Аритмогенна кардіоміопатія правого шлуночка — захворювання м’яза серця, що характеризується частковим або повним прогресуючим фіброзно-жировим заміщенням міокарда ПШ, пізніше — залученням у процес ЛШ з відносною інтактністю перегородки.
Епідеміологія
Захворювання недавно ідентифіковане та важко діагностується, тому його поширеність точно невідома, але вважається, що може варіювати в межах від 1:3000 до 1:10 000, співвідношення чоловіки : жінки становить 2,5:1. Перші клінічні прояви можуть виникнути в юнацькому віці, рідко — старше 40 років.
Етіологія
Точна причина захворювання невідома, однак у деяких родинах існують безсумнівні докази його успадковування. У більшості родин, де більше одного хворіючого, найбільш імовірним типом успадкування є аутосомно-домінантний. Описаний також один добре відомий варіант аритмогенної кардіоміопатії ПШ, що успадковується за аутосомно-рецесивним типом.
Генетичними дослідженнями ідентифіковано 7 локусів генів, відповідальних за розвиток захворювання. З аритмогенною кардіоміопатією ПШ також асоціюються мутації генів, що кодують білки вставних дисків (десмоплакін, плакоглобін зі специфічним фенотипом, плакофілін, десмоглеїн, десмоколін). Ознаки захворювання можуть варіювати навіть у членів однієї родини, і патологія може виявитися через покоління. Вважається, що заняття спортом не можуть викликати аритмогенну кардіоміопатію ПШ, проте захворювання частіше реєструють серед спортсменів. Мутації генів ріанодинових рецепторів серця (RyR2) асоціюються з поліморфною шлуночковою тахікардією, викликаною фізичними навантаженнями і ювенільною раптовою смертю.
Патологічна анатомія
При морфологічному дослідженні серця залученим виявляється частіше ПШ, що має плямистий вигляд: змінені ділянки можуть бути оточені нормальними тканинами. Залучення ПШ може бути регіонарним (20%) або дифузним (80%). Міокард ПШ прогресивно редукується, заміщаючись жировою і фіброзною тканиною, що відрізняється від нефіброзної жирової інфільтрації, що виникає в ПШ з віком. На ранніх стадіях захворювання стінки правих відділів серця товщають, але надалі через нагромадження жирової тканини та появи ділянок дилатації вони стають тоншими (рис. 12.1а, б).

Рис. 12.1. Аритмогенна кардіоміопатія ПШ: а) ділянки жирової тканини призводять до послаблення та вибухання м’язової стінки; б) ПШ збільшується
Жирове переродження міокарда поширюється частіше від епікардіальних шарів до ендокарда. Міокард уражується переважно в області виносного тракту, верхівки та субтрикуспідальної зони, які розглядаються як «трикутник дисплазії».
При аритмогенній кардіоміопатії ПШ ліпоматоз супроводжується переважно дилатацією виносного тракту, ПШ або генералізованою дилатацією. Фіброліпоматоз характеризується наявністю фокальної аневризми ПШ і випинанням в області верхівки, нижньої стінки, субтрикуспідальної та інфундибулярної зони.
У міру прогресування фіброзно-жирова дистрофія уражує також ЛШ та передсердя.
Патогенез
Серед молекулярних механізмів аритмогенної кардіоміопатії ПШ розглядаються генетично детермінуючі мутації в десмосомальних протеїнах, а також інгібування сигнальних шляхів. Стрес-індукований розрив десмосомальних зв’язків клітин може запускати процес апоптоза, викликати атрофію міокарда і заміщення його жировою тканиною.
Вогнища жирового переродження і інтерстиціального фіброзу при аритмогенній кардіоміопатії ПШ не проводять електричні імпульси, внаслідок чого дезорганізована структура серця зумовлює виникнення безладної електричної активності, електричні імпульси можуть ставати невпорядкованими, внаслідок чого, крім порушень ритму серця, можуть виникати порушення його скоротності (рис. 12.2а, б).
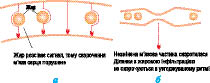
Рис. 12.2. Серце при аритмогенній кардіоміопатії ПШ: а) до скорочення; б) після скорочення
Клінічна картина
Основними клінічними симптомами аритмогенної кардіоміопатії ПШ є:
- відчуття серцебиття, перебоїв у роботі серця, напади шлуночкової тахікардії;
- підвищена стомлюваність, запаморочення, непритомність;
- симптоми СН;
- раптова зупинка кровообігу.
Описані 4 клінічні стадії захворювання:
- субклінічна, незначні шлуночкові аритмії можуть відзначатися або бути відсутні;
- стадія явних електричних порушень, правошлуночкові аритмії і ризик зупинки серця пов’язані з морфофункціональними змінами ПШ;
- стадія правошлуночкової недостатності з прогресуючим залученням ПШ і наступною його глобальною систолічною дисфункцією;
- стадія кінцевої бівентрикулярної СН.
Діагностика
На ЕКГ визначаються:
- спонтанні шлуночкові тахікардії зі зміною комплексу QRS по типу блокади лівої ніжки пучка Гіса;
- негативні зубці Т у відведеннях V1—4 на фоні синусового ритму;
- розширення комплексу QRS;
- неповна блокада правої ніжки пучка Гіса;
- ектопічні тяжкі аритмії: шлуночкова екстрасистолія, фібриляція шлуночків, передсердна тахікардія, фібриляція передсердь.
Приблизно у ⅓ пацієнтів реєструється характерна епсілон-хвиля і ППШ.
Методом холтерівського моніторингу можна діагностувати епізоди шлуночкової тахіаритмії. Для оцінки прогресування захворювання важливо проводити реєстрацію ЕКГ у динаміці.
При ехоКГ-обстеженні виявляються:
- дилатація ПШ і порушення його скоротності (асинергія, дифузна гіпокінезія, зниження ФВ);
- локальна аневризма ПШ;
- підвищена трабекулярність;
- трикуспідальна регургітація;
- емболія ЛА;
- збільшення правого передсердя;
- ліві відділи серця частіше не змінені.
За допомогою допплєр-ехоКГ визначається порушення діастолічної функції ПШ і трикуспідальна регургітація. Для більш точної візуалізації ПШ застосовують контрастну ехоКГ міокарда.
Методом МРТ візуалізують ділянки заміщення міокарда жировою тканиною, фокальне стоншення стінки та локальні аневризми. Продемонстровано гарну кореляцію між результатами цього методу і результатами морфологічного дослідження міокарда.
Для підтвердження діагнозу використовують рентгенконтрастну вентрикулографію, при якій виявляють дилатацію ПШ з сегментарними порушеннями його скорочення, випинання контуру в ділянці дисплазії і підвищення трабекулярності.
При ендоміокардіальній біопсії визначають фіброзно-жирову інфільтрацію міокарда ПШ.
Через труднощі та ризик проведення біопсії для підтвердження діагнозу «аритмогенна кардіоміопатія ПШ», а також неточностей в оцінці структури і функції ПШ за допомогою неінвазивних тестів Європейським кардіологічним товариством та Міжнародним товариством і кардіологічною федерацією розроблені критерії, згідно з якими діагноз встановлюють при наявності 2 великих або 1 великого + 2 малих або 4 малих діагностичних критеріїв (Corrado D. et al., 2000).
Великі діагностичні критерії:
- сімейний характер захворювання, підтверджений даними аутопсії або при хірургічному втручанні;
- епсилон-хвиля або локалізоване розширення комплексу QRS (>110 мс) у правих грудних відведеннях (V1–V3);
- фіброліпоматозне заміщення міокарда за даним ендоміокардіальної біопсії;
- значна дилатація і зниження ФВ ПШ при відсутності або мінімальному залученні ЛШ;
- локалізована аневризма ПШ;
- виражена сегментарна дилатація ПШ.
Малі діагностичні критерії:
- наявність у сімейному анамнезі випадків передчасної раптової смерті (у осіб віком молодше 35 років) внаслідок передбачуваної аритмогенної кардіоміопатії ПШ;
- ППШ на усередненій ЕКГ;
- інвертований зубець Т у правих грудних відведеннях у осіб віком старше 12 років при відсутності блокади правої ніжки пучка Гіса;
- шлуночкова тахікардія з ознаками блокади лівої ніжки пучка Гіса, документована за даними ЕКГ або результатами холтерівського моніторування або під час навантажувального тесту;
- часті шлуночкові екстрасистоли (>1000/24 год при холтерівському моніторуванні ЕКГ);
- помірна глобальна дилатація або зниження ФВ ПШ при незміненому ЛШ;
- помірна сегментарна дилатація ПШ;
- регіонарна гіпокінезія ПШ.
Лікування
Для вибору антиаритмічної терапії необхідне проведення інвазивного ЕФД і проб з дозованим фізичним навантаженням. Серед антиаритмічних засобів ефективні аміодарон і соталол. Дигоксин застосовують при тахісистолічній формі фібриляції передсердь для зниження ЧСС. Для відновлення синусового ритму проводять кардіоверсію.
Діуретики застосовують при СН у хворих із затримкою рідини.
З хірургічних методів лікування застосовують абляцію, якщо джерело порушеної електричної активності ідентифіковано за допомогою електрофізіологічних тестів. У випадках, якщо аритмія не контролюється за допомогою лікарських засобів або абляції (велике ураження або наявність множинних аритмогенних вогнищ), імплантують кардіовертер-дефібрилятор, у деяких випадках потрібна імплантація водія ритму. Трансплантацію серця застосовують рідко, якщо неможливе проведення контролю ритму іншими методами.
Прогноз
Результати нещодавно проведеного дослідження, яке включало 130 пацієнтів з аритмогенною кардіоміопатією ПШ, показали, що серцево-судинна смертність становила 16% (n=24), найбільш частою причиною була раптова смерть (29%) і СН (59%). Аналіз чинників ризику виявив найбільш несприятливі — наявність дисфункції ПШ або ЛШ і шлуночкову тахікардію.
ЛІТЕРАТУРА
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практ. руководство. Морион, Киев, 480 с.
- Коваленко В.Н. (ред.) (2008) Руководство по кардиологии. Морион, Киев, 1424 с.
- Пархоменко А.Н. (1998) Аритмогенная кардиомиопатия правого желудочка: диагностика, лечение, прогноз. Кардиология, 2: 59–64.
- Angelini A., Thiene G., Boffa G.M. et al. (1993) Endomyocardial biopsy in right ventricular cardiomyopathy. Int. J. Cardiol., 40: 274–282.
- Auffermann W., Wichter T., Breithardt G. et al. (1993) Arrhythmogenic right ventricular disease: MR imaging versus angiography. Amer. J. Radiol., 161: 549–555.
- Beffagna G., Occhi G., Nava А. et al. (2005) Regulatory mutations in transforming growth fаctог-β3 gene cause агrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res., 65: 366–373.
- Blake L.M., Scheinman M.M., Higgins C.B. (1994) MR features of arrhythmogenic right ventricular dysplasia. Amer. J. Radiol., 162: 809–812.
- Bomma C., Rutberg J., Tandri H. et al. (2004) Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Cardiovasc. Electrophysiol., 15: 300–306.
- Breithardt G., Wichter T., Haverkamp W. et al. (1994) Implantable cardioverter defibrillator therapy in patients with arrhythmogenic right ventricular cardiomyopathy? Long QT syndrome or no structural heart disease. Amer. Heart J., 127: 1151–1158.
- Castillo E., Tandri H., Rodriguez E.R. et al. (2004) Arrhythmogenic right ventricular dysplasia: ex vivo and in vitro fat detection with black-blood MR imaging. Radiology, 232: 38–48.
- Cooper L.T., Baughman K.L., Feldman A.M. et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease. Eur. Heart J., 28: 3076–3093.
- Corrado D., Fontaine G., Marcus F.I. et al. (2000) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation, 101: E101–E106.
- Danieli G.A., Rampazzo A. (2002) Genetics of arrhythmogenic right ventricular cardiomyopathy. Curr. Opin. Cardiol.,17: 218–221.
- Elliott P., Andersson B., Arbustini E. et al. (2008) Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J., 29: 270–276.
- Galderisi M., Mondillo S. (2007) Echocardiography in clinical practice. One Way S.r.l., 120 p.
- Haverkamp W., Rolf S., Osterziel K. et al. (2005) Die arrhythmogene rechtsventriculare kardiomyopathie. Herz., 30: 565–570.
- Hodgkinson К.А., Parfrey Р.S., Bassett А.S. et al. (2005) The impact of implantabIe cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J. Amer. Coll. Cardiol., 45: 400–408.
- Hulot J.S., Jouven X., Empana J.P. et al. (2004) Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation, 110: 1879–1884.
- Kinoshita O., Fontaine G., Rosas F. et al (1995) Time- and frequency domain analysis of the signal-averaged ECG in patients with arrhythmogenic right ventricular dysplasia. Circulation., 91: 715–721.
- Leclercq J.F., Coumel P. (1993). Late potentials in arrhythmogenic right ventricular dysplasia. Europ.Heart J.,14, (H):67–93.
- Mallat Z., Tedgui A., Fontaliran F. et al. (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. New Engl. J. Med., 335: 1224–1226.
- Maron B.J., Towbin J.A., Thiene G. et al. (2006) Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation., 113: 1807–1816.
- Paul М., Schulze-Bahr Е., Breithardt G. et al. (2003) Genetics of arrhythmogenic right ventricular cardiomyopathy — status quo and future perspectives. Z. Kardiol. 92: 128–136.
- Peters S., Trummel М. (2003) Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: value of standard ECG revisited. Апп. Noninvasive Electrocardiol., 8: 238–245.
- Piccini J.P., Nasir K., Bomma C. et al. (2005) Electrocardiographic findings over time in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Amer. J. Cardiol., 96:122–126.
- Sen-Chowdhry S., Syrris P., McKenna W.J. (2005) Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol., 16: 927–935.
- Tansey D.K., Aly Z., Sheppard M.N. (2005) Fat in the right ventricle of the normal heart. Histopathology, 46: 98–104.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Lippincott Williams&Wilkins, 1628 p.
- Turrini Р., Corrado О., Basso С. et al. (2003) Noninvasive risk stratification in arrhythmogenic right ventricular cardiomyopathy. Аnn. Noninvasive Electrocardiol., 8: 161–169.
- Wichter Т., Paul М., Wollmann С. et al. (2004) lmplantabIe cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center ехреrience of long-term follow-up and complications in 60 patients.Circulation., 109: 1503–1508.
- Yoerger D.M., Marcus F., Sherill D. et al. (2005) Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insight from multidisciplinary study of right ventricular dysplasia. J. Amer. Coll. Cardiol., 45: 860–865.