
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
СПЕЦИФІКАЦІЇ: КОНТРОЛЬНІ ВИПРОБУВАННЯ ТА КРИТЕРІЇ ПРИЙНЯТНОСТІ
Настанова 42-3.2:2004
Видання офіційне
Київ
Міністерство охорони здоров’я України
2012
1 РОЗРОБЛЕНО: ДП «Державний науковий центр лікарських засобів» (ДП «ДНЦЛЗ»)
РОЗРОБНИКИ: В. Георгієвський, доктор фарм. наук (керівник розробки); М. Ляпунов, доктор фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; А. Піотровська; О. Гризодуб, доктор хім. наук; О. Кричевська; Ю. Підпружников, доктор фарм. наук; О. Антипова, канд. фарм. наук; Н. Крупа; Т. Матвієнко; І. Юдіна
ВНЕСЕНО: Державною службою лікарських засобів і виробів медичного призначення Міністерства охорони здоров’я України
2 ПРИЙНЯТО І НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 31 грудня 2003 р. № 637
3 Ця настанова відповідає:
СРМР/ІСН/367/96 corr (Q6A) Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 1999 (CPMP/ICH/367/96 corr (Q6A) Керівні вказівки зі специфікацій: методики випробувань і критерії прийнятності для нових лікарських речовин і нових лікарських препаратів: хімічні речовини, 1999) у частині підрозділу 5.1
Guideline 3AQ1 la Specifications and control tests on the finished product (Настанова 3AQ1 la Специфікації та контрольні випробування готової продукції) у частині підрозділу 5.2
Ступінь відповідності — модифікований (MOD)
Переклад з англійської мови (en)
4 ВВЕДЕНО ВПЕРШЕ
© Державна служба лікарських засобів і виробів медичного призначення, 2012
© Колектив розробників, 2012
© МОРІОН, 2012
У фармацевтичному секторі України відбувається гармонізація законодавчої та нормативної бази з відповідними нормами Європейського Союзу (ЄС). У цей час в Україні вже набули чинності такі нормативні документи:
Частиною належної виробничої практики є контроль якості, пов’язаний із відбором проб, специфікаціями та проведенням випробувань вихідної сировини і матеріалів, проміжної, нерозфасованої та готової продукції. Незалежний контроль якості також є частиною системи сертифікації лікарських засобів. Необхідною документацією для контролю якості є специфікації. Загальні вимоги до специфікацій установлені в Настанові 42-01-2001.
ДФУ встановлює спеціальні загальні і/або конкретні мінімальні технічні вимоги (або вимоги на вибір) щодо:
Однак ДФУ, як і Європейська Фармакопея, не встановлює методології розробки специфікацій для застосування на різних етапах обігу лікарських засобів.
Загальний технічний документ (Common Technical Document — CTD), прийнятий в ЄС, Сполучених Штатах Америки (США) та Японії, вимагає подання в складі реєстраційного досьє специфікацій і методик випробувань готових лікарських засобів, діючих і допоміжних речовин. У CTD наведені посилання на спеціальні настанови, відповідно до яких слід розробляти специфікації на різні групи продукції. Ці настанови становлять методичну основу розробки специфікацій, включаючи обґрунтування критеріїв прийнятності та вибір методик випробувань.
Примітка. В реєстраційному досьє у форматі CTD специфікації наводять у розділі 3.2.S.4 «Control Drug Substance* («Контроль лікарської речовини»), який складається з таких пунктів:
3.2.5.4.1 «Specification» («Специфікація»);
3.2.5.4.2 «Analytical Procedures* («Аналітичні методики»);
3.2.5.4.3 «Validation Analytical Procedures* («Валідація аналітичних методик»); 3.2.S.4.4«Batch Analyses* («Аналізи серій»);
3.2.S.4.5 «Justification Specification* («Обґрунтування специфікації»).
Специфікації на допоміжні речовини наводять у розділі 3.2.P.4«Control Excipients» («Контроль допоміжних речовин»), який складається з таких пунктів:
3.2.Р.4.1 «Specifications» («Специфікації»);
3.2.Р.4.2 «Analytical Procedures* («Аналітичні методики»);
3.2.Р.4.3 «Validation Analytical Procedures* («Валідація аналітичних методик»);
3.2.Р.4.4 «Justification Specification* («Обґрунтування специфікацій»);
3.2.Р.4.5 «Excipients Human or Animal Origin* («Допоміжні речовини людського і тваринного походження»); 3.2.Р.4.6 «Novel Excipients» («Нові допоміжні речовини»).
Специфікації на лікарський препарат наводять у розділі 3.2.Р.5 «Control Drug Product» («Контроль лікарського препарату»), який складається з таких пунктів: 3.2.Р.5.1 «Specification(s)» («Специфікація(ї)»); 3.2.Р.5.2 «Analytical Procedures» («Аналітичні методики»); 3.2.Р.5.3 «Validation Analytical Procedures* («Валідація аналітичних методик»); 3.2.Р.5.4 «Batch Analyses* («Аналізи серій»);
3.2.Р.5.5 «Characterisation Impurites* («Характеристика домішок»); 3.2.Р.5.6 «Justification Specification(s)» («Обґрунтування специфікації(ій)»).
Ця настанова розроблена на основі двох настанов із якості, прийнятих у ЄС, які присвячені питанням розробки специфікацій на діючі речовини та лікарські засоби. У настанову внесені такі редакційні зміни:
а) дві настанови (СРМР/ІСН/367/96 і 3AQ11а) об’єднані в одну без зміни їх обсягу та змісту;
б) додатково введені розділи «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Позначення та скорочення» і додаток Б «Бібліографія». Основні положення викладені в розділі 5 і додатку А. Відповідно до цього номери пунктів і підпунктів додатково включають номер розділу 5, а також номер підрозділу 1, текст якого відповідає Настанові СРМР/ІСН/367/96, або номер підрозділу 2, текст якого відповідає Настанові ЗAQ11 а. Схеми рішень наведені в додатку А;
в) посилання на нормативні документи, що згадуються в тексті, наведені в повному обсязі в розділі 2 «Нормативні посилання». Замість посилань на деякі настанови з якості ЄС у тексті зроблені посилання на прийняті МОЗ України відповідні гармонізовані настанови. При згадуванні у тексті нормативного документа, прийнятого у рамках ІСН або у ЄС, у виносках наприкінці відповідних сторінок зазначено: «Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку»;
г) у настанові замість «Європейський Союз» або «Європейський Союз, Японія та США» зазначено «Україна»; слова «заявка на отримання торгової ліцензії» («аррlісаtіоn for a marketing authorisation*) замінені на «заявка на реєстрацію», а слова «досьє для торгової ліцензії» («dossier for marketing authorisation*) — на «реєстраційне досьє»;
д) замість «Європейська Фармакопея або фармакопея держави ЄС» зазначено «Державна Фармакопея України або Європейська Фармакопея, або інша відповідна фармакопея». Це пов’язано з тим, що ДФУ гармонізована з Європейською Фармакопеєю, а встановлені в ній національні додаткові вимоги або вимоги на вибір більш жорсткі. Під «іншою відповідною фармакопеєю» слід розуміти фармакопею держави ЄС, що гармонізована з Європейською Фармакопеєю, або Фармакопею США, або Фармакопею Японії;
е) тексти, що стосуються регуляторних положень ЄС, Японії та США, як інформаційний матеріал наведені у виносках;
ж) посилання на настанови СРМР/ІСН з валідації аналітичних методик доповнені посиланням на відповідну загальну статтю ДФУ;
и) інші незначні доповнення виділені іншим шрифтом і літерою N.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Специфікації: контрольні випробування та критерії прийнятності
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Спецификации: контрольные испытания и критерии приемлемости
MEDICINAL PRODUCTS
Specifications: test procedures and acceptance criteria
Чинна від 2004-04-01
Ця настанова поширюється на лікарські засоби для людини та встановлює рекомендації щодо специфікацій на діючі речовини та лікарські засоби.
Ця настанова рекомендується для підприємств, організацій і установ, що розробляють і/або серійно виробляють діючі речовини та лікарські засоби на території України, незалежно від відомчого підпорядкування та форми власності, для науково-експертних організацій і регуляторних органів, а також експертів та інспекторів, які здійснюють експертизу на етапі реєстрації (перереєстрації) лікарських засобів та інспектування їх виробництва.
Цю настанову рекомендується застосовувати при розробці специфікацій на діючі речовини та лікарські засоби, а також при складанні відповідних розділів реєстраційного досьє.
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 р.
Настанова 42-01-2001 Лікарські засоби. Належна виробнича практика
Настанова 42-3.1:2004 Настанови з якості. Лікарські засоби. Фармацевтична розробка
Настанова 42-3.3:2004 Настанови з якості. Лікарські засоби. Випробування стабільності
European Pharmacopoeia. 4th Edition. 2002 (Європейська Фармакопея. 4е видання. 2002)
СРМР/ІСН/381/95 (Q2A) Note for guidance on validation of analytical methods: definitions and terminology, 1995
(CPMP/ICH/381/95 (Q2A) Керівні вказівки з валідації аналітичних методик: терміни та визначення, 1995) СРМР/ІСН/281/95 (Q2B) Note for guidance on validation of analytical procedures: methodology, 1996
(CPMP/ICH/281/95 (Q2B) Керівні вказівки з валідації аналітичних методик: методологія, 1996) СРМР/ІСН/365/96 (Q6B) Note for guidance on specifications — Test procedures and acceptance criteria for biotechnological/biological products, 1999
(CPMP/ICH/365/96 (Q6B) Керівні вказівки зі специфікацій — Методики випробувань і критерії прийнятності для біотехнологічних/біологічних препаратів, 1999)
СРМР/ІСН/2737/99 (Q3A) Note for guidance in impurities testing: Impurities in new drag substances, 2002 (CPMP/ICH/2737/99 (Q3A) Керівні вказівки з випробувань на домішки: Домішки в нових лікарських речовинах, 2002).
СРМР/ІСН/282/95 (Q3B) Note for guidance on impurities in new drag products, 1996 (CPMP/ICH/282/95 (Q3B) Керівні вказівки щодо домішок у нових лікарських препаратах, 1996)
СРМР/ІСН/283/95 (Q3C) Note for guidance on impurities: residual solvents, 1997 (CPMP/ICH/283/95 (Q3C) Керівні вказівки щодо домішок: залишкові розчинники, 1997).
3.1 У цій настанові використано терміни, встановлені у Настанові 42-01-2001: валідація; готова продукція; забезпечення якості; контроль у процесі виробництва (виробничий контроль); лікарський засіб (лікарський препарат); нерозфасована продукція; проміжна продукція; серія; технологічний процес (виробничий процес).
3.2 У цій настанові використано термін, встановлений у Настанові 42-3.3:2004: вихідна серія.
3.3 Нижче наведено визначення термінів, додатково використаних у цій настанові. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1–3].
3.3.1 аналітична валідація (analytical validation, [2,3])
Поняття визначається змістом документа «Note for guidance validation of analytical procedures: methodology»* і загальної статті ДФУ «Валідація аналітичних методик і випробувань»N.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
3.3.2 випробування в процесі виробництва (in-process tests, [I])
Випробування, що можуть бути проведені у ході виробництва діючої речовини або лікарського препарату, а не як частина офіційного набору випробувань, що здійснюють перед випуском.
3.3.3 відстрочене вивільнення (delayed release, [1])
Вивільнення лікарської речовини (або лікарських речовин), що починається не відразу після орального введення лікарського препарату.
3.3.4 домішка (impurity, [І])
Будь-який компонент діючої речовини, що не є за хімічною структурою цією діючою речовиною (1), або будь-який компонент лікарського препарату, що не є за хімічною структурою діючою речовиною або допоміжною речовиною, що входить до складу лікарського препарату (2).
3.3.5 енантіомери (enantiomers, [І])
Сполуки з такою ж молекулярною формулою, що і діюча речовина, але такі, що відрізняються від неї просторовим розташуванням атомів у молекулі та що є не співпадаючими дзеркальними відображеннями.
3.3.6 загальне випробування (universal test, [1])
Випробування, яке вважається потенційно застосовним до всіх нових діючих речовин або всіх нових лікарських препаратів (наприклад, зовнішній вигляд, ідентифікація, кількісне визначення та випробування на домішки).
3.3.7 звичайне вивільнення; негайне вивільнення (immediate release, [I])
Вивільнення, при якому лікарський засіб розчиняється у вмісті шлунково-кишкового тракту без відстрочки початку процесу розчинення, пролонгування процесу розчинення або пролонгування всмоктування лікарської речовини.
3.3.8 ідентифікована домішка (identified impurity, [1])
Домішка, структура якої відома.
3.3.9 комбінований лікарський препарат (combination product, [1])
Лікарський препарат, що містить більше однієї діючої речовини.
3.3.10 критерії прийнятності (acceptance criteria, [1])
Числові межі, інтервали чи інші підхожі межі прийнятності результатів аналітичних процедур.
3.3.11 лікарські засоби, що добре розчиняються у воді (highly water soluble drugs, [1])
Лікарські засоби, для яких співвідношення доза/об’єм для розчинення менший або дорівнює 250 мл в інтервалі рН від 1,2 до 6,8.
Приклад.
Найнижча розчинність сполуки А при температурі (37±0,5) °С і рН 6,8 становить 1,0 мг/мл, а її вміст у лікарському засобі як діючої речовини становить 100 мг, 200 мг і 400 мг. Такий лікарський засіб слід вважати таким, що має низьку розчинність, оскільки для нього співвідношення доза/об’єм для розчинення більше за 250 мл (400 мг : 1,0 мг/мл = 400 мл).
3.3.12 модифіковане вивільнення (modified release, [1])
Вивільнення з лікарських форм, для яких характеристики вивільнення лікарської речовини (а саме часові рамки і/або місце) вибрані таким чином, щоб досягти терапевтичних цілей або зручності застосування. Ці цілі не можуть «бути досягнуті при використанні звичайних лікарських форм, таких як розчин або лікарська форма зі звичайним вивільненням. До твердих лікарських форм для орального застосування з модифікованим вивільненням належать лікарські препарати як із відстроченим, так і з пролонгованим вивільненням.
3.3.13 надлишок (overage, [2])
Додаткова кількість речовини (зазвичай діючої речовини), що перевищує кількість, зазначену в складі одиниці лікарської форми і яку додають при виготовленні серії продукції з метою компенсації втрат у ході виробництва і/або під час зберігання в остаточному пакованні. Звичайно виражають у відсотках.
3.3.14 неідентифікована домішка (unidentified impurity, [1])
Домішка, для якої визначені тільки якісні аналітичні характеристики (наприклад, час утримування при хроматографічному визначенні).
3.3.15 нова діюча речовина (new drug substance, [1])
Раніше не зареєстрована речовина, що має певну терапевтичну дію; також може називатися новою молекулярною речовиною або новою хімічною речовиною. Вона може являти собою комплексну сполуку, простий ефір або сіль раніше схваленої (дозволеної до медичного застосування)N діючої речовини.
3.3.16 новий лікарський препарат (new drug product, [1])
Лікарський препарат у такій лікарській формі (наприклад, таблетка, капсула, розчин, крем тощо), в якій він не був раніше зареєстрований. Він містить діючу речовину, як правило, але не обов’язково, в поєднанні з допоміжними речовинами.
3.3.17 періодичні випробування (periodic tests, [2])
Спеціальні випробування, якими за необхідності доповнюють рутинні випробування; їх проводять із певною періодичністю, встановленою для кожного конкретного випадку. Такі випробування зазначають окремо.
3.3.18 поліморфізм (polymorphism, [1])
Наявність різних кристалічних форм однієї і тієї ж діючої речовини. Це поняття може поширюватися на продукти сольватації чи гідратації (відомі також як псевдополіморфні форми) і на аморфні форми.
3.3.19 продукт розкладу (degradation product/decomposition product, [1])
Молекула, утворена в результаті хімічної зміни в молекулі лікарської речовини, що відбулася протягом часу і/або викликана впливом світла, температури, рН, води, або внаслідок реакції з допоміжною або іншою діючоюN речовиною і/або із системою первинне паковання/закупорювальний елемент.
3.3.20 пролонговане вивільнення (extended release, [1])
Вивільнення діючої речовини протягом тривалого періоду після застосування лікарського препарату завдяки спеціально підібраному складу.
3.3.21 рацемат (racemate, [1])
Суміш (тверда, рідка, газоподібна або розчин) еквімолекулярних кількостей двох видів енантіомерів, яка не наділена оптичною активністю.
3.3.22 реактив (reagent, [1])
Речовина, що використовується при виробництві діючої речовини і що не є вихідною сировиною або розчинником.
3.3.23 розчинник (solvent, [1])
Неорганічна або органічна рідина, що використовується як носій при приготуванні розчинів або суспензій у ході синтезу діючої речовини або при виробництві лікарського препарату.
3.3.24 рутинні випробування (routine tests, [2])
Випробування, які проводять для кожної промислової серії проміжної, нерозфасованої або готової продукції.
3.3.25 специфікація (specification, [1,2])
Перелік випробувань, посилань на аналітичні методики та відповідних критеріїв прийнятності, що являють собою числові межі, інтервали або інші критерії для випробувань, що описуються. У специфікації встановлюють набір критеріїв, яким повинні відповідати діюча речовина або лікарський препарат для того, щоб вони вважалися придатними для їх передбачуваного застосування. «Відповідність специфікаціям» означає, що діюча речовина і/або лікарський препарат будуть відповідати наведеним критеріям прийнятності за умови, що випробування проведені згідно з зазначеними в цих специфікаціях аналітичними методиками. Специфікації становлять собою необхідні стандарти якості, котрі пропонує та обґрунтовує виробник, а погоджують компетентні уповноважені органи [1].
Якісні і/або кількісні характеристики, яким має відповідати дана продукція, із зазначенням методик випробувань і допустимих меж [2].
3.3.26 специфікація на готову продукцію, що застосовується при випуску (specification of the finishedproduct (at release), [2])
Монографія, що визначає якісні та кількісні характеристики, яким має відповідати готова продукція на момент її виробництва (при випуску), із зазначенням методик випробувань і допустимих меж.
3.3.27 специфікація на готову продукцію, що застосовується протягом терміну зберігання (specification of the finished product (up to the end of shelf life), [2])
Монографія, що визначає якісні та кількісні характеристики, яким має відповідати лікарський препарат (що знаходиться на ринку) протягом встановленого терміну зберігання, із зазначенням методик випробувань і допустимих меж.
3.3.28 специфікована домішка (specified impurity, [1])
Ідентифікована чи неідентифікована домішка, що вибрана для включення в специфікацію на діючу речовину або лікарський препарат. Щоб гарантувати якість діючої речовини або лікарського препарату, цю домішку наводять у специфікації окремо із зазначенням граничного вмісту.
3.3.29 специфічне випробування (specific test, [1])
Випробування, яке вважають застосовним для конкретної діючої речовини або конкретного лікарського препарату залежно від їх специфічних властивостей і/або призначення.
3.3.30 факторизація (factorisation, [2])
Коректування маси взятої для виробництва серії продукції речовини (як правило, діючої речовини) на підставі розрахунку, проведеного з урахуванням фактичної активності цієї речовини, встановленої для його конкретної серії шляхом кількісного визначення.
Приклад.
Якщо теоретична кількість діючої речовини в серії готової продукції повинна становити 100 г, а при кількісному визначенні встановлено, що в даній серії речовини міститься 92,0 % мас., то факторизована маса, яку необхідно взяти, становитиме 100 г • 100 : 92,0 = 108,7 г.
3.3.31 хіральний (chiral, [1])
Не співпадаючий із дзеркальним відображенням; термін застосовується до молекул, конформацій і макроскопічних об’єктів, таких як кристали. Цей термін поширюється і на зразки речовин, молекули яких є хіральними, навіть у тому випадку, коли макроскопічні агрегати таких молекул є рацемічними.
3.3.32 швидкорозчинні лікарські препарати (rapidly dissolving products, [1])
Швидкорозчинним вважається твердий лікарський препарат для орального застосування із звичайним (негайним) вивільненням, якщо не менше 80% від зазначеної на етикетці кількості діючої речовини розчиняється протягом 15 хв у кожному з наступних середовищ: (1) рН 1,2, (2) рН 4,0 і (3) рН 6,8.
3.3.33 якість (quality, [1])
Відповідність діючої речовини або лікарського препарату його призначенню. Це поняття включає такі показники як ідентифікація, сила дії (вміст діючої речовини в кількісному вираженні)N i чистота.
3.4 Наведені в розділі 3 визначення понять застосовують до термінів, що використовуються у цій настанові. Визначення цих термінів можуть відрізнятися в інших нормативних документах, або терміни можуть мати інші значення.
ВЕРХ — високоефективна рідинна хроматографія
ВООЗ — Всесвітня організація охорони здоров’я
ГХ — газова хроматографія
ДСК — диференційна скануюча калориметрія
ДТА — диференційний термічний аналіз
ДФУ — Державна Фармакопея України
ЄС — Європейський Союз
МС — мас-спектрометрія
СІЛА — Сполучені Штати Америки
TГA — термічний гравіметричний аналіз
УФ — ультрафіолетовий
ЯМР — ядерний магнітний резонанс
СРМР — Committee for Proprietary Medicinal Products (Комітет із патентованих лікарських засобів)
GMP — Good Manufacturing Practice (належна виробнича практика)
ІСН — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for’Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
LAL-тест — Limulus amoebocyte lysate test (випробування з використанням лізату амебоцитів мечохвоста Limulus polyphenols)
PDG — Pharmacopoeial Discussion Group (фармакопейна дискусійна група)
Положення підрозділу 5.1 призначені для встановлення, наскільки це можливо, єдиного набору показників для включення в специфікації на нові діючі речовини та нові лікарські засоби. У підрозділі 5.1 наведені керівні вказівки щодо встановлення й обґрунтування критеріїв прийнятності та вибору методик випробувань для нових діючих речовин, отриманих шляхом хімічного синтезу, і вироблених із них нових лікарських препаратів, які не були раніше зареєстровані в Україні.
Специфікація — це перелік випробувань, посилань на аналітичні методики та відповідних критеріїв прийнятності, що являють собою числові межі, інтервали або інші критерії для випробувань, що описуються. У специфікації встановлюють набір критеріїв, яким мають відповідати діюча речовина або лікарський препарат для того, щоб вони вважалися придатними для їх передбачуваного застосування. «Відповідність специфікаціям» означає, що діюча речовина і/або лікарський препарат будуть відповідати наведеним критеріям прийнятності за умови, що випробування проведені згідно з аналітичними методиками, зазначеними в цих специфікаціях. Специфікації становлять собою необхідні стандарти якості, котрі пропонує і обґрунтовує виробник, а погоджують компетентні уповноважені органи, що є умовою для реєстрації лікарських засобів.
Специфікації є одним з елементів загальної стратегії контролю діючих речовин та лікарських засобів, розробленої з метою гарантії якості та постійності характеристик продукції. Інші елементи цієї стратегії включають доскональне визначення всіх характеристик продукції в ході її розробки (на цьому ґрунтуються специфікації), а також суворе дотримання правил належної виробничої практики (GMP): наприклад, відповідні приміщення й обладнання, валідований технологічний процес, валідовані методики випробувань, випробування сировини, випробування в процесі виробництва, випробування стабільності тощо.
Вибір набору показників і методів випробувань для специфікацій здійснюється більшою мірою для підтвердження якості діючої речовини та лікарського препарату, ніж для їх повної характеристики; особлива увага має приділятися тим показникам, стосовно яких встановлено, що вони є найбільш важливими для забезпечення безпеки та ефективності діючої речовини та лікарського препарату.
Якість діючих речовин і лікарських препаратів визначається їх призначенням, рівнем розробки, контролем у процесі виробництва, контролем за дотриманням правил GMP і валідацією процесу, а також специфікаціями, що застосовуються до них у ході розробки та виробництва. У підрозділі 5.1 встановлені положення щодо специфікацій, тобто таких випробувань, методик і критеріїв прийнятності, які відіграють провідну роль у забезпеченні якості нової діючої речовини та нового лікарського препарату при їх випуску та протягом терміну зберігання. Специфікації є важливою, але не єдиною складовою забезпечення якості. Для гарантії стабільного виробництва діючих речовин і лікарських препаратів високої якості необхідні всі елементи системи забезпечення якості.
Положення підрозділу 5.1 стосуються лише нових лікарських засобів (включаючи комбіновані лікарські препарати) на етапі реєстрації та, якщо це прийнятно, нових діючих речовин; вони не стосуються діючих речовин, що розробляються, або лікарських препаратів, які знаходяться на стадіях клінічних випробувань. Положення цього підрозділу можуть застосовуватися до синтетичних і напівсинтетичних антибіотиків і низькомолекулярних синтетичних пептидів; однак їх недостатньо для адекватного встановлення специфікацій для високомолекулярних пептидів і поліпептидів, а також біологічних/біотехнологічних препаратів. Вимоги до специфікацій, випробувань і методик для біологічних/біотехнологічних препаратів викладені в настанові «Note for guidance on specifications — Test procedures and acceptance criteria for biotechnological/biological products»*. Положення підрозділу 5.1 також не поширюються на радіофармацевтичні лікарські засоби, лікарські препарати, отримані шляхом ферментації, олігонуклеотиди, лікарські препарати з рослинної сировини, а також сировину тваринного чи рослинного походження.
У цьому підрозділі наведено критерії прийнятності, які слід встановлювати для всіх нових діючих речовин і нових лікарських засобів (тобто загальні критерії прийнятності), а також критерії прийнятності, які слід розглядати як специфічні критерії для конкретних діючих речовин і/або лікарських форм. Положення підрозділу 5.1 не треба розглядати як всеосяжні. Безперервно продовжується розробка нових і модифікація існуючих аналітичних технологій. Слід використовувати такі технології за наявності відповідного обґрунтування.
Положення цього підрозділу стосуються таких лікарських форм, як тверді та рідкі лікарські форми для орального застосування, а також лікарські форми для парентерального введення (малого та великого об’єму). Це не означає, що наведений перелік є повним, або що слід обмежуватися лише тими лікарськими формами, відносно яких застосовані положення підрозділу 5.1. Наведені лікарські форми служать моделями, які можуть бути застосовані до інших лікарських форм, що не розглядаються в цьому підрозділі. Бажане застосування положень підрозділу 5.1 і до інших лікарських форм, наприклад, лікарських форм для інгаляцій (порошків, розчинів тощо), лікарських засобів для місцевого застосування (кремів, мазей, гелів) і трансдермальних систем.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Наведені нижче підходи важливі для розробки та встановлення гармонізованих специфікацій. Їх застосування не є універсальним, але за певних обставин слід враховувати кожний із цих підходів. У цьому підрозділі наведено стислий виклад кожного підходу та зазначені обставини, за яких його слід використовувати. Як правило, пропозиції щодо застосування цих підходів мають бути обґрунтовані заявником і перед введенням узгоджені з компетентним уповноваженим органом.
Періодичні (вибіркові) випробування — це проведення певних випробувань при випуску щодо заздалегідь обраних серій і/або через заздалегідь встановлені проміжки часу (а не щодо кожної серії). При цьому серії, що не зазнають випробувань, повинні відповідати всім критеріям прийнятності, встановленим для цієї продукції. Таке проведення випробувань являє собою неповну програму випробувань і, таким чином, має бути подане до компетентного уповноваженого органу для узгодження перед його введенням. Цей підхід може бути застосовний, наприклад, до випробувань на залишкові кількості органічних розчинників і мікробіологічну чистоту щодо твердих лікарських форм для орального застосування. Визнано, що на момент подання заявки на реєстрацію лікарського засобу заявник може мати тільки обмежені дані (див. п. 5.1.2.5). Отже, даний підхід, як правило, необхідно застосовувати після реєстрації лікарського засобу. При проведенні випробувань про кожний випадок невідповідності критеріям прийнятності, встановленим для періодичних випробувань, слід у встановленому порядку повідомляти відповідний(і) компетентний(і) уповноважений(і) орган (органи). Якщо ці дані свідчать про необхідність поновлення рутинних (постійних) випробувань, слід ввести випробування при випуску щодо кожної серії.
Підхід, пов’язаний із відмінністю критеріїв прийнятності при випуску та протягом терміну зберігання, застосовний лише до лікарських препаратів. Цей підхід передбачає встановлення суворіших критеріїв при випуску лікарського препарату порівняно з критеріями, що застосовуються протягом терміну зберігання*. Прикладами показників для застосування цього підходу можуть служити кількісне визначення та рівні вмісту домішок (продуктів розкладу).
*У Японії та США цей підхід може бути застосовний лише щодо «внутрішніх» критеріїв, а не щодо критеріїв при випуску, що схвалюються уповноваженим органом. Тобто, у цих регіонах критерії прийнятності, схвалені уповноваженим органом, однакові на момент випуску та протягом усього терміну зберігання; однак заявник може встановити більш суворі «внутрішні» межі для критерію на момент випуску, щоб забезпечити більшу впевненість у тому, що лікарський препарат відповідатиме схваленому уповноваженим органом критерію прийнятності протягом терміну зберігання. У ЄС встановлена регуляторна вимога — мати окремі специфікації, що застосовуються при випуску та протягом терміну зберігання, якщо вони неоднакові.
Випробування в процесі виробництва в контексті підрозділу 5.1 — це випробування, що можуть бути проведені в ході виробництва діючої речовини або лікарського засобу за умови, що це краще, ніж проводити їх як частину офіційного набору випробувань, здійснюваних перед випуском продукції.
Випробування в процесі виробництва, які проводять лише для регулювання параметрів процесу в рамках робочого діапазону (наприклад, випробування твердості та крихкості ядер таблеток, на які має бути нанесено покриття, і маси окремих таблеток), не включають у специфікацію.
Певні випробування, що проводяться в ході виробничого процесу, можуть бути достатніми для підтвердження відповідності вимогам специфікації, якщо таке саме випробування включене в специфікацію та якщо критерій прийнятності ідентичний вимозі, встановленій у специфікації, що застосовується при випуску, або є більш суворим (наприклад, рН розчину). Однак такий підхід має пройти валідацію для доказу того, що функціональні характеристики продукції чи результати її випробувань у процесі виробництва не змінилися по відношенню до готової продукції.
Специфікації слід складати на підставі досвіду і даних, накопичених у ході розробки нової діючої речовини або лікарського засобу. На підставі цих даних можна запропонувати виключити деякі випробування зі специфікації чи замінити їх, наприклад:
а) виключити випробування на мікробіологічну чистоту діючих речовин і твердих лікарських форм, які, як було показано в ході розробки, не підтримують життєздатність і ріст мікроорганізмів (див. схеми рішень № 6 і № 8 у додатку А);
б) виключити визначення речовин, що екстрагуються з паковання, якщо було доведено з відтворюваними результатами, що в лікарському препараті не виявлено речовин, які екстрагуються, або їх кількість відповідає прийнятим стандартам щодо безпеки;
в) визначення розміру часток може бути проведене як випробування в процесі виробництва або виконане як випробування при випуску залежно від впливу цього показника на функціональні характеристики продукції;
г) випробування на розчинення щодо твердих лікарських форм для орального застосування зі звичайним вивільненням, виготовлених із діючих речовин, що добре розчиняються у воді, може бути замінене випробуванням на розпадання, якщо для цих лікарських препаратів у ході розробки було доведене швидке вивільнення діючої речовини (див. схеми рішень № 7(1) — № 7(2) у додатку А).
Визнано, що на момент подання заявки на реєстрацію заявник може мати в своєму розпорядженні лише обмежену кількість даних; це може вплинути на процес встановлення критеріїв прийнятності. В результаті, у міру накопичення додаткового досвіду виробництва конкретної діючої речовини або лікарського препарату, може виникнути необхідність переглянути критерії прийнятності (наприклад, допустимі межі вмісту певної домішки). Критерії прийнятності на момент подання заявки слід встановлювати обов’язково на підставі міркувань безпеки та ефективності.
Якщо була лише обмежена кількість даних, то спочатку затверджені випробування та критерії прийнятності слід переглядати в міру накопичення інформації з урахуванням того, чи можливе внесення змін. Залежно від ситуації можуть бути встановлені як менш, так і більш суворі критерії прийнятності.
У певних випадках, за умови схвалення компетентним уповноваженим органом, як робоча альтернатива рутинним випробуванням лікарського препарату при випуску може бути використаний випуск за параметрами. Одним із прикладів є випробування на стерильність лікарських препаратів, що стерилізуються в остаточному первинному пакованні на заключній стадії (що піддаються кінцевій стерилізації)N. У цьому випадку випуск кожної серії здійснюється на підставі задовільних результатів контролю певних параметрів, наприклад, температури, тиску й тривалості фази (фаз) кінцевої стерилізації при виробництві лікарського препарату. Ці параметри, як правило, можна контролювати й вимірювати з великою точністю, внаслідок чого вони надійніші для прогнозування забезпечення стерильності, ніж випробування на стерильність готової продукції. У програму випуску за параметрами можуть бути включені відповідні лабораторні випробування (наприклад, використання хімічного чи фізичного індикатора). Важливо зазначити, що перед тим, як запропонувати випуск за параметрами, процес стерилізації має бути належним чином валідований; через встановлені проміжки часу за допомогою проведення ревалідації слід також підтверджувати збереження валідованого стану. При здійсненні випуску за параметрами показник, що контролюється посередньо (наприклад, стерильність), разом із посиланням на відповідну методику випробувань слід включити в специфікації.
Альтернативними є такі методики, котрі можна використовувати для визначення якого-небудь показника, якщо вони дозволяють контролювати якість діючої речовини чи лікарського препарату так само, як офіційна методика чи краще за неї. Наприклад, для таблеток, що, як було доведено, не розкладаються в ході виробничого процесу, може бути допустимим використання при випуску спектрофотометричної методики як альтернативи офіційній хроматографічній методиці. Однак хроматографічна методика може використовуватися для доказу відповідності критеріям прийнятності протягом терміну зберігання лікарського препарату.
У ДФУ, Європейській Фармакопеї чи іншій відповідній фармакопеї наведено певні методики або посилання на них. У всіх випадках, коли це доцільно, слід використовувати фармакопейні методики*.
*Оскільки існують відмінності між фармакопейними методиками і/або критеріями прийнятності, наведеними у фармакопеях сторін ІСН (ЄС, США та Японії), складання гармонізованої специфікації можливе лише в тому випадку, якщо методики та критерії прийнятності вважаються придатними з погляду компетентних уповноважених органів усіх цих регіонів.
Застосовність документа ІСН Q6A у повному обсязі залежить від успішного завершення гармонізації фармакопейних методик щодо деяких показників, що звичайно включаються у специфікації на нові діючі речовини або нові лікарські препарати. Фармакопейна дискусійна група (PDG) Європейської Фармакопеї, Фармакопеї Японії та Фармакопеї США взяла зобов’язання гармонізувати методики.
Утих випадках, коли досягнуто гармонізації, прийнятним у всіх трьох регіонах є наведення в специфікації відповідного посилання на гармонізовану методику та критерії прийнятності. Наприклад, після гармонізації дані про стерильність, отримані при використанні методики Фармакопеї Японії, сама методика та критерії прийнятності вважаються придатними при реєстрації в усіх трьох регіонах. Із метою позначення гармонізованого статусу таких методик досягнута домовленість про включення у відповідні тексти формулювання, яке зазначає, що методики та критерії прийнятності всіх трьох фармакопей вважаються еквівалентними й отже взаємозамінними.
Оскільки загальне значення документа ICHQ6A пов’язане зі ступенем гармонізації аналітичних методик і критеріїв прийнятності, встановлених у фармакопеях, члени робочої групи експертів Q6A прийняли рішення про те, що жодна з трьох фармакопей не повинна вносити зміни в гармонізовану монографію в односторонньому порядку. Відповідно до методики PDG3 перегляду гармонізованих монографій і розділів «жодна фармакопея не може в односторонньому порядку переглядати будь-яку монографію або розділ після їх прийняття чи публікації».
Безперервно розробляються нові аналітичні технології та відбуваються зміни в існуючих технологіях. Слід використовувати такі технології, якщо це дає додаткову гарантію якості, або їх застосування обґрунтоване іншими міркуваннями.
Як правило, немає необхідності проводити випробування лікарського препарату щодо показників якості, що однозначно стосуються діючої речовини. Наприклад, звичайно не слід проводити випробування лікарського препарату на наявність домішок, які контролюють у діючій речовині, та які пов’язані з процесом синтезу, а не є продуктами розкладу. Для отримання більш докладної інформації слід звертатися до настанови «Note for guidance on impurities in new drug products»*.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Стандартний зразок (або стандартна речовина) — це речовина, приготована для використання як стандарт при кількісному визначенні, ідентифікації чи випробуванні на чистоту. Якість стандартного зразка має відповідати його призначенню. Стандартну речовину часто характеризують і оцінюють відповідно до її призначення за допомогою додаткових процедур, відмінних від тих, що використовуються при проведенні рутинних випробувань. Для стандартних зразків нових діючих речовин, призначених для використання при кількісних визначеннях слід у достатній мірі ідентифікувати і/або контролювати домішки та визначати чистоту за допомогою методики кількісного визначення.
Специфікація — це перелік випробувань, посилань на аналітичні методики та відповідних критеріїв прийнятності, що являють собою числові межі, інтервали чи інші критерії для випробувань, що описуються. У специфікації встановлюють набір критеріїв, яким повинні відповідати діюча речовина або лікарський препарат для того, щоб вони вважалися придатними для їх передбачуваного застосування. «Відповідність специфікаціям» означає, що діюча речовина і/або лікарський препарат відповідатимуть наведеним критеріям прийнятності за умови, що випробування проведені згідно з аналітичними методиками, зазначеними в цих специфікаціях. Специфікації становлять собою необхідні стандарти якості, котрі пропонує та обґрунтовує виробник, а погоджують компетентні уповноважені органи, що є умовою для реєстрації лікарських засобів.
Крім випробувань, що проводяться при випуску, специфікація може включати перелік випробувань, здійснюваних у процесі виробництва (як описано в п. 5.1.2.3), періодичних (вибіркових) випробувань та інших випробувань, які не завжди проводять щодо кожної серії. У таких випадках заявник повинен встановити, які випробування постійно проводять щодо кожної серії, а які випробування не є такими, з зазначенням та обґрунтуванням фактичної періодичності проведення випробувань. У цьому випадку діюча речовина і/або лікарський препарат все одно повинні відповідати всім критеріям прийнятності, якщо будуть проведені відповідні випробування.
Слід зазначити, що внесення змін у специфікацію після реєстрації вимагає попереднього узгодження з компетентним уповноваженим органом.
Якщо специфікацію пропонують вперше, слід подати обґрунтування кожної методики та кожного критерію прийнятності, включених у специфікацію. Залежно від ситуації таке обґрунтування має містити дані з розробки, що стосуються справи, фармакопейні стандарти, результати випробувань діючих речовин і лікарських препаратів, що використовувалися при токсикологічних і клінічних випробуваннях, а також результати прискорених і довгострокових випробувань стабільності. Крім того, слід брати до уваги прийнятний діапазон вірогідної варіабельності аналітичної методики та виробничого процесу. Слід врахувати всю цю інформацію.
Можуть бути застосовними та прийнятними також інші підходи, відмінні від викладених у цьому підрозділі. Альтернативні підходи заявник повинен обґрунтувати. Таке обґрунтування має базуватися на даних, отриманих на підставі досвіду синтезу нової діючої речовини і/або процесу виробництва нового лікарського препарату. При цьому обґрунтуванні можна розглядати теоретичні допустимі межі для конкретної методики чи конкретного критерію прийнятності, однак незалежно від застосовного підходу основоположними мають бути отримані фактичні результати.
При складанні й обґрунтуванні специфікацій слід враховувати результати випробувань серій, включених у програму випробувань стабільності, а також серій, отриманих при масштабуванні та валідації процесу; особлива увага має бути приділена вихідним серіям, що використовуються для випробувань стабільності. Якщо планується використання декількох виробничих дільниць, то при первинному встановленні випробувань і критеріїв прийнятності рекомендується розглянути дані, отримані на цих дільницях. Це необхідно, якщо первинний досвід виробництва діючої речовини або лікарського препарату на будь-якій конкретній дільниці обмежений. Якщо при встановленні випробувань і критеріїв прийнятності використовуються дані, отримані на одній репрезентативній виробничій дільниці, то продукція, вироблена на всіх інших дільницях, також повинна відповідати цим критеріям.
При обґрунтуванні певних критеріїв прийнятності рекомендується подання результатів випробувань у графічній формі, зокрема, значень кількісного вмісту та рівнів домішок. При такому наданні результатів випробувань слід включити дані, отримані в ході розробки, разом із даними про стабільність, що наявні для серій нової діючої речовини чи нового лікарського препарату, отриманих за допомогою таких процесів, які запропоновані для промислового виробництва. Обґрунтування пропонованого виключення випробування зі специфікації має базуватися на даних, отриманих у ході розробки, а також даних валідації процесу (якщо це доцільно).
При застосуванні рекомендацій, викладених у п. 5.1.3.2, слід враховувати положення настанов «Note for guidance validation of analytical methods: definitions and terminology» і «Note for guidance validation of analytical procedures: methodology»*, а також загальної статті ДФУ «Валідація аналітичних методик і випробувань»N.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Наведені нижче випробування та критерії прийнятності, як правило, застосовні до всіх нових діючих речовин.
а) Опис
Має бути подана якісна характеристика щодо фізичного стану (наприклад, тверда речовина, рідина) і кольору нової діючої речовини. Якщо які-небудь з цих характеристик змінюються під час зберігання, то таку зміну слід вивчити та вжити належних заходів.
б) Ідентифікація
Випробування при ідентифікації повинні давати можливість найкращим чином розрізнювати близько споріднені за структурою сполуки, які, можливо, можуть міститися у діючій речовині. Випробування при ідентифікації мають бути специфічними для нової діючої речовини, наприклад, абсорбційна спектрофотометрія в інфрачервоній області. Ідентифікація лише методом хроматографії за часом утримування не вважається специфічною. Однак, як правило, є допустимим використання двох хроматографічних методик, якщо розділення грунтується на різних принципах, або є комбінація випробувань в одній методиці, наприклад, ВЕРХ з УФ-спектрофотометричним детектором, ВЕРХ/МС або ГХ/МС. Якщо нова діюча речовина являє собою сіль, то випробування для її ідентифікації мають бути специфічними щодо кожного з іонів. Може бути достатнім випробування, специфічне щодо самої солі.
Для оптично активних нових діючих речовин може бути необхідним специфічне випробування для їх ідентифікації або проведення кількісного визначення, специфічного щодо хіральної сполуки. Більш докладну інформацію з цього питання див. у п. 5.1.3.3.1 (г).
в) Кількісне визначення
Для визначення вмісту нової діючої речовини має бути включена специфічна методика, що дозволяє отримувати стабільні результати. У багатьох випадках можливе застосування однієї й тієї ж методики (наприклад, ВЕРХ) як для кількісного визначення нової діючої речовини, так і для визначення вмісту домішок.
У тих випадках, коли обґрунтоване використання неспецифічної методики кількісного визначення, для досягнення специфічності слід використовувати інші підтверджувальні аналітичні методики. Наприклад, якщо для кількісного визначення діючої речовини обране титрування, слід використовувати поєднання кількісного визначення та підхожого випробування на домішки.
г) Домішки
До цієї категорії включені органічні та неорганічні домішки, а також залишкові кількості органічних розчинників. Для отримання повнішої інформації слід звертатися до настанов «Note for guidance on impurities in new drug substances» і «Note for guidance on impurities: residual solvents»*.
Схема рішень № 1 (див. додаток А) присвячена екстраполяції значущих меж вмісту домішок із набору даних, отриманих у ході розробки. Малоймовірно, щоб на момент подання заявки на реєстрацію була достатня кількість даних для оцінки стабільності процесу. Тому вважається недоцільним на момент подання заявки на реєстрацію встановлювати суворо обмежені критерії прийнятності на підставі наявних даних, що отримані при аналізі серій (див. п. 5.1.2.5).
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Наведені нижче випробування та критерії прийнятності, як правило, застосовні до всіх нових лікарських препаратів:
а) Опис
Слід подати якісний опис лікарської форми (наприклад, розмір, форма й колір). Якщо які-небудь з цих характеристик змінюються в ході виробництва чи під час зберігання, таку зміну слід вивчити та вжити належних заходів. Критерії прийнятності повинні включати остаточний прийнятний зовнішній вигляд. Якщо під час зберігання спостерігається зміна забарвлення, то може бути доцільним включення кількісної методики.
б) Ідентифікація
Випробування при ідентифікації повинні встановлювати справжність нової(их) діючої(их) речовини (речовин) у новому лікарському препараті та давати можливість розмежувати близько споріднені за структурою сполуки, які, можливо, можуть бути присутніми. Випробування при ідентифікації повинні бути специфічними для нової діючої речовини, наприклад, абсорбційна спектрофотометрія в інфрачервоній області. Ідентифікація лише методом хроматографії за часом утримування не вважається специфічною. Однак, як правило, вважається допустимим використання двох хроматографічних методик, коли розділення грунтується на різних принципах, або є комбінація випробувань в одній методиці, наприклад, ВЕРХ з УФ-спектрофотометричним детектором, ВЕРХ/МС або ГХ/МС.
в) Кількісне визначення
Із метою встановлення сили дії (кількісного вмісту діючої речовини) для всіх нових лікарських препаратів слід включити специфічну методику кількісного визначення, що дозволяє отримувати стабільні результати. У багатьох випадках можливе застосування однієї і тієї ж методики (наприклад, ВЕРХ) як для кількісного визначення нової діючої речовини, так і для визначення вмісту домішок. Для кількісного визначення в нових лікарських препаратах можуть бути використані результати випробувань на однорідність вмісту, якщо методи, що використовуються для визначення однорідності вмісту, є також прийнятними для кількісних визначень.
У тих випадках, коли обґрунтовано використання неспецифічної методики кількісного визначення, для досягнення специфічності слід використовувати інші підтверджувальні аналітичні методики. Наприклад, якщо для кількісного визначення діючої речовини обрано титрування, то слід використати поєднання кількісного визначення і підхожого випробування на домішки. Якщо у разі використання неспецифічної методики кількісного визначення є дані про вплив допоміжної речовини на результати аналізу, то слід використати специфічну методику.
г) Домішки
До цієї категорії включені органічні та неорганічні домішки (продукти розкладу), а також залишкові кількості органічних розчинників. Для отримання повнішої інформації слід звертатися до настанов «Note for guidance on impurities in new drug substances» і «Note for guidance on impurities: residual solvents»*.
У новому лікарському препараті необхідно контролювати органічні домішки, що утворюються внаслідок розкладу нової діючої речовини, а також домішки, що виникають у ході процесу виробництва лікарського засобу. Слід встановити прийнятні межі вмісту для індивідуальних специфікованих (включених у специфікацію) продуктів розкладу, які можуть являти собою як ідентифіковані, так і неідентифіковані продукти розкладу (залежно від конкретної ситуації), а також межі сумарного вмісту продуктів розкладу. Домішки, що утворюються в процесі синтезу нової діючої речовини, як правило, контролюють при випробуваннях діючої речовини і, отже, не включають до межі сумарного вмісту домішок. Однак якщо домішка, що утворюється в ході синтезу, є також і продуктом розкладу, рівень її вмісту слід контролювати та включати до межі сумарного вмісту домішок. Якщо за допомогою відповідних аналітичних методів переконливо доведено, що діюча речовина не розкладається в складі даного лікарського препарату при встановлених умовах зберігання, що пропонуються в заявці на реєстрацію, то випробування щодо продуктів розкладу можна скоротити або виключити за відповідним дозволом компетентного уповноваженого органу.
Схема рішень № 2 (див. додаток А) присвячена екстраполяції значущих меж вмісту продуктів розкладу з набору даних, отриманих у ході розробки. Малоймовірно, щоб на момент подання заявки на реєстрацію була достатня кількість даних для оцінки стабільності процесу. Тому вважається недоцільним на момент подання заявки на реєстрацію встановлювати суворо обмежені критерії прийнятності на підставі наявних даних, що отримані при аналізі серій (див. п. 5.1.2.5).
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Залежно від ситуації для діючих речовин і/або лікарських препаратів додатково до перерахованих вище загальних випробувань можуть бути передбачені наведені нижче випробування. Окремі випробування/критерії слід включати в специфікацію для контролю серії, якщо вони тісно пов’язані з якістю діючої речовини та лікарського препарату. В особливих випадках або в міру отримання нової інформації можуть бути потрібними випробування, які відрізняються від перерахованих нижче випробувань.
а) Фізико-хімічні властивості
До них належать такі показники, як рН водного розчину, температура чи діапазон температур плавлення та коефіцієнт заломлення. Методики, що використовуються для визначення цих показників, як правило, є уніфікованими та не вимагають значної доробки, наприклад, капілярний метод визначення температури плавлення, рефрактометрія за методом Аббе. Випробування, що виконуються в цій категорії, слід включати до специфікації, виходячи з фізичної природи нової діючої речовини та її призначення.
б) Розмір часток
Для деяких нових діючих речовин, призначених для використання в складі твердих лікарських форм або суспензій, розмір часток може значним чином впливати на швидкість розчинення, біодоступність і/або стабільність. У таких випадках слід встановити критерії прийнятності та проводити випробування з визначення розміру часток, використовуючи відповідну методику.
Схема рішень № 3 (див. додаток А) надає додаткові вказівки щодо випадків, коли слід проводити випробування із визначення розміру часток.
в) Поліморфні форми
Деякі нові діючі речовини існують у різних кристалічних формах, які відрізняються за фізичними властивостями. Поліморфізм також може бути пов’язаний із продуктами сольватації чи гідратації (які відомі також як псевдополіморфні форми) і аморфними формами. Відмінності між такими формами можуть у деяких випадках вплинути на якість і функціональні характеристики нових лікарських препаратів. Якщо існують відмінності, і було показано, що вони можуть впливати на функціональні характеристики лікарського препарату, біодоступність або стабільність, то слід зазначити відповідний стан твердої речовини.
Щоб визначити, чи існують множинні форми, звичайно використовують визначення фізико-хімічних властивостей за допомогою відповідних методів. Прикладами таких методів є: визначення температури плавлення (включаючи мікроскопію при високій температурі), абсорбційна спектрофотометрія в інфрачервоній області для речовин в твердому стані, дифракція рентгенівських променів на поверхні порошку, методики термічного аналізу, такі як диференційна скануюча калориметрія (ДСК), термічний гравіметричний аналіз (ТГА) і диференційний термічний аналіз (ДТА), а також спектроскопія комбінаційного розсіювання, оптична мікроскопія і ЯМР-спектроскопія для речовин в твердому стані.
Схеми рішень №№ 4( 1 )-4(3) (див. додаток А) надають додаткові вказівки щодо того, коли і як слід контролювати та перевіряти поліморфні форми.
Примітка. Ці схеми рішень необхідно розглядати послідовно. За допомогою схем рішень № 4(1) і №4(2) можна визначити, чи виявляє діюча речовина поліморфізм, і чи можуть вплинути різні поліморфні форми на функціональні характеристики лікарського препарату. Схему рішень № 4(3) слід застосовувати лише в тому випадку, якщо доведено, що і діючої речовини характерний поліморфізм, і показаний вплив на функціональні характеристики. За допомогою схеми рішень № 4(3) можна проаналізувати можливість зміни поліморфних форм у лікарському препараті, а також можливість впливу такої зміни на функціональні характеристики лікарського препарату.
Як правило, в технічному відношенні дуже важко виміряти поліморфні зміни в лікарських препаратах. Звичайно для контролю функціональних характеристик лікарського препарату можна використати посереднє випробування (наприклад, випробування на розчинення) (див. схему рішень № 4(3) у додатку А), а визначати вміст поліморфних форм і встановлювати критерій прийнятності для такого випробування слід лише в крайньому випадку.
г) Випробування для хіральних нових діючих речовин
Якщо нова діюча речовина являє собою переважно один енантіомер, а вміст протилежного енантіомеру нижчий кваліфікаційних та ідентифікаційних порогових значень, наведених у настановах «Note for guidance on impurities in new drug substances» і «Note for guidance on impurities in new drug products»*, то у зв’язку з практичними труднощами кількісного визначення при низьких рівнях вмісту протилежний енантіомер не визначають. Однак, з іншого боку, щодо такої домішки в хіральній новій діючій речовині й отриманому з неї лікарському препараті слід чинити відповідно до положень, викладених у цьому підрозділі.
За допомогою схеми рішень № 5 (див. додаток А) як для нових діючих речовин, так і для нових лікарських препаратів можна визначити, коли можуть знадобитися випробування з ідентифікації для xipaльних сполук, випробування на домішки та кількісні визначення відповідно до наступних положень:
1) Діюча речовина
Для хіральних діючих речовин, що розроблені як одиночний енантіомер, контроль протилежного енантіомеру проводять таким же чином, як і щодо інших домішок. Однак технічні обмеження можуть заважати введенню таких самих меж кількісного вмісту чи кваліфікаційних меж. Слід також показати надійність контролю за допомогою відповідних випробувань вихідної сировини чи проміжної продукції, навівши відповідне обґрунтування.
У специфікації має бути кількісне визначення діючої речовини, що дозволяє селективно встановлювати вміст енантіомерів. Для досягнення цієї мети вважається прийнятним використання специфічної для хіральної сполуки методики кількісного визначення чи комбінації неспецифічної методики кількісного визначення з відповідними методами контролю енантіомеру-домішки.
Для діючої речовини, розробленої як одиночний енантіомер, випробування з ідентифікації повинне(і) забезпечувати можливість розрізнювати обидва енантіомери та рацемічну суміш. Для діючої речовини, що являє собою рацемічну суміш, як правило, існують два випадки, коли доцільне проведення випробування з ідентифікації стереоізомерів у разі випробувань при випуску/випробувань на придатність: 1) коли існує велика ймовірність того, що рацемат може бути заміщений енантіомером, або 2) коли є дані про те, що вибіркова кристалізація може призвести до ненавмисного отримання нерацемічної суміші.
2) Лікарський препарат
Якщо не доведено, що під час виробництва лікарської форми та її зберігання рацемізація є незначною, вважається обов’язковим контроль іншого енантіомеру в лікарському препараті.
Якщо доведено, що під час виробництва лікарської форми та її зберігання рацемізація є незначною, то може бути достатнім кількісне визначення за допомогою методики, неспецифічної щодо хіральної сполуки. В іншому випадку слід проводити кількісне визначення за допомогою специфічної щодо хіральної сполуки методики або, як альтернативу, можна використовувати неспецифічну методику кількісного визначення в комбінації з валідованою методикою контролю присутності протилежного енантіомеру.
Випробування з стереоспецифічної ідентифікації, як правило, не є необхідним для включенння в специфікацію, що застосовується при випуску лікарського препарату. Якщо рацемізація в ході виробництва лікарської форми та при зберіганні незначна, то випробування з стереоспецифічної ідентифікації доцільніше включити в специфікацію на діючу речовину. Якщо рацемізація в лікарській формі відбувається, то для підтвердження ідентичності може служити кількісне визначення за допомогою специфічної щодо хіральної сполуки методики чи випробування на енантіомер-домішку.
д) Вміст води
Це випробування є важливим, якщо відомо, що нова діюча речовина є гігроскопічною чи руйнується під впливом вологи, або якщо відомо, що діюча речовина є стехіометричним гідратом. Критерії прийнятності можуть бути обґрунтовані за допомогою даних про ефекти гідратації чи про поглинання вологи. У деяких випадках можна вважати достатнім визначення втрати в масі при висушуванні; однак переважне використання методики визначення, специфічної щодо води (наприклад, титрування за методом К. Фішера).
е) Неорганічні домішки
Необхідність включення в специфікацію випробувань і критеріїв прийнятності щодо неорганічних домішок (наприклад, каталізаторів) має бути вивчена в ході розробки та ґрунтуватися на знаннях виробничого процесу. Методики визначення та критерії прийнятності для сульфатної золи чи загальної золи мають відповідати фармакопейним; інші неорганічні домішки можуть бути визначені за допомогою інших відповідних методик, наприклад, за допомогою атомно-абсорбційної спектрометрії.
ж) Мікробіологічна чистота
У специфікації може бути необхідним зазначення загальної кількості аеробних мікроорганізмів, загальної кількості дріжджових і плісеневих грибів, а також встановлення вимоги відсутності певних бактерій (наприклад, Staphylococcus aureus, Escherichia coli, Salmonella, Pseudomonas aeruginosa). Цю кількість слід відповідним чином визначати, використовуючи фармакопейні методики. При виборі виду випробування (випробувань) на мікробіологічну чистоту і критеріїв прийнятності слід виходити з природи діючої речовини, способу виробництва та призначення лікарського препарату. Наприклад, випробування на стерильність може бути доцільним для діючих речовин, вироблених як стерильна продукція, а випробування на ендотоксини — для діючих речовин, що використовуються для отримання ін’єкційного лікарського препарату.
Схема рішень № 6 (див. додаток А) надає додаткові вказівки щодо того, коли слід включати в специфікацію межі вмісту мікроорганізмів.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
У специфікації на певні нові лікарські препарати, як правило, слід включати додаткові випробування та критерії прийнятності. Наведений нижче перелік є репрезентативним прикладом лікарських препаратів, а також видів випробувань і критеріїв прийнятності, які може бути доцільним включати в специфікацію. Конкретні лікарські форми, що обґоворюються, охоплюють тверді й рідкі лікарські засоби для орального застосування, а також парентеральні лікарські засоби (малого та великого об’єму). Положення, наведені в п. 5.1.3.3.2, можуть бути застосовні й до інших лікарських форм. Слід зазначити, що питання, які стосуються оптично активних діючих речовин і твердого стану, розглянуті в п. 5.1.3.3.1.
Деякі з цих випробувань можуть бути застосовні також до м’яких капсул і гранул.
а) Розчинення
Специфікація на тверді лікарські форми для орального застосування, як правило, має містити визначення вивільнення діючої речовини з лікарського препарату. Вимірювання в одній точці, як правило, вважають застосовними до лікарських форм зі звичайним (негайним) вивільненням. Для лікарських форм з модифікованим вивільненням слід встановити відповідні умови випробувань і методики відбору проб. Наприклад, для лікарських форм із пролонгованим вивільненням слід проводити відбір проб через кратні проміжки часу, а для лікарських форм із відстроченим вивільненням слід проводити двостадійне випробування (із використанням різних середовищ послідовно чи паралельно, залежно від ситуації). У таких випадках при плануванні випробувань і встановленні критеріїв прийнятності важливо взяти до уваги групи населення, для яких призначений цей лікарський препарат (наприклад, особи похилого віку, які страждають ахлоргідрією). У деяких випадках (див. п. 5.1.3.3.2.1(б) «Розпадання») випробування на розчинення може бути замінене випробуванням на розпадання (див. схему рішень № 7(1) у додатку А).
Якщо показано, що при зміні швидкості розчинення значно змінюється біодоступність, то для лікарських препаратів зі звичайним (негайним) вивільненням бажано розробити умови випробування, які дозволяли б виявити серії з неприйнятною біодоступністю. Якщо на розчинність істотно впливають зміни в складі чи параметрах процесу, і такі зміни не контролюють за допомогою іншого встановленого в специфікації випробування, то також доцільно вибрати такі умови випробування на розчинення, які дозволяли б виявити ці зміни (див. схему рішень № 7(2) у додатку А).
Якщо розчинення істотно впливає на біодоступність, то слід встановити критерії прийнятності для випробування на розчинення, щоб відбраковувати серії з неприйнятною біодоступністю. Однак встановлені умови випробування та критерії прийнятності повинні дозволяти схвалювати серії, прийнятні в клінічному відношенні (див. схему рішень № 7(2) у додатку А).
Щоб встановити критерії прийнятності для лікарських препаратів із пролонгованим вивільненням, можна використати кореляцію результатів випробувань in vitro й in vivo за умови наявності даних про біодоступність у людини для складів, що характеризуються різною швидкістю вивільнення. Якщо такі дані відсутні, і не може бути доведено, що вивільнення лікарського засобу не залежить від умов випробування in vitro, критерії прийнятності слід встановити на підставі наявних результатів випробувань серій. Як правило, допустимі відхилення середньої швидкості вивільнення на заданий момент часу не повинні перевищувати ±10% від зазначеного на етикетці вмісту діючої речовини (тобто, загальне відхилення може становити 20%; таким чином, вимога (50+10)% означає, що допустима межа складає від 40% до 60%) за винятком випадків, коли за допомогою результатів дослідження біоеквівалентності обґрунтовані ширші межі (див. схему рішень № 7(3) у додатку А).
б) Розпадання
Для лікарських препаратів із високою швидкістю розчинення (розчинення >80% протягом 15 хв при рН 1,2, 4,0 і 6,8), що містять добре розчинні у фізіологічному інтервалі рН лікарські речовини (доза розчиняється в об’ємі, меншому за 250 мл при рН від 1,2 до 6,8), випробування на розчинення можна замінити випробуванням на розпадання. Проведення випробування на розпадання найдоцільніше, якщо був встановлений взаємозв’язок із розчиненням або було показано, що визначення розпадання є більш адекватним випробуванням, ніж визначення розчинення. У таких випадках випробування на розчинення можна не проводити. Що стосується вибору випробувань на розчинення або розпадання, то передбачається, що для підтвердження надійності складу та виробничого процесу слід надавати інформацію про розробку (див. схему рішень № 7(1) у додатку А).
в) Стійкість до роздавлювання та стираність
Як правило, доцільно проводити випробування стійкості до роздавлювання і/або стираності як контроль у процесі виробництва (див. п. 5.1.2.3). У таких випадках звичайно немає необхідності включати ці показники в специфікацію. Якщо показники стійкості до роздавлювання та стираність мають велике значення для якості лікарського препарату (наприклад, у разі жувальних таблеток), у специфікацію слід включити відповідні випробування та критерії прийнятності.
г) Однорідність
Це поняття включає як однорідність маси одиниць дозованого лікарського засобу, так і однорідність вмісту діючої речовини в одиницях дозованого лікарського засобу; при випробуваннях слід використовувати фармакопейні методики. Як правило, специфікація повинна містити одне з цих випробувань, але не обидва одночасно. Якщо доцільно, то такі випробування можуть проводитися в процесі виробництва; при цьому критерії прийнятності слід включити в специфікацію. Якщо для нових лікарських препаратів відхилення в масі перевищують ту допустиму межу, при якій можна визначати однорідність вмісту шляхом визначення відхилення в масі, заявники при розробці лікарських препаратів повинні пересвідчитися в тому, що лікарський препарат є достатньо однорідним.
д) Вміст води
Якщо доцільно, в специфікацію слід включати визначення вмісту води. Критерії прийнятності можуть бути обґрунтовані за допомогою даних про ефекти гідратації або про абсорбцію води лікарським препаратом. У деяких випадках можна вважати достатнім визначення втрати в масі при висушуванні; однак переважним є використання методики визначення, специфічної щодо води (наприклад, титрування за методом К. Фішера).
е) Мікробіологічна чистота
Визначення вмісту мікроорганізмів є невід’ємним атрибутом належної виробничої практики та забезпечення якості. Як правило, рекомендується проводити такі випробування щодо лікарського препарату за винятком тих випадків, коли його компоненти зазнавали випробувань до початку виробництва, а сам виробничий процес за результатами валідаційних досліджень не представляє значного ризику мікробної контамінації чи розмноження мікроорганізмів. Хоча ця настанова не має безпосереднього відношення до допоміжних речовин, слід зазначити, що принципи, які розглядаються в ній, можуть бути застосовні й до допоміжних речовин так само, як і до нових лікарських препаратів. В обох випадках доцільний підхід, пов’язаний із проведенням вибіркових (періодичних) випробувань, якщо це допустимо. (Щодо випробувань мікробіологічної чистоти допоміжних речовин див. схему рішень № 6 у додатку А).
Слід встановити критерії прийнятності щодо загальної кількості аеробних мікроорганізмів, загальної кількості дріжджових і плісеневих грибів, а також встановити вимогу відсутності певних бактерій (наприклад, Staphylococcus aureus, Escherichia coll, Salmonella, Pseudomonas aeruginosa). Мікробіологічну чистоту слід визначати, використовуючи фармакопейні методики за допомогою відповідних процедур при такій частоті відбору проб або в таких часових точках виробничого процесу, які обґрунтовані наявними даними та досвідом. При виборі виду випробування (випробувань) на мікробіологічну чистоту та критеріїв прийнятності слід враховувати природу діючої речовини, спосіб виробництва та призначення лікарського препарату. За наявності задовільного наукового обґрунтування можна запропонувати не проводити визначення мікробіологічної чистоти щодо твердих лікарських форм для орального застосування.
Схема рішень № 8 (див. додаток А) надає додаткові вказівки щодо випробувань на мікробіологічну чистоту.
Деякі з цих спеціальних випробувань, як правило, застосовні до рідких лікарських засобів для орального застосування, а також до порошків, що призначені для розведення з метою отримання таких рідких лікарських засобів.
а) Однорідність
Це поняття включає як однорідність маси одиниць дозованого лікарського засобу, так і однорідність вмісту діючої речовини в одиницях дозованого лікарського засобу; при випробуваннях слід використовувати фармакопейні методики. Як правило, специфікація повинна містити одне з цих випробувань, але не обидва одночасно. Якщо для нових лікарських препаратів відхилення в масі перевищують ту допустиму межу, при якій можна визначати однорідність вмісту шляхом визначення відхилення в масі, заявники при розробці лікарських препаратів повинні пересвідчитися в тому, що лікарський препарат є достатньо однорідним.
Якщо доцільно, випробування можуть бути проведені в процесі виробництва; однак критерії прийнятності слід включити в специфікацію. Цей принцип може бути застосований щодо рідких лікарських засобів як в однодозових, так і в багатодозових пакованнях.
Одиницею дозованого лікарського засобу вважають звичайну дозу, що застосовується хворим. Якщо контролюють фактичну дозу, яку отримує пацієнт, це можна зробити або за допомогою прямого вимірювання, або за допомогою розрахунку — шляхом ділення загальної виміряної маси чи об’єму лікарського препарату на передбачувану загальну кількість доз. Якщо пристрій для дозування (такий як медичні піпетки або пробки-крапельниці для флаконів) є невід’ємною частиною паковання, то для вимірювання дози слід використати цей пристрій. В іншому випадку слід використовувати стандартні одиниці вимірювання об’єму. Вибір пристрою для дозування, як правило, здійснюють в ході розробки.
Для порошків, призначених для розведення, як правило, вважається прийнятним випробування на однорідність маси.
б) рН
Якщо доцільно, то слід передбачити критерії прийнятності щодо рН і обґрунтувати пропоновані межі.
в) Мікробіологічна чистота
Визначення вмісту мікроорганізмів є невід’ємним атрибутом належної виробничої практики та забезпечення якості. Як правило, щодо лікарського препарату рекомендується проводити такі випробування за винятком тих випадків, коли його компоненти зазнавали випробувань до початку виробництва, а сам виробничий процес за результатами валідаційних досліджень не представляє значного ризику мікробної контамінації або розмноження мікроорганізмів. Хоча ця настанова не має безпосереднього відношення до допоміжних речовин, слід зазначити, що принципи, які розглядаються в ній, можуть бути застосовні й до допоміжних речовин так само, як і до нових лікарських препаратів. В обох випадках доцільний підхід, пов’язаний із проведенням вибіркових (періодичних) випробувань, якщо це допустимо. За наявності задовільного наукового обґрунтування можна запропонувати не проводити визначення мікробіологічної чистоти щодо порошків, призначених для розведення з метою отримання рідких лікарських засобів для орального застосування.
Слід встановити критерії прийнятності щодо загальної кількості аеробних мікроорганізмів, загальної кількості дріжджових і плісеневих грибів, а також встановити вимогу відсутності певних бактерій (наприклад, Staphylococcus aureus, Escherichia coli, Salmonella, Pseudomonas aeruginosa). Цю кількість слід визначати, використовуючи фармакопейні методики, за допомогою відповідних процедур при такій частоті відбору проб або в таких часових точках виробничого процесу, які обґрунтовані наявними даними та досвідом.
Схема рішень № 8 (див. додаток А) надає додаткові вказівки щодо випробувань мікробіологічної чистоти.
г) Вміст антимікробного консерванту
Якщо до складу рідких лікарських засобів для орального застосування необхідне введення антимікробного консерванту, то слід встановити критерії прийнятності щодо його вмісту. Критерії прийнятності для вмісту консерванту мають ґрунтуватися на значеннях вмісту антимікробного консерванту, що забезпечують збереження мікробіологічної чистоти лікарського препарату протягом всього періоду його застосування та протягом терміну зберігання. Використовуючи фармакопейну методику визначення ефективності антимікробного консерванту, слід довести його ефективність щодо затримки росту мікроорганізмів при найнижчій концентрації, що зазначена у специфікації.
Випробування з визначення вмісту антимікробного консерванту, як правило, слід проводити при випуску серії. У деяких випадках замість випробування при випуску може бути достатньо проведення випробування в процесі виробництва. Якщо випробування з визначення вмісту антимікробного консерванту проводять у процесі виробництва, критерії прийнятності все одно мають бути включені в специфікацію.
Хоча у специфікацію звичайно включають кількісне визначення антимікробного консерванту методом аналітичної хімії, під час розробки, в ході масштабування технологічного процесу, а також протягом терміну зберігання (наприклад, при випробуваннях стабільності відповідно до Настанови 42-3.3:2004) має бути доведена ефективність антимікробного консерванту.
д) Вміст антиоксиданту
Як правило, випробування з визначення вмісту антиоксиданту слід проводити при випуску серії. За певних обставин, якщо це обґрунтоване отриманими в ході розробки даними та результатами випробувань стабільності, може не знадобитися проведення випробувань протягом терміну зберігання, а замість випробувань при випуску може бути достатнім проведення випробувань у процесі виробництва (за наявності відповідного дозволу). Якщо визначення вмісту антиоксиданту проводять у процесі виробництва, критерії прийнятності все одно мають бути включені в специфікацію. Якщо випробування проводять тільки при випуску, то при внесенні змін у технологічний процес або систему контейнер/закупорювальний елемент для застосування такого підходу слід провести повторні дослідження.
е) Речовини, що екстрагуються
Якщо отримані в ході розробки та вивчення стабільності дані свідчать про те, що кількість речовин, які екстрагуються із системи контейнер/закупорювальний елемент, постійно нижчі таких рівнів вмісту, які є прийнятними та безпечними, то, як правило, допустиме виключення цього випробування зі специфікації. Якщо в систему контейнер/закупорювальний елемент або в склад лікарського засобу вносять зміни, то ці дослідження слід провести повторно.
Якщо дані свідчать про необхідність проведення випробувань щодо речовин, що екстрагуються із системи контейнер/закупорювальний елемент (наприклад, із гумових пробок, прокладок, пластикових флаконів тощо), то введення випробувань і критеріїв прийнятності слід вважати доцільним у разі розчинів для орального застосування, первинне паковання (контейнер і закупорювальний елемент) яких виготовлене не зі скла, або які вміщують у скляні флакони із закупорювальними елементами, виготовленими не зі скла. Слід навести перелік складових частин системи контейнер/закупорювальний елемент і надати дані щодо цих складових частин, починаючи з якомога більш ранньої стадії розробки.
ж) Вміст спирту
Якщо на етикетці зазначають кількісний вміст спирту згідно з вимогами ДФУ або інших відповідних нормативних документів, то його слід наводити в специфікації. Вміст спирту можна встановлювати за допомогою кількісного визначення або розрахункового методу.
и) Розчинення
У разі суспензій для орального застосування та сухих порошків, призначених для отримання суспензій, крім показників, рекомендованих вище, може бути доцільним (наприклад, у разі нерозчинної діючої речовини) включити в специфікацію випробування на розчинення та критерії прийнятності. Випробування на розчинення слід проводити при випуску. Таке випробування може бути проведене в процесі виробництва, якщо це обґрунтовано даними, отриманими в ході розробки лікарського препарату. Прилад для проведення випробування, середовища та умови випробування мають бути, по можливості, фармакопейними; в іншому випадку необхідне відповідне обґрунтування. Методики проведення випробування на розчинення з використанням як фармакопейних, так і нефармакопейних приладів, а також умови випробувань слід валідувати.
Вимірювання в одній точці контролю, як правило, застосовують до лікарських форм зі звичайним (негайним) вивільненням. Для лікарських форм із модифікованим вивільненням слід проводити відбір проб через певні відрізки часу. Критерії прийнятності необхідно встановлювати на підставі інтервалу відхилень, що спостерігається; при цьому слід враховувати профілі розчинення серій, для яких встановлені прийнятні функціональні характеристики in vivo. При вирішенні питання про необхідність уведення або випробування на розчинення, або визначення розміру часток слід брати до уваги дані, отримані в ході розробки.
к) Розмір часток (розподіл часток за розмірами)
У разі суспензій для орального застосування може бути доцільним включення в специфікації кількісних критеріїв прийнятності та методики визначення розміру часток (розподілу часток за розмірами). При вирішенні питання про необхідність уведення для таких складів або випробування на розчинення, або визначення розміру часток слід брати до уваги дані, отримані в ході розробки.
Визначення розміру часток (розподілу часток за розмірами) слід здійснювати при випуску. Таке випробування може бути проведене в процесі виробництва, якщо це обґрунтовано даними, отриманими в ході розробки лікарського препарату. Якщо в ході розробки доведено, що цей лікарський препарат постійно характеризується швидким вивільненням лікарської речовини, можна запропонувати виключення зі специфікації визначення розміру часток (розподілу часток за розмірами).
Визначення розміру часток (розподілу часток за розмірами) може бути також запропоноване замість випробування на розчинення; у цьому випадку необхідно подати відповідне обґрунтування. Критерії прийнятності повинні включати розподіл часток за розмірами, який виражають як відсоток часток, що мають розмір у даному діапазоні, від загального числа часток. Необхідно чітко встановити граничні значення для середнього, найбільшого і/або найменшого розміру часток.
Критерії прийнятності слід встановлювати на підставі діапазону значень, що спостерігається, з урахуванням профілів розчинення серій, для яких встановлені прийнятні функціональні характеристики in vivo, і призначення лікарського препарату. У ході розробки лікарського засобу необхідно вивчити можливість збільшення розмірів часток; результати цих досліджень слід враховувати при встановленні критеріїв прийнятності.
л) Редиспергованість
У разі суспензій для орального застосування, що характеризуються осіданням часток дисперсної фази при зберіганні (седиментацією), може бути доцільним встановити критерії прийнятності щодо редиспергованості. Відповідною процедурою може бути збовтування.
У специфікації має бути вказана методика (що виконується автоматично або вручну). Слід чітко визначити час, необхідний для ресуспендування при використанні зазначеної методики. Щоб обґрунтувати можливість вибіркових (періодичних) випробувань серій або запропонувати виключити даний показник зі специфікації, може бути достатньо даних, отриманих у ході розробки лікарського препарату.
м) Реологічні властивості
Для відносно в’язких розчинів або суспензій може бути доцільним включення у специфікацію реологічних характеристик (в’язкість) із визначенням, за необхідності, відносної щільності. Слід зазначити методику(и) випробувань та критерії прийнятності. Щоб обґрунтувати можливість вибіркових (періодичних) випробувань серій або запропонувати виключити ці показники зі специфікації, може бути достатньо даних, отриманих у ході розробки лікарського препарату.
н) Час підготовки до застосування
У разі сухих порошків, призначених для розведення перед застосуванням, слід передбачити критерії прийнятності щодо часу підготовки до застосування. Необхідно обґрунтувати вибір рідини для розведення. Щоб обґрунтувати можливість вибіркових (періодичних) випробувань серій або запропонувати виключити цей показник зі специфікації, може бути достатньо даних, отриманих у ході розробки лікарського препарату.
п) Вміст води
У разі лікарських препаратів для орального застосування, призначених для розведення, за необхідності слід запропонувати випробування та критерій прийнятності щодо вмісту води. Якщо вплив абсорбційної або гідратної води достатньо охарактеризований під час розробки лікарського препарату, то вважається достатнім визначення втрати в масі при висушуванні. У певних випадках може бути переважним застосування більш специфічної методики (наприклад, титрування за методом К. Фішера).
Для парентеральних лікарських препаратів можуть бути застосовні наступні випробування.
а) Однорідність
Це поняття включає як однорідність маси одиниць дозованого лікарського засобу, так і однорідність вмісту діючої речовини в одиницях дозованого лікарського засобу; при випробуваннях слід використовувати фармакопейні методики. Як правило, специфікація має містити одне з цих випробувань, але не обидва одночасно. Якщо для нових лікарських препаратів відхилення в масі перевищують ту допустиму межу, при якій можна визначати однорідність вмісту шляхом визначення відхилення в масі, заявники в ході розробки лікарських препаратів повинні пересвідчитися в тому, що лікарський препарат є достатньо однорідним.
Якщо доцільно (див. п. 5.1.2.3), випробування можуть бути проведені в процесі виробництва; однак критерії прийнятності слід включити в специфікацію. Це випробування може бути застосовне щодо лікарських засобів як в однодозових, так і в багатодозових пакованнях.
У разі порошків, призначених для розведення, як правило, вважається прийнятним випробування на однорідність маси.
б) рН
Якщо доцільно, слід передбачити критерії прийнятності щодо рН та обґрунтувати пропоновані межі.
в) Стерильність
Для всіх парентеральних лікарських засобів у специфікацію мають бути включені методика випробування та критерії прийнятності для оцінки стерильності. Якщо отримані в ході розробки та валідації дані дають змогу обґрунтувати випуск за параметрами, то цей підхід може бути запропонований для лікарських засобів, що стерилізуються в остаточному первинному пакованні на заключній стадії (див. п. 5.1.2.6).
г) Бактеріальні ендотоксини/пірогени
У специфікацію слід включити методику випробувань і критерії прийнятності щодо бактеріальних ендотоксинів; необхідно застосовувати-методику з використанням лізата амебоцитів мечохвоста Limulus polyphemus (LAL-тест). За наявності відповідного обґрунтування як альтернатива випробуванню на бактеріальні ендотоксини може бути запропоноване випробування на пірогени.
д) Механічні включення
Для парентеральних лікарських засобів слід встановити відповідні критерії прийнятності щодо механічних включень. Як правило, до них належать критерії прийнятності для видимих часток і/або прозорості розчину, а також, за необхідності, для невидимих часток.
е) Вміст води
За необхідності, для неводних парентеральних лікарських засобів і лікарських препаратів для парентерального застосування, призначених для розведення, слід запропонувати методику випробування та критерій прийнятності щодо вмісту води. Якщо вплив абсорбційної чи гідратної води достатньо охарактеризований під час розробки лікарського препарату, то вважається достатнім визначення втрати в масі при висушуванні. У певних випадках може бути переважним застосування більш специфічної методики (наприклад, титрування за методом К. Фішера).
ж) Вміст антимікробного консерванту
Якщо до складу парентерального лікарського засобу необхідне введення антимікробного консерванту, то слід встановити критерії прийнятності для його вмісту. Критерії прийнятності для вмісту консерванту мають ґрунтуватися на значеннях вмісту антимікробного консерванту, що забезпечують збереження мікробіологічної чистоти лікарського препарату протягом усього періоду його застосування та протягом терміну зберігання. Використовуючи фармакопейну методику визначення ефективності антимікробного консерванту, слід довести його ефективність щодо затримки росту мікроорганізмів при найнижчій зазначеній у специфікації концентрації.
Випробування з визначення вмісту антимікробного консерванту, як правило, слід проводити при випуску серії. У деяких випадках (за наявності відповідного дозволу) замість випробування при випуску може бути достатньо випробування в процесі виробництва. Якщо визначення вмісту антимікробного консерванту проводять у процесі виробництва, критерії прийнятності все одно мають бути включені в специфікацію.
Хоча в специфікацію звичайно включають кількісне визначення антимікробного консерванту методом аналітичної хімії, під час розробки, у ході масштабування технологічного процесу, а також протягом терміну зберігання (наприклад, при випробуваннях стабільності відповідно до Настанови 42-3.3-2004) має бути доведена ефективність антимікробного консерванту.
и) Вміст антиоксиданту
Як правило, визначення вмісту антиоксиданту слід проводити при випуску серії. За певних обставин, якщо це обґрунтовано отриманими в ході розробки даними та результатами випробувань стабільності, можна не проводити випробування протягом терміну зберігання, а замість випробувань при випуску може бути достатньо випробувань у процесі виробництва. Якщо вміст антиоксиданту визначають у процесі виробництва, критерії прийнятності все одно мають бути включені в специфікацію. Якщо випробування проводять лише при випуску, то при внесенні змін у технологічний процес або систему контейнер/закупорювальний елемент для застосування такого підходу слід провести повторні дослідження.
к) Речовини, що екстрагуються
Для парентеральних лікарських засобів контроль речовин, що екстрагуються із систем контейнер/закупорювальний елемент, вважається більш важливим, ніжу у разі рідких лікарських засобів для орального застосування. Однак якщо отримані в ході розробки та вивчення стабільності дані свідчать про те, що кількість речовин, які екстрагуються із системи контейнер/закупорювальний елемент, постійно нижча за такий рівень вмісту, який є прийнятним та безпечним, то може бути допустиме виключення цього випробування зі специфікації. Якщо в систему контейнер/закупорювальний елемент або в склад лікарського засобу вносять зміни, то ці дослідження слід провести повторно.
Якщо дані свідчать про необхідність проведення випробувань щодо речовин, які екстрагуються із системи контейнер/закупорювальний елемент, то введення випробувань і критеріїв прийнятності вважається доцільним для парентеральних лікарських засобів, первинне паковання (контейнер і закупорювальний елемент) яких виготовлене не зі скла, або які вміщують у скляні контейнери з гумовими закупорювальними елементами. Слід навести перелік складових частин системи контейнер/закупорювальний елемент (наприклад, гумова пробка тощо) і надати дані щодо цих складових частин, починаючи з якомога більш ранньої стадії розробки.
л) Випробування функціональних характеристик систем доставки
Для парентеральних лікарських засобів, пакування яких передбачене в шприци, що заповнюються заздалегідь, картриджі для автоматичних інжекторів або подібні пристрої, у специфікацію слід включати методики випробувань і критерії прийнятності, що стосуються функціональних характеристик систем доставки. До них можуть належати контроль можливості введення за допомогою шприца, тиску та герметичності закупорювання (витоку) і/або таких параметрів, як зусилля для видалення ковпачка з наконечника, зусилля для переміщення поршня, зусилля для приведення в дію інжектора тощо. За певних обставин ці випробування можуть бути проведені в процесі виробництва. Щоб обґрунтувати проведення вибіркових (періодичних) випробувань серій або запропонувати виключити деякі або всі такі характеристики зі специфікації, може бути достатньо даних, отриманих у ході розробки лікарського препарату.
м) Осмолярність
Якщо на етикетці лікарського препарату зазначена його тонічність, слід проводити відповідний контроль осмолярності. Щоб обґрунтувати проведення цього випробування в процесі виробництва, проведення вибіркових (періодичних) випробувань серій або визначення цього показника шляхом розрахунку, може бути достатньо даних, отриманих у ході розробки лікарського засобу та при валідації.
н) Розмір часток (розподіл часток за розмірами)
Для ін’єкційних лікарських засобів у вигляді суспензій може бути доцільним встановлення кількісних критеріїв прийнятності та методики визначення розміру часток (розподілу часток за розмірами). При вирішенні питання про необхідність введення або випробування на розчинення, або визначення розміру часток (розподілу часток за розмірами), слід брати до уваги дані, отримані в ході розробки.
Визначення розміру часток (розподілу часток за розмірами) необхідно здійснювати при випуску. Таке випробування може проводитися в процесі виробництва, якщо це обґрунтовано даними, отриманими в ході розробки лікарського препарату. Якщо в ході розробки доведено, що лікарський препарат постійно характеризується швидким вивільненням лікарської речовини, то можна запропонувати виключити зі специфікації визначення розміру часток (розподілу часток за розмірами).
Визначення розміру часток (розподілу часток за розмірами) може бути також запропоноване замість випробування на розчинення, якщо дослідження в ході розробки показали, що розмір часток є основним чинником, що впливає на розчинення; у цьому випадку необхідно надати відповідне обґрунтування. Критерії прийнятності повинні включати розподіл часток за розмірами, який виражають як відсоток часток, що мають розмір у даному діапазоні, від загального числа часток. Необхідно чітко встановити граничні значення для середнього, найбільшого і/або найменшого розміру часток.
Критерії прийнятності слід встановлювати на підставі діапазону значень, що спостерігається, з урахуванням профілів розчинення серій, для яких встановлені прийнятні функціональні характеристики in vivo, а також призначення лікарського препарату. У ході розробки лікарського засобу необхідно вивчити можливість збільшення розмірів часток; результати цих досліджень слід враховувати при встановленні критеріїв прийнятності.
п) Редиспергованість
Для ін’єкційних лікарських засобів у вигляді суспензій, що характеризуються осіданням часток при зберіганні (седиментацією), може бути доцільним встановити критерії прийнятності щодо редиспергованості. Підхожою процедурою може бути збовтування. У специфікації має бути зазначена методика (що виконується автоматично або вручну). Слід чітко визначити час, необхідний для ресуспендування при використанні зазначеної методики. Щоб обґрунтувати можливість вибіркових випробувань серій або запропонувати виключити цей показник зі специфікації, може бути достатньо даних, отриманих у ході розробки лікарського препарату.
р) Час підготовки до застосування
У разі всіх парентеральних лікарських засобів, призначених для розведення перед застосуванням, слід передбачити критерії прийнятності щодо часу підготовки до застосування. Необхідно обґрунтувати вибір рідини для розведення. Для швидкорозчинних лікарських препаратів може бути достатньо даних, отриманих у ході розробки, щоб обґрунтувати можливість проведення вибіркових випробувань серій або запропонувати виключення цього показника зі специфікації.
Положення підрозділу 5.2 присвячені складанню специфікацій, що необхідні для включення в реєстраційне досьє, яке подається до компетентних уповноважених органів із метою реєстрації/перереєстрації лікарського засобу, а також для виробництва лікарських засобів відповідно до правил GMP і контролю якості в офіційних лабораторіях.
У специфікації включають такі показники якості:
а) загальні характеристики лікарської форми, зокрема, описи та фармако-технологічні характеристики, що стосуються функціональних або технологічних властивостей продукції (наприклад, твердість, стираність звичайної таблетки), які визначають, як правило, за допомогою фізичних випробувань із встановленням допустимих меж;
б) ідентифікацію діючої(их) речовини (речовин);
в) кількісне визначення діючих речовин (для лікарських препаратів рослинного походження кількісне визначення компонентів із відомою терапевтичною активністю);
г) за необхідності, ідентифікацію та кількісне визначення допоміжних речовин, а саме:
д) випробування на чистоту (за необхідності, дослідження продуктів розкладу, залишкових кількостей розчинників або інших супровідних домішок, а також мікробіологічної чистоти);
е) фармако-технологічні випробування (наприклад, розчинення);
ж) випробування безпеки, включаючи, за необхідності, випробування аномальної або специфічної токсичності, зокрема, для біологічних препаратів.
Щоб встановити специфікації на готовий лікарський засіб, необхідно врахувати показники якості, пов’язані з виробничим процесом.
У ході розробки та валідації виробничого процесу слід встановити відповідний показник для кожного аспекту якості. Принаймні, ті показники якості, які вважаються критичними, мають бути об’єктом рутинного контролю відповідно до специфікацій.
Загальні зауваження ДФУ та Європейської Фармакопеї описують і визначають зміст, а також офіційне значення різних розділів фармакопейних монографій і загальних статей.
У попередніх частинах цього підрозділу перераховані ті показники якості, що мають бути включені у відповідні специфікації. За необхідності мають бути зроблені посилання на загальні статті ДФУ або Європейської Фармакопеї чи (за відсутності в них таких статей) на загальні статті інших відповідних фармакопей.
Що стосується монографій на лікарські препарати, то ДФУ, Європейська Фармакопея чи інша відповідна фармакопея визначають стандартний рівень якості. У заявці на реєстрацію лікарського препарату заявник має визначити найбільш відповідні засоби для досягнення заданої мети і додати відповідні дані аналітичної валідації. У будь-якому випадку при випуску готового лікарського засобу рутинні випробування якості мають відповідати умовам п. 5.2.1.4.
Крім того, оскільки фармакопейні монографії є регуляторними специфікаціями, у них встановлені граничні значення показників якості лікарського препарату на момент закінчення терміну його зберігання (за винятком положень, пов’язаних із виробництвом). Специфікації на готову продукцію, що застосовуються при її випуску, повинні містити більш суворі критерії, тобто відхилення від кількісного вмісту діючої(их) речовини (речовин) не повинне перевищувати ±5%, якщо не обґрунтовано інше.
а) Мета реєстраційного досьє — встановити рівень якості лікарського препарату, призначеного для розміщення на ринку. Цей рівень якості є підставою для складання специфікацій, тобто якісних і кількісних показників (із зазначенням методик випробувань і допустимих меж), яким має відповідати лікарський препарат протягом терміну зберігання.
У відповідних частинах реєстраційного досьє заявник пропонує термін зберігання лікарського препарату, головним чином, на підставі рівня вмісту діючих речовин (ефективність), допустимого рівня вмісту будь-яких продуктів розкладу або домішок (безпека) і постійності фармако-технологічних властивостей. На підставі поведінки лікарського препарату заявник встановлює підхожі умови зберігання, при яких буде підтримуватися відповідність лікарського препарату специфікаціям.
б) Для готового лікарського засобу заявник має встановити такі межі в специфікації, що застосовується при випуску (на момент виробництва) серії, які гарантуватимуть відповідність лікарського препарату пропонованій специфікації, що застосовується протягом терміну зберігання. Ці межі встановлюють на підставі критичного докладного огляду даних, отриманих при аналізі серій. Проте дуже важливим чинником із погляду принципів належної виробничої практики є набутий досвід. Однією з основних вимог GMP (див. Настанову 42–01–2001) є систематичний огляд усіх виробничих процесів у світлі набутого досвіду. Таким чином, заявник має приводити у відповідність або вдосконалювати специфікації, що застосовуються при випуску, на підставі досвіду, набутого виробником(ами) лікарського препарату.
в) Специфікація на готовий лікарський засіб, що застосовується при випуску, може відрізнятися від специфікації, що застосовується протягом терміну зберігання.
У деяких випадках показники якості лікарського препарату можуть змінюватися під час зберігання при затверджених умовах. При цьому критерії прийнятності для таких показників у відповідних специфікаціях, що застосовуються при випуску, слід встановлювати з урахуванням якості лікарського препарату, що вимагається протягом терміну зберігання (наприклад, надлишки для забезпечення стабільності).
Бажано, щоб усі специфікації (показники якості та допустимі межі), що застосовуються при випуску та протягом терміну зберігання, були наведені у вигляді зведеної таблиці (таблиця 1). У такій таблиці мають бути зазначені межі вмісту будь-яких вірогідних продуктів розкладу, які можуть утворитися під час зберігання при затверджених умовах.
Зведена таблиця може бути подана в такому вигляді:
Таблиця 1 — Зведена таблиця специфікацій, що застосовуються при випуску та протягом терміну зберігання
|
Показники |
Методи випробувань та допустимі межі для лікарського препарату |
|
| протягом терміну зберігання | при випуску | |
|
1. Характеристики лікарської форми |
||
|
2. Ідентифікація та кількісне визначення діючих речовин |
||
|
3.Випробування на чистоту |
||
|
4. Допоміжні речовини:
|
||
|
Примітка. Слід позначити зірочкою межі, для яких може знадобитися актуалізація у зв’язку з досвідом, набутим після випуску перших промислових серій числом п. |
||
Необхідно також вказати номер, присвоєний специфікації, поставити підпис і зазначити дату затвердження.
Таке подання повних наборів специфікацій не впливає на вибір і частоту випробувань, які будуть дійсно проводитися стосовно готового лікарського засобу (або, можливо, стосовно нерозфасованої чи проміжної продукції) (див. п. 5.2.1.4).
Відповідність кожної серії готового лікарського засобу специфікаціям, що застосовуються при випуску, має бути гарантована дотриманням правил GMP (див. Настанову 42–01–2001). Однак певні частини документації серії, які потрібні відповідно до GMP, можуть не входити до реєстраційного досьє.
У реєстраційному досьє має бути показано, що за допомогою конкретного виробничого процесу, який проводиться відповідно до правил GMP, можна отримувати готову продукцію, що постійно відповідає вибраним специфікаціям. При розробці таких специфікацій враховують:
а) описані у відповідній частині реєстраційного досьє дослідження з розробки (див. Настанову 42–3.1:2004), на підставі яких визначають особливості складу та виробничого процесу, які мають важливе значення для якості продукції;
б) описані у відповідній частині реєстраційного досьє результати валідації критичних стадій виробничого процесу, за допомогою яких можна контролювати масштабування процесу та відтворюваність серій.
Існують різні види випробувань:
а) випробування, що проводяться для кожної серії готової чи, можливо, нерозфасованої продукції;
б) випробування, проведення яких під час виробничої стадії (випробування проміжної продукції чи контроль у процесі виробництва) більшою мірою гарантує відповідність якості готової продукції, ніж проведення цих випробувань щодо готової чи нерозфасованої продукції;
в) періодичні випробування (випробування, що проводяться не для кожної серії готової продукції або, можливо, проміжної чи нерозфасованої продукції), частота проведення яких великою мірою залежить від дотримання інших правил GMP (наприклад, випробування на мікробіологічну чистоту);
г) випробування, проведення яких щодо певного показника якості готової продукції чи, можливо, нерозфасованої продукції на момент виробництва можуть бути замінені перевіркою іншого дуже залежного показника (наприклад, заміна випробування однорідності маси випробуванням на однорідність вмісту);
д) випробування, які не проводяться рутинно, оскільки гарантії відповідності у вигляді винятку надає виробник; такі специфічні випадки є винятковими (наприклад, ідентифікація барвників);
е) випробування для контролю відповідних критичних точок виробничого процесу, які проводять практично лише під час виготовлення перших серій продукції числом п і тимчасово в ході будь-яких суттєвих змін (наприклад, зміна виробничої дільниці, заміна матеріалів тощо). Згодом, внаслідок набутого досвіду й особливо після валідації технологічного процесу можна не проводити такі випробування для кожної серії (наприклад, щодо залишкових кількостей розчинників).
Можливе існування й інших ситуацій, відмінних від ситуацій, описаних вище.
У реєстраційному досьє мають бути чітко викладені окремі випробування, які необхідно проводити при випуску готової продукції; слід встановити, чи будуть вони проводитися рутинно для кожної серії готової продукції, або зазначити періодичність проведення випробувань (для кожної серії, для кожної n-ої серії, для перших п серій тощо).
Залежно від того, є лікарські препарати відомими чи ні, і чи має виробник відповідний реальний досвід виробництва, можливі два підходи.
У першому випадку випробування, що проводяться рутинно та періодично, можуть бути встановлені відразу на момент подання заявки на реєстрацію. Такий підхід застосовний, наприклад, для нового лікарського препарату з іншою силою дії, ніж у вже зареєстрованого лікарського препарату, або для нової звичайної лікарської форми (розчин, пресована таблетка тощо), що містить добре відому фармакопейну діючу речовину.
У другому випадку слід зазначити, періодичність проведення яких випробувань підлягає коректуванню після набуття досвіду роботи з лікарським препаратом. Такий підхід застосовний, наприклад, для лікарських засобів із новою діючою речовиною або для нової системи доставки такої, як таблетки з модифікованим вивільненням. Про результати досліджень і відповідні коректування слід поінформувати регуляторні уповноважені органи.
Випробування, запропоновані в обох випадках, мають бути наведені у вигляді зведених таблиць (таблиці 2 і 3). За необхідності, випробування, що пропонуються для перших п серій (таблиця 2) мають бути розмежовані з тими випробуваннями, що передбачені для рутинного промислового виробництва (таблиця 3).
Таблиця 2 — Пропонована схема проведення випробувань у початковий період промислового виробництва
|
Випробування |
Частота проведення випробувань (спостереження за першими п серіями) |
У цьому випадку заявник повинен зазначити кількість (п) серій.
Таблиця 3 — Схема проведення випробувань при стабільному промисловому виробництві
|
Випробування |
Частота проведення випробувань |
Кожна серія лікарського препарату, що знаходиться на ринку, має відповідати всім специфікаціям, що визначають очікуваний рівень його якості, незважаючи на те, що плани проведення випробувань формують або остаточно затверджують після набуття досвіду промислового виробництва.
У схеми проведення випробувань можуть бути включені хімічні, фізичні, фармако-технологічні, мікробіологічні або біологічні випробування.
Допустимі межі для різних показників якості в специфікаціях (див. п. 5.2.1.1) встановлюють з урахуванням усіх суттєвих елементів, що стосуються якості лікарського препарату (постійність його характеристик), його сили дії (рівень вмісту діючої речовини) і, за необхідності, його безпеки (ризик мікробної контамінації, продукти розкладу тощо).
Для більшості фармако-технологічних параметрів опис загальних методик випробувань і в деяких випадках стандартів або максимальних меж наведені в ДФУ або Європейській Фармакопеї, або (за відсутності в них таких статей) у інших відповідних фармакопеях. З урахуванням цього в специфікаціях необхідно встановити конкретні мінімальні і/або максимальні межі, адаптовані для лікарського препарату, що реєструється, щоб гарантувати відтворюваність готової продукції при виробництві.
Максимально допустиме відхилення у вмісті діючої речовини в готовому лікарському засобі на момент його виробництва не повинне перевищувати +5% за винятком відповідним чином обґрунтованих випадків. На підставі проведених випробувань стабільності виробник має запропонувати й обґрунтувати допустимі межі вмісту діючої речовини в готовому лікарському засобі протягом рекомендованого терміну зберігання аж до його закінчення.
а) Передбачені при випуску межі ±5% є прийнятними без подальшого обґрунтування.
б) Передбачені при випуску межі, що перевищують ±5%, необхідно обґрунтувати в розділі реєстраційного досьє, де описують фармацевтичну розробку, із наданням експериментальних результатів, як правило, при рівні довірчої імовірності 95%. Ширші межі також можуть бути обґрунтовані варіабельністю як технологічного процесу, так і методики кількісного визначення.
в) Застосування неадекватних виробничих процедур або неадекватних методик випробувань (із низькою точністю) не є обґрунтуванням для ширших меж при випуску.
г) Якщо виробник застосовує коректування кількості діючої речовини при виробництві готової продукції (факторизація), то його обов’язком є виконання вимог щодо меж ±5%. У такому випадку у відповідній частині реєстраційного досьє має бути чітко зазначений надлишок. Межі при випуску мають залишатися ±5% від установленого вмісту.
д) Посилання на фармакопеї, як правило, не приймаються як обґрунтування для ширших меж, оскільки в монографіях не зазначений точний склад лікарського препарату, і вони не є специфікаціями, що застосовуються при випуску продукції, а призначені для контролю лікарських препаратів офіційними лабораторіями протягом терміну зберігання.
е) У виняткових випадках для певних лікарських препаратів із відомим процесом розкладу, який не впливає на безпеку, може допускатися надлишок при випуску (вітаміни і т.п.). Метою такого надлишку є гарантування відповідного рівня вмісту діючої речовини протягом терміну зберігання. Цей надлишок не повинен спричинити надмірний рівень вмісту діючої речовини при випуску продукції, у зв’язку з чим слід адаптувати межі при випуску.
ж) Вимога щодо меж +5%, що застосовуються при випуску, головним чином стосується виробників продукції; межі вмісту діючих речовин можуть бути ширшими при контролі в сторонніх лабораторіях. Межі, що використовуються при повторному контролі в офіційних лабораторіях, можуть не бути ідентичними встановленим виробником межам при випуску (наприклад, внаслідок міжлабораторної варіабельності), хоча при такому контролі межі при випуску, встановлені виробником, слід брати до уваги. Якщо випробування при повторному контролі проводяться іншою лабораторією, то методики випробувань мають бути валідовані в цій лабораторії.
и) Надмірне наповнення для гарантії доставки теоретичної дози в одиниці лікарської форми має бути обґрунтоване у відповідній частині реєстраційного досьє, у якій описують фармацевтичну розробку. Як наслідок, мають бути адаптовані допустимі межі для визначення вмісту діючої речовини в одиниці дозованої лікарської форми.
а) Допоміжні речовини, що впливають на біодоступність діючої речовини, слід кількісно визначати в кожній серії за винятком випадків, коли біодоступність гарантується іншими відповідними випробуваннями, що встановлюються для кожного окремого випадку на підставі результатів досліджень із розробки.
б) Для консервантів межі вмісту при випуску від 90% до 110% є прийнятними без подальших обґрунтувань за винятком особливих випадків. Нижня межа вмісту антимікробних консервантів протягом терміну зберігання може бути знижена за умови отримання задовільних результатів випробувань ефективності антимікробних консервантів. Для антиоксидантів (хімічних консервантів) нижня межа протягом терміну зберігання може бути значно нижчою за 90% внаслідок їх розкладу для збереження стабільності інших речовин.
Методики випробувань мають бути описані досить детально, щоб надати можливість будь-якій офіційній лабораторії контролювати якість лікарського препарату протягом терміну його зберігання. Методики контролю мають бути валідовані відповідно до настанови «Note for guidance on validation of analytical procedures: methodology»* або загальної статті ДФУ «Валідація аналітичних методик і випробувань»N. Результати такої аналітичної валідації мають бути включені до реєстраційного досьє.
Таким чином, виклад методик(и) випробувань за необхідності має включати опис стандартного зразка з його специфікаціями, формулу розрахунку чи приклад розрахунку (незалежно від того, виконуються чи ні ці розрахунки автоматично), хроматограми та спектри (і т.п.) для підтвердження отриманих результатів.
Немає необхідності описувати методики відбору проб, які знаходяться в сфері дії GMP та інспекційних служб; винятком є методики відбору проб для випробувань, офіційно внесених у ДФУ чи Європейську Фармакопею (наприклад, випробування на стерильність).
У процедурі випробувань може бути використаний або офіційний стандартний зразок (ДФУ, Європейська Фармакопея, ВООЗ), або робочий стандартний зразок за умови, що останній стандартизований за офіційним стандартним зразком (див. настанову «Note for guidance on validation of analytical procedures: methodology»* або загальну статтю ДФУ «Валідація аналітичних методик і випробувань»N).
Необхідно пам’ятати, що аналітичний результат залежить від використаного методу. Тому передбачається, що специфікації, прийняті для випробувань і кількісного визначення, розробляють на підставі описаних у фармакопеї методів шляхом адаптації фармакопейних монографій.
Методи, відмінні від фармакопейних, можуть бути використані для контролю за умови, що вони валідовані щодо офіційного методу, та що при використанні цих методів можна зробити однозначний висновок про відповідність вимогам монографії так само, як і при застосуванні офіційних методів (див. загальні зауваження ДФУ і Європейської Фармакопеї).
Крім того, загальні фармакопейні методи можуть бути використані для лікарських препаратів, не описаних у фармакопеї, або для показників якості, не наведених у фармакопейній монографії. Використання цих методів вимагає відповідної валідації для кожного конкретного випадку.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
Відповідний розділ реєстраційного досьє, присвячений аналізу серій, повинен містити результати, отримані щодо всіх показників якості, які включені в специфікацію, що застосовується при випуску, незалежно від того, чи будуть вони контролюватися для кожної серії, чи ні.
За можливості, розмір послідовних серій має відповідати об’єму промислових серій, що випускаються всіма виробниками та на всіх виробничих дільницях, зазначених у заявці на реєстрацію у відповідній частині реєстраційного досьє. Якщо дані для промислових серій відсутні, то вони мають бути подані до компетентних уповноважених органів якнайшвидше після реєстрації лікарського препарату.
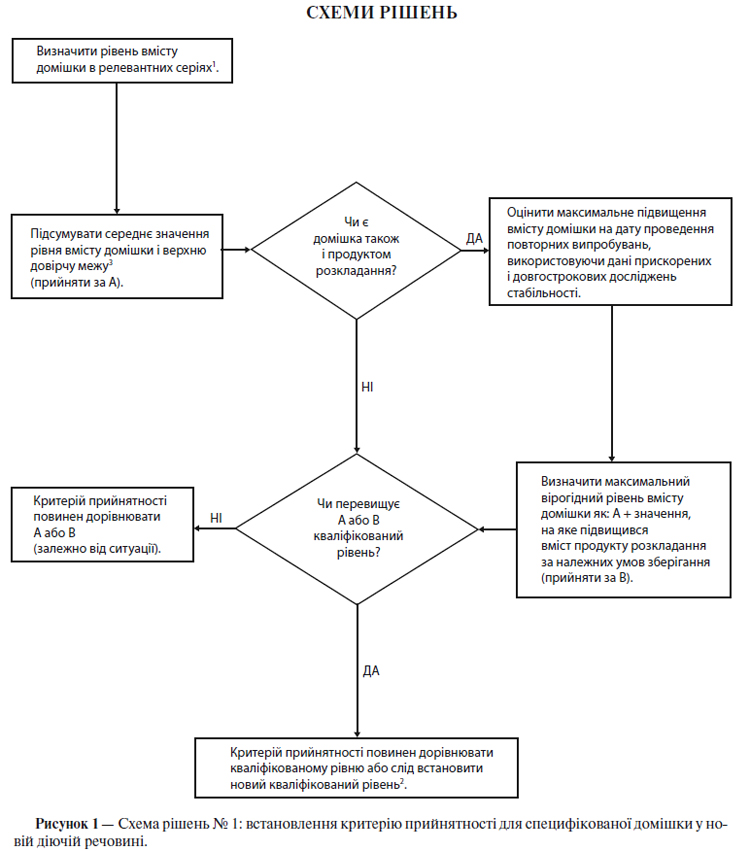
1Релевантні серії — серії, одержані в ході досліджень на етапах розробки, дослідно-промислового і промислового виробництва.
2Див. Настанову СРМР/ІСН/2737/99 (Q3A) «Note for guidance on impurities testing: Impurities in new drug substances».
3Верхня довірча межа дорівнює стандартному відхиленню для даних аналізу серії, помноженому на 3.
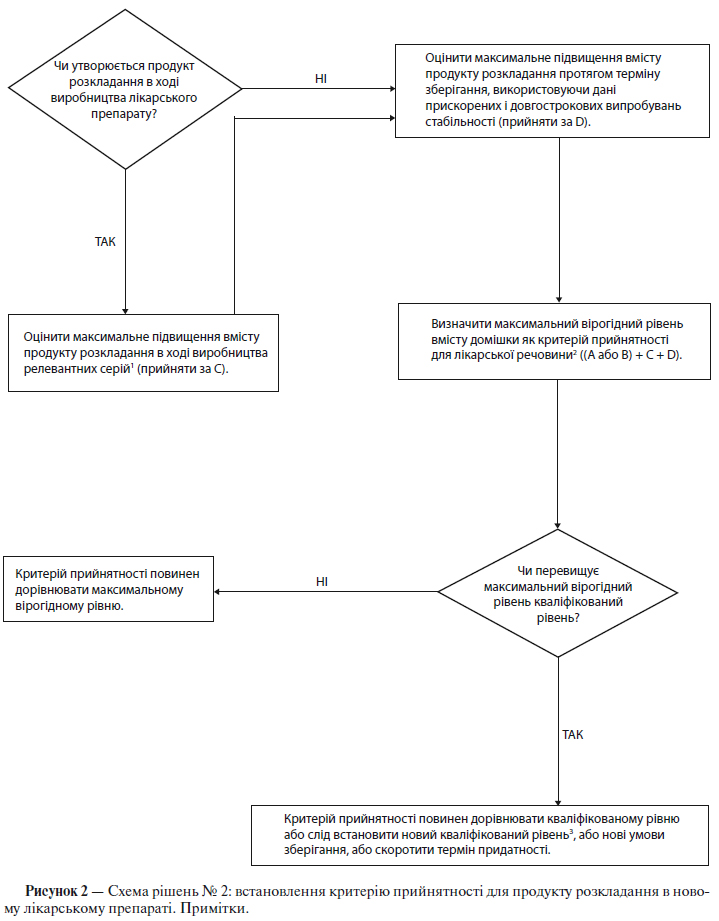
1Релевантні серії — серії, одержані в ході досліджень на етапах розробки, дослідно-промислового і промислового виробництва.
2Порядок визначення А і В див. на рисунку 1.
3Див. Настанову СРМР/ІСН/2737/99 (Q3A) «Note for guidance on impurities testing: Impurities in new drug substances».
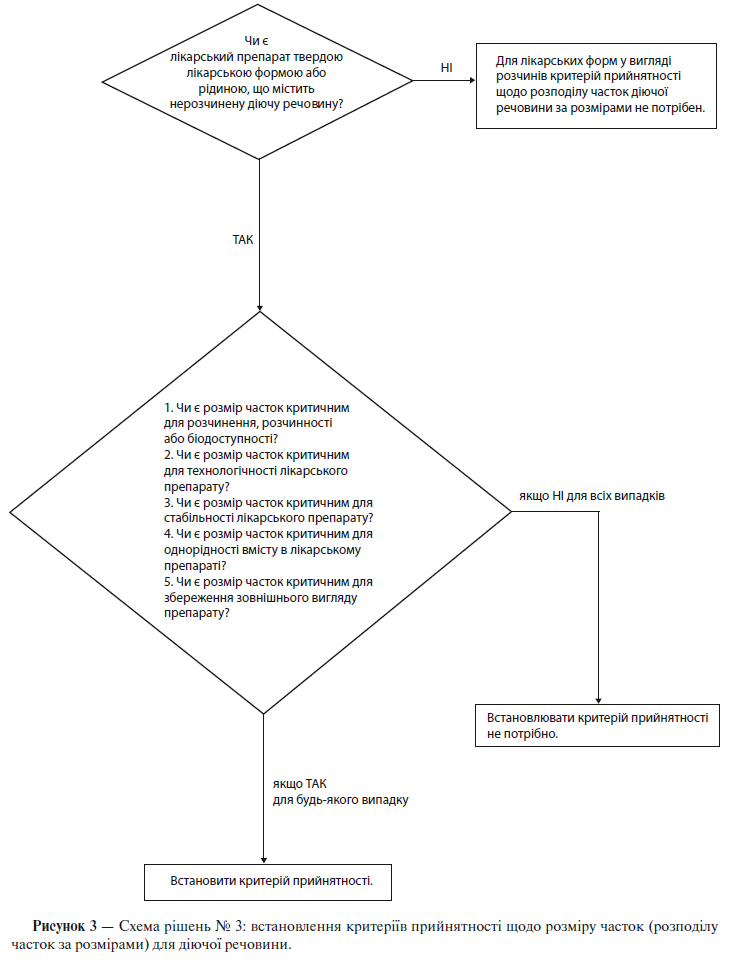
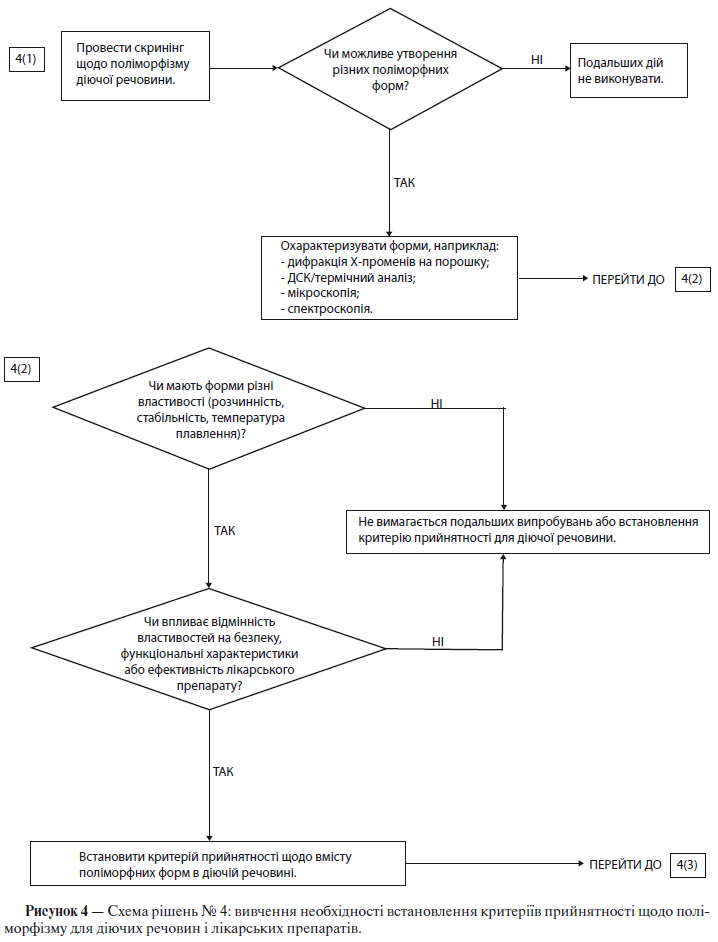
Лікарський препарат — тверда або рідка лікарська форма, що містить нерозчинену лікарську речовину1
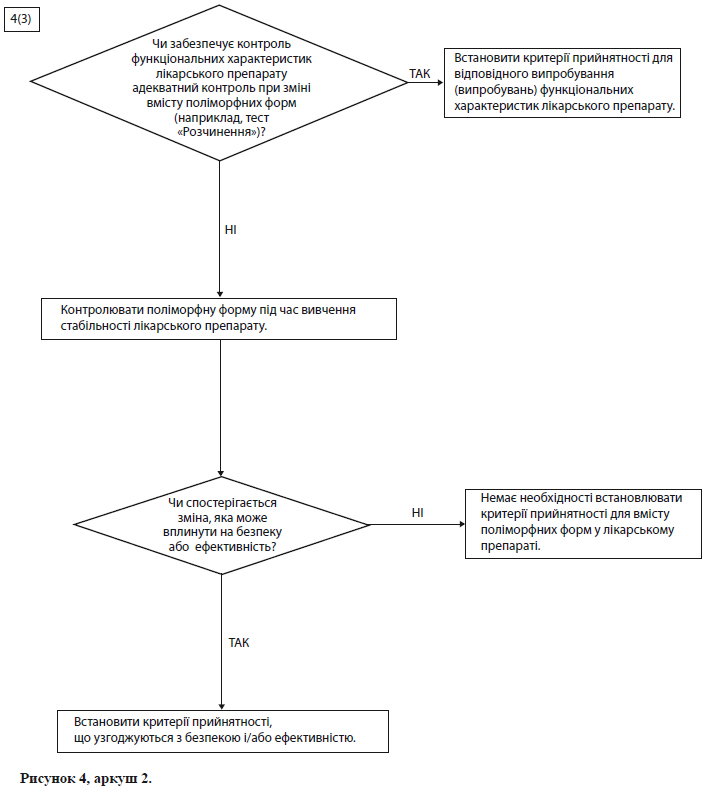
1Процедури, вказані на схемі, виконуються тільки у разі наявності технічної можливості визначення вмісту поліморфних форм у лікарському препараті.
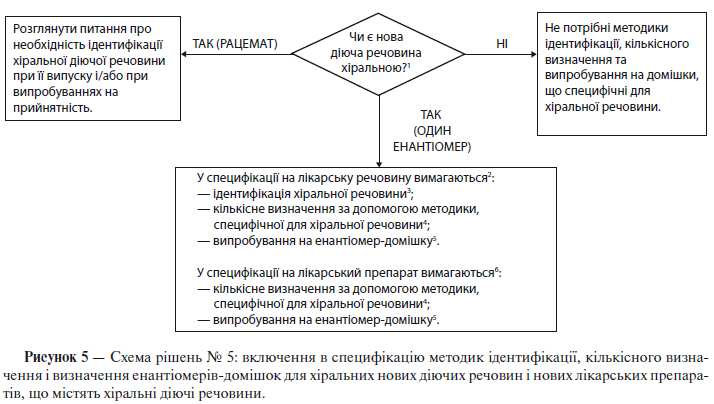
1Хіральні субстанції природного походження не розглядаються в цій настанові.
2Як і щодо інших домішок, джерелом яких є вихідна сировина, що використовується при синтезі лікарської речовини, слід передбачити контроль якості хіральних речовин, встановивши межі для відповідної вихідної сировини або проміжної продукції, якщо це обґрунтовано результатами досліджень, проведених в ході розробки. В основному це будуть випадки, коли є множинні хіральні центри (наприклад, три або більше), або коли бажаний контроль на стадії, що передує кінцевій стадії виробництва лікарської речовини.
3Замість методики ідентифікації хіральної сполуки може бути прийнятною методика кількісного визначення, специфічна щодо хіральної сполуки, або методика випробування на енантіомери-домішки.
4Замість методики кількісного визначення, специфічної щодо хіральної сполуки, прийнятна методика кількісного визначення, неспецифічна щодо хіральної сполуки, в поєднанні з методом контролю протилежного енантіомеру.
5Рівень вмісту протилежного енантіомеру в лікарській речовині можна визначити на основі даних, одержаних за допомогою методики кількісного визначення, специфічної щодо хіральної сполуки, або за допомогою застосування окремої методики.
6Випробування лікарського препарату, специфічні щодо стереоізомерів, можна не проводити, якщо показано, що рацемізація в процесі виробництва лікарського препарату і підчас зберігання готової лікарської форми є незначною.
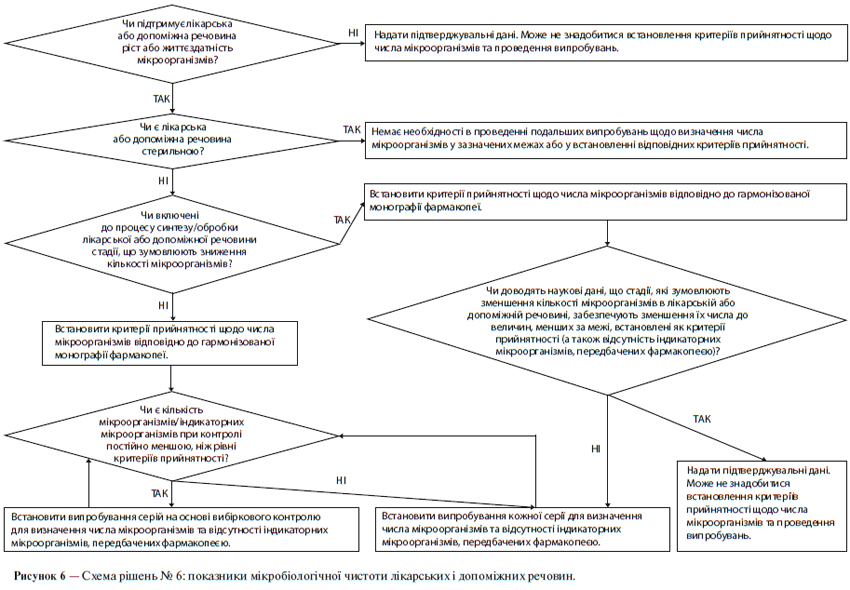
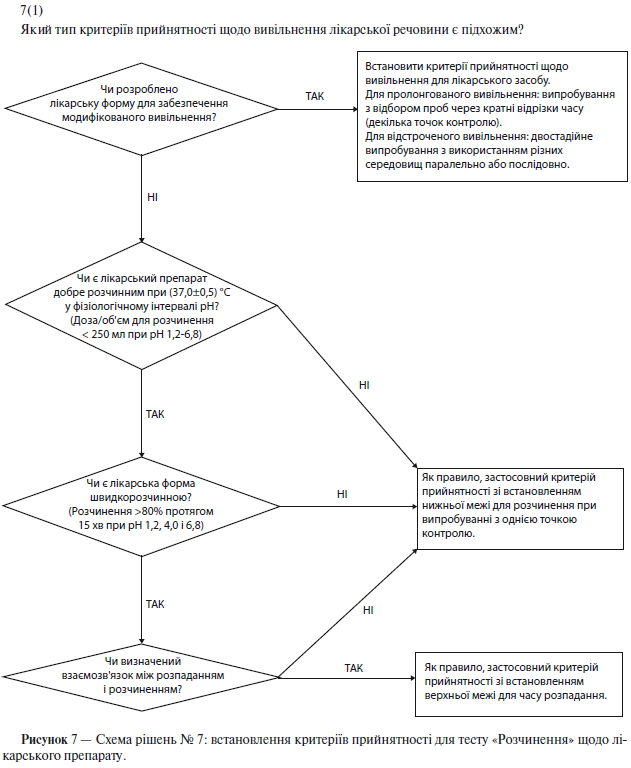
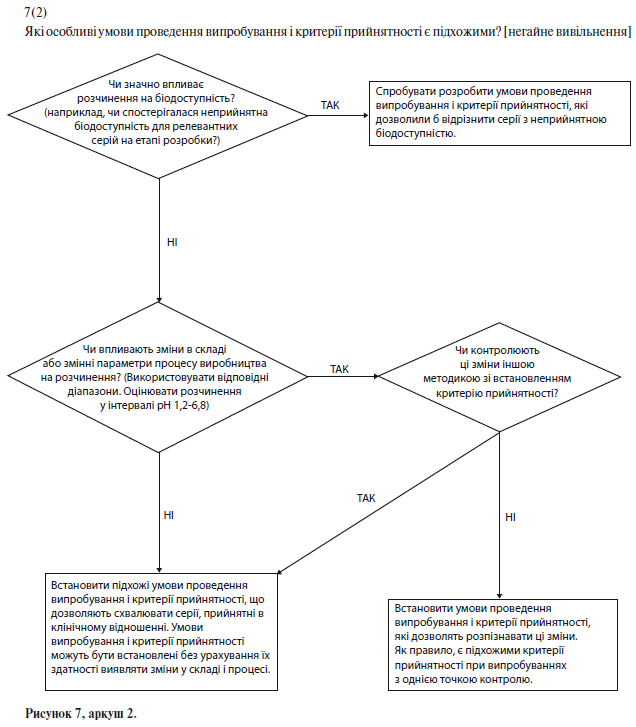
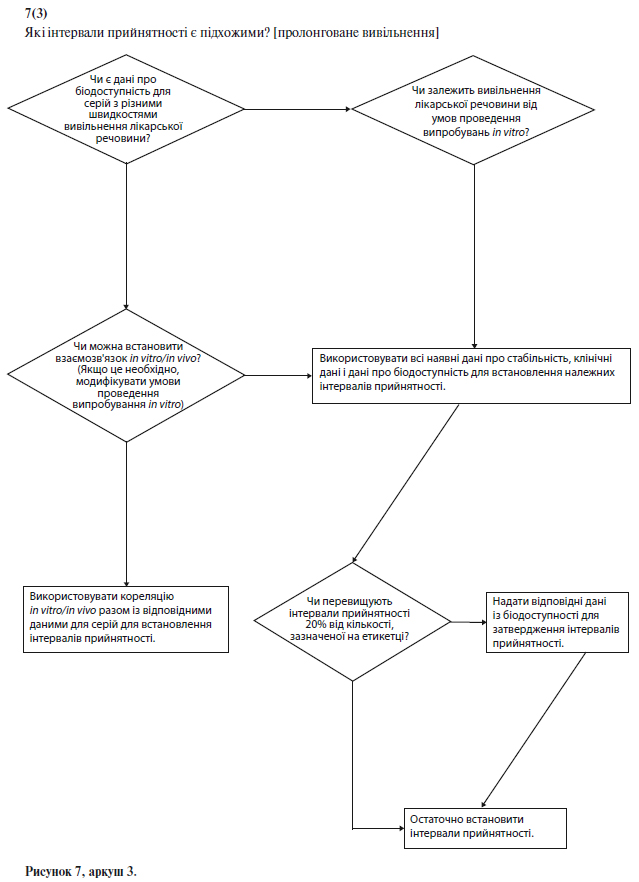
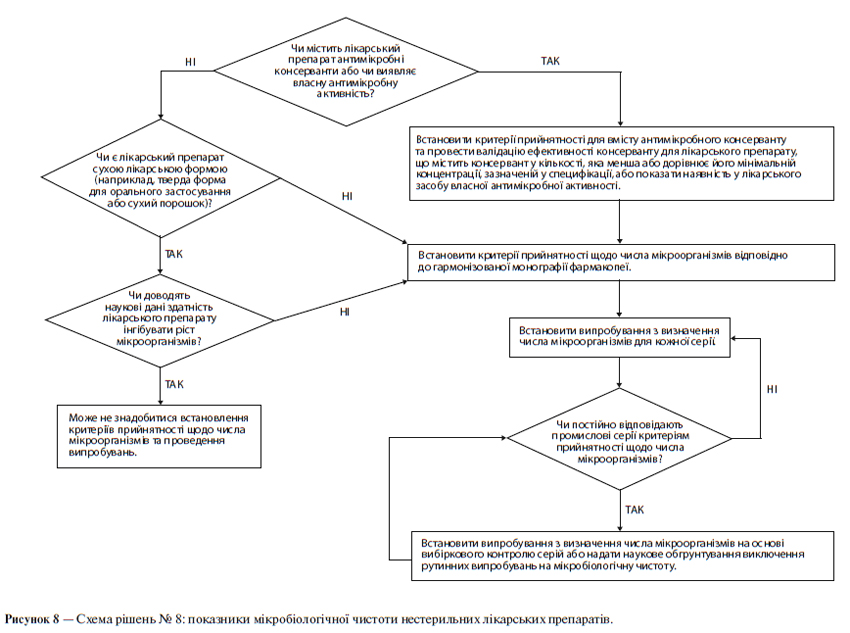
1 СРМР/ІСН/367/96 corr (Q6A) Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for New Drag Substances and New Drug Products: Chemical Substances, 1999
(CPMP/ICH/367/96 corr (Q6A) Керівні вказівки зі специфікацій: методики випробувань і критерії прийнятності для нових лікарських речовин і нових лікарських препаратів: хімічні речовини, 1999)
2 The rules governing medicinal products in the European Union. — V. ЗА. — Guidelines. — Medicinal products for human use. Quality and biotechnology. 3AQlla (revision 1991). Specifications and control tests on the finished product
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т ЗА. — Настанова. — Лікарські засоби для застосування в людини. Якість і біотехнологія. 3AQ11а (переглянуто в 1991). Специфікації та контрольні випробування готової продукції)
3 СРМР/ІСН/281/95 (Q2B) Note for guidance on validation of analytical procedures: methodology, 1996 (CPMP/ICH/281/95 (Q.2B) Керівні вказівки з валідації аналітичних методик: методологія, 1996)
Ключові слова: випробування, діюча речовина, контроль, критерій прийнятності, лікарська форма, лікарський засіб, методика, реєстраційне досьє, специфікація, якість.