
НАСТАНОВА
НАСТАНОВА З ЯКОСТІ
ЛІКАРСЬКІ ЗАСОБИ
ФАРМАЦЕВТИЧНА РОЗРОБКА
Настанова 42-3.1:2004
Видання офіційне
Київ
Міністерство охорони здоров’я України
2012
Передмова
1 РОЗРОБЛЕНО: ДП «Державний науковий центр лікарських засобів» (ДП «ДНЦЛЗ»)
РОЗРОБНИКИ: М. Ляпунов, доктор фарм. наук (керівник розробки); В. Георгієвський, доктор фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; М. Пасічник; О. Кричевська; А. Піотровська; К. Жемерова; Л. Алмакаєва, канд. фарм. наук
ВНЕСЕНО: Державною службою лікарських засобів і виробів медичного призначення Міністерства охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 31 грудня 2003 р. № 637
3 Ця настанова відповідає:
CPMP/QWP/155/96 Note for Guidance on Development Pharmaceutics, 1998 (CPMP/QWP/155/96 Керівні вказівки з фармацевтичної розробки, 1998) у частині розділу 5
CPMP/QWP/054/98 Decision Trees for the Selection of Sterilisation Methods. Annex to Note for Guidance on Development Pharmaceutics (CPMP/QWP/155/98), 2000 (CPMP/QWP/054/98 Схема рішень для вибору методів стерилізації. Додаток до керівних вказівок із фармацевтичної розробки (CPMP/QWP/155/96), 2000) у частині додатка А
Ступінь відповідності — модифікований (MOD)
Переклад з англійської мови (en)
4 ВВЕДЕНО ВПЕРШЕ
© Державна служба лікарських засобів і виробів медичного призначення, 2012
© Колектив розробників, 2012
© МОРІОН, 2012
Національний вступ
У фармацевтичному секторі України відбувається гармонізація законодавчої та нормативної бази з відповідними нормами Європейського Союзу (ЄС). На даний час в Україні вже введено в дію такі нормативні документи:
- Державна Фармакопея України (ДФУ), гармонізована з Європейською Фармакопеєю;
- Настанова 42–01–2001 «Лікарські засоби. Належна виробнича практика», гармонізована з настановою з належної виробничої практики (Good Manufacturing Practice — GMP) ЄС;
- Настанова 42–02–2002 «Лікарські засоби. Належна виробнича практика активних фармацевтичних інгредієнтів», гармонізована з додатком 18 до настанови з GMP ЄС, та ін.
Принципи забезпечення якості, викладені в Настанові 42–01–2001 (5.1.2а), вимагають здійснення заходів, які гарантують, що лікарські засоби розроблені й досліджені з урахуванням вимог GMP та належної лабораторної практики. Однак настанови з GMP не регламентують принципи і правила розробки лікарських засобів.
Загальні статті ДФУ передбачають проведення певних досліджень на етапі розробки готових лікарських засобів з метою дотримання фармакопейних вимог. При цьому в ДФУ наведено методи випробувань, за допомогою яких можна дослідити лікарські засоби та діючі речовини. Однак у ДФУ, як і в Європейській Фармакопеї, не визначена методологія організації цих досліджень.
Загальний технічний документ (Common Technical Document — CTD), прийнятий у ЄС, Сполучених Штатах Америки та Японії, встановлює структуру подання в реєстраційному досьє даних із фармацевтичної розробки. У CTD наведені посилання на спеціальні настанови, згідно з якими слід проводити дослідження з фармацевтичної розробки лікарських засобів, і які складають методичну основу організації цих досліджень і надання інформації в реєстраційному досьє.
Ця настанова розроблена на підставі двох настанов з якості Комітету з патентованих лікарських засобів (Committee for Proprietary Medicinal Products — CPMP), що стосуються питань створення лікарських засобів. Перша з цих настанов (CPMP/QWP/155/96) присвячена фармацевтичній розробці та поданню результатів цих досліджень у реєстраційному досьє, а друга (CPMP/QWP/054/98) є додатком до першого документа і містить схеми рішень щодо вибору методів стерилізації.
У настанову внесено такі редакційні зміни:
а) два документи CPMP (CPMP/QWP/155/96 і CPMP/QWP/054/98) об’єднані в одну настанову без зміни їх обсягу та змісту;
б) додатково введені розділи «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Позначення і скорочення» і додаток Б «Бібліографія». Основні положення викладені в розділі 5 і додатку А. Відповідно до цього номери пунктів і підпунктів додатково включають номер розділу 5, а також відповідний номер пункту настанови CPMP/QWP/155/96;
в) посилання на нормативні документи, що згадуються в тексті, наведені в повному обсязі в розділі 2 «Нормативні посилання». Замість посилань на деякі настанови з якості ЄС у тексті містяться посилання на прийняті МОЗ України відповідні гармонізовані настанови. При згадуванні в тексті настанов із якості, прийнятих у ЄС, у виносках у кінці відповідних сторінок зазначено: «Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку». Додатково зроблене посилання на настанову CPMP/QWP/2845/OO, що була прийнята пізніше за настанову CPMP/QWP/155/96;
г) посилання на директиву 75/318/ЕЕС із тексту настанови виключені, а замість номерів частин реєстраційного досьє в форматі, встановленому в додатку до цієї директиви, у примітках зазначені номери та назви відповідних розділів і пунктів реєстраційного досьє у форматі CTD;
д) у настанові замінені слова: «досьє торгової ліцензії» («marketing authorisation dossier»), «досьє» («dossier») та «заявка на отримання торгової ліцензії» («application for a marketing authorisation») — на «реєстраційне досьє»;
є) замість слів «фармакопея»,чи «Європейська Фармакопея» зазначено «Державна Фармакопея України чи Європейська Фармакопея». Це пов’язано з тим, що Державна Фармакопея України гармонізована з Європейською Фармакопеєю, а встановлені в ній національні додаткові вимоги чи вимоги на вибір більш жорсткі. Відповідно поряд із посиланням на текст Європейської Фармакопеї «Methods of preparation of sterile products» («Методи приготування стерильних продуктів») наведене посилання на гармонізований із ним загальний текст ДФУ «Методи приготування стерильних продуктів»; окрім того, у тексті додатково подані посилання на відповідні загальні статті та загальні тексти ДФУ;
ж) оскільки CTD був прийнятий після настанови CPMP/QWP/155/96, у відповідних підрозділах і пунктах був наведений відповідний текст з урахуванням вимог CTD; він був виділений іншим шрифтом і літерою141. Окрім того, з урахуванням структури й вимог CTD у структуру даної настанови додатково були включені пункти 5.3.1 «Розробка складу» і 5.4.1 «Загальні положення щодо вибору паковальних матеріалів», а також підрозділи 5.6 «Мікробіологічні характеристики лікарського препарату» і 5.7 «Сумісність лікарського препарату»;
и) у зв’язку з вимогами додатка G «Виробництво рідин і м’яких лікарських засобів» до Настанови 42–01–2001 і загальної статті ДФУ «М’які лікарські засоби для місцевого застосування» додатково введений підпункт 5.3.3.3 «Однорідність»;
к) у розділі 3 зазначені використані терміни й надані посилання на нормативні документи, в яких наведені визначення понять щодо цих термінів;
л) інші незначні доповнення виділені іншим шрифтом і літерою N.
НАСТАНОВА
Настанови з якості
ЛІКАРСЬКІ ЗАСОБИ
Фармацевтична розробка
Руководства по качеству
ЛЕКАРСТВЕННЬІЕ СРЕДСТВА
Фармацевтическая разработка
Quality guidelines
MEDICINAL PRODUCTS
Pharmaceutical development
Чинна від 2004-04-01
1 Сфера застосування
Ця настанова поширюється на лікарські засоби для людини та встановлює рекомендації стосовно структури досліджень з фармацевтичної розробки та інформації з цих досліджень, яку слід включати в реєстраційне досьє на готовий лікарський засіб.
Ця настанова рекомендується для підприємств, організацій і установ, що розробляють і/або серійно виробляють лікарські засоби на території України, незалежно від відомчого підпорядкування та форми власності, для науково-експертних організацій і регуляторних органів, а також експертів та інспекторів, які здійснюють експертизу на етапі реєстрації (перереєстрації) лікарських засобів та інспектування їх виробництва.
Цю настанову рекомендується застосовувати при плануванні та проведенні наукових досліджень із розробки готових лікарських засобів і валідації процесів, а також при складанні реєстраційного досьє.
2 Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 р.
Настанова 42–01–2001 Лікарські засоби. Належна виробнича практика
Настанова 42–3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності
Настанова 42–3.3:2004 Настанови з якості. Лікарські засоби. Випробування стабільності
Настанова 42–3.4:2004 Настанови з якості. Лікарські засоби. Виробництво готових лікарських засобів
Настанова 42–3.6:2004 Настанови з якості. Лікарські засоби. Допоміжні речовини
The rules governing medicinal products in the European Union. — V. ЗА. — Guidelines. Medicinal products for human use. Quality and biotechnology. — 3AQ10a (revision February 1994). — Plastic primary packaging materials
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. ЗА. — Настанови. Лікарські засоби для застосування в людини. Якість і біотехнологія. — 3AQ 10а (переглянуто у лютому 1994). — Пластикові матеріали для первинного паковання)
The rules governing medicinal products in the European Union. — V. 3B. — Guidelines. Medicinal products for human use. Safety, environment and information. — 3BR2a. — Matters relating to the replacement of CFC’s in medicinal products
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. ЗВ. — Настанови. Лікарські засоби для застосування в людини. Безпека, навколишнє середовище та інформація. — 3BR2a. — Питання, що стосуються заміни хлорфторвуглеців у лікарських засобах)
The rales governing medicinal products in the European Union. — V. 3B. — Guidelines. Medicinal products for human use. Safety, environment and information. — 3BR3a. — Replacement of chlorofluorocarbons (CFC) in metered dose inhalation products
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. ЗВ. — Настанови. Лікарські засоби для застосування в людини. Безпека, навколишнє середовище та інформація. — 3BR3a. — Заміна хлорфторвуглеців (CFC) у дозованих інгаляційних препаратах)
ISO 11137:1995(Е) Sterilization of health care products. Requirements for validation and routine control. Radiation sterilization
(ISO 11137:1995(E) Стерилізація продукції для охорони здоров’я. Вимоги до валідації та рутинного контролю. Стерилізація за допомогою випромінювання)
CPMP/QWP/158/96 Note for Guidance on dry powder inhalers, 1998
(CPMP/QWP/158/96 Керівні вказівки щодо інгаляторів із сухими порошками, 1998)
CPMP/QWP/604/96 Note for Guidance on quality of modified release products: A. Oral dosage forms; B. Transdermal dosage forms; Section 1 (Quality), 1999
(CPMP/QWP/604/96 Керівні вказівки щодо якості препаратів із модифікованим вивільненням: А. Оральні лікарські форми; В. Трансдермальні лікарські форми; Розділ 1 (Якість), 1999)
CPMP/QWP/2845/OO Note for Guidance on requirements for pharmaceutical documentation for pressured metered dose inhalation products, 2002
(CPMP/QWP/2845/OO Керівні вказівки щодо вимог до фармацевтичної документації для дозованих інгаляційних препаратів, що знаходяться під тиском, 2002)
The rules governing medicinal products in the European Union. — V. 2B. — Notice to applicants. Medicinal products for human use. Common Technical Document
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. 2В. — Інформація для заявників. Лікарські засоби для застосування в людини. Загальний технічний документ)
European Pharmacopoeia. 4th Edition. 2002
(Європейська Фармакопея. 4е видання. 2002)
З Терміни та визначення понять
3.1 У цій настанові використані терміни, встановлені у Настанові 42–01–2001: валідація; виробник; виробництво; вихідна сировина; готова продукція; лікарський засіб (лікарський препарат); паковальний матеріал; проміжна продукція; серія; технологічний процес (виробничий процес).
3.2 У цій настанові використані терміни, встановлені у Державній Фармакопеї України (ДФУ): ефективність антимікробних консервантів; ступінь надійності стерилізації.
3.3 У цій настанові використані терміни, встановлені у Настанові 42–3.2:2004: критерії прийнятності; надлишок; специфікація; специфікація на готову продукцію, що застосовується при випуску; специфікація на готову продукцію, що застосовується протягом терміну зберігання; якість.
3.4 У цій настанові використані терміни, встановлені у Настанові 42–3.3:2004: первинне паковання (первинний паковальний матеріал); термін зберігання (термін придатності).
3.5 У цій настанові використаний термін, встановлений у Настанові 42–3.4:2004: готовий лікарський засіб.
3.6 Нижче наведені визначення термінів, додатково використаних у цій настанові. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1–5].
3.6.1 допоміжна речовина (excipient, [I])
Будь-яка речовина лікарської форми за винятком діючої речовини.
3.6.2 діюча речовина; лікарська речовина; активний фармацевтичний інгредієнт (active substance; drug substance; active pharmaceutical ingredient, [2,3])
Будь-яка речовина (або суміш речовин), що призначена для використання у виробництві лікарського засобу і яка при використанні у виробництві лікарського засобу стає його активним інгредієнтом. Такі речовини виявляють фармакологічну чи іншу безпосередню дію; їх застосовують для лікування, діагностики чи профілактики захворювання, для зміни стану, структур або фізіологічних функцій організму, для догляду, обробки, а також для полегшення симптомів.
3.6.3 лікарська форма (pharmaceutical form; dosage form, [4])
Поєднання форми, в якій лікарський засіб представлений виробником (форма випуску), а також форми, в якій лікарський засіб призначений для застосування, включаючи фізичну форму (форма застосування).
3.6.4 сила дії лікарського засобу (strength of the medicinal product, [5])
Вміст діючих речовин у кількісному вираженні на одиницю дози чи одиницю об’єму, чи одиницю маси відповідно до лікарської форми.
4 Позначення і скорочення
ДФУ — Державна Фармакопея України
ЄС — Європейський Союз
ПАР — поверхнево-активна речовина
СНС — ступінь надійності стерилізації
CFC — chlorofluorocarbon (хлорфторвуглець)
СРМР — Committee for Proprietary Medicinal Products (Комітет із патентованих лікарських засобів)
CTD — Common Technical Document (Загальний технічний документ)
GMP — Good Manufacturing Practice (належна виробнича практика)
ІСН — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
ISO — International Standardization Organization (Міжнародна організація зі стандартизації)
QWP — Quality Working Party (робоча група з якості)
5 Рекомендації щодо інформації з фармацевтичної розробки в реєстраційному досьє на готові лікарські засоби
5.1 Загальна інформація
Реєстраційне досьє має містити інформацію про дослідження з розробки лікарського засобу, проведені для встановлення того, що лікарська форма, склад, виробничий процес, первинне паковання (контейнер і закупорювальний елемент), мікробіологічні характеристики й інструкції для застосування відповідають меті, зазначеній у заявці на реєстрацію. Слід розмежовувати такі дослідження і рутинні контрольні випробування, що проводяться відповідно до специфікацій. Крім того, у реєстраційному досьє необхідно зазначати й описувати склад, а також характеристики процесу (критичні параметри), що можуть впливати на відтворюваність від серії до серії, функціональні характеристики та якість лікарського засобу. Підтверджувальні дані та результати спеціальних досліджень або публікації в літературі можуть бути включені безпосередньо в реєстраційне досьє чи додані до нього. Додаткові підтверджувальні дані можуть бути наведені у вигляді посилань на відповідні частини реєстраційного досьє, що містять матеріали доклінічних або клінічних дослідженьN.
Примітка. У реєстраційному досьє у форматі CTD інформацію з фармацевтичної розробки готового лікарського засобу наводять у розділі 3.2.Р.2 «Pharmaceutical Development» («Фармацевтична розробка»), що складається з наступних пунктів і підпунктів:
3.2.Р.2.1 «Components of the Drug Product» («Компоненти лікарського препарату»):
3.2.P.2.1.1 «Drug Substance» («Лікарська речовина»);
3.2.Р.2.1.2 «Ехсіріеnts» («Допоміжні речовини»);
3.2.P.2.2 «Drug Product» («Лікарський препарат»):
3.2.Р.2.2.1 «Formulation Development» («Розробка складу»);
3.2.P.2.2.2 «Overages» («Надлишки»);
3.2.P.2.2.3 «Physicochemical and Biological Properties» («Фізико-хімічні та біологічні властивості»);
3.2.P.2.3 «Manufacturing Process Development» («Розробка виробничого процесу»);
3.2.P.2.4 «Container Closure System» («Система контейнер/закупорювальний елемент»);
3.2.P.2.5 «Microbiological Attributes» («Мікробіологічні характеристики»);
3.2.P.2.6 «Compatibility» («Сумісність»).
Щоб установити, що вибраний тип лікарської форми та пропонований склад відповідають меті, зазначеній у заявці на реєстрацію, мають бути проведені належні дослідження з фармацевтичної розробки. Метою цих досліджень є також встановлення тих аспектів складу й технологічного процесу, які є найбільш значущими для відтворення серій і які, отже, необхідно контролювати при рутинному виробництві.
З огляду на різноманітність діючих речовин і лікарських форм цю настанову слід розглядати лише як приклад вказівок щодо проведення наукових досліджень, результати яких можуть бути корисними для встановлення чинників, що впливають на якість готового лікарського засобу. З дотриманням загальних принципів, наведених у цій настанові, можуть бути розроблені окремі нормативні документі щодо специфічних видів продукції. Ця настанова розроблена в основному для лікарських засобів, що містять діючі речовини, отримані шляхом хімічного синтезу, але вона може бути застосована також до інших видів продукції. У разі розробки біологічних препаратів, таких як вакцини та лікарські засобі з донорської крові, можуть бути прийнятними альтернативні підходи.
5.2 Компоненти лікарського засобу
5.2.1 Діючі речовини
Вибір діючої речовини (діючих речовин), її (їх) характеристики та кількісний вміст (сила дії) мають відповідати передбачуваній меті; цей вибір необхідно обгрунтувати (експериментально і/або шляхом посилань на відповідні наукові джерела літератури)N.
5.2.1.1 Сумісність компонентів
Слід обговорити сумісність лікарської речовини з допоміжними речовинамиN. За необхідності мають бути надані результати досліджень сумісності діючої речовини (діючих речовин) із допоміжними речовинами. У разі комбінованих лікарських засобів також необхідно розглянути сумісність діючих речовин одна з одною.
Як підтверджувальні дані мають бути надані результати попередніх досліджень стабільності (якщо такі наявні).
5.2.1.2 Фізико-хімічні характеристики
Випробування діючих речовин на етапі, що передує вибору складу, можуть надати корисну інформацію.
Додатково слід обговорити ключові фізико-хімічні характеристики діючих речовин, що можуть вплинути на функціональні характеристики лікарського препарату, у зв’язку з пропонованою лікарською формою та шляхом уведення.
Якщо показано, що фізичний параметр діючої речовини мінливий і критичний для якості лікарського засобу, то його необхідно контролювати відповідним методом із встановленням критеріїв прийнятності в специфікації на діючу речовину чи іншими належними способами. Це може призвести до додаткових випробувань фізичних характеристик діючих речовин, що використовуються в певних лікарських формах (наприклад, тверді лікарські форми), у доповнення до тих випробувань, які проводять для більш простих лікарських форм (наприклад, розчинів), або до тих випробувань, що викладені в фармакопейній монографії.
Прикладами фізичних характеристик, які слід контролювати, є розчинність, вміст води, розподілу часток за розмірами, властивості кристалів, поліморфізм тощо, оскільки:
а) розчинність може впливати на вибір складу й вибір аналітичного методу;
б) вміст води може впливати на інші параметри, такі як властивості кристалів і розмір часток також на стабільність;
в) розподіл часток за розмірами може впливати на біодоступність, однорідність вмісту, властивості суспензії, розчинність, стабільність;
г) властивості кристалів і поліморфізм можуть впливати на розчинність, біодоступність або стабільність.
Очевидно, що ці параметри взаємопов’язані й можуть вимагати комплексного розгляду. Для ключових параметрів, що впливають на біодоступність, мають бути встановлені відповідні межі на підставі результатів досліджень тих серій лікарського засобу, що виявили прийнятні функціональні характеристики в дослідах in vivo.
5.2.2 Допоміжні речовини та інші неактивні компоненти
Вибір допоміжних речовин, включаючи їх концентрацію та характеристики, що можуть вплинути на функціональні характеристики лікарського препарату, має відповідати передбачуваній меті; його слід обговорити беручи до уваги функції допоміжних речовин.
5.2.2.1 Має бути надане пояснення функції кожного компонента в складі лікарського засобу з обгрунтуванням його включення. У деяких випадках можуть бути потрібні експериментальні дані для обгрунтування такого включення, наприклад, для антимікробних консервантів (див. Настанову 42–3.6:2004). При виборі якості допоміжної речовини слід орієнтуватися на її роль у складі лікарського засобу та передбачуваний виробничий процес. У деяких випадках слід розглянути й обгрунтувати кількісний вміст певних допоміжних речовин у складі лікарського препарату.
5.2.2.2 У відповідних випадках має бути встановлена сумісність і раціональність поєднанняN одних допоміжних речовин з іншими (наприклад, комбінація консервантів у подвійній консервуючій системі, тип і концентрація емульгаторівN та ін.). Може бути достатнім надання підтверджувальних даних зі стабільності.
5.2.2.3 Якщо при виробництві лікарського засобу використовуються нові (невідомі) компоненти, наприклад, матриця в лікарській формі з пролонгованим вивільненням, новий пропелент або речовина, що посилює проникність, має бути надана повна інформація про склад і функції цього компонента в складі лікарського препарату разом із документацією, що підтверджує його безпеку.
Примітка. Для нової допоміжної речовини (речовин), що використовується(ються) вперше в лікарському препараті чи при новому шляху введення, у реєстраційному досьє у форматі CTD у п. 3.2.Р.4.6 «Novel Excipients» («Нові допоміжні речовини») розділу 3.2.Р.4 «Control of Excipients» («Контроль допоміжних речовин») необхідно надати повний докладний опис виробництва та контролю, а також характеристику з перехресними посиланнями на дані, що підтверджують безпеку (доклінічні і/або клінічні дослідження) відповідно до тієї ж форми, що й для лікарської речовини. Докладна інформація про нові допоміжні речовини наводиться в розділі 3.2. А. З «Novel Excipients» («Нові допоміжні речовини»).
Нова допоміжна речовина, що вводиться до складу лікарського засобу, підлягає такій само процедурі розгляду, що й нова діюча речовина. При цьому в реєстраційному досьє рекомендується подати всю підтверджувальну інформацію (див. Настанову 42–3.6:2004). Винятком є речовини, раніше дозволені для використання в харчових продуктах (у разі лікарських засобів для орального застосування) або для використання в косметичних препаратах (у разі лікарських засобів для місцевого застосування). Однак стосовно таких допоміжних речовин, що вводяться нетрадиційним шляхом або у високих дозах, можуть знадобитися додаткові дані.
5.3 Лікарські форми
5.3.1 Розробка складу
Слід подати стисле резюме, в якому описується розробка лікарського препарату з урахуванням пропонованого шляху введення чи застосування. Необхідно обговорити відмінності між кожним складом, що використовувався при клінічних випробуваннях, і складом, що реєструється. За необхідності слід обговорити результати порівняльних досліджень in vitro (наприклад, розчинення) або порівняльних досліджень in vivo (наприклад, біоеквівалентність)N. При розробці складу та виборі лікарської форми слід приділити увагу терапевтичній дії, нозології, шляху введення діючої речовини, а також передбачуваному застосуванню лікарського засобу.
5.3.2 Надлишки
Використання надлишків у складі лікарських засобів може бути небажаною практикою внаслідок ризику передозування. Повинні бути обгрунтовані будь-які надлишки в кожному складі лікарського препарату, наведеному у реєстраційному досьєN.
Примітка. У реєстраційному досьє у форматі CTD склад лікарського препарату наводять у розділі 3.2.Р.1 «Description and Composition of the Drug Product» («Опис і склад лікарського препарату»).
Надлишки в основному використовують для заповнення втрат діючих речовин або ключових допоміжних речовин під час виробництва (виробничий надлишок) і/або для продовження терміну зберігання лікарського препарату (надлишок для стабільності). Ці поняття необхідно розмежовувати, оскільки в першому випадку ймовірність того, що пацієнту буде введена підвищена доза, досить мала, тоді як надлишок для стабільності може призвести до передозування, якщо серії лікарського засобу потраплять до пацієнта відразу після випуску. Включення будь-якого надлишку має бути обгрунтоване. Не слід використовувати надлишки у великій кількості (наприклад, понад 10%), щоб приховати нестабільність складу — краще зменшити термін зберігання, ніж піддавати пацієнта ризику отримання лікарського засобу у надмірних дозах. Так само надлишки не слід використовувати, щоб приховати недостатню точність або недостатню правильність аналітичних методик, а також неоптимальний виробничий процес. Уведення надлишку діючої речовини до складу лікарського засобу і/або у виробничу рецептуру слід завжди обґрунтовувати з погляду безпеки та ефективності лікарського препарату. Також необхідно пам’ятати, що надмірна доза може бути зумовлена пристроєм для доставки, наприклад, дозуючим клапаном інгаляційного лікарського засобу.
5.3.3 Фізико-хімічні параметри та біологічні властивостіN
Мають бути описані параметри, що стосуються функціональних характеристик лікарського препарату, такі як рН, іонна сила, розчинення, редиспергування, підготовка до застосування, розподіл часток за розмірами, агрегація, поліморфізм, реологічні властивості, біологічна активність або сила дії і/або імунологічна активністьN.
а) рН
Має бути належним чином вивчений вплив рН у встановлених для даного складу межах з наданням результатів експериментальних досліджень. Слід розглянути вплив рН на діючі речовини та за необхідності на такі допоміжні речовини, як, наприклад, антимікробні консерванти та ін. Результати вивчення рН щодо діючої речовини можуть знадобитися при дослідженні біодоступності лікарських засобів для орального застосування.
Під час вивчення стабільності лікарського засобу необхідно дослідити будь-які ефекти, що залежать від рН, незалежно від часу їх виявлення. Також необхідно розглянути фізіологічні впливи лікарського засобу, пов’язані з рН. Якщо необхідно підтримувати рН у вузькому діапазоні, може знадобитися використання буферних речовин.
б) інші параметри
Під час досліджень із фармацевтичної розробки залежно від складу лікарського засобу також мають бути розглянуті такі параметри, як розчинення та редиспергування, розподіл часток за розмірами, агрегація, реологічні властивості тощо. У складі кожного лікарського засобу для парентерального введення слід приділити увагу таким чинникам, як коригування тонічності, розмір часток дисперсної фази емульсій, розмір і форма твердих часток, включаючи зміну форми кристалів, в’язкість і/або можливість уведення за допомогою шприца* тощо.
*Під можливістю введення за допомогою шприца мають на увазі можливість успішного введення препарату за допомогою шприца та відповідної голки; у певних випадках цю можливість необхідно чітко довести.
5.3.4 Рідкі тa м’які лікарські форми
5.3.4.1 Компоненти лікарської форми
Результати експериментальних досліджень повинні свідчити про те, що кількісний вміст ключових компонентів у складі лікарського засобу є оптимальним для його передбачуваного застосування. Такими компонентами можуть бути:
а) антимікробні консерванти;
б) антиоксиданти;
в) інші речовини, включаючи поверхнево-активні речовини (ПАР), розчинники, комплексоутворювачі, речовини, що підвищують проникність, модифікатори вивільнення тощо.
5.3.4.1.1 У багатодозові лікарські засоби, що не самостерилізуються, може знадобитися введення антимікробних консервантів (див. Настанову 42–3.6:2004), але, як правило, їх не слід включати в стерильні лікарські засоби в пакованнях для разового застосування. При виборі меж кількісного вмісту підхожих консервантів слід приділити увагу таким чинникам, як умови зберігання, підготовка до застосування, розчинення перед застосуванням і частота відкривання паковання. Відповідно до методу випробування, викладеного в ДФУ чи Європейській Фармакопеї, необхідно провести дослідження ефективності консервуючої дії. Вважається, що ефективність такої дії має відповідати критерію А, якщо не обгрунтовано інше (див. ДФУ, загальний текст 5.1.3 «Ефективність антимікробних консервантів», або Європейську Фармакопею, загальний текст 5.1.3 «Efficacy of antimicrobial preservation» («Ефективність антимікробних консервантів»)). Випробування має пройти належну валідацію, включаючи використання відповідного негативного та позитивного контролю, а також вибір підхожих мікроорганізмів для доказу відповідної антибактеріальної та антифунгальної активності.
Для великих паковань, призначених для розділення (розфасування), можуть бути необхідними більш жорсткі випробування. Програма випробувань має дозволяти встановити для лікарського засобу той «термін зберігання під час застосування», який потім буде зазначений на етикетці. Цей період має бути настільки коротким, наскільки це можливо, особливо для стерильних лікарських засобів, наприклад, лікарських засобів для парентерального застосування або очних лікарських засобів.
Більш тривалі терміни зберігання для лікарських засобів у великих пакованнях повинні бути обгрунтовані, при цьому можуть бути необхідні додаткові випробування, що моделюють унесення мікроорганізмів під час використання (див. Настанову 42–3.6:2004).
Мають бути ретельно вибрані розміри паковань; вони повинні відповідати передбачуваній меті й частоті застосування лікарського засобу. Слід вивчити кількісний вміст консервантів протягом терміну зберігання, контролюючи його за відповідними специфікаціями на готовий лікарський засіб.
5.3.4.1.2 Антиоксиданти можуть розкладатися під час виробництва чи протягом терміну зберігання лікарського засобу. Рівень вмісту таких антиоксидантів має бути обгрунтований і підтверджений відповідними експериментальними даними з метою гарантії того, що протягом усього пропонованого терміну зберігання лікарського засобу (включаючи період застосування) підтримується їх достатня активність.
5.3.4.2 Сумісність з іншими лікарськими препаратами
Дослідження сумісності особливо важливе для лікарських засобів, призначених для внутрішньовенного введення.
Якщо в інструкціях, наведених у стислій характеристиці лікарського засобу, в інструкції для застосування та листку-вкладишіN надані вказівки про розчинення і/або розбавлення перед застосуванням, то мають бути наведені дані, що доводять фізичну та хімічну сумісність із рекомендованими розчинниками та матеріалами пристрою для введення протягом рекомендованого чи передбачуваного періоду застосування. Якщо в інструкції для застосування пропонується змішувати лікарський засіб з іншим визначеним лікарським препаратом перед уведенням, то мають бути наведені дані про їх повну сумісність після змішування протягом рекомендованого терміну зберігання у разі застосування при рекомендованій температурі зберігання та вірогідних граничних значеннях кількісного вмісту.
5.3.4.3 ОднорідністьN
Однорідність особливо критична для рідких і м’яких лікарських засобів, що становлять собою гетерогенні дисперсні системи. Слід показати, що склад лікарського засобу і пропонована технологія забезпечують однорідність розподілу діючих і (за необхідності) допоміжних речовин. Збереження однорідності рідких і м’яких лікарських засобів необхідно враховувати при обгрунтуванні пропонованих умов зберігання.
Однорідність, що досягається за допомогою процесу гомогенізації, має бути розглянута на стадії розробки і підтверджена дослідженнями з валідації, що надаються у відповідній частині реєстраційного досьє. Дослідження, проведені на стадії розробки, можуть служити для попередньої підготовки протоколів валідації, що використовуються для повномасштабних процесів гомогенізації при виробництві рідких і м’яких лікарських засобів. Особливу увагу при цьому слід приділити валідації процесу дозування рідких гетерогенних дисперсних систем у зв’язку з можливістю неоднорідності дозування внаслідок розшарування.
Примітка. У реєстраційному досьє у форматі CTD інформацію про валідацію процесу наводять уп. 3.2.Р.3.5 «Process Validation and/or Evaluation» («Валідація процесу і/або його оцінка»).
Для рідких лікарських засобів, що становлять собою гетерогенні дисперсні системи (наприклад, суспензії чи емульсії), для яких згідно зі специфікацією допускається розшарування під час зберігання, мають бути наведені дані з відновлення однорідності розподілу діючих і (за необхідності) допоміжних речовин перед застосуванням, а також дані про збереження однорідності після підготовки до застосування (наприклад, після збовтування).
Для лікарських засобів, що становлять собою гетерогенні дисперсні системи, однорідність і фізична стабільність яких залежить від реологічних параметрів, слід навести дані реологічних досліджень зразків із допустимими відхиленнями в кількісному вмісті ключових допоміжних речовин за рекомендованих умов зберіганняN.
5.3.5 Тверді лікарські форми
У твердих середовищах можливість хімічної несумісності або нестабільності безсумнівно менш значна, ніж у рідких або м’яких. Проте, якщо в стислій характеристиці лікарського засобу, а також в інструкції для застосування та листку-вкладишіN рекомендується розчинення або змішування (наприклад, із напоями) твердих лікарських форм перед застосуванням, необхідно провести відповідні дослідження з їх сумісності.
Відмінності у фізичних властивостях діючих і допоміжних речовин можуть також призводити до їх нерівномірного розподілу та зміни доставки до ділянки-мішені. Отже, необхідні дослідження однорідності та функціональних характеристик твердих лікарських форм стосовно проміжної продукції чи окремих одиниць лікарської форми.
5.3.5.1 Однорідність
Для забезпечення рівномірного розподілу діючої речовини, як правило, необхідні процеси змішування. Відмінності у властивостях поверхні, кристалічності, розмірі часток тощо можуть призвести до розділення порошків у сухій суміші. Однорідність, що досягається за допомогою процесу змішування, має бути розглянута на етапі розробки та підтверджена дослідженнями з валідації, що наводяться у відповідній частині реєстраційного досьє.
Примітка. У реєстраційному досьє у форматі CTD інформацію з валідації процесу наводять у п. 3.2.Р.3.5 «Process Validation and/or Evaluation» («Валідація процесу і/або його оцінка»).
Дослідження, проведені на етапі розробки, можуть служити для попередньої підготовки протоколів валідації, що використовуються для повномасштабних процесів змішування. Для одиниці твердої лікарської форми необхідно довести однорідність розподілу діючої речовини як в різних серіях, так і в одній серії, оскільки визначення вмісту в змішаній пробі не описуватиме розподіл діючої речовини між окремими одиницями лікарської форми. Отже, у специфікації на готовий лікарський засіб, що наведена у відповідній частині реєстраційного досьє, на підставі послідовних досліджень різних серій необхідно передбачити показник «Однорідність вмісту» (див. ДФУ, загальні статті «Таблетки» та «Однорідність вмісту діючої речовини в одиниці дозованого лікарського засобу», а також Настанову 42–3.2:2004).
Примітка. У реєстраційному досьє у форматі CTD специфікацію на готовий лікарський засіб наводять у п. 3.2.Р.5.1 «Specification(s)» («Специфікація(ї)»).
Рутинні випробування повинні підтверджуватися дослідженнями, проведеними на етапі розробки, особливо для лікарських засобів із низьким вмістом сильнодіючих речовин.
Як правило, не слід підтримувати практику застосування половини таблетки; однак якщо такий підхід обгрунтований у реєстраційному досьє, важливо довести збереження однорідності вмісту в половинках таблетки. Необхідно провести випробування ламкості.
5.3.5.2 Випробування функціональних характеристик
Функціональні характеристики можна розглядати як індикатор доставки діючої речовини з лікарської форми до ділянки-мішені; ці характеристики будуть залежати від виду лікарської форми та шляху введення. Вивільнення діючої речовини з лікарської форми може бути швидким (наприклад, із супозиторіїв і таблеток із нерегульованим вивільненням) або модифікованим; модифікація може бути пов’язана зі зміною рівня чи місця вивільнення (пролонговане чи відстрочене вивільнення).
Контроль функціональних характеристик одиниці твердої лікарської форми звичайно пов’язаний із визначенням розпадання лікарського засобу та розчинення діючої речовини у підхожому середовищі (див. ДФУ, загальні статті 2.9.1 «Розпадання таблеток і капсул», 2.9.2 «Розпадання супозиторіїв песаріїв» і «Тест «Розчинення» для твердих дозованих форм»).
5.3.5.2.1 Випробування на розпадання
Випробування на розпадання звичайно слід застосовувати або для кожної готової серії твердих лікарських форм для орального застосування та супозиторіїв, або на проміжній стадії для непокритих ядер таблеток чи інших форм перед нанесенням остаточної оболонки. Таке випробування призначене для доказу ефективного розпадання твердої лікарської форми після застосування (характеристика розпадання). Оскільки багато твердих лікарських форм розпадаються швидко, слід установити конкретне валідоване граничне значення, яке має знаходитися в межах, зазначених у ДФУ чи Європейській Фармакопеї. Рутинні випробування на розпадання можна не проводити, якщо в специфікацію на готовий лікарський засіб включений тест «Розчинення» із прийнятною вибірковістю. Методика випробування має відповідати методиці, описаній у ДФУ чи Європейській Фармакопеї.
5.3.5.2.2 Розчинення
Дійсна кількість діючої речовини, що вивільнилася з лікарської форми в резервуар із водним середовищем in vitro, призначена відображати поведінку лікарського засобу in vivo. На практиці поведінка in vivo залежить від різних чинників, що ускладнюють кореляцію in vivo — in vitro. Проте тест «Розчинення» надає ряд корисних даних; на етапі розробки для всіх твердих лікарських форм у плановому порядку необхідно проводити дослідження характеристик розчинення. На підставі результатів таких досліджень слід прийняти рішення про значущість тесту «Розчинення» стосовно прогнозування поведінки лікарського засобу in vivo.
Прилад для тесту «Розчинення», що використовується при випробуваннях твердих лікарських форм для орального застосування як із нерегульованим, так і з модифікованим вивільненням, має бути одним із тих, які описані в ДФУ чи Європейській Фармакопеї. Якщо доведено, що ці прилади не підходять, то для тесту «Розчинення» може бути вибране інше обладнання. У цьому випадку має бути наведене обгрунтування використання методу, що відрізняється від методу, описаного в ДФУ чи Європейській Фармакопеї.
а) Лікарські препарати з нерегульованим вивільненням.
Під час розробки й досліджень стабільності мають бути проведені дослідження з використанням тесту «Розчинення», щоб встановити, чи є необхідність включати такий тест у специфікацію на готовий лікарський засіб.
б) Лікарські препарати з модифікованим вивільненням.
Необхідно обгрунтувати вибір умов проведення тесту «Розчинення» і ступінь вивільнення для оцінки відтворюваності серій. При цьому необхідно враховувати дослідження in vivo, проведені для встановлення вивільнення та профілю абсорбції лікарського засобу; за можливості має бути проведена кореляція між показниками вивільнення in vitro та результатами in vivo, що дозволить встановити обгрунтовані норми для випробування in vitro.
Така кореляція особливо важлива для лікарських засобів, що містять діючі речовини з вузьким діапазоном терапевтичної дії.
Більш докладна інформація викладена в документі «Note for Guidance on quality of modified release products: A. Oral dosage forms. B. Transdermal dosage forms. Section 1 (Quality)»[2].
5.3.6 Інші лікарські форми
5.3.6.1 Трансдермальні пластири
Трансдермальні пластири — це еластичні лікарські препарати різних розмірів, що містять одну діючу речовину (або декілька діючих речовин) та призначені для аплікації на непошкоджену шкіру з метою доставки діючої(их) речовини (речовин) в системний кровотік.
Такі системи призначені забезпечити доставку діючої речовини крізь непошкоджену шкіру з постійним рівнем системної абсорбції. Для діючих речовин, призначених для введення в трансдермальні системи, вимагається відповідне поєднання фізико-хімічних властивостей, сили дії, біологічної сумісності та клінічної необхідності. Ці аспекти мають бути вивчені та розглянуті під час досліджень із розробки.
Зокрема, необхідно зосередити увагу на матриці-резервуарі та клейких матеріалах, щоб виключити можливу несумісність із діючою речовиною. Мають бути визначені характеристики вивільнення діючої речовини з пластиру з використанням спеціальних дифузійних камер із відповідною та обгрунтованою мембраною-бар’єром, наприклад, використовуючи один з описаних в Європейській Фармакопеї тестів (див. загальну монографію 2.9.4 «Dissolution test for transdermal patches» («Тест розчинення для трансдермальних пластирів»)*). Як у специфікацію на готову продукцію, що застосовують при випуску, так і в специфікацію на готову продукцію, що застосовують протягом терміну зберігання, може бути необхідним включення показників, що характеризують ступінь проникнення крізь мембрану.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.3.6.2 Дозовані лікарські препарати для інгаляцій, що знаходяться під тиском
З погляду бажаної дії необхідно ретельно дослідити розмір часток діючої речовини, що використовується у складі суспензій, а також якість пропонованого пропеленту-співрозчинника та ПАР.
Пропелент може взаємодіяти з діючою речовиною, змінюючи фізичні/хімічні властивості, наприклад, такі як розмір часток, сольватація, кристалічна форма тощо. Отже, на стадії розробки необхідно ретельно дослідити комбінацію цих речовин.
Спеціальні випробування та допустимі критерії прийнятності стосовно дозованих лікарських препаратів для інгаляцій, що знаходяться під тиском, які необхідно ввести в специфікацію на готову продукцію, що застосовують протягом терміну зберігання, визначаються загальною статтею ДФУ «Лікарські засоби, що знаходяться під тиском» і, як правило, включають: перевірку контейнера на герметичність, вимірювання тиску всередині контейнера, визначення відсотка виходу вмісту контейнера, визначення середньої маси лікарського засобу в одній дозі, кількість доз, що витягаються, визначення розміру часток і однорідність дозування. Розмір часток визначають для суспензійних лікарських засобів, призначених для введення в бронхи та легені, а однорідність дозування — для аерозолів у вигляді суспензій або емульсій, що чинять загальну дію на організмN.
Характеристикою лікарської форми, що підлягає дослідженню, може бути також вміст вологи. Нарівні з визначенням середньої маси лікарського засобу в одній дозі, що видається внаслідок дії на механізм дозуючого клапана, і за необхідності однорідності дозування слід визначити кількість діючої речовини, що надходить з клапана та розпилювача. За допомогою приладу, описаного в Європейській Фармакопеї, має бути досліджене осідання випущеної дози з урахуванням можливого осідання номінальної дози лікарського засобу.
Виходячи з результатів цих досліджень, може виникнути необхідність ввести у специфікацію на готовий лікарський засіб показники, що характеризують осідання, і визначати їх під час вивчення стабільності лікарського препарату. Необхідно здійснити спробу виявити кореляцію між результатами випробувань in vitro та in vivo для тих серій, для яких встановлені прийнятні функціональні характеристики в дослідженнях in vivo. Може також знадобитися дослідити осідання лікарського засобу в розпилювачі.
Примітка. Керівні вказівки щодо дозованих лікарських препаратів для інгаляцій, що знаходяться під тиском, можна знайти в документах «Matters relating to the replacement of CFS’s in medicinal products», «Replacement of chlorofluorocarboni (CFS) in metered dose inhalation products», «Note for Guidance on requirements for pharmaceutical documentation for pressured metered dose inhalation products», а також у відповідних загальних монографіях Європейської Фармакопеї, зокрема, «Preparation for Inhalation» («Препарати для інгаляцій») і 2.9.18 «Preparation for Inhalation: Aerodynamic Assessment of Fin; Particles» («Препарати для інгаляцій: аеродинамічна оцінка кінцевих часток»)*.
*Рекомендується користуватися зазначеними документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.3.6.3 Сухі порошки для інгаляцій
Сухі порошки для інгаляцій можуть бути або однодозовими, або багатодозовими. Необхідно розглянути характеристики часток, такі як розмір, форма, шорсткість і заряд, а також реологічні властивості суміші «діюча речовина — допоміжна речовина». Оскільки доставка дози може залежати від ступеня плинності, необхідно дослідити це як in vitro, так і in vivo. Доцільно спробувати провести кореляцію між результатами випробувань in vitro і in vivo тих серій, для яких встановлені прийняті функціональні характеристики in vivo. Потрібно також дослідити осідання лікарського засобу і розпилювачі. Іншим параметром, який доцільно розглянути, є вміст води в суміші «діюча речовина – допоміжна речовина».
Примітка. Керівні вказівки щодо цих лікарських препаратів можна знайти в документі «Note for Guidance on dry powder inhalers», а також у відповідних загальних монографіях Європейської Фармакопеї, зокрема, «Preparation for Inhalation» («Препарати для інгаляцій») та 2.9.18 «Preparation for Inhalation: Aerodynamic Assessment of Fine Particles» («Препарати для інгаляцій: аеродинамічна оцінка кінцевих часток»)*.
*Рекомендується користуватися зазначеними документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.4 Паковальні матеріали
5.4.1 Загальні положення щодо вибору паковальних матеріалів
Слід обговорити придатність первинного паковання (контейнера та закупорювального елемента), описаного у відповідній частині реєстраційного досьє та використовуваного для зберігання, транспортування (перевезення) і застосування лікарського препарату. У цьому обговоренні слід розглянути, наприклад, вибір матеріалів, захист від вологи та світла, сумісність матеріалів конструкції з лікарською формою (включаючи сорбцію контейнером і виділення з контейнера), безпеку матеріалів конструкції та експлуатаційні властивості (такі як відтворюваність дози, що видається пристроєм, який є частиною лікарського препарату)N.
Примітка. У реєстраційному досьє у форматі СТD первинне паковання описується в розділі 3.2.Р.7 «Container Closure System» («Система контейнер/закупорювальний елемент»).
Має бути обгрунтований вибір матеріалів для первинного паковання, включаючи відповідний аналіз безпеки медичного персоналу та пацієнтів при використанні лікарського препарату. У відповідних випадках мають бути проведені необхідні дослідження, що доводять цілість контейнера та закупорювального елемента, беручи до уваги необхідність упаковки, недоступної для дітей, або інші види ізоляції. Необхідно розглянути можливу взаємодію між лікарським засобом і контейнером (див. документ «Plastic primary packaging materials»*). Це стосується також змішування чи розведення лікарських засобів перед уведенням, наприклад, при додаванні лікарського засобу в контейнери великого об’єму з розчинами для інфузій.
При виборі первинних паковальних матеріалів слід також враховувати пропонований метод виробництва. Зокрема, для стерильних лікарських препаратів контейнери слід вибирати такі, що дозволять застосовувати оптимальний метод стерилізації лікарського засобу (див. підрозділ 5.5).
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.4.2 Сорбція контейнером
Мають бути представлені дані, які свідчать про те, що була вивчена можливість сорбції діючих речовин і певних допоміжних речовин із рідких або м’яких лікарських форм, якщо це стосується безпеки чи стабільності. Таке явище відоме для гумових закупорювальних елементів, а також для скляних і пластикових контейнерів і пристроїв для введення. В екстремальних випадках сорбція може призводити до просочення крізь стінки контейнера. Дослідження слід проводити в умовах, що моделюють умови застосування, наприклад, шляхом аналізу лікарського засобу у найвіддаленішій частині контейнера з розчином для інфузій, забезпеченого пристроєм для введення.
5.4.3 Виділення
Мають бути представлені дані, що свідчать про відсутність значного виділення будь-якого компонента первинного паковального матеріалу в рідину чи упаковані окремо по одній одиниці тверді лікарські засоби протягом терміну зберігання, якщо таке виділення може стосуватися безпеки або стабільностіN.
5.4.4 Відтворюваність дози
Якщо використовується дозуючий пристрій (наприклад, краплинна піпетка, дозувальний пристрій для ін’єкцій тощо), то має бути наданий доказ доставки відтворюваної та правильної дози в умовах випробування, які (наскільки можливо) відповідають застосуванню лікарського засобу пацієнтом. Особливу увагу слід приділити правильній підготовці ліофілізованих лікарських засобів у змінних картриджах і можливості отримання в картриджах однорідних суспензій після ресуспендування за умови дотримання рекомендацій, що містяться у листках-вкладишах для пацієнта чи інструкціях для медичного застосуванняN. Особлива увага також має бути приділена правильній підготовці ліофілізованих лікарських засобів у двокамерних картриджах і можливості однорідного ресуспендування суспензій у картриджах за умови дотримання рекомендацій, що містяться у листках-вкладишах для пацієнта чи інструкціях для медичного застосуванняN.
5.5 Виробничий процес
Слід пояснити й обгрунтувати вибір та оптимізацію виробничого процесу, описаного у відповідній частині реєстраційного досьє, зокрема, з урахуванням критичних аспектів. У відповідних випадках має бути пояснений і обгрунтований метод стерилізації.
Необхідно обговорити ті відмінності між процесом (процесами), за допомогою якого вироблені дослідні серії для клінічних випробувань, і виробничим процесом, описаним у реєстраційному досьє, що можуть впливати на функціональні характеристики лікарського препаратуN.
Примітка. У реєстраційному досьє у форматі CTD опис обраного виробничого процесу наводять у п. 3.2.Р.З.З «Description of Manufacturing Process and Process Controls» («Опис виробничого процесу та контролю процесу»), а його критичні етапи — у п. 3.2. Р. 3.4 «Controls of Critical Steps and Intermediates» («Контроль критичних етапів і проміжної продукції»).
Необхідно довести, що вибраний метод придатний для виготовлення даного лікарського засобу, при використанні вихідної сировини відповідної якості. Технологічний процес має давати можливість встановити відповідні специфікації таким чином, щоб можна було забезпечити якість готової продукції. Отже, дослідження з розробки технологічного процесу повинні бути підставою для його оптимізації та встановлення вимог до валідації. Такі дослідження мають бути спрямовані на мікробіологічні, а також на фізичні та хімічні параметри; на підставі результатів цих досліджень слід установлювати необхідність відповідного мікробіологічного контролю лікарського засобу. Особливу важливість має розробка виробничого процесу для біологічних лікарських засобів.
Для лікарських засобів, що мають бути стерильними (наприклад, лікарські засоби для парентерального застосування, очні лікарські засоби чи стерильні лікарські засоби для місцевого застосування), слід вибрати відповідний метод стерилізації; цей вибір повинен бути обгрунтований. Необхідно пам’ятати, що за можливості лікарські засоби слід стерилізувати в остаточному первинному пакованні на заключній стадії, використовуючи цілком валідований метод стерилізації вологим жаром (парою), сухим жаром або іонізуючим випромінюванням відповідно до вимог ДФУ (5.1.1 «Методи приготування стерильних продуктів») або Європейської Фармакопеї (5.1.1 «Methods of preparation sterile products» («Методи приготування стерильних продуктів»)). Якщо стерилізація в остаточному первинному пакованні на заключній стадії неможлива, то при всебічному науковому обгрунтувані можуть бути розглянуті стерилізуюча фільтрація чи проведення процесу в асептичних умовах.
Якщо вибраний метод стерилізації в остаточному первинному пакованні на заключній стадії, відмінний від описаного в ДФУ чи Європейській Фармакопеї, то у реєстраційному досьє слід подати належне пояснення та обгрунтування. Це обгрунтування має включати доказ того, що даний лікарські засіб чутливий до нагрівання — тобто діюча речовина чи певний ключовий компонент лікарського засобу у разі застосування цих умов стерилізації значною мірою розкладається. Проте чутливість до нагрівання паковального матеріалу — недостатнє обгрунтування для відмови від стерилізації в остаточному первинному пакованні термостабільного лікарського засобу. Перш ніж приймати будь-яке рішення про використання методу стерилізації, відмінного від такого в остаточних первинних пакованнях, має бути ретельно вивчене використання альтернативного первинного паковального матеріалу.
5.6 Мікробіологічні характеристики лікарського препаратуN
За необхідності мають бути обговорені мікробіологічні характеристики лікарської форми, включаючи, наприклад, обгрунтування відсутності випробування на мікробіологічну чистоту для нестерильних лікарських препаратів, а також вибір складу та ефективність антимікробних консервантів у лікарських препаратах, які їх містять. Для стерильних лікарських засобів необхідно розглянути цілісність системи контейнер — закупорювальний елемент для запобігання мікробній контамінаціїN.
5.7 Сумісність лікарського препаратуN
Слід обговорити сумісність лікарського препарату з розчинником (ами), який (і) використовується (ються) при його підготовці до застосування, або пристроями для дозування (наприклад, випадання осаду лікарської речовини в розчині, сорбція на ємностях для ін’єкційних лікарських засобів, стабільність), щоб надати відповідну допоміжну інформацію для маркуванняN.
5.8 Висновок
Результати досліджень із фармацевтичної розробки є підставою гарантії того, що лікарський засіб має якість, що відповідає його передбачуваному застосуванню. Належним чином розроблений лікарський засіб, вироблений згідно з принципами та правилами належної виробничої практики (GMP) (див. Настанову 42–01–2001) із використанням належним чином валідованих процесів і методі випробувань, має постійно відповідати необхідній специфікації на готову продукцію. Хоча дослідження з розробки звичайно не входять у сферу контролю інспекцій із GMP, проте вони мають бути проведені відповідно до принципів належної виробничої практики.
На підставі належним чином проведених досліджень із фармацевтичної розробки мають бути складені відповідні специфікації на готову продукцію, що застосовують при випуску і протягом терміну зберігання. Застосування цих специфікацій повинне гарантувати, що лікарський засіб буде мати потрібні показники якості як на момент випуску, так і протягом усього терміну зберігання (див. Настанову 42–3.2:2004).
Додаток А (обов’язковий). Вибір методів стерилізації
Лікарські засоби, що мають бути стерильними, слід стерилізувати в остаточному первинному пакованні на завершальній стадії (кінцева стерилізаціяN), як це зазначено в ДФУ та Європейській Фармакопеї. Якщо неможливо здійснити стерилізацію на завершальній стадії шляхом нагрівання внаслідок нестабільності лікарського препарату, має бути прийняте рішення про використання альтернативного методу стерилізації на завершальній стадії, стерилізуючої фільтрації та/або виробництво в асептичних умовах. Визнано, що для забезпечення ступеня надійності стерилізації (СНС), еквівалентного ступеню, що досягається за допомогою існуючих офіційних методів, можуть бути розроблені нові процеси стерилізації на завершальній стадії, відмінні від процесів, що описані у ДФУ та Європейській Фармакопеї; для таких процесів за умови їх належної валідації можуть бути запропоновані альтернативні підходи.
При просуванні вниз по кожній схемі рішень (рисунки А.1 і А.2) зрозуміло, що використання альтернативних методів, як правило, призводить до зниження ступеня надійності стерилізації. Отже, для якості та безпеки лікарського засобу дуже важливо забезпечити найвищий ступінь надійності стерилізації при найменшому рівні мікробного навантаження до стерилізації. Ці дві схеми рішень (рисунки А.1 і А.2) призначені для допомоги у виборі оптимального методу стерилізації за різних ускладнюючих обставин. (Аналогічний підхід слід застосовувати при виборі методів стерилізації проміжної продукції, яку вводять у готову продукцію при провадженні процесу в асептичних умовах).
Використання невідповідного чутливого до нагрівання паковального матеріалу не може саме по собі бути єдиною причиною вибору виробництва в асептичних умовах. Швидше виробники повинні вибрати найкращий метод стерилізації, що підходить для даного лікарського засобу, а потім підібрати відповідний паковальний матеріал. Однак при виборі паковального матеріалу для даного лікарського засобу можуть враховуватися чинники, не пов’язані з методом стерилізації. У таких випадках ці чинники слід чітко документувати, пояснювати й науково обґрунтовувати в реєстраційному досьє. Звичайно такі чинники, як тип контейнера, шлях уведення та вигідність для пацієнта враховують при виборі конкретного виду контейнера, що не буде витримувати термічної стерилізації на завершальній стадії (наприклад, для деяких очних лікарських засобів) і, отже, такі лікарські засоби виробляють за допомогою валідованих процесів в асептичних умовах. У таких випадках виробники в рамках відповідного часового графіка зобов’язані продовжувати пошук підхожих альтернативних контейнерів, що дозволили б ввести переважний метод стерилізації на завершальній стадії. Комерційні міркування не слід наводити як обгрунтування для відмови від стерилізації на завершальній стадії з можливим найвищим ступенем надійності стерилізації.
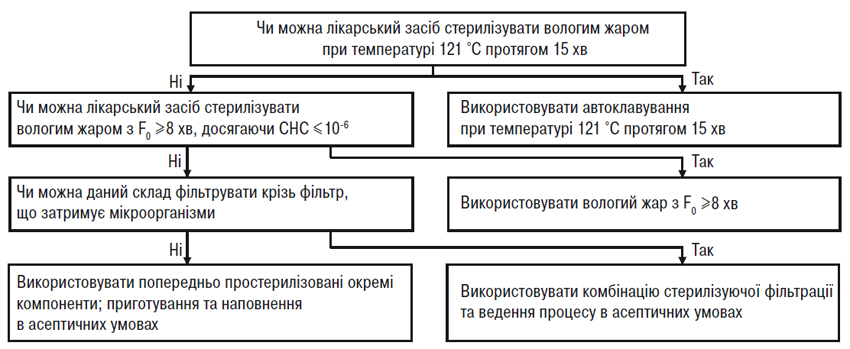
Рисунок А.1 — Схема рішень для вибору методу стерилізації лікарських засобів, що являють собою водні розчини.
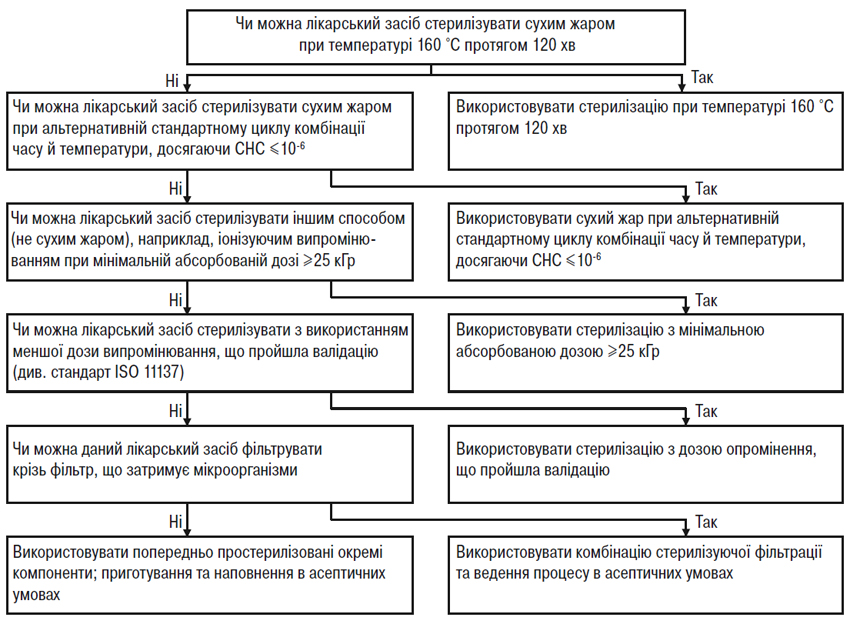
Рисунок А. 2 — Схема рішень для вибору методу стерилізації неводних рідких лікарських засобів, м’яких лікарських засобів та лікарських препаратів у вигляді сухих порошків.
Додаток Б (довідковий). Бібліографія
1 СРМР/ІСН/2736/99 corr (Q1A R) Note for guidance on stability testing: stability testing of new drug substances and products, 2000
(CPMP/ICH/2736/99 corr (Q1A R) Керівні вказівки з випробувань стабільності: випробування стабільності нових лікарських речовин і препаратів, 2000)
2 Good Manufacturing Practice for Active Pharmaceutical Ingredients: Annex 18 to the EU Guide to Good Manufacturing Practice. — Brussel: European Comission, 2001
(Належна виробнича практика для активних фармацевтичних інгредієнтів: Додаток 18 до Настанови ЄС з належної виробничої практики. — Брюссель: Європейська Комісія, 2001)
3 Настанова 42–02–2002 Лікарські засоби. Належна виробнича практика активних фармацевтичних інгредієнтів. — Київ: МОЗ України, 2002)
4 The rules governing medicinal products in the European Union. — V. 2. — Notice to Applicants. — V. 2C. — Regulatory Guidelines. — Guideline on the Categorisation of new Applications (NA) versus Variations Applications (V). — European Comission. — January 2002
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. 2. — Інформація для заявників. — Т. 2С. — Регулятивні настанови. — Настанова стосовно віднесення заявок до категорії нових заявок (НЗ) або заявок на внесення змін (ЗМ). — Брюссель: Європейська Комісія, 2002)
5 Directive 2001/83/ЕС of the European Parliament and of the Council, of 6 November 2001, on the Community code relating to medicinal products for human use//Official Journal of the European Communities, №L311, 28.11.2001, p. 67–128
(Директива 2001/83/EC Європейського парламенту і Ради ЄС від 6 листопада 2001 р. про звід законів Співтовариства відносно лікарських препаратів для людини//Official Journal of the European Communities, № L311, 28.11.2001,p. 67–128)
Ключові слова: діюча речовина, допоміжна речовина, лікарський засіб, лікарська форма, реєстраційне досьє, специфікація, технологічний процес, фармацевтична розробка.