
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
УПРАВЛІННЯ РИЗИКАМИ ДЛЯ ЯКОСТІ (ICH Q9)
СТ-Н МОЗУ 42-4.2:2011
Видання офіційне
Київ
Міністерство охорони здоров’я України
2012
Передмова
1 РОЗРОБЛЕНО: Державне підприємство «Державний науковий центр лікарських засобів і медичної продукції» (ДП «ДНЦЛЗ»)
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: М. Ляпунов, д-р фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; О. Соловйов, канд. мед. наук; Н. Тахтаулова; Ю. Підпружников, д-р фарм. наук; Н. Литвиненко
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Державна служба України з лікарських засобів
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 03.10.2011 року № 634
3 Настанова відповідає документу Європейського агентства з лікарських засобів (European Medicines Agency):
EMA/INS/GMP/79766/2011 Quality Risk Management (ICH Q9), 31 January 2011 (EMA/INS/GMP/79766/2011 Управління ризиками для якості (ICH Q10), 31 січня 2011)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 НА ЗАМІНУ:
Додатка 20 «Управління ризиком для якості» до Настанови СТ-Н МОЗУ 42-4.0:2011 «Лікарські засоби. Належна виробнича практика»
© Міністерство охорони здоров’я України, 2012
© Державна служба України з лікарських засобів, 2012
Передмова до документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)»
Документ ICH Q9 щодо управління ризиками для якості було узгоджено на етапі 4 на засіданні Керівного комітету ICH 9 листопада 2005 року.
Управління ризиками для якості може застосовуватись не тільки у виробництві, але й стосовно фармацевтичної розробки, а також при підготовці частини досьє торгової ліцензії щодо якості. Цю настанову застосовують також регуляторні органи у питаннях фармацевтичної оцінки частини досьє торгової ліцензії щодо якості, інспектування на відповідність GMP та роботи із передбачуваними дефектами якості. Проте для забезпечення зв’язку у березні 2008 року текст було долучено до Настанови з GMP як додаток 20. Після створення частини III Настанови з GMP було визнано, що частина III є більш прийнятною для розміщення цього тексту.
У рамках впровадження документа ICH Q9 у ЄС, у лютому 2008 року було опубліковано поправки до розділу 1 «Управління якістю» Настанови з GMP, які введено в дію з липня 2008 року. Ці поправки, що містять принципи управління ризиками для якості, внесено у зазначений розділ.
Текст цього документа, що був додатком 20, залишається необов’язковим; він надає приклади процесів та застосування управління ризиками для якості.
Національний вступ
Ця настанова є прийнятим зі змінами (версії en) нормативним документом Європейського агентства з лікарських засобів (European Medicines Agency) EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» (EMA/INS/GMP/79766/2011 «Управління ризиками для якості (ICH Q9)» [1], який включено до частини 3 чинного нормативного документа Європейського Союзу (ЄС) «EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use» («Європейські правила з належної виробничої практики лікарських засобів для людини та застосування у ветеринарії») (далі Настанова з GMP ЄC) [2]. Відповідно до цього Настанову СТ-Н МОЗУ 42-4.2:2011 введено до частини 3 Настанови СТ-Н МОЗУ 42-4.0:2011 «Лікарські засоби. Належна виробнича практика» [3], що гармонізована з Настановою з GMP ЄC [2].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, які відповідають чинному законодавству.
Цю настанову введено на заміну додатка 20 «Управління ризиком для якості» до Настанови СТ-Н МОЗУ 42-4.0:2011 «Лікарські засоби. Належна виробнича практика».
До цієї настанови було внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо у пункти, до яких вони відносяться; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [4], а позначення — відповідно до вимог стандарту СТ МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [6];
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені згідно з вимогами державних стандартів України: ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [4] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [5]; ці структурні елементи не позначені номерами, щоб зберегти у цій настанові нумерацію структурних елементів і правил документа EMA/INS/GMP/79766/2011. «Зміст» цієї настанови викладено з урахуванням додаткових структурних елементів;
- розділ «Нормативні посилання» не позначено номером та викладено за розділом «Сфера застосування»; у виносці зазначено, що розділ «Нормативні посилання» відповідає розділу 8 «References» документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» у частині нормативних документів. Бібліографічний опис стандартів ISO 7870-1:2007 та ISO 14971:2007, а також джерел літератури з розділу 8 «References», на які відсутні посилання у тексті документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)», наведено у національному додатку «Бібліографія»;
- розділ «Терміни та визначення понять» не позначено номером та викладено слід за розділом «Нормативні посилання»; у виносці зазначено, що розділ «Терміни та визначення понять» відповідає розділу 7 «Definitions» документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)». Усі терміни у розділі «Терміни та визначення понять» наведено за абеткою;
- додатково наведено терміни простір проектних параметрів (design space) та процесно-аналітична технологія (process analytical technology — PAT), а також їх визначення з документа EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8 (R2)) «Note for Guidance on Pharmaceutical Development» [12];
- у визначенні терміна якість (quality) замість примітки «див. ICH Q6А щодо визначення терміну «якість» спеціально для лікарської речовини та лікарських препаратів» наведено це визначення з посиланням на документ ICH Q6А, який внесено в розділ «Нормативні посилання»;
- додатково до посилань на деякі стандарти ISO та ISO/IEC в тексті цієї настанови наведені посилання на ідентичні стандарти ДСТУ ISO та ДСТУ ISO/IEC, що введені в Україні; ці стандарти зазначені також в розділі «Нормативні посилання»;
- в розділах «Нормативні посилання» та «Бібліографія» актуалізовано бібліографічний опис деяких стандартів ISO, IEC та ISO/IEC, а у тексті цієї настанови актуалізовано відповідні посилання на стандарти; замінено:
• ISO 7870:1993 на ISO 7870-1:2007;
• ISO 7871:1997 на ISO 7870-4:2011;
• ISO 8258:1991 на ISO 8258:1991/Cor 1:1993;
• ISO 14971:2000 на ISO 14971:2007;
• ISO/IEC Guide 73:2002 на ISO Guide 73:2009;
• IEC 60812 на IEC 60812:2006;
• IEC 61025 на IEC 61025:2006;
• IEC 61882 на IEC 61882:2001;
- в розділі «Нормативні посилання» додатково дано бібліографічний опис стандарту ISO 7873:1993, на який є посилання в тексті документа ICH Q9 та цієї настанови;
- у розділі 1 «Вступ» поряд з документами ICH щодо якості дано посилання на документи МОЗ України у наступному реченні: «Це основоположний або вихідний документ, який є незалежним від інших документів МОЗ України таN ICH щодо якості»;
- при посиланні у тексті на додаток, або розділ або пункт поряд з його номером додатково зазначали, що він стосується цієї настанови, наприклад, «… (див. додаток II до цієї настанови)» або «… (п. 4.4 цієї настанови)»;
- у п. І.9 додатка І при посиланні на стандарт ISO 7870-4:2011 зроблено таку виноску: «В документі ICH Q9 дано посилання на стандарт ISO 7871, який замінено на стандарт ISO 7870-4:2011»;
- у п. ІІ.5 додатка ІІ замість слів: «… з урахуванням інших настанов ICH» зазначено: «…з урахуванням відповідних чинних документівN, а також настанов ICH».
Ця настанова придатна для управління ризиками для якості стосовно фармацевтичної розробки, підготовки модуля «Якість» реєстраційного досьє, виробництва та дистрибуції лікарських засобів для людини (активних фармацевтичних інгредієнтів та лікарських препаратів, включаючи біотехнологічні та біологічні препарати) протягом їх життєвого циклу, а також експертизи реєстраційних досьє, аудиту та інспектування.
Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вноситимуть в документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» [1].
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Управління ризиками для якості (ICH Q9)
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Управление рисками для качества (ICH Q9)
MEDICINAL PRODUCTS
Quality Risk Management (ICH Q9)
Чинна від 2011-10-03
Сфера застосування
Ця настанова установлює принципи і положення (рекомендації) щодо системного підходу до управління ризиками для якості в рамках фармацевтичної системи якості та управління якістю у фармацевтичній промисловості. В цій настанові представлений ряд визнаних у міжнародному масштабі альтернативних методів та інструментів управління ризиками для якості разом із переліком можливих сфер застосування.
Ця настанова застосовна до різних аспектів фармацевтичної якості. Вона поширюється на управління ризиками для якості стосовно фармацевтичної розробки, підготовки модуля «Якість» реєстраційного досьє, виробництва та дистрибуції лікарських засобів для людини (активних фармацевтичних інгредієнтів та лікарських препаратів, включаючи біотехнологічні та біологічні препарати) протягом їх життєвого циклу, а також експертизи реєстраційних досьє, аудиту та інспектування. Ця настанова має полегшити виконання принципів та правил GMP, а також інших вимог до якості.
Ця настанова не призначена для створення нових регуляторних вимог.
Цю настанову рекомендується застосовувати суб’єктам господарювання (далі організаціям), які займаються фармацевтичною розробкою, підготовкою модуля «Якість» реєстраційного досьє, виробництвом та дистрибуцією лікарських засобів (лікарських препаратів та активних фармацевтичних інгредієнтів), незалежно від відомчого підпорядкування та форми власності, а також регуляторним органам у питаннях фармацевтичної оцінки модуля «Якість» реєстраційного досьє, інспектування на відповідність GMP та роботи із передбачуваними дефектами якості.
Цю настанову застосовують у фармацевтичній промисловості та регуляторній діяльності під час аудиту та інспектування організацій з боку регуляторних органів, а також для сертифікації фармацевтичної системи якості виробників лікарських засобів на добровільних засадах.
Нормативні посилання1
У цій настанові є посилання на такі нормативні документи:
ДСТУ ISO 7966:2001 Статистичний контроль. Карти приймального контролю (ISO 7966:1993, IDT)
ДСТУ ISO 8258:2001 Статистичний контроль. Контрольні карти Шухарта (ISO 8258:1991, IDT)
ДСТУ ISO/IEC Guide 51-2002 Аспекти безпеки. Настанови щодо їх включення до стандартів (ISO/IEC Guide 51:1999, IDT)
ДСТУ ISO 7873:2004 Статистичний контроль. Контрольні карти для арифметичного середнього з попереджувальними межами (ISO 7873:1993, IDT)
ДСТУ ISO 7870-1:2010 Статистичний контроль. Карти контрольні (ISO 7870-1:2007, IDT)
ISO 7966:1993 Acceptance control charts
ISO 8258:1991/Cor 1:1993 Shewhart control charts
ISO 7870-4:2011 Control charts — Part 4: Cumulative sum charts
ISO 7873:1993 Control charts for arithmetic average with warning limits
ISO/IEC Guide 51:1999 Safety aspects — Guideline for their inclusion in standards
ISO Guide 73:2009 Risk management — Vocabulary
IEC 61025:2006 Fault tree analysis (FTA)
IEC 60812:2006 Analysis techniques for system reliability — Procedure for failure mode and effects analysis (FMEA)
IEC 61882:2001 Hazard and operability studies (HAZOP studies) — Application guide
ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: Chemical substances
ICH Q8 Pharmaceutical development
WHO Expert Committee on Specifications for Pharmaceutical Preparations. Thirty-seventh Report. Geneva, World Health Organization, 2003 (WHO Technical Report Series № 908). — Annex 7 Application of hazard analysis and critical control point (HACCP) methodology to pharmaceuticals. — P. 99-112.
Довідкові джерела інформації наведено в національному додатку «Бібліографія».
1Розділ «Нормативні посилання» містить посилання на нормативні документи, наведені у розділу 8 «References» документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)».
Терміни та визначення понять1
Нижче подано терміни, вжиті в цій настанові, та визначення позначених ними понятьN.
Аналізування ризику (risk analysis)
Оцінка ризику у зв’язку з встановленою небезпекою [1].
Вимоги (requirements)
Явні або неявні потреби або очікування пацієнтів чи захисників їх інтересів (наприклад, медичних працівників, працівників регуляторних та законодавчих органів). У цьому документі «вимоги» означають не тільки встановлені законом, законодавчі або регуляторні вимоги, а також зазначені вище потреби та очікування [1].
Життєвий цикл препарату (product lifecycle)
Всі фази життя продукції від початкової розробки, знаходження на ринку і до припинення виробництва і медичного застосуванняN продукції [1].
Загальне оцінювання ризику (risk assessment)
Систематичний процес формування інформації для забезпечення прийняття рішення щодо ризику в рамках процесу управління ризиками. Він складається з ідентифікації небезпеки, а також аналізування та оцінювання ризиків, пов’язаних з впливом цієї небезпеки [1].
Здатність до виявлення (detectability)
Можливість виявити або встановити наявність, присутність або факт небезпеки [1].
Зниження ризику (risk reduction)
Заходи, вжиті для зменшення ймовірності випадків шкоди та серйозності цієї шкоди [1].
Ідентифікація ризику (risk identification)
Систематичне використання інформації відносно питання ризику або опису проблеми для визначення потенційних джерел шкоди (небезпеки) [1].
Інформування про ризик (risk communication)
Розподіл інформації про ризик та управління ризиком між особою, відповідальною за прийняття рішення, та іншими учасниками [1].
Контроль ризику (risk control)
Дії щодо впровадження рішень з управління ризиком (ISO Guide 73).
Небезпека (hazard)
Потенційне джерело шкоди (ISO/IEC Guide 51 та ДСТУ ISO/IEC Guide 51-2002N).
Огляд ризиків (risk review)
Огляд або моніторинг результатів процесу управління ризиками з урахуванням (при необхідності) нових знань та досвіду стосовно ризиків [1].
Особа(и), відповідальна(і) за прийняття рішення (decision maker(s))
Особа(и), що має відповідну компетенцію та повноваження для прийняття належних та своєчасних рішень щодо управління ризиком для якості [1].
Оцінювання ризику (risk evaluation)
Порівняння передбачуваного ризику з даними критеріями ризику з використанням кількісної та якісної шкали з метою визначення значущості ризику [1].
Прийняття ризику (risk acceptance)
Рішення прийняти ризик (ISO Guide 73).
Простір проектних параметрів (design space)
Багатофакторна комбінація та взаємодія вхідних перемінних (наприклад, характеристик матеріалу), а також параметрів процесу, при яких доведено забезпечення якості. Робота в рамках простору проектних параметрів не вважається зміною. Вихід за простір проектних параметрів розглядається як зміна і, як правило, є початком регуляторного процесу внесення змін після реєстрації. Простір проектних параметрів пропонує заявник; він є об’єктом оцінки і затвердження з боку регуляторних органів [12]N.
Процесно-аналітична технологія (process analytical technology — PAT)
Система планування, аналізу і контролю виробництва за допомогою періодичних вимірювань (тобто, під час технологічного процесу) критичних показників якості і функціональних характеристик сировини, оброблюваних матеріалів і процесів з метою забезпечення якості готового препарату [12]N.
Ризик (risk)
Комбінація ймовірності заподіяння шкоди та тяжкості цієї шкоди (ISO/IEC Guide 51 та ДСТУ ISO/IEC Guide 51-2002N).
Система якості (quality system)
Сукупність всіх аспектів системи, що впроваджує політику якості та забезпечує досягнення цілей щодо якості [1].
Тенденція (trend)
Статистичний термін, що позначає напрямок або ступінь зміни перемінної(их) [1].
Тяжкість (severity)
Міра можливих наслідків небезпеки [1].
Управління ризиками (risk management)
Систематичне здійснення політики управляння якістю, застосування методик та правил з метою загального оцінювання, контролювання, огляду ризиків та відповідного інформування [1].
Управління ризиками для якості (quality risk management)
Систематичний процес для загального оцінювання, контролювання, інформування та огляду ризиків для якості лікарського засобу протягом життєвого циклу препарату [1].
Учасник (stakeholder)
Будь-яка особа, група або установа, що можуть впливати на ризик, на яких може впливати ризик, або які вважають себе під впливом ризику. Особи, відповідальні за прийняття рішення, також можуть бути учасниками. У цьому документі первинними учасниками є пацієнт, медичний працівник, регуляторний уповноважений орган та промисловість [1].
Шкода (harm)
Збитки, заподіяні здоров’ю людини, у тому числі збитки, що є наслідком втрати якості продукції або придатності [1].
Якість (quality)
Ступінь, до якого сукупність властивостей, притаманних продукції, системі або процесу, відповідає вимогам [1].
Відповідність діючої речовини або лікарського препарату його призначенню. Це поняття включає такі показники, як ідентичність, сила дії та чистота (ICH Q6А)N.
1Розділ «Терміни та визначення понять» відповідає розділу 7 «Definitions» документа EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)».
Познаки та скорочення
АФІ — активний фармацевтичний інгредієнт
ДСТУ — національний стандарт України
DOE — Design of Experiments (план експериментів)
ЕМА — European Medicines Agency (Європейське агентство з лікарських засобів)
FMEA — Failure Mode Effects Analysis (аналіз характеру наслідків відмов)
FMECA — Failure Mode, Effects and Criticality Analysis (аналіз характеру, наслідків та критичності відмов)
FTA — Fault Tree Analysis (аналіз дерева помилок)
GMP — Good Manufacturing Practice (належна виробнича практика)
HACCP — Hazard Analysis and Critical Control Points (аналіз експлуатаційної безпеки та критичні контрольні точки)
HAZOP — Hazard Operability Analysis (аналіз експлуатаційної безпеки та працездатності)
HVAC — heating, ventilation and air conditioning (нагрівання, вентиляція та кондиціонування повітря)
ICH — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
ISO — International Organization for Standardization (Міжнародна організація зі стандартизації)
РАТ — Process Analytical Technologies (процесно-аналітична технологія)
PHA — Preliminary Hazard Analysis (попередній аналіз експлуатаційної безпеки)
SOP — Standard Operational Procedure (стандартна робоча методика)
WHO — World Health Organization (Всесвітня організація охорони здоров’я)
1. Вступ
Принципи управління ризиками ефективно застосовуються в багатьох галузях економічної діяльності та державного управління, включаючи фінанси, страхування, безпеку при виробництві, охорону здоров’я, фармаконагляд, а також установами, що забезпечують регуляторну діяльність у цих сферах. Хоча на сьогодні є декілька прикладів застосування управління ризиками для якості в фармацевтичній промисловості, вони мало чисельні та не відповідають усім вимогам управління ризиками. Крім того, важливість систем якості є визнаною в фармацевтичній промисловості, й стає очевидним, що управління ризиками для якості є важливим компонентом ефективної системи якості.
Зрозуміло, що ризик можна визначити як комбінацію ймовірності випадку завдання шкоди та тяжкості цієї шкоди. Однак, досягнення однозначного розуміння щодо застосування управління ризиками між різними учасниками є складним, оскільки кожний учасник може бути об’єктом різної потенційної шкоди; ймовірність виникнення будь-якої шкоди та характеристики її тяжкості для кожного учасника будуть різними. У випадку фармацевтичної продукції, хоча існують різні учасники, у тому числі пацієнти, медичні працівники, а також уряд та промисловість, найважливіше значення має надаватися захисту пацієнта через управління ризиками для якості.
При виробництві та застосуванні лікарського засобу, включаючи його компоненти, неодмінно у певному ступені присутній ризик. Ризик для якості є лише однією складовою загального ризику. Важливо усвідомлювати, що якість продукції слід підтримувати протягом життєвого циклу препарату таким чином, щоб характеристики, важливі для якості лікарського засобу, залишалися такими самими, як у препаратів, які застосовувалися при клінічних випробуваннях. Ефективний підхід до управління ризиками для якості може у подальшому гарантувати пацієнтові високу якість лікарського засобу шляхом встановлення превентивних заходів для ідентифікації та контролю можливих питань щодо якості у ході розробки та виробництва. Крім того, застосування управління ризиками для якості може сприяти прийняттю кращих та більш обґрунтованих рішень, може надати працівникам регуляторних органів більшу гарантію щодо можливостей компанії вирішувати питання з потенційними ризиками, а також може сприятливо вплинути на масштаб та рівень безпосереднього контролю з боку регуляторних органів.
Мета цього документу — запропонувати системний підхід до управління ризиками для якості. Це основоположний або вихідний документ, який є незалежним від інших документів МОЗ України таN ICH щодо якості (хоча й пов’язаний з ними) та який доповнює практики, вимоги, стандарти та правила стосовно якості, що існують в фармацевтичній промисловості та регуляторній діяльності. Документ надає спеціальні вказівки щодо принципів та деяких інструментів управління ризиками для якості, що сприяє прийняттю більш ефективних та послідовних рішень щодо ризику зі сторони працівників як регуляторних органів, так і промисловості стосовно якості лікарських речовин та лікарських препаратів протягом життєвого циклу продукції. Документ не призначений встановлювати будь-які нові обов’язки на додаток до чинних регуляторних вимог.
Не завжди доцільним та необхідним є офіційний процес управління ризиками (із використанням визнаних інструментів та/або внутрішніх методик, наприклад, стандартних робочих методик). Вважається прийнятним застосування неофіційних процесів управління ризиками (із використанням емпіричних інструментів та/або внутрішніх методик). Належне застосування управління ризиками для якості може полегшити виконання, але не скасовує обов’язки промисловців щодо дотримання регуляторних вимог, а також не замінює відповідний обмін інформацією між представниками промисловості та регуляторних органів.
2. Загальні положення
В цій настанові представлені принципи та приклади інструментів управління ризиками для якості, що можуть бути застосованими до різних аспектів фармацевтичної якості. Ці аспекти включають розробку, виробництво, дистрибуцію, а також інспектування та процеси подання заявок/оглядів протягом життєвого циклу лікарських речовин, лікарських препаратів, біологічних та біотехнологічних препаратів (у тому числі використання вихідної сировини, розчинників, допоміжних речовин, пакувальних та маркувальних матеріалів для лікарських засобів, біологічних та біотехнологічних препаратів).
3. Принципи управління ризиками для якості
Існують два основоположних принципи управління ризиками для якості:
- оцінювання ризику для якості має базуватися на наукових даних та бути безпосередньо пов’язаним із захистом пацієнта; та
- рівень зусиль, формалізації та документування процесу управління ризиками для якості має відповідати рівню ризику.
4. Загальний процес управління ризиками для якості
Управління ризиками для якості — це систематичний процес для загального оцінювання, контролю, інформування та огляду ризиків для якості лікарського засобу протягом його життєвого циклу. Модель управління ризиками для якості наведена на діаграмі (рис. 1). Можуть застосовуватись інші моделі. Значення кожного компоненту цієї структури може бути різним в різних випадках, однак надійний процес має враховувати всі компоненти, деталізовані до такого ступеня, який відповідає окремому ризику.
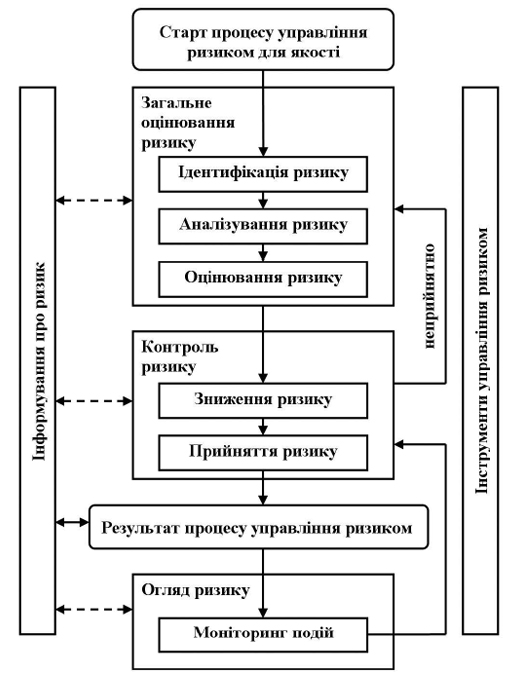
Рисунок 1 — Загальна схема типового процесу управління ризиками для якості.
На наведеній діаграмі не зазначено точки прийняття рішень, оскільки рішення можуть бути прийняті в будь-якій точці процесу. Ці рішення можуть повертати на попередній етап з метою пошуку подальшої інформації, щоб скоригувати моделі ризиків або навіть припинити процес управління ризиками на підставі інформації, що є основою такого рішення.
Примітка. «Неприйнятно» стосується не тільки законодавчих, адміністративних або регуляторних вимог, а також необхідності переглянути процес загального оцінювання ризику (рис. 1).
4.1. Обов’язки
Діяльність щодо управління ризиками для якості, як правило, але не завжди, здійснюється мультидисциплінарними групами. При формуванні груп до них слід включати експертів у відповідних галузях (наприклад, відділ якості, розробка бізнесу, інжиніринг, регуляторна діяльність, технологічні операції, продаж та маркетинг, юридична служба, статистика та клініка) на додаток до осіб, які обізнані щодо процесу управління ризиками для якості.
Особи, відповідальні за прийняття рішень, повинні:
- нести відповідальність за координацію управління ризиками для якості між різними функціями та відділами їх установи; а також
- гарантувати, що процес управління ризиками для якості є визначеним, приведеним у дію та перевірюваним, а також, що наявні достатні ресурси.
4.2. Старт процесу управління ризиками для якості
Управління ризиками для якості має включати систематичні процеси, що призначені для координації, полегшення та покращення прийняття науково обгрунтованих рішень стосовно ризику. Можливі етапи, використовувані для старту та планування процесу управління ризиками для якості, можуть включати наступне:
- визначення проблемного питання та/або питання, що становить собою ризик, у тому числі відповідні припущення, що визначають можливість ризику;
- збір вихідної інформації та/або даних стосовно потенційної небезпеки, шкоди або впливу на здоров’я людини, що мають відношення до загального оцінювання ризику;
- призначення керівника та визначення необхідних ресурсів;встановлення графіку, очікувані результати та відповідний рівень прийняття рішення щодо процесу управління ризиками.
4.3. Загальне оцінювання ризику
Загальне оцінювання ризику полягає у ідентифікації небезпеки та аналізуванні й оцінюванні ризиків, що пов’язані із цією небезпекою (як зазначено далі). Загальне оцінювання ризиків для якості розпочинають з чіткого опису проблеми або аспекту ризику. Якщо ризик, що розглядають, є чітко визначеним, легше встановити відповідний інструмент управління ризиками (див. приклади у розділі 5 цієї настанови), а також види необхідної інформації щодо аспекту ризику. Для чіткого визначення ризику(ів) з метою загального оцінювання ризику часто є корисними три основоположних питання:
- Що може відбуватися неправильно?
- Яка ймовірність (можливість) того, що це буде відбуватися неправильно?
- Які наслідки (їх тяжкість)?
Ідентифікація ризику — це систематичне використання інформації, щоб встановити небезпеку стосовно аспекту ризику або для опису проблеми. Інформація має включати історичні дані, теоретичний аналіз, висновки на основі інформації, а також інтереси учасників. Ідентифікація ризику пов’язана з питанням «Що може відбуватися неправильно?», а також з визначенням можливих наслідків. Це забезпечує основу для подальших етапів процесу управління ризиками для якості.
Аналізування ризику — це оцінка ризику, пов’язана з ідентифікацією небезпеки. Це процес встановлення якісного та кількісного зв’язку між ймовірністю випадку та тяжкістю шкоди. Для деяких інструментів управління ризиками можливість визначити шкоду (здатність до виявлення) також є фактором оцінки ризику.
Оцінювання ризику — це є порівняння встановленого та проаналізованого ризику із заданими критеріями ризику. При оцінюванні ризиків розглядають обгрунтованість доказу щодо всіх трьох основоположних питань.
При загальному оцінюванні ризику важлива надійність набору даних, оскільки це визначає якість результату. Припущення, що виявляють сутність проблеми, та обґрунтовані причини невизначеності будуть підвищувати правильність цього результату та/або допоможуть визначити його обмеження. Невизначеність є наслідком неповних знань про процес у поєднанні з його очікуваною або неочікуваною варіабельністю. Звичайними причинами невизначеності є нестаток знань із фармацевтичної науки та недостатнє розуміння процесу, підстави для шкоди (наприклад, неправильні режими процесу, причини варіабельності), а також недостатня можливість визначення проблем.
Результатом загального оцінювання ризику є або кількісна оцінка ризику або якісний опис діапазону ризику. Якщо ризик виражений кількісно, використовують числову ймовірність. Як альтернатива, ризик може бути виражений з використанням якісних ознак, таких як «високий», «середній» або «низький», які мають бути визначені настільки детально, наскільки це можливо. Іноді використовують «шкалу» ризиків для подальшого визначення ознак при ранжируванні ризиків. При кількісному загальному оцінюванні ризику оцінювання ризику передбачає ймовірність специфічного наслідку, представленого як сукупність обставин, що сприяють виникненню ризику. Таким чином, кількісна оцінка є корисною стосовно одного конкретного наслідку за один раз. Як альтернативу, деякі інструменти управління ризиками використовують відносну міру ризику у поєднанні з множинними рівнями тяжкості та ймовірності для загальної оцінки відносного ризику. На проміжних етапах процесу визначення шкали іноді можна застосовувати кількісну оцінку ризику.
4.4. Контроль ризику
Контроль ризику передбачає прийняття рішення щодо зниження та/або прийняття ризиків. Метою контролю ризику є зниження ризику до прийнятного рівня. Кількість зусиль, прикладених для контролю ризику, має бути пропорційною важливості ризику. Для розуміння оптимального рівня ризику особи, відповідальні за прийняття рішення, можуть застосовувати різні процеси, у тому числі аналіз витрат та прибутків.
Контроль ризику має зосередитись на таких питаннях:
- Чи є ризик понад прийнятний рівень?
- Що має бути зроблено для зниження або усунення ризику?
- Яким є прийнятний баланс між прибутками, ризиками та ресурсами?
- Чи виникають нові ризики як результат контролювання встановлених ризиків?
Зниження ризику зосереджене на процесах зменшення або уникнення ризику для якості при перевищенні встановленого (прийнятного) рівня (див. рис. 1). Зниження ризику може включати заходи, що приймають для зменшення тяжкості та ймовірності шкоди. Як частина стратегії контролю ризику можуть застосовуватись процеси, що покращують здатність до виявлення небезпеки та ризиків для якості. Впровадження заходів із зниження ризику може призводити до внесення нових ризиків до системи або до збільшення важливості інших існуючих ризиків. Таким чином, після впровадження процесу зниження ризику може бути доцільним переглянути загальне оцінювання ризику для встановлення та оцінки будь-якої можливої зміни ризику.
Прийняття ризику — це рішення прийняти ризик. Прийняття ризику може бути офіційним рішенням прийняти остаточний ризик або може бути пасивним рішенням, якщо остаточний ризик не встановлений. Стосовно деяких видів шкоди навіть найкращі практики управління ризиками для якості не в змозі зовсім усунути ризик. За таким умов може бути вирішено, що застосовується відповідна стратегія управління ризиками для якості, та що ризик для якості знижений до встановленого (прийнятного) рівня. Такий (встановлений) прийнятний рівень буде залежати від багатьох параметрів та має визначатися у кожному окремому випадку.
4.5. Інформування про ризик
Інформування про ризик — це розподіл інформації щодо ризику та управління ризиками між особами, відповідальними за прийняття рішення, та іншими особами. Сторони можуть бути поінформовані на будь-якій стадії процесу управління ризиками (див. рис. 1: пунктирні стрілки). Слід належним чином інформувати про результати процесу управління ризиками для якості, які мають бути задокументовані (див. рис. 1: безперервна стрілка). Має бути обмін інформацією між усіма заінтересованими сторонами; наприклад, між представниками регуляторних органів та промисловості, між представниками промисловості та пацієнтом, між внутрішнім персоналом компанії, представниками промисловості або регуляторного органу тощо. Включені відомості можуть стосуватися існування, характеру, форми, ймовірності, тяжкості, прийнятності, контролю, розгляду, здатності до виявлення або інших аспектів ризиків для якості. Немає необхідності інформувати про кожний випадок прийняття ризику. Інформування про рішення щодо управління ризиками для якості між промисловістю та регуляторними органами може ефективно відбуватися через існуючі канали, що встановлені відповідно до регуляторних документів та настанов.
4.6. Огляд ризиків
Управління ризиками має бути частиною діючого процесу управління якістю. Слід впровадити механізм огляду або моніторингу подій.
Результати процесу управління ризиками слід переглядати з урахуванням нових знань та досвіду. Якщо процес управління ризиками для якості був розпочатий, його слід продовжувати, щоб розглядати події, які можуть вплинути на попереднє рішення в рамках процесу управління ризиками для якості, незалежно від того, чи є ці події запланованими (наприклад, огляд препарату, інспекції, аудити, контроль змін), чи незапланованими (наприклад, основна причина при розслідуванні невідповідності, при відкликанні). Частота будь-якого огляду має ґрунтуватися на рівні ризику. Огляд ризику може включати перегляд рішення про прийняття ризику (п. 4.4 цієї настанови).
5. Методологія управління ризиками
Управління ризиками для якості ґрунтується на науковому та практичному підході до прийняття рішень. Воно передбачає документовані, прозорі та відтворювані методи по завершенню етапів процесу управління ризиками для якості на підставі наявних знань стосовно оцінювання ймовірності, тяжкості та іноді здатності до виявлення ризику.
Традиційно оцінку ризику для якості та управління ним здійснювали за допомогою різних неофіційних способів (наприклад, емпіричних та/або внутрішніх методик), що базувалися, наприклад, на комбінації спостережень, тенденцій та іншої інформації. Ці підходи продовжують забезпечувати корисною інформацією, що може надати допомогу у таких питаннях, як обробка рекламацій, дефекти якості, відхилення та розподіл ресурсів.
Крім того, представники фармацевтичної промисловості та регуляторних органів можуть оцінювати ризик та управляти ним за допомогою визнаних інструментів управління ризиками та/або внутрішніх методик (наприклад, стандартних робочих методик). Нижче наведений невичерпний перелік деяких таких інструментів (подальшу інформацію див. у додатку I до цієї настанови та у розділі 8 цієї настанови):
- Основні допоміжні методи управління ризиками (блок-схеми, контрольні карти тощо)
- Аналіз характеру наслідків відмов (Failure Mode Effects Analysis — FMEA)
- Аналіз характеру, наслідків та критичності відмов (Failure Mode, Effects and Criticality Analysis — FMECA)
- Аналіз дерева помилок (Fault Tree Analysis — FTA)
- Аналіз експлуатаційної безпеки та критичні контрольні точки (Hazard Analysis and Critical Control Points — HACCP)
- Аналіз експлуатаційної безпеки та працездатності (Hazard Operability Analysis — HAZOP)
- Попередній аналіз експлуатаційної безпеки (Preliminary Hazard Analysis — PHA)
- Ранжирування та фільтрація ризиків
- Відповідні статистичні методи
6. Впровадження управління ризиками для якості у промисловість та регуляторну діяльність
Управління ризиками для якості є процесом, що сприяє прийняттю науково обгрунтованих та практичних рішень при його інтеграції в системи якості (див. додаток II до цієї настанови). Як зазначено у вступі, належне застосування управління ризиками для якості не усуває обов’язків промисловців дотримуватись регуляторних вимог. Однак, ефективне управління ризиками для якості може сприяти прийняттю кращих та більш обґрунтованих рішень, що надасть представникам регуляторних органів більшої гарантії щодо здатності компанії вести справи з потенційними ризиками, а також може вплинути на масштаб та рівень безпосереднього контролю з боку регуляторного органу. Крім того, управління ризиками для якості може сприяти кращому використанню ресурсів усіма сторонами.
Навчання як працівників промисловості, так і персоналу регуляторних органів щодо процесів управління ризиками для якості забезпечує краще розуміння процесів прийняття рішень та створює довіру щодо результатів управління ризиками для якості.
Управління ризиками для якості слід інтегрувати в існуючу діяльність та належним чином задокументувати. У додатку II до цієї настанови представлено приклади ситуацій, коли застосування процесу управління ризиками для якості може забезпечити інформацією, що може бути використаною при різних фармацевтичних роботах. Ці приклади наведені тільки з метою ілюстрації та не можуть розглядатися як остаточний та вичерпний перелік. Ці приклади не призначені для встановлення будь-яких нових обов’язків на додаток до вимог, встановлених чинним законодавством.
Приклади промислової та регуляторної діяльності (див. додаток II до цієї настанови):
- Управління якістю
- Приклади промислової діяльності та робіт (див. додаток II до цієї настанови):
- Розробка
- Приміщення, обладнання та системи постачання
- Управління матеріалами
- Виробництво
- Лабораторний контроль та випробування стабільності
- Пакування та маркування
- Приклади регуляторної діяльності (див. додаток II до цієї настанови):
- Інспектування та оцінка діяльності
Оскільки регуляторні рішення приймають на регіональній основі, загальне розуміння та застосування принципів управління ризиками для якості може сприяти взаємній довірі та прийняттю більш послідовних рішень представниками різних регуляторних органів на підставі однакової інформації. Таке співробітництво може бути важливим при розробці політики та керівних документів, що вводять практики управління ризиками для якості та сприяють їх впровадженню.
Додаток I (довідковий). Методи та інструменти управління ризиками
Мета цього додатка — надати загальний огляд та посилання на деякі основні інструменти, що можуть бути використані при управлінні ризиком для якості в промисловості та регуляторній діяльності. Ці посилання наведені з метою розширення знань та надання більш детальної інформації щодо конкретного інструменту. Цей перелік не є вичерпним. Важливо зазначити, що жодний інструмент або набір інструментів не може бути застосовним до всіх випадків, коли використовують управління ризиками для якості.
I.1. Основні допоміжні методи управління ризиками
Деякими з простих засобів, що широко застосовуються для структурування управління ризиками шляхом упорядкування даних та для сприяння прийняттю рішень, є:
- Блок-схеми
- Контрольні карти
- Складання карти процесу (маппінг процесу)
- Діаграми причин та наслідків (що також називають діаграмами Ішикави (Ishikava diagram) або діаграмами «риб’ячий скелет»)
I.2. Аналіз характеру наслідків відмов (Failure Mode Effects Analysis — FMEA)
FMEA (див. IEC 60812) призначений для оцінювання характеру потенційних відмов для процесу, а також їх можливих наслідків на результат процесу та/або характеристики продукції. Якщо встановлені види відмов, слід застосовувати зниження ризику з метою усунення, обмеження, зменшення або контролю потенційних відмов. FMEA ґрунтується на розумінні продукції та процесу. FMEA систематично поділяє аналіз складних процесів на стадії, якими можна управляти. Це є потужний інструмент для сумарного розгляду характеру важливих відмов, чинників, що сприяють таким відмовам, та можливих наслідків таких відмов.
Можливі сфери застосування
FMEA можна застосовувати для визначення ступеня важливості ризиків та для перевірки ефективності заходів щодо контролю ризиків.
FMEA можна застосовувати до обладнання та виробничих дільниць, а також для аналізу виробничої операції та її результату стосовно продукції або процесу. FMEA визначає елементи/операції системи, що роблять її вразливою. Результати FMEA можуть бути використані як основа для планування або подальшого аналізу або для рекомендацій щодо використання ресурсів.
I.3. Аналіз характеру, наслідків та критичності відмов (Failure Mode, Effects and Criticality Analysis — FMECA)
FMEA може бути розширений, щоб включити дослідження ступеня тяжкості наслідків, відносної ймовірності інцидентів, а також їх здатності до виявлення; таким чином, FMEA стає аналізом характеру, наслідків та критичності відмов (Failure Mode, Effects and Criticality Analysis — FMECA, див. IEC 60812). Для проведення такого аналізу мають бути встановлені специфікації на продукцію та процес. За допомогою FMECA можуть бути встановлені точки, де необхідні додаткові запобіжні заходи, щоб звести ризики до мінімуму.
Можливі сфери застосування
Застосовувати FMECA у фармацевтичній промисловості слід переважно для відмов та ризиків, пов’язаних з виробничими процесами; хоча застосування FMECA цим не обмежується. Результатом FMECA є відносна «шкала» ризику для кожного виду відмови, за допомогою якої проводять ранжирування режимів на підставі відносного ризику.
I.4. Аналіз дерева помилок (Fault Tree Analysis — FTA)
Аналіз дерева помилок (FTA, див. IEC 61025) — це підхід, що припускає невідповідність функціональних характеристик продукції або процесу. За допомогою цього інструменту оцінюють одноразові помилки системи (або частини системи), але можуть бути поєднані численні чинники відмови шляхом встановлення причинних ланцюжків. Результати представляють у вигляді ілюстрації в формі дерева видів відмов. На кожному рівні дерева комбінації видів відмов можуть бути описані за допомогою логічних операторів («Та», «Або» тощо). FTA залежить від розуміння експертами процесу щодо встановлення причинних факторів.
Можливі сфери застосування
FTA можна застосовувати для встановлення шляху до основної причини відмови. FTA може бути застосовний для розслідування рекламацій або відхилень, щоб досягти повного розуміння їх основних причин та гарантувати, що заплановані удосконалення дозволять повністю вирішити проблему та не призведуть до виникнення інших проблем (тобто вирішення однієї проблеми вже є причиною іншої проблеми). Аналіз дерева помилок є ефективним інструментом для оцінки того, як численні фактори впливають на дану проблему. Результатом FTA є візуальне вираження видів відмов. FTA є корисним як для загального оцінювання ризику, так і для програм моніторингу розробки.
I.5. Аналіз експлуатаційної безпеки та критичні контрольні точки (Hazard Analysis and Critical Control Points — HACCP)
HACCP є системним, превентивним та запобіжним інструментом для забезпечення якості, надійності та безпеки продукції (див. WHO Technical Report Series № 908, 2003, Annex 7). Це структурований підхід із застосовуванням технічних та наукових принципів для аналізування, оцінювання, попередження та контролю ризику або несприятливих наслідків небезпеки, які є результатом планування, розробки, виробництва та застосування препаратів.
HACCP складається з семи наступних етапів:
1) проведення аналізу безпеки та визначення запобіжних заходів для кожної стадії процесу;
2) визначення критичних контрольних точок;
3) встановлення критичних меж;
4) введення системи перевірки критичних контрольних точок;
5) визначення коригувальних заходів, які мають бути прийняті, якщо при моніторингу встановлено, що критичні контрольні точки є неконтрольованими;
6) введення системи підтвердження, що система HACCP працює ефективно;
7) введення системи зберігання протоколів.
Можливі сфери застосування
HACCP можна застосовувати, щоб визначити ризики, пов’язані із фізичною, хімічною та біологічною небезпекою (у тому числі мікробною контамінацією), та управляти ними. HACCP найбільш корисний, коли розуміння продукції та процесу є достатньо повним для того, щоб забезпечити ідентифікацію критичних контрольних точок. Результатом HACCP є інформація щодо управління ризиками, яка полегшує моніторинг критичних точок не тільки у ході виробничого процесу, але й на інших етапах життєвого циклу.
I.6. Аналіз експлуатаційної безпеки та працездатності (Hazard Operability Analysis — HAZOP)
HAZOP (див. IEC 61882) заснований на теорії, яка припускає, що випадки ризику є наслідком відхилення від запланованих або робочих параметрів. Це є системна техніка «мозкового штурму» для ідентифікації небезпеки з використанням так званих «спрямовуючих слів». «Спрямовуючі слова» (наприклад, «ні», «більше», «інший ніж», «частина…» тощо) застосовують до відповідних параметрів (наприклад, контамінація, температура), щоб допомогти встановити можливі відхилення від звичайних або запланованих параметрів. Часто використовують групу людей зі знаннями та досвідом, що охоплюють розробку процесу або препарату та його застосування.
Можливі сфери застосування
HAZOP може застосовуватись щодо виробничих процесів, у тому числі щодо виробництва сторонніми виробниками, а також щодо постачальників, обладнання та технічних засобів для виробництва діючих речовин та лікарських засобів. Також HAZOP переважно застосовується у фармацевтичній промисловості для оцінки безпеки процесу. Як і у випадку HACCP, результатом аналізу HAZOP є перелік критичних операцій для управління ризиками. Це полегшує регулярний моніторинг критичних точок у ході виробничого процесу.
I.7. Попередній аналіз експлуатаційної безпеки (Preliminary Hazard Analysis — PHA)
PHA є інструментом аналізу, заснованого на використанні попереднього досвіду або знань щодо небезпеки або відмови, з метою виявлення інших факторів небезпеки в майбутньому, небезпечних ситуацій та випадків, що можуть бути причиною шкоди, а також з метою оцінювання їх ймовірності стосовно даної діяльності, даних технічних засобів, продукції або системи. Інструмент полягає у: 1) ідентифікації можливостей того, що станеться випадок, пов’язаний з ризиком; 2) якісній оцінці ступеня можливого ушкодження або шкоди для здоров’я, що є наслідком; 3) відносному ранжируванні небезпеки з використанням комбінації тяжкості та ймовірності випадку; а також 4) визначенні можливих коригувальних дій.
Можливі сфери застосування
PHA може бути корисним при аналізі існуючих систем або при визначенні небезпеки, якщо обставини не дозволяють застосовувати більш масштабний спосіб. PHA може бути застосовним до продукції, процесу та проекту виробничої дільниці, а також для оцінювання видів небезпеки для загального виду продукції, потім для класів продукції та, врешті решт, для окремого препарату. PHA найбільш часто застосовується на ранніх етапах розробки проекту, коли мало інформації щодо деталей плану або робочих методик; таким чином, PHA часто є попереднім інструментом для подальших досліджень. Як правило, небезпеку, встановлену при застосуванні PHA, у подальшому оцінюють за допомогою інших інструментів управління ризиками, що зазначені в даному розділі.
I.8. Ранжирування та фільтрація ризиків
Ранжирування та фільтрація ризиків є інструментом для порівняння та ранжирування ризиків. Ранжирування ризиків складних систем, як правило, вимагає оцінки численних різноманітних кількісних та якісних факторів щодо кожного ризику. Інструмент полягає у поділі основної проблеми, пов’язаної з ризиком, на багато компонентів, що необхідно для фіксування факторів, пов’язаних з ризиком. Ці фактори поєднують в одну відносну шкалу ризиків, яку можна застосовувати для ранжирування ризиків. «Фільтри», що являють собою значущі фактори або межі рівнів ризику, можуть бути використані для градації або ранжирування ризиків стосовно завдань управління або політики.
Можливі сфери застосування
Ранжирування та фільтрацію ризиків можна застосовувати для визначення пріоритетів щодо інспектування/аудиту виробничих дільниць зі сторони регуляторних органів або самих промисловців. Методи ранжирування ризиків є корисними, зокрема, в ситуаціях, коли ризики та наслідки, якими необхідно управляти, є різноманітним та представляють труднощі для порівняння при застосуванні тільки одного інструмента. Ранжирування ризиків доцільне, якщо для управління необхідно в рамках тієї самої організаційної схеми оцінити як кількісно оцінювані, так і якісно оцінювані ризики.
I.9. Допоміжні статистичні методи
Статистичні методи можуть допомагати управлінню ризиками для якості та полегшувати його. Вони забезпечують можливість ефективної оцінки даних, допомагають при визначенні важливості набору(ів) даних, а також сприяють прийняттю більш правильних рішень. Перелік деяких основних статистичних методів, широко застосовуваних у фармацевтичній промисловості, включає:
і) контрольні карти, наприклад:
- Карти приймального контролю (див. ISO 7966та ДСТУ ISO 7966:2001N)
- Контрольні карти для арифметичного середнього з попереджувальними межами (див. ISO 7873 та ДСТУ ISO 7873:2004N)
- Контрольні карти кумулятивних сум (див. ISO 7870-4:20111)
- Контрольні карти Шухарта (див. ISO 8258 та ДСТУ ISO 8258:2001N)
- Зважене рухоме середнє значення
іі) план експериментів (Design of Experiments — DOE)
ііі) гістограми
iv) діаграми Парето
v) аналіз придатності процесу
1В документі ICH Q9 дано посилання на стандарт ISO 7871, який замінено на стандарт ISO 7870-4:2011.
Додаток II (довідковий). Потенційне застосування управління ризиками для якості
Цей додаток призначений для визначення можливого застосування принципів та інструментів управління ризиками для якості промисловцями та представниками регуляторної діяльності. Однак, вибір конкретних інструментів управління ризиками повністю залежить від специфічних фактів та обставин.
Наведені приклади представлені для ілюстрації; вони є тільки рекомендаціями щодо можливого застосування управління ризиками для якості. Цей додаток не призначений для встановлення будь-яких нових обов’язків у доповнення до чинних регуляторних вимог.
II.1. Управління ризиками для якості, як частина інтегрованого управління якістю
Документація
Для огляду чинних версій та дотримання регуляторних вимог.
Для визначення необхідності та/або розробки змісту стандартних робочих методик (SOPs), настанов тощо.
Навчання та освіта
Для визначення відповідності попереднього навчання та/або подальших навчальних сесій на підставі освіти, досвіду та трудових навиків персоналу, а також для періодичної оцінки проведеного навчання (наприклад, його ефективності).
Для визначення знань, досвіду, кваліфікаційних характеристик та фізичних можливостей, що дозволяють персоналу виконувати роботу правильно та не чинити негативного впливу на якість продукції.
Дефекти якості
З метою забезпечення основи для визначення й оцінки можливого впливу на якість очікуваного дефекту якості, рекламації, тенденції, відхилення, розслідування, результатів, що не відповідають специфікації тощо, а також інформування про них.
Для сприяння інформуванню про ризик та визначення відповідного заходу щодо значних дефектів якості у співпраці з регуляторним уповноваженим органом (наприклад, відкликання).
Аудит/інспектування
Для встановлення частоти та сфери аудитів, як внутрішніх, так і зовнішніх, з урахуванням таких факторів:
- Вимоги чинного законодавства
- Загальний статус відповідності та історія компанії або виробничої дільниці
- Надійність діяльності компанії щодо управління ризиками для якості
- Складність дільниці
- Складність виробничого процесу
- Складність продукції та її терапевтичне значення
- Кількість та значимість дефектів якості (наприклад, відкликань)
- Результати попередніх аудитів/інспекцій
- Значні зміни будівель, обладнання, процесів, ключового персоналу
- Досвід виробництва препарату (наприклад, частота виробництва, об’єм та кількість серій)
- Результати випробувань в офіційних контрольних лабораторіях
Періодичний огляд
Для вибору, оцінки та інтерпретації даних, що свідчать про тенденцію в рамках огляду якості продукції.
Для інтерпретації даних моніторингу (наприклад, для систематичної оцінки належного проведення ревалідації або змін методики відбору проб).
Управління змінами / контроль змін
Для управління змінами на підставі знань та інформації, отриманої під час фармацевтичної розробки та виробництва.
Для оцінки впливу змін на відповідність готової продукції.
Для оцінки впливу на якість продукції змін, внесених до приміщень, обладнання, матеріалів та виробничого процесу, або переносів (трансферів) виробництва.
Для визначення відповідних заходів, що передують внесенню зміни, наприклад, додаткові випробування, (ре)кваліфікація, (ре)валідація або інформування регуляторних органів.
Постійне удосконалення
Для сприяння постійному удосконаленню процесів протягом життєвого циклу препарату.
II.2. Управління ризиками для якості, як частина регуляторної діяльності
Інспектування та систематична оцінка діяльності
Для сприяння розподілу ресурсів, у тому числі, наприклад, планування інспекцій та їх частоти, а також ретельність оцінювання (див. «Аудит» у п. II.1 додатка II до цієї настанови).
Для оцінки значущості, наприклад, дефектів якості, можливих відкликань та даних, отриманих при інспектуванні.
Для визначення необхідності та виду регуляторних заходів за результатами інспекції.
Для оцінки інформації, наданої промисловцями, у тому числі інформації щодо фармацевтичної розробки.
Для оцінки впливу пропонованих варіацій або змін.
Для визначення ризиків, які слід обговорювати з інспекторами та експертами для сприяння кращому розумінню того, як ризик можна контролювати, або як він контролюється (наприклад, випуск за параметрами, процесно-аналітична технологія (PAT)).
II.3. Управління ризиками для якості, як частина розробки
Для планування якості препарату та виробничого процесу, щоб постійно отримувати препарат із функціональними характеристиками, які відповідають його призначенню (див. документ ICH Q8).
Для розширення знань щодо функціональних характеристик препарату залежно від зміни характеристик матеріалів в широкому діапазоні (наприклад, розподіл часток за розмірами, вміст вологи, характеристики плинності), експлуатаційних характеристик та параметрів процесу.
Для оцінки критичних характеристик вихідної сировини, розчинників, вихідної сировини для активних фармацевтичних інгредієнтів, активних фармацевтичних інгредієнтів, допоміжних речовин або пакувальних матеріалів.
Для встановлення відповідних специфікацій, визначення критичних параметрів процесу та організації виробничого контролю (наприклад, на підставі інформації, отриманої на етапі фармацевтичної розробки при дослідженнях клінічної значущості показників якості та можливості контролювати їх у ході процесу).
Для зниження варіабельності показників якості:
- зниження дефектів препарату та матеріалів;
- зниження дефектів виробництва.
Для оцінки необхідності додаткових досліджень (наприклад, біоеквівалентності, стабільності) при масштабуванні та переносі технології.
Для використання концепції «простору проектних параметрів» (див. документ ICH Q8).
II.4. Управління ризиками для якості щодо технічних засобів, обладнання та систем постачання
Дизайн приміщень / обладнання
Для визначення відповідних зон, при проектуванні будівель та технічних засобів, наприклад:
- напрямок потоку матеріалів та персоналу;
- зведення до мінімуму контамінації;
- заходи щодо контролю паразитів;
- попередження плутанини;
- відкрите обладнання порівняно з закритим;
- чисті приміщення порівняно з ізолюючою технологією;
- спеціально призначені приміщення / обладнання або приміщення / обладнання.
Для визначення відповідних матеріалів обладнання, що контактує з препаратом, а також контейнерів (наприклад, вибір марки неіржавіючої сталі, сальників, змащувальних речовин).
Для визначення відповідних систем постачання (наприклад, пара, гази, джерело живлення, стиснуте повітря, система нагрівання, вентиляції та кондиціонування повітря (HVAC), вода).
Для визначення відповідного профілактичного обслуговування для з’єднаного обладнання (наприклад, перелік необхідних запасних частин).
Аспекти гігієни у приміщеннях
Для захисту препарату від небезпеки з боку навколишнього середовища, у тому числі хімічних, мікробіологічних та фізичних факторів небезпеки (наприклад, визначення належного одягу та організація гардеробної, аспекти гігієни).
Для захисту навколишнього середовища (наприклад, персонал, можливість перехресної контамінації) від небезпеки, пов’язаної із препаратом, що виробляється.
Кваліфікація приміщень / обладнання / систем постачання
Для визначення сфери та масштабу кваліфікації приміщень, будівель та технологічного обладнання, а також лабораторних приладів (у тому числі належних методів калібрування).
Очищення обладнання та контроль навколишнього середовища
Для розподілу зусиль та прийняття рішення з огляду на призначення (наприклад, багатоцільове чи спеціально призначене обладнання, серійне виробництво або безперервний технологічний процес).
Для визначення прийнятних меж для валідації очищення.
Калібрування / профілактичне обслуговування
Для встановлення відповідних графіків калібрування та профілактичного обслуговування.
Комп’ютерні системи та обладнання, контрольоване за допомогою комп’ютерів
Для вибору конфігурації комп’ютерів та програмного забезпечення (наприклад, модульна, структурована, стійка до збоїв система).
Для визначення масштабу валідації:
- ідентифікація критичних функціональних параметрів;
- вибір вимог та дизайну;
- огляд кодів;
- масштаб випробувань та методи випробувань;
- правильність електронних протоколів та підписів.
II.5. Управління ризиками для якості, як частина управління матеріалами
Систематична оцінка та оцінювання постачальників та виробників за контрактом
Для забезпечення всебічної оцінки постачальників та виробників за контрактом (наприклад, проведення аудиту, угоди з постачальниками щодо якості).
Вихідна сировина
Для оцінки відмінностей та можливих ризиків для якості, пов’язаних з варіабельністю вихідної сировини (наприклад, термін зберігання, схема синтезу).
Використання матеріалів
Для визначення того, чи є прийнятними для використання матеріали, що знаходяться у карантині (наприклад, для подальшої внутрішньої обробки).
Для визначення належного здійснення повторної обробки, переробки, використання поверненої продукції.
Умови зберігання, логістики та дистрибуції
Для оцінки адекватності угод щодо забезпечення відповідних умов зберігання та транспортування (наприклад, температура, вологість, дизайн контейнера).
Для визначення впливу на якість препарату невідповідностей щодо умов зберігання та транспортування (наприклад, холодовий ланцюг) з урахуванням відповідних чинних документівN, а також настанов ICH.
Для функціонування інфраструктури (наприклад, можливість забезпечувати належні умови відвантаження, тимчасового зберігання, поводження з небезпечними матеріалами та субстанціями, що підлягають контролю, митне очищення).
Для надання інформації щодо забезпечення придатності фармацевтичної продукції (наприклад, ранжирування ризиків для ланцюга постачання).
II.6. Управління ризиками для якості, як частина виробництва
Валідація
Для визначення сфери та масштабу діяльності щодо підтвердження, кваліфікації та валідації (наприклад, аналітичні методи, процеси, обладнання та методи очищення).
Для визначення масштабу подальших дій (наприклад, відбір проб, моніторинг та ревалідація).
Для розмежування критичних та некритичних стадій процесу з метою полегшення планування валідаційних досліджень.
Відбір проб та випробування у ході виробництва
Для оцінки частоти та масштабу випробувань в процесі виробництва (наприклад, для обґрунтування зменшення випробувань при умовах доведеного контролю).
Для оцінки та обґрунтування використання процесно-аналітичної технології (PAT) разом із випуском за параметрами та випуском у реальному часі.
Планування виробництва
Для встановлення належного плану виробництва (наприклад, відокремлене виробництво, виробництво кампаніями або порядок одночасного проведення технологічних процесів).
II.7. Управління ризиками для якості, як частина лабораторного контролю та випробувань стабільності
Результати невідповідності специфікаціям
Для встановлення можливих основних причин та коригувальних заходів у ході розслідування результатів, що не відповідають специфікаціям.
Період до проведення повторних випробувань / дата закінчення терміну придатності
Для оцінки правильності зберігання та випробування проміжної продукції, допоміжних речовин та вихідної сировини.
II.8. Управління ризиками для якості, як частина пакування та маркування
Дизайн паковань
Для дизайну вторинного паковання, призначеного для захисту первинного паковання препарату (наприклад, щоб забезпечити автентичність препарату, розбірливий напис на етикетці).
Вибір системи контейнер / закупорювальний засіб
Для визначення критичних характеристик системи контейнер / закупорювальний засіб.
Контроль етикеток
Для планування процедур контролю етикеток з огляду на можливість переплутування етикеток різних препаратів, у тому числі різних версій тієї самої етикетки.
Національний додаток (довідковий). Бібліографія
1. EMA/INS/GMP/79766/2011 Quality Risk Management (ICH Q9), 31 January 2011
2. EudraLex. — The Rules Governing Medicinal Products in the European Union. — Volume 4. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use
http://ec.europa.eu/health/documents/eudralex/vol-4/index en.htm
3. Настанова СТ-Н МОЗУ 42-4.0:2011 Лікарські засоби. Належна виробнича практика / М. Ляпунов, О. Безугла, О. Соловйов та ін. — Київ, МОЗ України, 2011.
4. ДСТУ 1.5-2003 Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів / І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003. — 91 с.
5. ДСТУ 1.7-2001 Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів / О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003. — 32 с.
6. Настанова СТ-Н МОЗУ 42-1.0:2005 Фармацевтична продукція. Система стандартизації. Основні положення / М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005. — 14 с.
7. Process Mapping by the American Productivity & Quality Center 2002, ISBN 1928593739
8. What is Total Quality Control; The Japanese Way, Kaoru Ishikawa (Translated by David J. Liu, 1985, ISBN 0139524339
9. Failure Mode and Effect Analysis, FMEA from Theory to Execution, 2nd Edition 2003, D.H. Stamatis, ISBN 0873895983
10. The Basics of FMEA, Robin McDermott, Raymond J. Mikulak, Michael R. Beauregard 1996 ISBN 0527763209
11. Guidelines for Failure Modes and Effects Analysis (FMEA) for Medical Devices, 2003 Dyadem Press ISBN 0849319102
12. EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8 (R2)) Note for Guidance on Pharmaceutical Development, June 2009
13. ISO 7870-1:2007 Control charts
14. ISO 14971:2007 Medical devises — Application of risk management to medical devices
Ключові слова: аналізування ризику, загальне оцінювання ризику, інформування про ризик, контроль ризику, лікарський засіб, належна виробнича практика, огляд ризиків, оцінювання ризику, ризик, система якості, управління ризиками для якості, якість.