
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ДОСЛІДЖЕННЯ БІОЕКВІВАЛЕНТНОСТІ
СТ-Н МОЗУ 42-7.1:2016
Видання офіційне
Київ
Міністерство охорони здоров’я України
2016
Передмова
1 РОЗРОБЛЕНО: ДП «Державний експертний центр Міністерства охорони здоров’я України».
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: Т. Талаєва, д-р мед. наук, професор; М. Головенко, д-р біол. наук, професор, академік НАМН України; І. Зупанець, д-р мед. наук, професор, М. Ляпунов, д-р фарм. наук, Л. Ковтун, канд. мед. наук; Н. Жукова.
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Міністерство охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 12.01.2017 р. № 22
3 Ця настанова відповідає документам:
- CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» (Настанова щодо досліджень біоеквівалентності), опублікована ЕМА 01.08.2010 р.
- EMA/CHMP/600958/2010/Corr.* «Appendix IV of Guideline on the Investigation of Bioequivalence (CPMP/QWP/EWP/1401/98 Rev. 1): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1», 2011 (Додаток 4 до Настанови щодо досліджень біоеквівалентності (CPMP/QWP/EWP/1401/98 Rev. 1): Представлення даних біофармацевтичних та біоаналітичних досліджень у Модулі 2.7.1, 2011)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО на заміну Настанови 42-7.1:2014 «Лікарські засоби. Дослідження біоеквівалентності»
© Міністерство охорони здоров’я України, 2016
© Державний експертний центр МОЗ України
Національний вступ
Визначення біодоступності та оцінка біоеквівалентності препарату є важливими елементами при розробці та державній реєстрації лікарського засобу. Біодоступність означає швидкість і ступінь, з якою діюча речовина чи її активний компонент абсорбуються з препарату і стають доступними в місці дії. За допомогою даних із біодоступності забезпечується оцінка абсорбованої фракції діючої речовини, а також надається інформація, пов’язана із фармакокінетикою препарату. Біоеквівалентність означає, що за результатами належним чином проведеного дослідження підтверджено відсутність суттєвої різниці у швидкості та ступені, з якими діюча речовина або її активний компонент у фармацевтично еквівалентних чи фармацевтично альтернативних препаратах стають доступними в місці дії препарату при введенні в тій же молярній дозі в аналогічних умовах. Дослідження доказу біоеквівалентності двох препаратів мають важливе значення як на етапі розробки нового препарату, так і в післяреєстраційний період.
Загальний технічний документ (CTD) [1], прийнятий у Європейському Союзі (ЄС), Сполучених Штатах Америки (США), Японії, встановлює вимоги щодо представлення даних досліджень біодоступності та біоеквівалентності у складі реєстраційного досьє на лікарський засіб. Конкретні вимоги щодо представлення цих даних у складі реєстраційного досьє [2, 13] залежать від типу реєстраційної форми лікарського засобу, що подається на державну реєстрацію [3].
Порядком проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення, затвердженого наказом Міністерства охорони здоров’я (МОЗ) України від 26 серпня 2005 р. № 426 (у редакції наказу МОЗ України від 23 липня 2015 р. № 460) (далі — наказ МОЗ № 460), передбачено надання у складі реєстраційного досьє на лікарський засіб даних випробувань, проведених для оцінки біодоступності та/чи біоеквівалентності лікарських засобів [3].
В ЄС введено окрему настанову CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» [4], що містить положення загального методичного підходу до організації та оцінки досліджень біодоступності та біоеквівалентності, визначає вимоги щодо дизайну, проведення та оцінки досліджень біоеквівалентності для лікарських засобів негайного вивільнення системної дії та містить рекомендації щодо процедури біовейвер на підставі біофармацевтичної системи класифікації (далі — БСК). На додаток до загальної настанови в ЄС введено доповнення EMA/CHMP/600958/2010/Corr.* «Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1» [5], в якому описано вимоги щодо представлення біофармацевтичних та біоаналітичних даних у розділі 2.7.1 Модуля 2 реєстраційного досьє на лікарський засіб.
Крім того, в контексті процедур оцінки даних Робоча група з фармакокінетики (Pharmacokinetics Working Party (PKWP)) Європейського агентства з лікарських засобів (ЕМА) вирішує специфічні питання стосовно фармакокінетичних досліджень та відповідні рішення відображає в документі EMA/618604/2008 Rev. «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» [6]. Такі рішення формуються на підставі сучасних наукових знань і нормативних вимог. Питання, що розглядаються в документі [6], у деяких випадках доповнюють інформацію, викладену в настановах [4, 5]. Рішення PKWP необхідно розглядати разом із вимогами настанов [4–6], враховуючи пріоритет останнього документа, що висвітлює конкретну проблему.
З огляду на вищевикладене є необхідність введення в Україні настанови, що містить гармонізовані з положеннями відповідних настанов ЄС та актуалізовані рекомендації щодо планування, проведення та оцінки досліджень біоеквівалентності, а також представлення отриманих даних і результатів дослідження у реєстраційних досьє на лікарські засоби.
Ця настанова розроблена на підставі настанов із клінічної фармакології та фармакокінетики, прийнятих в ЄС, а саме:
- CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» (Настанова щодо досліджень біоеквівалентності) [4].
- EMA/CHMP/600958/2010/Corr.* «Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/Corr.**): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1» (Додаток IV до Настанови щодо досліджень біоеквівалентності (CPMP/EWP/QWP/1401/98 Rev. 1/Corr.**): Представлення біофармацевтичних та біоаналітичних даних у Модулі 2.7.1) [5].
- EMA/618604/2008 Rev. «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» (Питання та відповіді: Позиція щодо специфічних питань, направлених до PKWP) [6].
Ця настанова замінює Настанову 42-7.1:2014 «Лікарські засоби. Дослідження біоеквівалентності».
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я (МОЗ) України.
Ця настанова регулярно переглядатиметься відповідно до змін і доповнень, що вносяться до «Guideline on the Investigation of Bioequivalence» та «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» [4–6].
Настанова містить положення, що відповідають чинному законодавству.
До цієї настанови внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни внесено безпосередньо у пункти, яких вони стосуються; ці зміни позначено іншим шрифтом та літерою N. Зміни і доповнення наведені у національному додатку «Перелік редакційних змін та доповнень» цієї настанови.
Ця настанова є методичними рекомендаціями для планування та проведення досліджень біодоступності та біоеквівалентності, а також оцінки результатів досліджень та представлення відповідних результатів.
Правовий статус цієї настанови відповідає правовому статусу відповідної настанови в ЄС та інших регіонах International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ІСН — Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини), з яким гармонізовано розроблену настанову. Цю настанову слід розглядати як технічний документ, що містить рекомендації для заявників — власників реєстраційних посвідчень, розробників, дослідників, виробників лікарських препаратів, експертних та компетентних уповноважених регуляторних органів та/чи інших заінтересованих осіб щодо виконання положень, визначених законодавством України у сфері обігу лікарських засобів. Ця настанова стосується специфічних наукових питань щодо досліджень біодоступності та біоеквівалентності. Положення цієї настанови відображають гармонізований (у рамках ЄС та ІСН) підхід; вони базуються на останніх наукових досягненнях у цій галузі знань.
У рамках чинного законодавства у сфері обігу лікарських засобів ця настанова не має сили нормативного правового акта, її положення є рекомендаціями. Дотримання положень цієї настанови заінтересованими сторонами сприятиме проведенню досліджень належним чином та полегшить проведення експертизи реєстраційних досьє, а також підвищить якість і безпеку лікарських засобів в Україні. Однак можуть бути застосовані альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових настанов викладено у документі EMA Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005) [10]. Вказаний підхід відповідає позиції Всесвітньої торгової організації (ВТО) відносно застосування стандартів.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Дослідження біоеквівалентності
MEDICINAL PRODUCTS
Investigation of Bioequivalence
Чинна від 2017-01-12
Сфера застосування
Ця настанова визначає положення щодо досліджень біодоступності та біоеквівалентності лікарських засобів. Ця настанова не поширюється на лікарські засоби для ветеринарії.
Ця настанова застосовується для лікарських засобів, що розробляються, досліджуються, реєструються та виробляються в Україні для продажу на внутрішньому ринку та з метою експорту або розробляються, досліджуються, реєструються поза межами України та імпортуються в Україну.
Ця настанова поширюється на планування, проведення та оцінку випробувань і досліджень біодоступності та біоеквівалентності препаратів на етапах фармацевтичної розробки, виробництва, формування реєстраційних досьє та реєстрації, а також внесення змін до матеріалів реєстраційного досьє та під час аудиту або інспектування установ, що проводять відповідні дослідження.
Ця настанова рекомендується для суб’єктів господарювання (далі — організацій), що займаються розробкою препаратів, проведенням досліджень біодоступності та біоеквівалентності, формуванням реєстраційного досьє та проведенням процедур державної реєстрації лікарських засобів на території України, незалежно від відомчого підпорядкування та форми власності, для відповідних заявників і підприємств-виробників, продукція яких реєструється та імпортується в Україну, для науково-експертних організацій та регуляторних органів, а також експертів, аудиторів та інспекторів, що проводять експертизу протоколів клінічних випробувань та відповідних матеріалів, представлених у складі реєстраційного досьє, при реєстрації (перереєстрації) та внесенні змін до реєстраційного досьє лікарських засобів, здійснюють аудит та інспектування.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
- Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use // Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128 (Директива 2001/83/EC Європейського Парламенту та Ради від 6 листопада 2001 р. про кодекс Співтовариства відносно лікарських препаратів, призначених для споживання людьми // Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128).
- The Rules Governing Medicinal Products in the European Union. Notice to Applicants — Vol. 2B. — Common Technical Document. — May 2008. — European Commission. Enterprise and Industry Directorate-General (Правила, що регулюють лікарські препарати в Європейському Союзі. — Т. 2В. — Загальний технічний документ. Європейська комісія. Головний директорат з підприємництва та промисловості).
- The Rules Governing Medicinal Products in the European Union. Notice to Applicants Vol. 2A — Procedures for marketing authorisation. Chapter 1 Marketing authorisation (Правила, що регулюють лікарські препарати в Європейському Союзі. — Т. 2А. — Процедури реєстрації. Розділ 1. Реєстрація).
- EMA/618604/2008 Rev. «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» (Питання та відповіді: Позиція щодо специфічних питань, направлених до PKWP).
- EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
- The Rules Governing Medicinal Products in the European Union. — Vol. 4. — EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Правила, що регулюють лікарські препарати в Європейському Союзі. — Т. 4. — Правила ЄС з належної виробничої практики лікарських препаратів для людини та застосування у ветеринарії).
- CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» (Настанова щодо досліджень біоеквівалентності).
- EMA/CHMP/600958/2010/Corr.* «Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1» (Додаток IV до Настанови щодо досліджень біоеквівалентності (CPMP/EWP/QWP/1401/98 Rev. 1): Представлення біофармацевтичних та біоаналітичних даних у Модулі 2.7.1).
- ICH topic E8, CPMP/ICH/291/95 «General Considerations for Clinical Trials» (Загальні міркування щодо клінічних досліджень).
- ICH E6 (R1), CPMP/ICH/135/95 «Guideline for Good Clinical Practice» (Настанова щодо належної клінічної практики).
- ICH E9, CPMP/ICH/363/96 «Statistical Principles for Clinical Trials» (Статистичні основи клінічних досліджень).
- ICH E3, CPMP/ICH/137/95 «Structure and Content of Clinical Study Reports» (Структура та зміст звіту клінічного дослідження).
- EMEA/CHMP/225411/2006 «CHMP guidance for users of the centralised procedure for generics/hybrid applications» (Настанова комісії з медичних продуктів для людини для використання при заявах на генерик/гібрид за централізованою процедурою).
- Eudralex, Vol. 3, 3CC3a «Pharmacokinetic studies in man» (Фармакокінетичні дослідження на людях).
- СРМР/EWP/280/96 Corr. 1 «Guideline on the pharmacokinetic and clinical evaluation modified release dosage forms» (Настанова з оцінки фармакокінетики та клініки лікарських форм з модифікованим вивільненням).
- CPMP/EWP/240/95 Rev. 1 «Guideline on clinical development of fixed combination medicinal products» (Настанова з клінічної розробки фіксованих комбінацій лікарських препаратів).
- CPMP/EWP/4151/00 Rev. 1 «Requirements for clinical documentation for orally inhaled products (OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of Asthma and Chronic Obstructive Pulmonary Disease (COPD)» (Вимоги до клінічної документації оральних інгаляційних препаратів, включаючи вимоги для доведення терапевтичної еквівалентності двох інгаляційних препаратів, що використовуються для лікування астми та хронічної обструктивної хвороби легень).
- CPMP/EWP/239/95 «Clinical Requirements for Locally Applied, Locally Acting Products containing Known Constituents» (Клінічні вимоги для препаратів місцевої дії та місцевого використання, що містять відомі компоненти).
- CPMP/ICH/367/96 (ICH Topic Q6A) «Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000» (Специфікації: процедури випробувань та критерії прийнятності, 2000).
- European Pharmacopoeia. 8th Ed. European Directorate for the Quality of Medicines (EDQM). — Council of Europe, 67075 Strasbourg Cedex, France 2014. (Європейська Фармакопея, восьме видання).
- Закон України «Про лікарські засоби» від 4 квітня 1996 р.
- Наказ МОЗ України від 26 серпня 2005 р. № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення» (у редакції наказу МОЗ України від 23 липня 2015 р. № 460, що зареєстрований у Міністерстві юстиції України 7 жовтня 2015 р. за № 1210/27655).
- Наказ МОЗ України від 23 вересня 2009 р. № 690 «Про затвердження Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань», що зареєстрований у Міністерстві юстиції України 29 жовтня 2009 р. за № 1010/17026 (зі змінами).
- Наказ МОЗ України від 8 грудня 2015 р. № 830 «Про затвердження і введення в дію Державної Фармакопеї України (ІІ видання)».
Терміни та визначення понять
Нижче наведено терміни, вжиті у цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведено на підставі [4, 12, 13] (див. Національний додаток «Бібліографія»). Визначення цих термінів можуть відрізнятися в інших нормативних документах або терміни можуть мати інші значенняN.
Діюча речовина (активний фармацевтичний інгредієнт, субстанція) (active substance, drug, drug substance, substance [3, 13, 17])
Будь-яка речовина або суміш речовин, що призначена для використання у виробництві лікарського засобу і під час цього використання, стає його активним інгредієнтом. Такі речовини мають фармакологічну чи іншу безпосередню дію на організм людини. У складі готових форм лікарських засобів їх застосовують для лікування, діагностики чи профілактики захворювання, для зміни стану, структур або фізіологічних функцій організму, для догляду, обробки та полегшення симптомів.
Енантіомери (enantiomers, [12])
Сполуки, що мають таку ж молекулярну формулу, як і діюча речовина, але відрізняються від неї просторовим розміщенням атомів у молекулі та не є їх дзеркальним відображенням.
Критерії прийнятності (межі прийнятності) (acceptance criteria, acceptance limits, [12])
Числові межі, інтервали чи інші прийнятні межі прийнятності результатів аналітичних процедур.
Лікарський засіб (medicinal product, [13, 17])
Будь-яка речовина чи комбінація речовин (одного або декількох активних фармацевтичних інгредієнтів (АФІ) та допоміжних речовин), що має властивості та призначена для лікування чи профілактики захворювань у людей, чи будь-яка речовина або комбінація речовин (одного чи декількох АФІ та допоміжних речовин), яка може бути призначена для запобігання вагітності, відновлення, корекції чи зміни фізіологічних функцій у людини шляхом здійснення фармакологічної, імунологічної чи метаболічної дії або для встановлення медичного діагнозу.
До лікарських засобів належать: АФІ, продукція in bulk; готові лікарські засоби (лікарські препарати, ліки, медикаменти); гомеопатичні засоби; засоби, які використовуються для виявлення збудників хвороб, а також боротьби зі збудниками хвороб або паразитами; лікарські косметичні засоби та лікарські домішки до харчових продуктів.
Готові лікарські засоби (лікарські препарати, ліки, медикаменти) (drug product, product, drug, medicine [17])
Дозовані лікарські засоби у вигляді та стані, в якому їх застосовують, що пройшли всі стадії виробництва (виготовлення), включаючи остаточне пакування.
Модифіковане вивільнення (modified release, [12])
Вивільнення з лікарських форм, для яких характеристики вивільнення діючої речовини (а саме часові рамки і/або місце) вибрані таким чином, щоб досягти терапевтичних цілей або зручності застосування. Ці цілі не можуть бути досягнуті при використанні лікарських форм із негайним вивільненням, таких як розчин або лікарська форма з негайним вивільненням. До твердих лікарських форм для орального застосування з модифікованим вивільненням належать лікарські препарати як із відстроченим, так і з пролонгованим вивільненням.
Негайне вивільнення (immediate release, [12])
Вивільнення, при якому лікарський засіб розчиняється у вмісті шлунково-кишкового тракту без відстрочки початку процесу розчинення, пролонгування процесу розчинення чи пролонгування всмоктування діючої речовини.
Сила дії лікарського засобу (дозування) (strenght of the medicinal product, [13])
Вміст діючих речовин у кількісному вираженні на одиницю дози чи одиницю об’єму, чи одиницю маси відповідно до лікарської форми.
Специфікація (specification, [12])
Перелік випробувань, посилань на аналітичні методики та відповідні критерії прийнятності, що становлять числові межі, інтервали чи інші критерії для випробувань, що описуються. У специфікації встановлюють набір критеріїв, яким повинні відповідати діюча речовина чи лікарський препарат для того, щоб вони вважалися придатними для їх передбачуваного застосування. «Відповідність специфікаціям» означає, що діюча речовина і/або лікарський препарат будуть відповідати наведеним критеріям прийнятності за умови, що випробування проведені згідно із зазначеними в цих специфікаціях аналітичними методиками. Специфікації становлять необхідні стандарти якості, які пропонує та обґрунтовує виробник, а погоджують компетентні уповноважені органи.
Фармацевтична еквівалентність (pharmaceutical equivalence, [4])
Лікарські засоби є фармацевтично еквівалентними, якщо вони містять ту ж саму кількість тієї ж самої діючої речовини (діючих речовин) у тих же лікарських формах та відповідають вимогам однакових або порівнянних стандартів.
Фармацевтична еквівалентність не обов’язково передбачає біоеквівалентність, оскільки відмінності у допоміжних речовинах та/чи у процесі виробництва можуть призвести до більш швидкого чи більш повільного розчинення та/чи абсорбції.
Фармацевтичні альтернативи (pharmaceutical alternatives, [4])
Лікарські засоби є фармацевтично альтернативними, якщо вони містять ту саму діючу речовину у вигляді різних солей, ефірів, складних ефірів, ізомерів, сумішей ізомерів, комплексів або похідних діючої речовини або відрізняються лікарською формою чи силою дії.
Познаки та скорочення
|
ANOVA |
– |
analysis of variance (дисперсійний аналіз) |
|
CPMP |
– |
Committee for Medicinal Products for Human Use (Комітет з лікарських засобів для людини) |
|
CRF |
– |
case report form (індивідуальна реєстраційна форма) |
|
CTD |
– |
Common Technical Document (загальний технічний документ) |
|
CV |
– |
coefficient of variation (коефіцієнт варіації) |
|
EEA |
– |
European Economic Area (Європейська економічна зона) |
|
EMA |
– |
The European Medicine Agency (Європейське агентство з лікарських засобів) |
|
EU (ЄС) |
– |
European Union (Європейський Союз) |
|
FCs |
– |
fixed combinations (фіксовані комбінації, лікарські засоби з фіксованою комбінацією) |
|
GLP |
– |
Good Laboratory Practice (належна лабораторна практика) |
|
GMP |
– |
Good Manufacturing Practice (належна виробнича практика) |
|
HVDP |
– |
highly variable drug products (високоваріабельні лікарські препарати) |
|
ICH |
– |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини) |
|
IS |
– |
internal standard (внутрішній стандарт) |
|
MF |
– |
matrix factor (матричний фактор) |
|
NTID |
– |
narrow therapeutic index drug (лікарський препарат із вузьким терапевтичним індексом) |
|
Ph.Eur. |
– |
European Pharmacopeia (Європейська Фармакопея) |
|
PKWP |
– |
Pharmacokinetics Working Party (Робоча група з фармакокінетики) |
|
QC |
– |
quality control sample (зразок контролю якості) |
|
QC-media (medium) |
– |
quality control media (середовище, призначене для контролю якості лікарського засобу) |
|
RSD |
– |
relative standard deviation (відносне стандартне відхилення) |
|
SmPC |
– |
summary of product characteristics (коротка характеристика лікарського засобу) |
|
Within-subject CV |
– |
within-subject coefficient of variation (внутрішньосуб’єктна варіабельність) |
|
АФІ |
– |
активний фармацевтичний інгредієнт |
|
БСК |
– |
біофармацевтична система класифікації |
|
ДФУ |
– |
Державна Фармакопея України |
|
МОЗ |
– |
Міністерство охорони здоров’я |
|
СТ-Н МОЗУ |
– |
Настанова МОЗ України |
|
США |
– |
Сполучені Штати Америки |
Фармакокінетичні параметри
|
Ae(0–t) |
– |
загальна екскреція з сечею незміненої діючої речовини лікарського засобу з моменту введення лікарського засобу до останньої точки, що визначається (t) |
|
AUC(0–t) |
– |
площа під кривою концентрація/час з моменту введення лікарського засобу до останньої точки, що визначається (t) |
|
AUC(0–∞) |
– |
площа під кривою концентрація/час, екстрапольована до нескінченності |
|
AUC(0–τ) |
– |
площа під кривою концентрація/час у стаціонарному стані |
|
AUC(0–72 ч) |
– |
площа під кривою концентрація/час з моменту введення лікарського засобу до 72-ї години |
|
Сmах |
– |
максимальна концентрація аналітуN |
|
Сmах, ss |
– |
максимальна концентрація аналітуN у стаціонарному стані |
|
Rmax |
– |
максимальна швидкість екскреції з сечею |
|
tmax |
– |
час досягнення Сmах |
|
tmax, ss |
– |
час досягнення Сmах, ss |
|
T½ (t½) |
– |
період напіввиведення |
|
λz |
– |
константа швидкості елімінації |
|
Залишкова площа (Residual area) |
– |
екстрапольована площа (AUC(0–∞) — AUC(0–t))/AUC(0—∞) |
Загальні положення
Ця настанова встановлює вимоги до дизайну, проведення та оцінки досліджень біоеквівалентності для лікарських форм негайного вивільнення системної дії.
1. Вступ
1.1. Основні положення
Два лікарських засоби, що містять однакову діючу речовину, вважаються біоеквівалентними, якщо вони є фармацевтично еквівалентними або фармацевтично альтернативними, а їх біодоступність (швидкість і ступінь абсорбції) після введення в однаковій молярній дозі знаходиться в попередньо встановлених критеріях прийнятності. Ці критерії встановлюються для забезпечення порівняння характеристик лікарських засобів in vivo, тобто для встановлення подібності щодо безпеки та ефективності.
Для оцінки величини та ступеня абсорбції діючої речовини в дослідженнях біоеквівалентності зазвичай використовують криву залежності концентрації від часу. Вибрані фармакокінетичні параметри та попередньо встановлені критерії прийнятності дають змогу зробити заключний висновок про біоеквівалентність порівнюваних лікарських засобів. AUC (площа під кривою концентрація/час) відображає ступінь впливу. Сmax (максимальна концентрація в плазмі крові або максимум впливу) та tmax (час досягнення максимальної концентрації в плазмі крові) є параметрами, на які впливає швидкість абсорбції.
Мета цієї настанови — встановлення вимог до дизайну, проведення та оцінки досліджень біоеквівалентності. Також розглядається можливість використання даних in vitro замість досліджень in vivo.
1.2. Генеричні лікарські засоби
Концепція біоеквівалентності є основною для генеричних лікарських засобів відповідно до статті 10(1) Директиви 2001/83/ЕС [13] та розділу ІІІ наказу № 460 [3]N. Метою встановлення біоеквівалентності лікарських засобів є доведення еквівалентності біофармацевтичної якості генеричного та референтного лікарських засобів, для того щоб екстраполювати результати доклінічних і клінічних досліджень, проведених для референтного лікарського засобу, на генеричний лікарський засіб. Генеричним лікарським засобом є лікарський засіб, що має такий самий якісний та кількісний склад діючих речовин і таку саму лікарську форму, що й референтний лікарський засіб, та чия біоеквівалентність із референтним лікарським засобом доведена відповідними дослідженнями біодоступності [13]. Різні солі, прості та складні ефіри, ізомери, суміші ізомерів, комплекси або похідні діючої речовини вважаються тією ж діючою речовиною за умови, що вони суттєво не відрізняються за властивостями щодо безпеки та/чи ефективності. Крім того, різні лікарські форми негайного вивільнення орального застосування можуть розглядатися як одна й та ж сама лікарська форма.
1.3. Інші типи заяв
Представлення даних досліджень біоеквівалентності у реєстраційному досьє на лікарський засіб може вимагатися для лікарських засобів із фіксованою комбінацією, гібридних лікарських засобів, а також представлення даних досліджень біоеквівалентності може бути необхідне при внесенні змін до матеріалів реєстраційного досьє на лікарський засіб протягом дії реєстраційного посвідчення, включаючи зміни, що потребують нової реєстрації лікарського засобу [3].
Наведені в цій настанові рекомендації щодо дизайну та проведення досліджень біоеквівалентності можуть також застосовуватися до порівняльних досліджень біодоступності при дослідженні різних лікарських форм (складів) під час розробки нового лікарського засобу, що містить нову хімічну сполуку, а також до порівняльних досліджень із біодоступності при заявах на зміни, що потребують нової реєстрації лікарського засобу, або для гібридних лікарських засобів [3], що ґрунтуються не лише на даних із біоеквівалентності.
2. Сфера застосування
Ця настанова містить рекомендації щодо досліджень біоеквівалентності лікарських засобів негайного вивільнення системної дії. Цією настановою також передбачаються умови, при дотриманні яких не вимагається проведення дослідження біодоступності (у разі додаткового дозування лікарського засобу, див. розділ 4.1.6 цієї настанови; у разі певних типів лікарських форм, див. Додаток ІІ до цієї настанови; у разі можливості застосування процедури біовейвер на підставі БСК, див. Додаток ІІІ до цієї настанови).
Особливі рекомендації щодо досліджень біоеквівалентності препаратів із модифікованим вивільненням, трансдермальних препаратів та оральних інгаляційних препаратів наведено в інших настановах (див. розділ 3 та розділ «Нормативні посилання» цієї настанови).
Сфера застосування цієї настанови обмежується хімічними сполуками. Рекомендації щодо порівняння біологічного лікарського засобу з референтним лікарським засобом наведено в інших настановах для подібних біологічних лікарських засобів.
Ця настанова не застосовується у тому разі, якщо біоеквівалентність не можна продемонструвати, використовуючи концентрації діючої речовини чи в разі обґрунтування її метаболітуN. У таких виняткових випадках проводять дослідження лікарського засобу з фармакодинамічними або клінічними кінцевими точками згідно з вимогами відповідних настанов щодо терапевтичного застосування.
Хоча концепція біоеквівалентності, ймовірно, може бути застосовна до рослинних лікарських засобів, основні принципи, викладені в цій настанові, не застосовуються до рослинних лікарських засобів, діючі речовини яких не так добре охарактеризовані, як хімічні сполуки.
Крім того, ця настанова не включає аспекти, пов’язані із заміною генеричного лікарського засобу, оскільки це є питанням національного регулювання. В Україні вимоги щодо доведення біоеквівалентності генеричних лікарських засобів викладено в наказі № 460 [3]N.
3. Законодавча база
Ця настанова стосується матеріалів реєстраційного досьє для лікарських засобів для людини, поданих на державну реєстрацію як генеричні лікарські засоби [3]. Ця настанова також може застосовуватися при підготовці матеріалів реєстраційного досьє для лікарських засобів для людини, поданих на державну реєстрацію як лікарський засіб за повним досьє, лікарський засіб із фіксованою комбінацією, гібридний лікарський засіб, та, крім того, може застосовуватися у разі змін, що потребують нової реєстрації лікарського засобу, та змін, можливих протягом дії реєстраційного посвідчення [3].
Розглядаючи цю настанову, необхідно враховувати вимоги до реєстраційного досьє у форматі CTD [3], а також європейські та настанови ICH щодо проведення клінічних випробувань та гармонізовані з ними настанови СТ-Н МОЗN.
Також ця настанова розглядається в поєднанні з відповідними настановами з якості та чинними рекомендаціями ЄС стосовно досліджень біодоступності та біоеквівалентності [6]N. Лікарський засіб, що використовувався для дослідження біоеквівалентності, має бути виготовлений відповідно до норм GMP [11, 18].
Дослідження біоеквівалентності, що проводилися в EU/EEA (European Economic Area — Європейська економічна зона), мають бути проведені відповідно до Директиви 2001/20/ЕС [14]. Дослідження, що проводилися за межами ЄС та призначені для реєстрації в EU/EEA, мають бути проведені відповідно до стандартів, викладених у Додатку І до Директиви 2001/83/ЕС зі змінами [13].
Компанії можуть також звертатися за науковою консультацією до EMA для специфічних запитів, не охоплених чинними настановами. В Україні передбачено консультації відповідно до вимог наказу № 460 [3] та наказу № 690 [15]N.
4. Основний текст настанови
4.1. Дизайн, проведення та оцінка досліджень біоеквівалентності
Кількість досліджень та їх дизайн мають бути відповідно обґрунтовані та залежати від фізико-хімічних характеристик діючої речовини, її фармакокінетичних властивостей та пропорційності складу лікарського засобу. Зокрема, слід враховувати лінійність фармакокінетики, необхідність проведення дослідження після вживання їжі та натще, потребу проведення енантіоселективного аналізу, а також відмови від проведення дослідження біоеквівалентності in vivo для додаткових дозувань (див. розділи 4.1.4, 4.1.5 та 4.1.6 цієї настанови).
У розділі 2.7.1 Модуля 2 реєстраційного досьє слід навести перелік усіх основних досліджень, проведених для заявленого лікарського засобу, наприклад досліджень біоеквівалентності заявленого лікарського засобу (тобто однаковий склад лікарського засобу і виробничий процес) із референтним лікарським засобом, що є на ринку ЄС або іншим обґрунтованим, відповідно до вимог наказу № 460 [3], референтним лікарським засобомN. Інформацію щодо дослідження слід включати в перелік усіх основних досліджень, незалежно від їх результатів. Для всіх проведених досліджень необхідно надавати повні звіти досліджень, за винятком пілотних досліджень, для яких достатньо надати короткий огляд дослідження (відповідно до настанови ICH E3 [19] та чинного законодавства України [15]N). Повний звіт пілотних досліджень слід надавати за запитом. До розділу 2.7 Модуля 2 реєстраційного досьє має бути також включено короткий огляд досліджень біоеквівалентності або порівняльних досліджень біодоступності, проведених протягом періоду розробки лікарського засобу. Дослідження біоеквівалентності заявленого лікарського засобу з не схваленим в ЄС референтним лікарським засобом (non-EU reference products [2]N) не слід подавати та включати у список досліджень (у разі подачі заявки в ЄСN).
4.1.1. Дизайн дослідження
Дослідження має бути сплановано таким чином, щоб ефект, зумовлений лікарським засобом, можна було відрізнити від впливу інших факторів.
Стандартний дизайн
Якщо порівнюються два препарати, рекомендований рандомізований перехресний дизайн з однократним введенням дози, що включає два періоди та дві послідовності. Періоди введення лікарського засобу мають бути розділені періодом відмивання, достатнім для забезпечення концентрації діючої речовини, нижчої межі кількісного визначення біоаналітичної методики у всіх суб’єктів на початку другого періоду дослідження. Як правило, для цього необхідно не менше п’яти періодів напіввиведення (T½).
Альтернативний дизайн
За певних обставин та за умови, що дизайн дослідження і статистичні аналізи науково обґрунтовані, як альтернативу можна розглянути інші добре вивчені дизайни дослідження, такі як паралельний дизайн — для речовин із дуже тривалим T½, та повторний дизайн, наприклад, для речовин із високоваріабельними фармакокінетичними характеристиками (див. розділ 4.1.10 цієї настанови).
Якщо дослідження з однократним введенням дози лікарського засобу не може бути проведене за участю здорових добровольців у зв’язку з переносимістю та якщо проведення такого дослідження неможливе для пацієнтів, допустиме проведення дослідження з багатократним введенням дози препарату пацієнтам.
У рідкісних випадках, коли недостатня чутливість аналітичної методики перешкоджає досить точним визначенням концентрації аналіту в плазмі крові після однократного введення дози препарату і коли концентрації у стаціонарному стані досить високі, щоб бути достовірно виміряними, як альтернатива дослідженню з однократним введенням дози препарату може бути прийнятним дослідження з багатократним введенням дози препарату. Однак, оскільки дослідження з багатократним введенням дози препарату є менш чутливим для визначення відмінностей у Cmax, такий дизайн дослідження може буде прийнятним тільки у разі, якщо заявник може адекватно обґрунтувати, що чутливість аналітичної методики не може бути поліпшена, і неможливо достовірно виміряти концентрацію вихідної сполуки після однократного введення дози препарату, враховуючи також, що у дослідженнях біоеквівалентності допустимо застосовувати дози, які перевищують терапевтичні (див. також розділ 4.1.6 цієї настанови). Враховуючи останні розробки у біоаналітичній методології, малоймовірно, що вихідна речовина не може бути виміряна точно і правильно. Тому використання дизайну дослідження з багатократним введенням дози препарату замість дослідження з однократним введенням дози у зв’язку з недостатньою чутливістю аналітичної методики може бути прийнятним лише у виняткових випадках.
При дослідженнях у стаціонарному стані період відмивання після введення дози препарату у перший період може накладатися на період встановлення стаціонарного стану при введенні другої дози препарату за умови, що цей період є досить тривалим і становить не менше п’яти T½.
4.1.2. Референтний та досліджуваний лікарські засоби
Референтний лікарський засіб
У реєстраційних досьє на генеричні та гібридні лікарські засоби обов’язково має бути посилання на референтний лікарський засіб, зареєстрований на підставі повного досьє за одним із таких типів форм про державну реєстрацію лікарського засобу відповідно до вимог наказу № 460 [3]N: лікарський засіб за повним досьє, лікарський засіб із добре вивченим медичним застосуванням, лікарський засіб із фіксованою комбінацією, за інформованою згодою (статті 8(3), 10а, 10b та 10с Директиви 2001/83/ЕС зі змінами [13]). Лікарський засіб, що застосовується як референтний лікарський засіб для дослідження біоеквівалентності, має відповідати вимогам наказу № 460 [3]N з урахуванням вимог пункту 6(1) другого параграфу Директиви 2001/83/ЕС зі змінами [13]. Відповідно до пункту 6(1) другого параграфу Директиви 2001/83/ЕС, лікарський засіб, що застосовується як референтний лікарський засіб, має бути частиною основної ліцензії на референтний лікарський засіб, включаючи його додаткові дози, лікарські форми, шляхи введення, маркування, які були проведені як зміни до реєстраційного досьє або зміни, що потребували нової реєстраціїN. Вибраний референтний лікарський засіб, що використовувався при доведенні біоеквівалентності шляхом проведення відповідних досліджень біодоступності, зазначається заявником в пункті 1.2 Модуля 1 (форма заяви про державну реєстрацію лікарського засобу), а обґрунтування його вибору надається в розділі 1.5.2 «Інформація щодо генеричних, гібридних або подібних біологічних лікарських засобів» Модуля 1 реєстраційного досьє.
У заяві про державну реєстрацію генеричного чи гібридного лікарських засобу або при змінах, що потребують нової реєстрації лікарського засобу, який було спочатку зареєстровано як генеричний або гібридний лікарський засіб [3], досліджувані препарати, як правило, порівнюються з відповідною лікарською формою референтного лікарського засобу, що доступний на ринках світу.
При змінах, що потребують нової реєстрації лікарського засобу, зареєстрованого за повним досьє [3]N (стаття 8(3) Директиви 2001/83/ЕС), якщо на ринку є декілька лікарських форм цього лікарського засобу, рекомендується, щоб лікарська форма, що використовувалася для первинної реєстрації лікарського засобу (яка використовувалась у клінічних дослідженнях ефективності та безпеки), використовувалась би як референтний лікарський засіб за умови, що вона доступна на ринку.
Вибір референтного лікарського засобу для дослідження біоеквівалентності є обов’язком заявника і має базуватися на результатах аналізу кількісного вмісту діючої речовини та даних розчинення лікарського засобу. Якщо не обґрунтовано інше, кількісний вміст діючої речовини в серії досліджуваного лікарського засобу не повинен відрізнятися більше ніж на 5% від кількісного вмісту діючої речовини в серії референтного лікарського засобу. Кількісний вміст діючої речовини в серії досліджуваного і референтного лікарських засобів має визначатися за допомогою методики аналізу, що використовується для рутинного контролю якості досліджуваного лікарського засобу. Заявник за допомогою кількісного аналізу вмісту діючої речовини і даних розчинення лікарських засобів повинен документально підтвердити вибір репрезентативної серії референтного лікарського засобу. При виборі серії референтного лікарського засобу для використання у дослідженні біоеквівалентності рекомендується дослідити більше ніж одну серію референтного лікарського засобу.
Досліджуваний лікарський засіб
Досліджуваний лікарський засіб, що використовується для дослідження біоеквівалентності, повинен бути репрезентативним до лікарського засобу, який буде реалізовуватися на ринку, що має бути вивчено та обґрунтовано заявником.
Наприклад, для оральних твердих лікарських форм системної дії:
а) досліджуваний лікарський засіб, як правило, слід відбирати із серії, розмір якої становить щонайменше 1/10 від промислової серії або 100 тис. одиниць, залежно від того, який із об’ємів більший, якщо заявником не обґрунтовано інше.
b) процес виробництва серій досліджуваного лікарського засобу, що використовуватимуться в дослідженні, має забезпечувати високий рівень гарантії того, що препарат і процес виробництва будуть відтворені у промисловому масштабі. Якщо об’єм промислової серії становить менше 100 тис. одиниць, для дослідження біоеквівалентності необхідно використовувати промислову серію повного обсягу.
c) характеристика та вимоги специфікації щодо критичних показників якості лікарського препарату, таких як розчинення, мають бути встановлені для досліджуваної серії, тобто серії, для якої була доведена біоеквівалентність.
d) додаткові пілотні та/чи промислові серії лікарського засобу, що подаються з метою проведення відповідної реєстраційної процедури, необхідно порівнювати зі зразками серії досліджуваного лікарського засобу, для якої доведена біоеквівалентність; вони мають демонструвати подібність профілів розчинення in vitro за прийнятних умов проведення випробування на розчинення (див. Додаток І до цієї настанови).
Порівняльні дослідження профілів розчинення необхідно провести для перших трьох промислових серій.
Якщо промислові серії в повному обсязі недоступні на момент подачі матеріалів досьє, заявник не має реалізовувати серію, поки не буде завершено порівняльне дослідження профілів розчинення.
Результати порівняльних досліджень профілів розчинення перших трьох промислових серій лікарського засобу мають бути надані заявником до компетентного органу за вимогою чи при отриманні результатів дослідження профілів розчинення, що не демонструють їх подібність (разом із запропонованим планом заходів).
Для інших лікарських форм системної дії з негайним вивільненням обґрунтування репрезентативності серії досліджуваного лікарського засобу необхідно проводити аналогічно.
Упаковка досліджуваних лікарських засобів
Референтний та досліджуваний лікарські засоби слід упаковувати індивідуально для кожного суб’єкта та періоду дослідження. Препарати можуть бути упаковані до їх відправки у місце дослідження чи безпосередньо у місці проведення дослідження. Пакування (включаючи маркування) має бути проведено відповідно до вимог належної виробничої практики (GMP), включаючи Додаток 13 до відповідної настанови ЕС [11] та гармонізованої з ним настанови СТ-Н МОЗУ 42-4.0:2016 [18]N. За необхідності та відповідно до національних стандартів місце виробництва повинно мати ліцензію на виробництво досліджуваних лікарських засобів, за винятком ситуацій, коли необхідна підготовка до застосування препарату або пакування та цей процес здійснюються безпосередньо в місці проведення дослідження фахівцями, що мають повноваження [14–16]. Усі країни, в яких проводяться дослідження, повинні мати стандарти не нижче вимог настанови ЕС [11] та гармонізованої з нею настанови СТ-Н МОЗУ 42-4.0:2016 [18]N.
Слід гарантувати можливість безпомилкової ідентифікації препарату, введеного кожному суб’єкту в кожний період дослідження. Тому упаковку, маркування і введення препаратів суб’єктам необхідно детально документувати. Така документація має включати опис усіх застережних заходів для запобігання та виявлення потенційних помилок із дозуванням. Рекомендується використовувати етикетки з відривною частиною.
4.1.3. Суб’єкти дослідження
Кількість суб’єктів
Кількість суб’єктів, залучених у дослідження, має ґрунтуватися на відповідному розрахунку розміру вибірки. Мінімальна кількість суб’єктів, що оцінюються у дослідженні біоеквівалентності, повинна становити не менше 12 осіб.
Вибір суб’єктів
Група суб’єктів для дослідження біоеквівалентності має бути відібрана таким чином, щоб була забезпечена можливість виявлення відмінностей між препаратами. Для зменшення варіабельності, не пов’язаної з відмінностями між препаратами, дослідження, як правило, слід проводити за участю здорових добровольців, крім випадків, коли введення препарату неприйнятне для здорових добровольців із причин безпеки і призводить до того, що дослідження стає неетичним. У більшості випадків для виявлення відмінностей між препаратами модель дослідження in vivo за участю здорових добровольців є прийнятною та дає змогу екстраполювати результати на інші популяції, для яких схвалено застосування референтного лікарського засобу (люди літнього віку, діти, пацієнти з нирковою або печінковою недостатністю тощо).
Критерії включення/виключення суб’єктів мають бути чітко зазначені у протоколі дослідження. Суб’єкти дослідження мають бути віком 18 років та старші й переважно мати індекс маси тіла від 18,5 до 30 кг/м2.
Скринінг суб’єктів дослідження щодо можливості їх участі у дослідженні слід проводити за даними клінічних лабораторних тестів, історії хвороб та медичного обстеження. Залежно від фармакотерапевтичної групи, до якої належить лікарський засіб, та його профілю безпеки може знадобитися проведення спеціальних медичних досліджень та вжиття застережних заходів до, під час і після завершення дослідження. Суб’єкти дослідження можуть бути будь-якої статі; однак у кожному конкретному випадку слід враховувати ризик для жінок репродуктивного віку. Бажано, щоб суб’єкти дослідження не палили і не мали в анамнезі алкогольної залежності чи залежності від лікарських засобів. У деяких випадках із міркувань безпеки або з фармакокінетичних причин необхідно передбачати фенотипування та/чи генотипування суб’єктів дослідження.
При паралельному дизайні дослідження групи суб’єктів мають порівнюватися за всіма відомими показниками, що можуть вплинути на фармакокінетику діючої речовини (наприклад, вік, маса тіла, стать, етнічне походження, тютюнопаління, прискорений/уповільнений метаболізм). Це є важливою передумовою для отримання достовірних результатів у таких дослідженнях.
Якщо відомо, досліджувана діюча речовина має побічні ефекти, а фармакологічні ефекти або ризики вважаються неприйнятними для здорових добровольців, як суб’єкти в дослідження можуть бути включені пацієнти (хворі), за умови вжиття необхідних застережних заходів та встановлення відповідного нагляду.
4.1.4. Проведення дослідження
Стандартизація
Щоб звести до мінімуму варіабельність усіх пов’язаних із випробуванням факторів, за винятком тих, що зумовлені досліджуваними лікарськими засобами, умови проведення дослідження необхідно стандартизувати. Тому рекомендовано стандартизувати дієту, вживання рідини і фізичне навантаження.
Має бути встановлено час доби прийому їжі. Суб’єкти дослідження не повинні вживати їжу щонайменше за 8 год до прийому лікарських засобів, якщо не обґрунтовано інше. Оскільки споживання рідини може вплинути на проходження через шлунок лікарських форм для орального застосування, досліджуваний та референтний лікарські засоби слід запивати стандартизованим об’ємом рідини (не менше 150 мл). Рекомендовано пити воду за бажанням, за винятком 1 год до і після прийому лікарського засобу; вживання їжі заборонено щонайменше протягом 4 год після прийому лікарського засобу. Їжа, що вживається після прийому препарату, має бути стандартизована за складом і часом прийому протягом достатнього періоду (наприклад, протягом 12 год).
Якщо дослідження має проводитися після вживання їжі, прийом лікарського засобу щодо їжі рекомендується здійснювати відповідно до SmPC референтного лікарського засобу. Якщо в SmPC референтного лікарського засобу відсутні специфічні вимоги, рекомендується, щоб суб’єкти почали вживати їжу за 30 хв до прийому лікарського засобу і тривалість вживання цієї їжі має становити 30 хв.
Оскільки біодоступність діючої речовини при застосуванні лікарської форми може залежати від тривалості проходження через шлунково-кишковий тракт та від інтенсивності регіонарного кровотоку, може знадобитися стандартизування положення тіла і фізичного навантаження суб’єкта дослідження.
Протягом відповідного періоду до і під час дослідження суб’єкти мають утримуватися від вживання їжі та напоїв, що можуть вплинути на функції серцево-судинної системи та шлунково-кишкового тракту, функцію печінки або нирок (наприклад, алкогольних напоїв або певних фруктових соків, таких як грейпфрутовий сік). Суб’єктам не слід приймати будь-які інші супутні лікарські засоби (включаючи рослинні лікарські засоби) протягом відповідного періоду до і під час дослідження. Проте застосування протизаплідних засобів дозволено. Якщо не можна уникнути прийому супутніх лікарських засобів і суб’єкти приймають ці засоби, наприклад, для усунення таких побічних ефектів, як головний біль, їх застосування обов’язково необхідно зареєструвати (із зазначенням дози та часу введення) та дослідити можливий їх вплив на результат дослідження. У рідкісних випадках із міркувань безпеки або переносимості усім суб’єктам дослідження призначають супутні лікарські засоби (наприклад опіоїдні антагоністи, протиблювотні засоби). У цьому разі обов’язково слід дослідити ризик впливу потенційної лікарської взаємодії чи біоаналітичної інтерференції на результати дослідження.
Лікарські засоби, що, відповідно до SmPC референтного лікарського засобу, застосовують виключно в комбінації з іншим препаратом (наприклад, певні інгібітори протеази приймають виключно у комбінації з ритонавіром), можуть досліджуватися або у зазначеній комбінації, або без застосування супутнього лікарського засобу.
У дослідженнях біоеквівалентності ендогенних речовин слід по можливості контролювати фактори, що можуть вплинути на базовий рівень ендогенних речовин (наприклад, суворий контроль дотримання дієти).
Час відбору проб
Для адекватного опису профілю концентрація/час необхідно відбирати достатню кількість проб. Для забезпечення достовірної оцінки максимального впливу (піка експозиції) слід передбачати частий відбір проб у період, максимально наближений до прогнозованого tmax. Зокрема, графік відбору проб має бути побудований таким чином, щоб уникнути ситуації, при якій Сmax стане першою точкою на кривій концентрація/час. Також графік відбору проб повинен досить тривало охоплювати криву концентрація/час, щоб забезпечити надійну оцінку ступеня впливу. Це досягається, коли AUC(0–t) охоплює не менше 80% AUC(0–∞). Для достовірної оцінки константи швидкості елімінації (що необхідна для надійної оцінки AUC(0–∞)) протягом періоду лог-лінійної фази елімінації слід відбирати не менше трьох-чотирьох проб. Оскільки для лікарських форм із негайним вивільненням фаза абсорбції не перевищує 72 год, то для порівняння ступеня впливу як альтернативу AUC(0–t) можна використовувати AUC, визначену для 72 год (AUC(0–72 год)). Тому для будь-якої лікарської форми з негайним вивільненням незалежно від T½, здійснювати відбір проб протягом періоду більше 72 год не вважається потрібним.
У дослідженнях із багатократним введенням дози для правильного визначення AUC(0–τ) слід відбирати пробу безпосередньо (протягом 5 хв) перед введенням лікарського засобу, а останню пробу — протягом 10 хв від номінального часу інтервалу дозування, тобто інтервалу між введеннями лікарського засобу.
Якщо як біологічна рідина використовується сеча, її, як правило, збирають протягом не менше трьох T½. Проте, відповідно до рекомендацій з відбору проб плазми крові, збір сечі протягом більше ніж 72 год проводити не слід. Якщо необхідно визначити швидкість виведення, інтервали між заборами проб впродовж фази абсорбції мають бути, по можливості, якомога коротшими (див. також розділ 4.1.5 цієї настанови).
Для ендогенних речовин графік відбору проб повинен дати змогу характеризувати їх базовий профіль для кожного суб’єкта в кожний період дослідження. Як правило, базовий профіль визначають на 2–3 зразках, відібраних до введення лікарського засобу. В інших випадках, щоб врахувати циркадні коливання базового вмісту ендогенної речовини, відбір проб слід проводити через регулярні періоди протягом 1–2 днів до введення лікарського засобу (див. розділ 4.1.5 цієї настанови).
Умови натще або після прийому їжі
Дослідження біоеквівалентності, як правило, слід проводити натще, оскільки ця умова вважається найбільш чутливою до виявлення потенційної відмінності між препаратами. Якщо в SmPC референтного лікарського препарату рекомендується його приймати натще або незалежно від вживання їжі, дослідження біоеквівалентності потрібно проводити в умовах натще. Якщо в SmPC референтного лікарського препарату рекомендується його приймати тільки після вживання їжі, дослідження біоеквівалентності слід проводити після вживання їжі.
Проте для препаратів зі специфічними властивостями лікарської форми (наприклад, мікроемульсії, тверді дисперсії) дослідження біоеквівалентності проводять як натще, так і після вживання їжі, крім випадків, коли препарат слід приймати виключно натще або виключно після вживання їжі.
Якщо потрібна інформація як у стані натще, так і після вживання їжі, прийнятно проводити або два окремі перехресні дослідження з двома періодами, або одне перехресне дослідження з чотирма періодами.
У дослідженнях, коли прийом лікарського засобу здійснюється після вживання їжі, склад їжі повинен відповідати рекомендаціям SmPC для референтного лікарського засобу. Якщо в SmPC референтного лікарського засобу відсутні особливі рекомендації з цього приводу, то їжа має бути висококалорійною (приблизно від 800 до 1000 ккал), з високим вмістом жирів (приблизно 50% загального вмісту калорій). На білки має відводитися близько 150 ккал, на вуглеводи — 250 ккал і на жири — 500–600 ккал. Склад їжі відносно вмісту білків, вуглеводів і жирів має бути описаний (у грамах, абсолютному і відносному (%) вмісту калорій).
4.1.5. Досліджувані характеристики
Фармакокінетичні параметри
Для оцінки фармакокінетичних параметрів необхідно використовувати фактичний час відбору проб. У дослідженнях біоеквівалентності після однократного введення дози лікарського засобу слід визначати AUC(0–t), AUC(0–∞), залишкову площу, Сmax та tmax. У дослідженнях біоеквівалентності з періодом відбору проб протягом 72 год і з визначенням концентрації у точці 72 год немає необхідності розраховувати AUC(0–∞) і залишкову площу; досить вказати у звіті AUC, усічену в точці 72 год, AUC(0–72 год). Додатково можуть бути представлені такі параметри, як константа швидкості елімінації (λz) і T½.
У дослідженнях визначення біоеквівалентності у стаціонарному стані лікарських форм із негайним вивільненням слід визначати AUC(0–τ), Сmax, ss, tmax, ss. При використанні сечі як біологічної рідини необхідно визначати Ae(0–t) і, якщо можливо, Rmax.
Для визначення фармакокінетичних параметрів у дослідженнях біоеквівалентності необхідно використовувати некомпартментальні методи. Застосування компартментальних методів для оцінки параметрів неприйнятне.
Вихідна сполука чи її метаболіти
Загальні рекомендації
Загалом оцінка біоеквівалентності має базуватися на вимірюванні концентрацій вихідної сполуки, оскільки Сmax вихідної сполуки зазвичай більш чутлива до виявлення відмінностей ступеня абсорбції між лікарськими засобами, ніж Сmax метаболіту.
Неактивні пролікарські засоби (inactive pro-drugs)
Для неактивних пролікарських засобів також рекомендується проводити дослідження біоеквівалентності вихідної сполуки. Немає необхідності визначати концентрацію активного метаболіту. Проте деякі пролікарські засоби можуть мати низькі концентрації в плазмі крові та швидко виводитись із кровотоку, що ускладнює доведення біоеквівалентності вихідної сполуки. У цьому разі допускається доводити біоеквівалентність основного активного метаболіту без вимірювання концентрацій вихідної сполуки. У контексті цієї настанови вихідна сполука може вважатися неактивним пролікарським засобом, якщо внесок такої сполуки у клінічну ефективність відсутній або дуже низький.
Використання даних метаболітів на заміну даним активної вихідної сполуки
Використання даних метаболітів на заміну даним активної вихідної сполуки не рекомендується. Така заміна можлива лише у разі, якщо заявник може належним чином обґрунтувати, що чутливість аналітичної методики вимірювання вихідної сполуки не може бути покращена та що після введення однократної дози лікарського засобу надійно виміряти концентрацію вихідної сполуки неможливо, беручи до уваги, що у дослідженнях біоеквівалентності дозволяється використання вищої однократної дози (див. також розділ 4.1.6 цієї настанови). Зважаючи на останні розробки в біоаналітичній методології, малоймовірно, що вихідну сполуку не можна виміряти достовірно і точно. Тому використання даних метаболіту на заміну даним активної вихідної сполуки може бути прийнятним лише у виняткових випадках. При використанні такої заміни заявник зобов’язаний надати усі дані, які підтверджують, що вплив метаболіту відображає вплив вихідної сполуки і що в терапевтичних дозах утворення метаболіту не є насичуваним процесом.
Енантіомери
Для визначення енантіомерів, як правило, прийнятне використання ахіральних біоаналітичних методик. Проте концентрацію індивідуальних енантіомерів слід вимірювати, коли виконуються всі нижчезазначені умови:
1) енантіомери виявляють різну фармакокінетику;
2) енантіомери виявляють виражені відмінності у фармакодинаміці;
3) відмінність у ступені впливу (AUC) енантіомерів зумовлена різницею у швидкості абсорбції.
Концентрацію кожного енантіомера необхідно вимірювати, якщо всі вищезазначені умови виконуються чи відомості про них відсутні. Якщо фармакологічно активним є тільки один із енантіомерів, а фармакологічна активність іншого енантіомера низька чи повністю відсутня, достатньо підтвердити біоеквівалентність тільки активного енантіомера.
Використання даних про екскрецію з сечею
Якщо неможливо достовірно виміряти профіль концентрація/час вихідної сполуки у плазмі крові, то для визначення ступеня впливу як заміна даних концентрації в плазмі крові можуть бути використані дані про екскрецію з сечею. Проте використання даних про екскрецію з сечею для оцінки максимуму впливу має бути ретельно обґрунтоване. Якщо можна визначити достовірне значення Сmax у плазмі крові, то для оцінки біоеквівалентності ці дані слід надати з даними про ступінь впливу, отриманими при використанні сечі. При використанні даних про екскрецію з сечею заявник має надати всі наявні дані, які підтверджують, що екскреція із сечею відображає вплив у плазмі крові.
Ендогенні речовини
Якщо досліджувана діюча речовина є ендогенною, розрахунок фармакокінетичних параметрів слід проводити із використанням поправки на її базовий рівень, щоб розраховані фармакокінетичні параметри стосувалися додаткових концентрацій, отриманих внаслідок прийому лікарського засобу. За умови прийнятної переносимості дози лікарського засобу і якщо додаткову концентрацію ендогенної речовини, що перевищує базову концентрацію, яка досягається після прийому лікарського засобу, можна надійно визначити, в дослідженні біоеквівалентності ендогенних лікарських засобів допускається введення доз, що перевищують терапевтичні дози. Якщо після введення різних доз конкретної ендогенної речовини різниця у впливі раніше не встановлена, її слід визначати або в ході пілотного дослідження, або в рамках (як частина) основного дослідження біоеквівалентності, використовуючи різні дози референтного лікарського засобу для забезпечення того, щоб доза, яка використовувалася для дослідження біоеквівалентності, давала змогу виявити потенційні відмінності між лікарськими засобами.
Точний метод для корекції базового рівня ендогенної речовини має бути заздалегідь визначений і обґрунтований у протоколі дослідження. Переважно слід застосовувати стандартний метод віднімання: віднімається або середня концентрація ендогенної речовини, визначена до прийому лікарського засобу, або AUC ендогенної речовини, визначена до прийому лікарського засобу. Рідко поправка на базовий рівень може не знадобитися, коли концентрація ендогенної речовини, визначена після прийому лікарського засобу, суттєво перевищує базову концентрацію.
У дослідженнях біоеквівалентності ендогенних речовин не можна безпосередньо оцінити наявність впливу попередньої терапії (ефект переносу), тому з обережністю слід підходити до вибору періоду відмивання, який має бути достатньо тривалим.
4.1.6. Дозування, що досліджуються
Якщо на державну реєстрацію лікарського засобу подається кілька дозувань досліджуваного лікарського засобу, то залежно від пропорційності складу в різних дозуваннях та інших властивостей препарату, описаних нижче, може бути достатньо проведення дослідження біоеквівалентності тільки на одному або двох дозуваннях. Вибір досліджуваного(-их) дозування(-нь) залежить від лінійності фармакокінетики діючої речовини.
У разі нелінійної фармакокінетики (тобто непропорційне збільшення AUC з підвищенням дози лікарського засобу) між різними дозуваннями препарату може бути різниця у чутливості визначення потенційних відмінностей між порівнюваними лікарськими засобами. У контексті цієї настанови фармакокінетика вважається лінійною, якщо різниця між відкоригованими відносно дози середніми AUC при порівнянні досліджуваного дозування (тобто дозування, що планується для дослідження біоеквівалентності) і дозування, для якого розглядається можливість не проводити дослідження еквівалентності in vivo, становить не більше 25%. Для оцінки лінійності заявник повинен критично оцінити всі доступні для загального користування опубліковані дані, що стосуються пропорційності дозування. Лінійність підтверджується, якщо відмінності між скоригованими відносно дози AUC знаходяться в межах ±25%.
Якщо доведено біоеквівалентність для дозування(-нь), найбільш чутливого(-их) для виявлення потенційних відмінностей між порівнюваними лікарськими засобами, то для іншого(-их) дозування(-нь) дослідження еквівалентності in vivo можна не проводити.
Загальні критерії відмови від проведення дослідження біоеквівалентності in vivo
У разі заяви про відмову від проведення дослідження еквівалентності in vivo для заявленого(-их) додаткового(-их) дозування(-нь) мають бути виконані такі вимоги:
а) технологічний процес для різних дозувань препарату однаковий;
b) якісний склад різних дозувань препарату однаковий;
c) склад різних дозувань препарату кількісно пропорційний, тобто співвідношення між кількістю кожної допоміжної речовини та кількістю діючої(-их) речовини(-ин) однакове для всіх дозувань (для препаратів негайного вивільнення ця вимога не стосується компонентів покриття, оболонок капсул, барвників та ароматизаторів).
У разі наявності деяких відхилень у кількісній пропорційності складу, умова с) вважається виконаною, якщо для досліджуваного дозування і дозування(-нь), для якого(-их) розглядається питання відмови у проведенні дослідження еквівалентності in vivo, виконуються умови i) та ii) або виконуються умови i) та iii):
і) вміст діючої(-их) речовини(-ин) становить менше 5% маси ядра таблетки або маси вмісту капсули;
іі) вміст різних допоміжних речовин ядра таблетки або вмісту капсули є однаковим для розглянутих дозувань, і змінюється лише вміст діючої речовини;
ііі) вміст наповнювача змінюється залежно від вмісту діючої речовини; кількість інших допоміжних речовин ядра таблетки або вмісту капсули залишається незмінним;
d) відповідні дані розчинення in vitro мають підтверджувати відсутність необхідності проведення досліджень біоеквівалентності in vivo для додаткового дозування (див. розділ 4.2 цієї настанови).
Лінійна фармакокінетика
Для лікарських засобів, що відповідають умовам, зазначеним, достатньо підтвердити біоеквівалентність лише для одного дозування лікарського засобу.
Як правило, дослідження біоеквівалентності слід проводити для найвищого дозування. Для лікарських засобів із лінійною фармакокінетикою і за умови високої розчинності діючої речовини (див. Додаток ІІІ до цієї настанови) прийнятно проводити дослідження біоеквівалентності на дозуванні, нижчому за максимальне. Вибір нижчого дозування також може бути обґрунтований із причин безпеки/переносимості, коли введення максимального дозування неприйнятне для здорових добровольців. Крім того, якщо чутливість аналітичної методики перешкоджає досить точному вимірюванню концентрації аналіту в плазмі крові після однократного введення максимального дозування лікарського засобу, може бути вибрана вища доза (перевага надається використанню декількох таблеток максимального дозування). Вибрана доза може бути вищою за максимальну терапевтичну дозу тільки у разі, якщо така доза добре переноситься здоровими добровольцями і відсутні обмеження з абсорбції чи розчинності при цій дозі.
Нелінійна фармакокінетика
Для лікарських засобів із нелінійною фармакокінетикою, що характеризується більше ніж пропорційним збільшенням AUC з підвищенням дози лікарського засобу в діапазоні терапевтичних доз, дослідження біоеквівалентності, як правило, слід проводити для максимального дозування. Як і у разі лікарських засобів із лінійною фармакокінетикою, вибір нижчого дозування може бути обґрунтований, якщо максимальне дозування не можна вводити здоровим добровольцям із причин безпеки/переносимості. У разі проблем із чутливістю аналітичної методики аналогічно лікарським засобам з лінійною фармакокінетикою також дозволяється застосування лікарських засобів із нелінійною фармакокінетикою у вищих дозах.
Дослідження біоеквівалентності лікарських засобів, для яких характерне менше ніж пропорційне збільшення AUC із підвищенням дози лікарського засобу в діапазоні терапевтичних доз, у більшості випадків слід проводити як для максимального, так і для мінімального дозування (або для дозування, фармакокінетика якого знаходиться в лінійному діапазоні), тобто у цьому разі слід проводити два дослідження біоеквівалентності. Якщо нелінійність фармакокінетики не зумовлена обмеженою розчинністю діючої речовини, а спричинена, наприклад, насиченням транспортерів, за умови виконання вищенаведених умов у (а–d) та якщо досліджуваний і референтний лікарські засоби не містять будь-яких допоміжних речовин, що можуть вплинути на моторику шлунково-кишкового тракту або транспортні білки, достатньо провести дослідження біоеквівалентності для найнижчого дозування (або дозування, фармакокінетика якого знаходиться в лінійному діапазоні).
Вибір інших дозувань лікарського засобу може бути обґрунтований, якщо у зв’язку із проблемами із чутливістю аналітичної методики не можна провести дослідження для найнижчого дозування, а застосування найвищого дозування у здорових добровольців неможливе з причин безпеки/переносимості.
Підхід брикетингу
Якщо дослідження біоеквівалентності необхідно провести більше ніж на двох дозуваннях, наприклад, через відхилення від пропорційності складу, можна використовувати так званий підхід брикетингу. У цьому разі може бути прийнятне проведення двох досліджень біоеквівалентності, за умови, що вибрані дозування є екстремумами (крайні точки), наприклад, найвище і найнижче дозування або два дозування, що найбільше відрізняються за складом, і тому будь-які відмінності у складі інших дозувань охоплюються двома проведеними дослідженнями.
Якщо оцінку біоеквівалентності необхідно провести у стані натще і після вживання їжі та для двох дозувань через нелінійну абсорбцію чи відхилення від пропорційності складу, достатньо провести дослідження натще і після вживання їжі лише для одного дозування. Відсутність необхідності проведення дослідження біоеквівалентності натще або після вживання їжі для інших дозувань лікарського засобу може бути обґрунтована попередніми результатами та/чи фармакокінетичними даними досліджуваного дозування із дослідження, проведеного натще і після вживання їжі. При виборі дизайну (натще або після вживання їжі) для дослідження іншого(-их) дозування(-нь) перевага надається умовам, які є найчутливішими для виявлення відмінностей між порівнюваними лікарськими засобами.
Лікарський засіб із фіксованою комбінацією
Умови пропорційності складу повинні виконуватися відносно всіх діючих речовин фіксованих комбінацій. При розрахунку вмісту кожної діючої речовини у фіксованій комбінації інша(-і) діюча(-і) речовина(-и) розглядається(-ються) як допоміжна(-і) речовина(-и). У разі двошарових таблеток кожний шар може розглядатися окремо.
4.1.7. Біоаналітична методологія
Біоаналітичну частину досліджень біоеквівалентності необхідно проводити згідно з принципами належної лабораторної практики (GLP). Водночас, оскільки біоаналітичні дослідження за участю людини не входять у сферу дії принципів GLP, місця проведення біоаналітичної частини досліджень не підлягають моніторингу в рамках національної програми відповідності принципам GLP.
Для отримання достовірних результатів, що можуть бути задовільно інтерпретовані, необхідно належним чином охарактеризувати біоаналітичні методики, що використовуються, повністю їх валідувати і задокументувати. У кожній серії аналітичних циклів у рамках дослідження слід проводити валідацію методики з використанням зразків контролю якості.
До характеристик біоаналітичної методики, необхідних для забезпечення прийнятності її виконання і достовірності отриманих результатів аналізу, належать: селективність, нижня межа кількісного визначення, функція відгуку (характеристика калібрувальної кривої), правильність, прецизійність і стабільність.
Оскільки концентрації до прийому лікарського засобу мають визначатися при ≤5% від Сmax, нижня межа кількісного визначення методики має становити ≤1/20 від Сmax або нижче (див. розділ 4.1.8 «Ефекти переносу» цієї настанови).
Проведення повторного аналізу досліджуваних зразків має бути заздалегідь передбачено протоколом дослідження (та/чи у стандартних операційних процедурах) до фактичного початку аналізу зразків. Як правило, повторний аналіз досліджуваних зразків через фармакокінетичні причини неприйнятний. Це особливо важливо для дослідження біоеквівалентності, оскільки може вплинути на результат цього дослідження.
Аналіз зразків слід проводити за умови відсутності інформації щодо схеми рандомізації досліджуваних препаратів.
4.1.8. Оцінка
У дослідженнях біоеквівалентності фармакокінетичні параметри, як правило, не можуть бути скориговані з урахуванням відмінностей у кількісному визначенні вмісту діючої речовини між серіями досліджуваного та референтного лікарських засобів. Однак у виняткових випадках, коли неможливо знайти серію референтного лікарського засобу, для якої кількісний вміст діючої речовини відрізняється на <5% кількісного вмісту діючої речовини в серії досліджуваного лікарського засобу (див. розділ 4.1.2 цієї настанови), може бути прийнятною поправка на вміст. Використання поправки на вміст діючої речовини має бути заздалегідь передбачено у протоколі дослідження та обґрунтовано включенням до протоколу дослідження результатів кількісного визначення вмісту діючої речовини у серіях досліджуваного та референтного лікарських засобів.
Облік суб’єктів дослідження
В ідеалі всі суб’єкти дослідження, які приймали лікарський засіб, мають бути включені у статистичний аналіз, за винятком суб’єктів перехресного дослідження, для яких відсутні прийнятні для оцінки дані як для досліджуваного, так і для референтного лікарських засобів (або суб’єктів у дослідженні з паралельними групами, для яких відсутні прийнятні для оцінки дані одного періоду).
Дані, отримані від усіх суб’єктів дослідження, слід обробляти однаково. Неприпустимо передбачати протоколом дослідження включення в аналіз даних «запасних» суб’єктів для заміни даних суб’єктів, які були виключені. Навіть якщо в ході дослідження суб’єкти не вибували із дослідження, необхідно передбачити включення в аналіз усіх суб’єктів дослідження, які приймали лікарський засіб.
У дослідженні з більше ніж двома введеннями (наприклад, дослідження із трьома періодами з двома референтними лікарськими засобами (один із ЄС, інший — зі США), або дослідження із чотирма періодами, що включає прийом досліджуваного і референтного лікарських засобів натще та після вживання їжі) слід проводити аналіз для кожного порівняння, за винятком даних, що не стосуються цього порівняння.
Причини для виключення суб’єкта з аналізу
Для об’єктивної оцінки результатів рандомізованих досліджень необхідно, щоб нагляд за всіма суб’єктами дослідження та введення їм препаратів здійснювали за однаковими правилами. Ці правила не повинні залежати від досліджуваного лікарського засобу або результату, тому рішення про виключення суб’єкта дослідження зі статистичного аналізу необхідно приймати перед початком біоаналізу.
Будь-яка причина може бути критерієм виключення суб’єкта дослідження з аналізу, якщо вона заздалегідь передбачена протоколом дослідження, а рішення про виключення суб’єкта прийняте перед початком біоаналізу зразків. Однак виключення даних суб’єкта з аналізу слід уникати, оскільки потужність дослідження зменшуватиметься, а для оцінки необхідно мати дані мінімум 12 суб’єктів дослідження.
Прикладами причин для виключення результатів, отриманих у суб’єкта дослідження, з аналізу можуть бути: блювання чи діарея у ході проведення окремого періоду дослідження, що можуть зробити профіль концентрація/час недостовірним. У виняткових випадках причиною для виключення суб’єкта з аналізу може бути поєднане застосування інших лікарських засобів.
Критерії виключення суб’єкта дослідження з аналізу мають бути заздалегідь описані у протоколі дослідження. Якщо виникає ситуація, що підпадає під критерій виключення суб’єкта дослідження з аналізу, про це має бути зазначено в індивідуальній реєстраційній формі (Сase Report Form — CRF) під час проведення дослідження. Виключення суб’єктів дослідження з аналізу на підставі цих заздалегідь визначених критеріїв має бути чітко відображено у звіті дослідження та надано перелік таких випадків.
Оскільки неможливо відрізнити ефекти препарату від інших ефектів, що впливають на фармакокінетику, виключення даних лише на підставі статистичного аналізу або тільки з фармакокінетичних причин неприйнятне.
Винятки з цього правила
1) суб’єкти дослідження, у плазмі крові яких концентрація референтного лікарського засобу не піддається вимірюванню чи вимірюється лише в дуже малих кількостях. Концентрація референтного лікарського засобу в плазмі крові суб’єкта дослідження вважається дуже низькою, якщо його AUC становить <5% середнього геометричного значення AUC для референтного лікарського засобу (розрахованого без врахування даних суб’єкта дослідження, виключеного з аналізу). Виключення даних з цієї причини може бути прийнятним лише у виняткових випадках і в цілому може ставити під сумнів достовірність проведеного дослідження;
2) суб’єкти з ненульовою базовою концентрацією, що становить >5% від Сmax. Такі дані слід виключити з розрахунку біоеквівалентності (див. пункт «Ефект переносу»).
Для лікарських форм із негайним вивільненням вищезазначене може бути результатом недотримання суб’єктом умов дослідження та недостатнього періоду відмивання відповідно. Ситуації, зазначеної у пункті 1, можна уникнути, якщо перевіряти ротову порожнину суб’єктів після прийому лікарського засобу для контролю проковтування суб’єктами препарату. Ситуації, зазначеної у пункті 2, можна уникнути, якщо передбачити достатній період відмивання дизайном дослідження. Біологічні зразки суб’єктів, які були виключені зі статистичного аналізу, потрібно аналізувати, а їх результати представити у звіті дослідження (див. пункт «Представлення даних»).
Як зазначено в розділі 4.1.4 цієї настанови, AUC(0–t) має охоплювати не менше 80% AUC(0–∞). Якщо це правило не виконується, суб’єкти не повинні виключатися зі статистичного аналізу. Однак, якщо AUC(0–t) охоплює <80% AUC(0–∞) для >20% випадків, необхідно розглянути питання правильності такого дослідження. Цей підхід не застосовується до досліджень, в яких тривалість періоду відбору біологічних зразків становить ≥72 год, а замість AUC(0–t) використовується AUC(0–72 год).
Параметри, що аналізуватимуться, і межі прийнятності
При дослідженні біоеквівалентності з однократним введенням дози параметри, які необхідно аналізувати, це AUC(0–t) або, за необхідності, AUC(0–72 год) та Cmax. Для цих параметрів 90% довірчий інтервал для співвідношення досліджуваного та референтного лікарських засобів має бути в діапазоні прийнятності від 80,00 до 125,00%. Щоб потрапити в діапазон прийнятності, потрібно, щоб нижня межа була 80,00% при округленні до двох знаків після коми, а верхня межа була 125,00% при округленні до двох знаків після коми.
При дослідженні біоеквівалентності форм із негайним вивільненням у стаціонарному стані AUC(0–τ) і Сmax, ss потрібно аналізувати з використанням такого ж самого діапазону прийнятності.
В окремих випадках, коли використовуються дані екскреції з сечею, Ае(0–t) (загальну екскрецію з сечею незміненої діючої речовини лікарського засобу з моменту введення лікарського засобу до останньої точки, що визначається (t)) слід аналізувати з використанням вищезазначеного діапазону прийнятності для AUC(0–t). Максимальну швидкість екскреції з сечею (Rmax) слід аналізувати, використовуючи такий же діапазон прийнятності, як і для Сmax.
Статистична оцінка tmax не вимагається. Однак, якщо швидке вивільнення заявляється як клінічно значиме і важливе для початку дії лікарського засобу, або воно пов’язане з побічними ефектами, то не має бути ніякої очевидної різниці в медіані tmax та у варіабельності цього показника між досліджуваним і референтним лікарськими засобами.
В особливих випадках для лікарських засобів із вузьким терапевтичним діапазоном може знадобитися звуження діапазону прийнятності для AUC (див. розділ 4.1.9 цієї настанови). Крім того, в особливих випадках для високоваріабельних лікарських засобів діапазон прийнятності для Сmax може бути розширений (див. розділ 4.1.10 цієї настанови).
Статистичний аналіз
Оцінка біоеквівалентності ґрунтується на 90% довірчих інтервалах для співвідношення середніх геометричних розглянутих параметрів (досліджуваний/референтний засіб). Цей метод еквівалентний для двох односторонніх критеріїв при нульовій гіпотезі біонееквівалентності при рівні значимості 5%.
Фармакокінетичні параметри після розгляду необхідно аналізувати з використанням моделі ANOVA. Аналіз слід проводити після логарифмічного перетворення даних. Довірчий інтервал для різниці між препаратами в логарифмічно перетвореній шкалі отримують із використанням моделі ANOVA. Цей довірчий інтервал потім назад перетворюється для отримання очікуваного довірчого інтервалу для співвідношення за вихідною шкалою. Застосування непараметричного аналізу не прийнятне.
Точну модель, що використовуватиметься для аналізу, необхідно заздалегідь зазначити у протоколі. При статистичному аналізі необхідно брати до уваги джерела змінних, що, як можна об’єктивно припустити, впливають на кінцеву змінну. Умови, що використовуються в моделі ANOVA, це зазвичай послідовність, суб’єкт дослідження в послідовності, період і препарат. Для всіх умов бажано використовувати фіксовані, а не випадкові ефекти.
Ефект переносу
Тест на вплив ефекту переносу не вважається значимим, і на підставі такого тесту не слід приймати будь-які рішення щодо аналізу (наприклад, аналізу лише першого періоду). Можливий вплив ефекту переносу можна безпосередньо розглядати при вивченні концентрацій у плазмі крові до введення у другий період (і пізніше — за потреби).
Якщо є будь-які суб’єкти, для яких концентрація до введення препарату >5% значення Сmax для суб’єкта в цей період, то статистичний аналіз варто проводити без даних цього суб’єкта, що випадають для цього періоду. У другий період дослідження це приведе до вилучення цього суб’єкта з аналізу. Дослідження не буде вважатися прийнятним, якщо ці виключення призведуть до участі <12 суб’єктів дослідження, яких можна оцінювати. Цей підхід не застосовується до ендогенних лікарських засобів.
Двохетапний дизайн
Допустимо використовувати двохетапний дизайн дослідження при спробі довести біоеквівалентність. Дослідження буде проведено для початкової групи суб’єктів дослідження, та їх дані аналізуватимуться. Якщо біоеквівалентність не буде доведена, додаткову групу можна набрати, а результати, отримані в обох групах, об’єднати в остаточному аналізі. Якщо цей підхід приймається, обов’язково необхідно вжити відповідних заходів для запобігання загальній похибці типу I та до початку дослідження слід чітко визначити критерії припинення експерименту. Аналіз даних першого етапу слід розглядати як проміжний аналіз, а аналіз обох етапів слід проводити при скоригованих рівнях значимості (при довірчих інтервалах відповідно з використанням скоригованої імовірності покриття, яка перевищуватиме 90%). Наприклад, використання 94,12% довірчого інтервалу для першого етапу і об’єднаних даних, отриманих у перший та другий етап, може бути прийнятним, але є багато інших варіантів довірчого інтервалу, і вибір рівня значущості (α) для проміжного аналізу є відповідальністю спонсора. План використання підходу двохетапного дизайну має бути заздалегідь визначений у протоколі поряд зі скоригованими рівнями значущості, що використовуватимуться для кожного з аналізів.
Коли аналізуються об’єднані дані двох етапів, умову етапу слід включити в модель ANOVA.
Представлення даних
Усі дані індивідуальних концентрацій і фармакокінетичних параметрів мають бути представлені для кожного препарату разом із загальною статистикою, такою як середнє геометричне, медіана, середнє арифметичне, стандартне відхилення, коефіцієнт варіації, мінімальне та максимальне значення. Індивідуальні криві концентрація/час у плазмі крові слід представити в лінійній та логарифмічній шкалі. Необхідно зазначати метод, що використовується для отримання фармакокінетичних параметрів із первинних даних. Слід зазначати кількість точок кінцевої логарифмічно/лінійної фази, що використовуються для оцінки константи елімінації (що необхідна для надійної оцінки AUC∞).
Для фармакокінетичних параметрів, що піддаються статистичному аналізу, слід представити точкову оцінку і 90% довірчий інтервал для співвідношення досліджуваного і референтного лікарських засобів.
Необхідно представити таблиці ANOVA, що включають відповідні статистичні тести всіх ефектів у моделі.
Звіт має бути достатньо докладним для можливості повторення фармакокінетичного та статистичного аналізу, наприклад, необхідно надати дані щодо фактичного часу відбору зразків крові після дозування, концентрації лікарських засобів, дані щодо отриманих показників фармакокінетичних параметрів для кожного суб’єкта в кожний період та представити схему рандомізації.
Необхідно детально описувати випадки вибування чи виключення суб’єктів із дослідження. Якщо можливо, дані концентрацій та фармакокінетичних параметрів таких суб’єктів необхідно представити в окремих списках, але не слід включати в загальну статистику.
Біоаналітичну методику необхідно задокументувати в валідаційному звіті перед дослідженням. Біоаналітичний звіт також необхідно надавати. Біоаналітичний звіт має включати короткий опис біоаналітичної методики, що використовується у дослідженні, і результати всіх калібрувальних стандартів і зразків контролю якості. Необхідно представити репрезентативну кількість хроматограм або інших первинних даних, що включають весь діапазон концентрацій для всіх зразків калібрувальних стандартів і зразків контролю якості, а також аналізованих зразків. Дані мають включати всі хроматограми аналітичних циклів принаймні для 20% суб’єктів зі зразками контролю якості та калібрувальними стандартами.
Якщо для певного препарату при певному дозуванні проведено ряд досліджень, деякі з яких продемонстрували біоеквівалентність, а деякі з яких ні, обсяг даних слід розглядати як єдине ціле. Необхідно розглядати тільки відповідні дослідження, як зазначено в розділі 4.1 цієї настанови. Наявність дослідження, що доводить біоеквівалентність, не означає, що ті дослідження, які не доводять біоеквівалентності, можуть відкидатися. Заявник має ретельно проаналізувати результати та обґрунтувати, що заявлена біоеквівалентність була доведена. Крім того, за необхідності комбінований аналіз всіх досліджень може бути наданий як додаток до аналізу окремого дослідження. Неприйнятно об’єднувати разом дослідження, що не доводять біоеквівалентності за відсутності дослідження, що доводило б біоеквівалентність.
4.1.9. Діючі речовини з вузьким терапевтичним індексом
В окремих випадках для діючих речовин із вузьким терапевтичним індексом (NTID) діапазон прийнятності для AUC має бути звужений до 90,00–111,11%. Діапазон прийнятності 90,00–111,11% також необхідно застосовувати для параметра Сmах, якщо значення Сmах особливо важливе для безпеки, ефективності або моніторингу рівня лікарського препарату. Неможливо визначити ряд критеріїв для класифікації NTID, тому це обов’язково потрібно вирішувати в кожному конкретному випадку на підставі клінічних даних, якщо діюча речовина характеризується вузьким терапевтичним індексом.
4.1.10. Високоваріабельні діючі речовини або препарати
Високоваріабельні лікарські препарати (HVDP) — це такі, в яких внутрішньосуб’єктна варіабельність параметра становить >30%. Якщо заявник припускає, що препарат може бути розглянутий як високоваріабельний у швидкості та/чи ступені абсорбції, може бути проведено дослідження з перехресним повторним дизайном.
HVDP, для яких більша різниця у Cmax вважається клінічно незначущою на підставі переконливого клінічного обґрунтування, можуть розглядатися з розширеним діапазоном прийнятності. Якщо це так, діапазон прийнятності для Сmах може бути розширений максимально до 69,84–143,19%. Для розширення діапазону прийнятності дослідження біоеквівалентності обов’язково проводять із повторним дизайном, де має бути продемонстровано, що внутрішньосуб’єктна варіабельність для Сmах референтного лікарського засобу становить >30%. Заявник має обґрунтувати, що розрахована внутрішньосуб’єктна варіабельність надійно оцінена і не є результатом викидів. Вимога щодо розширення інтервалу обов’язково попередньо наводиться у протоколі.
Ступінь розширення визначається на підставі внутрішньосуб’єктної варіабельності, яка спостерігається у дослідженні біоеквівалентності з використанням масштабованої середньої біоеквівалентності (scaled-averagebioequivalence), що визначається за формулою:
[U,L] = exp [±k·swr],
де U — верхня межа діапазону прийнятності; L — нижня межа діапазону прийнятності; k — константа, що становить 0,760; і swr — внутрішньосуб’єктне стандартне відхилення логарифмічно перетворених величин Сmах референтного лікарського засобу. У нижченаведеній таблиці представлено приклади того, як різні рівні варіабельності приводять до різних меж прийнятності із застосуванням цієї методології.
|
Внутрішньосуб’єктна |
Нижня межа |
Верхня межа |
|
30 |
80,00 |
125,00 |
|
35 |
77,23 |
129,48 |
|
40 |
74,62 |
134,02 |
|
45 |
72,15 |
138,59 |
|
≥50 |
69,84 |
143,19 |
![]()
Співвідношення середнього геометричного (GMR) має бути в межах звичайного діапазону прийнятності 80,00–125,00%.
Можливість розширити критерії прийнятності на підставі високої внутрішньосуб’єктної варіабельності не застосовується до AUC, для AUC діапазон прийнятності має залишатися 80,00–125,00% незалежно від варіабельності.
Припустимо застосовувати трьох- або чотирьохперіодну перехресну схему в дослідженні з повторним дизайном (додаткові умови наведено в Доповненні 2 настанови)N.
4.2. Тести на розчинення in vitro
Загальні аспекти досліджень розчинення in vitro стисло описані в Додатку І, включаючи основні вимоги використання фактора подібності (f2-тест).
4.2.1. Тести на розчинення in vitro як доповнення до дослідження біоеквівалентності
Результати тестів розчинення in vitro для серій досліджуваного і референтного лікарських засобів, що використовувалися в дослідженнях біоеквівалентності у трьох різних буферах (зазвичай рН 1,2; 4,5 та 6,8) та середовищі, призначеному для контролю якості лікарського засобу (QC-media), необхідно відобразити у звіті. Для деяких лікарських форм, таких як таблетки, що диспергуються в ротовій порожнині, можуть знадобитися дослідження з використанням різних експериментальних умов. Результати слід надавати у вигляді профілів відсотків вивільненої речовини від часу, вказуючи середні значення та підсумкову статистику.
Якщо інше не обґрунтовано, специфікації тестів розчинення in vitro, що будуть використовуватися для контролю якості препарату, мають бути отримані на підставі профілю розчинення серії, що визначена як біоеквівалентна референтному лікарському засобу (див. Додаток І до цієї настанови).
У разі, якщо результати тестів розчинення in vitro біосерій не відображають біоеквівалентності, продемонстрованої in vivo, дані, отримані в дослідженнях in vivo, переважають. Проте можливі причини розбіжності необхідно розглянути й обґрунтувати.
4.2.2. Тести на розчинення in vitro як підтвердження відсутності необхідності проведення дослідження in vivo для додаткових дозувань
Відповідні тести на розчинення in vitro мають підтвердити адекватність відмови від проведення дослідження біоеквівалентності in vivo. Таким чином, розчинення необхідно проводити при різних значеннях рН, як зазначено у попередньому розділі (зазвичай рН 1,2; 4,5 і 6,8), якщо не обґрунтовано інше. Подібність профілів розчинення in vitro (див. Додаток І до цієї настанови) має бути продемонстрована за всіх умов у межах заявленого ряду дозувань, тобто між додатковими дозуваннями і дозуванням(-ями) (тобто серією(-ями)), що використовувалась для дослідження біоеквівалентності).
При значеннях рН, коли умови розчинення не можуть бути виконані для всіх дозувань, розчинення in vitro може відрізнятися у різних дозувань. Проте у цьому разі порівняння з відповідним дозуванням референтного лікарського засобу має потім підтвердити, що цей факт стосується діючої речовини, а не препарату. Також заявник може продемонструвати подібність профілів при однаковому дозуванні (наприклад, можливість порівняння двох таблеток по 5 мг і однієї таблетки по 10 мг).
4.3. Звіт про результати дослідження
4.3.1. Звіт про результати дослідження біоеквівалентності
Звіт дослідження біоеквівалентності має містити повну документацію щодо протоколу, проведення та оцінки. Він має бути складений відповідно до норм чинного законодавства УкраїниN [15] та відповідної настанови CPMP/ICH/137/95 (ICH E3) «Structure and Content of Clinical Study Reports» [19] і підписаний дослідником згідно з вимогами чинного законодавства України [15, 17]N.
Слід зазначити повне ім’я та посаду відповідального дослідника (дослідників), місце і період проведення дослідження. Сертифікати аудитів, якщо наявні, потрібно включати у звіт.
Звіт дослідження біоеквівалентності має містити доказ того, що вибір референтного лікарського засобу проведений відповідно до вимог наказу № 460 [3]N. Слід включити у звіт дані про назву референтного лікарського засобу, дозування, лікарську форму, номер серії, виробника, термін придатності та країну закупівлі.
Необхідно надавати назву і склад досліджуваного(-них) лікарського(-их) засобу(-ів), що використовується в дослідженні. Слід зазначити розмір і номер серії, дату виробництва і, якщо можливо, термін придатності досліджуваного лікарського засобу.
Сертифікати аналізу серій референтного і досліджуваного лікарських засобів, що використовуються в дослідженні, мають міститися в додатку до звіту дослідження.
Отримані дані концентрацій та фармакокінетичних параметрів, а також результати статистичного аналізу мають бути представлені детально, як описано вище (див. «Представлення даних»).
4.3.2. Інші дані для включення в реєстраційне досьє
Заявник зобов’язаний подати підписану заяву, яка підтверджує, що досліджуваний лікарський засіб має той самий кількісний склад та виробляється за тим самим виробничим процесом, що і лікарський засіб, який поданий для одержання реєстраційного посвідчення. Необхідно надати інформацію, чи досліджуваний лікарський засіб вже масштабовано у виробництві. Необхідно представити дані порівняння профілів розчинення (див. розділ 4.2 цієї настанови).
Валідаційний звіт біоаналітичної методики необхідно включити в Модуль 5 реєстраційного досьє.
Для надання за вимогою мають бути доступні у відповідному електронному форматі (наприклад, відокремлені комою і пробілом текстові файли або у форматі Excel) досить детальні дані для можливого повторення фармакокінетичного та статистичного аналізу, наприклад, дані про фактичний час відбору проб крові, отримані концентрації діючої речовини, показники фармакокінетичних параметрів для кожного суб’єкта в кожний період та схема рандомізації.
4.4. Заяви на зміни, що можуть бути протягом дії реєстраційного посвідчення
Якщо змінюється склад лікарського засобу порівняно із затвердженим або метод виробництва був змінений таким чином, що можливий вплив на біодоступність, рекомендовано проведення дослідження біоеквівалентності in vivo, якщо інше не обґрунтовано. Будь-яке представлене обґрунтування має базуватися на загальних міркуваннях, наприклад, згідно з Додатком ІІІ, або на прийнятному рівні кореляції А in vitro/in vivo, що була встановлена (див. СРМР/EWP/280/96 Corr. 1 «Guideline on the pharmacokinetic and clinical evaluation modified release dosage forms» [20]).
У разі якщо біодоступність препарату, що змінюється, була досліджена і прийнятний рівень кореляції А між дією in vivo і розчиненням in vitro було встановлено, немає необхідності доведення біоеквівалентності in vivo, якщо профіль розчинення in vitro нового препарату подібний до профілю розчинення вже затвердженого лікарського засобу за однакових умов дослідження, що використовувалися для встановлення кореляції (див. Додаток І до цієї настанови).
У разі змін, які можливі протягом дії реєстраційного посвідчення [3, 13] для лікарських засобів, що були затверджені за повною і незалежною заявою, заявою на добре вивчене застосування, заявою на фіксовану комбінацію, заявою інформованої згоди відповідно до чинного законодавства УкраїниN [3, 13], препаратом порівняння для дослідження біоеквівалентності й розчинення in vitro зазвичай є затверджений лікарський засіб, присутній на ринку, з чинним на цей час виробничим процесом, пакуванням тощо.
Якщо зміни, які можливі протягом дії реєстраційного посвідчення [3, 13], вносяться для генеричного або гібридного лікарського засобу, препаратом порівняння для дослідження біоеквівалентності має, як правило, бути доступна на ринку серія референтного лікарського засобу. Якщо цей референтний лікарський засіб недоступний на ринку, порівняння з попереднім складом (генеричного або гібридного лікарського засобу) може бути прийнятним, якщо це обґрунтовано. Для змін, що не потребують дослідження біоеквівалентності, необхідно дотримуватися рекомендацій та вимог, зазначених в інших чинних регуляторних настановах.
Додаток І. Тести на розчинення і подібність профілів розчинення
1. Загальні аспекти тесту на розчинення, що мають відношення до біодоступності
У ході розробки лікарського засобу тести на розчинення використовуються як спосіб ідентифікації факторів препарату, що впливають та можуть мати вирішальний вплив на біодоступність препарату. Після визначення складу і розробки виробничого процесу тести на розчинення використовуються для контролю якості серій, виготовлених при масштабуванні процесу, і промислових серій, щоб забезпечити як відтворюваність від серії до серії, так і те, що профілі розчинення залишаються подібними до серій, що використовувались в основних клінічних випробуваннях. Крім того, тести на розчинення в деяких випадках можуть використовуватися для відмови від проведення досліджень біоеквівалентності in vivo. Таким чином тести на розчинення можуть служити для декількох цілей:
і — контроль якості продукту:
- для отримання інформації про серії досліджуваного лікарського засобу, що використовуються в дослідженнях біодоступності/біоеквівалентності та основних клінічних дослідженнях, для обґрунтування специфікацій контролю якості;
- для використання інструменту контролю якості, щоб продемонструвати відтворюваність у виробництві;
- для отримання інформації про референтний лікарський засіб, що використовується в дослідженнях біодоступності/біоеквівалентності та основних клінічних дослідженнях;
ii — заміна досліджень біоеквівалентності:
- для демонстрації в деяких випадках подібності між різними складами препаратів та референтним лікарським засобом (відмова від проведення досліджень біоеквівалентності in vivo, наприклад, зміни, що можуть бути протягом дії реєстраційного посвідчення, зміни препарату під час розробки та зміни генеричних лікарських засобів; див. розділ 4.2 та Додаток ІІІ до цієї настанови);
- для дослідження відтворюваності від серії до серії препарату (досліджуваного і референтного), що використовується як основа при виборі прийнятних серій для дослідження in vivo.
Методики випробування мають бути розроблені стосовно препарату на загальних та/чи спеціальних фармакопейних вимогах. Якщо такі вимоги є неприйнятними та/чи не відображають кінетики розчинення in vivo (тобто біорелевантність), можуть бути розглянуті альтернативні методики, якщо обґрунтовано, що вони є дискримінаційними та дають змогу розрізнити серії з прийнятними і неприйнятними характеристиками препарату in vivo. Має завжди розглядатися поточний стан інформації, включаючи взаємодію характеристик, отриманих на підставі БСК — класифікації та лікарської форми.
Має бути достатньо точок відбору для отримання значущих профілів розчинення, принаймні через кожні 15 хв. Рекомендується частіший відбір проб у період найбільших змін у профілі розчинення. Може бути необхідним отримання адекватного профілю шляхом відбору зразків з інтервалом 5 або 10 хв для швидкорозчинних препаратів, якщо повне розчинення відбувається протягом 30 хв.
Можна очікувати, що не буде будь-яких проблем із біодоступністю, якщо діюча речовина вважається добре розчинною та, крім того, це дозування швидко розчиняється у фізіологічному діапазоні рН, і відомо, що допоміжні речовини не впливають на біодоступність. І навпаки, розчинення лікарської форми може бути мірою обмеження швидкості абсорбції, якщо вважається, що діюча речовина має обмежену чи погану розчинність, а також, якщо допоміжні речовини впливають на вивільнення і подальше розчинення діючої речовини. У таких випадках рекомендується оцінити різні умови дослідження та проводити відповідний відбір зразків.
2. Подібність профілів розчинення
Дослідження подібності профілів розчинення і будь-які висновки, отримані за їх результатами (наприклад, обґрунтування для відмови від проведення досліджень біоеквівалентності in vivo), можуть вважатися прийнятними, тільки якщо профіль розчинення був задовільно охарактеризований із використанням достатньої кількості точок відбору.
Для препаратів із негайним вивільненням додатково до рекомендацій, наданих вище у пункті 1, порівняння у точці «15 хв» є важливим, щоб знати чи досягається повне розчинення до спорожнення шлунка.
Якщо більше ніж 85% лікарського засобу розчиняється протягом 15 хв, профілі розчинення можуть прийматися як подібні без подальшої математичної оцінки.
У разі якщо більше ніж 85% не розчиняється за 15 хв, а розчиняється протягом 30 хв, то необхідні принаймні три точки відбору: перша точка до 15 хв, друга — точно у 15 хв і третя, коли вивільнення досягає 85%.
Для лікарських засобів із модифікованим вивільненням необхідно дотримуватися рекомендацій, що наводяться у відповідній настанові.
Подібність профілів може бути встановлена за допомогою обчислення коефіцієнта подібності f2, як описано нижче:
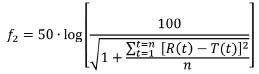
де: f2 — фактор подібності; n — кількість точок відбору; R(t) — середнє значення у відсотках розчиненого референтного препарату в час t після початку дослідження; T(t) — середнє значення у відсотках розчиненого досліджуваного препарату в час t після початку дослідження.
Для референтного та досліджуваного лікарських засобів необхідно визначати кількість розчиненої діючої речовини (у %).
Оцінка фактора подібності ґрунтується на таких умовах:
- мінімум три точки відбору (не враховуючи нульову);
- точки відбору для досліджуваного і референтного лікарських засобів мають бути однаковими;
- по 12 зразків у кожній точці відбору для досліджуваного і референтного лікарських засобів;
- не більше ніж одне середнє значення після розчинення 85% для будь-якого із препаратів;
- відносне стандартне відхилення або коефіцієнт варіації для будь-якого препарату мають бути менше 20% у першій точці і не більше 10% — з другої і до останньої точки.
Профілі розчинення вважаються подібними, якщо значення фактора подібності (f2) становить від 50 до 100. Якщо коефіцієнт подібності f2 не є задовільним, тоді подібність може бути встановлена шляхом порівняння результатів, отриманих модельно-залежними і модельно-незалежними методами, наприклад, за допомогою статистичного порівняння параметрів функції Вейбулла чи шляхом розрахунку кількості речовини (у %), розчиненої в певні моменти.
Альтернативні методи обчислення фактора подібності f2 для демонстрації подібності розчинення вважаються прийнятними, якщо вони статистично коректні й задовільно обґрунтовані.
Межі прийнятності необхідно попередньо визначити і обґрунтувати, а різниця має становити не більше 10%. Крім того, варіабельність даних розчинення досліджуваного і референтного лікарських засобів також має бути подібною, проте нижча варіабельність досліджуваного лікарського засобу може бути допустимою.
Слід надати доказ того, що проведено валідацію статистичного програмного забезпечення.
Необхідно представити чіткий опис і пояснення всіх етапів із наданням відповідних узагальнених таблиць.
Додаток ІІ. Вимоги до дослідження біоеквівалентності різних лікарських форм
Хоча ця настанова стосується лікарських засобів негайного вивільнення, Додаток ІІ забезпечує загальні рекомендації щодо вимог до біоеквівалентності для інших видів лікарських форм та специфічних лікарських форм негайного вивільнення.
Коли досліджуваний лікарський засіб містить іншу сіль, простий ефір, складний ефір, ізомер, суміш ізомерів, комплекс або похідну діючої речовини порівняно з референтним лікарським засобом, біоеквівалентність необхідно продемонструвати дослідженнями біоеквівалентності in vivo. Проте дослідження біоеквівалентності in vivo в деяких випадках може не вимагатися, як описано нижче в Додатку ІІІ, коли діюча речовина для досліджуваного та референтного лікарських засобів однакова (або містить солі з подібними властивостями, як визначено в розділі ІІІ Додатку ІІІ до цієї настанови).
Оральні лікарські форми з негайним вивільненням системної дії
Для лікарських форм, таких як таблетки, капсули та оральні суспензії, дослідження біоеквівалентності необхідне, крім випадків, коли прийнятний підхід процедури біовейвер на підставі БСК [3] (див. Додаток ІІІ до цієї настанови). Для таблеток, що диспергуються у ротовій порожнині, та оральних розчинів застосовують спеціальні вимоги, наведені нижче.
Таблетки, що диспергуються в ротовій порожнині
Таблетки, що диспергуються у ротовій порожнині, призначені для швидкого диспергування у ротовій порожнині. Перебування у ротовій порожнині та час контакту можуть бути критичними у випадках, коли діюча речовина також розчиняється в ротовій порожнині й може абсорбуватися безпосередньо через слизову оболонку ротової порожнини. Залежно від препарату, наприклад, таблеток, вкритих оболонкою, ковтання та подальша абсорбція також відбуватиметься у шлунково-кишковому тракті. Якщо може бути продемонстровано, що діюча речовина не абсорбується в ротовій порожнині, а має обов’язково проковтуватися та абсорбуватися в шлунково-кишковому тракті, тоді препарат може розглядатися за процедурою біовейвер на підставі БСК [3] (див. Додаток ІІІ до цієї настанови). Якщо це неможливо продемонструвати, біоеквівалентність обов’язково має оцінюватися за допомогою досліджень за участю людей.
Якщо запропонована нова лікарська форма — таблетки, що диспергуються у ротовій порожнині, — це зміни, що потребують нової реєстрації лікарського засобу, рекомендується трьохперіодне дослідження з метою оцінки застосування таблетки, що диспергується в ротовій порожнині, з/без супутнього прийому рідини. Однак, якщо у двохперіодному дослідженні доведено біоеквівалентність таблеток, що диспергуються у ротовій порожнині, прийнятих без води та референтного лікарського засобу, прийнятого з водою, біоеквівалентність таблеток, що диспергуються у ротовій порожнині, прийнятих із водою, можна припустити.
Якщо таблетки, що диспергуються у ротовій порожнині, є генеричним або гібридним лікарським засобом до вже затвердженого референтного лікарського засобу у формі таблеток, що диспергуються у ротовій порожнині, застосовуються такі рекомендації щодо дизайну дослідження:
- якщо референтний лікарський засіб може прийматися з водою або без неї, біоеквівалентність має бути продемонстрована без води, оскільки такі умови краще всього відображають застосування препарату. Це особливо важливо, якщо діюча речовина може бути розчинена і частково абсорбована в ротовій порожнині. Якщо біоеквівалентність продемонстровано при застосуванні препаратів без води, можна припустити біоеквівалентність, коли препарати приймаються з водою;
- якщо референтний лікарський засіб приймається лише одним способом (наприклад, тільки з водою), біоеквівалентність необхідно продемонструвати в таких же умовах (зазвичай двохперіодний перехресний дизайн);
- якщо референтний лікарський засіб приймається лише одним способом (наприклад, тільки з водою), а досліджуваний препарат призначений для додаткових умов прийому (наприклад, без води), звичайний та новий метод необхідно порівняти з референтним при звичайному шляху застосування (дизайн із трьома введеннями і трьома періодами у 6 послідовностях).
У дослідженнях оцінки таблеток, що диспергуються у ротовій порожнині, що приймаються без води, рекомендовано зволожувати ротову порожнину, ковтаючи 20 мл води безпосередньо перед розміщенням препарату на язиці. Рекомендовано не дозволяти приймати рідину раніше ніж через 1 год після прийому препарату.
Для інших оральних лікарських форм, таких як плівки, що диспергуються в ротовій порожнині, букальні таблетки або плівки, сублінгвальні таблетки та жувальні таблетки, необхідно використовувати підходи, подібні до тих, як для таблеток, що диспергуються у ротовій порожнині. Дослідження біоеквівалентності необхідно проводити відповідно до рекомендацій щодо застосування препарату.
Розчини для орального застосування
Якщо досліджуваний лікарський засіб є водним розчином для орального застосування на час прийому та містить діючу речовину в такій же, як і у референтного лікарського засобу, концентрації, дослідження біоеквівалентності може бути відхилене. Проте, якщо допоміжні речовини можуть впливати на проходження в шлунково-кишковому тракті (наприклад, сорбіт, маніт тощо), на абсорбцію (наприклад, поверхнево-активні речовини або допоміжні речовини, що можуть вплинути на транспортні білки), розчинність in vivo (наприклад, співрозчинники) або на стабільність діючої речовини in vivo, дослідження біоеквівалентності необхідно проводити, якщо тільки відмінності в кількості цих допоміжних речовин можуть бути адекватно обґрунтовані шляхом посилання на інші дані. Вимоги аналогічні, як при процедурі біовейвер на підставі БСК [3] (див. розділ IV.2 «Допоміжні речовини» Додатку ІІІ до цієї настанови), застосовують до подібності допоміжних речовин оральних розчинів.
У тих випадках, коли досліджуваний лікарський засіб є оральним розчином, що має бути біоеквівалентним іншій лікарській формі для орального застосування з негайним вивільненням, необхідно проводити дослідження біоеквівалентності.
Лікарські засоби з фіксованою комбінацією
Вимоги до дослідження біоеквівалентності лікарських засобів із фіксованою комбінацією викладені в настанові CPMP/EWP/240/95 Rev. 1 «Guideline on clinical development of fixed combination medicinal products» [21] та в наказі №460 [3]N. Умови можливості проведення процедури біовейвер для лікарських засобів із фіксованою комбінацією представлені в розділі V Додатку III.
Неоральні лікарські форми негайного вивільнення системної дії
Цей розділ застосовують, наприклад, для ректальних лікарських засобів. В основному дослідження біоеквівалентності необхідні. Процедура відмови від проведення дослідження біоеквівалентності in vivo може розглядатися у разі розчину, що містить активну діючу речовину у тій же концентрації, що і затверджений розчин, та має однаковий якісний і подібний кількісний склад допоміжних речовин (умови для оральних розчинів можна використовувати в цих випадках).
Парентеральні розчини
Дослідження біоеквівалентності, як правило, не є необхідними, якщо досліджуваний лікарський засіб вводиться як водний внутрішньовенний розчин, що містить таку ж активну речовину, як і затверджений на даний час лікарський засіб (референтний лікарський засіб)N. Проте дослідження біоеквівалентності необхідне, якщо будь-які допоміжні речовини взаємодіють із діючою речовиною (наприклад, утворюють комплекс) або інакше впливають на розподіл діючої речовини, якщо тільки не доведено, що досліджуваний і референтний лікарські засоби містять однакові допоміжні речовини в дуже схожій кількості, та може бути адекватно обґрунтовано, що будь-які відмінності в кількості не впливають на фармакокінетику діючої речовини.
У разі інших парентеральних шляхів введення, наприклад, внутрішньом’язовому або підшкірному, та якщо досліджуваний лікарський засіб належить до одного типу розчину (водний чи олійний), містить однакову концентрацію тієї ж діючої речовини та однакові допоміжні речовини в подібній кількості, що і затверджений на даний час лікарський засіб (референтний лікарський засіб)N, дослідження біоеквівалентності не є необхідними. Крім того, дослідження біоеквівалентності не є необхідними для водних парентеральних розчинів із порівнянними допоміжними речовинами у подібній кількості, коли може бути продемонстровано, що допоміжні речовини не впливають на в’язкість.
Ліпосомальні, міцелярні лікарські засоби та лікарські засоби у формі емульсій для внутрішньовенного введення
- Ліпосомальні лікарські форми. Фармакокінетичні питання, пов’язані з ліпосомальними препаратами для внутрішньовенного введення, потребують спеціального розгляду, що не представлений у цій настанові.
- Емульсії. Зазвичай для емульсій не розглядають відмову від проведення досліджень біоеквівалентності in vivo.
Проте для препаратів у формі емульсій може бути прийнятною відмова від проведення досліджень біоеквівалентності in vivo, якщо:
а) лікарський засіб не призначений для контролю вивільнення чи розподілу;
б) шлях та швидкість введення є такими ж, як і у затвердженого на даний час лікарського засобу (референтного лікарського засобу)N.
У цьому разі склад має бути якісно та кількісно однаковим із таким, як у затвердженій на даний час емульсії (референтному лікарському засобі)N. Необхідно представляти достатні дані для демонстрації дуже схожих фізико-хімічних характеристик, включаючи розподіл часток дисперсної ліпідної фази за розміром. На підтримку інших характеристик емульсії розглядають, наприклад, поверхневі властивості емульсії, такі як дзета-потенціал та реологічні властивості.
- Ліпіди для внутрішньовенного парентерального харчування можуть розглядатися в разі відмови проведення досліджень біоеквівалентності in vivo, якщо представлено задовільні дані порівняльних фізико-хімічних характеристик. Відмінності у складі можуть бути обґрунтовані з урахуванням природи і терапевтичних цілей таких лікарських форм.
- Міцелоутворювальні препарати. Розчини міцел для внутрішньовенного введення можна вважати «складними» розчинами, і тому зазвичай вони не розглядаються для відмови від проведення досліджень біоеквівалентності in vivo.
Проте для міцелоутворювальних препаратів може розглядатися відмова від проведення досліджень біоеквівалентності in vivo, якщо:
а) відбувається швидке руйнування міцел при розведенні та препарат не призначений для контролю вивільнення або доставки;
б) шлях і швидкість введення є такими ж, як і у затвердженого на даний час препарату (референтного лікарського засобу)N;
в) допоміжні речовини не впливають на розподіл діючої речовини.
У цих випадках якісний та кількісний склад міцелярної інфузії безпосередньо перед застосуванням має бути таким же, як і у затвердженого на даний час (референтного лікарського засобу)N. Необхідно представляти задовільні дані для демонстрації подібності фізико-хімічних характеристик, такі як критична концентрація міцел, здатність складу до розчинення (таких як максимальна концентрація добавки — maximum additive concentration), вільна та зв’язана діюча речовина та розмір міцел.
Це також застосовується у разі незначних змін у кількісному чи якісному складі за умови, що відсутні будь-які зміни в кількості чи типі поверхнево-активних речовин.
Лікарські форми з модифікованим вивільненням системної дії
Оральні лікарські форми з модифікованим вивільненням і трансдермальні лікарські форми
Необхідні дослідження біоеквівалентності відповідно до настанови СРМР/EWP/280/96 Corr. 1 «Guideline on the pharmacokinetic and clinical evaluation modified release dosage forms» [20].
Внутрішньом’язові та підшкірні лікарські форми з модифікованим вивільненням
Для суспензій або комплексів чи будь-якого виду матриці, призначених для відстроченого чи пролонгованого вивільнення діючої речовини, для внутрішньом’язового чи підшкірного введення, демонстрацію біоеквівалентності необхідно проводити, дотримуючись правил для лікарських форм із модифікованим вивільненням для позасудинного застосування, наприклад, для трансдермальних лікарських форм, відповідно до діючої настанови.
Лікарські засоби місцевої дії та місцевого застосування
Для лікарських засобів місцевого застосування (оральне, назальне, інгаляційне, офтальмологічне, у дерматології ректальне, вагінальне тощо), що діють у місці застосування, рекомендації можна знайти в інших настановах (CPMP/EWP/4151/00 Rev. 1, CPMP/EWP/239/95) [22, 23].
Відмова від проведення дослідження біоеквівалентності in vivo може бути прийнятною у разі розчинів, наприклад, очних крапель, назальних спреїв або нашкірних розчинів, якщо досліджуваний лікарський засіб належить до однакового типу розчину (водний чи олійний) та містить однакову концентрацію такої ж діючої речовини, що і затверджений на даний час лікарський засіб (референтний лікарський засіб)N. Незначні відхилення у складі допоміжних речовин можуть бути прийнятними, якщо відповідні фармацевтичні властивості досліджуваного та референтного лікарських засобів є ідентичними або суттєво схожими. Будь-які кількісні та якісні відмінності в допоміжних речовинах мають обов’язково бути задовільно обґрунтовані щодо впливу на терапевтичну еквівалентність. Шлях та спосіб введення мають бути також такими ж, як і для затвердженого на даний час лікарського засобу (референтного лікарського засобу)N, якщо інше не обґрунтовано.
Щоразу, коли системний вплив у результаті місцевого застосування препаратів місцевої дії призводить до ризику виникнення системних побічних явищ, його має бути визначено. Необхідно продемонструвати, що системний вплив досліджуваного лікарського засобу не більший, ніж у референтного лікарського засобу, тобто верхня межа 90% довірчого інтервалу не повинна перевищувати верхньої межі прийнятності, біоеквівалентної 125,00.
Гази
Якщо лікарський засіб — це газ для інгаляцій, не вимагається проведення дослідження біоеквівалентності.
Додаток ІІІ. Процедура біовейвер на підставі БСК
І. Вступ
Підхід процедури біовейвер на підставі БСК призначений для зменшення кількості досліджень біоеквівалентності in vivo, тобто він може розглядатися як заміна (surrogate) досліджень біоеквівалентності in vivo. Дослідження біоеквівалентності in vivo можуть не проводитися, якщо припущення про еквівалентність in vivo може бути обґрунтоване задовільними даними in vitro.
Застосування процедури біовейвер на підставі БСК обмежується високорозчинними діючими речовинами з відомою абсорбцією у людини, що не мають вузького терапевтичного індексу (див. розділ 4.1.9 цієї настанови). Процедура біовейвер на підставі БСК застосовується для лікарських засобів у твердих дозованих формах системної дії з негайним вивільненням для орального застосування, що мають однакову лікарську форму. Водночас процедура біовейвер на підставі БСК не застосовується для сублінгвальних, букальних лікарських засобів і лікарських засобів із модифікованим вивільненням. Для лікарських засобів, що диспергуються у ротовій порожнині, підхід процедури біовейвер на підставі БСК може застосовуватися лише тоді, коли абсорбція в ротовій порожнині може бути виключена.
Процедура біовейвер на підставі БСК призначена для розгляду питання біоеквівалентності між відповідним генеричним препаратом та референтними лікарськими засобами. Ці принципи можуть використовуватися для встановлення біоеквівалентності в заявках на генеричні лікарські засоби, на зміни, що потребують нової реєстрації інноваційних лікарських засобів, при змінах, які можливі протягом дії реєстраційного посвідчення, що потребують проведення досліджень біоеквівалентності, а також між лікарськими засобами, що використовувалися на етапі клінічних досліджень, та лікарськими засобами, що виробляються у промислових масштабах та наявні на ринку.
II. Загальні вимоги
Процедуру біовейвер на підставі БСК застосовують до лікарських засобів із негайним вивільненням, якщо:
- доведено, що діюча речовина проявляє високу розчинність і повну абсорбцію (I клас за БСК, див. розділ ІІІ цієї настанови); та
- або дуже швидкорозчинні (>85% діючої речовини за 15 хв), або швидкорозчинні (85% протягом 30 хв) характеристики розчинення in vitro для досліджуваного та референтного лікарських засобів продемонстровані з урахуванням специфічних вимог (див. розділ IV.1 цієї настанови); та
- допоміжні речовини, що можуть вплинути на біодоступність, є якісно і кількісно однаковими. В основному використання однакових допоміжних речовин у подібних кількостях є переважним (див. розділ IV.2 цієї настанови).
Процедуру біовейвер на підставі БСК також застосовують до лікарських засобів з негайним вивільненням, якщо:
- доведено, що діюча речовина проявляє високу розчинність і обмежену абсорбцію (III клас за БСК, див. розділ ІІІ цієї настанови); та
- дуже швидкорозчинні (>85% протягом 15 хв) характеристики розчинення in vitro для досліджуваного та референтного лікарських засобів продемонстровані з урахуванням специфічних вимог (див. розділ IV.1 цієї настанови); та
- допоміжні речовини, що можуть вплинути на біодоступність, є якісно і кількісно однаковими, та інші допоміжні речовини є якісно однаковими і кількісно дуже подібними (див. розділ IV.2 цієї настанови).
В основному ризики невідповідного рішення щодо можливості проведення процедури біовейвер на підставі БСК (наприклад, абсорбція, специфічна для певної ділянки, ризик взаємодії транспортних білків у ділянці абсорбції, склад допоміжних речовин і терапевтичні ризики) мають бути більш критично розглянуті для лікарських засобів, діюча речовина яких належить до III класу за БСК.
III. Діюча речовина
Як правило, переконливі опубліковані дані з рецензованих джерел наукової літератури можуть бути прийнятними для відомих сполук, для того щоб описати характеристики діючої речовини, важливі для проведення процедури біовейвер на підставі БСК.
Процедура біовейвер на підставі БСК може застосовуватися, коли діюча речовина у досліджуваному та референтному лікарських засобах є тією самою. Процедура біовейвер на підставі БСК також може застосовуватися, якщо досліджуваний та референтний лікарські засоби містять різні солі, за умови, якщо обидві належать до I класу за БСК (висока розчинність та повна абсорбція, див. розділи ІІІ.1 та ІІІ.2 цієї настанови). Процедура біовейвер на підставі БСК не застосовується, якщо досліджуваний лікарській засіб містить інший ефір, складний ефір, ізомер, рацемат, комплекс або продукт дериватизації діючої речовини порівняно з референтним лікарським засобом, оскільки означені відмінності можуть призвести до різної біодоступності, що не може бути встановлено на підставі досліджень, які використовуються в концепції процедури біовейвер на підставі БСК.
Діюча речовина не має належати до групи лікарських засобів із вузьким терапевтичним індексом (див. розділ 4.1.9 цієї настанови про лікарські засоби з вузьким терапевтичним індексом).
III.1. Розчинність
Необхідно встановити та обговорити профіль pH-залежної розчинності діючої речовини. Діюча речовина вважається високорозчинною, якщо найвища одноразова доза препарату негайного вивільнення повністю розчиняється у 250 мл буферних розчинів у діапазоні pH 1–6,8 при 37±1 °C. Для доказу необхідно провести дослідження принаймні у трьох буферних розчинах в межах цього діапазону (бажано при pH 1,2; 4,5 та 6,8) і також додатково при рКа, якщо її значення знаходиться у вищезазначеному діапазоні pH. Повторні визначення при кожному значенні pH можуть бути необхідними для досягнення чіткої класифікації розчинності діючої речовини (наприклад, методом струшування колби або іншим обґрунтованим методом). Значення pH кожного буферного розчину необхідно перевіряти до та після введення діючої речовини в буферний розчин.
III.2. Абсорбція
При процедурі біовейвер на підставі БСК надається перевага даним повної абсорбції у людини. В цьому контексті абсорбція вважається повною, якщо вимірюваний рівень абсорбції становить ≥85%. Повна абсорбція в основному пов’язана з високою проникністю. Повна абсорбція препарату має бути обґрунтована на підставі достовірних досліджень у людини. Дані досліджень абсолютної біодоступності або визначення масобалансу можуть бути використані на підтримку цього твердження.
Якщо дані дослідження масобалансу використовуються для підтвердження повної абсорбції, обов’язково має гарантуватися, що метаболіти, враховані при визначенні абсорбованої фракції, утворюються після абсорбції. Тому, коли визначають загальну радіоактивність сечі, має бути гарантовано, що не існує розпаду чи метаболізму незміненої діючої речовини в шлунковій чи кишковій рідині. Оксидативний (І фаза) і кон’югований (ІІ фаза) метаболізм може відбуватися лише після абсорбції (тобто не може відбуватися у шлунковій чи кишковій рідині). Отже, дані дослідження масобалансу підтверджують повну абсорбцію, якщо загальний вміст у сечі вихідної речовини, а також вміст у сечі та калі її метаболітів, що пройшли І оксидативну та ІІ кон’юговану фазу метаболізму, становить ≥85% дози.
Крім того, високорозчинні діючі речовини з неповною абсорбцією, тобто сполуки III класу за БСК, можуть мати право на процедуру біовейвер на підставі БСК за умови, що певні передумови відносно складу препарату і розчинення in vitro виконані (див. також розділ IV.2 «Допоміжні речовини» цієї настанови). Більш жорсткі вимоги також застосовуватимуться до препаратів, що пропонуються до І класу за БСК, якщо повна абсорбція не може бути переконливо продемонстрована.
Як підтвердження можна використовувати дані про біоеквівалентність препаратів для перорального застосування, що є водним розчином і твердою лікарською формою, оскільки це свідчить про те, що обмеження абсорбції зумовлені характеристиками препарату (із негайним вивільненням) та можуть вважатися незначними. Проведені належним чином дослідження проникності in vitro, включаючи стандартні зразки, можуть також вважатися підтверджувальними для даних in vivo.
IV. Лікарський засіб
IV.1. Розчинення in vitro
IV.1.1. Загальні положення
Дослідження мають гарантувати властивості негайного вивільнення і підтверджувати подібність між досліджуваними лікарськими засобами, тобто досліджуваний та референтний лікарські засоби мають виявляти подібність розчинення in vitro при фізіологічно відповідних експериментальних умовах рН. Проте це не встановлює кореляції in vitro/in vivo. Розчинення in vitro має досліджуватися в діапазоні рН 1–6,8 (принаймні рН 1,2; 4,5 та 6,8). Додаткові дослідження можуть бути необхідними при значеннях рН, в яких діюча речовина виявляє мінімальну розчинність. Використання будь-яких поверхнево-активних речовин неприйнятне.
Досліджуваний та референтний лікарські засоби мають відповідати вимогам розділу 4.1.2 основного тексту настанови. Відповідно до цих вимог доцільно досліджувати більше однієї серії досліджуваного та референтного лікарських засобів.
Порівняльні дослідження розчинення in vitro мають відповідати діючим фармакопейним стандартам. Таким чином, необхідно надати детальний опис умов проведення експериментів і аналітичних методик, включаючи валідаційні дані. Рекомендується використовувати 12 одиниць лікарського засобу для кожного дослідження для проведення статистичної оцінки. Звичайними умовами дослідження є:
- прилади: прилад із кошиком або прилад із лопаттю;
- об’єм середовища розчинення — ≤900 мл;
- температура середовища розчинення — 37±1 °C;
- перемішування:
- прилад із кошиком — як правило 100 об./хв;
- прилад із лопаттю — як правило 50 об./хв;
- графік відбору проб: наприклад, 10, 15, 20, 30 і 45 хв;
- буфер: рН 1,0–1,2 (зазвичай 0,1 М розчин кислоти хлористоводневої або штучний шлунковий сік без ферментів), pH 4,5 і 6,8 (або штучна кишкова рідина без ферментів); (необхідно забезпечити відповідний рівень рН протягом усього експерименту; рекомендовані буферні розчини описані в Європейській Фармакопеї або Державній Фармакопеї УкраїниN);
- інші умови: відсутність поверхнево-активних речовин; у разі желатинових капсул або таблеток із желатиновим покриттям може допускатися використання ферментів.
Необхідно представити повний комплект документів щодо результатів порівняльних досліджень in vitro, включаючи протокол випробовування, інформацію щодо серій досліджуваного та референтного лікарських засобів, детальні умови експерименту, валідаційні дані методик проведення експериментів, індивідуальні і середні результати та відповідну підсумкову статистику.
IV.1.2. Оцінка результатів розчинення in vitro
Лікарські засоби вважаються дуже швидкорозчинними, коли не менше 85% зазначеної на етикетці кількості діючої речовини розчиняється за 15 хв. У разі, коли це виконується для генеричного і референтного лікарських засобів, їх подібність може бути прийнята без будь-якого математичного обчислення.
Відсутність значущих відмінностей (подібність) має бути продемонстрована у випадках, коли за більше ніж 15 хв, але не більше ніж за 30 хв досягається практично повне розчинення (принаймні, 85% від зазначеної на етикетці кількості діючої речовини). Фактор подібності (див. Додаток І до цієї настанови) або інші відповідні тести слід використовувати для демонстрації профілю подібності досліджуваного та референтного лікарських засобів. Проте обговорення відмінності профілів розчинення щодо їх клінічної/терапевтичної відповідності вважається недоречним, оскільки дослідження не відображають будь-якої кореляції in vitro/in vivo.
IV.2. Допоміжні речовини
Хоча вплив допоміжних речовин у лікарських формах із негайним вивільненням на біодоступність швидкорозчинних діючих речовин, що повністю абсорбуються (тобто I клас за БСК) вважається малоймовірним, він не може бути повністю виключений. Тому навіть у разі діючих речовин I класу за БСК рекомендується використовувати подібну кількість тих самих допоміжних речовин у складі досліджуваного лікарського засобу як у референтного лікарського засобу.
Якщо процедуру біовейвер застосовують до діючих речовин III класу за БСК, допоміжні речовини мають бути якісно однаковими і кількісно дуже схожими, щоб виключити різні впливи на мембранні транспортери.
Як правило, для діючих речовин I та III класу за БСК слід використовувати добре вивчені допоміжні речовини у звичайній кількості та необхідно розглянути і обговорити можливі взаємодії, що впливають на біодоступність лікарського засобу та/чи характеристики розчинності. Необхідно надати опис функцій допоміжних речовин з обґрунтуванням того, що кількість кожної допоміжної речовини у межах звичайного діапазону. Допоміжні речовини, що можуть впливати на біодоступність, наприклад, сорбіт, маніт, лаурилсульфат натрію чи інші сурфактанти, мають бути визначені, а також оцінено їх можливий вплив на:
- моторику шлунково-кишкового тракту;
- схильність до взаємодії з діючою речовиною (наприклад, комплексоутворення);
- проникність лікарського засобу;
- взаємодію з мембранними транспортерами.
Допоміжні речовини, що можуть впливати на біодоступність, мають бути якісно та кількісно однаковими у випробовуваному та референтному лікарських засобах.
V. Лікарські засоби з фіксованою комбінацією (FCs)
Процедура біовейвер на підставі БСК застосовується до фіксованих комбінацій із негайним вивільненням, якщо всі діючі речовини в FCs належать до I чи III класу за БСК та для допоміжних речовин виконують умови, описані в розділі IV.2.
Інакше необхідно проводити дослідження біоеквівалентності in vivo.
Додаток IV. Представлення біофармацевтичних та біоаналітичних даних у розділі 2.7.1 Модуля 2
І. Вступ
Метою розділу 2.7.1 Модуля 2 реєстраційного досьє є узагальнення всієї відповідної інформації щодо біофармацевтичних досліджень і пов’язаних з ними аналітичних методів.
Цей додаток містить набір шаблонів таблиць для допомоги заявникам у підготовці розділу 2.7.1 Модуля 2, наведено вказівки щодо подачі результатів. Крім того, передбачається що стандартизоване представлення даних сприятиме процесу оцінки. Тому заявникам рекомендується використання цих шаблонів таблиць при підготовці розділу 2.7.1 Модуля 2. Цей додаток призначений для заяв на генеричні лікарські засоби відповідно до статті 10 (1) Директиви 2001/83/ЕС та чинного законодавства України [3]N. Крім того, у разі потреби, також рекомендується використовувати ці шаблони таблиць і в інших випадках, таких як заяви на зміни до матеріалів реєстраційного досьє, заяви на фіксовані комбінації, заяви на зміни, що потребують нової реєстрації, та заяви на гібридні лікарські засоби.
ІІ. Інструкції щодо заповнення та подачі таблиць
Таблиці необхідно заповнювати лише для основних досліджень, що зазначені в досьє, відповідно до розділу 6.1 цієї настанови. Якщо є більше, ніж одне основне дослідження, індивідуальні таблиці мають бути підготовлені для кожного дослідження. Крім того, мають бути дотримані такі вказівки для таблиць:
- Інформація щодо не схваленого в ЄС референтного лікарського засобу (non-EU reference products) не вимагається (в разі подачі заяви в ЄС).
- Таблиці в розділі 3 мають бути заповнені окремо для кожного аналіту в дослідженні. Якщо існує більше ніж один досліджуваний лікарський засіб, то структура таблиці має бути скоригована.
- Таблиці в розділі 4 мають заповнюватися для методики, що використовується у підтверджувальному (основному) дослідженні біоеквівалентності. Якщо визначається більше ніж один аналіт, то табл. 4.1 і потенційно табл. 4.3 мають заповнюватися для кожного аналіту окремо.
Загалом заявникам рекомендується використовувати перехресні посилання та примітки, якщо додається додаткова інформація. Поля, що не застосовуються, мають бути підписані як «не застосовуються» разом із пояснювальним посиланням — за необхідності.
Крім того, в кожному розділі шаблону має бути перехресне посилання на розміщення підтверджувальної документації або на вихідні дані в реєстраційному досьє.
Таблиці не мають бути відсканованими копіями, і інформація в них має бути зручна для пошуку. Настійно рекомендується, щоб заявники подавали розділ 2.7.1 Модуля 2 також у WORD (.doc) або RTF-форматі.
ІІІ. Вказівки щодо документації для процедури біовейвер на підставі БСК
Відповідні дані для обґрунтування застосування процедури біовейвер на підставі БСК мають бути включені в розділ 5.3.1 «Звіти дослідження порівняльної біодоступності та біоеквівалентності» Модуля 5. Короткий огляд даних повинен бути наданий в розділі 2.7.1 з обґрунтуванням проведення процедури біовейвер на підставі БСК та списком відповідних посилань.
1. Відмова проведення досліджень in vivo для різних дозувань
Таблиця 1.1. Якісний та кількісний склад досліджуваного лікарського засобу
|
Компонент |
Функція |
Діюча речовина, мг (інформація на етикетці) |
|||||
|
ХХ мг |
ХХ мг |
ХХ мг |
|||||
|
Ядро |
Кількість на одиницю |
%* |
Кількість на одиницю |
%* |
Кількість на одиницю |
%* |
|
|
Усього |
100% |
100% |
100% |
||||
|
Оболонка |
|||||||
|
Усього |
100% |
100% |
100% |
||||
*Кожний компонент виражається у відсотках (w/w) від загальної маси ядра або маси з оболонкою чи w/v для розчинів.
Інструкція
Включити склад усіх дозувань. Зробити додаткові колонки — за необхідності.
Таблиця 1.2. Дані розчинення іn vitro
|
Місце проведення тесту |
Розміщення звіту дослідження (том, сторінка, посилання) |
|
|
Умови розчинення |
Апаратура |
|
|
Оберти за хвилину |
||
|
Середовище |
||
|
Об’єм |
||
|
Температура |
||
|
Сурфактант |
||
|
Середовище розчинення |
Час відбору проб |
f2 |
||||||
| 5 | 10 | 15 | 20 | |||||
|
Дозування № кількість одиниць № серії |
pH |
|||||||
|
pH |
||||||||
|
pH |
||||||||
|
QC-середовище1 |
||||||||
|
Дозування № кількість одиниць № серії |
pH |
|||||||
|
pH |
||||||||
|
pH |
||||||||
|
QC-середовище1 |
||||||||
|
Дозування № кількість одиниць № серії |
pH |
|||||||
|
pH |
||||||||
|
pH |
||||||||
|
QC-середовище1 |
||||||||
1Тільки якщо середовище розчинення, призначене для оцінки вивільнення лікарського засобу, відрізняється від буферних.
Інструкція
Заповнювати цю таблицю потрібно, тільки якщо необхідна відмова від проведення дослідження біоеквівалентності in vivo для додаткових дозувань на додаток для дозування, що досліджується in vivo. Лише середнє значення відсотка розчинення має бути представлене, але необхідно позначити (*) результат, якщо RSD вище 10%, окрім першої точки, де ліміт становить 20%. Доповніть таблицю додатковими колонками відповідно до часу відбору проб. Якщо заявлено більше трьох діючих речовин, потрібно зробити додаткові рядки. Значення f2 мають бути розраховані відносно дозування, для якого доведено біоеквівалентність. Необхідно обґрунтувати альтернативний метод, використаний для розрахунку, якщо підхід f2 не використовується.
2. Інформація про дослідження біоеквівалентності
Таблиця 2.1. Інформація про досліджуваний та референтний лікарські засоби
|
Характеристика препарату |
Досліджуваний |
Референтний |
|
Назва |
||
|
Дозування |
||
|
Лікарська форма |
||
|
Виробник |
||
|
№ серії |
||
|
Розмір серії (біосерія) |
||
|
Кількісний вміст1 (% від зазначеного на упаковці) |
||
|
Розмір промислової серії |
||
|
Термін придатності (дата повторного тестування) |
||
|
Розміщення сертифіката на препарат |
Том/сторінка, посилання |
Том/сторінка, посилання |
|
Назва країни, в якій було закуплено референтний препарат |
||
|
Цей продукт був використаний у таких дослідженнях |
Код досліджень |
Код досліджень |
1Вноситься інформація для кожної активної субстанції фіксованої комбінації.
Інструкція
Якщо більше ніж одну серію досліджуваного чи референтного лікарського засобу використано для досліджень біоеквівалентності, необхідно заповнювати табл. 2.1 для кожної досліджуваної/референтної серії.
Таблиця 2.2. Місце(-я) проведення <дослідження>
|
Показник |
Назва організації |
Адреса організації |
Інспекція |
|
| Рік | Організація | |||
|
Місце клінічної частини дослідження |
||||
|
Місце біоаналітичної частини дослідження |
||||
|
Місце фармакокінетичного і статистичного аналізу |
||||
|
Спонсор дослідження |
||||
Таблиця 2.3. Інформація щодо <дослідження>
Назва дослідження:
|
Розміщення звіту том/сторінка, посилання Період дослідження
Дизайн Доза:
Кількість суб’єктів:
|
Інструкція
Необхідно заповнювати табл. 2.2 і 2.3 для кожного дослідження.
3. Результати
Таблиця 3.1. Фармакокінетичні дані для <аналіт> в <Код дослідження>
|
Фармакокінетичні параметри |
4Середнє арифметичне (± стандартне відхилення SD) |
|
| Досліджуваний лікарський засіб | Референтний лікарський засіб | |
|
|
||
|
AUC(0–t)1 |
||
|
AUC(0–∞)2 |
||
|
Cmax |
||
|
tmax3 |
||
1Можна вказувати AUC(0–72 год) замість AUC(0–t) у дослідженнях із періодом відбору зразків 72 год, коли концентрація при 72 год вимірюється. Тільки для препаратів негайного вивільнення.
2Немає необхідності вказувати AUC(0–∞), якщо AUC(0–72 год) вказується замість AUC(0–t).
3Медіана (Min, Max).
4Середнє арифметичне (±SD) може бути замінене на середнє геометричне (±CV%).
Таблиця 3.2. Додаткові фармакокінетичні дані для <аналіт> в <Код дослідження>
|
Концентрація в біологічній рідині, де |
Споріднена інформація |
|
AUC(0–t)/AUC(0–∞)<0,81 |
<Ідентифікаційний номер суб’єкта, # періоду, F2> |
|
Cmax є першою точкою |
<Ідентифікаційний номер суб’єкта, # періоду, F> |
|
Зразок до введення дози >5% Cmax |
<Ідентифікаційний номер суб’єкта, # періоду, F, концентрація до введення дози> |
1Тільки якщо остання точка відбору AUC(0–t) менше ніж 72 год.
2F=Т для досліджуваного препарату та F=R для референтного препарату.
Таблиця 3.3. Оцінка біоеквівалентності для <аналіт> в <Код дослідження>
|
Фармакокінетичні |
Середнє геометричне співвідношення досліджуваний/референтний лікарський засіб |
Довірчі інтервали |
CV%1 |
|
AUC2(0–t) |
|||
|
Cmax |
1Pозраховане за середньоквадратичною різницею (Residual Mean Squares). Для досліджень із повторним дизайном зазначають внутрішньосуб’єктну варіабельність (%), використовуючи дані тільки референтного лікарського засобу.
2У деяких випадках AUC(0–72 год).
Інструкція
Необхідно заповнювати табл. 3.1–3.3 для кожного відповідного аналіту.
4. Біоаналітика
Таблиця 4.1. Валідація біоаналітичної методики
|
Звіт щодо валідації аналітичного методу Місцезнаходження |
<Код дослідження> <Том/сторінка, примітка> |
|
|
Ця аналітична методика використовувалася в таких дослідженнях: |
<Коди досліджень> |
|
|
Короткий опис методики |
<Наприклад, HPLC/MS/MS, GC/MS, метод зв’язування лігандів> |
|
|
Біологічна матриця |
<Наприклад, плазма крові, кров, сеча тощо> |
|
|
Аналіт Місцезнаходження сертифіката на препарат |
<Назва> <Том/сторінка, примітка> |
|
|
Внутрішній стандарт (IS)1 Місцезнаходження сертифіката на препарат |
<Назва> <Том/сторінка, примітка> |
|
|
Калібрувальні концентрації (одиниці виміру) |
||
|
Нижня межа кількісного визначення (одиниця виміру) |
<Нижня межа кількісного визначення> <Правильність %> <Прецизійність %> |
|
|
QC концентрації (одиниці виміру) |
||
|
Міжсерійна правильність |
<Діапазон або отримані значення QC> |
|
|
Міжсерійна прецизійність |
<Діапазон або отримані значення QC> |
|
|
Внутрішньосерійна правильність |
<Діапазон або отримані значення QC> |
|
|
Внутрішньосерійна прецизійність |
<Діапазон або отримані значення QC> |
|
|
Матричний фактор (MF) (всі QC)1 IS нормалізований матричний фактор (всі QC)1 Коефіцієнт варіації (%) IS нормалізованого матричного фактора (всі QC)1 Відсоток отриманих значень зразків контролю якості (QCs) в діапазоні 85–115% від н.з.1, 4 Відсоток серій матриці з отриманим середнім значенням <80 та >120% від н.з.1, 4 |
Низький QC <Середнє значення> <Середнє значення> <CV%> <%> <%> |
Високий QC <Середнє значення> <Середнє значення> <CV%> <%> <%> |
|
Довгострокова стабільність основного розчину і робочих розчинів² (зміни, що спостерігаються, %) |
<Час> при <˚С> <%, діапазон або отримані значення QC> |
|
|
Короткострокова стабільність у біологічній матриці при кімнатній температурі або при температурі процесу обробки зразків (зміни, що спостерігаються, %) |
<Час> при <˚С> <%, діапазон або отримані значення QC> |
|
|
Довгострокова стабільність у біологічній матриці (зміни, що спостерігаються, %) Розміщення |
<Час> при <˚С> <%, діапазон або отримані значення QC> <Том/сторінка, примітка> |
|
|
Стабільність зберігання в автоматичному пробовідбірнику (зміни, що спостерігаються, %) |
<Час> при <˚С> <%, діапазон або отримані значення QC> |
|
|
Стабільність після пробопідготовки (зміни, що спостерігаються, %) |
<Час> при <˚С> <%, діапазон або отримані значення QC> |
|
|
Стабільність при заморожуванні та розморожуванні (зміни, що спостерігаються, %) |
<Температура, кількість циклів> <Діапазон або отримані значення QC> |
|
|
Допустимість розведення |
Концентрація розбавлення <у скільки разів> Правильність <%> Прецизійність <%> |
|
|
Часткова валідація3 Розміщення |
<Описати коротко причину ревалідації> <Том/сторінка, примітка> |
|
|
Перехресна валідація3 Розміщення |
<Описати коротко причину перехресної валідації> <Том/сторінка, примітка> |
|
1Може не застосовуватися для даної аналітичної методики.
2Допустимо повідомляти результати короткострокової стабільності, якщо немає даних довгострокової стабільності основного і робочого розчинів.
3Ці рядки необов’язкові. Повідомте про будь-яку валідацію, що була проведена після початкової валідації.
4н.з. — номінальне значення.
Інструкція
Деякі підходи в табл. 4.1 застосовують тільки для хроматографічних методів і не застосовують для ліганд-зв’язуючих методів. Вказувати «не застосовується», якщо підходи не є відповідними для даних аналізів. Необхідно заповнювати табл. 4.1 для кожного відповідного аналіту.
Таблиця 4.2. Період зберігання зразків дослідження
|
Код дослідження1 і аналіту |
Найдовший період зберігання |
|
<кількість> днів при температурі <˚С> |
|
|
<кількість> днів при температурі <˚С> |
1Тільки для основних досліджень.
Таблиця 4.3. Аналіз зразків <код дослідження>
|
Аналіт |
<Назва> |
|
Загальна кількість відібраних зразків |
<кількість> |
|
Загальна кількість зразків із прийнятними результатами |
<кількість> |
|
Загальна кількість повторно проаналізованих зразків1, 2 |
<кількість> |
|
Загальна кількість аналітичних циклів1 |
<кількість> |
|
Загальна кількість прийнятних аналітичних циклів1 |
<кількість> |
|
Повторний аналіз досліджуваних зразків з метою оцінки правильності визначення аналіту у клінічних зразкахN (incurred-samples) |
|
|
Кількість зразків |
<кількість> |
|
Відсоток зразків, у яких різниця між двома показниками була менше ніж 20% середнього значення хроматографічних аналізів або менше ніж 30% для методу зв’язування лігандів |
<%> |
1Без повторно проаналізованих досліджуваних зразків з метою оцінки правильності визначення аналіту в клінічних зразкахN (incurred-samples).
2З інших причин, окрім неприйнятності аналітичного циклу.
Інструкція
Необхідно заповнити табл. 4.3 для кожного відповідного аналіту.
Доповнення 1
Рекомендації щодо визначення абсолютної та відносної біодоступності
Абсолютна біодоступність
Інформація щодо абсолютної біодоступності важлива при загальній оцінці фармакокінетики лікарської речовини. Для ряду хімічних сполук інформація щодо абсолютної біодоступності полегшує оцінку для досліджень масобалансу та дає можливість робити висновки щодо впливу різних шляхів виведення на кліренс речовини. Ця інформація важлива при визначенні необхідності досліджень за участю пацієнтів із нирковою та печінковою недостатністю, а також необхідності досліджень міжлікарської взаємодії за рівнем екскреції жовчі. Інформація також корисна при прогнозуванні наслідків пресистемної міжлікарської взаємодії, а також рівнів абсорбції та метаболізму.
Таким чином, якщо це можливо, для нових лікарських речовин системної дії абсолютна біодоступність має бути визначена шляхом порівняння біодоступності лікарської форми невнутрішньовенного шляху введення з біодоступністю лікарської форми внутрішньовенного шляху введення. Для речовин із нелінійною фармакокінетикою увага має бути приділена дозі(-ам), що використовувалася(-ися) для оцінки абсолютної біодоступності. Крім того, дані з абсолютної біодоступності є важливими при проведенні оцінки за процедурою біовейвер на підставі БСК [4].
Відносна біодоступність
Рекомендується отримувати інформацію щодо відносної біодоступності різних лікарських форм (або складів), що використовувалися у процесі розробки препарату. За визначенням, відносна біодоступність — це порівняння біодоступності різних лікарських форм (або їх різних складів), що вводяться однаковим або різним невнутрішньовенним шляхом (наприклад, таблетки порівняно з оральним розчином).
Дослідження біоеквівалентності необхідні, крім випадку процедури біовейвер на підставі БСК, якщо зміни складу, що можуть вплинути на швидкість і ступінь абсорбції, проведені під час розробки препарату, а саме змінено склад препарату, що використовувався у ІІІ фазу клінічних досліджень, порівняно з кінцевим складом. Дослідження відносної біодоступності (або порівняльні дослідження біодоступності) рекомендовані між різними складами препарату протягом І, ІІ та ІІІ фази. Не вимагається демонстрація біоеквівалентності складів препарату, що використовувалися у ІІ та ІІІ фазі. Передбачається, що будь-які відмінності у швидкості чи ступені абсорбції між цими складами враховуються в дизайні досліджень ІІІ фази. Клінічна значущість відмінностей складів лікарського засобу, використовуваних у I–III фазах дослідження, має бути обговорена у додатках для нових хімічних діючих речовин у розділах 2.5 і 2.7.1 і врахована в оцінці фармакокінетичних даних у розділі 2.7.2 Модуля 2.
Рекомендації щодо супрабіодоступних препаратів
Супрабіодоступність досліджуваного препарату виявляється у суттєво вищому ступені абсорбції, ніж у референтного лікарського засобу.
У разі виявлення супрабіодоступності необхідно розглянути можливість розробки лікарської форми з меншою силою дії лікарського засобу. У цьому випадку має бути представлена біофармацевтична розробка і має бути подане остаточне порівняльне дослідження біодоступності зміненого нового продукту порівняно з референтним лікарським засобом. Можливість відмінностей у впливі їжі на швидкість та/чи ступінь абсорбції або можливість відмінностей в абсорбції взаємодії між зміненим новим продуктом та референтним лікарським засобом має бути обговорена, коли це доречно оцінювати в дослідженнях in vivo.
У разі якщо не було розроблено лікарської форми з меншою силою дії, властивості супрабіодоступного продукту мають бути вивчені у клінічних дослідженнях.
Доповнення 2
Можливість використання схеми трьохперіодного перехресного повторного дизайну для демонстрації внутрішньосуб’єктної варіабельності Сmax
Вимагається використовувати повторний дизайн, при якому референтний лікарський засіб прийнято більше ніж один раз, для демонстрації, що внутрішньосуб’єктна варіабельність для Сmax референтного лікарського засобу вища 30%. Найбільш доцільною в разі обґрунтування використання трьохперіодного повторного дизайну для розширення критеріїв прийнятності для Сmax була б рандомізація суб’єктів, які застосовують препарати, у такому порядку: RRT, RTR та TRR (де R — референтний лікарський засіб, Т — досліджуваний лікарський засіб). Такий дизайн — найбільш доцільний, оскільки всі суб’єкти отримують референтний лікарський засіб двічі, і, таким чином, оцінка внутрішньосуб’єктної варіабельності базується на даних усіх суб’єктів.
Рандомізація суб’єктів, в разі обґрунтування використання трьохперіодного повторного дизайну для розширення критеріїв прийнятності Сmax, які отримують препарати у порядку TRT та RTR, не вважається оптимальною. Однак такий дизайн міг би забезпечити оцінку внутрішньосуб’єктної варіабельності досліджуваного та референтного лікарських засобів. Оскільки ця оцінка базується лише на половині всіх суб’єктів дослідження, пов’язана з цим невизначеність буде більш вираженою, ніж така при використанні дизайну RRT/RTR/TRR. Таким чином, якщо використовується TRT-/RTR-дизайн, вищий шанс некоректного висновку щодо високої варіабельності референтного лікарського засобу.
Відповідно до вимог цієї настанови та вимог настанови ЄС [4], принаймні 12 суб’єктів необхідно для забезпечення розгляду валідності даних дослідження біоеквівалентності та для оцінки усіх ключових параметрів. Таким чином, якщо використовується трьохперіодний повторний дизайн для розширення критеріїв прийнятності Сmax із порядком введення TRT/RTR, то вважається, що принаймні 12 суб’єктів буде необхідно для забезпечення даних із послідовності RTR. Це передбачає дослідження, в якому вимагатиметься в загальній кількості принаймні 24 суб’єкти, якщо однакова кількість суб’єктів задіяна для двох прийомів у двох послідовностях.
Національний додаток (довідковий). Перелік редакційних змін та доповнень
Ця настанова розроблена на підставі настанов із клінічної фармакології та фармакокінетики, прийнятих в ЄС, а саме:
- CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» (Настанова щодо досліджень біоеквівалентності) [4];
- EMA/CHMP/600958/2010/Corr.* «Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/Corr.**): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1» (Додаток IV до Настанови щодо досліджень біоеквівалентності (CPMP/EWP/QWP/1401/98 Rev. 1/Corr.**): Представлення біофармацевтичних та біоаналітичних даних в Модулі 2.7.1) [5];
- EMA/618604/2008 Rev. «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» (Питання та відповіді: Позиція щодо специфічних питань, направлених до PKWP) [6].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я (МОЗ) України.
Ця настанова регулярно переглядатиметься відповідно до змін і доповнень, що вносяться до «Guideline on the Investigation of Bioequivalence» та «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» [4–6].
Настанова містить положення, що відповідають чинному законодавству.
До цієї настанови внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни внесено безпосередньо у пункти, яких вони стосуються; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5:2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [7], а позначення — відповідно до вимог стандарту СТ МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [8];
- додатково введено такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Познаки та скорочення», а також Доповнення 1 та 2 відповідно до інформації, викладеної в документі [6], та національний додаток «Бібліографія», оформлені відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладення, оформлення та вимоги до змісту нормативних документів» [7] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [9]. Розділ «Зміст» цієї настанови подано з урахуванням додаткових структурних елементів;
- основні положення викладено у розділі «Загальні положення». Кожний структурний елемент та його номер у цій настанові відповідають таким у настанові CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** [4], за винятком розділу «Definition». Кожний структурний елемент та його номер у цій настанові відповідають таким у настанові EMA/CHMP/600958/2010/Corr.* [5]; питання, викладені у розділах «Recommendations on determination of absolute and relative bioavailability» та «Suitability of a 3-period replicate design scheme for the demonstration of within-subject variability for Cmax» настанови EMA/618604/2008 Rev. [6], стосовно напрямку сфери дії настанові CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** [4], викладено у Доповненнях 1 та 2;
- розділ «Терміни та визначення понять» викладено на підставі розділу «Definition» настанови CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** [4]. Цей розділ не позначено номером та викладено після розділу «Нормативні посилання». Усі терміни у розділі «Терміни та визначення понять» наведено в алфавітному порядку, вони супроводжуються посиланням на нормативні документи, бібліографічний опис яких наведено у національному додатку «Бібліографія»;
- у розділі «Нормативні посилання» додатково наведено бібліографічний опис нормативних документів, зазначених у цій настанові;
- у національному додатку «Бібліографія» додатково наведено бібліографічний опис нормативних документів, на які є посилання у цій настанові;
- у розділі «Терміни та визначення понять» додатково наведено визначення термінів, що використовуються у цій настанові: «діюча речовина (активний фармацевтичний інгредієнт, субстанція)», «критерії прийнятності (межі прийнятності)», «енантіомери», «лікарський засіб», «лікарські препарати (готові лікарські засоби, ліки, медикаменти)», «негайне вивільнення», «модифіковане вивільнення», «сила дії лікарського засобу (дозування)», «специфікація» — з посиланням на відповідні нормативні документи, в яких вони наведені;
- розділ «Познаки та скорочення» складено на підставі матеріалів розділу «Definition» настанови CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** [4], а також у цьому розділі додатково наведено позначення скорочень, що використовуються у цій настанові;
- у цій настанові слова «заявка на торгову ліцензію» («marketing аuthorisation аpplication, аpplication») замінено на «реєстраційне досьє»; «торгова ліцензія» («marketing аuthorisation») — на «реєстрація»; «зміни» («variations») замінено на «зміни, можливі протягом дії реєстраційного посвідчення»; «розширення» («extension») замінено на «зміни, що потребують нової реєстрації лікарського засобу»; «що доступний на ринках» («if available on the market») замінено на «що доступний на ринках світу», «Ph.Eur. buffers recommended» замінено на «рекомендовані Європейською Фармакопеєю буфери»; «BCS based Biowaiver» замінено на «процедура біовейвер на підставі БСК»;
- по всьому тексту цієї настанови внесено редакційні зміни у посиланнях на структурні елементи цієї настанови, наприклад, замість «(see also section 4.1.6)» вказано «(див. також розділ 4.1.6 цієї настанови)»; замість «(see section 4.1.6)» вказано «(див. розділ 4.1.6 цієї настанови)»; замість «(see Appendix II)» вказано «(див. Додаток ІІ до цієї настанови)»;
- після слів «Європейська Фармакопея» додано слова «або Державна Фармакопея УкраїниN»;
- додатково до посилань на настанови ЄС зроблено посилання на відповідні гармонізовані документи, затверджені в Україні;
- додатково до посилань на конкретні документи ЄС зроблено посилання на чинне законодавство ЄС;
- внесено уточнення у визначення фармакокінетичних параметрів Ae(0–t), AUC(0–τ), Сmах, Сmах,ss;
- в розділи 1.3 «Інші типи заяв», 3 «Законодавча база», 4.1.2 «Референтний та досліджуваний лікарські засоби», «Лікарський засіб із фіксованою комбінацією» Додатку ІІ, 1 «Вступ» Додатку IV внесено уточнення щодо назв типів лікарських засобів, що подаються на державну реєстрацію, та вимог до них відповідно до вимог наказу № 460 [3];
- в абзац четвертий розділу 2 «Сфера застосування» внесено доповнення щодо можливості визначення, в разі обґрунтування, концентрації метаболіту;
- в останній абзац розділу 2 «Сфера застосування» внесено доповнення щодо регуляторного документа, прийнятого в Україні (наказ № 460 [3]), в якому викладено вимоги щодо доведення біоеквівалентності генеричних лікарських засобів;
- в розділ 3 «Законодавча база» внесено додатково посилання на настанови СТ-Н МОЗУ та настанови ЕМА [6]. Внесено доповнення щодо проведення консультацій відповідно до чинного законодавства України;
- в розділ 4.1 «Дизайн, проведення та оцінка досліджень біоеквівалентності» внесено доповнення щодо регуляторного документа, прийнятого в Україні (наказ № 690 [15]), та уточнення щодо вибору референтного лікарського засобу;
- в перший абзац розділу 4.1.2 «Референтний та досліджувані лікарські засоби» внесено уточнення щодо вимог пункту 6 (1) другого параграфу Директиви 2001/83/ЕС зі змінами [13];
- до пункту «Упаковка досліджуваних лікарських засобів» розділу 4.1.2 «Референтний та досліджувані лікарські засоби» внесено посилання на настанову СТ-Н МОЗУ 42-4.0:2016 [18]N;
- в розділі 4.1.7 «Біоаналітична методологія» замість позначення «SOP» використано «стандартна операційна процедура»;
- 4.1.10 «Високоваріабельні діючі речовини або препарати» внесено посилання на Доповнення 2 настанови.
- в перший абзац розділу 4.3.1 «Звіт про результати дослідження біоеквівалентності» внесено доповнення щодо регуляторного документа, прийнятого в Україні (наказ № 690 [15] та Закон України про лікарські засоби [17]);
- в третій абзац розділу 4.3.1 «Звіт про результати дослідження біоеквівалентності» внесено доповнення щодо регуляторного документа, прийнятого в Україні (наказ № 460 [3]);
- в розділі 4.4 «Заяви на зміни, що можуть бути протягом дії реєстраційного посвідчення» та пункті «Лікарські форми з модифікованим вивільненням системної дії» Додатку ІІ вказано актуальну версію настанови СРМР/EWP/280/96 Corr. 1 «Guideline on the pharmacokinetic and clinical evaluation modified release dosage forms» [20];
- в пунктах «Парентеральні розчини», «Ліпосомальні, міцелярні лікарські засоби та лікарські засоби у формі емульсій для внутрішньовенного введення», «Лікарські засоби місцевої дії та місцевого застосування» Додатку ІІ внесено уточнення щодо поняття «затверджений на даний час лікарський засіб» — референтний лікарський засіб;
- по тексту замінено «Модуля 2.7» або «Модуля 2.7.1» на «розділ 2.7 Модуля 2» або «розділ 2.7.1 Модуля 2»;
- по тексту замінено «ODT» на «таблетки, що диспергуються в ротовій порожнині»;
- по тексту внесено уточнення, в разі використання терміну «biowaiver» зазначається «відмова від проведення досліджень біоеквівалентності in vivo», та в разі використання терміну «BCS-based biowaiver» зазначається «процедура біовейвер на підставі БСК»;
- по тексту Додатку IV замість «Study ID» зазначено «Код дослідження»;
- в табл. 2.2. Додатку IV замість «EU Authority Inspection» зазначено «Інспекція»;
- в табл. 4.2. та 4.3 Додатку IV замість «#» зазначено «кількість»;
- в табл. 4.3 Додатку IV внесено уточнення: «Повторний аналіз досліджуваних зразків (incurred-samples)» замінено на «Повторний аналіз досліджуваних зразків з метою оцінки правильності визначення аналіту в клінічних зразкахN (incurred-samples)».
Національний додаток (довідковий). Бібліографія
1. The Rules Governing Medicinal Products in the European Union. Notice to Applicants — Vol. 2B. — Common Technical Document. — May 2008. — European Commission. Enterprise and Industry Directorate-General (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 2В. — Загальний технічний документ. Європейська комісія. Головний директорат з підприємництва та промисловості).
2. The Rules Governing Medicinal Products in the European Union. Notice to Applicants Vol. 2A — Procedures for marketing authorisation. Chapter 1 — Marketing authorisation (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 2А. — Процедури реєстрації. Розділ 1 — Реєстрація).
3. Наказ МОЗ України від 26 серпня 2005 р. № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення» у редакції наказу МОЗ України від 23 липня 2015 р. № 460, що зареєстрований у Міністерстві юстиції України 7 жовтня 2015 р. за № 1210/27655.
4. CPMP/QWP/EWP/1401/98 Rev. 1/Corr.** «Guideline on the Investigation of Bioequivalence» (Настанова щодо досліджень біоеквівалентності).
5. EMA/CHMP/600958/2010/Corr.* «Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1» (Додаток IV до Настанови щодо досліджень біоеквівалентності (CPMP/EWP/QWP/1401/98 Rev. 1): Представлення біофармацевтичних та біоаналітичних даних в Модулі 2.7.1).
6. EMA/618604/2008 Rev. «Questions & Answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP)» (Питання та відповіді: Позиція щодо специфічних питань, направлених до PKWP).
7. ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів. — Київ, Держспоживстандарт України, 2003.
8. Настанова СТ-Н МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації», що затверджена наказом МОЗ України від 14 вересня 2005 р. № 471 «Про затвердження документів з питань стандартизації фармацевтичної продукції».
9. ДСТУ 1.7-2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів — Київ, Держспоживстандарт України, 2003.
10. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
11. The Rules Governing Medicinal Products in the European Union. — Vol. 4. — EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 4. — Правила ЄС з належної виробничої практики лікарських препаратів для людини та застосування у ветеринарії).
12. CPMP/ICH/367/96 (ICH Topic Q6A) «Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000» (Керівні вказівки. Специфікації: контрольні випробування та критерії прийнятності, 2000).
13. Directive 2001/83/EC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use (Директива 2001/83/EC Європейського Парламенту та Ради від 6 листопада 2001 р. про кодекс Співтовариства відносно лікарських препаратів, призначених для споживання людьми).
14. Directive 2001/20/EC of the European Parliament and of the Council, of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use (Директива Європейського Парламенту та Ради 2001/20/EC «Про наближення законів, підзаконних актів та адміністративних положень держав-членів стосовно запровадження належної клінічної практики при проведенні клінічних випробувань лікарських засобів для вживання людиною» від 4 квітня 2001 р.).
15. Наказ МОЗ України від 23 вересня 2009 р. № 690 «Про затвердження Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань», що зареєстрований у Міністерстві юстиції України 29 жовтня 2009 р. за № 1010/17026 (зі змінами).
16. COMMISSION DIRECTIVE 2005/28/EC, of 8 April 2005, laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products (Директива Комісії 2005/28/EC від 8 квітня 2005 р., що встановлює принципи та детальні настанови належної клінічної практики, які стосуються досліджуваних лікарських засобів для вживання людиною, а також вимог надання дозволу на виготовлення або імпорт таких продуктів).
17. Закон України «Про лікарські засоби» від 4 квітня 1996 р.
18. Настанова СТ-Н МОЗУ 42-4.0:2016 «Лікарські засоби. Належна виробнича практика», що затверджена наказом МОЗ України від 29 липня 2016 р. № 798 «Про внесення змін до наказу МОЗ України від 16 лютого 2009 р. № 95».
19. ICH E3, CPMP/ICH/137/95 «Structure and Content of Clinical Study Reports» (Структура та зміст звіту клінічного дослідження).
20. СРМР/EWP/280/96 Corr. 1 «Guideline on the pharmacokinetic and clinical evaluation modified release dosage forms» (Настанова з оцінки фармакокінетики та клініки лікарських форм з модифікованим вивільненням).
21. CPMP/EWP/240/95 Rev. 1 «Guideline on clinical development of fixed combination medicinal products» (Настанова з клінічної розробки фіксованих комбінацій лікарських препаратів).
22. CPMP/EWP/4151/00 Rev. 1 «Requirements for clinical documentation for orally inhaled products (OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of Asthma and Chronic Obstructive Pulmonary Disease (COPD)» (Вимоги до клінічної документації оральних інгаляційних препаратів, включаючи вимоги для доведення терапевтичної еквівалентності двох інгаляційних препаратів, що використовуються для лікування астми та хронічної обструктивної хвороби легень).
23. CPMP/EWP/239/95 «Clinical Requirements for Locally Applied, Locally Acting Products containing Known Constituents» (Клінічні вимоги для препаратів місцевої дії та місцевого використання, що містять відомі компоненти).
Ключові слова: біоеквівалентність, фармакокінетика, процедура біовейвер за БСК, генерик, in vitro, розчинення.