
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ФАРМАЦЕВТИЧНА СИСТЕМА ЯКОСТІ (ICH Q10)
СТ-Н МОЗУ 42-4.3:2011
Видання офіційне
Київ
Міністерство охорони здоров’я України
2012
Передмова
1 РОЗРОБЛЕНО: Державне підприємство «Державний науковий центр лікарських засобів і медичної продукції» (ДП «ДНЦЛЗ»)
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: М. Ляпунов, д-р фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; О. Соловйов, канд. мед. наук; Н. Тахтаулова; Ю. Підпружников, д-р фарм. наук; Н. Литвиненко
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Державна служба України з лікарських засобів
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 3 жовтня 2011 року № 634
3 Настанова відповідає документу Європейського агентства з лікарських засобів (European Medicines Agency):
EMA/INS/GMP/79818/2011 Pharmaceutical Quality System (ICH Q10), 31 January 2011 (EMA/INS/GMP/79818/2011 Фармацевтична система якості (ICH Q10), 31 січня 2011)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2012
© Державна служба України з лікарських засобів, 2012
Передмова до документа EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)»
Документ ICH Q10 щодо фармацевтичної системи якості було узгоджено на етапі 4 на засіданні Керівного комітету ICH в червні 2008 року.
Відповідно до статті 6 Директиви 2003/94/ЕС та Директиви 91/412/ЕЕС власники ліцензії на виробництво наразі зобов’язані розробити та впровадити ефективну фармацевтичну систему забезпечення якості, щоб відповідати вимогам належної виробничої практики (GMP) та дотримуватись положень, викладених у розділі 1 Настанови з GMP.
Документ ICH Q10 надає приклад фармацевтичної системи якості, що призначена для повного життєвого циклу препарату й, таким чином, виходить поза чинні вимоги GMP, які (за винятком виробництва досліджуваних лікарських засобів для людини) не поширюються на ту частину життєвого циклу, що пов’язана з розробкою. На момент впровадження документа ICH Q10 у ЄС також визнано, що розділи 1, 2 та 7 Настанови з GMP мають бути актуалізовані для узгодження термінології та понять, застосованих у документі ICH Q10.
Зміст документа ICH Q10, що є доповненням до сфери GMP, не є обов’язковим. Його застосування має сприяти інноваціям, постійному вдосконаленню та посиленню взаємозв’язку фармацевтичної розробки та виробничої діяльності.
Національний вступ
Ця настанова є прийнятим зі змінами (версії en) нормативним документом Європейського агентства з лікарських засобів (European Medicines Agency) EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» (EMA/INS/GMP/79818/2011 «Фармацевтична система якості (ICH Q10)», прийнятим 31 січня 2011 р. [1], що є трибічно гармонізованою настановою ICH, яку включено до частини 3 чинного нормативного документа Європейського Союзу (ЄС) «EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use» («Європейські правила з належної виробничої практики лікарських засобів для людини та застосування у ветеринарії») (далі Настанова з GMP ЄC) [4]. Відповідно до цього Настанову СТ-Н МОЗУ 42–4.3:2011 введено до частини 3 Настанови СТ-Н МОЗУ 42-4.0:2011 «Лікарські засоби. Належна виробнича практика» [5], що гармонізована з Настановою з GMP ЄC [4].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, які відповідають чинному законодавству.
Цю настанову введено вперше.
До цієї настанови було внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо у пункти, до яких вони відносяться; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5–2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [6], а позначення — відповідно до вимог стандарту СТ МОЗУ 42–1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [9];
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені згідно з вимогами державних стандартів України: ДСТУ 1.5–2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [6] та ДСТУ 1.7–2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [7]; ці структурні елементи не позначені номерами, щоб зберегти у цій настанові нумерацію структурних елементів і правил документа EMA/INS/GMP/79818/2011. «Зміст» цієї настанови викладено з урахуванням додаткових структурних елементів;
- розділ «Терміни та визначення понять» не позначено номером та викладено слід за розділом «Нормативні посилання»; у виносці зазначено, що розділ «Терміни та визначення понять» відповідає розділу 5 «Glossary» документа EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)». Усі терміни у розділі «Терміни та визначення понять» наведено за абеткою; в посиланні на інші нормативні документи, що встановлюють терміни та визначення, замість зазначення категорії та номеру документа дано його порядковий номер у національному додатку «Бібліографія»; додатково наведено термін життєвий цикл (lifecycle) та його визначення з документа EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8 (R2)) «Note for Guidance on Pharmaceutical Development» [10];
- у цій настанові замість посилання на деякі стандарти ISO наведені посилання на гармонізовані з ними національні стандарти ДСТУ ISO, прийняті в Україні;
- у цій настанові слова «marketing authorisation» («торгова ліцензія») замінили на «реєстраційне досьє»; у деяких випадках слова «настанова ICH Q10» замінили на «цю настанову» або дали їх разом;
- у п. 1.1 (перший абзац) замість речення «Даний документ вводить в дію нову настанову ICH…» зазначено «Ця настанова гармонізована з настановою ICH Q10 …». Крім того, у п. 1.1 поряд з посиланнями на документ ICH Q10 дали посилання на цю настановуN; відповідно цьому при згадуванні регіональних вимог GMP дали посилання на вимоги чинної настанови «Лікарські засоби. Належна виробнича практика»N, оскільки прийнята в Україні настанова гармонізована з Настановою з GMP ЄС;
- далі у тексті цієї настанови слова «регіональні вимоги GMP» замінили на слова «вимоги GMP» або «вимоги чинної настанови «Лікарські засоби. Належна виробнича практика»N»;
- у п. 1.2 замість слів «– розробка складу (у тому числі системи контейнер / закупорювальний засіб)» зазначено «– розробка складу, а також системи контейнер / закупорювальний засіб)», оскільки згідно з настановою ICH Q8 це різні етапи фармацевтичної розробки;
- у п. 1.3 замість посилання на настанову ICH Q7 дали посилання на частину 2 чинної настанови «Лікарські засоби. Належна виробнича практика», а у виносці зазначили: «Частина 2 чинної настанови «Лікарські засоби. Належна виробнича практика» гармонізована з документом ICH Q7, на який дається посилання у настанові ICH Q10». Документ ICH Q7 не зазначили також у заголовку п. 1.3, оскільки його як частину 2 включено в Настанову з GMP ЄC та відповідно у гармонізовану з нею Настанову 42-4.0:2011 «Лікарські засоби. Належна виробнича практика»;
- у п. 1.4 замість речення «Регуляторні процеси будуть визначені на регіональному рівні» зазначено «Регуляторні процеси будуть визначені на рівні Міністерства охорони здоров’я УкраїниN»;
- у п. 1.5 замість його назви «1.5. Цілі ICH Q10» зазначено «1.5. Цілі цієї настановиN»;
- у п. 1.6.2 та п. 3.2.2 замість посилання на документ (настанову) ICH Q9 дали посилання на чинну настанову «Лікарські засоби. Управління ризиками для якості (ICH Q9)», гармонізовану з документом ICH Q9N;
- у п. 3.1.1 та 3.2.3 замість посилання на документ ICH Q8 дали посилання на чинну настанову «Лікарські засоби. Фармацевтична розробка (ICH Q8)», гармонізовану з документом ICH Q8N;
- у таблиці 1 замість слів «Згідно з регіональними регуляторними вимогами» зазначено «Згідно з регуляторними вимогами»;
- у п. 3.2.2 та таблиці 2 замість слів «CAPA methodology» зазначено «Методологія системи коригувальних та запобіжних дій (CAPA)»;
- у таблиці 2 та п. 3.2.3 замість скорочення «CAPA» зазначено «коригувальні та запобіжні дії (CAPA)»;
- у примітці до заголовка додатка 1 замість речення «Фактичний регуляторний процес буде визначений на регіональному рівні» зазначено «Фактичний регуляторний процес буде визначений на рівні Міністерства охорони здоров’я УкраїниN»;
- у таблиці 1.1 наведені посилання на настанови СТ-Н МОЗУ 42-3.0:2011, СТ-Н МОЗУ 42–4.2:2011 та СТ-Н МОЗУ 42–4.3:2011 замість посилань на документи ICH Q8, ICH Q9 та ICH Q10 відповідно, а у примітці до таблиці 1.1 додатково зазначено, що ці настанови МОЗ України гармонізовані з відповідними документами ICH;
- у назві додатка 2 поряд з посиланням на документ ICH Q10 наведено посилання на Настанову СТ-Н МОЗУ 42–4.3:2011;
- схему, наведену у додатку 2, зазначено як рисунок 2.1; у тексті додатка 2 слова «На цій схемі…» замінено на слова «На схемі, зображеній на рисунку 2.1 …».
Ця настанова придатна для побудови систем управління якістю для фармацевтичної промисловості (фармацевтичних систем якістю), що застосовні для фармацевтичної розробки та виробництва лікарських речовин (активних фармацевтичних інгредієнтів) та лікарських препаратів, включаючи біотехнологічні та біологічні препарати, протягом життєвого циклу продукції.
Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вноситимуть в документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» [1].
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Фармацевтична система якості (ICH Q10)
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Фармацевтическая система качества (ICH Q10)
MEDICINAL PRODUCTS
Pharmaceutical Quality System (ICH Q10)
Чинна від 2011-10-03
Сфера застосування
Ця настанова установлює положення (рекомендації) щодо системи управління якістю для фармацевтичної промисловості (фармацевтичної система якості).
Ця настанова поширюється на фармацевтичні системи якості, застосовні для фармацевтичної розробки та виробництва лікарських речовин (активних фармацевтичних інгредієнтів) та лікарських препаратів, включаючи біотехнологічні та біологічні препарати, протягом життєвого циклу продукції.
Цю настанову рекомендується застосовувати суб’єктам господарювання (далі — організаціям), які займаються розробкою та виробництвом лікарських засобів (лікарських препаратів та активних фармацевтичних інгредієнтів), незалежно від відомчого підпорядкування та форми власності.
Цю настанову застосовують для оцінки ефективності фармацевтичної системи якості під час аудиту та інспектування організацій з боку регуляторних органів, а також для сертифікації фармацевтичної системи якості виробників лікарських засобів на добровільних засадах.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
ДСТУ ISO 9000–2007 Системи управління якістю. Основні положення та словник термінів
ДСТУ ISO 9001–2009 Системи управління якістю. Вимоги
ДСТУ ISO 9004–2001 Системи управління якістю. Настанови щодо поліпшення діяльності
Настанова СТ-Н МОЗУ 42–3.0:2011 Лікарські засоби. Фармацевтична розробка (ICH Q8)
Настанова СТ-Н МОЗУ 42–4.0:2011 Лікарські засоби. Належна виробнича практика
Настанова СТ-Н МОЗУ 42–4.2:2011 Лікарські засоби. Управління ризиками для якості (ICH Q9)
ICH Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
ICH Q8 Pharmaceutical Development
ICH Q9 Quality Risk Management
ICH Q10 Pharmaceutical Quality System
Довідкові джерела інформації наведено в національному додатку «Бібліографія».
Терміни та визначення понять1
В цій настанові використані існуючі терміни ICH та ДСТУ ISON. У контексті цієї настанови слова «вимога», «вимоги» або «необхідно», що є присутніми у визначеннях ДСТУ ISON, не обов’язково означають регуляторну вимогу. У скобках після визначення наведено посилання на відповідне джерело літератури (див. національний додаток «Бібліографія»)N. За відсутності відповідного визначення у документах ICH або ДСТУ ISON наведено розроблені визначення для документа ICH Q10.
Нижче подано терміни, вжиті в цій настанові, та визначення позначених ними понятьN.
Досягнення якості продукції (product realisation)
Досягнення виробляння продукції, показники якості якої задовольняють потреби пацієнтів, фахівців у сфері охорони здоров’я та регуляторних органів (у тому числі, відповідають реєстраційному досьєN), а також внутрішні вимоги споживачів [1].
Життєвий цикл (lifecycle)
Всі фази життя продукції від початкової розробки, знаходження на ринку і до припинення виробництва і медичного застосування продукції [10]N.
Запобіжна дія (preventive action)
Дія, яку виконують для усунення причини потенційної невідповідності або іншої потенційно небажаної ситуації.
Примітка. Запобіжну дію виконують для попередження події, тоді як коригувальну дію — для попередження повторної події [8].
Зворотній зв’язок / прямий зв’язок (feedback / feed-forward)
Зворотній зв’язок — це модифікація або контроль процесу або системи за їх результатами або ефектами.
Прямий зв’язок — це модифікація або контроль процесу із застосуванням передбачуваних результатів або ефектів [12].
Зворотній зв’язок / прямий зв’язок можна технічно застосовувати у стратегії контролю процесу та концептуально — в управлінні якістю [1].
Зовнішні (аутсорсингові) роботи (outsourced activities)
Роботи, здійснювані виконавцем за письмовою угодою з замовником [1].
Контрольований стан (state of control)
Умова, за якої комплекс контрольних заходів постійно забезпечує стабільні функціональні характеристики процесу та якість продукції [1].
Коригувальна дія (corrective action)
Дія, яку виконують для усунення причини виявленої невідповідності або іншої небажаної ситуації.
Примітка. Коригувальну дію виконують для попередження повторного виникнення події, тоді як запобіжну дію — для попередження виникнення події [8].
Найвище керівництво (senior management)
Особа або група осіб, яка спрямовує та контролює діяльність компанії або дільниці на найвищому рівні, а також має повноваження та обов’язки, щоб задіяти ресурси компанії або дільниці [1,8].
Настанова з якості (quality manual)
Документ, який регламентує систему управління якістю організації [8].
Нововведення (інновація) (innovation)
Впровадження нових технологій або методологій [1].
Планування якості (quality planning)
Складова частина управління якістю, зосереджена на встановленні цілей у сфері якості і на визначенні операційних процесів та відповідних ресурсів, необхідних для досягнення цілей у сфері якості [8].
Показники функціональності (performance indicators)
Вимірювані значення, що використовують для кількісного вираження цілей у сфері якості з метою відображення функціональних характеристик організації, процесу або системи; в деяких регіонах використовують термін «метрика функціональності» («performance metrics» ) [1].
Політика в сфері якості (quality policy)
Загальні наміри та спрямованість організації, пов’язані з якістю, офіційно сформульовані найвищим керівництвом [8].
Постійне поліпшення (continual improvement)
Повторювана діяльність щодо збільшення можливості виконати вимоги [8].
Простір проектних параметрів (design space)
Багатофакторна комбінація та взаємодія вхідних перемінних (наприклад, характеристик матеріалу), а також параметрів процесу, при яких доведено забезпечення якості [10].
Спроможність процесу (capability of a process)
Здатність процесу створювати продукцію, яка відповідатиме вимогам до цієї продукції. Поняття спроможності процесу може бути визначене як термін з галузі статистики [1,8].
Стратегія контролю (control strategy)
Запланований комплекс контрольних заходів, заснований на розумінні продукції та процесу, що забезпечує функціональні характеристики процесу та якість продукції. Цей комплекс може включати контроль параметрів та характеристик, пов’язаних з діючою речовиною, матеріалами та компонентами для лікарського засобу, умовами функціонування приміщень та обладнання, контроль в процесі виробництва, специфікації на готову продукцію, а також пов’язані з цим методи та частоту моніторингу і контролю [1].
Управління змінами (change management)
Систематичний підхід до пропозиції, оцінки, узгодження, впровадження та огляду змін [1].
Управління знаннями (knowledge management)
Систематичний підхід до набування, аналізу, зберігання та розповсюдження інформації щодо продукції, виробничих процесів та компонентів [1].
Управління ризиками для якості (quality risk management)
Систематичний процес для загального оцінювання, контролювання, інформування та огляду ризиків для якості лікарського засобу протягом життєвого циклу препарату [11].
Фактор поліпшення (enabler)
Інструмент або процес, що забезпечує засоби для досягнення мети [1].
Фармацевтична система якості (pharmaceutical quality system — PQS)
Система управління, що спрямовує та контролює діяльність фармацевтичної компанії щодо якості [1,8].
Цілі в сфері якості (quality objectives)
Засоби для перетворення політики в сфері якості та стратегії у вимірювані дії [1].
Якість (quality)
Ступінь, до якого сукупність властивостей, притаманних продукції, системі або процесу, відповідає вимогам [11].
1Розділ «Терміни та визначення понять» відповідає розділу 5 «Glossary» документа EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)»
Познаки та скорочення
АФІ — активний фармацевтичний інгредієнт
ДСТУ — національний стандарт України
CAPA — corrective action and preventive action (коригувальна дія та запобіжна дія)
ЕМА — European Medicines Agency (Європейське агентство з лікарських засобів)
GMP — good manufacturing practice (належна виробнича практика)
ICH — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
ISO — International Organization for Standardization (Міжнародна організація зі стандартизації)
PQS — pharmaceutical quality system (фармацевтична система якості)
1. Фармацевтична система якості
1.1. Вступ
Ця настанова гармонізована з настановою ICH Q10N, що описує модель ефективної системи управління якістю для фармацевтичної промисловості (далі — фармацевтична система якості). У контексті цієї настанови термін «фармацевтична система якості» стосується моделі, описаної в документі ICH Q10.
В документі ICH Q10 описано одну комплексну модель ефективної фармацевтичної системи якості, що ґрунтується на концепції якості Міжнародної організації зі стандартизації (ISO), включає відповідні положення належної виробничої практики (GMP), а також доповнює документи ICH Q8 «Pharmaceutical Development» та ICH Q9 «Quality Risk Management». У документі ICH Q10 та у цій настановіN представлено модель фармацевтичної системи якості, яку можна застосовувати на всіх стадіях життєвого циклу продукції. Більша частина положень документа ICH Q10 та цієї настановиN щодо виробничих дільниць наразі встановлена регіональними вимогами GMP та вимогами чинної настанови «Лікарські засоби. Належна виробнича практика» відповідноN. Документ ICH Q10 та ця настановаN не призначені встановлювати будь-які нові положення, що виходять за рамки чинних регуляторних вимог. Таким чином, положення документа ICH Q10 та цієї настановиN, що доповнюють сучасні регіональні вимоги GMP та вимоги чинної настанови «Лікарські засоби. Належна виробнича практика» відповідноN, не є обов’язковими.
Документ ICH Q10 та ця настановаN свідчать про підтримку ефективної фармацевтичної системи якості з боку промисловості та регуляторних уповноважених органів з метою підвищення якості та доступності ліків у всьому світі та в УкраїніN в інтересах охорони здоров’я. Застосування положень документа ICH Q10 та цієї настановиN протягом життєвого циклу продукції має полегшити нововведення та постійне поліпшення, а також посилити зв’язок фармацевтичної розробки з виробничою діяльністю.
1.2. Сфера застосування
Ця настанова поширюється на системи, застосовні для фармацевтичної розробки та виробництва лікарських речовин (тобто, активних фармацевтичних інгредієнтів — АФІ) та лікарських препаратів, включаючи біотехнологічні та біологічні препарати, протягом життєвого циклу продукції.
Елементи цієї настановиN слід застосовувати у такий спосіб, щоб адаптувати та пропорційно розподіляти їх для кожної стадії життєвого циклу продукції, усвідомлюючи різницю між цими стадіями, та різні цілі кожної стадії.
У контексті цієї настанови життєвий цикл продукції включає таку технічну діяльність щодо нових та існуючих препаратів:
• Фармацевтична розробка:
- розробка лікарської речовини;
- розробка складу, а також системи контейнер / закупорювальний засіб;
- виробництво досліджуваного лікарського засобу;
- розробка системи доставки (якщо необхідно);
- розробка виробничого процесу та масштабування;
- розробка аналітичних методів.
• Перенос (трансфер) технології:
- переноси (трансфери) технології нової продукції з розробки у виробництво;
- переноси (трансфери) технології наявної на ринку продукції всередині або між виробничими та випробувальними дільницями.
• Промислове виробництво:
- придбання та контроль матеріалів;
- забезпечення технічними засобами, системами постачання та обладнанням;
- технологічний процес (включаючи пакування та маркування);
- контроль якості та забезпечення якості;
- видача дозволу на випуск;
- зберігання;
- дистрибуція (за винятком діяльності оптових торговців).
• Припинення виробництва продукції:
- збереження документації;
- зберігання зразків;
- подальша оцінка продукції та звітність.
1.3. Зв’язок цієї настановиN з вимогами GMP та стандартами ДСТУ ISON
Вимоги чинної настанови «Лікарські засоби. Належна виробнича практика», у тому числі вимоги її частини 2 стосовно діючих речовин2, а також керівні принципи щодо системи управління якістю, викладені в стандартах ДСТУ ISO, формують основу для цієї настановиN. Для досягнення цілей, зазначених нижче, ця настановаN доповнює GMP шляхом опису специфічних елементів системи якості та обов’язків керівництва. У цій настановіN представлено гармонізовану модель для фармацевтичної системи якості протягом життєвого циклу продукції; ця настановаN призначена для використання разом з вимогами GMP.
Вимоги GMP не направлені конкретно на всі стадії життєвого циклу продукції (наприклад, на розробку). Елементи системи якості та обов’язки керівництва, описані у цій настанові, призначені заохочувати до застосування наукових підходів та підходів, заснованих на оцінці ризиків, на кожній стадії життєвого циклу, таким чином сприяючи постійному поліпшенню протягом повного життєвого циклу продукції.
2Частина 2 чинної настанови «Лікарські засоби. Належна виробнича практика» гармонізована з документом ICH Q7, на який дається посилання у настанові ICH Q10.
1.4. Взаємозв’язок цієї настановиN та регуляторних підходів
Регуляторні підходи до конкретної продукції або виробничої дільниці мають бути відповідними ступеню розуміння продукції або процесу, результатам управління ризиками для якості та ефективності фармацевтичної системи якості. Після впровадження ефективність фармацевтичної системи якості можна, як правило, оцінити під час інспектування виробничої дільниці з боку регуляторних органів. Потенційні можливості для розширення застосування наукових підходів та підходів, заснованих на оцінці ризиків, вказано в додатку 1. Регуляторні процеси будуть визначені на рівні Міністерства охорони здоров’я УкраїниN.
1.5. Цілі цієї настановиN
Впровадження моделі Q10 має привести до досягнення трьох основних цілей, що доповнюють або посилюють вимоги GMP.
1.5.1. Досягти якості продукції
Створити, впровадити та підтримувати систему, що забезпечить поставку продукції з показниками якості, які задовольняють потреби пацієнтів, фахівців у сфері охорони здоров’я та регуляторних органів (включаючи відповідність вимогам, що затверджені регуляторними органами), а також інших внутрішніх та зовнішніх споживачів.
1.5.2. Встановити та підтримувати контрольований стан
Розробити та використовувати ефективні системи моніторингу та контролю функціональних характеристик процесів та якості продукції, таким чином забезпечуючи гарантію постійної придатності та спроможності процесів. Для визначення систем моніторингу та контролю може бути корисним управління ризиками для якості.
1.5.3. Сприяти постійному поліпшенню
Визначати та запроваджувати відповідні поліпшення якості продукції, вдосконалення процесів, зниження варіабельності, нововведення та посилення фармацевтичної системи якості, таким чином послідовно підвищуючи можливість задовольняти потреби у сфері якості. Для визначення та встановлення пріоритетних сфер для постійного поліпшення може бути корисним управління ризиками для якості.
1.6. Фактори поліпшення: управління знаннями та управління ризиками для якості
Застосування управління знаннями та управління ризиками для якості надасть компанії можливість ефективно та успішно впровадити цю настановуN. Ці фактори сприятимуть досягненню описаних у п. 1.5 цілей, надаючи можливість для прийняття рішень щодо якості продукції на основі наукових підходів та оцінки ризиків.
1.6.1. Управління знаннями
Знаннями про продукцію та процеси можна управляти при розробці, протягом присутності продукції на ринку і аж до припинення її виробництва і медичного застосування. Наприклад, діяльність при розробці з використанням наукових підходів забезпечує знання про продукцію та розуміння процесу. Управління знаннями — це систематичний підхід до набування, аналізу, зберігання та розповсюдження інформації щодо продукції, виробничих процесів та компонентів. Джерела знань включають (але не обмежуються ними): раніш відомі знання (що є загальнодоступним набутком або даними у внутрішніх документах); дослідження з фармацевтичної розробки; діяльність щодо переносу (трансферу) технології; дослідження з валідації процесу протягом життєвого циклу продукції; досвід виробництва; нововведення; постійне поліпшення; а також діяльність з управління змінами.
1.6.2. Управління ризиками для якості
Управління ризиками для якості є невід’ємним від ефективної фармацевтичної системи якості. Воно може забезпечити превентивний підхід до визначення, наукової оцінки та контролювання потенційних ризиків для якості. Це сприятиме постійному поліпшенню функціональних характеристик процесу та якості продукції протягом її життєвого циклу. У чинній настанові «Лікарські засоби. Управління ризиками для якості (ICH Q9)», гармонізованої з документом ICH Q9N, представлено принципи та приклади інструментів управління ризиками для якості, що можуть бути застосовані до різних аспектів якості у фармації.
1.7. Положення щодо структури та змісту
a) Щоб полегшити загальне розуміння та постійне застосування фармацевтичної системи якості її структура, організація та документація мають бути ретельно впорядкованими та зрозумілими.
b) Елементи цієї настановиN слід застосовувати у такий спосіб, щоб адаптувати та пропорційно розподіляти їх для кожної стадії життєвого циклу продукції, беручи до уваги різні цілі кожної стадії та доступні знання щодо кожної стадії.
c) При розробці нової фармацевтичної системи якості або модифікації існуючої системи якості слід враховувати масштаб та складність діяльності компанії. У структурі фармацевтичної системи якості мають бути передбачені відповідні принципи управління ризиками. Хоча деякі аспекти фармацевтичної системи якості можуть поширюватись на всю компанію, а інші бути специфічними для дільниці, ефективність фармацевтичної системи якості, як правило, має бути доведена на рівні дільниці.
d) Фармацевтична система якості має включати відповідні процеси, ресурси та відповідальність для забезпечення якості зовнішніх (аутсорсингових) робіт та закуповуваних матеріалів, як описано у п. 2.7.
e) Має бути визначена відповідальність керівництва у сфері фармацевтичної системи якості, як описано у розділі 2.
f) Фармацевтична система якості має включати такі елементи (описані у розділі 3): моніторинг функціональних характеристик процесу та якості продукції, коригувальні та запобіжні дії, управління змінами та аналізування з боку керівництва.
g) Для моніторингу ефективності процесів у сфері фармацевтичної системи якості слід визначити та використовувати показники функціональності, що описані у розділі 4.
1.8. Настанова з якості
Необхідно розробити настанову з якості або еквіваленту документацію, що має містити опис фармацевтичної системи якості. Такий опис має включати:
а) політику в сфері якості (див. розділ 2);
b) сферу застосування фармацевтичної системи якості;
c) визначення процесів фармацевтичної системи якості, а також їх послідовності, взаємодії та залежності один від одного. Корисними інструментами, що сприяють візуальному сприйманню процесів фармацевтичної системи якості, можуть бути карти та схеми процесів;
d) відповідальність керівництва в межах фармацевтичної системи якості (див. розділ 2).
2. Відповідальність керівництва
Для встановлення та підтримування зобов’язання щодо якості та для функціонування фармацевтичної системи якості на рівні всієї компанії особливо важливим є керівництво.
2.1. Зобов’язання керівництва
a) Щоб досягти цілей в сфері якості, найвище керівництво несе основну відповідальність за забезпечення наявності ефективної фармацевтичної системи якості, а також за визначення, доведення до відома та впровадження обов’язків, відповідальності та повноважень на рівні всієї компанії.
b) Керівництво має виконувати такі обов’язки:
1) брати участь у розробці, впровадженні, моніторингу та підтримуванні ефективної фармацевтичної системи якості;
2) виявляти сильну та очевидну підтримку фармацевтичної системи якості, а також забезпечити її впровадження на всіх рівнях очолюваної організації;
3) забезпечити наявність механізму своєчасної та ефективної передачі та поширення інформації щодо пов’язаних з якістю проблем до відома керівників відповідного рівня;
4) визначити персональні та колективні обов’язки, відповідальність та повноваження, а також взаємодію всіх структурних підрозділів, пов’язаних з фармацевтичною системою якості. Гарантувати, що взаємодії доведено до відома та зрозуміло на всіх рівнях організації. Відповідно до регіональних вимог необхідним є незалежний відділ/структурний підрозділ, уповноважений виконувати певні обов’язки в сфері фармацевтичної системи якості;
5) здійснювати аналіз з боку керівництва функціональних характеристик процесів та якості продукції, а також фармацевтичної системи якості;
6) підтримувати постійне поліпшення;
7) надавати відповідні ресурси.
2.2. Політика в сфері якості
а) Найвище керівництво має розробити політику в сфері якості, що визначає загальні наміри та спрямованість компанії стосовно якості.
b) Політика в сфері якості має містити намір виконувати регуляторні вимоги, а також сприяти постійному поліпшенню фармацевтичної системи якості.
c) Політика в сфері якості має бути доведена до відома персоналу усіх рівнів компанії та усвідомлена ним.
d) Політику в сфері якості слід періодично переглядати, щоб підтримувати її ефективність.
2.3. Планування якості
a) Найвище керівництво має забезпечити, щоб цілі в сфері якості, необхідні для впровадження політики в сфері якості, були визначені та доведені до відома.
b) Цілі в сфері якості мають бути підтримані на всіх відповідних рівнях компанії.
c) Цілі в сфері якості мають відповідати стратегії компанії та узгоджуватись з політикою в сфері якості.
d) Для досягнення цілей в сфері якості керівництво має забезпечити відповідні ресурси та навчання.
e) Слід встановити показники ефективності, що служать мірою досягнення цілей в сфері якості; їх слід піддавати моніторингу із регулярним інформуванням та подальшими заходами, як описано у п. 4.1 цієї настанови.
2.4. Управління ресурсами
а) Керівництво має визначити та забезпечити достатні та відповідні ресурси (людські, фінансові, матеріальні, приміщення та обладнання) для впровадження та підтримки фармацевтичної системи якості та постійного підвищення її ефективності.
b) Керівництво має забезпечити належне використання ресурсів щодо певної продукції, процесу або дільниці.
2.5. Внутрішній обмін інформацією
а) Керівництво має забезпечити розробку та впровадження належних процесів обміну інформацією в межах організації.
b) Процеси обміну інформацією мають забезпечувати передачу відповідної інформації між усіма рівнями компанії.
c) Процеси обміну інформацією мають забезпечувати відповідне та своєчасне поширення інформації стосовно проблем щодо якості продукції та фармацевтичної системи якості.
2.6. Аналізування з боку керівництва
а) Вище керівництво через аналізування зі свого боку несе відповідальність за керування фармацевтичною системою якості, щоб забезпечити її постійну придатність та ефективність.
b) Керівництво має оцінювати висновки періодичних оглядів функціональних характеристик процесів та якості продукції, а також фармацевтичної системи якості, як описано у розділах 3 та 4.
2.7. Управління зовнішніми (аутсорсинговими) роботами та закуповуваними матеріалами
Фармацевтична система якості, включаючи описану у цьому розділі відповідальність керівництва, поширюється на контроль та аналіз будь-яких зовнішніх (аутсорсингових) робіт та якість закуповуваних матеріалів. Фармацевтична компанія несе виключну відповідальність за гарантування того, що наявні процеси для забезпечення контролю зовнішніх (аутсорсингових) робіт та якості закуповуваних матеріалів. У таких процесах має бути задіяне управління ризиками для якості; процеси мають включати:
а) оцінювання перед здійсненням зовнішніх (аутсорсингових) робіт або вибором постачальників матеріалів придатності та компетенції іншої сторони виконувати такі роботи або постачати матеріал із використанням визначеного ланцюга постачання (наприклад, аудити, оцінка матеріалів, проведення кваліфікації);
b) визначення сфер відповідальності та процесів інформування щодо дій зацікавлених сторін, пов’язаних з якістю. Стосовно зовнішніх (аутсорсингових) робіт це має бути відображено у письмовій угоді між замовником та виконавцем;
c) моніторинг та аналіз діяльності виконувача або якості матеріалу від постачальника, а також визначення та впровадження будь-яких необхідних поліпшень;
d) моніторинг вхідних інгредієнтів та матеріалів для гарантії того, що вони надходять із затверджених джерел із використанням узгодженого ланцюга постачання.
2.8. Управління змінами у праві власності на продукцію
Якщо відбувається зміна права власності на продукцію (наприклад, через придбання) керівництво має прийняти до уваги складність цього процесу та гарантувати, що:
а) поточна відповідальність визначена для кожної задіяної компанії;
b) передано всю необхідну інформацію.
3. Постійне поліпшення функціональних характеристик процесу та якості продукції
У цьому розділі описано цілі на стадіях життєвого циклу продукції, а також чотири специфічні для фармацевтичної системи якості елементи, що розширюють вимоги GMP з метою досягнення цілей, визначених у цій настановіN (див. п. 1.5). Цей розділ не змінює вимоги GMP.
3.1. Цілі на стадіях життєвого циклу
Нижче описано цілі на кожній стадії життєвого циклу продукції.
3.1.1. Фармацевтична розробка
Ціль діяльності з фармацевтичної розробки — це створення продукції та виробничого процесу для постійного забезпечення запланованих функціональних характеристик та задоволення потреб пацієнтів і фахівців в сфері охорони здоров’я, а також для дотримання вимог регуляторних органів та внутрішніх споживачів. Підходи до фармацевтичної розробки описано у чинній настанові «Лікарські засоби. Фармацевтична розробка (ICH Q8)», гармонізованої з документом ICH Q8N. Вихідними даними для фармацевтичної розробки є результати пошукових експериментальних та клінічних досліджень, на які не поширюється ця настанова.
3.1.2. Перенос (трансфер) технології
Ціль діяльності з переносу (трансферу) технології — це передача знань щодо продукції та процесу від розробки до виробництва, а також всередині однієї виробничої дільниці або між різними дільницями для впровадження продукції у виробництво. Такі знання формують основу для виробничого процесу, стратегії контролю, підходу до валідації процесу, а також поточного постійного поліпшення.
3.1.3. Промислове виробництво
Цілі промислового виробництва полягають у впровадженні продукції, створенні та підтримці контрольованого стану та сприянні постійному поліпшенню. Фармацевтична система якості має забезпечити, щоб було досягнуто бажаної якості продукції та відповідних функціональних характеристик процесу, щоб комплекс контрольних заходів був відповідним, щоб були встановлені та оцінені можливості для поліпшення, а також щоб постійно збільшувався обсяг знань.
3.1.4. Припинення виробництва продукції
Ціль діяльності з припинення виробництва продукції — це ефективне управління заключною стадією її життєвого циклу. Для припинення виробництва слід застосовувати заздалегідь встановлений підхід до здійснення такої діяльності, як зберігання документації та зразків, а також подальше оцінювання продукції (наприклад, робота з рекламаціями і випробування стабільності) та подання звітності відповідно до регуляторних вимог.
3.2. Елементи фармацевтичної системи якості
Можливо, що вимоги GMP частково містять елементи, описані нижче. Однак, модель, представлена у цій настановіN, призначена для посилення цих елементів з метою сприяння підходу до якості продукції з урахуванням її життєвого циклу. Такими чотирма елементами є:
- система моніторингу функціональних характеристик процесу та якості продукції;
- система коригувальних та запобіжних дій (corrective action and preventive action (CAPA) system);
- система управління змінами;
- аналізування з боку керівництва функціональних характеристик процесу та якості продукції.
Ці елементи слід застосовувати у такий спосіб, щоб адаптувати та пропорційно розподіляти їх для кожної стадії життєвого циклу продукції, усвідомлюючи різницю між цими стадіями, та різні цілі кожної стадії. Протягом життєвого циклу продукції компанії заохочуються до оцінювання можливості інноваційних підходів до поліпшення якості продукції.
Далі опис кожного елементу завершується таблицею із прикладами застосування цього елементу на стадіях життєвого циклу фармацевтичної продукції.
3.2.1. Система моніторингу функціональних характеристик процесу та якості продукції
Щоб забезпечити контрольований стан, фармацевтичним компаніям слід планувати та мати в дії систему моніторингу функціональних характеристик процесів та якості продукції. Ефективна система моніторингу надає гарантію постійної спроможності процесів та контрольних заходів для виробництва продукції бажаної якості і визначення сфер для постійного поліпшення. Система моніторингу функціональних характеристик процесів та якості продукції має забезпечувати:
a) застосування управління ризиками для якості для визначення стратегії контролю. Така стратегія може включати параметри та характеристики стосовно лікарської речовини та вихідної сировини і компонентів для лікарських засобів, робочі параметри приміщень та обладнання, контроль у процесі виробництва, специфікації на готову продукцію, а також пов’язані із цим методи та частота моніторингу і контролю. Стратегія контролю має сприяти своєчасному зворотному/прямому зв’язку та відповідним коригувальним і запобіжним діям;
b) надання інструментів для вимірювання та аналізу параметрів та характеристик, визначених у стратегії контролю (наприклад, управління даними та статистичні інструменти);
c) аналізування параметрів та показників, визначених у стратегії контролю, для підтвердження постійної роботи у контрольованому стані;
d) визначення джерел варіабельності, яка впливає на функціональні характеристики процесу та якість продукції, з метою можливих дій щодо постійного поліпшення для зниження варіабельності або для її контролю;
e) зворотний зв’язок щодо якості продукції як із внутрішніми, так і з зовнішніми джерелами інформації, наприклад, такими як рекламації, відбракування продукції, невідповідності, відкликання, відхилення, аудити та інспектування з боку регуляторних органів та їх висновки;
f) надання знань для посилення розуміння процесу, розширення простору проектних параметрів (якщо вони встановлені), а також можливість інноваційних підходів до валідації процесів.
Таблиця 1 — Застосування системи моніторингу функціональних характеристик процесів та якості продукції протягом її життєвого циклу
|
Фармацевтична розробка |
Перенос (трансфер) технології |
Промислове виробництво |
Припинення виробництва |
|
Отримують знання щодо процесу та продукції. Моніторинг процесу та продукції, здійснюваний протягом розробки, можна застосовувати для визначення стратегії контролю на етапі виробництва. |
Моніторинг під час робіт з масштабування може забезпечити попередню оцінку функціональних характеристик процесу та успішне впровадження у виробництво. Знання, отримані під час переносу (трансферу) та масштабування, можуть бути корисними для подальшої розробки стратегії контролю. |
Слід застосовувати чітку систему моніторингу функціональних характеристик процесу та якості продукції для забезпечення функціонування у контрольованому стані та для визначення сфер для поліпшення. |
Коли виробництво призупиняється, такий моніторинг як випробування стабільності має тривати до завершення досліджень. Згідно з регуляторними вимогами слід виконувати відповідні дії щодо продукції, розміщеної на ринку. |
3.2.2. Система коригувальних та запобіжних дій (CAPA)
Фармацевтичній компанії необхідно мати систему здійснення коригувальних та запобіжних дій, що є результатом розслідування рекламацій, бракування продукції, невідповідностей, відхилень, відкликань, аудитів, інспектувань з боку регуляторних органів та їх висновків, а також тенденцій, виявлених при моніторингу функціональних характеристик процесу та якості продукції. Щоб виявити основну причину, до процесу розслідування слід застосовувати структурований підхід. Відповідно до чинної настанови «Лікарські засоби. Управління ризиками для якості (ICH Q9)», гармонізованої з документом ICH Q9N, ступінь зусиль, офіціальності та документування розслідування має відповідати ступеню ризику. Методологія коригувальних та запобіжних дій (CAPA) має призводити до поліпшення продукції та процесу, а також покращувати їх розуміння.
Таблиця 2 — Застосування системи коригувальних та запобіжних дій протягом життєвого циклу продукції
|
Фармацевтична розробка |
Перенос (трансфер) технології |
Промислове виробництво |
Припинення виробництва |
|
Досліджують варіабельність продукції або процесу. Методологія коригувальних та запобіжних дій (CAPA) може бути корисною, якщо коригувальні та запобіжні дії включено у повторюваний процес проектування та розробки. |
Коригувальні та запобіжні дії (CAPA) можна застосовувати для зворотного зв’язку, прямого зв’язку та постійного поліпшення. |
Слід застосовувати коригувальні та запобіжні дії (CAPA), а також оцінювати ефективність цих дій. |
Коригувальні та запобіжні дії (CAPA) мають тривати після припинення виробництва продукції. Слід враховувати вплив на продукцію, що залишилася на ринку, а також можливий вплив на іншу продукцію. |
3.2.3. Система управління змінами
Нововведення, постійне поліпшення, результати моніторингу функціональних характеристик процесів та якості продукції, а також коригувальні та запобіжні дії (CAPA) призводять до змін. З метою належної оцінки, затвердження та впровадження таких змін компанії необхідно мати ефективну систему управління змінами. Як правило, існує різниця у формальній стороні процесів управління змінами до першої подачі заявки до регуляторних органів та після подачі такої заявки, коли відповідно до регуляторних вимог може знадобитися внесення змін до поданої документації.
Система управління змінами гарантує, що постійне поліпшення відбувається вчасно та в ефективний спосіб. Вона забезпечує високий ступінь впевненості у тому, що зміна не призведе до непередбачуваних наслідків.
Система управління змінами залежно від стадії життєвого циклу має включати:
a) управління ризиками для якості, яке слід використовувати для оцінки пропонованих змін. Рівень зусиль та ступінь формальності оцінки має відповідати рівню ризику;
b) оцінку пропонованих змін, що має бути проведена стосовно реєстраційного досьєN, включаючи простір проектних параметрів (якщо він встановлений) та/або поточне розуміння продукції та процесу. Як зазначено у чинній настанові «Лікарські засоби. Фармацевтична розробка (ICH Q8)», гармонізованій з документом ICH Q8N, робота у рамках простору проектних параметрів не вважається зміною (з огляду на регуляторні вимоги). З точки зору фармацевтичної системи якості, всі зміни мають бути оцінені за допомогою системи управління змінами компанії;
c) оцінку пропонованих змін для гарантії технічного обгрунтування, що має бути проведена групами експертів, які мають належний досвід та знання у відповідних областях (наприклад, у сфері фармацевтичної розробки, виробництва, якості, регуляторних вимог та медицини). Для пропонованої зміни слід встановити майбутні критерії оцінки;
d) оцінку зміни після впровадження, яка має бути проведена для підтвердження того, що цілі зміни були досягнуті, а також що не відбулося негативного впливу на якість продукції.
Таблиця 3 — Застосування системи управління змінами протягом життєвого циклу продукції
|
Фармацевтична розробка |
Перенос (трансфер) технології |
Промислове виробництво |
Припинення виробництва |
|
Зміни є невід’ємною частиною процесу розробки; їх слід документувати. Ступінь формальності процесу управління змінами має відповідати стадії фармаце-втичної розробки. |
Система управління змінами має забезпечити управління поправками до процесу, внесеними під час переносу (трансферу) технології, та їх документування. |
При промисловому виробництві має бути в наявності офіційна система управління змінами. Нагляд з боку відділу якості має забезпечувати наукову, засновану на аналізі ризиків оцінку. |
Будь-які зміни після припинення виробництва продукції слід здійснювати відповідно до системи управління змінами. |
3.2.4. Аналізування з боку керівництва функціональних характеристик процесів та якості продукції
Аналізування з боку керівництва має надавати гарантію того, що протягом життєвого циклу здійснюється управління функціональними характеристиками процесів та якістю продукції. Залежно від розміру компанії та складності її діяльності аналізування з боку керівництва може являти собою ряд аналізів на різних рівнях управління і включати своєчасний та ефективний обмін інформацією, а також її поширення з метою доведення відповідних висновків щодо якості до найвищого керівництва для аналізування з його боку.
а) Система аналізування з боку керівництва має включати:
1) результати інспектувань з боку регуляторних органів та їх висновки, аудити та інші оцінювання, а також зобов’язання перед регуляторними органами;
2) періодичні огляди якості, що включають:
i) оцінки задоволення потреб споживачів через рекламації щодо якості продукції та відкликання;
ii) висновки з моніторингу функціональних характеристик процесів та якості продукції;
iii) ефективність змін щодо процесів та продукції, у тому числі тих, що є наслідками коригувальних та запобіжних дій;
3) контроль виконання після попередніх аналізувань з боку керівництва.
b) Система аналізування з боку керівництва має визначати відповідні дії, такі як:
1) поліпшення виробничих процесів та продукції;
2) надання та/або перерозподіл ресурсів і навчання;
3) набування та поширення знань.
Таблиця 4 — Застосування аналізування з боку керівництва функціональних характеристик процесів та якості продукції протягом її життєвого циклу
|
Фармацевтична розробка |
Перенос (трансфер) технології |
Промислове виробництво |
Припинення виробництва |
|
Підходи аналізування з боку керівництва можна застосовувати для забезпечення адекватності планування продукції та процесу. |
Підходи аналізування з боку керівництва слід застосовувати для забезпечення можливості серійного виробництва розробленої продукції та процесу. |
Аналізування з боку керівництва має бути структурованою системою, як описано вище, та має сприяти постійному поліпшенню. |
Аналізування з боку керівництва має поширюватись на такі елементи як стабільність продукції та рекламації щодо її якості. |
4. Постійне поліпшення фармацевтичної системи якості
У цьому розділі описано заходи, необхідні для управління фармацевтичною системою якості та її постійного поліпшення.
4.1. Аналізування фармацевтичної системи якості з боку керівництва
У керівництва має бути офіційний процес для періодичного аналізування фармацевтичної системи якості. Такий аналіз має включати:
a) оцінювання досягнення цілей фармацевтичної системи якості;
b) оцінку показників функціональності, що можуть бути застосовні для нагляду за ефективністю процесів у межах фармацевтичної системи якості, таких як:
1) рекламації, відхилення, коригувальні та запобіжні дії (CAPA), а також процеси управління змінами;
2) зворотній зв’язок щодо зовнішніх (аутсорсингових) робіт;
3) процеси самооцінки, у тому числі оцінки ризиків, аналіз тенденцій та аудити;
4) зовнішня оцінка, така як інспектування з боку регуляторних органів та висновки, а також аудити з боку споживачів.
4.2. Моніторинг внутрішніх та зовнішніх факторів, що впливають на фармацевтичну систему якості
Фактори, що піддають моніторингу з боку керівництва, можуть включати:
a) нові регуляторні вимоги, настанови та публікації щодо якості, що можуть вплинути на фармацевтичну систему якості;
b) нововведення (інновації), що можуть вдосконалити фармацевтичну систему якості;
c) зміни умов бізнесу та його цілей;
d) зміни щодо права власності на продукцію.
4.3. Результати аналізування та моніторингу з боку керівництва
Результати аналізування з боку керівництва фармацевтичної системи якості, а також моніторингу внутрішніх та зовнішніх факторів можуть включати:
a) поліпшення фармацевтичної системи якості та пов’язаних з нею процесів;
b) розміщення або перерозподіл ресурсів та/або навчання персоналу;
c) перегляд політики та цілей у сфері якості;
d) документування результатів аналізу з боку керівництва та дій, а також своєчасне ефективне сповіщення про них, включаючи інформування найвищого керівництва про відповідні наслідки.
Додаток 1 (довідковий). Потенційні можливості для посилення наукових та заснованих на оцінці ризиків підходів
з боку регуляторних органів*
*У цьому додатку відображено потенційні можливості для посилення регуляторних підходів. Фактичний регуляторний процес буде визначений на рівні Міністерства охорони здоров’я УкраїниN.
Таблиця 1.1
|
План дій |
Потенційна можливість |
|
1. Відповідність правилам GMP |
Відповідність — статус-кво |
|
2. Доказ ефективної фармацевтичної системи якості, включаючи ефективне застосування принципів управління ризиками для якості (наприклад, настанови: СТ-Н МОЗУ 42–4.2:2011 та СТ-Н МОЗУ 42–4.3:2011N). |
Можливість для:
|
|
3. Доказ розуміння продукції та процесів, включаючи ефективне застосування принципів управління ризиками для якості (наприклад, настанови: СТ-Н МОЗУ 42–3.0:2011 та СТ-Н МОЗУ 42–4.2:2011N). |
Можливість для:
|
|
4. Доказ ефективної фармацевтичної системи якості та розуміння продукції й процесів, включаючи ефективне застосування принципів управління ризиками для якості (наприклад, настанови: СТ-Н МОЗУ 42–3.0:2011, СТ-Н МОЗУ 42–4.2:2011 та СТ-Н МОЗУ 42–4.3:2011N). |
Можливість для:
|
|
Примітка. Настанови СТ-Н МОЗУ 42–3.0:2011, СТ-Н МОЗУ 42–4.2:2011 та СТ-Н МОЗУ 42–4.3:2011 гармонізовані відповідно з документами ICH Q8, ICH Q9 та ICH Q10N. |
|
Додаток 2 (довідковий). Схема моделі фармацевтичної системи якості за ICH Q10 та настановою СТ-Н МОЗУ 42–4.3:2011N
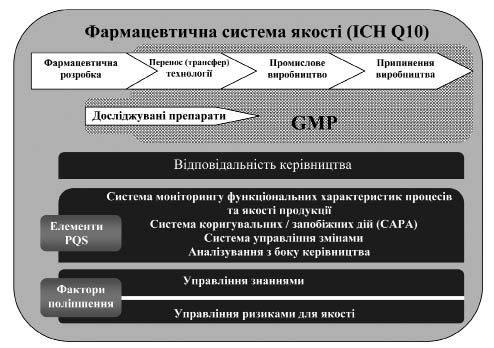
Рисунок 2.1 — Схема моделі фармацевтичної системи якості за документом ICH Q10 та цією настановоюN.
На схемі, зображеній на рисунку 2.1, показано основні особливості моделі фармацевтичної системи якості (Pharmaceutical Quality System — PQS). Фармацевтична система якості охоплює весь життєвий цикл продукції, включаючи фармацевтичну розробку, перенос (трансфер) технології, промислове виробництво та припинення виробництва, як показано у верхній частині схеми. Фармацевтична система якості розширює вимоги GMP, як зазначено на схемі. Також схема свідчить про те, що вимоги GMP поширюються на виробництво досліджуваних лікарських засобів.
Наступний горизонтальний блок ілюструє важливість відповідальності керівництва (поясненої у розділі 2) стосовно всіх стадій життєвого циклу продукції. У наступному за цим горизонтальному блоці наведений перелік елементів фармацевтичної системи якості, які є опорами моделі PQS. Ці елементи слід відповідно та пропорційно застосовувати до кожної стадії життєвого циклу, усвідомлюючи можливості визначення сфер для постійного поліпшення.
У наборі горизонтальних блоків у нижній частині схеми показано фактори поліпшення: управління знаннями та управління ризиками для якості, які застосовують протягом всіх стадій життєвого циклу. Ці фактори поліпшення допомагають у досягненні цілей фармацевтичної системи якості щодо впровадження продукції, встановлення та підтримки контрольованого стану, а також сприяють постійному поліпшенню.
Національний додаток (довідковий). Бібліографія
1. EMA/INS/GMP/79818/2011 Pharmaceutical Quality System (ICH Q10), 31 January 2011
2. Commission Directive 2003/94/EC of 8 October 2003 laying down the principles of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use
3. Commission Directive 91/412/EEC of 23 July 1991 laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products
4. EudraLex. — The Rules Governing Medicinal Products in the European Union. — Volume 4. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use
http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm
5. Настанова СТ-Н МОЗУ 42–4.0:2011 Лікарські засоби. Належна виробнича практика / М. Ляпунов, О. Безугла, О. Соловйов та ін. — Київ, МОЗ України, 2011.
6. ДСТУ 1.5–2003 Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів / І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003. — 91 с.
7. ДСТУ 1.7–2001 Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів / О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003. — 32 с.
8. ДСТУ ISO 9000–2007 Системи управління якістю. Основні положення та словник термінів
9. Настанова СТ-Н МОЗУ 42–1.0:2005 Фармацевтична продукція. Система стандартизації. Основні положення / М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005. — 14 с.
10. EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8 (R2)) Note for Guidance on Pharmaceutical Development, June 2009
11. EMA/INS/GMP/79766/2011 Quality Risk Management (ICH Q9), 31 January 2011
12. The Oxford English Dictionary / Ed. by J. Simpson and E. Weiner. — Oxford University Press, 2003
Ключові слова: відповідальність керівництва, лікарський засіб, належна виробнича практика, постійне поліпшення, припинення виробництва, промислове виробництво, перенос (трансфер) технології, фармацевтична розробка, фармацевтична система якості, якість.