Гіпертрофічна кардіоміопатія (ГКМП) — первинне ураження міокарда, зумовлене генетичною неповноцінністю скорочувальних білків, характеризується ГЛШ при відсутності серцевої або системної причини.
Епідеміологія
Широко поширена серед багатьох расових груп у країнах Європи, США, Канаді, Ізраїлі, Південній Америці і Далекому Сході. У загальній популяції її поширеність становить 0,2% і відзначається переважно у чоловіків.
Етіологія
На сьогодні за даними численних досліджень підтверджена роль генетичних порушень у розвитку ГКМП. Причиною хвороби є різні мутації генів, які кодують протеїни саркомерів, що дозволяє визначити захворювання як порушення контрактильного апарату міоцита. У більшості пацієнтів захворювання успадковується по аутосомно-домінантному типу, приблизно у 50–60% хворих виявляють мутації в одному з восьми генів, які кодують різні компоненти серцевого саркомера та асоційованих білків: важкого ланцюга β-міозину (14-та хромосома), серцевого тропоніну-Т (1-ша хромосома), серцевого тропоніну-І (19-та хромосома), α-тропоміозину (15-та хромосома), серцевого протеїну С, що зв’язує міозин (11-та хромосома), легких ланцюгів есенціального та регуляторного міозину (3-тя і 12-та хромосоми відповідно), а також серцевого актину (15-та хромосома). За останній час також виявлені мутації трьох інших генів протеїнів саркомера: титіну, тропоніну С і важкого ланцюга α-міозину.
У виникненні захворювання може також мати значення порушення взаємодії серця плода, що розвивається, з катехоламінами, тиреоїдними гормонами, соматотропіном, аденозином.
Розповсюдженість і локалізація гіпертрофії значно варіює, навіть серед родичів. Однакова мутація може призвести до тяжкої гіпертрофії шлуночка у одного члена родини і помірної гіпертрофії у іншого. Причини цих фенотипових розходжень у осіб з ідентичною генетичною мутацією не зовсім зрозумілі, але можуть бути наслідком інших генетичних чинників, які відіграють роль в експресії гіпертрофії серця, таких як DD-генотип АПФ, НУП та інші детермінанти росту міоцитів.
Патогенез
До основних патогенетичних чинників ГКМП належать зниження еластичності і скоротної здатності гіпертрофованого міокарда ЛШ з погіршенням його діастолічного наповнення, в результаті чого в перерахунку на одиницю маси міокарда робота серця істотно зменшується, коронарний кровотік не відповідає ступеню гіпертрофії міокарда. Порушується швидкість проведення збудження в шлуночках з асинхронним скороченням різних відділів міокарда, що знижує пропульсивну здатність ЛШ.
У результаті діастолічної дисфункції виникає хронічне підвищення кінцево-діастолічного тиску ЛШ, тиску заклинювання капілярів ЛА, застій у легенях, прогресуюча гіпертрофія передсердь. Систолічна функція не порушена або навіть посилена внаслідок гіпердинамічності ЛШ (ФВ досягає 80–90%), при цьому КДО зменшений (нерідко <100 мл, іноді навіть <70 мл).
Патологічна анатомія
Захворювання характеризується гіпертрофією міокарда, найчастіше в області міжшлуночкової перегородки, дезорганізацією кардіоміоцитів і міофибрил, фіброзом міокарда та ураженням дрібних судин.
Макроскопічно розрізняють три варіанти ГКМП: асиметрична (60–95%) — ізольована гіпертрофія міжшлуночкової перегородки (ізольований гіпертрофічний субаортальний стеноз); гіпертрофія різних відділів ЛШ, частіше апікальної частини (рис. 10.1); симетрична — тотальна концентрична гіпертрофія (5%) (рис. 10.2).
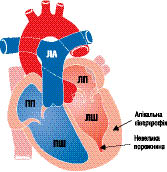
Рис. 10.1. ГКМП (апікальна гіпертрофія)
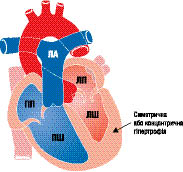
Рис. 10.2. ГКМП (симетрична гіпертрофія)
У 25% хворих визначають обструкцію виносного тракту ЛШ у стані спокою. Рівномірна концентрична гіпертрофія міокарда супроводжується значним збільшенням маси серця, але обструкція шлуночків не відзначається. ПШ залучається до патологічного процесу приблизно в 50% випадків, що значно погіршує перебіг захворювання.
Залежно від вираженості потовщення міокарда виділяють три ступені гіпертрофії: помірна (15–20 мм), середнього ступеня (21–25 мм), виражена гіпертрофія (>25 мм).
Характерною анатомічною ознакою ГКМП є структурна зміна мітрального клапана, передня стулка якого розміщена під кутом до площини клапана, потовщена і «випадає» в просвіт виносного тракту ЛШ, що утворює додаткову перешкоду кровотоку. Порожнина ЛШ невеликих розмірів, ліве передсердя часто гіпертрофоване і дилатоване.
Типові патогістологічні зміни включають гіпертрофію кардіоміоцитів і порушення взаємної орієнтації м’язових волокон (як найбільш частий результат мутацій саркомерів), а також вогнища фіброзу і рубцеві зміни внаслідок некрозу міокарда. Волокна розташовуються короткими рядами, мають схильність до закручування при відсутності змін інтрамуральних судин. Ядра клітин змінені, мають потворну форму, часто оточені світлою зоною («перинуклеарним німбом»), у якій відзначається накопичення глікогену.
Класифікація
Залежно від ступеня вираженості перешкоди відтоку крові виділяють дві основні форми ГКМП: обструктивна (рубрика І42.1 за МКХ-10), характеризується наявністю градієнта тиску між порожниною ЛШ і аортою (рис. 10.3) і необструктивна (рубрика І42.2 за МКХ-10) — без градієнта тиску (рис. 10.4).
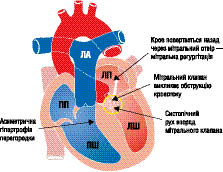
Рис. 10.3. ГКМП (асиметрична гіпертрофія перегородки з обструкцією)

Рис. 10.4. ГКМП (асиметрична гіпертрофія перегородки без обструкції)
Клінічна картина
Ступінь обструкції і ГЛШ не корелює з наявністю клінічних симптомів. Захворювання може бути повністю безсимптомним або маніфестувати в будь-якому віці, найбільш часто симптоми проявляються у пацієнтів віком 40–50 років .
Класична тріада симптомів включає біль у серці, задишку при навантаженні та синкопальні стани. Біль у ділянці грудної клітки відзначають 75% хворих, класичну стенокардію напруження — 25%. У багатьох хворих відзначається післяобідня стенокардія. Непритомність найчастіше виникає у хворих молодого віку. У деяких випадках раптова смерть може бути першим проявом захворювання, наступає з частотою приблизно 1% на рік.
При необструктивній формі фізикальне обстеження відхилень від норми може не виявити, проте іноді визначають збільшення тривалості верхівкового поштовху, аускультативно IV тон серця.
При обструктивній формі при фізикальному обстеженні виявляється пульсація сонних артерій, швидкий «уривчастий» пульс при пальпації сонних артерій; посилений тривалий верхівковий поштовх, що займає всю систолу аж до II тону.
Аускультативно тони серця глухі, визначається IV тон, а також систолічний шум (crescendo-diminuendo), що не проводиться або слабко проводиться на сонні артерії і ділянку спини, посилюється при зменшенні наповнення серця і зниженні ЗПСО (підйом із положення сидячи навпочіпки, натужування, прийом нітрогліцерину) і слабшає при збільшенні наповнення серця, підвищенні ЗПСО (у положенні лежачи, сидячи навпочіпки, при стисканні кулаків).
ГКМП має повільно прогресуючий перебіг, тяжкість залежить від локалізації та ступеня гіпертрофії міокарда в зоні потовщення, а також від обструкції виносного тракту ЛШ.
Погіршують перебіг захворювання серйозні ускладнення. Раптову смерть реєструють у 2–4% дорослих і 4–6% дітей. Іншими ускладненнями є передсердні аритмії, тромбоемболії, інфекційний ендокардит (5–9%) і застійна СН (10–15%).
Діагностика
Зміни на ЕКГ виявляють у 95% хворих. Найбільш частими з них є збільшення лівого передсердя, порушення реполяризації ЛШ у вигляді депресії сегмента ST та інверсії зубця Т і патологічні зубці Q (25–30%), ознаки ГЛШ. При апікальній гіпертрофії можливі глибокі «гігантські» (до 4 см) симетричні негативні зубці Т. Добове моніторування ЕКГ дозволяє виявити порушення ритму серця: шлуночкові екстрасистоли (88%), пароксизми шлуночкової тахікардії (25–30%), суправентрикулярні тахіаритмії (30–40%), а також порушення провідності.
Рентгенологічну картину багато в чому визначає ступінь вираженості захворювання. Можливі наступні рентгенологічні зміни: в першій косій проекції з’являється вибухання ЛШ, зумовлене гіпертрофією шляхів відтоку, відсутність талії серця та заокруглення дуг, розширення лівого передсердя.
ЕхоКГ є методом вибору і дозволяє виявити ГЛШ — потовщення стінки >1,5 мм в період діастоли (рис. 10.5) без збільшення його порожнини, збільшення лівого передсердя, порушення діастолічної функції при допплєрівській ехоКГ.

Рис. 10.5. ГКМП, необструктивна форма, В-режим, парастернальна позиція, довга вісь
За даними ехоКГ-обстеження в М-режимі найбільш часто можна виявити асиметричний характер гіпертрофії перегородки, систолічний передній рух мітрального клапана, невеликий розмір порожнини ЛШ, зменшення рухливості перегородки і передчасне закриття аортального клапана.
При двомірному зображенні визначаються різні варіанти локалізації гіпертрофії міокарда. Систолічна функція зазвичай не порушена, ФВ збільшена (часто >80%).
Приблизно у 25% хворих відзначають градієнт тиску між порожниною і виносним трактом ЛШ у стані спокою. Хоча клінічне значення градієнта виносного тракту у хворих з ГКМП інтенсивно обговорюється багато років, нині градієнт розглядається як показник справжньої обструкції викиду ЛШ. Прийнято угоду, за якою обструкція виносного тракту ЛШ визначається при наявності градієнта не менше 30 мм рт. ст. (Maron B.J. et al., 2003). Обструкція клінічно важлива (від середнього ступеня до тяжкого) тільки у випадках, якщо градієнт виносного тракту становить >50 мм рт. ст.
Залежно від величини градієнта тиску відповідно до класифікації NYHA виділяють наступні стадії захворювання:
I стадія — градієнт тиску до 25 мм рт. ст., як правило, скарг хворі не пред’являють;
II стадія — градієнт тиску до 36 мм рт. ст., самопочуття погіршується при фізичному навантаженні;
III стадія — градієнт тиску до 44 мм рт. ст., виражені клінічні симптоми — стенокардія, задишка та порушення гемодинаміки;
IV стадія — градієнт тиску 80 мм рт. ст. і вище, є значні порушення гемодинаміки.
При проксимальній формі ГКМП (субаортальному стенозі) найбільш характерними ехоКГ-ознаками є потовщення міжшлуночкової перегородки і зниження її екскурсії в базальному сегменті, збільшення співвідношення товщини міжшлуночкової перегородки і задньої стінки >1,3 (1,5–2) (рис. 10.6), наявність градієнта тиску при допплєрівській ехоКГ (рис. 10.7).
Рис. 10.6. ГКМП з обструкцією виносного тракту ЛШ, В-режим, парастернальна позиція, довга вісь

Рис. 10.7. ГКМП з обструкцією виносного тракту ЛШ. Апікальна п’ятикамерна позиція. Режим постійнохвильового допплєра
Для дистальної форми (апікальної ГКМП) при ехоКГ-дослідженні найбільш характерне потовщення та зменшення амплітуди руху міжшлуночкової перегородки у верхівковому сегменті по довгій осі у двовимірному зображенні, порожнина ЛШ у лівій апікальній чотирикамерній позиції пікоподібної форми за рахунок гіпертрофії дистальних відділів міжшлуночкової перегородки і відділів задньої стінки ЛШ, що прилягають до неї.
Концентрична (симетрична) форма характеризується потовщенням міжшлуночкової перегородки і задньої стінки ЛШ у діастолу при значному збільшенні загальної маси міокарда, зменшенням систолічного і діастолічного об’ємів ЛШ, підвищенням індексу співвідношення розмірів лівого передсердя і устя аорти.
Ізольована гіпертрофія ПШ при ехоКГ- дослідженні має наступні ознаки: збільшення діастолічної товщини і зменшення амплітуди руху міжшлуночкової перегородки у верхівковому сегменті, потовщення передньої стінки і зменшення діастолічного розміру ПШ.
При біопсії міокарда виявляють хаотичне розташування та вкорочення волокон міокарда, дегенеративні зміни із зникненням міофібрил, деформацію ядер клітин, фіброзне заміщення міокарда.
Катетеризацію порожнин серця зазвичай проводять при клінічно вираженій мітральній регургітації для оцінки можливості хірургічного лікування. Внутрішньошлуночкові градієнти тиску виявляють у ЛШ і рідше в ПШ. Градієнт підвищується після екстрасистол, під час проби Вальсальви і після інгаляції амілнітриту. Кінцево- діастолічний тиск підвищений внаслідок зниженої піддатливості шлуночка.
За даними вентрикулографії виявляють характерну деформованість камери, що залежить від форми ГКМП і також іноді підтверджують мітральну регургітацію. Коронарні артерії зазвичай широкі з адекватним кровотоком.
За допомогою МРТ можна найбільш точно оцінити морфологічні зміни, поширеність і вираженість гіпертрофії міокарда, особливо при діагностиці верхівкової форми і гіпертрофії нижньої частини міжшлуночкової перегородки і ПШ, визначити систолічну і діастолічну функцію ЛШ.
При підозрі на ГКМП діагноз встановлюють за даними генетичного дослідження (аналіз ДНК), що дозволяє виявити характерні мутації генів, відповідальних за синтез скорочувальних білків кардіоміоцитів.
Лікування
Повинне бути спрямоване на зменшення діастолічної дисфункції, гіпердинамічної функції ЛШ і усунення порушень ритму серця.
Загальні заходи включають обмеження фізичних навантажень, які збільшують гіпертрофію міокарда, підвищують внутрішньошлуночковий градієнт тиску і ризик раптової смерті.
Блокатори β-адренорецепторів є препаратами першої лінії у хворих незалежно від наявності або вираженості градієнта внутрішньошлуночкового тиску, що мають симптоми задишки або непереносимості фізичних навантажень, із зниженою скоротністю ЛШ, обмеженим латентним градієнтом виносного тракту, зниженим споживанням кисню міокардом і ішемією (схема 10.1). Блокатори β-адренорецепторів покращують симптоми у 70% хворих, знижуючи ЧСС і у такий спосіб покращуючи пасивне наповнення шлуночків і зменшуючи потребу міокарда в кисні.
Альтернативою може бути застосування верапамілу, який у дозі до 480 мг/добу у хворих як з необструктивною, так і обструктивною ГКМП зменшує вираженість симптоматики, особливо біль у ділянці серця, покращує розслаблення і наповнення шлуночків, зменшує ішемію міокарда і скоротність ЛШ. При застосуванні верапамілу може виникати погіршення гемодинаміки, збільшення обструкції виносного тракту, підвищення тиску в ЛА.
З обережністю слід давати навантаження хворим з обструкцією виносного тракту ЛШ у стані спокою.
При наявності порушень серцевого ритму доцільно призначати блокатори β-адренорецепторів і антиаритмічні засоби, проте слід зазначити, що застосування останніх не знижує ризик раптової смерті. У симптоматичних пацієнтів з обструкцією дизопірамід діє як антиаритмічний засіб (по відношенню як до суправентрикулярних, так і шлуночкових аритмій) і як засіб з негативною інотропною дією викликає зменшення вираженості симптомів. У дозах 300–600 мг/добу може зменшувати обструкцію виносного тракту і обсяг мітральної регургітації. Для зменшення вираженості побічних ефектів можна застосовувати в комбінації з блокаторами β-адренорецепторів у низьких дозах. Не слід застосовувати дизопірамід з соталолом/аміодароном внаслідок ризику проаритмогенної дії.
Наявність фібриляції передсердь зазвичай добре переноситься, проте у хворих з тяжкою діастолічною дисфункцією втрата передсердного «внеску» внаслідок аритмії може мати необоротні гемодинамічні наслідки, що вимагає невідкладного відновлення синусового ритму шляхом електричної або медикаментозної кардіоверсії за допомогою аміодарону. Останній ефективний для попередження пароксизмів фібриляції передсердь. Контроль ритму за допомогою блокаторів β-адренорецепторів або верапамілу покращує клінічний статус пацієнтів. Застосування варфарину показано як при пароксизмальній, так і при постійній формі фібриляції передсердь.
При лікуванні СН у хворих з ГКМП терапевтична стратегія повинна бути спрямована на стимуляцію регресії ГЛШ і усунення симптомів СН шляхом зниження тиску наповнення ЛШ без зменшення величини серцевого викиду. У цих випадках препаратами вибору можуть бути інгібітори АПФ і антагоністи рецепторів ангіотензину II у зв’язку з їх здатністю блокувати РААС і викликати зворотний розвиток ГЛШ.
Клінічні дослідження, проведені за останні роки, продемонстрували сприятливу дію інгібіторів АПФ на ряд важливих показників діастолічної функції, включаючи діастолічне наповнення, ізоволюмічне розслаблення і взаємозв’язок тиск — об’єм ЛШ і можливість зворотного розвитку процесів ремоделювання міокарда. При цьому покращання діастолічної функції (діастолічної розтяжності і здатності до розслаблення міокарда, зниження кінцево- діастолічного тиску наповнення ЛШ) було більш виражене у хворих з попередньо більш тяжким ступенем дисфункції.
Лікувальні заходи при ГКМП і СН у певній мірі мають парадоксальний характер. Діуретики слід застосовувати з обережністю, переважно при відсутності значної обструкції виносного тракту.
Засоби з інотропним ефектом, спрямовані на стимуляцію систолічного викиду (серцеві глікозиди і пресорні аміни), можуть спричинити несприятливий гемодинамічний ефект — вони посилюють обструкцію виносного тракту і не знижують підвищений кінцево-діастолічний тиск, можуть викликати розвиток асистолії. При збереженій систолічній функції може виникнути негативний ефект у зв’язку з посиленням скоротності шляхом підвищення внутрішньоклітинної концентрації іонів кальцію. Таким чином, при ГКМП «чистий» ефект від інотропних засобів, що мають позитивний ефект (як збільшення жорсткості міокарда, так і підвищення тиску наповнення ЛШ) призводить до погіршення діастолічної функції. Проте дигоксин можна застосовувати у хворих з діастолічною дисфункцією і фібриляцією передсердь для зниження ЧСС і/ або для відновлення синусового ритму.
Показання до проведення немедикаментозної терапії:
- значна обструкція виносного тракту ЛШ (максимальний градієнт ≥50 мм рт. ст.) і симптоми вираженої задишки;
- біль у ділянці серця і пресинкопальні або синкопальні стани;
- рефрактерність до максимальної медикаментозної терапії.
Останнім часом успішно апробований новий метод для зменшення обструкції виносного тракту у хворих, рефрактерних до медикаментозної терапії — перкутанна алкогольна абляція міжшлуночкової перегородки. Успішна алкогольна абляція супроводжується прогресивним зменшенням градієнта в період від 6 до 12 міс у 80% хворих, що супроводжується покращанням клінічного статусу, зменшенням вираженості симптомів і діастолічної дисфункції та збільшенням переносимості фізичних навантажень.
Метою хірургічного втручання при обструкції виносного тракту є усунення систолічного переднього руху мітрального клапана і септально-мітрального контакту шляхом розширення виносного тракту ЛШ. Найчастіше виконується септальна міотомія-міектомія, в результаті якої у 95% хворих визначають значне зменшення градієнта виносного тракту, мітральної регургітації, у 70% поліпшуються клінічні симптоми. Приблизно у 5% пацієнтів операція ускладнюється аортальною регургітацією, що зазвичай гемодинамічно незначима.
Попередження раптової смерті включає ідентифікацію маркерів ризику (включаючи синкопальні стани, раптову смерть родичів, наявність градієнта виносного тракту). Наявність множинних клінічних факторів ризику підвищує ризик раптової смерті; таким хворим необхідна імплантація кардіовертера-дефібрилятора.
Прогноз
Несприятливий; найчастіше хворі вмирають раптово, на фоні важкого фізичного навантаження, у тому числі при безсимптомному перебігу захворювання. ХСН розвивається не так часто.
Встановлені чинники ризику раптової смерті при ГКМП: маніфестація захворювання в молодому віці (до 16 років), наявність в сімейному анамнезі епізодів раптової смерті, часті синкопальні стани, нетривалі епізоди шлуночкової тахікардії, виявлені при 24-годинному моніторуванні ЕКГ, патологічна зміна рівня АТ при фізичному навантаженні. Ступінь ГЛШ або наявність обструкції виносного тракту ЛШ прогностичного значення не мають.
ЛІТЕРАТУРА
- Амосова Е.Н. (1999) Кардиомиопатии. Книга плюс, Киев, 422 с.
- Коваленко В.М., Лутай М.І., Сіренко Ю.М. (ред.) (2007) Серцево-судинні захворювання. Класифікація, стандарти діагностики та лікування кардіологічних хворих. Київ, 122 с.
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практ. руководство. МОРИОН, Киев, 480 с.
- Коваленко В.Н. (ред.) (2008) Руководство по кардиологии. Морион, Киев, 1424 с.
- Целуйко В.И., Белостоцкая Е.А. (2008) Генетические основы гипертрофической кардиомиопатии. Укр. кардиол. журн., 4: 118–122.
- Целуйко В.И., Максимова Н.А., Кравченко Н.А. и др. (1998) Генетический аспект гипертрофической кардиомиопатии. Кардиология, 6: 63–65.
- Шиллер Н., Осипов М.А. (2005) Клиническая эхокардиография. 2-е изд. Практика, Москва, 344 с.
- Adabag A.S., Casey S.A., Kuskowski M.A. et al. (2005) Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J. Amer. Coll. Cardiol., 45: 697–704.
- Arad M., Maron B.J., Gorham J.M. et al. (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med., 352: 362–372.
- Cambronero F., Marín F., Roldán V. et al. (2009) Biomarkers of pathophysiology in hypertrophic cardiomyopathy: implications for clinical management and prognosis. Eur. Heart J., 30: 139–151.
- Cooper L.T., Baughman K.L., Feldman A.M. et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease. Sur. Heart J., 28: 3076–3093.
- Crilley J.G., Boehm E.A., Blair E. et al. (2003) Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertroph. J. Amer. Coll. Cardiol., 41: 1776–1782.
- Dorn G.W., Force T. (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest., 115: 527–537.
- Elliott P., Anderson B., Arbustini E. et al. (2007) Classification of the cardiomyopathy position: statement from the European society of cardiology working group on myocardial and pericardial deseases. Sur. Heart J., 29: 270–276.
- Fassbach M., Schwartzkopff B. (2005) Elevated serum markers for collagen synthesis in patients with hypertrophic cardiomyopathy and diastolic dysfunction. Z. Kardiol., 94: 328–335.
- Galderisi M., Mondillo S. (2007) Echocardiography in clinical practice. One Way S.r.l., 120 p.
- Gietzen F.H., Leuner C.J., Obergassel L. et al. (2004) Transcoronary ablation of septal hypertrophy for hypertrophic obstructive cardiomyopathy: Feasibility, clinical benefit, and short-term results in elderly patients. Heart, 90: 638–644.
- Gomes A.V., Potter J.D. (2004) Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol. Cell. Biochem., 263: 99–114.
- Hughes S.E. (2004) The pathology of hypertrophic cardiomyopathy. Histopathology, 44: 412–427.
- Lips D.J., deWindt L.J., Kraaij D.J.W. et al. (2003) Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur. Heart J., 24: 883–896.
- Maron B.J., McKenna W.J., Danielson G.K. et al. (2003) American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J. Amer. Coll. Cardiol., 42: 1687–1713.
- Megevand A., Ingles J., Richmond D.R. et al. (2005) Long-term follow-up pf patients with obstructive hypertrophic cardiomyopathy treated with dual chamber pacing. Amer. J. Cardiol., 95: 991–993.
- Minamisawa S., Sato Y., Tatsuguchi Y. et al. (2003) Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun., 304: 1–4.
- Montgomery J.V., Harris K.M., Casey S.A. et al. (2005) Relation of electrocardiographic patterns to phenotypic expression and clinical outcome in hypertrophic cardiomyopathy. Amer. J. Cardiol., 96: 270–275.
- Moon J.C., Reed E., Sheppard M.N. et al. (2004) The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J. Amer. Coll. Cardiol., 43: 2260–2264.
- Morioka N., Shigematsu Y., Hamada M. et al. (2005) Circulating levels of heart-type fatty acid-binding protein and its relation to thallium-201 perfusion defects in patients with hypertrophic cardiomyopathy. Amer. J. Cardiol., 95: 1334–1337.
- Morita H., Seidman J., Seidman C.E. (2005) Genetic causes of human heart failure. J. Clin. Invest., 115: 518–526.
- Richard P., Charron P., Carrier L. et al. (2003) Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations and implications for a molecular diagnosis strategy. Circulation, 107: 2227–2232.
- Rogers D.P.S., Marazia S., Chow А.W. et al (2008) Effect of biventricular pacing on symptoms and cardiac remodelling in patients with end-stage hypertrophic cardiomyopathy. Eur. J. Heart Fail., 10: 507–513.
- Saumarez R.C., Pytkowski M., Sterlinski M. et al. (2008) Paced ventricular electrogram fractionation predicts sudden cardiac death in hypertrophic cardiomyopathy. Eur. Heart J., 29: 1653–1661.
- Sherrid M.V., Barac I., McKenna W.J. et al. (2005) Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Amer. Coll. Cardiol., 45: 1251–1258.
- Shirani J., Pick R., Roberts W.C. et al. (2000) Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J. Amer. Coll. Cardiol., 35: 36–44.
- Teraoka K., Hirano M., Ookubo H. et al. (2004) Delayed contrast enhancement of MRI in hypertrophic cardiomyopathy. Magn. Reson. Imaging., 22: 155–161.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Lippincott Williams&Wilkins, 1628 p.
- Zen K., Irie H., Doue T. et al. (2005) Analysis of circulating apoptosis mediators and proinflammatory cytokines in patients with idiopathic hypertrophic cardiomyopathy. Int. Heart J., 46: 231–244.