
СТАНДАРТ
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ОЦІНКА ВІРУСНОЇ БЕЗПЕКИ БІОТЕХНОЛОГІЧНИХ ПРОДУКТІВ, ОТРИМАНИХ З КЛІТИННИХ ЛІНІЙ ЛЮДСЬКОГО АБО ТВАРИННОГО ПОХОДЖЕННЯ (Q5А (R1))
СТ-Н МОЗУ 42-6.1:2016
Видання офіційне
Київ
Міністерство охорони здоров’я України
2016
Передмова
1 РОЗРОБЛЕНО: ДП «Державний експертний центр МОЗ України»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: Т. Талаєва, д-р мед. наук, професор; О. Баула, канд. хім. наук; В. Широбоков, д-р мед. наук, професор, академік НАМН України, академік НАН України, І. Кудрявцева, д-р фарм. наук; Л. Дорошук, М. Шилов, канд. мед. наук
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Міністерство охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 19.10.2016 р. № 1120
3 Ця настанова відповідає документу:
CPMP/ICH/295/95 (ICH Q5А (R1)) «Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin» (Оцінка вірусної безпеки біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2016
© Державний експертний центр МОЗ України
Національний вступ
Біотехнологічні лікарські препарати, отримані з клітинних ліній тваринного та людського походження, можуть представляти небезпеку зараження пацієнтів вірусними інфекціями, незважаючи на ретельний відбір та дослідження донорів клітин.
Видалення або інактивація кожного вірусу теоретично необхідні, однак на практиці це може бути нездійсненно та не потрібно. Реальним є зниження патогенного вірусного навантаження до остаточного рівня, який не є інфекційним. Методів інактивації вірусів відомо достатньо багато, хоча тільки деякі з них застосовуються при виробництві біологічних/біотехнологічних лікарських препаратів.
Інактивація вірусів — вкрай відповідальна процедура, ефективність та безпека якої мають бути переконливо підтверджені. Вирішальне значення має точне дотримання процесів виробництва, що регулюються відповідно до вимог Належної виробничої практики [3, 4].
Оцінка вірусної безпеки продуктів біотехнологічного походження, отриманих з клітинних ліній тваринного та людського походження, на етапі їх розробки, інактивація вірусів під час виробничого процесу є обов’язковою вимогою для підтвердження безпеки таких продуктів при реєстрації, і ці дані повинні міститися у реєстраційному досьє [1].
В Європейському Союзі (ЄС) діє спеціальна настанова CPMP/ICH/295/95 (ICH Q5А (R1)) «Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin» [2], у якій описані усі необхідні випробування, що мають бути проведені для встановлення належного рівня вірусної безпеки лікарського засобу.
Ця настанова розроблена на підставі настанови, прийнятої в ЄС:
CPMP/ICH/295/95 (ICH Q5А (R1)) «Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin» (Оцінка вірусної безпеки біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження) [2].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству України.
До цієї настанови було внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо у пункти, яких вони стосуються; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [5], а позначення — відповідно до вимог стандарту СТ-Н МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [6];
- додатково введено такі структурні елементи настанови, як «Національний вступ», «Сфера застосування», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [5] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [7]. Зміст цієї настанови подано з урахуванням додаткових структурних елементів;
- у національному додатку «Бібліографія» додатково наведено бібліографічний опис нормативних документів, посилання на які наведено у цій настанові;
- у розділі «Познаки та скорочення» додатково наведено позначення скорочень, що використовуються у цій настанові;
- у цій настанові словосполучення «заявка на торгову ліцензію/реєстраційний пакет» («marketing application/registration package») замінено на «реєстраційне досьє»;
- по всьому тексту внесено редакційні зміни у посилання на структурні елементи цієї настанови, наприклад замість «Section VI» вказано «(розділ VI цієї настанови)».
Ця настанова застосовна як методичні рекомендації для планування, розробки, організації досліджень щодо встановлення належного рівня вірусної безпеки продуктів біологічного походження.
Юридична сила цієї настанови відповідає юридичній силі відповідної настанови в ЄС та інших регіонах ІСН, з якою гармонізовано розроблену настанову. Цю настанову слід розглядати як технічний документ для надання консультацій заявникам та власникам реєстраційних посвідчень, компетентним уповноваженим органам та/або іншим зацікавленим особам щодо найкращого та найбільш прийнятного способу виконання положень, визначених фармацевтичним законодавством України. Положення цієї настанови відображають гармонізований (у рамках ЄС та ІСН) підхід; вони базуються на останніх наукових досягненнях у цій галузі знань.
У рамках чинного фармацевтичного законодавства ця настанова не має сили нормативно-правового акта, її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану позицію європейського фармацевтичного сектору; дотримання її положень зацікавленими сторонами (такими як заявники, власники реєстраційних посвідчень, розробники та виробники лікарських препаратів, експертні та регуляторні органи) полегшить оцінку реєстраційних досьє в Україні, а також допоможе у плануванні та проведенні досліджень з фармацевтичної розробки. Однак можуть бути використані також альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових настанов викладений у документі Європейського агентства з лікарських засобів (EMA) Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005) [8]. Вказаний підхід відповідає позиції Всесвітньої торгової організації (ВТО) відносно застосування стандартів.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Оцінка вірусної безпеки біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження (Q5А (R1))
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Оценка вирусной безопасности биотехнологических продуктов, полученных из клеточных линий человеческого или животного происхождения (Q5А (R1))
MEDICINAL PRODUCTS
Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (Q5А (R1))
Чинна від 2016-10-19
Сфера застосування
Ця настанова регламентує тестування та оцінку вірусної безпеки біологічних/біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження (ссавці, птахи, комахи тощо). Дані щодо вірусної безпеки повинні міститися у комплекті реєстраційних документів, що подаються при реєстрації лікарського засобу. У цій настанові не розглядаються нетрадиційні інфекційні агенти, що спричиняють такі захворювання, як губчата енцефалопатія (ГЕ) великої рогатої худоби.
Ця настанова не поширюється на лікарські засоби для ветеринарії.
Ця настанова застосовна до біотехнологічних/біологічних лікарських препаратів та їх активних речовин, що розробляються, реєструються та виробляються в Україні для продажу на внутрішньому ринку та з метою експорту або імпортуються в Україну.
Ця настанова поширюється на планування та проведення досліджень з оцінки вірусної безпеки біотехнологічних/біологічних продуктів, у тому числі біосимілярів, складання реєстраційних досьє та реєстрації, а також виробництво, його аудит та інспектування.
Ця настанова рекомендується для суб’єктів господарювання (далі — організацій), які займаються розробкою, реєстрацією та/або виробництвом біотехнологічних/біологічних продуктів, у тому числі біосимілярів, на території України, незалежно від відомчого підпорядкування та форми власності, для відповідних заявників та підприємств-виробників, продукція яких реєструється та імпортується в Україну, для науково-експертних організацій та регуляторних органів, а також експертів, аудиторів та інспекторів, що проводять експертизу при реєстрації (перереєстрації) біотехнологічних/біологічних продуктів, у тому числі біосимілярів, аудит та інспектування виробництва.
Терміни та визначення понять
Нижче наведені терміни, вжиті у цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [2] (див. національний додаток «Бібліографія»). Визначення цих термінів можуть відрізнятися в інших нормативних документів, або терміни можуть мати інші значенняN.
Видалення вірусу (virus removal)
Фізичне відділення вірусних часток від очікуваного продукту.
Випадковий вірус (adventitious virus)
Див. Вірус.
Вік клітин in vitro (in vitro cell age)
Період від розморожування флаконів з головного банку клітин (ГБК) до збору продукту у виробничу ємність, виміряний за часом, що минув у культурі клітин, числом подвоєння популяції клітин або кількістю пасажів клітин при субкультивуванні за визначеною процедурою для розведення культури.
Вірус (virus)
Потенційно патогенні інфекційні агенти, що реплікуються внутрішньоклітинно, які містять тільки один тип нуклеїнової кислоти (РНК або ДНК), не здатні зростати та розмножуватися бінарним діленням та розмножуються у вигляді свого генетичного матеріалу.
- Випадковий вірус (adventitious virus)
Ненавмисно внесений інфекційний вірус.
- Відповідний вірус (relevant virus)
Вірус, що застосовується в оціночних дослідженнях та є ідентичним або належить до того самого виду, що й вірус, наявність якого відома або передбачувана у клітинному субстраті або будь-якому іншому реагенті чи матеріалі, що використовуються у процесі виробництва.
- Ендогенний вірус (endogenous virus)
Вірусний організм, геном якого є частиною зародкової лінії виду, від якої походить клітинна лінія, та ковалентно інтегрований у геном тварини, від якої була отримана батьківська клітинна лінія. У контексті цієї настанови навмисно введені неінтегровані віруси, такі як вірус Епштейна — Барр, що застосовується для іморталізації клітинних субстратів, або вірус бичачої папіломи, підпадають під цю категорію.
- Неендогенний вірус (non-endogenous virus)
Віруси із зовнішніх джерел, наявні у ГБК.
- Неспецифічний модельний вірус (non-specific model virus)
Вірус, що застосовується у процесі характеристики вірусного кліренсу, метою якого є демонстрація здатності виробничого процесу видаляти та/або інактивувати віруси у цілому, тобто стійкості процесу очищення.
- Специфічний модельний вірус (specific model virus)
Вірус, близькоспоріднений з відомим або передбачуваним вірусом (того самого роду або сімейства), який має подібні фізичні або хімічні властивості.
Вірусний кліренс (viral clearance)
Елімінація цільового вірусу шляхом видаленням вірусних часток або інактивації його вірусної здатності.
Вірусоподібні частки (virus-like particles)
Структури, видимі в електронному мікроскопі та морфологічно подібні з відомими вірусами.
Головний банк клітин (ГБК) (master cell bank (MCB))
Аліквота єдиного пулу клітин, зазвичай підготовленого з вибраного клону клітин за певних умов, що розподілена у декілька контейнерів та зберігається за певних умов. ГБК застосовується для формування усіх робочих банків клітин. Випробування, що виконуються для нового ГБК (отриманого з попереднього первинного клону клітин, ГБК або робочого банку клітин — РБК), мають бути такими самими, як для ГБК, якщо не обґрунтовано інше.
Ендогенний вірус (endogenous virus)
Див. Вірус.
Інактивація (inactivation)
Зменшення інфекційної здатності вірусу, спричинене хімічною або фізичною модифікацією.
Клітини-продуценти (production cells)
Клітинний субстрат, що використовується для виробництва продукту.
Клітинний субстрат (cell substrate)
Клітини, що використовуються для виробництва продукту.
Мінімальний час експозиції (minimum exposure time)
Мінімальний період, протягом якого триває обробка.
Неендогенний вірус (non-endogenous virus)
Див. Вірус.
Необроблена маса клітин (unprocessed bulk)
Один чи більше пулів зборів клітин та поживних середовищ. Коли клітини важкодоступні, необроблена маса клітин являє собою рідину, зібрану з ферментеру.
Дослідження процесу з оцінки вірусного кліренсу (process evaluation studies of viral clearance)
Дослідження вірусного кліренсу, у яких застосовуються «відповідні» та/або специфічні «модельні» віруси для оцінки здатності виробничого процесу видаляти та/або інактивувати віруси.
Характеристика процесу вірусного кліренсу (process characterisation of viral clearance)
Дослідження вірусного кліренсу, у якому неспецифічні «модельні» віруси застосовуються для оцінки здатності виробничого процесу видаляти та/або інактивувати віруси.
Робочий банк клітин (РБК) (working cell bank (WCB))
РБК готується з аліквот гомогенної суспензії клітин, що отримані при культивуванні ГБК за встановлених умов.
Познаки та скорочення
|
ВНК |
— |
ниркові фібробласти хом’яка |
|
С127 |
— |
клітини пухлини молочної залози миші |
|
СНО |
— |
клітини яєчників китайського хом’яка |
|
CPMP або CHMP |
— |
Committee for Medicinal Products for Human Use (Комітет з лікарських препаратів для людини) |
|
CTD |
— |
Common Technical Document (Загальний технічний документ) |
|
EMA |
— |
European Medicines Agency (Європейське агентство з ліків) |
|
GMP |
— |
Good Manufacturing Practice (належна виробнича практика) |
|
ICH |
— |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини) |
|
ВІЛ |
— |
вірус імунодефіциту людини |
|
ГЕ |
— |
губчата енцефалопатія |
|
ГБК |
— |
головний банк клітин |
|
ДНК |
— |
дезоксирибонуклеїнова кислота |
|
ЕМ |
— |
електронна мікроскопія |
|
ЄС |
— |
Європейський Союз |
|
МА |
— |
антитіла мишей |
|
ПА |
— |
антитіла пацюків |
|
ПЛР |
— |
полімеразна ланцюгова реакція |
|
РБК |
— |
робочий банк клітин |
|
РНК |
— |
рибонуклеїнова кислота |
|
ТЕМ |
— |
трансмісійна електронна мікроскопія |
|
ХА |
— |
антитіла хом’яків |
Оцінка вірусної безпеки біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження
І. Вступ
Ця настанова стосується випробувань та оцінки вірусної безпеки біотехнологічних продуктів, отриманих з охарактеризованих клітинних ліній людського або тваринного походження (тобто ссавців, птахів, комах), та описує дані, які повинні подаватися у матеріалах реєстраційного досьє. У рамках цієї настанови термін «вірус» не включає нетрадиційні інфекційні агенти, такі як збудники губчатої енцефалопатії (ГЕ) корів та скрепі (пріонна хвороба дрібної худоби). Питання, пов’язані з ГЕ, заявникам рекомендується обговорювати з регуляторними органами.
Ця настанова поширюється на продукти, отримані з клітинних культур охарактеризованих клітинних банків. Положення настанови можуть застосовуватися до продуктів, отриманих з клітинних культур методами in vitro, таких як інтерферони, моноклональні антитіла та продукти, отримані за допомогою технологій рекомбінантної ДНК, включаючи рекомбінантні субодиничні вакцини; а також продуктів, отриманих з гібридомних клітин методами in vivo, як асцити. У останньому випадку необхідно розглянути спеціальні питання, а додаткова інформація щодо випробування клітин, які вирощуються із застосуванням методів in vivo, міститься у додатку 1 до цієї настанови.
Ця настанова не поширюється на інактивовані вакцини, усі живі вакцини, що містять агенти, які самореплікуються, та генно-інженерні живі вектори.
Ризик вірусного забруднення є особливістю, загальною для усіх біотехнологічних продуктів, отриманих з клітинних ліній. Таке забруднення можливе у разі забруднення власне джерела клітинних ліній (клітинних субстратів) або внаслідок випадкового внесення вірусу під час виробництва і може мати серйозні клінічні наслідки. До сьогодні, проте, біотехнологічні продукти, отримані з клітинних ліній, не були причиною зараження вірусами. Однак передбачається, що безпека біотехнологічних продуктів щодо вірусного забруднення може бути адекватно забезпечена лише шляхом застосовування програми випробування на наявність вірусів, а також оцінки ефективності видалення та інактивації вірусів під час процесу виробництва, як описано нижче.
Виділяють три принципові взаємодоповнювальні підходи до контролю потенційної вірусної контамінації біотехнологічних продуктів:
а) відбір та тестування клітинних ліній та іншої сировини, включаючи компоненти середовища, на відсутність небажаних вірусів, які можуть бути інфекційними та/або патогенними для людини;
б) оцінка здатності процесів виробництва видаляти інфекційні віруси;
в) тестування продукту на відповідних стадіях виробництва на відсутність забруднення щодо інфекційних вірусів.
Усі тести мають властиві обмеження кількісних аналізів на наявність вірусів, тобто здатність виявляти низькі концентрації вірусів через статистичні причини залежить від розміру вибірки. Тому жодним з вищевказаних підходів не можна абсолютно точно встановити безпеку продукту. У багатьох випадках відсутність вірусних інфекцій у кінцевому продукті не може бути гарантовано встановлена на підставі тільки безпосереднього їх виявлення, але також необхідно продемонструвати, що схема очищення здатна видалити та/або інактивувати віруси.
Тип та масштаб тестів на віруси та досліджень вірусного кліренсу, які вимагаються на різних етапах виробництва, будуть залежати від різних факторів та повинні розглядатися у кожному конкретному випадку системно та послідовно. Фактори, які необхідно враховувати, включають ступінь характеристики та кваліфікації банку клітин, природу будь-яких виявлених вірусів, складові поживного середовища, методи культивування, дизайн приміщення та обладнання, результати тестів на наявність вірусів після культивування клітин, результативність процесу очищення від вірусів, тип продукту та його заплановане клінічне застосування.
Метою даної настанови є опис загальної структури тестування продукту на наявність вірусів, експериментів з оцінки вірусного кліренсу та надання рекомендацій щодо підходу до планування випробувань на наявність вірусів і проведення досліджень вірусного кліренсу. Відповідна інформація описана у додатках до цієї настанови, а вибіркові терміни містяться у розділі «Терміни і визначення понять».
Виробники повинні адаптувати представлені у цій настанові рекомендації до певного продукту та процесу його виробництва. Виробники мають пояснювати та обґрунтовувати підхід, який вони використовують у загальній стратегії гарантування вірусної безпеки. Додатково до наданих детальних даних необхідно представити загальне резюме з оцінки вірусної безпеки для спрощення розгляду регуляторним органом. Це резюме повинно містити короткий опис усіх аспектів досліджень вірусної безпеки та стратегій, що використовуються для запобігання контамінації вірусами.
ІІ. Потенційні джерела вірусної контамінації
Контамінація біотехнологічних продуктів вірусами може походити з першоджерела клітинних ліній або бути наслідком випадкового внесення вірусів під час виробничого процесу.
А. Віруси, які можуть бути наявними у головному банку клітин
Клітини можуть містити латентну або персистуючу вірусну інфекцію (наприклад, вірус герпесу) або ендогенний ретровірус, який може передаватися вертикально від одного покоління клітин до іншого, оскільки вірусний геном зберігається у клітинах. Такі віруси можуть постійно експресуватися конститутивно, або вони можуть зненацька проявитися у формі інфекційного вірусу.
Віруси можуть потрапляти у ГБК декількома шляхами:
1) виділення клітинної лінії від інфікованих тварин;
2) використання вірусів для створення клітинних ліній;
3) використання забруднених біологічних реагентів, таких як компоненти сироватки крові тварин;
4) контамінація під час обробки клітин.
Б. Випадкові віруси, які можуть бути внесені під час технологічного процесу виробництва
Випадкове інфікування кінцевого продукту може відбуватися декількома шляхами, у тому числі при використанні:
1) забруднених біологічних реагентів, таких як компоненти сироватки крові тварин;
2) вірусів для індукції експресії специфічних генів, що кодують цільовий протеїн;
3) забрудненого реагенту, такого як афінна колонка для моноклональних антитіл;
4) забруднених допоміжних речовин у композиції;
5) забруднених клітин та поживного середовища.
Моніторинг параметрів культивування клітин може бути корисним для раннього виявлення потенційного випадкового вірусного забруднення.
ІІІ. Кваліфікація клітинних ліній: випробування на наявність вірусів
Важливою частиною кваліфікації клітинної лінії для використання у виробництві біотехнологічного продукту є відповідне випробування на наявність вірусу.
А. Випробування на віруси, рекомендовані для головного банку клітин, робочого банку клітин та клітин, що знаходяться у граничному для виробництва клітинному віці in vitro
У табл. 1 наведені приклади тестів на віруси, які повинні виконуватися лише один раз для кожного з різних джерел клітин, включаючи ГБК, РБК та клітини, що знаходяться у граничному клітинному віці in vitro для виробництва.
1. Головний банк клітин
Для ГБК необхідно провести розширені тестування на ендогенне та неендогенне вірусне забруднення. Для гетерогібридних клітинних ліній, у яких один або більше батьків походять від людини або нелюдиноподібних приматів, необхідно провести тестування на віруси, характерні для людини або нелюдиноподібних приматів, оскільки контамінація клітин такими вірусами може становити особливу небезпеку.
Для виявлення можливої вірусної контамінації випробування на наявність неендогенних вірусів повинні включати інокуляційні тести in vivo та in vitro та будь-які інші специфічні випробування, у тому числі видоспецифічні тести, такі як тестування утворення антитіл у мишей (МА), які підходять для виявлення потенційних забруднювальних вірусів з урахуванням історії пасажів клітинної лінії.
2. Робочий банк клітин
Кожний РБК як початковий клітинний субстрат для виробництва продукту повинен тестуватися на випадкові віруси шляхом прямого випробування або аналізу клітин, що знаходяться у граничному клітинному віці in vitro, які отримані з РБК. Якщо тести на неендогенні віруси проводили на ГБК, та клітини, що культивувалися в межах або поза межами граничного для виробництва клітинного віку in vitro, отримували з РБК та використовували для тестування на наявність випадкових вірусів, подібні випробування не потрібно проводити для початкового РБК. Тести на продукцію антитіл, як правило, не потрібно проводити для РБК. Також може бути прийнятним альтернативний підхід, за якого проводиться повний набір випробувань на РБК, а не на ГБК.
3. Граничний для виробництва клітинний вік in vitro
Межі граничного для виробництва клітинного віку in vitro повинні ґрунтуватися на даних, отриманих для клітин, що використовуються у дослідно-промисловому або промисловому виробництві, та мають бути рівними або перевищувати пропонований за цих умов клітинний вік in vitro. Як правило клітини, що використовуються у виробництві, отримують шляхом збільшення РБК; також для отримання клітин може використовуватися ГБК. Клітини, що знаходяться у граничному для виробництва віці in vitro, повинні оцінюватися один раз на ендогенні віруси, які могли бути не виявлені у ГБК або РБК. Проведення відповідних випробувань (наприклад in vitro та in vivo) принаймні один раз для клітин, що знаходяться у граничному для виробництва віці in vitro, надасть подальшу гарантію того, що процес виробництва не піддається випадковій вірусній контамінації. Якщо на цьому рівні виявлені будь-які відповідні віруси, процес повинен бути ретельно перевірений для встановлення джерела контамінації та, у разі необхідності — повністю перепланований.
Б. Рекомендовані випробування для виявлення та ідентифікації вірусів
Для виявлення ендогенних та випадкових вірусів можуть використовуватися численні випробування. Приклади цих випробувань наведені у табл. 2 цієї настанови. Ці випробування повинні розглядатися як рекомендовані на даний час, але наведений список не є вичерпним або остаточним.
Оскільки внаслідок наукового прогресу більшість відповідних методик може змінюватися, можуть бути прийнятними пропозиції щодо альтернативних методик, які супроводжуються адекватними підтверджувальними даними. Виробники повинні обговорити ці альтернативні підходи з регуляторними органами. Залежно від конкретного випадку можуть бути задіяні інші методики. Протоколи випробувань повинні гарантувати відповідну чутливість та специфічність методик. Якщо існує висока вірогідність вірусного забруднення вихідного клітинного субстрату, можуть бути використані специфічні тести та/або підходи.
Якщо клітинна лінія отримана від людини або нелюдиноподібного примата, необхідно також провести додаткові тести на наявність вірусів людини, таких як ВІЛ та гепатит. Для виявлення цих вірусів, а також інших специфічних вірусів слід застосовувати методи на основі полімеразної ланцюгової реакції (ПЛР).
Нижче наводиться короткий опис загальних принципів та основні вихідні дані, за допомогою яких виробник повинен обґрунтувати свої дії.
1. Випробування на наявність ретровірусів
Для ГБК та клітинної культури, що знаходяться у граничному для виробництва клітинному віці in vitro, необхідно проводити випробування на наявність ретровірусів, включаючи інфекційні випробування на чутливих клітинних культурах та електронну мікроскопію (ЕМ). Якщо інфекційні випробування не дали позитивного результату та жодних ретровірусних або ретровірусоподібних структур не виявлено за допомогою ЕМ, необхідно провести випробування зворотної транскриптази та інші відповідні випробування для виявлення ретровірусів, які можуть бути неінфекційними. Індуктивні дослідження у даному разі неефективні.
2. Випробування in vitro
Випробування in vitro проводять шляхом інокуляції випробуваних зразків (див. табл. 2 цієї настанови) у різних тестових клітинних культурах, здатних виявити широкий спектр вірусів людини та тварин. Вибір клітинних тестових культур залежить від видового походження випробовуваного банку клітин, однак у дослідження необхідно включати клітинні лінії людини та/або нелюдиноподібних приматів, що виявляють чутливість до вірусів людини. Характер випробувань та відбір випробовуваного зразка зумовлюються типом вірусу, який, ймовірно, буде наявний, з урахуванням походження або обробки клітин. Слід проводити аналіз на цитопатичні та гемадсорбуючі віруси.
3. Випробування in vivo
Для того щоб виявити віруси, які не проявляють ріст у клітинних культурах, випробувані зразки (див. табл. 2 цієї настанови) вводяться в організм тестових тварин, включаючи мишенят та дорослих мишей або запліднені яйцеклітини. Можуть використовуватися інші види тварин залежно від природи та джерела випробуваних клітинних ліній. Необхідно проводити моніторинг стану здоров’я піддослідних тварин та невідкладно виявляти та ідентифікувати причини будь-яких відхилень від норм стану здоров’я.
4. Випробування на утворення антитіл
Видоспецифічні віруси, наявні у клітинних лініях гризунів, можуть бути визначені шляхом введення випробуваних зразків (див. табл. 2 цієї настанови) тваринам, попередньо вільним від вірусів. Далі через певний період досліджуються рівень антитіл у сироватці або активність ферментів. Прикладами таких випробувань є тест на утворення МА, тест на утворення антитіл у пацюків (ПА) та тест на утворення антитіл у хом’яків (ХА). Віруси, що на даний час виявляються у такий спосіб, перелічені у табл. 3 цієї настанови.
В. Прийнятність клітинних ліній
Слід визнати, що деякі клітинні лінії, які використовуються для виробництва продукту, будуть містити ендогенні ретровіруси, інші віруси або вірусні послідовності. Для таких випадків план дій, рекомендований для організації виробництва, описано в розділі V цієї настанови. Прийнятність клітинних ліній, що містять інші віруси, відмінні від ендогенних ретровірусів, повинна розглядатися у кожному конкретному випадку регуляторними органами. При цьому враховується аналіз співвідношення ризик/користь, встановленого з інформації про користь продукту та його заявлене клінічне застосування, природу забруднювальних вірусів та їх здатність інфікувати людей або спричиняти у них захворювання, процес очищення препарату (наприклад дані оцінки вірусного кліренсу) та обсяг вірусних випробувань, проведених на очищеному балк-матеріалі.
ІV. Вірусні випробування у необробленому балк-матеріалі
Необроблений балк-матеріал — це один або декілька зборів клітин та поживне середовище. Якщо клітини не є легкодоступними (наприклад, при використанні порожнистих волокон або подібних систем), необроблений балк-матеріал являтиме собою рідину, зібрану на стадії ферментації. Репрезентативний зразок необробленого балк-матеріалу, відібраного безпосередньо з промислового реактора перед подальшою обробкою, є одним з найбільш гарантованих рівнів, на якому з високою вірогідністю можна визначити випадкову вірусну контамінацію. Відповідне випробування на наявність вірусів необхідно проводити на рівні необробленого балк-матеріалу, якщо тільки випробування на наявність вірусів не буде більш чутливим на початковому етапі обробки клітинної маси (наприклад, необроблений балк-матеріал може бути токсичним для тестових клітинних культур, а частково оброблений балк-матеріал — нетоксичний).
У деяких випадках може бути більш прийнятною для випробування суміш, що містить як живі, так і зруйновані клітини та супернатанти, відібрана з промислового реактора перед подальшою її обробкою. До реєстраційного досьє на лікарський засіб, поданий на реєстрацію, необхідно включити дані випробувань принаймні для 3 серій необробленого балк-матеріалу дослідно-промислового або промислового масштабу.
Виробникам слід розробити програми проведення регулярних випробувань на можливу наявність випадкових вірусів у виробничих серіях необробленого балк-матеріалу. Обсяг, кількість та частоту проведення тестування на наявність вірусів у необробленому балк-матеріалі необхідно встановлювати з урахуванням природи клітинних ліній, що використовуються для виробництва цільового продукту, результатів та обсягу випробувань на віруси під час кваліфікації клітинних ліній, способу культивування, джерела сировини та результатів досліджень вірусного кліренсу. Як правило, для випробування необробленого балк-матеріалу застосовуються скринінг-тести in vitro з використанням однієї або декількох клітинних ліній. За необхідності можна використовувати ПЛР-тести або інші відповідні методи.
Зібраний матеріал, у якому були виявлені випадкові віруси, не повинен використовуватися для виробництва продукту. Якщо будь-які випадкові віруси виявлені на будь-якій стадії, виробничий процес слід ретельно перевірити для встановлення причини забруднення та вжити відповідних заходів для їх усунення.
V. Оптимальний план дій для досліджень вірусного кліренсу та випробувань на наявність вірусів очищеного балк-матеріалу
Виробник повинен розробити найбільш ефективний та раціональний протокол досліджень на наявність вірусів, на різних етапах виробництва, від стадії ГБК до отримання кінцевого продукту, включаючи оцінку та характеристику вірусного кліренсу необробленого балк-матеріалу. Оцінка та характеристика вірусного кліренсу є важливим підтвердженням того, що кінцевий продукт повністю вільний від вірусів.
При виборі вірусів для використання у дослідженні кліренсу необхідно визначитися щодо необхідності оцінювати процеси, під час яких видаляються найбільш ймовірні специфічні для продукту віруси, та щодо можливостей оцінити спроможність процесу забезпечити видалення неспецифічних «модельних» вірусів (описано далі). Підходи до визначення «відповідних», специфічних та неспецифічних «модельних» вірусів надаються у розділі «Терміни та визначення понять» цієї настанови. При оцінці процесу необхідно мати інформацію про те, яка кількість вірусу може бути наявною у процесі, такому як отримання необробленого балк-матеріалу, та яка кількість вірусу може бути елімінована у процесі очищення для гарантії безпеки продукту. Для гарантії ефективності процесу вірусної інактивації слід знати час, необхідний для повної інактивації вірусів. При оцінці кліренсу відомих вірусних забруднень необхідні докладні, залежні від часу інактиваційні дослідження, демонстрація відтворюваності інактивації/видалення забруднювачів та оцінка параметрів процесу. Коли технологічний процес характеризується щодо надійності кліренсу з використанням неспецифічних «модельних» вірусів, особливу увагу необхідно приділити вірусам, які не були враховані при плануванні дослідження. На обсяг досліджень щодо характеристики вірусного кліренсу можуть впливати результати досліджень клітинних ліній та необробленого балк-матеріалу. Ці дослідження слід проводити, як описано у розділі VI цієї настанови.
У табл. 4 цієї настанови наводиться приклад плану дій при оцінці та характеристиці вірусного кліренсу, а також тестів на віруси в очищеному балк-матеріалі залежно від результатів тестів на віруси у клітинах та/або необробленому балк-матеріалі. У даному розділі розглядаються різні ситуації. В усіх представлених випадках необхідна характеристика кліренсу з використанням неспецифічних «модельних» вірусів. Найбільш типовими ситуаціями є випадки А та Б. Виробничі системи, забруднені вірусом, за винятком ретровірусу гризунів, як правило, не використовуються. Якщо існують переконливі та добре обґрунтовані підстави для виробництва препарату з використанням клітинної лінії з випадків В, Г або Ґ, їх необхідно обговорити з регуляторними органами.
Для випадків В, Г та Ґ важливо визначити ефективні заходи, що пройшли валідацію, з інактивації/видалення даного вірусу у виробничому процесі.
Випадок А. Якщо жодних вірусів, вірусоподібних часток або ретровірусоподібних часток не виявлено у клітинах або необробленому балк-матеріалі, необхідно провести дослідження з видалення та інактивації вірусів з неспецифічними «модельними» вірусами, як вказано вище.
Випадок Б. Якщо наявний тільки ретровірус гризунів (або ретровірусоподібні частки, які вважаються непатогенними, такі як частки А- та R-типу гризунів), слід провести оцінку процесу з використанням специфічних «модельних» вірусів, таких як вірус лейкемії мишей. Очищений балк-матеріал необхідно протестувати з використанням відповідних високоспецифічних та високочутливих для виявлення даного вірусу методів. У реєстраційному досьє необхідно надавати дані принаймні для 3 серій очищеного балк-матеріалу дослідно-промислового або промислового масштабу. Клітинні лінії, такі як СНО, С127, ВНК та мишачі гібридоми, часто використовуються як субстрати для виробництва готової продукції як такі, що не мають жодних проблем щодо вірусної контамінації одержаних продуктів. Для цих клітинних ліній, у яких ендогенні частки були детально охарактеризовані та був продемонстрований вірусний кліренс, проведення аналізу на наявність неінфекційних часток в очищеному балк-матеріалі зазвичай не вимагається. Необхідно проводити дослідження з неспецифічними «модельними» вірусами, як у випадку А.
Випадок В. Якщо відомо, що клітини або необроблений балк-матеріал містять вірус, за винятком ретровірусу гризунів, для якого відсутні дані щодо здатності інфікувати людей (наприклад, віруси, що зазначені у примітці 2 до табл. 3, за винятком ретровірусів гризунів (випадок Б)), дослідження з оцінки видалення та інактивації вірусів слід проводити з використанням ідентифікованого вірусу. Якщо неможливо використовувати ідентифікований вірус, необхідно використовувати «відповідні» або специфічні «модельні» віруси для демонстрації прийнятного вірусного кліренсу. Дані залежності часу інактивації ідентифікованого (або «відповідного», або специфічного «модельного») вірусу від ефективності процедури на критичних етапах інактивації повинні бути отримані у рамках оцінки процесу для цих вірусів. Очищений балк-матеріал необхідно протестувати з використанням відповідних високоспецифічних та високочутливих для виявлення даного вірусу методів. У реєстраційному досьє необхідно надавати дані принаймні для 3 серій очищеного балк-матеріалу пілотного або промислового масштабу.
Випадок Г. Якщо виявлений вірус патогенний для людини, наприклад вказаний у примітці 1 до табл. 3, продукт може бути прийнятним лише у виняткових випадках. У такій ситуації рекомендується використовувати ідентифікований вірус у дослідженнях з оцінки видалення або інактивації вірусу з використанням відповідних високоспецифічних та високочутливих для виявлення даного вірусу методів. Якщо неможливо використовувати ідентифікований вірус, слід використовувати «відповідні» та/або специфічні «модельні» віруси (описані далі). Необхідно продемонструвати, що у процесі досягаються видалення та інактивація вибраних вірусів під час процесів очищення та інактивації. Дані залежності часу інактивації ідентифікованого вірусу від ефективності задіяних процедур на критичних етапах інактивації повинні бути отримані у рамках оцінки процесу для цих вірусів. Очищений балк-матеріал необхідно протестувати з використанням відповідних високоспецифічних та високочутливих для виявлення даного вірусу методів. У реєстраційному досьє необхідно надавати дані принаймні для 3 серій очищеного балк-матеріалу пілотного або промислового масштабу.
Випадок Ґ. Якщо у клітинах або необробленому балк-продукті виявлено вірус, який не можна класифікувати за наявними в даний час методами, продукт, як правило, вважається неприйнятним, оскільки виявлений вірус може виявитися патогенним. Дуже рідко, якщо існують переконливі та добре обґрунтовані підстави для виробництва препарату з використанням такої клітинної лінії, їх необхідно обговорити з регуляторним органами.
VI. Оцінка та характеристика методів елімінації вірусів
Оцінка та характеристика методів елімінації та/або інактивації вірусів відіграють важливу роль у встановленні безпеки біотехнологічних продуктів. Багато випадків контамінації у минулому були пов’язані з агентами, про наявність яких не знали і навіть не підозрювали. Для біологічних продуктів, отриманих з різних вихідних матеріалів, за винятком повністю охарактеризованих клітинних ліній, оцінка елімінації вірусів дасть змогу з деяким ступенем впевненості стверджувати, що будь-які невідомі, неочікувані та небезпечні віруси можуть бути видалені. Дослідження слід проводити таким чином, щоб вони були добре задокументованими та контрольованими.
Мета досліджень вірусного кліренсу — оцінка етапу(-ів) процесу, який може вважатися ефективним при інактивації/видаленні вірусів, та кількісна оцінка загального рівня елімінації вірусів, досягнутого у даному процесі. Це досягається навмисним додаванням («метод добавок») значної кількості вірусів до сировини та/або до різних фракцій, які отримані на різних етапах процесу, та демонстрацією їх видалення або інактивації під час подальших етапів. Немає необхідності оцінювати або характеризувати кожний етап технологічного процесу, якщо відповідний кліренс досягається при застосуванні меншого числа етапів. Слід брати до уваги, що інші етапи процесу можуть опосередковано впливати на інактивацію/видалення вірусів. Виробники повинні пояснити та обґрунтувати підхід, що використовується в дослідженнях з оцінки вірусного кліренсу.
Зниження інфекційної здатності вірусу може досягатися шляхом видалення вірусних часток або інактивацією його інфекційності. Для кожного етапу виробництва, що оцінюється, повинен описуватися можливий механізм втрати інфекційної здатності вірусу з зазначенням того, чи є це результатом інактивації або видалення вірусу. Для етапів інактивації дослідження необхідно планувати таким чином, щоб відбір зразків проводився у різний час та була побудована крива інактивації (див. розділ VI.Б.5 цієї настанови).
Дослідження з оцінки вірусного кліренсу проводяться з метою демонстрації видалення вірусу, який, як відомо, наявний у ГБК, та/або надання певного рівня гарантії, що занесені віруси, які могли бути не виявлені раніше або могли отримати доступ при реалізації технологічного процесу виробництва, будуть еліміновані. Коефіцієнт зниження вірусного навантаження зазвичай виражається за логарифмічною шкалою, яка передбачає, що хоча залишкова вірусна інфекційність ніколи не буде знижена до нуля, вона може бути суттєво знижена математично.
Додатково до досліджень елімінації вірусів, про наявність яких відомо, слід проводити дослідження характеристики здатності видалення та/або інактивації інших вірусів. Мета досліджень з вірусами, які демонструють ряд біохімічних та біофізичних властивостей та про наявність яких невідомо або про неї не підозрюють, полягає в тому, щоб охарактеризувати надійність процедури, а не досягти конкретних інактивації або видалення вірусу. Бажано продемонструвати здатність організації процесу виробництва до інактивування або видалення вірусів (див. розділ VI.В цієї настанови). Такі дослідження не проводяться для оцінки специфічного ризику безпеки. Саме тому не вимагається досягнення конкретного рівня кліренсу.
А. Вибір вірусів для оцінки та характеристики вірусного кліренсу
Віруси для досліджень з оцінки кліренсу та характеристики процесу повинні вибиратися таким чином, щоб вони були подібні до вірусів, які можуть забруднювати продукт, та мали широкий спектр фізико-хімічних властивостей, що дають змогу перевірити здатність системи видаляти віруси у цілому. Виробник повинен обґрунтовувати вибір вірусів відповідно до мети досліджень з оцінки та характеристики вірусного кліренсу та положень цієї настанови.
1. «Відповідні» віруси та «модельні» віруси
Головним завданням досліджень вірусного кліренсу є визначення вірусів, які повинні використовуватися у дослідженнях. Виділяють три категорії таких вірусів: «відповідні» віруси, специфічні «модельні» віруси та неспецифічні «модельні» віруси.
«Відповідні» віруси — це віруси, що використовуються при оцінці вірусного кліренсу, які є або ідентифікованими вірусами, або вірусами того самого виду, що й виявлені віруси, або вірусами, які ймовірно забруднять клітинний субстрат, або будь-які інші реактиви чи матеріали, що використовуються у процесі виробництва. Процес очищення та/або інактивації повинен продемонструвати здатність видаляти та/або інактивувати такі віруси. Якщо «відповідний» вірус недоступний або він не досить добре адаптується до досліджень з оцінки вірусного кліренсу (наприклад, він не може бути вирощений in vitro у достатній кількості), замість нього повинен використовуватися специфічний «модельний» вірус. Відповідний специфічний «модельний» вірус може бути вірусом, який є близькоспорідненим з відомим або потенційним вірусом (того ж роду або сімейства) та має подібні до виявленого або потенційного вірусу фізичні та хімічні властивості.
Клітинні лінії, отримані від гризунів, зазвичай містять частки ендогенного ретровірусу або ретровірусоподібні частки, які можуть бути інфекційними (частки С-типу) або неінфекційними (цитоплазматичні частки А- та R-типу). Необхідно визначити здатність технологічного процесу видаляти та/або інактивувати ретровіруси гризунів у продуктах, отриманих з використанням таких клітин. Цього можна досягти, використовуючи вірус лейкемії мишей, специфічний «модельний» вірус для клітин мишей. Якщо клітинна лінія людини, що секретує моноклональні антитіла, була отримана шляхом іморталізації В-лімфоцитів вірусом Епштейна — Барр, слід визначити здатність технологічного процесу видаляти та/або інактивувати вірус герпесу. Як специфічний «модельний» вірус можна також використовувати вірус псевдосказу.
Якщо мета полягає у тому, щоб охарактеризувати здатність технологічного процесу видаляти та/або інактивувати віруси загалом, тобто охарактеризувати надійність процесу елімінації, дослідження характеристики вірусного кліренсу необхідно проводити на неспецифічних «модельних» вірусах з різними властивостями. Дані, отримані у дослідженнях з «відповідними» та/або специфічними «модельними» вірусами, можуть також сприяти такій оцінці. Немає необхідності тестувати усі типи вірусів. Перевагу слід надавати вірусам, які демонструють значну резистентність до фізичної та/або хімічної обробки. Результати, отримані на таких вірусах, надають корисну інформацію про здатність технологічного процесу видаляти та/або інактивувати віруси загалом. Вибір і кількість вірусів, що використовуються, залежать від якості та характеристики клітинних ліній і процесу виробництва.
Приклади придатних «модельних» вірусів, що представляють ряд фізико-хімічних структур, та приклади вірусів, які використовувалися в дослідженнях вірусного кліренсу, зазначені у додатку 2 до цієї настанови та табл. 1.
2. Інші питання
Додатковими питаннями, які слід розглянути, є:
а) використання вірусів, ріст яких може досягти високого титру і які є необхідними, хоча це не завжди може бути можливим;
б) наявність ефективного та надійного методу аналізу для виявлення кожного вірусу, який використовується на кожній стадії технологічного процесу, що має контролюватися;
в) урахування загрози для здоров’я персоналу, який проводить дослідження вірусного кліренсу, яку можуть представляти певні віруси.
Б. Розробка та проведення досліджень з оцінки та характеристики вірусного кліренсу
1. Приміщення та персонал
Вимоги Належної виробничої практики (GMP) містять обмеження на наявність будь-якого вірусу на поверхнях виробничого обладнання. Саме тому дослідження вірусного кліренсу мають проводитися в окремій лабораторії, оснащеній для проведення вірусологічних досліджень, та виконуватися персоналом, який має досвід роботи з вірусами, разом з персоналом, який займається розробкою та підготовкою процесу очищення обладнання у дослідно-промисловому масштабі.
2. Виробництво у дослідно-промисловому масштабі
Необхідно продемонструвати валідність дослідно-промислового масштабу виробництва. Метод очищення, що застосовується для дослідно-промислового масштабу, повинен якомога точніше відповідати методу виробництва у промисловому масштабі. Необхідно продемонструвати, що хроматографічне обладнання, висота шару сорбенту у колонці, лінійність швидкості потоку, відношення швидкості потоку до об’єму шару сорбенту (тобто час контакту), типи буфера та гелю, рН, температура та концентрація протеїну, солі та продукти репрезентативні для виробництва в промисловому масштабі. У результаті повинен бути отриманий подібний профіль елюювання. До інших процедур застосовуються подібні принципи. Відхилення, яких не можна уникнути, слід обговорити з точки зору їх впливу на процес виробництва.
3. Аналіз поетапної елімінації вірусів
При проведенні досліджень вірусного кліренсу бажано оцінити вплив критичних етапів виробництва на процес елімінації вірусу. Етапи, на яких, ймовірно, відбуватиметься елімінація вірусу, необхідно окремо оцінити щодо їх здатності видаляти або інактивувати вірус. Слід приділяти особливу увагу чіткому визначенню кожного окремого етапу. У матеріалі для дослідження кожного етапу повинна бути наявна достатня кількість вірусів, які будуть тестуватися, щоб можна було адекватно оцінити ефективність кожного етапу. Зазвичай вірус повинен додаватися до матеріалу, що знаходиться в процесі виробництва кожного етапу, який буде тестуватися. У деяких випадках може бути достатньо простого додавання вірусу високого титру до неочищеного балк-матеріалу та визначення його концентрації при переході від одного етапу до іншого. Якщо видалення вірусу відбувається внаслідок фракціонування продукту, рекомендується (за можливості та у разі необхідності) дослідити розподіл вірусного навантаження у різних фракціях. Якщо віроцидні буфери використовуються на різних етапах технологічного процесу, альтернативні стратегії, такі як паралельний метод добавок (spiking) у менш віроцидні буфери, може проводитися як частина загального процесу оцінки. Необхідно визначити титр вірусу до та після кожного етапу, що тестується. Кількісні аналізи інфекційності вірусу повинні бути достатньо чутливими та відтворюваними: їх потрібно проводити з такою кількістю повторів, яка дозволить гарантувати відповідну статистичну значимість результатів. Можна проводити кількісні аналізи, не пов’язані з визначенням інфекційності вірусу, якщо це обґрунтовано. Відповідний контроль вірусу слід включити в усі аналізи інфекційності вірусу для гарантії чутливості методу. Крім того, слід розглянути статистичні дані відбору проб вірусу при його низьких концентраціях (див. додаток 3 до цієї настанови).
4. Зіставлення фізичного видалення вірусу з інактивацією
Зниження інфекційності вірусу можна досягти при його видаленні або інактивації. Для кожного оцінюваного етапу виробництва потрібно описати можливий механізм втрати інфекційності вірусу з урахуванням того, чи буде це результатом його інактивації або видалення. Якщо у виробничому процесі досягається незначне зниження інфекційності вірусу, а елімінація вірусу вважається основним фактором безпеки продукту, необхідно ввести специфічні або додаткові етапи інактивації/видалення. Для певного етапу може бути необхідним розрізняти видалення та інактивацію, наприклад, коли існує вірогідність, що буфер, який використовується на більше ніж одному етапі елімінації вірусу, може сприяти інактивації під час кожного етапу; тобто інактивація вірусу, що здійснюється за допомогою буфера, та видалення вірусу, яке досягається на кожному з етапів хроматографічного очищення, мають розрізнятися.
5. Оцінка інактивації
Для оцінки інактивації вірусу у необроблену сировину або проміжний матеріал необхідно додати інфекційний вірус та вирахувати коефіцієнт зниження вірусного навантаження. Необхідно визнати, що інактивація вірусу не є простою реакцією першого порядку і зазвичай є більш складною, зі швидкою «фазою 1» та повільною «фазою 2». Саме тому дослідження необхідно планувати таким чином, щоб зразки відбиралися у різний час і будувалася крива інактивації. Рекомендується, щоб дослідження інактивації додатково до мінімального часу експозиції включало принаймні одну часову точку, яка повинна бути меншою, ніж мінімальний час експозиції, але більшою за нуль. Додаткові дані особливо важливі, якщо вірус є «відповідним» вірусом, про який відомо, що він є патогенним для людини, у зв’язку з чим розробляється ефективний процес інактивації. Однак для досліджень інактивації, у яких використовуються неспецифічні «модельні» віруси, або специфічні «модельні» віруси використовуються як сурогати вірусних часток, такі як внутрішньоцитоплазматичні ретровірусоподібні частки CHO, необхідно продемонструвати відтворювану елімінацію вірусу принаймні у двох незалежних дослідженнях. Завжди, коли це можливо, початкове вірусне навантаження слід визначати у вихідному матеріалі з додаванням вірусу, який може виявлятися. Якщо це неможливо, початкове вірусне навантаження можна розрахувати з титру вірусу, який додано до препарату. Якщо інактивація є дуже швидкою, щоб побудувати криву інактивації за даних умов процесу, необхідно проводити відповідні контрольні заходи для демонстрації того, що інфекційна здатність вірусу дійсно втрачається внаслідок інактивації.
6. Використання та інактивація колонок
У зв’язку з тим, що після багаторазового використання хроматографічних колонок та іншого обладнання, задіяного у схемі очищення, їх здатність до елімінації вірусу може змінюватися, оцінка стабільності вірусного кліренсу після декількох використань таких колонок може бути підтвердженням можливості їх повторного використання. Необхідно надати гарантію того, що будь-які віруси, які потенційно утримуються у системі виробництва, будуть відповідно знищені або видалені перед повторним використанням системи. Наприклад, такий доказ може бути наданий шляхом демонстрації того, що процедури очищення та регенерації дійсно інактивують або видаляють вірус.
7. Спеціальні запобіжні заходи
а) Слід дотримуватися обережності при підготовці вірусу високого титру, щоб не допустити агрегації, яка може посилити його фізичне видалення та зменшити інактивацію, спотворюючи таким чином кореляцію з фактичним виробництвом;
б) слід встановити мінімальну кількість вірусу, на якій може бути достовірно проведений аналіз;
в) дослідження повинно включати паралельні контрольні аналізи для оцінки втрати вірусом інфікуючої здатності внаслідок розведення, концентрації, фільтрації або зберігання зразків до титрування;
г) добавку вірусу необхідно вводити у продукт у невеликій кількості, щоб не розбавляти продукт та не змінювати його характеристику. Розведений зразок випробовуваного протеїну не вважається ідентичним продукту, отриманому у промисловому масштабі;
ґ) невеликі відмінності у, наприклад, буферах, середовищі або реактивах можуть значно вплинути на вірусний кліренс;
д) процес інактивації вірусу є залежним від часу, тому тривалість періоду, протягом якого продукт з добавкою вірусу залишається у певному буферному розчині або на певній хроматографічній колонці, повинна відтворювати умови процесу у промисловому масштабі;
е) буфери та продукт слід оцінювати окремо на токсичність або взаємний вплив на результати аналізу, що використовуються для визначення титру вірусу, оскільки ці компоненти можуть негативно вплинути на клітини-індикатори. Якщо розчини токсичні для клітин-індикаторів, можуть бути задіяні розведення, корекція рН або діаліз буфера, що містить введений вірус. Якщо сам продукт має антивірусну активність, дослідження вірусного кліренсу необхідно провести без продукту в холостому контролі, хоча невключення продукту або заміна подібним протеїном, який не має антивірусної активності, може вплинути на поведінку вірусу на деяких етапах виробництва. Необхідний належний контроль для демонстрації дієвості процедур, які використовуються лише для підготовки зразка для аналізу (наприклад, діаліз, зберігання) на видалення/інактивацію введеного вірусу;
є) у багатьох схемах очищення повторно використовують один і той самий або подібний буфери або колонки. Результати цього підходу слід враховувати при аналізі даних. Ефективність елімінації вірусів певним процесом може варіювати залежно від етапу виробництва, на якому він використовується;
ж) загальні фактори зниження елімінації вірусів можуть недооцінюватися, якщо умови виробництва або буфери є дуже цитотоксичними або віроцидними, тому їх слід розглядати у кожному конкретному випадку. Загальні фактори зниження елімінації вірусів також можуть бути надмірно переоцінені внаслідок наявних обмежень або невідповідного дизайну досліджень вірусного кліренсу.
В. Інтерпретація результатів досліджень вірусного кліренсу
Прийнятність результатів
Мета досліджень інактивації/видалення вірусу полягає в оцінці та характеристиці етапів процесу, які можуть вважатися ефективними для інактивації/видалення вірусів, та кількісній оцінці загального рівня зниження вірусного навантаження, що досягається у процесі виробництва. Для вірусних контамінантів, як вказано у випадках Б–Ґ, важливо продемонструвати, що не лише вірус видаляється або інактивується, але й те, що вірусний кліренс, який є складовою процесу очищення, може гарантувати відповідний рівень безпеки кінцевого продукту. Кількість вірусів, що видаляються або інактивуються в процесі виробництва, необхідно порівнювати з кількістю вірусів, що можуть бути наявні у необробленому балк-матеріалі.
Для проведення цього порівняння важливо оцінити кількість вірусу в необробленому балк-матеріалі. Цю оцінку необхідно провести, застосовуючи аналізи інфекційності вірусу або інші методи, такі як, трансмісійна електронна мікроскопія (ТЕМ). Весь процес очищення має бути здатним видаляти значно більшу кількість вірусів, ніж та, що, як очікується, наявна в однократній дозі необробленого балк-матеріалу. Для розрахунку коефіцієнтів зниження вірусного навантаження застосовують інформацію, викладену у додатку 4 до цієї настанови, а для розрахунку передбаченої кількості часток у дозі застосовують інформацію, викладену у додатку 5 до цієї настанови.
Виробники повинні враховувати, що механізми елімінації різних класів вірусів можуть відрізнятися. При оцінці даних, що підтверджують ефективність процедур інактивації/видалення вірусів, необхідно враховувати сукупність факторів, які включають:
а) придатність вірусів для використання у тестах;
б) дизайн досліджень вірусного кліренсу;
в) логарифмічне зниження, що досягнуте;
г) залежність інактивації вірусу від часу;
ґ) потенційний вплив змін у параметрах процесу на інактивацію/видалення вірусів;
д) межі чутливості аналізу;
е) можливу селективність методів інактивації/видалення для певних класів вірусів.
Ефективного вірусного кліренсу можна досягти за допомогою будь-якого з таких підходів: проведення численних етапів інактивації, численних додаткових етапів сепарації або комбінації етапів інактивації та сепарації. Оскільки методи сепарації можуть залежати від надто специфічних фізико-хімічних властивостей вірусу, які впливають на його взаємодією з гелевими матрицями та на здатність до преципітації, «модельні» віруси можна сепарувати в інший спосіб, ніж цільовий вірус. Необхідно належним чином визначати та контролювати параметри виробництва, які впливають на сепарацію. Відмінності можуть виникати внаслідок змін у поверхневих властивостях, наприклад, глікозилювання. Проте, незважаючи на цю потенційну змінність, ефективного видалення можна досягти за рахунок комбінації додаткових етапів сепарації або комбінації етапів сепарації та інактивації. Саме тому якісно сплановані етапи сепарації, такі як хроматографічні процедури, етапи фільтрації та екстракції, можуть бути ефективними етапами видалення вірусу, якщо їх виконують у належно контрольованих умовах. Етап ефективного видалення вірусу повинен надавати у результаті відтворюване зниження вірусного навантаження, яке продемонстроване принаймні двома незалежним дослідженнями.
Загальний коефіцієнт зниження вірусного навантаження, як правило, виражається сумою окремих коефіцієнтів. Однак зниження титру вірусу до рівня 1 log10 або менше буде вважатися незначним та не буде враховуватися, якщо не обґрунтовано інше.
Якщо незначне зниження інфекційності вірусу досягається у процесі виробництва, а видалення вірусу вважається основним фактором безпеки продукту, необхідно вжити специфічних додаткових заходів з інактивації/видалення. Для усіх вірусів виробники повинні обґрунтувати прийнятність отриманих коефіцієнтів зниження інфекційності. Результати необхідно оцінювати з урахуванням факторів, які перераховані вище.
Г. Обмеження досліджень вірусного кліренсу
Дослідження вірусного кліренсу сприяють підтвердженню того, що досягається прийнятний рівень безпеки кінцевого продукту, але самі по собі ці дослідження не визначають безпеку продукту. Однак ряд факторів при розробці та проведенні досліджень вірусного кліренсу може призвести до неправильної оцінки здатності процесу видаляти вірусну інфекційність. До цих факторів належать:
1. Вірусні препарати, що використовуються в дослідженнях кліренсу для технологічного процесу, можуть бути отримані на культурі тканин. Поведінка вірусу з культури тканин на етапі виробництва може бути відмінною від поведінки нативного вірусу; наприклад, якщо нативний та культивований віруси відрізняються за чистотою або ступенем агрегації.
2. Інактивацію інфекційності вірусу часто можна охарактеризувати двофазною кривою, у якій за швидкою початковою фазою настає більш повільна фаза. Можливо, що вірус, який не був видалений на першому етапі інактивації, може бути більш резистентним до наступних етапів. Наприклад, якщо резистентні частки приймають форму агрегатів вірусів, інфекційність може бути резистентною до ряду різних хімічних обробок та до нагрівання.
3. Здатність усього процесу усувати інфекційність виражається як сума логарифму зниження на кожному етапі. Отримана сума коефіцієнтів зниження інфекційності на різних етапах, особливо на етапах з незначним зниженням (наприклад нижче 1 log10), може призвести до переоцінки дійсного потенціалу елімінації вірусу. Більше того, показники зниження вірусного навантаження, які досягаються при повторенні ідентичних або майже ідентичних процедур, не слід враховувати, якщо не обґрунтований інший підхід.
4. Вираження коефіцієнтів зниження у вигляді логарифмічних знижень титру передбачає, що, хоча залишкова інфекційність вірусу може бути значно знижена, вона ніколи не дійде до нуля. Наприклад, зменшення інфекційності препарату, що містить 8 log10/мл інфекційних одиниць на коефіцієнт 8 log10, зберігає 0 log10/мл або 1 інфекційну одиницю на 1 мл, враховуючи межі виявлення методу.
5. Обробка процесу дослідно-промислового масштабу може відрізнятися від обробки процесу промислового масштабу, незважаючи на детальне планування процесу зменшеного масштабу.
6. Додавання індивідуальних факторів зниження вірусного навантаження в результаті подібних механізмів інактивації впродовж процесу виробництва може призвести до переоцінки загального вірусного кліренсу.
Ґ. Статистична обробка
Дослідження вірусного кліренсу повинні включати статистичний аналіз даних при оцінці результатів. Результати аналізів повинні бути статистично значущими для підтримки зроблених висновків (див. додаток 3 до цієї настанови).
Д. Повторна оцінка вірусного кліренсу
Кожного разу при внесенні значних змін у технологічний процес виробництва або стадію очищення необхідно розглянути прямий та опосередкований вплив такої зміни на вірусний кліренс та провести повторну оцінку системи, якщо необхідно. Наприклад, зміни у технологічному процесі виробництва можуть спричинити значні зміни кількості вірусу, що утворюється клітинною лінією; зміни на стадіях технологічного процесу можуть змінити ступінь вірусного кліренсу.
VII. Резюме
У цій настанові представлено підходи до оцінки ризику вірусного забруднення та до методів видалення вірусів з препарату. Впровадження таких підходів сприяє виробництву безпечних біотехнологічних продуктів, отриманих з клітинних ліній тваринного або людського походження, та підкреслює важливість багатьох стратегій, включаючи:
а) ретельну характеристику/скринінг клітинного субстрату вихідного матеріалу з метою визначення, які вірусні контамінанти наявні, якщо такі є;
б) оцінку ризику шляхом визначення тропізму контамінантів до тканин людини;
в) встановлення відповідної програми тестування на наявність сторонніх вірусів у необробленому балк-матеріалі;
г) ретельне планування досліджень вірусного кліренсу з використанням різних методів інактивації або видалення вірусу в конкретному технологічному процесі виробництва з метою досягнення максимального вірусного кліренсу;
ґ) виконання досліджень з оцінки інактивації та видалення вірусу.
Таблиця 1. Тести на наявність вірусів, які проводяться одноразово на клітинах різних рівнів
|
ГБК |
РБКa |
КЛІТИНИ НА МЕЖІб |
|
|
Тести на ретровіруси та інші ендогенні віруси |
|||
|
Інфекційність |
+ |
— |
+ |
|
ЕМв |
+в |
— |
+в |
|
Зворотна транскриптазаг |
+г |
— |
+г |
|
Інші вірусоспецифічні тестиґ |
Якщо застосовнийґ |
— |
Якщо застосовнийґ |
|
Тести на неендогенні або занесені віруси |
|||
|
In vitro випробування |
+ |
—д |
+ |
|
In vivo випробування |
+ |
—д |
+ |
|
Тести на утворення антитіле |
+е |
— |
— |
|
Інші вірусоспецифічні тестиє |
+є |
— |
— |
aДив. розділ III.A.2.
бКлітини на межі — клітини, що знаходяться у граничному для виробництва клітинному віці in vitro (див. розділ III.A.3).
вМожливе також виявлення інших збудників.
гНе є необхідним, якщо тест на ретровірусну інфекційність позитивний.
ґЯкщо застосовний, то застосовується для клітинних ліній, про які відомо, що вони були інфіковані такими агентами.
дДля першого РБК цей тест слід проводити на клітинах, що знаходяться у граничному для виробництва клітинному віці in vitro, отриманих з цього РБК; для наступних РБК один тест in vitro та in vivo слід проводити або безпосередньо на РБК, або на клітинах, що знаходяться у граничному для виробництва клітинному віці in vitro.
еНаприклад, тести на утворення МА, ПА, ХА зазвичай застосовуються до клітинних ліній гризунів.
єНаприклад, тести для клітинних ліній, отриманих від людини, нелюдиноподібних приматів або інших клітинних ліній, залежно від обставин.
Таблиця 2. Приклади застосування та обмеження випробувань на наявність вірусів
|
ТЕСТ |
ДОСЛІДЖУВАНИЙ ЗРАЗОК |
ЗДАТНІСТЬ ВИЯВЛЕННЯ |
ОБМЕЖЕННЯ ВИЯВЛЕННЯ |
|
Утворення антитіл |
Лізат клітин та їх культуральне середовище |
Специфічні вірусні антигени |
Антитіла, неінфекційні для тваринних тест-систем |
|
In vivo скринінг вірусу |
Лізат клітин та їх культуральне середовище |
Широкий діапазон вірусів, патогенних для людини |
Збудники, що не реплікуються або не викликають хвороб у тест-системах |
|
In vitro скринінг вірусу для: |
Широкий діапазон вірусів, патогенних для людини |
Збудники, що не реплікуються або не викликають хвороб у тест-системах |
|
|
1. Характеристики банку клітин 2. Скринінгу виробництва |
1. Лізат клітин та їх культуральне середовище (для співкультивування інтактні клітини повинні входити до досліджуваного зразка). 2. Необроблений балк-матеріал або лізат клітин та їх культуральне середовище з промислового реактора |
||
|
ТЕМ на: |
Віруси та вірусоподібні частки |
Кількісний аналіз з оцінкою ідентичності |
|
|
1. Клітинному субстраті 2. Супернатанти клітинної культури |
1. Життєздатні клітини 2. Безклітинний культуральний супернатант |
||
|
Зворотна транскриптаза |
Безклітинний культуральний супернатант |
Ретровіруси та експресована ретровірусна зворотна транскриптаза |
Виявляє тільки ензими з оптимальною активністю за переважних умов. Інтерпретація може ускладнюватися через наявність клітинних ензимів; фон у деяких концентрованих зразках |
|
Інфекційність ретровірусу |
Безклітинний культуральний супернатант |
Інфекційні ретровіруси |
Ретровіруси, нездатні до реплікації або утворення дискретних осередків або плям у вибраній тест-системі |
|
Співкультивування 1. Кінцева точка інфекційності 2. Кінцева точка ТЕМ 3. Кінцева точка зворотної транскриптази |
Життєздатні клітини |
Інфекційні ретровіруси |
Ретровіруси, не здатні до реплікації 1. Див. вище «Інфекційність ретровірусу» 2. Див. вище «ТЕМ»а 3. Див. вище «Зворотна транскриптаза» |
|
ПЛР |
Клітини, культуральна рідина та інші матеріали |
Специфічні послідовності вірусів |
Повинні бути наявними послідовності праймерів. Не показує, чи є вірус інфекційним |
аДодатково важко відрізнити досліджуваний зразок від індикаторних клітин.
Таблиця 3. Віруси, що виявляються за допомогою тестів утворення антитіл
|
МИШАЧІ АНТИТІЛА |
АНТИТІЛА ХОМ’ЯКІВ |
АНТИТІЛА ПАЦЮКІВ |
|
Вірус ектромелії2, 3 |
Вірус лімфоцитарного хоріоменінгіту (LCM)1, 3 |
Хантавірус1, 3 |
|
Хантавірус1, 3 |
Вірус пневмонії мишей (PVM)2,3 |
Вірус пацюків Кілхама2, 3 |
|
К-вірус2 |
Реовірус типу 3 (Reo3)1, 3 |
Вірус енцефаломієліту мишей (Theilers, GDVII)2 |
|
Вірус молочної дегідрогенази (лактатдегідрогенази) (LDM)1, 3 |
Вірус Сендай1, 3 |
Вірус пневмонії мишей (PVM)2, 3 |
|
Вірус лімфоцитарного хоріоменінгіту (LCM)1, 3 |
SV5 |
Коронавірус пацюків (RCV)2 |
|
Дрібний вірус мишей2, 3 |
Реовірус типу 3 (Reo3)1, 3 |
|
|
Аденовірус мишей (МАV)2, 3 |
Вірус Сендай1, 3 |
|
|
Цитомегаловірус мишей (МСМV)2, 3 |
Вірус сіалоакреоаденіту (SDAV)2 |
|
|
Вірус енцефаломієліту мишей (Theilers, GDVII)2 |
Вірус Тулана (HI)2, 3 |
|
|
Вірус гепатиту мишей (МНV)2 |
||
|
Ротавірус мишей (EDIM)2, 3 |
||
|
Вірус пневмонії мишей (PVM)2, 3 |
||
|
Вірус поліоми2 |
||
|
Реовірус типу 3 (Reo3)1, 3 |
||
|
Вірус Сендай1, 3 |
||
|
Тимічний вірус2 |
1Віруси, для яких доведена їх здатність інфікувати людину або приматів.
2Віруси, для яких відсутні докази здатності інфікувати людину.
3Вірус, що здатний до реплікації in vitro у клітинах людини або приматів.
Таблиця 4. План дій для оцінки процесу вірусного кліренсу та виявлення вірусів в очищеному балк-матеріалі
|
Випадок А |
Випадок Б |
Випадок В2 |
Випадок Г2 |
Випадок Ґ2 |
|
|
СТАТУС |
|||||
|
Наявність вірусу1 |
– |
– |
+ |
+ |
(+)3 |
|
Вірусоподібні частки1 |
– |
– |
– |
– |
(+)3 |
|
Ретровірусоподібні частки1 |
– |
+ |
– |
– |
(+)3 |
|
Ідентифікований вірус |
Незастосовно |
+ |
+ |
+ |
– |
|
Вірус, патогенний для людини |
Незастосовно |
–4 |
–4 |
+ |
Невідомо |
|
ВИКОНАННЯ |
|||||
|
Характеристика процесу вірусного кліренсу із застосуванням неспецифічних «модельних» вірусів |
Так5 |
Так5 |
Так5 |
Так5 |
Так7 |
|
Оцінка процесу вірусного кліренсу із застосуванням «відповідних» або специфічних «модельних» вірусів |
Ні |
Так6 |
Так6 |
Так6 |
Так7 |
|
Тест на наявність вірусу в очищеному балк-матеріалі |
Незастосовно |
Так8 |
Так8 |
Так8 |
Так8 |
1Результати тестів на наявність вірусів на клітинному субстраті та/або на необробленому балк-матеріалі. Культури клітин, що застосовуються для виробництва, інфіковані вірусами, як правило, неприйнятні. Ендогенні віруси (такі як ретровіруси) або віруси, які є невід’ємною частиною ГБК, можуть бути прийнятними, якщо застосовуються належні процедури оцінки вірусного кліренсу.
2Використання вихідного матеріалу, контамінованого вірусами, незалежно від того, чи є вони інфекційними та/або патогенними для людини, дозволяється тільки за виняткових обставин.
3Вірус виявляли прямим або непрямим методом.
4Вважається непатогенним.
5Слід проводити характеристику кліренсу, застосовуючи неспецифічні «модельні» віруси.
6Слід проводити оцінку процесу для «відповідних» вірусів або специфічних «модельних» вірусів.
7Див. опис випадку Ґ.
8Відсутність вірусу, що виявляється, слід підтвердити для очищеного балк-матеріалу з використанням високоспецифічних та високочутливих методів виявлення вірусу. У реєстраційному досьє мають бути представлені дані принаймні на 3 серії очищеного балк-матеріалу дослідно-промислового або промислового масштабу. Однак для клітинних ліній, таких як лінія CHO, для яких ендогенні частки були широко охарактеризовані та продемонстрований їх адекватний кліренс, зазвичай не потрібний кількісний аналіз наявних неінфекційних часток в очищеному балк-матеріалі.
Додаток 1 (обов’язковий). Продукти, отримані від охарактеризованих банків клітин, які були згодом вирощені in vivo
Для продуктів, що виробляються з рідин, отриманих від тварин, які було інокульовано клітинами з охарактеризованих банків, необхідно надавати додаткову інформацію про тварин.
Коли можливо, тварин, які використовувалися при виробництві біотехнологічних/біологічних продуктів, слід обирати з чітко охарактеризованих колоній без специфічних патогенів. Необхідно провести адекватне тестування на наявність відповідних вірусів, таких як ті, що зазначені у табл. 3 цієї настанови. Мають бути описані карантинні процедури для вперше прибулих та хворих тварин. Також слід надати підтвердження, що усі методи ізоляції, очищення та дезінфекції, які застосовуються на об’єкті, адекватні для стримування розповсюдження занесених збудників. Цього можна досягти за допомогою застосування програми сигналізування (sentinel program). Слід також включити перелік збудників, для яких проводиться тестування. На об’єкті або у межах доступності мають бути організовані ветеринарні служби. Слід описати, якою мірою віварій ізольований від інших зон виробничого об’єкту. Діяльність персоналу у виробничому процесі має бути адекватною для забезпечення безпеки.
Процедури утримання тварин повинні бути повністю описані. Сюди можна включити раціон харчування, графіки прибирання та годування, положення про регулярний ветеринарний догляд, якщо застосовне, а також докладну інформацію про особливий догляд за тваринами, яким інокульовано збудник. Слід також включити опис режиму попередньої імунізації тварин, підготовку інокуляту та зазначити місце та спосіб інокуляції.
Первинний матеріал, зібраний від тварин, можна вважати еквівалентом стадії виробництва необробленого балк-матеріалу з біореактора. Отже, до нього застосовують усі положення щодо тестування, описані у розділі IV цієї настанови. Крім того, виробник повинен оцінити біонавантаження необробленого балк-матеріалу, визначити, чи вільний матеріал від мікоплазми, а також провести видоспецифічні кількісні аналізи та тестування in vivo на дорослих та новонароджених мишах.
Додаток 2 (обов’язковий). Вибір вірусів для досліджень вірусного кліренсу
А. Приклади придатних «модельних» вірусів
Неспецифічні «модельні» віруси, що представляють ряд фізико-хімічних структур:
- SV40 (поліомавірус мавп 1), поліовірус людини 1 (Себін), парвовірус тварин або деякі дрібні безоболонкові віруси;
- вірус парагрипу або вірус грипу, вірус Синдбіс або деякі РНК-віруси в оболонці від середніх до великих;
- вірус герпесу (наприклад вірус простого герпесу та/або вірус псевдосказу) або деякі інші ДНК-віруси від середніх до великих.
Ці віруси наведені як приклади, та їх застосування не є обов’язковим.
В. Приклади вірусів, які застосовувалися у дослідженнях вірусного кліренсу
Декілька вірусів, що застосовувалися у дослідженнях вірусного кліренсу, представлені у табл. А-1. Однак оскільки це тільки приклади, застосування будь-якого з вірусів, зазначених у таблиці, не є обов’язковим, а виробники можуть обирати інші віруси, особливо ті, що відповідають їх власним процесам виробництва. Як правило, процес слід оцінювати на його здатність елімінації принаймні трьох різних вірусів з характеристиками, що відрізняються.
Таблиця А-1. Приклади вірусів, які застосовуються у дослідженнях вірусного кліренсу
|
Вірус |
Сімейство |
Рід |
Природний хазяїн |
Геном |
Ген протеїну оболонки (вірусу) |
Розмір, нм |
Форма |
Резистентність1 |
|
Вірус везикулярного стоматиту |
Rhabdo- |
Везикуловірус |
Кінь, бик |
РНК |
Так |
70х150 |
Кулеподібна |
Низька |
|
Вірус парагрипу |
Paramyxo- |
Параміксовірус |
Різні види |
РНК |
Так |
100–200+ |
Плеоморфна/сферична |
Низька |
|
Вірус лейкемії мишей (MuLV) |
Retro- |
Онковірус типу С |
Миша |
РНК |
Так |
80–110 |
Сферична |
Низька |
|
Вірус Cиндбіс |
Toga- |
Альфавірус |
Людина |
РНК |
Так |
60–70 |
Сферична |
Низька |
|
Вірус діареї великої рогатої худоби (BVDV) |
Flavi- |
Пестивірус |
Бик |
РНК |
Так |
50–70 |
Плеоморфна/сферична |
Низька |
|
Вірус псевдосказу |
Herpes- |
Свиня |
ДНК |
Так |
120–200 |
Сферична |
Середня |
|
|
Поліовірус Себін (Тип 1) |
Picorna- |
Ентеровірус |
Людина |
РНК |
Ні 2 |
5–30 |
Ікосаедральна |
Середня |
|
Вірус енцефаломіокардиту (ЕМК) |
Picorna- |
Кардіовірус |
Миша |
РНК |
Ні |
25–30 |
Ікосаедральна |
Середня |
|
Реовірус 3 |
Reo- |
Ортореовірус |
Різні види |
РНК |
Ні |
60–80 |
Сферична |
Середня |
|
Поліовірус мавп (SV40) |
Papova- |
Поліомавірус |
Мавпа |
ДНК |
Ні |
40–50 |
Ікосаедральна |
Дуже висока |
|
Парвовіруси (собак, свиней) |
Parvo- |
Парвовірус |
Собака, свиня |
ДНК |
Ні |
18–24 |
Ікосаедральна |
Дуже висока |
1Стійкість до фізико-хімічної обробки за даними досліджень процесу виробництва. Резистентність пов’язана з певними видами обробки та застосовується у контексті розуміння біології вірусу та характеру виробничого процесу. Фактичні результати можуть розрізнятися залежно від виду обробки. Ці віруси наведені як приклади, їх застосування не є обов’язковим.
Додаток 3 (обов’язковий). Статистичні розрахунки при оцінці аналізів на віруси
При титруванні вірусів виникають проблеми варіабельності, що є загальними для усіх біологічних систем аналізу. Для встановлення надійності дослідження необхідно проводити оцінку точності титрування вірусів та отриманих у результаті цього знижувальних коефіцієнтів, а також визначити обґрунтованість кількісних аналізів. Метою статистичної оцінки є встановлення факту, що випробування проводилися з достатнім рівнем достовірності.
1. Методи аналізу можуть бути дискретними або кількісними. Дискретні методи включають кількісні аналізи інфекційності у тварин або в культурі тканин з інфекційною дозою (TCID), за результатами яких визначається, інфікована тварина або культура тканин чи ні. За титрами інфекційності потім вимірюється число інфікованих тварин або клітин у культурі. У кількісних методах вимірювана інфекційність змінюється постійно у міру введення вірусу. Кількісні методи включають метод бляшок, коли кожна нарахована бляшка відповідає окремій інфекційній частці. Як дискретний, так і кількісний аналізи підлягають статистичній оцінці.
2. Варіабельність може виникнути в ході аналізу як результат помилок розведення, статистичних ефектів або відмінностей у системі аналізу, які або невідомі, або їх важко контролювати. Ці ефекти можуть бути більше виражені, коли порівнюються результати різних аналізів (міжаналітична варіабельність), ніж коли порівнюються результати, отримані у межах одного аналізу (внутрішньоаналітична варіабельність).
3. Межі 95% довірчого інтервалу для результатів внутрішньоаналітичної варіабельності мають бути близько ±0,5 log10 від середнього значення. Внутрішньоаналітичну варіабельність можна оцінювати за допомогою стандартних аналітичних методів. Міжаналітичну варіабельність можна контролювати за допомогою використання референтного препарату, ступінь активності якого має становити 0,5 log10 від середнього значення, встановленого у лабораторії, що можна вважати прийнятним. Аналізи з меншою точністю можуть бути прийнятними за умови відповідного обґрунтування.
4. Межі 95% довірчого інтервалу для коефіцієнта зниження інфекційності, що відмічається, слід розраховувати завжди, коли це можливо, у дослідженнях кліренсу «відповідних» та специфічних «модельних» вірусів. Якщо межі 95% довірчого інтервалу для вірусних аналізів у вихідному матеріалі становлять +s, а для вірусних аналізів матеріалу після цього етапу +а, межі 95% довірчого інтервалу для коефіцієнта зменшення розраховують за формулою:
![]()
Вірогідність виявлення вірусів при низьких концентраціях
При низьких концентраціях вірусів (тобто у діапазоні 10–1000 інфекційних часток на 1 л) зразок об’ємом у декілька мілілітрів може як містити, так і не містити інфекційних часток. Вірогідність (р), що цей зразок не містить інфекційних вірусів, становить:
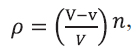
де:
V — загальний об’єм матеріалу, який необхідно дослідити, л;
v — об’єм зразка, л;
n — абсолютне число інфекційних часток, статистично розподілених у V.
Якщо V>v, це рівняння може бути приблизно виражене через розподіл Пуассона:
![]()
де с — концентрація інфекційних часток у літрі, або:
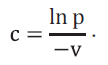
Наприклад, об’єм зразка становить 1 мл, вірогідність р при концентраціях вірусу від 10 до 1000 інфекційних часток на 1 л становить:
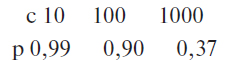
Це означає, що для концентрації 1000 вірусів на 1 л у 37% вибірки 1 мл не буде містити часток вірусів.
Якщо на наявність вірусу тестується частина зразків і тест негативний, слід розраховувати кількість вірусу, яка має міститися у загальному зразку для отримання позитивного результату. Це значення слід враховувати при обчисленні коефіцієнта зменшення. Визначення меж 95% довірчого інтервалу є бажаним. Однак у деяких випадках це може бути непрактичним через обмеження матеріалу.
Додаток 4 (обов’язковий). Розрахунок коефіцієнтів зниження у дослідженнях для виявлення вірусного кліренсу
Коефіцієнт зниження вірусного навантаження окремого етапу очищення або інактивації визначається як log10 співвідношення вірусного навантаження у матеріалі перед очищенням та вірусного навантаження у матеріалі після очищення, готовому для використання на наступному етапі процесу. Застосовуються такі абревіатури:
Вихідний матеріал:
об’єм — vʹ; титр — 10аʹ;
вірусне навантаження: (vʹ)(10аʹ).
Кінцевий матеріал:
об’єм — vʹʹ;
титр — 10аʹʹ;
вірусне навантаження: (vʹʹ)(10аʹʹ),
Індивідуальні коефіцієнти зниження вірусного навантаження Ri розраховуються за формулою:
![]()
Ця формула враховує титри та об’єми матеріалу перед та після етапу очищення.
Через неточність, характерну для деяких титрувань вірусів, для розрахунку загального коефіцієнта зниження приймаються індивідуальні коефіцієнті зниження вірусного навантаження більше ніж 1.
Загальний коефіцієнт зниження для всього процесу виробництва розраховується як сума логарифмів коефіцієнтів зниження на окремих етапах. Він представляє логарифм співвідношення вірусного навантаження на початку першого етапу очищення та наприкінці останнього етапу очищення. Коефіцієнти зниження вірусного навантаження зазвичай виражаються за логарифмічною шкалою, що засвідчує, що, незважаючи на неможливість зниження залишкової інфекційності вірусу до нуля, вона може бути суттєво зменшена математично.
Додаток 5 (обов’язковий). Розрахунок передбаченої кількості часток у дозі
Це стосується тих вірусів, для яких можна визначити вихідні кількості, наприклад ендогенних ретровірусів.
Приклад:
I. Умова
Виміряна або розрахована концентрація вірусу у зборах клітинних культур = 106/мл.
Розрахований коефіцієнт вірусного кліренсу = >1015.
Об’єм отриманої культури, що необхідний для приготування дози продукту, = 1 л (103 мл).
II. Обчислення розрахункової кількості часток у дозі
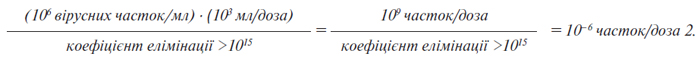
Таким чином, слід очікувати менше ніж одну частку на мільйон доз.
Національний додаток (довідковий). Бібліографія
1. Наказ МОЗ України від 26.08.2005 р. № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення», зареєстрований у Міністерстві юстиції України 19 вересня 2005 року за № 1069/11349 (із змінами).
2. CPMP/ICH/295/95 (ICH Q5А (R1)) «Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin» (Оцінка вірусної безпеки біотехнологічних продуктів, отриманих з клітинних ліній людського або тваринного походження).
3. The Rules Governing Medicinal Products in the European Union. — Volume 4. — EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 4. — Правила ЄС з належної виробничої практики лікарських препаратів для людини та застосування у ветеринарії).
4. Настанова СТ-Н МОЗУ 42-4.0:2011. — Лікарські засоби. Належна виробнича практика.
5. ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів/І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
6. Настанова СТ-Н МОЗУ 42-1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення/М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005.
7. ДСТУ 1.7-2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів/О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
8. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
Ключові слова: біотехнологічний продукт, вірусна безпека, контамінація, біотехнологічне виробництво, віруси, «модельні» віруси, «відповідні» віруси, вірусний кліренс.