
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
ФАРМАЦЕВТИЧНА РОЗРОБКА (ICH Q8)
СТ-Н МОЗУ 42-3.0:2011
Видання офіційне
Київ
Міністерство охорони здоров’я України
2011
Передмова
1 РОЗРОБЛЕНО: Державне підприємство «Державний науковий центр лікарських засобів і медичної продукції» (ДП «ДНЦЛЗ»)
ПЕРЕКЛАД І НАУКОВО-ТЕХНІЧНЕ РЕДАГУВАННЯ: М. Ляпунов, д-р фарм. наук (керівник розробки); О. Безугла, канд. фарм. наук; Ю. Підпружников, д-р фарм. наук; К. Жемерова, канд. фарм. наук; О. Соловйов, канд. мед. наук; Н. Тахтаулова
ВНЕСЕНО ДО ПРИЙНЯТТЯ: Державна служба України з лікарських засобів
2 ПРИЙНЯТО ТА НАДАНО ЧИННОСТІ: наказ Міністерства охорони здоров’я України від 03.10.2011 № 634
3 Настанова відповідає документу Європейського агентства з лікарських засобів (European Medicines Agency) та ICH:
EMEA/CHMP/167068/2004 — ICH. — Part I: Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 (R2) Pharmaceutical Development). — Part II: Annex to Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 Annex Pharmaceutical Development), June 2009
(EMEA/CHMP/167068/2004 — ICH. — Частина I: Керівні вказівки з фармацевтичної розробки (ICH Topic Q 8 (R2) Фармацевтична розробка). — Частина II: Додаток до керівних вказівок з фармацевтичної розробки (ICH Topic Q 8 Додаток до фармацевтичної розробки), квітень 2009)
Ступінь відповідності — модифікований (MOD)
Переклад з англійської (en)
4 ВВЕДЕНО ВПЕРШЕ
© Міністерство охорони здоров’я України, 2011
© Державна служба України з лікарських засобів
Національний вступ
Ця настанова є прийнятим зі змінами (версії en) нормативним документом Європейського агентства з лікарських засобів (European Medicines Agency) EMEA/CHMP/167068/2004 — ICH. — Part I: Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 (R2) Pharmaceutical Development). — Part II: Annex to Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 Annex Pharmaceutical Development), June 2009 (EMEA/CHMP/167068/2004 — ICH. — Частина I: Керівні вказівки з фармацевтичної розробки (ICH Topic Q 8 (R2) Фармацевтична розробка). — Частина II: Додаток до керівних вказівок з фармацевтичної розробки (ICH Topic Q 8 Додаток до фармацевтичної розробки), квітень 2009) [1,2].
Організація, відповідальна за цю настанову, — Міністерство охорони здоров’я України.
Настанова містить положення, які відповідають чинному законодавству.
Цю настанову введено вперше. Документ EMEA/CHMP/167068/2004 — ICH не замінює в Європейському Союзі (ЄС) настанов CPMP/QWP/155/96 «Note for Guidance on Development Pharmaceutics» («Керівні вказівки з фармацевтичної розробки») та CPMP/QWP/054/98 «Decision Trees for the Selection of Sterilisation Methods. Annex to Note for Guidance on Development Pharmaceutics (CPMP/QWP/155/98)» («Схема рішень для вибору методів стерилізації. Додаток до керівних вказівок із фармацевтичної розробки (CPMP/QWP/155/98)» [3,4]. Зазначені настанови залишаються в ЄС чинними поряд з документом EMEA/CHMP/167068/2004 — ICH [1,2]. Тому ця настанова також не замінює Настанову 42-3.1:2004 «Настанови з якості. Лікарські засоби. Фармацевтична розробка» [5], що розроблена на підставі двох настанов: CPMP/QWP/155/96 та CPMP/QWP/054/98 [3,4]. Рекомендується користуватися як цією настановою, так і Настановою 42-3.1:2004 [5].
До цієї настанови було внесено окремі зміни, зумовлені правовими вимогами та прийнятими в Україні гармонізованими нормативними документами. Деякі редакційні зміни було долучено безпосередньо у пункти, до яких вони відносяться; ці зміни позначено іншим шрифтом та літерою N.
До настанови внесено такі редакційні зміни та додаткову інформацію:
– назву цієї настанови наведено відповідно до вимог ДСТУ 1.5–2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [6], а позначення — відповідно до вимог стандарту СТ МОЗУ 42–1.0:2005 «Фармацевтична продукція. Система стандартизації. Основні положення» [7]; додатково в назві зазначено номер документа ICH Q8;
– додатково введені такі структурні елементи настанови, як «Передмова», «Національний вступ», «Сфера застосування», «Нормативні посилання», «Познаки та скорочення», а також національний додаток «Бібліографія», які оформлені згідно з вимогами державних стандартів України: ДСТУ 1.5–2003 «Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів» [6] та ДСТУ 1.7–2001 «Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів» [8]; ці структурні елементи не позначені номерами, щоб зберегти у цій настанові нумерацію структурних елементів і правил документа EMEA/CHMP/167068/2004 — ICH [1,2]. «Зміст» цієї настанови викладено з урахуванням додаткових структурних елементів;
– розділ «Терміни та визначення понять» складено на підставі розділу 3 «Glossary» частини I та розділу 4 «Glossary» частини II документа EMEA/CHMP/167068/2004 — ICH [1,2]. Цей розділ не позначено номером та викладено слід за розділом «Нормативні посилання»; у виносці зазначено, що розділ «Терміни та визначення понять» відповідає розділу 3 «Glossary» частини I та розділу 4 «Glossary» частини II документа EMEA/CHMP/167068/2004 — ICH [1,2]. Усі терміни у розділі «Терміни та визначення понять» наведено за абеткою; вони супроводжуються посиланнями на нормативні документи, бібліографічний опис яких наведено в національному додатку «Бібліографія»;
– замість посилання на номери документів ICH після визначень термінів «стратегія контролю» та «якість» наведені примітки, де дані посилання на відповідні документи ЄС, що пройшли етап 5 ICH, а також на гармонізовані з ними настанови МОЗ України;
– у розділі «Терміни та визначення понять» додатково наведені визначення понять таких застосованих термінів, як «лікарський препарат» [9,10], «управління ризиками для якості» [11,18], а також «матеріал» (з частини 2 Настанови 42-4.0:2011 «Лікарські засоби. Належна виробнича практика») [12,17];
– у розділі «Нормативні посилання» дано бібліографічний опис згадуваних в тексті нормативних документів; при цьому замість деяких настанов ICH приведений опис відповідних настанов CPMP/ICH, що пройшли етап 5 ICH. У тексті також замість посилань на номери документів ICH дані посилання на відповідні документи ЄС, що пройшли етап 5 ICH, а також додатково на гармонізовані з ними настанови МОЗ України;
– при посиланні в тексті цієї настанови на документи ICH та/або ЄС у виносках в кінці відповідних сторінок вказано: «Рекомендується користуватися цим документом до прийняття МОЗ України гармонізованої з ним настанови»;
– замість виділення деяких специфічних термінів знаком * або 1 в тексті цієї настанови ці терміни виділили іншим шрифтом; стосовно першого виділеного терміну дано виноску: «Позначення термінів, що виділені тут і далі цим шрифтом, наведені у розділі «Терміни та визначення понять»».
– у цій настанові замінені наступні слова:
- «заявка на отримання торгової ліцензії» («marketing арplication») — на «заявка на реєстрацію» (частина І);
- «досьє для торгової ліцензії» («MA dossier» — «dossier for marketing authorisation») — на «реєстраційне досьє» (частина І);
- «заявка» («арplication») — на «реєстраційне досьє» (частина ІІ);
– замість «Європейська фармакопея» вказано «Європейська фармакопея або інша відповідна фармакопея, або Державна Фармакопея України, гармонізована з Європейською фармакопеєюN». Під словами «інша відповідна фармакопея» мається на увазі фармакопея держави ЄС або фармакопея іншої країни, гармонізована з Європейською фармакопеєю, або Фармакопея США;
– у тексті цієї настанови замість посилань на номери пунктів реєстраційного досьє у форматі CTD (наприклад, 3.2.Р.1) вказано «в п. 3.2.Р.1 модуля 3 реєстраційного досьє у форматі CTD»;
– у п. 1.1 частини I цієї настанови стосовно реєстраційного досьє у форматі загального технічного документа (CTD) зроблено виноску, де зазначено наказ МОЗ України, яким введено в Україні такий формат реєстраційного досьє;
– у п. 1.2 частини I цієї настанови замість посилання на настанову ICH M4 зазначено (див. 4 документа СРМР/ICH/2887/99 (ICH Topic M 4) в розділі «Нормативні посилання»), оскільки вони пройшли етап 5 ICH та прийняті в ЄС;
– у п. 2.2.3 частини I цієї настанови замість слів: «… наприклад, результати випробування відносно фракцій, що вдихаються, для інгаляційного препарату» згідно з загальною статтею 2.9.18 Державної Фармакопеї України зазначено: «… наприклад, результати аеродинамічного визначення дрібнодисперсних часток препарату для інгаляцій»;
– у розділі «Передмова» частини II цієї настанови замість речення «Ця настанова є додатком до настанови Фармацевтична розробка (ICH Q8)» зазначено «Частина II є додатком до частини I цієї настанови»; замінені слова: «цей додаток» на «частина II цієї настанови», «основна настанова» та «основний документ ICH Q8» на «частина I цієї настанови»;
– у частині II цієї настанови замість деяких скорочень (наприклад, CQA) наводили назву терміну та скорочення у дужках: критичний показник якості (CQA);
– у п. 2.1 частини II цієї настанови один з аспектів стосовно цільового профілю якості препарату викладено таким чином: «• вивільнення або доставка терапевтично активної частини, а також властивості, що впливають на параметри фармакокінетики, ефективності та безпекиN (наприклад, розчинність, аеродинамічні характеристики)», оскільки це відповідає визначенню поняття цільового профілю якості препарату, а від розчинності та аеродинамічних характеристик може залежати як ефективність, так і безпека відповідного лікарського препарату;
– у п. 2.5 частині II цієї настанови при згадуванні правил GMP додатково дано посилання на Настанову СТ-Н МОЗУ 42–4.0:2011 «Лікарські засоби. Належна виробнича практика»;
– у п. 2.6 частині II цієї настанови замість речення «Зміна простору проектних параметрів є предметом регіональних вимог» зазначено «Зміна простору проектних параметрів є предметом чинного законодавстваN»;
– у тексті настанови уточнено нумерацію структурних елементів реєстраційного досьє в форматі CTD згідно з документами ICH та ЄС; так, наприклад, замість скорочених номерів «Р.2.1, Р.2.2, and P.2.3» зазначено «3.2.Р.2.1, 3.2.Р.2.2 та 3.2.P.2.3» (див. п. 3.1 частини ІІ цієї настанови та ін.);
– у додатку 1 додатково проставлений номер таблиці 1.1; у додатку 2 додатково проставлені номери: діаграми 2.1, рисунка 2.0 та рисунка 2.3; у додатках 1 та 2 номери таблиці, діаграми та рисунків доповнено номером додатка (наприклад, у додатку 2 замість рисунка 1а зазначено рисунок 2.1а); в тексті додатків наведені посилання на таблицю, діаграму та рисунки з вказівкою їх номерів;
– до рисунка 2.0 та рисунка 2.3 додатково надані примітки з перекладом позначень;
– інші незначні доповнення виділені іншим шрифтом і буквою N.
Ця настанова придатна для організації фармацевтичної розробки лікарських препаратів для людини, підготовки модуля 3 «Якість» реєстраційного досьє в форматі CTD, для побудови систем управління якістю для фармацевтичної промисловості (фармацевтичних систем якості), а також експертизи реєстраційних досьє, аудиту та інспектування на відповідність GMP.
У рамках чинного фармацевтичного законодавства ця настанова не має сили нормативно-правового акта; її положення є рекомендаціями. Цю настанову слід розглядати як гармонізовану (в рамках ICH) позицію світового фармацевтичного сектора. До фармацевтичної розробки можуть бути застосовані також альтернативні підходи за умови їх відповідного наукового обґрунтування. Такий підхід до правового статусу більшості наукових настанов прийнятий в документі Європейського агентства з лікарських засобів Doc. Ref. EMEA/P/24143/2004 «Procedure for European Union guidelines and related documents within pharmaceutical legislative framework» («Процедура щодо настанов і супутніх документів Європейського Союзу в рамках фармацевтичного законодавства») [13].
Ця настанова буде регулярно переглядатися відповідно до змін і доповнень, що вноситимуть в документ EMEA/CHMP/167068/2004 — ICH. — Part I: Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 (R2) Pharmaceutical Development). — Part II: Annex to Note for Guidance on Pharmaceutical Development (ICH Topic Q 8 Annex Pharmaceutical Development) [1,2].
НАСТАНОВА
ЛІКАРСЬКІ ЗАСОБИ
Фармацевтична розробка (ICH Q8)
ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Фармацевтическая разработка (ICH Q8)
MEDICINAL PRODUCTS
Pharmaceutical Development (ICH Q8)
Чинна від 2011-10-03
Сфера застосування
Ця настанова установлює положення (рекомендації) щодо системного підходу до фармацевтичної розробки лікарських препаратів для людини та інформації щодо фармацевтичної розробки, яку необхідно включати до модуля 3 «Якість» реєстраційного досьє в форматі CTD.
Ця настанова застосовна до лікарських препаратів, що розробляють, реєструють та виробляють в Україні для продажу на внутрішньому ринку та з метою експорту або що імпортують до України.
Ця настанова поширюється на дослідження з фармацевтичної розробки лікарських препаратів для людини, підготовку та експертизу модуля 3 «Якість» реєстраційного досьє в форматі CTD, побудову та оцінку фармацевтичних систем якості, застосовних для фармацевтичної розробки та виробництва лікарських препаратів протягом їх життєвого циклу.
Цю настанову рекомендується застосовувати суб’єктам господарювання (далі — організаціям), які займаються фармацевтичною розробкою, підготовкою модуля 3 «Якість» реєстраційного досьє в форматі CTD та подачею заявок на реєстрацію, виробництвом лікарських препаратів та побудовою фармацевтичної системи якості, незалежно від відомчого підпорядкування та форми власності, а також експертним та регуляторним органам у питаннях фармацевтичної оцінки модуля 3 «Якість» реєстраційного досьє в форматі CTD при реєстрації (перереєстрації) лікарських препаратів, проведенні аудиту та інспектування на відповідність GMP, а також для оцінки або сертифікації фармацевтичної системи якості виробників лікарських засобів на добровільних засадах.
Ця настанова придатна для організації фармацевтичної розробки лікарських препаратів для людини, підготовки модуля 3 «Якість» реєстраційного досьє в форматі CTD, побудови фармацевтичних систем якості, а також експертизи реєстраційних досьє, аудиту та інспектування на відповідність GMP.
Нормативні посилання
У цій настанові є посилання на такі нормативні документи:
Державна Фармакопея України. Перше видання. 2001 р.
Державна Фармакопея України. Перше видання. Доповнення 1. 2004 р.
Державна Фармакопея України. Перше видання. Доповнення 2. 2008 р.
Державна Фармакопея України. Перше видання. Доповнення 3. 2009 р.
Державна Фармакопея України. Перше видання. Доповнення 4. 2011 р.
Настанова 42–3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності
СТ-Н МОЗУ 42–4.0:2011 Лікарські засоби. Належна виробнича практика
Настанова 42–4.2:2011 Лікарські засоби. Управління ризиками для якості (ICH Q9)
Настанова СТ-Н МОЗУ 42–4.3:2011 Лікарські засоби. Фармацевтична система якості (ICH Q10)
European Pharmacopoeia. 7th Edition. European Directorate for the Quality of Medicines (EDQM). — Council of Europe, 67075 Strasbourg Cedex, France 2010
СPMP/ICH/365/96 (ICH Topic Q6В) Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Рroducts, 1999
СРМР/ICH/367/96 (ICH Topic Q6A) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000
СРМР/ICH/2887/99 (ICH Topic М 4) Common Technical Document for the Registration of Pharmaceuticals for Human Use: Organisation of Common Technical Document, 2004
СРМР/ICH/2887/99 — Quality (ICH Topic М 4 Q) Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality Overall Summary of Module 2 and Module 3: Quality, 2003
СРМР/ICH/2887/99 — Safety (ICH Topic М 4 S) Common Technical Document for the Registration of Pharmaceuticals for Human Use: Nonclinical Oberview and Nonclinical Summaries of Module 2 and Organisation of Module 4, 2003
СРМР/ICH/2887/99 — Efficacy (ICH Topic М 4 E) Common Technical Document for the Registration of Pharmaceuticals for Human Use: Clinical oberview and Clinical summary of Module 2 and Module 5: Study reports, 2003
CPMP/EWP/QWP/1401/98 Guideline on the Investigation of Bioequivalence, 2010
EudraLex. — The Rules Governing Medicinal Products in the European Union. — Volume 4. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use
http://ec.europa.eu/health/documents/eudralex/vol-4/index en.htm
EMA/INS/GMP/79766/2011 Quality Risk Management (ICH Q9), 2011
EMA/INS/GMP/79818/2011 Pharmaceutical Quality System (ICH Q10), 2011
Довідкові джерела інформації наведено в національному додатку «Бібліографія».
Терміни та визначення понять1
Нижче подано терміни, вжиті в цій настанові, та визначення позначених ними понять. Терміни англійською мовою, що відповідають стандартизованим у цьому розділі термінам, наведені на підставі [1,2,9,10,17,18,19] (див. національний додаток «Бібліографія»)N.
1Розділ «Терміни та визначення понять» відповідає розділу 3 «Glossary» частини I та розділу 4 «Glossary» частини II документа EMEA/CHMP/167068/2004 — ICH [1, 2].
Випробування при випуску у реальному часі (real time release testing, [2])
Можливість оцінки та гарантування якості в ході процесу виробництва та/або готової продукції на основі даних щодо процесу, які зазвичай включають обґрунтовану комбінацію визначених характеристик матеріалу та контролю параметрів процесу.
Доведений діапазон прийнятності (proven acceptable range, [2])
Визначений діапазон параметра процесу, для якого робота у цьому діапазоні (за умови зберігання інших параметрів постійними) буде приводити до вироблення продукції, що задовольняє відповідним критеріям якості.
Життєвий цикл (product lifecycle, [1])
Всі фази життя продукції від початкової розробки, знаходження на ринку і до припинення виробництва і медичного застосуванняN продукції [1].
Критичний параметр процесу (critical process parameter — CPP, [2])
Параметр процесу, варіабельність якого може вплинути на критичний показник якості, та який внаслідок цього має бути об’єктом моніторингу та контролю, щоб забезпечити необхідну якість отриманої в результаті процесу продукції.
Критичний показник якості (critical quality attribute — CQA, [2])
Фізична, хімічна, біологічна чи мікробіологічна властивість або характеристика, яка для забезпечення необхідної якості продукції має знаходитися у відповідних межах, відповідному діапазоні або мати відповідний розподіл.
Лікарський препарат (drug product; medicinal product, [9,10])
Лікарський засіб у певній лікарській формі, вміщений в остаточне паковання та призначений для розміщення на ринку [9].
Будь-яка речовина або комбінація речовин (у певній лікарській формі)N, що призначена для лікування чи профілактики захворювань у людини або може бути призначена для встановлення діагнозу або для відновлення, корекції чи зміни її фізіологічних функцій [10].
Примітка. В цій настанові терміну «лікарський препарат» відповідає термін «готовий препарат» («finished product»)N.
Матеріал (material, [12,17])
Загальне поняття, що означає сировину (вихідна сировина, реактиви, розчинники), допоміжні речовини та матеріали, проміжну продукцію, активні фармацевтичні інгредієнти та матеріали для пакування і маркування.
Офіційні експериментальні плани; план експериментів (formal experimental designs; design of experiments, [1])
Структурований, організований метод визначення взаємозв’язку між факторами, що впливають на процес, і продукцією, яку одержують у результаті цього процесу. (Також називають «планування експериментів»).
Постійне підтвердження процесу (continuous process verification, [1])
Альтернативний підхід до валідації процесу, при якому робочі характеристики виробничого процесу постійно контролюють і оцінюють.
Простір проектних параметрів (design space, [1])
Багатофакторна комбінація та взаємодія вхідних перемінних (наприклад, характеристик матеріалу), а також параметрів процесу, при яких доведено забезпечення якості. Робота в рамках простору проектних параметрів не вважається зміною. Вихід за простір проектних параметрів розглядається як зміна і, як правило, є початком регуляторного процесу внесення змін після реєстрації. Простір проектних параметрів пропонує заявник; він є об’єктом оцінки і затвердження з боку регуляторних органів.
Процесно-аналітична технологія (process analytical technology — PAT, [1])
Система планування, аналізу і контролю виробництва за допомогою періодичних вимірювань (тобто, під час технологічного процесу) критичних показників якості і функціональних характеристик сировини, оброблюваних матеріалів і процесів з метою забезпечення якості готового препарату.
Стійкість процесу (process robustness [1])
Здатність процесу витримувати мінливість в матеріалах, а також зміни процесу та обладнання без негативного впливу на якість.
Стратегія контролю (control strategy, [2,16,19])
Запланований комплекс контрольних заходів, заснований на розумінні продукції та процесу, що забезпечує функціональні характеристики процесу та якість продукції. Цей комплекс може включати контроль параметрів та характеристик, пов’язаних з діючою речовиною, матеріалами та компонентами для лікарського засобу, умовами функціонування приміщень та обладнання, контроль в процесі виробництва, специфікації на готову продукцію, а також пов’язані з цим методи та частоту моніторингу і контролю.
Примітка. Визначення терміну взято з документа EMA/INS/GMP/79818/2011 Pharmaceutical Quality System (ICH Q10). Див. також Настанову 42-4.3:2004 Лікарські засоби. Фармацевтична система якості (ICH Q10)N.
Управління ризиками для якості (quality risk management, [11,18])
Систематичний процес для загального оцінювання, контролювання, інформування та огляду ризиків для якості лікарського засобу протягом життєвого циклу препарату.
Цільовий профіль якості препарату (quality target product profile — QTPP, [2])
Очікуваний набір показників якості лікарського препарату, який в ідеалі буде досягнутий для забезпечення необхідної якості лікарського препарату з урахуванням його безпеки та ефективності.
Якість (quality, [1,14,15])
Відповідність діючої речовини або лікарського препарату його призначенню. Це поняття включає такі показники, як ідентичність, сила дії та чистота.
Примітка. Визначення терміну взято з документа СРМР/ICH/367/96 (ICH Topic Q6A) Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Див. також Настанову 42-3.2:2004 Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятностіN.
Якість шляхом розробки (quality by design — QbD, [2])
Системний підхід до розробки, заснований на надійних наукових даних та управлінні ризиками для якості, який починається з попереднього визначення цілей і приділяє особливу увагу розумінню продукції та процесу, а також контролю процесу.
Познаки та скорочення
ЄС — Європейський Союз
CPMP або CHMP — Committee for Medicinal Products for Human Use (Комітет із лікарських засобів для застосування людиною)
CTD — Common Technical Document (загальний технічний документ)
CPP — critical process parameter (критичний параметр процесу)
CQA — critical quality attribute (критичний показник якості)
ЕМЕА або ЕМА — European Medicines Agency (Європейське агентство з лікарських засобів)
FMEA — failure mode effects analysis (аналіз характеру наслідків відмов)
GMP — good manufacturing practice (належна виробнича практика)
ICH — International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Міжнародна конференція з гармонізації технічних вимог до реєстрації лікарських препаратів для людини)
NIR — near infrared (ближня інфрачервона область)
РАТ — process analytical technologies (процесно-аналітична технологія)
QbD — quality by design (якість шляхом розробки)
QTPP — quality target product profile (цільовий профіль якості препарату)
Частина I. Рекомендації з фармацевтичної розробки
1. Передмова
1.1. Ціль настанови
У цій настанові описаний рекомендований вміст п. 3.2.Р.2 («Фармацевтична розробка») реєстраційного досьє у форматі загального технічного документа (CTD) ICH M4 (див. розділ «Нормативні посилання»)N.2
Розділ реєстраційного досьє «Фармацевтична розробка» дає можливість представити відомості, одержані при використовуванні наукових підходів і управління ризиками для якості (див. документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» або гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.2:2011N) для розробки препарату і процесу його виробництва. Вперше це роблять при подачі первинної заявки на реєстрацію, а потім актуалізують у міру отримання нових знань протягом життєвого циклу3 препарату. Розділ «Фармацевтична розробка» призначений для того, щоб надати експертам і інспекторам можливість вичерпного розуміння препарату і виробничого процесу. В цій настанові також вказані області, де демонстрація більш глибокого розуміння фармацевтичної науки і технології може служити основою для більш гнучкого підходу з боку регуляторних органів. Ступінь гнучкості з боку регуляторних органів має ґрунтуватися на рівні відповідних наданих наукових знань.
2Реєстраційне досьє у форматі загального технічного документа (CTD) введено в Україні Наказом Міністерства охорони здоров’я України від 26.08.2005 № 426, який зареєстровано в Міністерстві юстиції України 19.09.2005 за № 1069/11349.
3Позначення термінів, що виділені тут і далі цим шрифтом, наведені у розділі «Терміни та визначення понять».
1.2. Сфера застосування
Ця настанова призначена, щоб надати вказівки щодо вмісту п. 3.2.Р.2 («Фармацевтична розробка») для лікарських препаратів в модулі 3 реєстраційного досьє в форматі загального технічного документа (CTD) (див. 4 документа СРМР/ICH/2887/99 (ICH Topic M 4) в розділі «Нормативні посилання»). Ця настанова не розповсюджується на вміст заявок на лікарські препарати, що знаходяться на стадіях клінічних досліджень в ході розробки ліків. Проте принципи цієї настанови також важливо враховувати на цих стадіях. Ця настанова може бути застосовна до інших видів продукції. Щоб визначити застосовність цієї настанови до конкретного виду продукції, заявники можуть консультуватися з відповідними регуляторними уповноваженими органами.
2. Фармацевтична розробка
Ціль фармацевтичної розробки — розробити якісний препарат і процес його виробництва, щоб постійно випускати продукцію із заданими функціональними характеристиками. Інформація і знання, одержані при дослідженнях в ході фармацевтичної розробки, а також досвід виробництва надають наукове розуміння, яке служить основою для встановлення простору проектних параметрів, специфікацій і виробничого контролю.
Інформація, одержана при дослідженнях в ході фармацевтичної розробки, може служити основою для управління ризиками для якості. Важливо визнати, що якість не може бути перевірена в препаратах; тобто, якість має бути закладена при розробці. Зміни у складі і виробничих процесах під час розробки і протягом життєвого циклу слід розглядати як можливості отримання додаткових знань для подальшого встановлення простору проектних параметрів. Так само може бути корисним включення відповідних відомостей, одержаних при експериментах, які привели до неочікуваних результатів. Простір проектних параметрів пропонується заявником і є об’єктом оцінки та затвердження з боку регуляторних органів. Робота в межах простору проектних параметрів не розглядається як зміна. Вихід за простір проектних параметрів розглядається як зміна і, як правило, є початком регуляторного процесу внесення змін після реєстрації.
У розділі «Фармацевтична розробка» слід описувати відомості, які доводять, що вибраний вид лікарської форми і запропонований склад відповідають передбачуваному призначенню. Кожна частина цього розділу має містити достатню інформацію, щоб забезпечити розуміння розробки лікарського препарату і виробничого процесу. Бажано приводити зведені таблиці і графіки, якщо вони вносять ясність і полегшують огляд інформації.
Як мінімум, необхідно визначити ті аспекти лікарських речовин, допоміжних речовин, систем контейнер/закупорювальний засіб і виробничих процесів, які є критичними для якості препарату; також слід обґрунтувати стратегію контролю. Критичні характеристики складу і параметри процесу, як правило, визначають за допомогою оцінки ступеня, з яким їх зміни можуть вплинути на якість лікарського препарату.
Крім того, заявник може приділити увагу тим дослідженням з фармацевтичної розробки, які можуть розширити знання про функціональні характеристики препарату в широкому діапазоні властивостей матеріалів, режимів обробки і параметрів процесу. Включення такої додаткової інформації в цей розділ надає можливість довести глибоке розуміння властивостей матеріалів, виробничих процесів і їх контролю. Це наукове розуміння полегшує встановлення розширеного простору проектних параметрів. В таких ситуаціях існують можливості для розробки більш гнучких підходів з боку регуляторних органів, щоб сприяти, наприклад:
- рішенням регуляторних органів, пов’язаним з ризиком (експертиза та інспекції);
- удосконаленням виробничого процесу в рамках описаного в досьє затвердженого простору проектних параметрів без подальшої експертизи з боку регуляторних органів;
- скороченню постреєстраційних заявок;
- контролю якості, що проводиться в реальному часі, що приводить до скорочення випробувань готового препарату при випуску.
Для реалізації таких гнучких підходів заявник повинен продемонструвати широкі знання функціональних характеристик препарату в діапазоні властивостей матеріалів, режимів і параметрів виробничого процесу. Таке розуміння має бути одержано заявником, наприклад, за рахунок офіційних експериментальних планів, процесно-аналітичної технології (PAT) та/або попередніх знань. Для отримання таких знань при виборі пріоритетності додаткових досліджень з фармацевтичної розробки може бути корисним використання відповідних принципів управління ризиками для якості.
Планування і проведення досліджень з фармацевтичної розробки має відповідати передбачуваній науковій цілі. Слід визнати, що основу для науково обґрунтованої заявки та її оцінки з боку регуляторних органів забезпечує не об’єм даних, а рівень одержаних знань.
2.1. Компоненти лікарського препарату
2.1.1. Лікарська речовина
Мають бути визначені та обговорені фізико-хімічні і біологічні властивості лікарської речовини, які можуть впливати на функціональні характеристики лікарського препарату і можливість його виробництва, або такі характеристики лікарської речовини, які спеціально для неї встановлені (наприклад, властивості для твердих речовин). Прикладами фізико-хімічних і біологічних властивостей, які може знадобитися дослідити, є розчинність, вміст води, розмір частинок, властивості кристалів, біологічна активність, а також проникність. Ці властивості можуть бути взаємозв’язані, що може вимагати їх розгляду в поєднанні.
Щоб оцінити потенційний вплив фізико-хімічних властивостей лікарської речовини на функціональні характеристики лікарського препарату, мають бути проведені дослідження відносно лікарського препарату. Наприклад, в Настанові СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та в гармонізованій з нею Настанові 42–3.2:2004N описані деякі обставини, при яких рекомендується проводити дослідження відносно лікарського препарату (наприклад, схема рішень № 3 і схема рішень № 4 (частина 2)). Такий же підхід застосовується і в Настанові СPMP/ICH/365/96 (ICH Topic Q6В)4 «Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products». Знання, отримані при дослідженнях з вивчення потенціального впливу властивостей лікарської речовини на функціональні характеристики лікарського препарату, можуть бути використані (за необхідності) для обґрунтування розділів специфікації на лікарську речовину (п. 3.2.S.4.5 модуля 3 реєстраційного досьє у форматі CTD).
Слід оцінити сумісність лікарської речовини з допоміжними речовинами, які вказані в п. 3.2.Р.1 модуля 3 реєстраційного досьє у форматі CTD. Для препаратів, що містять більш однієї лікарської речовини, слід оцінити також сумісність лікарських речовин одна з одною.
4Рекомендується користуватися цим документом до прийняття МОЗ України гармонізованої з ним настанови.
2.1.2. Допоміжні речовини
Вибір допоміжних речовин, їх концентрації і характеристики, які можуть вплинути на функціональні властивості лікарського препарату (наприклад, стабільність, біодоступність) або на можливість його виробництва, необхідно обговорити з урахуванням відповідної функції кожної допоміжної речовини. Мають бути включені всі речовини, що використовуються при виробництві лікарського препарату, незалежно від того, присутні вони в готовому препараті чи ні (наприклад, речовини, що використовуються в цілях обробки). Має бути встановлена сумісність одних допоміжних речовин з іншими, якщо це має відношення до справи (наприклад, комбінація консервантів в подвійній консервуючій системі). Також слід довести необхідність присутності допоміжних речовин для забезпечення їх передбачуваної функції (наприклад, антиоксидантів, підсилювачів проникності, дезінтегрантів, речовин для управління вивільненням), а також збереження цієї функції протягом передбачуваного терміну зберігання лікарського препарату. Інформацію про функціональні властивості допоміжної речовини можна використовувати, якщо необхідно, для обґрунтування вибору і показників якості допоміжної речовини, а також для обґрунтування специфікації на лікарський препарат (п. 3.2.Р.5.6 модуля 3 реєстраційного досьє в форматі CTD).
При необхідності, слід зробити перехресні посилання на інформацію, що підтверджує безпеку допоміжних речовин (п. 3.2.Р.4.6 модуля 3 реєстраційного досьє в форматі CTD).
2.2. Лікарський препарат
2.2.1. Розробка складу
Має бути підготовлено резюме з описом розробки складу і вказівкою тих характеристик, які є критичними для якості лікарського препарату, беручи до уваги передбачуване застосування та шлях введення. При встановленні критичних або взаємозв’язаних перемінних, які можуть бути важливими для забезпечення якості лікарського препарату, може бути корисною інформація щодо офіційних експериментальних планів.
У резюме має бути висвітлений хід розробки складу від початкової ідеї до остаточної композиції. В резюме також слід взяти до уваги вибір компонентів лікарського препарату (наприклад, властивості лікарської речовини, допоміжних речовин, системи контейнер/закупорювальний засіб, будь-яких значущих пристроїв для дозування), виробничий процес, а також, якщо необхідно, відомості, одержані при розробці подібного(их) лікарського(их) препарату(ів).
У цьому пункті реєстраційного досьє мають бути обґрунтовані будь-які межі вмісту допоміжних речовин, вказані в складі на серію (п. 3.2.Р.3.2 модуля 3 реєстраційного досьє у форматі CTD); це обґрунтування часто може бути засновано на досвіді, одержаному в ході розробки, або при виробництві.
Необхідно надати короткий опис складів, що використовувалися при клінічному вивченні безпеки і ефективності та при будь-яких значущих дослідженнях біодоступності або біоеквівалентності. Будь-які зміни складу, пропонованого для виведення на ринок, в порівнянні зі складами дослідних серій, що були використані при клінічних дослідженнях, а також первинних серій, на яких вивчалася стабільність, мають бути чітко описані; необхідно представити логічне обґрунтування змін.
Слід підготувати в узагальненому вигляді з перехресними посиланнями на дослідження (з вказівкою номерів досліджень) інформацію про порівняльні дослідження in vitro (наприклад, розчинення) або про порівняльні дослідження in vivo (наприклад, біоеквівалентність), яка дозволяє зіставити склади, що були використані при клінічних дослідженнях, і склад, пропонований для розміщення на ринку та описаний в п. 3.2.Р.1 модуля 3 реєстраційного досьє у форматі CTD. Якщо були спроби провести кореляцію між дослідженнями in vitro/in vivo, в цьому пункті слід надати результати таких досліджень з перехресними посиланнями на дослідження (з вказівкою номерів досліджень). Успішна кореляція може допомогти у виборі відповідних критеріїв прийнятності для тесту «Розчинення», що потенційно може усунути необхідність подальших досліджень біоеквівалентності при внесенні змін в препарат або виробничий процес.
Мають бути вказані будь-які особливості лікарського препарату (наприклад, риска на таблетці, надлишок при наповненні, заходи проти фальсифікації, які стосуються безпосередньо лікарського препарату) за умови логічного обґрунтування їх застосування.
2.2.2. Надлишки
Як правило, не схвалюється використання надлишку лікарської речовини для компенсації її розкладання під час виробництва або протягом терміну зберігання препарату, а також для збільшення терміну придатності.
Будь-які надлишки при виробництві лікарського препарату, незалежно від того, присутні вони в готовому препараті чи ні, необхідно обґрунтувати з урахуванням безпеки і ефективності препарату. Має бути представлена інформація щодо: 1) кількості надлишку, 2) причини його використання (наприклад, для компенсації очікуваних і документованих виробничих втрат) та 3) обґрунтування кількості надлишку. Надлишок необхідно включати в кількість лікарської речовини, приведену в складі на серію (п. 3.2.Р.3.2 модуля 3 реєстраційного досьє у форматі CTD).
2.2.3. Фізико-хімічні та біологічні властивості
Необхідно визначити та обговорити фізико-хімічні та біологічні властивості, що мають відношення до безпеки, функціональних характеристик лікарського препарату або можливості його виробництва. До них відносяться фізіологічні ефекти лікарської речовини і властивості складу. Дослідження мають включати, наприклад, результати аеродинамічного визначення дрібнодисперсних частокN препарату для інгаляцій. Так само, в цьому пункті слід представити інформацію щодо обґрунтування вибору тесту «Розчинення» замість тесту «Розпадання» або інших засобів для гарантії вивільнення ліків, а також щодо розробки та придатності вибраного тесту. Див. також Настанову СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та гармонізовану з нею Настанову 42–3.2:2004N (схема рішень № 4 (частина 3) та схема рішень № 7 (частина 1)) або Настанову СPMP/ICH/365/96 (ICH Topic Q6В)5 «Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological / Biological Products». Обговорювання має містити перехресні посилання на будь-які значущі дані про стабільність у п. 3.2.Р.8.3 модуля 3 реєстраційного досьє у форматі CTD.
5Рекомендується користуватися цим документом до прийняття МОЗ України гармонізованої з ним настанови.
2.3. Розробка виробничого процесу
Вибір, контроль і будь-яке удосконалення виробничого процесу, описаного в п. 3.2.Р.3.3 реєстраційного досьє у форматі CTD (тобто, передбачуваного для виробництва промислових серій), слід пояснити. Важливо розглянути критичні параметри складу, а також існуючі режими виробничого процесу, щоб пояснити вибір виробничого процесу і підтвердити придатність компонентів. Слід обговорити придатність обладнання, що використовується для виробництва передбачуваних препаратів. Дослідження з розробки процесу мають служити основою для удосконалення процесу, валідації процесу, постійного підтвердження процесу (якщо необхідно), а також для будь-яких вимог до контролю процесу. Якщо можливо, ці дослідження мають проводитися стосовно мікробіологічних, а також фізичних і хімічних характеристик. Знання, одержані при дослідженнях з розробки процесу, можуть бути використані (якщо необхідно) для обґрунтування специфікації на лікарський препарат (п. 3.2.Р.5.6 модуля 3 реєстраційного досьє у форматі CTD).
У програмі розробки виробничого процесу або в програмі удосконалення процесу мають бути зазначені критичні параметри процесу, за якими слід спостерігати або які слід контролювати (наприклад, закінчення грануляції), щоб гарантувати необхідну якість продукції.
Для продукції, що має бути стерильною, необхідно вибрати підхожий метод стерилізації лікарського препарату і первинного пакувального матеріалу; вибір слід обґрунтувати.
Необхідно обговорити значні відмінності між процесами виробництва дослідних серій для клінічних випробувань (безпека, ефективність, біодоступність, біоеквівалентність) або серій для первинних досліджень стабільності та процесом, описаним в п. 3.2.Р.3.3 модуля 3 реєстраційного досьє у форматі CTD. В обговоренні слід стисло описати вплив змін на функціональні характеристики, можливість виробництва і якість препарату. Інформацію необхідно представити так, щоб полегшити порівняння процесів і відповідних даних щодо аналізу серій (п. 3.2.Р.5.4 модуля 3 реєстраційного досьє у форматі CTD). Інформація має містити, наприклад, 1) ідентифікацію (наприклад, номер серії) і призначення виробленої серії (наприклад, номер серії для дослідження біоеквівалентності), 2) зазначення виробничої дільниці, 3) розмір серії, а також 4) будь-які значні відмінності в обладнанні (наприклад, різна конструкція, принцип роботи, розмір).
Для забезпечення гнучкого підходу до подальшого поліпшення процесу при описі розробки виробничого процесу корисно описати систему заходів, що дозволяє контролювати критичні параметри процесу або кінцеві точки процесу. Накопичення даних від моніторингу процесу під час розробки виробничого процесу може забезпечити корисну інформацію для поліпшення розуміння процесу. Має бути описана стратегія контролю процесу, яка забезпечує можливості для регулювання процесу з метою контролю всіх критичних характеристик.
Слід представити оцінку можливості процесу дійсно приводити до отримання продукції з передбачуваною якістю (наприклад, здійсненність виробничого процесу за різних умов роботи, при різному розмірі серії або на різному обладнанні). Розуміння стійкості процесу може бути корисним при загальному оцінюванні ризику та зниженні ризику (пояснення термінів див. в документі EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» та в гармонізованій з ним Настанові СТ-Н МОЗУ 42–4.2:2011N), а також для сприяння подальшому поліпшенню виробництва та процесу особливо в зв’язку з використанням засобів та методів управління ризиками (див. документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.2:2011N).
2.4. Система контейнер/закупорювальний засіб
Для препарату, пропонованого для розміщення на ринку, слід обговорити вибір та обґрунтувати підбір системи контейнер/закупорювальний засіб, описаної в п. 3.2.Р.7 модуля 3 реєстраційного досьє у форматі CTD. Слід надати увагу передбачуваному призначенню лікарського препарату і придатності системи контейнер/закупорювальний засіб для зберігання і транспортування (поставки), включаючи, при необхідності, контейнер для зберігання та транспортну тару для нерозфасованого лікарського препарату.
Слід обґрунтувати вибір первинних пакувальних матеріалів. Обговорення має містити відомості про дослідження, проведені для доказу цілості контейнера та закупорювального засобу. Слід надати увагу можливій взаємодії між продукцією та контейнером або етикеткою.
При підборі первинних пакувальних матеріалів слід врахувати, наприклад, вибір матеріалів, захист від вологи та світла, сумісність матеріалів пристрою з лікарською формою (включаючи сорбцію контейнером і виділення речовин з пакувального матеріалу), безпеку матеріалів пристрою. Якщо це має відношення до справи, слід включити обґрунтування для вторинних пакувальних матеріалів.
Якщо використовується дозуючий пристрій (наприклад, піпетка для крапель, шприц-ручка, інгалятор для сухих порошків), важливо довести, що за умов випробування, які, наскільки можливо, моделюють застосування препарату, видається відтворювана та правильна (точна) доза препарату.
2.5. Мікробіологічні властивості
Якщо доречно, в цьому підпункті (п. 3.2.Р.2.5 модуля 3 реєстраційного досьє у форматі CTD) слід обговорити мікробіологічні властивості лікарського препарату. Обговорення, наприклад, має містити:
– обґрунтування для проведення або виключення випробування на мікробіологічну чистоту для нестерильних лікарських препаратів (наприклад, схема рішень № 8 в Настанові СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» або гармонізованій з нею Настанові 42–3.2:2004N та Настанові СPMP/ICH/365/96 (ICH Topic Q6В)6 «Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products»;
– інформацію про вибір і ефективність консервуючих систем в препаратах, що містять антимікробний консервант, або антимікробну ефективність препаратів, які самі по суті є антимікробними;
– для стерильних препаратів інформацію про цілість системи контейнер / закупорювальний засіб з огляду запобігання мікробній контамінації.
Хоча показником, який зазвичай включають в специфікацію на лікарський препарат, є визначення вмісту консерванту за допомогою хімічних методів, в ході розробки слід довести ефективність антимікробного консерванту. За допомогою тесту на ефективність антимікробного консерванту має бути доведено, що антимікробний консервант при якнайменшому вмісті, вказаному в специфікації, є ефективним в плані управління кількістю мікроорганізмів. Концентрація, що використовується, має бути обґрунтована з огляду на ефективність і безпеку так, щоб використовувалася мінімальна концентрація консерванту, при якій досягається необхідний рівень ефективності протягом передбачуваного терміну зберігання препарату. Якщо це доречно, в ході розробки слід провести і задокументувати в цьому підпункті випробування з введенням мікроорганізмів за умов, які, наскільки можливо, моделюють застосування препарату пацієнтом.
6Рекомендується користуватися цим документом до прийняття МОЗ України гармонізованої з ним настанови.
2.6. Сумісність
З метою забезпечення відповідної супровідної інформації для маркування слід надати увагу сумісності лікарського препарату з розчинниками, що використовуються для підготовки до застосування (наприклад, випадання осадку, стабільність). Ця інформація має містити рекомендований термін зберігання під час застосування при рекомендованій температурі зберігання та при можливих межах концентрації. Також може знадобитися розглянути змішування або розведення препаратів перед застосуванням (наприклад, для препаратів, що додаються в контейнери великого об’єму з інфузійними лікарськими засобами).
Частина II. Додаток до рекомендацій з фармацевтичної розробки
1. Передмова
Частина II є додатком до частини I цієї настанови; в ній надано подальше роз’яснення основних концепцій, викладених у частині I цієї настанови. Крім того, у частині II описано принципи якості шляхом розробки (QbD). Частина II цієї настанови не призначена для встановлення нових стандартів або для введення нових регуляторних вимог; проте, у ній показано, яким чином описані у частині I цієї настанови концепції та засоби (наприклад, простір проектних параметрів) можуть бути введені заявником у практику стосовно всіх лікарських форм. Якщо вибором компанії є застосування якості шляхом розробки та управління ризиками для якості (див. документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.2:2011N) у поєднанні з відповідною фармацевтичною системою якості (див. документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.3:2011N), виникають можливості для поліпшення регуляторних підходів, що ґрунтуються на наукових засадах та загальному оцінюванні ризиків.
Підходи до фармацевтичної розробки
У будь-якому випадку, препарат має бути розроблений таким чином, щоб він задовольняв потреби пацієнтів та виконував передбачувану функцію. Стратегії розробки препаратів відрізняються від компанії до компанії та від препарату до препарату. Підхід до розробки та її масштаб також можуть бути різними; їх необхідно викласти у реєстраційному досьє. Заявник може обрати емпіричний підхід або більш систематизований підхід до розробки препарату, або комбінацію обох підходів. Додаток 1 ілюструє потенційні відмінності цих підходів. Більш систематизований підхід до розробки (що також називають якістю шляхом розробки) може включати, наприклад, залучення до розробки наявних наукових даних, результатів досліджень із використанням планування експериментів, використання управління ризиками для якості, а також управління знаннями (див. документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.3:2011 «Лікарські засоби. Фармацевтична система якості (ICH Q10)»N) протягом життєвого циклу продукції. Такий систематизований підхід може сприяти досягненню необхідної якості препарату та допомогти кращому розумінню стратегії компанії з боку регуляторних органів. Розуміння продукції та процесу може бути актуалізоване за допомогою знань, отриманих протягом життєвого циклу продукції.
Глибше розуміння продукції та процесу її виробництва може створити основу для більш гнучких підходів з боку регуляторних органів. Ступінь гнучкості з боку регуляторних органів визначається відповідним науковим рівнем, представленим у реєстраційному досьє. Це отримані та представлені наукові знання (але не обсяг зібраних даних), що формують основу для реєстраційного досьє, заснованого на наукових даних та загальному оцінюванні ризиків, а також для його оцінювання з боку регуляторних органів. Тим не менш, у кожному реєстраційному досьє мають бути представлені відповідні дані, які доводять, що ці знання ґрунтуються на надійних наукових принципах.
Фармацевтична розробка має включати, як мінімум, такі елементи:
- визначення цільового профілю якості препарату (QTPP) стосовно якості, безпеки та ефективності з урахуванням, наприклад, шляху введення, лікарської форми, біодоступності, сили дії та стабільності;
- визначення потенційних критичних показників якості (CQAs) лікарського препарату; таким чином, ті характеристики препарату, які впливають на якість продукції, мають бути об’єктом вивчення та контролю;
- визначення критичних показників якості лікарської речовини, допоміжних речовин тощо, а також вибір виду та кількості допоміжних речовин для випуску продукції необхідної якості;
- вибір відповідного виробничого процесу;
- визначення стратегії контролю.
Більш глибокий підхід до розробки препарату, заснований на якості шляхом розробки, може додатково включати такі елементи:
- систематичне оцінювання, переосмислення та поліпшення складу та процесу виробництва, у тому числі:
- визначення за допомогою, наприклад, наявних наукових даних, експериментальних досліджень, а також загального оцінювання ризиків характеристик матеріалів та параметрів процесу, що можуть впливати на критичні показники якості (CQAs) продукції;
- визначення функціональних взаємозв’язків, що пов’язують характеристики матеріалів та параметри процесу з критичними показниками якості (CQAs) продукції;
- використання глибокого розуміння препарату та процесу у комбінації із управлінням ризиками для якості для встановлення відповідної стратегії контролю, що може включати, наприклад, пропозиції щодо простору(ів) проектних параметрів та/або випробувань при випуску у реальному часі.
У результаті такий систематизований підхід може полегшити постійне поліпшення та інновації протягом життєвого циклу продукції (див. документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.3:2011 «Лікарські засоби. Фармацевтична система якості (ICH Q10)»N).
2. Елементи фармацевтичної розробки
В наступному розділі докладно розглядаються можливі підходи до отримання більш систематичного, більш глибокого розуміння препарату і процесу на стадії розробки. Наведені приклади носять тільки ілюстративний характер і не призначені для введення нових регуляторних вимог.
2.1. Цільовий профіль якості препарату
Цільовий профіль якості препарату формує основу для планування розробки препарату.
Стосовно цільового профілю якості препарату слід врахувати такі аспекти:
- передбачуване застосування у клінічних умовах, шлях введення, лікарська форма, системи доставки;
- сила(и) дії дози;
- система контейнер/закупорювальний засіб;
- вивільнення або доставка терапевтично активної частини, а також властивості, що впливають на параметри фармакокінетики, ефективності та безпекиN (наприклад, розчинення, аеродинамічні характеристики) відповідно до тієї лікарської форми препарату, що має бути розроблена;
- критерії якості лікарського препарату (наприклад, стерильність, чистота, стабільність та вивільнення лікарської речовини), відповідні призначеній для розміщення на ринку продукції.
2.2. Критичні показники якості
Критичний показник якості (CQA) — це фізична, хімічна, біологічна чи мікробіологічна властивість або характеристика, яка для забезпечення необхідної якості продукції має знаходитися у відповідних межах, відповідному діапазоні або мати відповідний розподіл. Критичні показники якості (CQAs), як правило, пов’язані з лікарською речовиною, допоміжними речовинами, проміжною продукцією (матеріалами в ході процесу) та лікарським препаратом.
Критичними показниками якості (CQAs) для твердих лікарських форм для орального застосування, як правило, є ті характеристики, що впливають на чистоту, силу дії, вивільнення лікарської речовини та стабільність препарату. Критичні показники якості (CQAs) для інших систем доставки можуть додатково включати більш специфічні характеристики препарату, такі як аеродинамічні властивості для інгаляційних препаратів, стерильність для парентеральних препаратів, адгезійні властивості для трансдермальних пластирів. Для лікарських речовин, вихідної сировини та проміжної продукції критичні показники якості додатково можуть включати ті характеристики, що впливають на критичні показники якості лікарського препарату (наприклад, розподіл часток за розмірами, насипна густина).
Для розробки препарату та процесу слід керуватися потенційними критичними показниками якості (CQAs) лікарського препарату, визначеними на підставі цільового профілю якості препарату та/або попередніх наукових даних. Перелік потенційних критичних показників якості (CQAs) може бути змінений після вибору складу та виробничого процесу, а також у міру накопичення знань щодо препарату та поглиблення розуміння процесу. Для вибору пріоритетів серед переліку критичних показників якості (CQAs) з метою їх подальшої оцінки може застосовуватися управління ризиками для якості. Відповідні критичні показники якості (CQAs) можуть бути ідентифіковані за допомогою повторюваного процесу управління ризиками для якості та експерименту, що дає можливість оцінити ступінь, до якого варіабельність показників може впливати на якість лікарського препарату.
2.3. Загальне оцінювання ризиків: взаємозв’язок характеристик матеріалів та параметрів процесу з критичними показниками якості (CQAs) лікарського препарату
Загальне оцінювання ризиків є цінним науково обґрунтованим процесом, використовуваним в управлінні ризиками для якості (див. документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.2:2011 «Лікарські засоби. Управління ризиками для якості (ICH Q9)»N), що може допомогти визначити, які з характеристик матеріалів та параметрів процесу мають потенційний вплив на критичні показники якості (CQAs) препарату. Загальне оцінювання ризиків, як правило, виконують на початку процесу фармацевтичної розробки і повторюють в міру надходження інформації та поглиблення знань.
Засновуючись на попередніх знаннях та вихідних експериментальних даних, інструменти загального оцінювання ризиків можуть бути використані для виявлення і класифікації параметрів (наприклад, процес, обладнання, вхідні матеріали), які потенційно можуть вплинути на якість продукції. Для наочного прикладу див. додаток 2. Попередній перелік потенційних параметрів може бути досить великим, але при подальших дослідженнях він може бути змінений, а також можуть бути змінені пріоритети (наприклад, за допомогою поєднання планування експериментів, механістичних моделей). Перелік можна у подальшому доопрацювати за допомогою експериментів, щоб визначити значимість окремих перемінних і потенційних взаємодій. Після ідентифікації важливих параметрів, їх можна далі вивчати (наприклад, за допомогою поєднання планування експериментів, математичних моделей або досліджень, які приводять до механістичного розуміння), щоб досягти більш високого рівня розуміння процесу.
2.4. Простір проектних параметрів
Взаємозв’язок між вхідними даними процесу (характеристиками матеріалу та параметрами процесу) і критичними показниками якості може бути описаний у просторі проектних параметрів (див. приклади у додатку 2).
2.4.1. Вибір перемінних
Загальне оцінювання ризиків та експерименти з розробки процесу, описані у п. 2.3, можуть привести до розуміння зв’язку параметрів технологічного процесу та характеристик матеріалів з критичними показниками якості препарату (CQAs) та їх впливу на CQAs, а також допомогти у визначенні перемінних та їх діапазонів, в яких можна досягти постійної якості. Ці параметри процесу і характеристики матеріалів можуть, таким чином, бути відібрані для включення у простір проектних параметрів.
У реєстраційному досьє має бути наданий опис параметрів процесу та характеристик матеріалів, визначених для простору проектних параметрів (тих, що були до нього включені), а також їх впливу на якість продукції. Має бути надано обґрунтування їх включення до простору проектних параметрів. У деяких випадках було б корисно надати також обґрунтування того, чому деякі параметри були виключені. У реєстраційному досьє мають бути описані відомості, отримані в результаті досліджень. Слід вказати ті параметри процесу та характеристики матеріалів, які не були змінені у ході розробки.
2.4.2. Опис простору проектних параметрів у реєстраційному досьє
Простір проектних параметрів може бути описаний у вигляді діапазонів характеристик матеріалів та параметрів процесу або за допомогою більш складних математичних співвідношень. Можна описати простір проектних параметрів як залежну від часу функцію (наприклад, циклічність температури та тиску для циклу ліофілізації) або як комбінацію перемінних, таку як компоненти багатомірної моделі. Також можуть бути включені фактори масштабування, якщо простір проектних параметрів призначений охоплювати декілька робочих масштабів. Вклад у визначення простору проектних параметрів може внести аналіз попередніх даних. Незалежно від того, як розробляється простір проектних параметрів, очікується, що робота в межах простору проектних параметрів буде приводити до вироблення продукції необхідної якості.
У додатку 2 наведені приклади різних можливих підходів до наведення простору проектних параметрів.
2.4.3. Простір (простори) проектних параметрів для операції
Заявник може обрати: чи встановлювати незалежні простори проектних параметрів для кожної операції або кількох операцій, чи встановлювати єдиний простір проектних параметрів, який охоплює багато операцій. Хоча часто простіше розробляти окремий простір проектних параметрів для кожної окремої операції, простір проектних параметрів, що охоплює весь процес може забезпечити більш оперативну гнучкість. Наприклад, у разі лікарського препарату, який зазнає розкладу в розчині перед ліофілізацією, простір проектних параметрів (наприклад, концентрація, час, температура) для контролю ступеня розкладу може бути виражений для кожної окремої операції або як сума цілком по всіх операціях.
2.4.4. Взаємозв’язок простору проектних параметрів з масштабуванням та обладнанням
При описі простору проектних параметрів заявник має враховувати вид бажаної операційної гнучкості. Простір проектних параметрів може бути розроблений у будь-якому масштабі. Заявник має обґрунтувати відповідність простору проектних параметрів, розробленого у малому або пілотному масштабі, для передбачуваного промислового виробництва, а також обговорити потенційні ризики у ході масштабування.
Якщо заявник пропонує застосовувати простір проектних параметрів до кількох робочих масштабів, то простір проектних параметрів має бути описаний у вигляді параметрів, що не залежать від масштабу. Наприклад, якщо було встановлено, що препарат є чутливий до зсуву при операції змішування, простір проектних параметрів має включати швидкість зсуву, а не швидкість перемішування. Як частина опису простору проектних параметрів можуть бути включені безрозмірні показники та/або моделі для масштабування.
2.4.5. Простір проектних параметрів порівняно з доведеними діапазонами прийнятності
Комбінація доведених діапазонів прийнятності не являє собою простір проектних параметрів. Тим не менш, доведені діапазони прийнятності, засновані на експериментах з однією перемінною, можуть надати корисну інформацію про процес.
2.4.6. Простір проектних параметрів та межі неприйнятності
Для параметрів процесу або характеристик матеріалів може бути корисним визначити межі неприйнятності, за якими не можуть бути досягнуті відповідні показники якості. Однак визначення меж неприйнятності або демонстрація невідповідностей не є неодмінною частиною встановлення простору проектних параметрів.
2.5. Стратегія контролю
Стратегія контролю призначена для забезпечення постійного виробництва продукції необхідної якості. При обговоренні елементів стратегії контролю в п. 3.2.Р.2 модуля 3 реєстраційного досьє у форматі CTD слід описати та обґрунтувати, яким чином контроль в процесі виробництва та контроль вихідної сировини (лікарської речовини та допоміжних речовин), проміжної продукції (матеріалів в процесі виробництва), системи контейнер / закупорювальний елемент і лікарського препарату сприяють якості кінцевої продукції. Такі контролі мають бути засновані на розумінні препарату, складу та процесу і включати, як мінімум, контроль критичних параметрів процесу і характеристик матеріалів.
Всеохоплюючий підхід до фармацевтичної розробки буде сприяти розумінню процесу і препарату та визначенню джерел варіабельності. Джерела варіабельності, що можуть впливати на якість продукції, слід ідентифікувати, належним чином осмислювати та у подальшому контролювати. Розуміння джерел варіабельності та їх впливу на подальші процеси або обробку, оброблювані матеріали та якість лікарського препарату може надати можливість перенести контролі на попередні етапи і звести до мінімуму необхідність випробування готового препарату. Розуміння препарату і процесу у поєднанні з управлінням ризиками для якості (див. документ EMA/INS/GMP/79766/2011 «Quality Risk Management (ICH Q9)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.2:2011 «Лікарські засоби. Управління ризиками для якості (ICH Q9)»N) буде сприяти контролю процесу таким чином, щоб для досягнення постійної якості продукції варіабельність (наприклад, сировини) можна було компенсувати шляхом адаптації.
Таке розуміння процесу може зробити можливим альтернативний процес виробництва, для якого варіабельність вихідних матеріалів може бути менш жорстко обмежена. Або замість цього може бути можливим розробити адаптивний етап процесу (етап, чутливий до вхідних матеріалів) з відповідним контролем процесу, щоб забезпечити постійну якість продукту.
Поглиблення розуміння функціональних характеристик препарату може виправдати використання альтернативних підходів для визначення того, що матеріал за показниками якості відповідає вимогам. Використання таких альтернатив може сприяти проведенню випробувань при випуску у реальному часі. Наприклад, тест «Розпадання» може служити заміною тесту «Розчинення» для твердих лікарських форм, що швидко розпадаються та містять добре розчинні лікарські речовини. Випробування однорідності дозованих одиниць в процесі виробництва (наприклад, використовуючи відхилення маси разом із аналізуванням в ближній інфрачервоній області (NIR)) може зробити можливим випробування при випуску у реальному часі та надати більш високу ступінь гарантії якості порівняно з традиційними випробуваннями готового препарату з використанням фармакопейних вимог щодо однорідності вмісту. Випробування при випуску у реальному часі можуть замінити випробування готової продукції, але не замінюють етапи огляду та контролю якості, передбачені правилами GMP для видачі дозволу на випуск серії (див. Настанову СТ-Н МОЗУ 42–4.0:2011 «Лікарські засоби. Належна виробнича практика»N).
Стратегія контролю може включати (але не обмежуватись цим):
- контроль характеристик вхідних матеріалів (наприклад, лікарської речовини, допоміжних речовин, первинних пакувальних матеріалів), заснований на розумінні їх впливу на технологічність або якість продукції;
- специфікацію(ї) на продукцію;
- контролі окремих операцій, що впливають на подальший процес або якість продукції (наприклад, вплив висушування на розклад, вплив розміру часток гранул на розчинення);
- випробування у процесі виробництва або випробування при випуску у реальному часі замість випробувань готової продукції (наприклад, вимірювання та контроль критичних показників якості (COAs) під час ведення процесу);
- програму моніторингу (наприклад, повне випробування продукції через рівні відрізки часу) для підтвердження багатомірних моделей прогнозування.
Стратегія контролю може включати в себе різні елементи. Наприклад, один з елементів стратегії контролю може базуватися на випробуванні готової продукції, в той час як інший може залежати від випробувань при випуску у реальному часі. В реєстраційному досьє необхідно навести обґрунтування щодо використання цих альтернативних підходів.
Прийняття принципів, викладених в цій настанові, може допомогти обґрунтувати альтернативні підходи до встановлення показників якості та критеріїв прийнятності у специфікаціях, як це описано у документі СРМР/ICH/367/96 (ICH Topic Q6A) «Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances» та гармонізованій з ним Настанові 42–3.2:2004 «Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності»N, а також у документі СPMP/ICH/365/96 (ICH Topic Q6В) «Note for Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Рroducts»7.
7Рекомендується користуватися цим документом до прийняття МОЗ України гармонізованої з ним настанови.
2.6. Управління життєвим циклом продукції та постійне поліпшення
Протягом усього життєвого циклу продукції компанії мають можливість оцінювати інноваційні підходи до поліпшення якості продукції (див. документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.3:2011 «Лікарські засоби. Фармацевтична система якості (ICH Q10)»N).
Можна піддавати моніторингу функціональні характеристики процесу, щоб гарантувати, що він працює, як і очікувалось, для виробництва продукції з показниками якості, передбаченими у просторі проектних параметрів. Як додатковий досвід, накопичений в ході рутинного виробництва, цей моніторинг може включати аналіз тенденцій виробничого процесу. Для деяких просторів проектних параметрів з використанням математичних моделей може бути корисним періодичне підтвердження для гарантування належного функціонування моделі. Підтвердження моделі є прикладом діяльності, якою можна управляти в рамках власної внутрішньої системи якості компанії за умови, що простір проектних параметрів не змінився.
При отриманні додаткових знань про процес може бути бажаним розширення, скорочення або перегляд простору проектних параметрів. Зміна простору проектних параметрів є предметом чинного законодавстваN.
3. Подання фармацевтичної розробки та пов’язаної з нею інформації у реєстраційному досьє в форматі загального технічного документа (CTD)
Інформація про фармацевтичну розробку подається у п. 3.2.Р.2 модуля 3 реєстраційного досьє в форматі CTD. Інша інформація, отримана в результаті досліджень з фармацевтичної розробки, може бути розміщена у реєстраційному досьє в форматі CTD у кілька різних способів; нижче наведені деякі конкретні пропозиції. Однак заявник має чітко вказати, де знаходиться різна інформація. На додаток до того, що представляють у реєстраційному досьє, деякі окреслені в цій настанові аспекти (наприклад, управління життєвим циклом продукції, постійне поліпшення) мають регулюватися фармацевтичною системою якості заявника (див. документ EMA/INS/GMP/79818/2011 «Pharmaceutical Quality System (ICH Q10)» та гармонізовану з ним Настанову СТ-Н МОЗУ 42–4.3:2011 «Лікарські засоби. Фармацевтична система якості (ICH Q10)»N).
3.1. Управління ризиками для якості та розробка препарату і процесу
Управління ризиками для якості може бути використане на різних етапах розробки препарату та процесу, а також впровадження у виробництво. У відповідні підпункти п. 3.2.Р.2 модуля 3 реєстраційного досьє в форматі CTD можуть бути включені оцінки, використані для спрямування та обґрунтування рішень в ході розробки. Наприклад, аналіз ризиків та функціональних взаємозв’язків, що пов’язують характеристики матеріалів і параметри процесу з критичними показниками якості продукції (COAs), можуть бути включені до підпунктів 3.2.Р.2.1, 3.2.Р.2.2 та 3.2.P.2.3. Аналіз ризиків, що пов’язує розробку виробничого процесу із якістю продукції, може бути включений до підпункту 3.2.P.2.3.
3.2. Простір проектних параметрів
Як основа пропонованого виробничого процесу, простір (простори) проектних параметрів може(уть) бути описаний(і) в підпункті реєстраційного досьє, що містить опис виробничого процесу і контролю процесу (3.2.P.3.3). При необхідності додаткова інформація може бути надана в підпункті реєстраційного досьє, де описано контроль критичних стадій і проміжної продукції (3.2.Р.3.4). Підпункти реєстраційного досьє щодо розробки препарату та виробничого процесу (3.2.Р.2.1, 3.2.Р.2.2 та 3.2.P.2.3) є придатними для розміщення резюме та опису досліджень з розробки препарату і процесу, які служать основою для простору(ів) проектних параметрів. В підпункті реєстраційного досьє, що містить обґрунтування специфікації(й) на лікарський препарат (3.2.P.5.6), може бути обговорений взаємозв’язок простору(ів) проектних параметрів із загальною стратегією контролю.
3.3. Стратегія контролю
Підпункт реєстраційного досьє, що містить обґрунтування специфікації на лікарський препарат (3.2.P.5.6), є хорошим місцем, щоб підсумувати загальну стратегію контролю лікарського препарату. Однак детальна інформація про контроль вхідних матеріалів та конт-роль процесу все одно має бути надана у відповідних структурних елементах реєстраційного досьє в форматі CTD (наприклад, лікарська речовина (підрозділ S), контроль допоміжних речовин (3.2.Р.4), опис виробничого процесу та контролю процесу (3.2.Р.3.3), контроль критичних стадій і проміжної продукції (3.2.Р.3.4)).
3.4. Інформація щодо лікарської речовини
Якщо критичні показники якості (COAs) лікарської речовини мають потенційну можливість впливати на критичні показники якості (COAs) лікарського препарату або на процес його виробництва, в пункті реєстраційного досьє щодо фармацевтичної розробки може бути доречним деяке обговорення критичних показників якості (COAs) лікарської речовини (наприклад, у підпункті 3.2.Р.2.1).
Додаток 1 (довідковий). Різні підходи до фармацевтичної розробки
Наведену нижче таблицю 1.1 було розроблено, щоб проілюструвати деякі потенційні протилежності між тим, що можна було б вважати мінімальним підходом та більш поглибленим підходом, заснованим на якості шляхом розробки, стосовно різних аспектів фармацевтичної розробки та управління життєвим циклом. Порівняння наведені лише для того, щоб допомогти зрозуміти ряд можливих підходів до фармацевтичної розробки; їх не слід вважати всеохоплюючими. Таблиця 1.1 не призначена для того, щоб спеціально визначити єдиний підхід, який компанія може вибрати для використання. При більш поглибленому підході встановлення простору проектних параметрів або використання випробувань при випуску в реальному часі не є обов’язковим. Існуюча у фармацевтичній промисловості практика є різноманітною і зазвичай лежить між цими двома підходами, представленими в таблиці 1.1.
Таблиця 1.1
|
Аспекти |
Мінімальні підходи |
Поглиблені підходи, засновані на якості шляхом розробки |
|
Повна фармацевтична розробка |
— Переважно емпіричний. — Дослідження з розробки частіше відбувається з однією перемінною в одному дослідженні. |
— Системний, що встановлює зв’язок між механістичним розумінням характеристик матеріалів та параметрів процесу з критичними показниками якості лікарського препарату. — Експерименти з багатьма перемінними, щоб зрозуміти препарат і процес. — Встановлення простору проектних параметрів. — Використання інструментів процесно-аналітичної технології (PAT). |
|
Виробничий процес |
— Незмінний. — Валідація переважно базується на перших промислових серіях. — Фокус на оптимізацію та відтворюваність. |
— Регулюється в межах простору проектних параметрів. — Підхід до валідації протягом життєвого циклу та, в ідеалі, безперервна веріфікація процесу. — Фокус на стратегію контролю та стійкість. — Використання статистичних методів контролю процесу. |
|
Контроль процесу |
— Випробування в процесі виробництва переважно для прийняття рішень «відповідає/не відповідає вимогам». — Автономні аналізи (поза лінією). |
— Використання інструментів РАТ з відповідним контролем прямого та зворотного зв’язку. — Технологічні операції відстежуються та спрямовуються для постійного поліпшення після затвердження. |
|
Специфікації на препарат |
— Основні засоби контролю. — Засновані на даних щодо серій, наявних на момент реєстрації. |
— Частина загальної стратегії контролю якості. — Засновані на необхідних функціональних характеристиках препарату з відповідними підтверджувальними даними. |
|
Стратегія контролю |
— Якість лікарського препарату контролюють переважно шляхом випробувань проміжної продукції (матеріалів в ході процесу) та готового препарату. |
— Якість лікарського препарату гарантована стратегією контролю, заснованою на оцінці ризиків для повного розуміння препарату і процесу. — Контроль якості зміщується на попередні етапи з можливістю випробування при випуску в реальному часі або з можливістю зменшення випробувань готової продукції. |
|
Управління життєвим циклом |
— Реагуюче (тобто, рішення проблем і коригувальні дії). |
— Запобіжні дії. — Постійне поліпшення полегшене. |
Додаток 2 (довідковий). Ілюстративні приклади
А. Використання інструменту оцінки ризиків
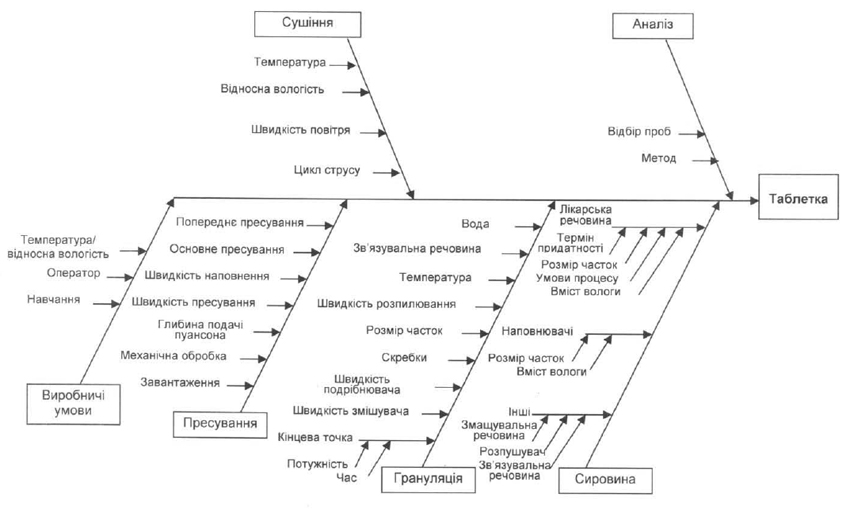
Наприклад, щоб розробити діаграму Ішикави (діаграму зв’язку причин та наслідків або «риб’ячий скелет»), що ідентифікує потенційні перемінні, які можуть вплинути на необхідний показник якості, могла б працювати разом багатофункціональна команда експертів. Далі команда могла б розташувати перемінні, базуючись на ймовірності, тяжкості і можливості виявлення невідповідностей із використанням аналізу характеру наслідків відмов (FMEA) або аналогічних інструментів, використовуючи за основу попередні знання і попередні експериментальні дані. Щоб оцінити вплив розміщених у верхній частині перемінних, щоб отримати більш глибоке розуміння процесу, а також щоб розробити правильну стратегію контролю, можна було б використати планування експериментів або інші експериментальні підходи.
Діаграма Ішикави (Ishikava Diagram)
В. Зображення взаємодій
Наведені нижче на рисунку 2.0 графіки відображають наявність або відсутність взаємодії між трьома параметрами процесу та вплив на рівень продукту розкладу Y. На рисунку 2.0 показана серія з двомірних діаграм, що ілюструють результат взаємодії між трьома параметрами процесу сушіння (початкового вмісту вологи, температури, середнього розміру часток) грануляту (проміжної продукції для лікарського препарату) та вплив на продукт розкладу Y. Різниця відповідних кутів нахилу прямих або кривих у межах діаграми вказує на присутність взаємодії. У цьому прикладі початковий вміст вологи і температура взаємодіють; але початковий вміст вологи та середній розмір часток не взаємодіють, також як і температура та середній розмір часток.
Рисунок 2.0
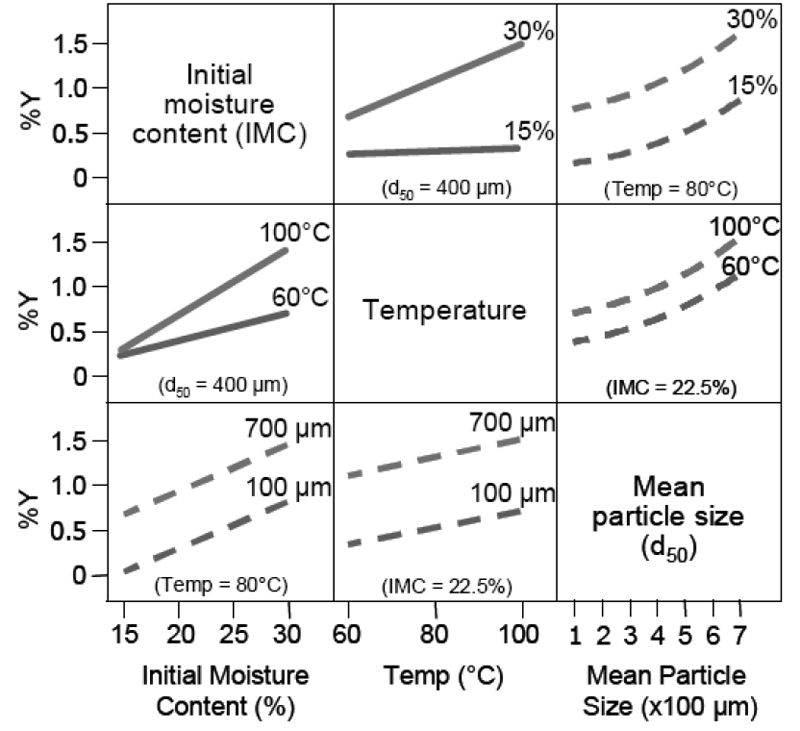 Initial Moisture Content (IMC) — початковий вміст вологи.
Initial Moisture Content (IMC) — початковий вміст вологи.
Mean Particle Size — середній розмір часток.
Temperature (Temp) — температура.
С. Презентації простору проектних параметрів
Приклад 1: Графіки розчинення зображені як поверхня ділянки (рис. 2.1а) і як контур ділянки (рис. 2.1b). Параметри 1 і 2 є факторами операції грануляції, які впливають на швидкість розчинення таблетки (наприклад, характеристика допоміжної речовини, кількість води, розмір гранул).
Рисунок 2.1
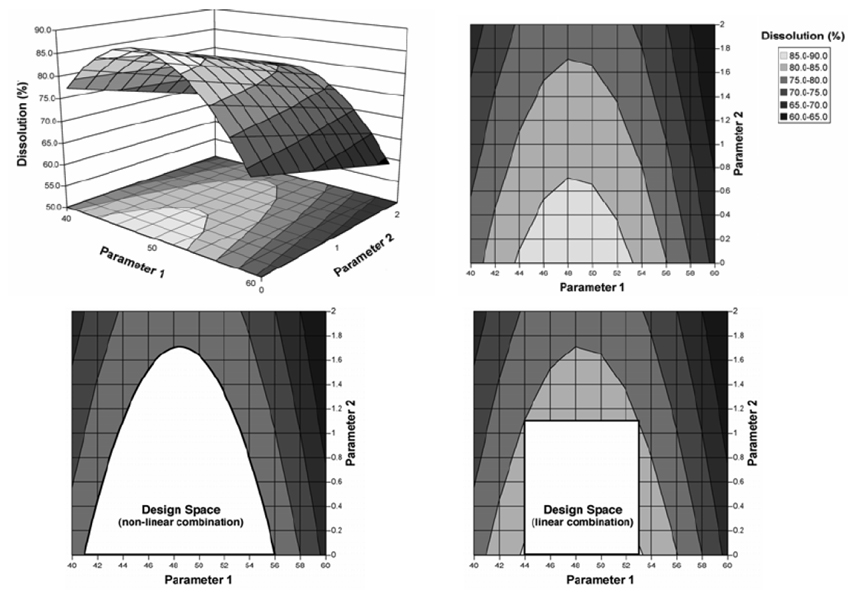
Рисунок 2.1а — Поверхня діаграми розчинення (dissolution), як функція двох параметрів операції грануляції. Необхідна величина розчинення 80 %.
Рисунок 2.1b — Контур діаграми розчинення (dissolution) з прикладу 2.1а.
Рисунок 2.1с — Простір проектних параметрів (Design Space) для параметрів грануляції, визначений шляхом нелінійного сполучення їх діапазонів, у межах яких досягнуто задовільне розчинення (тобто, > 80 %).
Рисунок 2.1d — Простір проектних параметрів (Design Space) для параметрів грануляції, визначений шляхом лінійного сполучення їх діапазонів, у межах яких досягнуто задовільне розчинення (тобто, > 80 %).
Два приклади надання можливих просторів проектних параметрів. На рис. 2.1с простір проектних параметрів визначений шляхом нелінійного сполучення діапазонів параметрів, при яких досягається відповідний показник якості — розчинення. У цьому прикладі простір проектних параметрів виражений зрівнюванням поверхні розчинення на межі задовільного значення (тобто, розчинення — 80 %). Діапазон прийнятності одного параметра залежить від значення іншого. Наприклад:
– якщо параметр 1 має значення 46, то діапазон для параметра 2 має бути від 0 до 1,5
– якщо параметр 2 має значення 0,8, то діапазон для параметра 1 має бути від 43 до 54
Підхід на рис. 2.1c ілюструє максимальний дозволений діапазон роботи для досягнення необхідного ступеня розчинення. На рис. 2.1d простір проектних параметрів визначений як менший діапазон, засновуючись на лінійному сполученні параметрів.
– параметр 1 має діапазон від 44 до 53
– параметр 2 має діапазон від 0 до 1,1
Хоча підхід на рис. 2.1d дає більш жорсткі обмеження, заявник для спрощення роботи може віддати йому перевагу.
У даному прикладі розглядається тільки два параметри і, таким чином, можна легко надати графічне зображення. Коли залучено багато параметрів, простір проектних параметрів може бути наданий для двох параметрів подібно до наведених вище прикладів при різних значеннях (наприклад, високому, середньому, низькому) у межах діапазону третього параметра, четвертого параметра і так далі. Як альтернатива, простір проектних параметрів може бути пояснений математично через рівняння, що описують відношення між параметрами для успішної роботи.
Приклад 2: Простір проектних параметрів визначають із загальної області успішних робочих діапазонів для декількох критичних показників якості. Відношення двох критичних показників якості, тобто стираності таблетки та розчинення, до двох технологічних параметрів операції грануляції показані на рис. 2.2а і 2.2b. Параметри 1 і 2 є факторами операції грануляції, які впливають на швидкість розчинення таблетки (наприклад, характеристика допоміжної речовини, вміст води, розмір гранул). Рис. 2.2c ілюструє перекриття цих областей і максимальний діапазон пропонованого простору проектних параметрів. Заявник може обрати використання всієї області як простору проектних параметрів, або будь-якої її частини.
Рисунок 2.2
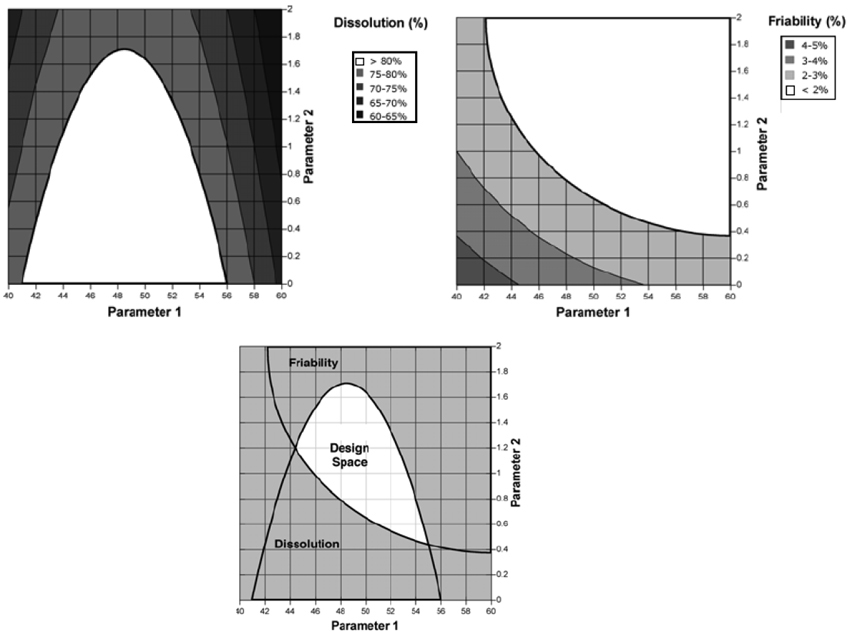
Рисунок 2.2а — Контур діаграми розчинення (Dissolution), як функція параметрів 1 та 2.
Рисунок 2.2b — Контур діаграми стираності (Friability), як функція параметрів 1 та 2.
Рисунок 2.2с — Пропонований простір проектних параметрів (Design Space), що складається з області перекриття діапазонів для стираності (Friability) і розчинення (Dissolution).
Приклад 3: Простір проектних параметрів для операції сушіння залежить від зміни температури та/або тиску у часі. Кінцева точка вмісту вологи становить 1–2 %. Робота за верхньою межею простору проектних параметрів може призвести до надмірного утворення домішки, а робота за нижньою межею простору проектних параметрів — до надмірного стирання часток.
Рисунок 2.3
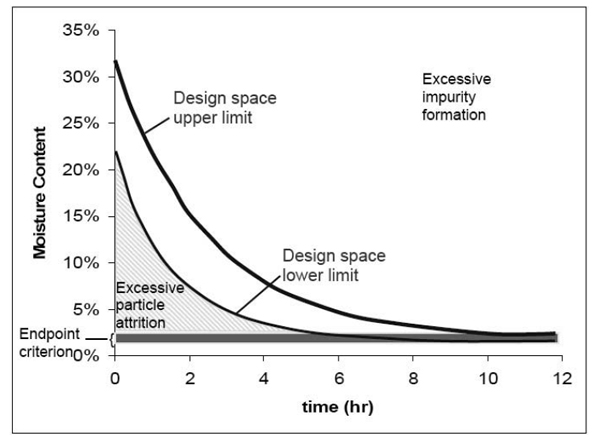
Moisture Content — вміст вологи.
Time (hr) — час (ч).
Design space lower limit — нижня межа простору проектних параметрів.
Design space upper limit — верхня межа простору проектних параметрів.
Excessive impurity formation — надмірне утворення домішок.
Excessive particle attrition — надмірне стирання часток.
Endpoint criterion — критерій кінцевої точки.
Національний додаток (довідковий). Бібліографія
1. EMEA/СHМР/167068/2004 — ICH. — Part I. — ICH Topic Q 8 (R2). — Step 5: Note for Guidance on Pharmaceutical Development, 2009
2. Annex to EMEA/СHМР/167068/2004 — ICH. — Part II. — ICH Topic Q 8 Annex. — Step 5: Annex to Note for Guidance on Pharmaceutical Development (EMEA/СHМР/167068/2004), 2009
3. CPMP/QWP/155/96. — Note for Guidance on Development Pharmaceutics, 1998
4. CPMP/QWP/054/98 Corr. — Decision Trees for the Selection of Sterilisation Methods. Annex to Note for Guidance on Development Pharmaceutics (CPMP/QWP/155/96), 2000
5. Настанова 42-3.1:2004. — Настанови з якості. Лікарські засоби. Фармацевтична розробка / М. Ляпунов, В. Георгієвський, О. Безугла та ін. — Київ, МОЗ України, 2004
6. ДСТУ 1.5–2003. — Національна стандартизація. Правила побудови, викладання, оформлення та вимоги до змісту нормативних документів / І. Аширова, О. Брянська, Є. Козир, Я. Юзьків. — Київ, Держспоживстандарт України, 2003
7. Настанова СТ-Н МОЗУ 42–1.0:2005. — Фармацевтична продукція. Система стандартизації. Основні положення / М. Ляпунов, В. Георгієвський, Т. Бухтіарова та ін. — Київ, МОЗ України, 2005
8. ДСТУ 1.7–2001. — Національна стандартизація. Правила і методи прийняття та застосування міжнародних і регіональних стандартів / О. Одноколов, В. Тетера, Я. Юзьків. — Київ, Держспоживстандарт України, 2003
9. CPMP/ICH/2736/99 (ICH Topic Q1A (R2)). — Note for Guidance on Stability Testing: Stability Testing of New Drug Substances and Products, 2003
10. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. — OJ L 311, 28.11.2001. — P. 67–128
11. Настанова 42–4.2:2011. — Лікарські засоби. Управління ризиками для якості (ICH Q9) / М. Ляпунов, О. Безугла, О. Соловйов та ін. — Київ, МОЗ України, 2011
12. Настанова СТ-Н МОЗУ 42–4.0:2011. — Лікарські засоби. Належна виробнича практика / М. Ляпунов, О. Безугла, О. Соловйов та ін. — Київ, МОЗ України, 2011
13. Doc. Ref. EMEA/P/24143/2004. — Procedure for European Union guidelines and related documents within pharmaceutical legislative framework, 2005
14. СРМР/ICH/367/96 (ICH Topic Q6A). — Note for Guidance Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, 2000
15. Настанова 42–3.2:2004. — Настанови з якості. Лікарські засоби. Специфікації: контрольні випробування та критерії прийнятності
16. Настанова 42–4.3:2011. — Лікарські засоби. Фармацевтична система якості (ICH Q10) / М. Ляпунов, О. Безугла, О. Соловйов та ін. — Київ, МОЗ України, 2011
17. EudraLex. — The Rules Governing Medicinal Products in the European Union. — Volume 4. EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use
http://ec.europa.eu/health/documents/eudralex/vol-4/index en.htm
18. EMA/INS/GMP/79766/2011. — Quality Risk Management (ICH Q9), 2011
19. EMA/INS/GMP/79818/2011. — Pharmaceutical Quality System (ICH Q10), 2011
20. EMEA/CHMP/CVMP/QWP/17760/2009 Rev 1 (Draft). — Guideline on the Use of Near Infrared Spectroscopy by the Pharmaceutical Industry and the Data Requirements for New Submissions and Variations, 2009
Ключові слова: виробничий процес, допоміжна речовина, лікарська речовина, лікарська форма, лікарський препарат (готовий препарат), мікробіологічні властивості, простір проектних параметрів, реєстраційне досьє, система контейнер/закупорювальний засіб, специфікація, стратегія контролю, управління ризиками для якості, фармацевтична розробка, якість, якість шляхом розробки.