
НАСТАНОВА
НАСТАНОВА З ЯКОСТІ
ЛІКАРСЬКІ ЗАСОБИ
ВИПРОБУВАННЯ СТАБІЛЬНОСТІ
Настанова 42-3.3:2004
Видання офіційне
Київ
Міністерство охорони здоров’я України
2012
Передмова
1 РОЗРОБЛЕНО: ДП «Державний науковий центр лікарських засобів» (ДП «ДНЦЛЗ»)
РОЗРОБНИКИ: В. Георгієвський, доктор фарм. наук (керівник розробки); М. Ляпунов, доктор фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; А. Піотровська; О. Гризодуб, доктор хім. наук; О. Кричевська; Ю. Підпружников, доктор фарм. наук; О. Антипова, канд. фарм. наук; Н. Крупа; Т. Матвієнко; І. Юдіна; К. Жемерова
ВНЕСЕНО: Державною службою лікарських засобів і виробів медичного призначення Міністерства охорони здоров’я України
2 ПРИЙНЯТО І НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 31 грудня 2003 р. № 637
3 Ця настанова відповідає:
СРМР/ІСН/2736/99 соrr (Q1A R) Note for guidance on stability testing: stability testing of new drug substances and products (revision), 2000 (CPMP/ICH/2736/99 corr (Q1A R) Керівні вказівки з випробувань стабільності: випробування стабільності нових лікарських речовин та препаратів (переглянуто), 2000) у частині підрозділу 5.1
СРМР/ІСН/279/95 (Q1В) Note for guidance on the photostability testing of new active substances and medicinal products, 1996 (CPMP/ICH/279/95 (Q1B) Керівні вказівки з випробувань світлостабільності нових діючих речовин та лікарських засобів, 1996) у частині підрозділу 5.2
СРМР/ІСН/280/95 (Q1C) Note for guidance on stability testing: requirements for new dosage form, 1996 (CPMP/ICH/280/95 (Q1C) Керівні вказівки з випробувань стабільності: вимоги щодо нових лікарських форм, 1996) у частині підрозділу 5.3
CPMP/QWP/122/02 Rev 1 Guideline on stability testing: stability testing of existing active substances and related finished products, 2003 (CPMP/QWP/122/02 Rev 1 Настанова з випробувань стабільності: випробування стабільності існуючих діючих речовин та відповідних готових лікарських засобів, 2003) у частині підрозділу 5.4
СРМР/ІСН/4104/00 (Q1D) Note for guidance on bracketing and matrixing for stability testing of drag substances and drug products, 2002(CPMP/ICH/4104/00 (Q1D) Керівні вказівки щодо брекетингу та побудови матриць для випробувань стабільності лікарських речовин та лікарських засобів, 2002) у частині підрозділу 5.5
СРМР/ІСН/420/02 (Q1E) Note for guidance on evaluation of stability data, 2003 (CPMP/ICH/420/02 (Q1E) Керівні вказівки щодо оцінки даних зі стабільності, 2003) у частині підрозділу 5.6
CPMP/QWP/2934/99 Note for guidance on in-use stability testing of human medicinal products, 2001 (CPMP/QWP/2934/99 Керівні вказівки з випробувань стабільності лікарських засобів для людини під час застосування, 2001) у частині підрозділу 5.7
CPMP/QWP/159/96 corr Note for guidance on maximum shelf-life for sterile products for human use after first opening or following reconstitution, 1998 (CPMP/QWP/159/96 corr Керівні вказівки щодо максимального терміну зберігання стерильних препаратів для людини після першого розкриття або після підготовки до застосування, 1998) у частині підрозділу 5.8
CPMP/QWP/576/96 Note for guidance on stability testing for a type II variation to a marketing authorization, 1998 (CPMP/QWP/576/96 Керівні вказівки з випробувань стабільності при зміні типу II у торговій ліцензії, 1998) у частині підрозділу 5.9
СРМР/ІСН/421/02 (Q1F) Note for guidance on stability data package for registration applications in climatic zones III and IV, 2003 (CPMP/ICH/421/02 (Q1F) Керівні вказівки щодо даних зі стабільності для заявок на реєстрацію у кліматичних зонах III і IV, 2003) у частині підрозділу 5.10
CPMP/QWP/609/96/Rev 2 Guideline on declaration of storage conditions: A: in the product information of medicinal products; B: for active substances. Annex to Note for guidance on stability testing of new drug substances and products. Annex to Guideline on stability testing of existing active substances and related finished products, 2003 (CPMP/QWP/609/96/Rev 2 Настанова з декларування умов зберігання: А: в інформаційних матеріалах для лікарських засобів; В: для діючих речовин. Додаток до керівних вказівок з випробувань стабільності нових лікарських речовин та препаратів. Додаток до настанови з випробувань стабільності існуючих діючих речовин та відповідних готових лікарських засобів, 2003) у частині додатка А
Ступінь відповідності — модифікований (MOD)
Переклад з англійської мови (en)
4 ВВЕДЕНО ВПЕРШЕ
© Державна служба лікарських засобів і виробів медичного призначення, 2012
© Колектив розробників, 2012
© МОРІОН, 2012
ПЕРЕДМОВА ДО ДОКУМЕНТА СРМР/ІСН/2736/99 соrr (Q1A R) «Stability testing guidelines: stability testing of new drug substances and products. Note for guidance on stability testing: stability testing of new drug substances and products (revision)»
Ця настанова є переглянутою версією настанови ІСН Q1A, в ній визначено набір даних зі стабільності для нової діючої речовини чи лікарського препарату, що є достатнім для заявки на реєстрацію в межах трьох регіонів: ЄС, Японії та США. Вона не має на меті обов’язково охопити всі випробування, необхідні для реєстрації в інших регіонах світу чи експорту в ці регіони.
У настанові як приклад наведено набір основних даних зі стабільності, необхідних для нових діючих речовин і лікарських препаратів; однак допускається достатня гнучкість щодо різних ситуацій, які виникають на практиці, у тому числі зумовлених особливими науковими уявленнями та характеристиками речовин, що оцінюються. Можуть бути використані альтернативні підходи, якщо є відповідні науково обґрунтовані причини.
ПЕРЕДМОВА ДО ДОКУМЕНТА СРМР/ІСН/279/95 (Q1B) «Photostability testing of new active substances and medicinal products. Note for guidance on the photostability testing of new active substances and medicinal products»
У керівних вказівках «Stability testing guidelines: stability testing of new drag substances and products. Note for guidance on stability testing: stability testing of new drag substances and products (revision)», спершу опублікованих як гармонізована тристороння настанова ІСН (далі в даному документі на неї посилаються як на «основну настанову»), констатовано, що випробування світлостабільності мають бути невід’ємною частиною стресових випробувань. Цей документ є доповненням до основної настанови та надає рекомендації щодо випробувань світлостабільності.
ПЕРЕДМОВА ДО ДОКУМЕНТА СРМР/ІСН/280/95 (Q1C) «Stability testing: requirements for new dosage form. Note for guidance on stability testing: requirements for new dosage form»
Цей документ є доповненням до керівних вказівок «Stability testing guidelines: stability testing of new drag substances and products. Note for guidance on stability testing: stability testing of new drug substances and products (revision)»; він містить рекомендації стосовно даних про стабільність нових лікарських форм, які мають бути надані заявником після подання первинної заявки для ліцензування нової діючої речовини та лікарського препарату.
ПЕРЕДМОВА ДО ДОКУМЕНТА CPMP/QWP/122/02 Rev 1 «Note for guidance on stability testing: stability testing of existing active substances and related finished products»
Перегляд у грудні 2003 року:
Настанову CPMP/QWP/122/02 corr було переглянуто на відповідність вимогам документів «Note for Guidance on Evaluation of Stability Data (CPMP/ICH/420/02)» і «Note for Guidance on Stability testing of New Drag Substances and Products (CPMP/ICH/2736/99 corr)».
Були внесені такі зміни:
1. У додатку II з посиланням на відповідні керівні вказівки описано, як за допомогою екстраполяції можна збільшити період до повторних випробувань або термін зберігання понад період, для якого є одержані в реальному часі дані.
2.У п. 2.2.7.3 «Лікарські засоби, упаковані в напівпроникні контейнери» як прийнятну альтернативу для умов зберігання — температура (25±2) °С і відносна вологість (40±5)% — були додатково включені умови: температура (30±2) °С і відносна вологість (35±5)%.
3. До передмови, що стосується перегляду в грудні 2002 року, було внесено уточнення про надання чинності зміні щодо проміжних умов зберігання.
Внесення поправок в січні 2003 року:
У п. 2.1.7 документа CPMP/QWP/122/02 були внесені правки, щоб надати керівні вказівки щодо тривалості довгострокових випробувань стабільності.
Перегляд в грудні 2002 року:
Настанові CPMP/QWP/556/96 був присвоєний новий номер CPMP/QWP/122/02; документ було переглянуто на відповідність вимогам документів «Note for Guidance on Stability Data Package for Registration in Climatic Zone III and IV(CPMP/ICH/421/02)», «Note for Guidance on Stability testing of New Drug Substances and Products (CPMP/ICH/2736/99 corr)», а також положенням Загального технічного документа (Common Technical Document) (CPMP/ICH/2287/99).
Внаслідок цього проміжні умови випробувань (температура (30±2) °С і відносна вологість 60%) будуть змінені на такі умови: температура (30±2) °С і відносна вологість 65%. Дата надання чинності цій зміні залежатиме від дати надання чинності настанові «Note for Guidance on Stability testing of New Drug Substances and Products (CPMP/ICH/2736/99 corr)».
У ЄС результати досліджень в нових умовах прийматимуться негайно. Більше того, результати випробувань стабільності, при яких відносна вологість була змінена (з 60% на 65%) в ході дослідження відповідно до нових вимог, також прийматимуться, якщо в матеріалах заявки будуть чітко вказані відповідні умови зберігання і дата їх зміни.
Рекомендується, щоб до лютого 2006 р. по можливості в усіх заявках на отримання торгових ліцензій були наведені результати завершених випробувань за проміжних умов зберігання (температура (30±2) °С і відносна вологість 65%).
Національний вступ
У фармацевтичному секторі України відбувається гармонізація законодавчої та нормативної бази з відповідними нормами Європейського Союзу (ЄС). У цей час в Україні вже надано чинності таким гармонізованим документам:
- Державна Фармакопея України (ДФУ), гармонізована з Європейською Фармакопеєю;
- Настанова 42-01-2001 «Лікарські засоби. Належна виробнича практика», гармонізована з настановою з GMP ЄС;
- Настанова 42-02-2002 «Лікарські засоби. Належна виробнича практика активних фармацевтичних інгредієнтів», гармонізована з додатком 18 до настанови з GMP ЄС;
- Настанова 42-01-2002 «Лікарські засоби. Належна практика дистрибуції», гармонізована з настановою з GDP ЄС, та ін.
Правила GMP активних фармацевтичних інгредієнтів передбачають контроль їх стабільності (5.11.5) та встановлення дати закінчення терміну придатності або повторних випробувань (5.11.6), що повинна ґрунтуватися на результатах оцінки даних, отриманих при вивченні стабільності. Правила GMP вимагають здійснення заходів, які гарантують, що якість лікарських засобів підтримується протягом усього терміну придатності при їх зберіганні, розповсюдженні та наступному обігу (5.1.2), а також випробувань продукції в процесі зберігання (5.1.4). Відповідно до правил GMP у специфікаціях на вихідну сировину має бути встановлений максимальний період зберігання до повторного контролю, а в специфікаціях на готову продукцію — термін придатності (5.4.11, 5.4.13). В обов’язки виробника входить дослідження стабільності продукції (5.6.2), результати якого визначають термін та умови її зберігання (5.6.14).
Загальні статті та монографії фармакопеї, а також специфікації виробників передбачають певні умови зберігання діючих і допоміжних речовин, яких відповідно до правил GDP повинні дотримуватися при їх зберіганні та дистрибуції. За правилами GDP лікарські засоби з терміном придатності, що минув, не підлягають продажу та поставці. Термін придатності й умови зберігання діючих речовин і лікарських засобів можна встановити лише на підставі результатів дослідження їх стабільності.
Дослідження стабільності необхідно здійснювати вже на етапі розробки діючих речовин і лікарських засобів. При державній реєстрації (перереєстрації) лікарських засобів потрібно надати дані зі стабільності у складі реєстраційного досьє. Однак ні у ДФУ, ні в настановах із GМР не наведена досить повна методологія організації цих досліджень.
Загальний технічний документ (Common Technical Document — CTD), прийнятий у ЄС, США та Японії, встановлює конкретні вимоги щодо надання даних зі стабільності діючих речовин і лікарських засобів у складі реєстраційного досьє і дає посилання на спеціальні настанови з якості та біотехнології, відповідно до яких слід проводити ці дослідження. Ці настанови й складають методичну основу досліджень стабільності діючих речовин і лікарських засобів, визначення періодів до проведення повторних випробувань і термінів придатності відповідно, а також умов зберігання.
Ця настанова розроблена на підставі декількох настанов із якості ІСН і GPMP, в яких розглядаються питання вивчення стабільності діючих речовин та готових лікарських засобів.
У настанову внесено такі редакційні зміни:
а) ряд документів СРМР та ІСН, присвячених дослідженням стабільності, об’єднано в одну настанову без зміни їх обсягу та змісту;
б) додатково введені розділи «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Позначення та скорочення», а також додаток В «Бібліографія». Основні положення викладені в розділі 5, а кожен підрозділ відповідає певному документу СРМР/ІСН. Відповідно до цього номери пунктів і підпунктів включають додатково номер розділу 5 і порядковий номер підрозділу (наприклад, 5.1.2.1; при цьому цифри 2.1 відповідають номеру пункту у відповідній настанові СРМР/ІСН). Усі терміни за абеткою наведені в розділі 3 «Терміни та визначення понять»;
в) посилання на нормативні документи, що згадуються в тексті, в повному обсязі наведені в розділі 2 «Нормативні посилання». При згадуванні у тексті нормативного документа, прийнятого у рамках ІСН або у ЄС, у виносках наприкінці відповідних сторінок зазначено: «Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку»;
г) у настанові замінено наступні слова:
- «Європейський Союз» — на «Україна»,
- «заявка на одержання торгової ліцензії» («application for a marketing authorisation») — на «заявка на реєстрацію»,
- «досьє для торгової ліцензії» («dossier for marketing authorisation») — на «реєстраційне досьє»;
д) замість «Європейська Фармакопея чи фармакопея держави ЄС» зазначено «Державна Фармакопея України чи Європейська Фармакопея, або інша відповідна фармакопея». Це пов’язано з тим, що Державна Фармакопея України гармонізована з Європейською Фармакопеєю, а встановлені в ній національні додаткові вимоги жорсткіші. Під словами «інша відповідна фармакопея» слід розуміти фармакопею держави ЄС, гармонізовану з Європейською Фармакопеєю, а також Фармакопею США та Фармакопею Японії;
є) додатково в розділі 3 «Терміни та визначення понять» наведені терміни, прийняті в інших настановах МОЗ України;
ж) у п. 5.1.1 зазначено, що Україна розташована в кліматичних зонах І і II;
и) «перевірка можливості об’єднання даних для кількох серій» додатково названа «перевіркою однорідності вибірки»;
к) у тексті настанови як примітка зазначено номери та назви відповідних пунктів реєстраційного досьє у форматі CTD, в яких слід наводити необхідну інформацію про стабільність;
л) інші незначні доповнення виділено іншим шрифтом та літерою N.
НАСТАНОВА
Настанови з якості
ЛІКАРСЬКІ ЗАСОБИ
Випробування стабільності
Руководства по качеству
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Испытания стабильности
Quality guidelines
MEDICINAL PRODUCTS
Stability testing
Чинна від 2004-04-01
1 Сфера застосування
Ця настанова поширюється на лікарські засоби для людини та містить рекомендації щодо вивчення їх стабільності.
Ця настанова рекомендується для підприємств, організацій та установ, що розробляють і/або серійно виготовляють діючі речовини та лікарські засоби на території України, незалежно від відомчого підпорядкування та форми власності, для науково-експертних організацій та регуляторних органів, а також експертів та інспекторів, що здійснюють експертизу на етапі реєстрації (перереєстрації) лікарських засобів та інспектування їх виробництва.
Цю настанову рекомендується застосовувати при плануванні та проведенні досліджень із вивчення стабільності діючих речовин і лікарських засобів, а також при складанні реєстраційного досьє.
2 Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 р.
Настанова 42-3.1:2004 Настанови з якості. Лікарські засоби. Фармацевтична розробка Настанова 42-3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності
Настанова 42-3.4:2004 Настанови з якості. Лікарські засоби. Виробництво готових лікарських засобів
European Pharmacopoeia. 4th Edition. 2002
(Європейська Фармакопея. 4е видання. 2002)
СРМР/ІСН/2737/99 (Q3A) Note for guidance in impurities testing: Impurities in new drug substances, 2002
(CPMP/ICH/2737/99 (Q3A) Керівні вказівки з випробувань на домішки: Домішки у нових лікарських речовинах, 2002)
СРМР/ІСН/282/95 (Q3B) Note for guidance on impurities in new drug products, 1996 (CPMP/ICH/282/95 (Q3B) Керівні вказівки щодо домішок у нових лікарських препаратах, 1996) СРМР/ІСН/283/95 (Q3C) Note for guidance on impurities: residual solvents, 1997 (CPMP/ICH/283/95 (Q3C) Керівні вказівки щодо домішок: залишкові розчинники, 1997) СРМР/ІСН/1507/02 (Q3C(M)) Maintenance of document for note for guidance on impurities: residual solvents, 2002
(CPMP/ICH/1507/02 (Q3C(M)) Додатковий документ до керівних вказівок щодо домішок: залишкові розчинники, 2002)
СРМР/ІСН/138/95 (Q5C) Note for guidance on quality of biotechnological products: Stability testing of biotechnological/biological products, 1995
(CPMP/ICH/138/95 (Q5C) Керівні вказівки з якості біотехнологічних препаратів: випробування стабільності біотехнологічних/біологічних препаратів, 1995)
СРМР/ІСН/365/96 (Q6B) Note for guidance on specifications — Test procedures and acceptance criteria for biotechnological/biological products, 1999
(СРМР/ІСН/365/96 (Q6B) Керівні вказівки із специфікацій — Методики випробувань та критерії прийнятності для біотехнологічних/біологічних препаратів, 1999)
CPMP/QWP/2819/00 (EMEA/CVMP/814/00) Note for Guidance on Quality of Herbal Medicinal Products, 2001
(CPMP/QWP/2819/00 (EMEA/CVMP/814/00) Керівні вказівки з якості лікарських засобів із рослинної сировини, 2001)
CPMP/QWP/297/97 Note for Guidance on Summary of Requirements for Active Substances in Part II of the Dossier, 1997
(CPMP/QWP/297/97 Керівні вказівки щодо вимог до діючих речовин для складання частини II досьє, 1997)
CPMP/QWP/2820/00 (EMEA/CVMP/815/00) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for Herbal Drugs, Herbal Drags Preparations and Herbal Medicinal Products, 2001
(CPMP/QWP/2820/00 (EMEA/CVMP/815/00) Керівні вказівки зі специфікацій: контрольні випробування та критерії прийнятності для лікарської рослинної сировини, препаратів з лікарської рослинної сировини, що призначені для виготовлення лікарських засобів, а також лікарських засобів з рослинної сировини, 2001)
Stability testing of pharmaceutical product containing well established drag substances in conventional dosage forms (WHO Technical Report Series, № 863. — Annex 5)
(Випробування стабільності лікарських засобів, що містять добре вивчені лікарські речовини у складі традиційних лікарських форм (Серія технічних доповідей ВООЗ, № 863. — Додаток 5)
ISO 10977:1993 Photography — Processed photographic colour films and paper prints. Methods for measuring image stability
(ISO 10977:1993 Фотографія — Оброблені фотографічні кольорові плівки та відбитки на папері. Методи визначення стабільності зображення)
Довідкові джерела інформації наведені в додатку В.
3 Терміни та визначення понять
3.1 У цій настанові використані терміни, встановлені в Настанові 42-3.1:2004: діюча речовина (лікарська речовина, активний фармацевтичний інгредієнт); допоміжна речовина; лікарська форма; сила дії лікарського засобу.
3.2 У цій настанові використаний термін, встановлений у Настанові 42-3.2:2004: специфікація.
3.3 У цій настанові використаний термін, встановлений у Настанові 42-3.4:2004: готовий лікарський засіб.
3.4 Нижче наведені визначення термінів, додатково використаних у цій настанові. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [7,8].
3.4.1 баланс маси (mass balance, [7])
Процес підсумовування маси, отриманої при кількісному визначенні, і маси продуктів розкладу для того, щоб побачити, наскільки близькою є отримана сума до 100% від вихідного значення з урахуванням допустимої помилки аналітичної методики.
3.4.2 брекетинг (bracketing, [7])
Складання плану досліджень стабільності таким чином, щоб випробуванню піддавались зразки лише з граничними значеннями певних показників у ряду (наприклад, сили дії, розміру паковання і/або наповнення) в усіх тих точках контролю, що й при проведенні повних досліджень. План передбачає, що стабільність зразків із проміжними значеннями показників у ряду ототожнюється зі стабільністю зразків із граничними значеннями. Якщо випробування повинне бути проведене для препаратів з різною силою дії, то брекетинг може бути застосований, зокрема, якщо зміна сили дії не пов’язана зі зміною складу (наприклад, для ряду таблеток різної маси, виготовлених із того самого основного гранулята, або для ряду капсул з оболонками різних розмірів, вміст яких має той самий склад, але різну масу). Брекетинг може бути застосований для лікарського препарату з різним розміром первинних паковань або з різним об’ємом наповнення, якщо матеріал паковання й тип закупорювання однакові.
3.4.3 вихідна серія (primary batch, [7])
Використовувана при офіційних дослідженнях стабільності серія діючої речовини або лікарського засобу, дані про стабільність якої подані в заявці на реєстрацію для встановлення періоду до проведення повторних випробувань або терміну зберігання відповідно. Вихідною серією діючої речовини має бути, принаймні, дослідно-промислова серія. Для лікарського засобу дві з трьох серій мають бути, як мінімум, дослідно-промисловими, а третя, може бути меншою за розміром, якщо вона є репрезентативною стосовно критичних стадій виробництва. Однак вихідною серією може бути й промислова серія.
3.4.4 герметичні контейнери (impermeable containers, [7])
Контейнери, що забезпечують постійний бар’єр для проникнення газів або розчинників, наприклад, герметизовані туби для м’яких лікарських засобів, запаяні скляні ампули для розчинів.
3.4.5 дані, що додатково підтверджують стабільність (supporting stability data, [7])
Дані, що не є основними даними зі стабільності, отриманими в ході офіційних досліджень; вони додатково підтверджують аналітичні методики, пропонований період до проведення повторних випробувань або термін зберігання, а також вказівки щодо умов зберігання, наведених на етикетці. Такі дані включають: (1) дані про стабільність для серій синтезованої діючої речовини на етапі розробки; дані для лабораторних і дослідно-промислових серій речовин; дані для досліджуваних складів, не призначених для реєстрації; дані для подібних складів; дані для лікарського препарату в контейнерах і/або із закупорювальними елементами, що відрізняються від запропонованих для розміщення на ринку; (2) інформацію про результати випробувань контейнерів та (3) інші наукові обґрунтування.
3.4.6 дата закінчення терміну придатності (expiration date, [7])
Дата, вказана на етикетці контейнера лікарського препарату, що визначає час, до якого серія цього препарату, як очікується, буде відповідати затвердженій специфікації (специфікації, що застосовується протягом терміну зберігання) при зберіганні у визначених умовах; після закінчення цієї дати дана серія препарату не повинна застосовуватися.
3.4.7 дата повторних випробувань (re-test date, [7])
Дата, що вказує, коли слід повторно оцінити зразки діючої речовини, щоб переконатися в тому, що вона все ще відповідає вимогам специфікації і, отже, придатна для використання при виробництві даного лікарського засобу.
3.4.8 довгострокові випробування (long term testing, [7])
Дослідження стабільності при рекомендованих умовах зберігання протягом періоду до проведення повторних випробувань або терміну зберігання, пропонованих (або затверджених) для зазначення на етикетці.
3.4.9 допустимі відхилення в умовах зберігання (storage condition tolerances, [7])
Прийнятні коливання температури та відносної вологості в технічних засобах для зберігання при офіційних дослідженнях стабільності. Обладнання має давати можливість регулювати умови зберігання в межах, зазначених у цій настанові. Під час зберігання при дослідженні стабільності слід контролювати фактичну температуру та відносну вологість. Короткочасні різкі коливання внаслідок відчинення дверей обладнання для зберігання приймаються як неминучі. Заявник має повідомити про вплив відхилень, викликаних ушкодженням обладнання, і описати такий вплив, якщо він вважає, що це може вплинути на результати випробувань стабільності. Відхилення, що перевищують протягом більше 24 год установлені межі (а саме, для температури ±2 °С і/або для відносної вологості ±5%), слід описати в протоколі дослідження; їх вплив необхідно оцінити.
3.4.10 дослідження при примусовому розкладі (forced degradation testing studies, [8])
Дослідження, що проводять для навмисного руйнування зразка. Їх проводять стосовно діючої речовини, як правило, на етапі розробки, а результати використовують для оцінки загальної світлостабільності речовини з метою розробки методу і/або з’ясування шляху розкладу.
3.4.11 дослідно-промислова серія (pilot scale batch, [7])
Серія діючої речовини або лікарського засобу, вироблена за допомогою технологічного процесу, який цілком відображає або моделює той, що здійснюватиметься при випуску серій промислового масштабу. Розмір дослідно-промислової серії твердих лікарських форм для орального застосування складає, як мінімум, одну десяту відносно серії промислового масштабу, або 100 000 таблеток/капсул, залежно від того, яка величина більша. Якщо розмір промислової серії складає 100 000 одиниць або менше, то розмір дослідно-промислової серії відповідає розміру промисловоїN.
3.4.12 кліматичні зони (climatic zones, [7])
Концепція розподілу світу на чотири зони, заснована на переважаючих щорічних кліматичних умовах (див. додатки Б і В, [29]).
3.4.13 лікарський засіб/лікарський препарат (medicinal product/drug product, [7,8])
Будь-яка речовина чи комбінація речовин (у певній лікарській формі)N, що призначені для лікування чи профілактики захворювань у людини, або які можуть бути призначені для встановлення діагнозу чи для відновлення, корекції або зміни її фізіологічних функцій.
Примітка. У цій настанові під таким терміном розуміють лікарський засіб у певній лікарській формі, вміщений в остаточне паковання та призначений для розміщення на ринку.
3.4.14 напівпроникні контейнери (semi-permeable containers, [7])
Контейнери, що дозволяють проникати розчиннику (звичайно воді), але перешкоджають втраті розчиненої речовини. Механізм транспорту розчинника полягає в абсорбції однією поверхнею контейнера, дифузії крізь матеріал контейнера та десорбції розчинника з іншої поверхні. Транспорт розчинника відбувається відповідно до градієнта парціального тиску. Приклади напівпроникних контейнерів: пластикові мішки та м’які мішки з поліетилену низької щільності для парентеральних лікарських засобів великого об’єму, а також ампули, флакони та пляшечки з поліетилену низької щільності.
3.4.15 нова діюча речовина (new molecular entity, [7])
Активний фармацевтичний інгредієнт, що раніше не був включений до складу будь-якого зареєстрованого лікарського препарату. Нова сіль, ефір або похідне (з нековалентним зв’язком) дозволеної для застосування діючої речовини вважається новою діючою речовиною, що вимагає випробування стабільності відповідно до цієї настанови.
3.4.16 офіційні дослідження стабільності (formal stability studies, [7])
Довгострокові, прискорені (і проміжні) дослідження, що проводяться стосовно вихідних серій (або серій, що зазнають випробувань стабільності відповідно до зобов’язання) згідно із запропонованим протоколом досліджень стабільності. Їх проводять із метою встановлення чи підтвердження періоду до проведення повторних випробувань для діючої речовини або терміну зберігання для лікарського засобу.
3.4.17 первинне паковання/паковання, що безпосередньо контактує з продукцією (primary/immediate pack, [8])
Компонент системи паковання, що безпосередньо контактує з діючою речовиною або лікарським засобом, включаючи будь-яке відповідне маркування.
3.4.18 період до проведення повторних випробувань (re-test period, [7])
Період, протягом якого діюча речовина вважається відповідною специфікації та, отже, може бути використана при виробництві даного лікарського засобу, якщо ця діюча речовина зберігалася при встановлених умовах. Після закінчення цього періоду серія діючої речовини, призначена для використання при виробництві лікарського засобу, має бути повторно випробувана на відповідність специфікації і потім негайно використана. Серія діючої речовини може бути випробувана багаторазово, і після кожного повторного випробування можна використовувати певну частину серії; це можна здійснювати доти, доки серія відповідатиме специфікації. Для більшості явно нестабільних біологічних/біотехнологічних речовин доцільніше встановлювати термін зберігання, а не період до проведення повторних випробувань. Це ж може бути прийнятним і для деяких антибіотиків.
3.4.19 підтверджувальні дослідження (confirmatory studies, [8])
Дослідження, що проводяться для визначення характеристик світлостабільності при стандартизованих умовах. Такі дослідження використовують, щоб визначити запобіжні заходи, потрібні для зменшення впливу світла при виробництві діючої речовини або відповідного лікарського засобу, а також необхідність використання світлостійкого паковання і/або спеціального маркування. Для підтверджуючих досліджень серію (серії) слід обирати відповідно до вказівок щодо вибору серій для довгострокових і прискорених випробувань, які описані в підрозділі 5.1.
3.4.20 побудова матриць (matrixing, [7])
Розробка плану досліджень стабільності таким чином, що у визначеній точці контролю відбирають і випробовують лише частину загальної кількості можливих зразків для всіх комбінацій факторів. У наступній точці контролю необхідно випробовувати інший комплект зразків із загальної кількості для всіх комбінацій факторів. План припускає, що стабільність випробуваних зразків ототожнюється зі стабільністю всіх зразків на даний момент часу. Повинні бути зазначені розбіжності в зразках для того самого лікарського засобу, наприклад, розбіжності, пов’язані з різними серіями, різною силою дії, різним розміром системи контейнер/закупорювальний елемент однакового типу і, можливо, у деяких випадках з різними системами контейнер/закупорювальний елемент.
3.4.21 прискорені випробування (accelerated testing, [7])
Дослідження, що є частиною офіційних досліджень стабільності, сплановані таким чином, щоб збільшити швидкість хімічного розкладу чи фізичної зміни діючої речовини або лікарського засобу за допомогою створення особливо несприятливих умов зберігання. Дані таких випробувань як додаток до результатів довгострокових досліджень стабільності можуть використовуватися для оцінки більш віддалених хімічних ефектів за умов неприскорених випробувань, а також для оцінки впливу короткочасних відхилень від умов зберігання, зазначених на етикетці, що можуть виникнути при транспортуванні. Результати, отримані при прискорених випробуваннях, не завжди дозволяють прогнозувати фізичні зміни.
3.4.22 промислова серія (production batch, [7])
Серія діючої речовини або лікарського засобу промислового масштабу, вироблена з використанням виробничого обладнання та у виробничому приміщенні таким чином, як зазначено в реєстраційному досьє.
3.4.23 проміжні випробування (intermediate testing, [7])
Дослідження, що проводяться при температурі 30 °С та відносній вологості 65% і призначені для помірного підвищення швидкості/ступеня хімічного розкладу чи фізичних змін діючої речовини або лікарського засобу, які планується зберігати тривалий час при температурі 25 °С.
3.4.24 середня кінетична температура (mean kinetic temperature, [7])
Розрахована температура, що (за умови підтримання її протягом певного періоду) забезпечує такий самий тепловий вплив на діючу речовину або лікарський засіб, як вплив у діапазоні вищих і нижчих температур протягом еквівалентного заданого періоду. Середня кінетична температура вища середньої арифметичної та визначається з урахуванням рівняння Арреніуса.
При встановленні середньої кінетичної температури для заданого періоду можна використовувати формулу J.D. Haynes (див. додаток В, [31]).
3.4.25 серії, що зазнають випробувань стабільності відповідно до зобов’язання (commitment batches, [7])
Промислові серії діючої речовини або готового лікарського засобу, дослідження стабільності яких розпочато або завершено після реєстрації відповідно до зобов’язання, вказаного в заявці на реєстрацію.
3.4.26 система контейнер/закупорювальний елемент (container closure system, [7])
Сукупність компонентів паковання, що містить лікарський засіб і забезпечує його захист. Система включає компоненти первинного паковання, а також компоненти вторинного паковання, якщо останнє призначено для забезпечення додаткового захисту лікарського препарату.
Примітка. Термін «система контейнер/закупорювальний елемент» еквівалентний поняттю «система паковання».
3.4.27 специфікація, що застосовується при випуску (specification — release, [7])
Набір фізичних, хімічних, біологічних і мікробіологічних випробувань і критеріїв прийнятності, за допомогою яких визначають, що лікарський засіб має необхідну якість на момент його випуску.
3.4.28 специфікація, що застосовується протягом терміну зберігання (specification — shelf life, [7])
Набір фізичних, хімічних, біологічних і мікробіологічних випробувань і критеріїв прийнятності, за допомогою яких визначають, що діюча речовина є придатною протягом періоду до дати проведення повторних випробувань, або яким має відповідати лікарський засіб протягом терміну зберігання.
3.4.29 стресові випробування лікарського засобу (stress testing of drug product, [7])
Випробування, що проводяться для оцінки впливу жорстких умов на лікарський засіб. Такі дослідження включають випробування світлостабільності (див. підрозділ 5.2) і спеціальні випробування для певних лікарських форм (наприклад, дозованих аерозолів для інгаляцій, кремів, емульсій, рідких лікарських препаратів, що містять воду, призначених для зберігання в холодильнику).
3.4.30 стресові випробування лікарської речовини (stress testing of drug substance, [7])
Випробування, що проводяться для з’ясування характеристик стабільності, властивих діючій речовині. Такі випробування є частиною стратегії розробки і, як правило, проводяться за більш несприятливих умов, ніж умови прискорених випробувань.
3.4.31 термін зберігання/термін придатності (shelflife/expiration dating period, [7])
Інтервал часу, протягом якого лікарський препарат, як очікується, відповідає затвердженій специфікації (специфікації, що застосовується протягом терміну зберігання), якщо він зберігається в умовах, зазначених на етикетці контейнера.
3.4.32 торгове паковання (marketingpack, [8])
Комбінація первинного та вторинного (наприклад, пачка з картону) паковання.
3.5 Наведені в даному розділі визначення понять застосовують до термінів, що використовуються в цій настанові. Визначення цих термінів можуть відрізнятися в інших нормативних документах або терміни можуть мати інші значення.
4 Позначення та скорочення
ВООЗ — Всесвітня організація охорони здоров’я
ЄС — Європейський Союз
ПВХ — полівінілхлорид
СРМР — Committee for Proprietary Medicinal Products (Комітет із патентованих лікарських засобів)
EMEA — European Medicines Evaluation Agency (Європейське агентство з оцінки лікарських засобів)
ІСН — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
ISO — International Standardization Organization (Міжнародна організація зі стандартизації)
5 Рекомендації з випробувань стабільності
5.1 Випробування стабільності нових діючих речовин і лікарських засобів
У цьому підрозділі розглядається інформація про стабільність (головним чином, нових діючих речовин, а також лікарських засобів, що містять їх), яку слід подавати у реєстраційних досьє. У цьому підрозділі не регламентується інформація, яка потрібна для скорочених заявок на реєстрацію, заявок на внесення змін, заявок на клінічні випробування тощо.
У цьому підрозділі не наведені докладні відомості про відбір проб і проведення випробувань для окремих лікарських форм із пропонованими для них системами контейнер/закупорювальний елемент.
Більш докладна інформація про нові лікарські форми та біологічні/біотехнологічні лікарські засоби наведена в підрозділі 5.3 і настанові ІСН Q5C* відповідно.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку
Примітка. В реєстраційному досьє у форматі CTD інформацію про стабільність діючої речовини наводять в розділі 3.2.S.7 «Stability» («Стабільність»), який складається з таких пунктів:
3.2.S.7.1 «Stability Summary and Conclusions» («Резюме зі стабільності і висновки»);
3.2.S.7.2 «Post-approval Stability Protocol and Stability Commitment» («Протокол постреєстраційного вивчення стабільності і зобов’язання стосовно стабільності»);
3.2.S.7.3 «Stability Data» («Дані про стабільність»).
Інформацію про стабільність готового лікарського засобу наводять в розділі 3.2.Р.7 «Stability» («Стабільність»), який складається з таких пунктів:
3.2.Р.7.1 «Stability Summary and Conclusions» («Резюме зі стабільності і висновки»);
3.2.Р.7.2 «Post-approval Stability Protocol and Stability Commitment» («Протокол постреєстраційного вивчення стабільності і зобов’язання стосовно стабільності»);
3.2.Р.7.3 «Stability Data» («Дані про стабільність»).
5.1.1 Загальні принципи
Мета проведення випробувань стабільності — це одержання даних про зміну якості діючої речовини або лікарського препарату з часом під впливом різних факторів навколишнього середовища, таких як температура, вологість і світло, а також встановлення рекомендованих умов зберігання та періоду до проведення повторних випробувань для діючої речовини або терміну зберігання для лікарського препарату.
Вибір умов проведення випробувань, описаних у цьому підрозділі, ґрунтується на аналізі впливу кліматичних умов у трьох регіонах світу: Європі, Японії та США. Середня кінетична температура в будь-якому регіоні світу може бути розрахована на підставі кліматичних даних, і світ може бути поділений на чотири кліматичні зони І–IV (див. додаток Б). Положення підрозділу 5.1 застосовні при випробуваннях стабільності для реєстрації лікарських засобів у державах, розташованих у кліматичних зонах І і II (у тому числі в Україні»)*.
*В рамках ІСН інформація зі стабільності, отримана в будь-якому з трьох регіонів — ЄС, Японії та США — має бути взаємно визнана в двох інших регіонах, за умови, що вона відповідає вимогам відповідних настанов, амаркування продукції відповідає національним/регіональним вимогам.
5.1.2 Керівні вказівки
5.1.2.1 Діюча речовина (активний фармацевтичний інгредієнт)
5.1.2.1.1 Загальна інформація
Інформація про стабільність діючої речовини є невід’ємною частиною систематизованого підходу до оцінки стабільності.
5.1.2.1.2 Стресові випробування
Стресові випробування можуть допомогти ідентифікувати ймовірні продукти розкладу, що, в свою чергу, дозволяє визначити шляхи розкладу та стабільність, властиву молекулі, а також підтвердити придатність аналітичних методик, що використовуються для вивчення стабільності. Характер стресових випробувань залежатиме від конкретної діючої речовини та виду лікарської форми.
Стресові випробування, наскільки можливо, слід проводити на одній серії діючої речовини. Мають бути вивчені вплив на діючу речовину температур, що перевищують температури при прискорених випробуваннях (з підвищеннями по 10 °С, наприклад, 50 °С, 60 °С тощо), та вологості (наприклад, відносної вологості 75% або вище), а також, за необхідності, окислення та фотоліз. У ході випробувань слід також оцінити здатність діючої речовини до гідролізу в широкому інтервалі рН, якщо речовина перебуває у вигляді розчину або суспензії. Невід’ємною частиною стресових випробувань мають бути випробування світлостабільності. Стандартні умови випробувань світлостабільності описано в підрозділі 5.2.
Вивчення продуктів розкладу в стресових умовах корисне при визначенні шляхів розкладу, а також при розробці та валідації відповідних аналітичних методик. Однак окреме вивчення певних продуктів розкладу може не знадобитися, якщо показано, що ці продукти не утворюються в умовах прискорених або довгострокових випробувань.
Результати таких досліджень мають складати невід’ємну частину інформації, що надається компетентному уповноваженому органу.
5.1.2.1.3 Вибір серій
Необхідно надати інформацію про дослідження стабільності як мінімум для трьох вихідних серій діючої речовини. Серії мають бути щонайменше дослідно-промисловими; вони повинні бути виготовленими з використанням того шляху синтезу, а також способу виробництва й методик, що моделюють остаточний процес, який планується для промислового виробництва. Якість діючої речовини в цілому для серій, що зазнають випробувань на стабільність, має бути репрезентативною щодо якості речовини, яка вироблятиметься в промисловому масштабі.
Може бути наведена й інша додаткова інформація.
5.1.2.1.4 Система контейнер/закупорювальний елемент
Дослідження стабільності слід проводити стосовно діючої речовини, що упакована з використанням такої системи контейнер/закупорювальний елемент, яка ідентична чи моделює паковання, пропоноване для зберігання та дистрибуції.
5.1.2.1.5 Специфікація
Інформація про специфікацію, що являє собою перелік випробувань, посилань на аналітичні методики, а також пропонованих критеріїв прийнятності, наведена в настановах 42-3.2:2004 та ІСН Q6B*. Крім того, у настанові ІСН Q3A* обговорюється специфікація щодо продуктів розкладу діючої речовини.
Дослідження стабільності мають включати випробування таких характеристик діючої речовини, що чутливі до змін у процесі зберігання та, як передбачається, можуть впливати на якість, безпеку і/або ефективність. Залежно від конкретної ситуації випробування мають поширюватися на фізичні, хімічні та біологічні властивості. Необхідно застосовувати валідовані аналітичні методики, що дозволяють охарактеризувати стабільність. Необхідність проведення повторних випробувань і їхній обсяг залежатимуть від результатів валідаційних досліджень.
*Рекомендується користуватися зазначеними документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.1.2.1.6 Частота випробувань
При довгострокових дослідженнях частота проведення випробувань має бути достатньою для визначення характеристик стабільності діючої речовини. Якщо пропонований період до проведення повторних випробувань діючої речовини становить 12 місяців і більше, випробування в умовах довгострокових досліджень, як правило, слід проводити кожні 3 місяці протягом першого року, кожні 6 місяців протягом другого року і потім щорічно протягом усього пропонованого періоду до проведення повторних випробувань.
При прискорених дослідженнях тривалістю 6 місяців рекомендується застосовувати не менше трьох точок контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 3 та 6 місяців). Якщо передбачається (на підставі досвіду розробки), що результати прискорених випробувань можуть впритул наблизитися до критеріїв «значної зміни», необхідно провести розширені випробування, збільшивши кількість зразків у кінцевій точці контролю або включивши четверту точку контролю в план проведення досліджень.
Якщо через «значну зміну» при прискорених дослідженнях необхідні дослідження при проміжних умовах зберігання, рекомендується проводити випробування як мінімум у чотирьох точках контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 3, 6 і 12 місяців), якщо тривалість дослідження становить 12 місяців.
5.1.2.1.7 Умови зберігання
Як правило, діючу речовину слід оцінювати при умовах зберігання (з відповідними припустимими відхиленнями), що дозволяють перевірити її термічну стабільність і, за необхідності, її чутливість до впливу вологи. Тривалість досліджень і умови зберігання слід вибирати таким чином, щоб вони відповідали періоду та умовам при зберіганні, постачанні та наступному застосуванні.
На момент подання заявки на реєстрацію довгострокові випробування мають бути проведені протягом не менше 12 місяців, принаймні, стосовно трьох вихідних серій; вони мають тривати й надалі протягом часу, достатнього для того, щоб охопити пропонований період до проведення повторних випробувань. Додаткові дані, зібрані протягом часу проведення експертизи реєстраційного досьє, мають бути подані до компетентного уповноваженого органу за його запитом. Дані, отримані при проведенні прискорених випробувань і, якщо необхідно, випробувань при проміжних умовах зберігання, можуть використовуватися для оцінки впливу короткочасних відхилень від умов зберігання, зазначених на етикетці; такі відхилення можуть виникнути, наприклад, при транспортуванні.
Умови зберігання діючих речовин при довгострокових, прискорених і, за необхідності, проміжних випробуваннях наведені в таблицях 5.1.1–5.1.3. Умови, описані в п. 5.1.2.1.7.1 «Загальний випадок», використовують, якщо до діючої речовини не застосовуються умови, описані нижче в пп. 5.1.2.1.7.2, 5.1.2.1.7.3, 5.1.2.1.7.4. За наявності відповідного обґрунтування можуть бути застосовані інші умови зберігання.
5.1.2.1.7.1 Загальний випадок
Таблиця 5.1.1
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(25±2) °С та (60±5)%
|
12
|
|
Проміжні
|
(30±2) °С та (60±5)%
|
6
|
|
Прискорені
|
(40±2) °С та (75±5)%
|
6
|
Якщо в умовах прискорених випробувань у будь-який момент часу протягом 6 міс досліджень спостерігається «значна зміна», то необхідно здійснити додаткові випробування при проміжних умовах зберігання, а також провести їх порівняльну оцінку щодо критеріїв «значної зміни». Дослідження при проміжних умовах зберігання має включати всі випробування, якщо не обґрунтовано інше. Первинна заявка на реєстрацію має містити дані досліджень при проміжних умовах зберігання, отримані як мінімум протягом 6 місяців (при загальній тривалості досліджень 12 місяців).
«Значна зміна» для діючої речовини означає зміну, при якій вона перестає відповідати специфікації.
5.1.2.1.7.2 Діюча речовина, призначена для зберігання в холодильнику
Таблиця 5.1.2
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(5±3) °С
|
12
|
|
Прискорені
|
(25±2) °С та (60±5)%
|
6
|
Дані, отримані для діючої речовини при її зберіганні в холодильнику, слід оцінювати відповідно до вказівок, наведених у п. 5.1.2.1.9 «Оцінка даних», за винятком випадків, докладно описаних нижче.
Якщо в умовах прискорених випробувань у проміжок часу з третього по шостий місяць зберігання спостерігається «значна зміна», пропонований період до проведення повторних випробувань має бути встановлений на підставі даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань.
Якщо «значна зміна» спостерігається протягом перших 3 місяців зберігання в умовах прискорених випробувань, слід подати дані про вплив короткочасних відхилень від умов зберігання, зазначених на етикетці, наприклад, при транспортуванні чи обробці. Якщо доцільно, ці матеріали можуть супроводжуватися даними наступних випробувань для одиничної серії діючої речовини протягом періоду, меншого ніж 3 місяці, але при більшій, ніж звичайно, частоті проведення випробувань.
Вважається, що немає необхідності продовжувати прискорені випробування діючої речовини протягом 6 місяців, якщо протягом перших 3 місяців зберігання спостерігається «значна зміна».
5.1.2.1.7.3 Діюча речовина, призначена для зберігання в морозильній камері
Таблиця 5.1.3
|
Дослідження (випробування)
|
Умови зберігання (температура)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
–(20±5) °С
|
12
|
Для діючих речовин, призначених для зберігання в морозильній камері, період до проведення повторних випробувань має ґрунтуватися на даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань. Оскільки умови для прискорених випробувань діючих речовин, призначених для зберігання в морозильній камері, відсутні, слід провести випробування на одиничній серії при підвищеній температурі (наприклад, (5±3) °С або (25±2) °С) протягом підхожого відрізка часу, щоб вивчити вплив короткочасних відхилень від умов зберігання, зазначених на етикетці, наприклад, при транспортуванні чи обробці.
5.1.2.1.7.4 Діюча речовина, призначена для зберігання при температурі нижче –20 °С
Випробування діючих речовин, призначених для зберігання при температурі нижче –20 °С, слід проводити, обґрунтовуючи в кожному окремому випадку.
5.1.2.1.8 Зобов’язання продовжувати випробування стабільності
Якщо наявні на момент подання заявки на реєстрацію дані довгострокових випробувань стабільності для вихідних серій не охоплюють пропонованого періоду до проведення повторних випробувань, слід взяти зобов’язання продовжувати дослідження стабільності, щоб остаточно встановити тривалість періоду до проведення повторних випробувань.
Якщо реєстраційне досьє містить дані довгострокових випробувань стабільності для трьох промислових серій, що охоплюють період до проведення повторних випробувань, зобов’язання продовжувати випробування стабільності не потрібне. У протилежному випадку слід взяти одне з наступних зобов’язань:
1) якщо реєстраційне досьє містить дані довгострокових досліджень стабільності щонайменше для трьох промислових серій, необхідно взяти зобов’язання про продовження цих досліджень протягом пропонованого періоду до проведення повторних випробувань;
2) якщо реєстраційне досьє містить дані досліджень стабільності менше ніж для трьох промислових серій, слід взяти зобов’язання продовжувати ці дослідження протягом пропонованого періоду до проведення повторних випробувань і ввести в довгострокові дослідження стабільності додаткові промислові серії (щоб загальна кількість серій становила не менше трьох); ці серії необхідно досліджувати протягом пропонованого періоду до проведення повторних випробувань;
3) якщо реєстраційне досьє не містить даних про стабільність промислових серій, слід взяти зобов’язання провести довгострокові дослідження стабільності перших трьох промислових серій протягом пропонованого періоду до проведення повторних випробувань.
Протокол довгострокового дослідження стабільності, що додається при взятті зобов’язання про подальше випробування стабільності, має бути таким самим, як і для вихідних серій, якщо науково не обґрунтований інший підхід.
5.1.2.1.9 Оцінка даних
Мета досліджень стабільності полягає в тому, щоб установити період до проведення повторних випробувань, застосовний для всіх серій діючої речовини, що будуть вироблені при однакових умовах. Цей період установлюють на підставі випробувань як мінімум трьох серій діючої речовини та оцінки інформації про стабільність (що охоплює, залежно від необхідності, результати фізичних, хімічних, біологічних і мікробіологічних випробувань). Ступінь варіабельності окремих серій забезпечує впевненість у тому, що вироблені надалі промислові серії відповідатимуть специфікації протягом заданого періоду до проведення повторних випробувань.
Дані можуть свідчити про такий незначний розклад і таку невелику варіабельність, що вже при їх розгляді очевидно, що необхідний період до проведення повторних випробувань буде затверджений. У таких випадках, як правило, немає необхідності в проведенні формального статистичного аналізу; достатньо надати повне обґрунтування відсутності такого аналізу.
Якщо очікується зміна кількісної характеристики в часі, то прийнятний підхід, що полягає у визначенні часу, після закінчення якого усереднена крива розкладу (при довірчій імовірності 95%) перетинається з допустимою нижньою межею, встановленою у специфікації. Якщо аналіз показує, що варіабельність від серії до серії невелика, корисно об’єднати дані для однієї загальної оцінки; це може бути зроблено шляхом відповідної статистичної обробки нахилів ліній регресій і точок їх перетинання з віссю ординат для окремих серій (наприклад, значення р для рівня значущості відбракування, що перевищує 0,25). Якщо недоцільно поєднувати дані для кількох серій, то загальний період до проведення повторних випробувань залежатиме від мінімального часу, протягом якого, як передбачається, характеристики серії залишатимуться в рамках критеріїв прийнятності.
Характер будь-якого взаємозв’язку з розкладом речовин визначатиме необхідність перетворення даних для аналізу лінійної регресії. Як правило, взаємозв’язок може бути поданий у вигляді лінійної, квадратичної або кубічної функції на арифметичній або логарифмічній шкалі. Щоб перевірити придатність даних, отриманих щодо кожної серії та об’єднаних даних для серій (якщо доцільно) для побудови прямої чи кривої розкладу слід застосовувати статистичні методи.
За наявності відповідного обґрунтування може бути проведена обмежена екстраполяція отриманих у реальному часі даних за межі терміну спостереження, щоб збільшити період до проведення повторних випробувань на момент його затвердження. Таке обґрунтування має враховувати відомі дані про механізм розкладу, результати прискорених випробувань, придатність математичної моделі, розмір серії, наявність підтверджувальних даних зі стабільності тощо. Однак при такій екстраполяції передбачається, що й далі (за рамками даних, що спостерігаються) існуватиме такий же взаємозв’язок із розкладом.
При будь-якій оцінці слід враховувати не лише кількісне визначення діючої речовини, але й рівні вмісту продуктів розкладу, а також інші відповідні характеристики.
5.1.2.1.10 Вказівки/маркування
Вказівки в маркуванні щодо зберігання необхідно формулювати згідно з відповідними національними/регіональними вимогами. Ці вказівки повинні ґрунтуватися на оцінці стабільності діючої речовини. За необхідності мають бути зазначені спеціальні вимоги, зокрема, для діючих речовин, замороження яких не допускається. Неприйнятне використання таких термінів, як «умови навколишнього середовища» або «кімнатна температура».
Період до проведення повторних випробувань має бути визначений на підставі інформації про стабільність, а дата проведення повторних випробувань, за необхідності, має бути зазначена на етикетці контейнера.
5.1.2.2 Лікарський засіб
5.1.2.2.1 Загальна інформація
Розробка офіційної програми дослідження стабільності для лікарського засобу має ґрунтуватися на знанні поведінки та властивостей діючої речовини, а також на досвіді, набутому при клінічних дослідженнях лікарського препарату. У програмі випробувань мають бути зазначені можливі зміни при зберіганні лікарського засобу та надане логічне обґрунтування вибору характеристик, що підлягають випробуванням при офіційних дослідженнях стабільності.
5.1.2.2.2 Випробування світлостабільності
Випробування світлостабільності (якщо вони необхідні) мають бути проведені як мінімум для однієї вихідної серії лікарського засобу. Стандартні умови випробування світлостабільності наведені в підрозділі 5.2.
5.1.2.2.3 Вибір серій
Необхідно надати інформацію про дослідження стабільності як мінімум для трьох вихідних серій із таким же складом і в тій же лікарській формі, як і лікарський засіб, що планується для розміщення на ринку, а також у таких контейнерах із закупорювальними елементами, у яких лікарський засіб надійде на ринок. Технологічний процес, що застосовується при виготовленні цих серій, має моделювати процес, що планується для виробництва промислових серій; цей процес повинен забезпечувати одержання лікарського засобу такої ж якості (відповідаючого такій же специфікації), як і лікарський засіб, призначений для надходження на ринок. Дві з цих трьох серій мають бути як мінімум дослідно-промисловими; третя серія, за наявності відповідного обґрунтування, може бути меншою. Якщо можливо, серії лікарського засобу мають бути вироблені з використанням різних серій діючої речовини.
Дослідження стабільності мають бути проведені окремо для лікарських засобів із різною силою дії та з різним розміром паковання, за винятком тих випадків, коли застосовують брекетинг або побудову матриць.
Може бути надана й інша підтверджувальна інформація.
5.1.2.2.4 Система контейнер/закупорювальний елемент
Випробування стабільності необхідно проводити щодо лікарської форми в пакованні, призначеному для розміщення на ринку (включаючи, за необхідності, будь-яке вторинне паковання та етикетки для контейнерів). Будь-які випробування щодо лікарського засобу без первинного паковання або в пакованні з інших пакувальних матеріалів можуть складати корисну частину стресових випробувань лікарської форми або, відповідно, розглядатися як додаткова підтверджувальна інформація.
5.1.2.2.5 Специфікація
Інформація про специфікації, що є переліком випробувань, посилань на аналітичні методики та пропонованих критеріїв прийнятності (включаючи принцип різних критеріїв прийнятності на момент випуску та протягом терміну зберігання) надана в настановах 42-3.2-2004 та Q6B*. Крім того, у настанові Q3B* розглядається специфікація щодо продуктів розкладу лікарського засобу.
Дослідження стабільності мають включати випробування таких характеристик лікарського засобу, що зазнають змін при зберіганні та, можливо, можуть впливати на якість, безпеку і/або ефективність. Необхідно випробовувати (залежно від конкретної ситуації) фізичні, хімічні, біологічні та мікробіологічні властивості, визначати вміст консервантів (наприклад, антиоксидантів, антимікробних консервантів), а також перевіряти функціональні характеристики (наприклад, для системи доставки дози).
Аналітичні методики мають бути цілком валідовані; вони повинні дозволяти характеризувати стабільність. Необхідність проведення повторних випробувань та їх обсяг залежатимуть від результатів валідаційних досліджень.
Критерії прийнятності в специфікації, що застосовується протягом терміну зберігання, слід встановлювати на підставі всієї наявної інформації про стабільність. Допускаються прийнятні та обґрунтовані відхилення критеріїв прийнятності в специфікації, що застосовується протягом терміну зберігання, та у специфікації, що застосовується при випуску, які ґрунтуються на оцінці стабільності та змін, що спостерігаються при зберіганні. Будь-які розбіжності між критеріями прийнятності, що застосовуються при випуску і протягом терміну зберігання, щодо вмісту антимікробних консервантів мають супроводжуватися обґрунтованою кореляцією хімічного складу та ефективності консервантів, доведеною на етапі фармацевтичної розробки для остаточного складу лікарського засобу (за винятком концентрації консерванту), призначеного для розміщення на ринку. З метою перевірки необхідно провести випробування стабільності однієї вихідної серії лікарського засобу щодо ефективності антимікробних консервантів (на додаток до кількісного визначення консервантів) протягом пропонованого терміну зберігання; це дослідження необхідно проводити незалежно від того, чи існують розбіжності між критеріями прийнятності щодо вмісту консерванту, що застосовуються при випуску і протягом терміну зберігання.
*Рекомендується користуватися зазначеними документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.1.2.2.6 Частота проведення випробувань
При довгострокових дослідженнях частота проведення випробувань має бути достатньою для встановлення параметрів стабільності лікарського засобу. Якщо пропонований термін зберігання лікарського засобу становить 12 місяців і більше, випробування при довгострокових дослідженнях, як правило, слід проводити кожні 3 місяці протягом першого року, кожні 6 місяців протягом другого року і потім щорічно протягом пропонованого терміну зберігання.
В умовах прискореного дослідження при його тривалості 6 місяців рекомендується проводити випробування не менше ніж у трьох точках контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 3 і 6 місяців). Якщо передбачається (на підставі досвіду розробки), що результати прискорених випробувань можуть впритул наблизитися до критеріїв «значної зміни», необхідно провести розширені дослідження шляхом збільшення кількості зразків в останній точці контролю або включення четвертої точки контролю в план досліджень.
Якщо в результаті прискореного дослідження відбулася «значна зміна», внаслідок чого знадобилося проведення досліджень при проміжних умовах зберігання, то рекомендується проводити випробування як мінімум у чотирьох точках контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 6, 9, 12 місяців) при тривалості дослідження 12 місяців.
При відповідному обґрунтуванні можуть бути застосовані скорочені плани досліджень (тобто, побудова матриць або брекетинг), коли частота проведення випробувань зменшена чи не проводяться випробування комбінацій певних критеріїв у повному обсязі.
5.1.2.2.7 Умови зберігання
Як правило, лікарський засіб слід оцінювати в умовах зберігання (з відповідними допустимими відхиленнями), що дозволяють перевірити його термічну стабільність і, за необхідності, чутливість до впливу вологи чи можливість втрати розчинника. Обрані умови досліджень мають відповідати умовам зберігання, постачання та наступного застосування; тривалість досліджень має бути достатньою, щоб охопити всі ці періоди.
За необхідності, як частина офіційних досліджень стабільності, мають бути проведені випробування стабільності лікарського засобу після його підготовки до застосування чи розведення. Вони необхідні, щоб одержати інформацію для маркування лікарського препарату щодо підготовки до застосування, умов зберігання та періоду використання після підготовки чи розведення. Такі випробування підготовленого до застосування чи розведеного лікарського препарату мають бути проведені на вихідних серіях у початковій і кінцевій точках пропонованого періоду його застосування. Якщо на момент подання заявки немає даних довгострокових випробувань, що охоплюють весь період терміну зберігання, мають бути подані дані на момент закінчення терміну 12 місяців або на останній момент часу, для якого є дані зі стабільності. Як правило, ці випробування немає необхідності повторювати на серіях, що досліджуються відповідно до зобов’язання про продовження випробувань стабільності.
На момент подання заявки на реєстрацію тривалість довгострокових випробувань як мінімум для трьох вихідних серій має складати не менше 12 місяців; випробування слід продовжувати протягом часу, достатнього для того, щоб охопити пропонований термін зберігання. Додаткові дані, зібрані протягом часу проведення експертизи реєстраційного досьє, мають бути надані уповноваженому органу за його запитом. Дані, отримані при проведенні прискорених випробувань і, якщо необхідно, при проміжних умовах зберігання, можуть використовуватися для оцінки впливу короткочасних відхилень від умов зберігання, зазначених на етикетці (такі відхилення можуть виникнути, наприклад, при транспортуванні).
Довгострокові, прискорені та, якщо необхідно, проміжні умови зберігання для лікарських засобів наведені у таблицях 5.1.4, 5.1.5, 5.1.7 та 5.1.8. Умови, описані в п. 5.1.2.2.7.1 «Загальний випадок», використовують, якщо до лікарського засобу не застосовуються умови, описані нижче в пп. 5.1.2.2.7.2, 5.1.2.2.7.3, 5.1.2.2.7.4, 5.1.2.2.7.5. За наявності відповідного обґрунтування можуть бути застосовані інші умови зберігання.
5.1.2.2.7.1 Загальний випадок
Таблиця 5.1.4
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(25±2) °С та (60±51%
|
12
|
|
Проміжні
|
(30±2) °С та (60±5)%
|
6
|
|
Прискорені
|
(40±2) °С та (75±5)%
|
6
|
Якщо в умовах прискорених випробувань протягом 6 місяців зберігання спостерігається «значна зміна», то необхідно провести додаткове випробування при проміжних умовах зберігання та оцінити його результати порівняно з критеріями «значної зміни». В реєстраційне досьє для первинної заявки на реєстрацію необхідно включати дані досліджень, отримані як мінімум протягом 6 місяців при проміжних умовах зберігання (при загальній тривалості таких досліджень 12 місяців).
Як правило, «значна зміна» для лікарського засобу означає:
1) зміну кількісного вмісту на 5% порівняно з початковим; або невідповідність критеріям прийнятності за силою дії при використанні біологічних або імунологічних методик визначення;
2) збільшення вмісту будь-якого продукту розкладу понад критерій прийнятності;
3) невідповідність критеріям прийнятності щодо зовнішнього вигляду, фізичних властивостей і функціональних характеристик (наприклад, колір, поділ фаз, здатність до ресуспендування, спікливість, твердість, доставка дози при одному натискуванні клапана); однак в умовах прискорених випробувань можна припускати, що відбудуться деякі зміни фізичних властивостей (наприклад, розм’якшення супозиторіїв, розплавлення кремів);
крім того, залежно від лікарської форми:
4) невідповідність критеріям прийнятності щодо рН;
5) невідповідність критеріям прийнятності щодо розчинення для 12 одиниць дозованого лікарського засобу.
5.1.2.2.7.2 Лікарські засоби, упаковані в герметичні контейнери
Чутливість до впливу вологи чи можливість втрати розчинника не є проблемою для лікарських засобів, упакованих у герметичні контейнери, які забезпечують постійний бар’єр, що перешкоджає проникненню вологи чи втраті розчинника. Таким чином, дослідження стабільності для лікарських засобів, що зберігають в герметичних контейнерах, можуть бути проведені в будь-яких контрольованих умовах або в умовах вологості навколишнього середовища.
5.1.2.2.7.3 Лікарські засоби, упаковані в напівпроникні контейнери
Для лікарських засобів, що містять воду, упакованих у напівпроникні контейнери, на додаток до досліджень фізичної, хімічної, біологічної та мікробіологічної стабільності необхідно проводити оцінку можливої втрати води. Таку оцінку слід проводити в описаних нижче умовах із низькою відносною вологістю. Зрештою, необхідно довести, що лікарські засоби, які зберігаються в напівпроникних контейнерах, можуть витримувати умови низької відносної вологості. Можуть бути розроблені й інші порівнянні підходи для неводних лікарських засобів, що містять розчинники.
Таблиця 5.1.5
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(25±2) °С та (40±5)%
|
12
|
|
Проміжні
|
(30±2) °С та (60±5)%
|
6
|
|
Прискорені
|
(40±2) °С та не більше 25%
|
6
|
Якщо в умовах прискорених випробувань протягом 6 місяців зберігання спостерігається «значна зміна», не пов’язана з втратою води, слід провести додаткові випробування в проміжних умовах зберігання (описаних у п. 5.1.2.2.7.1 «Загальний випадок») з метою оцінки впливу температури 30 °С. Якщо в умовах прискорених випробувань «значна зміна» пов’язана винятково з втратою води, немає необхідності проводити випробування при проміжних умовах зберігання. Однак необхідно подати дані, які доводять, що в лікарському засобі не відбувається значної втрати води протягом пропонованого терміну зберігання при температурі 25 °С та відносній вологості 40%.
Для упакованого в напівпроникні контейнери лікарського засобу після зберігання протягом 3 місяців при температурі 40 °С та відносній вологості не більше 25% «значною зміною» вважається втрата води, що становить 5% від вихідного вмісту. Однак при відповідному обґрунтуванні для невеликих контейнерів (місткістю 1 мл або менше) або для однодозових лікарських засобів може бути прийнятною втрата води, що становить 5% або більше після зберігання протягом 3 місяців при температурі 40 °С та відносній вологості не більше 25%.
Альтернативою дослідженню при стандартній відносній вологості (в умовах, рекомендованих у таблиці 5.1.5) є проведення досліджень стабільності при більш високій відносній вологості та визначення за допомогою розрахунку втрати води при стандартній вологості. З цією метою експериментальним шляхом визначають коефіцієнт проникності для системи контейнер/закупорювальний елемент або, як показано в наведеному нижче прикладі, використовують розрахунковий коефіцієнт ступеня втрати води під час випробування при двох різних значеннях відносної вологості, але при одній і тій же температурі. Коефіцієнт проникності для системи контейнер/закупорювальний елемент може бути визначений експериментально з використанням умов «найгіршого випадку» для досліджуваного лікарського засобу (наприклад, у ряді лікарських засобів із різною концентрацією тієї самої діючої речовини для випробувань обирають лікарський препарат із найменшою концентрацією).
Приклад визначення втрати води
Для лікарського засобу з даною системою контейнер/закупорювальний елемент, із контейнером даного розміру та при даному об’ємі наповнення доцільне визначення втрати води при стандартній відносній вологості шляхом помноження ступеня втрати води (виміряного при альтернативній відносній вологості, але при такій же температурі) на коефіцієнт ступеня втрати води, що зазначений у таблиці 5.1.6. При цьому необхідно довести лінійність ступеня втрати води при альтернативній відносній вологості протягом періоду зберігання.
Наприклад, при даній температурі 40 °С розрахунковим ступенем втрати води в процесі зберігання при відносній вологості не більше 25% є ступінь втрати води, виміряний при відносній вологості 75%, помножений на 3,0 — відповідний коефіцієнт ступеня втрати води.
Можна застосовувати також обґрунтовані коефіцієнти ступеня втрати води в умовах вологості, що відрізняються від умов, наведених у таблиці 5.1.6.
Таблиця 5.1.6
|
Альтернативна відносна вологість
|
Стандартна відносна вологість
|
Коефіцієнт ступеня втрати води
|
|
60%
|
25%
|
1,9
|
|
60%
|
40%
|
1,5
|
|
75%
|
25%
|
3,0
|
5.1.2.2.7.4 Лікарські засоби, призначені для зберігання в холодильнику
Таблиця 5.1.7
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(5±3) °С
|
12
|
|
Прискорені
|
(25±2) °С та(60±5)%
|
6
|
Якщо лікарський засіб упакований у напівпроникний контейнер, необхідно надати відповідну інформацію з метою оцінки втрати води.
Дані, отримані для лікарського засобу при його зберіганні в холодильнику, слід оцінювати відповідно до вказівок, наведених у п. 5.1.2.2.9 «Оцінка даних», за винятком особливих випадків, описаних нижче.
Якщо в умовах прискорених випробувань у період з третього по шостий місяць зберігання спостерігається «значна зміна», пропонований термін зберігання має ґрунтуватися на даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань.
Якщо «значна зміна» спостерігається протягом перших 3 місяців при зберіганні в умовах прискорених випробувань, слід подати матеріали вивчення впливу короткочасних відхилень від умов зберігання, зазначених на етикетці, наприклад, при транспортуванні та обігу. Такі матеріали, якщо доцільно, можуть супроводжуватися даними наступних випробувань одиничної серії лікарського засобу протягом періоду, що менше 3 місяців, але при більшій, ніж звичайно, частоті проведення випробувань. Якщо «значна зміна» спостерігається протягом перших 3 місяців зберігання, немає необхідності продовжувати прискорені випробування лікарського засобу протягом 6 місяців.
5.1.2.2.7.5 Лікарські засоби, призначені для зберігання в морозильній камері
Таблиця 5.1.8
|
Дослідження (випробування)
|
Умови зберігання (температура)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
–(20±5)°С
|
12
|
Для лікарських засобів, призначених для зберігання в морозильній камері, термін зберігання має ґрунтуватися на даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань. Оскільки відсутні умови для прискорених випробувань лікарських засобів, призначених для зберігання в морозильній камері, слід провести випробування одиничної серії при підвищеній температурі (наприклад, (5±3) °С або (25±2) °С) протягом підхожого періоду для того, щоб вивчити вплив короткочасних відхилень від пропонованих для розміщення на етикетці умов зберігання.
5.1.2.2.7.6 Лікарські засоби, призначені для зберігання при температурі нижче —20 °С
Випробування лікарських засобів, призначених для зберігання при температурі нижче –20 °С, слід проводити, обґрунтовуючи в кожному окремому випадку.
5.1.2.2.8 Зобов’язання продовжувати випробування стабільності
Якщо наявні на момент подання заявки на реєстрацію дані довгострокових випробувань стабільності для вихідних серій не охоплюють пропонованого терміну зберігання, слід взяти зобов’язання продовжувати дослідження стабільності, щоб точно встановити термін зберігання.
Якщо реєстраційне досьє містить дані довгострокових випробувань стабільності стосовно трьох промислових серій за період, що дорівнює пропонованому терміну зберігання, то немає необхідності брати зобов’язання проводити подальші випробування стабільності. У протилежному випадку необхідно взяти одне з наступних зобов’язань:
- якщо реєстраційне досьє містить результати досліджень стабільності як мінімум для трьох промислових серій, необхідно взяти зобов’язання про продовження довгострокових досліджень протягом пропонованого терміну зберігання, а також про проведення прискорених досліджень протягом 6 місяців;
- якщо реєстраційне досьє містить дані дослідження стабільності менше ніж для трьох виробничих серій, слід взяти зобов’язання продовжувати довгострокові дослідження протягом пропонованого терміну зберігання та прискорені випробування протягом 6 місяців, а також провести довгострокові дослідження протягом пропонованого терміну зберігання та прискорені випробування протягом 6 місяців додаткової кількості промислових серій (щоб загальна кількість серій була не менше трьох);
- якщо реєстраційне досьє не містить даних про стабільність промислових серій, слід взяти зобов’язання провести довгострокові дослідження стабільності перших трьох промислових серій протягом пропонованого терміну зберігання; а також прискорені дослідження протягом 6 місяців.
Протокол, що використовується для довгострокових досліджень стабільності відповідно до даного зобов’язання, має бути таким же, як і для вихідних серій, якщо науково не обґрунтований інший підхід.
Якщо для вихідних серій унаслідок «значної зміни» при прискорених дослідженнях стабільності знадобилося проведення випробувань при проміжних умовах зберігання, то випробування стабільності серій, досліджуваних відповідно до прийнятого зобов’язання, можуть бути проведені або в проміжних, або в прискорених умовах зберігання. Однак якщо при прискорених дослідженнях стабільності серій, що зазнають випробувань відповідно до взятого зобов’язання, спостерігається «значна зміна», слід також провести дослідження при проміжних умовах зберігання.
5.1.2.2.9 Оцінка даних
Необхідно систематизовано підходити до подання й оцінки інформації про стабільність; ця інформація має включати (залежно від ситуації) результати фізичних, хімічних, біологічних і мікробіологічних випробувань, у тому числі специфічних характеристик лікарської форми (наприклад, швидкості розчинення лікарських форм для орального застосування).
Мета досліджень стабільності полягає в тому, щоб на підставі випробувань як мінімум трьох серій лікарського засобу встановити термін зберігання та сформулювати розміщувані на етикетці вказівки щодо зберігання, застосовні для всіх серій, які надалі будуть вироблені й упаковані при однакових умовах. Ступінь варіабельності окремих серій забезпечує впевненість у тому, що вироблені надалі промислові серії відповідатимуть специфікації протягом усього терміну зберігання.
Якщо дані таких випробувань свідчать про настільки незначний розклад та настільки низьку варіабельність, що вже при їх розгляді очевидно, що потрібний термін зберігання буде затверджений, то, як правило, немає необхідності в проведенні формального статистичного аналізу; достатньо подати повне обґрунтування відсутності такого аналізу.
Якщо очікується зниження кількісної характеристики в часі, то до аналізу даних прийнятний підхід, що полягає у визначенні часу, після закінчення якого усереднена крива розкладу (при довірчій імовірності 95%) перетинається з критерієм прийнятності, встановленим у специфікації. Якщо при аналізі встановлено, що варіабельність від серії до серії невелика, корисно поєднувати дані для однієї загальної оцінки; це може бути зроблено шляхом відповідної статистичної обробки нахилів ліній регресій і точок їх перетинання з віссю ординат для окремих серій (наприклад, значення р для рівня значущості відбракування, що перевищує 0,25). Якщо недоцільно поєднувати дані для декількох серій, то загальний термін зберігання залежатиме від мінімального часу, протягом якого передбачається, що характеристики серії залишатимуться в рамках критеріїв прийнятності.
Характер взаємозв’язку з розкладом визначатиме необхідність у перетворенні даних для аналізу лінійної регресії. Як правило, взаємозв’язок може бути представлений у вигляді лінійної, квадратичної або кубічної функції на арифметичній або логарифмічній шкалі. Щоб перевірити придатність даних, отриманих щодо кожної серії, та об’єднаних даних для серій (залежно від того, що доцільно) для побудови прямої чи кривої розкладу слід застосовувати статистичні методи.
За наявності відповідного обґрунтування може бути проведена обмежена екстраполяція отриманих у реальному часі даних за межі терміну спостереження для збільшення терміну зберігання на момент затвердження. Таке обґрунтування має враховувати відомі дані про механізм розкладу, результати прискорених випробувань, придатність математичної моделі, розмір серії, наявність підтверджувальних даних зі стабільності тощо. Однак при такій екстраполяції передбачається, що й далі (за рамками даних, які спостерігаються) буде мати місце той самий взаємозв’язок із розкладом.
При будь-якій оцінці слід враховувати не лише кількісне визначення, але й рівні вмісту продуктів розкладу, а також інші підхожі характеристики. За необхідності слід приділити увагу перевірці балансу мас, а також різних характеристик стабільності та розкладу.
5.1.2.2.10 Вказівки/маркування
Необхідно сформулювати вказівки для маркування щодо умов зберігання згідно з національними/регіональними вимогами. Ці вказівки повинні ґрунтуватися на оцінці стабільності лікарського засобу. За необхідності мають бути передбачені спеціальні вказівки, зокрема, для лікарських засобів, які не можна заморожувати. Неприйнятне використання таких термінів, як «умови навколишнього середовища» або «кімнатна температура».
Має існувати прямий зв’язок між вказівкою на етикетці та доведеною стабільністю лікарського засобу. На етикетці контейнера слід зазначати дату закінчення терміну придатності.
5.2 Випробування світлостабільності нових діючих речовин і лікарських засобів
5.2.1 Загальна інформація
5.2.1.1 Для доказу того, що вплив світла не призводить до неприпустимих змін, слід оцінити характеристики світлостабільності, властиві новим діючим речовинам і лікарським засобам. Як правило, випробування світлостабільності проводять на одній серії продукції, відібраній відповідно до вказівок у пп. 5.1.2.1.3 і 5.1.2.2.3 «Вибір серій». За деяких обставин такі дослідження слід повторювати, якщо були внесені певні зміни (наприклад, до складу, паковання). Необхідність повторення таких досліджень залежить від характеристик світлостабільності, визначених на момент першого подання заявки на реєстрацію, і від типу внесеної зміни.
У цьому підрозділі, у першу чергу, розглядається одержання інформації про світлостабільність діючих речовин та відповідних лікарських засобів, що необхідна для складання реєстраційних досьє. Положення підрозділу 5.2 не стосуються світлостабільності лікарських засобів в умовах їх застосування, а також таких заявок на реєстрацію, на які не поширюються положення підрозділу 5.1. Можуть бути використані альтернативні підходи за умови, що надане їх наукове обґрунтування.
Рекомендується застосовувати систематизований підхід до випробувань світлостабільності, що охоплює, за необхідності, такі дослідження:
а) випробування діючої речовини;
б) випробування лікарського засобу без первинного паковання;
в) випробування лікарського засобу в первинному пакованні (за необхідності);
г) випробування лікарського засобу в торговому пакованні (за необхідності).
Обсяг випробувань продукції необхідно встановити, оцінивши, чи спостерігається допустима зміна після закінчення випробування під впливом світла (див. рисунок 5.2.1). Допустима зміна — це зміна в межах, обґрунтованих заявником.
Офіційні вимоги щодо маркування діючих речовин і лікарських засобів, нестійких до впливу світла, встановлюють у відповідних нормативних документах.
5.2.1.2 Джерела світла
Для випробувань світлостабільності можуть використовуватись описані нижче джерела світла. Заявник повинен або підтримувати відповідний контроль температури для зведення до мінімуму впливу її локальних змін, або за тих самих умов навколишнього середовища паралельно проводити контрольні випробування в темряві, якщо не обґрунтовано інше. Для варіантів 1 і 2 виробник лікарського засобу чи заявник може покладатися на специфікацію спектрального розподілу, надану виробником джерела світла.
Варіант 1
Будь-яке джерело, призначене для вироблення світла, подібне до стандарту випромінювання D65/ID65, наприклад, флуоресцентна лампа штучного денного світла, що випромінює у видимому та ультрафіолетовому діапазонах, ксенонова чи галогенова лампи. D65 — це стандарт для зовнішнього денного освітлення, визнаний на міжнародному рівні, та визначений у стандарті ISO 10977. ID65 є еквівалентним стандартом для віддзеркаленого денного освітлення всередині приміщення. Джерело світла, значна частина випромінювання якого знаходиться в області менше 320 нм, може бути оснащене відповідним(и) фільтром(ами) для усунення цього випромінювання.
Варіант 2
При другому варіанті той самий зразок слід піддати впливу як лампи з холодною білою флуоресценцією, так і лампи з випромінюванням у близькій ультрафіолетовій області.
1) Лампа з холодною білою флуоресценцією для вироблення світла, подібна зазначеній у стандарті ISO 10977.
2) Лампа з випромінюванням у близькій ультрафіолетовій області зі спектральним розподілом від 320 до 400 нм із максимальним випромінюванням енергії в діапазоні довжини хвиль від 350 до 370 нм; значна частина ультрафіолетового випромінювання має бути в діапазоні від 320 до 360 нм і від 360 до 400 нм.
5.2.1.3 Методика
При підтверджувальних дослідженнях зразки піддають впливу світла за умови загальної світлової експозиції не менше 1,2 млн лк•ч і енергетичної експозиції в близькій ультрафіолетовій області не менше 200 Вт•ч/м2 так, щоб можна було провести безпосереднє порівняння діючої речовини та лікарського засобу.
Зразки можуть піддаватися впливу світла з усіх боків при застосуванні валідованої хімічної актинометричної системи, що підтверджує одержання певного впливу світла, або протягом відповідного відрізка часу, якщо умови контролюють за допомогою каліброваних радіометрів/люксметрів. Приклад актинометричної методики наведений у п. 5.2.4 (див. також додаток В, [18]).
Якщо для оцінки внеску термоіндукованих змін до загальних змін, що спостерігаються, використовують захищені від світла контрольні зразки (наприклад, зразки, загорнуті в алюмінієву фольгу), то їх слід розміщувати поруч зі зразком, досліджуваним на світлостабільність.
5.2.2 Діюча речовина (активний фармацевтичний інгредієнт)
Випробування світлостабільності діючих речовин мають складатися з двох частин: досліджень при примусовому розкладі та підтверджувальних досліджень.
Метою випробувань при примусовому розкладі є оцінка загальної світлочутливості речовини для розробки методу і/або з’ясування шляху розкладу. Для валідації аналітичних методик, що застосовуються при цих випробуваннях, може бути використана сама діюча речовина і/або її прості розчини/суспензії. При проведенні таких досліджень зразки мають знаходитися в хімічно інертних і прозорих контейнерах. Для досліджень при примусовому розкладі можуть бути використані різні умови впливу світла залежно від світлочутливості конкретної діючої речовини та інтенсивності використовуваних джерел світла. З метою розробки та валідації методики доцільно обмежити час впливу світла і завершити дослідження, якщо спостерігається значний розклад. Для світлостійких речовин дослідження можуть бути завершені після одержання підхожого рівня впливу. Планування таких експериментів здійснюється за розсудом заявника, але кожен використовуваний рівень впливу має бути обґрунтований.
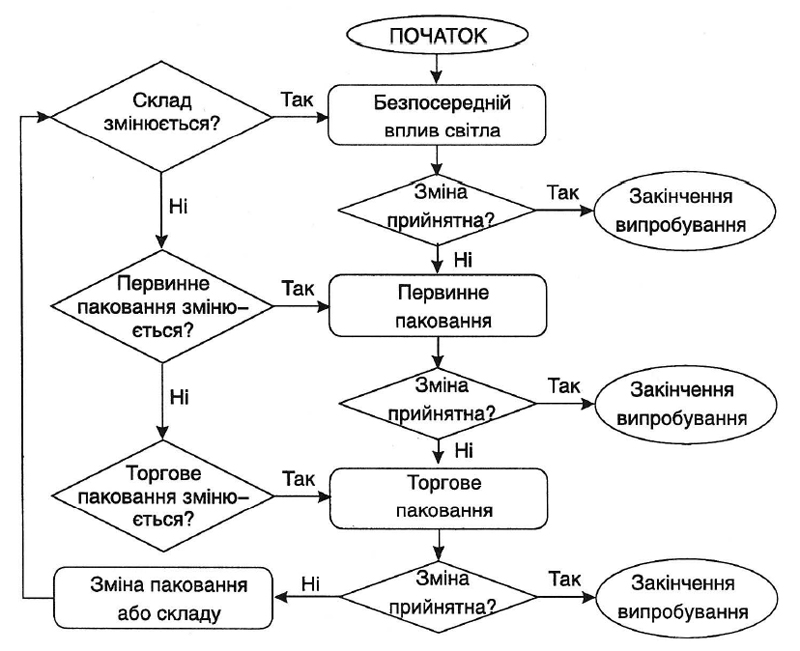
Рисунок 5.2.1 — Схема рішень при випробуваннях світлостабільності лікарських засобів.
В умовах примусового розкладу можуть виявлятися продукти розкладу, утворення яких малоймовірне в умовах підтверджувальних досліджень. Такі дані можуть використовуватися при розробці та валідації відповідних аналітичних методів. Якщо на практиці було доведено, що ці продукти розкладу не утворюються при підтверджувальних випробуваннях, то немає необхідності проводити їх подальше вивчення.
Потім мають бути проведені підтверджувальні дослідження для одержання інформації, необхідної для поводження з продукцією, її пакування та маркування (інформацію з планування таких досліджень див. у пп. 5.2.1.3 «Методика» та 5.2.2.1 «Надання зразків»).
Як правило, на етапі розробки випробувань зазнає лише одна серія діючої речовини, а потім характеристики світлостабільності мають бути підтверджені на одній серії, обраній відповідно до вимог підрозділу 5.1, якщо діюча речовина є явно стійкою чи нестійкою до впливу світла. Якщо результати підтверджувальних досліджень неоднозначні, то мають бути проведені випробування не менше ніж на двох додаткових серіях. Зразки мають бути обрані відповідно до вимог підрозділу 5.1.
5.2.2.1 Надання зразків
Слід гарантовано врахувати фізичні характеристики випробуваних зразків; потрібно вжити необхідних заходів, таких як охолодження і/або вміщення зразків у герметичні контейнери, щоб звести до мінімуму ефекти, зумовлені змінами фізичного стану, такими, як сублімація, випаровування чи плавлення. Мають бути обрані всі запобіжні заходи, щоб звести до мінімуму перешкоди, котрі заважають впливу світла на випробувані зразки. Слід врахувати й оцінити також можливі взаємодії між зразками та будь-яким матеріалом, що використовується для контейнерів або для загального захисту зразка, якщо вони стосуються проведених випробувань.
Пряма вимога у разі зразків твердих діючих речовин полягає в тому, що необхідно відібрати відповідну кількість зразка, вмістити у підхожу скляну або пластмасову чашку та (за необхідності) накрити підхожою прозорою кришкою. Тверді діючі речовини слід розподілити в контейнері шаром, товщина якого, як правило, не перевищує 3 мм. Діючі речовини, що є рідинами, слід випробовувати в хімічно нейтральних і прозорих контейнерах.
5.2.2.2 Аналіз зразків
Після закінчення впливу світла необхідно дослідити зразки на наявність будь-яких змін фізичних властивостей (наприклад, зовнішнього вигляду, прозорості або кольору розчину); слід також провести кількісне визначення та визначення вмісту продуктів розкладу за допомогою методу, відповідним чином валідованого відносно тих продуктів, що можуть утворитися при фотохімічному розкладі.
У разі зразків твердих діючих речовин відбір проб має забезпечувати використання в окремих випробуваннях репрезентативних порцій. Подібні міркування при відборі проб застосовують і до інших речовин, що можуть бути неоднорідними після впливу світла; для цього, наприклад, проводять гомогенізацію всього зразка. Аналіз зразків, що зазнали впливу світла, слід проводити одночасно з аналізом усіх захищених зразків, що були використані як засоби контролю, які знаходяться в темряві (якщо вони використовувалися при випробуванні).
5.2.2.3 Оцінка результатів
Дослідження при примусовому розкладі мають бути сплановані таким чином, щоб забезпечити необхідну інформацію для розробки та валідації методів випробувань, що застосовуються при підтверджувальних дослідженнях. Ці методи випробувань мають забезпечувати розподіл і виявлення продуктів фотолізу, що з’являються в ході підтверджувальних досліджень. Оцінюючи результати цих досліджень важливо усвідомлювати, що вони є частиною стресових випробувань і, отже, не призначені для встановлення якісних і кількісних меж змін.
Підтверджувальні дослідження мають визначати запобіжні заходи, потрібні при виробництві або виготовленні препарату, а також необхідність використання стійкого до впливу світла паковання. При оцінці результатів підтверджувальних досліджень щодо того, чи є зміна внаслідок впливу світла прийнятною, важливо враховувати результати інших офіційних досліджень стабільності, щоб гарантувати якість діючої речовини в обґрунтованих межах під час її використання (див. підрозділи 5.1 і 5.3–5.10 цієї настанови та настанови ІСН Q3A, Q3B і Q3C*).
*Рекомендується користуватися зазначенимими документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.2.3 Лікарський засіб
Як правило, дослідження лікарських засобів слід проводити послідовно, починаючи з випробувань зразка, що цілком знаходиться під впливом світла, і переходячи (за необхідності) до випробувань лікарського препарату в первинному пакованні, а потім у торговому пакованні. Випробування повинні проводитися доти, доки не будуть отримані результати, які доводять, що лікарський засіб адекватно захищений від впливу світла. Лікарський засіб слід піддавати впливу світла за умов, описаних у п. 5.2.1.3.
Як правило, на етапі розробки випробувань зазнає лише одна серія лікарського засобу, а потім характеристики світлостабільності мають бути підтверджені ще на одній серії, обраній відповідно до вимог підрозділу 5.1, якщо лікарський препарат є явно стійким або нестійким до впливу світла. Якщо результати підтверджувальних досліджень неоднозначні, то мають бути проведені випробування не менше, ніж ще на двох додаткових серіях.
Якщо було показано, що первинне паковання, таке як алюмінієві туби чи банки, цілком непроникне для світла, випробування, як правило, слід проводити лише стосовно лікарського засобу, що знаходиться під прямим впливом світла.
Може бути доцільним випробування певних лікарських засобів, таких як інфузійні розчини, креми для застосування в дерматології тощо, для підтвердження їх світлостабільності при застосуванні. Обсяг такого випробування встановлюється за розсудом заявника; він має залежати від інструкцій для застосування та узгоджуватися з ними.
Використовувані аналітичні методики мають бути належним чином валідовані.
5.2.3.1 Надання зразків
Слід гарантовано врахувати фізичні характеристики випробуваних зразків; мають бути вжиті необхідні заходи, наприклад, охолодження і/або вміщення зразків у герметичні контейнери, щоб звести до мінімуму ефекти, зумовлені змінами фізичного стану, такими як сублімація, випаровування чи плавлення. Мають бути обрані всі запобіжні заходи, щоб звести до мінімуму перешкоди, що заважають опромінюванню випробуваних зразків. Слід також врахувати й оцінити можливі взаємодії між зразками та будь-яким матеріалом, що використовується для контейнерів або для загального захисту зразка, якщо вони стосуються проведених випробувань.
Якщо це можна практично здійснити, то зразки лікарського засобу без первинного паковання слід випробовувати в таких же умовах, як і зразки діючої речовини. Зразки слід розташовувати так, щоб забезпечити максимальну площу для впливу джерела світла. Наприклад, таблетки, капсули тощо мають бути розкладені одним шаром.
Якщо прямий вплив світла на лікарський засіб неприйнятний (наприклад, внаслідок окислювання лікарського препарату), зразок слід вмістити у відповідний захисний інертний прозорий контейнер (наприклад, кварцовий).
Якщо необхідно випробувати лікарський засіб в первинному або торговому пакованні, зразки слід розміщувати горизонтально чи в поперечному напрямку відносно джерела світла, щоб забезпечити рівномірний вплив світла на зразки. При випробуванні контейнерів великого об’єму (наприклад, паковань, що надалі підлягають розподілу) може знадобитися деяке коригування умов випробувань.
5.2.3.2 Аналіз зразків
Після закінчення впливу світла необхідно дослідити зразки на наявність будь-яких змін фізичних властивостей (наприклад, зовнішнього вигляду, прозорості або кольору розчину, розчинення/розпадання для таких лікарських форм як таблетки, капсули тощо); слід також провести кількісне визначення та визначення вмісту продуктів розкладу за допомогою методу, відповідним чином валідованого стосовно продуктів, що можуть утворитися при фотохімічному розкладі.
У разі зразків порошків відбір проб має забезпечувати використання в окремих випробуваннях репрезентативних порцій. У разі твердих лікарських форм для орального застосування випробування слід проводити для відповідної кількості зразків, наприклад, для 20 таблеток або капсул. Подібні міркування при відборі проб застосовують і до інших зразків, що можуть бути неоднорідними після впливу світла; для цього, наприклад, проводять гомогенізацію або розчинення всього зразка (наприклад, кремів, мазей, суспензій тощо). Аналіз зразків, що зазнали впливу світла, слід проводити одночасно з аналізом будь-яких захищених від світла зразків, які були використані як засоби контролю, що знаходяться в темряві (якщо вони використовувалися при випробуванні).
5.2.3.3 Оцінка результатів
Залежно від ступеня зміни може знадобитися спеціальне маркування чи паковання, що дозволяє послабити вплив світла. При оцінці результатів підтверджувальних досліджень відносно того, чи є зміна внаслідок впливу світла прийнятною, важливо враховувати результати інших офіційних досліджень стабільності, щоб гарантувати якість лікарського засобу згідно зі специфікацією протягом терміну зберігання (див. підрозділи 5.1 і 5.3–5.10 цієї настанови та настанови ІСН Q3A, Q3B і Q3C*).
*Рекомендується користуватися зазначеними документами. Вони набудуть чинності в Україні з моменту їх прийняття в установленому порядку.
5.2.4 Хінінова хімічна актинометрія
Нижче наведено докладний опис актинометричної методики для контролю впливу світла від флуоресцентної лампи, що генерує випромінювання в близькій ультрафіолетовій області. Для інших джерел світла/актинометричних систем може бути використаний аналогічний підхід, але кожна актинометрична система має бути відкалібрована для використовуваного джерела світла.
Готують масооб’ємним способом достатню кількість 2% водного розчину хініну моногідрохлориду дигідрату (якщо необхідно, розчиняють при нагріванні).
Варіант 1
10 мл розчину вміщують у безбарвну ампулу місткістю 20 мл, герметично закупорюють і використовують як зразок. Окремо вмішують 10 мл розчину в безбарвну ампулу місткістю 20 мл (див. рис. 5.2.2), герметично закупорюють, загортають в алюмінієву фольгу для повного захисту від світла й використовують як контрольний зразок. Зразок і контрольний зразок піддають впливу джерела світла протягом заданого часу (годин). Після припинення впливу джерела світла визначають оптичну густину зразка (Аτ) і контрольного зразка (Ао) за довжини хвилі 400 нм. Розраховують різницю показників оптичної густини (А):
А=Аτ — Ао.
Тривалість впливу світла має бути достатньою для забезпечення зміни оптичної густини не менше ніж на 0,9.
Рисунок 5.2.2 — Форма та розміри ампули.
Варіант 2
Розчином заповнюють кварцову кювету з товщиною шару 1 см і використовують як зразок. Окремо заповнюють кварцову кювету з товщиною шару 1 см, загортають в алюмінієву фольгу для повного захисту від світла та використовують як контрольний зразок. Зразок і контрольний зразок піддають впливу джерела світла протягом заданого часу (годин). Після припинення впливу джерела світла визначають оптичну густину зразка (Аτ) і контрольного зразка (Ао) за довжини хвилі 400 нм. Розраховують різницю показників оптичної густини (А):
А=Аτ — Ао.
Тривалість впливу світла має бути достатньою для забезпечення зміни оптичної густини не менше ніж на 0,5.
Можуть використовуватися альтернативні форми паковання за умови відповідної валідації. Допускається використання альтернативних валідованих хімічних актинометрів.
5.3 Випробування стабільності нових лікарських форм
Нова лікарська форма визначається як лікарський засіб, що є іншим видом фармацевтичної продукції, але містить ті самі діючі речовини, що й зареєстрований у встановленому порядку лікарський засіб, який знаходиться на ринку.
До таких видів фармацевтичної продукції відносять:
а) лікарські засоби з іншим шляхом введення (наприклад, оральний на відміну від парентерального);
б) лікарські засоби з тим же шляхом введення, але в іншій лікарській формі (наприклад, капсули на відміну від таблеток, розчин на відміну від суспензії);
в) лікарські засоби з новими особливими функціональними властивостями чи системою доставки (наприклад, таблетки з негайним вивільненням на відміну від таблеток із модифікованим вивільненням).
Протоколи дослідження стабільності нових лікарських форм у принципі мають складатися з урахуванням положень підрозділу 5.1. Однак на момент подання заявки на реєстрацію в певних обґрунтованих випадках допускається скорочений обсяг даних зі стабільності (наприклад, дані за 6 місяців прискорених і 6 місяців довгострокових досліджень, проведення яких триває).
5.4 Випробування стабільності існуючих діючих речовин і відповідних лікарських засобів
5.4.1 Загальна інформація
5.4.1.1 Мета
Положення цього підрозділу доповнюють положення підрозділу 5.1 і стосуються випробувань стабільності існуючих діючих речовин і лікарських засобів, що їх містять. У цьому підрозділі під терміном «існуюча діюча речовина» розуміють діючу речовину, що була дозволена до застосування в Україні в складі відповідного лікарського засобу, зареєстрованого в установленому порядку.
Положення підрозділу 5.4 поширюються на діючі речовини, отримані шляхом хімічного синтезу, і відповідні лікарські засоби, на рослинну сировину, на препарати з рослинної сировини, призначені для виробництва готових лікарських засобів, і на самі готові лікарські засоби з рослинної сировини. Положення цього підрозділу не поширюються на радіоактивні лікарські засоби, біологічні лікарські засоби, а також лікарські препарати, отримані за допомогою біотехнології.
У цьому підрозділі наводяться приклади основних даних зі стабільності для зазначених діючих речовин і лікарських засобів. У той же час передбачено достатню гнучкість, що дозволяє охопити безліч різних практичних ситуацій, зумовлених певними науковими поглядами та властивостями досліджуваних речовин. За наявності науково обґрунтованих причин можуть застосовуватися й інші підходи.
5.4.1.2 Сфера застосування
У підрозділі 5.4 розглядається інформація про стабільність існуючих діючих речовин і відповідних лікарських засобів, що має бути надана в реєстраційних досьє.
Щодо рослинної сировини, препаратів із рослинної сировини, призначених для виробництва готових лікарських засобів, і готових лікарських засобів із рослинної сировини додатково слід керуватися розділом зі стабільності документа «Note for Guidance on Quality of Herbal Medicinal Products»*.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.4.1.3 Загальні принципи
Мета випробувань стабільності — одержання даних про зміну з часом якості діючої речовини або лікарського засобу під впливом різних факторів навколишнього середовища, таких, як температура, вологість і світло, а також установлення періодичності проведення повторних випробувань діючої речовини або терміну зберігання лікарського засобу та рекомендованих умов зберігання.
Вибір умов випробувань, встановлених у цьому підрозділі, зумовлений положеннями підрозділу 5.1.
5.4.2 Керівні вказівки
5.4.2.1 Діюча речовина (активний фармацевтичний інгредієнт)
5.4.2.1.1 Загальна інформація
Інформація про стабільність діючої речовини є невід’ємною частиною систематизованого підходу до оцінки стабільності.
Для діючих речовин, не описаних в офіційних фармакопейних монографіях (монографіях ДФУ, Європейської Фармакопеї чи іншої відповідної фармакопеї), вимагаються дослідження стабільності.
Для діючих речовин, описаних в офіційних фармакопейних монографіях (монографіях ДФУ, Європейської Фармакопеї чи іншої відповідної фармакопеї), для яких зазначені продукти розкладу та встановлені відповідні межі їх вмісту, але не визначений період до проведення повторних випробувань, прийнятні два варіанти:
а) заявник має зазначити, що діюча речовина повинна відповідати фармакопейній монографії безпосередньо перед виробництвом лікарського засобу. У цьому випадку не вимагається проведення досліджень стабільності за умови, що доведено придатність фармакопейної монографії для діючої речовини конкретного постачальника*;
б) заявник має встановити період до проведення повторних випробувань на підставі результатів довгострокових досліджень стабільності.
Що стосується лікарських засобів із рослинної сировини, то діючі речовини, включаючи рослинну сировину та препарати з рослинної сировини, що використовують як вихідну сировину у виробничому процесі, перед використанням (наприклад, перед екстракцією) мають відповідати специфікації.
*Див. настанову «Note for Guidance on Summary of Requirements for Active Substances in Part II of the Dossier». Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.4.2.1.2 Стресові випробування
Стресові випробування діючої речовини допомагають ідентифікувати можливі продукти розкладу, що у свою чергу, дозволяє встановити шляхи розкладу та властиву молекулі стабільність, а також валідувати застосовувані аналітичні методики щодо їх придатності для оцінки стабільності.
Для діючої речовини можна застосувати наступні підходи:
а) якщо діюча речовина описана в офіційній фармакопейній монографії (монографії ДФУ, Європейської Фармакопеї чи іншої відповідної фармакопеї) і цілком відповідає встановленим в ній критеріям прийнятності, не вимагається наводити дані про продукти розкладу, якщо вони перераховані у відповідних розділах цієї монографії;
б) якщо діючі речорини не описані в офіційних фармакопейних монографіях, існують два варіанти:
- якщо є опубліковані в літературі дані, то можна їх надати як обґрунтування шляхів розкладу;
- якщо такі дані в науковій літературі (включаючи офіційні фармакопеї) відсутні, необхідно провести стресові випробування. Результати цих досліджень складатимуть невід’ємну частину інформації, що подається компетентному уповноваженому органу.
Стресові випробування, за можливості, слід проводити на одній серії діючої речовини. Вони повинні включати дослідження впливу на діючу речовину температури при її послідовних підвищеннях на 10 °С вище температури прискорених випробувань (наприклад, 50 °С, 60 °С тощо) та вологості, якщо це доцільно (наприклад, відносної вологості 75% і вище), а також окислення та фотолізу. Якщо діюча речовина знаходиться у вигляді розчину або суспензії, у ході випробувань слід також визначити її здатність до гідролізу в широкому діапазоні рН. Невід’ємною частиною стресових випробувань мають бути випробування світлостабільності. Стандартні умови випробувань світлостабільності наведені в підрозділі 5.2.
Вивчення продуктів розкладу в стресових умовах є корисним для встановлення шляхів розкладу, а також для розробки і валідації відповідних аналітичних методик. Однак ці дослідження щодо певних продуктів розкладу можуть бути необов’язкові, якщо показано, що такі продукти розкладу не утворюються в умовах прискорених або довгострокових випробувань.
5.4.2.1.3 Вибір серій
Допустимі два варіанти:
а) необхідно надати інформацію про стабільність, що ґрунтується на результатах прискорених і довгострокових випробувань щонайменше двох промислових серій, виготовлених за допомогою такого ж способу виробництва (того ж шляху синтезу) і процедури, які описані у відповідній частині реєстраційного досьє. На момент подання заявки на реєстрацію тривалість довгострокових і прискорених випробувань має становити як мінімум 6 місяців;
або
б) необхідно надати інформацію про стабільність, що ґрунтується на результатах прискорених і довгострокових випробувань щонайменше трьох дослідно-промислових серій, виготовлених за допомогою такого ж способу виробництва (того ж шляху синтезу) і процедури, що описані у відповідній частині реєстраційного досьє. На момент подання заявки тривалість довгострокових і прискорених випробувань має становити як мінімум 6 місяців.
5.4.2.1.4 Система контейнер/закупорювальний елемент
Дослідження стабільності слід проводити з використанням діючої речовини, упакованої за допомогою системи контейнер/закупорювальний елемент, що ідентична пакованню, запропонованому для зберігання та дистрибуції, або моделює його.
5.4.2.1.5 Специфікація
Дослідження стабільності повинні охоплювати ті характеристики діючої речовини, що зазнають змін під час зберігання та можуть впливати на якість, безпеку і/або ефективність. За можливості, слід проводити випробування щодо фізичних, хімічних, біологічних та мікробіологічних властивостей. Необхідно застосовувати валідовані методики випробувань, що дозволяють характеризувати стабільність.
Критерії прийнятності — це числові межі, діапазони та інші критерії для конкретних випробувань. Вони повинні включати верхні межі вмісту окремих домішок і продуктів розкладу, а також межі їх сумарного вмісту, встановлені виходячи з міркувань безпеки і/або ефективності. Для діючих речовин, описаних в офіційних фармакопейних монографіях (монографіях ДФУ, Європейської Фармакопеї чи іншої відповідної фармакопеї), випробування слід проводити відповідно до монографії, або за допомогою методу, що валідований у порівнянні з фармакопейним методом. Слід навести обґрунтування того, що всі можливі домішки (домішки й продукти розкладу, що утворюються в ході процесу) достатньо контролюються.
Відносно специфікацій для рослинної сировини, препаратів із рослинної сировини та готових лікарських засобів, що містять їх, слід керуватися документом «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for Herbal Drags, Herbal Drags Preparations and Herbal Medicinal Products»*.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.4.2.1.6 Частота випробувань
При довгострокових дослідженнях частота випробувань має бути достатньою для встановлення профілю стабільності діючої речовини. В умовах довгострокового дослідження частота випробувань, як правило, складає кожні 3 місяці протягом першого року, кожні 6 місяців протягом другого року, а потім щорічно протягом запропонованого періоду до проведення повторних випробувань.
При прискорених випробуваннях тривалістю 6 місяців для проведення випробувань рекомендується мінімум три точки контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 3 і 6 місяців). Якщо в результаті прискорених випробувань очікується (на підставі досвіду розробки) «значна зміна», необхідно розширити обсяг випробувань за рахунок уведення додаткової кількості зразків у кінцевій точці контролю або введення в план дослідження четвертої точки контролю.
Якщо через «значну зміну» при прискорених випробуваннях виникла потреба провести випробування при проміжних умовах зберігання, то в ході таких досліджень тривалістю 12 місяців рекомендується як мінімум чотири точки контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 6, 9 і 12 місяців).
Для серій рослинної сировини та препаратів із рослинної сировини, щодо яких заявник має дані зі стабільності за певний відрізок часу, при наявності обґрунтування частота випробувань може бути меншою.
5.4.2.1.7 Умови зберігання
Як правило, оцінку діючої речовини слід проводити в умовах зберігання (з відповідними допустимими відхиленнями), що дозволяють досліджувати її термічну стабільність і, за необхідності, чутливість до вологи. Обрані умови досліджень мають відповідати умовам зберігання, транспортування та подальшого застосування; тривалість досліджень має бути достатньою, щоб охопити всі ці періоди.
На момент подання заявки на реєстрацію тривалість довгострокових випробувань для варіантів (а) і (б) має становити як мінімум 6 місяців; випробування слід продовжувати протягом часу, достатнього для того, щоб охопити запропонований період до проведення повторних випробувань. Додаткові дані, зібрані під час проведення експертизи реєстраційного досьє, мають бути подані до уповноважених органів за їх запитом. Дані прискорених випробувань і, за необхідності, випробувань при проміжних умовах зберігання можуть бути використані для оцінки впливу короткочасних відхилень від умов зберігання, зазначених на етикетці (які можуть виникнути, наприклад, при транспортуванні).
Умови зберігання при довгострокових і прискорених випробуваннях, а також проміжні умови зберігання для діючих речовин докладно описані в таблицях 5.4.1–5.4.3. Умови, описані в п. 5.4.2.1.7.1 «Загальний випадок» використовують, якщо до діючої речовини не застосовуються умови, описані нижче в пп. 5.4.2.1.7.2, 5.4.2.1.7.3, 5.4.2.1.7.4. За наявності відповідного обґрунтування можуть бути застосовані інші умови зберігання.
5.4.2.1.7.1 Загальний випадок
Таблиця 5.4.1
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові1
|
(25±2) °С та (60±5)% або (30±2) °С та (65±5)%
|
6 (варіанти а і б)
|
|
Проміжні2
|
(30±2) °С та (65±5)%
|
6
|
|
Прискорені2
|
(40±2) °С та (75±5)%
|
6
|
1Заявник самостійно вирішує, чи проводити довгострокові випробування при температурі (25±2) °С та відносній вологості (60±5)%, чи при температурі (30±2) °С та відносній вологості (65±5)%. В останньому випадку не вимагається надання ніяких додаткових даних зі стабільності при проміжних умовах зберігання.
2Для рослинної сировини та препаратів із рослинної сировини прискорені випробування та випробування при проміжних умовах можуть не проводитися, якщо це буде обґрунтоване заявником, і якщо в маркуванні чітко зазначено умови зберігання при температурі нижче 25 °С.
Якщо протягом 6 місяців при прискорених випробуваннях спостерігається «значна зміна», необхідно провести додаткові випробування при проміжних умовах зберігання та оцінити результати щодо критеріїв «значної зміни». Якщо не обґрунтовано інше, під час досліджень при проміжних умовах зберігання слід проводити всі випробування. Реєстраційне досьє, що подається вперше, має включати дані досліджень як мінімум протягом 6 місяців (при загальній тривалості досліджень при проміжних умовах зберігання 12 місяців).
«Значна зміна» для діючої речовини означає її невідповідність специфікації.
5.4.2.1.7.2 Діючі речовини, призначені для зберігання в холодильнику
Таблиця 5.4.2
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(5±3) °С
|
6 (варіанти а і б)
|
|
Проміжні
|
(25±2) °С та (60±5)%
|
6
|
Оцінку даних, отриманих при зберіганні діючої речовини у холодильнику, слід проводити відповідно до вказівок пункту 5.4.2.1.9 «Оцінка даних» цього підрозділу за винятком випадків, окремо обговорених нижче.
Якщо при прискорених випробуваннях у період з третього по шостий місяць спостерігається «значна зміна», пропонований період до проведення повторних випробувань має ґрунтуватися на даних довгострокових випробувань, отриманих у реальному часі.
Якщо при прискорених випробуваннях «значна зміна» спостерігається протягом перших 3 місяців, то слід всебічно дослідити вплив короткочасних відхилень від умов зберігання, зазначених на етикетці (які можуть виникнути, наприклад, при транспортуванні чи обробці). Це дослідження можна підтвердити (якщо це доцільно) додатковими випробуваннями однієї серії діючої речовини протягом 3 місяців, але з більшою частотою проведення випробувань, ніж звичайно. Вважається, що немає необхідності продовжувати прискорені випробування діючої речовини протягом 6 місяців, якщо протягом перших 3 місяців відбулася «значна зміна».
5.4.2.1.7.3 Діючі речовини, призначені для зберігання в морозильній камері
Таблиця 5.4.3
|
Дослідження (випробування)
|
Умови зберігання (температура)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
–(20+5) °С
|
6 (варіанти а і б)
|
Для діючих речовин, що призначені для зберігання в морозильній камері, період до проведення повторних випробувань установлюють на підставі даних довгострокових випробувань, отриманих у реальному часі. Оскільки відсутні умови для проведення прискорених випробувань таких діючих речовин, необхідно провести дослідження щодо однієї серії при підвищеній температурі (наприклад, (5±3) °С або (25±2) °С) протягом відповідного відрізка часу, щоб врахувати вплив короткочасних відхилень від умов зберігання, зазначених на етикетці (наприклад, при транспортуванні діючих речовин або поводженні з ними).
5.4.2.1.7.4 Діючі речовини, призначені для зберігання при температурі нижче –20 °С
Випробування діючих речовин, призначених для зберігання при температурі нижче –20 °С, слід проводити, обґрунтовуючи в кожному окремому випадку.
5.4.2.1.8 Зобов’язання продовжувати випробування стабільності
Якщо отримані при довгострокових випробуваннях дані про стабільність для вихідних серій не охоплюють запропонований період до проведення повторних випробувань, то слід взяти зобов’язання продовжувати дослідження стабільності, щоб остаточно встановити період до проведення повторних випробувань.
Якщо матеріали заявки містять дані довгострокових випробувань стабільності як мінімум для трьох промислових серій, що охоплюють запропонований період до проведення повторних випробувань, то зобов’язання продовжувати дослідження стабільності не потрібне. У протилежному випадку заявникові необхідно взяти одне з наступних зобов’язань:
1) якщо реєстраційне досьє містить дані досліджень стабільності щонайменше для трьох промислових серій, то слід взяти зобов’язання продовжувати ці дослідження протягом усього пропонованого періоду до проведення повторних випробувань;
2) якщо реєстраційне досьє містить дані досліджень стабільності менше ніж для трьох промислових серій, то слід взяти зобов’язання продовжувати довгострокові дослідження протягом пропонованого періоду до проведення повторних випробувань і ввести в дослідження додаткові промислові серії (щоб загальна кількість серій становила не менше трьох);
3) якщо реєстраційне досьє не містить даних про стабільність щодо промислових серій, то слід взяти зобов’язання провести довгострокові випробування стабільності на перших трьох промислових серіях протягом запропонованого періоду до проведення повторних випробувань.
Протокол довгострокового дослідження стабільності, доданий при взятті зобов’язання про подальше випробування стабільності, має бути таким же, як і для вихідних серій, якщо науково не обґрунтований інший підхід.
5.4.2.1.9 Оцінка даних
Мета досліджень стабільності полягає в тому, щоб установити період до проведення повторних випробувань, застосовний для всіх серій діючої речовини, що надалі будуть вироблятися за однакових умов. Цей період установлюють на підставі випробувань як мінімум двох або трьох серій діючої речовини та оцінки інформації про стабільність (що включає, при необхідності, результати фізичних, хімічних, біологічних і мікробіологічних випробувань). Ступінь варіабельності окремих серій забезпечує впевненість у тому, що вироблені надалі промислові серії відповідатимуть специфікації протягом заданого періоду до проведення повторних випробувань.
Дані можуть свідчити про такий незначний розклад і таку невелику варіабельність, що вже при їх розгляді очевидно, що необхідний період до повторних випробувань буде затверджено. У таких випадках, як правило, немає необхідності у формальному статистичному аналізі; досить надати обґрунтування відсутності такого аналізу.
Якщо очікується зміна кількісної характеристики в часі, то прийнятний підхід, що полягає у визначенні часу, після закінчення якого усереднена крива розкладу (при довірчій імовірності 95%) перетинається з допустимою нижньою межею, встановленою в специфікації. Якщо аналіз показує, що варіабельність від серії до серії невелика, корисно поєднувати дані для однієї загальної оцінки. Це може бути зроблено шляхом відповідної статистичної обробки нахилів ліній регресій і точок їх перетинання з віссю ординат для окремих серій (наприклад, значення р для рівня значущості відбракування, що перевищує 0,25). Якщо недоцільно поєднувати дані для декількох серій, то загальний період до проведення повторних випробувань залежатиме від мінімального часу, протягом якого, як передбачається, характеристики серії залишатимуться в рамках критеріїв прийнятності.
Характер будь-якого взаємозв’язку з розкладом речовин визначатиме необхідність у перетворенні даних для аналізу лінійної регресії. Як правило, взаємозв’язок може бути представлений у вигляді лінійної, квадратичної або кубічної функції на арифметичній або логарифмічній шкалі. Щоб перевірити придатність даних, отриманих щодо кожної серії, і об’єднаних даних для серій (якщо доцільно) для побудови прямої чи кривої розкладу слід застосовувати статистичні методи.
За наявності відповідного обґрунтування може бути проведена обмежена екстраполяція даних, отриманих у реальному часі в умовах довгострокових випробувань, за межі терміну спостереження, щоб збільшити період до проведення повторних випробувань на момент його затвердження. Таке обґрунтування має враховувати відомості про механізм розкладу, результати прискорених випробувань, придатність математичної моделі, розмір серії, наявність підтверджувальних даних про стабільність тощо. Однак при такій екстраполяції передбачається, що й далі (за рамками даних, які спостерігаються) матиме місце такий самий взаємозв’язок із розкладом.
Примітка. Якщо отримані в реальному часі дані підтверджені результатами прискорених або проміжних випробувань стабільності, то може бути встановлений період до проведення повторних випробувань/термін зберігання, що перевищує тривалість досліджень у реальному часі. Як правило, шляхом екстраполяції допускається встановлювати період до проведення повторних випробувань/термін зберігання, що дорівнює подвійній тривалості досліджень у реальному часі. Однак не можна подовжувати період до проведення повторних випробувань/термін зберігання більш ніж на 12 місяців понад тривалість досліджень у реальному часі. При цьому слід враховувати зміни, що відбулися, варіабельність отриманих даних, пропоновані умови зберігання та обсяг проведеного статистичного аналізу. Різні ситуації з цього питання наведені на схемі рішень (див. рис. 5.4.1). Більш докладна інформація викладена у підрозділі 5.6 цієї настанови.
При будь-якій оцінці слід враховувати не лише кількісне визначення, але й рівні вмісту продуктів розкладу, а також інші підхожі характеристики.
5.4.2.1.10 Вказівки/маркування
При зазначенні умов зберігання (температура, світло, вологість) слід керуватися додатком А.
Неприйнятне використання таких термінів, як «умови навколишнього середовища» або «кімнатна температура».
5.4.2.2 Лікарський засіб
5.4.2.2.1 Загальна інформація
Розробка офіційної програми дослідження стабільності для готового лікарського засобу має ґрунтуватися на знанні поведінки та властивостей діючої речовини й лікарської форми.
5.4.2.2.2 Випробування світлостабільності
Випробування світлостабільності (якщо вони необхідні) мають бути проведені щонайменше для однієї вихідної серії готового лікарського засобу. Стандартні умови випробування світлостабільності наведені в підрозділі 5.2.
5.4.2.2.3 Вибір серій
На момент подання заявки на реєстрацію необхідно надати дані досліджень стабільності для серій із таким же складом і в тій же лікарській формі, як і планований для розміщення на ринку лікарський препарат, а також у таких контейнерах із закупорювальними елементами, у яких цей препарат надійде на ринок.
Допустимі два варіанти:
а) щодо традиційних лікарських форм (наприклад, тверді лікарські форми з негайним вивільненням, розчини) і діючих речовин, про які відомо, що вони стабільні*, допускається надання інформації про стабільність щонайменше для двох дослідно-промислових серій;
б) щодо критичних лікарських форм або діючих речовин, про які відомо, що вони нестабільні, необхідно надати інформацію про стабільність для трьох вихідних серій. Дві з трьох серій мають бути, щонайменше, дослідно-промисловими, третя серія може бути меншою.
Технологічний процес, що використовується для виготовлення вихідних серій, повинен моделювати процес виробництва промислових серій; він має забезпечувати одержання лікарського засобу, що має таку ж якість і відповідає такій же специфікації, як і призначений для розміщення на ринку лікарський препарат. Якщо можливо, серії готового лікарського засобу мають бути виготовлені з використанням різних серій діючої речовини.
Дослідження стабільності мають бути проведені окремо для лікарських засобів із різною силою дії та з різним розміром контейнера, за винятком тих випадків, коли застосовують брекетинг або побудову матриць.
Може бути надана й інша підтверджувальна інформація.
*Діюча речовина вважається стабільною, якщо вона відповідає специфікації, встановленій заявником, або офіційній (фармакопейній) специфікації при зберіганні протягом 2 років при температурі 25 °С та відносній вологості 60% або при альтернативних умовах — при температурі 30 °С та відносній вологості 65%, а також при зберіганні протягом 6 місяців при температурі 40 °С та відносній вологості 75%.
5.4.2.2.4 Система контейнер/закупорювальний елемент
Випробування стабільності необхідно проводити стосовно лікарської форми в пакованні, призначеному для розміщення на ринку (включаючи, за необхідності, будь-яке вторинне паковання та етикетки для контейнерів). Будь-які випробування щодо лікарського засобу без первинного паковання або такого, що знаходиться в пакованні з інших пакувальних матеріалів, можуть складати корисну частину стресових випробувань лікарської форми або, відповідно, розглядатися як додаткова підтверджувальна інформація.
5.4.2.2.5 Специфікація
Дослідження стабільності мають включати випробування таких характеристик готового лікарського засобу, що зазнають змін при зберіганні та, можливо, можуть впливати на якість, безпеку і/або ефективність. Необхідно випробовувати (залежно від конкретної ситуації) фізичні, хімічні, біологічні та мікробіологічні властивості, визначати вміст консервантів (наприклад, антиоксидантів, антимікробних консервантів), а також перевіряти функціональні характеристики (наприклад, для системи доставки дози). Аналітичні методики мають бути цілком валідовані; вони повинні дозволяти характеризувати стабільність. Необхідність проведення повторних випробувань та їх обсяг залежатимуть від результатів валідаційних досліджень.
Критерії прийнятності у специфікації, що застосовується протягом терміну зберігання, слід встановлювати на підставі всієї наявної інформації про стабільність. Допускаються прийнятні та обґрунтовані відхилення критеріїв прийнятності в специфікації, що застосовується протягом терміну зберігання, та у специфікації, що застосовується при випуску, засновані на оцінці стабільності та змін, що спостерігаються при зберіганні. Будь-які розбіжності між критеріями прийнятності в цих двох специфікаціях щодо вмісту антимікробних консервантів повинні супроводжуватися обґрунтованою кореляцією хімічного складу та ефективності цих консервантів, доведеною на етапі фармацевтичної розробки для остаточного складу (за винятком концентрації антимікробного консерванту) лікарського засобу, призначеного для розміщення на ринку. З метою перевірки необхідно провести випробування стабільності однієї вихідної серії лікарського засобу щодо ефективності антимікробних консервантів (на додаток до їх кількісного визначення) протягом пропонованого терміну зберігання незалежно від того, чи є розбіжності між критеріями прийнятності щодо вмісту антимікробного консерванту в специфікаціях, що застосовуються при випуску та протягом терміну зберігання.
5.4.2.2.6 Частота проведення випробувань
При довгострокових дослідженнях частота проведення випробувань має бути достатньою для встановлення параметрів стабільності лікарського засобу. При довгострокових дослідженнях, як правило, випробування слід проводити кожні 3 місяці протягом першого року, кожні 6 місяців протягом другого року і потім щорічно протягом пропонованого терміну зберігання.
При прискорених дослідженнях тривалістю 6 місяців рекомендується проводити випробування не менше ніж у трьох точках контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 3 і 6 місяців). Якщо передбачається (на підставі досвіду розробки), що при прискорених випробуваннях результати можуть впритул наблизитися до критеріїв «значної зміни», необхідно провести розширені дослідження шляхом збільшення кількості зразків в останній точці контролю та включення четвертої точки контролю в план досліджень.
Якщо в результаті прискореного дослідження відбулася «значна зміна» і, як наслідок, знадобилося проведення досліджень при проміжних умовах зберігання, рекомендується проводити випробування як мінімум у чотирьох точках контролю, включаючи контроль на початку та при завершенні досліджень (наприклад, 0, 6, 9 і 12 місяців), при тривалості дослідження 12 місяців. При відповідному обґрунтуванні заявник може знизити частоту випробувань для лікарських засобів із рослинної сировини, якщо він має дані зі стабільності серій за певний час.
При відповідному обґрунтуванні можуть бути застосовані скорочені плани дослідження (тобто, побудова матриць або брекетинг), коли частота проведення випробувань зменшена або не проводяться випробування комбінацій певних критеріїв у повному обсязі.
5.4.2.2.7 Умови зберігання
Як правило, оцінку лікарського засобу слід проводити в умовах зберігання (з відповідними допустимими відхиленнями), що дозволяють досліджувати його термічну стабільність і, за необхідності, чутливість до вологи або можливість втрати розчинника. Обрані умови досліджень повинні відповідати умовам зберігання, транспортування та подальшого застосування; тривалість досліджень має бути достатньою, щоб охопити всі ці періоди.
За необхідності слід провести випробування стабільності лікарського засобу після його підготовки до застосування чи розведення, щоб одержати інформацію для маркування лікарського препарату щодо підготовки до застосування, умов зберігання та періоду використання після підготовки чи розведення. Такі випробування щодо підготовленого чи розведеного лікарського препарату мають бути проведені в початковий і кінцевий моменти пропонованого періоду його застосування; ці дослідження слід проводити на вихідних серіях як частину офіційних досліджень стабільності. Якщо на момент подання заявки на реєстрацію немає даних довгострокових випробувань, що охоплюють весь період терміну зберігання, мають бути подані дані на момент закінчення 6 місяців або на останній момент часу, для якого є дані про стабільність. Як правило, ці випробування немає необхідності повторювати на серіях, досліджуваних відповідно до зобов’язання про продовження випробувань стабільності.
На момент подання заявки на реєстрацію тривалість довгострокових випробувань має становити як мінімум 6 місяців для варіанта (а) або 12 місяців для варіанта (б); випробування слід продовжувати протягом часу, достатнього для того, щоб охопити запропонований термін зберігання. Додаткові дані, зібрані під час проведення експертизи реєстраційного досьє, мають бути подані до уповноваженого органу за його запитом. Дані прискорених випробувань і, за необхідності, випробувань при проміжних умовах зберігання можуть бути використані для оцінки впливу короткочасних відхилень від умов зберігання, зазначених на етикетці (такі відхилення можуть виникнути, наприклад, при транспортуванні).
Умови зберігання при довгострокових і прискорених випробуваннях, а також проміжні умови зберігання для лікарських засобів докладно описані в таблицях 5.4.4, 5.4.5, 5.4.7 та 5.4.8. Умови, описані в п. 5.4.2.2.7.1 «Загальний випадок» використовують, якщо до лікарського засобу не застосовуються умови, описані нижче в пп. 5.4.2.2.7.2, 5.4.2.2.7.3, 5.4.2.2.7.4, 5.4.2.2.7.5 і 5.4.2.2.7.6. За наявності відповідного обґрунтування можуть бути застосовані інші умови зберігання.
5.4.2.2.7.1 Загальний випадок
Таблиця 5.4.4
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові1
|
(25±2) °С та (60±5)% або (30±2) °С та (65±5)%
|
6 (варіант а)
12 (віріант б)
|
|
Проміжні
|
(30±2) °С та (65±5)%
|
6
|
|
Прискорені
|
(40±2) °С та (75±5)%
|
6
|
1Заявник самостійно вирішує, чи проводити довгострокові випробування при температурі (25±2) °С і відносній вологості (60±5)%, або при температурі (30±2) °С і відносній вологості (65±5)%. В останньому випадку не вимагається надання ніяких додаткових даних зі стабільності при проміжних умовах зберігання.
Якщо протягом 6 місяців при прискорених випробуваннях спостерігається «значна зміна», необхідно провести додаткові випробування при проміжних умовах зберігання та оцінити результати щодо критеріїв «значної зміни». Реєстраційне досьє, що подається вперше, має включати дані досліджень як мінімум протягом 6 місяців (при загальній тривалості досліджень при проміжних умовах зберігання 12 місяців).
Як правило, «значна зміна» для лікарського засобу означає:
- зміну кількісного вмісту на 5% порівняно з початковим; або невідповідність критеріям прийнятності за силою дії при використанні біологічних або імунологічних методик визначення;
- збільшення вмісту будь-якого продукту розкладу понад критерій прийнятності;
- невідповідність критеріям прийнятності щодо зовнішнього вигляду, фізичних властивостей і функціональних характеристик (наприклад, колір, поділ фаз, здатність до ресуспендування, спікливість, твердість, доставка дози при одному натисканні клапана); однак в умовах прискорених випробувань можна припускати, що відбудуться деякі зміни фізичних властивостей (наприклад, розм’якшення супозиторіїв, розплавлення кремів);
крім того, залежно від лікарської форми:
- невідповідність критеріям прийнятності стосовно рН;
- невідповідність критеріям прийнятності стосовно розчинення для 12 одиниць дозованого лікарсь кого засобу.
5.4.2.2.7.2 Лікарські засоби, упаковані в герметичні контейнери
Чутливість до впливу вологи або можливість втрати розчинника не є проблемою для лікарських засобів, упакованих у герметичні контейнери, що забезпечують постійний бар’єр, який перешкоджає проникненню вологи чи виходу розчинника. Таким чином, дослідження стабільності для лікарських засобів, що зберігають у герметичних контейнерах, можуть бути проведені в будь-яких контрольованих умовах або в умовах вологості навколишнього середовища.
5.4.2.2.7.3 Лікарські засоби, упаковані в напівпроникні контейнери
Для лікарських засобів, що містять воду, упакованих у напівпроникні контейнери, на додаток до досліджень фізичної, хімічної, біологічної та мікробіологічної стабільності необхідно проводити оцінку можливої втрати води. Таку оцінку слід проводити в описаних в таблиці 5.4.5 умовах із низькою відносною вологістю. Зрештою, необхідно довести, що лікарські засоби, які зберігаються в напівпроникних контейнерах, здатні витримувати умови низької відносної вологості. Можуть бути розроблені й інші порівнянні підходи для неводних лікарських засобів, що містять розчинники.
Таблиця 5.4.5
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(25±2) °С та (40±5)% або (30±2) °С та (35±5)%
|
6 (варіант а)
12 (віріант б)
|
|
Проміжні
|
(30±2) °С та (35±5)%
|
6
|
|
Прискорені
|
(40±2) °С та не більше 25%
|
6
|
Якщо в умовах прискорених випробувань протягом 6 місяців зберігання спостерігається «значна зміна», не пов’язана з втратою води, слід провести додаткові випробування в проміжних умовах зберігання (в умовах, описаних у п. 5.4.2.2.7.1 «Загальний випадок») із метою оцінки впливу температури 30 °С. Якщо в умовах прискорених випробувань «значна зміна» пов’язана винятково із втратою води, немає необхідності проводити випробування при проміжних умовах зберігання. Однак необхідно подати дані, які доводять, що в лікарському засобі не відбувається значної втрати води протягом пропонованого терміну придатності при його зберіганні при температурі 25 °С та відносній вологості 40%.
Для упакованого в напівпроникні контейнери лікарського засобу після зберігання протягом 3 місяців при температурі 40 °С та відносній вологості не більше 25% «значною зміною» вважається втрата води, що складає 5% від початкового вмісту. Однак при відповідному обґрунтуванні для невеликих контейнерів (місткістю 1 мл або менше) або для однодозових лікарських препаратів може бути прийнятною втрата води, що складає 5% або більше після зберігання протягом 3 місяців при температурі 40 °С та відносній вологості не більше 25%.
Альтернативою дослідженню при стандартній відносній вологості (в умовах, рекомендованих у таблиці 5.4.5) є проведення досліджень стабільності при більш високій відносній вологості та визначення за допомогою розрахунків втрати води при стандартній вологості. Для цього експериментальним шляхом визначають коефіцієнт проникності для системи контейнер/закупорювальнй елемент або, як показано в наведеному нижче прикладі, використовують розрахунковий коефіцієнт ступеня втрати води під час випробування при двох різних значеннях відносної вологості, але при одній і тій же температурі. Коефіцієнт проникності для системи контейнер/закупорювальний елемент може бути визначений експериментально з використанням умов «найгіршого випадку» для досліджуваного лікарського засобу (наприклад, у ряді лікарських засобів із різною концентрацією тієї самої діючої речовини для випробувань обирають лікарський препарат із найменшою концентрацією).
Приклад визначення втрати води
Для лікарського засобу з даною системою контейнер/закупорювальний елемент, із контейнером даного розміру та при даному обсязі наповнення, доцільне визначення втрати води при стандартній відносній вологості шляхом помноження ступеня втрати води (виміряного при альтернативній відносній вологості, але при такій самій температурі) на коефіцієнт ступеня втрати води, що зазначений у таблиці 5.4.6. При цьому необхідно довести лінійність ступеня втрати води при альтернативній відносній вологості протягом періоду зберігання.
Таблиця 5.4.6
|
Температура та стандартна відносна вологість
|
Загальні умови випробувань (температура та відносна вологість)
|
Коефіцієнт ступеня втрати води при даній температурі
|
|
25 °С та 25%
|
25 °С та 60%
|
1,9 = (100–25): (100–60)
|
|
25 °С та 40%
|
25 °С та 60%
|
1,5 = (100–40): (100–60)
|
|
40 °С та 25%
|
40 °С та 75%
|
3,0 = (100–25): (100–75)
|
Наприклад, при даній температурі 40 °С розрахунковим ступенем втрати води в процесі зберігання при відносній вологості не більше 25% є ступінь втрати води, виміряний при відносній вологості 75%, помножений на 3,0 — відповідний коефіцієнт ступеня втрати води.
Можна використовувати також обґрунтовані коефіцієнти ступеня втрати води в умовах вологості, що відрізняються від наведених у таблиці.
5.4.2.2.7.4 Лікарські засоби, призначені для зберігання в холодильнику
Таблиця 5.4.7
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
(5±3) °С
|
6 (варіант а)
12 (варіант б)
|
|
Прискорені
|
(25±2) °С та (60±5)%
|
6
|
Якщо лікарський засіб упакований у напівпроникний контейнер, необхідно надати відповідну інформацію з метою оцінки втрати води.
Дані, отримані для лікарського засобу при його зберіганні в холодильнику, слід оцінювати відповідно до вказівок, наведених у п. 5.4.2.2.9 «Оцінка даних», за винятком описаних нижче особливих випадків.
Якщо при прискорених випробуваннях у період з третього по шостий місяць зберігання спостерігається «значна зміна», пропонований термін зберігання має ґрунтуватися на даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань.
Якщо «значна зміна» спостерігається протягом перших 3 місяців при прискорених випробуваннях, слід подати матеріали про вплив короткочасних відхилень від умов зберігання, зазначених на етикетці, наприклад, при транспортуванні та обігу. Такі матеріали, якщо доцільно, можуть супроводжуватися даними наступних випробувань одиничної серії лікарського засобу протягом періоду менш ніж 3 місяці, але при більшій, ніж звичайно, частоті проведення випробувань. Якщо «значна зміна» спостерігається протягом перших 3 місяців зберігання, немає необхідності продовжувати прискорені випробування лікарського засобу протягом б місяців.
5.4.2.2.7.5 Лікарські засоби, призначені для зберігання в морозильній камері
Таблиця 5.4.8
|
Дослідження (випробування)
|
Умови зберігання (температура)
|
Мінімальний час вивчення стабільності на момент подання заявки (місяці)
|
|
Довгострокові
|
–(20±5) °С
|
6 (варіант а)
12 (варіант 6)
|
Для лікарських засобів, призначених для зберігання в морозильній камері, термін зберігання має ґрунтуватися на даних, отриманих у реальному часі при зберіганні в умовах довгострокових випробувань. Оскільки відсутні умови прискорених випробувань для лікарських засобів, призначених для зберігання в морозильній камері, слід провести випробування на одиничній серії при підвищеній температурі (наприклад, (5±3) °С або (25±2) °С) протягом підхожого періоду часу; це необхідно, щоб вивчити вплив короткочасних відхилень від умов зберігання, пропонованих для розміщення на етикетці.
5.4.2.2.7.6 Лікарські засоби, призначені для зберігання при температурі нижче –20 °С
Випробування лікарських засобів, призначених для зберігання при температурі нижче –20 °С, слід проводити, обґрунтовуючи в кожному окремому випадку.
5.4.2.2.8 Зобов’язання продовжувати випробування стабільності
Якщо наявні на момент подання заявки на реєстрацію дані довгострокових випробувань стабільності для вихідних серій не охоплюють пропонованого терміну зберігання, слід взяти зобов’язання продовжувати дослідження стабільності, щоб точно встановити термін зберігання.
Якщо реєстраційне досьє включає дані довгострокових випробувань стабільності щодо трьох промислових серій за період, що дорівнює пропонованому терміну зберігання, немає необхідності брати зобов’язання проводити подальші випробування стабільності. У протилежному випадку необхідно взяти одне з таких зобов’язань:
1) якщо в реєстраційному досьє є дані досліджень стабільності як мінімум для трьох промислових серій, необхідно взяти зобов’язання про продовження довгострокових досліджень протягом пропонованого терміну зберігання, а також про проведення прискорених досліджень протягом 6 місяців;
2) якщо в реєстраційному досьє є дані дослідження стабільності менше ніж для трьох промислових серій, слід взяти зобов’язання продовжувати довгострокові дослідження протягом пропонованого терміну зберігання та прискорені випробування протягом 6 місяців, а також зобов’язання провести довгострокові дослідження протягом пропонованого терміну зберігання та прискорені випробування протягом 6 місяців додаткової кількості промислових серій (щоб загальна кількість серій була не менше трьох);
3) якщо в реєстраційному досьє немає даних про стабільність промислових серій, слід взяти зобов’язання провести довгострокові дослідження стабільності перших трьох промислових серій протягом пропонованого терміну зберігання, а також прискорені дослідження протягом 6 місяців.
Протокол, що використовується для довгострокових досліджень стабільності відповідно до даного зобов’язання, має бути таким же, як і для вихідних серій, якщо науково не обґрунтований інший підхід.
Якщо для вихідних серій унаслідок «значної зміни» при прискорених дослідженнях стабільності знадобилося проведення випробувань при проміжних умовах зберігання, то випробування стабільності серій, досліджуваних відповідно до взятого зобов’язання, можуть бути проведені або в проміжних, або в прискорених умовах зберігання. Однак якщо при прискорених дослідженнях стабільності серій, що зазнають випробувань відповідно до взятого зобов’язання, спостерігається «значна зміна», слід також провести дослідження при проміжних умовах зберігання.
5.4.2.2.9 Оцінка даних
Необхідно застосовувати систематизований підхід до надання й оцінки інформації про стабільність; ця інформація повинна включати (залежно від ситуації) результати фізичних, хімічних, біологічних і мікробіологічних випробувань, у тому числі специфічних характеристик лікарської форми (наприклад, швидкості розчинення твердих лікарських форм для орального застосування).
Мета досліджень стабільності полягає в тому, щоб на підставі випробувань як мінімум двох або трьох серій лікарського засобу встановити термін зберігання та сформулювати розмішувані на етикетці вказівки зі зберігання, застосовні для всіх серій, що будуть у подальшому вироблені та упаковані при однакових умовах. Ступінь варіабельності окремих серій забезпечує впевненість у тому, що вироблені в подальшому промислові серії відповідатимуть специфікації протягом усього терміну зберігання.
Якщо дані таких випробувань свідчать про настільки незначний розклад та настільки низьку варіабельність, що вже при їх розгляді очевидно, що необхідний термін зберігання буде затверджено, то, як правило, немає необхідності в проведенні формального статистичного аналізу; достатньо надати повне обґрунтування відсутності такого аналізу.
Якщо очікується зниження кількісної характеристики в часі, то до аналізу даних прийнятний підхід, що полягає у визначенні часу, після закінчення якого усереднена крива розкладу (при довірчій імовірності 95%) перетинається з критерієм прийнятності, встановленим у специфікації. Якщо аналіз показує, що варіабельність від серії до серії невелика, корисно поєднувати дані для однієї загальної оцінки; це може бути зроблено шляхом відповідної статистичної обробки нахилів ліній регресій і точок їх перетинання з віссю ординат для окремих серій (наприклад, значення р для рівня значущості відбракування, що перевищує 0,25). Якщо недоцільно поєднувати дані для кількох серій, то єдиний термін зберігання залежатиме від мінімального часу, протягом якого передбачається, що характеристики серії залишатимуться в рамках критеріїв прийнятності.
Характер взаємозв’язку з розкладом визначатиме необхідність у перетворенні даних для аналізу лінійної регресії. Як правило, взаємозв’язок може бути представлений у вигляді лінійної, квадратичної або кубічної функції на арифметичній або логарифмічній шкалі. Щоб перевірити придатність даних, отриманих щодо кожної серії, та об’єднаних даних для серій (залежно від того, що доцільно) для побудови прямої чи кривої розкладу слід застосовувати статистичні методи.
За наявності відповідного обґрунтування може бути проведена обмежена екстраполяція отриманих у реальному часі даних за межі терміну спостереження для збільшення терміну зберігання на момент затвердження. Таке обґрунтування має враховувати відомі дані про механізм розкладу, результати прискорених випробувань, придатність математичної моделі, розмір серії, наявність підтверджувальних даних зі стабільності тощо. Однак при такій екстраполяції передбачається, що й далі (за рамками даних, що спостерігаються) матиме місце той самий взаємозв’язок із розкладом.
Примітка. Якщо отримані в реальному часі дані підтверджені результатами прискорених або проміжних випробувань стабільності, то може бути встановлений період до проведення повторних випробувань/термін зберігання, що перевищує тривалість досліджень у реальному часі. Як правило, шляхом екстраполяції допускається встановлювати період до проведення повторних випробувань/термін зберігання, що дорівнює подвійній тривалості досліджень у реальному часі. Однак не можна подовжувати період до проведення повторних випробувань/термін зберігання більш ніж на 12 місяців понад тривалість досліджень у реальному часі. При цьому слід враховувати зміни, що відбулися, варіабельність отриманих даних, пропоновані умови зберігання та обсяг проведеного статистичного аналізу. Різні ситуації з цього питання наведені на схемі рішень (див. рис. 5.4.1). Більш докладна інформація викладена у підрозділі 5.6 цієї настанови.
При будь-якій оцінці слід враховувати не лише кількісне визначення, але й рівні вмісту продуктів розкладу, а також інші підхожі характеристики. За необхідності слід приділити увагу перевірці балансу мас, а також різних характеристик стабільності та розкладу.
5.4.2.2.10 Вказівки/маркування
При зазначенні умов зберігання (температура, світло, вологість) слід керуватися додатком А.
Неприйнятне використання таких термінів, як «умови навколишнього середовища» або «кімнатна температура».
5.5 Плани випробувань стабільності діючих речовин і лікарських засобів: брекетинг і побудова матриць
5.5.1 Загальні відомості
У цьому підрозділі містяться рекомендації із застосування брекетингу та побудови матриць для досліджень стабільності, що проводяться відповідно до принципів, викладених у підрозділі 5.1.
У підрозділі 5.1 зазначено, що за наявності обґрунтування для випробувань стабільності нових діючих речовин і лікарських засобів можна застосовувати побудову матриць і брекетинг, але додаткових вказівок щодо цього питання не надано.
Цей підрозділ є настановою зі складання планів досліджень стабільності, що включають брекетинг і побудову матриць. У ньому визначені спеціальні принципи щодо випадків, коли можна застосовувати брекетинг або побудову матриць. Вибіркові приклади планів носять ілюстративний характер і не повинні розглядатися як єдино можливі чи найбільш підхожі для всіх випадків.
5.5.2 Керівні вказівки
5.5.2.1 Загальні принципи
План повних досліджень передбачає випробування зразків за кожним із передбачених показників у кожній точці контролю. План скорочених досліджень передбачає випробування зразків за кожним із передбачених показників не в кожній точці контролю. План скорочених досліджень може бути підхожою альтернативою плану повних досліджень, якщо використовується багатофакторне планування. Будь-який план скорочених досліджень має дозволяти адекватно прогнозувати період до проведення повторних випробувань або термін зберігання. Перед обґрунтуванням плану скорочених досліджень необхідно оцінити та підтвердити певні припущення. Слід врахувати потенційний ризик — установити менший період до проведення повторних випробувань або термін зберігання, ніж вони могли б бути встановлені при проведенні повних досліджень, внаслідок меншої кількості зібраних даних.
Під час випробувань за планом скорочених досліджень може розглядатися можливість переходу до плану повних досліджень або до плану менш скорочених досліджень, якщо наведене відповідне обґрунтування та дотримані принципи складання планів повних і скорочених досліджень. Однак при застосуванні статистичного аналізу слід унести належні корективи, щоб врахувати збільшення розміру вибірки в результаті такого переходу. Як тільки відбувається зміна плану досліджень стабільності, у точках контролю, що залишилися, необхідно проводити випробування за планом повних або менш скорочених досліджень.
5.5.2.2 Застосовність планів скорочених досліджень
Плани скорочених досліджень можуть застосовуватися при офіційних дослідженнях стабільності більшості типів лікарських засобів, хоча й для деяких складних систем доставки лікарського засобу з великою кількістю потенційних взаємодій «ліки — пристрій доставки» має бути подане додаткове обґрунтування. При дослідженнях діючих речовин побудова матриць має обмежену користь, а брекетинг взагалі незастосовний.
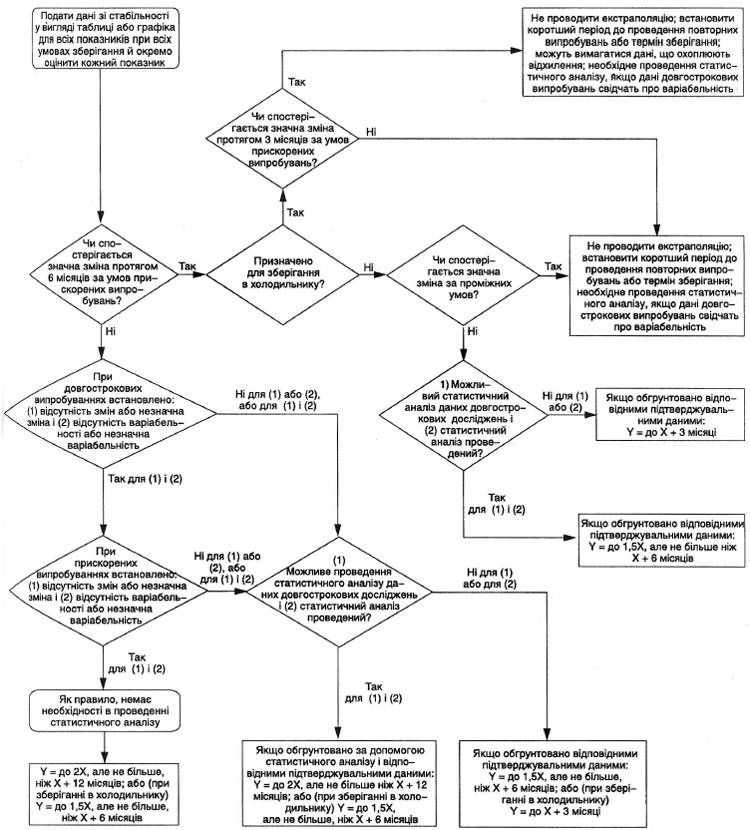
Рисунок 5.4.1 — Схема рішень при оцінці даних для встановлення періоду до повторних випробувань або терміну зберігання діючих речовин чи лікарських засобів (за винятком лікарських препаратів, призначених для зберігання в морозильній камері), де: Y — запропонований період до проведення повторних випробувань або термін зберігання; X — тривалість довгострокових випробувань, для яких є дані.
Використання брекетингу чи побудови матриць залежить від обставин, що докладно розглянуті нижче. Застосування будь-якого плану скорочених досліджень має бути обґрунтоване. У певних випадках достатнім обґрунтуванням є умови, описані в цій настанові, у той час як в інших випадках має бути наведене додаткове обґрунтування. Вид і рівень обґрунтування в кожному з цих випадків залежатимуть від наявних додаткових даних, що підтверджують стабільність. Якщо застосовується план, що включає побудову матриць, слід врахувати варіабельність даних і стабільність препарату, встановлену за допомогою таких додаткових даних.
Брекетинг і побудова матриць — це плани скорочених досліджень, засновані на різних принципах. Отже, застосуванню брекетингу чи побудови матриць мають передувати серйозний аналіз і наукове обґрунтування.
5.5.2.3 Брекетинг
Брекетинг (відповідно до визначення, наведеного в розділі 3) передбачає складання плану досліджень стабільності таким чином, що випробовують зразки лише з граничними значеннями певних показників у ряді (наприклад, сили дії, розміру упаковки і/або наповнення) в усіх тих точках контролю, що й при проведенні повних досліджень. План припускає, що стабільність зразків із проміжними значеннями показників у ряді ототожнюється зі стабільністю зразків із граничними значеннями.
Застосування плану, що передбачає брекетинг, вважається недоцільним, якщо не можна показати, що значення сили дії чи розміри упаковки і/або об’єми наповнення, обрані для випробувань, є дійсно граничними.
5.5.2.3.1 Критерії брекетингу
Критерії брекетингу — це змінні фактори (наприклад, сила дії, розмір паковання і/або об’єм наповнення), вплив яких на стабільність лікарського засобу має бути оцінений відповідно до плану досліджень.
5.5.2.3.1.1 Сила дії
Брекетинг може застосовуватися при дослідженнях лікарських засобів із різною силою дії, але які мають однаковий або дуже подібний склад. Прикладами можуть бути (1) капсули з різною силою дії, виготовлені шляхом заповнення оболонок однією й тією ж сумішшю порошків, але вміст яких має різну масу, (2) таблетки з різною силою дії, виготовлені шляхом пресування різних кількостей того самого гранулята, та (3) розчини для орального застосування з різною силою дії, склад яких дещо відрізняється лише використаними допоміжними речовинами (наприклад, барвниками, коригентами смаку) тощо.
За наявності обґрунтування брекетинг може застосовуватися при дослідженнях лікарських засобів із різною силою дії, у складі яких змінюється відносна кількість діючої речовини та допоміжних речовин. Таке обґрунтування може включати демонстрацію порівнянних профілів стабільності, отриманих для серій препарату з різними значеннями сили дії, що використовували для клінічних випробувань і при фармацевтичній розробці.
Як правило, брекетинг не слід застосовувати у разі використання різних допоміжних речовин в лікарських засобах із різною силою дії.
5.5.2.3.1.2 Розміри системи контейнер/закупорювальний елемент і/або об’єми наповнення
Брекетинг може застосовуватися при дослідженнях, у яких випробування проводяться для однієї і тієї ж системи контейнер/закупорювальний елемент, за умови: якщо один критерій (розмір контейнера або об’єм наповнення) варіює, то інший залишається незмінним. Однак якщо розглядається питання застосування брекетингу, коли й розмір контейнера й об’єм наповнення варіюють, то не можна стверджувати, що максимальний і мінімальний розміри контейнера є граничними значеннями критерію для всіх конфігурацій паковання. Необхідно подбати, щоб граничні значення критеріїв були обрані шляхом порівняння різних характеристик системи контейнер/закупорювальний елемент, що можуть вплинути на стабільність лікарського засобу. До таких характеристик залежно від ситуації належать: товщина стінок контейнера, геометрія закупорювального елемента, відношення площі поверхні до об’єму, відношення вільного простору до об’єму, проникність для водяних парів або кисню в розрахунку на одиницю лікарської форми чи на об’єм наповнення одиниці продукції.
За наявності обґрунтування брекетинг може застосовуватися при дослідженнях лікарських засобів з однаковим пакованням, але різними закупорювальними елементами. Обґрунтування може полягати в розгляді відносної проникності систем контейнер/закупорювальний елемент, що підлягають брекетингу.
5.5.2.3.2 Аналіз плану та потенційні ризики
Якщо після початку досліджень лікарський засіб з одним із граничних значень критерію більше не планується для розміщення на ринку, дослідження за планом продовжують, щоб підтвердити стабільність лікарських засобів із проміжними значеннями цього критерію. Слід взяти зобов’язання продовжувати дослідження стабільності щодо зареєстрованих лікарських засобів із граничними значеннями цього критерію.
До початку досліджень за планом, що передбачає брекетинг, слід оцінити, які наслідки це матиме для встановлення періоду до проведення повторних випробувань або терміну зберігання. Якщо буде встановлено, що стабільність лікарських засобів із граничними значеннями критерію різна, то лікарські засоби з проміжними значеннями критерію слід вважати не стабільнішими, ніж найменш стабільний лікарський засіб із граничним значенням критерію. Тобто, термін зберігання для лікарських засобів із проміжними значеннями критерію не має перевищувати термін зберігання найменш стабільного лікарського засобу з граничним значенням критерію.
5.5.2.3.3 Приклад плану
У таблиці 5.5.1 наведено приклад плану, що передбачає брекетинг. Він стосується лікарського засобу з трьома значеннями сили дії (50, 75 і 100 мг) і контейнерами трьох розмірів (15, 100 і 500 мл). У даному прикладі має бути показано, що контейнери з поліетилену високої щільності місткістю 15 мл і 500 мл дійсно представляють граничні значення критерію. Серії для кожної обраної комбінації повинні пройти випробування в кожній точці контролю, як передбачено в плані повних досліджень.
Таблиця 5.5.1 — Приклад плану, що передбачає брекетинг
|
Сила дії
|
50 мг
|
75 мг
|
100 мг
|
|
Серія
|
1
|
2
|
3
|
1
|
2
|
3
|
1
|
2
|
3
|
|
Розмір контейнера
|
15 мл
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
100 мл
|
|
500 мл
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Т — зразок, що підлягає випробуванням
|
5.5.2.4 Побудова матриць
Побудова матриць (відповідно до визначення, наведеного в розділі 3) — це розробка плану досліджень стабільності таким чином, що у певній точці контролю відбирають і випробовують лише частину загальної кількості можливих зразків для всіх комбінацій значень критеріїв. У наступній точці контролю слід випробовувати інший комплект зразків із загальної кількості для всіх комбінацій значень критеріїв. План припускає, що стабільність випробуваних зразків ототожнюється зі стабільністю всіх зразків у даний момент часу. Мають бути зазначені відмінності в зразках для того самого лікарського засобу, наприклад, відмінності, пов’язані з різними серіями, різною силою дії, різним розміром системи контейнер/закупорювальний елемент однакового типу і, можливо, у деяких випадках із різними системами контейнер/закупорювальний елемент.
Якщо стабільність лікарського засобу залежить від вторинного паковання, то можна будувати матриці паралельно для всіх систем паковання.
Кожна умова зберігання має оброблятися окремо за своїм власним планом, що передбачає побудову матриць. Побудову матриць не слід застосовувати для всіх випробуваних показників. Однак за наявності обґрунтування для різних випробуваних показників можуть застосовуватися альтернативні плани, що передбачають побудову матриць.
5.5.2.4.1 Фактори плану
Плани, що передбачають побудову матриць, можуть застосовуватися при дослідженнях лікарських засобів із різною силою дії, але однаковим або дуже подібним складом. Прикладами можуть бути (1) капсули з різною силою дії, виготовлені шляхом заповнення оболонок однією й тією ж сумішшю порошків, але вміст яких має різну масу, (2) таблетки з різною силою дії, виготовлені шляхом пресування різних кількостей того самого гранулята, та (3) розчини для орального застосування з різною силою дії, склад яких дещо відрізняється лише використаними допоміжними речовинами (наприклад, барвниками, коригентами смаку) та ін.
Іншими прикладами факторів плану, за якими можуть бути побудовані матриці, є різні серії лікарського засобу, виготовлені з використанням того самого процесу та обладнання, а також різні розміри контейнерів і/або різний об’єм наповнення для однієї і тієї ж системи контейнер/закупорювальний елемент.
За наявності обґрунтування плани, що передбачають побудову матриць, можуть застосовуватися, наприклад, при дослідженнях лікарських засобів із різною силою дії, у складі яких або змінюється відносна кількість діючої та допоміжної речовин або використовуються різні допоміжні речовини, а також лікарських засобів із різною силою дії, що упаковані з використанням різних систем контейнер/закупорювальний елемент, Як правило, обґрунтування має базуватися на додаткових даних, що підтверджують стабільність. Наприклад, якщо матриці будують відносно двох різних закупорювальних елементів або систем контейнер/закупорювальний елемент за допомогою додаткових даних, що підтверджують стабільність, необхідно навести значення відносного рівня переносу вологи чи довести однакову захисну дію від впливу світла. Як альтернатива можуть бути подані підтверджувальні дані, що свідчать про відсутність впливу на лікарський засіб кисню, вологи чи світла.
5.5.2.4.2 Аналіз плану
План, що передбачає побудову матриць, наскільки можливо має бути збалансований таким чином, щоб випробування зразків для будь-якої комбінації факторів були проведені в однаковому обсязі протягом усієї встановленої тривалості дослідження та у кінцевій часовій точці контролю перед поданням заявки. Однак внаслідок того, що проводити повний комплекс випробувань рекомендується лише у певних часових точках контролю (як описано нижче), може бути важко досягти повного збалансування в плані, що передбачає побудову матриць щодо часових точок контролю.
При використанні плану, що передбачає побудову матриць відносно часових точок контролю, слід передбачити, щоб у початковій і кінцевій точках контролю були випробувані зразки, що відповідають усім комбінаціям обраних факторів, а в проміжних точках контролю були випробувані зразки лише з певними наборами обраних комбінацій факторів. Якщо на момент подання реєстраційного досьє відсутні дані повних довгострокових випробувань протягом запропонованого терміну зберігання, то перед його поданням зразки з усіма комбінаціями факторів, таких як серія, сила дії, розмір контейнера та об’єм наповнення (поряд з іншими показниками), мають бути випробувані на момент закінчення терміну 12 місяців або на останній момент часу. Крім того, для зразків кожної обраної комбінації факторів мають бути представлені дані, отримані при випробуваннях як мінімум у трьох точках контролю протягом перших 12 місяців дослідження, включаючи контроль на початку випробувань. При побудові матриць для прискорених досліджень або досліджень при проміжних умовах зберігання необхідно гарантувати проведення випробувань для зразків усіх обраних комбінацій факторів як мінімум у трьох точках контролю, включаючи контроль на початку та при завершенні досліджень.
Якщо застосовується матриця факторного планування, і лікарський засіб з одним із значень сили дії або з одним із розмірів контейнера і/або об’ємів наповнення більше не планується для розміщення на ринку, то випробування стабільності щодо лікарського засобу з такою силою дії, розміром контейнера і/або об’ємом наповнення можуть бути продовжені. Отримані дані можуть використовуватися для підтвердження стабільності лікарського засобу з іншими значеннями сили дії або розмірами контейнера і/або іншим об’ємом наповнення, що передбачені в плані.
5.5.2.4.3 Приклади планів
Приклади матричного планування точок контролю для лікарського засобу з двома значеннями сили дії (S1 і S2) наведені в таблицях 5.5.2(а) і 5.5.2(б). Вирази «скорочення наполовину» і «скорочення на третину» означають стратегію скорочення випробувань відносно початкового плану повного дослідження. Наприклад, «скорочення наполовину» — це виключення однієї з кожних двох точок контролю, передбачених планом повного дослідження, а «скорочення на третину» — усунення однієї з кожних трьох точок контролю. У прикладах, наведених у таблицях 5.5.2(а) і 5.5.2(б), скорочення складає менше ніж половину й одну третину, оскільки включені повні дослідження всіх комбінацій критеріїв у деяких точках контролю, як описано в п. 5.5.2.4.2. Ці приклади включають повний комплекс випробувань у початковій і кінцевій точках контролю та на момент закінчення терміну 12 місяців від початку досліджень. Отже, максимальне скорочення становить менше половини (24/48) або однієї третини (16/48), тобто реально становить 15/48 або 10/48 відповідно.
Таблиця 5.5.2(а) — Приклад матричного планування точок контролю для лікарського засобу з двома значеннями сили дії
«Скорочення наполовину»
|
Точки контролю (місяці)
|
0
|
3
|
6
|
9
|
12
|
18
|
24
|
36
|
|
Сила дії
|
S1
|
Серія 1
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 3
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
S2
|
Серія 1
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 3
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Т — зразок, що підлягає випробуванням
|
Таблиця 5.5.2(б) — Приклад матричного планування точок контролю для лікарського засобу з двома значеннями сили дії
«Скорочення на третину»
|
Точки контролю (місяці)
|
0
|
3
|
6
|
9
|
12
|
18
|
24
|
36
|
|
Сила дії
|
S1
|
Серія 1
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 3
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
S2
|
Серія 1
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Серія 3
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Т —зразок, що підлягає випробуванням
|
У таблицях 5.5.3 (а) і 5.5.3(б) наведені додаткові приклади матричного планування для лікарського засобу з трьома значеннями сили дії та трьома розмірами контейнера. У таблиці 5.5.3 (а) наведене матричне планування лише точок контролю, а в таблиці 5.5.3(б) — матричне планування точок контролю та факторів. Згідно з таблицею 5.5.3(а) випробувань зазнають зразки, що відповідають усім комбінаціям факторів, включаючи серії, значення сили дії та розміри контейнера, у той час як згідно з таблицею 5.5.3(б) зразки з певними комбінаціями значень цих факторів випробуванню не підлягають.
Таблиця 5.5.3(а) — Приклад матричного планування точок контролю для лікарського засобу з трьома значеннями сили дії
|
Сила дії
|
S1
|
S2
|
S3
|
|
Розмір контейнера
|
А
|
B
|
C
|
А
|
B
|
C
|
А
|
B
|
С
|
|
Серія 1
|
Т1
|
Т2
|
ТЗ
|
Т2
|
Т3
|
Т1
|
ТЗ
|
Т1
|
Т2
|
|
Серія 2
|
Т2
|
ТЗ
|
Т1
|
ТЗ
|
Т1
|
Т2
|
Т1
|
Т2
|
ТЗ
|
|
Серія 3
|
ТЗ
|
Т1
|
Т2
|
Т1
|
Т2
|
ТЗ
|
Т2
|
ТЗ
|
Т1
|
|
Позначення:
S1, S2, S3 — різні значення сили дії; А, В, С — різні розміри контейнера; Т — зразок, що підлягає випробуванням;
Т1, Т2, ТЗ — точки контролю, де:
|
|
Точка контролю (місяці)
|
0
|
3
|
6
|
9
|
12
|
18
|
24
|
36
|
|
Т1
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Т2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
ТЗ
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
Таблиця 5.5.3(б) — Приклад матричного планування точок контролю та факторів для лікарського засобу з трьома значеннями сили дії
|
Сила дії
|
S1
|
S2
|
S3
|
|
Розмір контейнера
|
А
|
B
|
C
|
А
|
B
|
C
|
А
|
B
|
C
|
|
Серія 1
|
Т1
|
Т2
|
Т2
|
Т1
|
Т1
|
Т2
|
|
Серія 2
|
Т3
|
Т1
|
Т3
|
Т1
|
Т1
|
Т3
|
|
Серія 3
|
ТЗ
|
Т2
|
Т2
|
Т3
|
Т2
|
Т3
|
|
Позначення:
S1, S2, S3 — різні значення сили дії; А, В, С — різні розміри контейнера; Т — зразок, що підлягає випробуванням;
Т1, Т2, ТЗ — точки контролю, де:
|
|
Точка контролю (місяці)
|
0
|
3
|
6
|
9
|
12
|
18
|
24
|
36
|
|
Т1
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
Т2
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
|
ТЗ
|
Т
|
Т
|
Т
|
Т
|
Т
|
Т
|
5.5.2.4.4 Застосовність матричного планування та ступінь скорочення досліджень
Якщо складають план, що передбачає побудову матриць, то необхідно приділити увагу такому переліку питань (що не є вичерпним):
а) поінформованість про варіабельність даних;
б) очікувана стабільність лікарського засобу;
в) наявність додаткових даних, що підтверджують стабільність;
г) розбіжності стабільності лікарського засобу в рамках одного фактора та між різними факторами;
д) кількість комбінацій факторів при дослідженні.
Як правило, матричне планування застосовне, якщо додаткові дані свідчать про прогнозовану стабільність лікарського засобу. Матричне планування можливе, якщо додаткові дані, що підтверджують стабільність, свідчать про незначну варіабельність. Однак якщо додаткові дані підтверджують середню варіабельність, матричне планування має бути обґрунтоване статистично. Якщо додаткові дані свідчать про значну варіабельність, матричне планування незастосовне.
Статистичне обґрунтування має базуватися на оцінці запропонованого плану, що передбачає побудову матриць, або за здатністю знайти розбіжності ступеня розкладу лікарського засобу серед різних факторів, або за точністю при встановленні терміну зберігання.
Якщо вважають, що матричний план застосовний, слід визначити можливий ступінь скорочення плану повних досліджень залежно від кількості комбінацій факторів, що підлягають оцінці. Чим більше факторів, що стосуються лікарського засобу, і чим більше рівнів у кожному факторі, тим більший ступінь скорочення може бути обґрунтований. Однак будь-який скорочений план досліджень має дозволяти адекватно прогнозувати термін зберігання лікарського засобу.
5.5.2.4.5 Потенційний ризик
Внаслідок меншої кількості зібраних даних, дослідження за планом з побудовою матриць відносно факторів, а не точок контролю, як правило, характеризуються меншою точністю оцінки терміну зберігання та призводять до встановлення меншого терміну зберігання порівняно з тим, що був би встановлений відповідно до плану повних досліджень. Крім того, при встановленні терміну зберігання дослідження за планом із побудовою матриць можуть мати недостатню здатність виявляти певні головні ефекти або ефекти взаємодії, що призводить до неправильного об’єднання даних, отриманих для зразків, що відповідають різним факторам. При надмірному скороченні кількості випробуваних наборів факторів, а також якщо дані випробувань цих комбінацій факторів не можуть бути об’єднані для встановлення єдиного терміну зберігання, неможливо встановити терміни зберігання для лікарських засобів із такими комбінаціями факторів, випробування яких не проводилися.
Дослідження за планом, що передбачає побудову матриць лише щодо точок контролю, найчастіше мають таку ж здатність виявляти розбіжності ступеня змін для лікарських засобів із різними факторами, як і повні дослідження; з їх допомогою можна встановити такий же вірогідний термін зберігання, як і при повних дослідженнях. Ця властивість зумовлена передбачуваною лінійністю та проведенням випробувань зразків, що відповідають усім комбінаціям факторів як у початковій точці контролю, так і в кінцевій точці перед подачею заявки на реєстрацію.
5.5.2.5 Оцінка даних
Дані зі стабільності, отримані при дослідженнях за скороченим планом, слід обробляти таким же чином, як і дані повних досліджень.
5.6 Оцінка даних зі стабільності
5.6.1 Загальні відомості
У цьому підрозділі подані рекомендації з використання даних зі стабільності, отриманих відповідно до принципів, викладених у підрозділі 5.1, для того, щоб запропонувати період до проведення повторних випробувань або термін зберігання в реєстраційному досьє. У підрозділі 5.6 описано, коли і як можна застосовувати екстраполяцію, щоб запропонувати такий період до проведення повторних випробувань для діючої речовини або термін зберігання для лікарського засобу, що перевищує період, який охоплює «наявні дані, отримані в ході довгострокових досліджень стабільності в умовах довгострокового зберігання» (далі — «дані довгострокових випробувань»).
Керівні вказівки з оцінки та статистичного аналізу даних зі стабільності, що подані в підрозділі 5.1, є короткими за характером і обмеженими за сферою застосування. У підрозділі 5.1 зазначено, що підхожим підходом до аналізу кількісних даних зі стабільності для встановлення періоду до проведення повторних випробувань або оцінки терміну зберігання є регресійний аналіз, і рекомендується проводити статистичний аналіз для визначення можливості об’єднання даних для різних серій із використанням рівня значущості, що становить 0,25. Однак підрозділ 5.1 містить недостатньо докладну інформацію та не охоплює ситуацій, коли дослідження за повним або скороченим планом проводиться з включенням багатьох критеріїв.
Положення цього підрозділу доповнює вказівки, наведені в пп. 5.1.2.1.9 і 5.1.2.2.9.
Положення підрозділу 5.6 застосовні до оцінки даних зі стабільності, що мають бути подані в реєстраційному досьє для нових молекулярних сполук і відповідних лікарських засобів. У ньому містяться рекомендації зі встановлення періодів до проведення повторних випробувань і термінів зберігання для діючих речовин і лікарських засобів, призначених для зберігання при «кімнатній температурі» або при температурі нижче «кімнатної» (NB: термін «кімнатна температура» у даному випадку означає звичайні умови навколишнього середовища; його не слід застосовувати для зазначення умов зберігання в маркуванні). Положення цього підрозділу застосовні до досліджень стабільності, що проводять за планом з одним або багатьма критеріями, а також до досліджень, що проводять за повним або скороченим планом.
Для одержання рекомендацій із вибору та обґрунтування критеріїв прийнятності слід звертатися до Настанови 42-3.2:2004 та Настанови ІСН Q6B*, а для одержання рекомендацій із проведення досліджень за повним або скороченим планом — до підрозділу 5.5 цієї настанови.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в установленому порядку.
5.6.2 Керівні вказівки
5.6.2.1 Загальні принципи
Планування офіційних досліджень стабільності та їх виконання мають відповідати принципам, викладеним у підрозділі 5.1. Мета цих досліджень полягає в тому, щоб на підставі випробувань як мінімум трьох серій діючої речовини або лікарського засобу встановити період до проведення повторних випробувань або термін зберігання, застосовний для всіх серій, що будуть надалі вироблені й упаковані при однакових умовах, а також сформулювати інструкції щодо зберігання для розміщення на етикетці. Ступінь варіабельності окремих серій забезпечує впевненість у тому, що вироблені надалі промислові серії відповідатимуть специфікації протягом заданого періоду до проведення повторних випробувань або терміну зберігання.
Хоча слід очікувати звичайних змін у виробничому процесі й аналітичних методиках, важливо, щоб лікарський засіб був розроблений і вироблявся таким чином, щоб кількість діючої речовини на момент випуску серії становила 100% від зазначеної на етикетці. Якщо результати кількісного визначення, що використовуються для включення в реєстраційне досьє, на момент випуску серій перевищують 100% від зазначеного на етикетці значення, то запропонований термін зберігання (з огляду на зміни у виробничому процесі й аналітичних методиках) може бути перебільшений. З іншого боку, якщо результати кількісного визначення на момент випуску серій нижче 100% від зазначеного на етикетці значення, кількість діючої речовини може виявитися меншою нижньої межі прийнятності ще до закінчення пропонованого терміну зберігання.
Необхідно застосовувати систематизований підхід до подання й оцінки інформації про стабільність. Інформація про стабільність має включати (залежно від ситуації) результати фізичних, хімічних, біологічних і мікробіологічних випробувань, у тому числі специфічних характеристик лікарської форми (наприклад, швидкості розчинення твердих лікарських форм для орального застосування). Необхідно оцінити адекватність балансу маси. Слід розглянути фактори, що зумовлюють удавану відсутність балансу маси, у тому числі, наприклад, механізми розкладу, а також здатність аналітичних методик характеризувати стабільність і власну варіабельність аналітичних методик.
Основні концепції оцінки даних зі стабільності однакові для досліджень з одним і багатьма критеріями, а також для повних і скорочених досліджень. Дані, отримані при офіційних дослідженнях стабільності та, за необхідності, підтверджувальні дані, слід оцінити, щоб визначити критичні показники якості, які можуть впливати на якість і функціональні характеристики діючої речовини або лікарського засобу. Кожен показник необхідно оцінювати окремо; слід провести також загальну оцінку отриманих результатів для того, щоб запропонувати період до проведення повторних випробувань або термін зберігання. Пропонований період до проведення повторних випробувань або термін зберігання не повинні перевищувати прогнозованого для кожного окремо взятого показника.
У схемі рішень, наведеній на рисунку 5.6.1, визначено поетапний підхід до оцінки даних зі стабільності, а також указано, коли і якою мірою можна застосовувати екстраполяцію для встановлення пропонованого періоду до проведення повторних випробувань або терміну зберігання. У п. 5.6.3 наведені: (1) інформація про те, як слід аналізувати дані довгострокових випробувань щодо відповідних кількісних показників, отримані при дослідженні з багатьма критеріями за повним або скороченим планом, (2) інформація про те, як слід використовувати регресійний аналіз для оцінки періоду до проведення повторних випробувань або терміну зберігання, а також (3) приклади статистичних методик для визначення можливості об’єднання даних, отриманих для різних серій або інших критеріїв. Додаткові вказівки також наведені в літературі (див. додаток В [19–26]); однак ні наведені приклади, ні пропоновані джерела літератури не охоплюють усі застосовні статистичні підходи.
Як правило, можна припустити, що певні кількісні хімічні показники (наприклад, результати кількісного визначення, вміст продуктів розкладу, вміст консерванту) для діючої речовини або лікарського засобу при довгостроковому зберіганні мають нульову кінетику (див. додаток В, [19]). Таким чином, отримані для цих показників дані підлягають статистичному аналізу, описаному в п. 5.6.3, включаючи лінійну регресію та перевірку можливості об’єднання даних для різних серій. Хоча кінетика для інших кількісних показників (наприклад, рН, розчинність), як правило, невідома, може бути проведений такий самий статистичний аналіз, якщо це доцільно. Якісні характеристики та мікробіологічні властивості не підлягають цьому виду статистичного аналізу.
Наявність у цьому підрозділі рекомендацій щодо статистичних підходів не означає, що використання статистичного аналізу є кращим, якщо можна обґрунтувати, що він не є необхідним. Однак статистичний аналіз у певних випадках може бути корисний для підтвердження періодів до проведення повторних випробувань або термінів зберігання, отриманих за допомогою екстраполяції; він також може знадобитися для підтвердження пропонованих періодів до проведення повторних випробувань або термінів зберігання і в інших випадках.
5.6.2.2 Подання даних
Дані для всіх показників слід подавати у відповідній формі (наприклад, у вигляді таблиць, графіків, розповідного викладу); оцінка цих даних має бути включена в реєстраційне досьє. Необхідно навести всі значення кількісних показників у всіх точках контролю в тому вигляді, в якому вони були виміряні (наприклад, результат кількісного визначення у відсотках від значення, вказаного на етикетці). Якщо був проведений статистичний аналіз, необхідно зазначити й обґрунтувати використану методику та припущення, що лежать в основі прийнятої моделі. У реєстраційне досьє необхідно включити таблиці та/або графіки з результатами статистичного аналізу даних довгострокових випробувань.
5.6.2.3 Екстраполяція
Екстраполяція — це практика використання набору відомих даних для того, щоб зробити припущення стосовно даних, що будуть отримані в майбутньому. Щоб установити період до проведення повторних випробувань або термін зберігання, що перевищує тривалість довгострокових випробувань, у реєстраційному досьє може бути запропонована екстраполяція, особливо, якщо при прискорених випробуваннях не спостерігалося «значної зміни». Визначення доцільності екстраполяції даних зі стабільності, залежить від обсягу знань про характер зміни, придатності будь-якої математичної моделі та наявності відповідних підтверджувальних даних. Будь-яка екстраполяція має бути проведена таким чином, щоб розширений період до проведення повторних випробувань або термін зберігання був правомірним для випущеної в майбутньому серії за умови, що результати її випробувань будуть близькими до критеріїв прийнятності, встановлених на момент випуску.
При екстраполяції даних зі стабільності передбачається, що й далі за межами довгострокових випробувань (для яких наявні дані) зберігатиметься такий самий характер зміни. При застосуванні екстраполяції правильність припущення характеру зміни є критичною. При оцінці прямої чи кривої регресії для обробки даних довгострокових випробувань самі дані забезпечують перевірку правильності передбачуваного характеру зміни; щоб оцінити придатність даних для передбачуваної прямої чи кривої, можуть бути застосовані статистичні методи. Така внутрішня перевірка неможлива за межами проміжку часу, для якого наявні дані довгострокових випробувань. Таким чином, запропонований на підставі екстраполяції період до проведення повторних випробувань або термін зберігання завжди має бути підтверджений додатковими даними довгострокових випробувань стабільності в міру одержання таких даних. До протоколу дослідження стабільності серій, що випробовуються відповідно до прийнятого зобов’язання, слід включити часову контрольну точку на момент закінчення періоду до проведення повторних випробувань або терміну зберігання, встановленого за допомогою екстраполяції.
5.6.2.4 Оцінка даних при визначенні періоду до проведення повторних випробувань або терміну зберігання відповідно для діючих речовин або лікарських засобів, що підлягають зберіганню при кімнатній температурі
Систематизовану оцінку даних, отриманих у ході офіційних досліджень стабільності, слід проводити відповідно до вказівок, наведених у цьому підпункті. Дані зі стабільності слід оцінювати послідовно стосовно кожного показника. Для діючих речовин або лікарських засобів, призначених для зберігання при кімнатній температурі, проведення оцінки слід починати з будь-якої «значної зміни», що спостерігається в умовах прискорених випробувань і, за необхідності, в умовах проміжних випробувань. Потім розглядають тенденції і варіабельність даних довгострокових випробувань. Слід описати випадки, для яких може бути застосована екстраполяція для встановлення періоду до проведення повторних випробувань або терміну зберігання, що перевищує тривалість довгострокових випробувань, для яких наявні дані. На рисунку 5.6.1 наведена схема рішень для допомоги в цьому питанні.
5.6.2.4.1 Оцінка даних за відсутності «значної зміни» в умовах прискорених випробувань
Якщо в умовах прискорених випробувань не спостерігається «значної зміни», період до проведення повторних випробувань або термін зберігання залежить від характеру даних довгострокових і прискорених випробувань.
5.6.2.4.1.1 Оцінка даних довгострокових і прискорених випробувань, якщо вони свідчать про незначну зміну з часом або про відсутність зміни, а також про незначну варіабельність або її відсутність
Якщо дані довгострокових і прискорених випробувань стосовно певного показника свідчать про незначну зміну з часом або про її відсутність, а також про незначну варіабельність або її відсутність, то можна припустити, що цей показник для діючої речовини або лікарського засобу відповідатиме критеріям прийнятності протягом пропонованого періоду до проведення повторних випробувань або терміну зберігання. У таких випадках, як правило, немає необхідності в проведенні статистичного аналізу; однак слід надати обґрунтування відсутності такого аналізу. Це обґрунтування має включати обговорення характеру зміни чи її відсутності, значущість даних прискорених випробувань, баланс маси і/або інші підтверджувальні дані відповідно до вказівок підрозділу 5.1. При встановленні періоду до проведення повторних випробувань або терміну зберігання може бути запропонована екстраполяція за межі періоду, охоплюваного даними довгострокових випробувань. Пропонований період до проведення повторних випробувань або термін зберігання може перевищувати тривалість довгострокових випробувань (для яких наявні дані) не більше ніж удвічі; при цьому пропонований період до проведення повторних випробувань або термін зберігання не може перевищувати період, охоплюваний даними довгострокових випробувань, більше ніж на 12 місяців.
5.6.2.4.1.2 Оцінка даних довгострокових або прискорених випробувань, якщо вони свідчать про наявність зміни з часом і/або варіабельність
Якщо дані довгострокових або прискорених випробувань щодо певного показника свідчать про наявність зміни з часом і/або варіабельність в рамках одного критерію або для різних критеріїв, то для встановлення періоду до проведення повторних випробувань або терміну зберігання може бути корисним статистичний аналіз даних довгострокових випробувань. Якщо є розбіжності в стабільності для різних серій або зразків із різними значеннями критеріїв (наприклад, силою дії, розміром контейнера і/або об’ємом наповнення), або для зразків із різною комбінацією значень критеріїв (наприклад, сила дії — розмір контейнера і/або об’єм наповнення), що є перешкодою для об’єднання даних, пропонований період до проведення повторних випробувань або термін зберігання не повинен перевищувати найкоротшого періоду, підтвердженого даними для будь-якої серії, будь-якого значення критерію чи комбінації значень критеріїв. З іншого боку, якщо розбіжності пов’язані з конкретним критерієм (наприклад, із силою дії), то для лікарських засобів із різними значеннями в межах критерію (наприклад, для того самого лікарського засобу, що випускається з різною силою дії) можуть бути встановлені різні терміни зберігання. Необхідно надати всебічне обговорення причини таких розбіжностей і їх загальної значущості для лікарського засобу. Можна запропонувати екстраполяцію за межі періоду, охоплюваного даними довгострокових випробувань; однак ступінь екстраполяції залежатиме від того, чи можливий статистичний аналіз даних довгострокових досліджень для конкретного показника.
— Статистичний аналіз даних неможливий
Якщо статистичний аналіз даних довгострокових випробувань неможливий, але надані суттєві підтверджувальні дані, пропонований період до проведення повторних випробувань або термін зберігання може бути збільшений не більше ніж у 1,5 разу; при цьому пропонований період до проведення повторних випробувань або термін зберігання не повинен перевищувати більше ніж на 6 місяців періоду, охоплюваного даними довгострокових випробувань. Суттєві підтверджувальні дані — це дані достатніх довгострокових випробувань щодо серій на етапі розробки; тобто таких серій, що (1) мають дуже близький склад відносно складу вихідних серій для випробувань стабільності, (2) вироблені в меншому масштабі, ніж вихідні серії для випробувань стабільності або (3) упаковані за допомогою системи контейнер/закупорювальний елемент, що аналогічна пакованню вихідних серій для випробувань стабільності.
— Статистичний аналіз даних можливий
Якщо дані довгострокових випробувань піддаються статистичному аналізу, але аналіз не проведений, ступінь екстраполяції має бути таким самим, як у тому випадку, коли статистичний аналіз даних неможливий. Однак якщо статистичний аналіз проведений, може бути доцільно запропонувати період до проведення повторних випробувань або термін зберігання, що перевищує період, охоплюваний даними довгострокових випробувань, удвічі, але не більше ніж на 12 місяців; у такому випадку ця пропозиція має підтверджуватися результатами аналізу та відповідними даними.
5.6.2.4.2 Оцінка даних, якщо спостерігається «значна зміна» в умовах прискорених випробувань
Якщо в умовах прискорених випробувань спостерігається «значна зміна»*, період до проведення повторних випробувань або термін зберігання залежатимуть від результатів випробувань стабільності при проміжних умовах, а також при умовах довгострокових випробувань.
*В умовах прискорених випробувань можуть спостерігатися фізичні зміни, що не вважаються «значними змінами» та не потребують проведення випробувань при проміжних умовах, якщо немає іншої «значної зміни». Це такі зміни:
- розм’якшення супозиторію, що має плавитися при 37 °С, якщо температура плавлення безсумнівно доведена;
- невідповідність критеріям прийнятності при розчиненні 12 желатинових капсул або вкритих оболонкою таблеток, якщо таку невідповідність можна однозначно пояснити поперечною зшивкою. Однак якщо в умовах прискорених випробувань спостерігається фазовий поділ для м’якої лікарської форми, то випробування при проміжних умовах має бути проведене.
При встановленні факту відсутності «значної зміни» слід врахувати також можливі ефекти взаємодії.
5.6.2.4.2.1 Оцінка даних, якщо відсутня «значна зміна» при проміжних умовах випробувань
Якщо не спостерігається «значної зміни» при проміжних умовах випробувань, може бути запропонована екстраполяція за межі періоду, охоплюваного даними довгострокових випробувань; однак ступінь екстраполяції залежатиме від того, чи можливий статистичний аналіз даних довгострокових випробувань для оцінюваного показника.
— Статистичний аналіз даних неможливий
Якщо статистичний аналіз даних довгострокових випробувань неможливий, пропонований період до проведення повторних випробувань або термін зберігання може бути продовжений, але не більше ніж на три місяці понад період, охоплюваний даними довгострокових випробувань, за умови підтвердження відповідними даними.
— Статистичний аналіз даних можливий
Якщо дані довговтрокових випробувань для певного показника піддаються статистичному аналізу, але аналіз не проведений, ступінь екстраполяції має бути таким самим, як у тому випадку, коли статистичний аналіз даних неможливий. Однак, якщо статистичний аналіз проведений, може бути встановлений період до проведення повторних випробувань або термін зберігання, що перевищує період, охоплюваний даними довгострокових випробувань, у 1,5 разу, але не більше ніж на 6 місяців; це має підтверджуватися результатами статистичного аналізу та відповідними даними.
5.6.2.4.2.2 Оцінка даних, якщо спостерігається «значна зміна» при проміжних умовах випробувань
Якщо при проміжних умовах випробувань спостерігається «значна зміна», пропонований період до проведення повторних випробувань або термін зберігання не повинен перевищувати періоду, охоплюваного даними довгострокових випробувань. Крім того, може бути передбачений більш короткий період до проведення повторних випробувань або термін зберігання, ніж період, охоплюваний даними довгострокових випробувань.
5.6.2.5 Оцінка даних при визначенні періоду до проведення повторних випробувань або терміну зберігання для діючих речовин або лікарських засобів, що підлягають зберіганню при температурі нижче кімнатної
5.6.2.5.1 Діючі речовини або лікарські засоби, що підлягають зберіганню в холодильнику
Дані для діючих речовин або лікарських засобів, що підлягають зберіганню в холодильнику, слід оцінювати згідно з тими ж принципам, що описані в п. 5.6.2.4 для діючих речовин або лікарських засобів, що підлягають зберіганню при кімнатній температурі, за винятком тих випадків, коли є особливі вказівки, наведені нижче. Може бути використана схема рішень, наведена на рисунку 5.6.1.
5.6.2.5.1.1 Оцінка даних за відсутності «значної зміни» в умовах прискорених випробувань
Якщо в умовах прискорених випробувань не спостерігається «значної зміни», шляхом екстраполяції можна запропонувати збільшення періоду до проведення повторних випробувань або терміну зберігання за межі періоду, охоплюваного даними довгострокових випробувань, ґрунтуючись на викладених у п. 5.6.2.4.1 принципах, за винятком того, що ступінь екстраполяції має бути обмеженішим.
Якщо дані довгострокових і прискорених випробувань свідчать про незначну зміну з часом і про незначну варіабельність, пропонований період до проведення повторних випробувань або термін зберігання може перевищувати період, охоплюваний даними довгострокових випробувань, у 1,5 разу, але не більше ніж на 6 місяців, як правило, без підтвердження за допомогою статистичного аналізу.
Якщо дані довгострокових або прискорених випробувань для певного показника свідчать про наявність зміни з часом і/або варіабельності, пропонований період до проведення повторних випробувань або термін зберігання може бути продовжений; при цьому він не повинен перевищувати період, охоплюваний даними довгострокових випробувань, більше ніж на 3 місяці за наступних умов: (1) можливий статистичний аналіз даних довгострокових випробувань, але він не проведений або (2) дані довгострокових випробувань не піддаються статистичному аналізу, але надані відповідні підтверджувальні дані.
Якщо дані довгострокових або прискорених випробувань свідчать про наявність зміни з часом і/або варіабельності, пропонований період до проведення повторних випробувань або термін зберігання може перевищувати період, охоплюваний даними довгострокових випробувань, у 1,5 разу, але не більше ніж на 6 місяців за наступних умов: (1) можливий статистичний аналіз даних довгострокових випробувань і він проведений, і (2) пропозиція підтверджується результатами аналізу та відповідними даними.
5.6.2.5.1.2 Оцінка даних, якщо спостерігається «значна зміна» в умовах прискорених випробувань
Якщо в умовах прискорених випробувань у період з третього по шостий місяць спостерігається «значна зміна», пропонований період до проведення повторних випробувань або термін зберігання має ґрунтуватися на даних довгострокових випробувань. Екстраполяція вважається неприйнятною. Крім того, може бути передбачений коротший період до проведення повторних випробувань або термін зберігання, ніж період, охоплюваний даними довгострокових випробувань. Якщо дані довгострокових випробувань характеризуються варіабельністю, може бути доцільно підтвердити пропонований період до проведення повторних випробувань або термін зберігання за допомогою статистичного аналізу.
Якщо «значна зміна» спостерігається протягом перших 3 місяців випробувань в умовах прискорених випробувань, пропонований період до проведення повторних випробувань або термін зберігання має ґрунтуватися на даних довгострокових випробувань. Екстраполяція вважається неприйнятною. Може бути передбачений коротший період до проведення повторних випробувань або термін зберігання, ніж період, охоплюваний даними довгострокових випробувань. Якщо дані довгострокових випробувань характеризуються варіабельністю, може бути доцільно підтвердити пропонований період до проведення повторних випробувань або термін зберігання за допомогою статистичного аналізу. Крім того, слід обговорити вплив короткочасних відхилень від умов зберігання, зазначених на етикетці (наприклад, при транспортуванні чи обробці). Таке обговорення може бути підтверджене, за необхідності, проведенням додаткових випробувань на одній серії діючої речовини або лікарського засобу в умовах прискорених випробувань протягом періоду, меншого ніж 3 місяці.
5.6.2.5.2 Діючі речовини або лікарські засоби, що підлягають зберіганню в морозильній камері
Для діючих речовин або лікарських засобів, що підлягають зберіганню в морозильній камері, період до проведення повторних випробувань або термін зберігання має ґрунтуватися на даних довгострокових випробувань. Оскільки умови прискорених випробувань для таких діючих речовин або лікарських засобів відсутні, слід провести випробування на одній серії при підвищеній температурі (наприклад, (5±3) °С або (25±2) °С) протягом підхожого періоду для того, щоб вивчити вплив короткочасних відхилень від умов зберігання, запропонованих для зазначення на етикетці (наприклад, при транспортуванні чи обробці).
5.6.2.5.3 Діючі речовини або лікарські засоби, що підлягають зберіганню при температурі нижче –20 °С
Для діючих речовин або лікарських засобів, що підлягають зберіганню при температурі нижче –20 °С, період до проведення повторних випробувань або термін зберігання має бути оснований на даних довгострокових випробувань, а оцінка здійснена для конкретного випадку.
5.6.2.6 Загальні підходи до статистичної обробки даних
Якщо доцільно, при подачі первинної заявки на реєстрацію для аналізу даних зі стабільності в умовах довгострокових випробувань вихідних серій слід використовувати відповідний статистичний метод. Мета такого аналізу — встановити з високим ступенем вірогідності період до проведення повторних випробувань або термін зберігання, протягом якого кількісні показники залишатимуться в рамках критеріїв прийнятності для всіх серій, що будуть надалі вироблятися, пакуватися та зберігатися при тих самих умовах.
Якщо для оцінки даних довгострокових випробувань унаслідок зміни з часом і/або варіабельності був застосований статистичний аналіз, той же статистичний метод має бути використаний і для аналізу даних, отриманих для серій, що зазнають випробувань відповідно до зобов’язання продовжувати випробування стабільності, щоб підтвердити чи збільшити початково затверджений період до проведення повторних випробувань або термін зберігання.
Доцільним підходом до оцінки даних зі стабільності щодо кількісних показників і встановлення періоду до проведення повторних випробувань або терміну зберігання вважають регресійний аналіз. Характер взаємозв’язку між показником і часом визначатиме, чи слід виконувати перетворення даних для аналізу лінійної регресії. Взаємозв’язок може бути виражений лінійною або нелінійною функцією на арифметичній або логарифмічній шкалі. У деяких випадках нелінійна регресія може краще відобразити дійсний взаємозв’язок.
Прийнятний підхід до оцінки періоду до проведення повторних випробувань або терміну зберігання полягає в проведенні аналізу щодо кількісного показника (наприклад, кількісного визначення, вмісту продуктів розкладу) за допомогою визначення самого раннього моменту часу, при якому середнє значення (при довірчій імовірності 95%) перетинається з запропонованим критерієм прийнятності.
Якщо відомо, що значення оцінюваного показника зменшується в часі, то з критерієм прийнятності слід порівнювати нижнє граничне значення, обчислене з довірчою імовірністю 95%. Якщо відомо, що значення оцінюваного показника збільшується в часі, то з критерієм прийнятності слід порівнювати верхнє граничне значення, обчислене з довірчою імовірністю 95%. Якщо відомо, що значення оцінюваного показника може як збільшуватися, так і зменшуватися, або напрямок його зміни невідомий, слід обчислити з довірчою імовірністю 95% нижнє та верхнє граничні значення і порівняти їх з верхньою і нижньою межами критерію прийнятності.
Щоб забезпечити статистично обґрунтований висновок щодо оцінюваного періоду до проведення повторних випробувань або терміну зберігання, статистичний метод, що застосовується для аналізу даних, має враховувати план проведення досліджень стабільності. Описаний вище підхід може бути використаний для оцінки періоду до проведення повторних випробувань або терміну зберігання при випробуваннях на одній серії або на багатьох серіях, коли після відповідної статистичної перевірки дані об’єднали. Приклади статистичних підходів до аналізу даних зі стабільності для досліджень з одним або багатьма критеріями, що проводяться за повним або скороченим планом, наведені в п. 5.6.3. Посилання на відповідні джерела літератури наведені в додатку В (див. додаток В, [19–26]).
5.6.3 Приклади статистичних підходів до аналізу даних зі стабільності
Лінійна регресія, перевірка можливості об’єднання даних і статистичне моделювання, описані нижче, є прикладами статистичних методів і процедур. Їх можна використовувати при аналізі даних зі стабільності (для яких можлива статистична обробка) щодо кількісного показника, для якого пропонується критерій прийнятності.
5.6.3.1 Аналіз даних для однієї серії
Звичайно робиться припущення, що взаємозв’язок між певними кількісними показниками і часом є лінійним (див. додаток В, [19]). На рисунку 5.6.2 показана лінія регресії у випадку кількісного визначення лікарського засобу, верхній і нижній критерії прийнятності для якого становлять 105% і 95% від вказаного на етикетці значення, відповідно для даних довгострокових випробувань протягом 12 місяців і пропонованого терміну зберігання, що становить 24 місяці. У цьому прикладі застосовані верхнє та нижнє граничні значення, розраховані для середнього значення з довірчою імовірністю 95%, оскільки заздалегідь не було відомо, чи підвищиться результат кількісного визначення з часом чи знизиться (наприклад, у випадку лікарського препарату, що містить воду, у напівпроникному контейнері). Нижнє граничне значення перетинається з нижньою межею критерію прийнятності через 30 місяців, у той час як верхнє граничне значення не перетинається з верхньою межею критерію прийнятності і пізніше. Таким чином, пропонований термін зберігання (24 місяці) може бути підтверджений за допомогою статистичного аналізу даних кількісного визначення за умови виконання рекомендацій, викладених у пп. 5.6.2.4 і 5.6.2.5.
Якщо аналізуються дані для показника, що має лише верхню або нижню межу критерію прийнятності, рекомендується розраховувати з довірчою імовірністю 95% одне граничне значення для середнього результату. На рисунку 5.6.3 показана лінія регресії у випадку визначення вмісту продукту розкладу в лікарському засобі, коли наявні дані довгострокових випробувань протягом 12 місяців, запропонований термін зберігання 24 місяці та критерій прийнятності становить не більше 1,4%. Верхнє граничне значення для середнього результату, розраховане з довірчою імовірністю 95%, перетинається з критерієм прийнятності через 31 місяць. Отже, пропонований термін зберігання 24 місяці може бути підтверджений за допомогою статистичного аналізу даних щодо вмісту продукту розкладу за умови виконання рекомендацій, викладених у пп. 5.6.2.4 і 5.6.2.5.
Якщо використовується підхід, описаний вище, можна чекати, що середнє значення кількісного показника (наприклад, результат кількісного визначення, вміст продуктів розкладу) залишається в рамках критеріїв прийнятності до закінчення періоду до проведення повторних випробувань або терміну зберігання з довірчою імовірністю 95%.
Описаний вище підхід може бути використаний для оцінки періоду до проведення повторних випробувань або терміну зберігання для одиничної серії, окремих серій або багатьох серій у випадку об’єднання даних після відповідних статистичних перевірок, описаних у пп. 5.6.3.2–5.6.3.5.
5.6.3.2 Аналіз даних при повних однофакторних випробуваннях
Для діючої речовини або лікарського засобу з однією силою дії та при одному розмірі контейнера і/або об’ємі наповнення період до проведення повторних випробувань або термін зберігання, як правило, оцінюють на підставі даних зі стабільності як мінімум для трьох серій. При аналізі даних таких повних однофакторних досліджень, що проводяться окремо для кожної серії, можна розглянути два статистичних підходи.
Мета першого підходу — визначити, чи підтверджують дані, отримані окремо для всіх серій, пропонований період до проведення повторних випробувань або термін зберігання.
Мета другого підходу (перевірки однорідності вибірки) — визначити, чи можна дані для різних серій об’єднати для проведення загальної оцінки єдиного періоду до проведення повторних випробувань або терміну зберігання.
5.6.3.2.1 Оцінка того, чи підтверджують дані для всіх серій запропонований період до проведення повторних випробувань або термін зберігання
Мета цього підходу — оцінити, чи є встановлені періоди до проведення повторних випробувань або терміни зберігання для всіх серій тривалішими порівняно із запропонованими. Використовуючи процедуру, описану в п. 5.6.3.1, спочатку необхідно визначити періоди до проведення повторних випробувань або терміни зберігання для окремих серій за допомогою індивідуальних точок перетину, індивідуальних нахилів кривої та об’єднаної середньоквадратичної похибки, розрахованої для всіх серій. Якщо встановлений у такий спосіб для кожної серії період до проведення повторних випробувань або термін зберігання перевищує запропонований, то запропонований період до проведення повторних випробувань або термін зберігання, як правило, вважається прийнятним за умови виконання вказівок щодо екстраполяції, наведених у пп. 5.6.2.4 і 5.6.2.5. Як правило, немає необхідності перевіряти можливість об’єднання даних або встановлювати найбільш скорочену модель. Однак якщо один або кілька встановлених у такий спосіб періодів до проведення повторних випробувань або термінів зберігання менші запропонованого, може бути перевірена можливість об’єднання даних для різних серій із метою встановлення тривалішого періоду до проведення повторних випробувань або терміну зберігання.
З іншого боку, наведений вище підхід може бути застосований під час процесу об’єднання даних, описаного в п. 5.6.3.2.2. Якщо встановлено, що лінії регресії для серій мають загальний нахил, і всі встановлені періоди до проведення повторних випробувань або терміни зберігання, що ґрунтуються на загальному нахилі та індивідуальних точках перетину, є більш тривалими порівняно із запропонованим періодом до проведення повторних випробувань або терміном зберігання, то, як правило, немає необхідності продовжувати перевірку можливості об’єднання даних щодо точок перетину.
5.6.3.2.2 Перевірка можливості об’єднання даних для декількох серій (перевірка однорідності вибіркиN)
5.6.3.2.2.1 Коваріаційний аналіз
Перед об’єднанням даних для декількох серій з метою встановлення періоду до проведення повторних випробувань або терміну зберігання необхідно провести попередню статистичну перевірку, щоб визначити, чи мають лінії регресії для різних серій однаковий нахил і загальну точку перетину з віссю ординат. Якщо час вважається коваріантою, для перевірки розбіжностей у нахилах і точках перетину ліній регресії для різних серій може застосовуватись коваріаційний аналіз (ANCOVA). Кожну з цих перевірок слід проводити з використанням рівня значущості 0,25, щоб компенсувати очікувані низькі можливості плану досліджень внаслідок відносно обмеженого обсягу вибірки при звичайних офіційних дослідженнях стабільності.
Якщо при перевірці не підтверджується гіпотеза про рівність нахилів (тобто, якщо є значні розбіжності в нахилах для різних серій), вважається неприйнятним поєднання даних для всіх серій. Періоди до проведення повторних випробувань або терміни зберігання для окремих серій при дослідженнях стабільності можуть бути оцінені за допомогою підходу, описаного в п. 5.6.3.1, при використанні індивідуальних точок перетину, індивідуальних нахилів і об’єднаної середньоквадратичної похибки, розрахованої для всіх серій. Як період до проведення повторних випробувань або термін зберігання для всіх серій необхідно обрати найкоротший період із розрахованих для окремих серій.
Якщо при перевірці не підтверджується гіпотеза про однакові точки перетину, але підтверджується рівність нахилів (тобто, є значні розбіжності точок перетину, але відсутні значні розбіжності в нахилах для різних серій), дані можна об’єднати для розрахунку загального нахилу. Періоди до проведення повторних випробувань або терміни зберігання для окремих серій при дослідженні стабільності слід встановлювати за допомогою підходу, описаного в п. 5.6.3.1, використовуючи загальний нахил та індивідуальні точки перетину. Як період до проведення повторних випробувань або термін зберігання для всіх серій потрібно обрати найкоротший період із розрахованих для окремих серій.
Якщо перевірка при рівні значущості 0,25 підтверджує рівність нахилів і точок перетину (тобто, відсутні значні розбіжності в нахилі й точках перетину для різних серій), дані для всіх серій можна об’єднати. На підставі об’єднаних даних за допомогою підходу, описаного в п. 5.6.3.1, може бути розраховано єдиний період до проведення повторних випробувань або термін зберігання, що є застосовним для всіх серій. Установлений на підставі об’єднаних даних період до проведення повторних випробувань або термін зберігання, як правило, триваліший за період, встановлений на підставі даних для окремих серій, оскільки довірчий інтервал (інтервали) для середнього значення буде звужуватися в міру збільшення кількості даних при об’єднанні серій.
Описану вище перевірку можливості об’єднання даних для різних серій необхідно проводити в правильному порядку таким чином, щоб перевірка величин нахилів була проведена раніше, ніж перевірка значень точок перетину. Для встановлення періоду до проведення повторних випробувань або терміну зберігання може бути обрана найбільш скорочена модель (тобто, окремі нахили, загальний нахил з окремими точками перетину чи загальний нахил із загальною точкою перетину, залежно від ситуації).
5.6.3.2.2.2 Інші методи
При встановленні періоду до проведення повторних випробувань або терміну зберігання можуть використовуватися статистичні методи, що відрізняються від описаних вище (див. додаток В, [20–24]). Наприклад, якщо можна заздалегідь прийняти рішення щодо прийнятних розбіжностей у нахилі чи в середній величині періоду до проведення повторних випробувань або терміну зберігання для різних серій, то для визначення можливості об’єднання даних може бути використана відповідна методика оцінки еквівалентності нахилів або середньої величини періоду до проведення повторних випробувань або терміну зберігання. Однак таку методику необхідно заздалегідь установити, оцінити, обґрунтувати та, за необхідності, обговорити з компетентним уповноваженим органом. Щоб довести, що статистичні властивості обраної альтернативної методики є підхожими, може бути корисно провести дослідження шляхом моделювання (якщо це можливо) (див. додаток В, [24]).
5.6.3.3 Аналіз даних при повних багатофакторних дослідженнях
При повних багатофакторних дослідженнях стабільність лікарського засобу може деякою мірою відрізнятися для різних комбінацій факторів. При аналізі таких даних можуть бути використані два підходи.
Мета першого підходу — визначити, чи підтверджують дані для всіх комбінацій факторів пропонований термін зберігання.
Мета другого підходу (перевірки однорідності вибірки) — визначити, чи можна дані для різних комбінацій факторів об’єднати для проведення загальної оцінки єдиного терміну зберігання.
5.6.3.3.1 Оцінка того, чи підтверджений термін зберігання при всіх комбінаціях факторів
Мета цього підходу — оцінити, чи є встановлені терміни зберігання для всіх комбінацій факторів тривалішими, ніж пропонований термін зберігання. Статистична модель, що включає всі відповідні фактори та комбінації факторів, має бути побудована таким чином, як описано в п. 5.6.3.3.2.2.1, а термін зберігання слід розрахувати для кожного значення кожного фактора та кожної комбінації факторів.
Якщо всі терміни зберігання, встановлені за допомогою первинної моделі, перевищують запропонований термін зберігання, побудову наступної моделі не вважають необхідною, а запропонований термін зберігання, як правило, є підхожим за умови дотримання вказівок, наведених у пп. 5.6.3.2.4 і 5.6.3.2.5. Якщо один або кілька розрахованих термінів зберігання коротші запропонованого терміну зберігання, може бути проведена побудова моделі, як описано в п. 5.6.3.3.2.2.1. Однак не потрібно описувати остаточну модель перед оцінкою того, чи підтверджують дані запропонований термін зберігання. Терміни зберігання можуть бути розраховані на кожній стадії процесу побудови моделі, і, якщо всі терміни зберігання на будь-якій стадії триваліші, ніж запропонований, немає необхідності намагатися прийти до більш скороченої моделі.
Цей підхід може спростити аналіз даних складного багатофакторного дослідження стабільності порівняно з аналізом даних, описаним у п. 5.6.3.3.2.2.1.
5.6.3.3.2 Перевірка можливості об’єднання даних (перевірка однорідності вибіркиN)
Дані зі стабільності для різних комбінацій факторів об’єднувати не слід, якщо можливість такого об’єднання не підтверджена за допомогою статистичної перевірки.
5.6.3.3.2.1 Перевірка можливості об’єднання даних тільки для різних серій
Якщо кожну комбінацію факторів розглядати окремо, то дані зі стабільності можуть бути перевірені лише щодо можливості об’єднання даних для різних серій, а термін зберігання для кожної комбінації факторів може бути розрахований окремо з застосуванням процедури, описаної в п. 5.6.3.2. Наприклад, для лікарського засобу з двома дозуваннями, що може бути упакований у контейнери чотирьох розмірів, можуть бути проаналізовані вісім наборів даних, отриманих для 2×4 комбінацій «дозування — розмір паковання», і, відповідно, слід розрахувати вісім окремих термінів зберігання. Якщо бажано визначити єдиний термін зберігання, то терміном зберігання для лікарського засобу слід вважати найкоротший із термінів зберігання, розрахованих для всіх комбінацій факторів. Однак такий підхід не використовує перевагу, пов’язану з наявністю даних для всіх комбінацій факторів, і, як правило, призводить до встановлення коротших термінів зберігання, ніж підхід, описаний у п. 5.6.3.3.2.2.
5.6.3.3.2.2 Перевірка можливості об’єднання даних для всіх значень факторів і комбінацій факторів
Якщо перевіряють можливість об’єднання даних зі стабільності для всіх значень факторів і комбінацій факторів, і результати перевірки свідчать про можливість об’єднання даних, як правило, можна одержати єдиний термін зберігання, триваліший, ніж установлений на підставі даних для окремих комбінацій факторів. Термін зберігання триваліший, оскільки довірчий інтервал (інтервали) для середнього значення буде звужуватися в міру збільшення кількості даних при їх об’єднанні відносно різних серій, сил дії, розмірів контейнера і/або об’ємів наповнення тощо.
5.6.3.3.2.2.1 Коваріаційний аналіз
Для перевірки розбіжностей нахилу й точок перетину ліній регресії у випадку даних для різних значень критеріїв і комбінацій критеріїв може застосовуватись коваріаційний аналіз (див. додаток В, [25, 26]). Мета цієї процедури — визначити, чи можна об’єднувати дані, отримані для різних комбінацій критеріїв, для встановлення єдиного терміну зберігання.
Повна статистична модель має включати значення точок перетину та величину нахилів для всіх головних ефектів і ефектів взаємодії, а також значення, що відображають випадкову похибку виміру. Якщо може бути обґрунтовано, що взаємодії вищого порядку дуже малі, то, як правило, немає необхідності включати ці значення в модель. Якщо в початковій точці контролю лікарський засіб аналізували до його пакування в контейнер, то значення точки перетину для готового лікарського засобу в контейнері може бути виключене з повної моделі, оскільки результати цього аналізу є загальними для різних розмірів паковання і/або об’ємів наповнення.
Слід детально викласти результати перевірки можливості об’єднання даних для встановлення наявності статистично значущих розбіжностей для різних критеріїв і комбінацій критеріїв. Як правило, перевірку можливості об’єднання слід проводити в правильному порядку таким чином, щоб величини нахилів перевірити раніше, ніж точки перетину, а дані для ефектів взаємодії — раніше, ніж дані для головних ефектів. Наприклад, перевірку можна почати з нахилів і потім точок перетину для взаємодії вищого порядку, а продовжити перевіркою нахилів і потім точок перетину для простих головних ефектів. Для встановлення термінів зберігання може використовуватись найбільш скорочена модель, отримана за умови, що всі значення, які залишаються, є статистично значущими.
Усі перевірки слід проводити з використанням відповідних рівнів значущості. Рекомендується застосовувати рівень значущості 0,25 для даних, отриманих для різних серій, а рівень значущості 0,05 для даних, пов’язаних із різними значеннями критеріїв або комбінаціями критеріїв. Якщо така перевірка свідчить про можливість об’єднання даних для різних комбінацій критеріїв, термін зберігання може бути визначений відповідно до методики, описаної в п. 5.6.3.1, із використанням об’єднаних даних.
Якщо такі перевірки показали, що дані для певних значень критеріїв або комбінацій критеріїв об’єднувати не слід, може бути застосований один із двох підходів: (1) для зразків, що відповідають кожному значенню критерію, який залишився в моделі, або кожної комбінації критеріїв, може бути встановлений окремий термін зберігання; або (2) може бути встановлений єдиний термін зберігання на підставі найкоротшого з визначених для зразків, що відповідають усім значенням критерію, які залишилися в моделі, або всім комбінаціям критеріїв.
5.6.3.3.2.2.2 Інші методи
Можуть бути використані інші статистичні методи, що відрізняються від описаних вище (див. додаток В, [21–24]). Наприклад, для визначення можливості об’єднання даних може застосовуватися підхожа методика оцінки еквівалентності нахилів або середньої величини терміну зберігання. Однак таку методику необхідно заздалегідь установити, оцінити, належним чином обґрунтувати і, за необхідності, обговорити з компетентним уповноваженим органом. Щоб довести, що статистичні властивості обраної альтернативної методики є підхожими, може бути корисно провести дослідження шляхом моделювання (якщо це можливо) (див. додаток В, [24]).
5.6.3.4 Аналіз даних досліджень за планом, що передбачає брекетинг
Для аналізу даних зі стабільності, отриманих при дослідженні за планом, що передбачає брекетинг, можуть застосовуватися статистичні методи, описані в п. 5.6.3.3. Наприклад, для лікарського препарату, що має три сили дії (S1, S2 і S3) і три розміри паковання (Р1, Р2 і Р3) і досліджується згідно з планом, що передбачає брекетинг, коли випробовують лікарський засіб тільки в контейнерах із двома граничними розмірами (Р1 і РЗ), будуть отримані шість наборів даних для 3×2 комбінацій «сила дії — розмір паковання». Дані можуть бути проаналізовані окремо для кожної з шести комбінацій, щоб установити термін зберігання відповідно до вказівок п. 5.6.3.3.2.1, або може бути проведена перевірка можливості об’єднання даних перед установленням терміну зберігання згідно з п. 5.6.3.3.2.2.
План досліджень, що передбачає брекетинг, припускає, що стабільність лікарського засобу з проміжними значеннями сил дії або проміжними розмірами контейнера ототожнюється зі стабільністю зразків із граничними значеннями. Якщо статистичний аналіз показує, що стабільність лікарського засобу з граничними значеннями сил дії або граничними розмірами контейнера різна, лікарський засіб із проміжними значеннями сил дії або проміжними розмірами паковання слід вважати не стабільнішими, ніж найменш стабільний лікарський засіб із граничним значенням критерію. Наприклад, якщо встановлено, що лікарський засіб із розміром контейнера Р1 із наведеного вище плану брекетингу менш стабільний, ніж лікарський засіб із розміром контейнера Р3, то термін зберігання для лікарського засобу з розміром контейнера Р2 не повинен перевищувати термін зберігання для лікарського засобу з розміром контейнера Р1. Інтерполяцію даних для лікарських засобів із розмірами контейнерів Р1 і РЗ проводити не слід.
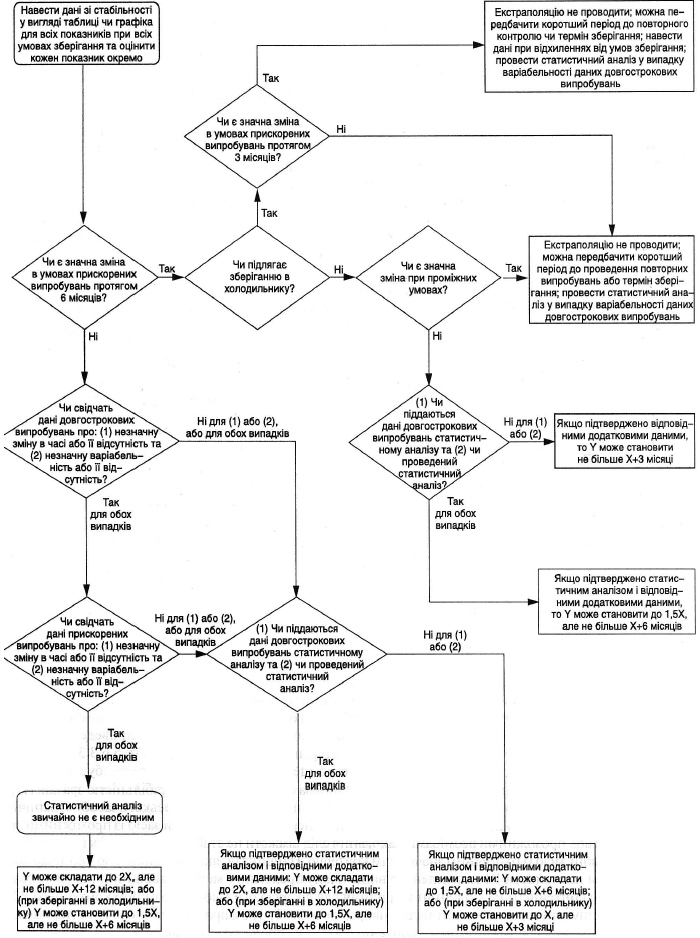
Рисунок 5.6.1 — Схема рішень для визначення періоду до проведення повторних випробувань або терміну зберігання для діючих речовин або лікарських засобів (за винятком лікарських препаратів, що підлягають заморожуванню), де: Y — запропонований період до проведення повторних випробувань або термін зберігання; X — тривалість довгострокових випробувань, для яких наявні дані.
5.6.3.5 Аналіз даних досліджень за допомогою матричного плану
Матричний план припускає, що в будь-якій визначеній точці контролю випробовують лише частину із загальної кількості зразків. Отже, важливо переконатися в тому, що всі зразки з усіма тими значеннями фактора чи з тими комбінаціями факторів, що можуть вплинути на оцінку терміну зберігання, були випробувані належним чином. Для достовірної інтерпретації результатів дослідження та встановлення терміну зберігання необхідно зробити й обґрунтувати деякі припущення. Наприклад, слід обґрунтувати припущення стосовно того, що стабільність випробуваних зразків характеризує стабільність усіх зразків. Крім того, якщо план досліджень не обміркований, деякі значення факторів або комбінації факторів можуть не піддаватися оцінці. Більше того, щоб можна було об’єднати дані для різних значень у комбінаціях факторів, може бути зроблене припущення, що взаємодії вищого порядку мізерно малі. Оскільки зазвичай неможливо статистично перевірити припущення стосовно того, що значення взаємодій вищого порядку мізерно малі, матричний план слід застосовувати лише в тому випадку, коли на підставі підтверджувальних даних справедливо припустити, що ці взаємодії дійсно мізерно малі.
Описана в п. 5.6.3.3 процедура статистичної обробки може застосовуватись до аналізу даних зі стабільності, отриманих при проведенні досліджень за матричним планом. При статистичному аналізі необхідно чітко зазначити використовувану методику та зроблені припущення. Наприклад, необхідно зазначити припущення, яке лежить в основі моделі стосовно того, що значення взаємодії мізерно малі. Якщо проводиться попередня перевірка з метою виключення з моделі взаємодій факторів, слід описати й обґрунтувати застосовувану процедуру. Слід зазначити остаточну модель, на якій ґрунтується оцінка терміну зберігання. Оцінку терміну зберігання слід проводити щодо кожного зі значень, що залишилися в моделі. Використання плану, що передбачає побудову матриць, може призвести до того, що встановлений термін зберігання буде коротший, ніж отриманий при проведенні повних досліджень.
Якщо один план досліджень передбачає і брекетинг, і побудову матриць, то може застосовуватися статистичний метод, описаний у п. 5.6.3.3.
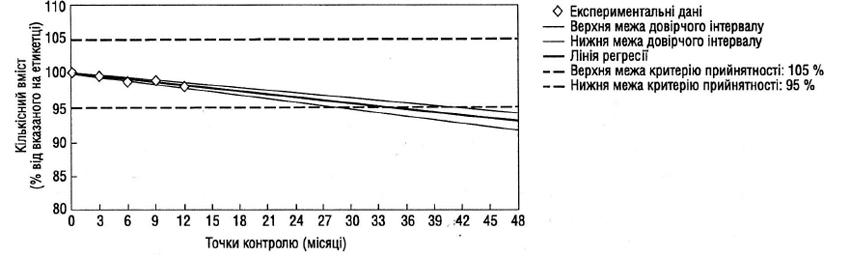
Рисунок 5.6.2 — Визначення терміну зберігання на підставі даних кількісного визначення з використанням верхньої та нижньої меж критерію прийнятності (довгострокові випробування стабільності при температурі 25 °С та відносній вологості 60%).
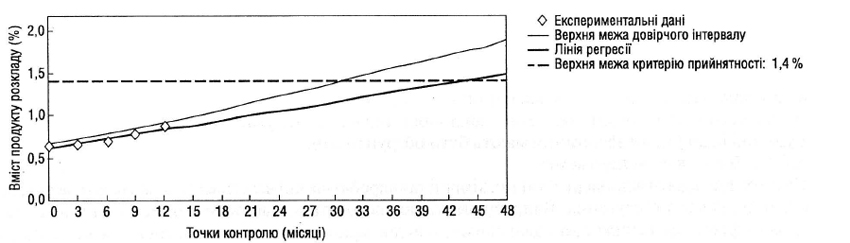
Рисунок 5.6.3 — Визначення терміну зберігання на підставі даних про вміст продукту розкладу з використанням верхньої межі критерію прийнятності (довгострокові випробування стабільності при температурі 25 °С та відносній вологості 60%).
5.7 Випробування стабільності лікарських засобів під час їх застосування
5.7.1 Загальні відомості
Мета випробувань стабільності лікарського засобу під час застосування — установити (за необхідності) період, протягом якого може застосовуватися лікарський препарат у багатодозовому контейнері після першого розкриття паковання зі збереженням якості, встановленої у специфікації.
Положення цього підрозділу застосовні до лікарських засобів у багатодозових контейнерах, що за своїм фізичним станом і хімічним складом після розкриття системи закупорювання внаслідок багаторазового відкривання та закривання можуть створювати ризик для вмісту, що знаходиться в них, через мікробіологічну контамінацію, проліферацію і/або фізико-хімічний розклад.
Зберігання якості лікарських засобів у багатодозових контейнерах після їх першого розкриття є важливим питанням. Даний принцип установлений у ДФУ та Європейській Фармакопеї. У підрозділі 5.7 сформульовані основні критерії вибору серій, плану випробувань, умов зберігання при проведенні випробувань, параметрів та методик випробувань тощо, беручи до уваги великий асортимент лікарських засобів.
Проте положення цього підрозділу слід розглядати разом із підрозділами 5.1 і 5.4, а також із Настановою 42-3.1:2004.
У реєстраційне досьє на лікарський засіб у багатодозовому контейнері слід включити або дані зі стабільності під час застосування, на яких ґрунтується визначення терміну зберігання під час застосування, або обґрунтування, чому не встановлюється термін зберігання під час застосування. Це обґрунтування може також ґрунтуватися на результатах експериментальних досліджень.
5.7.2 Керівні вказівки
5.7.2.1 Вибір серій
Випробування слід проводити як мінімум на двох серіях, щонайменше, дослідно-промислового масштабу. Термін зберігання, як мінімум, однієї з обраних серій має закінчуватися. Якщо результати таких досліджень відсутні, слід провести випробування однієї серії в кінцевій точці контролю при тих дослідженнях стабільності, результати яких подаються в реєстраційному досьє. Слід зазначити номер кожної серії, її розмір та дату виробництва. Тип контейнера та закупорювального елемента досліджуваного лікарського засобу, а також медичні пристрої (за їх наявності) мають бути ідентичні запропонованим для розміщуваного на ринку лікарського засобу.
Якщо лікарський засіб буде поставлятися в контейнерах декількох розмірів (більше одного) або з різною силою дії, то випробування стабільності під час застосування слід проводити щодо лікарського засобу, що найбільше зазнає змін. Слід завжди обґрунтовувати вибір лікарського засобу, що випробовується.
5.7.2.2 План випробувань
Наскільки можливо, випробування слід планувати таким чином, щоб моделювати застосування лікарського засобу на практиці, з огляду на об’єм наповнення контейнера та розведення/підготовку перед застосуванням. Через проміжки часу, порівнянні з проміжками, передбаченими для практичного застосування лікарського засобу, із контейнера необхідно видаляти відповідні кількості лікарського препарату, як правило, за допомогою методів, що викладені в інформаційних матеріалах (стислій характеристиці, листку-вкладиші або інструкції для медичного застосуванняN). Відбір проб слід проводити у звичайних умовах навколишнього середовища, при яких застосовується лікарський засіб.
Протягом запропонованого терміну зберігання під час застосування слід визначати відповідні фізичні, хімічні та мікробіологічні властивості лікарського засобу, що зазнають змін під час зберігання.
Якщо можливо, випробування слід проводити в проміжних точках контролю, а також наприкінці запропонованого терміну зберігання під час застосування, використовуючи кількість лікарського засобу, що залишилася в контейнері.
5.7.2.3 Умови зберігання під час випробувань
Лікарський засіб слід зберігати в умовах, рекомендованих у відповідних інформаційних матеріалах (стислій характеристиці, листку-вкладиші або інструкції для медичного застосуванняN) протягом усього періоду випробувань стабільності під час застосування.
Будь-які інші умови зберігання мають бути обґрунтовані.
5.7.2.4 Досліджувані параметри
Слід контролювати відповідні фізичні, хімічні та мікробіологічні властивості лікарського засобу, що зазнають змін під час застосування. Випробування, що проводяться, мають бути підхожими для конкретних лікарських форм; проте нижче наведені приклади видів параметрів, котрі може знадобитися досліджувати:
- фізичні: колір, прозорість, цілість закупорювальної системи, механічні включення, розмір часток;
- хімічні: кількісне визначення діючих речовин, антимікробних консервантів і антиоксидантів, визначення вмісту продуктів розкладу, рН;
- мікробіологічні: загальна кількість життєздатних мікроорганізмів, стерильність.
5.7.2.5 Аналітичні методики
Необхідно описати аналітичні методики, використовувані під час проведення дослідження; ці методики мають бути цілком валідовані. Слід застосовувати такі методи кількісного визначення, що дозволяють характеризувати стабільність.
5.7.2.6 Подання результатів
Результати слід узагальнити та подати у вигляді таблиць. Якщо це важливо, результати слід подати у вигляді графіків.
5.7.2.7 Оцінка
Необхідно навести зроблені висновки, що ґрунтуються на наданих даних. У випадку аномальних результатів їх наявність слід пояснити.
Щоб визначити необхідність зазначення терміну й додаткових умов зберігання під час застосування, необхідно враховувати дані досліджень стабільності в цей період.
5.7.2.8 Маркування первинного паковання
Термін зберігання під час застосування слід зазначати на етикетці. Крім того, слід залишати місце (якщо дозволяє розмір етикетки), де споживач може написати дату першого розкриття контейнера чи дату, до якої допускається використання лікарського засобу.
5.7.2.9 Стисла характеристика лікарського засобу, листок-вкладиш, інструкція з медичного застосуванняN та маркування вторинного паковання
Термін зберігання і рекомендації зі зберігання під час застосування (якщо вони необхідні) слід зазначати в стислій характеристиці лікарського засобу, листку-вкладиші, інструкції для медичного застосуванняN та в тексті маркування на вторинному (картонному) пакованні.
5.8 Максимальний термін зберігання стерильних лікарських засобів після першого розкриття паковання або після підготовки до застосування
5.8.1 Загальні відомості
Положення цього підрозділу застосовні до всіх стерильних лікарських засобів для людини, за винятком радіоактивних лікарських препаратів, екстемпоральних лікарських засобів, або модифікованих препаратів для негайного використання.
Оскільки важко прогнозувати всі можливі умови, при яких лікарський засіб будуть відкривати, розводити, готувати, зберігати тощо, споживач (наприклад, медичний персоналN) несе відповідальність за зберігання якості лікарського засобу, що вводиться пацієнтові. Щоб допомогти споживачеві зберегти якість лікарського засобу, заявник має провести необхідні дослідження та надати відповідну інформацію (наприклад, у стислій характеристиці лікарського засобу, інструкції для медичного застосуванняN, листку-вкладиші, на етикетці) з урахуванням прикладів, наведених нижче курсивом.
Заявникові слід враховувати також рекомендації ДФУ, Європейської Фармакопеї або іншої відповідної фармакопеї щодо часу та умов зберігання окремих категорій стерильних лікарських засобів після першого розкриття паковання.
У цьому підрозділі розглядається час між розкриттям паковання з лікарським засобом і введенням його пацієнтові; тривалість самого процесу введення до уваги не береться.
5.8.2 Стерильні лікарські засоби, що не містять консервантів
5.8.2.1 Загальні формулювання
Була доведена хімічна й фізична стабільність у процесі використання протягом х годин/днів при температурі у °С.
З мікробіологічної точки зору даний лікарський засіб слід застосовувати негайно, крім випадків, коли спосіб розкриття/підготування/розведення виключає можливість мікробної контамінації.
Якщо даний лікарський засіб не використовується негайно, то відповідальність за час і умови зберігання в процесі застосування покладається на споживача.
5.8.2.2 Спеціальний текст для інфузійних або ін’єкційних лікарських засобів
Була доведена хімічна й фізична стабільність у процесі застосування протягом х годин/днів при температурі у °С.
З мікробіологічної точки зору даний лікарський засіб слід застосовувати негайно. Якщо його не використовують негайно, то відповідальність за час і умови зберігання в процесі застосування покладається на споживача. Як правило, час зберігання не повинен перевищувати 24 год при температурі від 2 °С до 8 °С, крім випадків, коли підготування/розведення (тощо) проводиться в контрольованих асептичних умовах, що пройшли валідацію.
5.8.3 Водовмісні стерильні лікарські засоби, до складу яких входять антимікробні консерванти чи засоби, що самоконсервуються, а також безводні лікарські засоби (наприклад, масляні)
Була доведена хімічна й фізична стабільність у процесі застосування протягом х годин/днів при температурі у °С.
З погляду мікробіології після розкриття контейнера даний лікарський засіб може зберігатися протягом максимум z днів при температурі t °С. Відповідальність за зберігання під час застосування протягом іншого відрізка часу та при інших умовах покладається на споживача.
Заявник має обґрунтувати значення z і t для кожного окремого випадку; як правило, значення z не повинне перевищувати 28 днів.
5.9 Випробування стабільності при зміні типу II
У цьому підрозділі встановлені положення щодо проведення випробувань стабільності при змінах типу II у реєстраційному досьє після реєстрації лікарського засобу. Підрозділ 5.9 доповнює положення підрозділу 5.4.
Примітка. В ЄС зміни щодо первинної заявки діляться на зміни типу І і типу II. Зміни типу II — суттєві зміни, які включають зміни діючої речовини, сили дії, лікарської форми і шляхів введення; до них належать також інші зміни, специфічні для певних ветеринарних лікарських препаратів. При зміні типу II потрібна повна наукова оцінка. Зміни типу І (ІА і IB) — несуттєві зміни, при яких потрібно надавати повідомлення; вони можуть бути схвалені при дотриманні певних умов і наявності передбачених документів. Будь-яка зміна, яка класифікована як зміна типу І, але при внесенні якої не можуть бути дотримані встановлені умови, розглядається як зміна типу II (див. додаток В [6, 32, 33]).
У цьому підрозділі наведені приклади даних зі стабільності діючих речовин і/або лікарських засобів, необхідних при змінах типу II у реєстраційних досьє. Можна не дотримуватися наведених рекомендацій, якщо існують науково обґрунтовані причини використання альтернативних підходів.
У цьому підрозділі надані загальні вказівки щодо випробувань стабільності, однак на практиці допускається достатня гнучкість у разі особливих ситуацій, пов’язаних із різними науковими підходами чи характеристиками оцінюваної речовини.
Необхідні дослідження стабільності завжди слід продовжувати протягом усього затвердженого періоду до проведення повторних випробувань (для діючої речовини) або всього терміну зберігання (для лікарського засобу), а з появою будь-яких проблем щодо стабільності при зберіганні необхідно інформувати компетентні уповноважені органи.
Положення цього підрозділу застосовні до діючих речовин, отриманих шляхом хімічного синтезу, і відповідних лікарських засобів; вони не застосовні до радіоактивних лікарських засобів, біологічних лікарських засобів і лікарських препаратів, вироблених за допомогою біотехнології.
Мета цього підрозділу — вказати обсяг даних зі стабільності, які необхідно подати до компетентних уповноважених органів при певних видах змін типу II.
Зміни щодо діючих речовин і лікарських засобів представлені великим набором ситуацій.
У цьому підрозділі розглядається інформація, необхідна щодо діючих речовин і/або лікарських засобів при наступних змінах, що часто зустрічаються (якщо ці зміни не можуть бути віднесені до змін типу I)N:
- зміна в процесі виробництва діючої речовини;
- зміна в складі лікарського засобу;
- зміна первинного паковання лікарського засобу.
Обсяг і план випробувань стабільності при змінах і варіаціях має ґрунтуватися на знаннях і набутому досвіді щодо діючих речовин і лікарських засобів. Необхідно брати до уваги таку інформацію:
а) для діючих речовин:
1) дані про стабільність, включаючи результати стресових випробувань;
2) додаткові дані, що підтверджують стабільність;
3) первинні дані прискорених і довгострокових випробувань;
б) для лікарських засобів:
1) додаткові дані, що підтверджують стабільність;
2) первинні дані прискорених і довгострокових випробувань.
Заявник має дослідити, чи впливають зміни на характеристики якості діючих речовин і/або лікарських засобів і, отже, на їх стабільність.
Якщо потрібне подання даних зі стабільності, то обирати умови проведення випробувань слід відповідно до положень підрозділу 5.1.
5.9.1 Зміна в процесі виробництва діючої речовини
У випадку таких змін прийнятними можуть вважатися такі підходи:
а) Зміни, пов’язані з модифікаціями одного чи кількох етапів такого ж способу синтезу.
Якщо характеристики якості (наприклад, фізичні характеристики, профіль домішок) діючої речовини погіршуються, то потрібні дані порівняльних прискорених і довгострокових досліджень стабільності діючої речовини, отриманої у процесі синтезу до і після внесення зміни:
1) для діючої речовини, відомої як стабільна*: протягом 3 місяців щодо однієї серії, щонайменше, дослідно-промислового масштабу;
2) для діючої речовини, відомої як нестабільна: протягом 6 місяців щодо однієї серії, щонайменше, дослідно-промислового масштабу.
При змінах на заключній стадії процесу виробництва діючої речовини (наприклад, новий розчинник, умови кристалізації) можуть вимагатися додаткові дані зі стабільності готового лікарського засобу у випадку твердих лікарських форм, для яких фізичні характеристики діючої речовини можуть впливати на стабільність. Такі дослідження стабільності в умовах прискорених і довгострокових випробувань необхідно проводити протягом 3 місяців щодо двох дослідно-промислових серій.
б) Зміна, пов’язана зі зміною способу синтезу діючої речовини.
Потрібні дані порівняльних досліджень стабільності діючої речовини, отриманої за допомогою попереднього та нового способів синтезу, в умовах прискорених і довгострокових випробувань протягом 6 місяців щодо трьох серій, щонайменше, дослідно-промислового масштабу.
У випадку зміни в специфікації на діючу речовину, отриману за допомогою нового способу синтезу, а також якщо зміна може вплинути на стабільність лікарського засобу (наприклад, зміна фізичних характеристик, збільшення вмісту продуктів розкладу), можуть знадобитися додаткові дані дослідження стабільності відповідного(их) лікарського(их) засобу (засобів) в умовах прискорених і довгострокових випробувань протягом 3 місяців щодо двох серій, щонайменше, дослідно-промислового масштабу.
*Діюча речовина вважається стабільною, якщо вона відповідає специфікації, встановленій заявником, або офіційній (фармакопейній) специфікації при зберіганні протягом 2 років при температурі 25 °С та відносній вологості 60% і при зберіганні протягом 6 місяців при температурі 40 °С та відносній вологості 75%
5.9.2 Зміна в складі лікарського засобу
У випадку зміни в складі лікарського засобу, не віднесеної до зміни типу І, як прийнятні можуть розглядатися наведені нижче підходи.
Якщо діюча речовина відома як стабільна, то для традиційних лікарських форм (наприклад, тверді лікарські форми з нерегульованим вивільненням і розчини) потрібні дані порівняльних досліджень стабільності протягом 6 місяців в умовах довгострокових і прискорених випробувань щодо двох серій дослідно-промислового масштабу.
Якщо діюча речовина відома як нестабільна або лікарська форма є критичною (наприклад, форми з пролонгованим вивільненням), потрібні дані порівняльних досліджень стабільності протягом 6 місяців в умовах довгострокових і прискорених випробувань щодо трьох серій дослідно-промислового масштабу.
5.9.3 Зміна первинного паковання лікарського засобу
В усіх випадках зміни первинного паковання лікарського засобу, що не належить до зміни типу І, прийнятним є наведений нижче підхід.
Якщо паковання має менші захисні властивості чи існує ризик взаємодії (в основному для м’яких або рідких лікарських форм), потрібні дані порівняльних досліджень стабільності протягом 6 місяців в умовах довгострокових і прискорених випробувань щодо трьох серій лікарського засобу дослідно-промислового масштабу.
5.9.4 Наступні дослідження
При всіх описаних змінах (5.9.1–5.9.3.) перші три промислові серії, вироблені після затвердження зміни, мають пройти довгострокові випробування стабільності з використанням таких же протоколів випробування стабільності, що описані в попередніх підрозділах. Результати випробувань стабільності (за їх наявності) слід подати до компетентних уповноважених органів.
5.9.5 Додаткові вказівки
Якщо надані дані довгострокових (при температурі 25 °С та відносній вологості 60%) і прискорених (при температурі 40 °С та відносній вологості 75%) випробувань свідчать про відсутність несприятливого впливу зміни на стабільність лікарського засобу, як правило, може бути залишений початково встановлений термін зберігання; при цьому необхідно ґрунтуватися на порівнянні початково наданих і нових даних. Однак якщо дані свідчать про несприятливий вплив зміни на стабільність лікарського засобу, може бути встановлений новий термін зберігання. За таких обставин можливе застосування екстраполяції даних*.
*Якщо отримані в реальному часі дані підтверджені результатами прискорених випробувань стабільності, то може бути встановлений термін зберігання, що перевищує тривалість досліджень у реальному часі. Як правило, шляхом екстраполяції допускається встановлювати термін зберігання, що дорівнює подвійній тривалості досліджень у реальному часі. Однак максимальний термін зберігання, встановлений шляхом екстраполяції, не повинен перевищувати 3 років.
5.10 Дані зі стабільності для заявок на реєстрацію в державах, що розташовані в кліматичних зонах III і IV
5.10.1 Загальні відомості
У цьому підрозділі викладено підхід до більш широкого застосування положень підрозділу 5.1, а також стисло описано обсяг даних зі стабільності для нової діючої речовини або лікарського засобу. Цей обсяг даних вважається достатнім для включення в заявку на реєстрацію в державах, розташованих у кліматичних зонах III і IV.
У підрозділі 5.1 описано обсяг даних зі стабільності для реєстрації в державах, розташованих у кліматичних зонах І і II. Необхідно дотримуватися положень підрозділу 5.1 при одержанні даних зі стабільності для реєстрації лікарських засобів у різних країнах або регіонах, що знаходяться у зонах І і II, у тому числі в УкраїніN, державах ЄС, США, Японії. Для держав, розташованих у кліматичних зонах III і IV, обсяг даних, описаний у підрозділі 5.1, можна вважати прийнятним, за винятком даних для деяких умов зберігання. Підхід до класифікації країн відповідно до кліматичних зон І, II, III і IV наведений у літературі (див. додаток В, [29,30]) і в додатку Б до цієї настанови.
Всесвітня організація охорони здоров’я (ВООЗ) опублікувала настанову «Stability testing of pharmaceutical product containing well established drag substances in conventional dosage forms»*, що була актуалізована на 37-му засіданні Комітету експертів ВООЗ зі специфікацій на лікарські засоби (Женева, 22–26 жовтня 2001 p.). У настанові ВООЗ описані рекомендації з випробувань стабільності, включаючи умови зберігання для всіх чотирьох кліматичних зон.
Рекомендації з випробувань стабільності, наведені в даному підрозділі, ґрунтуються на положеннях підрозділу 5.1 і зазначеної настанови ВООЗ. Щоб гармонізувати умови зберігання при довгострокових випробуваннях для зон III і IV, у підрозділі 5.1 установлені наступні проміжні умови зберігання («Загальний випадок») для зон І і II: температура (30±2) °С та відносна вологість (65±5)%. Такі умови (температура (30±2) °С та відносна вологість (65±5)%) можуть бути також підхожою альтернативою при виборі умов довгострокових випробувань для зон І і II замість температури (25±2) °С та відносної вологості (60±5)%.
Цей підрозділ містить рекомендації щодо умов довгострокового зберігання при випробуванні стабільності нової діючої речовини або лікарського засобу для реєстрації в державах, що знаходяться в кліматичних зонах ІІІ і IV.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в у встановленому порядку.
5.10.2 Керівні вказівки
5.10.2.1 Зв’язок з іншими підрозділами цієї настанови
Положеннями цього підрозділу необхідно керуватися разом із підрозділами 5.1, 5.2, 5.3, 5.5 і 5.6, а також настановою ІСН Q5C*. Якщо в підрозділі 5.10 не описані конкретні альтернативи, необхідно дотримуватися рекомендацій підрозділу 5.1. У цьому підрозділі не наводяться повторно перераховані нижче частини підрозділу 5.1, які можна вважати загальними для реєстрації на будь-якій території світу: «Стресові випробування», «Вибір серій», «Система контейнер/закупорювальний елемент», «Специфікація», «Частота проведення випробувань», «Зобов’язання продовжувати випробування стабільності», «Оцінка даних», «Вказівки/маркування», а також умови зберігання для діючих речовин або лікарських засобів, призначених для зберігання в холодильнику та в морозильній камері.
*Рекомендується користуватися зазначеним документом. Він набуде чинності в Україні з моменту його прийняття в у встановленому порядку.
5.10.2.2 Умови зберігання
5.10.2.2.1 Загальний випадок
Рекомендовані умови зберігання для «загального випадку» при довгострокових і прискорених випробуваннях для кліматичних зон III і IV наведені у таблиці 5.10.1.
Таблиця 5.10.1
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подачі заявки (місяці)
|
|
Довгострокові
|
(30±2) °С та (65±5)%
|
12
|
|
Прискорені
|
(40±2) °С та (75±5)%
|
6
|
Для кліматичних зон III і IV не наведені рекомендації щодо проміжних умов зберігання. Таким чином, дані випробувань при проміжних умовах зберігання не важливі, якщо застосовуються принципи екстраполяції для встановлення періоду до проведення повторних випробувань або терміну зберігання, описані в підрозділі 5.6.
5.10.2.2.2 Лікарські засоби, упаковані в напівпроникні контейнери
Рекомендовані умови зберігання при довгострокових і прискорених випробуваннях для лікарських засобів, що містять воду, які знаходяться у напівпроникних контейнерах, для кліматичних зон III і IV наведені у таблиці 5.10.2.
Таблиця 5.10.2
|
Дослідження (випробування)
|
Умови зберігання (температура та відносна вологість)
|
Мінімальний час вивчення стабільності на момент подачі заявки (місяці)
|
|
Довгострокові
|
(30±2)°С та (35±5)%
|
12
|
|
Прискорені
|
(40±2) °С та (25±5)%
|
6
|
Як указано в підрозділі 5.1, прийнятний підхід для визначення ступеня втрати води при стандартній відносній вологості полягає в помноженні ступеня втрати води, визначеного при альтернативній відносній вологості при тій же температурі, на коефіцієнт ступеня втрати води (нижче в таблиці 5.10.3 наведено приклади). Коефіцієнти ступеня втрати води при даній температурі розраховують за загальною формулою:
(100 — стандартна відн. вологість, %)/(100 — альтернативна відн. вологість,%).
Таблиця 5.10.3
|
Альтернативна відносна вологість
|
Стандартна відносна вологість
|
Коефіцієнт ступеня втрати води
|
|
65%
|
35%
|
1,9
|
|
75%
|
25%
|
3,0
|
Можна використовувати також обґрунтовані коефіцієнти ступеня втрати води в умовах вологості, що відрізняються від умов, наведених у таблиці 5.10.3. При цьому необхідно довести лінійність ступеня втрати води при альтернативній відносній вологості протягом періоду зберігання.
5.10.2.2.3 Випробування при підвищеній температурі та/або екстремальних значеннях вологості
Особливі умови транспортування та особливі кліматичні умови, що виходять за рамки умов зберігання, які рекомендуються в підрозділі 5.10, необхідно підтвердити додатковими даними. Наприклад, такі дані можуть бути отримані при дослідженнях однієї серії лікарського засобу, проведених протягом 3 місяців при температурі 50 °С та вологості навколишнього середовища для того, щоб урахувати умови екстремально високої температури та екстремально низької вологості, а також при температурі 25 °С та відносній вологості 80% для того, щоб урахувати умови екстремально високої вологості.
Випробування стабільності в умовах високої вологості (наприклад, при температурі 25 °С та відносній вологості 80%) рекомендуються для твердих лікарських форм в пакованні, проникному для води й пари, наприклад, для таблеток у блістерах ПВХ/алюміній, призначених для розміщення на ринку на територіях з екстремально високою вологістю в зоні IV. Однак для твердих лікарських форм у первинному пакованні, що не пропускає водяної пари, наприклад, у блістерах алюміній/алюміній, випробування стабільності при зберіганні в умовах екстремально високої вологості вважаються необов’язковими.
5.10.2.3 Додаткові вказівки
Якщо не можна довести, що показники якості діючої речовини або лікарського засобу залишатимуться в межах критеріїв прийнятності при температурі (30±2) °С та відносній вологості (65±5)% протягом пропонованого періоду до проведення повторних випробувань або терміну зберігання, слід розглянути необхідність вживання наступних заходів: (1) скоротити період до проведення повторних випробувань або термін зберігання, (2) використовувати систему контейнер/закупорювальний елемент із сильнішими захисними властивостями або (3) навести додаткові застережливі вказівки в маркуванні.
Додаток А (обов’язковий). Декларування умов зберігання лікарських засобів
А. 1 Декларування умов зберігання готових лікарських засобів
А. 1.1 Загальні відомості
Відповідні умови зберігання, що узгоджуються із зазначеними в стислій характеристиці лікарського засобу, слід включати в інструкцію для медичного застосуванняN, листок-вкладиш і, за необхідності, зазначати на етикетці лікарського засобу. Умови зберігання лікарських засобів мають ґрунтуватися на оцінці досліджень стабільності, які проводяться щодо готового лікарського засобу. Умови зберігання діючих речовин мають базуватися на оцінці досліджень стабільності, які проводяться щодо діючої речовини. Докладні відомості про умови, що рекомендуються для таких досліджень стабільності, наведені в підрозділі 5.1. Для довгострокових досліджень обрані наступні умови: температура 25 °С та відносна вологість 60%; дані таких досліджень мають бути підтверджені результатами прискорених випробувань. Вибір умов для довгострокових досліджень ґрунтується на інформації про середню кінетичну температуру в різних кліматичних зонах. Середня кінетична температура включає щорічні зміни, тобто найнижчі та найвищі температури протягом зими й літа. Таким чином, зберігання при постійній температурі 25 °С під час довгострокових досліджень стабільності охоплює вплив фактичної температури, що відповідає умовам навколишнього середовища, включаючи відхилення від 25 °С.
Мета цього додатка — встановити уніфіковані формулювання для зазначення умов зберігання в маркуванні лікарських засобів і визначити випадки, коли їх слід застосовувати.
Цей додаток доповнює підрозділи 5.1–5.10 цієї настанови; його положення застосовні для всіх категорій лікарських засобів.
А. 1.2 Основні вказівки умов зберігання
Умови зберігання мають бути такими, щоб споживач міг їх дотримуватися; отже, необхідно обмежити вказівки умов зберігання такими, що досяжні на практиці. Підставою для вибору умов зберігання мають служити результати досліджень стабільності, подані на момент подачі реєстраційного досьє; таким чином, має існувати прямий зв’язок між зазначенням умов зберігання на етикетці та поданими показниками стабільності готового лікарського засобу. Неприйнятне використання таких термінів, як «кімнатна температура» або «умови навколишнього середовища».
Таблиця А.1
|
Умови проведення випробувань, при яких підтверджена стабільність (температура/відносна вологість)
|
Вказівка, що вимагається в маркуванні
|
Додаткова вказівка в маркуванні* (якщо необхідна)
|
Вказівка в короткій характеристиці, листку-вкладиші і інструкції для застосуванняN
|
|
25 °С/60% (довгострокові випробування) 40 °С/75% (прискорені випробування)
або 30 °С/65% (довгострокові випробування) 40 °С/75% (прискорені випробування)
|
Немає
|
«Не охолоджувати» або «Не заморожувати»
|
«Для лікарського препарату не потрібні спеціальні умови зберігання»
|
|
25 °С/60% (довгострокові випробування) 30 °С/60% або 65% (проміжні випробування) або 30 °С/65% (довгострокові випробування)
|
«Зберігати при температурі не вище 30 °С»
|
«Не охолоджувати» або «Не заморожувати»
|
«Зберігати при температурі не вище 30 °С»
|
|
25 °С/60% (довгострокові випробування)
|
«Зберігати при температурі не вище 25 °С»
|
«Не охолоджувати» або «Не заморожувати»
|
«Зберігати при температурі не вище 25 °С»
|
|
(5±3) °С (довгострокові випробування)
|
«Зберігати в холодильнику» або «Зберігати і транспортувати в холодильнику»**
|
«Не заморожувати»
|
«Зберігати в холодильнику (при температурі від +2 °С до +8 °С)» або «Зберігати і транспортувати в холодильнику (при температурі від +2 °С до+8 °С)»**
|
|
Нижче 0 °С
|
«Зберігати в морозильній камері» або «Зберігати і транспортувати в морозильній камері»***
|
«Зберігати в морозильній камері» (вказати діапазон температур) або «Зберігати і транспортувати в морозильній камері»*** (вказати діапазон температур)
|
|
*Залежно від лікарської форми і властивостей лікарського засобу може виникнути небезпека зниження якості внаслідок фізичних змін під дією низьких температур. Також в деяких випадках низькі температури можуть впливати на паковання. Щоб звернути увагу на таку можливість, може бути необхідною додаткова вказівка в маркуванні.
**При ухваленні рішення про необхідність, транспортування в холодильнику слід враховувати дані зі стабільності, отримані при довгострокових випробуваннях при температурі 25 °С і відносній вологості 60%. Таку вказівку слід використовувати у виняткових випадках.
***Таку вказівку слід використовувати, тільки якщо це необхідно.
|
У маркуванні лікарських засобів, розміщуваних на ринку України, необхідно застосовувати точні формулювання, наведені в таблиці А.1. Інші вказівки в маркуванні допускаються лише в тих випадках, якщо цього не можна уникнути, а також якщо документально підтверджено, що наведені вище основні умови зберігання є непідхожими. Альтернативна пропозиція має бути підтверджена відповідними даними; запропоновані умови зберігання мають бути досяжними на практиці.
А. 1.3 Інші спеціальні вказівки щодо зберігання
Лікарські засоби слід вміщувати в паковання, котре забезпечує стабільність і запобігає зниженню якості. Вказівку в маркуванні спеціальних умов зберігання не слід використовувати для компенсації неправильно обраного паковання чи паковання низької якості. Проте, щоб підкреслити необхідність спеціальних запобіжних заходів для споживача, можуть бути застосовані наступні вказівки, подані в таблиці А.2.
Таблиця А.2
|
Проблема при зберіганні
|
Додаткові вказівки в маркуванні* залежно від паковання
|
|
1
|
Чутливість до вологи
|
Зберігати в щільно закупореному контейнері***
|
|
2
|
Чутливість до вологи
|
Зберігати в оригінальному пакованні
|
|
3
|
Чутливість до світла**
|
Зберігати в оригінальному контейнері
|
|
4
|
Чутливість до світла**
|
Тримати контейнер*** в зовнішній пачці
|
|
*В інструкції для медичного застосуванняN, листку-вкладиші і на вторинному пакованні (якщо на ньому є місце) слід привести обґрунтування подібних вказівок в маркуванні (наприклад, «для захисту від дії світла»),
**Інформація про визначення чутливості до світла наведена в підрозділі 5.2.
***Слід використовувати назву конкретного контейнера (наприклад, флакон, блістер і т.п.).
|
У маркуванні лікарських засобів, розміщуваних на ринку України, необхідно застосовувати точні формулювання, наведені в таблиці А.2.
А.2 Декларування умов зберігання для діючих речовин
Для формулювання вказівок щодо умов зберігання діючих речовин слід керуватися наведеними вище принципами, розробленими відносно стандартних вказівок щодо умов зберігання готових лікарських засобів.
Для речовин, які слід зберігати/транспортувати в холодильнику або морозильній камері, в маркуванні необхідно зазначати діапазон температур.
Додаток Б (довідковий). Класифікація кліматичних зон
Таблиця Б.1 — Кліматичні зони та кліматичні умови*
|
Кліматичні умови
|
Зона І*
Помірний клімат
|
Зона II*
Середземноморський (субтропічний) клімат
|
Зона III
Жаркий сухий клімат або жаркий із помірною відносною вологістю
|
Зона IV
Дуже жаркий/вологий клімат
|
|
Середня річна температура
|
<20 °С
|
20,5–24,0 °С
|
>24 °С
|
>24 °С
|
|
Середня кінетична температура (фактична температура)
|
21 °С
|
26 °С
|
31 °С
|
31 °С
|
|
Середня річна відносна вологість
|
45%
|
60%
|
40%
|
70%
|
*Зони І і II — зони, в яких розташовані УкраїнаN, держави ЄС, США та Японія
Додаток В (довідковий). Бібліографія
1. Настанова 42-3.1:2004 Настанови з якості. Лікарські засоби. Фармацевтична розробка. — Київ, МОЗ України, 2004
2. Настанова 42-3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності. — Київ, МОЗ України, 2004
3. Настанова 42-3.4:2004 Настанови з якості. Лікарські засоби. Виробництво готових лікарських засобів. — Київ, МОЗ України, 2004
4. Фармацевтический сектор: Общий технический документ для лицензирования лекарственных средств в EC / Под ред. А.В. Стефанова (пред. редкол.) и др.; Авт.-сост.: В.А. Усенко, Н.А. Ляпунов,
Е.П. Безуглая и др. — К.: МОРИОН, 2002. — 256 с.
5. Надлежащая производственная практика лекарственных средств. Активнне фармацевтические ингредиенты. Готовые лекарственные средства. Руководства по качеству. Рекомендации PIC/S / Под
ред. Н.А. Ляпунова, В.А. Загория, В.П. Георгиевского, Е.П. Безуглой. — К.: МОРИОН, 2001. — 472 с.
6. The rules governing medicinal products in the European Union. — V. 2. — Notice to Applicants. — V. 2C. — Regulatory Guidelines. — Guideline on the Categorisation of New Applications (NA) versus Variations Applications (V). — Brussels: European Commission. — January 2002
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. 2. — Інформація для заявників. — Т. 2С. — Регулятивні настанови. — Настанова щодо віднесення заявок до категорії нових заявок (НЗ) або заявок на внесення змін (ЗМ). — Брюссель: Європейська Комісія. — Січень, 2002)
7. CPMP/ICH/2736/99 corr (Q1A R) Stability testing guidelines: stability testing of new drug substances and products. Note for guidance on stability testing: stability testing of new drug substances and products (revision), 2000
(CPMP/ICH/2736/99 corr (Q1A R) Керівні вказівки з випробувань стабільності: випробування стабільності нових лікарських речовин та препаратів (переглянуто), 2000)
8. СРМР/ІСН/279/95 (Q1В) Photostability testing of new active substances and medicinal products. Note for guidance on the photostability testing of new active substances and medicinal products, 1996
(CPMP/ICH/279/95 (Q1B) Керівні вказівки з випробувань світлостабільності нових діючих речовин та лікарських засобів, 1996)
9. СРМР/ІСН/280/95 (Q1C) Note for guidance on stability testing: requirements for new dosage form, 1996
(CPMP/ICH/280/95 (Q1C) Керівні вказівки з випробувань стабільності: вимоги щодо нових лікарських форм, 1996)
10. CPMP/QWP/122/02 Rev 1 Guideline on stability testing: stability testing of existing active substances and related finished products, 2003
(CPMP/QWP/122/02 Rev 1 Настанова з випробувань стабільності: випробування стабільності існуючих діючих речовин та відповідних готових лікарських засобів, 2003)
11. СРМР/ІСН/4104/00 (Q1D) Note for guidance on bracketing and matrixing for stability testing of drag substances and drug products, 2002
(CPMP/ICH/4104/00 (Q1D) Керівні вказівки щодо брекетингу та побудови матриць для випробувань стабільності лікарських речовин та лікарських засобів, 2002)
12. CPMP/QWP/2934/99 Note for guidance on in-use stability testing of human medicinal products, 2001
(CPMP/QWP/2934/99 Керівні вказівки з випробувань стабільності лікарських засобів для людини під час застосування, 2001)
13. CPMP/QWP/159/96 corr Note for guidance on maximum shelf-life for sterile products for human use after first opening or following reconstitution, 1998
(CPMP/QWP/159/96 corr Керівні вказівки щодо максимального терміну зберігання стерильних препаратів для людини після першого розкриття або після підготовки до застосування, 1998)
14. CPMP/QWP/576/96 Note for guidance on stability testing for a type II variation to a marketing authorization, 1998
(CPMP/QWP/576/96 Керівні вказівки з випробувань стабільності при змінах типу II у торговій ліцензії, 1998)
15. CPMP/QWP/609/96/Rev 2 Guideline on declaration of storage conditions: A: in the product information of medicinal products; B: for active substances. Annex to Note for guidance on stability testing of new drug substances and products. Annex to Guideline on stability testing of existing active substances and related finished products, 2003
(CPMP/QWP/609/96/Rev 2 Настанова з декларування умов зберігання: А: в інформаційних матеріалах для лікарських засобів; В: для діючих речовин. Додаток до керівних вказівок з випробувань стабільності нових лікарських речовин та препаратів. Додаток до настанови з випробувань стабільності існуючих діючих речовин та відповідних готових лікарських засобів, 2003)
16. СРМР/ІСН/420/02 (Q1E) Note for guidance on evaluation of stability data, 2003
(CPMP/ICH/420/02 (Q1E) Керівні вказівки щодо оцінки даних зі стабільності, 2003)
17. СРМР/ІСН/421/02 (Q1F) Note for guidance on stability data package for registration applications in climatic zones III and IV, 2003
(CPMP/ICH/421/02 (Q1F) Керівні вказівки щодо даних із стабільності для заявок на реєстрацію у кліматичних зонах III и IV, 2003)
18. Quinine Actinometry as a method for calibrating ultraviolet radiation intensity in light-stability testing of pharmaceuticals /Yoshioka S. et al. // Drug Development and Industrial Pharmacy. — 1994. — 20 (13). — P. 2049–2062.
19. Cartensen J.T. Stability and Dating of Solid Dosage Forms // Pharmaceuticals of Solids and Solid Dosage Forms. — 1977, Wiley-Interscience. — P. 182–185.
20. Ruberg S.J., Stegeman J.W. Pooling Data for Stability Studies: Testing the equality of Batch Degradation Slopes//Biometrics. — 1991. — 47. — 1059–1069.
21. Ruberg S.J., Hsu J.C. Multiple Comparison Procedures for Pooling Batches in Stability Studies Technometrics // 1992. — 34. — P. 465–472.
22. Shao J., Chow S.C. Statistical Inference in Stability Analysis//Biometrics. — 1994. — 50. — P. 753–763.
23. Murphy J.R., Weisman D. Using Random Slopes for Estimating Shelf-life // Proceedings of American Statistical Association of the Biopharmaceutical Section. — 1990. — P. 196–200.
24. Yoshioka S., Aso Y., Kojima S. Assesment of Shelf-life Equivalence of Pharmaceutical Products// Chem. Pharm. Bull. — 1997. — 45. — P. 1482–1484.
25. Chen J.J., Ahn H., Tsong Y. Shelf-life Estimation for Multifactor Stability Studies // Drug Inf. Journal. — 1997. — 31. — P. 573–587.
26. Fairweather W., Lin T.D., Kelly R. Regulatory, Design and Analysis Aspects of Complex Stability Studies // J. Phar. Sci. — 1995. — 84. — P. 1322–1326.
27. Schumacher P. Aktuella Fragen zur Haltbarkeit von Arzneimitteln [Current questions on drug stability] Pharmazeutische Zeitung. — 1974. — 119. — P. 321–324.
28. Grimm W. Storage conditions for Stability Testing — long term testing and stress tests // Drags made in Germany. — 1985 (Part I). — 28. — P. 196–202. — 1986 (Part II). — 29. — P. 39–47.
29. Dietz R., Feilner K., Gerst F., Grimm W. Drag Stability Testing — Classification of countries according to climatic zone // Drugs made in Germany. — 1993. — 36. — P. 99–103.
30. Grimm W. Extension of the International Conference on Harmonization Tripartite Guideline for Stability Testing of new Drug Substances and Products to Countries of Climatic Zones III and IV // Drag Developmentand Industrial Pharmacy. — 1998. — 24. — P. 313–325.
31. Haynes J.D. J. Pharm. Sci. — 1971. — 60. — P. 927–929.
32. The rules governing medicinal products in the European Union. — V. 2. — Notice to Applicants. — V. 2C. — Regulatory Guidelines. — Guideline on Dossier Requirements for Type IA and Type IB Notifications. — European Comission. — July 2003
(Правила, що регулюють лікарські засоби в Європейському Союзі. — Т. 2. — Інформація для заявників. — Т. 2С. — Регулятивні настанови. — Настанова щодо вимог до досьє при повідомленнях про зміни типу ІА та типу IB. — Європейська Комісія. — Липень, 2003)
33. Comission Regulation (EC) № 1085/2003 of 3 June 2003 concerning the examination of variations to the terms of a marketing authorization for medicinal products for human use and veterinary medicinal products falling within the scope of Council Regulation (EEC) № 2309/93 // Official Journal of the European Union. — L 159, 27.6.2003. — P. 24–45.
(Постанова Комісії (EC) № 1085/2003 від 3 червня 2003 p. щодо оцінки змін, які вносять в умови торгової ліцензії на лікарські засоби для застосування у людини та на ветеринарні лікарські засоби, що знаходяться у сфері дії Постанови Ради (EEC) № 2309/93 // Official Journal of the European Union. — L 159, 27.6.2003. — P. 24–45)
Ключові слова: випробування, діюча речовина, лікарський засіб, період до проведення повторних випробувань, реєстрація, специфікація, стабільність, термін зберігання, умови зберігання.