Метаболічна кардіоміопатія (раніше її визначали як дистрофію міокарда, міокардіодистрофію) — ураження міокарда без ознак запалення різної етіології, в основі якого лежить порушення обміну речовин, процесу утворення енергії і/або порушення її перетворення в механічну роботу, що призводить до дистрофії міокарда і недостатності скорочувальної та інших функцій серця.
Етіологія
Метаболічна кардіоміопатія розвивається в результаті впливу патогенних чинників при різних захворюваннях і станах (схема 8.1)
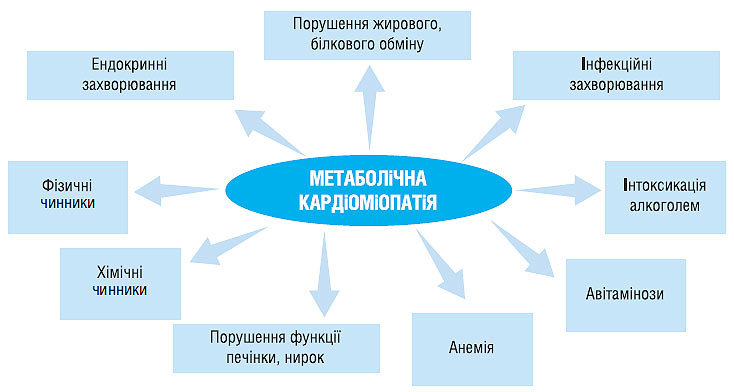 .
.
Схема 8.1. Захворювання і стани, які призводять до розвитку метаболічної кардіоміопатії
Серед фізичних чинників можуть розглядатися радіація, вібрація, перегрівання, переохолодження, гіперінсоляція.
До хімічних чинників відносяться лікарські засоби, токсичний вплив побутових і промислових отрут.
Патогенез
У виникненні і розвитку метаболічного ураження міокарда при різних захворюваннях суттєве значення має порушення іннервації, транспорту та утилізації енергії в кардіоміоцитах, тобто їх енергозабезпечення.
Напруженість регулюючих систем, функцій міокарда та метаболічних процесів у кардіоміоцитах обмежує резервні можливості серця. Тривала гіперфункція сама по собі, а особливо у несприятливих умовах на фоні основного захворювання, може призвести до виникнення енергетичного дефіциту і порушення пристосувальних змін у міокарді.
Механізми зниження продукції енергії в ураженому серці включають зниження щільності капілярів, збільшення міжкапілярної відстані, а також збільшений діаметр гіпертрофованих кардіоміоцитів, що погіршує дифузію кисню і зумовлює виникнення гіпоксії міокарда. Один із механізмів пов’язаний також з порушенням функції мітохондрій, яке зумовлене редукованим синтезом окисних ензимів. Зниження синтезу окисних ензимів відбувається внаслідок порушення проліферативної реакції, що частково опосередкована експресією рецепторів PPARα, які відіграють ключову роль у біогенезі мітохондрій. Ці рецептори регулюють транскрипцію багатьох ензимів і переносників (транспортерів), які беруть участь у транспорті і окисненні жирних кислот. Знижується також здатність серця відновлювати запаси макроергічних фосфатів. Зменшення окиснення жирних кислот викликає накопичення ліпідів, що сприяє некрозу ушкоджених мембран, при цьому вивільнення реактивних молекул (цитохромів, радикалів кисню) призводить до апоптозу. Прискорений гліколіз, викликаний порушенням процесів окисного фосфорилування, призводить до ацидозу, що в свою чергу інгібує багато реакцій, які включені в процес скорочення — розслаблення. Одним із таких процесів найбільш важливим є підвищення концентрації кальцію в цитозолі, що ініціює безліч реакцій, які призводять до некрозу міоцитів.
У прогресуванні метаболічної кардіоміопатії провідну роль відіграє посилення реакцій вільнорадикального перекисного окиснення ліпідів клітинних мембран. Пошкоджуючи мембрани, гідроперекиси і вільні радикали знижують активність ліпідозалежних ферментативних реакцій (до яких відносяться основні життєвоважливі ферменти іонного транспорту і дихального ланцюга мітохондрій), змінюють мембранорецепторні системи клітини, зумовлюючи розвиток медіаторного дисбалансу, активують протеолітичні та лізосомальні ферменти.
Патологічна анатомія
Метаболічні ураження міокарда охоплюють всі стадії порушення обміну серцевого м’яза — від функціональних розладів до грубих структурних змін. Морфологічні зміни відбуваються всередині клітин міокарда і не супроводжуються збільшенням їх кількості. Найбільш чутливі до патогенних впливів мітохондрії та ендоплазматична мережа. Для дегенеративних змін міокарда характерне мозаїчне порушення структури кардіоміоцитів: у тій самій клітині серед набряклих мітохондрій із частково або повністю зруйнованими внутрішніми перегородками можуть бути мітохондрії з нормальною структурою.
Як правило, усунення патогенної причини призводить до поступової нормалізації ультраструктур кардіоміоцита, яка зумовлена внутрішньоклітинними регенераторними процесами. Ушкоджені міофібрили відновлюються у результаті активної діяльності рибосом: поступово зменшується внутрішньоклітинний набряк, з’являються зерна глікогену, зменшується кількість жирових включень. При тривалому та інтенсивному впливі ушкоджуючих факторів на міокард дистрофічні зміни можуть призводити до глибоких морфологічних змін, що закінчуються розвитком міокардіофіброзу.
Втрата частини міокарда відновлюється шляхом збільшення маси специфічних структур у неушкоджених клітинах, відбувається гіперплазія мітохондрій, саркоплазматичного ретикулума, рибосом. У результаті розвивається гіпертрофія міокарда, що являє собою компенсаторну регенераторно-гіперпластичну реакцію, характерну для міокарда. Біохімічні процеси частіше порушуються у ЛШ.
Клінічна картина
Клінічні прояви різноманітні і не є специфічними. Початкові стадії можуть мати безсимптомний перебіг, згодом зниження скоротної функції міокарда може призвести до тяжкої СН.
Часто на фоні проявів основного захворювання визначають кардіалгію (частіше в області верхівки серця (92%), рідше — за грудиною (15%)), розширення границь серця, приглушеність тонів, невеликий систолічний шум на верхівці серця, порушення ритму (в основному екстрасистолічну аритмію).
Діагностика
ЕКГ є основним методом розпізнавання дистрофічних змін міокарда, які стосуються в основному процесу реполяризації і проявляються найчастіше змінами кінцевої частини шлуночкового комплексу: визначається депресія сегмента ST, що має висхідний характер до позитивного зубця Т. Зубець Т також може бути деформованим, низькоамплітудним, згладженим або негативним.
Також визначається зниження вольтажу комплексу QRS, особливо виражене при ожирінні і мікседемі, при тиреотоксикозі амплітуда зубців, як правило, збільшена. У деяких випадках можуть виникати уповільнення внутрішньопередсердної провідності, збільшення інтервалу Q–T, порушення внутрішньошлуночкової провідності. Серед порушень ритму найчастіше відзначають синусову тахікардію і екстрасистолічну аритмію.
При формулюванні діагнозу слід насамперед вказати основне захворювання або етіологічний чинник, характер перебігу кардіоміопатії та основні клінічні прояви (наявність порушень ритму і провідності, стадію СН).
У диференціальній діагностиці метаболічної кардіоміопатії мають значення навантажувальні та медикаментозні проби, при потребі проводиться коронарографія.
Лікування
Незалежно від пошкоджувального чинника, принциповими для метаболічної кардіоміопатії можуть бути наступні положення:
- порушення метаболізму міокарда при своєчасному лікуванні оборотні;
- виражена СН розвивається порівняно рідко, в основному в кінцевій стадії захворювання, але СН, що виникла, резистентна до серцевих глікозидів і успіх терапії цілком залежить від ступеня відновлення порушеного обміну речовин у міокарді.
Допомогу хворим слід починати з виявлення і усунення причини розвитку дистрофії міокарда. Велике значення має відмова від паління і зловживання алкоголем, виключення фізичного та психоемоційного перенапруження.
Поряд з лікуванням основного захворювання необхідне відновлення адекватного енергетичного обміну. На перший план виступає застосування комплексу лікарських засобів, спрямованих на поліпшення транспорту кисню в тканини та його утилізації.
На обмін речовин у клітині впливають дві групи лікарських засобів: регулятори екстрацелюлярної природи (гормональні препарати, блокатори і стимулятори центральної і периферичної нервової системи) і регулятори метаболізму інтрацелюлярної природи (ферменти і антиферменти, вітаміни, кофактори, різноманітні метаболіти), які впливають на різні шляхи обміну речовин.
При порушенні процесів окисного фосфорилування застосовують комплекс вітамінів, що включає вітаміни В1, В2, пантотенову і ліпоєву кислоти. Вітаміни групи В впливають на білковий, ліпідний, вуглеводно-енергетичний обмін, синтез амінокислот, нуклеотидів.
Серед препаратів з антиоксидантними властивостями широко застосовують токоферолу ацетат, його одночасне застосування з вітаміном РР (нікотиновою кислотою) сприяє покращанню енергетичного забезпечення скоротної функції міокарда. Активним антиоксидантом, що бере участь у окислювально-відновних процесах, є вітамін С.
Велике значення для нормалізації метаболізму міокарда має достатнє надходження в організм незамінних амінокислот; у тому числі метіоніну, лейцину, аланіну, валіну, лізину, трионіну, триптофану, що є пластичним матеріалом для синтезу білка, ферментів, коферментів. З метою покращання їх засвоєння рекомендується призначати ці засоби у комплексі з анаболічними стероїдами (метандієнон, нандролон).
При прогресуванні дистрофічного процесу показане застосування внутрішньо калію хлориду, калію і магнію аспарагинату з метою усунення закономірного дефіциту внутрішньоклітинного калію і порушення балансу кальцію та магнію, що призводить до відновлення регуляції всіх функцій міокарда (збудливості, провідності, автоматизму та скоротливості).
Для активації синтезу білків та нуклеїнових кислот застосовують солі оротової кислоти (оротат калію/магнію).
Проведена терапія повинна бути спрямована на підвищення генерації енергії та стійкості міокарда до гіпоксії. Останнім часом велику увагу приділяють ролі серотонінергічної системи в регуляції стресорної реакції. Специфічною особливістю нікотинаміду є його здатність стимулювати процеси аеробного окиснення і обмін глікогену, тим самим підвищуючи стійкість кардіоміоцитів до гіпоксії.
Тривалість інтенсивної метаболічної терапії на ранніх стадіях у хворих з переважно функціональними порушеннями становить 3 тиж. При прогресуванні дистрофії міокарда і виявленні органічного ураження серця курс терапії слід повторювати кілька разів на рік.
УРАЖЕННЯ СЕРЦЕВО-СУДИННОЇ СИСТЕМИ ПРИ ЕНДОКРИННИХ ПОРУШЕННЯХ
Серцево-судинна система часто залучається до патологічного процесу при захворюваннях залоз внутрішньої секреції. У клінічній картині можуть переважати функціональні зміни з боку серцево-судинної системи, і пацієнт з ендокринним захворюванням стає фактично «кардіальним» хворим. Ураження серця при ендокринних захворюваннях в основному зумовлено порушеннями обміну, викликаними нестачею або надлишком того або іншого гормону в організмі.
Ураження серця при цукровому діабеті
Термін «діабетична кардіоміопатія» вперше запропонований в 1954 р. для позначення кардіальних змін, що передують ІХС.
Патогенез
Патогенез метаболічної кардіоміопатії при цукровому діабеті багатофакторний, ураження серцево-судинної системи зумовлене складними порушеннями процесів обміну, що виникають у зв’язку з абсолютною або відносною недостатністю інсуліну та порушенням толерантності до глюкози.
Патогенез міокардіальних порушень включає кілька основних механізмів: пошкодження кардіоміоцитів, мікроциркуляторні та нейровегетативні порушення. Перший механізм пов’язаний з порушенням метаболізму кардіоміоцитів, зниженням ефективності енергетичних, пластичних процесів і зміною іонного метаболізму, в результаті чого знижуються компенсаторні можливості серцево-судинної системи, порушується скоротна функція міокарда, зменшується толерантність до фізичних навантажень. Другий механізм ґрунтується на мікроциркуляторних порушеннях у дрібних артеріях міокарда як локальний прояв генералізованої мікроангіопатії. Третій механізм включає ураження вегетативної нервової системи в результаті формування нейровегетодистонії.
Кардіоміопатія, що не зумовлена порушенням коронарного кровообігу, виникає у хворих молодого віку з ювенільним цукровим діабетом, для яких не характерний розвиток вираженого атеросклерозу, або у пацієнтів старшого віку без супутньої ІХС.
Інсулін чинить на серце пряму дію, що полягає у збільшенні надходження і стимуляції окиснення глюкози і лактату, підвищенні утворення глікогену в клітинах міокарда. Непрямий ефект інсуліну проявляється у зниженні вмісту вільних жирних кислот у плазмі крові, зменшенні їх надходження в серцевий м’яз.
Дефіцит інсуліну викликає порушення утилізації тканинами глюкози і підсилює розщеплення ліпідів і білків, а також призводить до виражених змін складу внутрішнього середовища організму — гіперглікемії, гіперкетонемії, гіперліпідемії з накопиченням у крові вільних жирних кислот, диспротеїнемії, метаболічного ацидозу, оксидантного стресу, що викликає апоптоз міоцитів. Ці порушення є визначальними факторами зміни структури і функції міокарда.
Патогенез і морфогенез діабетичного ураження серця зумовлені не тільки впливом гіперінсулінемії на ендотелій судин, енергетичні та метаболічні процеси в міокарді, але й безпосередньо пов’язані з токсико-метаболічним ушкодженням кардіоміоцитів.
Існує думка, що причиною руйнування структур кардіоміоцитів, порушення структури сарколеми і її дериватів, порушення іонної рівноваги та зниження активності актоміозинового комплексу кардіоміоцитів є пряма глюкозотоксичність.
У патогенезі кардіоміопатії важливу роль відіграє тканинна гіпоксія. Велике значення у розвитку гіпоксії має порушення транспорту кисню кров’ю, функції дихальних ферментів під впливом вираженого ацидозу. При цукровому діабеті потреба тканин, у тому числі міокарда, у кисні підвищена.
Важливим чинником розвитку міокардіодистрофії є порушення нейроендокринної регуляції функції серця, яке пов’язане з переважанням ефектів контрінсулярних гормонів. Доведено, що у пацієнтів підвищується продукція адренокортикотропного і соматотропного гормонів, а також глюкокортикоїдів, катехоламінів і глюкагону, що призводить до ініціації цілої групи метаболічних та ультраструктурних процесів, які викликають розвиток метаболічної кардіоміопатії.
Патогенез збільшення жорсткості міокарда пов’язаний з порушенням транспорту кальцію, електромеханічним дисбалансом, що супроводжується асинхронністю розслаблення та механічних факторів.
Патологічна анатомія
Фіброз міокарда, що відзначається при метаболічних порушеннях, пов’язаний з порушенням внутрішньоклітинного метаболізму оксиду азоту і кальцію, а також з проліферативними процесами, зумовленими дією інсуліну та ІФР. Морфологічною основою дистрофії міокарда при цукровому діабеті є мікроангіопатія, що характеризується інфільтрацією тучними клітинами і фібриноїдним набряком стінок дрібних судин. При морфологічному дослідженні виявляють розвиток апоптозної дегенерації, втрату синаптичних пухирців, появу великих вакуолей у цитоплазмі клітин симпатичних гангліїв. При гістохімічному дослідженні в стінках судин визначається відкладення глікопротеїнів, а на ультраструктурному рівні — потовщення базальної мембрани судинної стінки. Важливе значення надають дезорганізації м’язових волокон гіпертрофованого міокарда.
Клінічна картина і діагностика
Хворі з ювенільним цукровим діабетом іноді відзначають біль колючого характеру в ділянці серця. Виникнення тахікардії у стані спокою пов’язане з ураженням блукаючого нерва і відносним переважанням тонусу симпатичного відділу вегетативної нервової системи. Тахікардія супроводжується неефективними скороченнями міокарда, що призводить до виснаження енергоресурсів і, як наслідок, до зниження скоротної функції міокарда і розвитку СН.
Розміри серця в межах норми. Деяке приглушення тонів серця і систолічний шум на верхівці частіше відзначають у осіб, які хворіють цукровим діабетом >5 років. У подальшому гіперглікемія та інсулінорезистентність асоціюються із збільшенням маси ЛШ і появою симптомів СН.
На ЕКГ визначається синусова тахікардія або брадикардія, шлуночкова екстрасистолічна аритмія, порушення процесів реполяризації: зсув сегмента ST, зміна амплітуди, інверсія, сплощення, згладженість або двофазність зубця Т, порушення внутрішньошлуночкової провідності.
При ехоКГ-обстеженні найбільш ранньою ознакою ураження міокарда при цукровому діабеті є порушення діастолічної функції, що відзначають у 27–69% безсимптомних хворих.
Рівень глікемії у плазмі крові натще >7,0 ммоль/л.
Лікування
Одним із основних завдань лікування хворих з діабетичною кардіоміопатією є профілактика подальшого прогресування ураження міокарда і розвитку СН. Важливою є боротьба з факторами ризику: палінням, ожирінням, малорухомим способом життя, незбалансованим харчуванням. Рекомендації з оптимізації способу життя повинні бути обґрунтованими щодо необхідності дотримування відповідної низькокалорійної дієти для зменшення маси тіла, відмови від паління, регулярних фізичних навантажень.
Важливим завданням є нормалізація обміну речовин, що включає досягнення цільових рівнів глюкози, аглюкозурії, нормалізації рівня глікозильованого гемоглобіну. Регулярні фізичні навантаження дозволяють знизити резистентність до інсуліну, підвищити толерантність до глюкози, сприяють утилізації глюкози крові і вільних жирних кислот у м’язовій тканині, позитивно впливають на функціонування серцево-судинної системи.
Фармакотерапія цукрового діабету II типу спрямована на посилення секреції інсуліну, зниження інсулінорезистентності. До складу терапії включені препарати з різними механізмами дії: бігуаніди, похідні сульфонілсечовини, глітазони, глініди, інгібітори α-глюкозидази, інсулін. Застосування метформіну дозволяє покращити контроль глюкози крові у хворих на цукровий діабет і сприяє зниженню загальної смертності на 36%.
Для відновлення метаболічних порушень у міокарді призначають препарати α-ліпоєвої кислоти, що активує ферменти мітохондрій, збільшує окиснення глюкози, сповільнює глюконеогенез і кетогенез, як антиоксидант захищає клітини від ушкоджувальної дії вільних радикалів. Застосовують також препарати, що сприяють корекції порушень обміну речовин в міокарді: триметазидин, триметилгідразинію пропіонат.
Тиреотоксична хвороба серця
Патогенез
Порушення функції серцево-судинної системи є частим ускладненням тиреотоксикозу. Зміни серцево-судинної системи при тиреотоксикозі («тиреотоксичне серце») зумовлені впливом надлишкової кількості тиреоїдних гормонів (L-тироксину і 3,5,3-трийод-L-тироніну) на процеси обміну в міокарді, гемодинаміку і симпатичну нервову систему. Одним із важливих ефектів тиреоїдних гормонів є роз’єднання окисного фосфорилування, що призводить до зниження в серцевому м’язі вмісту АТФ і креатинфосфату. У результаті відбувається пригнічення анаболічних процесів: знижується синтез і посилюється розпад глікогену і білка, знижується вміст калію в еритроцитах та інших клітинах. Споживання кисню міокардом збільшується, але ефективність його утилізації в процесі біологічного окиснення знижується. При надмірній кількості тироксину порушується проникність мітохондріальних мембран.
Під впливом тиреоїдних гормонів відбувається посилення скоротної функції міокарда, вірогідно, внаслідок активізації стимулювального впливу на серце і прямої дії тироксину на серцевий м’яз. Унаслідок порушень енергетичних процесів і роботи калій-натрієвого насоса відбувається прискорення спонтанної деполяризації в клітинах синусного вузла, що призводить до утворення в ньому імпульсів з вищою частотою. Надлишок тиреоїдних гормонів змінює симпатичний і парасимпатичний вплив на міокард. При вираженому тиреотоксикозі в результаті різкого зниження ефективності біологічного окиснення, переважання розпаду білка над його синтезом знижується рівень енергетичних ресурсів і пластичних процесів, що в результаті призводить до пригнічення скоротної функції міокарда.
Гемодинаміка
В основі гіперфункції серця при тиреотоксикозі лежить підвищення скоротної здатності міокарда, яке зумовлене як підвищенням активності симпатичної нервової системи, так і безпосередньою дією тиреоїдних гормонів на міокард. При тиреотоксикозі відбуваються різкі зміни гемодинаміки: збільшується ХОК (в основному за рахунок підвищення ЧСС), швидкість кровотоку і ОЦК. Периферичний судинний опір у великому колі кровообігу знижується, а в малому підвищується. У результаті підвищується пульсовий тиск. Серцевий м’яз відчуває діастолічне перевантаження, а праві відділи серця ще і систолічне перевантаження, збільшена робота серця відбувається у вкрай несприятливому для нього режимі: внаслідок змін гемодинаміки ЛШ працює в умовах постійної ізотонічної гіперфункції, а правий — в умовах змішаного типу гіперфункції (навантаження об’ємом і опором), проте при цьому відсутні умови для розвитку компенсаторної гіпертрофії міокарда (посилений розпад і знижений синтез білка, зменшена кількість АТФ і креатинфосфата). Все це досить швидко призводить до розвитку СН.
Патологічна анатомія
Гістологічні зміни міокарда при тиреотоксикозі характеризуються запаленням і дегенерацією аж до розвитку вогнищ некрозу і фіброзу. Гістологічні зміни в міокарді непостійні і неспецифічні. Чинники, що зумовлюють ураження серцево-судинної системи у хворих з дифузним токсичним зобом, спочатку викликають дистрофічні зміни, а надалі дегенеративно-склеротичні. При тяжкому перебігу захворювання виникають дегенеративні зміни в мітохондріях і їх розпад.
Клінічна картина і діагностика
Хворі часто скаржаться на біль у ділянці серця ниючого, колючого, нерідко стенокардитичного характеру, а також серцебиття, що виникає в стані спокою, а при фізичному навантаженні неадекватно посилюється. Хворі відзначають підвищену збудливість, пітливість, м’язову слабкість, тремор рук, зменшення маси тіла. Істотним симптомом є постійна синусова тахікардія, вираженість якої зростає відповідно до ступеня тяжкості токсичного зоба. У 10–20% хворих діагностується тахісистолічна форма фібриляції передсердь. Характерне підвищення САТ, яке зумовлене збільшенням серцевого викиду. Задишка має місце як при навантаженні, так і у стані спокою. СН, в основному правошлуночкова, відзначається в 15–25% випадків. Ознаки лівошлуночкової недостатності звичайно виражені менше, оскільки дуже швидко виникає слабкість ПШ.
При огляді відзначається прекардіальна пульсація і пульсація артерій. Аускультативно — підвищення звучності тонів серця, особливо першого, майже завжди вислуховується систолічний шум на верхівці серця і ЛА.
На ЕКГ, крім синусової тахікардії або фібриляції передсердь, відзначається підвищення амплітуди зубця Р, іноді зміни комплексу QRS, зниження сегмента ST і вольтажу зубця Т.
При ехоКГ-дослідженні на ранній стадії захворювання виявляють помірну гіпертрофію — потовщення задньої стінки, міжшлуночкової перегородки і збільшення скорочувальної функції ЛШ. У подальшому розвивається дилатація порожнин серця, збільшується маса міокарда, зменшується систолічний об’єм крові і ХОК, знижується скоротна функція міокарда.
У сироватці крові визначається підвищення рівнів загального і вільного тироксину, трийодтироніну, зниження рівня тиреотропного гормону.
Лікування
Проводиться у трьох напрямках: нормалізація функції щитовидної залози (досягнення еутиреоїдного стану), усунення симптомів недостатності кровообігу і відновлення синусового ритму (при фібриляції передсердь).
З метою компенсації тиреотоксикозу застосовують антитиреоїдні препарати або проводиться оперативне лікування або радіойодотерапія.
Для зменшення синусової тахікардії застосування серцевих глікозидів недоцільне; широко призначають блокатори β-адренорецепторів. При тахісистолічній формі фібриляції передсердь проводять комбіновану терапію антиаритмічними засобами (пропафенон) і блокаторами β-адренорецепторів, досягаючи відновлення синусового ритму або переведення фібриляції передсердь у нормосистолічну форму.
Лікування СН не має специфічних особливостей і обов’язково повинно проводитися на фоні антитиреоїдної терапії. Необхідно враховувати, що чутливість міокарда до глікозидів наперстянки може бути підвищена.
КЛІМАКТЕРИЧНА (ДИСГОРМОНАЛЬНА) КАРДІОМІОПАТІЯ
Епідеміологія
Зміна демографічної структури суспільства призвело до збільшення в популяції частки жінок старшої вікової групи (на сьогодні у світі близько 500 млн жінок віком старше 50 років, тобто в період менопаузи).
Про існування зв’язку між розладом діяльності серця і зміною функції жіночих статевих органів відомо давно. Захворювання може розвиватися внаслідок дефіциту естрогенів не тільки в клімактеричний період, але й у жінок молодого віку з різними гінекологічними захворюваннями (міома матки, ендометріоз та ін.), при посткастраційному і передменструальному синдромах. Клімактеричну кардіоміопатію діагностують іноді і у чоловіків (клімакс визначають у 10–20% осіб чоловічої статі).
Патогенез
Менопауза, яка не є власне захворюванням, призводить до порушення ендокринної рівноваги в організмі та сприяє розвитку серцево-судинних захворювань.
Порушення активності естрогенів має основне значення у патогенезі обмінних порушень. Естрогени в нормі позитивно впливають на білковий і електролітний обмін у міокарді і регулюють симпатичний вплив на серце. При патологічному клімаксі в міокарді відбуваються метаболічні порушення, що призводять до дистрофічних змін, які у більшості випадків носять оборотний характер і лише іноді закінчуються розвитком міокардіофіброзу (кардіосклерозу) (схема 8.2).

Cхема 8.2. Метаболічні зміни в менопаузі
Збільшення кількості абдомінального жиру і розвиток абдомінального ожиріння пов’язані як з фізіологічними змінами, так і зі змінами способу життя. Серед причин абдомінального ожиріння в період менопаузи можна виділити зміну балансу енергії — зниження швидкості обмінних процесів наряду з підвищенням апетиту та збільшенням надходження енергії з їжею на фоні підвищення тонусу симпатичної нервової системи, посилення глюкокортикоїдної стимуляції і зниження рівня гормону росту.
В основі патогенезу клімактеричної АГ лежить гіпоестрогенія, що супроводжується підвищенням збудливості гіпоталамо-гіпофізарних структур, порушенням центральної та периферичної регуляції судинного тонусу. Одним із механізмів є відсутність в період менопаузи депрессорного ефекту фолікулярного гормону.
Клінічна картина
Найпоширенішими є скарги на тривалий, майже постійний біль у ділянці серця різноманітного характеру, що локалізується зліва від грудини, в області верхівки серця. Біль не провокується фізичним напруженням. Кардіалгія не припиняється після прийому нітрогліцерину. Характерне відчуття серцебиття при нормальному пульсі, не пов’язане з фізичним навантаженням, нерідко з’являється в стані спокою.
Хворі часто скаржаться на відчуття незадоволеності вдихом, неможливість вдихнути повними грудьми, що не пов’язане з фізичним навантаженням і часто виникає у стані спокою.
Типові порушення функції вегетативної нервової системи: гіперемія або блідість шкіри, пітливість, припливи крові, серцебиття, оніміння кінцівок, озноб, порушення ритму дихання, поліурія, запаморочення, порушення терморегуляції.
Велика кількість скарг зумовлена змінами психічного стану: емоційна лабільність, дратівливість, плаксивість, підвищена збудливість, нерідко пригнічений настрій, страхи, погіршення пам’яті. Посилення симптомів пов’язане з навантаженнями, особливо емоційними.
При патологічному клімаксі часто виникає симптоматична АГ. Надалі, після зникнення припливів крові та інших проявів клімактеричного синдрому, невротичний стан може стати причиною розвитку гіпертонічної хвороби.
У більшості чоловіків з клімактеричною кардіоміопатією визначають ті або інші симптоми патологічного клімаксу з боку сечостатевої системи: відсутність або зниження (зрідка підвищення) лібідо, зниження потенції. Хворі часто скаржаться на розлади сечовипускання, що звичайно пов’язане з доброякісною гіперплазією передміхурової залози.
Вазомоторний синдром проявляється у вигляді припливів крові, тобто відчуття жару, що виникає раптово, у верхній половині тулуба, шкірі обличчя, шиї, що послідовно змінюється гіперемією та потовиділенням. Поряд з припливами крові в окремих областях тіла періодично з’являються парестезії: відчуття оніміння, поколювання, повзання мурашок.
Клімактерична кардіоміопатія може виникнути гостро або розвиватися поступово. Характерна невідповідність між інтенсивністю і тривалістю синдрому серцевого болю та задовільним станом кровообігу.
При об’єктивному обстеженні характерна невідповідність між великою кількістю скарг і відсутністю клінічних ознак коронарної або СН.
Діагностика
На ЕКГ найбільш частими змінами є зниження сегмента ST і/або інверсія зубця Т, які в основному реєструють у правих і середніх грудних відведеннях (V1–4). Зубець Т може тривалий час бути негативним, потім позитивним, а через кілька днів знову негативним без будь-якого зв’язку з клінічною картиною хвороби на фоні задовільного стану хворого. Зміни на ЕКГ не відповідають клінічним проявам, фізичні навантаження практично не впливають на конфігурацію зубців. Часто виникають синусова аритмія, передсердна і шлуночкова екстрасистолія, пароксизмальна суправентрикулярна тахікардія. Іноді реєструють порушення передсердно-шлуночкової і внутрішньошлуночкової провідності.
На ранніх стадіях клімактерична кардіоміопатія, як правило, має ізольований перебіг і характеризується типовою клінічною картиною захворювання. У більш пізній період клінічна картина залежить від приєднання ІХС, наявності запального процесу в міокарді та супутніх захворювань, що безсумнівно обтяжує перебіг кардіоміопатії і погіршує прогноз.
Лікування
Повинне бути спрямоване на усунення всіх симптомів захворювання. Важливе значення має модифікація способу життя, що включає підвищення фізичної активності та дотримання дієти з обмеженням споживання насичених жирів і збільшенням у раціоні частки моно- і поліненасичених жирів і грубої клітковини. Для нормалізації діяльності нервової системи звичайно призначають седативні препарати, транквілізатори, іноді антидепресанти.
Для лікування АГ у період постменопаузи найбільш доцільне призначення інгібіторів АПФ і діуретиків, які не повинні суттєво впливати на показники вуглеводного та ліпідного обміну. Жінкам у постменопаузальний період повинні призначатися тільки високоселективні блокатори β-адренорецепторів нової генерації, що не впливають негативно на ліпідний та вуглеводний обмін (карведилол, небіволол).
Призначення замісної гормонотерапії є патогенетично обґрунтованим у лікуванні хворих з клімактеричною кардіоміопатією. Застосовують препарати, що містять естрогени та гестагени. Статеві гормони пригнічують підвищену активність гіпоталамо-гіпофізарних структур мозку і опосередковано впливають на серце, нормалізуючи вплив вегетативної нервової системи. Не виключено, що статеві гормони послаблюють підвищену активність САС і тим самим нормалізують метаболічні процеси в міокарді. Естрогени мають безпосередню вазоконстрикторну дію на коронарні судини, а також нормалізують електролітний і білковий обмін в міокарді. Дози і загальна тривалість лікування залежать від вихідного гормонального фону і рівня естрогенів, лікування слід проводити під наглядом ендокринолога. Необхідно відзначити, що клімактерична кардіоміопатія є захворюванням, що самовиліковується, при якому гормони мають лише допоміжну замісну дію, тому гормональну терапію слід призначати на тривалий період. Лікування гормонами усуває тяжкі прояви клімактеричного синдрому і після закінчення вікової перебудови ендокринної системи захворювання зникає.
Прогноз
Як правило, сприятливий. Зниження працездатності в більшості випадків носить тимчасовий характер. Повне виключення хворих зі звичного трудового середовища, як правило, відіграє негативну роль, призводить до зайвої концентрації уваги на тяжких відчуттях з боку серця.
УРАЖЕННЯ СЕРЦЯ ПРИ ПОРУШЕННІ ОБМІНУ РЕЧОВИН
Порушення обміну речовин в організмі завжди впливають на перебіг метаболічних процесів в міокарді, часто викликаючи порушення його функції і структури. При різних захворюваннях спочатку може порушуватись один або кілька шляхів метаболізму, що надалі обов’язково буде мати вплив на енергозабезпечення серцевого м’яза. При деяких порушеннях обміну у проміжній тканини міокарда та в коронарних судинах відкладаються патологічні продукти порушеного метаболізму білків, вуглеводів, мінералів або накопичуються надлишкові компоненти нормального обміну. До таких захворювань відносять амілоїдоз, глікогеноз, гемохроматоз та ін.
Порушення білкового обміну. Амілоїдоз
Визначення
Амілоїдоз — системне захворювання невизначеної етіології, що характеризується позаклітинним відкладенням в органах і тканинах (головним чином у медії артерій, периваскулярній сполучній і нервовій тканині, у ретикулоендотеліальній системі, а також міокарді, нирках, печінці, шкірі) особливого білка β-фібрилярної структури — амілоїда.
Етіологія і патогенез
Амілоїдоз є наслідком порушення білкового обміну і може бути набутим або успадкованим. Ряд авторів пов’язують розвиток захворювання зі зміною властивостей білків тканин внаслідок аутоімунних процесів під впливом комплексу антиген — антитіло. Диспротеїнемія з нагромадженням у плазмі крові грубодисперсних фракцій білка і аномальних білків (парапротеїнів) призводить до виходу останніх із судин у тканини з утворенням амілоїдних субстанцій.
В останні роки стала можливою більш точна біохімічна ідентифікація білків, що входять до складу амілоїдних фібрил, на підставі чого виділені типи амілоїду, визначений зв’язок окремих їх типів з клінічними формами амілоїдозу, вивчені білки-попередники, що вірогідно беруть участь у синтезі білків.
Виділяють чотири типи амілоїдозу: первинний (системний), вторинний, сімейний (спадковий) і сенільний (старечий).
Найбільш поширений первинний тип (85%) з переважним ураженням серця, при якому амілоїд утворений легкими ланцюгами молекул k і λ імуноглобуліну (AL-тип), часто асоційований з мієломною хворобою, який частіше відзначають у чоловіків, рідко віком молодше 30 років.
Вторинний амілоїдоз виникає в результаті утворення неімуноглобулінових білків, міофібрили містять амілоїдний протеїн А, що не належить до імуноглобулінів (АА-тип); це часто відбувається при хронічних запальних захворюваннях — ревматоїдному артриті, туберкульозі, хворобі Крона і сімейній середземноморській лихоманці.
Сімейний, або спадковий, амілоїдоз найчастіше є наслідком утворення мутантного білка преальбуміну (транстиретину). Встановлено аутосомно-домінантний тип успадкування. Виявлено гени, відповідальні за синтез цих білків й ідентифікований характер генних мутацій.
Сенільний кардіальний амілоїдоз, також відомий як амілоїд SSA, виникає внаслідок утворення патологічного транстиретину у осіб старшого віку. Виділяють дві форми пов’язаного з віком амілоїдозу — амілоїдоз передсердь, що охоплює тільки передсердя, і старечий аортальний амілоїдоз, обмежений аортою.
Патологічна анатомія
Міокард при амілоїдозі серця дуже щільний на дотик, потовщений, мало піддається розтягненню. Обєм порожнин серця істотно не змінений або незначно збільшений. Амілоїд відкладається у різних відділах серця, переважно в міокарді передсердь і шлуночків, ендокарді, клапанах, перикарді, нерідко в синусному і AV-вузлах, а також у дрібних артеріальних і венозних судинах, включаючи vasa vasorum коронарних артерій, звужуючи їх просвіт аж до повної обтурації. У результаті м’язові волокна серця виявляються «замурованими» в масах амілоїду, що призводить до атрофії скоротного міокарда.
Клінічна картина
Амілоїдне ураження серця не має специфічних симптомів, розвивається поступово і може тривалий час мати безсимптомний перебіг, навіть при виявленні відкладень амілоїду в міокарді при біопсії. Варто звернути увагу, що під час появи симптомів уже існує досить значна інфільтрація серця амілоїдом. У деяких пацієнтів виникає біль у ділянці серця, іноді стенокардичного характеру як наслідок нагромадження маси амілоїду в коронарних артеріях.
У 10–15% випадків визначається ортостатична гіпотензія, іноді з симптомами синкопального стану.
При аускультації на фоні глухих тонів серця можна вислухати систолічний шум мітральної регургітації, при розвитку СН — протодіастолічний ритм галопу.
Часто визначають різні порушення ритму, які можуть бути причиною раптової смерті. У деяких хворих відзначається виражена брадикардія.
СН виявляють у 45–56% хворих. Спочатку домінує правошлуночкова СН з підвищеним тиском у яремних венах, гепатомегалією, периферичними набряками, асцитом. У подальшому виникає систолічна дисфункція і застійна СН.
Діагностика
Зміни на ЕКГ неспецифічні, найбільш типова наявність брадикардії, зниження амплітуди зубців. Іноді наявність патологічного зубця Q і відсутність зубця R у відведеннях V1–3 симулюють картину ІМ. Накопичення відкладень амілоїду у провідній системі можуть зумовлювати різноманітні порушення утворення і проведення імпульсу; можливі різні порушення провідності, включаючи повну AV-блокаду серця, часто виявляються передсердні і шлуночкові порушення ритму (синдром слабкості синусного вузла, фібриляція передсердь (у 30% хворих), шлуночкова екстрасистолічна аритмія).
Двомірна ехоКГ і допплєрографія є основними методами неінвазивної діагностики. При обстеженні виявляють нормальні або зменшені розміри порожнини ЛШ зі значним потовщенням міокарда та характерним порушенням його структури з дифузним гранулярним блиском (рис. 8.1а, б). Відзначається також потовщення міжпередсердної перегородки і стулок клапанів, збільшення розмірів передсердь, наявність невеликого або помірного перикардіального випоту. Порушення діастолічної функції ЛШ і ПШ відбувається за рестриктивним типом порушення їх наповнення. У тяжких випадках виявляються ознаки різного ступеня порушення систолічної функції обох шлуночків.
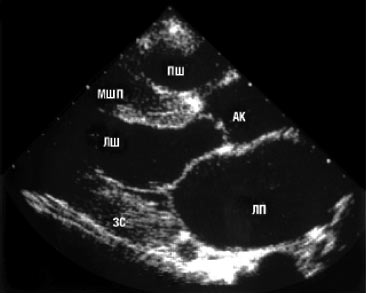
a
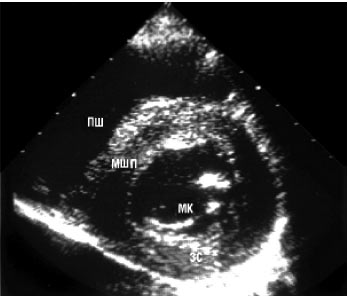
б
Рис. 8.1. Амілоїдоз серця. В-режим: а) парастернальна позиція, довга вісь; б) парастернальна позиція, коротка вісь. Тут і надалі на рисунках: МШП — міжшлуночкова перегородка; ЛП — ліве передсердя; АК — аортальний клапан; МК — мітральний клапан; ЗС — задня стінка
При рентгеноскопії визначають зменшення пульсації контуру серця, розміри серця збільшені (кардіомегалія) і, як правило, не відповідають ступеню тяжкості застійної СН.
Досягненням останніх років є застосування в клінічній практиці методу сцинтиграфії з міченим 123I сироватковим Р-компонентом (SАР) для оцінки розподілу амілоїду в організмі. Р-компонент міститься у невеликій кількості (5–10%) в амілоїді всіх типів; радіоактивний SАР, введений пацієнту з амілоїдозом, специфічно зв’язується з амілоїдними відкладеннями і може бути візуалізований і кількісно оцінений за серією сцинтиграм. Метод особливо корисний для контролю за динамікою тканинних відкладень амілоїду в процесі лікування.
Для діагностики також використовують сцинтиграфію з ізотопом технецію 99mТс-пірофосфатом, здатним зв’язуватися з амілоїдом багатьох типів, проте ця проба виявляється позитивною тільки при значних нашаруваннях амілоїду в серці, які можна виявити і методом ехоКГ.
МРТ використовується для ідентифікації потовщення міокарда і невеликого розміру порожнини ЛШ при амілоїдозі, що дозволяє проводити порівняння з даними ехоКГ.
Діагноз «амілоїдоз» повинен бути підтверджений ендоміокардіальною біопсією. При вивченні біоптатів тканин важливо не тільки виявити амілоїд, але і провести імуногістохімічне дослідження для ідентифікації його типу.
Діагноз «амілоїдоз серця» частіше встановлюється при аутопсії, оскільки прижиттєво у ряді випадків не виявляють об’єктивних причин, які б пояснювали виникнення патологічних ознак.
Лікування
Терапія при первинному амілоїдозі включає клітинну антиплазмову терапію, що зупиняє продукцію легких ланцюгів, а також застосування алкілуючих засобів (мелфалан) і преднізолону. Сприятливий ефект хіміотерапії доведений у двох рандомізованих випробуваннях. Перспективна трансплантація стовбурових клітин з органною ремісією у 50% випадків. Іншим підходом до лікування амілоїдозу серця може бути застосування талідоміду з дексаметазоном. Нещодавно показана ефективність застосування леналідоміду.
Для лікування пацієнтів з порушенням ритму серця призначають антиаритмічні препарати. При явищах повної поперечної блокади і слабкості синусного вузла ефективна імплантація штучного водія ритму. Кардіостимулятори застосовують для лікування пацієнтів з тяжкими клінічно вираженими порушеннями провідності.
СН часто рефрактерна до медикаментозної терапії. Для зменшення симптомів недостатності кровообігу основними препаратами є діуретики, які застосовують з обережністю і в низьких дозах, а також вазодилататори — інгібітори АПФ або блокатори рецепторів ангіотензину II, хоча вони погано переносяться і можуть викликати значну артеріальну гіпотензію або ортостатичні симптоми, особливо у пацієнтів з амілоїдіндукованою дисфункцією автономної нервової системи. Не рекомендується застосовувати дігоксин у зв’язку з його токсичністю і небезпекою розвитку аритмій, проте при ретельному ЕКГ-моніторуванні його можна застосовувати для контролю ритму у пацієнтів з фібриляцією передсердь.
Блокатори кальцієвих каналів неефективні при лікуванні амілоїдозу серця. Хворі можуть мати гіперчутливість до негативних інотропних ефектів блокаторів кальцієвих каналів, їх застосування може призвести до наростання симптомів декомпенсації.
Блокатори β-адренорецепторів іноді провокують порушення провідності, що може становити загрозу для життя.
При різкому зниженні скорочувальної здатності передсердь, що свідчить про масивність інфільтрації, навіть при синусовому ритмі показано застосування антиагрегантів або антикоагулянтів, що зумовлено підвищеним ризиком тромбоутворення.
Трансплантацію серця зазвичай не проводять, оскільки виникають рецидиви амілоїдозу в алотрансплантаті, а також неухильне прогресування його в інших органах, що скорочує тривалість життя хворих.
Прогноз
Перебіг амілоїдозу прогресуючий, прогноз несприятливий, хоча залежить від форми, часу встановлення діагнозу і ступеня залучення життєво важливих органів. Кожний із чотирьох основних типів амілоїдної хвороби має різні ступені ураження серця, клінічні симптоми і прогноз. Виживаність хворих зі старечим амілоїдозом набагато вища, ніж з первинним амілоїдозом, — у середньому відповідно 60,0 і 5,5 міс з часу встановлення діагнозу. Летальний кінець (приблизно через 1,5–2,5 року після появи перших ознак ураження серця) зазвичай настає внаслідок порушень ритму і провідності, а також позасерцевих ускладнень (легеневої або системної емболії). У хворих із залученням провідної системи нерідко виникає раптова смерть. Найнижча виживаність відзначена у хворих з рефрактерною до терапії застійною СН (у середньому 6 міс), особливістю якої є переважно правосерцевий або тотальний її тип з різким набряканням шийних вен і значним підвищенням венозного тиску, застійним збільшенням печінки і порожнинних набряків (гідроторакс, гідроперикард, асцит).
Порушення переважно ліпідного обміну
Голодування і кахексія
Патогенез
Голодування, тривале неповноцінне харчування, кахексія призводять до порушень діяльності серця, які супроводжуються зменшенням маси міокарда, зазвичай пропорційно менше, ніж зменшення маси тіла внаслідок атрофії м’язових волокон, дегенеративних змін у міокарді і розвитку СН.
Патологічна анатомія
При голодуванні мікроскопічно відзначається вакуолізація міофібрил, особливо навколо ядер, зміни хроматину ядер і мітохондрій. У випадках, що зайшли далеко, виявляють буру атрофію і дистрофічні зміни міокарда.
Клінічна картина і діагностика
Основними проявами порушення функції серцево-судинної системи при голодуванні є синусова брадикардія, зменшення ХОК, зниження венозного тиску і АТ (переважно систолічного), що часто супроводжується запамороченням, а при швидкій зміні положення з горизонтального у вертикальне — непритомністю. Часто виникають набряки, зумовлені гіпопротеїнемією і збільшенням ОЦК (але не СН).
На ЕКГ відзначаються відхилення осі серця вправо, синусова брадикардія, низький вольтаж зубців, іноді зміни зубця Т і комплексу QRS, які, очевидно, зумовлені порушенням обміну енергії і електролітів у міокарді.
Лікування полягає у відновленні повноцінного харчування.
Ожиріння
Епідеміологія
Епідеміологічні дослідження свідчать, що ожиріння пов’язане із серцево-судинними захворюваннями і передчасною смертністю. Ожиріння саме по собі призводить до комплексного і прогностично несприятливого ураження серця. Ймовірність розвитку ГЛШ у осіб з нормальною масою тіла становить 5,5%, а у осіб з ожирінням — 30%. За даними Фремінгемського дослідження встановлена наявність високого вірогідного зв’язку між ІМТ, розмірами порожнин серця і товщиною стінок ЛШ.
Базуючись на сучасних даних, ожиріння являє собою незалежний фактор ризику розвитку СН, що є причиною її розвитку у 11% чоловіків і 14% жінок у США. За даними Фремінгемського дослідження збільшення ІМТ на кожен 1 кг/м2 підвищує ризик розвитку СН на 5% у чоловіків і на 7% у жінок незалежно від інших факторів ризику.
Етіологія
Ожиріння може бути самостійним захворюванням, що виникає внаслідок надлишкового споживання їжі з високою калорійністю, або синдромом, що супроводжує різні захворювання і розвивається внаслідок ряду нейроендокринних, соціальних, поведінкових і генетичних факторів. Генетичні чинники відіграють важливу роль у розвитку ожиріння. Результати досліджень свідчать, що існує група генів, яка рідко ідентифікується, що викликають значне ожиріння, проте більш часто виявляються гени «сприйнятливості», які детермінують схильність до ожиріння і регулюють розподіл жирової маси в організмі, швидкість обмінних процесів і їх реакцію на фізичну активність і дієту, контролюють харчові звички. Ідентифіковано >41 сайта в геномі, які, можливо, пов’язані з розвитком ожиріння в популяції.
Патогенез
При ожирінні відбувається поступове збільшення розмірів клітин жирової тканини, що веде до зміни їх властивостей. Гормонально-метаболічні порушення, характерні для ожиріння, можуть прямо впливати на структуру і масу міокарда. У пацієнтів з ожирінням адипоцити жирової тканини вивільняють велику кількість біологічно активних субстанцій, що беруть участь у регуляції судинного тонусу: ангіотензин II, інтерлейкіни, простагландини, естрогени, ІФР, ФНП-α, інгібітор активатора плазміногена-1, лептин та інші, що підвищує ризик розвитку серцево-судинних ускладнень, при цьому знижується рівень адипонектину, специфічного циркулюючого білка жирової тканини, що бере участь у регулюванні метаболізму ліпідів і глюкози (схема 8.3). Синтезований у жировій тканині лептин, важливий маркер енергетичного балансу, стимулює гіперсимпатикотонію, сприяє підвищенню рівня АКТГ, кортизолу і альдостерону.

Схема 8.3. Нейрогуморальні ефекти адипоцитів
Провідне значення в розвитку різних форм ожиріння мають зміни функціонування гіпоталамо-гіпофізарної системи. Ендоканабіноїдна система, що розташована в мозку (гіпоталамусі) і периферично в жировій тканині (адипоцитах), печінці, скелетних м’язах і травному тракті, за допомогою канабіноїдних рецепторів 1-го типу (СВ1) бере участь у центральній та периферичній регуляції енергетичного балансу, а також у метаболізмі глюкози і ліпідів, відіграє роль у контролі споживання їжі і маси тіла. Гіперактивація цієї системи асоційована з мотивацією до збільшення споживання їжі та ожиріння і призводить до порушення механізмів зворотного зв’язку, які підтримують стійкий гомеостаз.
Приєднання АГ при ожирінні відбувається приблизно у 60% хворих, механізми її формування пов’язують з розвитком гормонально-метаболічних відхилень, викликаних накопиченням жирової тканини. Ключову роль серед них відіграє розвиток інсулінорезистентності і компенсаторної гіперінсулінемії, що посилює затримку натрію нирками і в свою чергу сприяє подальшому росту ОЦК. Гіпертензивну дію може чинити і лептин, що стимулює симпатичну нервову систему. Ожиріння, АГ, дисліпідемію і гіперглікемію, в основі яких лежить інсулінорезистентність, об’єднують у поняття «метаболічний синдром».
При високому ступені ожиріння не можна виключити певну роль гіпоксії в зміні нейроендокринної регуляції кровообігу і розвитку дистрофії міокарда. Включення гіпоксичного фактора в патогенез дистрофічної ураження серця може стати істотним механізмом не тільки її виникнення, але й розвитку СН.
Гемодинаміка
Серце у хворих на ожиріння перевантажене об’ємом. ОЦК і об’єм плазми крові збільшуються пропорційно ступеню збільшення маси тіла, що призводить до збільшення наповнення ЛШ і ударного об’єму, дилатації і росту маси ЛШ. Вважається, що зростання серцевого викиду при ожирінні фізіологічно пов’язане із задоволенням метаболічних потреб збільшеної тканинної маси тіла. Розвиток серцево-судинних ускладнень при ожирінні зумовлений виснаженням компенсаторних механізмів міокарда, збільшенням величини ОЦК, що формується пропорційно об’єму судинної мережі периферичних тканин. Вміст жирової тканини в організмі, що наростає, призводить до десинхронізації фізіологічних взаємозв’язків між серцем і кровообігом периферичних метаболічно активних тканин.
Серцевий викид у стані спокою у хворих з тяжким ступенем ожиріння досягає 10 л/хв, причому на забезпечення кровотоку в жировій тканині використовується від ⅓ до ½ цього об’єму. Збільшений об’єм крові у свою чергу збільшує венозне повернення в ПШ і ЛШ, викликаючи їх дилатацію, збільшуючи напруження стінки. Це призводить до ГЛШ, що супроводжується зниженням діастолічної піддатливості камери, призводячи до підвищення тиску наповнення ЛШ і його розширення.
Збільшення товщини міокарда знижує надмірне напруження його волокон, що дозволяє зберегти нормальну скорочувальну здатність ЛШ, одночасно створює передумови для діастолічної дисфункції, в основі якої лежить відносне зменшення кількості капілярів на одиницю об’єму м’язової тканини та погіршення умов дифузії кисню в гіпертрофованих м’язових волокнах. При подальшому прогресуванні дилатації ЛШ збільшення напруження стінки призводить до систолічної дисфункції.
Патологічна анатомія
При ожирінні відзначають збільшене відкладення жиру під епікардом обох шлуночків та у поверхневих шарах міокарда, що згодом призводить до атрофії м’язових волокон, заміщення їх жировою тканиною (cor adiposum). Міокард на розрізі має жовтуватий відтінок. Виявляють наявність дифузної м’язової гіпертрофії, що є найбільш характерним проявом ожиріння з боку серцево-судинної системи.
Клінічна картина
Ожиріння у дорослої людини встановлюють при ІМТ ≥30,0 кг/м2. Клінічно виражені розлади кровообігу розвиваються у хворих з ІМТ ≥40,0 кг/м2.
Скарги на біль у серці ниючого, колючого характеру, серцебиття і перебої в роботі серця при фізичних навантаженнях. При накопиченні надлишкової маси тіла поступово розвивається прогресуюча задишка при навантаженні, виникає ортопное і пароксизмальна нічна задишка, з’являються набряки нижніх кінцівок, можливе збільшення об’єму живота.
У багатьох проспективних дослідженнях встановлено, що збільшення маси тіла призводить до підвищення АТ. У хворих з ожирінням високий ризик приєднання ІХС, перебіг якої особливо агресивний і тяжкий.
Серце починає займати «поперечне» положення у зв’язку з високим стоянням діафрагми, зміщуючись вліво і дещо назад. Аускультативно визначається виражена глухість тонів, є схильність до збільшення частоти пульсу.
При крайньому ступені ожиріння іноді відзначають клінічний синдром, що проявляється поєднанням сонливості, альвеолярної гіповентиляції і легеневої гіпертензії з гіпертрофією ПШ — синдром Піквіка.
Діагностика
На ЕКГ звичайно синусова тахікардія, відхилення електричної осі серця вліво, зниження сегмента ST у I–II і V5–6 відведеннях, сплощений і негативний зубець Т. У деяких хворих реєструється низькоамплітудний зубець РІІІ і глибокий QІІІ. Відзначаються ознаки ГЛШ.
При ехоКГ-дослідженні виявляють гіпертрофію і дилатацію ЛШ, збільшення лівого передсердя, діаметра висхідної аорти. За допомогою допплєрівської ехоКГ виявляють ознаки діастолічної дисфункції, може визначатися аортальна регургітація. Надалі відбувається порушення і систолічної функції. Можливе розшарування листків перикарда за рахунок відкладення жиру. Проведення ехоКГ-дослідження часто утруднене в зв’язку з великою товщиною грудної клітки, звуженням міжреберних проміжків, зміщенням серця в задньому напрямку.
При вивченні гемодинамічних показників у всіх пацієнтів виявлене збільшення ОЦК, що супроводжується наростанням ригідності міокарда ЛШ, зростанням тиску його наповнення і ХОК. Із збільшенням ступеня ожиріння підвищується кінцевий діастолічний тиск у ПШ, середній тиск у ЛА, тиск заклинювання в легеневих капілярах і кінцевий діастолічний тиск у ЛШ. Ці зміни викликають розширення порожнин лівого передсердя, ПШ і правого передсердя. Тиск крові в ПШ, як правило, також підвищений.
Рентгенологічна картина завжди змінена внаслідок високого стояння діафрагми і скупчення жиру в області верхівки серця, що створює картину його уявного збільшення. Пульсація млява, тонус серця знижений.
Лікування
Початкові дистрофічні зміни міокарда при ожирінні є значною мірою оборотними при нормалізації маси тіла. Першочерговим етапом лікування є корекція харчових звичок і підвищення фізичної активності. Специфічні рекомендації включають 30 хв фізичної активності принаймні 5 разів на тиждень, зменшення калорійності їжі в середньому до 1500 ккал/добу, зниження споживання жирів до 30–35% денної енергетичної цінності (з включенням у раціон до 10% мононенасичених жирних кислот, наприклад маслинової олії), відмова від трансгенних жирів, збільшення споживання продуктів, що містять волокна, до 30 г/добу і відмова від рідких моно- і дисахаридів.
Для зменшення маси тіла застосовують медикаментозні та хірургічні методи лікування ожиріння. Призначають інгібітори ліпаз (засоби периферичної дії) і анорексигенні засоби (центральної дії).
Лікування серцево-судинних розладів у хворих з ожирінням залежить від характеру ураження серця. Для лікування АГ найбільш доцільне призначення інгібіторів АПФ і діуретиків, які повинні бути нейтральними відносно показників вуглеводного та ліпідного обміну. Слід призначати тільки високоселективні блокатори β- адренорецепторів нової генерації, що не мають негативного впливу на ліпідний та вуглеводний обмін (карведилол, небіволол).
При наявності ознак СН лікування проводять відповідно до сучасних рекомендацій.
АЛКОГОЛЬНА КАРДІОМІОПАТІЯ
Епідеміологія
Одна з форм алкогольного ураження серця, відзначається у 50% осіб, які протягом тривалого часу зловживають алкоголем.
Алкогольну кардіоміопатію виявляють приблизно у ⅓ всіх хворих з неішемічною кардіоміопатією, 40–50% хворих помирають протягом 3–6 років.
Етіологія
Етіологічним фактором є етанол і/ або його метаболіти. Розвиток алкогольної кардіоміопатії можуть зумовити стресові стани, недостатність харчування (дефіцит білків, вітамінів), спадкова схильність, вірусна інфекція на фоні зниження імунітету, зміни, що передують стану міокарда. Не завжди відзначається виразний паралелізм між кількістю етанолу, що вживається, тривалістю інтоксикації та ступенем ураження серця.
Патогенез
Основний метаболіт етанолу, ацетальдегід, чинить прямий ушкоджуючий вплив на клітинні та субклітинні мембрани кардіоміоцитів, що пов’язаний з його здатністю розчиняти ліпіди і збільшувати плинність біологічних мембран. На певному етапі інтоксикації це може викликати порушення обміну речовин у міокарді та інгібування основних шляхів утилізації енергії в клітинах серця; у результаті пригнічення функції дихального ланцюга мітохондрій виникає гіпоксія міокарда. Опосередкований вплив на міокард проявляється в результаті впливу алкоголю на різні відділи нервової системи і функцію надниркових залоз.
Патологічна анатомія
Тривале вживання алкоголю викликає жирову інфільтрацію міокарда, дегенеративні зміни в стінках коронарних артерій і нейронах, що розташовані в серці. При мікроскопічному дослідженні відзначають зникнення поперечної смугастості міофібрил, пікноз ядер, інтерстиціальний набряк, вакуольну і жирову дистрофію, іноді поодинокі або множинні вогнища некрозу, дрібні ділянки фіброзу.
Клінічна картина
Як правило, хворі впевнено заперечують зловживання алкоголем.
Розгорнуту клінічну картину з явищами СН, стійкими порушеннями ритму і провідності, тромбоемболічними ускладненнями, кардіомегалію виявляють рідко.
Першими клінічними проявами найчастіше бувають порушення ритму без ознак застійної СН. Розвиток захворювання має кілька стадій — від функціональних розладів, порушень ритму серця минущого характеру до стійкої гіпертрофії міокарда з наступним розвитком СН.
До найбільш частих і типових клінічних симптомів відносять:
- збудження, тремор рук, метушливість, багатослівність;
- відчуття нестачі повітря, кардіалгію, тахікардію;
- похолодіння кінцівок;
- відчуття жару у всьому тілі, гіперемію шкіри обличчя, ін’єкцію судин склер;
- пітливість;
- підвищення АТ.
Початковими ознаками захворювання прийнято вважати серцебиття і задишку при фізичному навантаженні. На більш пізніх стадіях захворювання стан пацієнтів поступово погіршується.
Діагностика
На ЕКГ характерними змінами є укорочення інтервалу Р–Q, подовження інтервалу Q–T у поєднанні з невеликою елевацією сегмента ST і загостреним високим з широкою основою зубцем Т, синусова аритмія, браді- або тахікардія. Часто порушення ритму (передсердна і шлуночкова екстрасистолічна аритмія, фібриляція передсердь) і провідності (AV- і внутрішньошлуночкова блокади) виникають після тривалого і/або одноразового вживання великої кількості алкоголю (синдром «святкового» серця).
Про наявність алкогольної кардіоміопатії може свідчити відсутність певної причини фібриляції передсердь (тиреотоксикоз, ревматична вада серця) у чоловіків молодого віку.
Діагностику утруднює і відсутність маркерів алкогольного ураження серця.
Діагностувати алкогольну кардіоміопатію легше у тому випадку, якщо в анамнезі є вказівки на тривале вживання алкоголю і визначаються клінічні ознаки кардіомегалії, аритмії або застійної СН при відсутності інших причин, здатних призвести до аналогічних порушень серцевої діяльності.
При ехоКГ-дослідженні відзначається дилатація порожнини ЛШ, зниження його скорочувальної здатності, можлива дифузна гіпокінезія. При допплєрівському дослідженні можуть виявлятися ознаки мітральної регургітації.
Лікування
При лікуванні обов’язково виключають вживання алкоголю. Повна абстиненція може зупинити прогресування ураження серця на ранніх етапах (звичайно в перші 2–6 міс).
На ранніх стадіях без проявів СН і при наявності кардіалгії, тахікардії, АГ і аритмії рекомендовано блокатори β-адренорецепторів. При вираженій кардіомегалії слід призначати серцеві глікозиди і відповідно контролювати їх прийом з метою попередження кардіотоксичного ефекту. У комплексне лікування включають сечогінні засоби, вітаміни, анаболічні гормони, солі калію і магнію.
Прогноз
При повній відмові від вживання спиртних напоїв і під впливом лікування розміри серця у хворих з алкогольною кардіоміопатією нерідко зменшуються. Відновлення основних функцій міокарда та покращання загального стану настають дуже повільно, терміни відносного видужання становлять місяці й роки.
ЛІТЕРАТУРА
- Александров А.А., Кухаренко С.С. (2006) Миокардиальные проблемы ожирения. Рос. кардиол. журн., 2: 11–17.
- Артемчук А.Ф. (2000) Клинические особенности и терапия сердечно-сосудистых нарушений при алкоголизме. Укр. кардіол. журн., 4: 68–71.
- Волков В.И., Исаева А.С. (2008) Эндокринное старение женщины и проблема сердечно-сосудистой патологии. Укр. кардіол. журн., 6: 85–93.
- Ефимов А.С., Соколова Л.К., Рыбченко Ю.Б. (2005) Сахарный диабет и сердце. Мистецтво лікування, 34: 44–49.
- Зубкова С.Т., Тронько Н.Д. (2006) Сердце при эндокринных заболеваниях. Библиотечка практикующего врача, Киев, 200 с.
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практ. руководство. Морион, Киев, 480 с.
- Коваленко В.Н. (ред.) (2008) Руководство по кардиологии. Морион, Киев, 1424 с.
- Моисеев В.С., Сумароков А.В. (2001) Болезни сердца. Универсум паблишинг, Москва, с. 369–378.
- Alpert M.A. (2001) Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome. Amer. J. Med. Sci., 321: 225–236.
- Bartnik M., Van der Berghe G., Betteridge J. et al. (2007) Guidelines on diabetes, pre-diabetes and cardiovascular diseases. Eur. Heart J., 28: 88–136.
- Cooper L.T., Baughman K.L., Feldman A.M. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease. Eur. Heart J., 28: 3077–3093.
- Côté M., Matias I., Lemieux I. et al. (2007) Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int. J. Obes. (Lond), 31: 692–699.
- Di Marzo V., Matias I. (2005) Endocannabinoid control of food intake and energy balance. Nature Neuroscience, 8: 585–589.
- Falk R.H. (2005) Diagnosis and management of the cardiac amyloidoses. Circulation, 112: 2047–2060.
- Fauchier L. (2003) Alcoholic cardiomyopathy and ventricular arrhythmias. Chest., 123: 1320–1325.
- Galinier M., Pathak K., Roncalli J. et al. (2005) Obesity and cardiac failure. Arch. Mal. Coeur. Vaiss., 98: 39–45.
- Gertz M.A., Blood E., Vesole D.H. et al. (2004) A multicenter phase 2 trial of stem cell transplantation for immunoglobulin light-chain amyloidosis (E4A97): An Eastern Cooperative Oncology Group Study. Bone Marrow Transplant., 34: 149–154.
- Hemery Y., Broustet H., Guiraude O. et al. (2000) Alcohol and rhythm disorders. Ann. Cardiol. Angeiol., 49: 473–479.
- Huss J.M., Kelly D.P. (2005) Mitochondrial energy metabolism in heart failure: a question of balance. J. Clin. Invest., 115: 547–555.
- Ingwall J.S., Weiss R.G. (2004) Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res., 95: 135–145.
- Katz A.M. (2006) Physiology of the heart. 4th ed. Williams&Wilkins, Lippincot, 644 p.
- Kholova I., Niessen H.W. (2005) Amyloid in the cardiovascular system: a review. J. Clin. Pathol., 58: 125–133.
- Maceira A.M., Joshi J., Prasad S.K. et al. (2005) Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation, 111: 186–193.
- Matias I., Gonthier M.P., Orlando P. et al. (2006) Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J. Clin. Endocrinol. Metab., 91: 3171–3180.
- Miller S.R., Sekijima Y., Kelly J.W. (2004) Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest., 84: 545–552.
- Murtagh B., Hammill S.C., Gertz M.A. et al. (2005) Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Amer. J. Cardiol., 95: 535–537.
- Poirier P., Giles T.D., Bray G.A. et al. (2006) Obesity and cardiovascular disease: pathophysiology, evaluation and effect of weight loss. Circulation, 113: 898–918.
- Rajkumar S.V., Dispenzieri A., Kyle R.A. (2006) Monoclonal gammopathy of undetermined significance, Waldenstrom macroglobulinemia, AL amyloidosis, and related plasma cell disorders: Diagnosis and treatment. Mayo Clin. Proc., 81: 693–703.
- Rutter M.K., Parise H., Benjamin E.J. et al. (2003) Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation, 107: 448–454.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Williams& Wilkins, Lippincott, 1628 p.
- Torp-Pedersen C., Caterson I., Coutinho W. et al. (2007) Cardiovascular responses to weight management and sibutramine in high-risk subjects: an analysis from the SCOUT trial. Eur. Heart J., 28: 2915–2923.
- Trayhurn P., Wood I.S. (2004) Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr., 92: 347–355.