
СТАНДАРТ
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ПЛАСТИКОВІ МАТЕРІАЛИ ДЛЯ ПЕРВИННОЇ УПАКОВКИ ЛІКАРСЬКИХ ЗАСОБІВ
СТ-Н МОЗУ 42-3.16:2014
Видання офіційне
Київ
Міністерство охорони здоров’я України
2014
Передмова
1 РОЗРОБЛЕНО: ДП «Державний експертний центр Міністерства охорони здоров’я України»
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: О. Нагорна, канд. мед. наук; О. Баула, канд. хім. наук; І. Кудрявцева, д-р фарм. наук; Л. Дорошук
РЕКОМЕНДОВАНО ДО ПРИЙНЯТТЯ: Міністерство охорони здоров’я України
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 22.07.2014 р. № 515
3 Ця настанова відповідає документам:
CPMP/QWP/4359/03 — EMEA/СVМР/205/04 Guideline on Plastic Immediate Packaging Materials, London, 19 May 2005 (Настанова із пластикових матеріалів для первинної упаковки, Лондон, 19 травня 2005 року)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2014
© Державний експертний центр МОЗ України
Національний вступ
Загальний технічний документ (Common Technical Document — СTD) [1], прийнятий ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use — Міжнародна конференція по гармонізації технічних вимог до реєстрації лікарських засобів для застосування людям), вимагає представлення у складі реєстраційного досьє певної інформації щодо пластикових матеріалів, що використовують як первинну упаковку для активних фармацевтичних інгредієнтів (АФІ) та готових лікарських засобів.
Порядком проведення експертизи реєстраційних матеріалів на лікарські засоби [2] передбачено у структурі реєстраційного досьє надання даних про упаковку (первинну і вторинну) для АФІ та готових лікарських засобів. Оскільки в України при державній реєстрації лікарських засобів до зарубіжних та вітчизняних заявників висуваються однакові вимоги [2], у складі реєстраційного досьє має бути наявна інформація про упаковку, що відповідає тій інформації, яку необхідно надавати у реєстраційному досьє у форматі Загального технічного документа (ЗТД).
В Європейському Союзі (ЄС) введено спеціальну настанову CPMP/QWP/4359/03 — EMEA/СVМР/205/04 «Guideline on Plastic Immediate Packaging Materials» щодо інформації, яка має бути представлена у реєстраційному досьє (формат ЗТД), щодо пластикових матеріалів, які використовують як первинну упаковку для АФІ та готових лікарських засобів. Від рівня досліджень на етапі фармацевтичної розробки та вибору пластикових матеріалів для системи контейнер/закупорювальний засіб можуть залежати якість і безпека лікарського засобу.
Виходячи з вищевикладеного, зазначимо, що актуальною проблемою є введення в Україні настанови, яка містить рекомендації щодо планування та проведення досліджень для вибору первинної упаковки для АФІ та готових лікарських засобів, а також складання реєстраційних досьє та їх експертизи при реєстрації. Такі рекомендації мають бути гармонізовані з положеннями відповідної настанови ЄС.
Ця настанова розроблена на підставі настанови з якості, прийнятої в ЄС:
- CPMP/QWP/4359/03 — EMEA/СVМР/205/04 Guideline on Plastic Immediate Packaging Materials, London, 19 May 2005 (CPMP/QWP/4359/03 — EMEA/СVМР/205/04 Настанови із пластикових матеріалів для первинної упаковки, Лондон, 19 травня 2005 року).
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, що відповідають чинному законодавству.
До цієї настанови внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни долучено безпосередньо у пункти, до яких вони відносяться; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
- назву цієї настанови наведено відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [3], а позначення — відповідно до вимог стандарту СТ-Н МОЗУ 42-1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [4].
- додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Терміни та визначення понять», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені відповідно до вимог ДСТУ 1.5-2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [3] та ДСТУ 1.7-2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [5]. «Зміст» цієї настанови викладено з урахуванням додаткових структурних елементів;
- основні положення викладені у розділі «Рекомендації щодо пластикових матеріалів для первинної упаковки» та у трьох додатках; при цьому кожний структурний елемент та його номер у цій настанові відповідають таким у настанові CPMP/QWP/4359/03 — EMEA/СVМР/205/04, за винятком номера «Glossary»;
- розділ «Терміни і визначення понять» складено на підставі розділу 7 настанови CPMP/QWP/4359/03 — EMEA/СVМР/205/04 «Glossary». Цей розділ не позначено номером та викладено після розділу «Нормативні посилання». Усі терміни у розділі «Терміни та визначення понять» наведено в алфавітному порядку, вони супроводжуються посиланням на нормативні документи, бібліографічний опис яких наведено у національному додатку «Бібліографія»;
- у розділі «Нормативні посилання» додатково наведений бібліографічний опис нормативних документів, зазначених у цій настанові;
- у національному додатку «Бібліографія» додатково наведений бібліографічний опис нормативних документів, посилання на які наведені у цій настанові;
- у розділі «Познаки та скорочення» додатково наведені позначення скорочень, що використовують у цій настанові;
- у цій настанові слова «заявка» («application») та «заявка на отримання торгової ліцензії» («marketing authorization application») замінені на «реєстраційне досьє»;
- по всьому тексту внесені редакційні зміни у посилання на структурні елементи цієї настанови, наприклад замість «(see section 4)» вказано «(див. розділ 4 цієї настанови)»;
- у п. 1.1 замість тексту «Ця настанова заміняє Настанову із пластикових матеріалів для первинної упаковки, опубліковану в документі Rules Governing Medicinal Products (3AQ10a)», вказано «Ця настанова гармонізована із настановою CPMP/QWP/4359/03 — EMEA/СVМР/205/04, яка замінила настанову 3AQ10a, опубліковану у томі 3А Правил, що регулюють лікарські засоби в Європейському Союзі»;
- у п. 1.1 додатково уточнено, що ця настанова стосується лікарських засобів для застосування у людейN;
- у п. 1.1 виключено посилання на відповідні пункти та їх назви у реєстраційному досьє європейського формату 1998 р., оскільки для реєстрації лікарських засобів в Україні передбачено надання досьє лише у форматі ЗТД. Пункти досьє, що відносяться до препаратів для застосування у ветеринарії виключені, оскільки ця настанова стосується лікарських засобів для застосування у людей;
- у п. 1.3 вказані структурні елементи досьє у двох форматах: ЗТД та ЄС-ЗТД, посилання на європейський формат попередньої версії 1998 р. виключено. Пункти досьє, що відносяться до препаратів для застосування у ветеринарії, виключені, оскільки ця настанова стосується лікарських засобів для застосування у людей;
- у п. 1.3 замість згадування у тексті настанови CPMP/QWP/155/96, що втратила чинність, указана діюча в ЄС настанова з фармацевтичної розробки EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8). Разом із настановами ЄС/ICH зазначені також гармонізовані з ними настанови МОЗ України. Виключено посилання на настанову CPMP/ICH/380/95, що втратила чинність, замість якої введено настанову CPMP/ICH/2736/99;
- замість слів «Європейська фармакопея або фармакопея держави ЄС» вказано «Європейська фармакопея або інша відповідна фармакопея, або Державна фармакопея УкраїниN». Під словами «інша відповідна фармакопея» мається на увазі фармакопея держави ЄС або фармакопея іншої країни, гармонізована з Європейською фармакопеєю, або Фармакопея США;
- замість назви додатку 1 «Дерево рішень для представлення документації» вказано «Схема рішень для представлення документації щодо пластикових пакувальних матеріалів для АФІ»;
- замість назви додатку 2 «Дерево рішень для представлення документації щодо пластикових пакувальних матеріалів» вказано «Схема рішень для представлення документації щодо пластикових пакувальних матеріалів для готових лікарських засобів»;
- замість назви додатку 3 «Кореляційна таблиця для представлення інформації» вказано «Кореляційна таблиця для представлення інформації щодо пластикових пакувальних матеріалів для АФІ і готових лікарських засобів у реєстраційних досьє різних форматів»;
- у таблиці, наведеній у додатку 3, додана колонка для формату досьє ЄС-ЗТД за версією 2008 р. та виключена колонка, що стосується реєстраційного досьє європейського формату 1998 р. У зв’язку з цим в таблиці представлені номери відповідних частин реєстраційних досьє у форматах ЗТД та ЄС-ЗТД. Інформація щодо реєстраційного досьє для ветеринарних препаратів виключена, оскільки ця настанова стосується лікарських засобів для застосування у людей.
Настанова має такі технічні відхилення:
- оскільки ця настанова встановлює положення щодо пластикових пакувальних матеріалів для АФІ і готових лікарських засобів для застосування у людей, з настанови виключені всі положення щодо препаратів для застосування у ветеринарії, а також посилання на відповідні директиви та настанови ЄС.
Ця настанова придатна для організації планування та проведення досліджень із вибору первинної упаковки з пластикових матеріалів для АФІ та готових лікарських засобів для застосування у людей, а також складання відповідних частин реєстраційного досьє та їх експертизи на етапі реєстрації.
У рамках чинного фармацевтичного законодавства ця настанова не має сили нормативно-правового акта, її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану позицію європейського фармацевтичного сектору; дотримання її положень заінтересованими сторонами (такими як заявники, власники реєстраційних посвідчень, розробники та виробники лікарських засобів, експертні та регуляторні органи) полегшить оцінку реєстраційних досьє, а також підвищить якість та безпеку АФІ та готових лікарських засобів в Україні. Тим не менш можуть бути застосовані альтернативні підходи за умови їх відповідного наукового обґрунтування.
Такий підхід до правового статусу більшості наукових настанов викладений у документі Європейського агентства з лікарських засобів (EMEA) Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005» («Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005») [9]. Зазначений підхід відповідає позиції ВТО щодо застосування стандартів.
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Пластикові матеріали для первинної упаковки лікарських засобів
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Пластиковые материалы для первичной упаковки лекарственных средств
MEDICINAL PRODUCTS
Plastic Immediate Packaging Materials
Чинна від 2014-07-22
Сфера застосування
Ця настанова встановлює положення (рекомендації) щодо досліджень пластикових матеріалів первинної упаковки для АФІ та готових лікарських засобів, а також відповідної інформації та документації, які слід включати в реєстраційне досьє.
Ця настанова застосовна до АФІ та готових лікарських засобів, що розробляються, реєструються та виробляються в Україні для медичного застосування в Україні та з метою експорту, або що імпортуються в Україну.
Ця настанова поширюється на планування та проведення досліджень із вибору первинної упаковки з пластикових матеріалів для АФІ та готових лікарських засобів, а також складання відповідних розділів реєстраційного досьє та їх експертизи на етапі реєстрації та при внесенні змін стосовно введення нової пластикової первинної упаковки, пластикових матеріалів для первинної упаковки АФІ та готових лікарських засобів.
Ця настанова рекомендується для суб’єктів господарювання (далі — організацій), які займаються розробкою, реєстрацією та/чи виробництвом АФІ та готових лікарських засобів для застосування у людей у пластикових упаковках на території України, незалежно від відомчого підпорядкування та форми власності, для відповідних заявників та підприємств-виробників, продукція яких реєструється та імпортується в Україну, для науково-експертних організацій та регуляторних органів, а також експертів, що проводять експертизу під час проведення процедури реєстрації АФІ та готових лікарських засобів.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 року
Державна Фармакопея України. Перше видання. Доповнення 1. 2004 року
Державна Фармакопея України. Перше видання. Доповнення 2. 2008 року
Державна Фармакопея України. Перше видання. Доповнення 3. 2009 року
Державна Фармакопея України. Перше видання. Доповнення 4. 2011 року
Настанова 42-3.1:2004. — Настанови з якості. Лікарські засоби. Фармацевтична розробка. — Київ, МОЗ України, 2004
Настанова 42-3.0:2011. — Настанови з якості. Лікарські засоби. Фармацевтична розробка (ІСН Q8). — Київ, МОЗ України, 2011
Настанова 42-3.3:2004. — Настанови з якості. Лікарські засоби. Випробування стабільності. — Київ, МОЗ України, 2004
The Common Technical Document for the Registration of Pharmaceuticals for Human Use. — ICH Harmonised Tripartite Guideline. — Brussels, February 6–7, 2002. (Загальний технічний документ для реєстрації лікарських засобів для застосування людям. — Гармонізована трьохстороння настанова ICH. — Брюссель, 6–7 лютого 2002 р.)
The Rules Governing Medicinal Products in the European Union. Notice to applicants. — Volume 2B. — Medicinal products for human use. Presentation and content of the dossier. — 1998 edition. — European Commission. Directorate General III — Industry (Правила, що регулюють лікарські препарати в Європейському Союзі. Інформація для заявників. — Том 2В. — Лікарські препарати для застосування людям. Представлення та зміст досьє. — Видання 1998 року. — Європейська комісія. Головний директорат III — Промисловість)
The Rules Governing Medicinal Products in the European Union. Notice to Applicants — Volumе 2B. — Common Technical Document. — May 2008. — European Commission. Enterprise and Industry Directorate-General. Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 2В. — Європейська комісія. Головний директорат по підприємництву та промисловості)
Directive 2001/83/ЄC of the European Parliament and of the Council, of 6 November 2001 on the Community code relating to medicinal products for human use//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128 (Директива 2001/83/ЄC Європейського Парламенту та Ради ЄС від 6 листопада 2001 року про зведення законів Спільноти у відношенні лікарських препаратів для застосування людям//Official Journal of the European Communities. — L 311, 28.11.2001. — P. 67–128)
Commission Directive 2002/72/ЄC of 6 August 2002 relating to plastic materials and articles intended to come into contact with foodstuffs//Official Journal of the European Communities. — L 220, 15.8.2002. — P. 1–77 (Директива Комісії 2002/72/ЄC від 6 серпня 2002 року у відношенні пластичних матеріалів та виробів, призначених для контакту з продуктами харчування//Official Journal of the European Communities. — L 220, 15.8.2002. — P. 1–77)
Commission Directive 2003/63/ЄC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use. Annex I: Analytical, Pharmacotoxilogical and Clinical Standards and Protocols in Respect of the Testing of Medicinal Products. — Official Journal of the European Union. — № L 159 of 27.6.2003 (Директива Комісії 2003/63/ЄC від 25 червня 2003 року, що доповнює Директиву 2001/83/ЄC Європейського Парламенту та Ради ЄС про зведення законів Спільноти у відношенні лікарських препаратів для застосування людям. Додаток I: Аналітичні, фармакотоксикологічні та клінічні стандарти і протоколи щодо досліджень лікарських засобів. — Official Journal of the European Union. — № L 159 of 27.6.2003)
The Rules Governing Medicinal Products in the European Union. — Volumе 3А. — Guidelines. Medicinal products for human use. Quality and biotechnology. — 3АQ10a. — Plastic Primary Packaging Materials (Правила, що регулюють лікарські препарати в Європейському Союзі. — Том 3А. — Настанови. Лікарські препарати для застосування людям. Якість та біотехнологія. — 3AQ10a. — Пластикові матеріали для первинної упаковки)
CPMP/QWP/4359/03 — EMEA/СVМР/205/04 Guideline on Plastic Immediate Packaging Materials, London, 19 May 2005 (Настанова з пластикових матеріалів для первинної упаковки, Лондон, 19 травня 2005 року)
CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) Note for Guidance on Stability Testing: Stability Testing of New Drug Substances and Products, August 2003 (Настанова з випробувань стабільності: дослідження стабільності нових лікарських речовин та препаратів, серпень 2003 року)
CPMP/QWP/122/02 Rev 1 corr. Guideline on stability testing: stability testing of existing active substances and related finished products, London, 17 December 2003 (Настанова з випробувань стабільності: дослідження стабільності існуючих діючих речовин та відповідних готових препаратів, Лондон, 17 грудня 2003 року)
EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8) Note for Guidance on Pharmaceutical Development, May 2006 (Настанови з фармацевтичної розробки, травень 2006 року)
European Pharmacopoeia. 7th Edition. European Directorate for the Quality of Medicines (EDQM). — Council of Europe, 67075 Strasbourg Cedex, France 2010
Терміни та визначення понять
Нижче наведені терміни, вжиті у цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [6–8](див. національний додаток «Бібілографія»). Визначення цих термінів можуть відрізнятися в інших нормативних документах або терміни можуть мати інші значенняN.
Дослідження взаємодій (interaction studies, [6])
Дослідження для виявлення будь-яких взаємодій між пластиковим компонентом упаковки та продуктом, що призводять до неприйнятних змін якості продукту або упаковки за нормальних умов зберігання/застосування.
Дослідження екстракції (extraction studies, [6])
Дослідження, при яких спеціально поміщають зразок компонента упаковки у відповідну систему розчинників за екстремальних умов з метою отримання максимальної кількості речовин, що екстрагуються з упаковки у розчинник.
Компонент упаковки (packaging component, [6])
Будь-яка окрема частина системи контейнер/закупорювальний засіб, включаючи контейнери (наприклад ампули, флакони, пляшечки), закупорювальні засоби (наприклад кришки, що загвинчуються, пробки), засоби контролю першого розкриття, внутрішні закупорювальні засоби для контейнера, пристрої для введення та етикетки на контейнери.
Міграція (migration, [6])
Переміщення речовин (речовин, що виділяються) із пластикового компонента упаковки у вміст контейнера за умов, які відтворюють умови передбачуваного застосування.
Пластиковий матеріал (plastic material, [6])
Матеріал, який містить одну чи більше органічну макромолекулярну сполуку, отриману шляхом полімеризації, поліконденсації або адитивної полімеризації, чи за допомогою іншого подібного процесу з молекул із меншою молекулярною масою або шляхом хімічного перетворення природних макромолекул, які є основним інгредієнтом. Еластоміри, а також природний та синтетичний каучук не входять у сферу застосування цієї настанови.
Придатність (suitability, [6])
Оцінка системи контейнер/закупорювальний засіб щодо його/її захисних властивостей, безпеки, сумісності та експлуатаційних властивостей (функціонування).
Система контейнер/закупорювальний засіб (container closure system, [6])
Сукупність компонентів упаковки, яка вміщує та захищає діючу речовину або лікарську форму. До неї входять компоненти первинної упаковки, а також компоненти вторинної упаковки, якщо останні призначені для забезпечення додаткового захисту АФІ або готового лікарського засобу.
Сумісність (compatibility, [6])
Доведення відсутності суттєвої взаємодії між матеріалом системи контейнер/закупорювальний засіб та його/її вмістом, яка могла би призводити до зміни ефективності й стабільності лікарського засобу або становити ризик для прояву токсичної дії.
Сорбція (sorption, [6])
Прикріплення розчиненої речовини до пластикового компонента упаковки внаслідок фізико-хімичних явищ, пов’язаних з властивостями пакувального матеріалу та хімічними властивостями діючої речовини або інших розчинних речовин, що входять до складу лікарського засобу.
Специфікація (specification, [6, 7, 8])
Перелік випробувань, посилань на аналітичні методики та відповідних критеріїв прийнятності, які можуть представляти собою числові межі, діапазони або інші критерії, що відповідають описаним випробуванням [6].
Перелік випробувань, посилань на аналітичні методики та відповідних критеріїв прийнятності, які представляють собою числові межі, інтервали або інші критерії для описуваних випробувань. У специфікації встановлюють набір критеріїв, яким мають відповідати лікарська речовина або лікарський препарат для того, щоб вони вважалися придатними для їх передбачуваного застосування. «Відповідність специфікаціям» означає, що лікарська речовина та/або лікарський препарат у випадку їх випробування відповідно до перелічених аналітичних методик будуть відповідати вказаним критеріям прийнятності. Специфікації — це необхідні стандарти якості, запропоновані та обґрунтовані виробником та затверджені регуляторними органами [7].
Якісні та/або кількісні характеристики з указанням методик випробувань та припустимих меж, яким має відповідати даний препарат [8].
Познаки та скорочення
|
ЄС |
— |
Європейський Союз |
|
CPMP або CHMP |
— |
Committee for Medicinal Products for Human Use (Комітет з лікарських препаратів для застосування людям) |
|
CTD (ЗТД) |
— |
Common Technical Document (Загальний технічний документ) |
|
EMEA |
— |
European Medicines Agency (Європейське агентство з ліків) |
|
EU-CTD (ЄС-ЗТД) |
— |
European Union Common Technical Document (Загальний технічний документ Європейського союзу) |
|
АФІ |
— |
активний фармацевтичний інгредієнт |
|
NTA |
— |
Notice to Applicants (Інформація для заявників) |
Рекомендації щодо пластикових матеріалів для первинної упаковки
1. Вступ
1.1. Мета настанови
Ця настанова гармонізована з настановами CPMP/QWP/4359/03 — EMEA/СVМР/205/04, які замінили настанову 3AQ10a, опубліковану у томі 3А Правил, що регулюють лікарські засоби у Європейському Союзі. Ця настанова стосується інформації, яка має бути представлена у реєстраційному досьє, щодо пластикових матеріалів, які використовуються у якості первинної упаковки для діючих речовин та готових лікарських засобів для застосування людямN.
У відношенні пластикових матеріалів для первинної упаковки ця настанова стосується розділів 3.2.S.6, 3.2.Р.2 та 3.2.Р.7 модуля 3 частини 1 реєстраційного досьє, що встановлено у додатку 1 до Директиви 2003/63/ЄС, яка доповнює Директиву 2001/83/ЄС.
1.2. Сфера застосування
Ця настанова містить специфічні вимоги до пластикових матеріалів для первинної упаковки. Ця настанова не призначена для опису загальних вимог, які також застосовні й до інших видів пакувальних матеріалів, або вимог до властивостей системи контейнер/закупорювальний засіб, наприклад, функціональним характеристикам.
Сфера застосування цієї настанови обмежується пластиковими матеріалами для первинної упаковки, тобто тільки пакувальними матеріалами, які перебувають у безпосередньому контакті з АФІ або готовим лікарським засобом. Матеріали можуть бути частиною контейнера, закупорювального засобу або герметизуючого елементу, або іншими частинами системи (систем) контейнер/закупорювальний засіб.
Ця настанова не застосовна для еластомірів, а також природного та синтетичного каучуку.
Ця настанова не застосовується ретроспективно до лікарських засобів, які вже виведені на ринок у затверджених упаковках. Проте, нові заяви на реєстрацію або заяви про внесення змін до реєстраційних матеріалів стосовно введення нової пластикової первинної упаковки, пластикових матеріалів для первинної упаковки АФІ і готових лікарських засобів мають відповідати положенням цієї настанови, незалежно від того, чи буде матеріал використовуватися вперше або він вже використовується для упаковки інших АФІ або готових лікарських засобів.
1.3. Загальні принципи
Дані, які мають бути представлені щодо пластикових пакувальних матеріалів, залежать від фізичного стану АФІ (див. схему рішень у додатку 1 до цієї настанови), а також від лікарської форми та шляху введення готового лікарського засобу (див. схему рішень у додатку 2 до цієї настанови). Дані потрібно представляти у відповідності зі стандартною формою, описаною у пунктах 3.2.S.6 та 3.2.Р.2 модуля 3 ЗТД (див. також інформацію для заявників у томі 2Б Правил, що регулюють лікарські засоби у Європейському Союзі, щодо лікарських засобів для застосування людям). Кореляційна таблиця щодо розміщення цих даних у досьє форматів ЗТД та ЄС-ЗТД представлена у додатку 3 до цієї настанови.
Цією настановою слід користуватися разом з чинними редакціями таких настанов:
- з фармацевтичної розробки: EMEA/СHМР/167068/2004 — ICH (ICH Topic Q8) «Note for Guidance on Pharmaceutical Development» та гармонізованими з ним Настановами 42-3.1:2004, СТ-Н МОЗУ 42-3.0:2011N;
- з випробування стабільності: CPMP/ICH/2736/99 (ICH Topic Q1A (R2)) «Note for Guidance on Stability Testing: Stability Testing of New Drug Substances and Products» и CPMP/QWP/122/02 Rev 1 corr. «Guideline on stability testing: stability testing of existing active substances and related finished products» та гармонізованою з ним Настановою СТ-Н МОЗУ 42-3.3:2004N.
Крім того, коли вказано у цій настанові, потрібно враховувати положення законодавства ЄС щодо пластикових матеріалів та виробів, що знаходяться у контакті з харчовими продуктами, зокрема Директиви Комісії 2002/72/ЄС щодо пластикових матеріалів та виробів, що знаходяться у контакті з харчовими продуктами.
2. Місце розміщення у реєстраційному досьє документації, яка має бути представлена
Нижче наведені вказання тільки для досьє у форматі ЗТД для полегшення сприйняття порядку розміщення у реєстраційному досьє інформації, що вимагається. Інформація про місце розміщення відомостей для АФІ та готових лікарських засобів у досьє, складених у форматі ЄС-ЗТД, наведена у додатку 3 до цієї настанови.
Система контейнер/закупорювальний засіб для АФІ [3.2.S.6]
У цій частині досьє має бути представлена інформація щодо пластикових матеріалів, які використовуються у системі контейнер/закупорювальний засіб для АФІ. Інформація має включати:
- загальні відомості щодо виду та природи матеріалу у відповідності з п. 3.1 цієї настанови;
- специфікації на пластикові матеріали (див. п. 3.2 цієї настанови);
- результати досліджень речовин, що екстрагуються, та взаємодій, якщо необхідно (див. розділи 4 та 5 цієї настанови), та/або токсикологічну документацію, якщо необхідно (див. розділ 6 цієї настанови).
Фармацевтична розробка. Система контейнер/закупорювальний засіб [3.2.P.2]
Мають бути представлені дані, накопичені під час розробки препарату, з метою обґрунтування вибору пластикових матеріалів з точки зору стабільності, цілісності та сумісності з готовим лікарським засобом, способу введення та будь- яких процедур стерилізації, якщо вони необхідні. Представлені дані повинні містити наступне:
- детальні відомості про сумісність пластикового матеріалу з готовим лікарським засобом, отримані при проведені досліджень щодо речовин, які екстрагуються, та взаємодій, якщо необхідно (див. розділи 4 та 5 цієї настанови), та/або токсикологічну документацію, якщо необхідно (див. розділ 6 цієї настанови);
- аналіз обговорення світлостабільності пластикового матеріалу, якщо продукти розпаду матеріалу, що з’являються під впливом світла, можуть значно впливати на сумісність контейнера та лікарського засобу;
- детальні відомості про вплив виробничого процесу лікарського засобу на пластиковий матеріал, якщо необхідно (наприклад, умов стерилізації).
Система контейнер/закупорювальний засіб для готового лікарського засобу [3.2.Р.7]
Інформація, представлена в даній частині модуля 3, має включати:
- опис системи контейнер/закупорювальний засіб з вказанням усіх компонентів з пластикових матеріалів;
- загальну інформацію про пластиковий матеріал, що обраний для компонента упаковки, як вказано у п. 3.1 цієї настанови;
- специфікації на кожний пластиковий матеріал, як вказано у п. 3.2 цієї настанови.
3. Дані, які мають бути представлені
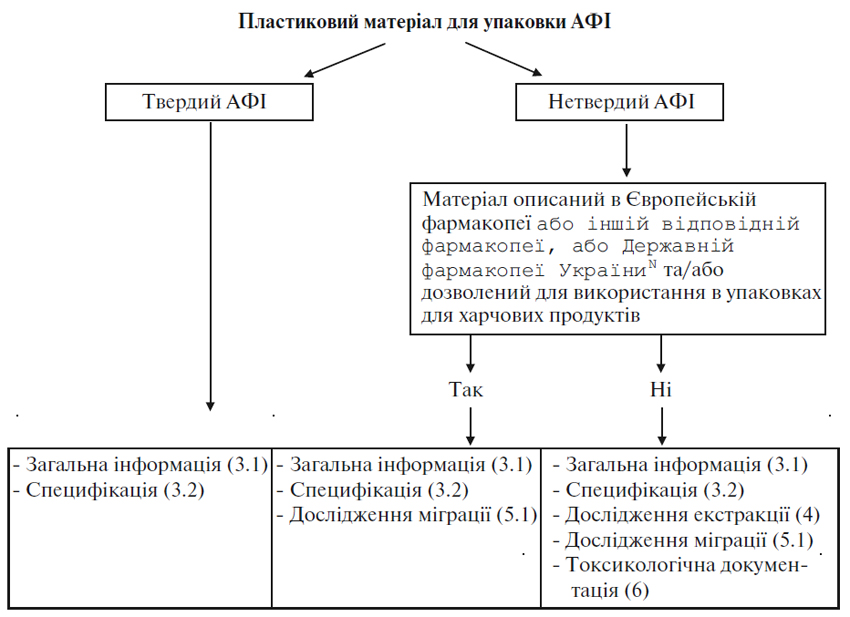
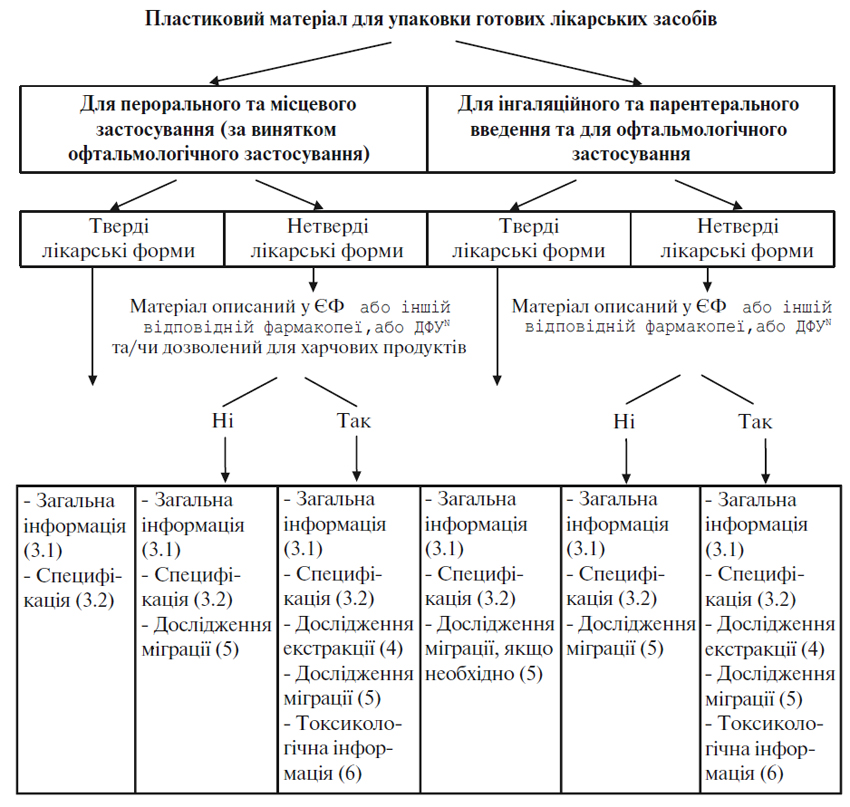
3.1. Загальна інформація
Для усіх пластикових матеріалів, які використовуються у якості матеріалу первинної упаковки для АФІ або готових лікарських засобів, слід указати:
- хімічну назву матеріалу;
- хімічну назву (назви) будь-якого мономеру, що використовується.
У випадках, коли нетверді АФІ та нетверді готові лікарські засоби контактують з пластиковими матеріалами, має бути вказана додаткова інформація, наведена нижче.
Для пластикових матеріалів, призначених для упаковки нетвердих АФІ:
- повний якісний склад пластикового матеріалу (включаючи добавки, такі як антиоксиданти, стабілізатори, пластифікатори, змащуючи речовини, розчинники та/або барвники), якщо матеріал упаковки не описаний у Європейській фармакопеї або іншій відповідній фармакопеї, або Державній фармакопеї УкраїниN та постачальник матеріалу не може надати підтвердження його відповідності законодавчим положенням щодо пакувальних матеріалів для харчових продуктів.
Для пластикових матеріалів, призначених для упаковки нетвердих готових лікарських засобів:
- назву постачальника матеріалів, якщо лікарський засіб призначений для інгаляційного або парентерального введення або для застосування у офтальмології;
- повний якісний склад пластикового матеріалу, як вказано вище, якщо лікарський засіб призначений для інгаляційного або парентерального введення або для застосування у офтальмології та матеріал упаковки не описаний у Європейській фармакопеї або іншій відповідній фармакопеї, або Державній фармакопеї УкраїниN, або, крім того, якщо фармакопейна монографія дозволяє використання декількох добавок, з який виробник може вибрати одну або декілька у певних межах. Якісний склад також має бути вказаний для матеріалів, не описаних у фармакопеях, якщо вони використовуються для нетвердих лікарських засобів, призначених для перорального або місцевого застосування (за виключенням препаратів для офтальмології), якщо постачальник матеріалу не може надати підтвердження його відповідності законодавчим положенням щодо пакувальних матеріалів для харчових продуктів.
3.2. Специфікації
При складанні специфікацій на пластикові матеріали для упаковки, яка буде контактувати з АФІ або готовими лікарськими засобами, необхідно зробити посилання на відповідні монографії Європейської фармакопеї або іншої відповідної фармакопеї, або Державної фармакопеї УкраїниN. Якщо наводиться посилання на монографію, то необхідно продемонструвати відповідність матеріалу цій монографії.
Якщо пластиковий матеріал не описаний у Європейській фармакопеї або іншій відповідній фармакопеї, або Державній фармакопеї УкраїниN, має бути складена внутрішня монографія відповідно до нижченаведеного та з урахуванням загальних фармакопейних методів:
- опис матеріалу;
- ідентифікація матеріалу;
- характерні властивості, наприклад, механічні, фізичні властивості.
Для пластикового матеріалу, призначеного для упаковки нетвердих АФІ та готових лікарських засобів, внутрішня монографія повинна містити таку додаткову інформацію:
- ідентифікація основних добавок, зокрема тих, які можуть мігрувати у вміст упаковки (таких як антиоксиданти, пластифікатори, каталізатори, активатори тощо);
- ідентифікація барвників;
- природа та кількість речовин, що екстрагуються, на підставі результатів дослідження екстракції (див. розділ 4 цієї настанови).
Немає необхідності включати цю інформацію до внутрішньої специфікації, якщо пластиковий пакувальний матеріал використовується для нетвердих готових лікарських засобів, призначених для перорального або місцевого застосування (за винятком офтальмологічного), або нетвердих АФІ, якщо постачальник матеріалу може надати підтвердження його відповідності положенням законодавства щодо харчових продуктів.
Для демонстрації відповідності встановленій внутрішній специфікації потрібно надати сертифікат аналізу однієї репрезентативної серії матеріалу.
4. Дослідження екстракції
Мета досліджень екстракції — визначити ті добавки у матеріалі, які можуть бути екстраговані готовим лікарським засобом або АФІ при контакті з матеріалом. Дослідження екстракції вважаються необхідними для пластикового матеріалу, що використовується для систем контейнер/закупорювальний засіб для нетвердих АФІ та для нетвердих лікарських форм для перорального або місцевого застосування (за виключенням офтальмологічного), коли матеріал не описаний у Європейській фармакопеї або іншій відповідній фармакопеї, або Державній фармакопеї УкраїниN та не затверджений для використання в упаковках для харчових продуктів. Для пластикового матеріалу, не описаного у фармакопеї, що використовується в системах контейнер/закупорювальний засіб для нетвердих готових лікарських засобів для інгаляційного або парентерального або для офтальмологічного введення, дослідження екстракції вимагаються, навіть якщо цей матеріал дозволений для використання в упаковках для харчових продуктів.
Зазвичай дослідження включають експозицію зразка матеріалу у відповідній системі розчинників за стресових умов для збільшення ступеня екстракції. Розчинник, який використовується для екстракції, має володіти такою ж здатністю до екстрагування речовин, як й АФІ/лікарська форма, за необхідності. У випадку лікарських засобів переважним розчинником може бути сам лікарський засіб або плацебо (основа-носій). У специфікації на пластиковий матеріал мають бути вказані природа та кількість усіх речовин, що екстрагуються.
5. Дослідження взаємодій
З метою оцінки придатності обраного пластикового пакувального матеріалу для його використання за призначенням, слід продемонструвати сумісність матеріалу з АФІ або готовим лікарським засобом. Дослідження можуть бути проведені при використанні пластикового матеріалу або пластикового компонента, або самого контейнеру. Масштаб та план дослідження взаємодій залежить від фізичного стану АФІ та готового лікарського засобу відповідно.
Для твердих АФІ та твердих лікарських форм: ризик взаємодії низький, та зазвичай вивчення взаємодії вмісту з контейнером не вимагається. Для твердих лікарських форм, призначених для інгаляційного чи парентерального введення (наприклад ліофілізовані препарати) можуть знадобитися дослідження взаємодій пакувального матеріалу та компонентів лікарського засобу.
Для нетвердих АФІ та рідких лікарських форм: ризик взаємодії зумовлює необхідність всебічних відповідних досліджень, специфічних для кожної діючої речовини/лікарського засобу. У дослідженнях необхідно оцінити критичні функціональні характеристики системи контейнер/пристрій для введення та переконатися, що не відбувається жодних значних змін, які призводять до погіршення якості АФІ чи готового лікарського засобу.
Дослідження взаємодій можуть включати дослідження міграції, щоб оцінити вимивання речовин із пластикового матеріалу в композицію/АФІ, та/чи дослідження сорбції, щоб оцінити можливу втрату якості внаслідок ефектів адсорбції чи абсорбції.
5.1. Дослідження міграції
На стадії розробки дослідження міграції мають бути проведені для АФІ/первинної композиції, щоб дати змогу вибрати прийнятний пакувальний матеріал для АФІ та готового лікарського засобу відповідно.
Дослідження міграції під час розробки необхідні, коли у результаті дослідження екстракції виявлені одна чи декілька речовин, що екстрагуються (див. розділ 4 цієї настанови). У таких випадках необхідно продемонструвати за умов, типових для запланованого застосування, що речовини не будуть мігрувати у таких кількостях, які можуть змінювати ефективність і стабільність АФІ/готового лікарського засобу або представляти токсикологічну небезпеку. Дослідження мають бути проведені на одній серії АФІ/готового лікарського засобу, за потреби. Моделюючі дослідження з використанням випробувальних середовищ (як у разі харчових продуктів) можуть розглядатися лише як попередні дослідження; їх проведення не виключає необхідності проведення досліджень для АФІ/готового лікарського засобу. Слід описати аналітичні методики з урахуванням загальних фармакопейних методів. Нефармакопейні аналітичні методи мають бути валідовані. Може виникнути необхідність запропонувати межі для максимального вмісту речовин, що виділяються.
Дослідження міграції можна не проводити лише тоді, коли на підставі даних, отриманих при вивченні екстракції, розрахована максимальна кількість індивідуальної речовини, що вимивається, наявність якої є допустимою у АФІ/готовому лікарському засобі, знаходиться на рівнях, для яких доведена токсикологічна безпека. Якщо дослідження міграції не є необхідним та не було проведено, потрібно надати обґрунтування цього.
Якщо пластиковий матеріал складається із шарів різних пластикових матеріалів, можливість міграції компонентів зовнішнього шару в лікарський засіб має бути оцінена залежно від природи продукту та його передбачуваного використання. Більше того, має бути продемонстровано, що компоненти типографської фарби чи клею, що застосовують на зовнішній поверхні системи контейнер/закупорювальний засіб, не будуть переміщуватися у лікарський засіб.
У тих випадках, коли дослідження міграції на етапі розробки не були проведені, речовини, що вимиваються, слід контролювати під час офіційних випробувань стабільності за нормальних та прискорених умов зберігання.
5.2. Дослідження сорбції
На етапі розробки можуть знадобитися дослідження вивчення взаємодії між пакувальним матеріалом та композицією внаслідок сорбції АФІ або допоміжної речовини на пакувальний матеріал, що призводить до змін у якості продукту. Дослідження сорбції мають бути проведені, якщо під час випробування стабільності спостерігаються зміни стабільності лікарського засобу, які можуть бути спричинені адсорбцією чи абсорбцією компонентів препарату пластиковим матеріалом.
Дослідження сорбції у разі пластикових матеріалів, призначених для упаковки нетвердих АФІ, не є обов’язковими.
6. Токсикологічна інформація/документація
У разі пластикового матеріалу, що використовується в системі контейнер/закупорювальний засіб для АФІ або готових лікарських засобів, необхідно надати токсикологічні дані для речовин, що екстрагуються та вимиваються, залежно від рівня їх вмісту та хімічної структури. Якщо використовуваний пластиковий матеріал або добавки описані у Європейській фармакопеї або іншій відповідній фармакопеї, або Державній фармакопеї УкраїниN, або дозволені для використання в упаковках для харчових продуктів, токсикологічні дані можуть не знадобитися. У разі пластикових матеріалів або добавок, не описаних у фармакопеї, які використовують у системах контейнер/закупорювальний засіб для лікарських засобів, призначених для інгаляційного чи парентерального або офтальмологічного введення, необхідно надавати токсикологічну інформацію, навіть якщо вони дозволені для використання в упаковках для харчових продуктів.
Додаток 1 (обов’язковий). Схема рішень для представлення документації щодо пластикових пакувальних матеріалів для АФІ
Додаток 2 (обов’язковий). Схема рішень для представлення документації щодо пластикових пакувальних матеріалів для готових лікарських засобів
Додаток 3 (обов’язковий). Кореляційна таблиця для надання інформації щодо пластикових пакувальних матеріалів для АФІ та готових лікарських засобів у реєстраційних досьє різних форматів
|
Інформація |
Формат CTD [1] NTA, том 2B, 2003 р. |
Формат EU-CTD [10] NTA, том 2B, 2008 р. |
|
Система контейнер/закупорювальний засіб для діючих речовин (АФІ) |
3.2.S.6 |
3.2.1.6 |
|
Система контейнер/закупорювальний засіб для готових лікарських засобів |
3.2.P.2 |
3.2.2.2 |
|
3.2.P.7 |
3.2.2.7 |
Додаток (довідковий). Бібліографія
1. The Common Technical Document for the Registration of Pharmaceuticals for Human Use. — ICH Harmonised Tripartite Guideline. — Brussels, February 6–7, 2002 (Загальний технічний документ для реєстрації лікарських засобів для застосування людям. — Гармонізована трьохстороння настанова ICH. — Брюсель, 6–7 лютого 2002 року).
2. Наказ МОЗ України від 26.08.2005 р. № 426 «Про затвердження Порядку проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення» (у редакції наказу МОЗ України від 04.01.2013 р. № 3), зареєстрований у Міністерстві юстиції України 15 вересня 2005 року за № 1069/11349.
3. ДСТУ 1.5-2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів/ І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
4. Настанова СТ-Н МОЗУ 42-1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення / М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005.
5. ДСТУ 1.7-2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів / О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003.
6. CPMP/QWP/4359/03 — EMEA/СVМР/205/04 Guideline on Plastic Immediate Packaging Materials, London, 19 May 2005 (Настанови з пластикових матеріалів для первинної упаковки, Лондон, 19 травня 2005 року).
7. СРМР/ICH/367/96 (ICH Topic Q6A) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000 (Настанови зі специфікацій: методики випробувань та критерії прийнятності для нових лікарських речовин та нових лікарських препаратів: хімічні речовини, 2000).
8. The Rules Governing Medicinal Products in the European Union. — Volume 3А. — Guidelines. — Medicinal Products for Human Use. Quality and Biotechnology. — 3AQ11a. — Specifications and Control Tests on the Finished Product (Настанови. Лікарські препарати для застосування людям. Якість та біотехнологія. — 3AQ11a. — Специфікації та контрольні випробування готового препарату).
9. EMEA/P/24143/2004 Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework, 2005 (Процедура щодо настанов та супутніх документів Європейського Союзу в рамках фармацевтичного законодавства, 2005).
10. The Rules governing Medicinal Products in the European Union. Notice to Applicants — Volume 2B. — Common Technical Document. — May 2008. — European Commission. Enterprise and Industry Directorate-General.
Ключові слова: активний фармацевтичний інгредієнт (АФІ), лікарська форма, готовий лікарський засіб, препарат, пластиковий матеріал, реєстраційне досьє, система контейнер/закупорювальний засіб, сорбція, специфікація, міграція, екстракція.