Если жалобы больного, анамнестические сведения, клинические исследования указывают на возможность поражения почек или мочевых путей, врач прежде всего должен обратить внимание на показатели мочевого осадка, наличие поли- или олигоурии. Повышение уровня креатинина (мочевины) в сыворотке крови является поздним признаком, а нарушения микции указывают скорее на поражение нижних мочевых путей, а не почек. При немотивированных повышениях температуры тела, признаках воспаления, боли в пояснице, отеках характерной локализации, нарушениях характеристик мочи и мочеиспускания первоначально проводят химическое, микроскопическое (мочевой осадок) и бактериологическое исследование мочи, исследование крови (признаки воспаления, анемия, гипоальбуминемия, креатинин, мочевина), УЗИ почек. Этот простой и быстро выполнимый набор манипуляций часто дает основные направления для дальнейших диагностических поисков, нередко сам по себе определяет диагноз. Сравнительно дешевый и быстрый первоначальный диагностический набор позволяет избежать инвазивных и радиологических методов. При наличии изменений хотя бы по данным одного исследования показаны расшифровка протеинурии (24-часовая потеря белка и каких именно фракций), определение калия, фосфора, креатинина в сыворотке крови, уточнение состава отходящих солей и конкрементов. Только после исчерпания возможностей неинвазивных методов исследования целесообразно применить радиологические и инвазивные методики.
Исследования мочи
Исследования мочи начинаются со стандартизации ее сбора. Крайне нежелательно хранение мочи перед отправкой ее в лабораторию. Для рутинных исследований обычно достаточно «случайной» порции. Это утренняя порция мочи, относительно концентрированная, высокой осмолярности, кислой среды (рН<7), так как именно при этих условиях наилучшим образом сохраняются форменные элементы. Измерение кислотности оправдано только в свежевыпущенной моче. Со временем мочевина расщепляется бактериями, освобождается аммиак, который ощелачивает мочу. Обычно pH мочи в пределах 4,5 и 8,0. Если при метаболическом ацидозе pH мочи при повторных определениях выше 5,5, то говорят о тубулярном ацидозе с нарушением выделения кислот почками.
Моча должна быть собрана в максимально чистых условиях. В период менструации исследование мочи проводить нецелесообразно. Использование тампонов не исключает контаминацию. В то же время выявление лейкоцитарных и эритроцитарных цилиндров никогда не может быть объяснено менструальными выделениями. При сборе суточной мочи необходимо тщательно следить за тем, чтобы сбор проводили в течение 24 ч, не больше и не меньше. Непосредственно перед сбором суточной мочи следует опорожнить мочевой пузырь, ни в коем случае не учитывая полученную порцию мочи. В период сбора мочи каждую, даже минимальную порцию, надо учитывать и собирать для исследования. Перед дефекацией опорожнить мочевой пузырь, особенно это касается женщин, которым труднее, чем мужчинам, задержать мочеиспускание при опорожнении кишечника. В завершение всего периода сбора мочи вновь опорожнить мочевой пузырь, полученную порцию необходимо присовокупить к общему исследуемому объему. При нарушениях сбора мочи лучше повторить всю процедуру заново, чем направить на исследование нестандартно собранную суточную мочу.
Для микробиологических исследований собирают среднюю порцию утренней мочи (особенно важно для диагностики туберкулеза почек). Катетеризация пузыря сама по себе способна дать ложноположительные результаты за счет «протаскивания» катетером флоры из мочеиспускательного канала. В исключительных случаях применяют пункцию мочевого пузыря. При наличии более 100 000 микробных тел в 1 мл вероятность инфекции мочевыводящей системы диагностически высока. Две подряд положительные пробы делают диагноз вероятным на 99%. При количестве микробных тел менее 10 000 прежде всего можно думать о случайном загрязнении. Монофлора типична для неосложненной инфекции мочевых путей. Разнообразные возбудители выявляют при микробном осложнении опухолей почек, мочекаменной болезни, энтеровезикальных свищах и т. д. Для экстренной диагностики возможно применение нитритного теста. Нитраты, выделяемые с мочой, переводятся микробами в нитриты. Последние улавливают тест-полоски. Количество ложноположительных результатов при тщательном соблюдении техники не превышает 0,4%. Но число ложноотрицательных результатов варьирует. При исследовании утренней мочи ложноотрицательных результатов 15–22%. При исследовании случайной порции собранной днем мочи ложноотрицательные результаты достигают 55–65%. При наличии бактериурии определить локализацию бактериального очага микробиологическими методами практически невозможно.
Критерии диагноза «инфекция мочевых путей» представлены в табл. 31.1.
Таблица 31.1
Клинико-лабораторные критерии диагноза «инфекция мочевых путей»
| Состояние | Клинические критерии | Лабораторные критерии |
| Асимптомная бактериурия | Нет мочевой симптоматики |
>10 лейкоцитов/мкл |
| Острая неосложненная инфекция мочевых путей у женщин | Дизурия, императивные позывы,частые мочеиспускания, боль над лобком; нет инфекции в ближайшие 2 нед; нет лихорадки; нет боли в пояснице | >102 КОЕ/мл микроорганизмов в культуре из средней порции мочи;
>10 лейкоцитов/мкл |
| Острый неосложненный пиелонефрит | Лихорадка; озноб; боль в пояснице; нет аномалии мочевых путей. Исключены иные состояния | >104 КОЕ/мл микроорганизмов в культуре из средней порции мочи;
>10 лейкоцитов/мкл |
| Рецидивирующая инфекция мочевых путей у женщин | Более чем 2 микробиологически подтвержденных эпизода острой неосложненной инфекции мочевых путей в последние 12 мес. Нет структурных или функциональных нарушений |
>10 лейкоцитов/мкл |
| Осложненная инфекция мочевых путей | Комбинация симптомов, представленных выше. Один или более факторов осложнения |
>10 лейкоцитов/мкл |
*КОЕ — колониеобразующая единица.
В норме свежевыпущенная моча прозрачна. Изменение прозрачности и окраски мочи — субъективный признак. Изменение окраски не всегда связано с существенными изменениями объективных признаков. Помутнение свежесобранной мочи частое, но не обязательное свидетельство инфекции мочевых путей и кристаллурии. Большее значение для диагноза имеет изменение цвета мочи, который в норме соломенно-желтая. Моча бесцветная, водянистая обусловлена разбавлением урохромов и типична для полиурической стадии почечной недостаточности, для осмотического диуреза. Концентрированная моча, интенсивно насыщенного цвета характерна при ограничении потребления жидкости, при применении антидиуретиков. Изменение цвета мочи зависит от употребления пищевых красителей (свекла — красный цвет) или окрашивающих лечебно-диагностических препаратов (метиленовый синий, индикан, рифампицин, сульфаниламиды, доксорубицин). Темножелтая, коричневая моча характерна при гипербилирубинемии. Серая или черная моча заставляет думать о метастазирующих меланомах (меланурия). Пурпурная моча или моча, приобретающая интенсивно-пурпурный цвет на свету, является результатом порфиринурии или порфобилиногенурии. Ярко-красная, прозрачная, «лаковая» моча своим цветом обязана миоглобину или гемоглобину. Пероксидазный тест при красной окраске мочи заставляет искать следующие причины: гемоглобинурия-миоглобинурия-гематурия. При условии отсутствия эритроцитов по данным микроскопии вероятность гематурии исключается. Для ДД мио- и гемоглобинурии применяют исследование крови. Миоглобин имеет низкий удельный вес, практически полностью уходит через мембранный фильтр и не изменяет цвет сыворотки крови (за исключением случаев почечной недостаточности). Однако будучи продуктом распада мышц, миоглобин сочетается с высокой активностью креатинкиназы. Гемоглобин дает красное окрашивание сыворотки крови. В целом изменение окраски мочи отражает изменение ее удельного веса, то есть объективными величинами являются плотность (удельный вес), осмолярность мочи. Осмолярность — количество молекул мочи на 1 кг растворителя, то есть на 1 л воды (ммоль/л = ммоль/кг). Осмолярность мочи зависит от гидратации организма, во многом определена привычками пациента, и концентрационной способности почек. После водной нагрузки осмолярность мочи может снизиться до 120 ммоль/кг воды, что соответствует удельному весу 1,002. При длительной жажде у молодых здоровых лиц концентрация мочи достигает 1300 ммоль/л (удельный вес — 1,040). «Случайное» определение удельного веса мочи малопродуктивно для диагностики. Так, оно может быть очередным аргументом (одним из аргументов) за или против диагноза «сахарный диабет», но очень и очень вспомогательным аргументом. Однако при точном учете поступающей и выделенной жидкости, в интенсивной медицине, при контроле водной нагрузки в случаях нефро- или уролитиаза динамическое определение осмолярности мочи имеет большое значение.
Протеинурия всегда заставляет врача думать о патологических состояниях. Протеинурия — выделение с мочой белка в количестве, превышающем нормальные значения (у взрослых — 30–50 мг/сут, в случаях интенсивных физических нагрузок — до 150 мг/сут, у детей — до 100 мг/сут по Лоури). Значительная часть белка мочи здорового человека состоит из уромукоида. Этот белок сепернируется в дистальных канальцах, ответственен за формирование гиалиновых цилиндров. Его физиологическое значение не известно.
Базальная мембрана клубочков — надежный фильтр для белков крови. Высокомолекулярные белки (альбумин, иммуноглобулины, трансферрин) ею задерживаются, а низкомолекулярные (миоглобин/гемоглобин, амилаза, свободные легкие цепи, пептиды) пропускаются. В проксимальных отделах канальцев абсорбируется 93% секретируемых белков. Фильтрующий эффект мембраны зависит не только от размеров молекулы белка, но и от количества данного белка в сыворотке крови, от состояния самой мембраны. Так, альбумин не секретируется не из-за размеров молекулы, а из-за наличия электрического заряда мембраны, отталкивающего данную молекулу.
При определении количества белка в моче следует помнить, что современные экспресс-методы с бумажными тест-полосками не регистрируют белок Бенса — Джонса и могут дать ложноотрицательные результаты, а рутинные коагуляционные пробы могут дать ложноположительные результаты при применении рентгенконтрастных средств или некоторых лекарственных препаратов. В диагностике парапротеинурий наилучшие результаты дает электрофорез концентрированной мочи. Если белок выявлен хотя бы в одном «общем» (случайном) исследовании мочи, то необходимо провести определение белка в 24-часовой пробе.
Надпочечная протеинурия (преренальная) развивается без предшествующего поражения почек в условиях высокой концентрации в крови белка с высоким клиренсом. Так, лизоцим выделяется при лейкозе, миоглобин — при поражении мышц, амилаза — при панкреатите, белок Бенса — Джонса при плазмоцитоме (80% всех случаев плазмоцитом), при болезни Вальденстрема (у 30% больных), при первичном амилоидозе (у 83% пациентов с этой формой парапротеиноза). Очень редко белок Бенса — Джонса выделяется при злокачественных лимфомах. При выделении миоглобина и белка Бенса — Джонса выраженность протеинурии может достигать 10 г/сут и выше.
Преренальная протеинурия возможна при заболеваниях, сопровождающихся гипертермией. Появление протеинурии связывают в этих случаях с повышенным катаболизмом, который происходит при лихорадке с температурой тела >38 °С. Однако, возможно, это явление — следствие скрытой почечной патологии, индикатором которой является повышенная температура тела.
Непосредственно почечная протеинурия является следствием нескольких причин.
Клубочковая протеинурия — итог повышенной проницаемости базальной мембраны клубочков при ее разнообразных поражениях. Для клубочковой протеинурии типичен уровень выделения белка >3 г/сут (при потере белка >3 г/сут, как правило, развивается гипопротеинемия и другие изменения, характерные для нефротического синдрома). До 3 г/сут причины протеинурии более разнообразны (включая надпочечные, ортостатические, или преходящие, или гемодинамические, тубулярные и подпочечные протеинурии). При нарушении клубочкового фильтра и канальцевой реабсорбции развивается смешанный тип протеинурии.
Электрофорез мочи позволяет разграничить низко- и высокомолекулярные фракции белка. Определение микроальбуминурии в 12-часовой ночной моче — ранний признак диабетической нефропатии. При патологической протеинурии при ортостазе она повышается значительно больше, чем в физиологических условиях. Последний вид протеинурии типичен для людей молодого возраста с синдромом гипермобильности, с гиперлордозом. Ортостатическая, или гемодинамическая, или преходящая, протеинурия невыражена, выявляют также после длительных переходов, у беременных за счет затруднения венозного возврата из почки, при опущенной почке, при застойной сердечной недостаточности.
Канальцевая протеинурия обусловлена неспособностью канальцев абсорбировать нормальный объем белка, проходящего через мембрану клубочков. Представлена низкомолекулярными белками (бета-2-микроглобулин). Отмечают при отравлениях свинцом, кадмием, синдроме Фанкони, тубулоинтерстициальном нефрите, дисметаболической нефропатии, пиелонефрите.
Постренальная протеинурия обусловлена воспалительными поражениями мочевых путей, не превышает 1 г/сут и легко распознается по клиническим данным и особенностям мочевого осадка.
ДД-схему протеинурии в зависимости от сочетания с другими признаками можно представить как протеинурию в сочетании с:
- отеками, гипертензией, гематурией = гломерулонефрит;
- дизурией, лейкоцитурией, абдоминальным синдромом = пиелонефрит или обструктивная уропатия;
- АГ = опухоль или врожденная аномалия сосудов или почечная дисплазия;
- дистрофией, костными дефектами = тубулопатия или хроническая почечная недостаточность;
- гематурией, лейкоцитурией = интерстициальный нефрит или дисметабоолическая нефропатия или гипопластическая дисплазия.
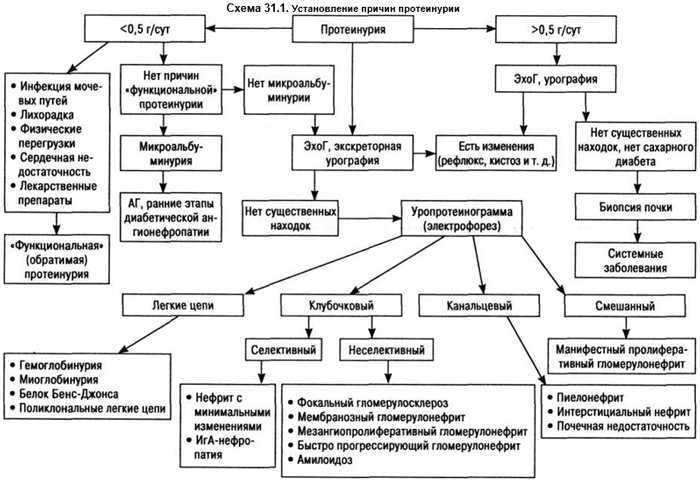
Возможный путь ДД-причин протеинурии представлен на схеме 31.1.
Гематурия — мочевой синдром, при котором в 5 исследованиях в поле зрения при микроскопии мочевого осадка находится >3 эритроцитов (для женщин как пограничное значение приемлема цифра 5). Указанное количество эритроцитов характеризует микрогематурию. Визуально выявляемая гематурия обозначается как макрогематурия. В зависимости от объема крови и кислотности мочи ее цвет при гематурии варьирует от красного до коричневого. Последнее было известно как «моча цвета мартовского пива». Но сейчас вряд ли кто может похвастаться тем, что он видел настоящее мартовское пиво. Образ умер. Вместо него в американской литературе появился другой: «моча цвета колы». Более точную характеристику дает исследование по Нечипоренко (>1000/мл) или по Аддису — Каковскому (>1 млн/мл). Причины, приводящие к гематурии, можно разделить на ренальные, внепочечные (связанные с нарушением системы свертывания и тромбообразования) и обусловленные патологией мочевыводящего тракта. Эритроциты — единственный форменный элемент, придающий моче тревожный красный цвет. Стандартизованно эритроциты ищут в поле объектива 40х и окуляра 10х. В норме в поле зрения находятся 0–2 эритроцита. Фазово-контрастная микроскопия, представляя исследователю морфологию эритроцитов, позволяет определить уровень гематурии. Эритроциты, прошедшие через мембрану клубочков, практически всегда деформированы. При наличии в препарате более 30% дисморфных эритроцитов можно с большой долей уверенности говорить о почечной гематурии. Более точным доказательством патологии клубочков является выявление акантоцитов. Если они составляют 5% и более общего количества эритроцитов в моче, то диагноз «гломерулярное кровотечение» чрезвычайно вероятен. Чувствительность пробы достигает 70%, специфичность — 98%. При постренальной гематурии количество дисморфных эритроцитов исчезающе мало.
ДД-схему при гематурии в зависимости от ее сочетания с другими признаками можно представить как гематурию в сочетании с:
- отеками, гипертензией, протеинурией = гломерулонефрит;
- дизурией, абдоминальным синдромом, лейкоцитурией = цистит или пиелонефрит или обструктивная уропатия;
- кожно-геморрагическим синдромом = коагулопатия или тромбоцитопатия, или системные заболевания;
- протеинурией, кристаллурией, стигмами дисэмбриогенеза, нефропатией в семейном анамнезе = наследственный нефрит или дисметаболическая нефропатия или гипопластическая дисплазия;
- изолированной гематурией = IgA-нефропатия или эссенциальная гематурия, связанная с артериовенозными шунтами.
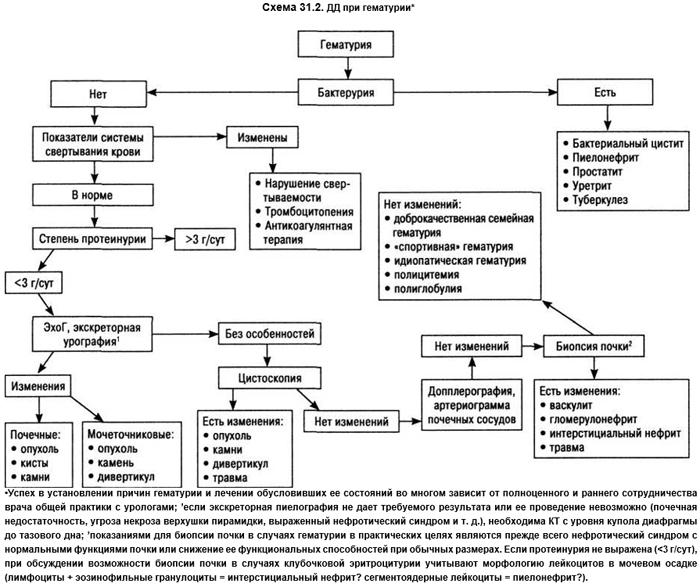
Один из возможных путей ДД при гематурии в зависимости от других симптомов и синдромов представлен на схеме 31.2.
В моче здоровых людей может быть выявлено до 5 лейкоцитов в поле зрения. Большее количество лейкоцитов в моче — явный признак воспалительного процесса в почках или мочевых путях. При этом наряду с лейкоцитами нередко определяют бактерии или грибы. Сочетание в моче большого количества лейкоцитов и бактерий принято называть пиурией. Хотя не во всех случаях лейкоцитурии выявляют бактериурию. Косвенным признаком наличия микробных тел является нейтрофильная лейкоцитурия. Изредка лейкоцитурия обусловлена туберкулезом и камнями почек, кистами, применением цитостатиков или воздействием ионизирующего излучения. Возможность возникновения лейкоцитурии при вирусных поражениях почек не общепризнана.
При установлении диагноза важно не только количественное определение лейкоцитов, но и характер лейкоцитурии. Определяя морфологию осадка мочи (нейтрофильный или лимфоцитарный характер лейкоцитурии) окраской по Романовскому — Гимзе, лейкоцитурию можно разделить на бактериальную и абактериальную. Первая характерна для инфекции мочевых путей. Нейтрофильная лейкоцитурия достигает при этом 95%. Абактериальная лейкоцитурия — признак интерстициального воспалительного процесса в почечной ткани, хотя в данном случае возможно присоединение микробного воспаления. При дисметаболической нефропатии в осадке могут определять большое количество эозинофильных гранулоцитов. Они же возможны при общих аллергозах. У больных гломерулонефритом в моче преобладают лимфоциты и макрофаги (мононуклеарный характер лейкоцитурии, отражающий иммунологическую активность процесса).
ДД-схему при лейкоцитурии в зависимости от сочетанных признаков можно представить как лейкоцитурию в сочетании с:
А. патологической бактерурией:
- дизурией, абдоминальным синдромом, гематурией = цистит или пиелонефрит или мочекаменная болезнь;
- протеинурией, гематурией = пиелонефрит;
- отеками, АГ, снижением функций почек = системные заболевания или сочетанная патология почек.
Б. абактериальной лейкоцитурией:
- гематурией, протеинурией, кристаллурией = дисметаболическая нефропатия;
- АГ, протеинурией, гематурией = интерстициальный нефрит или нефрит при системных васкулитах и системных заболеваниях соединительной ткани.
Цилиндры мочевые — белковые слепки дистальных канальцев почек. Белковую основу цилиндров составляют уропротеин Тамма — Хорсфалла и агрегированные белки в сыворотке крови. Цилиндры бывают чисто белковые (гиалиновые, восковидные) и содержащие в белковом матриксе различные включения (клетки, жир, клеточный детрит).
Эритроцитарные и гемоглобиновые цилиндры рассматривают как надежный признак гломерулярной или почечно-паренхиматозной гематурии. Выявляют не чаще чем в 1/2 всех случаев гематурий. Свойственны всем острым и хроническим гломерулопатиям, острому интерстициальному нефриту, травмам почек, сосудистым катастрофам, физическим перегрузкам.
Лейкоцитарные цилиндры при наличии лейкоцитурии являются доказательством пиелонефрита. Необходимо дифференцировать с тубулярными эпителиальными цилиндрами.
Тубулярные эпителиальные цилиндры выявляют после острой почечной недостаточности и других острых повреждениях канальцев самой разнообразной этиологии, особенно при интерстициальном нефрите. Дифференцируют от лейкоцитарных цилиндров методом фазовоконтрастной микроскопии.
Восковидные цилиндры — матрикс с белком в сыворотке крови. Образуются в дистальных отделах нефрона, выявляют при хронических заболеваниях почек.
Гиалиновые цилиндры — мукопротеиновый матрикс, секретируемый канальцами, — могут в небольшом количестве присутствовать в нормальной моче. В больших количествах выявляют при выраженной протеинурии, после обезвоживания, после применения диуретиков.
Зернистые цилиндры свойственны поражениям почек при общих тяжелых заболеваниях. Возможны при хроническом нефрите.
Жировые цилиндры, жировые тельца, жировые капли, «мальтийские кресты» определяют при выраженном нефротическом синдроме, значительной протеинурии, тяжелых поражениях канальцев почек. Наилучшим образом выявляют при микроскопии в поляризованном свете.
Кристаллы отражают как особенности питания, водного режима, так и генетически детерминированные особенности обмена веществ. Чаще всего определяют конвертообразные кристаллы оксалатов. Для цистинурии типичны гексагональные кристаллы цистина. Трипельфосфаты, которые могут свидетельствовать о хроническом воспалении почек и мочевых путей, имеют форму крышки гроба.
Микроорганизмы, отдельные клетки, эпителий. Подвижные микроорганизмы распознаются только в свежеприготовленном препарате. Но идентифицировать возбудителей по данным изучения мочевого осадка невозможно. Несложно выявить грибы и хламидии. Большие проблемы возникают при определении микобактерий. Важное значение имеет поиск опухолевых клеток, особенно при попытках установления причин микро- и макрогематурии. Если в препарате содержится большое количество эпителиальных клеток, то это свидетельствует о загрязнении, и препарат лучше не изучать и не интерпретировать.
Биопсия почек позволяет получить гистологическую характеристику нефропатии, представляет возможности для иммуногистохимических исследований. Это заключительный этап в диагностике заболеваний почек. Но необходимость его выполнения решается каждый раз индивидуально. После биопсии почки возможны гематома, гематурия, уринома, генерализация почечной инфекции, боль. Смертность даже при выполнении процедуры квалифицированными специалистами составляет 0,1%. Противопоказаниями к проведению биопсии почки являются маленькие, сморщенные почки, единственная (функционально или анатомически) почка, нарушения свертываемости крови, неконтролируемая АГ, острая почечная или паранефральная инфекция, гидронефроз, недостаточный контакт с пациентом.
Нарушения диуреза
Обычно в сутки взрослый выделяет 700–2000 мл мочи, ребенок — 200–1500 мл/м2 поверхности тела. Частота мочеиспусканий у взрослого составляет 4–6 раз в сутки, преимущественно в дневное время. У новорожденного мочеиспускание может быть 1 раз в час.
Олигурия (100–500 мл/сут, у ребенка <200 мл/м2 поверхности тела или <24 мл/кг массы тела) может быть обусловлена внеклеточным дефицитом воды, снижением эффективного кровотока при сердечной недостаточности или шоке (протекают с повышением удельного веса мочи, или осмолярности), острой или хронической почечной недостаточностью, обструкцией мочевых путей (протекают с нормальной или сниженной осмолярностью мочи). Анурия — снижение выделения мочи до 100 мл/сут и менее у взрослых или <50 мл/м2 поверхности тела у детей.
Путь неотложной ДД при олигоурии представлен на схеме 31.3.
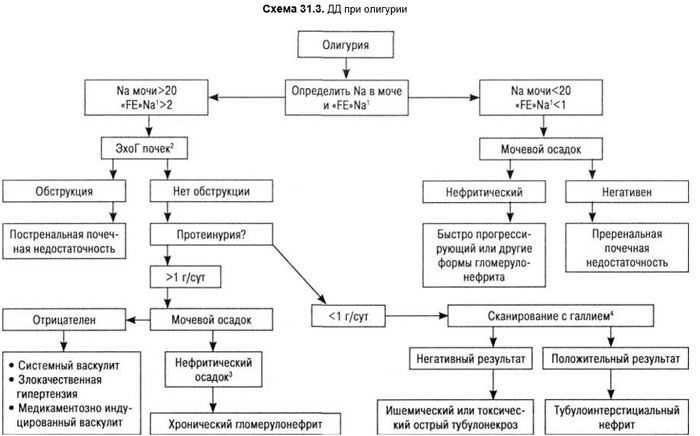
1Определение содержания натрия (Na) в моче — чувствительный показатель почечной перфузии. При ухудшении перфузии почки при ее неизмененной функции резорбция ионов натрия максимально возрастает, поэтому его концентрация в моче снижается до минимума. Еще более чувствительным параметром является фракционное выделение Na («FE»Na), определяющее экскрецию натрия во взаимосвязи с гломерулярной фильтрацией. Формула расчета: «FE»Na = (Na мочи : Na плазмы крови): (креатинин мочи : креатинин плазмы крови) • 100
ПОМНИТЬ: после применения диуретиков концентрация Na в моче никогда не будет показателем истинной перфузии почек!
2результаты эхоГ почек в первые 24–48 ч постренальной обструкции могут быть не изменены. Поэтому необходимо проведение исследований в динамике;
3нефритический мочевой осадок является показателем воспаления клубочков при наличии эритроцитов, лейкоцитов, эритроцитарных и гиалиновых цилиндров, клеточного детрита;
4сканирование с галлием положительно и в первые 48 любых почечных инфекций, но тем не менее признается высокодостоверной методикой в ДД тубулонекроза и тубулоинтерстициального нефрита.
Полиурия (>2500 мл/сут у взрослых или >1500 мл/м2 поверхности тела у детей) развивается при полидипсии, сахарном диабете (или других формах осмотического диуреза), при несахарном диабете вследствие дефицита антидиуретического гормона, при наследственном или приобретенном нефрогенном несахарном диабете (невосприимчивость к антидиуретическому гормону). В то время как при массивном осмотическом диурезе (сахарный диабет, приступ подагры, внутривенное введение маннитола) выделяется моча высокой осмолярности, при психогенной полидипсии, при несахарном диабете выделяется максимально разбавленная моча. Снижение концентрационной способности почек определяется при дефиците калия, гиперкальциемии, интоксикациях литием, при амилоидозе, при полиурической стадии острой почечной недостаточности, при частичной обструкции мочевых путей, при «реакции освобождения» (устранение препятствия оттоку мочи). Все неблагоприятно протекающие нефропатии ведут к снижению концентрационной и выделительной способности почек. В итоге развивается изостенурия.
Никтурия объясняется изостенурией и развивается при сердечной недостаточности, при почечной недостаточности, при неконтролироумой АГ. Необходимо дифференцировать от ночной поллакиурии.
Поллакиурия — частое выделение мочи небольшими порциями, указывающее на уменьшение емкости мочевого пузыря, его воспаление, опухолевое поражение, доброкачественную гипертрофию предстательной железы или ее опухоль, опухоль уретры.
Недержание мочи
Недержание мочи — ее непроизвольное поступление в уретру. Чаще развивается у женщин преимущественно пожилого возраста. По выраженности синдрома выделяют несколько вариантов:
- Стресс-недержание. Непроизвольное мочеиспускание при физической активности, приводящей к повышению внутрибрюшного давления (кашель, натуживание и т. д.). Обусловлено слабостью сфинктера. Объем теряемой мочи обычно небольшой. Чаше отмечают у женщин и при ожирении. Если уретра находится в естественной анатомической позиции, то повышение внутрибрюшного давления равномерно передается и на уретру, и на мочевой пузырь. При слабости мышц тазового дна проксимальная уретра и шейка мочевого пузыря в ответ на повышение внутрибрюшного давления грыжеподобно выпячиваются сквозь тазовое дно и давление в уретре снижается. Чаще всего этот механизм вторичен при гипермобильности уретры из-за слабости мышц, связок после родов, в менопаузальный период, на фоне приема психотропных препаратов или после оперативных вмешательств в этой области, анатомически укороченной уретре. Важным условием диагноза именно этой формы недержания является неспособность пациента произвольно прервать процесс мочеиспускания.
- Императивные позывы (ургентное недержание). Непроизвольное мочеиспускание при ощущении заполнения мочевого пузыря, так называемая нестабильность детрузора. Характеризуется как резко возникающий позыв, не оставляющий времени даже на стремительное движение к туалету. Типично сочетание с учащенным мочеиспусканием и никтурией. Для исключения инфекции необходимо осведомиться о наличии дизурии, неприятных ощущений над лобком, выделений из влагалища или уретры, ощущении неполного опорожнения. Признаками неврологически обусловленной нестабильности детрузора являются спастические парезы ног, мышечная слабость, онемение. Предшествующие катетеризации, жемчужные ванны, спринцевания могут указывать на раздражение или воспаление уретры или мочевого пузыря. Гематурия, дизурия или отхождение тканей предполагают опухоль мочевого пузыря. При ургентных позывах следует исключить гипертрофию предстательной железы.
- Парадоксальное недержание. Мочеиспускание при переполненном мочевом пузыре, когда внутрипузырное давление превышает сопротивление сфинктера.
- Полное недержание мочи. Постоянное подтекание мочи вне связи с наполнением мочевого пузыря или позывами на мочеиспускание.
- Отсутствие рефлекса. Периодическое непроизвольное мочеиспускание вне физического напряжения или позывов.
- Функциональное недержание. Мочеиспускание при невозможности посещения туалета (моральные запреты, прикованность к постели и т. п.).
- Смешанный тип недержания. Несколько причин недержания, свойственен лицам старческого возраста.
- Энурез. У взрослых энурез требует исключения обструкции, инфекции, нестабильности детрузора, неврологических нарушений. У детей это чаще психосоматическая проблема.
Энурез — недержание мочи после завершения 4-го года жизни, отмечающееся минимум 1 раз в неделю. Первичный энурез: ребенок никогда «не оставался сухим» с раннего возраста. Вторичный энурез — возобновление непроизвольного отхождения мочи после светлого промежутка длительностью не менее 6 мес. Ночной энурез составляет около 70% всех случаев энуреза и может быть следствием относительной недостаточности антидиуретического гормона. Дневной энурез (5% случаев) — дневное недержание с отхождением чаще всего небольшого объема мочи. Сочетанный вариант (дневное и ночное недержание) составляет около 25%. После исключения органических факторов следует вспомнить о таких причинах энуреза, как:
- Слишком раннее приучение к горшку, особенно детей с задержкой развития. Жесткость и нетерпеливость в воспитании гигиенических навыков.
- Психические нагрузки (рождение младшего ребенка в семье; конфликты; развод родителей и т. д.).
Внешне дети безразличны к своему состоянию, но внутри тяжело переживают его, формируется депрессия.
Дизурия (болезненное мочеиспускание, замедленное наступление микции) развивается при воспалительных или опухолевых поражениях задней уретры или льетодиевого треугольника.
Краткие перерывы в истечении мочи («заикание») свидетельствуют о незаметно отходящих мелких камнях или мягких тканях.
Пневматурия («кипящая струя») — признак кишечно-пузырного свища. В редких случаях пневматурия указывает на острую газообразующую инфекцию мочевого пузыря.
Двусторонние заболевания почек
Двусторонние поражения почек можно классифицировать по этиологическим, морфологическим, клиническим и функциональным критериям. В зависимости от первичного очага поражения они подразделяются на гломерулопатию, интерстициальные заболевания почек, кистозные поражения, сосудистую нефропатию и нарушения канальцевого аппарата. Все эти состояния могут протекать без нарушения функции почки, с острой и хронической почечной недостаточностью. Кистозные поражения почек, будучи объемным процессом, сравнительно неплохо и быстро диагностируют, используя методы неинвазивной интраскопии (эхоГ, экскреторная рентгенография, КТ). В то же время различия между гломерулопатиями и интерстициальными процессами, тубулопатиями не всегда клинически убедительны.
Гломерулопатии
Собирательное понятие, объединяющее заболевания, при которых основным объектом поражения являются почечные клубочки. Гломерулярные поражения могут быть обусловлены генетическими изменениями, отложением иммунных комплексов в клубочках или их первоначальным образованием здесь, наличием антител к базальной мембране, патологией обмена веществ, нарушением внутрисосудистого свертывания, а также определяются состоянием системы комплемента, характеристиками нейтрофильных гранулоцитов, моноцитов, тромбоцитов, факторов свертывания. Клиницисту важна ДД иммунных и неиммунных гломерулопатий. Классическим примером неиммунной генетически детерминированной гломерулопатии является наследственный нефрит. Возможно возникновение гломерулопатий при микробно-воспалительном процессе в почках. Не исключено возникновение иммунной гломерулопатии и при пиелонефрите. Некоторые клиницисты предпочитают выделять воспалительные гломерулопатии (нефритический синдром или острый гломерулонефрит; возможны и хронические варианты) и гемодинамические (нефротический синдром). Последний вариант обусловлен повышенной проницаемостью клубочковых капилляров. Но в практике различные варианты гломерулопатий нередко сосуществуют.
Все вышеуказанные факторы способны вызвать повреждение клубочков с гибелью нефрона. В итоге повышается АД, снижается фильтрационная способность, за счет повышения проницаемости мембран появляется гемато-, цилиндр- и протеинурия. В зависимости от выраженности отдельных патогенетических составляющих клинически это может выражаться как бессимптомная гематопротеинурия, острый нефритический синдром, быстро прогрессирующий гломерулонефрит, нефротический синдром, хронический гломерулонефрит. Достаточно надежным признаком гломерулопатий является протеинурия >2–3 г/сут (исключить протеинурию при моноклональной гаммапатии) и дисморфные эритроциты. Далеко непросто установить связи между морфологическими, гистохимическими и клиническими проявлениями гломерулопатий.
Хотя и говорится о идиопатических гломерулопатиях, но эта группа представляет собой еще не окончательно охарактеризованные и неклассифицированные состояния. Вторичными называют гломерулопатии при системных и инфекционных заболеваниях, на фоне опухолей, медикаментозного или лучевого поражения.
Неиммунные заболевания клубочков составляют не более 15% всех гломерулопатий. Они развиваются при сахарном диабете, гемолитико-уремическом синдроме, амилоидозе, синдроме Альпорта (?). Но в основном (85%) гломерулопатий обусловлены иммунными механизмами, прежде всего за счет отложения комплекса антитело — антиген в гломерулярных капиллярах или в мезангиуме. Реже это антитела против базальной мембране, как при синдроме Гудпасчера или быстро прогрессирующем нефрите. Еще реже капилляры разрушаются за счет клеточных иммунных реакций. При всех указанных механизмах развивается отек, экссудация, внутри- и внекапиллярная пролиферация, утолщение базальной мембраны, некроз и склероз.
Бессимптомные протеинурия и/или гематурия
Чаще выявляют случайно, реже сами пациенты обращают внимание на рецидивирующую гематурию. Возможна изолированная протеинурия без изменения мочевого осадка. Она может быть единственным выражением гломерулопатий, проявляясь как длительно сохраняющаяся не зависящая от положения тела протеинурия и как ортостатическая протеинурия. Оба варианта могут протекать как со структурными нарушениями гломерулярной сосудистой петли, так и без таковых. Но поскольку бессимптомная протеинурия без структурных нарушений прогностически благоприятна, то при наличии соответствующих клинических сомнений желательно проведение пункционной бипсии почек.
Клубочковая гематурия может проявляться как микро- и макрогематурия. Признаками клубочковой гематурии являются наличие эритроцитарных цилиндров, дисморфные эритроциты в мочевом осадке, протеинурия выше 2 г/сут. Если эти условия отсутствуют, то необходимо исключить другие источники кровотечения, в том числе с применением эхоГ и цистоскопии. Следует помнить, что с бессимптомной гематурии могут манифестировать многие системные заболевания, практически все гломерулопатии, ряд инфекций (особенно туберкулез), опухоль. Из гломерулопатий особого упоминания заслуживают фокально-сегментарный гломерулонефрит (прежде всего IgA-нефропатия), диффузный мезангиопролиферативный гломерулонефрит, наследственные заболевания.
Наиболее частой причиной изолированной гематурии является IgA-нефропатия (болезнь Бергера). Развивается чаще у детей и мужчин в возрасте 20–30 лет. Среди всех гематурических синдромов составляет 25–40% в Азии, 10–20% — в Европе, крайне редко возникает у лиц негроидной расы. Иногда выявляют случайно при лабораторных исследованиях как микрогематурию или как макрогематурию через несколько дней после неспецифических инфекций верхних дыхательных путей. У 85% больных повышен уровень IgA в крови. Признак неспецифичен, поскольку повышение IgA выявляют у больных алкоголизмом, при капилляротоксикозе, СКВ. Хотя, учитывая высокую вероятность поражения почек при указанных заболеваниях, повышение IgA вряд ли случайно. Аналогично отложениям IgA в почках, иммуноглобулин откладывается в коже. В динамике у больных с IgA-нефритом возможно развитие почечной недостаточности, АГ. Вероятность формирования нефротического синдрома невысокая. При начале заболевания в детском возрасте вне видимой связи с инфекцией, при минимальной или рекуррентной гематурии, небольшой протеинурии, отсутствии АГ прогноз хороший. В первую очередь необходимо провести ДД с постстрептококковым нефритом. В противоположность IgA-нефропатии при постстрептококковом нефрите патологические изменения развиваются не через 2–3 дня после перенесенной инфекции, а через 6–21 день, типичны отеки, АГ, снижение уровня комплемента, повышение титра антистрептококковых антител. Окончательный диагноз возможен после пункционной биопсии и выявлении IgA в мезангиуме.
Острый нефритический синдром
Характеризуется неожиданным началом, нередко с предшествующей инфекцией. В мочевом осадке типичны гематурия, эритроцитарные цилиндры, дисморфные эритроциты, протеинурия вариабельна. Гломерулярная фильтрация снижена, склонность к олигурии. Задержка натрия и соответственно воды ведут к отекам и повышению АД. При постинфекционных формах достаточно высока вероятность спонтанного выздоровления. Острый постинфекционный нефрит развивается после стрептококковых инфекций, при инфекционном эндокардите, при абсцессах, при заражении пневмококкоками, менингококками, микоплазмами, вероятно, при вирусах Эпштейна — Барр и эпидемического паротита.
Острый гломерулонефрит — негнойное диффузное, сегментарное или фокальное воспаление клубочков обеих почек. Неинфекционный острый гломерулонефрит развивается при СКВ, васкулите, криоглобулинемии, синдроме Гудпасчера, первичной гломерулопатии (мембранопролиферативный нефрит, IgA-нефрит, идиопатический быстропрогрессирующий гломерулонефрит).
Острый постстрептококковый нефрит — классическая форма эндокапиллярного острого гломерулонефрита с отложением иммунных комплексов субэпителиально на внешней стороне базальной мембраны. Развивается остро спорадически или эндемически через 7–30 дней после инфекции, вызванной бета-гемолитическим стрептококком группы А. Поражает в основном детей школьного возраста, но около 8% больных составляют лица в возрасте старше 50 лет. Для гломерулонефрита, развившегося после фарингита и тонзиллита, типичен латентный период в 6–21 день (чаще связан со стрептококком 12-го типа). После импетиго, инфицированных ран, гнойного отита латентный период составляет 14–30 дней (чаще стрептококк 29-го типа).
Анализ причин острого гломерулонефрита основывается на данных анамнеза и клиники. Полиорганное поражение типично для системных заболеваний (узелковый периартериит, СКВ). Лихорадка и острый нефритический синдром сочетаются при инфекциях или системных заболеваниях. К постинфекционным гломерулонефритам относится и поражение почек при инфекционном эндокардите, которое в современных условиях (на фоне леченного инфекционного эндокардита) чаще протекает как иммунокомплексное заболевание, а не результат эмболий. Наличие соответствующей аускультативной картины при выслушивании сердца, данные эхоГ), посева крови помогают выяснить причину острого нефритического синдрома. Клиническая картина острого гломерулонефрита варьирует от бессимптомных протеинурии и микрогематурии (у 50% больных) до макрогематурии, отеков, АГ, олигурии, головной боли, сомноленции и судорог (при энцефалопатии). В крови снижено содержание комплемента (табл. 31.2), повышены титры антистрептококковых антител, кретинина. В моче типичные нефритические изменения (эритроцитурия, зернистые/эритроцитарные/цилиндры; протеинурия, повышение осмолярности/повышение удельного веса). При исследовании биоптатов в световом микроскопе — пролиферация мезангиума, интерстициальный отек, инфильтрация гранулоцитами, реже экстракапиллярная пролиферация, в электронном микроскопе субэпителиальные депозиты, а при иммунофлюоресценции глыбчатые отложения IgG, СЗ, фибрина. По данным Вовси, классический острый постстрептококковый гломерулонефрит при соблюдении постельного режима, тепла и диеты в 80% случаев завершается спонтанным выздоровлением. Но у 1% детей и у 10% взрослых острый нефротический синдром трансформируется в быстро прогрессирующий гломерулонефрит.
Таблица 31.2
Динамика содержания СЗ и С4 фракций комплемента при различных вариантах поражения клубочков
| Болезнь или синдром | СЗ | С4 |
| Острый постинфекционный гломерулонефрит | ↓↓ | ↓↓ |
| Быстро прогрессирующий гломерулонефрит I типа (с антителами к базальной мембране) | N | N |
| Быстро прогрессирующий гломерулонефрит II типа (пауци-иммунный) | N | N |
| Мембранопролиферативный гломерулонефрит: | ||
| I тип
активация комплемента по классическому пути |
↓↓ | N или ↓ |
| II тип
активация комплемента по альтернативному пути |
↓↓ | N |
| Мезангиопролиферативный гломерулонефрит | N | N |
| Мембранозный гломерулонефрит; нефрит с минимальными изменениями*; фокально-сегментарный гломерулосклероз; IqA | N | N |
| Криоглобулинемия | N или ↓↓ | ↓↓ |
| Гломерунефрит при СКВ | ↓ или ↓↓↓ | ↓ или ↓↓↓ |
| СПИД-ассоциированная нефропатия** | ↓ | ↓↓ |
| Амилоидоз, сахарный диабет | N | N |
*Липоидный нефроз по прежней терминологии; **свойственна прежде всего ВИЧ-инфицированным, применявшим героин.
Быстро прогрессирующий гломерулонефрит (гломерулонефрит crescendo). Развивается нечасто. Синдром характеризуется фульминантным течением с почечной недостаточностью, протеинурией, гематурией; гистологически — фокальным и сегментарным некрозом и пролиферацией эпителиальных клеток большинства клубочков. Поражает лиц всех возрастов, но чаще всего молодых, внешне крепких. Возможны несколько вариантов. Идиопатический:
I тип обусловлен наличием антител к базальной мембране. По разным статистическим сводкам он составляет 8–22% всех случаев быстро прогрессирующего гломерулонефрита. Первичный антиген — компонент комплемента IV типа. Аутоагрессивные Т-лимфоциты запускают процесс образования антител, которые выявляют и в крови, и методом иммунофлюоресцентной микроскопии на базальной мембране гломерул. У 50–90% больных эти антитела перекрестно реагируют с базальной мембраной клубочков. Поэтому быстро прогрессирующий гломерулонефрит с антителами к базальной мембране клубочков может быть без легочных кровотечений (собственно гломерулонефрит) и с легочными геморрагиями (синдром Гудпасчера — см. ниже).
II тип — с циркулирующими иммунокомплексами. Составляет 35–40% всех случаев быстро прогрессирующего гломерулонефрита. Рассматривается как идиопатический, но отмечен при сифилисе, малярии, малигномах, криоглобулинемии, IgA-нефропатии, на фоне назначения D-пеницилламина.
III тип — патогенетический механизм не известен. Характеризуется и как пауци-иммунный быстро прогрессирующий гломерулонефрит (35–42%). Особенностью является отсутствие иммунных комплексов или отложений комплемента в клубочках. Серологические маркеры данного варианта гломерулонефрита — антинейтрофильные цитоплазматические аутоантитела ANCA (при системных васкулитах, внепочечных заболеваниях); антитела к протеиназе-3 лейкоцитов (цитоплазматические или C-ANCA), антимиелопероксидазные антитела (перинуклеарные или P-ANCA).
Быстро протекающий нефрит гистологически выглядит как мембранозный нефрит (наиболее частая причина нефротического синдрома у взрослых), в 20% завершающийся почечной недостаточностью, или мембранопролиферативный гломерулонефрит. Быстро прогрессирующий гломерулонефрит может начаться малосимптомно или дебютировать с тяжелых уремических симптомов или катастрофической задержки жидкости (отеки, застой в легких). Ведущими клинико-морфологическими признаками являются быстрое снижение клубочковой фильтрации, экстракапиллярная пролиферация с полулуниями, спонтанной ремиссии не наблюдается. Подобно вышеописанному острому постстрептококковому гломерулонефриту наблюдаются, но значительно выраженнее, общие симптомы, почечная недостаточность, гематурия, протеинурия. АГ может быть выражена меньше, чем при постстрептококковом нефрите. Надежный критерий диагноза «быстро прогрессирующий нефрит» — наличие антител к базальной мембране клубочков. Но их выявляют не всегда.
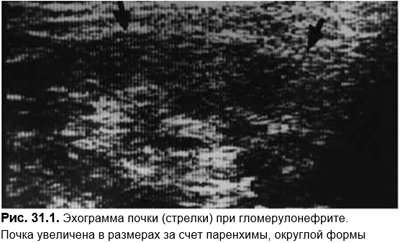
Для прогноза и выбора терапевтической тактики решающей является биопсия почек. Для быстро прогрессирующего гломерулонефрита характерны экстракапиллярная пролиферация более чем в 50% полученных клубочков. В ДД клубочковых механизмов почечной недостаточности быстро прогрессирующего гломерулонефрита необходимо учитывать сосудистые (прегломерулярные) механизмы (аневризма аорты, артериальные эмболии, гемолитико-уремический синдром, васкулит, злокачественная АГ, ССД и др.), постгломерулярные заболевания (острый тубулонекроз, острый интерстициальный нефрит, острая уратная нефропатия, обструктивная уропатия). Понятно, что ДД основывается на анамнезе (наличие васкулитов, ДБСТ, боль в животе при аневризме аорты, уратная нефропатия при цитостатической терапии лимфом и лейкозов, прием лекарственных препаратов при остром тубулонекрозе и т. д.), данных клинической картины, где определяющим и первоначальным является не нефритический синдром, а вышеуказанные состояния. Лабораторные исследования: картина крови при гемолитико-уремическом синдроме, воспалительные сдвиги и иммунные сдвиги при васкулитах, экскреция натрия и осмолярность мочи при тубулонекрозе, гиперурикемия при уратной нефропатии. Необходимо проведение эхоГ (рис. 31.1) и допплерографии почечных сосудов, позволяющей легко исключить обструктивную нефропатию, аневризму аорты, описать состояние почечной паренхимы, кровоток по сосудам почечной ножки и внутрипочечным сосудам.
Синдром Гудпасчера обусловлен циркуляцией антител к базальной мембране клубочков, протекает с быстро прогрессирующей почечной недостаточностью и гемоптоэ. Последнее не удивительно, если представить себе принцип строения и функции легочных альвеол и легочных клубочков. Упрощенно говоря, клубочек — это альвеола с сосудистым клубочком внутри и измененным растворителем (вода заменена на газ). Как указано выше, перекрестно реагирующие антитела к базальной мембране клубочков и альвеол выявляют у 50–90% пациентов с быстро прогрессирующим гломерулонефритом I типа. Но синдром Гудпасчера отмечают значительно реже. Это обусловлено тем, что для фиксации антител необходимо предшествующее повреждение альвеол (грипп, курение, промышленные токсические газы). Имеют значение и предрасполагающие генетические факторы. Синдром Гудпасчера развивается преимущественно у мужчин в возрасте 20–40 лет, особенно при гаплотипе HLA DR2. Диагностируют на основании быстро прогрессирующего экстракапиллярного гломерулонефрита с полулуниями с наличием циркулирующих антител к базальной мембране и гемоптоэ с быстро меняющимися легочными инфильтратами. АГ бывает не всегда. ДД основана на оценке легочно-почечного синдрома, исключении васкулита (узелковый периартериит, капилляротоксикоз), для которых не типично кровохарканье, гранулематоза Вегенера (васкулит верхних и нижних дыхательных путей с присоединением вакулита почек), легочной эмболии с нефротическим синдромом, застойной сердечной недостаточности с уремией, инфекционного эндокардита правых отделов сердца с инфарктом легкого и иммунокомплексным нефритом, силиконовой нефропатией, лекарственной реакции.
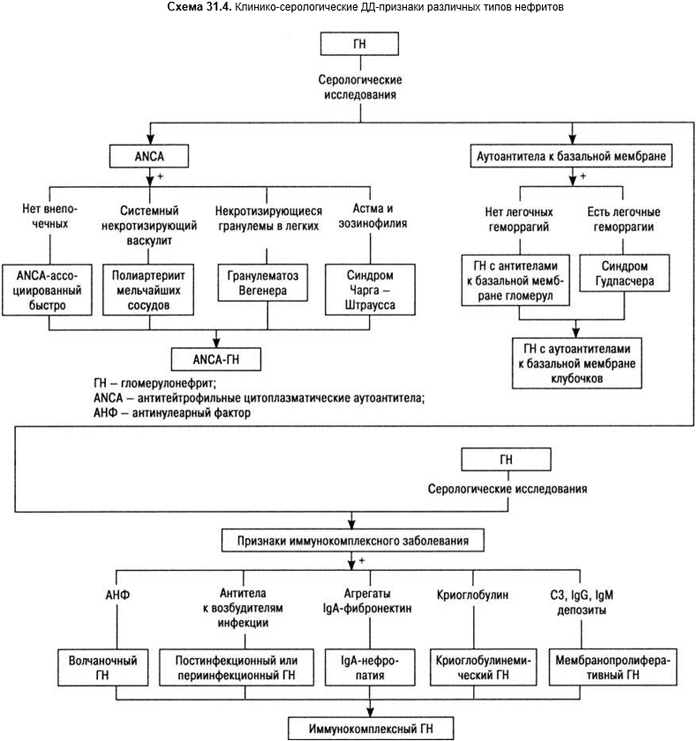
Клинико-иммунологические особенности различных типов нефритов суммированы в схеме 31.4.
Нефротический синдром
Обусловлен повышенной проницаемостью клубочковых капилляров для белка в плазме крови. Нет нефротического синдрома без генетической неполноценности почек. Как первичный идиопатический (первичное гломерулярное поражение) развивается при гломерулонефрите с минимальными изменениями (14%), при фокально-сегметарном гломерулосклерозе (15%), при мембранозном гломерулонефрите (45%), при мембранозно-пролиферативном нефрите (15%) и при других пролиферативных заболеваниях почек. Вторичный нефротический синдром (вторичное поражение клубочков) отмечают при аллергических реакциях на укус насекомых, при сывороточной болезни, пыльцевой аллергии, при постстрептококковом нефрите, дифтерии, инфекционном эндокардите, проказе, гепатите В, цитомегалии, инфекционном мононуклеозе, малярии (особенно 4-дневной), трипаносомозе, шистосомозе, филляриозе, при применении препаратов золота, пецилламина, НПВГТ, солей висмута, ртути, лития, каптоприла, употреблении героина, при СКВ (до 20% всех случаев нефротического синдрома), амилоидозе, неспецифическом язвенном колите, узелковом васкулите и капилляротоксикозе, при герпетиформном дерматите, синдроме Гудпасчера, саркоидозе, сахарном диабете, болезни Ходжкина и неходжкинских лимфомах, хроническом миелолейкозе, множественной миеломе, раке легкого, молочной железы, почек, щитовидной железы, яичников и шейки матки, феохромоцитоме, меланоме, IgA-нефрите, синдроме Альпорта, болезни Фабри, синдроме «желтых ногтей», серповидно-клеточной анемии, дефиците альфа-1-антитрипсина, преэклампсии.
Нефротический синдром можно диагностировать при потере белка более 3 г/сут у взрослых или 3,75 г/сут/1,73 м2 поверхности тела. Нефротический синдром — во многом проблема педиатрии. У детей чаще развивается в возрасте 1,5 года–4 лет, среди заболевших преобладают мальчики. У взрослых различий в заболеваемости по половому признаку нет.
Симптоматика нефротического синдрома разворачивается только в случае превышения потери белка над способностью к синтезу альбумина. Для нефротического синдрома типичны умеренная гипотензия, высокая чувствительность к мочегонным препаратам, редкое развитие острой почечной недостаточности, гипоальбуминемия и отеки. Отеки первоначально локализуются на веках и на ногах, в тяжелых случаях возможна анасарка. Клинически значимые отеки появляются только при уровне альбуминов в крови ниже 25 г/л. Признаками нефротического синдрома являются гиперлипопротеинемия с быстро развивающимися атеросклеротическими изменениями. Гиперлипидемия чаще (63%) разворачивается по ПА или НВ типу (по Фридериксену), реже (27%) V тип, еще реже (по 5% каждый) Ш и IV тип. Повышенная свертываемость крови со склонностью к тромбозам и тромбоэмболиям, чрезпочечная потеря антитромбина III, выраженная агрегация тромбоцитов обусловливают высокую вероятность развития венозных тромбозов, тромбозов почечной артерии и тромбэмболий. Группа высокого риска — пациенты с альбуминемией <20 г/л. Типичны потеря через почки транспортных протеинов и иммуноглобулинов (высокий риск инфекции вплоть до спонтанного перитонита), преренальная азотемия. АГ типична для вторичного нефротического синдрома на фоне СКВ и сахарного диабета. ДД нефротического и нефритического синдромов основывается на следующие опорные пункты. Для нефротического синдрома типичны отеки, протеинурия, повышение уровня холестерина и снижение альбумина в сыворотке крови. По данным эхоГ почки увеличены в размерах. Для нефритического синдрома характерны АГ, умеренная протеинурия, клеточный мочевой осадок (эритроцитурия, зернистые цилиндры), снижение фильтрационной способности почек, альбумин и холестерин в сыворотке крови не изменены. ДД различных форм и причин нефротического синдрома основывается прежде всего на уточнении его основного признака — отеков — и может быть, в зависимости от сочетания с другими проявлениями, представлена как нефротический (отечный) синдром в сочетании с:
- АГ, гематурией = гломерулонефрит;
- кожно-геморрагическим синдромом = системный васкулит;
- абдоминальным синдромом = геморрагический васкулит, или тромбоз почечных вен, или амилоидоз;
- токсическим синдромом при отравлениях = токсический нефроз;
- пиурией, обструктивной уропатией = вторичный нефротический синдром;
- азотемией, АГ = хроническая почечная недостаточность.
Или как отечный синдром иного характера:
- АГ, гематурией, протеинурией = гломерулонефрит;
- поражением сердечно-сосудистой системы = ревматизм или врожденные пороки сердца, или другие причины нарушения кровообращения;
- брадикардией, адинамией = гипотиреоз;
- энтеропатией, дистрофией = нарушение всасывания или переваривания;
- кожно-геморрагическим синдромом = аллергоз («атопический нефротический синдром»).
Хронический гломерулонефрит — вероятный исход всех первичных и вторичных нефритов. По секционным данным отмечают с частотой 0,2–0,8%. Не нашли подтверждения мысли о его возможной инфекционной, токсической или обменной этиологии. Выявления депозитов иммуноглобулинов и комплемента могут косвенно свидетельствовать об иммунном генезе. Признаком местной внутрипочечной гиперкоагуляции является определение фибринопептидов. Гистологически во многих гломерулах отмечают увеличение мезангиального матрикса, утолщение базальной мембраны, запустевание сосудистых петель, гломерулярные синехии. Интерстиций фиброзирован, тубулярный аппарат атрофирован. Характеризуется теми же признаками, что и острый гломерулонефрит. Но у большинства пациентов не удается выявить острого начала (первично хронический гломерулонефрит). Более того, значительная часть больных на протяжении многих лет остается бессимптомной. Гематурия и протеинурия на выражены, и заболевание манифестирует только на стадии почечной недостаточности. Значительную помощь в диагностике оказывает эхоГ, позволяющая выявить плотные, а впоследствии и уменьшенные (сморщенные) почки. При наличии такой картины и повышенного креатинина терминальную почечную недостаточность с необходимостью гемодиализа следует ожидать в ближайшие месяцы–год. Исходом хронического нефрита является почечная недостаточность.
Диабетическая нефропатия
Развивается в ближайшие 15–20 лет после начала сахарного диабета у 55% больных ювенильной формой сахарного диабета I типа и у 10% больных II типа. В случаях неадекватного лечения сроки манифестации диабетической нефропатии сдвигаются не в лучшую сторону. Заболевание является неиммунной гломерулопатией и частой причиной нефротического синдрома у взрослых. Первые признаки гломерулопатии появляются в ближайшие 1–4 года после начала сахарного диабета. Они проявляются бессимптомной невыраженной протеинурией и абактериальной лейкоцитурией. Позднее присоединяется микрогематурия. В моче также определяют жировые тельца и жировые цилиндры, что указывает на поражение канальцевого аппарата. Одновременно нередко выявляют диабетическую ретинопатию. При повышении протеинурии до 3 г/сут и более, а также азотемии клиническая симптоматика становится достаточно яркой. После этого в течение ближайших 3–8 лет прогрессирует снижение клубочковой фильтрации с развитием терминальной почечной недостаточности.
Врожденные заболевания клубочков протекают с теми же признаками, что и приобретенные гломерулопатии. К врожденным заболеваниям гломерул относят синдром Альпорта (наследственный нефрит), болезнь Фабри (диффузная ангиокератома), синдром «желтых ногтей» (наследственная онихоостеодистрофия) и синдром тонкой базальной мембраны.
Наследственный нефрит (синдром Альпорта)
Семейные случаи прогредиентно текущей нефропатии с микро- и макрогематурией, почечной глюкозурией, серин-, аланин-, таурин-, глицин-, оксипролин- и пролинурией, протеинурией вплоть до нефротического синдрома, с формированием в итоге почечной недостаточности. Показана генетическая гетерогенность наследственного нефрита. Хотя имеются и а/p варианты, наиболее частыми формами наследственного нефрита являются Х-сцепленные доминантные. Мутация возникает в гене COL 4А5, ответственного за синтез цепи альфа-5 коллагена 4-го типа.
I тип наследуется а/д и проявляется уже у детей. Из внепочечных проявлений известны внутренняя тугоухость, особенно на высокие частоты, катаракта, пигментный ретинит, периферическая нейропатия, нарушения функций тромбоцитов, лейкоцитопения с относительным лимфоцитозом. При офтальмологическом обследовании выявляют катаракту, лентиконус, помутнение хрусталика, изменения на глазном дне. Злокачественно текущие варианты свойственны мужчинам. При биопсии почек определяют расщепление базальной мембраны клубочков, утолщение гломерулярной и тубулярной мембраны и пенистые липидсодержащие клетки в интерстиции.
II тип наследуется Х-сцепленно. Выявляют у людей молодого возраста. Сочетается с глухотой, аномалиями глаз.
III тип. Х-сцепленный. Локализация гена — Xq21–Xq22. Нефропатия, тугоухость, аномалии глаз. Терминальная почечная недостаточность в возрасте 20–38 лет.
IV тип. Х-сцепленный. Тип взрослых, нет тугоухости, нет тромбоцитопатии. Аномалии глаз — у 15%.
V тип. А/д. Возраст формирования почечной недостаточности неизвестен. Тугоухость, тромбоцитопатия.
VI тип. А/д. Манифестирует рано. Тугоухость, аномалии глаз.
Болезнь Фабри (диффузная ангиокератома) передается а/p, Х-сцепленно. Является нарушением обмена гликосфинголипидов, обусловленным дефицитом альфа-галактозидазы-А. В итоге во всех органах накапливается тригексозилкерамид. Почечная патология проявляется умеренной протеинурией и гематурией. К 30–40-му году жизни формируется почечная недостаточность. В мочевом осадке выявляют пенистые клетки, в почечных биоптатах при электронной микроскопии — липидные включения в клетках клубочкового эпителия. Внепочечная симптоматика проявляется изменениями кожи (точечные или большей величины синюшные или черноватые образования преимущественно на губах, щеках, в подмышечных впадинах, на кончиках пальцев, около пупка, на мошонке, уменьшение потоотделения), неврологическими изменениями (раздражительность, головная боль, парестезии), ревматоидноподобная боль, помутнение роговицы, сосудисто обусловленная патология сердца, ЦНС.
Синдром «желтых ногтей» — наследственная онихоостеодисплазия, а/д. Нарушение связано с локусом групп крови системы АВО. Проявляется уже в детском и юношеском возрасте. Синдром является сочетанием дисплазии или гипоплазии ногтей рук и ног, отсутствием надколенника и другими дисплазиями костей, умеренной гемат- и протеинурией (в 10% случаев трансформирующейся в нефротический синдром). Гистологически в почках определяют неспецифический гломерулосклероз и повышение клеточности мезангиума.
Синдром тонкой базальной мембраны (доброкачественная семейная гематурия). Вероятен а/д тип наследования. Единственной патогистологической находкой является истончение базальной мембраны клубочков до 150–225 нм (в норме 300–400 нм). У большинства пациентов протекает бессимптомно. Состояние выявляют случайно по результатам анализов мочи или во время эпизодов макрогематурии. Очень редко отмечают боль в поясничной области, что является одним из ее отличий от IgA-нефропатии. Кроме того, при IgA-нефропатии обычно не прослеживаются семейные случаи гематурии.
В отличие от Х-сцепленно наследуемых наследственных нефритов нет передачи по линии отец–сын, нет внепочечных проявлений (глухота, нарушение зрения) и случаев почечной недостаточности.
Отдаленный прогноз при доброкачественной семейной гематурии благоприятен.
Тубулоинтерстициальные нефропатии
Тубулоинтерстициальная нефропатия может быть обусловлена инфекцией, обструкцией, лекарственными средствами, электролитными нарушениями (гиперкальциемическая, гипокальциемическая и уратная нефропатии) и другими более редкими состояниями (постлучевой нефрит, серповидно-клеточная анемия, балканская нефропатия, множественная миелома, саркоидоз, синдром Шегрена, кистозные поражения). Возможно острое или хроническое течение. Клиническая картина обусловлена синдромом поражения проксимальных канальцев (ацидоз, синдром Фанкони), дистальных канальцев (ацидоз, гиперкалиемия, потеря натрия), медуллярного слоя (снижение концентрационной способности почек), вторичным поражение клубочков (снижение фильтрационной способности, АГ, протеинурия). Чаще мочевой центрифугат беден клетками. Процесс характеризуется абактериальной лейкоцитурией, эритроцитурией. При бактериальном воспалении определяют лейкоциты (возможно в большом количестве), клетки канальцевого эпителия, отдельные эритроциты, при аллергическом интерстициальном нефрите — эозинофильные гранулоциты. Протеинурия — не более 1,5 г/сут, выделяются низкомолекулярные белки (лизоцим, бета-микроглобулин). Возможна глюкозурия.
Острый бактериальный пиелонефрит, являющийся в большинстве случаев результатом восходящей инфекции (у детей чаще гематогенный занос), без предшествующего изменения почек (тубулопатии), без нарушения естественного пассажа мочи — большая редкость. Такой пиелонефрит быстро излечивается и не представляет динамических проблем. Любой пиелонефрит затяжного течения (более 1 нед) требует исключения вторичности его генеза. Наиболее распространенные возбудители — грамотрицательная флора (кишечная палочка, энтеробактерии). Во многих случаях невозможно клинически отдифференцировать инфекцию нижних мочевых путей (уретрит, цистит) от инфекции верхних мочевых путей (пиелонефрит). Инструментальными исследованиями доказано, что у 28–52% людей с симптомами инфекции нижних отделов мочевых путей имеется и субклинический инфекционный процесс в почках.
Пиелонефрит диагностируют преимущественно у иммуносупремированных пациентов, у детей, женщин, а также у мужчин пожилого возраста с гипертрофией простаты. Среди этих групп существенную бактериурию чаще всего выявляют у мужчин пожилого возраста. И это связано не с пиелонефритическим процессом как таковым, но обусловлено нарушением мочеиспускания и большим объемом остаточной мочи.
Острый первичный пиелонефрит у мужчин молодого возраста типичен для фимоза, при половых контактах с женщинами с вагинитом, при гомосексуализме, СПИДе (СД4 Т-лейкоциты <200/мкл).
Острый пиелонефрит протекает с повышением температуры тела, иногда с ознобом, с общим плохим самочувствием, с тошнотой, рвотой, не исключены понос, боль в пояснице, миалгия. При лабораторных исследованиях отмечают лейкоцитоз вплоть до токсической зернистости лейкоцитов, повышение СОЭ. В анализах мочи пиурия, микрогематурия, следовая протеинурия. В острых случаях поданным эхоГ нечеткость рисунка паренхимы почек, стертость контуров органа, отек пирамидок. Но в целом картина неспецифична. При остром пиелонефрите экскреторная урография, как правило, не показана (угроза тубулонекроза или некроза сосочка пирамидки). Типичными осложнениями являются преходящая почечная недостаточность при двустороннем тяжелом пиелонефрите (крайне редко при одностороннем), сепсис, хронизация пиелонефрита. При ДД необходимо исключить аппендицит, дивертикулит, панкреатит, базальную пневмонию с синдромом боли в животе.
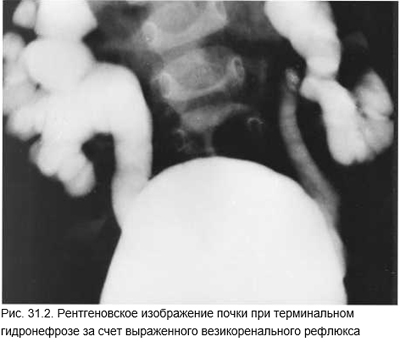
Острый неосложненный пиелонефрит через несколько дней при адекватной терапии стихает. Если этого не произошло или развиваются рецидивы, то следует исключать вторичный пиелонефрит, инфицированный гидронефроз, перинефритический абсцесс. Вторичный пиелонефрит должен быть исключен как можно быстрее при каждом остром пиелонефрите, потому что это решительно меняет тактику лечения и прогноз. Вторичный пиелонефрит развивается на фоне рефлюкса* (рис. 31.2), аномалии почек или мочеточника, камня почки. Методом выбора является эхоГ, быстро и без нагрузки для больного выявляющая обструкцию мочевых путей, забрюшинные, в том числе и перинефритические абсцессы.
Медикаментозное поражение почек может быть обусловлено разными механизмами. Повреждения клубочков чаще дозонезависимые. Имеют, вероятнее всего, иммунную природу. Достаточно часто вызываются D-пеницилламином, НПВП. Протекают как мембранозно-пролиферативный нефрит с нефротическим синдромом. Как дозозависимая хроническая токсическая тубулоинтерстициальная нефропатия протекают изменения почек при применении фенацетина и парацетамола. Острая дозозависимая почечная недостаточность развивается при неблагоприятной реакции на аминогликозиды, цефалоспорины и сульфаниламиды. (Подробнее дозозависимый медикаментозно обусловленный интерстициальный нефрит см. Тубулонекроз). Для дозонезависимой реакции гиперчувствительности типичен острый интерстициальный нефрит.
Острый медикаментозный дозонезависимый интерстициальный нефрит часто связан с внепочечными проявлениями непереносимости того или иного препарата. Наиболее частыми препаратами, вызывающими дозонезависимый интерстициальный нефрит, являются метациклин, ампициллин, оксациллин, цефалоспорины, рифампицин, сульфаниламиды, НПВП (ибупрофен, напроксен), противосудорожные (фенитоин), иммуносупрессанты, тиазидные диуретики, фуросемид, этакриновая кислота, антикоагулянты, аллопуринол, циметидин, каптоприл, клофибрат. Клиническая картина складывается из внепочечных симптомов гиперчувствительности (могут отсутствовать) в виде сыпи, артралгии, лихорадки, эозинофилии, повышения уровня IgE, выделения эозинофильных гранулоцитов с мочой и почечных симптомов. Острая олигурическая (55%) или неолигурическая (45%) почечная недостаточность с частичным выпадением тубулярных функций в 80% случаев благополучно завершается после отмены препарата. Гистологически в почках на высоте патологических изменений выявляют интерстициальные инфильтраты из лимфоцитов, плазмоцитов, отек при неизмененных клубочках и отрицательных иммунофлюоресцентных тестах.
ДД острого дозонезависимого интерстициального нефрита проводят с острым иммунокомплексным гломерулонефритом (в результате бактериальной инфекции, потребовавшей применения антибиотиков) и острым тубулонекрозом при сепсисе или на фоне применения тубулотоксичных препаратов. Изменения клеточного состава мочи, ее осмолярности, экскреции натрия позволяют отдифференцировать острый тубулонекроз от острого дозонезависимого интерстициального нефрита. Определение эритроцитарных цилиндров в моче свидетельствует в пользу гломерулонефрита. А эозинофильные гранулоциты в моче типичны для интерстициального нефрита. Окончательные сомнения могут быть разрешены только биопсией почек.
Хронический интерстициальный нефрит объединяет хронический пиелонефрит, нефропатию после применения анальгетиков, балканскую нефропатию, лучевой нефрит, саркоидоз почек и изменения почек при синдроме Шегрена, кистоз почек.
Хронический интерстициальный нефрит при применении анальгетиков развивается у лиц, длительное время принимавших комплексные обезболивающие препараты (особенно сочетание кислоты ацетилсалициловой и фенацетина, парацетамола, кофеина и кодеина) и нередко осложняется некрозом сосочков пирамидок или раком мочеточников. Повреждающий эффект препаратов объясняется образованием свободных радикалов в медуллярной ткани почек. Перед развитием почечной симптоматики могут манифестировать другие симптомы: боль самой разнообразной локализации, особенно головная (без достоверных клинико-инструментальных и лабораторных отклонений), симптоматика со стороны желудочно-кишечного тракта. Последняя во многом обусловлена кислотой ацетилсалициловой и характеризуется развитием эрозий и язв в желудочно-кишечном тракте. Скрытые и явные кровотечения при язвенно-эрозивных процессах приводят к развитию анемии. Вдобавок салицилаты блокируют агрегацию тромбоцитов, что не способствует гемостазу. Позднее анемия усугубляется почечной недостаточностью и побочным действием производных фенацетина (р-фенитидина), вызывающих образование мет- и сульфгемоглобина и гемолиз. Этот же препарат способен вызвать коричневато-грязную окраску кожи, типичную для пациентов, злоупотребляющих обезболивающими средствами. Требуется принять достаточно большое количество (1,5–2 кг) фенацетина, чтобы развился медикаментозный интерстициальный нефрит. Болеют им чаще женщины пожилого возраста (при всем старании требуется время, чтобы успеть съесть 2 кг таблеток), производящие впечатление не совсем психически уравновешенных (понятно, ни один вполне нормальный человек не осилит 2 кг фенацетина). Речь идет, видимо, о нераспознанной депрессии, выливающейся в страх за свое здоровье. Почечная симптоматика сводится на ранних этапах к стерильной лейкоцитурии, затем проявляется упорной рецидивирующей инфекцией мочевых путей, очень умеренной тубулярной протеинурией, медленно, подспудно нарастающей почечной недостаточностью, коликами, макрогематурией и дизурией за счет отхождения некротизированных почечных сосочков.
Диагноз «нефропатия, обусловленная приемом анальгетиков», основывается на данных анамнеза, на выявлении почечной патологии у людей (чаще женщин) зрелого и пожилого возраста с неустойчивой психикой, при сочетании почечной патологии и эрозивно-язвенных изменений желудочно-кишечного тракта и наличии анемии, тубулоацидоза, гиперкалиемии при низком уровне креатинина, определении производных фенацетина в моче, при почечной колике без признаков мочекаменной болезни (папиллонекроз), эхоГ- или рентгенологическом подтверждении некроза сосочков пирамидок (кальцинаты, гематурия, гистологические изменения).
До 15% больных с анальгетическим хроническим интерстициальным нефритом заболевают опухолями лоханок, мочеточника, мочевого пузыря. Поэтому каждый случай гематурии негломерулярного происхождения (отсутствие дисморфных эритроцитов и эритроцитарных цилиндров) у этих пациентов должен быть сигналом к экстренному исключению объемного злокачественного процесса (эхоГ, экскреторная урография, КТ, Я.МР, цистоскопия).
Хронический бактериальный пиелонефрит диагностируют при наличии бактериурии, клинико-лабораторного подтверждения тубулоинтерстициального нефрита, при таких типичных радиологических выявлениях, как деформации собирательной системы и изменений паренхимы. Хронический пиелонефрит не может быть первичным. Он развивается на фоне обструктивной нефропатии, прежде всего рефлюкса (выявляют у 35–45% детей с рецидивирующей инфекцией мочевых путей), генетически детерминированных тубулопатий, салурий и других состояний. Поэтому всегда требуются поиски основного состояния, приведшего к хроническому пиелонефриту.
Лучевой нефрит с поражением клубочков, канальцев, сосудов и интерстиция развивается при облучении в дозе более 20–25 Гр и завершается интерстициальным фиброзом. Латентный период колеблется от нескольких месяцев до 10 лет. Развивается после облучения ретроперитонеального пространства при лимфоме, опухоли Вильмса, раке яичников и яичек. Лучевой нефрит может начаться остро через 6–10 мес после облучения с гемато- и протеинурии, АГ или с самого начала протекать как первично-хронический. Современные протоколы химиотерапии уменьшили потребность облучения и вероятность развития лучевого нефрита в современных условиях значительно снизилась.
Балканскую нефропатию эндемически отмечают в Югославии, Румынии и Болгарии. Протекает как хронический интерстициальный нефрит с развитием почечной недостаточнсти к возрасту 30–60 лет. На этом фоне нередко развивается рак мочевых путей.
Интерстициальное образование гранулем выявляют у 35–45% больных саркоидозом. Однако такая высокая частота получена только по гистологическим данным, клинически или лабораторно изменения почек при саркоидозе определяют редко. Еще реже формируется почечная недостаточность. В первую очередь при саркоидозе следует исключать гломерулопатии и гиперкальциемическую нефропатию.
При болезни Шегрена тубулопатия может формироваться как результат лимфоплазмоцитарной инфильтрации интерстиция почки. Клиническая значимость этого процесса мала, но возможно развитие почечной формы несахарного диабета, проксимального канальцевого ацидоза, дистальный канальцевый ацидоз формируется очень редко.
Кистозные поражения почек
Обусловлены расширением трубочек и/или собирательных канальцев с образованием кист в паренхиме почек. Наряду с генетически детерминированными кистами известно кистообразование при хронической почечной недостаточности любого происхождения. Кистоподобные образования формируются после кровоизлияний, распада опухоли, образования каверн. Клиническая картина истинных кист вариабельна и определяется местными осложнениями, экстраренальными проявлениями и прогрессирующей почечной недостаточностью из-за сдавления паренхимы почки кистами. Кисты почек в работе практического врача выявляют либо при массовых обследованиях (группа риска — больные с гиперметропией, слепые, глухонемые), и в этих случаях они являются бессимптомными, либо сопровождающие их лихорадка, аллергические реакции, рецидивирующий неспецифический болевой синдром и некоторые другие расстройства служат основанием для первоначально ошибочных выводов. Но если заключение о наличии солитарной кисты воспринимается врачом с облегчением, как реализация «диагностического экстаза», то мнение о редкости поликистозной болезни отодвигает ее на одно из последних мест в цепи диагностического поиска, а в случае верификации диагноза последний иногда воспринимается с недоверием. Имеется не менее 23 синдромов, одним из проявлений которых является или может быть поликистоз. Поликистоз почек в научной литературе принято подразделять на два типа:
- поликистоз с аутосомно-рецессивным типом передачи, или инфантильный тип поликистоза;
- поликистоз с аутосомно-доминантным типом передачи, или поликистоз взрослого типа.
Но поликистоз с аутосомно-доминатным типом передачи и поликистоз с аутомно-рецессивным типом передачи могут проявляться на любом этапе жизни, от пренатального до подросткового периода и даже у взрослых. Клинико-радиологические проявления различных типов поликистоза сходны. Чаще отмечают поликистоз с аутосомно-доминатным типом передачи. Обычно при этом типе есть множество вариантов внепочечного поликистоза, но кисты печени чаще выявляют при поликистоз с аутосомно-рецессивным типом передачи.
Поликистоз с аутосомно-рецессивным типом передачи обусловлен мутацией гена PKDHD1, расположенного на коротком плече 6-й хромосомы. Ген ответственен за синтез белка фиброцестин/полидуктин, принципиально важного для сохранения нормальной структуры почечных канальцев и протоков печени. Первичный дефект при этом заключается в дисфункции ресничек. Общим анатомическим проявлением является не обструктивное двустороннее симметричное расширение собирательных канальцев почек.
Гены, ответственные за поликистоз с аутосомно-доминантным типом передачи, локализуются на коротком плече 16-й хромосомы (PKD1, 85% всех случаев поликистоз с аутосомно-доминантным типом передачи) и на длинном плече 4-й хромосомы (PKD2). Гены кодируют соответственно белки полицистин-1 и -2. Белки экспрессируются в формирующейся почке. При поликистозе с аутосомно-доминантным типом передачи, в отличие от поликистоза с аутосомно-рецессивным типом передачи, кисты развиваются в пределах нефрона.
Для практических целей сохраняет значение синдромальная классификация. Важнейшие из синдромов, основным проявлением которых является поликистоз, следующие:
- Поликистоз новорожденных (детский тип, синдром Поттера II тип I, а/p тип наследования. Кистозное перерождение почек и печени. Проявляется уремией и ведет к смерти ребенка вскоре после рождения или даже внутриутробно. Этот тип поликистоза характерен для населения некоторых районов Скандинавии. По данным эхоГ выявляет поликистоз почек уже у плодов, что позволяет прервать заведомо бесперспективную беременность.
- Синдром Поттер II тип II, характер наследования не установлен. Поликистозная дисплазия почки часто сочетается с пороками развития мочеточника или даже его гомолатеральной атрезией. Начало в позднем грудном или в детском возрасте. Первые признаки заболевания выявляют в возрасте старше 2 лет, иногда позже. Проявляются болью в животе, гепатомегалией. Возможна портальная гипертензия, прогрессирует почечная недостаточность.
- Поликистозная болезнь взрослых (наиболее известный тип заболевания), а/д тип наследования. Хотя первые неспецифические признаки могут появиться в детском или подростковом возрасте, манифестная картина разворачивается на 3-м десятилетии жизни, характеризуется гематурией и прогрессирующей почечной недостаточностью. Поликистоз почек нередко сочетается с кистами в печени, поджелудочной железе, селезенке, яичниках и семенных канальцах, а также мешотчатой аневризмой сосудов мозга (у 4% молодых взрослых и до 10–15% у стариков). С поликистозной болезнью взрослых могут сочетаться миотоническая дистрофия, синдром Пейтца — Егерса, сфероцитоз.
- Особо обсуждается группа состояний, носящих название «нефронофтиз» и «медуллярная кистозная болезнь». Тип наследования различный.
- Синдром Каччи — Риччи — «пенистая почка» (рис. 31.3а, б), а/p, кистозная дисплазия дистальных почечных канальцев (терминальный поликистоз).
- Синдром Цельвегера (церебро-гепаторенальный), а/p, сочетает характерный внешний вид, мышечную гипотонию, судорожные припадки, признаки геморрагического диатеза, кисты коры почек.
- Синдром Смита — Лемли, а/p. Врожденная карликовость, умственная отсталость, мышечная гипотония, микрогения, клинодактилия, поликистоз почек, генитальная дисплазия у мальчиков.
- Все аутосомные синдромы трисомии, особенно трисомии Д, Е.
- Синдром Мирауда (а/р) — семейная кистозная дисплазия почек с кистозной дисплазией печени и мозга.
- Синдром Кароли — врожденное сегментарное мешотчатое расширение внутрипеченочных желчных путей. Склонность к образованию внутрипеченочных камней, холангиту, абсцессам печени. Нередко множественные кисты почек, фиброхолангиоматоз желчных путей. Эхокартина достаточно специфична: от ворот печени вглубь паренхимы радиально распространяются тяжи. Возможна картина внутрипеченочного холелитиаза с феноменом «акустической тени».
- Синдром Гиппеля — Линдау, а/д. Рассматривается и в группе факоматозов. Ангиомы сетчатки и мозжечка, которые могут сочетаться с кистами почек, поджелудочной железы, с феохромоцитомой, гипернефромой.
- Синдром двуликого Януса. Поликистозная болезнь почек юношеского типа. Отмечают крайне редко, тип наследования а/p? Одна половина лица имеет грустное выражение, вторая — более веселое. Кожа дряблая, коричневато-грязная, гипермобильность суставов, плоскостопие.

Несмотря на врожденный характер заболеваний этой группы, период правильной диагностики поликистоза существенно отдален не только от этапа более или менее выраженных морфологических изменений органов, но и от первых клинических проявлений. Такая ситуация во многом обусловлена сложностью визуализации мелких кист, расположенных в глубине паренхиматозных органов. Требуется время, в течении которого, кисты, увеличиваясь в размерах, минуют барьер разрешающей способности того или иного метода исследования. Между тем внедрение новых неинвазивных методов интраскопии, прежде всего эхоГ, существенно снизило «возрастной ценз» диагностики. По данным эхоГ кисты выглядят как округлые эхонегативные образования с феноменом усиления звука. Однако при множественных мелких кистах этот феномен не определяется как из-за размера кист, так и за счет бесчисленного множества поверхностей раздела. Эхокартина определяется степенью поражения органа. Кисты, увеличиваясь, могут сливаться и сдавливать паренхиму, в этих случаях почка производит впечатление изрешеченного органа. При синдроме Каччи — Риччи («терминальный поликистоз») множество кист создает эхокартину «пенистой почки», слой паренхимы едва определяется.
Очень типична картина медуллярной дисплазии — «губчатой почки» (рис. 31.4а, б), а/д. Отмечают относительно часто. В 20% — спорадическая мутация. Этот тип дисплазии нередко сочетается с голубыми склерами, тугоухостью, ломкостью костей, синдромами Элерса — Данлоса, Видемана — Беквита, гемигипертрофии, оксалаткальциевой нефропатией (камни, плотно спаяны с паренхимой, при оперативном вмешательстве отделяются с кровотечением). Морфологическая основа — кистозное расширение собирательных канальцев в пирамидках. Обычно выявляют при рентгеновских исследованиях, выполненных по поводу гематурии. Пирамидки при этом выглядят плотными, с накоплениями контраста («гроздья винограда»), с кальцинатами на сосочках пирамид. Клинически проявляется резистентной к вазопрессину полиурией, рецидивирующей гематурией, инфекцией мочевых путей, гиперкальциурией с нефрокальцинозом. Клубочковая фильтрация снижается только при тяжелом нефрокальцинозе. ДД медуллярной дисплазии проводят с дистальным тубулярным ацидозом, гиперпаратиреоидизмом (при наличии нефрокальциноза). Почки (в противоположность другим синдромам поликистоза) не только не увеличены, но могут быть даже уменьшены в размерах. Пирамидки, обычно эхонегативные, становятся эхопозитивными (эффект отражения ультразвука от множества мелких кист) и напоминают лепестки цветка. Мы назвали этот признак «феномен ромашки».
При поликистозе почек кисты в других органах выявляют позже. Это понятно, если вспомнить, что киста — это патологическое образование, возникшее вследствие появления жидкости в патологически измененном месте, заполненное жидкостью и увеличивающееся в результате накопления жидкости. Ни один орган не продуцирует столько жидкости, сколько почка. Так, кисты в поджелудочной железе формируются через 1,5–2 года после появления кист в почках. При паталого-анатомическом исследовании пораженной почки видно, что орган утрачивает бобовидную форму, становится округлым, бугристым, увеличивается в размерах, пронизан кистозными полостями различной величины, заполненными асептической мочой. Микроскопически среди единичных дистрофированных клубочков определяют кисты, выстланные уплощенным эпителием. Кисты окружены незрелой соединительной тканью с участками клеточно-воспалительной инфильтрации.
Поликистоз почек взрослых
Передается а/д. Клинические симптомы появляются в возрасте 30–50 лет. Почечная недостаточность формируется в 50–70 лет. Кисты в паренхиме почки приводят к отчетливому увеличению размеров органа. Диагноз основывают на семейном анамнезе, выявлении плотных бугристых увеличенных почек, в ряде случаев сочетающихся с плотной увеличенной бугристой печенью, данных эхоГ. Осложнениями являются медленно прогрессирующая почечная недостаточность, гематурия, присоединение инфекции и уросепсис, АГ или синдром потери натрия с гипотензией. Внепочечные симптомы обусловлены кистами печени (40%), легких, аневризмами сосудов мозга (15%) с вероятностью субарахноидальных кровоизлияний. Разрыв аневризм возникает у 75% их носителей, особенно в случаях неконтролируемой АГ, как правило, в возрасте до 50 лет. У 1/3 больных отмечают пролапс митрального клапана и аортальную регургитацию.
Нефронофтиз
А/p. Морфологическая основа — врожденные кисты на границе коры и паренхимы с интерстициальным фиброзом и вторичным гломерулосклерозом. Возможны два варианта нефронофтиза:
1. Ювенильный нефронофтиз, ген на хромосоме 2q13, приводит к почечной недостаточности уже в детском или подростковом возрасте. Часто сочетается с пигментным ретинитом, дегенерацией сетчатки, колобомой. В этом случае ювенильный нефронофтиз известен как синдром ретинально-ренальной дисплазии.
2. Взрослый тип, а/д, по типу медуллярной кистозной дисплазии, протекает без глазной патологии, терминальная почечная недостаточность формируется в зрелом возрасте. По данным эхоГ почки уменьшены в размерах, кисты настолько малы, что визуализируются редко. Кальцинаты не характерны.
ДД нефронофтиза с другими вариантами поликистоза основывается на следующих критериях. При нефронофтизе боль в боку и пояснице не характерна. Гематурия и АГ развиваются редко, почки уменьшены в размерах, клубочковая фильтрация снижена. Терминальная почечная недостаточность формируется в возрасте 4–6 или в 30–50 лет. Около 80% всех случаев являются семейными. При кистозных почках взрослого типа часто отмечают боль, АГ и гематурию. Почки увеличены в размерах, клубочковая фильтрация может быть снижена. Четко прослеживается семейная отягощенность. При медуллярной дисплазии боль отмечают только при отхождении камней или при гематурии. Почки обычных размеров, выявляют нефрокальциноз, снижение клубочковой фильтрации — только при осложненном течении. Выраженная почечная недостаточность формируется редко. Семейные случаи составляют не более 25%.
Поликистозная трансформация почек при почечной недостаточности
Кисты развиваются вторично при хронической почечной недостаточности. Количество и объем кист хорошо коррелирует с длительностью и выраженностью почечной недостаточности. При хроническом гемодиализе кисты выявляют у 90% больных. Анамнез, определение небольших сморщенных почек с кистами позволяет правильно интерпретировать генез этих кист.
Солитарные кисты почек выявляют чаще случайно. Если они не достигают больших размеров, сдавливающих почку, то прогноз благоприятен. Достоверно связаны с общей дисплазией соединительной ткани.
Нефропатии сосудистого генеза (подробнее описаны в соответствующих разделах)
Важнейшими сосудистыми заболеваниями, ведущими к патологии почек, являются АГ, приводящая к нефросклерозу, стеноз почечной артерии, закупорка артерии (эмболия, тромбоз почечной артерии, аневризма аорты), системный васкулит, диабетическая ангиопатия, гемолитико-уремический синдром, ССД. Клиническая картина почечной патологии при этих состояниях варьирует в зависимости от заболевания, обусловившего патологию почек. Так, при стенозах почечной артерии ведущим симптомом является АГ. Протеинурия обычно очень умеренная, функциональная способность почки снижается относительно поздно. При злокачественной АГ у 20–25% больных активируется система ренин — ангиотензин с быстрым ограничением клубочковой фильтрации, поскольку формируются доброкачественный или злокачественный артериолярный нефросклероз (тяжесть определяется скоростью развития склероза).
При системных васкулитах и гемолитико-уремическом синдроме симптоматика полностью аналогичная таковой при заболеваниях клубочков: протеинурия, гематурия, снижение фильтрационной способности. Повышение уровня креатинина может быть настолько стремительным, в течение нескольких дней, что возникает необходимость в исключении быстро прогрессирующего нефрита. При гемолитико-уремическом синдроме нарушается кровоток по мелким сосудам за счет их закупорки фибрином и тромбоцитами. Признаками гемолитико-уремического синдрома являются: фрагментированные эритроциты в мазке крови, ретикулоцитоз, повышение ЛДГ, тромбопения, обильный мочевой клеточный осадок, острая почечная недостаточность.
При прогрессирующем системном склерозе (ССД) облитерирующая ангиопатия интерлобарных артерий ведет к снижению перфузии почечной ткани. Склеродермическая почка особенно типична для больных пожилого возраста. Быстро развивается сморщенная почка.
Иммуннообусловленные поражения почек достаточно близко примыкают к нефропатиям сосудистого генеза, поскольку всегда сопровождаются нарушением микроциркуляции, а иногда и изменением более крупных, чем капилляры, сосуды. Иммунные нефропатии нами рассмотрены выше в разных разделах. Эта глава посвящена общим закономерностям и в то же время особенностям различных вариантов иммунных нефропатий.
Антигены могут быть почечными эндогенными (базальная мембрана клубочков или канальцев) и экзогенными (пересаженная почка), а также внепочечными эндогенными (нуклеиновые кислоты, опухоли, криоглобулины) или экзогенными (бактерии, вирусы, грибы, вакцины, лекарственные препараты).
Для инициации иммунного воспаления антиген должен быть локализован собственно в почке. Это может быть при первичной его там локализации (например коллаген IV типа базальной мембраны) или осажден (депонирован) из крови. В последнем случае место фиксации антигена (циркулирующего иммунного комплекса) определяется его физикохимическими свойствами. Молекулярная масса и размеры иммунного комплекса объясняют его отложение мезангии, на базальной мембране клубочков, на эндотелии с внутренней стороны базальной мембраны или на эпителиальных клетках с ее наружной стороны. Так, при болезни Бергера (IgA-нефропатии) иммунные комплексы содержат IgA, велики по размеру и могут откладываться только в мезангии. Бактериальные антигенные комплексы содержат IgG, невелики по размерам и откладываются в базальной мембране или на эпителиальной стороне базальной мембраны клубочков. Иммунное воспаление развивается далее по классическому или альтернативному пути активации комплемента, IgE — опосредованному, врожденному комплемент-дефицитарному и т. д., то есть патогистологические и соответственно клинические характеристики зависят от локализации и типа иммунного ответа.
Характеристики иммунных нефропатий суммированы в табл. 31.3.
Таблица 31.3
Основные характеристики иммуннообусловленных поражений почек
| Тип иммунной реакции | Характер поражения почек | Этиология или антиген | Место отложения компонентов иммунного комплекса | ||
| IgЕ | Комплемент | Фибрин | |||
| I, IgЕ-опосредованный | Иммунные тубулоинтерстициальные поражения | Разнообразные | Интерстиций | ||
| II, цитотоксический | Анти-БМК болезни | Базальная мембрана клубочков | Линейно вдоль базальной мембраны | Линейно вдоль базальной мембраны; иногда отсутствует | Экстракапиллярно |
| Анти-БМК болезни | Канальцевые антигены | Линейно вдоль базальной мембраны канальцев | Линейно вдоль базальной мембраны канальцев | ||
| Сверхострое отторжение | I класс HLA антигенов | Диффузно по сосудам | — | По сосудам | |
| ГН при болезни Вегенера | Цитоплазматическая МПО нейтрофильных гранулоцитов | Пауци-иммунный | Пауци-иммунный | Полулуния | |
| III, иммунокомплексный | Иммунокомплексный ГН (постстрептококковый, сывороточный, СКВ, гепатит В) | Различные экзогенные и эндогенные антигены | Глыбчато в стенке капилляров или в интерстиции | Глыбчато в стенке капилляров или в интерстиции | Минимально |
| IV, клеточно-опосредованный, поздний | Хроническое отторжение почки | II класс HLA антигенов | Диффузно зернисто | Диффузно зернисто | Сосудистое отложение |
| Хронический ГН | Антигены почки | Различно | Различно | Сосудистое отложение | |
| Непосредственно комплементобусловлено (альтернативный путь) | Мембранопролиферативный ГН | Иммунный комплекс, с3-нефритический фактор | Иногда в мезангиуме, чаще нет | с3 субэндотелиально или интрамембранозно | Редко |
| СПИД | Фокальный сегментарный ГН | ВИЧ | Фокально и сегментарно IqM | Фокально и сегментарно | — |
| Врожденный дефицит комплемента | Волчаночно-подобный синдром | Дефицит комплемента | Фокально
мезангиально |
Фокально мезангиально | |
| Гемолитико-уремический синдром | Дефицит фактора Н | Ишемический и коагуляционный некроз | |||
БМГ — базальная мембрана клубочков; БМК — базальная мембрана канальцев; МПО — миелопероксидаза; ГН — гломерулонефрит.
- I тип иммунной нефропатии (IgE-опосредованный, анафилактический, немедленный). Т-лимфоциты, контактируя с аллергеном, высвобождают ИЛ-4 и ИЛ-5, активирующие базофильные гранулоциты, мастоциты и запускающие синтез IgE. Высвобождаются вазоактивные субстанции (гистамин), ИЛ-4. Сосуды спазмируются, активно вырабатывается простагландин, тромбоциты выделяют факторы свертывания, фибрин выпадает в осадок, развивается тромбоз.
- II тип иммунной нефропатии (цитотоксический антитело-опосредованный тип). Классический пример — синдром Гудпасчера. Поражение почек обусловлено линейными депозитами антител к коллагену IV типа базальной мембраны клубочков. Активация комплемента через c1-компонент (классический путь) или с3 (альтернативный путь) комплексом антиген — антитело образует белок (мембраноатакующий комплекс) из с5–с9-компонентов. Мембраноатакующий комплекс разрушает ткань непосредственно или косвенно, активируя провоспалительные клетки. Например, с5–с7 привлекает нейтрофильные гранулоциты в очаг воспаления. Они освобождают лизосомы, разрушая базальную мембрану, образуя перекисные соединения, свободные радикалы, эйкозаноиды, стимулируют полимеризацию фибрина. При болезнях с антителами к базальной мембране цитотоксические антитела с комплементом откладываются участками, более или менее равномерно. ANCA, обусловливая цитотоксическую иммунную реакцию, специфичны к компонентам клетки (миелопероксидаза, протеиназа 3, лактоферрин, катепсины В, D и G). ANCA активируют нейтрофильные гранулоциты, они прикрепляются к эндотелию сосудов почек, запуская воспалительный процесс.
- III тип иммунной нефропатии (иммунокомплексный нефрит). Иммунные комплексы выявляют в кровотоке, откладываются в мезангии, в интерстиции, в стенках капилляров. Особенность реакции состоит в том, что антиген первоначально содержится в крови, где на него и образуется антитело. Иммунный комплекс может формироваться в крови, откуда он удаляется ретикуло-эндотелиальными клетками или оседает в мезангии, стенках капилляров. Причем оседают и фиксируются прежде всего мелкие циркулирующие иммунные комплексы, крупные удаляются лимфатическими узлами, печенью и селезенкой. Но для III типа реакции характерно вначале оседание антигена на почечных структурах, только затем фиксируются антитело и комплемент. Фиксация антигена определяется многими факторами: аффинностью, размерами, соотношением антиген/антитело, проницаемостью сосудов, наличием на мезангиальных и интерстициальных клетках рецепторов к Fc-фрагменту IgG, а на эпителиальных клетках гломерул — к с3b. Иммунные комплексы откладываются субэпителиально. Тут же активируются нейтрофильные гранулоциты, выделяющие лизосомальные ферменты, и лейкоциты, выбрасывающие цитокины. Представленный механизм повреждения отмечают при нефрите с минимальными изменениями, мезангиопролиферативном, мембранозном, мембранопролиферативном, мезангиокапиллярном, некротизирующем, быстро прогрессирующем.
- IV тип иммунной нефропатии, клеточно-опосредованный, поздней гиперчувствительности. В качестве образца можно указать на пересаженную почку. Такая же реакция возникает при хроническом постстрептококковом нефрите.
Непосредственно комплементобусловленный нефрит возникает в отсутствии антигена и антитела. В мезангиальных и интерстициальных капиллярах откладывается с3 и пропердин. У больных с депозитами с3 в крови содержится белок, непосредственно переводящий с3 в активную форму с3b. Эта молекула называется с3 нефритический фактор и является термостабильным IgG-аутоантителом с молекулярной массой 150 000.
Непосредственная активация комплемента приводит к пролиферации клеток и утолщению стенок капилляров, что создает гистологическую картину мембранопролиферативного гломерулонефрита I–III типа.
ВИЧ-обусловленные нефропатии текут по двум вариантам: 1) протеинурическая форма с быстро прогрессирующим сегментарным гломерулосклерозом свойственна мужчинам, горожанам и лицам негроидной расы, применявшим наркотики инъекционным путем; 2) непротеинурическая медленно прогрессирующая ВИЧ-ассоциированная нефропатия развивается у серопозитивных гомосексуалистов европеоидной расы.
Гистологически определяют депозиты с3, IgM, фокальный гло.мерулосклероз, запустение гломерул, инфильтрацию интерстиция CD8+ и CD2+ Т-лимфоцитами, тубулоретикулярные структуры в эндотелии гломерул. Последнее позволяет говорить о вирусных включениях, являющихся антигенами и запускающими иммунный процесс.
Нефропатия при врожденном дефиците комплемента протекает по типу СКВ-подобного синдрома и гемолитико-уремического синдрома.
Тубулярный синдром
Может быть врожденным или приобретенным. Обусловлен частичным или полным выпадением одной или нескольких функций канальцев. Обычно изменений клубочкового аппарата не развивается. Синдром объединяет состояния с нарушением транспорта аминокислот, электролитов и воды, нарушения всасывания глюкозы, смешанные нарушения функций канальцев.
Основные клинические признаки тубулопатий представлены в табл. 31.4.
Таблица 31.4
Основные клинические признаки некоторых тубулопатий
| Ведущий синдром | Первичные тубулопатии | Фенотипически сходные состояния |
| Аномалии скелета (почечные остеопатии) | Фосфат-диабет, болезнь Де Тони — Дебре — Фанкони, почечный тубулярный ацидоз | Витамин D-зависимый рахит, гипофасфатазия, целиакия, псевдогипопаратиреоидизм |
| Полиурия | Почечная глюкозурия, почечный несахарный диабет, почечный солевой диабет (псевдогипоальдостеронизм) | Нефронофтиз Фанкони, пиелонефрит, тирозинемия, хроническая почечная недостаточность |
| Нефролитиаз | Цистинурия, глицинурия, иминоглицинурия | Оксалоз и вторичная гипероксалурия, ксантинурия, синдром Леша — Нихена |
Нарушения транспорта аминокислот в терапевтической практике имеют второстепенное значение. Эта группа пациентов отсеивается в детском возрасте. У взрослых прежде всего следует помнить о цистинурии, вызывающей рецидивирующий нефролитиаз. Заболевание передается а/p, обусловлено нарушением всасывания аминокислот в проксимальных канальцах. Одновременно с мочой выделяются аргинин, лизин, орнитин. При микроскопии мочевого осадка выявляют типичные гексагональные кристаллы. Камни видны уже при обзорном снимке брюшной полости, но плотность их значительно ниже, чем кальциевых.
Ренальная глюкозурия развивается достаточно редко (1 случай изолированной глюкозурии на 1000 случаев сахарного диабета), наследуется а/д. Обусловлена нарушением всасывания глюкозы в проксимальных канальцах. Уровень глюкозы в крови не меняется. Заболевание протекает бессимптомно. Сочетается с синдромом Фанкони и ренальным проксимальным ацидозом. При ДД следует исключить сахарный диабет и побочное действие рифампицина и тиазидных производных. Эти препараты способны вызвать развитие почечной глюкозурии.
Нарушения канальцевого транспорта фосфатов
Врожденные нарушения транспорта фосфатов проявляются уже в раннем детском возрасте симптомами рахита и известны как фосфат-диабет (витамин D-резистентный рахит), или семейный гипофосфатемический рахит. При этом страдает не только всасывание фосфатов в проксимальных отделах почечных канальцев, но и их транспорт из кишечника. Основные симптомы фосфат-диабета — рахит и нарушения роста, гипофосфатемия, повышение активности щелочной фосфатазы в сыворотке крови, усиленная экскреция фосфатов с мочой. Реакция на применение витамина D даже в высоких дозах слабая. Различают четыре типа фосфат-диабета:
- Х-сцепленная гипофосфатемия. Классический тип фосфатемического диабета. Характеризуется резким снижением реабсорбции фосфатов в проксимальных канальцах. Фенотип больных определяется степенью экспрессивности мутантного гена. Типичны выраженные изменения костей скелета, которые у детей служат показанием к ортопедическому лечению.
- А/д тип наследования, с Х-хромосомой не связан. Деформаций скелета практически не отмечают.
- А/p тип наследования. Зависимость от витамина D (гипокальциемический рахит, остеомаляция, гипофосфатемический витамин D-резистентный рахит с аминоацидурией).
- А/p тип наследования или возникает спорадически. Дефицит витамина D3. Болеют преимущественно девочки. Деформации скелета не обязательны.
При ДД учитывают витамин D-зависимый рахит (наследуется а/p) как результат нарушенного образования активной формы витамина D в почках и банальный гиповитаминоз D. Оба эти состояния протекают с деформациями костей, нарушениями роста, миалгией, тетанией. В крови гипокальциемия и гипофосфатемия. Уровень паратгормона повышен. При наследственном витамин D-зависимом рахите реакция на введение витамина D в высоких дозах положительная, а при банальном витамин D-дефицитном рахите положительный ответ отмечают уже на стандартные терапевтические дозы. При витамин D-резистентном рахите боль в мышцах возникает редко. Нетипичны тетания и гипокальциемия. Уровень паратгормона не изменен.
Дистальный тубулярный ацидоз (I тип, см. синдром Батлера — Олбрайта)
Характерны снижение pH мочи в сочетании с хроническим гиперхлоремическим метаболическим ацидозом, гипокалиемия, нарушения роста, нефролитиаз, повышенная экскреция калия с мочой. Может быть семейным (а/д) или спорадическим (возникает практически только у женщин), сочетаться с другими канальцевыми нарушениями. Клинически отмечают задержку роста, симптомы гипокалиемии, артралгии, миалгии, оссалгии. Вторичный спорадический синдром дистального тубулярного ацидоза развивается при аутоиммунных заболеваниях с гипергаммаглобулинемией, пересадке почек, нефрокальцинозе, медуллярной дисплазии, хронической обструкции почек, лечении препаратами лития или амфтеррицином В. Диагноз основывают на наличии системного ацидоза, изменении pH мочи >5,3, гипокалиемии. ДД проводят с проксимальным канальцевым ацидозом, медуллярной дисплазией, синдромом Шегрена, повреждением почек лекарственными препаратами (амфотерицин В, анальгетики), с хроническими интерстициальными заболеваниями почек и с обструктивной уропатией.
Проксимальный тубулярный ацидоз (II тип)
Обусловлен нарушением всасывания бикарбонатов в проксимальных канальцах. Потеря бикарбонатов и сопутствующих катионов обусловливает уменьшение эффективного объема крови и вторичный гиперальдостеронизм. Проксимальный тубулярный ацидоз развивается как семейная или спорадическая форма, при полном или неполном синдроме Де Тони — Дебре — Фанкони, при применении ингибиторов карбангидразы, при передозировке тетрациклиновых, интоксикации тяжелыми металлами, хронических интерстициальных поражениях почки, после трансплантации почки, при амилоидозе, множественной миеломе, при первичном и вторичном гиперальдостеронизме, рахите, непереносимости фруктозы, синдроме Вильсона — Коновалова. Клинически отмечают рахит у детей, остеомаляцию у взрослых. При значительной гипокалиемии развивается мышечная симптоматика. ДД с дистальным канальцевым ацидозом основывают на следующие опорные пункты. Для дистального канальцевого ацидоза типичны нормальная реабсорбция бикарбонатов, их потеря с мочой минимальна, уровень бикарбоната в сыворотке крови удерживается ниже 16–18 ммоль/л, ацидоз быстро устраняется при применении бикарбонатов, уровень калия в сыворотке крови низок, но легко восстанавливается после применения калия, никогда не отмечают синдром ДеТони — Дебре — Фанкони, но типичен нефрокальциноз. Для проксимального канальцевого ацидоза характерны снижение канальцевой реабсорбции бикарбонатов, их выраженная потеря с мочой, уровень бикарбонатов в сыворотке крови выше 16–18 ммоль/л, ацидоз слабо реагирует на вливание раствора бикарбоната, уровень калия в сыворотке крови крайне низок и плохо отвечает на применение калия. Нередко диагностируют синдром Фанкони, а нефрокальциноз крайне редко.
Синдром Де Тони — Дебре — Фанкони, часто ассоциируемый с проксимальным канальцевым ацидозом, может протекать как смешанная тубулопатия с нарушением обратного всасывания аминокислот, глюкозы, с фосфатурией, с повышенной реабсорбцией мочевой кислоты и развитием гиперурикемии. Наиболее тяжелая форма тубулопатии проксимального типа. Поражаются не только проксимальные канальцы, но и клубочки. Наследуется а/p. Экспрессивность гена значительно варьирует. У детей часто сочетается с цистинозом. При отсутствии отложений цистина в тканях болезнь протекает благополучно.
В младшем возрасте протекает с упорным рахитом и задержкой роста, у взрослых с остеомаляцией и болью в костях. Будучи у детей врожденным, у взрослых синдром ДеТони — Дебре — Фанкони может быть вторичным и отмечаться при цистинозе, гликогеновой болезни, синдроме Лоу или появиться после отравления свинцом, кадмием, ртутью, применения просроченных тетрациклинов, при болезни легких цепей при множественной миеломе, при болезни Вильсона.
Тубулярный ацидоз III типа — это комбинация I и II типа.
Тубулярный ацидоз IV типа — спорадически возникающее состояние на фоне умеренной почечной недостаточности при сахарном диабете, ВИЧ-нефропатии, интерстициальных процессах в почках, на фоне НПВП, ингибиторов АПФ, К+-сберегающих диуретиков, триметоприма. Все указанные препараты влияют на ось ренин — альдостерон — тубулярный аппарат. Возникает дефицит альдостерона или резистентность к нему, снижение экскреции К+. Появляется гиперкалиемия, снижается образование аммиака, падает экскреция кислот, pH мочи остается, как правило, в пределах нормы.
Острая почечная недостаточность
Развивается в течение дней–недель в связи с внезапным выключением функции почки с развитием олигоанурии и с повышением уровня мочевины в крови. Основной признак — быстрое снижение клубочковой фильтрации, но при этом отмечают и нарушение функции канальцев. В противоположность хронической почечной недостаточности эти показатели могут восстанавливаться. Первоначально клиническая картина определяется заболеванием, вызвавшим острую почечную недостаточность. По данным лабораторных методов исследования стремительно повышается содержание азотистых шлаков крови. Острая почечная недостаточность может протекать как не олигурическая (объем суточной мочи — более 400 мл/сут) с относительно обнадеживающим прогнозом и олигурическая (объем суточной мочи — менее 400 мл/сут) с плохим прогнозом. Острая почечная недостаточность может протекать как преренальная, острый тубулонекроз (острая почечная недостаточность в узком смысле слова), постренальная при обструкции мочевых путей и может быть обусловлена другими заболеваниями, обусловливающими снижение клубочковой фильтрации. К ним относятся острый и быстро прогрессирующий гломерулонефрит, васкулит, злокачественная АГ, острый интерстициальный нефрит, гемолитико-уремический синдром, острый тромбоз почечных артерий, острая почечная недостаточность при беременности, гиперкальциемия, мочекислая (уратная) нефропатия, острая почечная недостаточность при ранее существовавшей хронической.
Преренальная почечная недостаточность
Развивается приуменьшении эффективного объема кровообращения, что ведет к снижению кровотока в почках и повышению азотистых соединений в крови. Отмечают при кровопотерях, ожогах, усиленном потоотделении, диарее, жажде, дефиците солей, интенсивной потери воды через почки, при переходе воды в «третье» пространство (перитонит, илеус, асцит, отеки, панкреатит), сердечной недостаточности, сепсисе и феохромоцитоме. В моче снижено содержание натрия, осмолярность ее высока, мочевой осадок не изменен.
Гепаторенальный синдром — вариант острой преренальной почечной недостаточности. Развивается у пациентов с далеко зашедшим циррозом печени, фульминантным гепатитом, множественными массивными метастазами в печень, токсическим поражением печени и протекает с быстрым снижением клубочковой фильтрации. У таких больных до развития почечной недостаточности проявляются все признаки прогрессирующей печеночной недостаточности (желтуха, зуд, печеночная энцефалопатия, гипоальбуминемические отеки, увеличение протромбинового времени). Гепаторенальный синдром провоцируется применением лекарственных препаратов, поэтому он чаще развивается в клиниках. Может манифестировать после кровотечения из варикозных вен пищевода, после удаления асцитической жидкости из брюшной полости, массивной мочегонной терапии. Экскреция натрия с мочой снижается ниже 10 ммоль/л. Моча концентрированная. Осмолярность мочи выше осмолярности плазмы крови. Прогноз гепаторенального синдрома плохой. При ДД гепаторенального синдрома следует исключать острый тубулонекроз (шоковую почечную недостаточность) при кровотечениях из варикозных вен пищевода или сепсисе и обратимую преренальную азотемию при гиповолемии. При остром тубулонекрозе содержание натрия в моче 30–80 ммоль/л, соотношение осмолярности мочи к осмолярности плазмы крови составляет 0,9–1,05, соотношение кретинина мочи к креатинину плазмы крови <15. При преренальной азотемии концентрация натрия в моче < 10 ммоль/л, соотношение осмолярности мочи к осмолярности плазмы крови >1,1, соотношение креатинина мочи к креатинину плазмы крови >15. Изменения мочи при гепаторенальном синдроме и преренальной азотемии практически идентичны. Поэтому в ряде случаев ДД возможна только путем осторожного пробного введения жидкости под контролем диуреза, легочной симптоматики, АД и центрального венозного давления.
Острый тубулонекроз
Развивается при циркуляторно-септических нарушениях и как нефротоксический после применения некоторых лекарственных препаратов, рентген- контрастных препаратов, при гемолизе, миолизе, отравлениях тяжелыми металлами, четыреххлористым углеродом, этиленгликолем и др.
При циркуляторно-септическом остром тубулонекрозе разрешающими моментами являются снижение АД и сепсис. Иногда этот вид острого тубулонекроза развивается после оперативных вмешательств как итог потери жидкости и эндотоксикоза. Нефротоксический вариант некроза канальцев развивается при применении аминогликозидов, рифампицина, амфотеррицина В, метотрексата, доксорубицина, НПВП. Тубулонекроз после использования рентгенконтрастных средств с наибольшей вероятностью формируется у людей с низкой фильтрационной способностью почек, при диабетической нефропатии, при множественной миеломе. Рабдомиолиз выявляют у пациентов, страдающих алкоголизмом, при синдроме раздавливания, интоксикациях, нарушениях обмена веществ в мышцах и длительных судорогах. Признаками миолиза являются наличие миоглобина в моче, повышение мышечной фракции ЛДГ. Аналогичный механизм известен для гемолиза.
Постренальная почечная недостаточность (обструктивная нефропатия). Отмечают при врожденных стенозах мочеточника, при сдавлении мочеточника опухолями (забрюшинные опухоли, опухоли яичников) или при ретроперитонеальном фиброзе, закупорке мочеточника камнями, некротизированными сосочками или тромбами, раке мочевого пузыря, заболеваниях предстательной железы, стенозе уретры или ее закупорке камнями, при наличии клапанной уретры.
Алгоритм действий при острой олигурии/остро возникшей азотемии
Причинами могут быть все состояния, ответственные за развитие острой почечной недостаточности. Их ДД проводят на основании данных анамнеза, клинического, лабораторного и эхо- или (реже) радиологических обследований, морфологического исследования пункционных биоптатов. На основании весьма краткого анамнеза исключают не менее 50% причин острой почечной недостаточности: 45–55% всех случаев острой почечной недостаточности связаны с тяжелыми хирургическими ситуациями (постоперационный период, шок). На 2-м месте стоит применение нефротоксических средств или рентгенконтрастных препаратов, прежде всего у больных с диабетической нефропатией или у пациентов со сниженным функциональным резервом почек. К критическому снижению клубочковой фильтрации ведут и многочисленные системные заболевания соединительной ткани и сосудов. Их признаками являются боль в суставах, кожная сыпь, иммунные сдвиги.
Обязательно следует оценить характер мочеиспускания. Смена олигурии полиурией, эпизоды острой задержки мочи свидетельствуют в пользу обструкции мочевых путей. При полной анурии, но отстутствии эхопризнаков блокады оттока мочи, можно думать об остром двустороннем тромбозе почечных артерий. При физикальном обследовании необходимо обращать внимание на признаки дефицита внеклеточной жидкости (спавшиеся вены шеи, ортостатический коллапс), свидетельствующие о преренальной азотемии. Но объем мочи более 1,5 л/сут на фоне повышения содержания азотистых соединений свидетельствует против преренальной почечной недостаточности. При обследовании больного (увеличенный мочевой пузырь) легко исключают и такие причины постренальной почечной недостаточности, как затруднения опорожнения мочевого пузыря: объемные образования малого таза (доброкачественная гиперплазия и рак предстательной железы, опухоли женских половых органов, послеродовый отек и гипотония мочевого пузыря). У пациентов с острой почечной недостаточностью крайне важно провести анализ первой же полученной порции мочи. Внимание должно быть сосредоточено на ее осмолярности и характеристиках мочевого осадка. При преренальной почечной недостаточности осмолярность мочи выше 350 ммоль/л, протеинурия не определяется или минимальна. При остром тубулонекрозе осмолярность мочи приближается к осмолярности плазмы крови, а в мочевом осадке преобладает канальцевый эпителий и грубозернистые цилиндры. При острой преренальной почечной недостаточности экскреция натрия с мочой снижена, поскольку несмотря на снижение клубочковой фильтрации, канальцевый эпителий выполняет свои функции и реабсорбирует натрий. При остром тубулонекрозе натрий не абсорбируется, «уводит за собой» много воды, моча не концентрированная. При изучении мочевого осадка эритроцитарные цилиндры, дисморфные эритроциты свидетельствуют в пользу нефритического синдрома. При остром токсическом интерстициальном нефрите при специальной окраске мочи можно выявить эозинофильные гранулоциты. По данным эхоГ почек и мочевых путей четко определяют признаки гломерулонефрита, нефротического синдрома, нефросклероза, обструктивной нефропатии.
Хроническая почечная недостаточность
Хроническая почечная недостаточность — необратимое нарушение гомеостатической функции почек, возникающее в конечной стадии их тяжелого прогрессирующего заболевания. Только при потере 75% почечной ткани появляется клинически значимое снижение клубочковой фильтрации. Для тотальной почечной недостаточности характерно присутствие полного симптомокомплекса гомеостатических нарушений, связанных с включением в процесс всех элементов нефрона. Парциальная хроническая почечная недостаточность — изолированное (комбинированное) нарушение функции одного или нескольких механизмов, поддерживающих гомеостаз (чаще при наследственных и врожденных нефропатиях).
Выявление почечной недостаточности — начало интенсивного и по возможности стремительного диагностического поиска. Вся работа врача над этой проблемой строится на определении причин почечной недостаточности, попытках выявления обратимости изменений, в стремлении затормозить прогрессирование нарушений почечных функций. Хроническая почечная недостаточность развивается при целом ряде заболеваний клубочков, при обструктивной нефропатии, при интерстициальных заболеваниях почек и их врожденных заболеваниях. Является результатом длительного снижения клубочковой фильтрации, канальцевой реабсорбции, нарушения эндокринной функции обеих почек. Нарушение функции почек протекает по типу снижения выведения продуктов обмена, нарушенного выведения воды и солей, нарушения обмена активной формы витамина D и образования эритропоэтина.
Нарушение экскреторной функции почек (вначале определяют по снижению клиренса креатинина до 30–40 мл/мин и ниже, а затем и по повышению концентрации креатинина в крови) ведет к нарушению выведения натрия (задержка натрия или почечная потеря натрия) и воды, гиперурикемии, нарушению образования аммиака, нарушению секреции калия в дистальных канальцах, задержке азотистых соединений и гиперфосфатемии. Задержка натрия проявляется отеками, «обводненным легким» и АГ, а почечная потеря натрия — гипотензией и дальнейшим снижением клубочковой фильтрации. Гиперурикемия клинически проявляется как вторичная подагра. Нарушение образования аммиака и секреции калия ведет к ацидозу, гипервентиляции, гиперкалиемии, деминерализации костей. Задержка азотистых шлаков проявляется энцефалопатией и нейропатией, желудочно-кишечной симптоматикой, гемолизом, серозитами, нарушением функции тромбоцитов (геморрагический синдром). Гиперфосфатемия может протекать с повышенным образованием кальций-фосфата, что приводит к зуду и возможному кальцинозу мягких тканей. Гиперфосфатемия с гипокальциемией обусловливает вторичный гиперпаратиреоидизм, а он уже приводит к миопатии и фибропластическому оститу.
Нарушение эндокринной функции почек проявляется дефицитом активной формы витамина D, снижением продукции эритропоэтина, изменением синтеза ренина, неадекватным ответом на адиурекрин. Снижение активной формы витамина D приводит к снижению всасывания кальция в кишечнике (что усиливает эффект гипокальцемии при гиперфосфатемии), к остеомаляции. Дефицит витамина D и гиперфосфатемия обусловливают развитие так называемой почечной остеопатии. Снижение синтеза эритропоэтина приводит к торможению эритропоэза и является существенным компонентом «почечной анемии». Нарушение образования ренина может протекать по типу его усиленного синтеза (клинически манифестирует как АГ) и сниженного образования (гипоренинемический гипоальдостеронизм = гиперкалиемия + тубулярный ацидоз, а это — гипервентиляция, деминерализация костей). Неспособность собирательных трубочек и дистальных канальцев адекватно ответить на антидиуретический гормон проявляются полиурией и почечным вариантом несахарного мочеизнурения.
Клиническая картина хронической почечной недостаточности
Длительное время пациенты могут не предъявлять жалоб или отмечать повышенную утомляемость, слабость, снижение памяти. Иногда почечную недостаточность выявляют случайно при установлении причин АГ или патологического мочевого осадка.
При общем обследовании отмечают изменения кожи. Она теряет эластичность, приобретает грязно-коричневую окраску (меланоз). Повышенная кровоточивость проявляется многочисленными кровоизлияниями. У пациентов, находящихся на гемодиализе, появляется псевдопорфирия, буллезные изменения кожи. Более 1/2 больных мучает упорный кожный зуд.
Неврологические изменения проявляются прежде всего колебаниями настроения, раздражительностью, общим беспокойством, плохим сном, невозможностью длительное время концентрировать внимание. В запущенных случаях появляются судорожные подергивания, кома. Периферическую уремическую нейропатию определяют прежде всего в дистальных отделах ног как симметричные сенсомоторные расстройства, снижаются сухожильные рефлексы и вибрационная чувствительность, замедляется нервная проводимость. Парестезии ступней и их повышенная чувствительность приводят к появлению колющей боли (прежде всего по ночам). Выраженность боли уменьшается после ходьбы, что заставляет пациентов вставать и ходить по ночам. Следует сказать, что уремическая полинейропатия появляется в запущенных стадиях, при клиренсе креатинина ниже 10 мл/мин. Если полинейропатия развивается раньше, то следует прежде всего исключить заболевания, протекающие с одновременным поражением почек и нервов (сахарный диабет, амилоидоз, множественная миелома, узелковый периартериит, прием нейротоксических препаратов, например нитрофурантоина, при почечной недостаточности). Мышечный синдром (атрофия мышц, мышечная слабость преимущественно в проксимальных группах, мышечные подергивания) при хронической почечной недостаточности может быть признаком ренальной остеопатии при вторичном гиперальдостеронизме или дефиците витамина D, проявлением нейропатии, гипер- или гипокалиемии.
Сердце и сосуды неизменно поражаются при хронической почечной недостаточности. Признаком ренальной гипертензии является гипертрофия ЛЖ. Практически у всех больных отмечают уремический перикардит. Последний проявляется интенсивной болью за грудиной, лихорадкой, лейкоцитозом, эхопризнаками накопления жидкости в полости перикарда.
Желудочно-кишечная симптоматика включает анорексию, уремический запах изо рта, тошноту, рвоту, диарею. Характерным для пациентов с хронической почечной недостаточностью, особенно находящихся на гемодиализе, является формирование ангиодисплазии, приводящей к желудочно-кишечным кровотечениям.
Поражение костей и суставов известно как почечная остеопатия. Причинами является задержка фосфатов, ведущая к связыванию кальция, дефицит витамина D, вторичный гиперпаратиреоидизм, а также массивное поступление алюминия с диализными жидкостями. Это ведет не только к алюминиевой остеомаляции, но и к алюминиевой деменции. Признаками ренальной остеопатии являются диффузная плохо локализованная боль в костях, слабость мышц, спонтанные переломы ребер, позвонков, шейки бедренной кости. У половины всех детей с почечной недостаточностью отмечают низкорослость.
Практически у всех больных с креатинином в сыворотке крови выше 260–350 мкмоль/л развивается нормохромная нормоцитарная анемия. Она обусловлена дефицитом эритропоэтина и блокадой эритропоэза уремическими токсинами. Наряду с этим из-за уремии сокращается и продолжительность жизни эритроцитов. Анемия усиливается и за счет геморрагического синдрома, характерного для пациентов с уремией. При нефропатии, вызванной приемом анальгетиков, выраженная анемия формируется рано (гемолиз, спровоцированный анальгетиками и желудочно-кишечные кровотечения).
Водный баланс сохраняется у большинства пациентов до развития олигурии в терминальной стадии почечной недостаточности. Но вследствие ограничения функциональной способности почек он легко нарушается. Осмолярность мочи меняется в узком коридоре: 300 мосм/л (изостенурия). Поэтому при жестком ограничении поступления воды баланс становится отрицательным, а уменьшение при этом экстрацеллюлярного объема со снижением перфузии почек приводит к дальнейшему снижению клубочковой фильтрации. В то же время при избытке воды легко развивается гипергидратация. Она проявляется быстрым увеличением массы тела, появлением видимых отеков и, наконец, «обводненным» легким. Последнее проявляется одышкой, а на рентгенограммах — затемнением, распространяющимся бабочкообразно от ворот к периферии.
Гиперкалиемия развивается только при олигурии, в терминатной стадии хронической почечной недостаточности. При ДД следует помнить, что гиперкалиемия развивается при целом ряде состояний. Возможно избыточное поступление калия извне (погрешности диеты, применение калийсодержащих препаратов, например калиевой соли пенициллина, прием калийсодержащих солей взамен поваренной), снижение секреции калия в дистальных отделах канальцев (олигурия, гиперренинемический гипоальдостеронизм, прием калийсберегаюших диуретиков, НПВП, интоксикация препаратами наперстянки), выход калия из клеток (гемолиз, миолиз, катаболический тип обмена, метаболический ацидоз).
Кальциноз внутренних органов развивается при повышении уровня фосфата кальция в сыворотке крови >5,7 ммоль/л в результате снижения выделения фосфатов почками. Кальций откладывается в коже, сердце, скелетных мышцах, легких, роговице, в сосудах и периартикулярно.
Почечный метаболический ацидоз развивается при прогрессировании почечной недостаточности как итог сниженного аммониеобразования (NH4+) в канальцах. Поэтому нарушается выведение катионов водорода. В результате формируется ацидоз и снижается уровень бикарбонатных ионов в сыворотке крови. Вначале ацидоз компенсирован, поскольку гипервентиляция приводит к выдыханию углекислоты. Почечный метаболический ацидоз проявляется одышкой, анорексией, тошнотой, рвотой.
ДД хронической и острой почечной недостаточности основывают на данных анамнеза, клинических, лабораторных и инструментальных методов. Для хронической почечной недостаточности в анамнезе типичны семейные варианты нефропатий, наличие у больного аутоиммунного заболевания, полиартрита, криоглобулинемии, сахарного диабета, длительной АГ, злоупотребление анальгетиками, множественная миелома. При лучевых методах исследования выявляют сморщенные почки, кистоз почек, обструктивную уропатию. Типичны такие рентгенологические признаки, как остепороз. При лабораторных исследованиях определяют анемию (при исключении других причин), гипокальциемию, повышение активности щелочной фосфатазы. В ряде случаев при проведении ДД необходимо исключить одностороннее или преимущественно одностороннее поражение почки. Это особенно актуально, когда клиническая картина заболевания напоминает или полностью совпадает с клинической картиной симметричного поражения почек. Как правило, в этих случаях имеется урологическая патология с вовлечением в патологический процес паренхимы почек или мочевых путей (опухоль, туберкулез, кистозная трансформация, нефролитиаз), а также патология сосудистой системы почек. Аномалии развития и положения почек часто односторонние и становятся основой признаков, совпадающих с картиной двустороннего поражения почек.
Односторонние поражения почек
Основной признак одностороннего поражения почки — отсутствие почечной недостаточности. Уровень креатинина в сыворотке крови повышается только при снижении клубочковой фильтрации на 50% и более. Если при заболевании одной почки все-таки повышается уровень креатинина, то функциональная активность другой оказывается существенно сниженной. Односторонняя патология почек проявляется общей симптоматикой, болью, повышением температуры тела, АГ, изменениями объема и консистенции органа, изменениями мочевого осадка.
Особенно тревожно увеличение размеров почки. Его отмечают прежде всего при опухоли, при поликистозе почки, при гигантских солидных кистах, при гидронефрозе, при остром пионефрозе, при пиелонефрите.
Опухоли почек
Опухоли почек представлены у взрослых в основном злокачественными новообразованиями, а среди них на первом месте (80%) — гипернефрома (аденокарцинома). Опухоль Вильмса (нефробластома) отмечают в основном у детей в возрасте до 6 лет (часто ассоциируется с унилатеральным парциальным гигантизмом). Опухоли собирательной системы почек составляют не более 8–10% всех опухолей почек. Возможны лимфомы и очень редко саркомы почек. Доброкачественные опухоли (ангиомиолипомы, гемангиомы, фибромы, липомы, сравнительно часто малигнизирующиеся аденомы) четко ассоциируются с генетической патологией.
В клинической картине опухолей почек особое место занимает гематурия. На первых этапах она может быть непостоянной. Гематурия безболезненная, негломерулярная, морфология эритроцитов не изменена. Боль отмечают не чаще, чем в 1/4 случаев. Появляется при прорастании опухоли в околопочечную клетчатку и при увеличении размеров почки. В случаях увеличения размеров почки возможно и пальпаторное выявление опухоли. Боль может быть обусловлена и массивной гематурией, обструкцией мочеточника сгустками крови. Иногда опухоли почек, в том числе и доброкачественные, протекают с полиглобулией. Как проявление паранеопластического синдрома возникают лихорадка (у 15% больных), дерматоз, нейромиопатия. Нередко опухоли почек продуцируют гормоны или гормоноподобные вещества. Продукция простагландинов и антидиуретически подобного гормона приводят к гиперкальциемии и синдрому Шварца — Бартера. Выброс пролактина ведет к развитию галактореи, а гонадотропина к потере либидо и развитию гинекомастии. Образование АКТГ обусловливает синдром Кушинга, а эритропоэтинов — полиглобулию. Цитологическое исследование мочевого осадка является обязательным при наличии негломерулярной гематурии и подозрении на прорастание опухоли в собирательную систему почек.
Гипернефрома (аденокарцинома почки) составляет 1,5–2% всех случаев рака у взрослых. Мужчины болеют чаще женщин (3:2). Более чем в половине случаев манифестирует с гематурии. Первоначально это микрогематурия, не замечаемая пациентом. Затем появляются общие неспецифические симптомы: повышенная утомляемость, слабость, разбитость, уменьшение массы тела, интермиттирующая лихорадка. Практически одинаково часто выявляют анемию и полиглобулию. Для гипернефромы типичны паранеопластические реакции: эозинофилия, тромбоцитоз, повышение СОЭ, лейкемоидная реакция. Боль в поясничной области, в боку появляется поздно, когда опухоль вышла за пределы капсулы почки и широко инфильтрировала околопочечные ткани. Прорастание опухоли в почечную и нижнюю полую вены проявляется отеками ног и нижней части туловища. Диагноз «гипернефрома» устанавливается методами неинвазивной диагностики. Уже при обзорном рентгеновском снимке брюшной полости можно выявить очаги кальцинации в опухоли. По данным экскреторной пиелографии определяют увеличение размеров почки, деформацию собирательной системы. Методом выбора и экстренной диагностики является эхоГ. Опухоль выглядит как сравнительно однородное образование («шоколадная киста» при кровоизлиянии в ткань опухоли), при одновременной прицельной нефробиопсии можно получить материал для цитологической диагностики. ЭхоГ-картина может быть уточнена, особенно в диагностике поражения ЛУ, при проведении КТ или ЯМР. Отдаленные метастазы прежде всего выявляют в мозгу, костях, легких.
Нефробластома (опухоль Вильмса) у взрослых развивается крайне редко, но типична для детского возраста. Симптоматика очень скудна. Гематурия, боль, повышение температуры тела отступают на второй план. Чаще всего опухоль определяют при пальпации, когда она достигает очень больших размеров. Даже после радикального удаления опухоли вероятность появления нефробластомы в контрлатеральной почке высока. Прорастание опухоли в сосуды обусловливает ее стремительное метастазирование. Нефробластома нередко сочетается с гомолатеральным парциальным гигантизмом. Поэтому при выявлении непропорционально больших частей тела у ребенка (парциальный гигантизм) необходим тщательный эхоконтроль почек в динамике (рис. 31.5).
Другие злокачественные опухоли почек
Возможна инфильтрация почки опухолевыми клетками при лимфомах, что быстро приводит к развитию «выключенной» почки. На эхоГ она выглядит как гигантское образование, утерявшее типичную структуру и бобовидную форму.
Рак мочевых путей
Карциномы собирательной системы почек, мочеточника или мочевого пузыря ассоциируются прежде всего с занятостью пациентов на производстве ароматических аминов, красок и в резиновой промышленности, с приемом анальгетиков, фенацетина, циклофосфамида. Но самым частым фактором риска является курение, ответственное более чем за 50% всех случаев рака мочевого пузыря. Чаще выявляют плоскоклеточный рак, реже опухоль, происходящую из переходного эпителия. Последний вариант карциномы сочетается с уролитиазом и мочевой инфекцией. На первое место в клинической картине рака мочевых путей выдвигается микрогематурия, долгое время являющаяся единственным симптомом. Макрогематурия характерна при раке лоханки или мочеточника и в результате блокады мочеточника тромбом нередко сочетается с почечной коликой. Рак мочевого пузыря длительное время протекает с рецидивирующей макро-/микрогематурией, периодически болезненными позывами на мочеиспускание, под маской упорной мочевой инфекции. Если нет обструкции мочевых путей, то наилучшие результаты получают при экскреторной урографии, выявляющей деформацию мочевыводящей системы: пристеночный дефект наполнения, нередко с неправильными контурами. Хорошие результаты получают при цистоскопии (рак мочевого пузыря) или изучении цитологического состава промывных вод пузыря при раке пузыря и вышерасположенных отделов мочевыводящей системы. КТ выявляет инвазивный рост опухоли и рано определяет поражение регионарных ЛУ.
Гидронефроз
Гидронефроз — итог хронической обструкции мочевых путей. Односторонний гидронефроз возникает при нефролитиазе, аномалии развития одного мочеточника, при сдавлении одного мочеточника опухолью. Односторонний рефлюкс может быстро превратиться в двусторонний. Обструкция двух мочеточников, застой в мочевом пузыре или обструкция уретры приводят к двустороннему гидронефрозу. Частой причиной гидронефроза является пузырно-мочеточниковый рефлюкс тяжелой степени. Рефлюкс — гидродинамический феномен, связанный с несостоятельностью уретровезикального соустья. Причины могут быть различны. О «первичном» рефлюксе говорят при наличии сегментарной или тотальной дисплазии мышц дистального отдела мочеточника. «Вторичный» пузырно-мочеточниковый рефлюкс вызывается неполноценностью иннервации, что связано с нарушением формирования нервно-мышечного аппарата, а также циститом, дисфункцией мочевого пузыря гиперрефлекторного типа. Необходимо отметить, что далеко не каждый случай рефлюкса приводит к гидронефрозу. Частота рефлюкса во многом определяется возрастом и полом пациента. Дети и женщины болеют им чаще. Так, у девочек в возрасте до 1 года без клинической симптоматики рефлюкс определяют с частотой 50%, в 15 лет — 1%, у более старших рефлюкс не выявляют. Такое возрастное распределение частоты развития рефлюкса объясняется, видимо, анатомическими особенностями мочеточников у детей (широкие, прямые), несовершенством нервной регуляции перистальтики полых органов у младенцев. Связь рефлюкса с инфекцией подтверждается тем, что по некоторым данным, 75% рефлюксов 1 степени у детей благополучно излечиваются антибиотиками. Но сам рефлюкс создает условия для инфицирования вышерасположенных мочевых путей. При тяжелых степенях пузырно-мочеточникового рефлюкса, как правило, развивается рефлюкс-нефропатия. Ее развитие во многом определяется выраженностью и длительностью микробно-воспалительного процесса в почечной ткани. Этот процесс приобретает самостоятельное значение, так что антирефлюксная терапия не решает проблему прогноза рефлюкс-нефропатии.
Первыми симптомами гидронефроза могут быть односторонняя тупая боль, непостоянная, распространяющаяся в паховую область. Даже далеко зашедшие варианты гидронефроза редко выявляют пальпаторно. Обычно гидронефроз определяют при УЗИ. В связи с вероятностью заноса инфекции ретроградную пиелографию следует проводить только после эхоГ и экскреторной в/в урографии. При ангиографических исследованиях вначале регистрируют «немую» почку, а затем виден только «мешок с жидкостью». Большие возможности в установлении причин гидронефроза предоставляет КТ, особенно в случаях ретроперитонеального фиброза или сдавления мочеточников опухолью. Лабораторные изменения при неинфицированном гидронефрозе не имеют диагностического значения. Выраженный гидронефроз приводит к почечной недостаточности. Инфицированный гидронефроз (особенно его крайний вариант — пионефроз) проявляется потрясающими ознобами, болью, интоксикацией, лабораторными изменениями, свидетельствующими о гнойной инфекции.
Односторонний острый пиелонефрит сравнительно редко остается таковым. В интересах больного врач должен помнить о высокой частоте двустороннего острого пиелонефрита (см. выше).
Туберкулез почки. Является результатом гематогенного заноса инфекции, поэтому чаще протекает как двусторонний процесс, но возможно и одностороннее поражение. Туберкулез почки выявляют обычно не ранее 5-го года инфицирования. Нередко в анамнезе удается найти легочный процесс, плеврит, а при обследовании — рентгенологические признаки туберкулеза легких и ЛУ. Клиническая картина туберкулеза почек и мочевыводящей системы, в отличии от пиелонефрита, очень стерта и сочетается с признаками поражения мочевого пузыря и половых органов. Обычно это общие неспецифические симптомы, поллакиурия, дизурия, недержание мочи, безболезненное увеличение придатка яичка и утолщение семенного канатика. Туберкулезное поражение семенных пузырьков и предстательной железы протекают бессимптомно. При лабораторных исследованиях выявляют безболевую микрогематурию и «стерильную» пиурию, то есть лейкоцитурию без признаков бактериальной инфекции. Для определения микобактерий в моче требуется специальная окраска или посев мочи на микобактерии. Микроскопия центрифугата 24-часовой мочи после окраски на кислотоустойчивую флору дает положительные результаты в 40–60% случаев. Ложноположительные результаты могут быть при выявлении нетуберкулезных кислотоустойчивых микроорганизмов, в частности М. smegmatis. Для предотвращения ошибки сбор мочи у мужчин следует проводить после тщательной гигиенической обработки крайней плоти.
Из дополнительных методов исследования на первом месте стоит цистоскопия, позволяющая рано определить изъязвления и туберкулезные бугорки в мочевом пузыре. Эхо- и рентгенография на ранних этапах малоинформативны. Лучевая диагностика выявляет неправильной формы деформации чашечек, а затем кистообразные каверны в паренхиме почек (рис. 31.6). Возможно определение кальцинатов.
Нефролитиаз, уролитиаз
Патогенез камнеобразования заключается либо в пересыщении мочи камнеобразующими солями, появлении центров кристаллизации, либо в снижении концентрации ингибиторов роста кристаллов.
Камни почек проявляются как нефрокальциноз, при затруднении оттока мочи, при их отхождении или при наслоении вторичной инфекции. Нефрокальциноз — результат осаждения кристаллов кальция в паренхиме почек или в канальцах рядом с верхушками пирамидок. Выявляют при первичном гиперпаратиреоидизме, гипервитаминозе D, медуллярной дисплазии почек, молочно-щелочном синдроме и тубулярном ацидозе. Нередко сочетается с уролитиазом. Без уролитиаза протекает бессимптомно, пока не выявляют при эхоисследовании или при обзорной рентгенографии брюшной полости. Крупный конкремент остается в лоханке, проявляясь тупой болью в пояснице, гематурией, присоединением инфекции. Отхождение маленького камня может быть бессимптомным, проявляясь только кратковременными прерываниями струи мочи («мочевое заикание»). Но в большинстве случаев движение камня сопровождается типичной почечной коликой. Обычно боль начинается как умеренная, тупая, в течение ближайшего часа усиливается, становясь нестерпимой, иррадиирует в промежность, мошонку, половые губы. По мере продвижения камня боль спускает ниже, при локализации камня в пузыре или уретре боль отдает в головку полового члена, в яички. Наиболее болезненно продвижение «песка», камней с острыми краями, что сопровождается массивной гематурией за счет ранения мочеточников. Это характерно при выделении мочевой кислоты или фосфатов. Коралловидные камни-слепки длительное время вследствие своих размеров не могут проникнуть в мочеточники и поэтому не обусловливают колики. ДД почечнокаменной колики проводится с коликой при отхождении сгустка крови или детрита (туберкулез почки, опухоль, антикоагулянтная терапия), при некрозе почечного сосочка, гидронефрозе. Наличие камней обусловливает повышенную вероятность наслоения инфекции. В свою очередь хроническая мочевая инфекция приводит к образованию камней. Так, протей, обладающий уреазной активностью, приводит к образованию гексагональных кристаллов по типу «крышки гроба».
Редким осложнением мочекаменной болезни с хронической обструкцией и мочевой инфекцией Proteus mirabilis является ксантогранулематозный пиелонефрит. Заболевание очень часто одностороннее. Характерное проявление — имбибиция тканей почки и околопочечной клетчатки макрофагами, перегруженными липидами. Быстро формируется околопочечный фиброз. Значительно увеливаются почки, так что у 25% больных удается их прощупать. Характерны боль в поясничной области, животе, повышение температуры тела. При рентгенологическом исследовании в 1/3 случаев определяют «отключенную» почку. Неоднородность картины паренхимы напоминает кистозную почку или заставляет исключать опухоль. Еще реже возникает необходимость исключения крупноклеточного интерстициального нефрита.
Мочевой осадок при мочекаменной болезни характеризуется неизмененными (не гломерулярными) эритроцитами и лейкоцитами. Микроскопически удается выявить кристаллы, которые служат указанием на причину гемат- и лейкоцитурии. При инфекционных заболеваниях почки это чаще всего кристаллы по типу «гробовой крышки». Плоские гексагональные кристаллы типичны для цистинурии, кристаллы по типу почтовых конвертов (квадратные и прямоугольные) образуются из оксалата кальция. Мочевая кислота кристаллизируется в ромбовидные образования, а бесцветные пикообразные кристаллы или аморфные зерна образованы из фосфатов. Большинство (90–95%) конкрементов — рентгенконтрастны и хорошо визуализируются уже на обзорных снимках брюшной полости. Особенно хорошо проецируются оксалатно-кальциевые образования. При этом типе визуализации (без контрастирования) плохо видны карбонаты, фосфаты, ураты, цистиновые камни. ЭхоГ является высокочувствительной методикой. Если камень располагается в мочеточнике в области малого таза, то возникают сложности в его отличии от флеболита или кальцината ЛУ. При обструкции мочеточника и камне низкой плотности, расположенном дистально или превезикально, мочеточник виден на всем протяжении, что служит косвенным указателем на наличие уролитиаза. Но следует помнить, что мочеточник может визуализироваться на всем протяжении и после отхождения камня, что объясняется его послеболевой гипотонией. Нефро- или уролитиаз, выявление отходящего камня еще ни о чем не говорит. В современном урбанистическом обществе с высоким потреблением белка мочекаменная болезнь достаточно распространена. Для установления ее причин необходимо уточнить анамнез (климат, потоотделение, пищевые привычки, степень минерализации воды, объем потребляемой жидкости: при удельном весе мочи 1,010 и ниже камнеобразования не происходит), прием слабительных средств (приводят к концентрации мочи), наличие пиелонефрита, нарушений обмена веществ, аномалии развития почки. Большое значение для диагноза имеет непосредственный химический анализ камня или косвенный анализ по рентгеновской плотности, сопутствующей кристаллурии, наличию мочевой инфекции. Кальциевые камни (оксалатно-кальциевые, фосфатно-кальциевые, смешанные) составляют до 80% всех камней. Вторичные фосфаты составляют 10%, уратные камни — 8%, цистиновые камни — 1%. Еще 1% приходится на более редкие камни, например ксантиновые.
Кальциевые камни выявляют при гиперкальциурии, гипероксалурии, гиперурикозурии с/без гиперкальциемии (идиопатическая гиперкальциурия, дистальный тубулярный ацидоз, медуллярная дисплазия, интестинальная и наследственная гипероксалурия, гиперурикемия, саркоидоз, множественная миелома).
У 5% пациентов с кальциевыми камнями отмечают первичный гиперпаратиреоидизм.
Идиопатическая гиперкальциурия — наследственное состояние. Является причиной образования конкрементов у 75% женщин и 50% мужчин с уролитиазом. При идиопатической гиперкальциурии кальция выделяется >300 мг/сут (>7,5 ммоль/сут) у мужчин и >250 мг/сут (>6,2 ммоль/сут) у женщин.
Гипоцитратурия (цитраты мочи <350 мг/сут [<1820 мкмоль/сут]) сама по себе или в сочетании с другими расстройствами также ведет к камнеобразованию, поскольку цитраты образуют с кальцием растворимые соли.
Гипероксалурия (оксалаты мочи >40 мг/сут [>440 мкмоль/сут]) может быть первичной или вторичной. Причинами вторичной гипероксалурии являются:
- избыточное потребление оксалатов (шпинат, шоколад, орехи, чай);
- усиленное всасывание оксалатов в кишечнике (синдром бактериального обсеменения тонкой кишки; хронический панкреатит; болезни желчевыводящей системы);
- операции на тощей или подвздошной кишке.
При гиперуратурии (мочевая кислота в моче >750 мг/сут [>4 ммоль/сут] у женщин и >800 мг/сут [>5 ммоль/сут] у мужчин) острые игольчатые кристаллы служат основой для кристаллизации на них солей кальция.
Ураты типичны для гиперурикемии или сниженной растворимости мочевой кислоты в условиях закисления мочи. Выявляют у 60% пациентов с мочекислым диатезом, а также при миелопролиферативном и лимфобластном лейкозе, особенно в условиях цитостатической терапии.
Цистиновые и ксантиновые камни — свидетельство соответствующих вариантов наследственных нарушений обмена.
Рыхлые фосфатно-аммониевые камни (струвиты) образуются на фоне инфекции мочевых путей, вызванной микрофлорой с уреазной активностью.
Сосудистые катастрофы почек
Инфаркт почки может протекать незаметно или сопровождаться болью в поясничной области, продолжительность которой колеблется от нескольких часов до нескольких суток, гематурией и преходящей АГ. Отмечают при тупых травмах органов брюшной полости, при атеросклерозе, васкулите, при мерцании предсердий или инфекционном эндокардите. Двусторонний тромбоз почечных артерий крайне редок и ведет к острой почечной недостаточности. Полная односторонняя закупорка почечной артерии ведет к инфаркту всей почки и феномену «немой почки». Значительно чаще развиваются сегментарные инфаркты, что при экскреторной урографии выглядит как «выпадение» сегмента, а на поздних этапах — как рубцовое втяжение участка паренхимы.
Тромбоз почечных вен отмечают преимущественно при гиперкоагуляции на фоне нефротического синдрома (мембранозный гломерулонефрит или амилоидоз), при тупой травме живота, при тромбозе нижней полой вены или гипернефроме почки с прорастанием в почечную вену. Острый тромбоз почечных вен сопровождается болью в животе и поясничной области, протеинурией, гематурией (за счет венопочечной гипертензии). Двусторонний тромбоз быстро приводит к олигурии и почечной недостаточности. Хронический тромбоз малосимптомен, проявляется тромбэмболическим синдромом. Диагноз подтверждают данные эхоГ, допплерографии, кавографии и селективной венографии.
Заболевания предстательной железы и органов мошонки
Заболевания предстательной железы, яичек и их придатков проявляются нарушением микции, признаками воспаления (при простатите, орхите) или общими симптомами с поражением костей при раке предстательной железы. Протекают с болью внизу живота, в промежности или в мошонке.
Простатит
У мужчин молодого и зрелого возраста — достаточно частая причина дизурии, поллакиурии, боль перинеальной локализации. Очень часто не удается подтвердить бактериальную природу воспаления.
Острый бактериальный простатит характеризуется ознобом, лихорадкой, болью в области промежности. При осторожной пальпации через прямую кишку выявляют чувствительную болезненную увеличенную и горячую предстательную железу. Значительное увеличение железы в размерах приводит к острой задержке мочи. Массаж предстательной железы категорически запрещен в связи с угрозой развития бактеремии. Возбудители, чаще всего энтеробактерии, выявляют в моче, а в 25% случаев и в крови. На эхоГ острый простатит проявляется небольшим увеличением размеров органа с признаками его отека. Редким осложнением является абсцесс предстательной железы. Клинически он протекает со всеми признаками гнойного воспаления. На эхоГ выглядит как негативное образование с неровными контурами. Хронический простатит проявляется увеличением предстательной железы, повышением акустической плотности паренхимы, появлением внутри нее мелких конкрементов.
Хронический бактериальный простатит малосимптомен. Периодически беспокоит боль в надлобковой области, в промежности, в области крестца, учащенное и болезненное мочеиспускание. При пальцевом исследовании предстательная железа неравномерно уплотнена. В ее секрете большое количество лейкоцитов, иногда скоплениями, макрофаги с жировыми включениями (овальные жировые тельца), бактерии.
Чаще диагностируют хронический абактериальный простатит. Причины его возникновения достоверно не известны. Симптоматика аналогична бактериальному простатиту. В секрете точно так же выявляют лейкоциты и жировые тельца.
Гипертрофия предстательной железы
Доброкачественную гипертрофию предстательной железы (аденома по прежней терминологии) редко выявляют в возрасте до 50 лет. По данным аутопсии ее диагностируют у мужчин в возрасте 30–40 лет в 8%, в 50–60 лет — в 50%, старше 80 лет — в 85–90%. Критерием доброкачественной гипертрофии предстательной железы является увеличение ее объема >30 см3. Критерии Американской урологической ассоциации более строгие: 1) объем предстательной железы >30 см3; 2) максимальная скорость истечения мочи <10 мл/см3) объем остаточной мочи >50 мл. Если в качестве критерия диагноза брать только объем железы, то частота гипертрофии предстательной железы у мужчин в возрасте 55–74 лет составляет 19%. При использовании всех трех критериев частота гипертрофии предстательной железы составляет 4%.
Симптоматика сводится к обструктивной уропатии и проявлениям вторичной инфекции. Первые симптомы — увеличение периода натуживания до появления мочеиспускания при ощущении переполнения мочевого пузыря, уменьшение калибра и скорости истечения струи, учащение ночного мочеиспускания. Позднее мочеиспускание совершается буквально по каплям. Мочевой пузырь может пальпироваться внизу живота как тугоэластичное образование или даже обусловливать местное выбухание. В других случаях его не удается прощупать даже при значительных объемах остаточной мочи. Острая задержка мочи может развиться и без достаточно ярких первоначальных симптомов. Обструкция безболезненна, она приводит к гидронефрозу, наслоению инфекции и, в итоге, к почечной недостаточности. При ректальном исследовании определяется увеличенная эластичная железа, хорошо ограниченная от окружающих тканей, безболезненная. Плотные включения и узлы позволяют исключать рак предстательной железы. При доброкачественной гипертрофии предстательной железы в моче возможны лецитиновые зерна, но в целом лабораторные данные неспецифичны, только при наслоении вторичной инфекции появляются пиурия, микрогематурия. При урографии определяется выбухание дна мочевого пузыря, а в случаях существенной обструкции — расширение мочеточников и собирательной системы почек. Наиболее достоверным методом ранней диагностики является эхоГ. При доброкачественной гипертрофии контуры железы четкие, поверхность гладкая, структура железы однородная, в паренхиме можно выявить отдельные кальцинаты и лецитиновые зерна. Хорошо визуализируется остаточная моча.
Рак предстательной железы
Аденокарцинома предстательной железы — одна из наиболее частых опухолей у мужчин в возрасте старше 50 лет, и частота ее повышается с возрастом. Саркому предстательной железы диагностируют крайне редко и практически только у детей.
Рак предстательной железы растет очень медленно и длительное время никак не проявляется. В 90% случаев, при отсутствии ранней инструментальной диагностики, рак предстательной железы дебютирует с признаков обструкции. У 50% больных первыми симптомами рака предстательной железы являются признаки метастазирования опухоли: боль в пояснично-крестцовом сочленении, в позвоночнике, в костях, анемия, уменьшение массы тела, увеличение печени, появление увеличенного плотного ЛУ над левой ключицей.
Пальпаторно при раке предстательной железы на ранних этапах выявляют отдельные плотные узлы в ее паренхиме. Позднее предстательная железа становится диффузно плотной, бугристой, не смещаемой, ткани в окружности индурированы, границы органа не определяются.
При лабораторных исследованиях выявляется повышение активности кислой простатической фосфатазы в крови, свидетельствующей о переходе рака предстательной железы за пределы капсулы органа, об анемии, повышении СОЭ. Большую диагностическую ценность имеет определение простатспецифического антигена. Типично повышение активности щелочной фосфатазы в крови при метастазах в костях.
Наиболее доступным методом визуальной диагностики является эхоГ. На ранних стадиях предстательная железа асимметрична, по мере роста опухоли железа увеличивается, особенно в переднезаднем размере. Паренхима становится неоднородной, появляются очаги пониженной плотности, хотя у 10–15% больных раковые узлы оказываются более плотными, чем ткань железы. При выраженной инвазии масса опухоли определяется за пределами предстательной железы. Достоверность эходиагностики рака предстательной железы составляет 60–70%, поэтому при выявлении подозрительных диагностически неясных узлов в предстательной железе показана прицельная биопсия под контролем УЗИ. Экскреторная урография призвана уточнить степень обструкции, а КТ — выявить прорастание опухоли в семенные пузырьки и в окружающие ткани. Рентгеновские снимки поясничных позвонков и других частей скелета, сцинтиграфия показаны для раннего выявления метастазов.
Боль при заболеваниях органов мошонки
Сосредоточие в мошонке герминативных органов, сосудов, серозных оболочек, нервной ткани, возрастная этапность формирования при сравнительно нередкой травматизации, обусловливает большой спектр возможных патологических состояний.
На клиническом этапе важно убедиться в наличии яичек в мошонке, оценить соответствие их размеров возрасту, исключить грыжу, сперматоцеле и водянку. В любом возрасте возможно варикозное расширение вен семенного канатика (варикоцеле). У взрослых пальпаторное обследование мошонки должно быть обязательным скрининговым исследованием для раннего выявления опухолей. Метод самопальпации для выявления рака, подобно тому, как это проводят женщины для раннего выявления рака молочной железы, рассматривается зарубежными авторами как «первая линия онкологической обороны» и признан настолько важным, что рекомендуется как желательный при самообследовании и обязательный при диспансерном наблюдении.
При многих вирусных, онкологических и некоторых других врожденных и приобретенных состояниях изменения органов мошонки отражают течение основного процесса и являются только одним из признаков общего страдания организма. Как правило (80%), поражение органов мошонки, даже в случаях острой боли, протекает без повышения общей температуры тела. Лихорадка свойственна инфекциям с половым путем передачи, местным гнойным острым (не хроническим) процессам и системным заболеваниям.
Увеличение размеров яичек, кроме случаев опухолей, воспаления, в том числе и посттравматического, возможно при:
- Синдроме ломкой Х-хромосомы, или синдром Мартина — Белла, или синдром макроорхидии и связанного с полом слабоумия: 1) место поломки (вторичная констрикция) в Xq28 (вблизи конца длинного плеча Х-хромосомы) в 2–50% всех метафазных пластинок в культуре клеток на среде, обедненной фолатами или с добавлением антиметаболитов; 2) слабоумие у мужчин. Несоответствующая ему задержка речевого развития, дефектный символизм, аутизм, агрессивность, в детстве гипермобильность. Тонкие двигательные реакции страдают, грубая моторика не изменена; 3) эпилепсия у 1/3 пациентов; 4) большие оттопыренные уши; 5) дисморфия лица: удлиненное лицо с выступающим лбом, припухлыми веками, полными губами, широкими крыльями носа; 6) с пубертатного возраста — избыточный рост гонад; 7) частота — 1:1000–1:1500 новорожденных мальчиков. Не менее 3–5% мужчин с тяжелой умственной отсталостью оказываются носителями данного синдрома.
Следует сразу оговориться, что приведенные здесь и далее те или иные характерные проявления и/или критерии врожденных или приобретенных синдромов не исключают иной комбинации или неполного проявления указанных симптомов и/или стигм, аномалий, признаков. Приведенный ниже список врожденных синдромов развития не является полным, а включает наиболее частые состояния. Поэтому необходима содружественная работа клинициста, специалистов по ультразвуковой диагностике и клинической генетике.
Атрофию гонад (после исключения ишемических, посттравматических, радиационных, химических повреждений) отмечают при:
- Дистрофии миотонической Куршмана — Штайнера (Гидрамнион + сниженная двигательная активность плода + гипотония мышц, мышечная слабость + задержка умственного развития + катаракта + алопеция + миопатическое удлиненное лицо с бедной мимикой + слабость глотательных движений + сниженная перистальтика пищевода и его умеренное расширение + гипотония желудочно- кишечного тракта + мышечные атрофии + парезы + аномалии скелета + пороки диафрагмы + нарушение альвеолярной вентиляции + нарушения ритма сердца + косолапость).
- Синдроме Маккла — Уэлса (потрясающие ознобы + крапивница + прогрессирующая потеря слуха + нефротический синдром + глаукома + полая стопа).
Гипоплазию яичек отмечают при таких патологиях:
- Алкогольная эмбриофетопатия (пренатальная дистрофия с последующей общей дистрофией + дисморфия лица + блефарофимоз + гипопластический фильтр + узкая верхняя губа + гипоплазия верхней челюсти + микрогения + задержка психомоторного развития + двигательная гиперактивность + пороки сердца + онихогипоплазия).
- Синдром Альстрома — Ольсена (ожирение + сахарный диабет + глухота + тапеторетинальная дегенерация + катаракта + глаукома + нистагм + нарушения ЭКГ + акантоз + гиперурикемия + гипераминоацидурия + гиперлипидемия + аномалии скелета).
- Синдром Барде — Билля (задержка умственного развития + ожирение + тапеторетинальная дегенерация + полидактилия).
- Синдром Берлина (эктодермальная дисплазия + карликовость + тонкие ноги + задержка умственного развития).
- Анемия Блекфана — Даймонда (нормохромная арегенераторная анемия + микроцефалия + расщелина неба + микрофтальмия + аномалии большого пальца + сколиоз + пороки сердца).
- Синдром Бьерсона — Форсмана — Лемана (задержка психомоторного развития + микроцефалия + энофтальм + монголоидный разрез глаз + птоз).
- Синдром Вермера (повышение уровня гастрина в сыворотке крови + гастринома + струма + аденома паращитовидных желез + гиперпаратиреоидизм + гиперплазия коры надпочечников + гиперинсулинизм + инсулинома + липомы + полипоз толстого кишечника).
- Синдром Гемина (ригидная шея + спонтанно образующиеся келоидные рубцы + анорхидия + крипторхизм).
- CHARGE-синдромокомплекс (колобома + микрофтальмия + атрезия хоан + пороки сердца + нарушение слуха + задержка психомоторного развития).
- Синдром врожденного дискератоза (пойкилодермия + гиперкератоз + эритема + гипергидроз + ониходистрофия + лейкоплакия + конъюнктивит + анемия + панцитопения).
- Ихтиоз с мужским гипогонадизмом (ихтиоз + микропенис + аплазия клеток Лейдига + высокорослость).
- Синдром Кальмана (дефицит гонадотропина + гипоталамическое повышение секреции гонадотропин-рилизинг-фактора + аносмия + гипоплазия гениталий + стерильность).
- Синдром Клайнфельтера (высокорослость + гипоплазия гениталий + евнухоидизм + аномалия хромосом).
- Синдром Лоуренса — Муна (дегенерация тапеторетинальная + задержка психомоторного развития + спастические парапарезы).
- Синдром LEOPARD (лентигенез + ЭКГ-нарушения + гипертелоризм + стеноз легочной артерии + задержка умственного развития).
- Синдром Лешке (пигментные пятна на коже + задержка психомоторного развития + ожирение + недостаточность надпочечников).
- Синдром Менгеля — Конигсмарка— Берлина — Мак-Кьюсика (дисплазия ушных раковин + дисморфия лица + задержка умственного развития + слухового восприятия + низкорослость).
- Пойкилодермия врожденная типа Ротмунда — Томсона (эритема ретикулярная + пойкилодермия + катаракта + гипотрихоз + алопеция + акромикрия + гипоплазия большого пальца кистей + гипоплазия лучевой и локтевой костей + аномалии ногтей и зубов + инфантилизм).
- Синдром Прадера — Вилли (ожирение + гипотония мышц + акромикрия + задержка психомоторного развития + сниженная двигательная активность ребенка).
- Синдром Рихарда — Рундля (атаксия + мышечная гипотония + арефлексия + нистагм + глухота + кифосколиоз + деформация стоп).
- Синдром Рувалькаба (брахиметакарпия + микроцефалия + дисморфия лица + дисплазия позвонков + краниостеноз + низкорослость + гиперпигменгация кожи).
- Синдром Санктис — Катчионе (пигментная ксеродерма + задержка умственного развития + атаксия + микроцефалия).
- Ульнаромаммарный синдром (дефекты лучевой кости + гипоплазия сосков и апокриновых желез + гипотрихоз + ожирение + запоздалая/сниженная фертильность вплоть до стерильности).
- Синдром Уильямса — Бойрена (низкорослость + лицо фавна или эльфа + грубый низкий голос + стеноз) аорты + стеноз легочной артерии + дисплазия радужки + задержка умственного развития).
- Хондродисплазия точечная в результате Х-хромосомальной делеции (седловидный гипопластичный нос + задержка умственного развития + билатеральная симметричная кальцификация эпифизов + алопеция + низкорослость + катаракта + брахифалангия + укорочение концевых фаланг).
Только после клинического обследования можно переходить к инструментальным методам, ибо руки не должны идти впереди головы.
В современных условиях диагностический поиск начинают с УЗИ, нередко на этом он и завершается. Желательно присутствие при обследовании лечащего врача, а в диагностически сложных случаях его активное, заинтересованное участие обязательно.
ЭхоГ мошонки показана при ее травмах, любом увеличении размеров и изменении формы, местной температуры тела, окраски, болевом синдроме, наличии атипичных пальпируемых образований, уменьшении размеров гонад или их отсутствии, общей диспластичности строения ребенка и его стигмированности, синдромах «маленького Геркулеса», задержке умственного или моторного развития, гипертрофии или ожирении, высокорослости, карликовости, алопеции, гипотрихозе, гипертрихозе, сращении бровей (синофрис), стенозе аорты или легочной артерии, факоматозе, хромосомных болезнях и поликистозе. УЗИ потока крови по сосудам яичка и по его паренхиме показаны при подозрении на перекрут яичка, варикоцеле, воспаление, травму, опухоль, при «скользящем» и неопущенном яичке.
Крипторхизм
Невозможность пропальпировать яички в мошонке — едва ли не самая частая причина направления детей на УЗИ. Частота крипторхизма среди взрослых определяется в 0,1–0,5%. В детском возрасте по данным иностранных авторов частота одностороннего крипторхизма составляет 1:150–1:200, двустороннего — 1:600, анорхизма — 1:5000–1:20000. Особенно часто крипторхизм выявляют у недоношенных детей и новорожденных с недостаточной массой тела.
Яичко закладывается близ медиальной поверхности первичной почки и затем, к 7-му месяцу внутриутробного развития, перемещается в мошонку. Перемещение яичка из глубин тела с постоянно высокой температурой в мошонку с относительно низкой температурой обеспечивает стабильный, полноценный и постоянный сперматогенез. В животном мире для снижения температуры гонад и обеспечения размножения необходима сезонная миграция особей. Так, птицы летят на север, угри опускаются в глубокие холодные воды океана, лосось переходит в верховья холодных рек. Избавившись от фиксированной сезонности размножения, человек расплачивается (или награжден?) сексуальным видением мира.
Учитывая, что плод в большинстве случаев находится в матке вниз головой, движение яичек в мошонку направлено против гравитации. Механизм опускания яичек сложен, неоднозначен и изучен не полностью. Чаще (60–75%) задерживается в своем движении правое яичко. Это объясняют его размерами, превосходящими левое и затрудняющими движение по паховому каналу. Кроме того, движение левого яичка облегчается «выдавливанием» из брюшной полости растущей сигмовидной кишкой.
Различают истинный и ложный крипторхизм. Истинный крипторхизм — яичко находится в естественном тестикулярном тракте (от почки до мошонки). Неопущенное яичко чаще всего (80%) находится в паховом канале у входа в мошонку.
Клиническая картина крипторхизма (синдром «пустой мошонки») складывается из различного характера боли в промежности, паху или поясничной области, выраженность которых увеличивается в пубертатный период, нарушений фертильности, а также изменений психики и высокой вероятности разнообразных дистантных аномалий. Хотя до 50% больных бесплодны, ряд случаев крипторхизма наследуется вероятней всего аутосомно-доминантно с неполной пенетрантностью. Поэтому врач должен поинтересоваться наличием этой патологии у родственников больного. Но в семьях подобное состояние чаще скрывают, поэтому нельзя полагаться на расспросы, а желательно тактично настоять на полноценном обследовании.
Крипторхизм является одним из проявлений большой группы генетически детерминированных состояний. Кроме синдромов, указанных при обсуждении проблемы уменьшенных яичек, крипторхизм с высокой частотой отмечают при таких патологиях:
- Синдром Аарского (низкорослость + гипертелоризм + птоз + короткий широкий нос + брахифалангия + «кожа прачки» на ладонях + паховые грыжи + расщепленная мошонка).
- Синдром Мак-Доноу (умственная отсталость + пороки сердца + дисморфии лица + кифосколиоз + дефекты мышц брюшной стенки).
- Синдром Кросса (гипопигментация кожи и волос + долихоцефалия + судороги + атетоз + спастика + вторичные контрактуры + микрофтальмия + помутнение роговицы + микрокорнеа + вторичный эктропион + амавроз + нистагм).
- Х-связанный синдром Микрофтальмии-микроцефалии синдром (микрофтальмия + блефарофимоз + утолщенные веки + гидроцефалия + умственная неполноценность).
- М-синдром (задержка умственного развития + нарушения роста + слабое зрение + глухота + дисморфия лица + полиостическая дисплазия).
- Синдром Перельмана (высокорослость + асцит фетальный без обшей водянки + гигантские внутренние органы + почечные гамартомы + фокальная нефробластома + опухоль Вильмса + дисморфии лица).
- Синдром сливообразного живота (синдром Игла— Барретта). Состояние названо так потому, что сквозь дефектную брюшную стенку просвечиваются внутренности. Диагностируют на основании следующих признаков: гипоплазия или атрофия мышц брюшной стенки + дисплазия почек + гидронефроз + мегауретер + везикоуретероренальный рефлюкс + мегацистис + расширение проксимальной уретры + стеноз меатуса).
- Рецессивно наследуемый синдром множественных птеригиумов (тризм + птоз + контрактуры коленных и бедренных суставов + аномалии позвонков).
- Синдром Рубинштейна — Тейби (низкорослость + микроцефалия + задержка умственного развития + широкие большие пальцы кистей и стоп + антимонголоидный разрез глаз + крючковидный нос).
- Синдром Юберга — Марсиди (задержка умственного развития + низкорослость + снижение слуха вплоть до глухоты + микропенис + гипоплазия мошонки + седловидный нос + камподактилия).
Для ультразвуковой визуализации яичка в паховом канале длинную ось линейного трансдьюсера следует располагать по оси канала. Современные диагностические системы позволяют выявить яичко при его диаметре в 10 мм.
Случаи нахождения яичка в брюшной полости, где его эхолокация малореальна, составляют 20% истинного крипторхизма. Исключения представляют случаи паравезикального расположения яичка, когда оно хорошо визуализируется на фоне заполненного мочевого пузыря. Для выявления топики внутрибрюшинно расположенного яичка наиболее информативны ЯМР-исследования.
Ложный крипторхизм включает случаи «скользящего яичка» (втягивание яичка в брюшную полость при грубых манипуляциях, охлаждении), и эктопию яичка (его расположение в бедренном канале, в перианальной области, у корня полового члена). Попадание яичка в эти области тоже в определенной степени не случайность. Еще в конце прошлого века было доказано, что гунтеровский тяж имеет несколько пучков (хотя до сих пор спорят, реальны ли эти тяжи и какова цель их существования). Основной направлен в мошонку, где чаще всего и благополучно завершается миграция яичка. Остальные пять пучков (хвосты Локвуда) могут направить яичко в необычное для человека положение, которое является тем не менее нормой для представителей некоторых видов животного мира: промежностное яичко — у свиньи, утконоса, бобра; паховое — у верблюда, выдры; лобково-пенальное — у сумчатых.
Неопушенные яички уменьшены в размерах, плотность их вначале понижена, затем возрастает по сравнению с нормой, не определяется зернистость строения. Внутри яичка при исследовании цветным энергетическим допплером выявляют обеднение кровотока, а в запущенных случаях он совершенно не определяется. Конечно, это не значит, что кровотока там нет. Просто он настолько незначителен, что даже современные высокочувствительные ультразвуковые методики его не регистрируют. Сцинтиграфия с технецием более чувствительна, но при наличии энергетического допплера применение изотопных методик строго лимитировано и при данных показаниях в современных условиях имеет, скорее, историческое значение.
Из-за нарушения развития зародышевого эпителия развивается бесплодие. Поэтому низведение яичка должно быть проведено как можно раньше.
В любом случае неопущенное яичко остается однородным. Появление неоднородности внутри паренхимы неопущенного яичка требует незамедлительного исключения опухоли. Риск ее возникновения в неопущенном яичке в 40–50 раз выше, чем при естественном расположении. Единственным методом профилактики опухоли является раннее низведение яичка (с возраста 3 мес до 2 лет). Рядом автором получены сведения, что если низведение яичка проведено после 2-летнего возраста, то вероятность его малигнизации не существенно отличается от таковой в вообще не низведенном яичке. Но в любом случае низводить яичко необходимо (даже при условии невосполнимых изменений паренхимы с целью более легкого выявления возможной малигнизации).
Крипторхизм часто сочетается с иными аномалиями, прежде всего почек, поэтому некоторые исследователи рекомендуют обязательную реноэхографию и экскреторную рентгенографию пациентам с крипторхизмом. Поскольку отсутствие яичка в мошонке может быть объяснено его эктопией (нахождение яичка в бедренном канале, у корня полового члена и т. д.), то необходимо УЗИ соответствующих областей. Наиболее часто ошибкой при исследовании является ложноположительная диагностика расположения неопущенного яичка у корня полового члена, когда поперечное изображение кавернозного тела принимается за гонаду. Для исключения ошибки достаточно отведения кавернозных тел в контрлатеральную сторону.
Причинами боли в мошонке являются перекрут яичек, перекрут придатков яичек, острый эпидидимит, орхит, инфаркт яичек, синдром Фурнье (некротизирующее воспаление мошонки), опухоли и метастазы опухолей яичек, водянка, варикоцеле и сперматоцеле.
Опухоли яичек
Как правило, опухоли яичек злокачественны. Развиваются у мужчин в возрасте 25–35 лет. Высока вероятность выявления опухоли в ретенцированном яичке, поэтому у мужчин молодого возраста с крипторхизмом определение метастазов требует прежде всего исключения опухолей гонад. Прежде всего это семиномы (37%), эмбриональные карциномы и хорионэпителиомы (33%), тератомы (25%). Оставшиеся 5% составляют лимфомы, опухоли, исходящие из сертолиниевых клеток и клеток промежуточного эпителия. У детей новообразования яичка выявляют преимущественно до 3-летнего возраста (50%) и представлены тератомами. В школьном возрасте первичная опухоль яичек развивается редко. Обычно это опухоли смешанного эмбрионального характера, рост которых ускорился и ткань малигнизировалась с наступлением половой зрелости.
Избыточную секрецию гонадотропина, приводящую к гинекомастии, выявляют не чаще чем в 8–11 %. Из генетически детерминированных опухолей яичка известен синдром Карнея. Определяется как редкое состояние, развивающееся преимущественно у юношей, наследуемое и характеризующееся ассоциацией с мукокутанным лентигенезом, невусами, кардиальными миксомами и кушингоидным фенотипом. Клинические проявления: 1) многочисленные мелкие преимущественно центрофациальные лентиго, распространяющиеся на красную кайму губ, веки и конъюнктиву; 2) кожные миксомы; 3) у 60% больных выявляют миксомы сердца, чаще бессимптомные, обычно предсердной, но возможно и желудочковой локализации; 4) первичная узловатая пигментная гиперплазия коры надпочечников с более или менее выраженным кушингоидным фенотипом; 5) у 1/2 пораженных мужчин — различные типы двусторонних опухолей яичек — крупноклеточная кальцифицирующаяся опухоль сертоллиевых клеток или опухоль клеток Лейдига. Достаточно часто преждевременное половое созревание в анамнезе, 6) изредко аденома гипофиза с акромегалией или гигантизмом. Ни один из описанных в литературе пациентов не имел полный набор всех указанных признаков. При наличии 2 признаков диагноз «синдром Карнея» может быть установлен как вероятный. В качестве варианта синдрома Карнея известны синдромы MEN, NAME, LAMB. ДД проводят с синдромами Пейтца — Егерса, гастрокутанного, LEOPARD.
Сравнительно длительное время опухоли яичка, а тем более семенного канатика, протекают безболезненно. Непальпируемые образования в мошонке выявляют при эхоГ с частотой приблизительно 9 случаев на 1600 исследований. Но 8 из 9 образований оказываются злокачественными. Это объясняет, почему более 1/2 больных с опухолями яичек поступают в стационар уже с наличием забрюшинных и другой локализации метастазов, что проявляется (10–15% всех пациентов) болью в животе, наличием пальпируемых образований в брюшной полости, тошнотой, рвотой, аденопатией, дыхательной недостаточностью, кровохарканьем, отеками ног. Около 10% больных первоначально жалуются на недостаточность эякуляторной функции при сохраненных эрекции и либидо, поскольку вытеснение специфического герминативного эпителия опухолевыми клетками ведет к сниженной продукции спермы.
Возможны припухлость яичка, появление пальпируемого образования или каменистого уплотнения в паренхиме, неоднородность консистенции. (Отношение к выявленным образованиям в мошонке определяется следующим правилом: большинство образований внутри яичка злокачественны, большинство образований вне яичка доброкачественны). Яичко может быть бугристым. Боль в мошонке, тупая или острая, возникает поздно. Она может симулировать перекрут яичка или эпидидимит. Боль становится сильной и нестерпимой при инфаркте опухоли и прорастании серозной оболочки.
УЗИ яичек при подозрении на опухоль проводят для исключения опухоли или ее подтверждения как при наличии, так и при отсутствии пальпируемых изменений, при увеличении размеров яичка, при наличии эпидидимита или гидроцеле, при лейкозе, наличии забрюшинных опухолей, забрюшинных метастазов неизвестной опухоли, контроля эффективности химио- или лучевой терапии опухоли яичка, для динамического наблюдения пациентов с ранее леченными опухолями яичка, при наличии конкрементов в яичке для ранней диагностики опухоли и в редких случаях — для устранения тревоги у лиц с канцерофобией.
Любое изменение эхотекстуры яичка позволяет тут же исключить опухоль. На эхоГ опухоль выглядит как структура, отличающаяся от обычной однородной тестикулярной ткани. Опухоли яичка, как правило, гипоэхогенны. Первоначально опухоль обычно локализуется по задней части яичка, длительное время объем и форма яичка могут не изменяться. Наряду с общей гипоэхогенностью опухоли яичка в ней возможны мелкие беспорядочные внутренние эхосигналы (семинома, лимфома), но возможны гиперэхогенные (тератомы, эмбриональная карцинома, хорионэпителиома) и смешанные структуры. Наличие жидкости в пределах опухоли свидетельствует о ее распаде, кровоизлиянии или внутренней кисте. Последнюю отмечают при тератомах. Опухоль может выглядеть как локальное поражение с четкими ровными контурами и как массивное, разрушающее паренхиму, с неправильными очертаниями.
Данные эхоГ позволяют рано выявить даже не пальпируемые опухоли, оценить их распространенность, одно- или двусторонность поражения. Поэтому необходимо еще раз подчеркнуть, что исследование показано больным лейкозами, поскольку поражение яичек является проявлением рецидива изолированного (тестикулярного) или комбинированного (костномозгового + тестикулярного).
УЗИ само не «создает» диагноз, диагноз устанавливается клинически с привлечением иных методик, на совокупности которых и проводят ДД. Эпидермальная киста напоминает опухолевое поражение, но является доброкачественной, содержит внутри себя ороговевающие ткани, поэтому выглядит неоднородной. Плотность внутренних эхоструктур достаточно высока.
При диагностике опухолей яичка из неинвазивных методов визуализации большей чувствительностью и специфичностью обладает только ЯМР, но проведение этого исследования оправдано только после эхоГ. Окончательная ДД возможна только по данным гистологических исследований, но вероятный диагноз «тератома» важен для хирурга, ибо позволяет изменить тактику операции и при подтверждении наличия тератомы воздержаться от орхэктомии, ограничившись органосохраняющим вмешательством.
Нельзя говорить о стадии опухоли (Tl, Т2) по данным УЗИ яичка, так как часто не видно прорастание опухоли в белочную оболочку (Т2) или эпидидимис (Т2). Невозможно поданным эхоГ разграничить лейкемическую инфильтрацию и лимфому яичка, интерстициальную опухоль и опухоль, происходящую из герминативного эпителия. По данным эхоГ нельзя абсолютно уверенно говорить о доброкачественности или злокачественности образования, кровоизлиянии в опухоль или о кисте внутри нее.
Мелкие конкременты яичка
Микролитиаз яичка в большинстве случаев — доброкачественное образование, хотя может быть предраковым состоянием. Микролитиаз бывает одно- и двусторонним. Протекает бессимптомно. Возникает в неопущенных яичках, после химиотерапии, при синдроме Клайнфельтера, при ложном мужском гермафродитизме. В случаях односторонне неопущенного яичка выявление в нем микрокальцинатов требует немедленного исключения опухоли (по данным биопсии). На эхоГ кальцинаты видны как типичные эхопозитивные высокоинтенсивные мелкие структуры с феноменом нежной акустической тени.
Кисты яичек
Истинные кисты яичек выявляют крайне редко. Большинство кист, описываемых до внедрения в клиническую практику эхоГ мошонки, оказались ложными. Истинная врожденная киста яичка, как правило, односторонняя и одиночная. Выглядит как мелкая эхонегативная структура. Может располагаться внутри яичка или быть связанной с белочной оболочкой и тогда пальпируется как плотная мелкая структура диаметром 3–4 мм. При расположении внутри яичка она обычно располагается ближе к его середине. Гистологически истинная киста берет начало у корня яичка. Каждый случай выявления кисты яичка, особенно кист множественных или двусторонних, должен быть причиной исключения поликистозной болезни (провести УЗИ печени, селезенки, почек + рентгенологические исследования головного мозга), синдромов трисомий и факоматозов (искать пигментные пятна цвета «кофе с молоком», участки депигментации, ангиомы, фибромы, липомы; а у матери ребенка — миомы). Киста придатка яичка может быть признаком синдрома Гиппеля — Линдау (гемангиобластомы головного мозга, прежде всего продолговатого и мозжечка + умеренная полипитемия + феохромоцитома + кисты внутренних органов и костей + факомы).
Приобретенная семенная киста может достигать больших размеров, симулируя водянку оболочки. Однако в случае семенной кисты яичко оттеснено ею и не окружено жидкостью со всех сторон. При УЗИ внутреннее содержимое совершенно однородно. Но по данным эхоГ различить кисту яичка от его опухоли очень сложно.
Воспаление яичка
Орхит (воспаление яичка) развивается после травмы или как инфекционный процесс. Инфекция нередко распространяется ретроградно из предстательной железы или мочевого пузыря через семявыносящий проток. Процесс обычно начинается в придатке (эпидидимит), но достаточно быстро переходит собственно на яичко. Поэтому часто употребляется термин «орхоэпидидимит». Изолированный орхит развивается реже вследствие гематогенного заноса инфекции.
Наряду с вирусно-бактериальным генезом орхита (и орхоэпидидимита) возможно воспаление яичка как итог некротизирующего васкулита при облитерирующем эндартериите (болезни Винивартера — Бюргера), системном васкулите и аутоиммунных ревматических болезнях, особенно часто при болезни Бехчета (афты слизистой оболочки полости рта + эрозивно-язвенные поражения гениталий + гипопион радужной оболочки + узловатая эритема + тромбозы артериальные и/или венозные + ревматические изменения мягких тканей и суставов + желудочно-кишечные кровотечения + возможные аневризмы аорты и ее основных ветвей).
Орхит у детей препубертатного возраста нетипичен.
При орхите постепенно развивается диффузное напряжение и болезненность в области яичка и/или семенного канатика. Болевой приступ характеризуется постепенным началом, развивается в течение нескольких часов или даже суток, хотя достаточно много пациентов отмечают резкое начало боли, симулирующее перекрут яичка. При орхите характерны покраснение и отек кожи мошонки (20%), лихорадка (33%). Возможны изменения предстательной железы и пиурия (40%). Сканирование с технецием (в современных условиях проводится редко) выявляет характерные особенности: вначале накопление технеция повышено, при хроническом воспалении снижено. На эхоГ орхит выглядит как увеличение размеров яичка с сохранением однородности его внутренней структуры (важнейший ДД-признак с опухолями) или как смазанность, нечеткость рисунка паренхимы при нормальных очертаниях органа. При значительном воспалении за счет отека паренхима яичка может быть гомогенно сниженной эхоплотности (по сравнению со здоровым яичком с противоположной стороны) или неоднородной плотности. При остром орхите допплерографически определяют повышение кровоснабжения. Обычно очаг воспаления находится в верхнем полюсе яичка. Косвенными признаками воспаления являются расширение семенного канатика, реактивное умеренное по объему гидроцеле, отек кожи мошонки. Но эти же признаки возможны и при опухоли.
В динамике на фоне лечения эхоплотность яичка при орхите возвращается к норме, но при поствоспалительных атрофических процессах может оставаться пониженной. Атрофия яичка возможна при хроническом воспалении, тогда же допплерографически регистрируют обеднение кровотока.
Орхит может привести к формированию абсцесса. Абсцесс при проведении эхоГ выглядит как ограниченное круглое образование без внутренних эхоструктур в поле орхита. Прогрессирование абсцесса устанавливается на основании его распространения и появления неоднородности в очаге. Сложности возникают при выявлении абсцесса на фоне хронического орхита, когда могут отсутствовать лихорадка, боль и однородность эхоструктуры. Пальпируемое плотное образование позволяет исключать прежде всего опухоль. Диагноз проясняется при анамнестических сведениях, указывающих на орхит и эпидидимит. Окончательные сомнения снимают данные гистологического исследования (пункционной биопсии).
Воспаление придатка яичка (эпидидимит)
Эпидидимит — наиболее частая причина острого опухания и боли в мошонке у взрослых. Причины те же, что и при орхите. Примерно в 84% случаев эпидидимита высевают кишечную палочку. Клинически при воспалении придаток иногда плохо дифференцируется от яичка, но эта дифференциация доступна методикам УЗИ. На эхоГ придаток яичка увеличен, гипоэхогенен (при кровоизлиянии эхогенность повышается). По данным эхоГ возможна детальная диагностика места воспаления: тотальное или в области хвоста. Вторичными признаками эпидидимита являются реактивное гидроцеле, воспаление кожи мошонки. При благоприятном течении воспаления размеры и плотность придатка возвращаются к норме. При неэффективности терапии по данным эхоГ можно выявить вовлечение в процесс яичка (орхоэпидидимит) или формирование абсцесса.
Среди всех форм эпидидимита особого внимания заслуживает процесс туберкулезной этиологии. Его эхопризнаки полностью идентичны банальному воспалению, диагноз устанавливают клинически, по данным лабораторных и микроскопических исследований. Воспаление быстро распространяется на яичко, творожистый некроз выглядит как абсцесс, а хроническое течение туберкулезного воспаления приводит к неоднородности эхокартины. Свойственные туберкулезному эпидидимиту и орхиту малоболезненность, отсутствие ярких симптомов воспаления, неоднородность эхоплотности, заставляют исключать опухоли.
Эходиагностика орхита и эпидидимита в режиме серой шкалы, по крайней мере на начальных этапах, может быть малочувствительной. У 17–20% больных вообще не определяется четких эхоизменений тестикулярных структур. Значительно эффективней энергетическая допплерография. Хорошо выявляют гиперемию яичка, придатка или то и другое одновременно, степень выраженности признака и распространение воспаления.
Сперматоцеле
Сперматоцеле — ретенционная киста, протекает бессимптомно, часто выявляют случайно у мужчин среднего возраста. Выглядит как плотноэластичная киста, расположенная на семенном канатике, безболезненная, легко сдвигающаяся в стороны, но с большим ограничением — вверх и вниз. Клинический диагноз «сперматоцеле» редко нуждается в подтверждении эхоГ. Однако при небольших размерах сперматоцеле, при ДД этого доброкачественного образования с опухолью необходимой методикой может оказаться эхоГ. Крайне редко сперматоцеле достигает больших размеров, что создает сложности в его отличии от гидроцеле. При больших размерах сперматоцеле (>15 мм) необходимо дуплексное сканирование с целью исключения компрессии артерии яичка.
По данным эхоГ может быть сложно отличить сперматоцеле от гидроцеле семенного канатика. Это очень редкая аномалия, отражающая варианты закрытия влагалищного отростка брюшины. Гидроцеле семенного канатика может сообщаться с брюшной полостью и мошонкой или только с брюшной полостью и слепо завершаться на уровне нижних частей семенного канатика, в обоих этих случаях пациент напрягается при крике и натуживании. Возможно кистевидное гидроцеле семенного канатика, полностью изолированное от брюшной полости и мошонки. В этом случае оно не изменяется при повышении внутрибрюшинного давления, но может уменьшаться при вдавливании его в паховый канал. По данным эхоГ это образование совершенно эхонегативно. Изображение гидроцеле семенного канатика зависит от его локализации, наличия или отсутствия сообщения с брюшной полостью. Гидроцеле семенного канатика может смещать яичко, но никогда, в отличии от истинной водянки яичка, не окружает его.
Гидроцеле
Гидроцеле — водянка оболочки яичка, представляет собой патологическое скопление жидкости между листками влагалищной оболочки яичка.
У детей гидроцеле может сформироваться за счет отекания серозной внутрибрюшинной жидкости через необлитерированный вагинальный отросток брюшины, при избыточной секреции жидкости оболочками яичка.
Известен вариант симптоматического гидроцеле после операции пересадки почки. Осложнение прекрасно поддается склеротерапии тетрациклином.
У взрослых мужчин и у детей гидроцеле может образоваться при эпидидимите или орхите, при опухолях (10% всех случаев гидроцеле у взрослых), после травмы, вазэктомии, при лимфостазе, при перекруте яичка. Но в большинстве случаев точная причина остается не известной.
Объем жидкости в начальный период гидроцеле, особенно у детей, непостоянен, что может заставить лечащего врача думать о пахово-мошоночной грыже.
Клинически гидроцеле определяют как тугоэластичное образование без признаков воспаления, безболезненное. При диафаноскопии цвет розовый, ровный, прозрачность полная. При УЗИ содержимое эхонегативное, однородное, в нем четко визуализируются яичко и придаток. Хорошо видна перегородка между двумя половинками мошонки. При длительно существующем гидроцеле, особенно посттравматическом в просвете видны перегородки, нити фибрина.
Для ДД с петлей кишки, попавшей в мошонку при паховой грыже, необходимо длительное наблюдение с целью выявления перистальтики, типичной слоистой структуры стенки кишки, пузырьков газа в просвете.
При гидроцеле эхоГ позволяет четко установить диагноз в клинически сомнительных случаях, отдифференцировать образование от опухоли, оценить состояние яичка и придатка.
Варикоцеле
Варикоцеле — варикозное расширение вен семенного канатика — обусловливает по данным разных авторов от 10–15 до 40% случаев мужского бесплодия. Вена яичка является частью обходного ренокавального анастомоза, развивающегося компенсаторно при затруднении оттока по почечной вене функционального или первично анатомического характера. Указанный обходной путь состоит из вены яичка, гроздьевидного сплетения, наружной семенной вены. В качестве причин варикоцеле называют ретроаортальное расположение почечной вены, рубцовый процесс в забрюшинной клетчатке, острый угол отхождения верхней брыжеечной артерии, вследствие чего почечная вена оказывается зажатой между аортой и верхней брыжеечной артерией.
Варикоцеле может рассматриваться как одно из проявлений венопочечной гипертензии.
Остро возникшее варикоцеле требует исключения забрюшинной опухоли.
Варикоцеле может возникать без первичного повышения венозного давления как отражение мезенхимальной дисплазии стенки вены. Такой вариант сопутствует общей дисплазии соединительной ткани и часто наблюдается при синдромах гипермобильности, Марфана, Элерса — Данлоса.
В настоящее время частота варикоцеле у юношей и мужчин молодого возраста повышается, что можно объяснить неконтролируемым занятием спортом с отягощением.
В норме САД в почечной вене не превышает 10 мм рт. ст., при венозной гипертензии оно повышается до 20 мм рт. ст. и более, что приводит к снижению утилизации кислорода тканями органов, расположенных выше уровня препятствия оттоку. На экспериментальной модели застоя в органах мошонки животных и в клинике при исследовании мужчин с варикоцеле, состоящих в бесплодном браке, показано, что формирующаяся при этом заболевании очаговая неполная ишемия сопровождается циркуляторной, а затем и вторичной тканевой гипоксией. Кроме того, придаток яичка более чувствителен к ненормальному температурному режиму, чем само яичко, поэтому его нахождение в клубке варикозно трансформированных вен ведет к эффекту «гемодинамического термостата». Все это в совокупности обусловливает нарушение процессов сперматогенеза в яичках, уменьшение в них сперматозоидов и сперматид с компенсаторным увеличением сперматогоний. Одновременно нарушается транспортная и секреторная функции придатка яичка. В итоге не только снижается фертильность, но и увеличивается количество эпизодов невынашивания беременности, обусловленного увеличением количества аномальных гамет, участвующих в оплодотворении.
При обследовании 34 асимптомных молодых мужчин физикально варикоцеле выявлено у 15%, по данным эхоГ — у 18% (то есть при всей важности эхоГ результаты этой методики в аналогичных ситуациях не существенно отличаются от традиционных методов), а с применением цветного допплерографического изучения кровотока по венам яичка варикоцеле выявлено уже у 35% (по ретроградному кровотоку).
Варикоцеле возникает первоначально, как правило, слева. Расширение вен справа чаще вторично по сравнению с левой стороной. При обследовании ПО мужчин левостороннее варикоцеле выявлено у 50%, двустороннее — у 48%, изолированное правостороннее — всего у 2%.
Степень изменений спермограммы не коррелирует напрямую с выраженностью варикоцеле. Но есть мнение, что критический временной рубеж существования варикоцеле сравнительно невелик и составляет всего 1 год. Лечение улучшает качество спермы не более чем на 50%, поэтому своевременный точный диагноз клинически крайне важен.
При сканировании в серой шкале расширенные вены выявляются при увеличении их размера на 2–3 мм как тяж кзади от яичка. Признаками варикоцеле являются:
- степень — расширение вен в положении стоя и спадение их в положении лежа, увеличение диаметра на 1 мм при пробе Вальсальвы с усилением интенсивности цветовой индикации, инверсия и регургитация потока крови в вене левого яичка в покое и при пробе Вальсальвы.
- степень — сохранение расширения вен на уровне верхнего полюса яичка и ниже, а также в положении лежа, снижение линейной скорости кровотока в артерии яичка на 40%.
- степень — расширение вен ниже полюса с атрофией яичка, выраженный стаз крови.
УЗИ далеко не всегда способно выявить типы венозной системы. Так, в 75% случаев вена яичка представлена одним стволом, в 25% — двумя и более стволами. Поэтому, если оперирующий хирург не выявляет дополнительные стволы вены и оставляет их неперевязанными, варикоцеле рецидивирует. Более информативной методикой в этом случае является флебография, по итогам которой в ряде случаев возможно проведение окклюзии (склеротерапии).
Травма мошонки
При травмах, требующих неотложного хирургического вмешательства (разрыв яичка, обширная гематома), ранний диагноз предотвращает необратимые последствия инфекционных осложнений и ишемической атрофии. Травма мошонки на эхоГ проявляется как гематоцеле, посттравматический эпидидимит, эпидидимическая гематома, гематома яичка или инфаркт, гиперемия яичка, его опухание и разрыв. При разрыве яичка видна линия разлома, фрагментация яичка, изменение его контуров и эхоплотности. В случае обширной гематомы необходимо применение цветного допплеровского картирования для оценки состояния сосудов и определения тактики хирургического вмешательства.
Кровоизлияние в мошонку — гематоцеле — диагностируют по наличию в ближайшем анамнезе травмы, клинической картины (отек, болезненность, гиперемия), на основании данных эхоГ (скопление жидкости в мошонке). Но, в отличии от гидроцеле, диафаноскопия малорезультативна, а по результатам эхоГ жидкость неоднородна, в просвете определяют множество мелких перемещающихся эхоструктур.
ЭхоГ при травме мошонки показана для уточнения ущерба (подтверждение или исключение разрыва яичка определяют прогноз), дифференциации гематомы мягких тканей от гематоцеле, динамического наблюдения пациента после хирургического вмешательства или для выработки показаний к проведению консервативной терапии.
Минимальный размер гематомы, требующей хирургического вмешательства, не известен. Однако принято считать, что если размер субкапсулярной или внутрияичковой гематомы менее 1/3 диаметра яичка, то возможна выжидательная консервативная тактика. Точно также маленькое гематоцеле без признаков разрыва яичка свидетельствует против выбора хирургического метода лечения. Но комбинацию гематоцеле любого размера с интратестикулярной гематомой необходимо расценивать как признак разрыва яичка даже при отсутствии эхопризнаков разрыва. В то же время есть сведения, что эхоГ может оказаться недостаточно информативной. Так, при разрыве яичка точность УЗИ составила 56%. Поэтому рекомендуется раннее оперативное вмешательство при подозрении на разрыв яичка.
Но увеличение, болезненность и покраснение мошонки может быть и результатом поражений других органов, весьма далеко расположенных собственно от мошонки. Так, возможно затекание крови в мошонку при травме живота, а у новорожденных — при кровоизлиянии в надпочечник. Эхоисследование забрюшинного пространства и клиническая оценка результатов (перинатальная гипоксия в анамнезе, в основном изменение правой половины мошонки) разъясняют ситуацию.
Перекрут яичка
Обычно только небольшая часть стенки мошонки, не покрыта влагалищной оболонкой, что позволяет закрепить яичко внутри мошонки. Перекрут яичка — вращение его по длинной оси, продолжением которой является семенной канатик. Перекрут яичка может возникнуть, когда яичко свободно подвешено в мошонке из-за ненормального слияния влагалищной оболонки со стенкой мошонки. Из всех случаев «острой мошонки» по данным отделения неотложной помощи 16% составляет перекрут яичка, 46% — перекрут его аппендикса и 35% — перекрут придатка яичка.
Перекрут может быть спровоцирован травмой, сексуальным возбуждением, усиленной работой, но может быть и спонтанным, нередко возникает во сне. Описан перекрут у новорожденных, чаще имеющий в основе интранатальный механизм. Возможен подострый перекрут яичка, для которого, как это указано для младенцев, не типична острая интенсивная боль. При таком варианте перекрута роль эхоисследования особенно велика. Перекрут приводит к пережатию сосудов, проходящих в семенном канатике. При этом необходимо срочное хирургическое вмешательство, в противном случае происходят необратимые изменения тестикулярной ткани (по образному выражению — «смерть в мошонке»). Чаше всего (70%) перекрут выявляют у подростков в возрасте 12–18 лет. Это объясняет важность проблемы для прогноза дальнейшей фертильности.
Клинически перекрут характеризуется чаще острой нестерпимой, иногда шоковой болью с развитием признаков эпидидимита и орхита (увеличение размеров яичка), болью в животе. У новорожденных перекрут безболезненный, проявляется увеличением половины мошонки, пальпируемым образованием, отсутствием транслюминисценции и кремастерного рефлекса. У пациентов более старшего возраста клинически картина перекрута (заворота) яичка может напоминать орхит. В отличии от орхита напряженное болезненное яичко расположено горизонтально. Изменения односторонние. На пораженной стороне есть втяжение мошонки. Придатки яичка как отдельные структуры пропальпировать не удается. Подъем яичка кверху усиливает боль. Контрлатеральное яичко гипермобильно. Анализы мочи не изменены.
По данным эхоГ перекрут яичка характеризуется негомогенностью изображения паренхимы с беспорядочным чередованием гипер- и гипоэхогенных участков, утолщением покровных тканей мошонки, отечным гиперэхогенным придатком, очень небольшим по объему гидроцеле. На ранней стадии эхоГ при сканировании в режиме серой шкалы изменений можно и не выявить или они неспецифичны (изменение эхоплотности). Позднее регистрируют изменение структуры яичка (инфаркт и кровотечение). При сопоставительных исследованиях показано, что при неизмененной эхоплотности яичко при операции оказывается жизнеспособным, при гипоэхогенных или неоднородных по эхогенности яичках они нежизнеспособны. Все остальные эхопризнаки (размеры яичка, кровоснабжение и толщина кожи мошонки, наличие реактивного гидроцеле) прогностически незначимы.
Необходимо применение тканевого (энергетического) допплера. Методика сочетает возможности морфологической визуализации органа, что свойственно собственно эхоГ, и оценки нюансов кровотока, что ранее было возможно только сцинтиграфически. Исследование необходимо проводить симметрично, чтобы выявить минимальные изменения как, например при неполном перекруте или самопроизвольном разрешении. В пораженном яичке кровоток обедняется и даже полностью не определяется (при воспалении кровоток усиливается). Самопроизвольное устранение перекрута приводит к реактивно усилению кровотока, что четко видно по сравнению с предыдущими исследованиями.
Проведение эхоГ обязательно и с точки зрения ДД перекрута яичка с целым рядом состояний («острая» мошонка): ущемленная грыжа, ущемленное яичко, идиопатическое кровоизлияние в яичко, отек при болезни Шенляйна — Геноха или панкреатите и т. д.
Острые перекруты гидатид яичка и его придатка
В основе острого перекрута гидатид (в 96% случаев поражаются гидатиды яичка, в 4% — придатка и парадидимиса) лежит тромбоз вен гидатиды, сосудистая система которой не подготовлена к функциональным перестройкам. Причиной могут быть различные факторы, ухудшающие венозный отток из нижней полой вены. У 2/3 пациентов заболевание проявляется с левой стороны (в левом яичке условия венозного оттока в результате особенностей строения путей венозного оттока, по сравнению с правым яичком, затруднены). Клинически перекрут гидатид выглядит как орхоэпидидимит. Их дифференциация возможна только с помощью эхоГ.
Основным методом лечения является удаление гидатиды с перевязкой ее сосудистой ножки. В этом заключается профилактика восходящего тромбоза вен семенного канатика. Только хирургический метод лечения позволяет избежать атрофии яичка, которая раньше, при консервативном лечении, ошибочно диагностированного орхоэпидидимита (без применения эхоГ), под маской которого и протекает острый перекрут гидатид, достигала 46,5%.
Прочие состояния
Среди других состояний, включенных в так называемый синдром скротальных масс, следует сказать об аномально расположенных и сформированных органах, оказавшихся в мошонке. Из всех этих редких состояний прежде всего упоминается спленогонадная ассоциация. Аномалия, как считается, возникает на 6–7-й неделе внутриутробной жизни либо за счет минимальных воспалительных спаек между закладками яичка и селезенки с последующей транслокацией части селезеночной ткани в мошонку, либо в результате тесного контакта закладочных клеток селезенки и яичка. В большинстве случаев атипичная селезенка и яичко связаны в мошонке только соединительно-тканным тяжем без нарушения белочной оболочки гонады. В большинстве ситуаций ортотопно расположенная селезенка не изменена. Все случаи спленогонадной ассоциации левосторонние, 50% всех диагностированных случаев приходится на детей в возрасте младше 10 лет. Клинически аномалия проявляется безболезненным увеличением яичка без признаков отека мошонки, трактуется чаще всего как грыжевое выпячивание. По данным эхоГ выявляют дополнительное, чаще небольшое (диаметром 1–2 см) образование, расположенное в непосредственной близости от яичка и отличающееся от него по эхотекстуре. Возникают сложности ДД в отличии образования от полиорхидии, экстрагонадной опухоли, эпидидимита. Сцинтиграфия с технецием-99m, накапливающимся в селезенке, помогает заподозрить эктопированную в мошонку селезенку. Но окончательный диагноз возможен только в процессе операции. Однако важно, что эхоисследование позволяет отказаться от орхоидэктомии.
Синдром Фурнье — некротизирующее подкожное воспаление мужских гениталий, некротизирующий фасциит. Остро развивается интенсивная боль в гениталиях, повышение температуры тела, озноб. Кожа на гениталиях краснеет, опухает, возможно ее гангренозное изменение.
*Везикоуретеральный рефлюкс может быть как одно-, так и двусторонним. У пациентов с односторонним рефлюксом при наличии здоровой контрлатеральной почки риск развития почечной недостаточности минимальный. Но АГ и мочевая инфекция этот риск резко повышают и должны быть исключены априори.