуль VI) та, якщо необхідно, можна включити пропозиції щодо дій.
Про результати оцінки ризику, що включає нові або змінені ризики та ризики, що мають вплив на співвідношення користі/ризику діючої речовини (АФІ)/лікарських засобів, що розглядаються, слід повідомляти населення, включаючи працівників з медичною та/або фармацевтичною освітою та пацієнтів (див. IX.C.6), а також відповідних заявників (власників реєстраційних посвідчень).
IX.B.4. Вимоги до якості
IX.B.4.1. Відстеження
Усю валідацію, пріоритизацію, оцінку, терміни, рішення, дії, плани, подання повідомлень а також всі інші основні етапи слід реєструвати та систематично відстежувати. Системи відстеження слід використовувати для документування, вони мають включати також сигнали, для яких процес валідації, що проводився, не вказував на новий потенційно причинно-наслідковий зв’язок або новий аспект відомого зв’язку. Усі записи необхідно архівувати (ст. 24(1) ІП 520/2012 [6]) (див. модуль I).
IX.B.4.2. Системи якості та документація
Основною ознакою системи управління сигналом є те, що вона чітко задокументована для забезпечення її належного та ефективного функціонування, а ролі, обов’язки та необхідні завдання стандартизовані, виконуються персоналом з відповідною кваліфікацією та є зрозумілими всім зацікавленим сторонам, існує положення про відповідний контроль та, за необхідності, про покращення/вдосконалення системи. Тому на місцях слід мати системи забезпечення та контролю якості, що відповідають стандартам системи якості та застосовується до всіх процесів управління сигналом (див. модуль I). Слід розробити, задокументувати та впровадити детальні процедури для цієї системи якості. Слід розподілити та затвердити обов’язки та відповідальність за дії щодо ведення документації, контроль якості та огляд, а також забезпечення реалізації коригуючих та превентивних заходів. Сюди належать функції з аудиту забезпечення якості системи управління сигналом, включаючи аудит підрядників. Слід гарантувати конфіденційність даних та документів за допомогою відповідних нормативно-правових актів, безпеку їх зберігання, включаючи цілісність при передаванні.
За допомогою систем відстеження всі сторони повинні зберігати та проводити аудит записів щодо дій з управління сигналом та відповідних запитів та їх результатів, включаючи те, як сигнали були виявлені, валідовані, підтверджені та оцінені (ст. 24(2) ІП 520/2012 [6]).
Від заявників (власників реєстраційних посвідчень) може запитуватися документація, що демонструє відповідність цим положенням та розглядається до та після видачі реєстраційного посвідчення.
Персоналу слід пройти спеціальне навчання у сфері діяльності з управління сигналом відповідно до розподілу обов’язків та відповідальності. Систему навчання та місце знаходження записів щодо навчання слід документувати, а СV (curricula vitae) (відомості про освіту та професійний досвід) та функціональні обов’язки слід архівувати.
IX.C. Функціонування мережі ЄС/в УкраїніN
IX.C.1. Ролі та обов’язки
У контексті функціонування регуляторної мережі заявникам (власникам реєстраційних посвідчень), EMA та національним уповноваженими органам слід постійно проводити моніторинг даних, що наявні у базі даних EudraVigilance (в Україні — (АІСФ)N), для визначення, чи є нові ризики або чи ризики змінилися, та чи впливають ті ризики на співвідношення користі/ризику. Слід застосовувати визнану методологію виявлення сигналу та валідувати виявлені сигнали.
EMA та національні уповноважені органи повинні співпрацювати під час моніторингу даних, що наявні в базі даних EudraVigilance (стаття 18(1) ІП 520/2012 [6], (в Україні — (АІСФ)N).
Відносно лікарських засобів, що зареєстровані згідно з Регламентом (ЄС) № 726/2004[5] (лікарські засоби, що зареєстровані за централізованою процедурою (САР)), EMA в моніторингу даних в EudraVigilance повинен допомагати доповідач, що призначений PRAC відповідно до ст. 62(1) Регламенту (ЄС) № 726/2004 [5] (ст. 22(5) ІП 520/2012 [6]).
Для лікарських засобів, що зареєстровані відповідно до Директиви 2001/83/ЄС [1] у більш ніж одній країні ЄС, та для діючих речовин, що містяться у декількох лікарських засобах, коли принаймні одне реєстраційне посвідчення було видано згідно з Директивою 2001/83/ЄС [1], країни ЄС можуть домовитися в межах CMDh у співпраці з PRAC призначити провідну державу ЄС для моніторингу даних у базі даних EudraVigilance та для валідації та підтвердження сигналів від імені інших країн ЄС. Провідну державу ЄС може підтримувати співпровідна держава, яка має допомагати провідній державі ЄС у виконанні її завдань. Будь-яке таке призначення слід переглядати принаймні кожні чотири роки (ст. 22(1) ІП 520/2012 [6]). При призначенні провідної держави ЄС та, за необхідності, співпровідної держави, CMDh у співпраці з PRAC може враховувати, чи будь-яка держава ЄС діє як референтна держава ЄС відповідно до ст. 28(1) Директиви 2001/83/ЄС [1] або як доповідач щодо оцінки регулярно оновлюваних звітів з безпеки згідно зі ст. 107(е) Директиви 2001/83/ЄС [1] (ст. 22(2) ІП 520/2012 [6]).
Усі держави ЄС залишаються відповідальними за моніторинг даних бази даних EudraVigilance відповідно до ст. 107h(1)(c) та ст. 107h(3) Директиви 2001/83/ЄС [1] (ст. 22(4) ІП 520/2012 [6]).
Національні уповноважені органи та EMA повинні здійснювати валідацію та підтверджувати будь-який сигнал, що був виявлений ними під час постійного моніторингу даних у базі даних EudraVigilance (ст. 21(4) ІП 520/2012 [6]) (в Україні — (АІСФ)N).
У разі якщо для лікарських засобів або діючих речовин (АФІN) доповідач назначений PRAC, йому слід підтверджувати валідовані сигнали. У разі якщо для лікарських засобів або діючих речовин (АФІN) призначена провідна держава ЄС, цій провідній державі ЄС слід підтверджувати валідовані сигнали.
Підтвердження доповідачем PRAC або провідною державою ЄС означає надання повідомлення за допомогою Європейського інструменту відстеження питань з фармаконагляду (EPITT) (див. розділ IX.C.5) про те, що сигнал дійсний. Слід надати обґрунтування, коли сигнал не підтверджений. Усі підтверджені сигнали передаються до PRAC. Для таких лікарських засобів або діючих речовин, відносно яких була призначена провідна держава ЄС, такій провідній державі ЄС слід валідувати та підтверджувати одноразово сигнали, які вона виявила. Для таких лікарських засобів або діючих речовин (АФІN), відносно яких не була призначена провідна держава ЄС, національному уповноваженому органу слід здійснювати валідацію та підтверджувати одноразово сигнали, які він виявив.
IX.C.1.1. Ролі та обов’язки EMA
EMA повинне:
- опубліковувати на європейському вебпорталі, що присвячений лікарським засобам, перелік діючих речовин/лікарських засобів та уповноважений орган (провідну державу ЄС, співпровідну державу ЄС або EMA), що відповідальний за їх моніторинг у EudraVigilance (стаття 22(3) ІП 520/2012 [6]);
- після консультації з PRAC публікувати перелік медичних явищ, що слід враховувати для виявлення сигналу, за потребою (ст. 19(2) ІП 520/2012 [6]);
- підтримувати моніторинг даних у базі даних EudraVigilance, надаючи національним уповноваженим органам доступ до:
- вихідних даних та статистичних звітів, що дозволяють проводити огляд всіх побічних реакцій, повідомлення про які надані до EudraVigilance на діючу речовину або лікарський засіб;
- замовлених запитів (запитів за певними умовами) на підтримку здійснення оцінки повідомлень про індивідуальні випадки з безпеки та серії випадків (case series);
- замовленого згрупування та стратифікації даних, що дозволяють ідентифікувати групи пацієнтів з підвищеним ризиком розвитку побічних реакцій або ризиком розвитку більш серйозної побічної реакції;
- статистичних методів виявлення сигналу (ст. 23 ІП 520/2012 [6]);
- забезпечити відповідну підтримку для моніторингу даних у базі даних EudraVigilance заявниками (власниками реєстраційних посвідчень) (ст. 23 ІП 520/2012 [6]);
- підготовити технічний документ, що встановлює загальні вимоги до виявлення сигналу та описує вихідні дані та статистичні звіти EudraVigilance;
- бути адміністратором Європейського інструменту відстеження питань з фармаконагляду (EPITT) для валідованих сигналів, що потребують подальшої оцінки (ст. 21(5) ІП 520/2012 [6]);
- керувати моніторингом даних EudraVigilance, виявленням сигналу та валідацією сигналу для лікарських засобів, що зареєстровані за централізованою процедурою (САР), та діючих речовин, що містяться у декількох лікарських засобах, коли принаймні одне реєстраційне посвідчення видане відповідно до Регламенту (ЄС) № 726/2004 [5];
- вводити валідовані сигнали, що воно виявило в EPITT;
- валідувати (включаючи, за необхідності, до бази даних EudraVigilance) та вводити до EPITT будь-який інший сигнал, про який повідомлено третьою стороною (наприклад регуляторним органом із країни поза межами ЄС) та стосується лікарських засобів, що зареєстровані за централізованою процедурою (САР), або діючої речовини, щодо яких EMA здійснює моніторинг даних в EudraVigilance;
- підтвердити у співпраці з державами ЄС якнайшвидше та не пізніше 30 днів з дня одержання EMA будь-якого сигналу, про який повідомили заявники (власники реєстраційних посвідчень) та який стосується лікарських засобів, що зареєстровані за централізованою процедурою (САР), або діючих речовин, щодо яких EMA здійснює моніторинг даних в EudraVigilance. У цьому контексті, коли дійсність сигналу не підтверджується, особливу вагу слід приділити будь-якій додатковій інформації, що може дозволити підтвердити сигнал (ст. 21(3) ІП 520/2012 [6]), див. розділ IX.B.3.3;
- передавати підтверджені сигнали до PRAC для первинного аналізу та визначення пріоритетності відповідно до ст. 28а(2) Регламенту (ЄС) № 726/2004 [5] (ст. 21(5) ІП 520/2012 [6]);
- негайно поінформувати зацікавлених заявників (власників реєстраційних посвідчень) про висновки PRAC щодо оцінки будь-якого підтвердженого сигналу (ст. 21(5) ІП 520/2012 [6]);
- зберігати контрольні записи (записи щодо контрольного аналізу процесу/аудиту) про свої дії щодо виявлення сигналу (ст. 24(1) ІП 520/2012 [6]).
IX.C.1.2. Ролі та обов’язки провідної держави ЄС
Провідна держава ЄС повинна:
- керувати моніторингом даних EudraVigilance, виявленням сигналу, валідацією та підтвердженням сигналу для діючих речовин/лікарських засобів, відносно яких вона призначена провідною;
- підтверджувати сигнали, що виявлені або валідовані національним уповноваженим органом для цих речовин/лікарських засобів;
- валідувати (включаючи, за необхідності, у базі даних EudraVigilance) та ввести до EPITT будь-який інший сигнал, про який повідомлено третьою стороною (наприклад регуляторним органом із країни поза ЄС) для цих речовин/лікарських засобів;
- підтверджувати якнайшвидше та не пізніше 30 днів після одержання будь-якого валідованого сигналу, про який повідомили заявники (власники реєстраційних посвідчень) на ці діючи діючі речовини/лікарські засоби. У цьому контексті, коли дійсність сигналу не підтверджується, особливу увагу слід приділити будь-якій додатковій інформації, що може дозволити підтвердити сигнал (ст. 21(3) ІП 520/2012 [6]), див. розділ IX.B.3.3;
- зберігати контрольні записи (записи щодо контрольного аналізу процесу/аудиту) про свої дії щодо виявлення сигналу (ст. 24(1) ІП 520/2012 [6]).
IX.C.1.3. Ролі та обов’язки національних уповноважених органів
Національні уповноважені органи повинні особливо проводити моніторинг даних, що одержані на їх території (ст. 18(4) ІП 520/2012 [6], положення Порядку [2]N), включаючи дані, що надходять з джерел, що згадані у розділі IX.B.1.
Якщо провідна держава ЄС або EMA призначені для моніторингу діючої речовини/лікарського засобу, національним уповноваженим органам:
- слід вводити валідовані сигнали, що були виявлені, до EPITT провідної держави ЄС або доповідача, що призначений PRAC для підтвердження.
- якщо жодна провідна держава ЄС або EMA не була призначена для моніторингу діючої речовини/лікарського засобу, національні уповноважені органи повинні:
- проводити моніторинг даних у базі даних EudraVigilance для речовин/лікарських засобів, що зареєстровані на їх території;
- валідувати та підтвердити будь-який сигнал, який вони виявили у базі даних EudraVigilance для речовин/лікарських засобів, що зареєстровані на їх території;
- вводити валідований та підтверджений сигнал, який вони виявили, до EPITT для речовин/лікарських засобів, що зареєстровані на їх території;
- підтверджувати якнайшвидше та не пізніше 30 днів після одержання будь-якого валідованого сигналу, про який повідомив їм власник реєстраційного посвідчення на діючу речовину/лікарський засіб, що зареєстровані на їх території. У цьому контексті, коли дійсність сигналу не підтверджується, особливу вагу слід приділити будь-якій додатковій інформації, що може дозволити підтвердити сигнал (ст. 21(3) ІП 520/2012 [6]), див розділ IX.B.3.3.
Національні уповноважені органи повинні зберігати контрольні записи (записи щодо контрольного аналізу процесу/аудиту) про свої дії щодо виявлення сигналу (ст. 24(1) ІП 520/2012 [6]). В Україні Центр відповідно до повноважень Порядку [2]N проводить виявлення/управління сигналом, вимагаючи використання АІСФN.
IX.C.1.4. Ролі та обов’язки Комітету оцінки ризику у фармаконагляді
PRAC повинен:
- визначити пріоритетність валідованих та підтверджених сигналів для подальшої оцінки (ст. 28а Регламенту (ЄС) № 726/2004 [5]);
- призначити доповідача для оцінки валідованих та підтверджених сигналів у визначені для оцінки терміни;
- передати до СНМР або CMDh, за необхідності, будь-які рекомендації щодо заходів, яких необхідно вжити після оцінки сигналу;
- проводити регулярний перегляд методології управління сигналом для застосування та оприлюднення рекомендацій, за необхідності (ст. 20(3) ІП 520/2012 [6]);
- розглядати принаймні кожні чотири роки провідну та співпровідну держави ЄС, що відповідальні за моніторинг даних у базі EudraVigilance (ст. 22(1) ІП 520/2012 [6];
- переглядати перелік медичних явищ, що слід враховувати при виявленні сигналу до їх опублікування EMA (ст. 19(2) ІП 520/2012 [6]).
IX.C.1.5. Ролі та обов’язки заявника (власника реєстраційного посвідчення)
Заявнику (власнику реєстраційного посвідчення) слід постійно проводити моніторинг безпеки його лікарських засобів та інформувати уповноважені органи про будь-які зміни, що можуть впливати на співвідношення користі/ризику.
Заявник (власник реєстраційного посвідчення) повинен:
- проводити моніторинг даних у базі EudraVigilance (в Україні — АІСФN) у міру їх доступності (ст. 18(2) ІП 520/2012 [6]). Див. також розділ щодо прав доступу до EudraVigilance для групи зацікавлених сторін III у Політиці доступу до інформації про лікарські засоби для людини у базі даних EudraVigilance92. Частота моніторингу має становити принаймні один раз на місяць, вона повинна бути пропорційною ідентифікованому ризику, потенційному ризику та необхідності додаткової інформації (ст. 18(3) ІП 520/2012 [6]);
- валідувати будь-який сигнал, що виявлений в EudraVigilance (в Україні — АІСФN) та повинен негайно поінформувати відповідний уповноважений орган про виявлення сигналу відповідно до переліку, що опублікований EMA (ст. 21(2) ІП 520/2012 [6]). На етапі валідації слід враховувати елементи інформації, що представлені у розділі IX.B.3.3;
- повідомити письмово у вигляді Нового питання з безпеки уповноважені органи у державах ЄС та УкраїніN, де зареєстрований лікарський засіб, та EMA за допомогою електронної пошти ([email protected]) (див. також модуль VI), про будь-яке нове питання з безпеки, що виникає внаслідок його діяльності з виявлення сигналу, що може мати значний вплив на співвідношення користі/ризику лікарського засобу та/або має наслідки для громадського здоров’я;
- співпрацювати з PRAC для оцінки сигналів, надаючи додаткову інформацію на вимогу;
- зберігати контрольні записи (записи щодо контрольного аналізу процесу/аудиту) про свої дії щодо виявлення сигналу.
IX.C.2. Періодичність моніторингу даних в EudraVigilance (в Україні — АІСФN)
Національні уповноважені органи та EMA повинні забезпечити постійний моніторинг даних у базі даних EudraVigilance (в Україні — АІСФN) з частотою, що пропорційна ідентифікованому ризику, потенційному ризику та необхідності отримання додаткової інформації (ст. 18(3) ІП 520/2012 [6], положення Порядку [2]N). Моніторинг повинен базуватися на періодичних оглядах статистичних вихідних даних (наприклад повідомлення з моніторингу реакцій) для визначення, чи наявні нові або змінені ризики у профілі безпеки діючої речовини (АФІN)/лікарського засобу. Статистичні вихідні дані повинні містити інформацію про побічні реакції у структурованій ієрархії (наприклад ієрархія MedDRA) за діючою(ими) речовиною(ами) (АФІN)/лікарським (и) засобом (засобами) та дозволяти застосовувати, за необхідності, фільтри та граничні величини за кількома полями.
Базова частота для перегляду статистичних вихідних даних бази EudraVigilance (в Україні — АІСФN) має бути один раз на місяць. Рішення про збільшення базової частоти моніторингу даних у базі EudraVigilance (в Україні — АІСФN) можуть приймати провідна держава ЄС, національний уповноважений орган або EMA, якщо це обґрунтовано ідентифікованими або потенційними ризиками застосування лікарського засобу або необхідністю додаткової інформації. PRAC слід поінформувати про рішення та обґрунтування.
Для лікарських засобів, що підлягають додатковому моніторингу (див. модуль Х), частота огляду статистичних вихідних даних має бути кожні 2 тижні до закінчення додаткового моніторингу. 2-тижнева частота аналізу статистичних вихідних даних може також застосовуватися для будь-якого іншого лікарського засобу із врахуванням таких критеріїв:
- будь-який лікарський засіб, що вважається тим, що має ідентифікований або потенційний ризик, який може значно впливати на співвідношення користі/ризику або мати наслідки для громадського здоров’я. До таких можуть бути віднесені ризики, що пов’язані із суттєвими помилками у застосуванні, зловживанням або застосуванням не за затвердженими показаннями. До базової частоти моніторингу лікарського засобу можна повернутися, якщо ризики не підтверджені;
- будь-який лікарський засіб, інформація про безпеку якого обмежена у зв’язку з низькою експозицією пацієнтів під час розробки лікарського засобу, включаючи лікарські засоби, що зареєстровані на певних умовах або за виключних обставин, або для яких існують вразливі або незначною мірою вивчені популяції пацієнтів або важлива відсутня інформація (наприклад діти, вагітні, пацієнти з нирковою недостатністю), у той час як післямаркетингова експозиція вірогідно буде значною;
- будь-який лікарський засіб, що містить діючі речовини (АФІN), що вже схвалені в ЄС/в УкраїніN, але показаний для застосування у новій популяції пацієнтів або за допомогою нового шляху введення;
- будь-який лікарський засіб, реєстраційне посвідчення якого зазнало суттєвих змін (наприклад зміни до показань, нозології, лікарської форми або шляху введення), у зв’язку з чим вносяться зміни до популяції пацієнтів, які зазнають впливу лікарського засобу, або профілю безпеки.
Підтвердження сигналу, що виникає в результаті діяльності з моніторингу даних EudraVigilance (в Україні — АІСФN) не обов’язково означає, що лікарський засіб повинен більш часто контролюватися і повинен бути застосований пропорційний підхід до ризику.
Проведення моніторингу з більшою, ніж кожні 2 тижні, частотою має ґрунтуватися на пропозиції провідної держави ЄС, національного уповноваженого органу або EMA. Вона може бути націлена на питання безпеки, що викликає інтерес, особливо під час надзвичайних ситуацій, що загрожують громадському здоров’ю (наприклад пандемій) та може застосовуватися у контексті замовлених запитів або тривожних сигналів майже в реальному часі, що формуються у Системі аналізу даних EudraVigilance (EVDAS) (в Україні — АІСФN).
IX.C.3. Аналіз, визначення пріоритетності та оцінка сигналу Комітетом оцінки ризику у фармаконагляді
Коли EMA або національний уповноважений орган, що валідують або підтверджують сигнал, вважає за необхідне вжити невідкладних дій, ще перед наступним засіданням PRAC, їй або йому слід ініціювати процедуру Швидкого застереження (тривожного сигналу Rapid Alert) (див. модуль XII). Усі інші сигнали, що були виявлені, валідовані або підтверджені EMA або національним уповноваженим органом, слід надсилати до PRAC для розгляду на його наступному засіданні. При розгляді сигналу PRAC слід узгодити визначення пріоритетності на підставі впливу на окремого пацієнта або громадське здоров’я потенційної зміни у співвідношенні користі/ризику. Залежно від визначення пріоритетності аналіз необхідності у подальшій оцінці або будь-якій безпосередній рекомендації щодо необхідних заходів слід проводити із врахуванням строків, що запропоновані EMA або національним уповноваженим органом, що визначили сигнал.
Коли PRAC (в Україні — ЦентрN) вважає сигнал високопріоритетним на даному засіданні, рекомендацію(ї) слід надавати під час того самого засідання, а відповідну(і) процедуру(и) слід ініціювати EMA та/або національному уповноваженому органу разом із заявником (власником реєстраційного посвідчення).
Коли PRAC (в Україні — ЦентрN) вважає за необхідне здійснювати подальшу оцінку сигналу, йому слід призначити доповідача та визначити строк для цієї оцінки із врахуванням пріоритетності сигналу.
Доповідачу з оцінки сигналу слід передавати до PRAC оцінку із зазначенням, чи існують нові ризики, чи змінилися вже визначені ризики або чи існує зміна у співвідношенні користі/ризику діючої речовини або лікарського засобу, що розглядаються. Оцінка також має включати запропоновану рекомендацію щодо дій, якщо необхідно. PRAC (в Україні — ЦентрN) може також дійти висновку, що на час оцінки на рівні ЄС/УкраїниN не потрібно вживати жодних дій.
Після розгляду звіту з оцінки доповідача PRAC слід надати рекомендацію щодо дії(й), зазначаючи причини, на яких вона ґрунтується. Рекомендація має включати графік впровадження будь-якої дії (будь-якого заходу), яка (який) вимагається від заявника (власника реєстраційного посвідчення) залежно від ступеня та серйозності питання згідно зі ст. 107h(2) Директиви 2001/83/ЄС [1] та ст. 28а(2) Регламенту (ЄС) № 726/2004 [5].
IX.C.4. Процедури регуляторного контролю в ЄС/УкраїніN
Рекомендацію PRAC (в Україні — ЦентрN) щодо дій слід направити до СНМР у разі діючої речовини (АФІN), що зареєстрована за централізованою процедурою, та до CMDh у разі діючої речовини (АФІ), що зареєстрована за національною процедурою, включаючи реєстрацію за процедурою взаємного визнання.
СНМР або CMDh можуть вибрати будь-яку або комбінацію таких дій (заходів):
- заявнику (власнику реєстраційного посвідчення) слід проводити подальшу оцінку даних та надати результати цієї оцінки до встановленого строку;
- заявнику (власнику реєстраційного посвідчення) слід подавати (спеціальний або незапланований) регулярно оновлюваний звіт з безпеки;
- заявнику (власнику реєстраційного посвідчення) слід спонсорувати післяреєстраційне дослідження з безпеки відповідно до узгодженого протоколу та надавати заключні результати цього дослідження;
- у заявника (власника реєстраційного посвідчення) слід запитати подання Плану управління ризиком або останньої версії ПУР;
- заявнику (власнику реєстраційного посвідчення) слід впровадити будь-які заходи, які необхідні для забезпечення безпечного та ефективного застосування лікарського засобу;
- реєстраційне посвідчення має бути змінено, призупинено, відкликано або не поновлюватися;
- державам ЄС або Комісії слід ініціювати, за необхідності, процедуру, що передбачена у ст. 31 або у Розділі 4 Невідкладної союзної процедури, або ст. 31, за необхідності, Директиви 2001/83/ЄС [1];
- термінові обмеження щодо безпеки слід здійснити згідно зі ст. 22 Регламенту (ЄС) 1234/2008 [8];
- слід провести інспекцію у ЄС, аудит системи фармаконагляду уповноваженою установою в УкраїніN для перевірки відповідності власника реєстраційного посвідчення на лікарський засіб вимогам з фармаконагляду, що викладені у Розділах IX та XI Директиви 2001/83/ЄС [1], (положення Порядку [2], положення Порядку проведення експертизи [7]N);
- лікарський засіб слід включити до переліку лікарських засобів, що підлягають додатковому моніторингу в межах, що визначені ст. 23 Регламенту (ЄС) № 726/2004 [5].
Процедуру, якщо вона рекомендована PRAC та узгоджена з СНМР або CMDh (в Україні — ЦентромN), за необхідності, слід ініціювати відповідно до графіку, за яким реєстраційне посвідчення має бути змінено, призупинено, відкликано або не поновлюватися.
IX.C.5. Організація ведення записів у регуляторній мережі ЄС та України
EMA та уповноважені органи повинні зберігати контрольні записи (записи щодо контрольного аналізу процесу/аудиту) про свої дії щодо управління сигналом, що пов’язані з базою EudraVigilance (в Україні — АІСФN), та відповідні запити та їх наслідки.
Будь-який сигнал, що був виявлений та валідований EMA та національним уповноваженим органом відповідно до процедур, що описані в розділі IX.B, слід вводити до Європейського інструменту відстеження питань з фармаконагляду (EPITT) (що застосовує інтернет-технології), адміністратором якого є EMA. Усі наступні оцінки, строки, рішення, дії (заходи), плани, повідомлення та всі інші основні дії EMA або національному уповноваженому органу слід реєструвати та відстежувати систематично у ЕРІТТ відповідно до настанови «Обмін інформацією про сигнали за допомогою ЕРІТТ у Регуляторній мережі ЄС» (ЕМА/383041/2011).
IX.C.6. Прозорість
У ст. 26(1) Регламенту (ЄС) № 726/2004 [5] стверджується, що EMA повинно у співпраці з державами ЄС та Комісією створити та підтримувати Европейський вебпортал з питань лікарських засобів для розповсюдження інформації про лікарські засоби, що зареєстровані в ЄС. Ця інформація буде включати висновки PRAC після оцінки сигналів та будь-яких рекомендацій. В Україні інформація з питань безпеки представлена на вебсайті МОЗ, ЦентруN.
Ключові слова: безпека застосування лікарських засобів, належна практика фармаконагляду, настанова, сигнал, управління сигналом, управління ризиками, фармаконагляд.
Частина X
Модуль X — Додатковий моніторинг
X.A. Вступ
Фармаконагляд є важливим процесом системи охорони здоров’я, мета якого полягає у швидкому виявленні та реагуванні на потенційні загрози для безпеки, пов’язані з використанням лікарських засобів.
Лікарський засіб реєструється за умови наявності чітких показників того, що на момент реєстрації співвідношення користі/ризику оцінюється як позитивне для цільової групи населення. Проте не всі ризики можуть бути виявлені на момент реєстрації, деякі ризики, пов’язані з використанням лікарського засобу, можуть бути виявлені або додатково охарактеризовані у післяреєстраційний період життєвого циклу лікарського засобу. З метою посилення моніторингу безпеки лікарських засобів до законодавства ЄС щодо здійснення фармаконагляду 2010 р. було внесено додаткові зміни у 2012 р. Зокрема, були запроваджені рамкові вимоги стосовно посиленого післяреєстраційного збору даних, пропорційного до ступеня ризику, пов’язаного із застосуванням лікарських засобів, одним з елементів якого є концепція додаткового моніторингу для певних лікарських засобів.
Як визначено у ст. 23 Регламенту (ЄС) № 726/2004 [5] та статті 11 Директиви 2001/83/ЄС [1] EMA повинно у співпраці з державами-членами створити, підтримувати та оприлюднювати перелік лікарських засобів, що є об’єктом додаткового моніторингу (далі — перелік). Такі лікарські засоби легко ідентифікуються за допомогою перевернутого рівностороннього трикутника чорного кольору ▼, як це передбачено в Імплементаційній постанові (ЄС) № 198/2013 (ІП 198/2013 [30]). Цей трикутник буде супроводжуватися пояснювальною запискою в короткій характеристиці лікарського засобу (SmPC) (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N) наступним чином:
«Цей лікарський засіб підлягає додатковому моніторингу. Це дозволить швидко визначити нову інформацію щодо безпеки. Працівників з медичною та/або фармацевтичною освітою та пацієнтів просять повідомляти про будь-які підозрювані побічні реакції. Див. в розділі «Побічні реакції» про те, як повідомляти про побічні реакції.»
Аналогічний запис також буде включений до листка-вкладки (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N). Цей пояснювальний запис повинен заохочувати працівників з медичною та/або фармацевтичною освітою і пацієнтів повідомляти про всі підозрювані побічні реакції.
Положення щодо фармаконагляду Регламенту (ЄС) № 726/2004 [5] та Директиви 2001/83/ЄС [1] були змінені Постановою (ЄС) № 1027/2012 [31] та Директивою 2012/26/ЄС [32] відповідно. Ці поправки вплинули на зміст і сферу дії ст. 23 Регламенту (ЄС) № 726/2004 [5] і застосовуються для централізовано зареєстрованих лікарських засобів з 5 червня 2013 р. Ця ННПФ враховує нові положення, що стосуються переліку лікарських засобів, які вимагають додаткового моніторингу з питань безпеки.
Післяреєстраційні спонтанні повідомлення про випадки побічних реакцій залишаються основним елементом фармаконагляду. Дані про побічні реакції на лікарські засоби є основним джерелом інформації для заходів з виявлення сигналу (див. Модуль IX). Тому підвищення обізнаності працівників з медичною та/або фармацевтичною освітою та пацієнтів щодо необхідності повідомляти про підозрювані побічні реакції та заохочення їх до цього є потенційно важливим засобом моніторингу профілю безпеки лікарського засобу.
Концепція додаткового моніторингу базується, насамперед, на необхідності підвищення відсотку інформування про побічні реакції для нових зареєстрованих лікарських засобів, для яких профіль безпеки не може бути повністю охарактеризований на момент їх реєстрації, або у разі виникнення нових проблем з безпеки для певних лікарських засобів, які повинні бути краще охарактеризовані. Основними цілями посиленого моніторингу є збір додаткової інформацію якомога раніше для виявлення характеру ризику, пов’язаного з використанням лікарських засобів у клінічній практиці, і врахування її з метою безпечного та ефективного використання лікарських засобів у подальшому.
Даний модуль складається з двох розділів:
- у розділі X.B. наведені загальні принципи присвоєння лікарським засобам статусу таких, що підлягають додатковому моніторингу, а також розглядаються питання комунікації та прозорості;
- у розділі X.C. розглядаються діяльність мережі ЄС щодо нагляду за статусом додаткового моніторингу, комунікаційна стратегія та вплив на заходи в сфері фармаконагляду.
X.B. Структури та процеси
X.B.1. Загальні принципи присвоєння лікарському засобу статусу такого, що підлягає додатковому моніторингу
Усі лікарські засоби реєструються за умови, що користь від лікування оцінюється як така, що перевищує потенційний ризик. Щоб дійти такого висновку під час видачі реєстраційного посвідчення, оцінюються дані клінічних досліджень, що проводилися під час розробки лікарського засобу. Проте побічні реакції, що виникають рідко або в результаті тривалого застосування, можуть проявитися лише після широкого медичного використання лікарського засобу та/або після тривалого застосування. Остаточна оцінка користі та ризиків лікарського засобу може бути здійснена в умовах повсякденної медичної практики, що може відрізнятися від умов клінічних випробувань. Наприклад, з клінічних випробувань можуть бути виключені деякі групи пацієнтів з множинними супутніми захворюваннями або ті, що використовують певні супутні лікарські засоби. Тому після розміщення лікарського засобу на ринку його використання широкою популяцією вимагає продовження моніторингу. Заявники (власники реєстраційних посвідчень) та уповноважені органи безперервно контролюють лікарські засоби на наявність будь-якої інформації, що стає доступною, і оцінюють її вплив на співвідношення користі/ризику лікарського засобу. Для окремих лікарських засобів необхідно проводити розширений післяреєстраційний збір даних, щоб забезпечити виявлення будь-яких нових загроз для безпеки настільки швидко, наскільки це можливо, і забезпечити негайне здійснення відповідних заходів. Тому з метою посилення моніторингу певних лікарських засобів та, зокрема, для заохочення інформування про спонтанні побічні реакції, було запроваджено концепцію додаткового моніторингу.
Лікарському засобу може бути присвоєний статус такого, що підлягає додатковому моніторингу, під час отримання реєстраційного посвідчення або в деяких випадках на більш пізніх етапах життєвого циклу лікарського засобу, для якого була виявлена нова проблема з безпеки. Присвоєння статусу додаткового моніторингу є особливо важливим під час видачі реєстраційного посвідчення на лікарський засіб, що містить нову діючу речовину, а також для всіх лікарських засобів біологічного походження, які є пріоритетними для фармаконагляду. Уповноважені органи також можуть вимагати присвоєння лікарським засобам статусу таких, що підлягають додатковому моніторингу, якщо на ці лікарські засоби поширюються особливі вимоги, наприклад, проведення ПДБЛЗ, а також умови або обмеження щодо безпеки та ефективності застосування лікарського засобу.
X.B.2. Комунікація та прозорість
Статус додаткового моніторингу повинен бути доведений до відома працівників з медичною та/або фармацевтичною освітою і пацієнтів у такий спосіб, щоб заохотити їх повідомляти про підозрювані побічні реакції, не викликаючи при цьому надмірної тривоги. Це може бути досягнуто, наприклад, шляхом роз’яснення необхідності краще охарактеризувати профіль безпеки нового лікарського засобу завдяки виявленню додаткових ризиків, при цьому ці потенційні ризики слід представляти в контексті відомих переваг для цього лікарського засобу. Загальнодоступний перелік лікарських засобів зі статусом додаткового моніторингу повинен постійно оновлюватися EMA. Крім того, працівники з медичною та/або фармацевтичною освітою та пацієнти повинні мати можливість легко розпізнавати такі лікарські засоби за допомогою спеціального маркування. Опублікування цього переліку разом з поширенням відповідної інформації повинно заохочувати працівників з медичною та/або фармацевтичною освітою і пацієнтів повідомляти про всі підозрювані побічні реакції для всіх лікарських засобів, які є предметом додаткового моніторингу.
X.C. Функціонування мережі ЄС
X.C.1. Критерії включення лікарського засобу до переліку додаткового моніторингу
X.C.1.1. Обов’язкова сфера застосування
Згідно зі ст. 23(1) Регламенту (ЄС) № 726/2004 [5], включення наступних категорій лікарських засобів до переліку є обов’язковим:
- лікарські засоби, зареєстровані в ЄС, що містять нову діючу речовину, яка станом на 1 січня 2011 р. не була у складі жодного лікарського засобу, зареєстрованого в ЄС;
- будь-який біологічний лікарський засіб, що не потрапив до попередньої категорії, та був зареєстрований після 1 січня 2011 р.;
- лікарські засоби, для яких ПДБЛЗ запитується в момент реєстрації (пункт (сb) ст. 9(4) Регламенту (ЄС) № 726/2004 [5] та пункт (b) ст. 21а Директиви 2001/83/ЄC [1]);
- лікарські засоби, зареєстровані з конкретними зобов’язаннями або з підозрюваними побічними реакціями, що перевищують ті, про які йдеться в розділі 3 Директиви 2001/83/ЄC [1] (пункт (сb) ст. 9(4) Регламенту (ЄС) № 726/2004 [5] та пункт (b) ст. 21а Директиви 2001/83/ЄC [1]);
- лікарські засоби, для яких ПДБЛЗ було запропоновано після видачі реєстраційного посвідчення (ст. 10a (1) Регламенту (ЄС) № 726/2004 [5] та пункт (а) ст. 22а (1) Директиви 2001/83/ЄC [1]);
- лікарські засоби, на які були надані умовні реєстраційні посвідчення (ст. 14 (7) Регламенту (ЄС) № 726/2004 [5]);
- лікарські засоби, зареєстровані за виняткових обставин (ст. 14(8) Регламенту (ЄС) № 726/2004 [5] та ст. 22 Директиви 2001/83/ЄC [1]).
X.C.1.2. Опціональна сфера застосування
Згідно зі ст. 23(2) Регламенту (ЄС) № 726/2004 [5], також існує можливість включити до переліку лікарські засоби, які не відносяться до обов’язкової сфери застосування. Це може бути зроблено на прохання Європейської комісії або національного уповноваженого органу у встановленому порядку, після консультації з PRAC.
Як зазначено у ст. 23(2) Регламенту (ЄС) № 726/2004 [5], ситуації, що можуть бути підставою для подання запиту на внесення до переліку, включають таку, якщо реєстраційне посвідчення видається за умов виконання принаймні одного з наступних пунктів:
- дотримання умов або обмежень щодо безпечного та ефективного застосування лікарського засобу (ст. 9(4)(c) Регламенту (ЄС) №726/2004 [5], ст. 21a(d) Директиви 2001/83/ЄC) [1];
- здійснення певних заходів, спрямованих на убезпечення застосування лікарського засобу з метою включення до системи управління ризиками (ст. 9(4)(ca) Регламенту (ЄС) № 726/2004 [5], ст. 21a(a) Директиви 2001/83/ЄC [1]);
- як зобов’язання до проведення післяреєстраційного дослідження з ефективності лікарського засобу (ст. 9(4)(cb) Регламенту (ЄС) №726/2004 [5], ст. 21a(b) Директиви 2001/83/ЄC [1]);
- наявність адекватної системи фармаконагляду (ст. 21а(е) Директиви 2001/83/ЄC [1]).
Сфера застосування ст. 23(2) Регламенту (ЄС) № 726/2004 [5] включає не тільки лікарські засоби, що зареєстровані, або щодо яких були встановлені певні умови після набуття чинності новим законодавством з питань фармаконагляду, але й лікарські засоби, що були зареєстровані, або щодо яких були встановлені певні умови до настання цієї дати, за обставин, якщо має місце принаймні одна з описаних вище ситуацій для опціональної сфери застосування.
Правила фармаконагляду в цілому та додаткового моніторингу зокрема, враховують, що повний профіль безпеки лікарського засобу може бути підтверджений лише після його розміщення на ринку. Тому необхідно приділяти належну увагу цінності включення лікарського засобу до переліку з огляду на підвищення обізнаності щодо безпечного та ефективного застосування лікарського засобу та/або надання будь-якої додаткової інформації для оцінки такого лікарського засобу. У зв’язку з цим, рішення щодо включення лікарського засобу, що підпадає під дію умов переліку, має враховувати характер та зміст умов або зобов’язань, якими супроводжується видача реєстраційного посвідчення, включаючи потенційний вплив на громадське здоров’я. Таке рішення повинно також включати розгляд питання про доцільність статусу додаткового моніторингу по відношенню до інших додаткових видів діяльності з фармаконагляду, запропонованих в ПУР, наприклад, щодо цілей ПДБЛЗ.
X.C.2. Критерії визначення початкового періоду часу для внесення до переліку додаткового моніторингу
X.C.2.1. Обов’язкова сфера застосування
Для лікарських засобів, що містять нові діючі речовини, а також для всіх біологічних лікарських засобів, зареєстрованих після 1 січня 2011 р., початковий період часу для внесення до переліку становить п’ять років від базисної дати ЄС (Union Reference Date (URD)), зазначеної у ст. 107c(5) Директиви 2001/83/ЄC [1].
X.C.2.2. Опціональна сфера застосування
Період часу для включення до переліку лікарських засобів, дозволених на певних умовах, визначається за рішенням Європейської Комісії або національного уповноваженого органу та зумовлений можливістю виконання умов та зобов’язань, що є умовою видачі реєстраційного посвідчення.
Якщо такі умови видачі реєстраційного посвідчення діють протягом життєвого циклу лікарського засобу, передбачається, що лікарський засіб, що був раніше вилучений з переліку, може бути доданий до переліку ще раз, якщо, наприклад, критерії, передбачені в ст. 23 (2) Регламенту (ЄС) № 726/2004 [5], не будуть виконані.
X.C.3. Функції та обов’язки
X.C.3.1. Європейська Комісія
Європейська комісія приймає рішення на підставі рекомендації PRAC:
- у разі, коли лікарський засіб зареєстрований за централізованою процедурою, що підпадає під дію умов, передбачених у ст. 23(2) Регламенту (ЄС) 726/2004 [5], і повинні бути включені до переліку.
X.C.3.2. EMA
EMA:
- відповідає за опублікування переліку лікарських засобів, що підлягають додатковому моніторингу, на Європейському вебпорталі з електронним посиланням на вебсторінку, на якій інформація про лікарський засіб та резюме ПУР розміщені у відкритому доступі;
- координує збір інформації, що повинна надаватися уповноваженим органами в рамках мережі ЄС, для складання, ведення та опублікування переліку;
- відповідає за вилучення з переліку лікарських засобів після завершення заздалегідь встановленого періоду часу;
- враховує перелік лікарських засобів зареєстрованих за централізованою процедурою, що є предметом додаткового моніторингу при визначенні частоти і процесів його діяльності з виявлення сигналів;
- інформуватиме відповідних заявників (власників реєстраційних посвідчень), коли лікарський засіб, зареєстрований за централізованою процедурою, буде включено до переліку лікарських засобів, що вимагають додаткового контролю;
- підтримує процес консультацій з PRAC про включення лікарських засобів до переліку.
X.C.3.3. Національні уповноважені органи
Національні уповноважені органи:
- інформують EMA про те, які лікарські засоби, що зареєстровані на національному рівні, повинні бути включені до переліку, або з наданням електронного посилання на національну вебсторінку, де інформація про лікарський засіб і резюме ПУР є загальнодоступними;
- за рекомендацією PRAC приймають рішення щодо того, чи має певний лікарський засіб, зареєстрований на національному рівні, що підпадає під дію умов, передбачених у ст. 23(2) Регламенту (ЄС) 726/2004 [5], підлягати додатковому моніторингу та, відповідно, включатися до переліку;
- розміщують у відкритому доступі на своєму національному вебпорталі перелік лікарських засобів, зареєстрованих на їх території, що підлягають додатковому моніторингу. Перелік містить електронне посилання на вебсайт, де інформація про лікарський засіб та резюме ПУР розміщені у відкритому доступі;
- повідомляють EMA про будь-які оновлення, які необхідно зробити стосовно лікарських засобів, зареєстрованих на національному рівні, та включених до переліку, опублікованого EMA;
- враховувати список лікарських засобів, що зареєстровані на національному рівні, які є предметом додаткового моніторингу, при визначенні частоти і процесів для діяльності з виявлення сигналів;
- інформують відповідних заявників (власників реєстраційних посвідчень) у разі, коли їх лікарські засоби, що зареєстровані на національному рівні, внесені до переліку.
X.C.3.4. Комітет з оцінки ризиків у фармаконагляді (PRAC)
PRAC:
- надає рекомендації на вимогу Європейської комісії або національного уповноваженого органу залежності від обставин щодо того, чи має лікарський засіб, що підпадає під дію умов, передбачених у ст. 23(2) Регламенту (ЄС) 726/2004 [5], включатися до переліку.
X.C.3.5. Заявники (власники реєстраційних посвідчень)
Заявники (власники реєстраційних посвідчень):
- повинні включати до короткої характеристики лікарського засобу (SmPC) та листка-вкладки (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N) їх лікарських засобів, що підлягають додатковому моніторингу, символ трикутника чорного кольору (▼), і стандартизовану пояснювальну фразу про додатковий моніторинг;
- повинні включати інформацію про стан додаткового моніторингу до будь-яких матеріалів, що поширюються серед працівників з медичною та/або фармацевтичною освітою і пацієнтів, а також повинні докладати максимум зусиль, щоб заохотити їх повідомляти про побічні реакції, за узгодженням з національними уповноваженими органами;
- повинні надавати підтверджуючі дані про стан виконання будь-яких умов, що були прийняті національними уповноваженими органами або Європейською комісією;
- повинні повідомляти про відповідні зміни шляхом додавання/видалення символу чорного кольору, напису та стандартної пояснювальної фрази до/з короткої характеристики лікарського засобу та листка-вкладки (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N), за необхідності.
X.C.4. Складання та підтримка переліку
Як визначено у ст. 23 Регламенту (ЄС) № 726/2004 [5] EMA разом з державами-членами створює, підтримує та оприлюднює перелік лікарських засобів, що підлягають додатковому моніторингу. Цей перелік включає назви та діючі речовини всіх лікарських засобів, затверджених в ЄС, що підлягають додатковому моніторингу, незалежно від процедури реєстрації (тобто, чи були вони зареєстровані централізовано, або на національному рівні). Крім того, як визначено у ст. 106 Директиви 2001/83/ЄС [1], кожна держава-член повинна розміщувати у відкритому доступі на національному вебпорталі перелік лікарських засобів, зареєстрованих на її території, що підлягають додатковому моніторингу, і вживати всіх можливих заходів для стимулювання працівників з медичною та/або фармацевтичною освітою і пацієнтів до надання повідомлень про будь-які підозрювані побічні реакції на лікарські засоби.
X.C.4.1. Процес складання переліку
EMA за підтримки Європейської комісії відповідає за визначення лікарських засобів, зареєстрованих за централізованою процедурою, що вимагають додаткового моніторингу. Національні уповноважені органи відповідають за визначення лікарських засобів, зареєстрованих на національному рівні, що вимагають додаткового моніторингу.
Лише лікарські засоби, що належать до обов’язкової сфери застосування, відповідно до ст. 23(1) Регламенту (ЄС) 726/2004 [5] вносяться до переліку автоматично. Для прийняття рішення щодо лікарських засобів, що відносяться до опціональної сфери застосування, потрібна консультація PRAC.
EMA та національні уповноважені органи зберігатимуть інформацію, що є загальнодоступною, і забезпечуватимуть її оновлення. Тоді як EMA матиме безпосередній доступ до відповідних даних щодо лікарських засобів, зареєстрованих за централізованою процедурою, стосовно лікарських засобів, зареєстрованих на національному рівні, EMA спиратиметься на точну і своєчасну інформацію, надану національними уповноваженими органами щодо включення або вилучення лікарських засобів з переліку та надання електронного посилання на національні вебпортали, де інформація про такі лікарські засоби та резюме ПУР розміщені у відкритому доступі. EMA та країни-члени забезпечуватимуть вільний доступ громадськості до переліку.
X.C.4.2. Процес ведення переліку
Перелік оновлюється щомісяця після кожного засідання PRAC, за необхідності.
X.C.4.2.1 Внесення лікарських засобів до переліку
Обов’язкова сфера застосування
Відповідно до ст. 23(1) Регламенту (ЄС) 726/2004 [5] лікарський засіб, що відноситься до обов’язкової сфери застосування, вноситиметься до переліку автоматично на постійній основі. У випадку, коли лікарський засіб затверджується шляхом процедури взаємного визнання, референтна держава (RMS) повинна повідомити EMA одразу ж після завершення процедури. Крім того, кожен національний уповноважений орган протягом 15 днів повідомляє EMA про реєстрацію на національному рівні того лікарського засобу, який повинен бути внесений до переліку, та надає електронні посилання на національний вебпортал, де розміщена інформація про лікарський засіб та резюме ПУР у відкритому доступі. EMA вносить лікарські засоби до переліку під час наступного оновлення після отримання рішення Європейської комісії, у випадку, коли йдеться про лікарські засоби зареєстровані за централізованою процедурою, або після отримання повідомлення від національних уповноважених органів.
Опціональна сфера застосування
Відповідно до ст. 23(2) Регламенту (ЄС) № 726/2004 [5] перед внесенням до переліку лікарських засобів, що належать до опціональної сфери застосування, потрібна консультація PRAC.
У випадку застосування процедури взаємного визнання референтна країна (RMS) повинна взяти на себе ініціативу та звернутися за консультацією до PRAC, як тільки це буде потрібно, але до завершення процедури.
У випадку застосування виключно національної процедури національний уповноважений орган повинен проконсультуватися з PRAC, як тільки умови вважатимуться необхідними, але до завершення процедури.
EMA вносить лікарські засоби зареєстровані за централізованою процедурою до переліку протягом 15 днів з моменту отримання рішення Європейської комісії. Стосовно лікарських засобів, зареєстрованих нецентралізовано, після завершення процедури кожний національний уповноважений орган протягом 15 днів повинен повідомити EMA про ті лікарські засоби, що повинні бути внесені до переліку, та надати електронні посилання на національний вебпортал, де інформація про лікарський засіб та резюме ПУР розміщені у відкритому доступі.
X.C.5. Символ чорного кольору та пояснювальні написи
Для лікарських засобів, включених до переліку, коротка характеристика лікарського засобу (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N) повинна містити напис:
«Цей лікарський засіб підлягає додатковому моніторингу. Це дозволить швидко визначити нову інформацію з безпеки. Працівників з медичною та/або фармацевтичною освітою просять повідомляти про будь-які підозрювані побічні реакції. Див. розділ «Побічні реакції» про те, як повідомляти про побічні реакції», якому передує перевернутий рівносторонній чорний трикутник (ІП 198/2013 [30]). Аналогічний напис також буде включений до листка-вкладки (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N). Після того, як лікарський засіб додається або вилучається з переліку, заявник (власник реєстраційного посвідчення) повинен оновити коротку характеристику лікарського засобу і листок-вкладку (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N), щоб додати або видалити, залежно від обставин, чорний символ, напис і стандартну пояснювальну фразу.
Якщо рішення щодо внесення або вилучення лікарського засобу з переліку приймається під час оцінки в рамках регуляторної процедури (наприклад подання заяви на отримання реєстраційного посвідчення, розширення показань, перереєстрація), коротка характеристика лікарського засобу та листок-вкладка (в Україні — Інструкція для медичного застосування лікарського засобу (Порядок проведення експертизи реєстраційних матеріалів [7])N) повинні бути оновлені до завершення процедури, щоб у разі необхідності, додати або видалити символ чорного кольору, напис та стандартну пояснювальну фразу з інформації про лікарський засіб.
Якщо рішення щодо внесення або вилучення лікарського засобу з переліку приймається поза межами регуляторної процедури, заявник (власник реєстраційного посвідчення) повинен представити зміни, щоб оновити інформацію про лікарський засіб відповідним чином.
X.C.6. Прозорість
Відповідно до ст. 23 Регламенту 726/2004 [5] EMA оприлюднює список найменувань і діючих речовин усіх лікарських засобів, затверджених у ЄС, що підпадають під додатковий моніторинг та загальні критерії для внесення лікарських засобів до переліку. Національний уповноважений орган також оприлюднює перелік лікарських засобів, дозволених на їх території, що підлягають додатковому моніторингу.
Перелік має містити електронні посилання на відповідні вебпортали, де інформація про лікарський засіб та резюме ПУР розміщені у відкритому доступі.
Ключові слова: безпека застосування лікарських засобів, додатковий моніторинг, лікарський засіб, належна практика фармаконагляду, настанова, фармаконагляд.
Частина XI
Модуль XV — Процес комунікації з питань безпеки
XV.A. Вступ
Цей модуль є настановою для заявників (власників реєстраційних посвідчень), уповноважених органів в УкраїніN, країнах ЄС та ЕМА, як комунікувати та здійснювати координацію інформації з безпеки в УкраїніN та ЄС. Процес комунікації щодо інформації з безпеки між пацієнтами та медичними/фармацевтичними працівниками є обов’язком загальної системи охорони здоров’я і важливим елементом для досягнення цілей фармаконагляду, ґрунтуючись на підтримці раціонального, безпечного та ефективного використання лікарських засобів, запобігаючи шкоді побічних реакцій та сприяючи захисту здоров’я пацієнтів та населення (див. модуль I).
Комунікація з безпеки є широким поняттям, що об’єднує різні типи інформації щодо лікарських засобів, включаючи встановлену інформацію, яка міститься в інформації про лікарський засіб (наприклад ту, що міститься у короткій характеристиці лікарського засобу (SmPC)/інструкції для медичного застосування лікарського засобуN, листку-вкладці, етикетці на упаковці), та публічних звітах з оцінки. Хоча деякі принципи у цьому модулі (наприклад розділ ХV.В.1 та В.2.) застосовуються до всіх типів комунікації з безпеки, у модулі акцентовано увагу на комунікації, що стосується нової інформації з безпеки або тієї, що з’явилася відносно попередньо відомого або невідомого ризику лікарського засобу, що має або може мати вплив на співвідношення користі/ризику лікарського засобу та його умови застосування. Якщо не вказано інакше, термін «комунікації з питань безпеки» у цьому модулі необхідно розглядати як інформацію з безпеки, яка з’явилася.
Досвід на сьогодні продемонстрував необхідність координувати комунікацію з питань безпеки в рамках регуляторної системи УкраїниN, ЄС. Передбачається високий рівень зацікавленості населення, коли виникають нові питання з безпеки, та важливо, щоб своєчасно надавалася чітка та узгоджена інформація в рамках УкраїниN, ЄС. Тому нове законодавство з фармаконагляду включає ряд документів для підкріплення процесу комунікації з питань безпеки та його координації93.
Процес комунікації щодо важливої нової інформації з безпеки лікарських засобів повинен враховувати думку та припущення зацікавлених сторін, включаючи пацієнтів та медичних працівників з належним врахуванням відповідного законодавства.
Комунікація відрізняється від прозорості, метою якої є надати загальний доступ до інформації, яка пов’язана з оцінкою даних, прийняттям рішення та моніторингом безпеки, який проводять уповноважені органи. Нове законодавство ЄС з фармаконагляду передбачає безпрецедентний рівень безпеки. Положення прозорості, що застосовуються до кожного процесу з фармаконагляду, надаються у відповідних модулях ННПФ.
Розділ ХV.В. цього модулю описує принципи та засоби комунікації з питань безпеки. Розділ ХV.С. надає настанову щодо координації та розповсюдження повідомлень з безпеки в рамках системи ЄС, УкраїниN. Обидва розділи надають певні міркування щодо інформаційних листів-звернень до медичних працівників (DHPC) та надають спеціальну настанову для їх підготовки. Це спричинено основним значенням інформаційних листів-звернень до медичних працівників (DHPC) для їх спрямування, а також рівнем координації, який вимагається між власниками реєстраційних посвідчень та уповноваженими органами під час їх підготовки.
XV.B. Структури та процеси
XV.B.1. Цілі комунікації з питань безпеки
Метою комунікації з питань безпеки є:
- своєчасне надання підтвердженої інформації щодо ефективного та безпечного застосування лікарських засобів;
- сприяння змінам у практиках охорони здоров’я (включаючи практики самолікування) якщо необхідно;
- зміна ставлення, рішень та підходів відносно використання лікарських засобів;
- підтримка діяльності, спрямованої на мінімізацію ризику;
- сприяння обґрунтованим рішенням щодо раціонального використання лікарських засобів.
Додатково до наведеного вище ефективні комунікації з питань безпеки високої якості можуть підтримати впевненість громадськості в регуляторній системі.
ХV.B.2. Принципи комунікації з питань безпеки
Повинні застосовуватися наступні принципи комунікації з питань безпеки:
- необхідність комунікації щодо інформації з безпеки повинна відбуватися при здійсненні фармаконагляду та управління ризиком, включаючи оцінку ризику (див. модуль VІІ ННПФ);
- повинна бути відповідна координація та співпраця між різними сторонами, що комунікують з питань безпеки (наприклад уповноважені органи, інші громадські організації та заявники (власники реєстраційних посвідчень);
- комунікація з питань безпеки повинна надавати важливу, зрозумілу, точну та узгоджену інформацію та досягати цільової аудиторії в потрібний для неї час з метою вживання необхідних дій;
- комунікація з питань безпеки повинна бути результативною для відповідної аудиторії (наприклад пацієнтів чи медичних/фармацевтичних працівників), сприятиме цьому використання відповідної мови та врахування різних рівнів знань та потреби в інформації. При цьому точність та узгодженість наданої інформації не повинна страждати;
- інформація щодо ризиків повинна бути представлена в контексті користі лікарських засобів та включати наявну та важливу інформацію щодо серйозності, тяжкості, частотності, факторів ризику, часу початку, реверсивності потенційних побічних реакцій та, якщо це можливо, передбачений час видужання;
- при здійсненні комунікації з питань безпеки слід зважувати непевності щодо питання безпеки. Це особливо важливо для нової інформації, що часто розповсюджується в той час, коли уповноважений орган проводить її оцінку; прийнятність комунікації на цій стадії повинна зіставлятися з можливістю збентеження, якщо непевності неналежно представлені;
- інформація щодо конкуруючих ризиків, таких як ризик нелікування, повинна включатися, якщо необхідно;
- найбільш відповідні кількісні показники необхідно використовувати при описі та порівнянні ризиків, наприклад, використання абсолютних ризиків, а не лише відносних ризиків; для порівняння ризиків знаменники повинні бути однаковими за величиною. Використання інших засобів, таких як графічне представлення ризику та/або співвідношення користі/ризику, також може застосовуватися;
- пацієнти та медичні/фармацевтичні працівники повинні, якщо можливо, отримувати консультацію, а повідомлення попередньо тестуватися на початковій стадії підготовки комунікації з питань безпеки, зокрема щодо складних питань з безпеки (див. модуль VІІ ННПФ);
- якщо важливо, процес комунікації з питань безпеки повинен доповнюватися згодом додатковими даними, наприклад, про вирішення питання з безпеки або оновлені рекомендації;
- ефективність процесу комунікації з питань безпеки необхідно оцінювати, якщо це прийнятно та можливо (див. ХV.В.7.);
- процес комунікації з питань безпеки повинен здійснюватися відповідно до вимог щодо захисту персональних даних та конфіденційності.
ХV.B.3. Цільова аудиторія
Основна цільова аудиторія процесу комунікації з питань безпеки, яка виявлена регуляторними органами та/або заявниками (власниками реєстраційних посвідчень), може бути представлена пацієнтами та/або медичними працівниками, які використовують (наприклад прописують, контролюють, відпускають, вводять або приймають) лікарські засоби.
Медичні працівники представляють основну цільову аудиторію. Ефективна комунікація з питань безпеки з цією аудиторією дозволяє їм надавати чітку та корисну інформацію своїм пацієнтам, таким чином сприяючи безпеці пацієнтів та впевненості в регуляторній системі. Медичним працівникам, які працюють у клінічній практиці і тим, що залучені до клінічних досліджень, необхідно надати відповідну інформацію щодо будь-якого питання з безпеки одночасно.
Організації пацієнтів, споживачів та медичних/фармацевтичних працівників можуть відігравати роль так званих множників, оскільки вони можуть розповсюдити важливу інформацію з безпеки цільовій аудиторії.
Засоби масової інформації також є цільовою аудиторією процесу комунікації з питань безпеки. Здатність засобів масової інформації звертатися до пацієнтів, медичних/фармацевтичних працівників та широкої громадськості є критичним елементом поширення нової та важливої інформації про лікарські засоби. Спосіб, яким інформація з безпеки розповсюджується засобами масової інформації, буде впливати на її сприйняття громадськістю, а тому важливо, щоб засоби масової інформації отримували інформацію з безпеки безпосередньо від уповноважених органів додатково до інформації, яку вони отримуються з інших джерел, наприклад, від заявників (власників реєстраційних посвідчень).
ХV.B.4. Зміст процесу комунікації з питань безпеки
Враховуючи принципи, що описані в розділі ХV.B.2., при комунікації з питань безпеки потрібно, щоб усім учасникам цього процесу стали відомі:
- важлива інформація, що з’являється, щодо будь-якого зареєстрованого лікарського засобу, що впливає на співвідношення користі/ризику лікарського засобу при будь-яких умовах використання;
- причина ініціювання комунікації з питань безпеки, що чітко пояснена цільовій аудиторії;
- будь-які рекомендації медичним/фармацевтичним працівникам та пацієнтам, як розглядати питання з безпеки;
- якщо необхідно, заява про домовленість між заявником (власником реєстраційного посвідчення) та уповноваженим органом щодо наданої інформації з безпеки;
- інформація про будь-які зміни в інформації про лікарський засіб (наприклад короткій характеристиці лікарського засобу (SmPC)/інструкції для медичного застосування лікарського засобуN або листку-вкладці);
- перелік посилань на літературу, якщо необхідно, або посилання на джерело, де можна знайти більш детальну інформацію;
- якщо необхідно, нагадування про необхідність повідомляти про підозрювані побічні реакції відповідно до національних систем спонтанних повідомлень.
При здійсненні процесу комунікації з питань безпеки інформація, що надається, має бути об’єктивною і не повинна вводити в оману (ст. 106а(1) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Інформація з безпеки не повинна включати будь-які дані або заяву, що може бути рекламою в рамках сфери дії розділу VIII Директиви 2001/83/ЄС [1].
ХV.B.5. Засоби процесу комунікації з питань безпеки
Інструменти та канали94 комунікації з питань безпеки стали більш чисельними та змінюються з часом, пропонуючи громадськості більше інформації, ніж було раніше можливо. Необхідно розглянути використання цього ряду засобів, що збільшується, під час процесу комунікації з питань безпеки з метою досягнення цільової аудиторії та відповідності їх зростаючим очікуванням. Різні інструменти та канали комунікації будуть розглянуті нижче в розділах ХV.В.5.1.–ХV.В.5.9.
ХV.B.5.1. Інформаційний лист-звернення до медичних працівників (DHPC)
Інформаційний лист-звернення до медичних працівників (DHPC) визначено у цьому документі як повідомлення-втручання, шляхом якого важлива інформація з безпеки надається безпосередньо конкретним медичним/фармацевтичним працівникам заявниками (власниками реєстраційних посвідчень) або уповноваженим органом для їх інформування про необхідність вжити певних дій або адаптувати їх практики стосовно лікарського засобу. Інформаційний лист-звернення до медичних працівників не надає відповіді на запити від медичних/фармацевтичних працівників, вони також не вважаються освітніми матеріалами для виконання стандартних дій з мінімізації ризиків.
Підготовка інформаційних листів-звернень до медичних працівників вимагає співробітництва між заявником (власником реєстраційного посвідчення) та уповноваженим органом. Угоди між цими двома сторонами слід досягати до розповсюдження інформаційного листа-звернення до медичних працівників заявником (власником реєстраційного посвідчення). Угода повинна включати зміст інформації (див. розділ ХV.B.4) та план повідомлення, включаючи запланованих реципієнтів та графік розповсюдження інформаційних листів-звернень до медичних працівників (див. модуль VІІ ННПФ).
Якщо існує декілька власників реєстраційних посвідчень на одну і ту саму діючу речовину (АФІN), для якої повинно бути створено Інформаційний лист-звернення до медичних працівників, зазвичай повинне надаватися одне узгоджене повідомлення.
Якщо можливо, рекомендується, щоб організації медичних/фармацевтичних працівників або наукові товариства залучалися, якщо необхідно, під час підготовки інформаційного листа-звернення до медичних працівників для гарантії, що інформація, яку воно міститиме, є корисною та адаптованою для цільової аудиторії.
Інформаційний лист-звернення до медичних працівників може доповнюватися іншим засобами та каналами повідомлення, та повинен застосовуватися принцип надання узгодженої інформації (розділ ХV.В.2).
Інформаційний лист-звернення до медичних працівників може бути додатковим заходом з мінімізації ризиків та входити до ПУР (див. модулі V та ХV ННПФ).
Інформаційний лист-звернення до медичних працівників повинен розповсюджуватися в певних ситуаціях, особливо, якщо існує необхідність вжити негайних заходів або змінити існуючу практику відносно лікарського засобу:
- призупинення, відкликання або анулювання реєстраційного посвідчення з причин з безпеки;
- важлива зміна стосовно використання лікарського засобу внаслідок обмеження показання, нового протипоказання або зміни в рекомендованій дозі внаслідок причин з безпеки;
- обмеження в можливості використання або призупинення вживання препарату з потенційними шкідливим впливом на лікування пацієнта.
До інших причин, коли виникає необхідність у розповсюдженні інформаційних листів-звернень до медичних працівників, є:
- нові значні попередження або застереження щодо використання в інформації про лікарський засіб;
- нові дані, що визначають попередньо невідомий ризик або зміну в частоті або серйозності відомого ризику;
- обґрунтовані дані, що лікарський засіб не такий ефективний, як вважалося раніше;
- нові рекомендації щодо запобігання або лікування побічних реакцій, або уникнення зловживання або медичної помилки при застосуванні лікарського засобу;
- поточна оцінка важливого потенційного ризику, для якого наявні дані в певний момент часу, є недостатніми для прийняття регуляторної дії (у цьому випадку інформаційний лист-звернення до медичних працівників повинен сприяти пильному моніторингу питання з безпеки в клінічній практиці та сприяти підготовці повідомлень, та, можливо, надавати інформацію про те, як мінімізувати потенційний ризик).
Уповноважений орган може розповсюдити або попросити заявника (власника реєстраційного посвідчення) розповсюдити інформаційний лист-звернення до медичних працівників у будь-якому випадку, якщо уповноважений орган вважає це необхідним для подальшого безпечного та ефективного використання лікарського засобу.
XV.B.5.2. Документи непрофесійною мовою
Комунікаційні матеріали, викладені непрофесійною мовою (наприклад використання формату питання та відповіді), допомагають пацієнтам та пересічним громадянам зрозуміти науковий доказ та регуляторні дії стосовно питання безпеки. Документи, викладені непрофесійною мовою, повинні містити рекомендації уповноваженого органу та пораду щодо мінімізації ризиків для пацієнтів та медичних/фармацевтичних працівників стосовно питання з безпеки, та повинні супроводжуватися відповідною додатковою інформацією.
Документи, викладені непрофесійною мовою, як правило, є корисними для громадськості, та її представників, які зацікавлені в предметі розгляду, але не мають наукового або регуляторного досвіду. Також необхідно зробити посилання з приводу питання на те, де безпосередні читачі зможуть знайти інші матеріали з більш детальною інформацією.
Уповноважені органи публікують документи непрофесійною мовою на своїх національних вебпорталах лікарських засобів та можуть додатково розповсюджувати їх іншим зацікавленим сторонам, таким як організації пацієнтів та медичних/фармацевтичних працівників.
Якщо можливо, рекомендується, щоб пацієнти та медичні/фармацевтичні працівники залучалися під час підготовки документів непрофесійною мовою для гарантії, що інформація, яка буде міститися у цих документах, є корисною та адаптованою для цільової аудиторії.
ХV.B.5.3. Комунікації з питань безпеки з представниками преси
Комунікація з питань безпеки з пресою включає пресрелізи та пресбрифінги, які призначені головним чином для журналістів.
Уповноважені органи можуть надсилати пресрелізи безпосередньо журналістам, додатково оприлюднюючи їх на своїх вебсайтах. Це гарантує, що журналісти додатково до отриманої інформації з інших джерел отримують інформацію, що узгоджується з науковою оцінкою уповноваженого органу. Взаємодія із засобами масової інформації є важливим способом звернутися до більш широкої аудиторії, а також посилити довіру до регуляторної системи.
Пресрелізи можуть також створюватися та публікуватися власниками реєстраційних посвідчень. Їх пресрелізи можуть відображати позицію заявника (власника реєстраційного посвідчення) з питання безпеки, при цьому повинні містити посилання на будь-яку регуляторну дію, що вжита уповноваженим органом. Відповідні поточні перегляди повинні зазначатися у будь-якому повідомленні заявником (власником реєстраційного посвідчення).
Хоча пресрелізи призначені для журналістів, їх також буде читати решта аудиторії, наприклад, медичні/фармацевтичні працівники, пацієнти та широка громадськість. Тому необхідно зробити посилання на відповідні матеріали на цю тему. У випадках, якщо також готується Інформаційний лист-звернення до медичних працівників, медичні/фармацевтичні працівники в ідеалі повинні отримати його раніше або приблизно одночасно з оприлюдненням та розповсюдженням пресрелізу. Це потрібно для того, щоб вони були краще підготовлені до надання відповідей пацієнтам.
Пресбрифінги з журналістами повинні розглядатися уповноваженими органами з питань безпеки або інших тем стосовно безпеки лікарських засобів, до яких існує підвищений інтерес засобів масової інформації, або коли необхідно здійснити комунікацію з приводу складних або важливих для громадського здоров’я питань.
ХV.B.5.4. Вебсайт
Вебсайт є ключовим засобом для громадськості (включаючи пацієнтів та медичних працівників) для активного пошуку в інтернеті певної інформації про лікарські засоби. Уповноважені органи, а також заявники (власники реєстраційних посвідчень) повинні гарантувати, що важлива інформація з безпеки, що оприлюднена на вебсайтах, які вони контролюють, легкодоступна та зрозуміла громадськості. Інформація на вебсайтах повинна бути новою, а стара інформація відмічатися як така або видалятися.
Нове законодавство з фармаконагляду передбачає створення вебпорталу лікарських засобів ЄС, який буде містити інформацію про всі лікарські засоби, що зареєстровані в ЄС (ст. 26 Регламенту (ЄС) №1235/2010 [4]). Цей вебпортал стане ключовим засобом для повідомлення нової інформації з безпеки громадянам ЄС та буде містити інформацію всіма офіційними мовами ЄС. Кожна країна ЄС повинна встановлювати та підтримувати національний вебпортал лікарських засобів, який буде посилатися на вебпортал лікарських засобів ЄС (ст. 106а Директиви 2001/83/ЄС [1]). До тих пір, поки вебпортал повністю не встановлений та не введений у дію, вебсайт EMA буде діяти як внутрішня платформа для інформування цієї важливої нової інформації з безпеки.
ХV.B.5.5. Інші комунікації з питань безпеки завдяки інтернет-технологіям
Онлайн-інформація з безпеки може також розповсюджуватися через інші вебзасоби. При використанні більш нових та швидких каналів комунікації особливу увагу слід приділити гарантуванню, що точність інформації, що надається, не зазнає ризику. При здійсненні комунікації слід враховувати нові інструменти комунікації, що використовують різні цільові аудиторії.
ХV.B.5.6. Бюлетені та інформаційні листи
Завдяки бюлетеням та інформаційним листам на регулярній основі можна надавати нову інформацію про лікарські засоби та їх безпеку й ефективність. Уповноважені органи можуть охопити широку аудиторію завдяки цим інструментам, використовуючи інтернет-технології та інші існуючі засоби.
ХV.B.5.7. Комунікація між уповноваженими органами
Якщо один уповноважений орган приймає регуляторне рішення щодо певного питання з безпеки, інші уповноважені органи зазвичай повинні відповідати на запити або надавати повідомлення щодо того ж питання. Необхідно розглянути використання матеріалів комунікації іншими органами, наприклад, загальні підходи. Загальні підходи передбачають підготовку відповідних документів уповноваженим органом для допомоги своєму власному персоналу та персоналу уповноважених органів, з якими він співпрацює, при наданні відповіді на зовнішні запити або комунікації щодо певного питання з безпеки.
ХV.B.5.8. Відповідь на запити від громадськості
Уповноважені органи та заявники (власники реєстраційних посвідчень) повинні мати системи для відповіді на запити про лікарські засоби, що надходять від окремих членів громадськості. Відповіді повинні враховувати інформацію, що є загальнодоступною, та повинні включати відповідні рекомендації пацієнтам та медичним/фармацевтичним працівникам, які розроблені уповноваженими органами. Якщо питання стосуються окремої рекомендації щодо лікування, пацієнтам необхідно рекомендувати звернутися до лікаря.
Відносно цього до заявників (власників реєстраційних посвідчень) застосовуються ст. 86(2) та ст. 98(1) Директиви 2001/83/ЄС [1].
ХV.B.5.9. Інші засоби комунікації з питань безпеки
Додатково до описаних вище існують інші інструменти та канали, наприклад, публікації в наукових журналах та журналах професійних організацій.
Деякі інструменти та канали можуть використовуватися в контексті управління ризиками; заходи з мінімізації ризиків часто включають спеціальні програми комунікації з приводу ризиків. Інструменти, що використовуються в таких програмах, наприклад, повідомлення-попередження для пацієнтів або керівництва з безпеки для медичних/фармацевтичних працівників, не входять у сферу дії цього модуля та описуються більш детально в модулі ХVІ.
ХV.B.6. Ефективність комунікації з питань безпеки
Комунікації з питань безпеки вважаються ефективним, якщо надану інформацію, отримує та розуміє цільова аудиторія настільки, наскільки це було заплановано, підтвердженням чого є здійснення цією аудиторією відповідних дій. Необхідно застосувати відповідні механізми для визначення ефективності комунікацій, що орієнтовані на чітко визначені цілі. Вимірювання ефективності дозволяє врахувати недоліки комунікації та допомагає прийняти рішення щодо визначення пріоритетів та адаптування засобів та практик для задоволення потреб цільової аудиторії. Підхід, що базується на дослідженні є відповідним для встановлення, що комунікації з питань безпеки відповідають стандарту, представленому в розділі ХV.B.2. Цей підхід може оцінювати різні результати, включаючи підходи, ставлення та знання. При оцінці ефективності комунікації з питань безпеки сфера дії оцінки може бути розширена (див. модуль ХVІ).
У випадку безпосередньої комунікації з медичним/фармацевтичним працівником за допомогою Інформаційних листів-звернень до медичних працівників заявник (власник реєстраційного посвідчення) повинен відповідати за оцінку розповсюдження Інформаційних листів-звернень до медичних працівників, які він готує, та повинен інформувати уповноважені органи про результат такого розповсюдження та про будь-які виявлені складнощі (наприклад проблеми стосовно переліку реципієнтів або часу та механізму розповсюдження). Повинна вживатися відповідна дія, якщо необхідно, для коригування ситуації або запобігання подібним проблемам у майбутньому.
ХV.B.7. Вимоги системи якості до процесу комунікації з питань безпеки
Відповідно до вимог системи якості, що описані в модулі I ННПФ, повинні існувати процедури для гарантії, що комунікації з питань безпеки відповідають принципам, які описані в розділі ХV.В.2., у разі необхідності.
Зокрема процес комунікації повинен підлягати контролю якості для гарантії його точності та ясності. З цією метою необхідно дотримуватися та документувати процедури перевірки відповідно до обов’язків.
ХV.С. Функціонування регуляторної системи ЄС, УкраїниN
ХV.С.1. Координація сповіщення з питань безпеки в ЄС, УкраїніN
В ЄС, УкраїніN пацієнти та медичні/фармацевтичні працівники все більше і більше сприймають уповноважений орган як провайдера важливої інформації щодо лікарських засобів. Для того, щоб комунікація з питань безпеки була ефективною та адекватною, потрібна координація в рамках регуляторної системи УкраїниN, ЄС95. Належний рівень координації процесу комунікації з питань безпеки є особливо важливим для того, щоб медичні/фармацевтичні працівники та пацієнти отримали узгоджену інформацію про регуляторні рішення в ЄС, УкраїніN.
Для оприлюднення повідомлень з безпеки уповноважені органи можуть використовувати різні інструменти та канали, що описані в розділі ХV.В.5. До публікації повідомлення з безпеки країни ЄС, ЕМА або Європейська Комісія повинні інформувати одна одну не менш ніж за 24 години, якщо не існує потреби у терміновому публічному сповіщенні з метою захисту громадського здоров’я (ст. 106а(2) Директиви 2001/83/ЄС [1]).
Для діючих речовин (АФІN), що містяться в лікарських засобах, які зареєстровані більш ніж в одній країні ЄС, ЕМА буде відповідати за координацію комунікації між національними уповноваженими органами щодо їх сповіщення з питань безпеки (ст. 106а(3) Директиви 2001/83/ЄС [1]).
З практичної точки зору, зважаючи на можливість нашарування заходів прозорості й активні комунікації та з метою зосередження уваги на темах, що мають велике значення для здоров’я, не вся інформація з безпеки, яка публікується країною ЄС або ЕМА, буде підлягати систематичному обміну та координації. Тільки повідомлення з безпеки, що стосуються майбутнього лікарських засобів, та що стосуються діючих речовин (АФІN), що містяться у лікарських засобах, які зареєстровані більше ніж в одній країні ЄС, вимагають координації з боку регуляторної системи ЄС, а саме:
- призупинення, відкликання або анулювання реєстраційного посвідчення внаслідок зміни співвідношення користі/ризику лікарського засобу;
- початок або закінчення процедури передачі на розгляд ЄС з причин безпеки;
- обмеження показання або популяції лікування або додавання нового протипоказання;
- розповсюдження інформаційних листів-звернень до медичних працівників, що узгоджені відповідними уповноваженими органами країн ЄС або ЕМА (див. розділ ХV.С.2.1.);
- інші нові питання з безпеки, які розцінюються національним уповноваженим органом або ЕМА такими, що можуть викликати інтерес у громадськості або у засобів масової інформації більше ніж в одній країні ЄС (наприклад публікація важливих даних щодо безпеки в (наукових) журналах, регуляторна дія, що вживається з причин безпеки в країні ЄС або в країні за межами ЄС).
ХV.С.1.1. Процес обміну та координації сповіщень з питань безпеки
Уповноважені органи країни ЄС або ЕМА повинні інформувати регуляторну систему ЄС про оприлюднення сповіщення з питань безпеки, яке стосується діючих речовин (АФІN), що містяться в лікарських засобах, що зареєстровані більше ніж в одній країні ЄС, та яке стосується будь-якої з ситуацій, які визначені в розділі ХV.С.1. Воно повинно включати графік оприлюднення інформації (ст. 106а(3) Директиви 2001/83/ЄС [1]). Якщо можливо, повідомлення з безпеки повинно надсилатися до системи не менше ніж за 24 години до публікації (ст. 106а(2) Директиви 2001/83/ЄС [1]), щоб дозволити членам регуляторної системи ЄС підготувати або спланувати своє власне повідомлення, якщо необхідно. Під координацією ЕМА країни ЄС повинні докладати всіх необхідних зусиль для формування узгодження загального повідомлення (ст. 106а(3) Директиви 2001/83/ЄС [1]).
ЕМА має вирішити у кожному окремому випадку на основі значення для громадського здоров’я та терміновості питання з безпеки, популяції та кількості залучених країн ЄС та ймовірної уваги засобів масової інформації, чи потрібні у подальшому додаткові дії для розповсюдження повідомлення з безпеки, а саме:
- підготовка загальних підходів (див. розділ ХV.В.5.7), які повинні розповсюджуватися в регуляторній системі ЄС. Документ про загальний підхід повинен допомогти регуляторній системі ЄС дати відповідь на будь-який інформаційний запит, що може виникнути після публікації сповіщення з безпеки.
- підготовка сповіщення з безпеки ЕМА додатково до сповіщення країни ЄС, яке повинно також розповсюджуватися в регуляторній системі ЄС разом з графіком його публікації.
ЕМА має підготовити документи для внутрішнього використання (наприклад пресслужбою) відносно того, як подавати аудиторії офіційну позицію стосовно певного питання, та будь-яке повідомлення ЕМА стосовно безпеки разом з державою(ми) ЄС, що розпочала(ли) процес, та провідною державою ЄС, PRAC або доповідачем (залежно від ситуації). За необхідності слід проконсультуватися з PRAC, а також CHMP або CMDh.
Координація повідомлень з безпеки повинна проводитися разом із зацікавленим(и) заявником(ами) (власником(ами) реєстраційного(их) посвідчення(ь)). Якщо можливо, ЕМА та уповноважені органи в країнах ЄС повинні надати будь-яке повідомлення з безпеки до його публікації зацікавленому(им) заявнику(ам) (власнику(ам) реєстраційного(их) посвідчення(ь)) разом з графіком опублікування інформації. Будь-яка інформація персонального або комерційного чи конфіденційного характеру повинна видалятися, якщо надання такої інформації громадськості не сприятиме захисту громадського здоров’я (ст. 106а(4) Директиви 2001/83/ЄС [1]).
При обміні та координації повідомлень з безпеки в рамках регуляторної системи ЄС повинна використовуватися система ранніх повідомлень ЄС (ENS). Така система була розроблена для використання ЕМА для завчасного повідомлення уповноваженим органам в країнах ЄС та Європейській Комісії інформації з безпеки щодо лікарських засобів, зареєстрованих за централізованою процедурою. Ця система повинна також використовуватися уповноваженими органами в країнах ЄС з метою обміну та координації повідомлень з безпеки.
Система ранніх повідомлень включає керівників агенцій з лікарських засобів (НМА), членів PRAC, СНМР, CMDh, діючі точки контакту для повідомлень з безпеки при уповноваженому органі в країні ЄС, Європейській Комісії та ЕМА. Діючі точки контакту повинні гарантувати, що будь-яка інформація, якою обмінюються через систему, досягає своєчасно відповідного персоналу в кожному уповноваженому органі, включаючи відповідний персонал, що працює в відділах комунікації.
Повідомленнями з безпеки регуляторної системи ЄС необхідно обмінюватися з міжнародним партнерами відповідно до рекомендацій, представлених у модулі ХІV, особливо, коли йдеться про заборону застосування ліків чи вживання будь-яких спеціальних заходів з конфіденційності.
Додатково до координації повідомлень з безпеки в рамках регуляторної системи ЄС уповноважені органи в країнах ЄС та ЕМА повинні взаємодіяти із зацікавленими сторонами в ЄС (в основному з організаціями пацієнтів та спеціалістів-медиків), які можуть відігравати ключову роль у перевірці та розповсюдженні інформації кінцевим користувачам (пацієнтам та спеціалістам-медикам). Рекомендується, щоб національні органи та ЕМА зберігали контактні дані відповідних організацій пацієнтів та спеціалістів-медиків в оновленому вигляді.
ХV.С.1.2. Обмін інформацією з безпеки, яку надають треті сторони
Існують ситуації, коли нова інформація з безпеки повинна публікуватися або публікується стороною, яка не є уповноваженим органом країни ЄС або ЕМА (наприклад наукові журнали, наукові товариства). Уповноважені органи повинні повідомити в регуляторну систему ЄС будь-яку таку інформацію з безпеки, про яку вони дізналися, разом з визначенням часу публікації, якщо відома. Якщо необхідно та після оцінки інформації ЕМА повинна підготувати та розповсюдити документ про загальні положення або повідомлення з безпеки ЕМА для реагування на інформацію, що надійшла від третьої сторони (див. розділ ХV.С.1.1.).
У контексті співробітництва з органами за межами ЄС ЕМА або уповноваженому органу країни ЄС може стати відомо про повідомлення з безпеки, що опубліковані цими органами (див. модуль ХІV). У цих випадках ЕМА повинно, якщо необхідно, підготувати та розповсюдити загальні положення або повідомлення з безпеки в рамках регуляторної системи ЄС. В усіх випадках умови будь-яких відповідних угод про конфіденційність з регуляторним органами, які не входять в ЄС, та заборони на отриману інформацію не повинні порушуватися.
ХV.С.1.3. Вимоги до заявника (власника реєстраційного посвідчення) в ЄС, УкраїніN
Як тільки заявник (власник реєстраційного посвідчення) в ЄС, УкраїніN має намір зробити публічне повідомлення щодо інформації з питань фармаконагляду стосовно використання лікарського засобу, він одночасно або до публічного повідомлення повинен поінформувати про це уповноважені органи в країнах ЄС, УкраїніN, ЕМА та Європейську Комісію (ст. 106а Директиви 2001/83/ЄС [1], положення Порядку [2]N). Це стосується повідомлень, що призначені для ЄС, УкраїниN, а також для країн за межами ЄС (якщо повідомлення стосуються лікарських засобів, які зареєстровані в ЄС або для яких надана думка згідно зі ст. 58 Регламенту (ЄС) 726/2004 [5], УкраїниN. Інформування уповноважених органів одночасно з громадськістю (наприклад без попереднього повідомлення органів) повинно відбуватися лише у виняткових випадках та з обґрунтованих причин. Якщо можливо, інформацію необхідно надавати не менш, ніж за 24 години до її публікації.
Заявник (власник реєстраційного посвідчення) повинен гарантувати, що інформація для громадськості представлена об’єктивно та не вводить в оману (ст. 106а Директиви 2001/83/ЄС [1], положення Порядку [2]N).
Якщо заявнику (власнику реєстраційного) посвідчення стає відомо, що третя сторона (див. розділ ХV.С.1.2.) має намір зробити повідомлення, що може потенційно вплинути на співвідношення користі/ризику лікарського засобу, який зареєстрований в ЄС, УкраїніN заявник (власник реєстраційного посвідчення) повинен інформувати відповідні уповноважені органи в країнах ЄС та ЕМА, Україні та докладати всіляких зусиль для надання змісту повідомлення відповідним органам.
ХV.С.1.4. Врахування думки третіх сторін
Треті сторони (наприклад наукові журнали, наукові товариства, організації пацієнтів) заохочуються до інформування ЕМА та уповноважених органів в країнах ЄС про будь-яку відповідну нову інформацію з безпеки лікарських засобів, які зареєстровані в ЄС, та, якщо запланована публікація, до обміну інформацією перед публікацією.
ХV.С.1.5. Мови та переклади
Узгоджені повідомлення повинні досягати громадськості в рамках ЄС, УкраїниN своєчасно та офіційними мовами країн ЄС, УкраїниN, як вказано країнами, на ринку яких знаходиться лікарський засіб.
З метою координації ЕМА повинна використовувати англійську мову для інформування регуляторної системи ЄС про будь-яке повідомлення з безпеки. При інформуванні ЕМА уповноважені органи в країнах ЄС заохочуються до надання у перекладі англійською мовою їх повідомлень з безпеки з метою ініціації процесу координації. За відсутності перекладу усього тексту необхідно надати переклад резюме англійською.
ХV.С.2. Інформаційний лист-звернення до медичних працівників в ЄС, УкраїніN
В ЄС, УкраїніN інформаційний лист-звернення до медичних працівників (DHPC) (див. розділ ХV.В.5.1.), як правило, розповсюджуються одним або групою заявників (власників реєстраційних посвідчень) на відповідний(і) лікарський(і) засіб(засоби) або активну(і) субстанцію(ї) на вимогу національного уповноваженого органу або ЕМА, або за власною ініціативою заявника (власника реєстраційного посвідчення). Заявник (власник реєстраційного посвідчення) повинен отримати згоду відповідного національного уповноваженого органу або ЕМА щодо змісту DHPC (та плану комунікації) до його розповсюдження.
ХV.С.2.1. Обробка інформаційних листів-звернень до медичних працівників (DHPC)
Ситуації, коли інформаційний лист-звернення до медичних працівників (DHPC) необхідний або слід розглянути його необхідність, надаються в розділі ХV.В.5.1.
Роль та відповідальність уповноважених органів, ЕМА та заявників (власників реєстраційних посвідчень) при підготовці та обробці інформаційних листів-звернень до медичних працівників залежить від процедури реєстрації даних лікарських засобів:
- для лікарських засобів, зареєстрованих за централізованою процедурою та для лікарських засобів, що підлягають процедурі передачі на розгляд ЄС з причин безпеки відповідні заявники (власники реєстраційних посвідчень) повинні подавати проєкт інформаційних листів-звернень до медичних працівників та план комунікації (включаючи запланованих реципієнтів та графік розповсюдження інформаційних листів-звернень до медичних працівників) ЕМА, яке має координувати процес перегляду своїми науковими комітетами (наприклад PRAC, СНМР, CMDh);
- для лікарських засобів, що зареєстровані за процедурою взаємного визнання або децентралізованою процедурою, заявник (власник реєстраційного посвідчення) повинен подавати проєкт інформаційних листів-звернень до медичних працівників та будь-який план комунікації референтній країні ЄС, яка повинна координувати процес із заявником (власником реєстраційного посвідчення), інформуючи зацікавлені країни ЄС про будь-яку запропоновану дію;
- для лікарських засобів, зареєстрованих за національною процедурою, а не за процедурою взаємного визнання або децентралізованою процедурою, заявник (власник реєстраційного посвідчення) повинен подавати проєкт інформаційних листів-звернень до медичних працівників та будь-який план комунікації уповноваженим органам країн ЄС, УкраїниN, де лікарські засоби зареєстровані.
Заявник (власник реєстраційного посвідчення) повинен надавати мінімум два робочі дні для коментарів. Проте, якщо можливо, слід надати більше часу. Визначення часу може бути змінено відповідно до терміновості ситуації.
ЕМА повинне координувати перегляд інформаційних листів-звернень до медичних працівників у рамках своїх наукових комітетів/груп, якщо необхідно (наприклад залучення PRAC та кінцеве оформлення СНМР, CMDh). PRAC повинен завжди залучатися до перегляду DHPC щодо питання з безпеки, яке розглядається PRAC, та DHPC повинно входити в оцінку PRAC (див. модуль ХІІ). ЕМА може також вимагати рекомендації від PRAC щодо питань, які стосуються інших повідомлень з безпеки.
Якщо зміст інформаційних листів-звернень до медичних працівників та план комунікації від заявника (власника реєстраційного посвідчення) погоджені національними уповноваженими органами або ЕМА, національні уповноважені органи або ЕМА повинні обмінятися фінальною версією інформаційних листів-звернень до медичних працівників та планами комунікації, використовуючи систему ранніх повідомлень (див. розділ ХV.С.1.1). При цьому ЕМА повинна координувати будь-які послідовні повідомлення з безпеки, якщо необхідно, використовуючи процес, що описаний у розділі ХV.С.1.1. Система ранніх повідомлень використовується лише у тому випадку, якщо інформаційний лист-звернення до медичних працівників стосується активної субстанції, яка зареєстрована більше ніж в одній країні ЄС.
У випадках, якщо уповноважений орган за межами ЄС/УкраїниN вимагає розповсюдження DHPC на їх території для лікарського засобу, який також зареєстрований в ЄС/УкраїніN, заявник (власник реєстраційного посвідчення) повинен повідомити відповідні уповноважені органи в ЄС/УкраїніN. Це частина правової вимоги, згідно з якою власник реєстраційного посвідчення повинен повідомити уповноважені органи про будь-яку нову інформацію, яка може вплинути на співвідношення користі/ризику лікарського засобу (ст. 16(2) Регламенту (ЄС) 726/2004 [5] та 23 (2) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Необхідність будь-якої послідовної комунікації, наприклад, інформаційних листів-звернень до медичних працівників, в ЄС/УкраїніN необхідно розглянути та погодити в кожному конкретному випаду.
Блок-схема, що описує обробку інформаційних листів-звернень до медичних працівників, надається на рис. ХV.І наприкінці модуля.
ХV.С.2.2. Переклад інформаційних листів-звернень до медичних працівників (DHPC)
Для лікарських засобів, зареєстрованих за централізованою процедурою, що підлягають процедурі передачі на розгляд ЄС з причин безпеки, та у багатьох випадках для лікарських засобів, що зареєстровані за процедурою взаємного визнання або децентралізованою процедурою, робочою мовою для підготовки інформаційних листів-звернень до медичних працівників повинна бути англійська.
Якщо текст інформаційних листів-звернень до медичних працівників погоджений, заявник (власник реєстраційного посвідчення) повинен підготувати переклади офіційними мовами країн ЄС/УкраїниN, як зазначено країнами, у яких повинно розповсюджуватися DHPC. Проєкти перекладів необхідно подавати до країн ЄС/УкраїниN для перегляду протягом відповідного часу (не більше ніж два робочі дні).
Для лікарських засобів, зареєстрованих шляхом централізованої процедури та препаратів, що підлягають процедурі передачі на розгляд ЄС з причин безпеки, відповідний заявник (власник реєстраційного посвідчення) повинен надати ЕМА повний набір всіх кінцевих версій офіційними мовами ЄС, а також будь-які додаткові документи стосовно повідомлення.
ХV.С.2.3. Публікація інформаційних листів-звернень до медичних працівників (DHPC)
Уповноважені органи можуть оприлюднювати фінальну версію інформаційних листів-звернень до медичних працівників. Визначення часу для такого оприлюднення необхідно регулювати з врахуванням часу розповсюдження інформаційних листів-звернень до медичних працівників. Уповноважені органи можуть також випускати додаткове повідомлення з безпеки та розповсюджувати інформаційні листи-звернення до медичних працівників серед відповідних медичних організацій, якщо необхідно.

1ЕМА повинна координувати перегляд DHPC в рамках своїх наукових комітетів (наприклад PRAC, СНМР) та CMDh.
Рис. ХV.1: Блок-схема обробки DHPC в ЄС
Частина XII
Модуль XVI — Заходи з мінімізації ризику: відбір інструментів та показників ефективності
XVI.A. Вступ
Заходи з мінімізації ризику є втручаннями, що призначені для запобігання або зменшення частоти виникнення побічних реакцій, що пов’язані з тривалістю дії лікарського засобу, або для зменшення ступеня тяжкості або впливу на пацієнта у разі виникнення побічних реакцій. Планування та реалізація заходів з мінімізації ризику та оцінка їх ефективності є ключовими елементами управління ризиком.
Рекомендації, що надаються у цьому модулі, слід розглядати в контексті більш широкої настанови ННПФ, зокрема, що пов’язаної з модулем V ННПФ.
Заходи з мінімізації ризику можуть включати рутинні або додаткові заходи з мінімізації ризику. Рутинна діяльність з мінімізації ризику застосовна до усіх лікарських засобів та включає використання різних інструментів, що докладно описані в модулі V ННПФ. Додаткові заходи з мінімізації ризику описані докладно у цьому модулі XVI ННПФ. Тому обидва модулі необхідно читати разом для повного розуміння вибору інструментів з мінімізації ризику.
Проблеми безпеки лікарського засобу, як правило, адекватно вирішуються за допомогою рутинних заходів з мінімізації ризиків (див. модуль V ННПФ). Однак у виняткових випадках рутинних заходів з мінімізації ризиків може бути недостатньо для деяких ризиків, та додаткові заходи з мінімізації ризику будуть необхідні для управління ризиком та/або покращення співвідношення користі/ризику лікарського засобу. Цей модуль надає певні рекомендації із застосування додаткових заходів з мінімізації ризиків, включаючи вибір інструментів та оцінки їх ефективності. За певних обставин, однак, оцінка ефективності може також застосовуватися до рутинних заходів з мінімізації ризиків, що пов’язані з проблемою(ами) безпеки, які описані у короткій характеристиці лікарського засобу/інструкції для медичного застосування лікарського засобуN та листку-вкладці (наприклад коротка характеристика лікарського засобу/інструкція для медичного застосування лікарського засобуN надає рекомендації з клінічних дій понад рутинні стандарти клінічного лікування стосовно самого ризику або для управління цільовою популяцією). Зважаючи на зазначене вище, рекомендації, що надані у цьому модулі стосовно оцінки ефективності, також стосуються рутинних заходів з мінімізації ризиків.
На підставі проблем безпеки, що описані у специфікації з безпеки (див. модуль V ННПФ), слід визначити відповідні заходи з мінімізації ризиків. Кожну проблему безпеки слід розглядати індивідуально, а при виборі найпридатніших заходів з мінімізації ризиків слід враховувати серйозність потенційної(их) побічної(их) реакції(й) та її(їх) ступінь тяжкості (вплив на пацієнта), можливість запобігти її(їх) виникненню або клінічні заходи, що необхідно вжити для зниження ризику, показання, спосіб застосування, цільову популяцію та заклад охорони здоров’я, де застосовуватиметься лікарський засіб. Проблему безпеки можна вирішувати за допомогою більш ніж одного заходу з мінімізації ризиків, а захід з мінімізації ризиків може охоплювати більш ніж одну проблему безпеки.
Директива 2001/83/ЄС [1], Порядок [2]N вказують на те, що заявник (власник реєстраційного посвідчення) повинен «контролювати результати реалізації заходів з мінімізації ризику, що містяться у ПУР або ті, що встановлені як умови видачі реєстраційного посвідчення відповідно до ст. 21а, 22 або 22а» (ст. 104(2)(d) Директиви 2001/83/ЄС [1], положення Порядку [2]N). Директива 2001/83/ЄС [1] та Постанова (ЄС) № 726/2004 [5] також включають положення стосовно обов’язків ЕМА та національних уповноважених органів з моніторингу результатів вжиття заходів з мінімізації ризиків, що включені до ПУР, або заходів, що встановлені як умови видачі реєстраційного посвідчення.
Цей модуль містить рекомендації, що стосуються принципів:
- розробки та запровадження заходів з мінімізації ризиків, включаючи приклади інструментів з мінімізації ризиків;
- оцінки ефективності заходів з мінімізації ризиків.
Розділ XVI.B описує розробку, запровадження та координацію заходів з мінімізації ризиків, а також загальні принципи оцінки їх ефективності. У розділі XVI.С. розглядається застосування цих заходів та принципів в умовах регуляторної мережі ЄС та УкраїніN.
XVI.B. Структури та процеси
XVI.B.1. Загальні принципи
Метою заходів з мінімізації ризиків є оптимізація безпечного та ефективного застосування лікарського засобу протягом його життєвого циклу. Співвідношення користі/ризику лікарського засобу можна покращити шляхом зменшення впливу побічних реакцій або шляхом збільшення користі, наприклад, в результаті чіткого визначення та/або виключення цільових пацієнтів, чи управління лікуванням, наприклад, шляхом застосування особливого режиму дозування, відповідного тестування, наступного спостереження за пацієнтом. Заходи з мінімізації ризику спрямовані на оптимальне застосування лікарського засобу у клінічній практиці з метою належного його застосування, у належній дозі, у належний час, належному пацієнту, а також з належною інформацією та контролем.
Більшість проблем безпеки вирішуються за допомогою рутинних заходів з мінімізації ризиків (див. модуль V ННПФ). У виняткових випадках для окремих важливих ризиків рутинні заходи з мінімізації ризику можуть бути недостатніми, що потребуватиме застосування додаткових заходів з мінімізації ризику. При визначенні, чи потрібні додаткові заходи з мінімізації ризиків, слід оцінити проблему безпеки стосовно частоти її виникнення, серйозність, ступінь тяжкості, вплив на громадське здоров’я та можливість запобігання їй. У цьому разі особливу увагу слід приділити тому, чи можна досягнути мети за допомогою рутинних заходів з мінімізації ризиків, а, якщо вони будуть оцінені як недостатні, то, які додаткові заходи з мінімізації ризиків є найбільш доречними. Додаткові заходи з мінімізації ризиків слід спрямувати на найважливіші ризики, яким можна запобігти, а навантаження, пов’язане з додатковими заходами з мінімізації ризиків, слід співставити з користю для пацієнтів.
Станом на момент затвердження цього документа до додаткових заходів з мінімізації ризиків належить низка інструментів. Ця сфера постійно розвивається, ймовірно, що у майбутньому будуть розроблені нові інструменти. Технічні досягнення, такі як інтерактивні інструменти, що базуються на інтернет-технологіях, можуть стати широко застосовними у якості додаткових заходів з мінімізації ризиків до навчальних матеріалів у паперовому форматі.
Успішна реалізація додаткових заходів з мінімізації ризиків вимагає зусиль від усіх зацікавлених сторін, включаючи заявників (власників реєстраційних посвідчень), пацієнтів та медичних працівників. Реалізація цих заходів у системах охорони здоров’я вимагає оцінки для гарантії того, що їх цілі досягаються, а також, що заходи, які реалізуються на місцях, є пропорційними ризикам, враховуючи співвідношення користі/ризику лікарського засобу та зусилля, що вимагаються від медичних працівників та пацієнтів для реалізації цих заходів. Отже, важливо забезпечити, щоб додаткові заходи з мінімізації ризиків, включаючи оцінку їх ефективності, не створювали надмірного навантаження на систему охорони здоров’я, заявників (власників реєстраційних посвідчень), регуляторні органи та, найважливіше, на пацієнтів. Для цього вони повинні мати чітко визначені цілі, що має значення для мінімізації певних ризиків та/або оптимізації співвідношення користі/ризику. Чіткі цілі, визначені індикатори оцінки та основні етапи оцінки їх ефективності повинні застосовуватись при розробці додаткових заходів з мінімізації ризиків, а також повинен здійснюватися пильний контроль їх запровадження та ефективності. Суть проблеми безпеки у контексті оцінки співвідношення користі/ризику лікарського засобу, терапевтична потреба у лікарському засобі, цільова популяція та необхідні клінічні заходи для мінімізації ризиків є факторами, які слід враховувати при виборі інструментів з мінімізації ризиків та розробці стратегії реалізації для досягнення бажаного результату для охорони здоров’я. Оцінка ефективності повинна сприяти завчасному проведенню коригуючих дій, за їх необхідності, та може вимагати внесення змін з часом. Визнано, що ця сфера медичних наук постійно розвивається, при цьому вона не має одностайно узгоджених стандартів та підходів. Тому важливо скористатися будь-якими доречними елементами методів фармакоепідеміології та інших наук, таких як соціальні/поведінкові науки та якісні методи дослідження.
Введення додаткових заходів з мінімізації ризиків слід розглядати як «програму», де розробляються певні інструменти разом із запровадженням схеми та стратегії їх оцінки. Тому в описі заходів з мінімізації ризиків, що є невід’ємною частиною ПУР (див. модуль V ННПФ), слід приділити відповідну увагу таким питанням:
- обґрунтування: коли вводяться додаткові заходи з мінімізації ризиків, слід надати обґрунтування цим додатковим заходам;
- цілі: кожний запропонований додатковий захід з мінімізації ризиків повинен включати визначені цілі та чіткий опис того, як певну проблему безпеки будуть вирішувати за допомогою запропонованого додаткового заходу з мінімізації ризику;
- опис: цей розділ ПУР повинен описувати вибрані заходи з мінімізації ризиків, включаючи інструменти, що будуть застосовуватися, та основні елементи змісту;
- запровадження: цей розділ ПУР повинен надати додаткову пропозицію для запровадження додаткових заходів з мінімізації ризиків (наприклад умови та період часу або частота втручання, докладні дані про цільову аудиторію, план розповсюдження навчальних інструментів; як буде координуватися захід, якщо він стосується більш ніж одного заявника (власника реєстраційного посвідчення));
- оцінка: у цьому розділі ПУР слід надати докладний план із зазначенням основних етапів оцінки ефективності додаткових заходів з мінімізації ризиків у контексті процесу та у контексті показників загального результату для здоров’я (наприклад зменшення ризику).
XVI.B.2. Заходи з мінімізації ризику
Метою заходів з мінімізації ризиків є полегшення прийняття інформованого рішення на підтримку мінімізації ризику при призначенні, відпуску та/або застосуванні лікарського засобу. Тоді як рутинні заходи застосовуються до кожного лікарського засобу (див. модуль V НПФ), додаткові заходи з мінімізації ризику слід застосовувати тільки, коли вони вважаються необхідними для безпечного та ефективного застосування лікарського засобу (див. також XVI.C.), їх повинні розробляти та забезпечувати особи з відповідною кваліфікацією.
Додаткові заходи з мінімізації ризику можуть відрізнятися значною мірою за метою, дизайном, цільовою аудиторію та складністю. Ці заходи можна застосовувати з метою відповідного вибору пацієнтів за винятком пацієнтів, яким це протипоказано, для підтримки моніторингу під час лікування, що має відношення для важливих ризиків та/або управління побічною реакцією. Додатково можна розробляти особливі заходи для мінімізації ризику виникнення та реалізації медичної помилки (див. Посібник PRAC з належної практики мінімізації ризику та запобігання медичним помилкам96) та/або для забезпечення відповідного способу застосування лікарського засобу, коли їх не можливо реалізувати тільки за допомогою інформації про лікарський засіб та маркування.
У розділі XVI.B.2. описуються додаткові заходи з мінімізації ризику, що можна розглядати додаткового до рутинних заходів, включаючи:
- навчальні програми;
- програми контрольованого доступу;
- інші заходи з мінімізації ризику.
XVI.B.2.1. Навчальна програма
Навчальні програми базуються на цільовому наданні інформації з метою доповнення інформації, що міститься у короткій характеристиці лікарського засобу, листку-вкладці/інструкції для медичного застосування лікарського засобуN. У будь-якому навчальному матеріалі увагу слід зосередити на цілях, які можна здійснити. Він повинен забезпечувати якісні та чіткі повідомлення, що описують заходи, яких слід вжити для запобігання та мінімізації ідентифікованих ризиків.
Метою навчальної програми є покращення застосування лікарського засобу шляхом позитивного впливу на дії медичних працівників та пацієнтів, що спрямовані на мінімізацію ризику. Тому навчальні матеріали слід створювати, виходячи з того, щоб у них містилася практична рекомендація для цільового навчання та що вживання цього заходу вважається необхідним для мінімізації важливого ризику та/або для оптимізації співвідношення користі/ризику. У контексті навчальної програми інструменти можуть мати декілька різних цільових аудиторій, можуть вирішувати більш ніж одну проблему безпеки та можуть постачатися із застосуванням комбінації інструментів та засобів (наприклад паперовий, аудіо-, відео- та вебформат, навчання при персональному контакті). В ідеальному випадку навчальні матеріали повинні бути доступними у низці форматів, так, щоб забезпечити відсутність обмежень у доступі через недієздатність або відсутність доступу до інтернету. Коли можливо, відповідність інструменту та засобів цільовій аудиторії (наприклад доречна мова, малюнки, діаграми або інша графічна підтримка) слід заздалегідь тестувати на користувачах з метою оптимізації успіху на етапі впровадження.
Зміст будь-якого навчального матеріалу слід повністю узгодити з поточною схваленою інформацією про лікарський засіб, такою як коротка характеристика лікарського засобу, листок-вкладка/інструкція для медичного застосування лікарського засобуN, він повинен скоріше доповнювати, ніж дублювати інформацію у короткій характеристиці лікарського засобу, листку-вкладці/інструкції для медичного застосування лікарського засобуN. Не слід включати рекламні елементи, прямі або приховані (наприклад логотипи, кольори бренду продукту, асоціативно-сугестивні зображення та малюнки), навчальний матеріал слід зосереджувати на ризику (ах), що стосуються лікарського засобу, та управлінні тими ризиками, що вимагають додаткових заходів з мінімізації.
Будь-яку навчальну програму слід повністю відокремлювати від рекламної діяльності, а контактну інформацію про лікарів або пацієнтів, що зібрана за допомогою навчальних програм, не слід застосовувати для рекламної діяльності.
Навчальні інструменти, що описані нижче, можна розглядати в індивідуальному порядку або в комбінації під час розробки навчальної програми з метою додаткової мінімізації ризику.
XVI.B.2.1.1. Навчальні інструменти
Навчальний інструмент повинен мати чітку визначену сферу дії та повинен включати однозначне(і) формулювання стосовно важливого (их) ризику(ів), що викликає занепокоєність та який слід вирішити за допомогою запропонованого інструменту, суть такого(их) ризику(ів) та певні кроки, які слід зробити медичним працівникам та/або пацієнтам для мінімізації цих ризиків. Цю інформацію слід зосередити на чітко визначених діях, що стосуються певних проблем безпеки, що описані в ПУР. Також інформація не повинна містити даних, що не мають безпосереднього відношення до проблеми безпеки та що вже відповідно представлена у короткій характеристиці лікарського засобу або листку-вкладці/інструкції для медичного застосування лікарського засобуN. У навчальних матеріалах повинні міститися посилання на коротку характеристику лікарського засобу або листок-вкладку/інструкцію для медичного застосування лікарського засобуN. Додатково до вступу до навчального матеріалу про те, що він є необхідним для гарантії безпечного та ефективного застосування лікарського засобу, а також для відповідного управління визначеними важливими ризиками, елементами для включення до навчальних матеріалів можуть бути:
- інструкції з призначення, включаючи відбір, тестування та моніторинг пацієнта;
- інструкції з управління такими ризиками (для медичних працівників та пацієнтів або осіб, які їх доглядають);
- інструкції стосовно того, як та куди повідомляти про побічну реакцію, що представляє певний інтерес.
Подальші рекомендації стосовно обов’язків заявника або заявника (власника реєстраційного посвідчення), а також уповноважених органів надані у розділі XVI.C.
XVI.B.2.1.1.1. Навчальні інструменти, що спрямовані на медичних працівників
Метою будь-якого навчального інструменту, що спрямований на медичного працівника, повинно бути надання особливих рекомендацій стосовно застосування (що робити) та/або протипоказань (чого не робити), а також застережень (наприклад, як лікувати побічну реакцію), що пов’язані з лікарським засобом та певними важливими ризиками, що потребують додаткових заходів з мінімізації ризиків, включаючи:
- відбір пацієнтів;
- управління лікуванням, таке як дозування, тестування та моніторинг;
- особливі процедури введення лікарського засобу або відпуск лікарського засобу;
- деталі інформації, що потрібно надати пацієнтам.
Формат певного інструменту повинен залежати від інформації, яку слід донести. Наприклад, якщо низка заходів необхідна до виписування рецепта для пацієнта, найбільш доречним форматом може бути контрольний перелік.
Брошура може бути більш адекватною для посилення інформованості про певні важливі ризики з приділенням уваги ранньому розпізнаванню та управлінню побічними реакціями, тоді як плакати для демонстрації у певних клінічних умовах можуть включати корисні рекомендації (пам’ятку) стосовно лікування або дозування. Можна надати перевагу іншим форматам, залежно від цілей інструменту.
XVI.B.2.1.1.2. Навчальні інструменти, що спрямовані на пацієнтів та/або осіб, які їх доглядають
Метою будь-яких інструментів, що спрямовані на пацієнтів та/або осіб, які їх доглядають, повинно бути покращення їх інформованості про ранні ознаки та симптоми певних побічних реакцій, що викликають потребу в додаткових заходах з мінімізації ризиків, та про оптимальний порядок дій у разі виникнення будь-яких з цих ознак або симптомів. Якщо доцільно, навчальний інструмент, що спрямований на пацієнта, можна застосовувати для надання інформації про правильне введення лікарського засобу та для нагадування пацієнту про важливу діяльність, наприклад, ведення щоденника прийому лікарського засобу або діагностичні процедури, які пацієнт повинен вести або проходити, а також зрештою обговорювати із спеціалістами-медиками з метою забезпечення дотримання будь-яких дій, що необхідні для безпечного та ефективного застосування лікарського засобу.
Пам’ятка для пацієнтів
Метою цього інструменту є забезпечення того, що особлива інформація про лікування, що проводиться на теперішній час, та його важливі ризики (наприклад потенційні взаємодії з іншими видами лікування, що погрожують життю) зберігається у пацієнта протягом усього часу та може передаватися відповідному медичному працівнику, якщо необхідно. Інформація повинна зводитися до мінімуму, що необхідний для передачі ключової інформації стосовно мінімізації ризику та необхідних дій, за будь-яких обставин, включаючи екстрені випадки. Здатність до зручного носіння пам’ятки пацієнтом (наприклад її можна покласти до гаманця) повинна бути вирішальною особливістю її дизайну.
XVI.B.2.2. Програма контрольованого доступу
Програма контрольованого доступу складається із заходів, спрямованих на контроль доступу до лікарського засобу понад рівень контролю, що забезпечується рутинними заходами з мінімізації ризику, зокрема, правовим статусом/умовами відпуску. Оскільки програма контрольованого доступу має наслідки для усіх зацікавлених сторін, застосування такої програми повинно бути обмежене та повинно керуватися тільки чіткою терапевтичною потребою у лікарському засобі на підставі його продемонстрованої користі (наприклад, лікарський засіб лікує серйозну хворобу, що не має альтернативних видів лікування; він лікує пацієнтів, у яких існуючі види лікування не були результативними), характер пов’язаного ризику (наприклад ризик загрожує життю), а також ймовірністю того, що цей ризик можна вирішити за допомогою такої програми. Тому контрольований доступ можна розглядати тільки як інструмент для мінімізації важливого ризику із значним впливом на громадське здоров’я або окремого пацієнта для лікарського засобу з чітко продемонстрованою користю, але який не буде доступним поза програмою, при якій пацієнт може отримати доступ, за умови виконання однієї або більше вимог перед тим, як лікарський засіб буде призначений або відпущений, для гарантії його безпечного застосування.
Приклади вимог, що необхідно виконати до того, як лікарський засіб буде призначений та/або відпущений, та/або застосовуватиметься у програмі контрольованого доступу, перераховані нижче (вони можуть застосовуватися окремо або у комбінації):
- певне тестування та/або обстеження пацієнта для забезпечення відповідності строго визначеним клінічним критеріям;
- особа, яка призначає лікарський засіб, особа, яка відпускає лікарський засіб, та/або пацієнт, які документують отримання лікарських засобів та розуміють інформацію про серйозний ризик лікарського засобу;
- чітко визначені процедури систематичного спостереження за пацієнтом за допомогою включення до особливої системи збору даних, наприклад, реєстру пацієнтів;
- лікарські засоби можуть відпускатися тільки в аптеках, що мають ліцензію та дозвіл на відпуск даного лікарського засобу.
В окремих випадках вимога тестування або проведення моніторингу пацієнта у певний спосіб може також застосовуватися як інструмент контрольованого доступу. Наприклад, моніторинг стану здоров’я пацієнта, лабораторні показники або інші характеристики до та/або під час лікування, наприклад, проведення електрокардіограм, тестів функції печінки, регулярних аналізів крові, тестів на вагітність (що можуть бути складовою програми запобігання вагітності). Заходи слід вживати на місцях для забезпечення того, що має проводиться моніторинг відповідно до короткої характеристики лікарського засобу/інструкції для медичного застосування лікарського засобуN, коли це має критичне значення для співвідношення користі/ризику лікарського засобу.
XVI.B.2.3. Інші заходи з мінімізації ризиків
XVI.B.2.3.1. Система контрольованої дистрибуції/розповсюдження
Система контрольованої дистрибуції являє собою низку заходів, що запроваджуються для забезпечення спостереження за усіма стадіями ланцюга дистрибуції лікарського засобу до етапу призначення та відпуску лікарського засобу. Замовлення та відправлення лікарського засобу з однієї або декількох визначених дистриб’юторських точок полегшують відстеження руху лікарського засобу. Цей вид заходів можна розглянути для тих лікарських засобів, що контролюються у кожній країні за відповідними національними законами для запобігання зловживанню та неправомірному їх використанню.
XVI.B.2.3.2. Програма запобігання вагітності
Програма запобігання вагітності (ПЗВ) — це низка заходів, спрямованих на мінімізацію впливу на вагітність під час лікування лікарським засобом з відомими або потенційними тератогенними ефектами. Метою такої програми є забезпечення відсутності вагітності у пацієнток на момент початку лікування, ненастання вагітності під час лікування та/або незабаром після припинення лікування. ПЗВ також може бути спрямована на пацієнтів-чоловіків, коли застосування лікарського засобу біологічним батьком може мати негативний ефект на результат вагітності.
ПЗВ являє собою комбінацію застосування навчальних інструментів/матеріалів та здійснення контролю відповідного доступу до лікарського засобу. Тому слід розглянути окремо та/або в комбінації з розробкою ПЗВ такі елементи:
- навчальні інструменти/матеріали, що націлені на медичних працівників та пацієнтів для інформування про тератогенний ризик, та необхідні заходи для мінімізації цього ризику (наприклад рекомендації стосовно необхідності застосовувати більш ніж одного методу контрацепції, та рекомендації стосовно різних видів контрацепції, інформацію, що включена для пацієнтки відносно того, як довго уникати настання вагітності після припинення лікування, інформація на той випадок, якщо лікується партнер-чоловік);
- контрольований доступ на рівні призначення або відпуску для забезпечення проведення тесту на вагітність та констатація негативних результатів медичним працівником перед призначенням або відпуском лікарського засобу;
- термін дії рецепта обмежений, його слід використати протягом 30 днів;
- надання консультації у випадку настання незапланованої вагітності та оцінка наслідків будь-якої випадкової вагітності.
Розробку та запровадження реєстру вагітних (як окрему діяльність або як частину ПЗВ) слід також розглянути для загального включення пацієнток, які завагітніли під час лікування або протягом відповідного часу після припинення лікування (наприклад 3 місяці). Застосування цього систематичного інструменту для збору інформації про результат вагітності може бути корисним в оцінці ефективності програми запобігання вагітності та/або у полегшенні подальшої характеристики ризику, зокрема у ранній період після реєстрації, коли дані про вагітність у жінки можуть бути дуже обмеженими та/або коли потенційна занепокоєність щодо вагітності може ґрунтуватися тільки на доклінічних даних.
XVI.B.2.3.3. Інформаційний лист-звернення до медичних працівників (DHPC)
Інформаційний лист-звернення до медичних працівників (DHPC) — інформаційне втручання, завдяки якому заявник (власник реєстраційного посвідчення) або уповноважений орган надає важливу інформацію безпосередньо медичним працівникам, що повідомляє їх про необхідність вжити певних заходів, що стосуються застосування лікарського засобу (див. модуль I ННПФ). Наприклад, інформаційний лист-звернення до медичних працівників може стосуватися змін призначення лікарського засобу для мінімізації певних його ризиків та/або зменшення впливу побічних реакцій на ефективність лікарського засобу. Ситуації, у яких слід розглянути розповсюдження інформаційного листа-звернення до медичних працівників, докладно описані у модулі XV ННПФ.
XVI.B.3. Запровадження заходів з мінімізації ризиків
Додаткові заходи з мінімізації ризику можуть включати одне або більше втручань, що слід запровадити у сталий спосіб у визначеній цільовій групі. Особливу увагу слід приділити як визначенню часу, так і частоті будь-якого втручання та процедурам, спрямованим на охоплення цільової популяції. Наприклад, одноразове розповсюдження навчальних матеріалів може бути недостатнім для забезпечення охоплення усіх потенційних осіб, які призначають лікарський засіб, тобто лікарів, та/або тих, хто його використовує, включаючи нових лікарів та користувачів. У такому разі може знадобитися додаткове регулярне розповсюдження. Однак може бути і навпаки, коли навчальні матеріали можуть бути необхідними на початку застосування нового лікарського засобу, та непотрібними або нерелевантними згодом, після тривалого їх використання протягом низки років застосування лікарського засобу (див. модуль V ННПФ). Оскільки заходи з мінімізації ризиків мають різні специфічні цілі, деякі заходи, такі як пам’ятки для пацієнта, програми контрольованого доступу та програми запобігання вагітності, зазвичай, застосовуватимуться до усіх майбутніх процедур реєстрації одного й того ж самого лікарського засобу, тоді як інші, такі як DHPC та навчальні матеріали необов’язково можуть знадобитися для цього. Доречність кожного заходу та наявність потреби у них для майбутніх процедур реєстрації на один і той самий лікарський засіб слід ретельно розглядати під час реєстрації лікарського засобу (та пояснити це у ПУР). Особливу увагу слід приділити формату та змісту навчальних матеріалів для забезпечення чіткої відмінності від будь-якого рекламного матеріалу, що розповсюджується. Подання навчального матеріалу для оцінки уповноваженим органом слід відокремлювати від подання рекламного матеріалу, а у супровідному листі слід чітко зазначати, чи матеріали є рекламними, чи навчальними. Крім того, навчальні матеріали слід розповсюджувати окремо від рекламних матеріалів як «автономне» повідомлення, та слід чітко зазначити, що інструменти не є рекламним матеріалом і, що вони призначені для мінімізації ризику. Механізми забезпечення якості повинні гарантувати, що наявні системи дистрибуції відповідають меті та можуть піддаватися аудиту.
XVI.B.4. Ефективність заходів з мінімізації ризиків
Оцінка ефективності додаткових заходів з мінімізації ризику необхідна для встановлення, чи втручання було ефективним, чи ні, і, якщо ні, чому та які коригуючі дії необхідні. Оцінку слід проводити окремо для додаткових інструментів з мінімізації ризиків та для програми мінімізації ризиків у цілому.
Оцінку ефективності слід проводити у найбільш відповідний час, враховуючи час, необхідний для ініціації заходів з мінімізації ризику, очікуване застосування лікарського засобу системою охорони здоров’я та інші релевантні обставини.
Слід планувати регулярну перевірку ефективності одного або більше специфічних інструментів або загальної програми, якщо доцільно. Особливе значення мають такі періоди часу:
- після початкового запровадження програми мінімізації ризику (наприклад протягом 12–18 місяців), щоб забезпечити можливість внесення змін у разі їх необхідності;
- під час оцінки при продовженні дії реєстраційного посвідчення/перереєстрації.
У будь-який час, коли оцінюється ефективність заходу, ретельну увагу слід приділити необхідності продовження додаткового заходу з мінімізації ризику.
Оцінка ефективності повинна розглядати різні аспекти мінімізації ризику, тобто сам процес (тобто наскільки запровадження програми відповідає плану), його вплив на знання та поведінкові зміни у цільовій аудиторії (тобто показник (и) впливу на зміни у поведінці) та наслідки (тобто, до якого ступеню заздалегідь визначені цілі мінімізації ризику досягнуті, за короткий та тривалий термін). При плануванні стратегії оцінки належну увагу слід приділити тому, які аспекти процесу та результати можна реалістично виміряти для уникнення утворення неточних або дезорієнтуючих даних або неналежного надмірного навантаження на систему охорони здоров’я або на інші зацікавлені сторони. Період часу для оцінки кожного аспекту втручання, а також встановлення реалістичних систем показників, за якими оцінюється ефективність інструменту, слід також ретельно розглядати та планувати до введення.
Для оцінки ефективності додаткових заходів з мінімізації ризиків слід розглянути дві категорії показників:
- показники процесу;
- показники результату.
Показники процесу необхідні для збирання доказів успішності етапів запровадження додаткових заходів з мінімізації ризиків. Ці показники процесу повинні надати можливість зрозуміти, до якого ступеня програма виконана відповідно до запланованого, та чи спостерігається запланований вплив на поведінку. Систему показників запровадження слід визначити заздалегідь та відстежувати з часом. Оцінка процесу запровадження може також покращити розуміння процесу(ів) та причинно-наслідкових механізмів, завдяки яким додаткові заходи з мінімізації ризику призводять або не призводять до бажаного контролю визначених важливих ризиків.
Показники результату надають загальну оцінку рівня контролю ризику, що був досягнутий за допомогою оцінюваного заходу з мінімізації ризику. Наприклад, коли метою втручання є зменшення частоти та/або ступеня тяжкості побічної реакції, кінцевий показник успіху буде пов’язаний з цією метою.
У виняткових обставинах, коли обґрунтовано, що оцінка показників результату є недоцільною (наприклад невідповідне число пацієнтів, які зазнали впливу лікарського засобу, рідкісні побічні явища), оцінка ефективності повинна ґрунтуватися винятково на ретельній інтерпретації даних про показники процесу.
Висновком оцінки може бути те, що захід з мінімізації ризику повинен залишатися незмінним або потрібні зміни у існуючих заходах. В іншому випадку оцінка може вказувати на те, що мінімізація ризику є недостатньою та її слід посилити (наприклад шляхом змін у застереженні або рекомендацій у короткій характеристиці лікарського засобу, листку-вкладці/інструкції для медичного застосування лікарського засобуN, покращуючи чіткість рекомендації для мінімізації ризику, та/або шляхом додавання додаткових інструментів або покращення існуючих інструментів). Іншим рішенням може бути те, що мінімізація ризику є непропорційною або їй не вистачає чіткої направленості та її можна зменшити або спростити (наприклад шляхом зменшення кількості інструментів або частоти втручання, або шляхом видалення втручань, що виявилися такими, що не сприяють мінімізації ризику). За всіх обставин навантаженню на пацієнта та систему охорони здоров’я слід приділити особливу увагу.
Додатково до оцінки ефективності заходів з мінімізації ризиків в управлінні проблемами безпеки також важливо контролювати, чи втручання з мінімізації ризику має непередбачені (негативні) результати для охорони здоров’я, що розглядається, у короткотривалій та/або довготривалій перспективі. Приклади незапланованих результатів можуть включати надмірне навантаження на систему охорони здоров’я або припинення застосування лікарського засобу у пацієнтів, навіть якщо співвідношення користь/ризик було для них позитивним.
Законодавство визначає: «Будь-яке дослідження…, що оцінює ефективність заходів управління ризиком» як ПДБЛЗ (ст. 1(15) Директиви 2001/83/ЄС [1]). Отже, якщо дослідження проводиться для оцінки індикаторів результату стосовно поведінки або безпеки, слід притримуватися докладної настанови з проведення ПДБЛЗ, яке представлене у модулі VIII ННПФ. Ця настанова не застосовується до оцінки простих маркерів процесу (наприклад, розповсюдження інструментів, що охоплюють цільову популяцію). Якщо доцільно, слід застосовувати Настанову з методологічних стандартів у фармакоепідеміології, що розроблено Європейською мережею центрів фармакоепідеміології та фармаконагляду97.
XVI.B.4.1. Показники процесу
Показники процесу є індикаторами ступеня запровадження оригінального плану та/або змін у його реалізації. Показники процесу повинні доповнювати, але не заміняти оцінку досягнення цілей заходів з мінімізації ризиків (тобто індикатори результату). Залежно від характеру втручань різні показники процесу можна визначити для оцінки їх здійснення.
XVI.B.4.1.1. Охоплення цільової популяції
Коли заходи з мінімізації ризику включають надання інформації та рекомендацій медичним працівникам та/або пацієнтам за допомогою навчальних матеріалів, індикатори дистрибуції та отримання слід застосовувати для отримання базової інформації про запровадження. Ця система показників повинна бути спрямована на оцінку, чи були доставлені матеріали цільовій аудиторії та чи вони дійсно були отримані цільовою популяцією.
XVI.B.4.1.2. Оцінка клінічних знань
Для оцінки інформованості цільової аудиторії, її ставлення та рівня знань, що досягнутий навчальними діями або наданням іншої інформації (наприклад завдяки навчальній програмі, спрямованій на запобігання впливу лікарського засобу під час вагітності), слід застосовувати науково досконалі методи опитування. У додатку I модуля XVI підсумовуються основні методологічні аспекти, які слід розглянути для планування та запровадження опитування.
Анкета для опитування, як правило, включає набір стандартних питань, відповіді на які вносяться при телефонному контакті, при особистому інтерв’ю, або самостійно вносяться відповідачем до поштового/електронного повідомлення, що повторюються з часом. Такий підхід можна пристосувати до моніторингу ставлення та знань у різних вибірках, що включає представників з кожного сегменту аудиторії, що викликає інтерес у цільових групах медичних працівників та/або пацієнтів. Якщо доцільно, слід застосовувати психометричні показники. Коли можливо, слід вибрати рандомізовану вибірку та адекватний розмір вибірки. На відміну від цього, застосування груп із захисту прав пацієнтів або групи підтримки пацієнтів для перевірки знань можна вважати по суті необ’єктивним у зв’язку з самостійним відбором, отже, його слід уникати.
Відповідну увагу слід надавати цілям дослідження, дизайну дослідження, розміру та репрезентативності вибірки, робочому визначенню залежних та незалежних змінних, а також статистичному аналізу. Ретельну увагу слід приділити вибору найбільш доречних інструментів збору даних (наприклад опитувальників).
XVI.B.4.1.3. Оцінка клінічних дій
Для вимірювання ефективності навчальних дій та/або надання інформації слід оцінювати не тільки клінічне знання, але й клінічні дії, що випливають з них (тобто поведінку призначення). Дослідження використання лікарських засобів за допомогою вторинного застосування електронних записів або за допомогою витягів з медичних карток є корисними варіантами для кількісного визначення клінічних дій, якщо забезпечуються репрезентативність цільової популяції та відповідні бази даних. Аналіз записів про призначення, особливо, коли вони поєднані з іншими записами про пацієнтів (наприклад клінічними або демографічними даними), може дозволити здійснити оцінку поведінки призначення, включаючи одночасне призначення двох взаємодіючих лікарських засобів, дотримання рекомендації стосовно лабораторного контролю, а також відбір та моніторинг пацієнтів. Застосовуючи відповідні статистичні методи (наприклад аналіз часових рядів, аналіз виживаності, логістична регресія) до когорти користувачів лікарських засобів, можна аналізувати різні аспекти призначення та застосування, які можуть забезпечити розуміння понад винятково описових доказів. Особливу увагу слід приділити проведенню та тлумаченню досліджень використання лікарських засобів у різних країнах, включаючи правовий статус/умови відпуску лікарського засобу та те, як він призначається та відпускається, оскільки моделі призначення можуть відображати не тільки інформацію про лікарський засіб та будь-яке втручання з мінімізації ризиків, але також національні настанови, аспекти, що пов’язані з послугами, що надаються у системі охорони здоров’я, місцевою медичною практикою та обмеженнями відшкодування. Така різноманітність національних систем охорони здоров’я на території ЄС може бути підставою для проведення дослідження з однаковими цілями у декількох країнах.
Дослідження поведінки, що ґрунтуються на даних, що зібрані під час опитувань, слід розглядати тільки у випадку, коли відсутні будь-які дані для оцінки клінічних дій (тобто проведення дослідження використання лікарського засобу на підставі самостійно повідомлених даних, що зібрані при опитуванні медичних працівників та/або пацієнтів).
XVI.B.4.2. Показники результату
Кінцевими показниками успіху програми з мінімізації ризику є результати безпеки, тобто частота та/або серйозність побічних реакцій відносно експозиції пацієнтів, що зазнала впливу лікарського засобу за межами інтервенційного дослідження, та ці результати безпеки повинні бути показником(ами) результату. Така оцінка повинна включати порівняння епідеміологічних показників частоти результату, таких як рівень захворюваності або кумулятивна частота побічної реакції, що отримані, наприклад, у контексті ПДБЛЗ. Використання відповідних результатів, пов’язаних з безпекою, що представляють інтерес, повинно розглядатися (наприклад замісні кінцеві точки, такі як відповідний біомаркер як заміна клінічної кінцевої точки), якщо такий підхід полегшує оцінку ефективності. При будь-якому підході наукова точність та визнані принципи епідеміологічного дослідження повинні завжди бути визначальними для оцінки показника кінцевого результату, який представляє інтерес. Слід розглянути необхідність порівняння частоти до та після імплементації заходів з мінімізації ризиків (тобто дизайн «до та після»). Якщо дизайн «до та після» неможливий (наприклад, якщо заходи з мінімізації ризиків введені на момент отримання реєстраційного посвідчення), порівняння результуючих показників частоти, що отримані після лікування, у порівняні з попередньо визначеним референтним показником, отриманим після огляду літератури, історичних даних, очікуваної частоти в загальній популяції, було б прийнятним (тобто, аналіз наявних даних у порівняні з очікуваними даними), та повинне враховувати будь-яке стимульоване повідомлення, зміни у догляді за пацієнтами та/або заходах з мінімізації ризиків з часом. Вибір будь-якої відповідної референтної групи слід обґрунтувати.
Методи для вимірювання ефективності заходів з мінімізації ризиків повинні бути пропорційними ризику, що буде мінімізований. По суті, використання рівнів спонтанних повідомлень (тобто кількість повідомлень про підозрювані побічні реакції протягом фіксованого періоду часу) може бути прийнятним у контексті стандартної мінімізації ризиків. Спонтанне повідомлення слід розглядати з обережністю при оцінці частоти побічних явищ в популяції, яка отримує лікування, оскільки воно може використовуватися у дуже специфічних обставинах, наприклад, якщо побічна реакція на лікарський засіб є рідкісною, вихідна частота побічного явища в загальній популяції незначна та тісний зв’язок між лікуванням та побічним явищем. У таких обставинах, якщо прямий показник щодо ризику в популяції, яка отримує лікування, неможливо отримати, завдяки спонтанним повідомленням можна отримати приблизний показник частоти побічних реакцій в популяції, яка отримує лікування, за умови, що можна отримати обґрунтовано достовірні дані для оцінки рівня повідомлень у контексті застосування лікарського засобу. Проте добре відомі помилки, що впливають на процес надання повідомлень про підозрювані побічні реакції, можуть надати оманливі результати. Наприклад, застосування заходів з мінімізації ризиків у відповідь на проблему з безпеки, виявлену в післяреєстраційний період лікарського засобу, може підвищити усвідомлення виникнення побічних реакцій, пов’язаних із застосуванням лікарського засобу, що зрештою може призвести до підвищення рівня надходження повідомлень. За цих обставин аналіз спонтанних повідомлень може призвести до помилкового висновку, що лікування було неефективним. Зниження рівня повідомлень з часом може призвести до помилкового висновку, що лікування було ефективним.
XVI.B.5. Координація
Якщо декілька лікарських засобів, включаючи лікарські засоби, що зареєстровані відповідно до ст. 10(1) або 10(3) Директиви 2001/83/ЄС [1] (далі вказуються як «генерики» або «гібридні»), з однією діючою речовиною/АФІ наявні на ринку, повинен бути відповідний підхід щодо використання додаткових заходів з мінімізації ризиків, що координуються або передбачаються уповноваженими органами. Якщо необхідна координована дія для фармакотерапевтичної групи лікарських засобів, слід відповідно погодити гармонізований підхід. За цих умов перспективне планування повинно гарантувати, що ефективність заходів з мінімізації ризиків (див. XVI.В.4.) може розглядатися для кожного окремого лікарського засобу, а також для сукупності лікарських засобів.
XVI.B.6. Системи якості заходів з мінімізації ризиків
Хоча багато експертів може залучатися до розробки та імплементації заходів з мінімізації ризиків, кінцева відповідальність за якість, точність та наукову цілісність цих заходів та планування їх опису покладена на заявника (власника реєстраційного посвідчення) та його УОВФ/КОВФN.
Заявник (власник реєстраційного посвідчення) відповідає за оновлення ПУР, якщо нова інформація стає наявною та повинна бути врахована відповідно до принципів якості, що детально описані в модулі І ННПФ. Версії з відслідковуваними змінами ПУР повинні подаватися для спрощення регуляторної оцінки. Ці записи, ПУР та пов’язані системи управління ризиками, як і будь-які документи щодо заходів з мінімізації ризиків, можуть підлягати аудиту, інспекції у ЄС, аудиту системи фармаконагляду уповноваженою установоюN (див. модуль ІІІ ННПФ).
Заявник (власник реєстраційного посвідчення) повинен гарантувати відповідний контроль версій інструментів мінімізації ризиків з метою гарантування, що всі медичні працівники та пацієнти отримують найновіші інструменти мінімізації ризиків своєчасно, та що інструменти, що застосовуються, відповідають схваленій інформації про лікарський засіб. З цією метою власників реєстраційних посвідчень заохочують відслідковувати отримання будь-яких інструментів з мінімізації ризиків цільовою групою. Ці записи мають підлягати аудиту, інспекції у ЄС, аудиту системи фармаконагляду уповноваженою установоюN.
Заявник (власник реєстраційного посвідчення) повинен гарантувати, що механізми повідомлення результатів досліджень або аналізів для оцінки ефективності заходів з мінімізації ризиків, задокументовані. Вони можуть підлягати аудиту, інспекції у ЄС, аудиту системи фармаконагляду уповноваженою установоюN.
XVI.С. Функціонування мережі ЄС, УкраїниN
Для лікарських засобів, зареєстрованих шляхом централізованої процедури, додаткові заходи з мінімізації ризиків, що рекомендовані PRAC, погоджені СНМР та узгоджені Європейською Комісією шляхом рішення Комісії, стануть умовами для безпечного та ефективного використання лікарського засобу.
У додатку ІІ висновку СНМР будуть описані основні елементи будь-яких додаткових заходів з мінімізації ризиків, які встановлені для заявника (власника реєстраційного посвідчення), як умова для безпечного та ефективного використання лікарського засобу. Через специфічність систем охорони здоров’я в країнах ЄС/УкраїніN та того, як певний(і) ризик(и) управляється(ються) в рамках цих систем, в залежності від національних можливостей, можуть виникнути особливості та відмінності в імплементації деяких заходів з мінімізації ризиків, та вимагається додаткове погодження з країнами ЄС/УкраїноюN (наприклад програми профілактики вагітності, контрольований розподіл). Тому для лікарських засобів, зареєстрованих шляхом централізованої процедури, законодавство передбачає, що додатково до рішення Комісії, яке адресоване заявнику (власнику реєстраційного посвідчення) за ст. 127а Директиви 2001/83/ЄС [1], може бути рішення Комісії, яке адресоване країнам ЄС, що зобов’язує їх гарантувати, що певні умови та/або обмеження імплементовані заявником (власником реєстраційного посвідчення) на їх території.
Тому в додатку до рішенні Комісії стосовно ст. 127а Директиви 2001/83/ЄС [1] може описуватися зобов’язання національних уповноважених органів, гарантувати, що додаткові заходи з мінімізації ризиків імплементовані в країнах ЄС відповідно до ключових елементів. Подальші детальні дані або ключові елементи щодо будь-яких додаткових заходів з мінімізації ризиків повинні також включатися в додаток 6 ПУР (див. модуль V ННПФ).
Для лікарських засобів, зареєстрованих за процедурою взаємного визнання та децентралізованою процедурою додаткові заходи з мінімізації ризиків слід включити в додаток 6 ПУР, та можна також представити, як умови видачі реєстраційного посвідчення.
В усіх випадках імплементація додаткових заходів з мінімізації ризиків має місце на національному рівні та дозволяє країнам ЄС/Україні адаптувати необхідні умови та обмеження до будь-яких національних правових вимог та локальних систем охорони здоров’я.
XVI.С.1. Роль та відповідальність у рамках регуляторної мережі ЄС, УкраїниN
В цьому розділі описується відповідальність різних органів в процесі розробки, імплементації та оцінки додаткових заходів з мінімізації ризиків, спрямованих на безпечне та ефективне застосування лікарського засобу в ЄС/УкраїніN.
З урахуванням відмінностей різних систем охорони здоров’я в країнах ЄС/УкраїніN, ключові елементи додаткових заходів з мінімізації ризиків, які необхідно імплементувати скоординовано серед країн ЄС, слід погодити на рівні ЄС, забезпечуючи узгодження детальних даних локальної імплементації на національному рівні. За умови, що деякі ключові елементи специфічні лише для деяких країн ЄС/України (наприклад діяльність, пов’язана з системою охорони здоров’я однієї країни ЄС/УкраїниN) або якщо додаткові заходи з мінімізації ризиків не встановлюються як умова видачі реєстраційного посвідчення, їх слід включити в ПУР.
XVI.С.1.1. Європейське агентство з лікарських засобів
ЕМА спільно з країнами ЄС та за підтримки PRAC повинно контролювати ефективність заходів з мінімізації ризиків, які містяться в ПУР, та умов, на які посилаються в пунктах (с), (са), (сb), та (сс) ст. 9(4) або пунктах (а) та (b) ст. 10а(1) та в ст. 14(7) та (8) Регламенту (ЄС) №726/2004 [5] (ст. 28а(1)(а) Регламенту (ЄС) №726/2004 [5]).
При здійсненні моніторингу результатів заходів з мінімізації ризиків ЕМА повинна підтримати наукову оцінку PRAC результатів заходів з мінімізації ризиків, які включають додаткові заходи з мінімізації ризиків шляхом інтеграції даних, що надані згідно з ресурсами та дослідницькою діяльністю країни ЄС. PRAC зробить рекомендації СНМР або CMDh відносно будь-якої необхідної регуляторної дії.
XVI.С.1.2. Комітет з фармаконагляду та оцінки ризиків (PRAC)
PRAC повинен оцінити результати заходів з мінімізації ризиків, включаючи додаткові заходи з мінімізації ризиків, та надавати рекомендації відносно будь-якої необхідної регуляторної дії.
Додатково до консультування щодо досліджень та заходів, які описані в ПУР, PRAC буде оцінювати протокол та результати встановлених післяреєстраційних досліджень з безпеки, які мають на меті оцінити ефективність заходів з мінімізації ризиків (див. модуль VIІІ НПФ).
XVI.С.1.3. Уповноважені органи
Національні уповноважені органи відповідають на національному рівні за контроль імплементації встановлених додаткових заходів з мінімізації ризиків, що є умовою видачі реєстраційного посвідчення для безпечного та ефективного застосування лікарського засобу в ЄС, незалежно від типу заяви, згідно з якою лікарський засіб був зареєстрований.
Для таких заходів з мінімізації ризиків, які повинні бути виконані після реєстрації лікарського засобу, національні уповноважені органи повинні гарантувати швидкий розгляд та погодження втручання із заявником (власником реєстраційного посвідчення).
Національні уповноважені органи при підтримці PRAC та СНМР або CMDh можуть сприяти гармонізації імплементації інструментів з мінімізації ризиків для генериків з однією діючою речовиною/АФІN. Якщо додаткові заходи з мінімізації ризиків вважаються необхідними для генеричних лікарських засобів і пов’язані з питаннями з безпеки, що стосуються діючої речовини/АФІN, заходи з мінімізації ризиків, що застосовуються до генеричних лікарських засобів, повинні бути аналогічними заходам референтного лікарського засобу. Додаткові заходи з мінімізації ризиків для гібридних лікарських засобів можуть вимагатися за деяких обставин, додатково до тих, що передбачені для референтного лікарського засобу (наприклад інший склад або спосіб введення, або питання несумісності). Для сприяння цьому PRAC може рекомендувати імплементувати ключові елементи додаткових заходів з мінімізації ризиків для всіх відповідних лікарських засобів, зареєстрованих за національною процедурою (як умова видачі реєстраційного посвідчення на них) та за згоди може зробити ці загальні вимоги загальнодоступними для сприяння гармонізованій імплементації на національному рівні.
Додатково до зазначеного вище для лікарських засобів, зареєстрованих шляхом централізованої процедури, відповідальність національних уповноважених органів при гарантуванні імплементації заходів з мінімізації ризиків може передаватися їм шляхом рішення Комісії за ст. 127а Директиви 2001/83/ЄС [1].
Додатково національні уповноважені органи повинні погодити кінцевий зміст, формат та способи проведення заходів з мінімізації ризиків, включаючи друкований матеріал, платформи на основі вебтехнологій та інші аудіо-, відеозасоби, а також планування графіку впровадження із заявником (власником реєстраційного посвідчення) до того, як лікарський засіб буде виведений на їх ринок або у будь-який час пізніше, якщо необхідно (див. модуль XVI ННПФ додаток І).
Національний уповноважений орган винесе рішення щодо відповідних національних освітніх матеріалів та/або інших інструментів мінімізації ризиків, оскільки вони пов’язані з ключовими елементами, які погоджені на рівні ЄС, та описані в ПУР (див. модуль XVI ННПФ додаток І). Подібним чином визначення ефективності додаткових заходів з мінімізації ризиків може вимагатися в одній країні ЄС у межах спеціальних умов надання медичної допомоги або якщо результати досліджень ефективності не можуть екстраполюватися з досліджень, що проведені в інших країнах ЄС, внаслідок національної специфічності.
Національні уповноважені органи спільно з ЕМА за сприяння PRAC/уповноваженого органу в УкраїніN повинні на національному рівні проводити моніторинг результату заходів з мінімізації ризиків, які зазначені в ПУР, та умов, які вказані в ст. 21а, 22 або 22а Директиви 2001/83/ЄС [1] (ст. 107h(1)(а) Директиви 2001/83/ЄС [1], положеннях Порядку [2], положеннях Порядку проведення експертизи [7]N).
Якщо карти-повідомлення пацієнта (див. XVI.В.2.1.1.2.) включені в зовнішню упаковку лікарського засобу, вони вважаються частиною маркування, тому текст і формат повинні погоджуватися із відповідним уповноваженим органом (повний текст включений до додатку ІІІ торгової ліцензії).
Для лікарських засобів, зареєстрованих шляхом централізованої процедури, якщо існують певні національні умови (наприклад багатомовні документи), карти-повідомлення пацієнта можуть не відповідати формату гаманця. У таких випадках карта-повідомлення може не включатися в упаковку лікарського засобу та не повинна розглядатися як частина маркування. У цьому випадку національні уповноважені органи повинні узгодити кінцевий зміст та формат, як для іншої додаткової діяльності з мінімізації ризиків.
XVI.С.2. Роль та відповідальність заявника (власника реєстраційного посвідчення) в ЄС, УкраїніN
Заявник (власник реєстраційного посвідчення) в ЄС/УкраїніN несе відповідальність та гарантує дотримання умов реєстраційного посвідчення на їх лікарські засоби, де б вони не використовувалися в рамках ЄС/УкраїниN. Відповідальністю заявника (власника реєстраційного посвідчення) є імплементувати усі умови або обмеження, що сприяють безпечному використанню лікарського засобу на певній території.
Заявник (власник реєстраційного посвідчення) повинен чітко визначити цілі будь-якого запропонованого заходу з мінімізації ризиків та показники для оцінки його ефективності. Заявника (власника реєстраційного посвідчення) заохочують обговорювати плани з мінімізації ризиків з уповноваженими органами в країнах ЄС/УкраїніN якомога раніше, наприклад, коли вважається імовірним, що певна діяльність з мінімізації ризиків має адаптуватися до різних систем охорони здоров’я, що існують у різних країнах ЄС/УкраїніN.
Будь-який додатковий захід з мінімізації ризиків слід розробляти відповідно до загальних принципів, описаних в XVI.В.1. та XVI.В.2. та повністю документувати у ПУР (див. ННПФ модуль V).
Заходи, схвалені в ПУР, заявник (власник реєстраційного посвідчення) повинен імплементувати на національному рівні після погодження з національними уповноваженими органами.
Заявник (власник реєстраційного посвідчення) повинен надати інформацію стосовно статусу імплементації додаткових заходів з мінімізації ризиків, які погоджено з національними уповноваженими органами, та інформувати їх про будь-які зміни, проблеми або питання, з якими він зіткнувся під час імплементації додаткових заходів з мінімізації ризиків. Будь-які відповідні зміни до імплементації інструментів слід погодити з національними органами до імплементації.
При імплементації інструментів на основі вебтехнологій заявник (власник реєстраційного посвідчення) повинен застосовувати вимоги, які є специфічним для кожної країни ЄС/УкраїниN. Особливу увагу слід приділити розгляду питань, що пов’язані з доступністю, ідентифікуванням, відповідальністю, конфіденційністю і захистом даних.
Для генеричних лікарських засобів заявник (власник реєстраційного посвідчення) повинен розробити заходи з мінімізації ризиків відповідно до сфери дії, змісту та формату інструментів, що використовувалися для референтного лікарського засобу. Складання графіку та планування дій слід чітко координувати з метою мінімізації тягаря для систем охорони здоров’я.
Заявник (власник реєстраційного посвідчення) повинен проводити моніторинг результатів заходів з мінімізації ризиків, що містяться в ПУР, або які є умовою видачі реєстраційного посвідчення стосовно ст.ст. 21а, 22 або 22а Директиви 2001/83/ЄС [1] (ст. 104(3)(d) Директиви 2001/83/ЄС [1], відповідно до положень Порядку проведення експертизи [7]N). Загальні принципи оцінки ефективності надаються в XVI.В.3.
Заявник (власник реєстраційного посвідчення) повинен повідомити про оцінку впливу додаткової діяльності з мінімізації ризиків при оновленні ПУР (див. V.В.11.4).
Заявник (власник реєстраційного посвідчення) повинен повідомити в РОЗБ результати оцінки ефективності заходів з мінімізації ризиків, що можуть вплинути на безпеку або оцінку співвідношення користі/ризику (див. VІІ.В.5.16.5. та VІІ.С.5.5).
Для генеричних лікарських засобів ефективність заходів з мінімізації ризиків слід оцінювати заявникам (власникам реєстраційних посвідчень) у тісній співпраці з уповноваженими органами. Якщо у рамках додаткових заходів з мінімізації ризиків обґрунтована доцільність проведення досліджень, то для лікарських засобів з однією діючою речовиною/АФІN раціонально проводити спільні дослідження з метою мінімізації навантаження на системи охорони здоров’я. Наприклад, якщо схвалюється проведення когортного дослідження, то включення до нього учасників повинно залежати від того, яку торговельну назву має лікарський засіб, головне, щоб ці ліки мали одну діючу речовину/АФІN. Однак реєстрація певних детальних даних лікарського засобу є важливою для швидкого визначення будь-якої нової загрози щодо безпеки при застосуванні певного лікарського засобу.
Заявник (власник реєстраційного посвідчення) повинен гарантувати своєчасне повідомлення уповноважених органів для здійснення відповідної регуляторної оцінки та дій (див. також ХVІ.С.2. та модуль ННПФ V та VІІ).
ХVІ.С.3. Медичні працівники та пацієнти
Медичні працівники та пацієнти не мають жодних правових зобов’язань відносно імплементації законодавства з фармаконагляду. Проте співпраця медичних працівників та пацієнтів є важливою для успіху навчальних програм та/або для програм контрольованого доступу з метою оптимізації співвідношення користі/ризику Бажано, щоб вони уважно розглядали будь-які додаткові заходи з мінімізації ризиків, які можуть бути застосовані з метою безпечного та ефективного застосування лікарських засобів.
ХVІ.С.4. Вплив ефективності заходів з мінімізації ризиків на ПУР/РОЗБ в ЄС, УкраїніN
Оновлення РОЗБ та ПУР повинні включати оцінку резюме результату додаткових заходів з мінімізації ризиків, які імплементовані для зменшення важливих ризиків в ЄС/УкраїніN. В ПУР увага повинна зосереджуватися на тому, як буде здійснюватися мінімізація ризиків та/або планування фармаконагляду. У РОЗБ повинна також надаватися оцінка щодо того, як імплементовані заходи впливають на профіль безпеки та/або співвідношення користі/ризику лікарського засобу. Загалом увага повинна зосереджуватися на інформації, що стала відома протягом звітного періоду або з моменту імплементації останнього(іх) заходу(ів) з мінімізації ризиків в ЄС/УкраїніN. Якщо існує паралельне подання оновлення РОЗБ та ПУР до уповноважених органів регуляторної мережі ЄС/України, слід розглянути використання модуля загального змісту (див. ННПФ модуль V та VII). Для оцінки застосовується настанова в ХVІ.В.4.
Результати оцінки(ок) ефективності заходів з мінімізації ризиків повинні завжди відображатися у ПУР. В рамках цієї критичної оцінки заявник (власник реєстраційного посвідчення) повинен вивчати фактори, що сприяють успіху або послабленню заходів з мінімізації ризиків. Цей критичний аналіз може включати посилання на досвід за межами ЄС/УкраїниN.
Оцінка ефективності заходів з мінімізації ризиків повинна зосереджуватися на тому, чи були вони успішними щодо мінімізації ризиків. Це слід здійснювати, використовуючи комбінацію показників процесу та результату, як описано в ХVІ.В.3. Може бути доцільним розрізняти заходи з мінімізації ризиків, імплементовані на момент реєстрації, та заходи, введені пізніше, в післяреєстраційний період.
При представлені результатів оцінки ефективності заходів з мінімізації ризиків слід розглянути такі аспекти:
1. Оцінка повинна бути представлена у контексті: а) короткого описання імплементованого(них) заходу(ів) з мінімізації ризиків, б) визначення їх цілі(ей) та в) вибраних показників процесу та результату.
2. Оцінка повинна включати відповідний аналіз типу побічної(их) реакції(й), включаючи її серйозність та превентивність. Слід також включити відповідні логістичні фактори, що можуть вплинути на клінічну реалізацію заходу з мінімізації ризиків.
3. Оцінка повинна включати аналіз реалізації заходів з мінімізації ризиків у стандартній клінічній практиці, включаючи будь-яке відхилення від оригінального плану. Така оцінка може включати результати досліджень застосування лікарського засобу.
4. Показники результату повинні бути, як правило, ключовою кінцевою точкою при оцінці досягнених цілей заходів з мінімізації ризиків.
Пропозиції щодо змін для покращення управління ризиками слід представити в регіональному додатку РОЗБ (див. ННПФ модуль VII). ПУР слід оновити для врахування інформації, що виникає відносно ефективності заходів з мінімізації ризиків.
Загалом заявники (власники реєстраційних посвідчень) генеричних лікарських засобів не повинні на регулярній основі подавати РОЗБ в ЄС. В Україні заявники (власники реєстраційних посвідчень) на генеричні лікарські засоби повинні надавати РОЗБ відповідно до положень Порядку [2]N. Частота оновлень ПУР повинна бути пропорційною ризикам лікарського засобу. Загалом увага при оновлені ПУР повинна зосереджуватися на заходах з мінімізації ризиків та наданні оновлень щодо імплементації таких заходів. Якщо існує послідовна зміна до резюме ПУР, це слід описати в супровідному листі. Зміни до інформації про лікарський засіб не слід здійснювати через окреме оновлення ПУР, а краще подати заяву на зміни. РОЗБ може також викликати безпосередньо оновлення інформації про лікарський засіб (якщо РОЗБ подається заявником (власником реєстраційного посвідчення) на даний генеричний лікарський засіб).
ХVІ.С.5. Прозорість
Повинні існувати процедури для гарантування повної прозорості щодо надання відповідної інформації про заходи з мінімізації ризиків для відповідних лікарських засобів.
Відповідно до ст. 106 Директиви 2001/83/ЄС [1] та ст. 26 Регламенту (ЄС) № 726/2004 [5] ЕМА та національні уповноважені органи повинні робити загальнодоступними загальні звіти з оцінки лікарських засобів, а також резюме ПУР (ст. 31 ІП 520/2012 [6], положення Порядку [2]N), включаючи заходи з мінімізації ризиків, що описані у цьому документі.
Для лікарських засобів, зареєстрованих шляхом централізованої процедури, ЕМА повинно робити загальнодоступними:
- Резюме ПУР (ст. 26(1)(с) Регламенту (ЄС) №726/2004 [5]) з акцентом на діяльність з мінімізації ризиків, що описана в даному документі (ст. 31.1 ІП 520/2012 [6]);
- Європейський загальний звіт з оцінки (ЕРАR), який включає будь-які умови видачі реєстраційного посвідчення, такі як додаткові заходи з мінімізації ризиків (ст. 26(1)(j) Регламенту (ЄС) № 726/2004 [5]).
Шляхом розміщення на національних вебпорталах лікарських засобів країни ЄС/УкраїнаN повинні робити загальнодоступними принаймні наступні документи:
- Загальний звіт з оцінки; він повинен включати резюме, написане у спосіб, який зрозумілий широкому загалу (ст. 21(4), ст. 106(а) Директиви 2001/83/ЄС [1]);
- Коротку характеристику лікарського засобу та листок-вкладку (ст. 21(3), ст. 106(б) Директиви 2001/83/ЄС [1])/інструкцію для медичного застосування лікарського засобуN;
- Умови видачі реєстраційного посвідчення разом з будь-якими кінцевими термінами для виконання таких умов (ст. 21 (3) Директиви 2001/83/ЄС [1], положення Порядку проведення експертизи [7]N);
- Резюме ПУР (ст.106(с) Директиви 2001/83/ЄС [1]); із особливим зосередженням уваги на діяльності з мінімізації ризиків, що описана в даному документі (ст. 31.1 ІП 520/2012 [6], положення Порядку [2]N).
Для сприяння охороні здоров’я рекомендовано, щоб ЕМА та національні уповноважені органи зробили наступну інформацію доступною через свої вебсайти:
- Детальні дані додаткових заходів з мінімізації ризиків, що є умовою видачі реєстраційного посвідчення (наприклад, коли інструменти повідомлення про ризик складаються з друкованого матеріалу, надається копія або, якщо можливо, рекомендується надання електронного доступу до навчального матеріалу, картки пацієнта, контрольного переліку або інших інструментів мінімізації ризику);
- детальні дані про реєстри захворювань або речовин вимагаються в рамках системи обмеженого розповсюдження.
XVI. Додаток 1. Ключові елементи методології опитування
Опитування є методом систематичного збору первинних даних безпосередньо у відібраних учасників більшої популяції. Вони проводяться з метою характеристики більшої популяції та можуть бути перехресними (одноразовими) або пролонгованими (повторюються через деякий час).
В контексті оцінки ефективності заходів з мінімізації ризиків опитування може проводитися для оцінки розуміння, знань та дій після навчання у вказаній цільовій популяції відносно безпеки та управління ризиком лікарського засобу.
Опитування не завжди може бути найбільш відповідним способом для оцінки дій, оскільки зазвичай воно передбачає збір та аналіз інформації, самостійно наданої медичними працівниками та пацієнтами. Більше того, участь в опитуванні по суті може вносити зміни до дій. Слід також зважити на те, що при опитуванні вибірка може бути нерепрезентативною у контексті цільових користувачів, оскільки найбільш ймовірно у опитуванні беруть участь зацікавлені медичні працівники та/або більш мотивовані або освічені особи.
Як мінімум, зазначені нижче елементи повинні розглядатися при плануванні та імплементації опитування з метою мінімізації потенційних помилок та оптимізації узагальненості результатів для запланованої популяції:
1) процедури відбору опитуваних та стратегія їх залучення;
2) дизайн та управління інструментом(ами) збору даних;
3) аналітичні підходи;
4) етика, конфіденційність та загальна ймовірність здійснення дослідження.
ХVI. Додаток 1.1. Процедури відбору зразків та стратегія залучення
При здійсненні будь-якого опитування може відбутися зміщення щодо основи вибірки та залучення учасників. Це призводить до того, що досліджувана популяція не схожа або не представляє собою заплановану популяцію в одному або більшості аспектів. Більш того, слід врахувати, що помилки не можна виключити, лише збільшуючи основу вибірки, розмір вибірки та рівень відповіді. Помилки слід мінімізувати, обираючи оптимальну основу вибірки, враховуючи вік, стать, географічний розподіл та додаткові характеристики досліджуваної популяції. Помилку можна також мінімізувати, гарантуючи, що вибірка містить відповідне різноманіття, що дозволить класифікувати результати за ключовими характеристиками популяції (наприклад шляхом додаткової вибірки невеликої, але важливої підгрупи). Ключові елементи мають розглядатися на основі вибірки, включаючи вік, стать, географічний розподіл та додаткові характеристики досліджуваної популяції. Наприклад, при опитуванні лікарів стратегія рандомізованого відбору досліджуваної вибірки повинна врахувати, чи буде достатньо загальної рандомізованої вибірки, чи вибірка повинна бути класифікована за ключовими характеристиками, такими як спеціальність, тип практики (наприклад первинна допомога, допомога спеціаліста, академічна лікарня). При опитуванні пацієнта слід врахувати дохід та освіту, захворювання, застосування лікарських засобів у хронічному чи гострому стані.
Додатково до загальної репрезентативності цільової популяції при стратегії залучення слід уважно розглянути потенційні джерела залучення. Для залучення медичних працівників, переліки спонсорів, вебпанелі, професійні та наукові товариства можуть представляти можливі підходи. Проте їх репрезентативність для запланованої цільової популяції лікарів слід уважно переглянути для кожного дослідження. При залученні пацієнтів слід розглянути відповідні клінічні заклади та вебпанелі. Стратегію залучення слід спланувати, враховуючи можливість збору точних та повних даних. Слід докласти зусиль для документування частини нереспондентів та їх характеристики для оцінки потенційного впливу на репрезентативність вибірки.
ХVI. Додаток 1.2. Дизайн та управління інструментом(ами) збору даних
Підходи до збору даних при опитуванні можуть варіювати від особистого інтерв’ю, тестування, вивчення та збору біологічних зразків, що передбачено рамками звичайної клінічної практики, до телефонного інтерв’ю на основі вебтехнологій та паперових анкет. Самостійне аудіоопитування за допомогою комп’ютера (A-CASI), інтерактивна система голосової відповіді (IVRS) або підходи змішаного типу також можуть бути використані. Вибір найбільш відповідного підходу для збору даних буде залежати від характеристик цільової популяції, захворювання, характеристик лікування та типу даних, що будуть збирати.
Кожний підхід до збору даних буде вимагати спеціального дизайну одного або більше специфічних інструментів. Проте загальні міркування щодо дизайну, які можуть застосовуватися до всіх інструментів, включають наступне:
- тягар для учасників, тобто тривалість, когнітивне навантаження, чутливість до пацієнта;
- чіткість та послідовність питань; наприклад, використання чіткої мови, мінімізація припущень, починаючи з найбільш важливих питань та залишаючи чутливі питання на пізніше;
- повнота відповідей; наприклад, структурні питання з метою приведення до єдиної чіткої відповіді, що дозволяє вибрати «невідомо» або «не знаю»;
- планування інструменту для збору даних; наприклад, чіткий потік, керівництва з технологічною підтримкою (уникнення шаблонів, нагадування про відсутність відповіді та візуальні зображення);
- тестування та перегляд інструменту; наприклад, формальне тестування, використовуючи когнітивне попереднє тестування, таке як інтерв’ю тет-а-тет, зондуючі питання, настанова для проведення інтерв’ю або підготовлений інтерв’юер та процес «думати вголос»;
- стимули для покращання рівня відповідей; наприклад, зведені дані зворотного зв’язку відповідно до опитування учасників.
XVI. Додаток 1.3. Аналітичні підходи
Основні аналітичні елементи аналізу повинні включати:
- описова статистика, наприклад:
- процент учасників, які правильно відповіли на питання;
- класифікація за вибраною змінною;
- дані про відсутність відповіді або неповну відповідь;
- порівняння характеристик респондентів та нереспондентів (якщо наявні дані);
- порівняння характеристик респондентів та загальної цільової популяції.
Якщо оцінюються результати опитування, слід розглянути такі ключові пункти:
- відмінності у виборі ймовірностей (наприклад якщо для певних підгруп була зроблена додаткова вибірка);
- відмінності в рівнях відповіді;
- післякласифікаційне оцінювання відповідно до зовнішньої популяції;
- групування.
Приклади класифікаційних аналізів опитування лікарів включають наступне:
- спеціальність лікаря;
- географічне розміщення;
- отримання будь-яких навчальних матеріалів;
- обсяг прописуваних лікарських засобів.
XVI. Додаток 1.4. Етика, конфіденційність та загальна здійсненність дослідження
Етичні вимоги та вимоги до конфіденційності даних не гармонізовані серед країн ЄС та мають помітні відмінності в національних (або регіональних) процесах. Національні (або регіональні) відмінності можуть існувати щодо доцільності надання стимулів для учасників опитування. Можуть також виникати питання конфіденційності щодо дозволу контакту з лікарями на основі переліку лікарів, який міститься у заявника (власника реєстраційного посвідчення).
Загальна оцінка здійсненності дослідження є ключовим етапом в успішній імплементації опитування. Для збору клінічних даних ключові елементи такої оцінки включають:
- збір інформації на дільниці та характеристика досліджуваної популяції (пацієнти або медичні працівники);
- оцінку обґрунтованого розміру вибірки дослідження, необхідної кількості баз для досягнення розміру вибірки, приблизна тривалість періоду збору даних (наприклад на основі оцінюваної кількості пацієнтів, частоти візитів пацієнта та очікуваного рівня відповіді пацієнта);
- оцінку ресурсів баз та зацікавленості в дослідженні.
Ключові елементи оцінки здійсненності можуть бути різними в різних дизайнах дослідження (наприклад залучення та збір даних на основі вебтехнологій) та в оцінках лікарів.
XVI. Доповнення I.1. Вступ
Навчальні програми є додатковими заходами з мінімізації ризику (див. модуль XVI ННПФ) та зазвичай включають навчальний(і) матеріал(и), метою якого(их) є мінімізація важливого ризику та/або максимальне збільшення співвідношення користі/ризику лікарського засобу. Зміст будь-якого навчального матеріалу слід повністю привести у відповідність до діючої інформації про лікарський засіб, тобто короткої характеристики лікарського засобу/інструкції для медичного застосування лікарського засобуN, листка-вкладки та маркування, він повинен скоріше доповнювати, ніж дублювати дані в інформації про лікарський засіб, тобто короткій характеристиці лікарського засобу, листку-вкладці/інструкції для медичного застосування лікарського засобуN.
Коли розробка та розповсюдження навчального матеріалу рекомендована PRAC та підтримана СНМР або CMDh/уповноваженим органомN, основні елементи будь-якого навчального матеріалу узгоджуються на рівні ЄС/УкраїниN. Потім проєкти навчального(их) матеріалу(ів), в якому(их) розглядаються основні елементи, заявник (власник реєстраційного посвідчення) повинен надати до уповноважених органів держав ЄС/УкраїниN для оцінки, потім їх слід запровадити у державах ЄС/УкраїніN після схвалення уповноваженими органами.
У модулі V ННПФ надаються рекомендації щодо вимог до включення основних елементів навчального(их) матеріалу(ів) та/або навчального(их) матеріалу(ів), що розповсюджуються у державах ЄС/УкраїніN у додатку ПУР.
Це Доповнення до модулю XVI ННПФ надає подальшу настанову заявникам (власникам реєстраційних посвідчень) щодо подання проєкту навчальних матеріалів до уповноважених органів, а також настанову для цих органів у підтримку оцінки таких матеріалів, зокрема стосовно формату та змісту. Через особливості національних систем охорони здоров’я та через те, як здійснюється управління певними ризиками в межах цих систем, окремі держави ЄС/УкраїниN можуть мати додаткові вимоги. У цьому випадку слід дотримуватися настанови, представленої у цьому Доповненні до модулю XVI ННПФ, разом з національними настановами.
Це Доповнення застосовне до лікарських засобів, що зареєстровані як за централізованою, так і національною процедурою, включаючи лікарські засоби, що зареєстровані за процедурою взаємного визнання та децентралізованою процедурою.
XVI. Доповнення I.2. Принципи навчальних матеріалів
До навчальних матеріалів застосовуються такі принципи:
- потреба в навчальних матеріалах може узгоджуватися під час регуляторної процедури, під час видачі реєстраційного посвідчення або у післяреєстраційний період, наприклад, після введення нового ПУР або оновлення існуючого ПУР;
- будь-який навчальний матеріал слід ретельно розробляти для досягнення цілей з мінімізації ризику;
- слід звертати увагу на особливі проблеми безпеки та надавати чіткі формулювання та стислі повідомлення, що описують заходи, яких слід вжити для запобігання та мінімізації цих ризиків;
- національні версії навчального матеріалу повинні надаватися заявником (власником реєстраційного посвідчення) до відповідних уповноважених органів держав ЄС/УкраїниN тільки після завершення регуляторної процедури, якою узгоджений додатковий захід з мінімізації ризику;
- проєкт навчальних матеріалів слід готувати офіційною(ми) мовою(ми) відповідно до вимог держави ЄС/УкраїниN;
- навчальні матеріали не повинні включати або комбінуватися з рекламними елементами, прямими або прихованими (наприклад асоціативно-сугестивними зображеннями та малюнками);
- методи розповсюдження та цільова аудиторія у кожній державі ЄС/УкраїніN визначаються на національному рівні відповідним уповноваженим органом держави ЄС/УкраїниN;
- залежно від відповідної цільової аудиторії заявник (власник реєстраційного посвідчення) повинен надати до кожного національного уповноваженого органу пропозицію стосовно навчального(их) матеріалу(ів). Цільова популяція визначає, який інструмент, зміст, формат, тип мови та рівень читабельності є доречним для навчального матеріалу. При адаптації слід докладати певних зусиль, з метою задоволення потреби цільових пацієнтів (див. модуль XV ННПФ);
- уповноважені органи держав ЄС/УкраїниN, у яких лікарський засіб продається/буде продаватися, повинні оцінювати відповідну(і) національну(і) версію(і) навчального(их) матеріалу(ів);
- заявник (власник реєстраційного посвідчення) повинен розповсюджувати навчальний(і) матеріал(и) у державі ЄС/УкраїніN тільки після схвалення уповноваженими органами цієї держави ЄС/УкраїниN;
- якщо лікарський засіб не розміщується на ринку держави ЄС/УкраїниN, розповсюдження матеріалу у цій державі ЄС/УкраїніN, не вимагається. У будь-якому разі необхідність розповсюдження будь-якого навчального матеріалу слід обговорювати з уповноваженим органом кожної держави ЄС/УкраїниN;
- заявник (власник реєстраційного посвідчення) повинен проводити контроль версій та забезпечувати розповсюдження тільки останньої узгодженої версії навчального матеріалу. Дату схвалення уповноваженим органом держави ЄС/УкраїниN слід включати до навчального матеріалу в якості довідки для медичних працівників та/або пацієнтів;
- без шкоди для оригінальності формату навчального матеріалу в інтересах охорони здоров’я, щоб навчальний матеріал, що застосовується різними заявниками (власниками реєстраційних посвідчень) для однієї і тієї ж діючої речовини/АФІN, залишався якомога більш подібним для того, щоб доставляти одноманітні повідомлення та уникати дезорієнтування цільової популяції. Тому заявникам (власникам реєстраційних посвідчень) суворо рекомендується обмінюватися змістом своїх навчальних матеріалів на вимогу інших заявників (власників реєстраційних посвідчень).
XVI. Доповнення I.3. Подання навчальних матеріалів
Якщо інші національні вимоги не застосовуються, проєкт навчального матеріалу слід подавати до уповноважених органів держав ЄС/УкраїниN таким чином:
- з супровідним листом та/або формою-запитом, що включає таку інформацію:
- контактну інформацію про заявника (власника реєстраційного посвідчення) та, за необхідності, іншу організацію, що виступає підрядником під час подання (принаймні, назва та адреса електронної пошти)
- регуляторну процедуру, що призводить до необхідності навчального(их) матеріалу(ів) із допоміжними документами (наприклад висновком СНМР, позицією CMDh та рішенням Європейської Комісії, включаючи умови реєстраційного посвідчення та інші додатки, висновок національного уповноваженого органу/уповноваженого органу УкраїниN, схвалений ПУР, оціночний звіт, що визначає потребу у цьому додатковому засобі з мінімізації ризику);
- детальний план запровадження навчального матеріалу, що містить таку інформацію:
- цільова(і) популяція(ї);
- метод розповсюдження (наприклад паперовий формат, електронна пошта, соціальні мережі, наукові спільноти та/або асоціації пацієнтів, публікація на вебсайтах);
- момент часу, коли передбачається почати розповсюдження та частота подальшого розповсюдження;
- очікувана дата виходу на ринок або дата початку продажу лікарського засобу (у випадку видачі реєстраційного посвідчення);
- як документи в загальному відкритому електронному форматі для обробки тексту запропонованих матеріалів мовою, яка вимагається країною ЄС/УкраїноюN;
- запланований макет та, якщо необхідно, зображення та графічна презентація інформації (наприклад малюнки, схеми, діаграми, відео).
Якщо зміни в ризику та/або необхідності у додаткових заходах з мінімізації ризиків були визначені та зміни в ключових елементах та/або змісті навчального(них) матеріалу(ів) були погоджені на рівні ЄС/УкраїниN та/або національними уповноваженими органами, заявник (власник реєстраційного посвідчення) повинен подавати до уповноважених органів країни ЄС/УкраїниN переглянуті пропозиції щодо навчального матеріалу для оцінки та схвалення. В переглянутому навчальному матеріалі повинні висвітлюватися зміни до матеріалів, які раніше були схвалені уповноваженим органом.
XVI. Доповнення 1.4. Формат та макет навчальних матеріалів
Навчальні матеріали повинні мати відповідний формат та макет.
Рекомендується рядок «назва» для визначення типу навчального матеріалу, наприклад, адміністративна настанова, контрольний перелік для прописування лікарського засобу, картка попередження, навчальний листок для пацієнтів.
Формат навчальних матеріалів повинен включати таке:
- торговельну назву лікарського засобу, назву діючої(их) речовин(и)/АФІN та/або терапевтичну групу у дужках. Проте, якщо навчальний матеріал застосовується до декількох лікарських засобів різних заявників (власників реєстраційних посвідчень) в країнах ЄС/УкраїніN, навчальний матеріал повинен посилатися лише на діючу речовину/АФІN та повинен додаватися перелік торговельних назв, що застосовуються в країні ЄС/УкраїніN;
- темний символ поруч із торговельною назвою або назвою діючої речовини/АФІN разом із пояснювальним стандартним формулюванням для додаткового моніторингу, якщо лікарський засіб знаходиться під додатковим моніторингом (див. модульX ННПФ).
Матеріал слід форматувати таким чином:
- жирні крапки слід використовувати для чіткого представлення інформації;
- матеріали мають бути якомога стислішими; проте, якщо навчальний матеріал довгий, слід додати вступний текст з резюме ключової інформації та можна включити зміст;
- якщо з’являється логотип заявника (власника реєстраційного посвідчення) та/або лікарського засобу, заявник (власник реєстраційного посвідчення) повинен з’являтися тільки раз в кожному навчальному матеріалі, бажано, на першій або останній сторінці та не повинен бути більшим за назву документа;
- для контролю версій на кожній сторінці навчального матеріалу повинен використовуватися унікальний ідентифікатор документа та слід вказувати на першій та останній сторінці дату останнього перегляду тексту (тобто дата схвалення матеріалу відповідним національним уповноваженим органом) у форматі «(місяць) (рік)», якщо тип навчального матеріалу не вимагає виключень (наприклад відео повинно мати унікальний ідентифікатор документа, який з’являється спочатку та вкінці).
XVI. Доповнення 1.5. Зміст навчальних матеріалів
Референтними документами, що повинні використовуватися при підготовці навчальних матеріалів, є погоджені ПУР (включаючи його додатки), інформація про лікарський засіб та умови видачі реєстраційного посвідчення.
Навчальний матеріал повинен містити повідомлення про ключові елементи, що погоджені в залежності від регуляторної процедури на рівні ЄС/УкраїниN або уповноваженим органом країни ЄС/України та зазначені в умовах видачі реєстраційного посвідчення (як вказано в ст. 9(4) Регламенту (ЄС) № 726/2004 [5] та ст. 21а(а) Директиви 2001/83/ЄС [1], положеннях Порядку [2], положеннях Порядку проведення експертизи [7]N).
Навчальний матеріал може також містити посилання на вебсайт уповноваженого органу країни ЄС/УкраїниN, ЕМА або певний вебсайт заявника (власника реєстраційного посвідчення) (див. XVI, доповнення 1.7.), якщо коротка характеристика лікарського засобу та/або листок-вкладка/інструкція для медичного застосування лікарського засобуN є загальнодоступними на цих вебсайтах.
Посилання на інші вебсайти для отримання «більш детальної інформації», як правило, не будуть прийнятними, якщо вони не посилаються на коротку характеристику лікарського засобу та/або листок-вкладку/інструкцію для медичного застосування лікарського засобуN, або якщо не застосовуються спеціальні умови, наприклад, з метою посилання на тест на певні антитіла або посилання на відео, яке інструктує пацієнтів, як приймати лікарський засіб та/або використовувати пристрій, у разі погодження з уповноваженим(и) органом(ами) країн(и) ЄС/УкраїниN.
Зображення або графічна презентація інформації повинні використовуватися, якщо самого тексту недостатньо для відповідної передачі ключових елементів повідомлень, та не повинні бути промоційними (наприклад використання певного приладу для введення лікарського засобу).
Сфера дії інформації в навчальному матеріалі повинна обмежуватися погодженими ключовими елементами. Додаткова інформація, така як дані ефективності, порівняння безпеки з іншими лікарськими засобами або формулювання, які означають, що лікарський засіб добре переноситься, або що побічні реакції виникають з низькою частотою, не повинні включатися. Проте за певних обставин уповноважені органи країн ЄС/УкраїниN можуть розглядати включення даних про ефективність, за умови, що це належним чином обґрунтовано заявником (власником реєстраційного посвідчення). Посилання на інші лікарські засоби за межами сфери дії навчального матеріалу не дозволяється.
Формулювання, що спрямоване на заохочення до інформування про будь-які підозрювані побічні реакції, та інформацію про спосіб надання повідомлення в уповноважений орган країни ЄС/УкраїниN також слід включати.
XVI. Доповнення 1.6. Оцінка та публікація навчальних матеріалів уповноваженими органами країн ЄС/УкраїниN
Строки оцінки проєктів навчальних матеріалів різними уповноваженими органами країн ЄС/УкраїниN можуть варіювати залежно від, наприклад, додаткових заходів з мінімізації ризиків, типу запитуваних навчальних матеріалів або якості поданих проєктів. Проте для оцінки зазвичай, у середньому потрібно близько 60 днів. Такий підхід не передбачає відміни будь-яких інших строків, що визначені уповноваженими органами на національному рівні.
В інтересах громадського здоров’я уповноважені органи країн ЄС/УкраїниN відповідно до національного законодавства можуть публікувати погоджені навчальні матеріали у визначених розділах на своїх вебсайтах.
Заявники (власники реєстраційних посвідчень) виключно відповідальні за надання уповноваженим органам останніх погоджених версій навчальних матеріалів.
XVI. Доповнення 1.7. Публікація навчальних матеріалів на спеціальному вебсайті власника реєстраційного посвідчення
Заявник (власник реєстраційного посвідчення) може публікувати навчальний(і) матеріал(и) на спеціально призначеному (або іншому відповідному) вебсайті за умови, що заявник (власник реєстраційного посвідчення) дотримується наступного:
- спосіб, у який проводиться розповсюдження через вебсайт, слід погоджувати з уповноваженим органом країни ЄС/УкраїниN, тобто як первинний або як додатковий спосіб розповсюдження;
- адресу вебсайту слід надавати уповноваженому органу країни ЄС/УкраїниN;
- формулювання, що інформація вебсайту відповідає навчальному матеріалу, який схвалений уповноваженим органом, повинно подаватися уповноваженому органу країни ЄС/УкраїниN;
- спеціальний вебсайт не повинен включати будь-яке посилання на документи або на інші вебсайти/сторінки або вебпосилання, що не погоджені з уповноваженим органом країни ЄС/УкраїниN;
- усі елементи та інформація на спеціальному вебсайті повинна бути представлена офіційною(ими) мовою(ами), як вимагається законодавством країни ЄС/УкраїниN, або у виняткових випадках за погодженням уповноваженого органу країни ЄС англійською мовою в країнах ЄС;
- спеціальний вебсайт не повинен містити посилання або інформацію про лікарські засоби, що не продаються в такій країні ЄС/УкраїніN. Можна посилатися на інші відповідні документи, такі як коротка характеристика лікарського засобу та/або листок-вкладка/інструкція для медичного застосування лікарського засобуN та резюме ПУР.
Частина XІІІ
Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби
СП.ІІ.А. Вступ
Біологічний лікарський засіб (далі — біологічний препарат) є лікарським засобом, що містить активну субстанцію/діючу речовинуN, що вироблена або екстрагована з біологічного джерела, та яка вимагає для своєї характеристики та визначення своєї якості комбінації фізико-хімічних біологічних тестувань разом з процесом виробництва та його контролем (розділ 3.2.1.1.(б), частина І, додаток 1 Директиви 2001/83/ЄC [1], положення Порядку проведення експертизи реєстраційних матеріалів [7]N).
Біологічні препарати включають дуже широкий та різноманітний спектр лікарських засобів. Вони включають медичні речовини отримані з крові та плазми, біотехнологічні лікарські засоби (наприклад з використанням технології рекомбінантної ДНК), всі типи вакцин для профілактики та ВТЛЗ. Ця частина ННПФ не застосовується до вакцин та ВТЛЗ, оскільки вже існує окрема спеціальна настанова щодо цих препаратів (див. частину ННПФ «Спеціальні питання щодо препаратів або популяції І: Вакцини для профілактики інфекційних хвороб» (далі — СП І) та Керівництво щодо дослідження безпеки та ефективності, а також управління ризиком ВТЛЗ98).
Якщо не вказано інакше в певних розділах, ця частина ННПФ застосовується до всіх біологічних лікарських засобів незалежно від регуляторного шляху схвалення або статусу ексклюзивності на ринку, тобто вона застосується до референтних біологічних лікарських засобів (далі — референтний лікарський засіб), до «подібних біологічних лікарських засобів» (далі — біосиміляри) та до препаратів, що містять таку ж або дуже подібну активну субстанцію/діючу речовинуN, але не схвалені як біосиміляри (наприклад інші версії інтерферону бета-1а, фактор VIII або імуноглобулін людини нормальний) (далі — пов’язані препарати).
Біосиміляр є біологічним лікарським засобом, що містить версію активної субстанції/діючої речовиниN вже схваленого референтного лікарського засобу в ЄЕЗ/УкраїніN, та який продемонстрував подібність до референтного лікарського засобу, виходячи з характеристик якості, біологічної активності, безпеки та ефективності на основі загального порівняльного аналізу (див. Настанову щодо подібних біологічних лікарських засобів99).
Нормативно-правові вимоги з фармаконагляду та ННПФ застосовуються до біологічних лікарських засобів, так само як до інших лікарських засобів. Рекомендації цієї частини ННПФ не замінюють жодних з них. Проте, як описано нижче, біологічні лікарські засоби, пов’язані з декількома специфічними завданнями при здійсненні фармаконагляду. Тому ця частина «Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби» (далі — СП ІІ) слід розглядати та дотримуватися разом з модулями ННПФ, що пов’язані з процесом, розробкою та імплементацією фармаконагляду для біологічних лікарських засобів для гарантування, що ці вимоги дотримуються. Глава А СП ІІ описує декілька зі спеціальних питань та вимог, глава Б СП ІІ надає настанову щодо розгляду їх в контексті основних процесів з фармаконагляду, що описані в ННПФ, та глава В СП ІІ надає вказівки стосовно функціонування мережі ЄС/УкраїниN.
Хоча існує окрема настанова щодо відстеження донора медичних субстанцій, які отримані з крові та плазми (див. Настанову щодо лікарських засобів, отриманих з плазми100), загальні принципи фармаконагляду та відстежування пацієнта у цій частині ННПФ також застосуються до таких лікарських засобів. Відповідні настанови, що мають розглядатися, включають Настанову щодо оцінки імуногенності терапевтичних білків біотехнологічного походження, Настанову щодо порівнянності біотехнологічних лікарських засобів після зміни у виробничому процесі, Настанову щодо подібних біологічних лікарських засобів, що містять білки біотехнологічного походження як активну субстанцію: неклінічні та клінічні питання, Настанову щодо подібних біологічних лікарських засобів, які містять біотехнологічні білки як активну субстанцію: питання якості та Настанову щодо процесу валідації для виробника активних субстанцій біотехнологічного походження та Дані, які мають надаватися при регуляторному поданні документів101. Інші настанови з вимогами щодо здійснення фармаконагляду для біосимілярів слід також розглянути.
У цій частині ННПФ на всі застосовані нормативно-правові вимоги посилаються таким чином, який пояснюється у вступній записці до ННПФ та, як правило, визначаються модальним дієсловом «мають». Керівництво щодо імплементації нормативно-правових вимог надається з використанням модального дієслова «повинні».
СП.ІІ.А.1. Аспекти фармаконагляду специфічні для біологічних лікарських засобів
На відміну від хімічно синтезованих лікарських засобів, що можуть, як правило, легко характеризуватися та відтворюватися різними виробниками, біологічні активні субстанції є складними молекулами, що виробляються, як правило, з використанням складних виробничих процесів з багатьма висхідними та низхідними етапами, що формують загальний профіль безпеки, якості та ефективності. Виробничий процес (включаючи вибір лінії клітин, сировину або вихідні матеріали, процес ферментації та очищення, кінцеве формування) є настільки ж визначальним фактором якості препарату, як активна субстанція, та незначні зміни у будь-якому виробничому етапі можуть вплинути на якість препарату та, як наслідок, на безпеку та ефективність. Досягнення в біотехнологічних та аналітичних науках дозволить забезпечити кращу характеристику та контроль біологічних препаратів, але саме їх фундаментальна складність створює спеціальні вимоги до біологічних лікарських засобів при здійсненні фармаконагляду.
СП.ІІ.А.1.1. Імуногенність
Як у будь-якого лікарського засобу, профіль безпеки біологічного лікарського засобу визначається частково прямими або непрямими фармакологічними, включаючи імуногенні, властивостями активної субстанції (наприклад підвищена імуномодуляція або імуносупресія), допоміжних речовин та домішок, пов’язаних з процесом (наприклад, білки клітини хазяїна) або сприйнятливістю, пов’язаною з «хазяїном» або захворюванням (наприклад алергічні реакції, індуковані лікарським засобом, аутоімунні, запальні явища). Щодо біологічних та небіологічних лікарських засобів основні принципи оцінки співвідношення користі/ризику на основі процесу, що описані в модулях ННПФ І–ХVІ, застосовуються до потенційних або ідентифікованих ризиків. Проте внаслідок більш складної природи біологічні лікарські засоби представляють більший потенційний ризик імуногенності порівняно з небіологічними лікарськими засобами та вимагають спеціального розгляду. Це детально розглядається в Настанові щодо оцінки імуногенності терапевтичних білків біотехнологічного походження102.
У більшості випадків імуногенність до біологічного лікарського засобу не буде мати клінічного значення, наприклад, тимчасова поява антитіл, та не вплине на співвідношення користі/ризику. Проте, у деяких випадках імуногенність може призвести до серйозних загрозливих для життя реакцій.
В рамках цієї глави «імуногенність» стосується небажаної імунної відповіді, яка вважається потенційно клінічно значимою та може вимагати спеціального фармаконагляду та дій з управління ризиком для лікарського засобу.
Джерела імуногенності біологічних препаратів є багатофакторними та включають один або більше факторів, пов’язаних з лікарським засобом (наприклад, вибір лінії клітин, пост-трансляційні зміни та зміни в 3D-структурі під час низхідної обробки, домішки, вибір контейнерів лікарського засобу), фактори, пов’язані з лікуванням (наприклад шлях введення, частота прийому дози) та фактори, пов’язані з пацієнтом або захворюванням (наприклад генетичні дані, супутнє лікування, характер основного захворювання та імунний статус).
Клінічні наслідки імуногенності можуть включати часткову або повну втрату ефективності препарату внаслідок індукування нейтралізуючих антитіл, змінену фармакокінетику внаслідок зв’язування з антитілами, загальні імунні ефекти, такі як анафілаксія, створення імунних комплексів та потенційне індукування перехресної реактивності з ендогенними білками або іншими аутоантитілами.
Спеціальна оцінка імуногенності вимагається під час розробки та до реєстрації біотехнологічних препаратів (див. Настанову з оцінки імуногенності терапевтичних білків біотехнологічного походження103). Проте неклінічні моделі та аналітичні методи/біоаналізи не можуть, як правило, передбачити імуногенність у людини. Більш того, обмежений розмір вибірки дореєстраційних досліджень або рідкісність захворювання, що має лікуватися, може не дозволити оцінити рідкісні наслідки імуногенності до реєстрації. Невпевненість відносно імуногенності повинна відображатися в ПУР (див. СП.ІІ.В.1.) та вимагає певної діяльності або нагляду в післяреєстраційний період, якщо необхідно.
Зокрема, для біосимілярів, початкове реєстраційне посвідчення ґрунтується на продемонстрованій та прийнятній біоподібності якості, безпеки та ефективності відповідно до загального порівняльного дослідження. Це дослідження сплановане таким чином, щоб виключити будь-які відповідні відмінності між біосиміляром та референтним лікарським засобом. Проте в Настанові щодо подібних біологічних лікарських засобів, що містять білки біотехнологічного походження як активну субстанцію: неклінічні та клінічні питання104 зазначається, що «…даних, отриманих за результатами проведення дореєстраційних клінічних досліджень є, як правило, недостатньо для визначення рідкісних побічних ефектів. Тому слід проводити детальний моніторинг клінічної безпеки біосимілярів на постійній основі у післяреєстраційний період, включаючи безперервну оцінку співвідношення користі/ризику…». Після характеристики імуногенності на момент початкового реєстраційного посвідчення наступне завдання стосовно будь-якого біологічного лікарського засобу стосується змін у виробництві або якості, та факт, що імуногенність може потенційно бути внесена або змінена в будь-який час після реєстрації, потенційно призведе до зміненого профілю безпеки або ефективності лікарського засобу.
СП.ІІ.А.1.2. Варіабельність виробництва
Заявники (власники реєстраційних посвідчень) на лікарські засоби часто вносять зміни у виробничий процес своїх лікарських засобів після реєстрації. Це відбувається з багатьох причин, включаючи, наприклад, зміни у вихідних матеріалах, обладнанні або регуляторні вимоги.
Виробничі зміни можуть бути більш складними для біологічних лікарських засобів. Вони мають підтверджуватися порівняльним дослідженням та подаватися заявником (власником реєстраційного посвідчення) як зміна або як розширення до реєстраційного посвідчення для визначення, що лікарські засоби до та після зміни є порівнянними, та що якість, безпека та ефективність не зазнали негативних змін. Відповідно до Настанови щодо порівнянності лікарських засобів біотехнологічного походження після зміни у виробничому процесі105 демонстрація порівнянності є багатоетапним процесом, починаючи з досліджень якості. Якщо заявник (власник реєстраційного посвідчення) може надати доказ порівнянності шляхом фізико-хімічних/аналітичних та біологічних аналізів, тоді доклінічні або клінічні дослідження лікарського засобу після змін не вимагаються. У інших випадках процес зміни може вимагати підтверджувальних доклінічних та/або клінічних даних та спеціальних вимог з фармаконагляду. Пункт (17) постанови (ЄС) № 1235/2010 [4], вказує, що « … ПУР, як правило, вимагаються для нових активних субстанцій, біосимілярів, лікарських засобів для застосування у педіатрії та для лікарських засобів для застосування у людини, що включає значну зміну в реєстраційному посвідченні, включаючи новий виробничий процес лікарського засобу біотехнологічного походження». Положеннями Порядку [2] та Порядку проведення експертизи реєстраційних матеріалів [7] визначено вимоги до подання ПУРN. В Настанові щодо оцінки імуногенності терапевтичних білків біотехнологічного походження106 також посилаються на необхідність розглянути ПУР, якщо можливі зміни в імуногенності (див. СП ІІ.А.1.1.). Висновок стосовно того, що є «значною» зміною у виробничому процесі можна робити лише у кожному конкретному випадку на основі дослідження порівнянності.
Більшість виробничих змін повинні призводити до випуску порівнянного препарату та необхідність, масштаб та характер доклінічних та клінічних порівняльних досліджень буде визначатися у кожному конкретному випадку. Проте буде неможливо прогнозувати імуногенність лише на основі фізико-хімічних/аналітичних та біологічних аналізів, підтверджуючі клінічні дослідження (якщо вимагаються) не завжди зможуть виявити рідкісні наслідки будь-якої зміненої імуногенності до схвалення змін у виробництві. Тому біологічні лікарські засоби потенційно зумовлені цим динамічним профілем якості з потенціалом виникнення серйозних нових ризиків (безпека або ефективність) у будь-який момент під час життєвого циклу препарату внаслідок змін у якості препарату або характеристиках (що можуть також бути пов’язані з обробкою препарату та характеристиками пацієнта).
Ці потенційні зміни є важливими не лише щодо лікарського засобу (наприклад зміна в специфікаціях якості з часом), але також щодо лікарських засобів з тією самою МНН. Протягом тривалого післяреєстраційного періоду референтний лікарський засіб, біосиміляри та пов’язані препарати можуть потенційно проявляти різні профілі безпеки, оскільки ці препарати еволюціонують протягом їх життєвого циклу. Чи був імплементований оновлений ПУР (див. СП.ІІ.Б.1.2.) у підтримку затвердження даної виробничої зміни, це підкреслює важливість для біологічних лікарських засобів тривалого, протягом життєвого циклу, фармаконагляду та управління ризиками для швидкого відстеження будь-яких важливих змін у безпеці та ефективності препарату з часом.
СП.ІІ.А.1.3. Стабільність та холодовий ланцюг
Існують суворі контролі процесу для біологічних лікарських засобів для гарантування, що виробничі процеси та стандарти залишаються в рамках затверджених специфікацій. Після виробництва та випуску підтримується загальна стабільність препарату, дотримуючись відповідних умов зберігання та транспортування, холодового ланцюга та належних практик розповсюдження (див. Настанову з належної практики розповсюдження лікарських засобів для людини107).
Більше ніж для небіологічних препаратів недотримання цих процесів та стандартів може вплинути на стабільність та якість біологічних препаратів, що, в свою чергу, може внести або змінити імуногенність (див. СП.ІІ.А.1.1) або забруднення. Хоча дуже рідко, зокрема для препарату, який вже був випущений, такі дефекти та відхилення, як правило, вплинули б на певні серії.
Тому фармаконагляд протягом життєвого циклу на рівні препаратів та серій є важливим питанням для біологічних препаратів (див. СП.ІІ.А.1.4.).
СП.ІІ.А.1.4. Відстежуваність препарату
Внаслідок виробничої варіабельності з часом у післяреєстраційний період у межах та між препаратами з подібними активними субстанціями, основною вимогою для фармаконагляду біологічних лікарських засобів є необхідність гарантувати постійну відстежуваність лікарського засобу та серії при медичному застосуванні. Це особливо важливо для біологічних лікарських засобів порівняно з хімічно синтезованими внаслідок більш властивої варіабельності характеристик препарату.
Чи то референтний лікарський засіб, біосиміляр чи пов’язаний препарат, важливо, щоб різні лікарські засоби з тим самим МНН могли одразу розрізнятися з метою, щоб нові питання з безпеки, що виникли відносно лікарського засобу, та імуногенність (див. СП.ІІ.А.1.1.) швидко відстежувалися та оцінювалися протягом життєвого циклу лікарського засобу, та постачання могло відстежуватися до місць та пацієнтів, якщо необхідно. Оскільки будь-який такий лікарський засіб, як правило, зберігає ту саму торговельну назву після значної зміни у виробничому процесі, відстежуваність серії є важливим аспектом, який має розглядатися при будь-яких пов’язаних оновленнях ПУР (див. СП.ІІ.Б.1.).
Оскільки торговельна назва лікарського засобу та інформація про серію, включена в упаковку, ця інформація має записуватися та повідомлятися на всіх рівнях в мережі постачання від випуску виробником до виписування рецепта, відпуску та застосовування пацієнтом. Біологічні лікарські засоби складають дуже різноманітну сукупність препаратів для широкого спектру терапевтичних областей та клінічних умов для виписування рецептів, відпуску, постачання та застосування однаково різноманітні. Тому відстежуваність має бути повністю інтегрована в різних закладах охорони здоров’я та в інфраструктуру, що може варіювати між лікарськими засобами та між країнами, наприклад, інфраструктура для запису електронних даних та пов’язування записів. Більшість лікарських засобів буде постачатися в лікарні та, якщо не існує зв’язку записів, мають застосовуватися інші методи для збору інформації щодо експозиції, наприклад, сканування стандартного штрих-коду на всіх етапах в ланцюгу постачання. Національні органи з охорони здоров’я повинні також працювати у напрямку кращої інтеграції та автоматизації інформації щодо виписування рецептів.
Слід зазначити, що практика виписування рецептів та взаємозамінності лікарських засобів, та зокрема переведення та зміщення між біологічними лікарськими засобами, не входить до цієї частини ННПФ. Торговельна назва лікарського засобу та номер серії біологічного лікарського засобу, що застосовується, мають записувати медичні спеціалісти та надавати пацієнту. Це особливо важливо у випадку, коли різні лікарські засоби, що містять одні й ті ж активні субстанції, одночасно наявні на фармацевтичному ринку та сам пацієнт застосовує взаємозамінність.
СП.ІІ.Б. Структури та процеси
СП.ІІ.Б.1. Система управління ризиками
Всі заяви на реєстрацію, що подані в ЄС після 2 липня 2012 р. (за централізованою процедурою) або 21 липня 2012р. (за процедурою взаємного визнання або децентралізованою процедурою) повинні містити ПУР, що має бути схвалений уповноваженими органами до видачі реєстраційного посвідчення. Подання ПУР або його оновлення також вимагається для лікарських засобів, щодо яких початкова заява була подана до вищевказаних дат, якщо вноситься значна зміна до реєстраційного посвідчення, включаючи новий виробничий процес біотехнологічних лікарських засобів (пункт (17) Постанови (ЄС) № 1235/2010 [4]) (див. ННПФ модуль V). В Україні ПУР є складовою матеріалів реєстраційного досьє на лікарські засоби, що подаються на реєстрацію відповідно до положень Порядку проведення експертизи реєстраційних матеріалів [7]N.
Як загальний принцип будь-яке оновлення ПУР референтного лікарського засобу у післяреєстраційний період повинно подібним чином застосовуватися до відповідних біосимілярів та відповідних лікарських засобів і навпаки, окрім випадків, коли обґрунтовано, наприклад, якщо наявна інформація вказує, що клінічне питання, яке вимагає оновлення, стосується певного лікарського засобу (тобто не стосується активної субстанції або допоміжних речовин). Для біосиміляру вимагаються всі частини ПУР, за виключенням частини ІІ модуля СI «Епідеміологія цільової популяції» ПУР.
СП.ІІ.Б.1.1. Зміст плану управління ризиками
СП.ІІ.Б.1.1.1. ПУР частина І «Огляд лікарського засобу»
Походження активної субстанції/діючої речовиниN біологічного лікарського засобу слід включити як важливу інформацію про його склад (див. модуль V ННПФ щодо біологічних лікарських засобів).
СП.ІІ.Б.1.1.2. ПУР, частина ІІ «Специфікації з безпеки»
СП.ІІ.Б.1.1.2.1. ПУР, модулі СVII «Ідентифіковані та потенційні ризики» та СVIIІ «Резюме питань з безпеки»
Відповідно до вимог модуля V НППФ специфікації з безпеки повинні включати важливі ідентифіковані ризики, важливі потенційні ризики та відсутню інформацію.
Потенціал імуногенності та відповідні клінічні наслідки (див. СП.ІІ.А.1.1.) повинні повністю оцінюватися та обговорюватися в рамках заяви на реєстрацію (або внесення зміни) у відповідних розділах «Резюме клінічної безпеки» заяви на реєстрацію. Імуногенність може виникнути протягом життєвого циклу біологічного лікарського засобу, але це не є питанням з безпеки. Її слід включити до специфікації з безпеки ПУР лише, якщо зроблено висновок після обговорення щодо необхідності її класифікації як важливого ризику (ідентифікованого або потенційного) або як відсутньої інформації. У таких випадках це питання слід визначити якомога точніше (включаючи будь-які спеціальні потенційні клінічні ризики при визначенні випадку) для того, щоб могли розроблятися певні заходи з фармаконагляду для розгляду непевності (див. СП.ІІ.Б.1.1.3.). Настанова щодо оцінки імуногенності терапевтичних білків біотехнологічного походження108, а також будь-яка відповідна наявна Настанова щодо препарату/класу з оцінки імуногенності (наприклад Настанова щодо оцінки імуногенності моноклональних антитіл, які призначені для клінічного застосування in vivo109) повинні використовуватися з метою визначення найбільш відповідної стратегії для подальшої оцінки потенційного ризику.
У випадку значної зміни у виробничому процесі, що вимагає зміни в ПУР (див. СП.ІІ.Б.1.2.), потенційну імуногенність та клінічні наслідки слід включити до специфікації з безпеки. Якщо жодного спеціального потенційного клінічного питання не було визначено (на відміну від значної виробничої зміни з непевними клінічними наслідками), щодо відсутньої інформації, яка вказана в оновленій специфікації з безпеки, слід зробити посилання на «імуногенність після значної зміни у виробничому процесі». Для біосимілярів та відповідних лікарських засобів резюме питань з безпеки повинно принаймні бути таким, як у референтного лікарського засобу, якщо не обґрунтовано інакше. Таке обґрунтування може включати ситуації, коли певний ризик, що пов’язаний з референтним препаратом, як відомо, був пов’язаний з компонентом, виробничим процесом (на відміну від активної субстанції) або іншим фактором, що не пов’язаний з біосиміляром або відповідним лікарським засобом, або якщо елементи специфікації з безпеки є специфічними для певного застосовування (наприклад показання або шлях введення), що є відсутнім для деяких лікарських засобів (але слід розглянути можливість застосування не за показанням).
В ПУР слід включити важливі ризики або відсутню інформацію стосовно непевностей, що визначені в дослідженні порівнянності, відносно серйозності та частотності побічних реакцій біосимілярів порівняно з референтним лікарським засобом, та слід оцінити необхідність додаткових заходів з фармаконагляду або заходів з мінімізації ризиків.
Будь-які інші запропоновані відмінності в специфікаціях з безпеки біосиміляру порівняно з референтним лікарським засобом слід належним чином обґрунтувати на основі результату загального порівняльного дослідження.
СП.ІІ.Б.1.1.2.2. ПУР модуль СVI «Додаткові вимоги ЄС/Україні щодо специфікації з безпеки»
Для всіх біологічних лікарських засобів слід описати можливість внесення інфекції залишковими продуктами біологічного матеріалу, що використовується у виробничому процесі, а також внесення забруднення під час виробничого процесу та можливості перенесення інфекційних агентів.
СП.ІІ.Б.1.1.3. ПУР частина ІІІ «План з фармаконагляду»
Слід обговорити необхідність та плани постійного відстеження сигналу протягом життєвого циклу лікарського засобу та фармаконагляду, специфічного для лікарського засобу, включаючи питання специфічні для серії, зокрема після значної зміни у виробничому процесі. У цьому контексті план з фармаконагляду повинен включати обговорення клінічних умов застосування лікарського засобу, та як це може вплинути на стандартне записування та повідомлення торговельна назва лікарського засобу та його серії (наприклад, чи застосовується в первинній чи третинній медичній допомозі), та яка додаткова діяльність з фармаконагляду або заходи з мінімізації ризиків можуть вимагатися у підтримку відстежуваності лікарського засобу (наприклад надання наклейок/відривних етикеток, штрих-кодів).
СП.ІІ.Б.1.1.3.1. ПУР частина ІІІ розділ «Рутинна діяльність з фармаконагляду»
У цьому розділі заявник (власник реєстраційного посвідчення) повинен розглянути:
- клінічні умови застосування лікарського засобу та як це може вплинути на записування та повідомлення, торговельна назва лікарського засобу та його серії;
- заходи, що будуть вноситися до стандартного контролю повідомлень про випадки для отримання інформації про торговельну назву лікарського засобу та номер(и) серії (див. також ННПФ модуль VI);
- діяльність з виявлення сигналу, що проводиться для виявлення питань з безпеки, специфічних для серії;
- будь-які побічні явища, що представляють спеціальний інтерес, з визначеннями, що виявлені як важливі потенційні ризики, для яких буде введено спеціальний нагляд з безпеки (див. також ННПФ частину СП І).
СП.ІІ.Б.1.1.3.2. ПУР частина ІІІ розділ «Додаткова діяльність з фармаконагляду»
У цьому розділі заявник (власник реєстраційного посвідчення) повинен розглянути:
- будь-які додаткові заходи, введені разом з національними уповноваженими органами у підтримку відстеження лікарського засобу (наприклад, надання наклейок/відривних етикеток, штрих-коди);
- діяльність, що проводиться для вимірювання вихідних рівнів побічних явищ, що представляють спеціальний інтерес, бажано за показанням, у цільовій віковій групі за лікарським засобом;
- діяльність, що проводиться для постійного моніторингу частоти повідомлень або рівнів побічних явищ, що представляють спеціальний інтерес, на основі наявних даних експозиції, та порівняння таких рівнів з відповідними визначеними вихідними рівнями (використовуючи методи, такі як аналіз «спостережуваних/очікуваних явищ») (див. також частину ННПФ СП І);
- використання існуючих реєстрів пацієнтів або інших джерел даних (або встановлення нового реєстру, якщо існуючі джерела даних є невідповідними) (див. ННПФ модуль VІІI);
- для біосиміляру будь-який спеціальний моніторинг безпеки, що накладається на референтний лікарський засіб або клас лікарського засобу та його важливість для даного лікарського засобу.
Для значних змін у виробничому процесі, які вимагають оновлення ПУР (див. СП.ІІ.Б.1.2.), за умови, що назва препарату, як правило, не змінюється, слід зосередити увагу на фармаконагляді серії протягом узгодженого періоду часу на момент подання документів на внесення зміни у виробництво. Цей період нагляду повинен починатися після схвалення зміни, якщо нові серії з’явилися ринку.
Імуногенність
Якщо імуногенність включена до специфікації з безпеки (див. СП.ІІ.Б.1.1.2), відповідні стратегії для оцінки імуногенності та пов’язаних клінічних наслідків в післяреєстраційний період слід запропонувати як додаткову діяльність з фармаконагляду. При застосуванні принципів оцінки імуногенності слід дотримуватися Настанови з оцінки імуногенності терапевтичних білків біотехнологічного походження110, а також будь-якого відповідної наявної настанови щодо лікарського засобу або класу лікарських засобів щодо оцінки імуногенності (наприклад Настанова щодо оцінки імуногенності моноклональних антитіл, призначених для клінічного застосування in vivo111).
В залежності від характеру будь-якої потенційної імуногенності та даних, які створюють питання, або характеру відсутньої інформації, додаткова діяльність з фармаконагляду повинна мати чітко визначені цілі. План може включати біоаналітичні методи (наприклад аналізи in vitro, серологічні дослідження), доклінічні дослідження, інтервенційні клінічні дослідження або епідеміологічні підходи за даними спостережень. Будь-які аналітичні та клінічні кінцеві точки, що стосуються потенційного ризику, включаючи ті, що стосуються безпеки та ефективності (наприклад з метою оцінки потенційних ефектів нейтралізуючих антитіл), слід чітко визначити у підтримку їх характеристики при пасивному нагляді (наприклад шляхом цільового спостереження), додатковій діяльності з фармаконагляду або епідеміологічних дослідженнях.
З цих причин при визначенні оптимальної стратегії для оцінки імуногенності в ПУР слід застосувати багатодисциплінарний підхід із залученням експертів з якості, неклінічних та клінічних досліджень, фармаконагляду та епідеміології.
Якщо визначено новий клінічний ризик, що може мати імуногенну етіологію, це слід досконально дослідити при будь-якій наступній оцінці ризику. Якщо ризик є специфічним для певного лікарського засобу або серії, потенційна основна причина повинна оцінюватися з метою визначення можливості мінімізації ризику або елімінації (наприклад етапи виробництва).
Післяреєстраційні дослідження з безпеки
Повинен використовуватися найбільш оптимальний дизайн дослідження, зважаючи на мету ПДБЛЗ (див. ННПФ модуль VIII). Якщо має використовуватися існуючий реєстр або має встановлюватися новий реєстр, бажано включити порівняльну або неекспоновану групу. Заохочується створення об’єднаних реєстрів захворювань.
Біосиміляри та пов’язані препарати
Будь-який спеціальний моніторинг з безпеки, що встановлюється для референтного лікарського засобу або класу лікарських засобів, слід відповідно розглянути в плані з фармаконагляду, якщо не обґрунтовано інакше (наприклад, якщо питання з безпеки є специфічним для лікарського засобу порівняння та не включені до специфікації з безпеки біосиміляру або пов’язаного препарату). Якщо застосовується та можливо, уповноважені органи повинні заохочувати заявників (власників реєстраційних посвідчень) біосимілярів та пов’язаних препаратів брати участь у будь-якому фармакоепідеміологічному дослідженні, що проводиться для референтного препарату, якщо не обґрунтовано інакше (див. СП.ІІ.Б.1.1.12).
СП.II.Б.1.1.4. Частина V ПУР «Заходи з мінімізації ризику»
Оцінка будь-якого нового клінічного ризику, що пов’язаний з біологічним засобом, повинна включати аналіз основної причини з метою оцінки можливості мінімізації або усунення ризику шляхом, наприклад, аналітичних досліджень.
В якості загального принципу для покращення відстеження біологічних лікарських засобів усі короткі характеристики/інструкції для медичного застосуванняN лікарського засобу на біологічні засоби (також з відповідним формулюванням у листку-вкладишу) повинні включати твердження, що торговельна назва та номер серії лікарського засобу, що вводиться, слід чітко зазначати у картці пацієнта. Відповідне формулювання слід також включити до релевантного навчального матеріалу, безпосередніх повідомлень для медичних спеціалістів (див. СП.II.Б.6) та рекламного матеріалу щодо лікарського засобу, якщо необхідно. Застосування інших інструментів, таких як наклейки/відривні етикетки на пакуванні лікарських засобів слід також розглядати для полегшення чіткого запису у картці пацієнта та надання інформації пацієнтам. Якщо необхідно, слід також рекомендувати застосування доступних технологій та інфраструктури для сканування штрих-кодів.
Діяльність з мінімізації ризику, що має місце стосовно референтного лікарського засобу, у принципі повинна включатися до ПУР на біосиміляри та пов’язані засоби та навпаки. Будь-яке відхилення від цього (наприклад коли мінімізація ризику стосується конкретно референтного лікарського засобу) слід обґрунтувати.
СП.II.Б.1.2. Оновлення ПУР у зв’язку зі змінами у виробництві
СП.II.Б.1.2.1. Потенційний вплив змін у виробництві
Якщо при оцінці порівнянності визначають потенційний вплив змін у виробництві у контексті клінічної релевантності, зміна вимагає подання оновленого ПУР, якщо тільки інше не обґрунтовано. Це обґрунтування слід робити залежно від конкретної ситуації.
Неможливо надати певні інструкції стосовно того, що може становити клінічно релевантний вплив зміни у виробництві у кожній ситуації, таке рішення необхідно приймати на підставі результатів порівняння або іншої оцінки якості або клінічної оцінки, що підтверджує зміну у процесі, а також будь-які релевантні прецеденти або досвід.
Навіть незначні зміни у процесі виробництва можуть потенційно мати непередбачені значимі клінічні ефекти. У випадках, коли дослідження порівнянності або оцінка не визначають потенційний вплив клінічної релевантності, однак може бути доречним подання оновленого ПУР у зв’язку із зміною у процесі виробництва на підставі аналізу ризику або попереднього досвіду.
СП.II.Б.1.2.2. Аналіз ризику
На підтримку цього процесу та для забезпечення дотримання Декларативної частини (17) Постанови (ЄС) № 1235/2010 [4] усі заяви на внесення зміни до процесу виробництва біологічного лікарського засобу у рутинному порядку повинні включати оновлення ПУР, якщо заявник (власник реєстраційного посвідчення) вважає, що це є необхідним, або за результатами аналізу ризику потенційної значимості та потреби або її відсутності в оновленні ПУР. Цей процес відповідає концепціям, що передбачені у настанові ICH-Q5E (Порівнянність біотехнологічних/біологічних засобів, у процес виробництва яких вноситимуться зміни) та ICH-Q10 (Система фармацевтичної якості)112.
Аналіз ризику, що надає заявник (власник реєстраційного посвідчення), може бути короткою заявою з відповідними обґрунтуваннями або більш складним аналізом, що ґрунтується на доказах, якщо цього вимагає характер зміни (зокрема, якщо існує прецедент для типу зміни, що призводить до клінічно значимого впливу).
СП.II.Б.1.2.3. Оновлення ПУР
Якщо заявник (власник реєстраційного посвідчення) вважає, що оновлення ПУР є необхідним, його слід надати разом з заявою, що є підставою для такого оновлення. Інакше, якщо уповноважений орган робить висновок про необхідність оновлення ПУР, він повинен надавати заявнику (власнику реєстраційного посвідчення) рекомендації щодо характеру змін, що очікуються у ПУР. Оновлення ПУР слід надавати якомога раніше для уможливлення його схвалення у контексті внесення змін до процесу виробництва.
При оновленні ПУР слід звернути увагу на специфікацію з безпеки, план фармаконагляду та заходи з мінімізації ризику. Для випадків, коли при оцінці порівнянності виявляється потенційний вплив зміни у виробництві у контексті клінічної релевантності, особливу увагу слід приділяти опису в якості рутинної діяльності з фармаконагляду, як протягом відповідного періоду часу після зміни у виробництві можна проводити специфічну для певної серії оцінку для легкого відрізнення лікарських засобів до та після внесення зміни.
Після оновлення ПУР у наступних РОЗБ (див. СП.II.Б.3.) слід особливо оцінювати повідомлення та будь-яку іншу інформацію, що може вказувати на новий клінічний ризик, пов’язаний із зміною у процесі. Ця оцінка повинна стосуватися певної проблеми, що включена до будь-якої оновленої специфікації з безпеки ПУР на підставі внесення зміни у виробництво. Також можуть вноситися зміни (та повторно встановлюватися) до циклу подання РОЗБ згідно з оновлюваним ПУР.
СП.II.Б.2. Управління та подання повідомлень про побічні реакції
Вимоги до управління та подання повідомлень про побічні реакції, що описані у Модулі VI ННПФ, застосовуються рівною мірою до біологічних та небіологічних лікарських засобів. Крім того, за допомогою методів збору інформації та, коли необхідно, за допомогою наступного відстеження (follow up) повідомлень про підозрювані побічні реакції уповноважені органи повинні впевнитися, що вжиті всі відповідні заходи для чіткого визначення будь-якого призначеного, відпущеного або проданого на їх території біологічного засобу, який є предметом повідомлення про підозрювану побічну реакцію, приділяючи особливу увагу торговельній назві лікарського засобу та номеру серії (ст. 102(е) Директиви 2001/83/ЄC [1]) [6], положення Порядку [2]N). При поданні повідомлень про підозрювані побічні реакції уповноважені органи та заявники (власники реєстраційного посвідчення) повинні надавати усю наявну інформацію щодо кожного окремого випадку (див. Модуль VI ННПФ), включаючи торговельну назву лікарського засобу та номер(и) серії (ст. 28(3)h) ІП 520/2012 [6]), положення Порядку [2]N). Для цього держави ЄС/УкраїнаN та заявники (власники реєстраційних посвідчень) повинні заохочувати медичних спеціалістів до надання пацієнтам та особам, які за ними доглядають, інформації про торговельну назву та номер(и) серії будь-якого біологічного лікарського засобу, що вводиться, незалежно від місця призначення, постачання або введення, а також технічної інфраструктури, що може існувати. Уповноважені органи та заявники (власники реєстраційних посвідчень) повинні заохочувати репортерів до включення інформації щодо торговельної назви лікарського засобу та номера серії. На місцях слід запровадити процедуру відстеження для отримання інформації про номер серії, коли він не зазначений у первинному повідомленні. Слід дотримуватися карти бізнес-процесу, що включена до Модуля VI ННПФ.
Якщо у ПУР для біологічного лікарського засобу зазначаються певні заходи, що слід вжити з метою збору інформації про визначені клінічні кінцеві точки (наприклад кінцеві точки імуногенності), окрім зазначення торговельної назви лікарського засобу та інформації про серію, можна розробити та викласти певні лабораторні/аналітичні дані, визначення випадків та анкети та посилатися на них у ПУР для відстеження побічних явищ, що представляють спеціальний інтерес.
Коли заявники (власники реєстраційних посвідчень) та уповноважені органи вважають, що використання їх вебсайту полегшить збір повідомлень про підозрювані побічні реакції завдяки наданню форм повідомлень або відповідних контактних даних для безпосереднього інформування (див. Модуль VI ННПФ), будь-які такі заходи слід застосовувати для інформування, сприяння та полегшення отримання інформації щодо торговельної назви та серії лікарського засобу у повідомленнях про побічні реакції.
СП.II.Б.3. Регулярно оновлюваний звіт з безпеки
Вимоги до оцінки сигналу як частини РОЗБ у Модулі VII ННПФ застосовуються рівною мірою до біологічних та небіологічних лікарських засобів (див. СП.II.В.1.2. для оцінки РОЗБ на біосиміляри).
СП.II.Б.3.1. Розділ РОЗБ «Оцінка експозиції та схеми застосування»
Для підтримки процесів управління сигналом (див. СП.II.Б.4.) заявники (власники реєстраційного посвідчення) повинні докласти усіх зусиль для отримання даних про реальне використання лікарського засобу (а не покладатися виключно на зведені дані про реалізацію). Джерела реальних даних важливі для оцінки загальної експозиції та схем застосування.
СП.II.Б.3.2. Розділи РОЗБ «Огляд сигналів (нові, поточні та закриті)» та «Оцінка сигналів та ризиків»
Рекомендації, що надані у СП.II.Б.4., слід застосовувати до процесу оцінки сигналу у РОЗБ, тобто слід надати висновки щодо кожного конкретного випадку стосовно того, чи застосовується сигнал до окремого лікарського засобу або до усіх лікарських засобів з тією самою діючою речовиною. У зв’язку з положеннями СП.II.Б.1.2. та відповідно до Керівництва щодо порівнянності біотехнологічних лікарських засобів після внесення зміни до процесу виробництва113 після значної зміни у процесі виробництва (що, як правило, вимагатиме подання оновленого ПУР) у РОЗБ слід особливо оцінювати повідомлення та будь-яку іншу інформацію, що може вказувати на новий клінічний ризик, пов’язаний із зміною у процесі виробництва. Для проведення такої оцінки необхідні дані про специфічні для певної серії моделі експозиції. Це слід представляти у контексті певної проблеми, що включена до будь-якої оновленої специфікації з безпеки ПУР внаслідок зміни у виробництві.
Після значної зміни у процесі виробництва цикл подання РОЗБ може також змінюватися (або відновлюватися) згідно з оновлюваним ПУР (тільки у випадку, коли це важливіше за вимоги гармонізованого циклу щодо біосимілярів та пов’язаних засобів).
СП.II.Б.4. Управління сигналом
Вимоги для управління сигналом у Модулі IX ННПФ застосовуються рівною мірою до біологічних та небіологічних засобів. Так само, як усі інші лікарські засоби, біологічні засоби вимагають постійного фармаконагляду для виявлення та оцінки потенційних нових клінічних ризиків (безпеки або ефективності), що можуть виникнути під час життєвого циклу лікарського засобу. Однак це особливо важливо для біологічних лікарських засобів з причин, що описані у СП.II.A.1., зокрема завдяки внутрішній мінливості у процесі виробництва, що може потенційно змінити імуногенність лікарського засобу та викликати клінічні наслідки.
Тому виявлення сигналу для біологічних лікарських засобів повинне бути специфічним до лікарського засобу, а також до діючої речовини. Усі етапи управління сигналом слід здійснювати на рівні торговельної назви лікарського засобу, а також діючої речовини. У випадку сигналу слід докласти будь-яких зусиль для ідентифікації будь-якої загальної основної причини, такої як серія.
Процеси повинні бути, зокрема, чутливими для виявлення будь-яких гострих та серйозних нових ризиків, що можуть виникнути після зміни у процесі виробництва або якості біологічного лікарського засобу, а також важливих відмінностей між серіями одного й того самого засобу (це особливо важливо після внесення значної зміни до процесу виробництва, беручи до уваги, що торговельна назва лікарського засобу, зазвичай, не змінюється). Важливі відмінності між референтними засобами та біосимілярами або пов’язаними засобами слід виявляти під час життєвого циклу лікарського(их) засобу(ів) на підставі доступної інформації. Протягом життєвого циклу лікарського засобу слід проводити моніторинг будь-яких клінічних наслідків імуногенності, що може потенційно виникнути (як теоретичного ризику).
Інформація про експозицію у післяреєстраційний період необхідна для управління сигналом для біологічних лікарських засобів, але біологічні лікарські засоби часто призначаються або відпускаються у стаціонарах, та необхідна інформація про експозицію може бути недоступною у базах даних популяційного рівня. Заявники (власники реєстраційного посвідчення) повинні докласти усіх зусиль для отримання даних про актуальне використання, специфічне для лікарського засобу (див. СП.II.Б.3) та застосовувати усі методи та джерела даних для отримання надійної та оновленої інформації. Дані про чисельність групи ризику для підрахунку показника частоти виникнення побічних реакцій та дані про підозрювані побічні реакції (див. Модуль IX ННПФ) слід аналізувати у підтримку постійного виявлення сигналу та, зокрема, виявлення будь-яких очевидних змін у частоті повідомлень про підозрювані побічні реакції або тенденцій, що можуть вказувати на нові сигнали (зокрема після змін у виробництві). Деякі діючі речовини або лікарські засоби можуть також вимагати підвищеної частоти моніторингу даних та значна зміна у процесі виробництва біологічного засобу може, в індивідуальному порядку, обґрунтувати специфічні заходи з виявлення сигналу (див. Модуль IX ННПФ). Будь-які такі вимоги слід зазначити у ПУР (див. СП.II.Б.1.1.3 та СП.II.Б.1.2). За необхідності слід також брати до уваги постійний аналіз непропорційності та методи «порівняння того, що спостерігається, з тим, що очікується» (див. Настанову «Питання, специфічні для лікарського засобу або популяції, частина I: Вакцини для профілактики інфекційних хвороб», Модуль IX ННПФ та Настанову з методологічних стандартів у фармакоепідеміології Європейської мережі центрів фармакоепідеміології та фармаконагляду114).
Будь-який сигнал слід оцінювати у контексті даних про експозицію, специфічних для певної серії, включаючи номери/коди доставлених або проданих серій, їх розмір та регіони або країни, куди були доставлені відповідні серії. Запровадження посилених процесів для рутинного моніторингу полегшить більш раннє виявлення нових ризиків та змін у безпеці або якості лікарських засобів з часом.
Для нових сигналів необхідно приймати рішення в індивідуальному порядку стосовно того, може чи ні стосуватися сигнал лікарського засобу, що розглядається, або усіх лікарських засобів з однаковою діючою речовиною. Однак у якості запобіжного заходу, недостатній доказ специфічності сигналу, що виявлений стосовно біосиміляру або пов’язаного препарату, може обґрунтувати застосування регуляторного заходу стосовно референтного лікарського засобу та навпаки. Будь-який новий клінічний ризик, який, за підозрами, має імуногенну етіологію, слід повністю вивчити для визначення, чи ризик є характерним для лікарського засобу за торговельна назвою або серією, та оцінки його потенційної основної причини з метою визначення можливості мінімізації або усунення ризику (наприклад етапи виробництва).
СП.II.Б.5. Додатковий моніторинг
Біологічні засоби, що були зареєстровані після 1 січня 2011 р., слід включати до переліку лікарських засобів, що підлягають додатковому моніторингу (ст. 23(1)(b) Регламент (ЄС) № 726/2004 [5]). Вони видалятимуться з переліку в обов’язковому порядку через 5 років після референтної дати Союзу, якщо тільки період додаткового моніторингу не буде подовжений (ст. 23(3) Регламент (ЄС) № 726/2004 [5]).
СП.II.Б.6. Інформування щодо безпеки
У цій частині ННПФ розглядаються певні аспекти надання інформації щодо біологічних засобів завдяки складним процесам їх виробництва та складу, а також ефектам, що вони спричиняють на організм людини, включаючи можливі побічні реакції, що викликані імуногенністю (див. СП.II.A.1.). У настанові не розглядаються загальні принципи та методи інформування про безпеку. Вони описані у Модулі XV ННПФ та також застосовуються до біологічних засобів.
Слід усвідомлювати специфічні проблеми, які часто виникають у пацієнтів та медичних спеціалістів у зв’язку із застосуванням біологічних засобів, для можливого їх вирішення, інформування про ризики біологічних засобів для представлення складних питань, з наукової, технічної та медичної точки зору, мовою, зрозумілою для пацієнтів та громадськості, а також для спеціалістів-медиків різних спеціальностей. Деякі технічні терміни та концепції вимагають ретельного роз’яснення для забезпечення їх належного розуміння та уникнення соціального розповсюдження ризику115 завдяки, наприклад, біотехнологічним методам, головним чином, технології рекомбінантних ДНК, що не є загальновідомими для неспеціалістів, та які можуть сприйматися деякими особами або популяціями як ті, що є неприродними та негативно впливають на природу, людський організм або гени. Отже, інформацію про процес виробництва та його варіабельність, діючу речовину та її спосіб дії, а також про допоміжні речовини та можливі залишки слід повідомляти пацієнтам та медичним спеціалістам для їх належного розуміння.
Імуногенність є специфічним джерелом побоювань стосовно біологічних лікарських засобів, що призводить до потреб в інформації, які слід послідовно задовольняти. Також може виникати потреба в розгляді в інформаційних документах питань попереднього впливу (експозиції) того самого препарату або лікарських засобів з перехресною імуногенністю. Стосовно біосимілярів консультації з пацієнтами та медичними спеціалістами продемонстрували наявність потреб в інформації про якість, безпеку, ефективність, екстраполяцію, порівнянність та взаємозамінність. Документ ЕМА «Питання та відповіді стосовно біоподібних засобів (подібних біологічних лікарських засобів)»116, що розроблений за результатами консультацій з представниками пацієнтів та медичними спеціалістами, та документ, що містить узгоджену інформацію та розроблений Європейською Комісією «Що Вам необхідно знати про біоподібні лікарські засоби»117, можна застосовувати як джерело роз’яснень при розробці проєктів документів, що містять інформацію, специфічну для певного лікарського засобу.
Підвищення довіри при застосуванні біологічних засобів вимагає не тільки інформування про аспекти, специфічні для певного засобу, але також про існуючі механізми нагляду за безпекою. У інформаційних повідомленнях можна посилатися на відповідне резюме ПУР (див. Модуль V ННПФ). За необхідності можна надавати дані про порівнянність. Сприяння поданню повідомлень про підозрювані побічні реакції вимагає деякої специфічної інформації щодо біологічних лікарських засобів. Пацієнтів та спеціалістів-медиків слід інформувати про те, що побічні реакції можуть виникати, навіть якщо лікарський засіб раніше добре переносився, наприклад, завдяки варіабельності виробництва або змінам у виробництві, або довгостроковим ефектам або відстроченим у часі ефектам, та що важливо надавати повідомлення про побічні реакції, що виникають навіть після довготривалого застосування або з невідомими характеристиками. Відносно повідомлення про побічні реакції та ефективного управління ризиком відстеження є значним завданням в управлінні відповідним застосуванням та при здійсненні фармаконагляду за біологічними засобами (див. СП.II.A.1.4.) та, отже, становить особливе завдання для біологічних засобів по відношенню до пацієнтів та медичних спеціалістів. Тому при інформуванні слід підкреслювати важливість зазначення торговельної назви лікарського засобу (або МНН) та найменування заявника (власника реєстраційного посвідчення) та номера(ів) серії при поданні повідомлень про підозрювані побічні реакції.
Інші специфічні цілі інформування про безпеку біологічних лікарських засобів можуть бути спрямовані на уникнення помилок при зберіганні та транспортуванні, зокрема стосовно дотримання вимог холодового ланцюга (див. СП.II.A.1.3.) та введення, що часто вимагає особливих медичних пристроїв.
Для забезпечення належного розуміння слід проводити консультації з пацієнтами та медичними спеціалістами стосовно проєктів інформаційних документів (див. Модулі XI та XV ННПФ).
СП.II.В. Функціонування мережі ЄС/УкраїниN
СП.II.В.1. Ролі та обов’язки
СП.II.В.1.1. Заявник (власник реєстраційного посвідчення) в ЄС/УкраїніN
Лікарські засоби, що розроблені за допомогою одного з біотехнологічних процесів, що перераховані у Додатку до Регламенту (ЄС) № 726/2004 [5], або ті, що відповідають будь-яким іншим критеріям цього Додатку, слід реєструвати в ЄС за централізованою процедурою.
В Україні положеннями Порядку проведення експертизи реєстраційних матеріалів [7] визначені вимоги до надання матеріалів реєстраційного досьє на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію) в УкраїніN.
СП.II.В.1.1.1. План управління ризиками
Заявник (власник реєстраційного посвідчення) несе відповідальність за подання ПУР згідно з форматом та змістом, що представлені у Модулі V ННПФ та СП.II.Б.1.1., відповідно до положень Порядку [2]N. У випадку значних змін до процесу виробництва слід надавати аналіз ризику та оновлений ПУР (див. СП.II.Б.1.2.).
СП.II.В.1.1.2. Повідомлення про побічні реакції та управління сигналом
При поданні повідомлень про підозрювані побічні реакції заявники (власники реєстраційних посвідчень) повинні надати всю наявну інформацію щодо кожного окремого випадку, включаючи стосовно біологічних засобів, торговельну назва та номер(и) серії лікарського засобу, що вводиться (ст. 28(3)(h) ІП 520/2012) [6], положення Порядку [2]N).
Управління сигналом слід проводити, як описано у Модулі IX ННПФ. Процеси виявлення сигналу для біологічних лікарських засобів повинні бути особливо чутливими до виявлення будь-яких важливих та серйозних ризиків, що можуть виникнути після зміни у процесі виробництва або якості, та важливих відмінностей між серіями одного й того самого лікарського засобу. Будь-які важливі відмінності між референтними лікарськими засобами та біосимілярами або пов’язаними препаратами слід визначати протягом життєвого циклу лікарського(их) засобу(ів) на підставі наявної інформації. Протягом життєвого циклу лікарського засобу слід проводити моніторинг будь-яких клінічних наслідків імуногенності, що може потенційно виникнути (як теоретичний ризик).
СП.II.В.1.1.3. Регулярно оновлюваний звіт з безпеки
Якщо це має значення для інтерпретації даних про безпеку, включаючи новий сигнал про безпеку, що був виявлений за період, що охоплює РОЗБ, заявники (власники реєстраційних посвідчень) повинні включати до РОЗБ резюме релевантної інформації про серії, що постачалися протягом звітного періоду РОЗБ, включаючи номери серій, країни (держави ЄС/УкраїнаN) та регіони, куди такі серії постачалися, розмір серій та будь-яку доступну інформацію про кількість серій, що постачалися до кожної з країн. Слід надати усі припущення, що застосовувалися для розрахунків.
СП.II.В.1.1.4. Додатковий моніторинг
Для біологічних засобів, що включені до переліку лікарських засобів, що підлягають додатковому моніторингу в обов’язковому або факультативному порядку (ст. 23(1) та (1a) Регламент (ЄС) № 726/2004 [5]), обов’язком заявника (власника реєстраційного посвідчення) є здійснення заходів, що описані у Модулі Х ННПФ.
СП.II.В.1.1.5. Інформування про безпеку
Додатково до рекомендацій, що надані у СП.II.Б.6., інформування про безпеку є важливою діяльністю, яку заявник (власник реєстраційного посвідчення) повинен брати до уваги протягом життєвого циклу біологічних лікарських засобів, а також слід додержуватися додаткових рекомендацій, наданих у СП.II.В.2., специфічних для ЄС/УкраїниN.
СП.II.В.1.2. Уповноважені органи у державах ЄС/УкраїніN
СП.II.В.1.2.1. План управління ризиком
Під час оцінки ПУР та його оновлень стосовно біосимілярів до уваги слід брати специфікацію з безпеки, план фармаконагляду та план з мінімізації ризику, що включені до ПУР на референтний лікарський засіб (див. СП.II.Б.1.1.). Слід оцінювати аналіз ризику, що надається заявником (власником реєстраційного посвідчення) на біологічний лікарський засіб у випадку зміни у процесі виробництва, а у висновку слід розглянути необхідність в оновленні ПУР на основі цієї оцінки (див. СП.II.Б.1.2.).
СП.II.В.1.2.2. Повідомлення про побічні реакції
Держави ЄС/УкраїнаN повинні впевнитися за допомогою методів збору інформації та, якщо необхідно, за допомогою наступного відстеження (follow up) повідомлень про підозрювані побічні реакції, що усі відповідні заходи вжиті для чіткого визначення будь-якого біологічного лікарського засобу, що призначався, відпускався або продавався на їх території, що є предметом повідомлення про підозрювану побічну реакцію, звертаючи особливу увагу на торговельну назву лікарського засобу (як визначено у ст.1 (20) Директиви 2001/83/ЄC [1]) та номер серії (ст. 102 (е) Директиви 2001/83/ЄC [1]). Для виконання цього зобов’язання національні уповноважені органи повинні узгоджувати із заявниками (власниками реєстраційних посвідчень), якщо необхідно, систему для забезпечення відстеження біологічних лікарських засобів, що призначаються, відпускаються або реалізуються, для інформування медичних спеціалістів та пацієнтів про необхідність надавати торговельну назву лікарського засобу (або, якщо доцільно, МНН із зазначенням найменування заявника (власника реєстраційного посвідчення) та номер серії при поданні повідомлень про підозрювану побічну реакцію та робити цю інформацію доступною для виявлення сигналу та оцінки повідомлень про окремі випадки.
Держави ЄС/УкраїнаN повинні сприяти на своїй території поданню повідомлень про підозрювані побічні реакції за допомогою альтернативних систем повідомлення, доступних для медичних спеціалістів та споживачів, додатково до форматів, що використовують інтернет-технології (ст. 102 Директиви 2001/83/ЄC [1], положення Порядку [2]N). Якщо форми повідомлень в електронному форматі та форматі, що використовує інтернет-технології, а також інструменти отримання даних розроблені, увагу слід приділити оптимізації здатності цих форм сприяти наданню інформації про лікарський засіб та його серію. Це може включати автоматичні підказки, якщо торговельна назва або серія лікарського засобу не зазначена, або перелік (що випадає) усіх наявних лікарських засобів, коли вибирається певна діюча речовина.
СП.II.В.1.2.3. Регулярно оновлюваний звіт з безпеки
Для оцінки РОЗБ на біосиміляри критично, щоб дані можна було оцінювати паралельно до даних про безпеку, що зібрані для референтного лікарського засобу. Для оцінки РОЗБ на біологічні лікарські засоби, що містять однакову діючу речовину або однакову комбінацію діючих речовин, що мають різні реєстраційні посвідчення, що видані у більш ніж одній державі ЄС/УкраїніN, незалежно від того, чи належать вони одному й тому самому заявнику (власнику реєстраційного посвідчення), слід дотримуватися процедури єдиної оцінки ЄС/УкраїниN для РОЗБ додатково до гармонізації частоти та дат подання РОЗБ згідно з переліком референтних дат ЄС (EURD list)/переліком строків подання РОЗБ (ст. 107e-g Директиви 2001/83/ЄC [1], положення Порядку [2]N). Цю оцінку повинна проводити держава ЄС, яку призначає Координаційна група з процедури взаємного визнання та децентралізованої процедури — для людини (CMDh), якщо жодна з торгових ліцензій, що розглядаються, не видавалася за централізованою процедурою (див. Модуль VII НПФ). В Україні оцінку РОЗБ здійснює Державний експертний центр МОЗN.
СП.II.В.1.3. Європейське агентство з лікарських засобів
Так само, як відносно усіх інших лікарських засобів, ЕМА несе відповідальність за координацію існуючих наукових ресурсів для фармаконагляду за біологічними лікарськими засобами, такою як координація:
- оцінки аналізу ризику, який надає заявник (власник реєстраційного посвідчення) на біологічний лікарський засіб у випадку зміни до виробничого процесу та на підставі цієї оцінки — положення щодо рекомендації про необхідність оновити ПУР (див. СП.II.Б.1.5.);
- процедури єдиної оцінки ЄС для РОЗБ на біологічні лікарські засоби, що містять однакову діючу речовину або однакову комбінацію діючих речовин, коли принаймні одна з торгових ліцензій, що розглядаються, була видана за централізованою процедурою (див. Модуль VII ННПФ).
Для виявлення сигналу стосовно біологічних засобів ЕМА повинно надати доповідачам, провідним державам ЄС та національним уповноваженим органам електронні звіти про моніторинг реакцій та інші вихідні дані та статистичні звіти перш за все на рівні лікарського засобу, ніж на рівні речовини, а також надавати заявникам (власникам реєстраційних посвідчень) відповідну підтримку для моніторингу бази даних EudraVigilance на рівні лікарського засобу.
ЕМА повинно вести та публікувати перелік біологічних лікарських засобів, що підлягають додатковому моніторингу на обов’язковій або факультативній основі (ст. 23 Регламенту (ЄС) № 726/2004 [5]).
СП.II.В.1.3.1. Комітет оцінки ризику у фармаконагляді
PRAC:
- на вимогу Європейської Комісії або компетентного органу держави ЄС, якщо необхідно, надає рекомендацію стосовно необхідності (або її відсутності) включати до переліку лікарських засобів, що підлягають додатковому моніторингу, біологічний лікарський засіб, що повинен відповідати умовам, що встановлені у ст. 23(1а) Регламенту (ЄС) № 726/2004 [5];
- призначає доповідача щодо процедури єдиної оцінки ЄС для РОЗБ на біологічні лікарські засоби, що містять однакову діючу речовину, коли принаймні одне з реєстраційних посвідчень, що розглядаються, було видане за централізованою процедурою (ст. 107e-107g Директиви 2001/83/ЄC [1]) (див. Модуль VII ННПФ);
- приймає рекомендацію стосовно процедури єдиної оцінки ЄС для РОЗБ на біологічні лікарські засоби, як визначено у Переліку EURD list (ст. 107e Директиви 2001/83/ЄC [1]);
- надає рекомендацію стосовно ПУР (ст. 61а(6) Регламенту (ЄС) № 726/2004 [5]); для ПУР на біосиміляри PRAC повинен забезпечити, якщо необхідно, включення до плану фармаконагляду та плану з мінімізації ризику заходів, що подібні заходам, які передбачені для референтного лікарського засобу.
СП.II.В.2. Інформування щодо безпеки стосовно біологічних лікарських засобів в ЄС/УкраїніN
Додатково до рекомендацій, що надані у СП.II.Б.6., стосовно інформування про біологічні лікарські засоби в ЄС/УкраїніN слід враховувати таке.
Робочі деталі про процеси інформування можуть відрізнятися відповідно до різних сценаріїв, що існують у державах ЄС/УкраїніN стосовно застосування біологічних лікарських засобів, зокрема стосовно взаємозамінності та практик взаємозаміни біосимілярів. Ці відмінності слід враховувати під час координації інформування про безпеку в країнах ЄС/УкраїніN, одночасно зберігаючи загальну узгодженість наукових повідомлень щодо співвідношення користі/ризику на території держав ЄС/УкраїниN. Уповноважені органи у державах ЄС/УкраїніN повинні публікувати локальною мовою роз’яснення термінів та концепцій стосовно біологічних лікарських засобів та іншу інформацію для пацієнтів, зокрема оцінки порівнянності, а також надавати медичним спеціалістам інформаційний матеріал. Це повинно полегшити своєчасне інформування пацієнтів з метою забезпечення інформованого терапевтичного вибору (включаючи можливу зміну у лікуванні), адекватну мінімізацію ризику та повідомлення про підозрювані побічні реакції. В основі інформування в ЄС/УкраїніN повинна лежати прозорість стосовно того, як були досягнуті регуляторні рішення, а також стосовно ролей та відповідальності кожної зацікавленої сторони у ЄС/УкраїніN (див. СП.II.В.1.).
Бібліографія
1. Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128 (Директива 2001/83/ЄC Європейського Парламенту та Ради від 6 листопада 2001 року про кодекс Співтовариства відносно лікарських препаратів, призначених для застосування людям//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128).
2. Наказ Міністерства охорони здоров’я України від 27 грудня 2006 р. № 898 «Про затвердження Порядку здійснення фармаконагляду», зареєстровано в Міністерстві юстиції України 29 січня 2007 р. за № 73/13340 (у редакції наказу МОЗ України від 26 вересня 2016 р. № 996).
3. Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010 amending, as regards pharmacovigilance//Official Journal of the European Union. — L 348, 31.12.2010. (Директива 2010/84/ЄС Європейського парламенту та Ради від 15 грудня 2010 р. про внесення поправок в тому, що стосується фармаконагляду//Official Journal of the European Union. — L 348, 31.12.2010).
4. Regulation (EU) 1235/2010 of the European Parliament and of the Council of 15 December 2010 amending, as regards pharmacovigilance of medicinal products for human use, Regulation (EC) 726/2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, and Regulation (EC) 1394/2007 on advanced therapy medicinal products//Official Journal of the European Union. — L 348, 31.12.2010. (Регламент (ЄС)1235/2010 Європейського парламенту та Ради від 15 грудня 2010 р. про внесення змін, щодо фармаконагляду лікарських засобів для людини до Регламенту (EC) 726/2004, що встановлює процедури Співтовариства для вирішення і спостереження лікарських засобів для людини та застосування у ветеринарії та створення Європейського агентства з лікарських засобів, та Регламенту (ЄС) 1394/2007 щодо високотехнологічних лікарських засобів//Official Journal of the European Union. — L 348, 31.12.2010).
5. Regulation (EC) 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency//Official Journal of the European Union. — L 136, 30.04.2004. (Регламент (ЄС) 726/2004 Європейського Парламенту та Ради від 31 березня 2004 року встановлює процедури Співтовариства для вирішення і спостереження лікарських засобів для людини і ветеринарного використання і створення Європейське агентство з лікарських засобів//Official Journal of the European Union. — L 136, 30.04.2004).
6. Commission implementing regulation (EU) 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council//Official Journal of the European Union. — L 159, 20.06.2012. (Імплементаційна постанова Комісії (ЄС) 520/2012 від 19 червня 2012 р. на виконання діяльності фармаконагляду, передбаченої в Регламенті (ЄС) 726/2004 Європейського парламенту і Ради та Директиви 2001/83/EC Європейського парламенту і Ради//Official Journal of the European Union. — L 159, 20.06.2012).
7. Наказ Міністерства охорони здоров’я України від 26 серпня 2005 р. № 426 «Порядок проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення«, зареєстровано в Міністерстві юстиції України 19 вересня 2005 р. за № 1069/11349 (у редакції наказу МОЗ України від 23 липня 2015 р. № 460).
8. Commission Regulation (EC) 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products//Official Journal of the European Union.– L 334, 12.12.2008. (Регламент Комісії (EC) 1234/2008 від 24 листопада 2008 р. з обстеження варіацій за умовами торгової ліцензії для лікувальних продуктів для використання людиною і ветеринарних лікарських препаратів//Official Journal of the European Union. – L 334, 12.12.2008).
9. Commission Regulation (EC) 1085/2003 of 3 June 2003 concerning the examination of variations to the terms of a marketing authorization for medicinal products for human use and veterinary medicinal products falling within the scope of Council Regulation (EEC) № 2309/93//Official Journal of the European Union.– L 159, 27.6.2003. — р. 24–45 (Постанова Комісії (EC) 1085/2003 від З червня 2003 стосовно оцінки змін в умовах торгової ліцензії на лікарські препарати для людини та лікарські препарати для ветеринарії, що знаходяться у сфері дії Постанови Ради (EEC) 2309/93//Official Journal of the European Union.– L159, 27.6.2003. — р. 24–45).
10. Directive 2005/36/EC of the European parliament and of the council of 7 September 2005 on the recognition of professional qualifications//Official Journal of the European Union.– L 255, 30.09.2005 (Директива 2005/36/EC Європейського Парламенту та Ради від 7 вересня 2005 року про визнання професійних кваліфікацій//Official Journal of the European Union.– L 255, 30.09.2005).
11. Наказ Міністерства юстиції України від 12 квітня 2012 р. № 578/5 «Про затвердження Переліку типових документів, що створюються під час діяльності органів державної влади та місцевого самоврядування, інших установ, підприємств та організацій, із зазначенням строків зберігання документів», зареєстровано в Міністерстві юстиції України 17 квітня 2012 р. за № 571/20884.
12. Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data//Official Journal of the European Communities. — L 281, 23.11.95 (Директива 95/46/EC Європейського парламенту та Ради від 24 жовтня 1995 р. про захист фізичних осіб стосовно обробки персональних даних та про вільний рух таких даних//Official Journal of the European Communities. — L 281, 23.11.95).
13. Decision 2119/98/EC of the European Parliament and of the Council of 24 September 1998 setting up a network for the epidemiological surveillance and control of communicable diseases in the Community//Official Journal of the European Union.– L 268, 03.10.1998, р. 1–7 (Рішення 2119/98/ЄС Європейського Парламенту та Ради від 24 вересня 1998 р., що встановлює мережу епідеміологічного контролю та контролю за інфекційними захворюваннями у Співтоваристві//Official Journal of the European Union.– L 268, 03.10.1998, р. 1–7).
14. Commission regulation (EC) 658/2007 of 14 June 2007 concerning financial penalties for infringement of certain obligations in connection with marketing authorisations granted under Regulation (EC) 726/2004 of the European Parliament and of the Council//Official Journal of the European Union.– L 155, 15.06.2007 (Регламент комісії (EC) 658/2007 від 14 червня 2007 р. відносно фінансових санкцій за порушення певних зобов’язань у зв’язку з маркетинговими дозволами, що надані відповідно до Правил (EC) 726/2004 Європейського парламенту та Ради//Official Journal of the European Union.– L 155, 15.06.2007).
15. Directive № 2001/20/EC of the European Parliament and of the council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use//Official Journal of the European Communities. — L 121, 01.05.2001 (Директива № 2001/20/EC Європейського Парламенту та Ради від 4 квітня 2001 р. на зближення законів, правил та адміністративних положень держав — членів у зв’язку із здійсненням належної клінічної практики при проведенні клінічних випробувань на лікарські препарати для застосування у людини//Official Journal of the European Communities. — L 121, 01.05, 2001).
16. Наказ Міністерства охорони здоров’я України від 23 вересня 2009 р. № 690 «Про затвердження Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань і Типового положення про комісії з питань етики», зареєстровано в Міністерстві юстиції України 29 жовтня 2009 р. за № 1010/17026 (у редакції наказу МОЗ України 12 липня 2012 р. № 523).
17. Council Regulation (EC) № 297/95 of 10 February 1995 on fees payable to the European Agency for the Evaluation of Medicinal Products//Official Journal of the European Union.– L 35, 15.02.1995, р. 1 (Регламент Ради (ЄС) № 297/95 від 10 лютого 1995 року про збір, який сплачується до Європейського агентства з оцінки лікарських засобів//Official Journal of the European Union.– L 35, 15.02.1995, р. 1).
18. Regulation (EC) № 45/2001 of the European Parliament and of the Council of 18 December 2000 on the protection of individuals with regard to the processing of personal data by the Community institutions and bodies and on the free movement of such data//Official Journal of the European Communities. — L 8, 12.01.2001, Регламент (ЄС) № 45/2001 Європейського Парламенту та Ради від 18 грудня 2000 р. про захист осіб щодо обробки особистих даних інституціями та органами Співтовариства та про вільну передачу такої інформації//Official Journal of the European Communities. — L 8, 12.01.2001).
19. VOLUME 9A of the Rules Governing Medicinal Products in the European Union. Guidelines on Pharmacovigilance for Medicinal Products for Human Use (Правила нагляду за лікарськими засобами в Європейському Союзі. Директиви з фармаконагляду за лікарськими засобами для людини — Том 9А).
20. International Conference for Harmonisation (ICH) Guideline E2E on Pharmacovigilance Planning (Міжнародна конференція з гармонізації (ICH) Керівництво ІСН з планування фармаконагляду) — настанова ІСН E2E.
21. Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products (Регламент (ЄС) № 1394/2007 Європейського парламенту і Ради від 13 листопада 2007 р. про високотехнологічні (біотехнологічні) лікарські засоби) — Регламент (ЄC) № 1394/2007.
22. EMA/CHMP/ICH/309348/2008 ICH guideline E2F on development safety update report (Настанова ICH-E2F щодо оновлюваного звіту з безпеки препарату, що знаходиться в стадії розробки).
23. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use (Регламент (ЄC) № 1901/2006 Європейського парламенту і Ради від 12 грудня 2006 про лікарські засоби для педіатричного застосування) — Регламент (ЄC) № 1901/2006.
24. Наказ Міністерства охорони здоров’я України від 17 травня 2001 р. № 185 «Про затвердження критеріїв визначення категорій відпуску лікарських засобів», зареєстровано в Міністерстві юстиції України 31 травня 2001 р. за № 464/5655.
25. International Conference for Harmonisation (ICH) Harmonised Tripartite Guideline Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting E2D (Міжнародна конференція з гармонізації (ICH) Гармонізоване тристороннє Керівництво з післяліценційного управління даними з безпеки: Визначення та стандарти прискореної звітності E2D) — настанова ICH-E2D.
26. ICH E2B(R2) — Maintenance of the ICH Guideline on Clinical Safety Data Management: Data Elements for Transmission of Individual Case Reports (Настанова ICH з управління даними з клінічної безпеки: Елементи даних для надання повідомлення про індивідуальний випадок, пов’язаний з безпекою) — настанова ICH E2B(R2).
27. Directive 2002/98/EC of the European Parliament and of the Council of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC (Директива № 2002/98/ЄС Європейського парламенту та Ради від 27 січня 2003 р. про встановлення стандартів якості та безпеки для збору, тестування, обробки, зберігання і розподілу крові і компонентів крові людини, внесення правок до Директиви 2001/83/ЄС).
28. International Conference for Harmonisation (ICH) Guideline E2C(R2) Periodic Benefit-Risk Evaluation Report (Міжнародна конференція з гармонізації (ICH) Керівництво E2C(R2) Регулярний звіт з оцінки співвідношення користь/ризик) — настанова ICH-E2C(R2).
29. International Conference for Harmonisation (ICH) Harmonised Tripartite Guideline Clinical Safety Data Management: Definitions and Standards for Expedited Reporting E2A (Міжнародна конференція з гармонізації (ICH) Гармонізоване тристороннє Керівництво з управління даними з клінічної безпеки: Визначення та стандарти прискореної звітності E2A) — настанова — ICH-E2А.
30. Commission Implementing Regulation (EU) No 198/2013 of 7 March 2013 on the selection of a symbol for the purpose of identifying medicinal products for human use that are subject to additional monitoring (Імплементаційна Постанова (ЄС) № 198/2013 від 7 березня 2013 р. з вибору символу для цілей ідентифікації лікарських засобів для людини, які є предметом додаткового моніторингу) — ІП 198/2013.
31. Regulation (EU) No 1027/2012 of the European Parliament and of the Council of 25 October 2012 amending Regulation (EC) No 726/2004 as regards pharmacovigilance (Постанова (ЄС) № 1027/2012 Європейського парламенту та Ради від 25 жовтня 2012 року про внесення змін до Регламенту (EC) № 726/2004 щодо фармаконагляду) — Постанова (ЄС) № 1027/2012.
32. Directive 2012/26/EU of the European Parliament and of the council of 25 October 2012 amending Directive 2001/83/EC as regards pharmacovigilance (Директива № 2012/26/ЄС Європейського парламенту та Ради від 25 жовтня 2012 р. про внесення поправок в тому, що стосується фармаконагляду) — Директива 2012/26/ЄС.
33. EMA/541760/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module I — Pharmacovigilance systems and their quality systems» (Настанова з належної практики фармаконагляду (ННПФ). Модуль І — Фармаконагляд та його система якості. Перегляд 2*).
34. EMA/816573/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module II — Pharmacovigilance system master file» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІІ — Майстер-файл системи фармаконагляду. Перегляд 2*).
35. EMA/119871/2012 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module ІІI — Pharmacovigilance inspections» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІІІ — Інспекції системи фармаконагляду. Перегляд 1*).
36. EMA/228028/2012 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module IV — Pharmacovigilance audits» (Настанова з належних практик фармаконагляду (ННПФ). Модуль ІV — Аудит з фармаконагляду. Перегляд 1*).
37. EMA/838713/2011 Rev. 2* «Guideline on good pharmacovigilance practices (GVP) Module V — Risk management systems» (Настанова з належних практик фармаконагляду (ННПФ). Модуль V — Системи управління ризиками. Перегляд 2*).
38. EMA/873138/2011 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module VI — Management and reporting of adverse reactions to medicinal products» (Настанова з належних практик фармаконагляду (ННПФ). Модуль VI — Управління та звітування про побічні реакції лікарських засобів. Перегляд 1*).
39. EMA/816292/2011 Rev. 1* «Guideline on good pharmacovigilance practices (GVP). Module VII — Periodic safety update report» (Настанова з належних практик фармаконагляду (ННПФ) Модуль VII — Регулярно оновлюваний звіт з безпеки. Перегляд 1*).
40. EMA/102307/2017 «Explanatory Note to GVP. Module VII» (Пояснювальна записка до Модулю VII ННПФ).
41. EMA/813938/2011 Rev 2* Corr** «Guideline on good pharmacovigilance practices (GVP). Module VIII — Post-authorisation safety studies» (Настанова з належних практик фармаконагляду (ННПФ). Модуль VIІІ — Післяреєстраційне дослідження з безпеки. Перегляд 2* Corr**).
42. EMA/395730/2012 «Guideline on good pharmacovigilance practices (GVP). Module VIII Addendum I — Requirements and recommendations for the submission of information on non-interventional post-authorisation safety studies» (Настанова з належних практик фармаконагляду (ННПФ). Модуль VIІІ Доповнення І — Вимоги та рекомендації щодо подання інформації про неінтервенційні післяреєстраційні дослідження з безпеки).
43. EMA/827661/2011 «Guideline on good pharmacovigilance practices (GVP). Module IX — Signal management» (Настанова з належних практик фармаконагляду (ННПФ). Модуль IX — Управління сигналом).
44. EMA/169546/2012 «Guideline on good pharmacovigilance practices (GVP). Module X — Additional monitoring» (Настанова з належних практик фармаконагляду (ННПФ). Модуль X — Додатковий моніторинг).
45. EMA/118465/2012 «Guideline on good pharmacovigilance practices (GVP). Module XV — Safety communication» (Настанова з належних практик фармаконагляду (ННПФ). Модуль XV — Процес комунікації з питань безпеки).
46. EMA/204715/2012 Rev. 2* «Guideline on good pharmacovigilance practices (GVP). Module XVI — Risk minimisation measures: selection of tools and effectiveness indicators» (Настанова з належних практик фармаконагляду (ННПФ). Модуль XVI — Заходи з мінімізації ризику: Відбір інструментів та показників ефективності. Перегляд 2*).
47. EMA/168402/2014 Corr* «Guideline on good pharmacovigilance practices (GVP). Product- or Population-Specific Considerations II: Biological medicinal products (Настанова з належних практик фармаконагляду (ННПФ). Частина XIIІ — Спеціальні питання щодо препаратів або популяції ІІ: біологічні лікарські засоби).
48. EMA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо керівництв та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
49. Наказ Міністерства охорони здоров’я України від 19 липня 2005 р. № 360 «Про затвердження Правил виписування рецептів на лікарські засоби і вироби медичного призначення, Порядку відпуску лікарських засобів і виробів медичного призначення з аптек та їх структурних підрозділів, Інструкції про порядок зберігання, обліку та знищення рецептурних бланків», зареєстровано в Міністерстві юстиції України 20 липня 2005 р. за № 782/11062.
50. Постанова Кабінету Міністрів України від 26 травня 2005 р. № 376 «Про затвердження Порядку державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію)».
51. Піняжко О.Б. Методичні підходи до проведення оцінки технологій охорони здоров’я в Україні на основі європейської моделі/О.Б. Піняжко, О.М. Заліська//Соціальна фармація в охороні здоров’я. — 2015. — № 2. — С. 44–54.
52. EUnetHTA Guideline «Methods for health economic evaluations», 2015
53. EUnetHTA Joint Action 2, Work Package 8. HTA Core Model ® version 3.0 (Pdf); 2016. Available from www.htacoremodel.info/BrowseModel.aspx.
54. Guidelines for conducting Health Technology Assessment (HTA). AOTMiT. Version 3.0. [Електронний ресурс]. Warsaw 2016., 59 p.
55. Inception Impact Assessment «Strengthening of the EU cooperation on Health Technology Assessment (HTA)». [Електронний ресурс]. — 2016. — Режим доступу до інформації: http://ec.europa.eu/smartregulation/roadmaps/docs/2016_sante_144_health_ technology_assessments_en.pdf.
56. The Cochrane Handbook, Chapter 10, Version 5.1.0.
57. https://amstar.ca/Amstar_Checklist.php.
58. http://handbook-5-1.cochrane.org/.
Ключові слова: фармаконагляд, безпека застосування лікарських засобів, аудит, якість, план управління ризиками, регулярний звіт з безпеки, сигнал, побічні реакції лікарських засобів, післяреєстраційні дослідження з безпеки.
1Див. VI.A.2.3 для визначення поняття першоджерела.
2Всесвітня організація охорони здоров’я (ВООЗ). The importance of pharmacovigilance: safety monitoring of medicinal products. Geneve: WHO; 2002.
3Див. Volume 2C of the Rules Governing Medicinal Products in the EU; http://ec.europa.eu/health/documents/eudralex/vol-2/index_en.htm.
4Фізична особа – це реальна людина на відміну від юридичної, яка є організацією.
5ema.europa.eu. Вебсторінка стосовно подання даних щодо лікарських засобів (ст. 57 Регламенту (ЄС) № 726/2004 [5])
6ema.europa.eu. Вебсторінка Eudravigilance
7Більш докладну інформацію про Інститут внутрішніх аудиторів (IIA) можна знайти на вебсайті: www.theiia.org; про Міжнародну організацію стандартизації (ISO): www.iso.org; Асоціацію аудиту та контролю інформаційних систем: (ISACA) www.isaca.org; Міжнародний комітет з стандартів аудиту та забезпечення достовірності інформації (IAASB): www.ifac.org; Міжнародну організацію вищих контрольних органів (INTOSAI): www.issai.org.
8Див. www.ema.europa.eu.
9Дивись http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000429.jsp&mid=WC0b01ac0580029590.
10Див. www.ema.europa.eu, EMA/606103/2014.
11Див. www.ema.europa.eu.
12See www.ema.europa.eu
13Підхід «Проста мова» включає логічну організацію інформації (та надання пріоритету інформації про заходи, що підлягають виконанню), розподіляючи інформацію на частини, легкі для засвоєння, а також розміщуючи інформацію у спосіб, що покращує читабельність документа. Див. http://www.plainenglish.co.uk/campaigning/past-campaigns/legal/drafting-in-plain-english.html та Управління профілактики та сприяння охороні здоров’я. Проста мова: перспективна стратегія для зрозумілого повідомлення медичної інформації та підвищення освіченості у питаннях здоров’я. Роквіль. Доступно за адресою: http://health.gov/communication/literacy/plainlanguage/IssueBrief.pdf (доступно з 1 вересня 2015 р.).
14Див. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000683.jsp&mid=WC0b01ac 058067a113).
15Настанова щодо деталей різних категорій варіацій, про функціонування процедур, викладених у розділах II, IIa, III і IV Регламенту Комісії (ЄС) № 1234/2008 від 24 листопада 2008 р. щодо розгляду змін до умов маркетингових дозволів для лікарських засобів для людини і ветеринарних лікарських засобів і до документів, які подаються відповідно до цих процедур.
16Див. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000104.jsp&mid= WC0b01ac0580025b88.
17Див. VI. Додаток 2. для ознайомлення з детальною інструкцією з моніторингу медичної та наукової літератури.
18Приклади цифрових засобів комунікації включають, однак не обмежуються наступними: вебсайт, вебсторінка, блог, відеоблог, соціальні мережі, інтернет-форум, чат, медичний портал.
19Благодійний внесок (фінансовий чи інший) заявника (власника реєстраційного посвідчення) організації/сайту не дає право на власність, якщо власник реєстраційного посвідчення не контролює зміст сайту.
20Може застосовуватися національне законодавство про конфіденційність даних, які стосуються ідентифікації пацієнта і відправника повідомлення.
21Відсутня інформація про підозрювану побічну реакцію.
22Для подальшої інструкції щодо репортування про інші дублікати див. розділ А.1.11 «Інші ідентифікатори випадків у попередніх трансмісіях» настанови ICH-E2B(R2) [26].
23Для подальших інструкцій щодо надання таких повідомлень, див. настанову ICH-E2B(R2) [26], розділ A.1.14 «Чи був випадок медично підтверджений, якщо він не був наданий працівником з медичною та/або фармацевтичною освітою?».
24Guideline on the Exposure to Medicinal Products during Pregnancy: Need for Post-Authorisation Data.
25Див. примітку 16.
26Guideline on conduct of pharmacovigilance for medicines used by the paediatric population.
27Рада міжнародних організацій медичних наук (CIOMS). Визначення та застосування термінів фармаконагляду за вакцинами (повідомлення CIOMS/Робочої групи ВООЗ з розробки фармаконагляду вакцин). Женева: CIOMS; 2012. http://www.cioms.ch/.
28http://www.ich.org/.
29Для отримання рекомендацій щодо цих термінів див. Правила, що регулюють лікарські засоби в Європейському Союзі, Том 10, Настанови щодо застосування клінічних випробувань, Настанови щодо досліджуваних лікарських засобів і недосліджуваних лікарських засобів (NIMPs) (Ares (2011) 300458 — 18/03/2011).
30Див. ст. 3(3), третій абзац ст. 107(1) Директиви 2001/83/ЕС.
31Див. Правила, що регулюють лікарські засоби в Європейському Союзі, Том 10, докладна настанова щодо збору, перевірки та подання звітів про несприятливі події/реакції, що виникають у результаті клінічних випробувань лікарських засобів для людини (CT-3), (2011/C 172/01).
32Це не стосується лікарських засобів, що надаються в якості благодійних пожертвувань з науковою метою, якщо власник реєстраційного посвідчення не контролює таке дослідження.
33Для комбінованих досліджень, у яких використовується первинний і вторинний збір даних, необхідно дотримуватися вимог, що застосовуються до досліджень з первинним збором даних.
34Наприклад, спонсор, заявник, власник реєстраційного посвідчення, госпіталь чи оптовик.
35Власники реєстраційних посвідчень повинні надавати повідомлення про індивідуальний випадок, пов’язаний з безпекою до уповноваженого органу держав-членів відповідно до перехідних положень, викладених у ст. 2 (4) і ст. 2 (5) Директиви 2010/84/ЄС [3] та положень ПорядкуN [2] та більш детально в VI.C.4.1.
36Як зазначено в Commission Communication on the Community Marketing Authorization Procedures for Medicinal Products (98/C 229/03).
37Це не стосується добровільно пожертвованих лікарських засобів для науково-дослідних цілей, якщо власник реєстраційного посвідчення не контролює дослідження.
38Див. VI.С.6.2.3.6 рекомендації щодо електронного надання повідомлень.
39Остання версія. (Ref.: EMA/410/01).
40Ref.: EMEA/149995/2008.
41Див. також chapter 1, section 5.1.1 of Volume 2A (Notice to Applicants) of The Rules Governing Medicinal Products in the European Union. http://ec.europa.eu/health/documents/eudralex/vol-2/index_en.htm.
42http://www.ema.europa.eu.
43http://eudravigilance.ema.europa.eu.
44Note for guidance — EudraVigilance Human — Processing of Safety Messages and Individual Case Safety Reports (ICSRs) (EMA/H/20665/04/Final Rev.2).
45Див. примітку 37.
46Стосовно обнулення повідомлення див. VI.C.6.2.2.10.
47Під «якщо це можливо» слід розуміти випадки, коли від першоджерела отримано достатньо інформації для підготовки короткого послідовного опису випадку.
48Рада міжнародних організацій медичних наук (CIOMS). Існуючі проблеми фармаконагляду: Прагматичні підходи (CIOMS V). Женева: CIOMS; 2001. http://www.cioms.ch/.
49На практиці, оригінальний текст наданий першоджерелом офіційною мовою Союзу, крім англійської, повинен бути включений в ICSR, якщо він на прохання держави-члена, де сталася реакція або EMA.
50Ванкуверські рекомендації також доступні на вебсайті Міжнародного комітету редакторів медичних журналів: www.icmje.org.
51Як представлено в законодавстві ЄС (Директива 2011/62/ЄС).
52Див. EMA documents for electronic submission of information on medicines: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/ document_listing/document_listing_000336.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580410138&jsenabled=true.
53Як тільки функції бази даних EudraVigilance, зазначені в ст. 24(2) Регламенту (ЄС) № 726/2004 [5], встановляться.
54Як тільки функції бази даних EudraVigilance, зазначені в ст. 24(2) Регламенту (ЄС) № 726/2004 [5], встановляться
55Як тільки функції бази даних EudraVigilance, зазначені в ст.24(2) Регламенту (ЄС) № 726/2004 [5], встановляться.
56Обов’язково, якщо вони є предметом повідомлення про підозрювані побічні реакції (ст. 102(е) Директиви 2001/83/ЄС і ст. 28 (3) ІП № 520/2012 [6]).
57Див. детальну настанову щодо збору, перевірки та презентації звітів побічних явищ/реакцій, що виникають у ході клінічних випробувань лікарських засобів для людини; доступно на http://ec.europa.eu/health/documents/eudralex/vol-10/.
58Настанова ICH-E2C(R2) [28] не повинна обмежувати обсяг інформації, що надається при оцінці співвідношення користь/ризик лікарського засобу. Будь ласка, ознайомтесь з діючими нормативно-правовими актами у відповідних країнах та регіонах. Щодо вимог ЄС див. VII. C.5.
59http://www.ema.europa.eu.
60http://www.ich.org/.
61ICH-E2D Управління даними безпеки у післяреєстраційний період: визначення та стандарти для термінового повідомлення [25].
62Для подання РОЗБ в ЄС/Україні уповноважена особа, відповідальна за фармаконагляд, на власний розсуд визначає найбільш відповідну особу для підписування даного документа згідно зі структурою та зобов’язаннями заявника (власника реєстраційного посвідчення). Слід включити заяву, що підтверджує призначення уповноваженої особи, відповідальної за фармаконагляд. Не слід надавати листи про делегування повноважень.
63Для РОЗБ, що включають декілька лікарських засобів, з практичних причин цю інформацію можна надавати на титульній сторінці РОЗБ (див. Додаток ІІ)
64Приклади загальних описів можна знайти в ICH-E3: Структура та зміст звітів про клінічне випробування та CIOMS VII (Рада міжнародних організацій медичних наук). DSUR: гармонізація формату та змісту регулярних звітів з безпеки під час клінічних випробувань – Звіт Робочої групи CIOMS VII). Женева: CIOMS; 2006. http://www.cioms.ch/.
65Стандартні вимоги для рукописів, що подаються до біомедичних журналів. Міжнародний комітет редакторів медичних журналів. Медичний журнал Нової Англії 1997, січень 23; 336(4): 309–15. Доступні онлайн: http://www.nejm.org/doi/full/10.1056/NEJM199701233360422 67.
66Стандартні вимоги для рукописів, що подаються до біомедичних журналів: написання та редагування біомедичних публікацій (оновлено у квітні 2010 р.) Етичні принципи публікацій: спонсорство, авторство та звітність, Міжнародний комітет редакторів медичних журналів. http://www.icmje.org/urm_full.pdf.
67Сигнал — це інформація, отримана з одного чи декількох джерел, включаючи спостереження та експерименти, що вказує на новий потенційний причинно-наслідковий зв’язок або новий аспект відомого зв’язку між втручанням та явищем або низкою пов’язаних явищ, побічних або сприятливих, та яка вважається достатньою для обґрунтування контролюючих заходів (ст. 19(1) ІП 520/2012 [6]).
68У регуляторній мережі ЄС та в рамках регулярного звіту з безпеки термін «сигнал» у цьому розділі відповідає терміну «валідований сигнал», що описаний у модулі IX НПФ.
69Початковий перелік референтних дат ЄС був прийнятий СНМР/CMDh після консультації PRAC у вересні 2012 р. та опублікований 1 жовтня 2012 р.
70http://www.emea.europa.eu.
71Див. «Настанова щодо короткої характеристики лікарського засобу», що опублікована на вебсайті Європейської Комісії в Пам’ятці для заявників, том 2C: http://ec.europa.eu/health/files/eudralex.
72www.ema.europa.eu.
73www.ISO.org.
74www.ema.europa.eu.
75Див. глави ІІ, ІІа, ІІІ та ІV постанови Комісії (ЄС) №1234/2008 від 24 листопада 2008 р. відносно розгляду змін в умовах видачі торгових ліцензій на лікарські засоби для людини та ветеринарні препарати, та щодо документації, яка має подаватися відповідно до цих процедур.
76Дослідження проводяться з основною метою виявлення, характеристики або кількісного визначення загрози безпеки, підтверджуючи профіль безпеки лікарського засобу або вимірювання ефективності заходів управління ризиком.
77Ст. 34(4) Імплементаційної Постанови Комісії (ЄС) 520/2012 [6].
78Рада міжнародних організацій медичної науки.
79http://ec.europa.eu/health/documents/eudralex/vol-10/ctqa_v11.pdf.
80http://ex.europa.eu/health/documents/eudralex/vol-10/.
81http://www.encepp.eu/standards_and_guidances/methodologicalGuide.shtml.
82http://www.encepp.eu/standards_and_guidances/checkListProtocols.shtml.
83http://www.ema.europa.eu.
84http://www.pharmacoepi.org/resources/guidelines_08027.cfm.
85http://www.encepp.eu/code_of_conduct/documents/ENePPCodeofConduct_Rev3.pdf.
86http://www.encepp.eu/encepp_studies/indexRegister.shtml.
87www.ema.europa.eu.
88www.ema.europa.eu.
89http://ec.europa.eu/health/documents/eudralex/vol-10/.
90http://www.encepp.eu/encepp_studies/indexRegister.shtml.
91Політика доступу до інформації про лікарські засоби для людини у базі даних EudraVigilance, що опублікована 23 серпня 2011 р. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2011/07/WC500108538.pdf.
92Директива 2010/84/ЄС [3], що вносить зміни до Директиви 2001/83/ЄС [1], Регламент (ЄС) №1235/2010 [4], що вносить зміни до Регламенту (ЄС) № 726/2004 [5] та ІП № 520/2012 [6] про виконання дій з фармаконагляду, що передбачені в Регламенті (ЄС) № 726/2004 [5] та Директиві 2001/83/ЄС [1].
93У рамках цього розділу інструменти та канали представлені без розмежування, оскільки вони часто співпадають та немає загальної узгодженості щодо їх класифікації.
94Тобто уповноважені органи в країнах ЄС, EMA та Європейська Комісія.
95Комітет з оцінки ризику у фармаконагляді. Good practice guide on risk minimisation and prevention of medication errors (EMA/606103/2014). Лондон: EMA; 18 листопада 2015 р. Доступний на: www.ema.europa.eu.
96http://www.encepp.eu.
97Див. http://www.ema.europa.eu.
98Див. http://www.ema.europa.eu.
99Див. http://www.ema.europa.eu.
101Див. http://www.ema.europa.eu.
102Див. http://www.ema.europa.eu.
103Див. http://www.ema.europa.eu.
104Див. http://www.ema.europa.eu.
105Див. http://www.ema.europa.eu.
106Див. http://www.ema.europa.eu.
107Див. http://www.ec.europa.eu.
108Див. http://www.ema.europa.eu.
109Див. http://www.ema.europa.eu.
110Див. http://www.ema.europa.eu.
111Див. http://www.ema.europa.eu.
112Див. http://www.ema.europa.eu.
113Див. http://www.ema.europa.eu.
114Див. http://www.encepp.eu.
115Концепція соціального розповсюдження ризику описує зміни у сприйнятті ризику на різних стадіях розповсюдження інформації, наприклад, шляхом наукових дебатів або обговорення у загальних ЗМІ.
116Див. http://www.ema.europa.eu.
117Див. http://www.ec.europa.eu.