
СТАНДАРТ
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ФАРМАЦЕВТИЧНА РОЗРОБКА ЛІКАРСЬКИХ ЗАСОБІВ ДЛЯ ПЕДІАТРИЧНОГО ЗАСТОСУВАННЯ
СТ-Н МОЗУ 42-3.15:2014
Видання офіційне
Київ
Міністерство охорони здоров’я України
2014
Передмова
1 РОЗРОБЛЕНО: ДП «Державний експертний центр МОЗ України»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: Р. Богатирьова, д-р мед. наук, професор, член-кор. НАМН України; О. Баула, канд. хім. наук; О. Нагорна, канд. мед. наук; І. Кудрявцева, д-р фарм. наук; М. Денисова, д-р мед. наук, професор; О. Шадрін, д-р мед. наук, професор; Н. Зелінська, д-р мед. наук; Л. Дорошук, К. Кузьменко
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Міністерство охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 24.07.2014 р. № 521
3 Ця настанова відповідає документам:
EMA/CHMP/QWP/805880/2012 Rev. 1 «Guideline on Pharmaceutical Development of Medicines for Paediatric Use» (Настанова з фармацевтичної розробки лікарських препаратів для педіатричного застосування (перша переглянута версія))
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2014
© Державний експертний центр МОЗ України
Національний вступ
Фармацевтична розробка лікарських засобів для педіатричного застосування — це дослідження з планування та створення готових лікарських засобів, метою якого є виробництво препаратів, які можна застосовувати для лікування саме дітей. При розробці педіатричних лікарських засобів необхідно враховувати, що їх застосування має бути легким, безпечним та ефективним, добре сприйматися пацієнтами. Розробка педіатричних лікарських засобів, особливо тих, що призначені для лікування немовлят, є досить складним завданням, зважаючи на обмеженість інформації про переносимість різних лікарських форм, дозу, смакову прийнятність тощо у дітей різних цільових вікових груп. Недостатність відповідної інформації та відсутність придатних для дітей лікарських форм може призводити до передозування та непередбачуваних побічних реакцій або недодозування, і відповідно, відсутності очікуваної ефективності лікування.
В Європейському Союзі (ЄС) введено спеціальну настанову EMA/CHMP/QWP/805880/2012 Rev. 1 «Guideline on Pharmaceutical Development of Medicines for Paediatric Use» [1], де подано технічні підходи до планування та проведення досліджень із фармацевтичної розробки лікарських засобів для дітей у віковій категорії від народження до 18 років.
З огляду на вищевикладене є необхідність введення в Україні настанови, яка містить рекомендації щодо проведення досліджень із фармацевтичної розробки педіатричних лікарських засобів. Такі рекомендації мають бути гармонізовані з положеннями відповідної настанови ЄС.
Ця настанова розроблена на підставі настанови, прийнятої в ЄС:
EMA/CHMP/QWP/805880/2012 Rev. 1 «Guideline on Pharmaceutical Development of Medicines for Paediatric Use» (Настанова з фармацевтичної розробки медичних продуктів для педіатричного застосування (перша переглянута версія)) [1].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству.
До цієї настанови внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни долучено безпосередньо у пункти, яких вони стосуються; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [2], а позначення — відповідно до вимог стандарту СТ МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [3];
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [2] та ДСТУ 1.72001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [4]. «Зміст» цієї настанови подано з урахуванням додаткових структурних елементів;
- основні положення викладено у розділі «Рекомендації з фармацевтичної розробки», при цьому кожний структурний елемент та його номер у цій настанові відповідають таким у настанові EMA/CHMP/QWP/805880/2012 Rev. 1 [1];
- розділ «Терміни і визначення понять» укладено на основі розділу «Definitions» настанови EMA/CHMP/QWP/805880/2012 Rev. 1 [1]. Цей розділ не позначено номером та викладено слідом за розділом «Нормативні посилання». Усі терміни у розділі «Терміни та визначення понять» наведено в алфавітному порядку, вони супроводжуються посиланням на нормативний документ, бібліографічний опис якого наведено у національному додатку «Бібліографія»;
- у розділі «Нормативні посилання» додатково наведено бібліографічний опис нормативних документів, що зазначаються у цій настанові;
- у національному додатку «Бібліографія» додатково наведено бібліографічний опис нормативних документів, посилання на які наведено у цій настанові;
- у розділі «Познаки та скорочення» додатково наведено позначення скорочень, що використовуються у цій настанові;
- у цій настанові слова «торгова ліцензія» («marketing authorisations») замінено на «реєстраційне посвідчення»; «листок-вкладка» («package leaflet») замінено на «інструкція для медичного застосування»; «ліцензування» («аuthorisation») замінено на «реєстрація»; «заявник» («applicant») замінено на «виробник»;
- замість слів «Європейська Фармакопея» вказано «Європейська фармакопея або інша відповідна фармакопея, або Державна фармакопея УкраїниN». Під словами «інша відповідна фармакопея» мається на увазі фармакопея держави ЄС або фармакопея іншої країни, гармонізована з Європейською фармакопеєю або Фармакопеєю США;
- по всьому тексту внесено редакційні зміни у посилання на структурні елементи цієї настанови, наприклад замість «(see section 3)» вказано «(див. розділ 3 цієї настанови)»;
- Додаток до цієї настанови складено на основі рис. 1 (Figure 1) настанови EMA/CHMP/QWP/805880/2012 Rev. 1 [1]. Цей додаток подано слідом за розділом 12 цієї настанови;
- pозділ 6.1 цієї настанови доповнено текстом та таблицею згідно з розділом 3 документа EMEA/CHMP/PEG/194810/2005 [10];
- додатково до посилань на настанови ЄС зроблено посилання на відповідні гармонізовані документи, затверджені в Україні.
Ця настанова є рекомендаціями для планування та проведення досліджень із фармацевтичної розробки лікарських засобів для дітей.
Правовий статус цієї настанови відповідає правовому статусу відповідної настанови в ЄС та інших регіонах ІСН, з якою гармонізовано розроблену настанову. Цю настанову слід розглядати як технічний документ для надання рекомендацій заявникам та власникам реєстраційних посвідчень, компетентним уповноваженим органам та/чи іншим заінтересованим особам щодо найкращого та найбільш прийнятного способу виконання положень, визначених фармацевтичним законодавством України. Ця наукова настанова пов’язана зі специфічними питаннями щодо фармацевтичної розробки педіатричних лікарських засобів. Положення цієї настанови відображують гармонізований (у рамках ЄС та ІСН) підхід; вони базуються на останніх наукових досягненнях у цій галузі знань.
У рамках чинного фармацевтичного законодавства ця настанова не має сили нормативно-правового акта, її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану позицію європейського фармацевтичного сектора; дотримання її положень заінтересованими сторонами (такими як заявники, власники реєстраційних посвідчень, розробники та виробники лікарських препаратів, експертні та регуляторні органи) полегшить оцінку реєстраційних досьє, а також підвищить якість та безпеку педіатричних лікарських засобів в Україні. Однак можуть бути застосовані альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових настанов викладений у документі Європейського агентства з лікарських засобів (EMA) Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005) [5]. Зазначений підхід відповідає позиції ВТО відносно застосування стандартів.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Фармацевтична розробка лікарських засобів для педіатричного застосування
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Фармацевтическая разработка лекарственных средств для педиатрического применения
MEDICINAL PRODUCTS
Pharmaceutical Development of Medicines for Paediatric Use
Чинна від 2014-07-24
Сфера застосування
Ця настанова визначає положення (рекомендації) щодо фармацевтичної розробки педіатричних лікарських засобів.
Ця настанова застосовна до педіатричних лікарських засобів, що розробляються, реєструються та виробляються в Україні для медичного застосування в Україні та з метою експорту, або що імпортуються в Україну.
Ця настанова поширюється на планування та проведення досліджень на етапах фармацевтичної розробки педіатричних лікарських засобів, складання реєстраційних досьє та реєстрації, а також на виробництво, його аудит та інспектування.
Ця настанова рекомендується для суб’єктів господарювання (далі — організацій), які займаються розробкою, поданням заявок на реєстрацію та/чи виробництвом педіатричних лікарських засобів на території України, незалежно від відомчого підпорядкування та форми власності, для відповідних заявників та підприємств-виробників, продукція яких реєструється та імпортується в Україну, для науково-експертних організацій та регуляторних органів, а також експертів, аудиторів та інспекторів, що проводять експертизу при реєстрації (перереєстрації) педіатричних лікарських засобів, аудит та інспектування виробництва.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 р.
Державна Фармакопея України. Перше видання. Доповнення 1. 2004 р.
Державна Фармакопея України. Перше видання. Доповнення 2. 2008 р.
Державна Фармакопея України. Перше видання. Доповнення 3. 2009 р.
Державна Фармакопея України. Перше видання. Доповнення 4. 2011 р.
Наказ МОЗ України від 26.08.2005 р. № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення» (у редакції наказу МОЗ України від 04.01.2013 р. № 3), зареєстрований у Міністерстві юстиції України 19 вересня 2005 р. за № 1069/11349
Regulation EC № 1901/2006 of the European Parliament and of the Council, amending regulation EEC № 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation EC № 726/2004 (Регламент ЄС № 1901/2006 Європейського Парламенту та Ради, що доповнює Регламент ЄЄС № 1768/92, Директиву 2001/20/ЄС, Директиву 2001/83/ЄС та Регламент ЄС № 726/2004)
Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128 (Директива 2001/83/ЄC Європейського Парламенту та Ради від 6 листопада 2001 року про кодекс Співтовариства відносно лікарських препаратів, призначених для застосування людям//Official Journal of the European Communities. — L 311, 28.11.2001. — р. 67–128)
Commission Directive 2003/63/EC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use. Annex I: Analytical, Pharmacotoxilogical and Clinical Standards and Protocols in Respect of the Testing of Medicinal Products. — Official Journal of the European Union. — № L 159 of 27.6.2003 (Директива Комісії 2003/63/ЄC від 25 червня 2003 року, що доповнює Директиву 2001/83/ЄC Європейського Парламенту та Ради від 6 листопада 2001 року про кодекс Співтовариства відносно лікарських препаратів, призначених для застосування людям. Додаток I: Аналітичні, фармако-токсикологічні та клінічні стандарти і протоколи щодо досліджень лікарських засобів. — Official Journal of the European Union. — № L 159 of 27.6.2003)
ІСН Q8 (R2) «Pharmaceutical Development» (Фармацевтична розробка)
Настанова СТ-Н МОЗУ 42-3.0:2011. — Лікарські засоби. Фармацевтична розробка (ІСН Q8)
Настанова СТ-Н МОЗУ 42-3.1:2004. — Лікарські засоби. Фармацевтична розробка
СРМР/QWP/155/96 «Note for Guidance on Development Pharmaceutics» (Керівні вказівки з фармацевтичної розробки)
The Rules Governing Medicinal Products in the European Union. Notice to Applicants. — Volume 2C. — Guideline on Summary of Product Characteristics (SmPC). — (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 2C. — Настанова з Короткої характеристики лікарських засобів)
Терміни та визначення понять
Нижче наведені терміни, вжиті у цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1] (див. національний додаток «Бібліографія»). Визначення цих термінів можуть відрізнятися в інших нормативних документах або терміни можуть мати інші значенняN.
Педіатричний лікарський засіб, що відповідає віку (аge-appropriate paediatric medicine)
Лікарський засіб із фармацевтичною композицією, призначеною для застосування в цільовій віковій групі.
Попередня композиція (так звана адаптована композиція) (рreliminary formulation (as called enabling formulation))
Попередня композиція є відносно простою та легкою для приготування препарату з метою полегшення проведення доклінічних досліджень та/або ранніх клінічних стадій розробки, які в іншому випадку можуть бути відкладені до розробки кінцевого педіатричного лікарського засобу, що відповідає віку.
Педіатрична композиція (рaediatric formulation)
Склад конкретної лікарської форми лікарського засобу для педіатричного застосування.
Педіатричний препарат (рaediatric preparation)
Педіатрична композиція з конкретною силою дії (наприклад таблетки по 5 мг, розчин для ін’єкцій по 5 мг/мл), а у випадку педіатричних композицій для одноразового застосування — вміст контейнеру (наприклад розчин для ін’єкцій 5 мг/мл, 1 мл=5 мг або 2 мл=10 мг).
Педіатричний лікарський засіб (рaediatric medicine/paediatric medicinal product)
Педіатричний препарат у своєму контейнері із закупорювальною системою та, у разі наявності, разом з будь-яким дозувальним пристроєм або пристроєм для введення, а також інструкцією для медичного застосування.
Фармацевтична розробка (рharmaceutical development)
У контексті цієї настанови фармацевтична розробка стосується усіх складових розділу 3.2.Р. Модуля 3 ЗТД, інструкції для медичного застосування та короткої характеристики (розділ 6 цієї настанови). Вона визначається як процес перетворення активної фармацевтичної речовини у педіатричний лікарський засіб, який може бути введений дитиною самостійно або дорослим, включаючи усі відповідні фармацевтичні характеристики, наприклад контроль вихідних матеріалів, валідація аналітичних методів тощо.
Фармацевтичний дизайн лікарського засобу (рharmaceutical design of a medicinal product)
Склад, лікарська форма, шлях введення, частота дозування, упаковка, дозуючий пристрій та пристрій для введення, інструкція для медичного застосування лікарського засобу.
Познаки та скорочення
|
ЄС |
— |
Європейський Союз |
|
СНМР |
— |
Committee for Medicinal Products for Human Use (Комітет з лікарських препаратів для людини) |
|
ICH |
— |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини) |
|
ЕМА |
— |
European Medicines Agency (Європейське агентство з лікарських засобів) |
|
ВООЗ |
— |
Всесвітня організація охорони здоров’я |
|
КМУ |
— |
Кабінет Міністрів України |
|
SmPC |
— |
Summary of Рroduct Сharacteristics (Коротка характеристика лікарського засобу) |
|
ЗТД |
— |
Загальний технічний документ |
|
EFSA |
— |
The European Food Safety Scientific Opinions (Європейські наукові висновки щодо безпеки харчових продуктів) |
|
JECFA |
— |
Expert committee on food additives (Експертний комітет з харчових добавок) |
|
ВІЛ |
— |
вірус імунодефіциту людини |
|
ADI |
— |
Acceptable daily intake (щоденна допустима доза) |
Рекомендації з фармацевтичної розробки
Мета цієї настанови полягає у тому, щоб полегшити розробку та доступність педіатричних лікарських засобів, які відповідають віку дитини. Ця мета має бути досягнута без необґрунтованого проведення клінічних випробувань у дітей та без затримки реєстрації лікарських засобів для інших вікових груп.
Важливим завданням розробки педіатричних лікарських засобів з урахуванням вікових особливостей дітей є забезпечення доступності дітей у цільовій віковій групі до лікарських засобів відповідної якості з позитивним співвідношенням користь/ризик, забезпечення належного дотримання пацієнтами призначення без створення зайвих труднощів для пацієнта та/чи осіб, що здійснюють догляд за ними.
Ця настанова призначена для надання додаткових рекомендацій при плануванні та проведенні досліджень із фармацевтичної розробки лікарських засобів для дітей у віковій категорії від народження до 18 років. Цю настанову необхідно застосовувати разом з іншими відповідними чинними документами ЄС та ICH, а також законодавчими актами України у сфері обігу лікарських засобівN (див. розділ 3 цієї настанови). У цій настанові враховано сучасний науковий і технічний стан виробництва та контролю педіатричних лікарських засобів.
1. Вступ
26 січня 2007 р. набув чинності «Педіатричний Технічний Регламент» (Технічний Регламент ЄС № 1901/2006). Ціль «Педіатричного Технічного Регламенту» — «сприяти розробці та доступності лікарських засобів для застосування у педіатричній популяції, щоб до цих лікарських засобів застосовувалися дослідження високої якості та був відповідний дозвіл назастосування у педіатричній популяції, а також з метою удосконалення наявної інформації про застосування лікарських засобів у різних педіатричних популяціях». На підставі вищенаведеного очікується, що збільшиться кількість дозволених до застосування педіатричних лікарських засобів, а знання щодо параметрів якості, критичних для цих препаратів, будуть розширюватися.
Фізичні, метаболічні та психологічні процеси, властиві зростанню людини від народження до дорослого віку, показують, що дітей не можна розглядати ані як маленьких дорослих, ані як однорідну групу дітей. Унаслідок цього результати клінічних випробувань за участю дорослих не обов’язково будуть екстрапольовані на дітей. Таким чином, для підтвердження безпеки та ефективності лікарського засобу в усіх цільових вікових групах, для яких він розробляється, у багатьох випадках може потребуватися проведення клінічних досліджень за участю дітей різного віку.
Крім того, у дітей застосування лікарських засобів спричиняє певні фармацевтичні проблеми, які не спостерігаються тією ж мірою у дорослих та поява яких може залежати від віку. Наприклад, діти молодшого віку просто нездатні проковтнути таблетки традиційного розміру, новонародженим можуть знадобитися дуже малі об’єми лікарського засобу для парентерального введення, щоб уникнути об’ємного перевантаження тощо. Тому у дітей слід застосовувати лікарські засоби, фармацевтичний дизайн яких пристосований для застосування в цільовій віковій групі, тобто педіатричні лікарські засоби, що відповідають віку.
Прийнятність і перевага різних лікарських форм розрізняється у дітей. Вік дитини, індивідуальний стан здоров’я, поведінка, інвалідність, оточення та культура на сьогодні вважаються найбільш імовірними факторами, які визначають прийнятність і надання переваги дитиною тій чи іншій лікарській формі. Однак первинну фармацевтичну розробку педіатричного лікарського засобу слід зосередити на мінімальній кількості прийнятних лікарських форм, які відповідатимуть потребам більшості дітей у цільовій віковій групі. Отже, лікарські форми, які полегшать введення широкого діапазону доз, та які прийнятні для дітей різного віку, задовольнятимуть цілий комплекс їх потреб.
Метою цієї настанови є надання рекомендацій щодо особливостей розробки педіатричних лікарських засобів, їх промислового виробництва та щодо необхідності розробляти лікарські засоби, які найбільш пристосовані для дітей, замість практикувати застосування у дітей наявних препаратів у спосіб, відмінний від затвердженого, або застосування придатних для дітей лікарських форм, приготованих екстемпорально з лікарських засобів для дорослих.
2. Сфера застосування
Принципи, викладені у цій настанові, слід застосовувати при фармацевтичній розробці усіх педіатричних лікарських засобів, щодо яких подається заява нареєстрацію, на зміни, що потребують нової реєстрації, або при внесенні змін уреєстраційне досьє. Залежно від фази розробки принципи цієї настанови також доцільно застосовувати для плану педіатричного дослідження. Регуляторна політика щодо лікарських засобів має бути принципово спрямована на збереження здоров’я населення, а це може бути досягнуто шляхом усунення перешкод для вільного обігу безпечних лікарських засобів натериторії ЄС та УкраїниN.
Оскільки клінічна та фармацевтична інформація накопичується з часом при розробці та протягом подальшого життєвого циклу лікарського засобу, фармацевтичний дизайн педіатричного препарату на ранній стадії клінічного випробування може відрізнятися від такого на кінцевих фазах досліджень. Наранній стадії розробки важливо зосередитися на придатності та безпеці запропонованої композиції/препарату. Якщо виробник не може запропонувати педіатричний лікарський засіб, слід розглянути, принаймні, вибір шляху введення, лікарської форми, необхідного дозування (гнучкість) та допоміжних речовин у композиції/препараті, включаючи смакову привабливість, а також пристрою для введення. Застосування попередніх (так званих адаптованих) педіатричних композицій/препаратів на ранніх фазах клінічного випробування може вважатися прийнятним у разі відповідного обґрунтування, проте це незвільняє виробника від вимоги розробити композицію/препарат, який буде вироблятися у промислових масштабах та контролюватися, як це вимагає «Педіатричний Технічний Регламент». Таким чином, попередні композиції/препарати, що застосовуються відповідно до інструкцій з обробки зареєстрованих лікарських засобів, як правило, не будуть вважатися прийнятними для реєстрації, якщо тільки це належним чином не обґрунтовано та не підтверджено. Успішний перехід із попередньої композиції/препарату накомерційну (промислову) композицію/препарат слід, як правило, підтверджувати відповідними зведеними дослідженнями щодо різних композицій/препаратів, які використовувалися протягом розробки.
Оскільки з часом з’являються нові наукові дані, фармацевтичним виробникам необхідно переглядати придатність (практичність), якість, безпеку та ефективність зареєстрованих педіатричних лікарських засобів. Це відповідає положенням ст. 23 Директиви 2001/83/ЄС [6] та пункту 7 Порядку державної реєстрації (перереєстрації) лікарських засобів, затвердженого постановою Кабінету Міністрів України від 26.05.2005 р. № 376 [7]N, де зазначено, що виробники мають враховувати науково-технічний прогрес протягом усього життєвого циклу продукту та адаптувати або покращувати його на користь пацієнтів та задля підтримання позитивного співвідношення користь/ризик.
У цій настанові не описані загальні підходи до фармацевтичної розробки лікарських засобів. Ця настанова не містить вичерпної інформації та передбачає існування інших складових, які мають суттєве значення для фармацевтичної розробки педіатричних лікарських засобів. Будь-які відмінності від викладеного у цій настанові будуть прийнятні за умови їх відповідного наукового обґрунтування. Не слід розглядати нижченаведене як єдино можливі рішення для фармацевтичної розробки педіатричних лікарських засобів.
3. Законодавча база
Цю настанову слід застосовувати разом із Директивою 2001/83/ЄС, наказом МОЗ України від 26.08.2005 р. № 426N, «Педіатричним Технічним Регламентом», Європейською фармакопеєю або іншою відповідною фармакопеєю, або Державною фармакопеєю УкраїниN та іншими відповідними чинними документами ЄС та ICH, а також законодавством України у сфері обігу лікарських засобівN.
4. Загальні питання
Будь-який лікарський засіб призначений задовольняти потреби пацієнта та послідовно досягати цільового призначення. Для досягнення зазначених цілей при фармацевтичній розробці слід дотримуватися систематичного підходу відповідно до положень настанови ICH Q8 та гармонізованих з ним настанов СТ-Н МОЗУ 42-3.1-2004, СТ-Н МОЗУ 42-3.0-2011N. Занеобхідності цільовий профіль якості продукту слід встановлювати зурахуванням особливих потреб педіатричної популяції. На підставі цільового профілю продукту мають бути визначені критичні параметри його якості, композиція та параметри процесу, які можуть впливати на якість. Такий підхід допоможе визначити фармацевтичний дизайн педіатричного лікарського засобу.
Фармацевтичний дизайн лікарського засобу включає усі складові Модуля 3Загального технічного документа (ЗТД), короткої характеристики (SmPC) та інструкції для медичного застосування, наприклад склад препарату, вибір лікарської форми, вибір первинної та вторинної упаковки тощо.
Приймаючи рішення щодо прийнятності фармацевтичного дизайну педіатричного лікарського засобу, крім описаного у розділах 6–12 цієї настанови, також слід враховувати:
- мінімальний вік дітей у цільовій віковій групі, відповідну фізіологію розвитку та вікові характеристики дітей у цільовій віковій групі;
- стан, що підлягає лікуванню, стан, пов’язаний з особливостями дитини (наприклад дитина з фізичними або психічними розладами, зобмеженням прийому рідини, з великою кількістю супутніх захворювань, з нездатністю ковтати внаслідок критичного захворювання);
- «критичність» дози (тобто крута крива «доза—відклик», вузьке терапевтичне вікно), режим дозування (тобто розрахунок дози, титрування дози, гнучкість дозування);
- вікова активність дітей у цільовій віковій групі (наприклад у школі, дитячому садку);
- максимальна тривалість терапії та частота дозування;
- умови, в яких застосовується лікарський засіб (наприклад в умовах стаціонару чи амбулаторно);
- характеристики дитини й особи, яка здійснює догляд за дитиною, та їх поведінка.
5. Характеристика активної речовини
Фізико-хімічні характеристики певної діючої речовини бажано змінювати так, щоб під час використання у виробництві готового лікарського засобу діюча речовина ставала його активним інгредієнтом. Наприклад, у деяких випадках для виробництва рідкої лікарської форми може знадобитися речовина зпокращеною розчинністю, тобто інша сіль або сіль замість основи. Крім того, покращити прийнятність лікарського засобу дитиною можна шляхом вибору менш розчинної форми активної речовини для подолання проблем ізнеприємним смаком, наприклад, основи замість солі. Також безпеку лікарських засобів для пацієнтів педіатричного профілю можна посилити за рахунок уникнення використання певних солей, наприклад мезилатів.
У ранній фазі фармацевтичної розробки рекомендується, щоб при виборі форми активної речовини (кислота/основа, сіль, поліморфна модифікація, сольват тощо) враховувалися властивості, які впливають на розробку педіатричних лікарських засобів. Вибрана форма активної речовини має дозволити розробку педіатричного лікарського засобу, що відповідає віку, для застосування у цільовій віковій групі. Форма активної речовини, вибрана для розробки педіатричної композиції, може відрізнятися від форм, які використовують у лікарських засобах для дорослих.
6. Шлях введення та лікарська форма
6.1. Загальні питання
Обґрунтування вибору, а також переваги і недоліки певної педіатричної лікарської форми з конкретним шляхом введення слід обговорити для дітей укожній цільовій віковій групі. Питання, які потребують обговорення, включають, принаймні, стан(-и), що потребують лікування, тривалість лікування, властивості активної речовини, необхідність певних допоміжних речовин у педіатричному препараті (та їх безпека), будь-які дозуючі пристрої та пристрої для введення, питання стабільності, вимоги до дозування, ризик помилок із дозуванням та такі питання, як простота введення та сприйняття пацієнтами.
Для однієї й тієї ж активної речовини можуть знадобитися різні шляхи введення та/чи лікарські форми з метою забезпечення адекватного лікування дітей у цільовій віковій групі та, за необхідності, при різних станах здоров’я, розвитку хвороби або поведінкових характеристиках.
Смакову, кольорову привабливість лікарської форми педіатричного лікарського засобу слід ретельно співвідносити як із ризиком неадекватного прийняття пацієнтом, так і з випадковим застосуванням, та обговорити щодо всіх параметрів лікарського засобу, тобто лікарської форми, композиції, первинної та вторинної упаковок.
Одним із найважливіших питань при розробці педіатричних лікарських засобів є вибір найбільш прийнятної лікарської форми, що відповідає віку. Проведено невелику кількість досліджень щодо можливості застосування різних композицій пацієнтам педіатричного профілю. Зокрема, існують перестороги щодо віку, від якого маленькі діти можуть безпечно проковтнути звичайні таблетки та капсули.
Для допомоги у вирішенні цього питання розроблена матриця (таблиця), що поєднує різні вікові групи, шляхи введення та лікарські форми [10]. Ця матриця не є вичерпною доказовою розробкою, вона ґрунтується нарезультатах опитування лікарів-педіатрів, вчених-фармацевтів та батьків. Матриця не має характеру рекомендацій стосовно розробки особливої лікарської форми для цільової вікової групи, проте відображає деякі загальні питання прийнятності різних лікарських форм. Класифікація лікарських форм надається окремо для основних шляхів введення. Різні шляхи введення та відповідні лікарські форми обговорюються у інших розділах цієї настанови. Матриця в основному містить традиційні лікарські форми. Інші лікарські форми, які можуть мати особливе значення для пацієнтів педіатричного профілю, описані в інших розділах цієї настанови.
Матриця в основному зазначає переважні шляхи введення та композиції залежно від віку дітей.
Таблиця. Шляхи введення/лікарські форми залежно від віку
|
Шлях введення Лікарська форма |
(1) |
(2) |
(3) |
(4) |
(5) |
(6) |
|
Пероральний |
||||||
|
розчин/краплі |
2 |
4 |
5 |
5 |
4 |
4 |
|
емульсія/суспензія |
2 |
3 |
4 |
5 |
4 |
4 |
|
шипучі лікарські форми |
2 |
4 |
5 |
5 |
4 |
4 |
|
порошки/гранули |
1 |
2 |
2 |
4 |
4 |
5 |
|
таблетки |
1 |
1 |
1 |
3 |
4 |
5 |
|
капсули |
1 |
1 |
1 |
2 |
4 |
5 |
|
сублінгвальні таблетки |
1 |
2 |
3 |
4 |
5 |
5 |
|
жувальні таблетки |
1 |
1 |
1 |
3 |
5 |
5 |
|
Назальний |
||||||
|
назальні розчини/краплі |
3 |
4 |
4 |
4 |
4 |
4 |
|
м’які лікарські форми |
2 |
3 |
3 |
4 |
4 |
4 |
|
Ректальний |
||||||
|
супозиторії |
4 |
5 |
5 |
4 |
3 |
2 |
|
ректальні клізми |
5 |
4 |
4 |
3 |
3 |
2 |
|
ректальні капсули |
2 |
3 |
4 |
4 |
4 |
3 |
|
Нашкірний/трасдермальний |
||||||
|
мазі, креми, гелі |
4 |
4 |
4 |
5 |
5 |
5 |
|
рідкі лікарські форми |
4 |
4 |
4 |
5 |
4 |
4 |
|
трансдермальні пластирі |
1 |
2 |
2 |
4 |
4 |
5 |
|
Парентеральний |
||||||
|
внутрішньовенні ін’єкції/інфузії |
5 |
4 |
4 |
4 |
4 |
3 |
|
внутрішньом’язові ін’єкції/інфузії |
3 |
3 |
3 |
4 |
4 |
3 |
|
підшкірні ін’єкції |
4 |
4 |
4 |
5 |
5 |
5 |
|
насосні системи |
5 |
4 |
4 |
4 |
4 |
3 |
|
Інгаляційний |
||||||
|
небулайзери |
2 |
3 |
4 |
5 |
4 |
3 |
|
дозуючі інгалятори/спейсери |
1 |
3 |
4 |
5 |
4 |
4 |
|
сухі порошкові інгалятори |
1 |
1 |
3 |
4 |
5 |
5 |
|
Очний |
||||||
|
очні краплі |
3 |
4 |
4 |
4 |
5 |
5 |
|
м’які лікарські форми |
2 |
3 |
4 |
4 |
4 |
4 |
|
(1) недоношені новонароджені (2) доношені новонароджені (3) малюки і діти, що починають ходити (4) діти дошкільного віку (5) діти шкільного віку (6) підлітки 1 — не застосовується 2 — застосовується, але є певні складності 3 — можливе застосування 4 — належна прийнятність 5 — найкраще застосування, якому слід надавати перевагу |
||||||
6.2. Пероральний шлях введення
Пероральне введення можна здійснювати за допомогою декількох типів лікарських форм. Загалом основний вибір потрібно зробити між застосуванням рідкої та твердої лікарських форм для перорального застосування. Переваги та недоліки будь-якої лікарської форми для перорального застосування/композиції, пов’язані з особливостями певної цільової вікової групи, слід враховувати при виборі певної лікарської форми/композиції.
Тверді однодозові лікарські форми для перорального застосування можуть забезпечити точне та легке дозування. Однак у разі індивідуального підбору дози кількість дозувань лікарського засобу, необхідних для лікування пацієнтів у цільовій віковій групі, зростає. Альтернативні підходи, які можуть забезпечити гнучкість дозування для таблеток, включають нанесення ліній поділу, що дозволяють вводити частину дози, або маленьких таблеток, які містять лише частину необхідної дози і які для отримання повної дози необхідно приймати по декілька одночасно (див. розділ 6.2.1 цієї настанови).
Порошки, гранули та рідини для перорального застосування зазвичай забезпечують більшу гнучкість у дозуванні, ніж однодозові тверді лікарські форми. Деякі однодозові тверді лікарські форми для перорального застосування, такі як дисперговані або шипучі, призначені для диспергування, суспендування чи розчинення перед введенням. Як правило, не слід приймати частину рідини, приготованої з такої лікарської форми, для отримання дози, необхідної для цільової вікової групи. Проте такий підхід може бути виправданим у деяких випадках за умови, що процедура обробки належним чином підтверджена, включаючи, наприклад, простоту приготування рідкого препарату, гомогенність отриманої рідини та можливість відміряти певний об’єм. Багаторазові маніпуляції з лікарським засобом становлять підвищений ризик допущення помилок у дозуванні, тому таких маніпуляцій слід, поможливості, уникати.
Діти можуть не вміти або не бажати ковтати певну лікарську форму та/чи педіатричний препарат, навіть коли лікарська форма/композиція/препарат самі по собі вважаються такими, що в основному відповідають віку. Тому виробникам рекомендується вивчити можливість виведення нафармацевтичний ринок різних лікарських форм/композицій/препаратів (наприклад рідини для перорального застосування і таблетки). Якщо це неможливо, слід обговорити альтернативні підходи щодо застосування препарату (див. «Обробка твердих лікарських форм для полегшення введення» нижче та розділ 10 цієї настанови).
Введення через живильні трубки може знадобитися для дітей, які мають проблеми з ковтанням будь-якої лікарської форми/композиції/препарату (див. розділ 6.2.3 цієї настанови).
6.2.1. Тверді препарати для перорального застосування
Порошки та гранули
Порошки та гранули можна давати дітям від народження за умови, що їх можна застосовувати у формі рідини. У твердій формі їх зазвичай вводять разом із напівтвердою їжею. У такому вигляді їх застосування вважається прийнятним з моменту, коли дитина вже здатна вживати напівтверду їжу, що зазвичай відбувається у віці приблизно 6 міс.
Ризик аспірації, задухи та у відповідних випадках розжовування (див. розділ 8цієї настанови) порошків/гранул слід обговорити щодо цільової вікової групи, розміру, форми та кількості (об’єму) порошків/гранул, а також будь-яких специфічних характеристик активної речовини або композиції.
Введення порошків та гранул відбувається за допомогою дозатора, якщо тільки вони не упаковані у однодозові упаковки, такі як саше (див. розділ 11.3 цієї настанови).
Таблетки
Розмір та форма таблеток мають суттєве значення для здатності дитини проковтнути таблетку. Прийнятність розміру та форми таблеток для цільової вікової групи слід підтвердити та обґрунтувати шляхом відповідних досліджень або за допомогою клінічних даних, якщо застосовне (див. розділ 10 цієї настанови). Слід зазначити, що у літературі кількість наявних даних щодо впливу розміру, форми та кількості таблеток на прийнятність у різних вікових групах обмежена. При лікуванні хронічних захворювань прийнятність таблеток певного розміру та форми у дітей може бути поліпшена шляхом відповідної обробки. Прийнятність розміру та форми таблеток можна також покращити шляхом надання відповідних інструкцій щодо поєднаного застосування знапівтвердою їжею. Для таблеток, не призначених для проковтування у цілому вигляді, наприклад, для диспергованих, жувальних, шипучих таблеток, питання щодо розміру та форми мають менше значення. Однак питання привабливості можуть значно впливати на прийнятність цих типів таблеток.
Маленькі таблетки, що містять частину необхідної дози, можуть розглядатися як один із заходів для поліпшення прийнятності та/чи гнучкості дозування. Ці маленькі таблетки спроектовані таким чином, щоб необхідна доза для дітей урізних цільових вікових групах отримувалася шляхом прийому однієї чи декількох маленьких таблеток (інколи вони називаються «міні-таблетками»). Якщо для досягнення необхідної дози потрібно застосовувати одночасно декілька таблеток, прийнятність цієї кількості таблеток необхідно обговорити та обґрунтувати для відповідних цільових вікових груп.
Крім розміру та форми таблеток, необхідно додатково обґрунтувати придатність таблеток для дітей, пов’язану з різним станом здоров’я або розвитку захворювання, а також ризиками недостатнього дозування, задухи, аспірації та розжовування (див. розділ 8 цієї настанови). У коротку характеристику та інструкцію для медичного застосування лікарського засобу слід внести відповідні застереження щодо необхідності проковтувати таблетки у цілому вигляді без розжовування, тобто коли їх не можна жувати. Таблетки з негайним вивільненням зазвичай призначені для проковтування у цілому вигляді, якщо інше не вказано у короткій характеристиці та інструкції для медичного застосування, проте у багатьох випадках їх також можна розжовувати. Коли розжовування таблеток із негайним вивільненням прийнятне, необхідно обговорити потенційний вплив розжовування на таку характеристику продукту, як смакова привабливість.
Капсули
Зазвичай капсули призначені для прийому у цілому вигляді. За належного обґрунтування тверді капсули можна також відкривати та приймати лише їх вміст за умови, що була продемонстрована можливість розкриття капсули та вилучення її вмісту. Якщо тверду капсулу можна відкривати перед застосуванням, то її вміст має відповідати тим самим вимогам, що й порошки та гранули для перорального застосування. Придатність застосування капсул у цілому вигляді або після відкриття слід обговорити та обґрунтувати для усіх відповідних цільових вікових груп (див. «Обробка твердих лікарських форм для полегшення введення» цієї настанови).
Як і для таблеток, у літературі кількість наявної інформації щодо прийнятності капсул відповідного розміру у різних вікових групах обмежена. Коли капсули необхідно приймати у цілому вигляді, потрібно враховувати прийнятність розміру та форми капсули та будь-які супутні ризики, як описано для таблеток.
Дисперговані у ротовій порожнині та жувальні препарати
Дисперговані у ротовій порожнині та жувальні препарати включають тверді лікарські форми для перорального застосування, які не потрібно проковтувати уцілому вигляді та запивати рідиною. Дисперговані у ротовій порожнині таблетки можна приймати іншим способом, ніж заплановано, тобто можна диспергувати таблетки у рідині перед застосуванням або ковтати відразу, недиспергуючи їх у роті.
Якщо існує ризик, пов’язаний із безпосереднім ковтанням диспергованої уротовій порожнині або жувальної таблетки та/чи диспергована у ротовій порожнині композиція не має бути диспергована іншим способом перед застосуванням, це необхідно зазначити у короткій характеристиці та інструкції для медичного застосування.
Слід ретельно розглянути ризик задухи при застосуванні диспергованої або жувальної таблетки, оскільки дитина може бути нездатною чи не бажати застосовувати таблетки відповідно до зазначеного способу.
Обробка твердих лікарських форм для полегшення введення
Якщо тверді препарати для перорального застосування необхідно застосовувати дітям, слід враховувати, що деякі діти не вміють або не бажають приймати певну лікарську форму, хоча загалом вона вважається відповідною віку. Завідсутності будь-яких інших відповідних віку лікарських форм слід розглянути альтернативні методи введення твердих препаратів для перорального застосування (наприклад диспергування або подрібнення таблеток, змішування з їжею чи напоями). У разі використання альтернативних методів цей підхід слід підтвердити, а в короткій характеристиці та інструкції для медичного застосування необхідно надати чіткі вказівки щодо обробки лікарської форми перед введенням. Підтвердження обробки має включати такі аспекти, як прийнятність пацієнтом, точність дозування, сумісність із пропонованим пристроєм для введення, потенційний вплив на біодоступність, атакож будь-які ризики для людини, яка здійснює маніпуляції з лікарською формою (див. розділ 10 цієї настанови).
Лінії поділу (риски) використовують для надання можливості ввести частину необхідної дози або для зручності проковтування. Використання ліній поділу втаблетках для отримання частин необхідної дози не може бути прийнятним уразі критичних доз. Слід продемонструвати легкість розламування таблеток із рискою.
За відповідного обґрунтування та підтвердження, розділення чи подрібнення таблетки перед введенням може також бути альтернативним методом введення таблетки дітям, які мають труднощі з проковтуванням таблетки у цілому вигляді. Також може бути доцільним диспергувати або розчиняти таблетку урідині перед прийомом. Крім того, капсули можна відкривати та приймати їх вміст.
Поділені/подрібнені таблетки або вміст капсули можна вживати з їжею чи напоями (див. розділ 10 цієї настанови) і це має бути вказано у короткій характеристиці та інструкції для медичного застосування. Необхідно продемонструвати придатність маніпуляцій із лікарською формою, включаючи сумісність із будь-яким запропонованим пристроєм для введення.
Якщо характеристики активної речовини чи лікарської форми унеможливлюють будь-яку маніпуляцію з препаратом для полегшення введення, це має бути чітко вказано у короткій характеристиці та інструкції для медичного застосування лікарського засобу.
6.2.2. Рідкі препарати для перорального застосування
Загальні питання
Рідкі лікарські форми для перорального введення зазвичай вважаються прийнятними для доношених дітей від народження, а також і для недоношених новонароджених, які можуть ковтати та приймати ентеральне харчування. Зазвичай рідкі лікарські форми на водній основі у багатодозових контейнерах потребують консервантів, у той час як для твердих лікарських форм консерванти не потрібні. Це надає перевагу у застосуванні дітям твердих лікарських форм порівняно з рідкими. Проте, як і для будь-якого аспекту фармацевтичної розробки, використання консервантів не має бути єдиним фактором для прийняття рішення про вибір між рідкими та твердими лікарськими формами для перорального застосування.
Рідкі препарати для перорального застосування, які містять консерванти, зазвичай вважаються прийнятними для дітей від народження, за умови, що ці консерванти (та будь-які інші допоміжні речовини) можна вважати безпечними для дітей у цільовій віковій групі (див. розділ 9 цієї настанови). Що стосується рідких препаратів, які готуються із твердих лікарських форм, то у складі упаковки необхідно передбачити наявність розчинників, якщо вони інші, ніж вода. Рідкі лікарські форми для перорального застосування необхідно упаковувати разом із відповідним дозуючим пристроєм, якщо тільки виробником не продемонстровано, що наявні на ринку дозуючі пристрої придатні для точного дозування рекомендованих доз та що ці пристрої доступні (див. розділ 11.3 цієї настанови). Дозуючий пристрій має бути придатним для відмірювання усіх рекомендованих доз, цю придатність необхідно підтвердити щодо певної рідкої композиції/препарату. Це особливо критично для в’язких пероральних рідин. У короткій характеристиці та інструкції для медичного застосування лікарського засобу мають бути чіткі вказівки щодо правильного застосування дозуючого пристрою з метою забезпечення прийому необхідної дози. У разі необхідності застосовування наявних на ринку дозуючих пристроїв, необхідно вказати у короткій характеристиці та інструкції для медичного застосування лікарського засобу тип такого пристрою (включаючи будь-який перехідник).
Ризики неправильного дозування при використанні дозуючого пристрою слід обговорити та обґрунтувати щодо критичності дози для дітей у цільовій віковій групі та потенційних помилок у дозуванні педіатричного лікарського засобу. Коли неправильне дозування може призвести до потенційно серйозного ризику для дітей, слід розглянути запобіжні заходи, такі як використання спеціального дозуючого пристрою, застосування однодозової упаковки або вибір іншої лікарської форми.
Об’єм дози рідкого препарату для перорального застосування може впливати наприйнятність пацієнтом. Для препаратів із відомими смаковими проблемами маленькі об’єми зазвичай краще переносяться, тільки якщо більше розведення препарату не може краще замаскувати неприємний смак.
Суспензії для перорального застосування
Критичні параметри якості продукту, які слід розглянути для пероральних суспензій, включають такі фізико-хімічні властивості, як в’язкість, здатність допіноутворення, сорбція повітря, утворення осаду, прилипання суспендованої активної речовини до первинної упаковки та дозуючого пристрою. Якщо утворення осаду уникнути неможливо, то для зниження ризику недостатнього збовтування та уникнення помилок у дозуванні через неоднорідний розподіл активної речовини рекомендується забезпечити швидке ресуспендування припомірному збовтуванні.
Слід обговорити ризики недостатнього дозування чи передозування у тому разі, якщо вміст контейнера не був збовтаний належним чином або взагалі не був збовтаний. У короткій характеристиці та інструкції для медичного застосування слід надати чіткі вказівки щодо правильного відмірювання дози, включаючи застереження щодо неправильного дозування у разі недостатнього збовтування препарату. У випадках, коли неправильне збовтування препарату призводить допотенційного серйозного ризику для здоров’я дитини, слід вжити відповідних заходів. Це може включати застосування однодозової упаковки або вибір іншої лікарської форми.
Краплі для перорального застосування
Пероральні краплі можуть бути найбільш прийнятною лікарською формою для застосування лікарських засобів у низьких дозах або малих об’ємах. Ризик неправильного відмірювання кількості крапель, а також правильність і точність відміряного об’єму слід обґрунтовувати відповідно до критичності дози. Для уникнення помилок при відрахуванні кількості крапель препарату, коли доза становить більше 10 крапель, необхідно застосовувати альтернативні дозуючі пристрої. Лише пероральні краплі можуть вважатися прийнятною лікарською формою для педіатричних лікарських засобів, що містять активні речовини з широким терапевтичним вікном, якщо не обґрунтовано інше.
Вимірюваний об’єм (тобто розмір краплі) визначається конструкцією та фізичними характеристиками крапельниці, фізико-хімічними властивостями рідини та тим, як користуються крапельницею. Чіткі вказівки щодо правильного використання крапельниці слід включити до короткої характеристики та інструкції для медичного застосування лікарського засобу.
Шипучі, розчинні та дисперговані препарати
Ці препарати призначені для розчинення чи диспергування у рідині перед введенням. Застосування шипучих препаратів дітям може обмежуватися відносно великим об’ємом рідини, необхідної для їх розчинення, та високим вмістом електролітів.
Мінімальний об’єм рідини для розчинення або диспергування та будь-який об’єм для обполіскування необхідно обговорювати та обґрунтовувати відносно цільової вікової групи. У короткій характеристиці та інструкції для медичного застосування лікарського засобу слід надавати чіткі вказівки щодо правильного його розчинення чи диспергування. Ці вказівки мають включати інформацію про мінімальний об’єм рідини, необхідний для розчинення або диспергування препарату, включаючи будь-які об’єми для обполіскування, а також про особливі вимоги до збовтування чи змішування.
При введенні лікарського засобу без попереднього диспергування чи розчинення слід розглянути потенційні ризики, характерні для диспергованих у ротовій порожнині або жувальних лікарських форм. Будь-які альтернативні способи перорального введення лікарського засобу мають бути чітко описані у короткій характеристиці та інструкції для медичного застосування.
6.2.3. Введення через живильну трубку
Пероральні лікарські засоби можна вводити через зонд пацієнтам, які перебувають на штучному вигодовуванні з причин, пов’язаних з їх станом здоров’я або віком, наприклад недоношеним новонародженим, які не можуть ковтати, але можуть отримувати ентеральне харчування.
Слід розглянути можливість введення препарату через живильну трубку, коли таке введення застосовується як основний шлях введення чи розглядається як найімовірніший варіант. При фармацевтичній розробці слід враховувати розмір часток, в’язкість, об’єм дози та об’єм рідини, необхідний для промивання, хімічну сумісність перорального лікарського засобу з матеріалом живильної трубки та ризик фізичної закупорки трубки. Відтворення дози після проходження через трубку необхідно продемонструвати, використовуючи зонди для ентерального харчування та промивні об’єми, що відповідають цільовій віковій групі.
Крім того, за необхідності слід обговорити ризики, пов’язані з випадковим вдиханням препарату та можливим впливом на біодоступність, залежно від розміщення трубки.
Коли передбачається введення препарату через живильні трубки, то у короткій характеристиці та інструкції для медичного застосування слід вказати, чи можна вводити препарат цим шляхом, включаючи вказівки щодо правильного проведення процедури.
6.2.4. Оромукозні препарати
Правильне застосування та прийнятність оромукозних препаратів залежать від віку дитини та її здатності утримувати препарат у певній ділянці ротової порожнини протягом встановленого часу. Адгезивні властивості оромукозних препаратів слід обговорювати щодо певної ділянки ротової порожнини, в якій вони мають застосовуватися. Дітям молодшого віку для запобігання ризику проковтування обполіскувача для рота чи гелю для зубів ці лікарські форми слід застосовувати, використовуючи ватну паличку, губку або подібний аплікатор.
6.3. Назальні препарати
Зазвичай назальні препарати вважають придатними для застосування дітям усіх вікових груп. Придатність назального шляху введення для місцевого та системного застосування певного педіатричного лікарського засобу слід обговорити та обґрунтувати з урахуванням того, що активна речовина (та допоміжні речовини) можуть спричинити біль або подразнення. Використання будь-якого консерванту необхідно обґрунтувати відповідно до розділу 9 цієї настанови. Також слід обговорити прийнятність лікарського засобу пацієнтом, зважаючи на смакові відчуття, що виникають при його прийомі.
Для назальних препаратів місцевої дії необхідно обговорити ризики можливого небажаного системного впливу. Пристрої для назального введення мають бути адаптованими до розміру носової порожнини дітей у цільовій віковій групі, враховуючи об’єм, що доставляється.
6.4. Препарати для інгаляцій
Має бути підтверджена прийнятність пацієнтом педіатричних лікарських засобів для пероральної інгаляції (включаючи розчини для розпилення) та їх доцільність для застосування дітям певного віку.
Дозовані інгалятори під тиском можна застосовувати дітям від народження заумови використання спеціального спейсера та лицьової маски. Діти більш старшого віку можуть застосовувати інгалятор за допомогою спейсера чи без такого. Виробник має підтвердити придатність запропонованих пристроїв для застосування у цільовій віковій групі.
За відсутності відповідної конструкції порошкові інгалятори зазвичай використовують лише дітям старшого віку, оскільки дитина приймає дозу задопомогою повітря, що вдихається.
6.5. Препарати для ректального введення
Супозиторії
Розмір (довжина та діаметр) супозиторія має відповідати віку та параметрам дитини. Якщо конструкцією супозиторія не передбачено можливості введення частини необхідної дози, то його не слід розрізати у зв’язку з високим ризиком помилок у дозуванні, пов’язаних із неоднорідним розподілом активної речовини і труднощами відтворення дози при розрізанні.
Рідкі препарати для ректального введення
При виборі довжини ректального наконечника клізми та будь-якого об’єму для введення слід враховувати вік та параметри дитини. За необхідності слід розглянути можливість використання градуйованих пристроїв для введення (попередньо заповнених шприців із ректальним наконечником). У короткій характеристиці та інструкції для медичного застосування потрібно надати чіткі вказівки щодо методу введення дитині необхідної дози.
6.6. Нашкірні та трансдермальні препарати
При розробці нашкірних та трансдермальних препаратів для дітей слід брати доуваги вікові зміни у бар’єрній функції шкіри, такі як товщина дерми, гідратація та перфузія епідермісу, а також відношення площі поверхні тіла до маси тіла, що змінюється.
Застосування допоміжних речовин, які, як відомо, підвищують чутливість шкіри (наприклад деякі сурфактанти й адгезиви) необхідно ретельно зважити та обґрунтувати. Слід визначити необхідність або обмеження використання водонепроникних чи інших типів матеріалів як покриття для нашкірних лікарських засобів. У відповідних випадках слід обговорити вплив оклюзії, підвищеної температури або теплового нагріву на проникність шкіри та ризик передозування.
Розмір та форму трансдермальних пластирів слід адаптувати до розміру та форми тіла дитини, вони не повинні заважати повсякденній діяльності. Нашкірні лікарські форми слід застосовувати переважно на тих ділянках шкіри, які недоступні для самостійного видалення пластиру дитиною. Якщо пластирі необхідно застосовувати на легкодоступних для дітей ділянках шкіри, слід обговорити вплив навмисного видалення пластиру на клінічний результат.
При розробці пластирів слід надавати перевагу таким, які немає необхідності розрізати для отримання нижчої дози, тобто забезпечити діапазон розмірів та доз, які відповідають віку. Однак деякі типи пластирів (наприклад матричного типу) можна розробляти для отримання різних доз/сили дії шляхом розрізання. Розрізання вважається прийнятним лише за наявності чітко промаркованих ліній розрізу і за умови, що однорідність дози та постійність параметрів доставки були належним чином продемонстровані.
У коротку характеристику та інструкцію для медичного застосування лікарського засобу слід включити інформацію про можливість або неможливість розрізання пластиру для отримання нижчих доз із чіткими вказівками про процедуру отримання нижчих доз шляхом розрізання помаркованих лініях. Також слід надати вказівки щодо безпечного видалення частини розрізаного пластиру або щодо можливості застосовувати частини розрізаного пластиру, що залишилися.
6.7. Очні та вушні препарати
Очні та вушні препарати зазвичай розробляють для однієї групи пацієнтів, включаючи дітей, дорослих та людей літнього віку. Очні та вушні препарати можуть погано сприйматися деякими дітьми, однак за відсутності більш придатних їх слід вважати прийнятними лікарськими формами для дітей усіх вікових груп.
Щоб запобігти застосуванню консервантів із потенційною місцевою токсичною дією на рогівку та/чи слизову оболонку для дітей, особливо новонароджених, слід розглянути препарати для одноразового введення або препарати для багаторазового введення у спеціальній упаковці, що не потребує використання консервантів. Це особливо важливо при тривалому застосуванні.
Дітей молодшого віку ще не можна навчити тримати очі відкритими під час процедури. Важливо, щоб у інструкції для медичного застосування була інформація про те, як тримати контейнер із лікарським засобом та дитину для правильного введення педіатричного препарату.
6.8. Парентеральне введення
Загальні питання
Парентеральне введення є найбільш розповсюдженим шляхом для введення активних речовин тяжко хворим дітям і клінічно нестабільним доношеним та недоношеним новонародженим.
Вибір внутрішньовенного, підшкірного чи внутрішньом’язового шляху введення слід обґрунтувати з урахуванням передбачуваного клінічного ефекту, основних характеристик активної речовини та сприймання дитиною (болючі відчуття).
Шлях внутрішньовенного введення (центральний або периферичний), місце ін’єкції, об’єм ін’єкції, швидкість введення, в’язкість, рН, застосування буферів, осмолярність та за необхідності — товщину і довжину голки слід описати та обґрунтувати щодо характеристик лікарського засобу для парентерального введення, віку та маси тіла дитини, максимальної кількості ін’єкцій на добу і тривалості лікування. За необхідності слід розглянути застосування мікроголок або шприців без голок, особливо для лікарських засобів, що потребують частого введення чи тривалого лікування.
Багатократні розведення (для отримання необхідної дози) неприпустимі, оскільки вони можуть призвести до помилок у дозуванні, їх можна уникнути шляхом забезпечення відповідних концентрацій парентеральних лікарських засобів.
Мінімальний об’єм дозування лікарського засобу залежить від точності відповідного дозуючого пристрою. Коли необхідно, слід описати розмір шприца та градуювання, яке дозволяє точно вводити необхідну дозу. Об’єм ін’єкції слід обґрунтувати відповідно до віку дитини. Зазвичай об’єм підшкірних та внутрішньом’язових ін’єкцій не має перевищувати 1 мл, однак для новонароджених та немовлят передбачаються менші об’єми. Деякі препарати для парентерального введення можуть бути призначені для екстрених ситуацій, коли венозний доступ нелегко встановити (наприклад увідділеннях реанімації чи інтенсивної терапії). Слід обговорити придатність парентеральних лікарських засобів, які зазвичай застосовують в екстрених ситуаціях, для внутрішньокісткового введення та включити відповідну інформацію до короткої характеристики та інструкції для медичного застосування лікарського засобу.
Новонародженим слід вводити лише дуже маленькі об’єми лікарських засобів, щоб запобігти перевантаженню об’ємом та забезпечити достатній простір для необхідних живильних рідин. Інфузії не мають бути настільки концентрованими, щоб використання стандартного обладнання для введення унеможливлювало отримання відповідних дозувань. Зокрема це слід розглядати відносно лікарських засобів, призначених для введення у вигляді безперервної інфузії. Крім того, у ході розробки необхідно дослідити специфічні проблеми, пов’язані з несумісністю лікарських засобів в інфузійній системі, осмолярністю, непідходящим розріджувачем та потенційною можливістю передозування чи недостатнього дозування внаслідок ефектів затримки об’єму в системах для внутрішньовенного введення.
Амбулаторне застосування
У разі, якщо парентеральні лікарські засоби необхідно застосовувати дітям в амбулаторних умовах, слід продемонструвати, що лікарська форма для парентерального введення достатньою мірою придатна для введення самою дитиною чи особою, що здійснює за нею догляд. Це особливо важливо у випадках, коли парентеральне введення може знадобитися за відсутності кваліфікованої особи, що здійснює догляд.
6.9. Фіксовані комбінації доз
Фіксовані комбінації доз часто розробляють як альтернативну замісну терапію для пацієнтів, які вже раніше отримували лікування окремими препаратами, особливо при хронічних захворюваннях, таких як ВІЛ або туберкульоз. Вони можуть бути корисними для пацієнтів, щоб спростити лікування або поліпшити дотримання схеми лікування. При клінічній необхідності виробник має розглянути усі можливі варіанти для розробки фіксованої комбінації, що відповідає віку у всіх або окремих підгрупах педіатричної популяції, якщо тільки цьому не заважатиме складність отримання необхідних доз або відсутність гнучкості для належного підбору дози.
7. Частота дозування
Вибір частоти дозування має бути обґрунтований щодо характеристик активної речовини, фармакокінетичного профілю, показань для застосування та зручності застосування/дотримання схеми лікування дитиною та особою, яка здійснює догляд за нею. Враховуючи ці критерії, для амбулаторного застосування слід надавати перевагу схемі введення максимум двічі надобу. Стосовно педіатричних лікарських засобів, які можна застосовувати частіше ніж два рази на добу, слід приділити особливу увагу придатності застосування в амбулаторних умовах за відсутності кваліфікованого персоналу (дитячий садок, школа тощо).
8. Препарати з модифікованим вивільненням
За необхідності слід розглянути застосування у дітей лікарських засобів ізмодифікованим вивільненням. Розробку препаратів із модифікованим вивільненням не слід обмежувати пероральним шляхом введення. Альтернативні шляхи введення можна застосовувати залежно від характеристик активної речовини (наприклад трансдермальний).
Лікарські форми із пролонгованим вивільненням можуть знадобитися для дітей, яким необхідно приймати препарат у школі або вночі. Їх застосування може значно скоротити частоту дозування та сприяти дотриманню схеми прийому.
Для твердих пероральних препаратів із модифікованим вивільненням слід враховувати ризик розжовування. Тому необхідно обговорити вплив розжовування таких лікарських засобів на їх безпеку та ефективність. Пацієнт не має піддаватися серйозному ризику у зв’язку із розжовуванням препарату.
При розробці пероральних лікарських форм із модифікованим вивільненням для педіатричного застосування особливу увагу слід приділяти фізіологічним станам, пов’язаним із віком дитини, наприклад рН вмісту шлунка, шлунково-кишковій скорочувальній здатності (вивільнення шлунка, час транзиту) та їх мінливості, оскільки це може впливати на абсорбцію лікарського засобу.
9. Допоміжні речовини у композиції
9.1. Загальні питання
Вибір допоміжної речовини, придатної для використання у педіатричному лікарському засобі, є одним з основних елементів його фармацевтичної розробки.
Хоча основні принципи використання певної допоміжної речовини однакові для лікарських засобів для дорослих та для дітей, включення будь-якої допоміжної речовини у педіатричний лікарський засіб потребує додаткового розгляду питань безпеки. Вживання дитиною допоміжної речовини може призвести до впливу, відмінного від такого на дорослу людину, допоміжна речовина може мати відмінну дію на розвиток органів і систем. З огляду наобмеженість даних щодо безпеки застосування допоміжних речовин у певній віковій групі слід дотримуватися обережного підходу при їх використанні упедіатричних лікарських засобах.
Загалом при виборі відповідної допоміжної речовини для педіатричних лікарських засобів враховують таке:
- функція допоміжної речовини у композиції та можливі альтернативи;
- профіль безпеки допоміжної речовини у дітей усіх цільових вікових груп, отриманий на підставі разової чи добової дози (а не концентрації чи сили дії лікарського засобу);
- очікувана тривалість лікування, тобто короткострокова (разова доза/декілька днів) порівняно з довгостроковою (тижні, місяці, хронічна);
- тяжкість стану, що підлягає лікуванню (наприклад захворювання, що загрожують життю) та терапевтичні альтернативи;
- прийнятність пацієнтом, включаючи привабливість (наприклад смак, місцевий біль);
- алергія та сенсибілізація.
У разі, якщо використання допоміжних речовин зі встановленим ризиком не можна уникнути, слід належним чином зіставити додаткову користь від застосування вибраної лікарської форми (і шляхів введення) та можливість застосування інших лікарських форм та шляхів введення, які не потребують використання таких допоміжних речовин. Слід надати всебічне обґрунтування розробки з урахуванням відносних переваг та ризиків і можливих альтернатив.
Нові дані можуть дозволити зробити припущення щодо виникнення питань пробезпеку допоміжних речовин, які використовують у зареєстрованих педіатричних лікарських засобах, або у разі перевищення добової дози, або удеяких цільових вікових групах. У таких випадках у ролі запобіжного заходу виробникам рекомендується уникати використання у нещодавно розроблених педіатричних лікарських засобах допоміжних речовин із потенційно небезпечними властивостями доти, поки подальші дослідження недозволять зробити науково обґрунтовані висновки про їх безпеку.
Незважаючи на вищенаведене, слід визнати, що використання нової допоміжної речовини (використаної вперше у лікарському засобі або для нового шляху введення) є фармацевтичною інновацією і застосування цієї нової допоміжної речовини можна належним чином обґрунтувати шляхом відповідних доклінічних досліджень, проте необхідно усвідомлювати, що проблеми з безпекою можуть виникнути лише під час масштабнішого застосування лікарського засобу у дітей. Тому необхідно зіставити додаткову користь від нової допоміжної речовини у певному педіатричному лікарському засобі із використанням інших допоміжних речовин зі встановленим профілем безпеки та застосуванням інших лікарських форм або шляхів введення.
Алергія може розвиватися від раннього дитинства, а сенсибілізація у дітей може виникати легше, ніж у дорослих. Щоб запобігти виникненню сенсибілізації та розширити можливості лікування дітей з алергією, виробникам слід по можливості уникати використання у лікарських засобах допоміжних речовин, про які відомо, що вони спричиняють сенсибілізацію/алергію.
Для оцінки профілю безпеки кожної допоміжної речовини у педіатричній композиції (див. Додаток до цієї настанови), щоб зробити загальний висновок про необхідність додаткових даних, слід користуватися нижченаведеними джерелами інформації (подано за значимістю):
- Настанови Комісії, ICH та ЕМА.
- Наукові висновки СНМР (наприклад документ із викладенням позиції СНМР, висновок СНМР щодо процедури направлення).
- Якісний склад допоміжних речовин у лікарських засобах, дозволених для застосування дітям, а також їх кількісний склад, якщо він відомий.
- Законодавство, що регулює харчові продукти (Харчове законодавство):
- це джерело інформації має деякі обмеження, оскільки стосується тільки харчових продуктів (тобто постійного і тривалого перорального застосування);
- усі відповідні допоміжні речовини, описані у Харчовому законодавстві як придатні для педіатричної популяції, зазвичай вважаються прийнятними для використання у педіатричних лікарських засобах для перорального застосування, якщо в інших джерелах інформації немає додаткових відомостей щодо їх безпеки та якщо формулювання у Харчовому законодавстві самі пособі не викликають сумнівів. У разі виникнення сумнівів щодо безпеки допоміжну речовину необхідно або вилучити із композиції, або виробник має обґрунтувати прийнятність цієї допоміжної речовини;
- вищенаведене не застосовується до новонароджених, стосовно яких зазвичай вимагаються додаткові клінічні дані;
- безпека відповідних допоміжних речовини, описаних у Харчовому законодавстві, потребує подальшої оцінки для використання у лікарських засобах не для перорального застосування.
- Європейські наукові висновки щодо безпеки харчових продуктів (EFSA):
- це джерело інформації має деякі обмеження, оскільки стосується лише харчових продуктів (тобто постійного і тривалого перорального застосування), і ці дані не можна застосовувати до дітей. Однак застороги для дорослих можуть поставити питання щодо безпеки застосування допоміжної речовини для дітей.
- Інші джерела інформації, наприклад:
- Експертний комітет з харчових добавок (JECFA), який є спільним Комітетом ВООЗ та Організації з харчових продуктів та сільського господарства;
- інформація у відповідній літературі;
- інформація виробників (власна інформація), така як неопубліковані наукові дані.
Отримані дані про допоміжну речовину у заявленому педіатричному лікарському засобі слід узагальнити та обговорити їх значимість щодо вікових груп, показань для застосування, шляхів введення і типу лікарської форми, тривалості лікування, максимального добового споживання допоміжної речовини та її дози.
Слід підкреслити, що відповідальністю виробника є обґрунтування того, що кожна допоміжна речовина в педіатричному лікарському засобі безпечна для застосування за призначенням у цільовій віковій групі. Якщо використання допоміжної речовини у педіатричному лікарському засобі неможливо обґрунтувати, використовуючи вищенаведені джерела, можуть потребуватися нові токсикологічні дані щодо цієї допоміжної речовини.
9.2. Барвники
Використання будь-якого барвника у педіатричному лікарському засобі необхідно розглядати і обґрунтовувати щодо його алергічного потенціалу та мінімального токсикологічного впливу у цільових вікових групах, прийнятності пацієнтами педіатричного профілю, а також щодо необхідності уникати випадкових помилок у дозуванні. Якщо існує необхідність розрізняти схожі лікарські засоби з метою уникнення випадкових помилок у дозуванні, то до прийняття рішення про використання барвників необхідно розглянути питання використання, наприклад іншої форми, розміру чи тиснення. Обґрунтування має стосуватися необхідності забарвлення лікарського засобу та вибору певного барвника.
На відміну від інших допоміжних речовин, використання барвників улікарських засобах регулюється Директивою 2009/35/ЄС [8] та наказом МОЗ України від 15.01.2003 р. № 8 [9]N.
9.3. Ароматизатори
Приємний смак лікарського засобу має велике значення для прийнятності пацієнтом. Ароматизатори можуть бути особливо необхідними для пероральних рідких композицій. Необхідність використання певного ароматизатора у педіатричному лікарському засобі має бути чітко описана та обґрунтована. Необхідно надати відомості про якісний та кількісний склад будь-якого компонента ароматизатора, який, як відомо, має визнану дію чи ефекти. Слід розглянути питання безпеки, включаючи ризик виникнення алергії та сенсибілізації.
9.4. Консерванти
Зазвичай використання консервантів вважають прийнятним у багатодозових препаратах, однак для багатьох консервантів дані щодо рівнів безпечної дози для дітей все ще залишаються обмеженими. Необхідність використання консервантів у педіатричному лікарському засобі та вибір системи консервантів із найнижчою концентрацією необхідно обґрунтовувати, виходячи зі співвідношення ризик/користь.
Необхідно обговорити відповідність системи консервантів для цільових вікових груп. Якщо дані щодо безпеки для дітей відсутні, виробник має обґрунтовувати рівень дози консервантів (пропоновані межі безпеки) зурахуванням припустимих меж для дорослих, а також врахувати можливість застосування альтернативних лікарських форм.
Виробникам рекомендується розглядати новітні підходи, які дають можливість створити педіатричну композицію без використання консервантів.
9.5. Цукри та підсолоджувачі
Задовільна прийнятність пацієнтом пероральних педіатричних композицій має першорядне значення, і солодкий смак відіграє у цьому важливу роль.
Підстави для використання конкретного підсолоджувача у педіатричному лікарському засобі необхідно чітко описати та обґрунтувати. Слід обговорити питання безпеки, включаючи стани організму, які можуть обмежувати застосування певного виду цукру або підсолоджувача (наприклад діабет, тяжка ниркова недостатність).
Вибір підсолоджувачів та їх концентрація залежить від властивостей активної речовини та використання ароматизаторів.
Слід ретельно обґрунтувати використання цукрів, що спричиняють карієс. Упедіатричних композиціях, призначених для тривалого застосування, бажано уникати частого вживання та/чи високих доз підсолоджувачів. Слід враховувати потенційну проносну дію багатоатомних спиртів (наприклад сорбітолу, манітолу). Осмотичні властивості багатоатомних спиртів також можуть вливати на біодоступність. Слід зазначити, що наявні дані провідповідні граничні значення для застосування багатоатомних спиртів у дітей обмежені.
У відповідних випадках необхідно розглянути альтернативні підходи допокращення смаку лікарських засобів (нанесення покриття, утворення комплексів, вибір носія, регулювання в’язкості).
10. Прийнятність для пацієнта
Прийнятність може бути визначена як загальна здатність і готовність пацієнта застосовувати лікарський засіб за призначенням, а особи, що здійснює догляд — вводити його відповідно до призначення. Прийнятність пацієнтом може мати значний вплив на дотримання схеми прийому та, відповідно, на безпеку та ефективність препарату. Прийнятність визначається характеристиками лікарського засобу та користувачем. Це включає фармацевтичні характеристики продукту, такі як:
- смакова привабливість, розмір та форма;
- ступінь складності маніпуляцій, які має здійснити дитина чи особа, що здійснює догляд за нею, перед застосуванням лікарського засобу;
- необхідна доза, наприклад об’єм дозування, кількість таблеток тощо;
- необхідна частота дозування;
- вибраний пристрій для введення;
- первинна та вторинна системи упаковка/закупорювальний засіб;
- фактичний спосіб введення лікарського засобу та будь-який пов’язаний ізним біль або дискомфорт.
Оцінка прийнятності пацієнтом педіатричного лікарського засобу має бути невід’ємною частиною досліджень із фармацевтичної розробки. Прийнятність пацієнтом лікарського засобу слід вивчати на самих дітях у рамках будь-якого клінічного дослідження, до якого включений запропонований лікарський засіб. В обґрунтованих випадках, коли клінічні дослідження не проводяться або коли прийнятність пацієнтом не вивчається у клінічних дослідженнях, прийнятність лікарського засобу, запропонованого для продажу, має бути продемонстрована іншим способом, наприклад, посиланням на джерела літератури чи дослідженнями у відповідних дорослих групах. Рідкі пероральні препарати зазвичай вважаються прийнятними від народження, однак смакове сприйняття розвивається і стає суттєвим.
Стосовно зареєстрованих лікарських засобів, для яких прийнятність наявної композиції була вивчена під час розробки, слід упевнитися, що для них забезпечено належну прийнятність пацієнтом. У разі внесення змін доскладу зареєстрованих композицій виробник має оцінити вплив зміни та переконатися у тому, що забезпечується належна прийнятність пацієнтом.
Під належною прийнятністю пацієнтом не слід розуміти 100% прийняття лікарського засобу дітьми у цільовій віковій групі. Однак різні методи оцінки прийнятності пацієнтом можуть призвести до різних результатів. Тому придатність вибраного методу для перевірки прийнятності пацієнтом та відповідності застосовуваних меж слід обговорити та обґрунтувати щодо ризиків застосування препарату, включаючи ризики для популяції (наприклад розвиток мікробної резистентності через недостатню прийнятність різних препаратів, що містять антибіотики), а також слід враховувати характеристики цільової вікової групи, захворювання, характерні для педіатричної популяції, частоту застосування, супутню терапію.
Смакова привабливість
Смакова привабливість є одним з основних елементів прийнятності пацієнтом педіатричного лікарського засобу для перорального застосування. Це також може стосуватися назальних та інгаляційних лікарських засобів. Смакова привабливість визначається як загальна оцінка лікарського засобу (зазвичай для перорального застосування) щодо його запаху, смаку, післясмаку і текстури (відчуття у роті). Вона зумовлена характеристиками активної речовини та способу її введення до складу готової лікарської форми. Інформація про смакову привабливість активної речовини має бути отримана на ранній стадії розробки, наприклад для відповідних дорослих груп або злітературних джерел. Смакова привабливість активної речовини має сприяти правильному вибору готової лікарської форми та шляхів введення. Смакова привабливість педіатричного лікарського засобу має бути задовільною сама по собі (тобто без змішування з їжею чи напоями), якщо необґрунтовано інше.
Може бути розроблений педіатричний лікарський засіб із нейтральним смаком або з певним і загалом прийнятним смаком, їх вибір має бути обґрунтований. Зазвичай слід розробляти лікарський засіб із нейтральним смаком, особливо якщо його застосовують для лікування при хронічних станах, оскільки сильні ароматизатори можуть стати непривабливими при тривалому застосуванні. Розробка запланованої цільової смакової привабливості (нейтральний або специфічний смак) має бути чітко описана та включати інформацію щодо відповідних альтернативних складів або лікарських форм.
Прикладами заходів для покращення смакової привабливості лікарського засобу можуть бути: раціональний вибір допоміжних речовин (включаючи засоби для маскування смаку, підсолоджувачі та ароматизатори), зміна розміру часток активної речовини або допоміжних речовин, вибір іншої солі активної речовини, покриття активної речовини, покриття готової лікарської форми, застосування комплексоутворюючих речовин (наприклад циклодекстринів) або (для рідких препаратів) зменшення кількості вільного активного інгредієнта у розчині шляхом вибору іншої сили дії та подальшого зменшення об’єму. Однак педіатричні композиції/препарати для перорального застосування немають ставати дуже привабливими для дітей іншим способом (наприклад схожими на цукерку), тому що це, як відомо, може підвищити ризик випадкового отруєння.
Змішування з їжею
З різних причин може бути бажаним змішувати педіатричний лікарський засіб зїжею та напоями. Якою б не була причина, її слід пояснити та обґрунтувати. Змішування з їжею та напоями може мати призначення маскувати незадовільний смак лікарського засобу у випадках, коли продемонстровано, що смакова привабливість педіатричного лікарського засобу не може бути додатково поліпшена та неможливо розробити альтернативні лікарські форми.
Рекомендації щодо змішування з їжею чи напоями можуть також застосовуватися для лікарського засобу з уже наданою привабливістю як додатковий засіб поліпшення прийнятності та полегшення проковтування. Крім того, відсутність рекомендацій щодо змішування з їжею чи напоями негарантує, що особи, які здійснюють догляд за дітьми, не використовуватимуть цей спосіб для введення лікарського засобу. Тому ефект змішування препарату з певним типом звичних харчових продуктів або напоїв слід розглянути та/чививчити для кожного педіатричного лікарського засобу. У короткій характеристиці та інструкції для медичного застосування завжди має міститися чітка інформація, чи можна змішувати препарат з їжею чи напоями. Якщо змішування з їжею чи напоями не рекомендується, то у короткій характеристиці та інструкції для медичного застосування слід чітко вказати, що таке змішування не вивчалося, і за це несе відповідальність користувач.
Крім того, слід додавати відповідні попередження у випадках, коли припускається несумісність із певними типами харчових продуктів та напоїв. Якщо рекомендується змішування з їжею чи напоями, слід чітко вказувати типи харчових продуктів та напоїв, включаючи будь-які температурні умови, якщо необхідно. Користувач має бути проінструктований, що лікарський засіб слід змішувати з малою кількістю (наприклад однією ложкою) чи іншою обґрунтованою кількістю їжі або напоїв і що його слід застосувати у визначений період після змішування. У виняткових випадках для забезпечення адекватної смакової прийнятності або розчинення може знадобитися більша кількість їжі чи напоїв. Однак слід уникати великого об’єму їжі або напоїв (наприклад повна склянка/тарілка), тому що дитина не зможе або не схоче спожити усю кількість.
Різні харчові продукти чи напої можуть мати різні властивості або відрізнятися за їх впливом на лікарський засіб. Вибір будь-яких харчових продуктів або напоїв слід обґрунтувати щодо їх впливу на властивості педіатричного лікарського засобу. Зрозуміло, що їжа та напої зазвичай є нестандартизованими продуктами і дослідження прийнятності пацієнтом та сумісності не можуть охопити весь діапазон їх різноманітності. Незважаючи на це, у короткій характеристиці та інструкції для медичного застосування слід чітко зазначити, придатність яких харчових продуктів та напоїв для змішування з лікарськими засобами була продемонстрована. Слід надати інформацію простабільність лікарського засобу в рекомендованих харчових продуктах та напоях, якщо тільки не обґрунтовано інше. Ця інформація має включати відомості про будь-які температурні обмеження. Деякі лікарські засоби можуть розчинятися повністю або частково у харчових продуктах, і це може вливати наїх дію та фармакокінетичний профіль.
Коли пропонується змішування з їжею та напоями, слід розглянути їх можливий вплив на фармакокінетичні характеристики лікарського засобу. Залежно від інформації, отриманої у ході попередніх досліджень педіатричного лікарського засобу, може знадобитися визначення його біодоступності.
11. Система упаковка/закупорювальний засіб, дозуючий пристрій, пристрій для введення, пакування
11.1. Загальні питання
Система упаковка/закупорювальний засіб та пристрій для введення мають бути розроблені для використання у цільових вікових групах. Коли їх використовують разом, має бути забезпечене належне застосування лікарського засобу.
Якщо не обґрунтовано інше, система упаковка/закупорювальний засіб, що використовується підлітками, має бути роздільною і зручною для перенесення та, якщо обґрунтовано, має дозволяти брати окремі дози із собою до школи, на заняття спортом тощо. Якщо необхідно, у короткій характеристиці та інструкції для медичного застосування слід указати, що лікарський засіб можна застосовувати лише у комбінації з відповідним пристроєм для введення.
Виробникам рекомендується розглядати нову упаковку та підходи до способів введення лікарського засобу, які покращать застосування дитиною, дотримання рекомендацій із застосування, а також зручність для дитини та/чи особи, що здійснює догляд за нею, при цьому знижуючи ризик випадкових помилок удозуванні.
Система упаковка/закупорювальний засіб має дозволяти відрізнити лікарський засіб від кондитерського виробу та іграшки, щоб зменшити привабливість лікарського засобу для дітей.
Слід брати до уваги практичність системи упаковка/закупорювальний засіб. Наприклад, деякі пляшки для рідкого перорального препарату є достатньо маленькими, щоб можна було вилучити весь вміст за допомогою шприца для перорального введення відповідної довжини. Інші контейнери потребують «перехідника для шприца», який становить приєднану до горлечка пляшки пробку, до якої можна приєднати шприц для перорального введення. Перехідник для шприца дозволяє успішно видалити з пляшки увесь її вміст.
11.2. Розмір контейнера
Повний вміст контейнера слід обґрунтувати стосовно:
1) рекомендацій із дозування та тривалості застосування для кожної цільової вікової групи у короткій характеристиці та інструкції для медичного застосування;
2) випадкових помилок у дозуванні, особливо ризику 10-кратного передозування;
3) випадкового проковтування всього вмісту контейнера;
4) прийнятності пацієнтом.
11.3. Дозуючий пристрій
Особливу увагу слід приділити зручності й точності введення лікарського засобу. Також слід обговорити критичність дози (крута крива доза/відповідь, вузьке терапевтичне вікно).
Рідкий педіатричний лікарський засіб необхідно постачати разом із дозуючим пристроєм, якщо тільки не обґрунтовано інше. Фізичні властивості рідкого препарату мають значення для визначення точності дозування. Для забезпечення точності дозування слід вивчити поєднання педіатричного препарату з дозуючим пристроєм.
Якщо немає необхідності постачати лікарський засіб разом із дозуючим пристроєм, слід продемонструвати, що точність дозування досягається задопомогою широко доступних дозуючих пристроїв, таких як мірні ложки та мірні чашки. Інструкції для користувача мають містити інформацію продозуючі пристрої, які слід використовувати разом із лікарським засобом.
Слід обговорити відповідність дозуючого пристрою віку дитини. У групах наймолодшого віку шприц для перорального введення може бути більш надійним пристроєм для введення необхідної дози, ніж чашка чи ложка.
Номінальний об’єм дозуючого пристрою та його градуювання слід визначити зурахуванням рекомендованих доз, ризику передозування та недодозування, атакож доступності більш високої або низької сили дії лікарського засобу. Дозуючі пристрої можна використовувати повторно для перорального введення за умови відповідної очищення. Вказівки з очищення дозуючого пристрою слід включити до короткої характеристики або інструкції для медичного застосування.
Якщо пристрій для введення спеціально сконструйований для доставки необхідних доз певного продукту, наприклад чашка для відмірювання певної кількості гранул, то назву лікарського засобу слід зазначати на дозуючому пристрої для запобігання помилковому використанню для інших лікарських засобів.
Деякі дозуючі пристрої, такі як шприци для перорального введення, можуть мати певний «мертвий простір». Значимість «мертвого простору» збільшується у міру зменшення вимірюваних об’ємів. Необхідно продемонструвати, що це немає суттєвого значення для точності вимірювання запланованого об’єму. Неправильне промивання шприців та голок може призвести до значного передозування. Слід обговорити ризик такого передозування для здоров’я дитини. У певних випадках у коротку характеристику та інструкцію для медичного застосування можна внести відповідне застереження, тобто непромивати шприц або голку.
Точність дозуючих пристроїв для педіатричних лікарських засобів із крутою кривою доза–відгук або вузьким терапевтичним вікном може потребувати особливого розгляду. Точність таких дозуючих пристроїв слід обговорити та обґрунтувати.
11.4. Інші пристрої
Для введення лікарських засобів шляхами, що потребують використання певних пристроїв для введення, наприклад масок для обличчя, небулайзерів, необхідно обґрунтувати придатність пристроїв для цільових вікових груп.
Необхідно враховувати простоту введення лікарського засобу дитиною чи особою, що здійснює за нею догляд, труднощі при введенні дітям, які цього небажають, та надійність пристрою у повсякденній практиці. Якщо виробник неможе довести доступність на ринку пристрою для введення, то такий пристрій має постачатися разом із лікарським засобом.
12. Інформація для користувача (коротка характеристика лікарського засобу та інформація для пацієнта)
Виробники зобов’язані надавати чіткі інструкції для користувача, які допоможуть правильно та у повному обсязі ввести педіатричний лікарський засіб. Ці інструкції мають ураховувати різні способи введення лікарського засобу дітям від народження до дорослого віку. За необхідності рекомендується розробляти інструкції, призначені як для дитини, так і для особи, що здійснює за нею догляд. Інформація для пацієнта стосовно неухильного дотримання інструкцій має бути достатньо надійною щодо дітей, які не бажають приймати препарат, особливо якщо повне їх дотримання є вирішальним для результатів лікування.
Детальні вказівки щодо подання інформації наведено у Настанові з короткої характеристики лікарського засобу (SmPC).
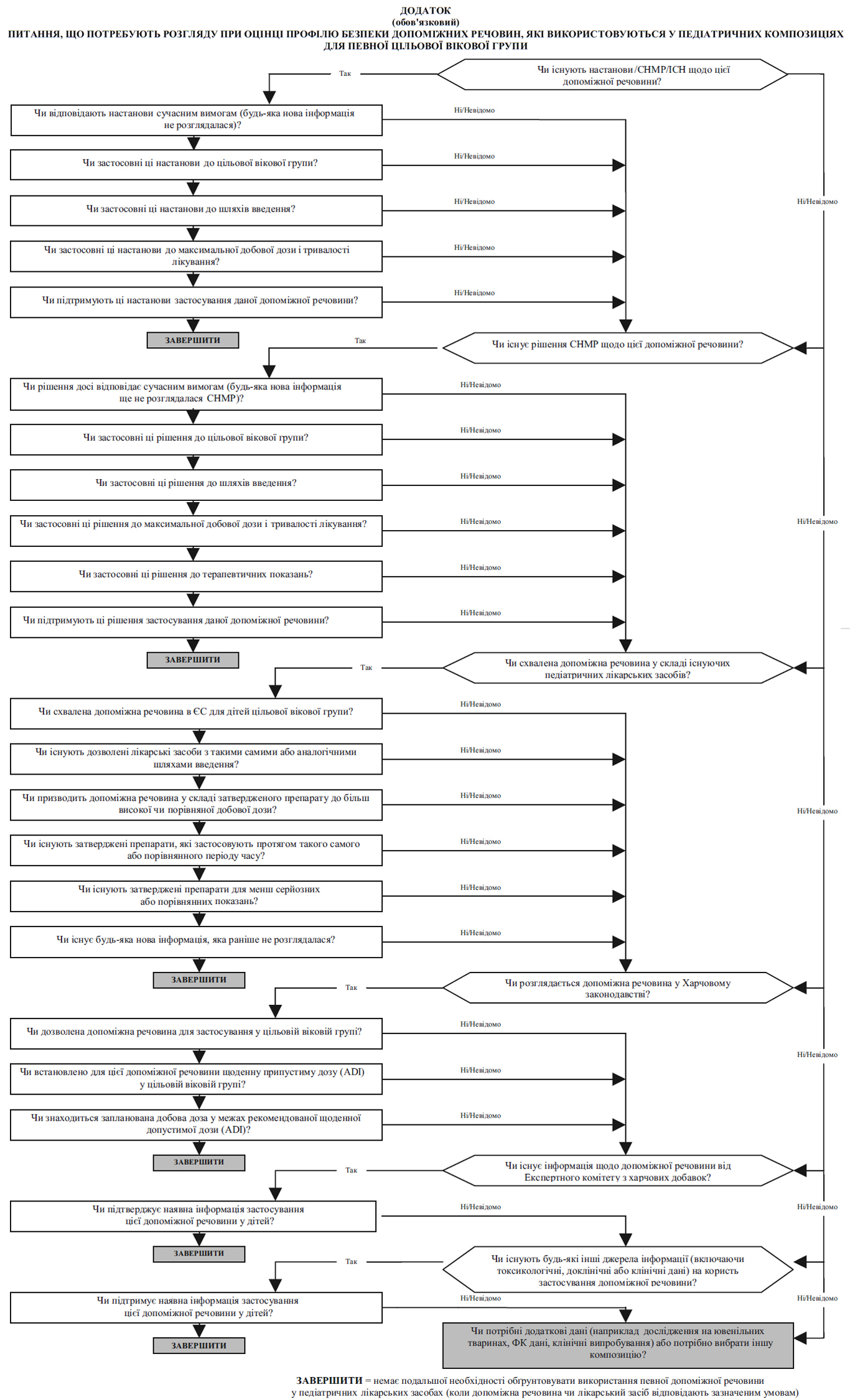
Національний додаток (довідковий). Бібліографія
1. EMA/CHMP/QWP/805880/2012 Rev. 1 «Guideline on Pharmaceutical Development of Medicines for Paediatric Use» (Настанова з фармацевтичної розробки лікарських препаратів для педіатричного застосування (перша переглянута версія)).
2. ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів/ І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
3. Настанова СТ-Н МОЗУ 42-1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення/ М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005.
4. ДСТУ 1.7-2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів/ О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
5. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
6. Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use//Official Journal of the European Communities. — L 311, 28.11.2001. — р. 67–128 (Директива 2001/83/ЄC Європейського Парламенту та Ради від 6 листопада 2001 року про кодекс Співтовариства відносно лікарських препаратів, призначених для застосування людям//Official Journal of the European Communities. — L 311, 28.11.2001. — р. 67–128).
7. Порядок державної реєстрації (перереєстрації) лікарських засобів і розмірів збору за їх державну реєстрацію (перереєстрацію), затверджений Постановою Кабінету Міністрів України від 26.05.2005 р. № 376.
8. Directive 2009/35/EC of the European Parliament and of the Council, of 23 April 2009 on the colouring matters which may be added to medicinal products (recast) //Official Journal of the European Communities. — L 109, 30.04.2009. — р. 11–13 (Директива 2009/35/ЄС Європейського Парламенту та Ради від 23 квітня 2009 року про барвники, які можуть додаватися до лікарських препаратів (нова редакція).
9. Наказ МОЗ від 15.01.2003 р. № 8 (зареєстрований у Мін’юсті України 29.01.2003 р. за № 69/7390) «Про затвердження Переліків допоміжних речовин та барвників, дозволених до застосування у виробництві лікарських засобів, що (лікарські засоби) реєструються в Україні та виготовляються в аптечних умовах за рецептами лікарів і замовленнями лікувально-профілактичних закладів».
10. EMEA/CHMP/PEG/194810/2005 «Reflection Paper: Formulations of Choice for the Paediatric Population» (Пояснювальна записка: Композиції вибору для педіатричної популяції).
Ключові слова: діти, фармацевтична розробка, якість.