
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ДОМІШКИ В НОВИХ ЛІКАРСЬКИХ РЕЧОВИНАХ ТА НОВИХ ЛІКАРСЬКИХ ПРЕПАРАТАХ
СТ-Н МОЗУ 42-3.9:2014
Видання офіційне
Київ
Міністерство охорони здоров’я України
2014
Передмова
РОЗРОБЛЕНО: Державна наукова установа «Науково-технологічний комплекс «Інститут монокристалів» НАН України» (ДНУ «НТК «Інститут монокристалів» НАН України»)
Державне підприємство «Державний експертний центр МОЗ України»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: М. Ляпунов, д-р фарм. наук; О. Антіпова, канд. фарм. наук; О. Баула, канд. хім. наук; О. Нагорна, канд. мед. наук; О. Безугла, канд. фарм. наук; І. Зінченко
ВНЕСЕНО ДО ПРИЙНЯТТЯ: Державне підприємство «Державний експертний центр МОЗ України»
ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 28 листопада 2014 р. № 903
Настанова відповідає наступним нормативним документам:
СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) Note for Guidance on Impurities Testing: Impurities in New Drug Substances, October 2006
(СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) Керівні вказівки щодо випробувань на домішки: домішки в нових лікарських речовинах, жовтень 2006) стосовно підрозділу А,
СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) Note for Guidance on Impurities in New Drug Products, June 2006
(СРМР/ICH/2738/99 (ICH Topic Q3B (R2)) Керівні вказівки щодо домішок в нових лікарських препаратах, червень 2006) стосовно підрозділу В
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2014
Національний вступ
У цій настанові поєднані прийняті зі змінами (версії en) два нормативні документи Комітету із лікарських засобів для людини (Committee for Medicinal Products for Human Use — CPMP) та Міжнародної конференції з гармонізації технічних вимог до реєстрації лікарських препаратів для людини (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use — ICH):
- СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) «Note for Guidance on Impurities Testing: Impurities in New Drug Substances», October 2006 (СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) «Керівні вказівки щодо випробувань на домішки: домішки в нових лікарських речовинах», жовтень 2006) [1];
- СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) «Note for Guidance on Impurities in New Drug Products», June 2006 (СРМР/ICH/2738/99 (ICH Topic Q3B (R2)) «Керівні вказівки щодо домішок в нових лікарських препаратах», червень 2006) [2].
Положення зазначених нормативних документів СРМР/ICH викладені відповідно у підрозділах А та В цієї настанови.
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству.
Загальний технічний документ (Common Technical Document — СTD) [3–5], що регламентує міжнародну структуру реєстраційного досьє, вимагає надати у модулі 3 інформацію стосовно домішок в лікарських речовинах (п. 3.2.S.3.2) та лікарських препаратах (п. 3.2.Р.5.5). Необхідно також навести специфікації на домішки, методики їх аналізу й обґрунтування цих специфікацій для проміжної й нерозфасованої продукції (п. 3.2.S.2.4 і п. 3.2.Р.3.4), лікарської речовини та лікарського препарату (п. 3.2.S.4 і п. 3.2.Р.5).
При фармацевтичній розробці слід визначити профіль домішок, що утворюються в ході виробничого процесу (п. 3.2.Р.2.3) [6–9]. Вміст і профіль домішок досліджують у ході зберігання лікарської речовини і лікарського препарату при випробуваннях їх стабільності (п. 3.2.S.7 і п. 3.2.Р.8) [10–12].
У СTD наведені посилання на спеціальні настанови, що складають методичну основу випробувань на домішки та надання про них інформації в реєстраційному досьє [1, 2, 8, 10, 11, 13, 14].
Порядок реєстрації (перереєстрації) лікарських засобів в Україні також передбачає структуру реєстраційного досьє у форматі загального технічного документа (СTD). В Україні введені в дію гармонізовані настанови, що є методичною основою фармацевтичної розробки лікарських препаратів [7], розробки специфікацій [9], випробувань стабільності [12] тощо. Але СTD потребує використання більш специфічних настанов, що стосуються домішок [1, 2]. Тому актуальною проблемою є введення в Україні настанови стосовно домішок в нових лікарських речовинах, що отримують шляхом хімічного синтезу, та продуктів розкладання в нових лікарських препаратах для людини, що містять одержані шляхом хімічного синтезу нові лікарські речовини. Така настанова має бути гармонізована з нормативними документами СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) та СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) [1, 2].
До цієї настанови були внесені окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо у пункти, до яких вони відносяться; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [15], а позначення — відповідно до вимог стандарту СТ МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [16];
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Познаки та скорочення», а також національний додаток «Бібліографія», що оформлені згідно з вимогами державних стандартів України: ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [15] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [17]; ці структурні елементи не позначені номерами, щоб зберегти у цій настанові нумерацію структурних елементів і правил документів СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) та СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) [1, 2]. Розділ «Зміст» цієї настанови викладено з урахуванням додаткових структурних елементів;
- у розділі «Зміст» цієї настанови після Додатка В.2 «Пояснення стосовно наведення в реєстраційному досьє результатів ідентифікації та кваліфікації продуктів розкладання» вилучено назви: «Приклад 1: Максимальна добова доза 50 мг» та «Приклад 2: Максимальна добова доза 1,9 г», оскільки ці назви не стосуються окремих пунктів додатка В.2, позначених номерами;
- основні положення наведено в розділі «Положення щодо випробувань на домішки», де підрозділ А відповідає документу СРМР/ICH/2737/99, а підрозділ В — документу СРМР/ICH/2738/99 (див. розділ «Передмова»). Ці підрозділи позначені буквами англійського алфавіту А і В, що використані в утворенні номерів окремих структурних елементів і правил; при цьому в настанові зберігається їх цифрова нумерація згідно з настановами СРМР/ICH/2737/99 та СРМР/ICH/2738/99. У розділі «Терміни та визначення понять» спочатку наведений словник спеціальних термінів (розділ 8 «Glossary») документа СРМР/ICH/2737/99, а потім словник спеціальних термінів (розділ 7 «Glossary») документа СРМР/ICH/2738/99. Бібліографічний опис нормативних документів, на які є посилання в цій настанові, наведено в розділі «Нормативні посилання» та/або в національному додатку «Бібліографія». Таким чином, ідентичні переклади двох нормативних документів (СРМР/ICH/2737/99 та СРМР/ICH/2738/99) поєднано в одну настанову без зміни їх об’єму та змісту;
- замість назви підрозділу А «Настанова з випробувань на домішки: домішки в нових лікарських речовинах» («Impurities Testing Guideline: Impurities in New Drug Substances») зазначено «Домішки в нових лікарських речовинах», оскільки розділ називається «Рекомендації щодо випробувань на домішки»;
- у кожному з підрозділів розділу «Терміни та визначення понять» усі терміни наведено за українською абеткою;
- при визначенні терміна «поріг інформування» («reporting threshold») у фразі: «Поріг інформування (reporting threshold) — це те саме, що й рівень інформування (reporting level) в настанові ICH Q2B», виключено посилання на настанову ICH Q2B, оскільки в зазначеній настанові не дано визначення такого терміна;
- визначення терміна «нова лікарська речовина» («new drug substance») дано тільки один раз у підрозділі 1 розділу «Терміни та визначення понять»; в підрозділі 2 замість цього визначення зазначено: «див. визначення терміна в п. 1 розділу «Терміни та визначення понять»». У визначенні терміна «нова лікарська речовина» та в п. А.1 замість слів «у регіоні або державі ЄС» зазначено «в УкраїніN або державі ЄС», оскільки в документі ICH термін «регіон» розповсюджується на ринки США, ЄС та Японії, а ця настанова призначена для застосування в Україні;
- у розділі «Терміни та визначення понять» до визначення кожного з термінів: «поріг інформування», «поріг ідентифікації» та «поріг кваліфікації» наведено примітку, де зазначено синонім цього терміна згідно з загальним текстом 5.10 «Контроль домішок у субстанціях для фармацевтичного застосування», включеним до Державної Фармакопеї України (Доповнення 2);
- у тексті поряд з посиланням на настанови ICH та/або ЄС додатково зроблено посилання на гармонізовані з ними настанови МОЗ України, а також на загальний текст 5.4 «Залишкові кількості органічних розчинників», включений до Державної Фармакопеї України (Доповнення 1), гармонізований з настановою CPMP/ICH/283/95 (ICH Topic Q3С (R3)) «Note for Guidance on Impurities: Residual Solvents»;
- замість посилань на дві настанови з валідації аналітичних методик CPMP/ICH/381/95 (ICH Topic Q2A) «Note for Guidance on Validation of Analytical Methods: Definitions and Terminology» та CPMP/ICH/281/95 (ICH Topic Q2B) «Note for Guidance on Validation of Analytical Procedures: Methodology» наведено посилання на настанову CPMP/ICH/381/95 (ICH Topic Q2 (R1)) «Note for Guidance on Validation of Analytical Procedures: Text and Methodology», що поєднала ці дві настанови, а також додатково на гармонізовану з ними загальну статтю «Валідація аналітичних методик і випробувань», включену до Державної Фармакопеї України;
- при згадуванні у п. А.2 «Класифікація домішок» належної виробничої практики (GMP) в кінці сторінки зроблено виноску, де зазначено: «Тут і далі під належною виробничою практикою (GMP) слід розуміти відповідні положення, що регламентовані в чинних версіях настанови МОЗ України «Лікарські засоби. Належна виробнича практика» (див. розділ «Нормативні посилання») та настанови з GMP ЄС (див. [19] у національному додатку «Бібліографія»)»;
- у тексті поряд з ЄС (Європейським Союзом) зазначена Україна;
- назву додатка А.1 «Thresholds» («Пороги») наведено відповідно до сфери застосування підрозділу А цієї настанови «Пороги для домішок в нових лікарських речовинах»;
- у додатку А.1 замість виноски в кінці сторінки наведено примітку до таблиці з тією ж інформацією, що й у виносці;
- у назві додатка А.3 додатково введено слово «…домішок»: «Схема рішень щодо ідентифікації та кваліфікації домішок»;
- у настанові замінено наступні слова:
- «заявка» («application»), «заявка на реєстрацію» («registration application»), «файл заявки» («application file») — на «реєстраційне досьє»,
- «цей документ» — на «підрозділ А (або В) цього документа», «ця настанова» — на «підрозділ А (або В) цієї настанови»;
- інші незначні доповнення виділені іншим шрифтом та літерою N.
Ця настанова придатна для застосування при організації випробувань стосовно домішок в нових лікарських речовинах, що отримують шляхом хімічного синтезу, та продуктів розкладання в нових лікарських препаратах для людини, що містять одержані шляхом хімічного синтезу нові лікарські речовини, для розробки відповідної реєстраційної документації, а також для експертизи реєстраційних досьє.
У рамках чинного фармацевтичного законодавства ця настанова не має сили нормативно-правового акта; її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану (в рамках ICH та ЄС) позицію світового фармацевтичного сектору. Стосовно домішок в нових лікарських речовинах та продуктів розкладання в нових лікарських препаратах можуть бути застосовані також альтернативні підходи за умови їх відповідного наукового обґрунтування. Такий підхід до правового статусу більшості наукових настанов прийнятий в документі Європейського агентства з ліків Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within pharmaceutical legislative framework» («Процедура щодо настанов і супутніх документів Європейського Союзу в рамках фармацевтичного законодавства») [18].
Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вноситимуть в нормативні документи СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) «Note for Guidance on Impurities Testing: Impurities in New Drug Substances» [1] та СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) «Note for Guidance on Impurities in New Drug Products» [2].
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Домішки в нових лікарських речовинах та нових лікарських препаратах
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Примеси в новых лекарственных веществах и новых лекарственных препаратах
MEDICINAL PRODUCTS
Impurities in New Drug Substances and New Drug Products
Чинна від 2014-11-28
Сфера застосування
Ця настанова встановлює положення (рекомендації) стосовно домішок в нових лікарських речовинах, що отримують шляхом хімічного синтезу, та продуктів розкладання в нових лікарських препаратах для людини, що містять одержані шляхом хімічного синтезу нові лікарські речовини.
Ця настанова застосовна до нових лікарських речовин та нових лікарських препаратів, що розробляють та реєструють в Україні з метою продажу на внутрішньому ринку, експорту до інших країн або імпорту до України.
Ця настанова поширюється на дослідження та стандартизацію домішок в нових лікарських речовинах та продуктів розкладання в нових лікарських препаратах для людини, а також підготовку та експертизу реєстраційних досьє у форматі загального технічного документа (CTD).
Цю настанову рекомендується застосовувати суб’єктам господарювання (далі — організаціям), які займаються стандартизацією лікарських речовин та лікарських препаратів для людини, підготовкою реєстраційних досьє у форматі CTD та подачею заявок на реєстрацію, незалежно від відомчого підпорядкування та форми власності. Цю настанову рекомендується застосовувати також експертним та регуляторним органам у питаннях оцінки реєстраційних досьє в форматі CTD при реєстрації (перереєстрації) лікарських речовин та лікарських препаратів для людини.
Ця настанова придатна для планування та проведення досліджень стосовно домішок у нових лікарських речовинах і нових лікарських препаратах для людини, для розробки відповідної реєстраційної документації, а також для експертизи реєстраційних досьє.
Нормативні посилання
У цій настанові наведено посилання на такі нормативні документи:
- Державна Фармакопея України. Перше видання. 2001 р.
- Державна Фармакопея України. Перше видання. Доповнення 1. 2004 р.
- Державна Фармакопея України. Перше видання. Доповнення 2. 2008 р.
- Настанова 42-3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності
- Настанова 42-3.3:2004 Настанови з якості. Лікарські засоби. Випробування стабільності
- Настанова СТ-Н МОЗУ 42-4.0:2014 Лікарські засоби. Належна виробнича практика
- CPMP/ICH/381/95 (ICH Topic Q2 (R1)) Note for Guidance on Validation of Analytical Procedures: Text and Methodology, 1995
- (CPMP/ICH/381/95 (ICH Topic Q2 (R1)) Руководящие указания по валидации аналитических методик: руководство и методология, 1995)
- CPMP/ICH/283/95 (ICH Topic Q3С (R3)) Note for Guidance on Impurities: Residual Solvents, 1998
- (CPMP/ICH/283/95 (ICH Topic Q3С (R3)) Руководящие указания по примесям: остаточные растворители, 1998)
- СРМР/ICH/367/96 Corr (ICH Topic Q6A) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000
- (СРМР/ICH/367/96 Corr (ICH Topic Q6A) Настанова щодо специфікацій: методики випробувань і критерії прийнятності для нових лікарських речовин і нових лікарських препаратів: хімічні речовини, 2000)
- CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) Note for guidance on stability testing: stability testing of new drug substances and products, 2003
- (CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) Руководящие указания по испытаниям стабильности: испытания стабильности новых лекарственных веществ и препаратов, 2003)
- СРМР/ICH/2738/99 (ICH Topic Q3A (R2)) Note for Guidance on Impurities Testing: Impurities in New Drug Substances, October 2006
- (СРМР/ICH/2738/99 (ICH Topic Q3A (R2)) Настанова щодо випробувань на домішки: Домішки в нових лікарських речовинах, жовтень 2006)
- СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) Note for Guidance on Impurities in New Drug Products, June 2006
- (СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) Настанова щодо домішок в нових лікарських препаратах, червень 2006)
Терміни та визначення понять
Нижче подано терміни, вжиті в цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1, 2] (див. національний додаток «Бібліографія»)N.
1. Терміни та визначення понять до підрозділу А цієї настанови [1]:
Вихідна речовина; вихідна сировина (starting material)
Речовина, що використовується при синтезі нової лікарської речовини і входить як фрагмент до структури проміжної продукції та/або нової лікарської речовини. Вихідні речовини, як правило, доступні у промисловому масштабі, а їх структура, хімічні та фізичні властивості встановлені.
Домішка (impurity)
Будь-який компонент нової лікарської речовини, який за своєю хімічною сутністю не є новою лікарською речовиною.
Домішка-енантіомер (enantiomeric impurity)
Сполука, що має однакову молекулярну формулу з лікарською речовиною, але відрізняється від неї просторовим розташуванням атомів в молекулі і є її дзеркальним відображенням.
Дослідження з хімічної розробки (chemical development studies)
Дослідження, що проведені з метою масштабування, оптимізації та валідації виробничого процесу для нової лікарської речовини.
Ідентифікована домішка (identified impurity)
Домішка, для якої встановлена структурна характеристика.
Кваліфікація (qualification)
Процес одержання й оцінки даних, на підставі яких визначають біологічну безпеку індивідуальної домішки або даного профілю домішок при встановленому(них) рівні(ях) вмісту.
Ліганд (ligand)
Агент із сильною спорідненістю до іонів металів.
Неідентифікована домішка (unidentified impurity)
Домішка, для якої не встановлені структурні характеристики, а визначені лише якісні аналітичні властивості (наприклад час утримування при хроматографуванні).
Неспецифікована домішка (unspecified impurity)
Домішка, для якої встановлюють межі в специфікації на нову лікарську речовину на підставі загальноприйнятих критеріїв прийнятності, але яку не вказують окремо з установленням спеціальних критеріїв прийнятності.
Нова лікарська речовина (new drug substance)
Компонент з установленою терапевтичною активністю, який раніше не було зареєстровано в УкраїніN або державі ЄС (також названий новою молекулярною сполукою або новою хімічною сполукою). Це може бути комплексна сполука, простий ефір або сіль раніше санкціонованої лікарської речовини.
Поліморфні форми (polymorphic forms)
Різні кристалічні форми однієї й тієї ж лікарської речовини. До них належать сольватовані або гідратовані продукти (їх називають ще псевдополіморфними формами), а також аморфні форми.
Поріг ідентифікації (identification threshold)
Межа, при перевищенні якої (>) домішку слід ідентифікувати.
Примітка. Синонім цього терміна — «порогове значення для ідентифікації» (див. загальний текст 5.10 «Контроль домішок у субстанціях для фармацевтичного застосування» у Доповненні 2 до Державної Фармакопеї України)N.
Поріг інформування (reporting threshold)
Межа, при перевищенні якої (>) про домішку необхідно інформувати. Поріг інформування — це те саме, що й рівень інформування.
Примітка. Синонім цього терміна — «порогове значення для інформування» (див. загальний текст 5.10 «Контроль домішок у субстанціях для фармацевтичного застосування» у Доповненні 2 до Державної Фармакопеї України)N.
Поріг кваліфікації (qualification threshold)
Межа, при перевищенні якої (>) домішку слід кваліфікувати.
Примітка. Синонім цього терміна — «порогове значення для кваліфікації» (див. загальний текст 5.10 «Контроль домішок у субстанціях для фармацевтичного застосування» у Доповненні 2 до Державної Фармакопеї України)N.
Потенційна домішка (potential impurity)
Домішка, що теоретично може утворитися при виробництві або зберіганні. Насправді вона може бути або наявною, або відсутньою у новій лікарській речовині.
Проміжна продукція (intermediate)
Речовина, яку отримують на стадіях синтезу нової лікарської речовини і яка піддається подальшому хімічному перетворенню, перш ніж стане новою лікарською речовиною.
Профіль домішок (impurity profile)
Опис ідентифікованих і неідентифікованих домішок, що наявні в новій лікарській речовині.
Реактив (reagent)
Речовина, що використовується при виробництві нової лікарської речовини і не належить до вихідної сировини, проміжної продукції або розчинників.
Розчинник (solvent)
Неорганічна або органічна рідина, що використовується як носій для приготування розчинів чи суспензій при синтезі нової лікарської речовини.
Рослинні препарати (herbal products)
Лікарські засоби, що містять діючі речовини, що є виключно рослинними речовинами та/або рослинними лікарськими препаратами. У деяких випадках також можуть бути наявні неорганічні речовини або матеріали тваринного походження.
Специфікована домішка (specified impurity)
Домішка, що в специфікації на нову лікарську речовину вказана окремо, і для якої встановлені межі на підставі спеціальних критеріїв прийнятності. Специфікована домішка може бути ідентифікованою або неідентифікованою.
Стороння забруднююча домішка (extraneous contaminant)
Домішка, що виникає внаслідок надходження зі стороннього джерела в процесі виробництва.
2. Терміни та визначення понять до підрозділу В цієї настанови [2]:
Домішка (impurity)
Будь-який компонент нового лікарського препарату, який не є лікарською речовиною або допоміжною речовиною в лікарському препараті.
Дослідження з розробки (development studies)
Дослідження, що проведені з метою масштабування, оптимізації та валідації виробничого процесу для нового лікарського препарату.
Ідентифікований продукт розкладання (identified degradation product)
Продукт розкладання, для якого установлена структурна характеристика.
Кваліфікація (qualification)
Процес одержання й оцінки даних, на підставі яких визначають біологічну безпеку індивідуального продукту розкладання або даного профілю продуктів розкладання при встановленому(них) рівні(ях) вмісту.
Неідентифікований продукт розкладання (unidentified degradation product)
Продукт розкладання, для якого не встановлені структурні характеристики, а визначені лише якісні аналітичні властивості (наприклад час утримування при хроматографуванні).
Неспецифікований продукт розкладання (unspecified degradation product)
Продукт розкладання, для якого встановлюють межі в специфікації на новий лікарський препарат на підставі загальноприйнятих критеріїв прийнятності, але який не вказують окремо з визначенням спеціальних критеріїв прийнятності.
Нова лікарська речовина (new drug substance)
Див. визначення терміна в п. 1 розділу «Терміни та визначення понять».
Поріг ідентифікації (identification threshold)
Межа, при перевищенні якої (>) продукт розкладання слід ідентифікувати.
Поріг інформування (reporting threshold)
Межа, при перевищенні якої (>) про продукт розкладання необхідно інформувати.
Поріг кваліфікації (qualification threshold)
Межа, при перевищенні якої (>) продукт розкладання слід кваліфікувати.
Продукт розкладання (degradation product)
Домішка, що виникає в результаті хімічної зміни лікарської речовини, що відбувається під час виробництва та/або зберігання нового лікарського препарату під впливом, наприклад, світла, температури, рН або внаслідок реакції з допоміжною речовиною та/або первинним пакувальним матеріалом (системою контейнер/закупорювальний засіб).
Профіль домішок (impurity profile)
Опис ідентифікованих і неідентифікованих домішок, що наявні в новому лікарському препараті.
Профіль продуктів розкладання (degradation profile)
Опис продуктів розкладання, що виявлені в лікарській речовині або лікарському препараті.
Специфікований продукт розкладання (specified degradation product)
Продукт розкладання, який в специфікації на новий лікарський препарат указано окремо, і для якого встановлені межі на підставі спеціальних критеріїв прийнятності. Специфікований продукт розкладання може бути ідентифікованим або неідентифікованим.
Познаки та скорочення
ЄС — Європейський Союз
МОЗ — Міністерство охорони здоров’я
СТ-Н МОЗУ — настанова МОЗ України
CPMP або CHMP — Committee for Medicinal Products for Human Use (Комітет з лікарських засобів для людини)
CTD — Common Technical Document (загальний технічний документ)
EMEA — European Medicines Agency (Європейське агентство з лікарських засобів)
GMP — Good Manufacturing Practice (належна виробнича практика)
ICH — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
TDI — Total Daily Intake (загальний добовий прийом)
Положення щодо випробувань на домішки
А. Домішки в нових лікарських речовинах
А.1. Вступ
Підрозділ А цього документа є настановою для складання реєстраційних досьє відносно вмісту та кваліфікації домішок у нових лікарських речовинах, які отримують шляхом хімічного синтезу, що раніше не були зареєстровані в УкраїніN або державі ЄС. Він не поширюється на нові лікарські речовини, що використовують при розробці на стадії клінічних досліджень. Підрозділ А цієї настанови також не поширюється на такі види лікарських речовин: біологічні/біотехнологічні, пептиди, олігопептиди, радіофармацевтичні, продукти ферментації й одержувані з них напівсинтетичні продукти, рослинні препарати та сировина тваринного або рослинного походження.
Домішки в нових лікарських речовинах слід розглядати з двох точок зору:
- хімічні аспекти, що включають класифікацію й ідентифікацію домішок, опис їх утворення, перелік домішок у специфікаціях, а також коротке обговорення аналітичних методик; та
- аспекти безпеки, що включають спеціальну настанову для кваліфікації тих домішок, що були відсутні або наявні (головним чином на низьких рівнях вмісту) в серіях нової лікарської речовини, використаних при вивченні безпеки та клінічних випробуваннях.
А.2. Класифікація домішок
Домішки можуть бути класифіковані за такими категоріями:
- органічні домішки (пов’язані з процесом і лікарським засобом);
- неорганічні домішки;
- залишкові розчинники.
Органічні домішки можуть виникати внаслідок виробничого процесу та/або при зберіганні нової лікарської речовини. Вони можуть бути ідентифікованими або неідентифікованими, леткими або нелеткими та включати:
- вихідні речовини (вихідну сировину);
- побічні продукти;
- проміжну продукцію;
- продукти розкладання;
- реактиви, ліганди та каталізатори.
Неорганічні домішки можуть з’являтися в результаті виробничого процесу. Як правило, вони є відомими й ідентифікованими та включають:
- реактиви, ліганди та каталізатори;
- важкі метали або інші залишкові метали;
- неорганічні солі;
- інші матеріали (наприклад допоміжний матеріал для фільтрації, вугілля).
Розчинники — це неорганічні або органічні рідини, що використовуються як носії для приготування розчинів чи суспензій при синтезі нової лікарської речовини. Оскільки їх токсичність, як правило, відома, легко вибрати відповідний контроль (див. CPMP/ICH/283/95 (ICH Topic Q3С (R3)) «Note for Guidance on Impurities: Residual Solvents» та/або загальний текст 5.4 «Залишкові кількості органічних розчинників» Державної Фармакопеї України (Доповнення 1)N).
До винятків з правил підрозділу А цього документа належать: (1) сторонні забруднюючі домішки, що не мають утворюватися в нових лікарських речовинах і можуть бути пов’язані, відповідно, з питаннями належної виробничої практики (GMP)1; (2) поліморфні форми; (3) домішки-енантіомери.
1Тут і далі під належною виробничою практикою (GMP) слід розуміти відповідні положення, що регламентовані в чинних версіях настанови МОЗ України «Лікарські засоби. Належна виробнича практика» (див. розділ «Нормативні посилання») та настанови з GMP ЄС (див. [19, 20] у національному додатку «Бібліографія»).
А.3. Обґрунтування щодо інформування про домішки та їх контролю
А.3.1. Органічні домішки
Заявник повинен навести узагальнений опис про дійсно наявні та можливі домішки, що імовірно можуть утворюватися під час синтезу, очищення та зберігання нової лікарської речовини. Цей узагальнений опис має ґрунтуватися на логічній науковій оцінці хімічних реакцій, що відбуваються у ході синтезу, домішок, які пов’язані з сировиною та роблять свій внесок у профіль домішок нової лікарської речовини, а також можливих продуктів розкладання. Це докладне обговорення може стосуватися тільки тих домішок, наявність яких логічно чекати на підставі знань про хімічні реакції та наявних умов.
Крім того, заявник повинен узагальнити результати лабораторних досліджень, що проведені для виявлення домішок у новій лікарській речовині. Такий узагальнений опис має містити результати випробувань серій, що виготовлені під час розробки процесу, та серій, вироблених за допомогою пропонованого промислового процесу, а також результати стресових випробувань (див. CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) «Note for guidance on stability testing: stability testing of new drug substances and products», а також Настанову 42-3.3:2004 «Настанови з якості. Лікарські засоби. Випробування стабільності»N), проведених для ідентифікації можливих домішок, що виникають під час зберігання. Профіль домішок у серіях лікарської речовини, призначених для розміщення на ринку, необхідно порівняти з профілем домішок тих серій, що були використані при розробці; слід обговорити будь-які відмінності.
Необхідно описати дослідження, проведені з метою характеристики дійсно наявних у новій лікарській речовині домішок, що мають рівень вмісту більший (>) за поріг ідентифікації, зазначений у додатку А.1 (наприклад розрахований із використанням фактора відгуку лікарської речовини). Слід звернути увагу на те, що в будь-якій серії, що вироблена за допомогою пропонованого промислового процесу, будь-яка домішка з рівнем вмісту, що більший (>) за поріг ідентифікації, має бути ідентифікована. Крім того, будь-який продукт розкладання, що виявляють при дослідженнях стабільності за рекомендованих умов зберігання, при рівні вмісту більше (>), ніж поріг ідентифікації, має бути ідентифікований. Якщо ідентифікація домішки не може бути здійснена, то в досьє необхідно включити узагальнений опис результатів лабораторних досліджень, що демонструють невдалу спробу. Якщо були спроби ідентифікації домішок з рівнями вмісту, що не перевищують (≤) порогів ідентифікації, то також доцільно повідомити про результати таких досліджень.
Ідентифікація домішок з рівнями вмісту, що явно не перевищують (≤) порогів ідентифікації, як правило, не вважається обов’язковою. Проте для таких потенційних домішок, що можуть володіти надзвичайною силою дії, призводити до токсичних або фармакологічних ефектів при рівнях вмісту, що не перевищують (≤) порогів їх ідентифікації, необхідно розробити аналітичні методики. Усі домішки слід кваліфікувати, як описано далі в підрозділі А цієї настанови.
А.3.2. Неорганічні домішки
Неорганічні домішки, як правило, виявляють і кількісно визначають за допомогою фармакопейних або інших відповідних методик. Протягом розробки слід оцінити перенесення каталізаторів у нову лікарську речовину. Слід обговорити необхідність включення неорганічних домішок у специфікацію на нову лікарську речовину або виключення їх з цієї специфікації. Критерії прийнятності мають базуватися на фармакопейних нормах або відомих даних про безпеку.
А.3.3. Розчинники
Необхідно обговорити й навести відповідно до настанови CPMP/ICH/283/95 (ICH Topic Q3С (R3)) «Note for Guidance on Impurities: Residual Solvents» та/або загального тексту 5.4 «Залишкові кількості органічних розчинників» Державної Фармакопеї України (Доповнення 1)N контроль залишкової кількості розчинників, що використовують в процесі виробництва нової лікарської речовини.
А.4. Аналітичні методики
До реєстраційного досьє необхідно включити документоване підтвердження того, що аналітичні методики є валідованими й придатними для виявлення та кількісного визначення домішок (див. CPMP/ICH/381/95 (ICH Topic Q2 (R1)) «Note for Guidance on Validation of Analytical Procedures: Text and Methodology» та/або загальну статтю «Валідація аналітичних методик і випробувань» Державної Фармакопеї УкраїниN). Частиною обґрунтування при виборі альтернативних порогів можуть стати технічні фактори (наприклад продуктивність виробництва і методологія контролю) з урахуванням досвіду виробництва за допомогою пропонованого промислового процесу. Використання двох десяткових розрядів для порогів (див. додаток А.1) не обов’язково відображає точність аналітичної методики, яку використовують для рутинного контролю якості. Таким чином, при обґрунтуванні та належній валідації може бути прийнятним використання методів з більш низькою точністю (наприклад тонкошарова хроматографія). У реєстраційному досьє необхідно обговорити відмінності між аналітичними методиками, що застосовували при розробці, та тими, що пропонують для аналізу промислової продукції.
Межа кількісного визначення для аналітичної методики не має перевищувати (≤) порога інформування.
Рівні вмісту органічних домішок можуть бути визначені за допомогою різних методів, включаючи такі, за допомогою яких можна порівняти аналітичний відгук домішки з таким відповідного стандартного зразка або відгуком найновішої лікарської речовини. Стандартні зразки, що використовують в аналітичних методиках для контролю домішок, мають бути охарактеризовані й оцінені відповідно до їх призначення. Як стандарт для розрахунку рівнів вмісту домішок може бути використана лікарська речовина. Якщо фактори відгуку лікарської речовини та відповідної домішки не близькі, ця практика все ж таки може бути прийнятною за умови застосування коефіцієнту похибки (correction factor) або фактичного завищення вмісту домішок. Критерії прийнятності й аналітичні методики, що використовують для визначення ідентифікованих і неідентифікованих домішок, можуть ґрунтуватися на аналітичних допущеннях (наприклад однаковий відгук детектора). У реєстраційному досьє необхідно обговорити ці допущення.
А.5. Інформування про домішки, що містяться в серіях
У реєстраційному досьє слід надати результати аналітичних досліджень усіх серій нової лікарської речовини, що були використані для клінічних випробувань, досліджень безпеки та випробувань стабільності, а також тих серій, що є репрезентативними для пропонованого промислового процесу. Кількісні результати мають бути наведені в числовому вираженні, а не із застосуванням таких загальних виразів, як «відповідає», «знаходиться в межах» тощо. Необхідно інформувати про будь-яку домішку, рівень вмісту якої перевищує (>) поріг інформування (див. додаток А.1), а також про загальний вміст домішок, що виявлено в цих серіях нової лікарської речовини, із зазначенням аналітичних методик. Результати, що менші за 1,0 %, слід указувати з двома знаками після коми (наприклад 0,06%, 0,13%); результати, що становлять 1,0 % і більше, слід указувати з одним знаком після коми (наприклад 1,3%). Результати слід округлювати із використанням загальноприйнятих правил (див. додаток А.2). Рекомендується надавати дані у вигляді таблиць (наприклад великоформатних таблиць). Домішки слід позначати за допомогою числового коду або відповідної ознаки, наприклад часу утримування. Якщо пропонується більш високий поріг інформування, це необхідно повністю обґрунтувати. Слід підсумовувати вміст усіх домішок, рівень вмісту кожної з яких перевищує (>) поріг інформування, й указувати у вигляді загального вмісту домішок.
Якщо аналітичні методики замінюють під час розробки, при повідомленні про результати необхідно зазначати використану методику із наданням відповідної інформації про її валідацію. Мають бути передбачені репрезентативні хроматограми. Хроматограми репрезентативних серій, одержані при дослідженнях з валідації аналітичних методик, що доводять розділення та можливість визначення домішок (наприклад хроматограми зразків із доданими домішками), разом із результатами будь-яких інших рутинних випробувань відносно домішок можуть служити для відображення профілів домішок. Заявник повинен гарантувати наявність повних профілів домішок в окремих серіях (наприклад відображених на хроматограмах) та їх надання, якщо буде така вимога.
Необхідно надати таблицю, що дозволяє встановити, яка окрема серія нової лікарської речовини була використана в кожному дослідженні безпеки та кожному клінічному випробуванні.
Звіт для кожної серії нової лікарської речовини має містити:
- ідентифікатор серії та її розмір;
- дату виробництва;
- виробничу дільницю;
- виробничий процес;
- вміст окремих домішок і загальний вміст домішок;
- призначення серій;
- посилання на застосовану аналітичну методику.
А.6. Перелік домішок у специфікаціях
Специфікація на нову лікарську речовину має містити перелік домішок. Для прогнозування тих домішок, що, ймовірно, будуть наявні в промисловій продукції, слід використовувати результати вивчення стабільності, досліджень з хімічної розробки та рутинних аналізів серій. Вибір домішок для включення у специфікацію на нову лікарську речовину має ґрунтуватися на домішках, виявлених у серіях, що були вироблені за допомогою пропонованого промислового процесу. Ті окремі домішки із спеціальними критеріями прийнятності, що включені у специфікацію на нову лікарську речовину, в підрозділі А цієї настанови зазначені як «специфіковані домішки». Специфіковані домішки можуть бути як ідентифікованими, так і неідентифікованими.
Необхідно надати обґрунтування щодо включення домішок у специфікацію або їх виключення зі специфікації. Таке обґрунтування має містити обговорення профілів домішок в тих серіях, що були використані при дослідженнях безпеки та клінічних випробуваннях; необхідно також враховувати профіль домішок для тих серій, що вироблені за допомогою пропонованого промислового процесу. Специфіковані ідентифіковані домішки необхідно включати разом зі специфікованими неідентифікованими домішками, якщо рівень їх вмісту більше (>) за поріг ідентифікації, зазначений в додатку А.1. Якщо відомо, що домішки володіють надзвичайно сильною дією або призводять до виникнення токсичних чи несподіваних фармакологічних ефектів, межа виявлення/кількісного визначення аналітичної методики має бути зіставною з рівнем, при якому домішки слід контролювати. Для неідентифікованих домішок мають бути чітко вказані методики та допущення, що зроблені для встановлення рівня домішки. Специфіковані неідентифіковані домішки мають бути указані за допомогою відповідних якісних аналітичних ознак (наприклад «неідентифікована домішка А», «неідентифікована домішка з відносним часом утримування 0,9»). Для будь-якої неспецифікованої домішки слід включити загальний критерій прийнятності, що не перевищує (≤) порога ідентифікації (див. додаток А.1), а також критерій прийнятності для загального вмісту домішок.
Критерії прийнятності слід встановлювати не вище рівня, що обґрунтований даними з безпеки; вони мають узгоджуватися з рівнем, що досягається після виробничого процесу, й аналітичними можливостями. Якщо це не пов’язано із безпекою, то критерії прийнятності для домішок мають ґрунтуватися на даних, одержаних для серій нової лікарської речовини, вироблених за допомогою пропонованого промислового процесу; допускається достатній запас з урахуванням звичайної виробничої й аналітичної варіабельності, а також показників стабільності нової діючої речовини. Хоча очікуються звичайні виробничі зміни, значна варіабельність рівнів домішок від серії до серії може свідчити про те, що процес виробництва нової лікарської речовини недостатньо контрольований та валідований (див. схему рішень № 1 щодо встановлення критеріїв прийнятності відносно специфікованої домішки в новій лікарській речовині в документі СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та/або в Настанові 42-3.2:2004 «Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності»N). Використання двох значущих цифр після коми для порогів (див. додаток А.1) не обов’язково вказує на точність критеріїв прийнятності для специфікованих домішок і загального вмісту домішок.
Отже, специфікація на нову лікарську речовину має містити, якщо необхідно, наступний перелік домішок:
- органічні домішки:
- кожна специфікована ідентифікована домішка;
- кожна специфікована неідентифікована домішка;
- будь-яка неспецифікована домішка з критерієм прийнятності, що не перевищує (≤) порога ідентифікації;
- загальний вміст домішок;
- залишкові розчинники;
- неорганічні домішки.
А.7. Кваліфікація домішок
Кваліфікація — це процес отримання та оцінки даних для встановлення біологічної безпеки окремої домішки або даного профілю домішок при заданому(их) рівні(ях) вмісту. Заявник повинен надати обґрунтування щодо встановлення критеріїв прийнятності для домішки з урахуванням питань безпеки. Рівень будь-якої домішки, наявної в новій лікарській речовині, що була достатньо вивчена при дослідженнях безпеки та/або клінічних випробуваннях, слід розглядати як кваліфікований. Наявні при дослідженнях на тваринах та/або людях домішки, що також є основними метаболітами, як правило, вважаються кваліфікованими. Також може бути обґрунтований більш високий рівень кваліфікованої домішки, ніж наявний у новій лікарській речовині, на підставі аналізу дійсної кількості домішки, яку вводили у попередніх дослідженнях безпеки, що стосуються справи.
Якщо перевищені звичайні пороги кваліфікації, наведені в додатку А.1, та якщо для домішки відсутні дані щодо кваліфікації пропонованого критерію прийнятності, можуть бути необхідні дослідження з отримання таких даних.
Для деяких окремих лікарських засобів можуть бути прийнятні більш високі або більш низькі пороги для кваліфікації домішок з урахуванням наукового обґрунтування та рівня вмісту, включаючи фармакологічні ефекти даної групи препаратів і досвід клінічного застосування. Наприклад, кваліфікація може бути особливо важливою, якщо є дані, що раніше такі домішки в певних лікарських засобах або фармакотерапевтичних групах були пов’язані з побічними реакціями у пацієнтів. У таких випадках може бути прийнятним менший поріг кваліфікації. Навпаки, більший поріг кваліфікації може бути прийнятним для окремих лікарських засобів, якщо рівень вмісту, пов’язаний з безпекою, менше звичайного на підставі аналогічних міркувань (наприклад популяція пацієнтів, ефекти даної групи лікарських засобів, аспекти клінічного застосування). Пропозиції щодо альтернативних порогів слід розглядати у кожному конкретному випадку.
У додатку A.3 («Схема рішень щодо ідентифікації та кваліфікації домішок») описані умови для кваліфікації домішок при перевищенні порогів. У деяких випадках зниження рівня вмісту домішки до значення, що не перевищує порога, може бути доцільнішим, ніж надання даних з безпеки. Альтернативою кваліфікації домішки можуть бути достатні дані наукової літератури. Якщо не підходить жоден з описаних вище випадків, слід розглянути додаткові випробування безпеки. Чи вважатимуться дослідження відповідними для кваліфікації домішки, залежатиме від кількості факторів, включаючи популяцію пацієнтів, добову дозу, шлях введення та тривалість застосування. Такі дослідження можуть бути проведені з використанням нової лікарської речовини, що містить домішки, які підлягають контролю; хоча іноді можуть бути придатними дослідження із використанням виділених домішок.
Хоча підрозділ А цієї настанови не призначений для застосування на етапі розробки, пов’язаному з клінічними випробуваннями, на пізніших етапах розробки надані в підрозділі А цієї настанови пороги можуть бути корисними при оцінці нових домішок, наявних у тих серіях лікарської речовини, що були вироблені за допомогою пропонованого промислового процесу. Будь-яка нова домішка, що виявлена на більш пізніх етапах розробки, має бути ідентифікована, якщо рівень її вмісту перевищує (>) поріг ідентифікації, наведений у додатку А.1 (див. «Схему рішень щодо ідентифікації та кваліфікації домішок» у додатку A.3). При дослідженнях щодо оцінки безпеки для кваліфікації домішки слід порівняти нову лікарську речовину, що містить репрезентативну кількість нової домішки, із заздалегідь кваліфікованою речовиною. Також можуть бути розглянуті дослідження щодо оцінки безпеки із використанням виділеної домішки.
Додаток А.1 (обов’язковий). Пороги для домішок в нових лікарських речовинах
|
Максимальна добова доза1 |
Поріг інформування2,3 |
Поріг ідентифікації3 |
Поріг кваліфікації3 |
|
≤2 г/доба |
0,05% |
0,10% або 1,0 мг/доба |
0,15% або 1,0 мг/доба |
|
>2 г/доба |
0,03% |
0,05% |
0,05% |
1Кількість лікарської речовини, що приймається протягом доби.
2Вищі пороги ідентифікації мають бути науково обґрунтовані.
3Можуть бути прийнятні нижчі пороги, якщо домішка є надзвичайно токсичною.
Додаток А.2 (обов’язковий). Пояснення стосовно наведення в реєстраційному досьє результатів ідентифікації та кваліфікації домішок
Цей додаток наведений тільки з метою пояснення; він не служить прикладом того, які саме результати відносно домішок слід представляти у реєстраційному досьє. Як правило, вихідні дані не представляють.
Приклад 1: Максимальна добова доза становить 0,5 г
Поріг інформування = 0,05%
Поріг ідентифікації = 0,10%
Поріг кваліфікації = 0,15%
|
«Вихідний» результат (%) |
Повідомлений результат (%) |
Розрахований загальний добовий |
Дія |
|
| Ідентифікація (чи перевищений поріг 0,10%?) | Кваліфікація (чи перевищений поріг 0,15%?) | |||
|
0,044 |
Не повідомляють |
0,2 |
Ні |
Ні |
|
0,0963 |
0,10 |
0,5 |
Ні |
Ні |
|
0,12 |
0,121) |
0,6 |
Так |
Ні1) |
|
0,1649 |
0,161) |
0,8 |
Так |
Так1) |
Приклад 2: Максимальна добова доза становить 0,8 г
Поріг інформування = 0,05%
Поріг ідентифікації = 0,10%
Поріг кваліфікації = 1,0 мг TDI
|
«Вихідний» результат (%) |
Повідомлений результат (%) |
Розрахований загальний добовий |
Дія |
|
| Ідентифікація (чи перевищений поріг 0,10%?) | Кваліфікація (чи перевищений поріг 1,0 мг TDI?) | |||
|
0,066 |
0,07 |
0,6 |
Ні |
Ні |
|
0,124 |
0,12 |
1,0 |
Так |
Ні1, 2 |
|
0,143 |
0,14 |
1,1 |
Так |
Так1 |
1Після ідентифікації, якщо певний фактор відгуку дуже відрізняється від первинного допущення, може бути доцільним провести повторне визначення кількості наявної домішки та повторну оцінку порога кваліфікації (див. додаток А.1).
2Для контролю, якщо поріг перевищений, повідомлені результати слід оцінити відносно порогів таким чином: якщо поріг зазначено в %, повідомлений результат, округлений до такого ж знака після коми, що й поріг, слід безпосередньо порівняти з порогом. Якщо поріг зазначено як TDI, повідомлений результат слід перевести у TDI, округлити до такого ж знака після коми, що й поріг, і безпосередньо порівняти з порогом. Наприклад, кількість домішки при рівні вмісту 0,12% відповідає TDI 0,96 мг (абсолютна кількість), що округлили до 1,0 мг; таким чином, поріг кваліфікації, зазначений як TDI (1,0 мг), не перевищений.
Додаток А.3 (обов’язковий). Схема рішень щодо ідентифікації та кваліфікації домішок
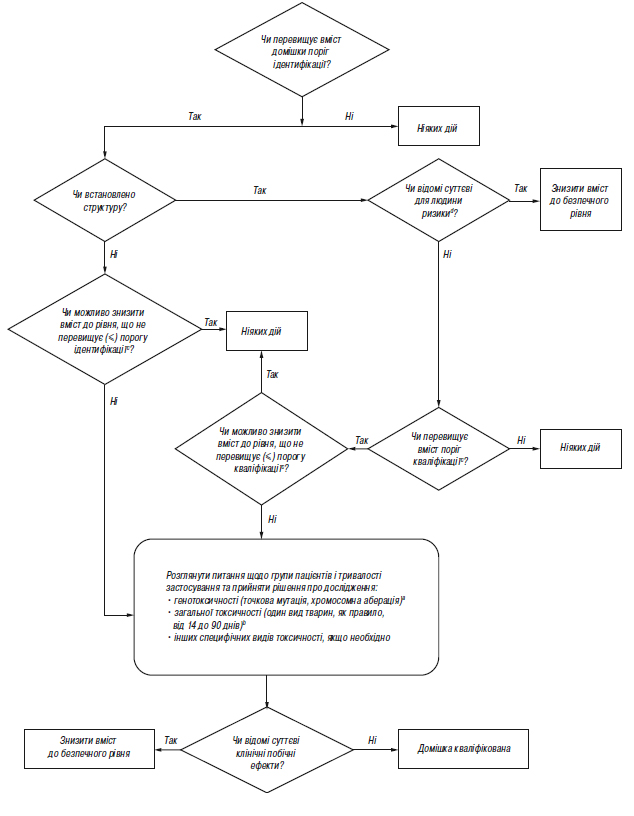
aЯкщо доцільно, то слід провести мінімальний скринінг (наприклад за генотоксичністю). Для мінімального скринінгу прийнятними вважаються дослідження з визначення точкової мутації та з визначення хромосомної аберації.
bЯкщо необхідним є вивчення загальної токсичності, одне дослідження або декілька досліджень мають дозволяти провести порівняння некваліфікованої та кваліфікованої речовини. Тривалість дослідження слід встановлювати на підставі наявної значущої інформації; дослідження необхідно проводити на таких видах тварин, на яких можливість визначити токсичність домішки є максимальною. З огляду на кожний конкретний випадок, можуть бути прийнятними дослідження гострої токсичності, особливо у разі однодозових ліків. Взагалі, прийнятними вважаються дослідження тривалістю мінімум 14 днів і максимум 90 днів.
cМожуть бути прийнятні нижчі пороги, якщо домішка є надзвичайно токсичною.
dНаприклад, чи відомі вам дані щодо безпеки цієї домішки, або чи заважає її структура застосуванню у людини при даній концентрації?
В. Домішки в нових лікарських препаратах
В.I. Вступ
В.1.1. Мета
У підрозділі В цього документа надано настанову для складання реєстраційних досьє відносно вмісту та кваліфікації домішок у нових лікарських препаратах, що містять одержані шляхом хімічного синтезу нові лікарські речовини, які раніше не були зареєстровані в УкраїніN або державі ЄС.
В.1.2. Передумови
У цій настанові підрозділ В доповнює підрозділ А, що гармонізовано з настановою СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) «Note for Guidance on Impurities Testing: Impurities in New Drug Substances», до якого слід звертатися відносно основних принципів. Якщо необхідно, слід також брати до уваги настанову CPMP/ICH/283/95 (ICH Topic Q3С (R3)) «Note for Guidance on Impurities: Residual Solvents» та/або загальний текст 5.4 «Залишкові кількості органічних розчинників» Державної Фармакопеї України (Доповнення 1)N.
В.1.3. Сфера застосування
Підрозділ В цієї настанови поширюється тільки на ті домішки в нових лікарських препаратах, що відносяться до продуктів розкладання лікарської речовини або продуктів реакції лікарської речовини з допоміжною речовиною та/або з первинним пакувальним матеріалом (системою контейнер/закупорювальний засіб); у цій настанові їх загалом називають «продукти розкладання». Як правило, домішки, що наявні в новій лікарській речовині, не вимагається контролювати або включати у специфікацію на новий лікарський препарат за винятком тих випадків, коли вони також є продуктами розкладання (див. СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та/або Настанову 42-3.2:2004 «Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності»N).
На домішки, що спричинені наявністю в новому лікарському препараті допоміжних речовин, або речовин, що екстрагуються чи виділяються з системи контейнер/закупорювальний засіб, підрозділ В цієї настанови не поширюється. Підрозділ В цієї настанови також не застосовний до нових лікарських препаратів, що знаходяться на стадії розробки, пов’язаної з клінічними випробуваннями. Ця настанова не поширюється на такі види препаратів: біологічні/біотехнологічні препарати, пептиди, олігонуклеотиди, радіофармацевтичні препарати, продукти ферментації й одержувані з них напівсинтетичні препарати, рослинні препарати, а також сировина тваринного та рослинного походження. Зі сфери застосування підрозділу В цього документа також виключені: (1) сторонні забруднюючі домішки, що відсутні в нових лікарських препаратах і, найбільш вірогідно, пов’язані з питаннями належної виробничої практики (GMP); (2) поліморфні форми; (3) домішки-енантіомери.
В.II. Обґрунтування щодо інформування про продукти розкладання та їх контролю
Заявник повинен узагальнити дані про продукти розкладання, що виявлені в ході виробництва та/або досліджень стабільності нового лікарського препарату. Такі узагальнені дані мають ґрунтуватися на ретельній науковій оцінці потенційних шляхів розкладання нового лікарського препарату та домішок, що виникають внаслідок його взаємодії з допоміжними речовинами та/або первинним пакувальним матеріалом (системою контейнер/ закупорювальний засіб). Крім того, заявник повинен узагальнити результати будь-яких лабораторних досліджень, проведених з метою визначення продуктів розкладання в новому лікарському препараті. Такі узагальнені дані мають також містити результати випробувань серій, вироблених в ході розробки, і серій, репрезентативних для пропонованого промислового процесу. Необхідно надати обґрунтування для виключення тих домішок, що не є продуктами розкладання (наприклад домішок, наявних в лікарській речовині, або домішок, що з’являються з допоміжних речовин). Профіль домішок для серій, репрезентативних для пропонованого промислового процесу, необхідно порівняти з профілями домішок для тих серій, що використовували при розробці; будь-які відмінності слід обговорити.
Будь-який продукт розкладання, виявлений при дослідженнях стабільності за рекомендованих умов зберігання, необхідно ідентифікувати, якщо рівень його вмісту перевищує (>) поріг ідентифікації, наведений у додатку В.1. Якщо ідентифікація продукту розкладання нездійсненна, в реєстраційне досьє необхідно включити узагальнені дані про лабораторні дослідження, що свідчать про невдалі спроби ідентифікації.
Якщо рівні вмісту продуктів розкладання не перевищують (≤) порога ідентифікації, як правило, їх не потрібно ідентифікувати. Проте слід розробити методики аналізу для таких продуктів розкладання, що можуть володіти надзвичайно сильною дією, призводити до токсичних або значних фармакологічних ефектів при рівнях вмісту, що не перевищують (≤) порога ідентифікації. У виняткових випадках можуть бути розглянуті технічні фактори (наприклад продуктивність, низький вміст лікарської речовини порівняно із вмістом допоміжних речовин або використання допоміжних речовин, що є сировиною тваринного чи рослинного походження) як частина обґрунтування для вибору альтернативних порогів з урахуванням досвіду виробництва за пропонованим промисловим процесом.
В.III. Аналітичні методики
До реєстраційного досьє необхідно включити документоване підтвердження того, що аналітичні методики є валідованими й придатними для виявлення та кількісного визначення продуктів розкладання (див. CPMP/ICH/381/95 (ICH Topic Q2 (R1)) «Note for Guidance on Validation of Analytical Procedures: Text and Methodology» та/або загальну статтю «Валідація аналітичних методик і випробувань» Державної Фармакопеї УкраїниN). Зокрема, аналітичні методики мають бути валідовані для доказу специфічності відносно специфікованих і неспецифікованих продуктів розкладання. Якщо необхідно, то при такій валідації слід використовувати зразки, що зберігалися у відповідних стресових умовах: світло, тепло, вологість, кислотний/лужний гідроліз й окиснення. Якщо при використанні аналітичної методики виявляється наявність інших піків додатково до піків продуктів розкладання (наприклад піки лікарської речовини, домішок, що виникають в ході синтезу лікарської речовини, допоміжних речовин і домішок з допоміжних речовин), то такі піки слід позначити на хроматограмах й обговорити їх походження у валідаційній документації.
Межа кількісного визначення для аналітичної методики не має перевищувати (≤) порога інформування.
Рівні вмісту продуктів розкладання можуть бути визначені за допомогою різних методів, включаючи такі, за допомогою яких можна порівняти аналітичний відгук продукту розкладання з таким відповідного стандартного зразка або відгуком найновішої лікарської речовини. Стандартні зразки, що використовують в аналітичних методиках для контролю продуктів розкладання, мають бути охарактеризовані й оцінені відповідно до їх призначення. Для розрахунку рівнів вмісту продуктів розкладання може бути використана лікарська речовина. Якщо фактори відгуку неблизькі, ця практика все ж таки може бути прийнятною за умови застосування коефіцієнту похибки (correction factor) або фактичного завищення вмісту продуктів розкладання. Критерії прийнятності й аналітичні методики, що використовують для визначення ідентифікованих і неідентифікованих продуктів розкладання, можуть ґрунтуватися на аналітичних допущеннях (наприклад однаковий відгук детектора). У реєстраційному досьє необхідно обговорити ці допущення.
Необхідно обговорити відмінності між аналітичними методиками, що застосовували при розробці, та тими, що пропонують для продукції, що виробляється серійно.
В.IV. Інформування про продукти розкладання, що містяться в серіях
У реєстраційному досьє слід надати результати аналітичних досліджень усіх серій нового лікарського препарату, що були використані для клінічних випробувань, досліджень безпеки та випробувань стабільності, а також тих серій, що є репрезентативними для пропонованого промислового процесу. Кількісні результати мають бути наведені в числовому вираженні, а не із застосуванням таких загальних виразів, як «відповідає», «знаходиться в межах» тощо. Необхідно інформувати про будь-який продукт розкладання, рівень вмісту якого перевищує (>) поріг інформування (див. додаток В.1), а також про загальний вміст продуктів розкладання, що виявляють у цих серіях нового лікарського препарату, із зазначенням аналітичних методик. Результати, що менші за 1,0%, слід указувати до такого знака після коми, як у разі відповідного порога інформування (наприклад 0,06%); результати, що становлять 1,0% і більше, слід указувати з одним знаком після коми (наприклад 1,3%). Результати слід округлювати із використанням загальноприйнятих правил (див. додаток В.2). Рекомендується надавати дані у вигляді таблиць (наприклад великоформатних таблиць). Продукти розкладання слід позначати за допомогою числового коду або відповідної ознаки, наприклад часу утримування. Якщо пропонується більш високий поріг інформування, це необхідно повністю обґрунтувати. Слід підсумовувати вміст усіх продуктів розкладання, рівень вмісту кожного з яких перевищує (>) поріг інформування, й указувати у вигляді загального вмісту продуктів розкладання.
Слід навести хроматограми з позначеними піками (або інші еквівалентні дані при використанні інших аналітичних методик) для репрезентативних серій, включаючи хроматограми, одержані при дослідженнях з валідації аналітичних методик, а також довгострокових і прискорених дослідженнях стабільності. Заявник повинен гарантувати наявність повних профілів продуктів розкладання в окремих серіях (наприклад відображених на хроматограмах) та їх надання, якщо буде така вимога.
Документація для кожної серії нового лікарського препарату, що описаний в реєстраційному досьє, має містити:
- ідентифікатор, силу дії та розмір серії;
- дату виробництва;
- виробничу дільницю;
- виробничий процес;
- первинний пакувальний матеріал (контейнер/закупорювальний засіб);
- вміст окремих продуктів розкладання та загальний вміст продуктів розкладання;
- призначення серій (наприклад клінічні випробування, дослідження стабільності);
- посилання на застосовану аналітичну методику;
- номер серії лікарської речовини, використаної в новому лікарському препараті;
- умови зберігання для досліджень стабільності.
В.V. Перелік продуктів розкладання у специфікаціях
Специфікація на новий лікарський препарат має містити перелік передбачуваних продуктів розкладання, що можуть виникати в ході серійного виробництва препарату та за рекомендованих умов зберігання. Для характеристики профілю продуктів розкладання слід використовувати результати вивчення стабільності, знання про шляхи розкладання, результати досліджень з розробки препарату, а також лабораторних досліджень. Вибір продуктів розкладання для включення у специфікацію на новий лікарський препарат має ґрунтуватися на продуктах розкладання, виявлених у серіях, що були вироблені за допомогою пропонованого промислового процесу. Ті окремі продукти розкладання із спеціальними критеріями прийнятності, що включені у специфікацію на новий лікарський препарат, в підрозділі В цієї настанови зазначені як «специфіковані продукти розкладання». Специфіковані продукти розкладання можуть бути як ідентифікованими, так і неідентифікованими.
Необхідно надати обґрунтування щодо включення продуктів розкладання у специфікацію або їх виключення зі специфікації. Таке обґрунтування має містити обговорення профілів продуктів розкладання в тих серіях, що були використані при дослідженнях безпеки, клінічних випробуваннях і дослідженнях стабільності; необхідно також враховувати профіль продуктів розкладання для тих серій, що вироблені за допомогою пропонованого промислового процесу. Специфіковані ідентифіковані продукти розкладання необхідно включати разом зі специфікованими неідентифікованими продуктами розкладання, якщо рівень їх вмісту більше (>) порога ідентифікації, зазначеного в додатку В.1. Якщо відомо, що продукти розкладання володіють надзвичайно сильною дією або призводять до виникнення токсичних чи несподіваних фармакологічних ефектів, межа виявлення/кількісного визначення аналітичної методики має бути зіставною з рівнем, при якому продукти розкладання слід контролювати. Для неідентифікованих продуктів розкладання мають бути чітко вказані методики та допущення, що зроблені для встановлення рівня продукту розкладання. Специфіковані неідентифіковані продукти розкладання мають бути указані за допомогою відповідних якісних аналітичних ознак (наприклад «неідентифікований продукт А», «неідентифікований продукт з відносним часом утримування 0,9»). Для будь-якого неспецифікованого продукту розкладання слід включити загальний критерій прийнятності, що не перевищує (≤) порога ідентифікації (див. додаток В.1), а також критерій прийнятності для загального вмісту продуктів розкладання.
Для даного продукту розкладання критерії прийнятності слід встановлювати з урахуванням критеріїв прийнятності, встановлених для нього в лікарській речовині (якщо необхідно), його кваліфікованого рівня, збільшення вмісту при дослідженнях стабільності, а також з урахуванням пропонованого терміну придатності й рекомендованих умов зберігання для нового лікарського препарату. Більше того, кожний критерій прийнятності має бути встановлений не вище, ніж кваліфікований рівень для даного продукту розкладання.
Якщо це не пов’язано з безпекою, то критерії прийнятності для продукту розкладання мають ґрунтуватися на даних, одержаних для серій нового лікарського препарату, вироблених за допомогою пропонованого промислового процесу; допускається достатній запас з урахуванням звичної виробничої й аналітичної варіабельності, а також показників стабільності нового лікарського препарату. Хоча очікуються звичайні виробничі зміни, значна варіабельність рівнів продуктів розкладання від серії до серії може свідчити про те, що процес виробництва нового лікарського препарату недостатньо контрольований та валідований (див. схему рішень № 2 щодо встановлення критеріїв прийнятності відносно специфікованого продукту розкладання в новому лікарському препараті в документі СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та/або в Настанові 42-3.2:2004 «Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності»N).
У підрозділі В цієї настанови використання двох значущих цифр після коми для порогів (див. додаток В.1) не обов’язково указує на точність критеріїв прийнятності для специфікованих продуктів розкладання та загального вмісту продуктів розкладання.
Отже, специфікація на новий лікарський препарат має містити, якщо необхідно, наступний перелік продуктів розкладання:
- кожний специфікований ідентифікований продукт розкладання;
- кожний специфікований неідентифікований продукт розкладання;
- будь-який неспецифікований продукт розкладання з критерієм прийнятності, що не перевищує (≤) порога ідентифікації;
- загальний вміст продуктів розкладання.
В.VI. Кваліфікація продуктів розкладання
Кваліфікація — це процес отримання та оцінки даних для встановлення біологічної безпеки окремого продукту розкладання або даного профілю продуктів розкладання при заданому(их) рівні(ях) вмісту. Заявник повинен надати обґрунтування щодо встановлення критеріїв прийнятності для продукту розкладання з урахуванням питань безпеки. Рівень будь-якого продукту розкладання, наявного в новому лікарському препараті, що був достатньо вивчений при дослідженнях безпеки та/або клінічних випробуваннях, слід розглядати як кваліфікований. Таким чином, доцільно включати будь-яку наявну інформацію про фактичний вміст продуктів розкладання в серіях, що були використані для досліджень безпеки та/або клінічних випробувань. Наявні при дослідженнях на тваринах та/або людях продукти розкладання, що також є основними метаболітами, як правило, вважаються кваліфікованими. Продукти розкладання можна вважати кваліфікованими при рівнях вмісту, вищих за рівні їх вмісту при дослідженнях безпеки, на підставі порівняння між фактичними дозами, що приймалися при дослідженнях безпеки, і передбачуваною дозою нового лікарського препарату. При обґрунтуванні таких більш високих рівнів вмісту слід враховувати такі фактори, як: (1) кількість продукту розкладання, що вводили в попередніх дослідженнях безпеки та/або клінічних випробуваннях і визнали безпечним; (2) збільшення кількості продукту розкладання; та (3) інші фактори, що стосуються безпеки, якщо необхідно.
Якщо перевищені звичайні пороги кваліфікації, наведені в додатку В.1, та якщо для продукту розкладання відсутні дані щодо кваліфікації пропонованого критерію прийнятності, можуть бути необхідні додаткові дослідження для отримання таких даних (див. додаток В.3).
Для деяких окремих нових лікарських препаратів можуть бути прийнятні більш високі або більш низькі пороги для кваліфікації продуктів розкладання з урахуванням наукового обґрунтування та рівня вмісту, включаючи фармакологічні ефекти даної групи ліків і досвід клінічного застосування. Наприклад, кваліфікація може бути особливо важливою, якщо є дані, що раніше такі продукти розкладання в певних нових лікарських препаратах або фармакотерапевтичних групах були пов’язані з побічними реакціями у пацієнтів. У таких випадках може бути прийнятним менший поріг кваліфікації. Навпаки, більший поріг кваліфікації може бути прийнятним для окремих нових лікарських препаратів, якщо рівень вмісту, пов’язаний з безпекою, менше звичайного на підставі аналогічних міркувань (наприклад популяція пацієнтів, ефекти даної групи ліків, аспекти клінічного застосування). Пропозиції щодо альтернативних порогів слід розглядати у кожному конкретному випадку.
У додатку В.3 («Схема рішень щодо ідентифікації та кваліфікації продуктів розкладання») описані умови для кваліфікації продуктів розкладання при перевищенні порогів. У деяких випадках зниження рівня вмісту продукту розкладання (наприклад за рахунок використання контейнера й закупорювального засобу з кращими захисними властивостями або зміни умов зберігання) до значення, що не перевищує (≤) порога, може бути більш доцільним, ніж надання даних щодо безпеки. Альтернативою кваліфікації продукту розкладання можуть бути достатні дані наукової літератури. Якщо не підходить жоден з описаних вище випадків, слід розглянути додаткові випробування безпеки. Чи вважаються дослідження відповідними для кваліфікації продукту розкладання, залежатиме від кількості факторів, включаючи популяцію пацієнтів, добову дозу, шлях введення і тривалість застосування нового лікарського препарату. Такі дослідження можуть бути проведені з використанням нового лікарського препарату або речовини, що містить продукти розкладання, які підлягають контролю; хоча іноді можуть бути придатними дослідження з використанням виділених продуктів розкладання.
Хоча підрозділ В цієї настанови не призначений для застосування на етапі розробки, пов’язаному з клінічними випробуваннями, на пізніших етапах розробки надані в підрозділі В цієї настанови пороги можуть бути корисними при оцінці нових продуктів розкладання, що наявні в тих серіях нового лікарського препарату, які були вироблені за допомогою пропонованого промислового процесу. Будь-який новий продукт розкладання, що виявлений на більш пізніх етапах розробки, має бути ідентифікований (див. «Схему рішень щодо ідентифікації та кваліфікації продуктів розкладання» у додатку В.3), якщо рівень його вмісту перевищує (>) поріг ідентифікації, наведений в додатку В.1. Аналогічно, слід розглянути питання про кваліфікацію продукту розкладання, якщо рівень його вмісту перевищує (>) поріг кваліфікації, наведений у додатку В.1.
Дослідження безпеки мають передбачати порівняння результатів випробувань безпеки нового лікарського препарату або лікарської речовини, що містить репрезентативну кількість продукту розкладання, із заздалегідь кваліфікованою речовиною; хоча також можуть бути розглянуті дослідження з використанням виділених продуктів розкладання.
Додаток В.1 (обов’язковий). Пороги для продуктів розкладання в нових лікарських препаратах
Пороги інформування
|
Максимальна добова доза, г1 |
Поріг, %2, 3 |
|
≤1 |
0,1 |
|
>1 |
0,05 |
Пороги ідентифікації
|
Максимальна добова доза1 |
Поріг, %2, 3 |
|
<1 мг |
1,0 або 5 μг TDI (залежно від того, що менше) |
|
1–10 мг |
0,5 або 20 μг TDI (залежно від того, що менше) |
|
>10 мг–2 г |
0,2 або 2 мг TDI (залежно від того, що менше) |
|
>2 г |
0,10 |
Пороги кваліфікації
|
Максимальна добова доза1 |
Поріг, %2, 3 |
|
<10 мг |
1,0 або 50 μг TDI (залежно від того, що менше) |
|
10–100 мг |
0,5 або 200 μг TDI (залежно від того, що менше) |
|
>100 мг–2 г |
0,2 або 3 мг TDI (залежно від того, що менше) |
|
>2 г |
0,15 |
1Кількість лікарської речовини, що приймається протягом доби.
2Пороги для продуктів розкладання виражені або як відсоток від кількості лікарської речовини, або як кількість продукту розкладання, що приймається протягом доби (TDI). Можуть бути прийнятні нижчі пороги, якщо продукт розкладання є надзвичайно токсичним.
3Вищі пороги мають бути науково обґрунтовані.
Наведення порогів інформування, ідентифікації та кваліфікації для продуктів розкладання в новому лікарському препараті у вигляді функції максимальної добової дози1
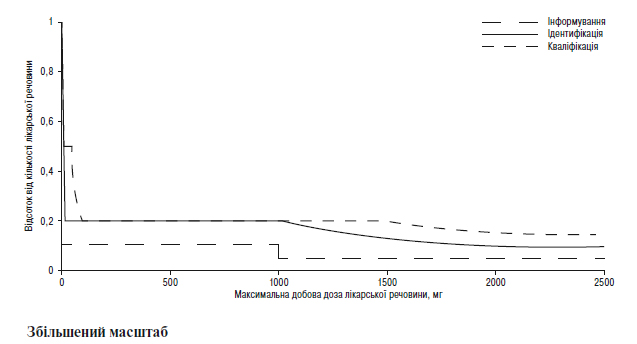
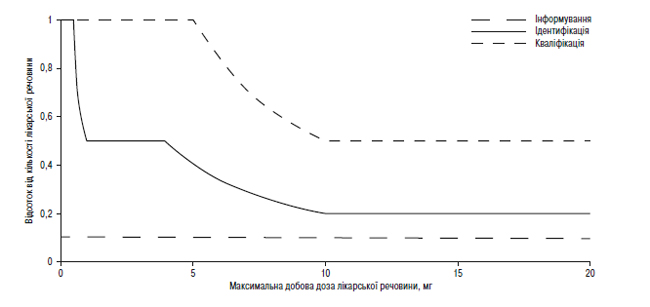
1Фактичні значення порогів слід вибирати з таблиць, наведених вище в цьому додатку В.1.
Додаток В.2 (обов’язковий). Пояснення стосовно наведення в реєстраційному досьє результатів ідентифікації та кваліфікації продуктів розкладання
Додаток наведений тільки з метою пояснення; він не служить прикладом того, які результати відносно продуктів розкладання слід наводити в реєстраційному досьє. Як правило, «вихідні» дані не наводять.
Приклад 1: Максимальна добова доза становить 50 мг
Поріг інформування: 0,1%
Поріг ідентифікації: 0,2%
Поріг кваліфікації: 200 μг
|
«Вихідний» результат, % |
Повідомлений результат, % (поріг інформування = 0,1%) |
Загальний добовий прийом (TDI) продукту |
Дія |
|
| Ідентифікація (чи перевищений поріг 0,2%?) | Кваліфікація (чи перевищений поріг 200 μг TDI?) | |||
|
0,04 |
Не повідомляють |
20 |
Ні |
Ні |
|
0,2143 |
0,2 |
100 |
Ні |
Ні |
|
0,349 |
0,31 |
150 |
Так |
Ні1 |
|
0,550 |
0,61 |
300 |
Так |
Так1 |
Приклад 2: Максимальна добова доза становить 1,9 г
Поріг інформування: 0,05%
Поріг ідентифікації: 2 мг
Поріг кваліфікації: 3 мг
|
«Вихідний» результат, % |
Повідомлений результат, % (поріг інформування = 0,05%) |
Загальний добовий прийом (TDI) продукту розкладання, мг |
Дія |
|
| Ідентифікація (чи перевищений поріг 2 мг TDI?) | Кваліфікація (чи перевищений поріг 3 мг TDI?) | |||
|
0,049 |
Не повідомляють |
1 |
Ні |
Ні |
|
0,079 |
0,08 |
2 |
Ні |
Ні |
|
0,183 |
0,181 |
3 |
Так |
Ні1, 2 |
|
0,192 |
0,191 |
4 |
Так |
Так1 |
1Після ідентифікації, якщо певний фактор відгуку значно відрізняється від первинного допущення, може бути доцільним провести повторне визначення фактичної кількості наявного продукту розкладання та повторну оцінку відносно порога кваліфікації (див. додаток В.1).
2Для контролю того, що поріг перевищений, повідомлені результати слід оцінити відносно порогів таким чином: якщо поріг зазначено в %, повідомлений результат, округлений до такого ж знака після коми, що й поріг, слід безпосередньо порівняти з порогом. Якщо поріг зазначено як TDI, повідомлений результат слід перевести у TDI, округлити до такого ж знака після коми, що й поріг, і безпосередньо порівняти з порогом. Наприклад, кількість при рівні розкладання 0,18% відповідає загальному добовому прийому (TDI) 3,4 мг домішки (абсолютна кількість), що округлили до 3,0 мг; таким чином, поріг кваліфікації, зазначений як TDI (3 мг), не перевищений.
Додаток В.3 (обов’язковий). Схема рішень щодо ідентифікації та кваліфікації продуктів розкладання
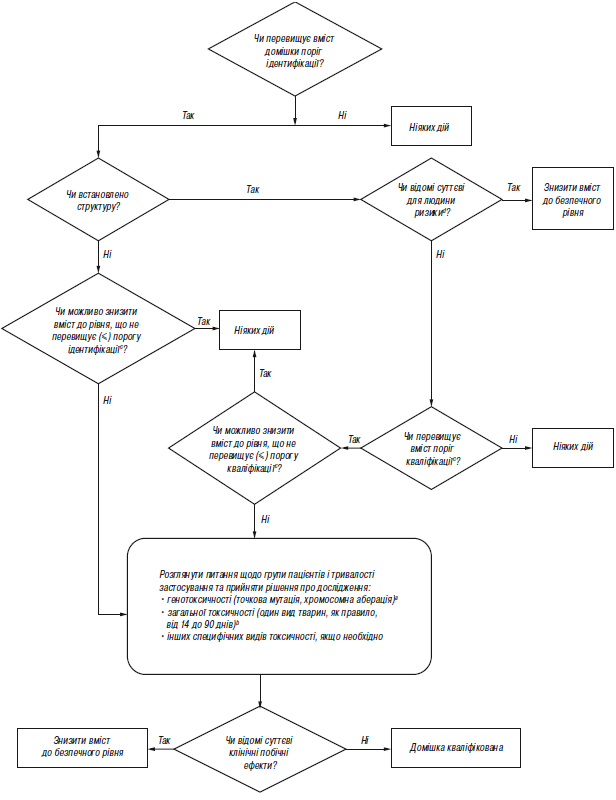
аЯкщо доцільно, слід провести мінімальний скринінг (наприклад за генотоксичністю). Для мінімального скринінгу прийнятними вважаються дослідження з визначення точкової мутації та хромосомної аберації.
bЯкщо необхідним є вивчення загальної токсичності, одне дослідження або декілька досліджень мають дозволяти провести порівняння некваліфікованої та кваліфікованої речовини. Тривалість дослідження слід встановлювати на підставі наявної значущої інформації; дослідження необхідно проводити на таких видах тварин, на яких можливість визначити токсичність продукту розкладання є максимальною. З огляду на кожний конкретний випадок, можуть бути прийнятними дослідження гострої токсичності, особливо у разі однодозових ліків. Взагалі, прийнятними вважаються дослідження тривалістю мінімум 14 днів і максимум 90 днів.
cМожуть бути прийнятні нижчі пороги, якщо продукт розкладання є надзвичайно токсичним.
dНаприклад, чи відомі вам дані щодо безпеки цього продукту розкладання, або чи заважає його структура застосуванню у людини при даній концентрації?
Національний додаток (довідковий). Бібліографія
1. СРМР/ICH/2737/99 (ICH Topic Q3A (R2)) Note for Guidance on Impurities Testing: Impurities in New Drug Substances, October 2006.
2. СРМР/ICH/2738/99 (ICH Topic Q3В (R2)) Note for Guidance on Impurities in New Drug Products, June 2006.
3. The Common Technical Document for the Registration of Pharmaceuticals for Human Use. — ICH Harmonised Tripartite Guideline. — Brussels, February 6–7, 2002.
4. Commission Directive 2003/63/EC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use. Annex I: Analytical, Pharmacotoxilogical and Clinical Standards and Protocols in Respect of the Testing of Medicinal Products. — Official Journal of the European Union. — № L 159/49 of 27.6.2003.
5. Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use // Official Journal of the European Communities. — L 311, 28.11.2001.
6. EMEA/CHMP/167068/2004 — ICH. — Part I: Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 (R2) Pharmaceutical Development). — Part II: Annex to Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 Annex Pharmaceutical Development), June 2009.
7. Настанова СТ-Н МОЗУ 42-3.0:2011. — Лікарські засоби. Фармацевтична розробка (ICH Q8) / М. Ляпунов, О. Безугла, Ю. Підпружников та ін. — Київ, МОЗ України, 2012.
8. СРМР/ICH/367/96 (ICH Topic Q6A) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000.
9. Настанова СТ-Н МОЗУ 42-3.2:2004. — Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності / В. Георгієвський, М. Ляпунов, О. Безугла та ін. — Київ, МОЗ України, 2004.
10. CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) Note for Guidance on Stability Testing: Stability Testing of New Drug Substances and Products, 2003.
11. CPMP/QWP/122/02 Rev 1 corr Guideline on stability testing: stability testing of existing active substances and related finished products, 2003.
12. Настанова СТ-Н МОЗУ 42-3.3:2004. — Настанови з якості. Лікарські засоби. Випробування стабільності / В. Георгієвський, М. Ляпунов, О. Безугла та ін. — Київ, МОЗ України, 2004.
13. СРМР/ICH/283/95 (ICH Topic Q3С (R3)) Note for Guidance on Impurities: Residual Solvents, March 1998.
14. CPMP/SWP/5199/02 — ЕМЕА/CHMP/QWP 251344/2006 Guideline on the Limits of Genotoxic Impurities, London, 28 June 2006.
15. ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів / І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
16. Настанова СТ-Н МОЗУ 42-1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення / М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005.
17. ДСТУ 1.7–2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів / О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
18. EMEA/P/24143/2004. — Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005.
19. EudraLex. — The Rules Governing Medicinal Products in the European Union. — Volume 4. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (http://ec.europa.eu/health/documents/eudralex/vol-4/index en.htm).
Ключові слова: домішка, ідентифікація, інформування, кваліфікація, лікарська речовина, лікарський препарат, продукт розкладання, поріг, профіль, реєстраційне досьє, специфікація.