Важнейшие субъективные и объективные симптомы. Современное представление о группах риска с точки зрения семейного врача как об одном из отправных условий диагностики
При встрече с больным врачу следует сразу нацеливаться на заболевания, наиболее вероятные для данного пола, возраста, профессии. Это позволяет не только диагностировать уже проявившиеся заболевания, когда пациент обратился к вам с теми или иными расстройствами, а целенаправленно на современном уровне провести диспансерное наблюдение. Ведь задачей семейного врача является владение универсальными знаниями при индивидуальном подходе, возможность лечения различных заболеваний и способность быть грамотным координатором консультаций.
По возрасту выделяются следующие группы риска (адаптировано по K.L. Kahn, H.L. Green, 1991).
- Новорожденные — грудные дети — дети в возрасте до 2 лет.
Дети с массой тела 2000 г и меньше при рождении (эта категория остается группой риска на всю жизнь, так как именно из нее во многом формируются группы пациентов с кардиальными и цереброваскулярными катастрофами, с АГ, сахарным диабетом); с перинатальной асфиксией; гипербилирубинемией, требующей гемотрансфузий; с мышечной гипотонией; с ранним искусственным вскармливанием (угроза развития колита, аллергических расстройств); перенесшие бактериальный менингит; с анамнестическими указаниями на внутриутробную инфекцию; при наличии в семье туберкулеза, сифилиса или другой хронической инфекции; дисморфичные; имеющие старших тугоухих сибсов; из семей беженцев и вынужденных переселенцев; асоциальных семей; живущие в домах, построенных до 1960 г.; живущие в непосредственной близости от фабрик, заводов, оживленных магистралей; из регионов с дефицитом фтора в питьевой воде.
- Дети в возрасте 2 года–6 лет.
То же самое плюс повышенная фоточувствительность.
- Дети в возрасте 7–12 лет (см. выше).
- Дети и подростки в возрасте 13–18 лет.
Дети из семей переселенцев; контактные по туберкулезу; безработные; не посещающие школу; не привитые от гепатита В; с ранним началом половой жизни; имеющие несколько половых партнеров; болевшие сифилисом, гонорреей или контактирующие с больным сифилисом или гонореей; би- и гомосексуалы; контактирующие с ВИЧ-инфицированным; депрессивные; употребляющие алкоголь и/или наркотики; проживающие в регионах с низким содержанием фтора и фотосенсибилизированные; мужчины с анамнестическими указаниями на крипторхизм, орхопексию, тестикулярную атрофию.
- Лица в возрасте 19–39 лет.
Заядлые курильщики, лица, злоупотребляющие алкоголем; с нарушениями и изменениями, выявленными при самоконтроле. Радиационное облучение верхней половины тела в анамнезе. Женщины в возрасте 35 лет и старше с анамнестическими указаниями на семейные случаи рака груди у родственниц в пременопаузальный период. Мужчины с анамнестическими указаниями на крипторхизм, орхопексию, тестикулярную атрофию. Лица с семейными формами рака кожи, диспластичными невусами, фотосенсибилизацией. Выраженное ожирение. Сахарный диабет в семейном анамнезе или женщины с диабетом беременных в семейном анамнезе. Проститутки и лица, имеющие нескольких половых партнеров. Лица с заболеваниями, передающимися половым путем или леченные от этих болезней. Контактирующие с туберкулезом, гемодиализными пациентами. Би- и гомосексуалы. Проживающие в одном доме со стариками. Фотосенсибилизированные. С повышенным риском развития пневмококковой инфекции (хронические заболевания легких и сердца, серповидноклеточная анемия, нефротический синдром, удаленная селезенка, сахарный диабет, лимфомы, алкоголизм, заболевания почек). Люди с метаболическими нарушениями, гемоглобинопатией и иммуносупрессией.
- Лица в возрасте 40 лет–64 года.
Пациенты с семейными формами рака кожи или диспластичными невусами. Злоупотребляющие алкоголем или заядлые курильщики. С изменениями, выявленными при самообследовании. С радиооблучением верхней половины тела в анамнезе. Пациенты с АГ, заболеваниями коронарных артерий, транзиторными нарушениями мозгового кровообращения, фибрилляцией предсердий, сахарным диабетом, с выраженным ожирением. Лица с клинической картиной заболеваний, передающихся половым путем, ведущие неупорядоченную сексуальную жизнь. Наркоманы. Гомо- и бисексуалы. Лица, контактирующие с больными туберкулезом или сифилисом. Подвергающиеся воздействию сильного шума. Лица в возрасте 50 лет и старше с наличием у родственников первой степени родства колоректального рака. Люди с семейным полипозом кишечника, с установленным диагнозом воспалительного поражения толстой кишки, аденоматозных полипов. Женщины в перименопаузальный период с повышенным риском развития остеопороза (уроженки Кавказа, после двусторонней ооэктомии до менопаузы). Безработные, депрессивные пациенты, алкоголики, наркоманы. Живущие в одном доме с детьми или со стариками. Реципиенты крови. Лица с высоким риском развития пневмококковой инфекции.
- Лица в возрасте 65 лет и старше.
Пациенты с риском развития цереброваскулярных или коронарных заболеваний. Рак кожи в семейном анамнезе, диспластичные невусы, фотосенсибилизация. Злоупотребление алкоголем и никотином. Ожирение, сахарный диабет. Лица, контактирующие с больными туберкулезом, сифилисом, с ВИЧ-инфицированными. Пациенты с раком молочной железы, эндометрия, яичников в анамнезе. Лица с воспалительными заболеваниями толстой кишки и полипозом кишечника в семейном анамнезе. Депрессивные пациенты, алкоголики и наркоманы. Женщины с высоким риском развития остеопороза (см. выше).
Умение выяснить историю заболевания, предшествующие факторы, время появления первых симптомов, их характер, развитие болезни — важнейшая составная часть врачебного искусства. Беседа с пациентом и/или с его родственниками требует такта, умения расположить к себе, создав доверительную атмосферу. Успех и прочность контакта определяют первые 10–15 мин встречи. Целесообразно дать пациенту возможность изложить первые жалобы, после чего направляющими вопросами профессионально уточнять их и регулировать беседу, поскольку представляемые сведения обычно неполны. Например, приходится уточнять время возникновения, длительность, периодичность, характер тех или иных расстройств, связь их с приемом пищи (и какой именно), физической нагрузкой и т. п. Одновременно врачу следует уточнить и состояние всех систем организма, эмоциональные, психические, нервные реакции. Уже по тому, как излагаются жалобы, можно много сказать о личности пациента, его взаимоотношениях с окружающими.
Абсолютно неприемлемы для индивидуальной врачебной работы анкеты, опросники и т. п. Они с большими оговорками и условностями применимы только на этапе массовых эпидемиологических работ, скрининговых исследований.
Успех диагноза — в личном общении. После изложения проблем, послуживших непосредственной причиной визита пациента, есть смысл переходить к беседе по анамнезу заболевания, который во многом переплетается с анамнезом жизни. Расстройства, беспокоящие больного в момент визита, могут быть только эпизодом общего хронического заболевания или его осложнением. Желателен поиск факторов риска, если они могут быть причиной данного заболевания, так как их устранение способствует благоприятной динамике. В этой же части беседы можно уточнить характер работы, профессиональные или семейные конфликтные ситуации. Не следует надеяться, что пациент изложит эти конфликты, хотя бы только потому, что не все их осознают. Пациент, как правило, не склонен излагать психологические конфликтные проблемы, считая, что все может быть решено лечением, направленным на соматические расстройства. Но с профессиональной точки зрения очень опасно ставить печать «психогенного» заболевания: органические причины всегда могут выйти на первый план. Да и органические заболевания могут манифестировать с психических, неврологических и эмоциональных расстройств. Здесь необходимы знания хотя бы основ психологии. Для того чтобы лечение было успешным, врачу необходимо ответить на вопрос: «Какую роль болезнь играет в структуре личности пациента? Какие потребности она обслуживает?». Это особенно важно в связи с тем, что болезнь зачастую играет функцию защиты от чрезмерных требований окружающих, от нерешаемых проблем на работе или в семье. В таком случае пациент будет испытывать бессознательное сопротивление, не захочет «отдать» болезнь из страха остаться незащищенным до тех пор, пока либо не будут выработаны другие способы защиты, либо не исчезнет источник опасности. На помощь интернисту может придти психолог или семейный психотерапевт. А решить вопрос о необходимости такого рода помощи практикующий врач может, наблюдая пациента, анализируя его взаимоотношения с окружающими. Источником информации о пациенте могут быть такие важные проявления в поведении:
- как пациент входит в ваш кабинет, а именно: какое сообщение он посылает вам своим видом: «посмотри, какой я несчастный»…, «я боюсь тебя»…, «вот теперь и ты обратишь на меня внимание» и т. д.;
- как пациент осваивает пространство, направляется ли прямо к стулу, осматривает или трогает предметы вокруг себя, сколько времени он на это отводит;
- как пациент владеет своим телом, скован или раскрепощен, насколько свободно движутся его руки, разно- или однообразные движения они выполняют. Нет ли тенденции сжаться, «уменьшиться в размерах», какая походка, осанка;
- насколько естественно больной раздевается, как реагирует на прикосновения: «раскрывается» навстречу вам или сжимается;
- как отвечает на вопросы о своих ощущениях в теле: задумываясь, прислушиваясь к себе или заученными «машинными» фразами, односложно или с удовольствием;
- смотрит ли вам в глаза открыто, спокойно и долго или просто в вашу сторону, блуждает взглядом по комнате;
- естественно ли звучит голос, что при этом происходит с его дыханием, кожными покровами, мышцами лица;
- соответствует ли эмоциональная окраска содержанию высказываний. Например, «все хорошо, у меня здесь ничего не болит» пациент может говорить плаксивым голосом, а «больно» — радостно хихикая. Это свидетельствует о конфликте между внутренним миром и тем, что ваш посетитель пытается продемонстрировать, чтобы соответствовать ожиданиям окружающих. Это также сигнал неоднозначных отношений пациента со своими болезнями, требующий дополнительного изучения и анализа;
- приходят ли на прием родственники, если да, то с какой целью: получить помощь больному или с бессознательной просьбой помощи себе, подтвердить свою правоту, оказать через вас давление? Таскание больного по врачам, настаивание на выполнении диагностических процедур может быть эквивалентом жестокого обращения, подсознательным стремлением продемонстрировать заботу о нежеланном ребенке, вытеснив тем самым представление о своей греховности. Мотивов может быть много, надо только разобраться в них.
Умение обращать внимание на такие нюансы позволяет врачу подойти к ответу на основной вопрос о том, насколько благополучно внутреннее «Я» пациента, а, следовательно, насколько его личность может «нуждаться» в болезни для самосохранения и решения важнейших для нее проблем отношений с социумом.
При дальнейшей беседе не приемлемы вопросы о предшествующих диагнозах, надо спрашивать о симптомах. Если же больной сослался на тот или иной диагноз, установленный до вашей встречи, то следует тут же уточнить, на основании каких сведений он был установлен, чтобы самому осмыслить прежнюю информацию в новых условиях.
Нельзя отвлекаться, решать какие-либо другие задачи во время беседы, это мешает создать цельное мнение и отрицательно сказывается на доверительности изложения. В этот момент нежелательно вести записи в истории болезни или в амбулаторной карточке. Единственно, что позволяется в присутствии пациента, — это набросать клиническую схему динамики симптомов. Отсутствие записей, сделанных при сборе анамнеза, создает (и обоснованно) у пациента впечатление, что его доверие к врачу не будет использовано другими.
Внешний вид врача, четкость и правильность речи, отсутствие панибратства, сертификаты и дипломы, представленные в вашем кабинете — все это располагает визитера к подробному и откровенному изложению сведений о себе и своих близких. Ведь никто из нас не будет беседовать о себе, если он не убедится, что перед ним сочетание высокого профессионализма и заинтересованности.
Полноценный анамнез включает и сбор семейного анамнеза. При этом получают информацию не только о семейных заболеваниях, но и о социальном окружении пациента. Не все семейные заболевания — наследственные. Например, ожирение и алкоголизм чаще обусловлены внешними факторами.
Умение вести беседу не преподается и не может преподаваться в лекционном зале, оно достигается многолетней практической работой, умением быстро, почти неосознанно, оценить личность больного. Чем больше врач знает, тем больше он получит в результате сбора анамнеза. Правильно собранные жалобы и анамнез позволяют с большой долей достоверности (минимум на 50%) диагностировать болезнь, 1/3 успеха заключается в осмотре. Сложно дать определение, но из практической работы хорошо известно, что по итогам опроса и первого впечатления о больном интуитивно устанавливается диагноз.
Интуиция (от латинского — пристально, внимательно смотреть) — способ постижения истины без логического обоснования, основанный на предшествующем опыте. В сущности, логическое мышление, безусловно, участвует в этом, но совершается оно настолько быстро, что цепь ассоциаций не контролируется сознанием. Таким образом, первоначально мы чаще и определяем степень тяжести и прогноз заболевания пациента. Опытный врач, лишь взглянув на больного, может установить синдром Марфана, хондродистрофию и многие другие состояния.
Какие же важнейшие группы субъективных и объективных симптомов учитывает врач?
Боль — важнейший и наиболее частый симптом. Острая боль обычно не является причиной диагностических проблем. Начало острой боли четко определено во времени. Часто причина объясняется достаточно просто, но хроническая боль представляет большие сложности в диагностическом и лечебном аспектах. Как субъективный симптом боль воспринимается и описывается пациентами по-разному. Но таксономически боль определяется как «неприятные сенсорные или эмоциональные ощущения, ассоциирующиеся с острым или потенциально возможным разрушением тканей и описываемые в терминах такого разрушения». Из определения следует, что боль — сложно организованный процесс. Для нее в сущности не важна причина, вызвавшая ее. Личностно-психологические особенности пациента, характер его воспитания и культуры, профессия значительно сказываются как на пороге болевой чувствительности, так и на экспрессивности жалоб.
При работе по проблеме боли необходимо прежде всего выяснить время ее возникновения, степень интенсивности (постоянная, флюктуирующая, позволяет ли спать), основное расположение, иррадиацию, предшествующие факторы (особое внимание — посещению пациентом таких «модных специалистов», как мануальных терапевтов, хиропрактиков, специалистов по акупунктуре). Каков был эффект предшествующих вмешательств? Какова динамика выраженности боли: уменьшилась, без изменений, усилилась.
Клинически необходимо выделять боль поверхностную и глубокую. Поверхностная чаще очень четко локализована, острая, связана с изменениями кожи или близко расположенных структур. Глубинная боль тупая, локальность ее выражена меньше, она иррадиирует в соседние регионы или по зонам Захарьина — Геда. С нею необходимо дифференцировать боль при заболеваниях нервной системы, поражении нервных стволов, например, невралгию, гиперэстезию или симметричную боль при нейропатии. Обычно эта боль еще менее локальна, мало поддается действию анальгетиков. Боль, связанная с нервными стволами, может быть представлена как:
- периферическая нейропатия (дегенеративная). Обычно двухсторонняя, прежде всего — в кистях и стопах, очень часто ассоциируются с дисэстезией. Нередко сопутствует сахарному диабету, гипотиреоидизму, сифилису, болезни Педжета, поступлению в организм токсинов (свинец, полициклические углеводороды);
- боль от сдавления (туннельный, карпальный синдром; перелом в анамнезе, торакотомия с последующей межреберной болью; грыжесечение с развитием позднего сдавления илиоингвинального нерва);
- радикулопатия. Наиболее типичное проявление — боль в спине с иррадиацией в соматом;
- каузалгия (симпатическая боль);
- невралгия. Может быть пароксизмальной и непароксизмальной. Возникает прежде всего вследствие повреждения V или X нерва. Рано формируются триггерные зоны.
Многие женщины жалуются на боль в грудных железах (масталгия, мастодиния). В 1/2 всех случаев это циклическая боль, двухсторонняя, связанная с менструальным циклом и предшествующая менструальным кровотечениям. Боль усиливается при психических перегрузках, злоупотреблении кофе, никотином, скудной растительной пище. В 9–12% случаев эта боль возникает в местах прежних травм, абсцессов, маститов и обусловлена, видимо, реакцией рубцовой ткани на периодически возникающие изменения гидратации тканей в ответ на колеблющийся уровень гормонов. При нециклической и персистирующей боли в молочной железе необходимо выполнить маммографию, даже если пальпаторно или эхографически ничего не выявлено. Врач обязан документально зафиксировать все изменения, в том числе (и прежде всего) трудно классифицируемые и повторить исследование не позже, чем через 1 мес. Диагностический алгоритм при боли в молочной железе представлен на схеме 2.1.
Но у 10% женщин с раком молочной железы первым симптомом является именно боль в железе. Боль (сравнительно поздно) и/или пальпируемое образование в молочной железе (рано) требуют исключения рака молочной железы. Рак молочной железы составляет 25–30% всех карцином у женщин и обусловливает 15–20% летальных исходов от злокачественных новообразований у женщин. Известно много работ по оценке диагностической значимости разных скрининговых методик, которые бы позволяли диагностировать рак молочной железы до появления пальпируемой массы. Но в практических условиях именно факт выявления врачом или пациенткой узла в молочной железе служит отправной точкой для исключения рака.
Факторами риска развития рака молочной железы являются:
- 1Возраст больной. Заболеваемость и смертность повышаются с возрастом. Только 1% всех случаев опухоли приходится на женщин в возрасте моложе 30 лет, а 75% — старше 40 лет.
- 1Предрасположенность к раку передается и по женской, и по мужской линии. Мать, сестра или дочь пациентки с односторонним раком заболевают им в 1,2–2 раза чаще, чем в среднем популяция. Если рак двусторонний и развился в пременопаузальный период, то риск повышается в 9 раз.
- 1Наличие предшествовавшего рака груди. Если ранее уже был рак молочной железы, то риск возрастает в 5–10 раз.
- Чем раньше женщина рожает первого ребенка, тем ниже риск развития рака молочной железы. У родившей ребенка в 30 лет более высокий риск, чем у вообще не рожавшей.
- Раннее менархе повышает риск развития рака. Ранняя (до 45 лет) менопауза, естественная или хирургически индуцированная, снижает риск развития рака.
- Другие неоплазмы в анамнезе, особенно рак яичников, матки, ободочной и прямой кишки, слюнных желез.
- Ожирение в постменопаузальный период.
- Высокий социально-экономический уровень жизни.
- Уроженка Северных регионов Европы.
- Употребление алкоголя.
- Гормональная заместительная терапия.
- Пероральные контрацептивы.
Наряду с определением факторов риска врачу необходимо выяснить, когда, как и кем был выявлен узел, его локализацию, изменение за период менструального цикла, консистенцию, поверхность, чувствительность, стяжение кожи над узлом или изменение ее цвета, смещение соска или наличие выделений из него, характер регионарных лимфатических узлов, отек руки (рис. 2.1).
Ни один из этих признаков не позволяет однозначно трактовать образование (узел) как доброкачественное или злокачественное. Так, при раке груди в 60% пациенток узел подвижный, в 40% — имеет гладкую поверхность, в 40% — мягкий или кистоподобный. Важно оценить состояние узла (узлов) в динамике. Обследование повторить на 3-й день после менструации, когда влияние эстрогенов и прогестерона минимально. Если узлы расположены в обеих молочных железах, уменьшаются в размерах и становятся в этот период менее плотными, исчезает чувство распирания, то больше оснований говорить о фиброкистозном поражении. То есть ДЦ во многом строится в зависимости от наличия одного или множественных узлов и возраста пациентки. В пременопаузальный период из каждых 6 случаев 5 узлов в молочной железе — доброкачественны; в постменопаузальный — 5 из 6 злокачественны. Важным этапом установления диагноза является аспирационная (тонкоигольная) биопсия. Ложно-негативные результаты выявляют с частотой 5–20%, а ложноположительные — 1–2%. ДД-путь представлен в схеме 2.2.
Из методов неинвазивной визуализации первое место занимает маммография. Частота ложно-негативных результатов составляет 8–14%. Эхографическая диагностика не получила абсолютного признания.
Частота рака молочной железы у мужчин составляет 0,5–1% случаев рака молочной железы у женщин. Исходы его значительно хуже, чем у женщин, но только потому, что о возможности данного вида рака у мужчин даже не задумываются, и диагностика, как правило, очень запоздалая.
Синдром Тейтца как причина ложно-положительной диагностики масталгии возникает достаточно редко.
Сильнейшую тревогу пациентов всегда вызывает боль в области сердца, но врачу всегда следует помнить, что в области сердца еще не значит в сердце.
Особой проблемой является хроническая боль у детей. Наряду с перечисленными причинами следует помнить о травмированном ребенке, глубокой невротизации и опухолях. Оценка боли у детей проводится с учетом их возраста. Большинство способов разработано для оценки острой боли. Используют такие критерии, как поза, специфический дистресс, выражение лица, вокализация/характер плача, рефлексы, сердцебиение, АД, дыхание, показатель рО2 (чрескожное измерение), лотовый тест. Применимы такие лабораторные показатели, как уровень катехоламинов, гормонов роста, кортикостероидов, глюкагона, инсулина, p-эндорфинов. Для детей в возрасте 3 лет и старше применимы визуально-аналоговая шкала, болевой термометр, проективные тесты, опросные листы, дневники, шкала качества жизни.
Слабость и утомляемость прежде всего требуют исключения физиологических причин (работа, недосыпание). Наблюдение пациентов с изолированными жалобами на усталость и утомляемость позволило убедиться, что через 1 мес у 85% жалобы самопроизвольно исчезли, у 6% выявлена депрессия, у 3% — сахарный диабет, у 3% — сердечная недостаточность, еще у 3% — другие причины плохого самочувствия. Таким образом, не стоит на основании одной жалобы назначать обследование или применять стимуляторы. Но после исключения преходящих причин при жалобах пациента на слабость и утомляемость следует проверить наличие хронического заболевания или интоксикации. Необходимо при первой встрече с пациентом решить, являются ли предъявляемые симптомы действительно слабостью или признаком миастении, миопатии, пареза. Колебания проявлений слабости и утомляемости по времени суток — важная подсказка для дальнейшего врачебного поиска.
Именно слабость и утомляемость как неспецифический симптом часто предшествует другим проявлениям опухолей или хронических инфекций (туберкулез, хронический гепатит), эндокринных (гипопитуитаризм) и неврологических (рассеянный склероз) заболеваний, а также сопровождает их, как и большинство острых состояний. При этом характерно нарастание слабости к вечеру.
Утомляемость может быть проявлением алкоголизма, интоксикации пестицидами, тяжелыми металлами, препаратами лития, антидепрессантами, а также при отмене кортикостероидов. Во всех этих случаях она достаточно постоянна.
Но очень часто слабость является проявлением психических состояний, прежде всего — депрессий (см. выше). Депрессивные состояния в практике интерниста часто остаются нераспознанными, и длительное время таких пациентов наблюдают по поводу заболеваний сердца, желудочно-кишечного тракта, гипотонического состояния. У пациентов с ларвированными депрессиями наряду с усталостью регистрируют многочисленные страхи, тревожность, частое просыпание по ночам, ощущение греховности и недооцененности, многочисленные психосоматические расстройства. Усталость, потеря интереса к жизни, вялость отмечает пациент уже с утра, даже не вставая с постели. Выделяют эндогенную депрессию (маниакально-депрессивный синдром), соматогенную (при тяжелых заболеваниях, прежде всего опухолевых) и реактивную (психогенную). В ряде случаев возможно сочетание причин: рак желудка, безусловно, весьма реальная причина и соматогенной, и психогенной депрессии. ДД депрессий может быть непростой и требует участия психиатра.
Наряду с этими состояниями и такое трудноуловимое понятие, как метеолабильность: вялость, слабость, утомляемость, сонливость и вегетовисцеральные дисфункции, связанные с прохождением метеофронтов. 8–12% всего населения в той или иной степени отмечают эти симптомы. Все они вегетативно лабильны. Здоровый отдохнувший человек обычно не подвержен метеолабильности.
Рассматривая проблему слабости и утомляемости, следует помнить о синдроме хронической усталости — СХУ. Выявляют относительно редко (37:100 000 населения). В 70% случаев поражаются женшины 25–49 лет, обычно молодые, активно работающие и быстро продвигающиеся по служебной лестнице. Заболевание связывают с хронической вирусной инфекцией (особенно герпетической или с неизвестным герпесподобным вирусом) или с первичным психическим заболеванием с последующим нарушением эндокринной, иммунной и других систем. Критерии диагноза СХУ:
- Большие (обязательные):
- постоянное снижение работоспособности на 50% и более, а также усталость у ранее здоровых людей, отмечающиеся не менее 6 мес;
- отсутствие заболевания или других причин, которые могут обусловить такое состояние.
- Дополнительные (малые) критерии: нарушение памяти или концентрации внимания;
- фарингит;
- болезненные шейные лимфатические узлы;
- мышечная боль;
- полиартралгия;
- необычная, новая для больного головная боль;
- неосвежающий сон;
- недомогание после физического напряжения.
Диагноз СХУ считается достоверным, если выявлены оба обязательных критерия и четыре признака из восьми малых, которые также отмечаются не менее 6 мес.
Диагностика СХУ включает следующие этапы:
- подробный анамнез и физикальное обследование;
- оценка психического статуса (при выявлении отклонений провести психиатрическое, неврологическое и психологическое обследование);
- биохимическое исследование крови и мочи;
- проведение дополнительных исследований при наличии клинических показаний в целях исключения других состояний.
ДД проводят исходя из конкретных симптомов больного.
Таким образом, утомляемость и слабость — симптом, сопровождающий многие состояния. Его расшифровка требует грамотного сбора анамнеза и физикального обследования. Объем и набор лабораторных и инструментальных исследований целиком определяются сопутствующими симптомами.
Аппетит — биологическое условие выживания особы, а в итоге — сохранения вида, и эта функция организма сохраняется достаточно долго даже при сравнительно тяжелых состояниях. Чаще сниженный аппетит — это результат психогенных факторов (насильственного кормления, семейных и профессиональных стрессов) или тяжелых психических расстройств (anorexia nervosa). Со снижением аппетита протекают заболевания желудочно-кишечного тракта (особенно рак желудка), гепатит и другие заболевания печени, семейный холестаз, сердечная и почечная недостаточности, недостаточность надпочечников, алкоголизм, наркотическая зависимость. Радиотерапия, цитостатики и дигиталис также отрицательно сказываются на аппетите. Обменные нарушения (абеталипопротеинемия, оксалоз) характеризуются резким угнетением аппетита.
Повышенный аппетит может быть при психологических и психических расстройствах, при гипертиреозе или в дебюте сахарного диабета.
Нарушения глотания разнообразны по причинам. Основными причинами дисфагии в широком смысле являются нарушения пищевода врожденные и приобретенные (атрезия, фистулы у маленьких детей, сдавление извне, инородные тела, онкологические процессы, ахалазиямегаэзофагус, грыжи диафрагмы, дисфагия при нейромышечных заболеваниях, крикофарингеальная дисфункция), дифтерия, полиомиелит, столбняк.
При орофарингеальной дисфагии пища тут же после проглатывания регургитируется, возможна аспирация и вытекание пиши через нос. Этот тип расстройства глотания возникает при бульбарном и псевдобульбарном парезе (дерматомиозит, миастения), могут быть и механические причины: опухоли средостения, струма, рубцы пищевода. При эзофагите и ульцерозных процессах отмечают боль при прохождении пищи. При эзофагеальной дисфагии пища либо задерживается в средней или дистальной части, либо медленно проталкивается по пищеводу. Причинами могут быть склеродермия, ахалазия, механические препятствия за счет опухолей, симптоматические эзофагеальные спазмы. Важным ДЦ-признаком считается затруднение При прохождении твердой пищи при органических стенозах, а твердой и жидкой — при моторных нарушениях. У женщин пожилого возраста возможно развитие синдрома Плуммер — Винсона: сидеропеническая дисфагия, сочетание железодефицитной анемии и сужения пищевода. Невротические жалобы на комок в горле, затруднения глотания эмоциональны, красочны, а при проверке пациенты свободно глотают и жидкую, и твердую пищу.
Нарушения вкуса и обоняния могут быть обусловлены центральными и периферическими причинами. Обоняние ухудшается при травмах черепа, рассеянном склерозе, болезни Паркинсона, синдроме Кушинга, синдроме Шегрена, аллергическом рините, после гриппа, при контакте с бензолом. Восприятие вкуса снижается при парезе лицевого нерва, гипотиреозе, синдроме Шегрена, злокачественной опухоли, стоматите. На восприятие вкуса и запаха влияют местные анестетики, ампициллин, амфотеррицин, цитостатики, опиаты, препараты лития, инсектициды, тяжелые металлы.
Икота представляет диагностическое значение при ее упорном рецидивировании. Икота центрального происхождения может быть при энцефалите, энцефаломаляции, опухоли мозга, уремии, спинной сухотке, приеме опиума и опиатов. Икоту периферического характера отмечают при холецистите, подциафрагмальном абсцессе, грыже диафрагмального отверстия диафрагмы, опущении и дилатации желудка. Икота в сочетании с дисфагией позволяет исключать рак дистального отдела пищевода, медиастинит, плеврит, перикардит, опухоль средостения и ворот легкого.
Отрыжка, срыгивание и рвота
Многие механизмы для этих состояний общие.
Отрыжка — прохождение газа из желудка через пищевод в полость рта. Отрыжка, пусть нечасто, но возникает практически у всех. Она представляет диагностическую проблему в случаях ее регулярности и сочетания с другими симптомами. Симптоматическая отрыжка не связана с органическими состояниями. Легко распознается отрыжка после употребления газированных напитков. Отрыжка отмечается у невротических пациентов, заглатывающих воздух во время еды. Но если отрыжка носит гнилостный характер, сочетается с болью в животе и другими признаками, то необходимо пристальное внимание к пациенту. Отрыжка может быть признаком стенозирования выходящего отдела желудка, наличия желудочно-толстокишечного свища, рака желудка или сопровождать холелитиаз.
Срыгивание — регургитация небольших по объему пищевых масс. Может возникать у невропатов, при банальном переедании. Но в практическом плане важнее исключить все органические причины, известные для отрыжки и рвоты.
Рвота — регургитация больших по объему масс желудочного содержимого. Может быть обусловлена поражением многих органов, быть спорадической или хронической. Рвотные массы кислые, чем отличаются от регургитационных. Из осложнений рвоты отмечают гипокалемический метаболический ацидоз, аспирацию и синдром Меллори — Вейсса (разрыв кардиального отдела слизистой оболочки пищевода).
Рвота центрального генеза возможна при повышении внутричерепного давления (часто без предшествующей тошноты) и при гипертоническом кризе, у невропатов, при anorexia nervosa, при мигрени и приступах головокружениях при болезни Меньера, после травм мозга, при опухолях мозга, энцефаломаляции и табетическом кризе, энцефалите и менингите, морской болезни, солнечных или тепловых ударах.
Рвота при инфекционных заболеваниях типична для отитов (новорожденные), инфекциях мочевыводящих путей, пищевых токсикоинфекций.
Необходимо помнить о рвоте при непереносимости ряда продуктов (белок коровьего молока, целиакия).
Острые заболевания органов брюшной полости, такие как холецистит, аппендицит, панкреатит, перитонит, гастроэнтерит, мезентериальная недостаточность (ишемия кишечника) могут быть причиной рвоты. В эту же группу могут быть отнесены случаи рвоты при гепатите и гепатопатии.
Утренняя рвота типична для алкогольного гастрита или повышения внутричерепного давления.
Инфаркт задней стенки левого желудочка (ЛЖ), застойная сердечная недостаточность также весьма часто сопровождаются рвотой.
Рвота метаболического характера типична для первого триместра беременности, уремии, диабетической и церебральной комы, адреногенительного синдрома с потерей солей, аминоацидурии, гиперпаратиреоидизме, болезни Адиссона, а также для ги- пертензионных кризов при феохромоцитоме.
Рвота характерна для лучевого поражения, для интоксикации препаратами наперстянки, солями тяжелых металлов, передозировки витаминов А или D, при приеме эстрогенов, леводопы, цитоста- тиков, некоторых антибиотиков, препаратов железа, калия и многих других.
Жажда (полидипсия) нередко обусловлена психогенными факторами и развивается при неврозах, реже — в дебюте психоза. Первичная полидипсия чаще возникает у молодых женщин, типичным является дневная жажда с колебаниями объема принятой жидкости как по часам, так и по дням. Ночь, в отличие от гипофизарного диабета, обычно проходит спокойно. Осмолярность сыворотки крови снижена. Крайне редко причиной жажды являются первичные органические поражения мозга. Такие лекарственные препараты, как антиконвульсанты, хлорпромазиновые производные и антихолинэргические средства вызывают сухость слизистой оболочки и в результате этого — жажду. Ощущение жажды возникает при дыхании через рот, например, при массивных аденоидах. Жажда отмечается при синдромах Конна, молочно-щелочном и нефронофтизе.
Жажда возникает при изотонической, гипертонической и реже — при гипотонической дегидратации. В терапевтической практике это сахарный и несахарный диабеты, меллитурия неглюкозовой этиологии.
По определению группы экспертов ВОЗ (ISPAD Consensus <…>, 2000) сахарный диабет — это нарушение обмена веществ множественной этиологии, характеризующееся хронической гипергликемией, связанной с недостаточностью секреции инсулина или недостаточной его активностью, или при сочетании этих двух факторов.
Сахарный диабет составляет 50% всех эндокринопатий, занимая 3-е место среди непосредственных причин смерти, уступая только сердечно-сосудистым и онкологическим заболеваниям. До 5% населения болеет сахарным диабетом (diabetus mellitus). Среди детей (0–17 лет) инсулинзависимый сахарный диабет возникает с частотой 0,01–0,2:1000 в России; 1,2:1000 — в США; 3,4:1000 — в Англии. Количество заболевших детей увеличивается каждый год на 16–30 случаев на 100 тыс. детей.
Выделяют инсулинзависимый (диабет 1-го типа) и инсулиннезависимый (диабет 2-го типа, более распространенный) варианты. Понятие «сниженная толерантность к глюкозе» является эквивалентом латентного, субклинического диабета.
Сахарный диабет следует исключить в таких группах и/или при нижеуказанных состояниях:
- группа риска по развитию сахарного диабета (дети от родителей с сахарным диабетом или имеющие сибсов с сахарным диабетом или ожирением). Пациенты с антителами к инсулину, глутамат-декарбоксилазе и к островковым клеткам. Пациенты с иными состояниями, характеризующимися высокой вероятностью развития сахарного диабета (см. ДД различных форм сахарного диабета);
- полиурия, полидипсия, полифагия, уменьшение массы тела или ее быстрое увеличение. Сонливость, ощущение усталости, снижение успеваемости в школе;
- энурез вторичный;
- гнойничковые инфекции кожи, грибковый вагинит;
- повторная рвота, запах ацетона изо рта, кетоацидоз, дыхание Куссмауля, кома, напряжение мышц брюшной стенки, боль в животе, лейкоцитоз;
- ожирение.
Иммунологические критерии инсулинзависимого сахарного диабета или преддиабета (аутоиммунный инсулит):
- аутоантитела к островковым клеткам поджелудочной железы (ІСА);
- антитела к глутамат-декарбоксилазе (GAD);
- антитела к инсулину (ІАА).
Биохимические критерии сахарного диабета:
- повторное слепое определение уровня глюкозы в крови натощак и после пищевой нагрузки, гликозилированного гемоглобина (НbА1C);
- при удовлетворительном состоянии и нормальном уровне глюкозы в крови после обычного приема пищи возможно проведение стандартного теста толерантности к глюкозе с нагрузкой глюкозой из расчета 1,75 г/кг массы тела, но не более 75 г.
О сахарном диабете (1-го или 2-го типа) говорят, если в двух независимо друг от друга изученных пробах венозной крови уровень глюкозы превышает 6,7 ммоль/л (120 мг/дл), или через 2 ч после приема 75 г глюкозы (у детей — 1,75 г/кг массы тела) ее уровень в крови выше 10 ммоль/л (180 мг/дл). О сниженной толерантности к глюкозе говорят, если ее уровень в венозной крови после нагрузки в пределах 6,7–10 ммоль/л (120–180 мг/дл).
Несколько иные и более детализированные критерии диагноза «сахарный диабет» предложены ВОЗ (табл. 2.1).
Таблица 2.1
Диагноз «сахарный диабет» и другие категории гипергликемии по рекомендациям ВОЗ (1999)
| Показатель | Концентрация глюкозы в ммоль/л (мг/дл) | |||
| Цельная кровь | Плазма крови | |||
| венозная | капиллярная | венозная | капиллярная | |
| Сахарный диабет | ||||
| Натощак | >6,1 (>110) | >6,1 (>110) | >7,0 (>126) | >7,0 (>126) |
| Через 2 ч после нагрузки глюкозой или оба показателя | >10,0 (>180) | >11,1 (>200) | >11,1 (>200) | >12,2 (>220) |
| Нарушенная толерантность к глюкозе | ||||
| Натощак (если определяется) | <6,1 (<110) | <6,1 (< 110) | <7,0 (<126) | <7,0 (<126) |
| Через 2 ч после нагрузки глюкозой | >6,7 (>120) и <10,0 (<180) | >7,8 (>140) и <11,1 (<200) | >7,8 (>140) и <11,1 (<200) | >8,9 (>160) и <12,2 (<200) |
| Нарушенная гликемия | ||||
| Натощак | >5,6 (>100) и <6,1 (<110) | >5,6 (>100) и <6,1 (<110) | >6,1 (>110) и <7,0 (<126) | >6,1 (>110) и <7,0 (<126) |
| Через 2 ч (если определяется) | <6,7 (<120) | <7,8 (<140) | <7,8 (<140) | <8,9 (<160) |
Показаниями для проведения теста на толерантность к глюкозе (глюкозный профиль) являются сахарный диабет у родственников (особенно важно изучение глюкозного профиля у ребенка от родителей больных сахарным диабетом), трудно классифицируемые ангиопатия, нейропатия и нарушения зрения, при необъяснимой глюкозурии. Пробу необходимо проводить после 10–12 ч голодания в спокойных условиях, без физической нагрузки. Проведение пробы противопоказано при ранее диагностированном сахарном диабете, нецелесообразно при стрессовых реакциях пациента, при одновременном применении тиазидов, фенотиазина и его производных, кортикостероидов, гормональных контрацептивов, трициклических антидепрессантов, лития, ГИНК2, индометацина, кислоты ацетилсалициловой, блокаторов β-адренорецепторов и, безусловно, антидиабетических препаратов.
В 80% случаев сахарный диабет диагностируют на такой стадии, когда проведение глюкозной нагрузки не обосновано. Диагноз «сахарный диабет» может быть также признан как достоверный, если в двух случайных дневных пробах венозной крови уровень глюкозы превышает 10 ммоль/л.
Сахарный диабет 2-го типа
Сахарный диабет 2-го типа ранее был известен как инсулиннезависимый сахарный диабет или «диабет полных». Последний термин (хотя и не принят в настоящее время) прекрасно отражает конституциональные особенности пациентов: до 85–90% больных на момент установления диагноза страдают ожирением. Если ожирения нет, то рассматривается вопрос о частичном дефекте (β-клеток. Сахарный диабет 2-го типа чаще возникает у взрослых, у которых он и является численно преобладающей формой сахарного диабета. У подростков развивается редко. Но в странах с высокими показателями ожирения сахарный диабет достаточно часто отмечают у детей и подростков. Предрасполагающие факторы — переедание и малоподвижный образ жизни. В анамнезе детей с сахарным диабетом 2-го типа нередко отмечают внутриутробную задержку развития. Последняя поэтапно влечет за собой нарушения постнатального питания, ожирение, гиперинсулинизм, развитие резистентности к инсулину, затем, вполне логично, появляется диабет. Высок удельный вес генетических факторов. Монозиготные близнецы, в отличие от диабета 1-го типа, в 100% случаев конкордантны по сахарному диабету 2-го типа. Вероятен положительный семейный анамнез, особенно если проводить у родителей стандартный глюкозотолерантный тест. В Японии сахарный диабет 2-го типа диагностируют в 8–9 раз чаще, чем 1-го типа. Высокому риску по развитию сахарного диабета 2-го типа подвержены коренные американцы и канадцы, афроамериканцы, мексиканские американцы, индийцы (даже после их переезда в Европу), австралийские аборигены.
Клиническая картина сахарного диабета 2-го типа, как правило, не резко выражена. По нашему мнению, острое тяжелое начало сахарного диабета 2-го типа свидетельствует больше в пользу упущенного врачом периода дебюта, чем об истинном начале болезни. Кетоацидоз даже без применения инсулина развивается очень редко, в основном при стрессах или инфекционных заболеваниях. Основные ДД-отличия сахарного диабета 2-го типа от 1-го типа приведены в табл. 2.2.
Таблица 2.2
ДД-признаки сахарного диабета 1 -го и 2-го типа
| Характеристика | Тип | |
| 1-й | 2-й | |
| Возраст | Все периоды детства | Пубертатный и старше |
| Начало | Часто острое, быстрое прогрессирование | Чаще медленное, нерезкое; реже — острое |
| Инсулиновая зависимость | Постоянная, общая, тяжелая | Не всегда. Инсулин требуется при неэффективности оральных препаратов |
| Секреция инсулина | Отсутствует или очень незначительная | Различна |
| Чувствительность к инсулину | Нормальная | Снижена |
| Генетика | Полигенный характер | Полигенный характер |
| Раса/этнические особенности | Все группы, частота заболеваемости широко варьирует | Некоторые этнические группы особенно подвержены риску заболевания |
| Частота (% сахарного диабета у людей молодого возраста) | Обычно 90% и выше | В большинстве стран — ниже 10%; в Японии — около 80% |
| Аутоиммунный процесс* | Да | Нет |
| Кетоз | Типичен | Не характерен |
| Ожирение | Нет | Более чем у 80% |
| Акантоз | Нет | Типичен |
*Едва ли не основной ДД-признак. Если при сахарном диабете 2-го типа у пациента нет ожирения, то обязательно следует провести исследование на наличие аутоантител к островковым клеткам.
Первым и основным методом лечения сахарного диабета 2-го типа (в отличие от 1-го типа) является нормализация массы тела за счет снижения энергетической ценности питания, физические упражнения и здоровый образ жизни.
О сахарном диабете 2-го типа следует помнить, наблюдая детей с избыточной массой тела и особенно подростков. В некоторых этнических группах целесообразно проведение периодических скрининговых определений уровня глюкозы в крови или (что проще и дешевле при массовых исследованиях) глюкозы мочи. В Японии из-за высокой вероятности развития сахарного диабета 2-го типа у детей в школах проводят регулярный скрининг на основании определения глюкозурии.
Инсулинзависимый сахарный диабет (1-го типа) характеризуется постепенным истощением секреции инсулина и склонностью к кетоацидозу. На фоне генетической предрасположенности (HLA-B8, -В15, -DR4, -Dw3) такие вирусные инфекции, как эпидемический паротит, краснуха, коксаки В4 и некоторые другие, а также токсины вызывают лимфоплазмоцитарный клинически стертый панкреатит с активацией аутоиммунного ответа и гибелью клеток островкового аппарата. Подтип ІА практически всегда начинается еще в детском возрасте, половыхразличий не выявлено. Антитела к островковому аппарату выявляют спорадически и в невысоком титре. При подтипе 1В В-лимфоциты начинают вырабатывать цитотоксические аутоантитела к клеткам островкового аппарата. Заболевание начинается обычно в возрасте после 30–35 лет, преимущественно у женщин, часто сочетается с тиреоидитом Хашимото, пернициозной анемией и базедовой болезнью. У таких пациентов значительно чаще, чем в популяции, выявляют НLАтипа В8, DR4 и Dw3.
Генетические дефекты функционирования бета-клеток, ранее известные как юношеский инсулинзависимый диабет (MODY— maturity—onset diabetes in the young)
При MODY-подтипах сахарного диабета гипогликемия развивается в основном у подростков (табл. 2.3). Заболевание передается а/д. Диабет, по крайней мере в течение ближайших 5 лет после установления диагноза, протекает как инсулиннезависимый. Секреция инсулина нарушена. Тяжело кетоза не отмечают. Некоторые формы MODY-подтипов могут начинаться с тяжелых осмотических расстройств, что служит причиной ошибочной диагностики сахарного диабета 1-го типа. Однако в противоположность сахарному диабету 1-го типа отмечается положительный семейный анамнез (а/д тип передачи), не бывает тяжелого кетоза и с применением инсулина в низкой дозе достигается хороший метаболический контроль.
Таблица 2.3
Классификация трех наиболее частых генетических дефектов без поражения бета-клеток, протекающих с явлениями сахарного диабета
| Дефект гена | HNF-1 альфа* | Глюкокиназа «глюкозо-чувствительный ген» | HNF-4 альфа* |
| Локализация на хромосоме | 12q | 7р | 20q |
| Мутации | Много | Много | Много |
| Частота каждого из подтипов среди всех форм MODY- диабета в больших сериях | 65% | 10–15% | 5% |
| Начало гипергликемии | Пубертатный и старше | Внутриутробно, врожденная гипергликемия | Пубертатный, юношеский возраст |
| Тяжесть гипергликемии | Прогрессирующая вплоть до тяжелой | Средняя, персистирующая | Прогрессирует до тяжелой |
| Патофизиология | Дисфункция бета-клеток | Дисфункция бета-клеток, нарушение толерантности к глюкозе | Дисфункция бета-клеток |
| Микрососудистые осложнения | Часто | Редко | Часто |
*HNF — hepatic nuclear trascription factor.
Наряду с выше охарактеризованными тремя подтипами MODY-диабета есть еще два варианта. Один из них связан с дефектами фактора содействия инсулину (IPF-1), передается а/p типом и в гомозиготном состоянии сочетается с агенезией поджелудочной железы. Другой (пятый вариант) — связан с HNF-16eTra и сочетается с кистозными почками. Причем почки при рождении не изменены.
Характер наблюдения пациента определяется генным дефектом. Дети и подростки с мутациями глюкокиназы или фактора транскрипции для хорошего контроля гликемии должны получать только рациональное питание. У некоторых детей с мутациями фактора транскрипции прогрессирующий дефицит бета-клеток в итоге приводит к необходимости приема гипогликемизирующих препаратов. HNF-1 альфа варианты могут контролировать сульфонилмочевиной в низких дозах (однако риск развития гипогликемии при этом достаточно высок). Около 30% детей с мутациями HNF-1 альфа уже в зрелом возрасте нуждаются в инъекциях инсулина.
Сахарный диабет в сочетании с ожирением и гипертрихозом у женщин носит название синдрома Ашара — Тьера (триада Морганьи). ДД проводится с синдромом Морганьи (внутренний гиперостоз лобной кости, вирилизация, гирсутизм, ожирение).
Сахарный диабет со сниженной толерантностью к глюкозе взрослого типа. Гломерулосклероз и тубулярная атрофия, значительно уменьшающие продолжительность жизни. Возможны сколиоз, гиперостоз лобной кости, гипогонадизм.
С сахарным диабетом протекает и синдром Альстрема — Ольсена — Хальгрена. А/p тип. Для него типичны ожирение, сахарный диабет, нейросенсорная тугоухость (развивается на первом десятилетии жизни), гипогонадизм, тапеторетинальная дегенерация с гемералопией, катаракта (обычно заднего полюса хрусталика), ювенильная глаукома, нистагм, плохое зрение с рождения, гиперурикемия, гипераминоацидурия, гипогенитализм, аномалии скелета.
Диабет беременных — снижение толерантности к глюкозе, выявленное в период беременности. У детей, рожденных от таких матерей, выше риск развития перинатальных осложнений, отмечается обратимая гипертрофическая (инфильтративная) кардиомиопатия с возможной обструкцией выносного тракта, в неонатальный период — гипогликемические состояния. У матерей через 5–8 лет после беременности обычно развивается типичный сахарный диабет.
Вторичный сахарный диабет развивается при поражении поджелудочной железы (резекция, хронический панкреатит, гемохроматоз, муковисцидоз), при эндокринных заболеваниях (болезнь Кушинга, соматостатома, глюкагонома, феохромоцетома, акромегалия, гипертиреоз, синдром Кона), при генетических синдромах (синдром Прадера — Вилли, атаксия Фридрейха), при нарушениях рецепторного аппарата (инсулиновая резистентность при черном акантозе (acantosis nigra) и осложнениях медикаментозной терапии (стероиды, антиовуляторные противозаточные средства, трициклические антидепрессанты).
Традиционно диагностику сахарного диабета врач начинаете исследования по поводу глюкозурии. Однако при сахарном диабете глюкозурия начинается при уровне глюкозы в крови 11,1–13,3 ммоль/л (200–240 мг/дл). При этом уровне в проксимальных канальцах возможна полная резорбция глюкозы. У пациентов пожилого возраста с нарушенными функциями почек «глюкозовый порог» еще выше.
Очень редко возникает наследственно обусловленная недостаточность резорбтивной функции почечных канальцев, вследствие чего выявляют стабильную глюкозурию без развития сахарного диабета. Транзиторная глюкозурия может появиться в последний триместр у 10–20% беременных, она обусловлена значительным увеличением кровотока через почки. В отличие от сахарного диабета, уровень глюкозы в крови при ней не меняется, проба с глюкозной нагрузкой нормальная.
На другие формы меллитурии, не обусловленные наличием глюкозы в моче, а связанные с потерей других сахаров, приходится всего 1%. Еще в детском возрасте выявляют пентозурию (прогностически благоприятную), фруктозурию, протекающую в двух формах: благоприятная эссенциальная фруктозурия и тяжелая а/p передающаяся недостаточность фруктозо-1-фосфат-альдолазы. Галактозурия наступает при галактоземии, обусловленной дефицитом галактоз- 1-фосфат-уридилтрансферазы. Крайне редко развивается мальтозурия и сахарозурия.
Несахарный диабет (diabetus insipidus) как первичное заболевание является расстройством гипофизарно-гипоталамической регуляции секреции антидиуретического гормона. Поступление анти- диуретического гормона (вазопрессина) в кровь регулируется афферентными импульсами от осмо- и волюморецепторов. В зависимости от степени выраженности дефекта за сутки выделяется от 3 до 40 л мочи с низким удельным весом (1001–1005). Осмолярность сыворотки крови высокая. Выраженная гипернатриемия. Клинически заболевание обычно проявляется остро, с полиурии и полидипсии. Пациент просыпается даже по ночам, чтобы попить и посетить туалет. Пока удается сохранять равновесие между принятой и выделенной жидкостью, состояние больных меняется незначительно, но при малейшей задержке с поступлением воды быстро развивается обезвоженность вплоть до повышения температуры, гемоконцентрации и шока с потерей сознания. Вот почему проба с ограничением жидкости очень опасна, применять ее можно только в стационаре, она противопоказана при подозрении на несахарное мочеизнурение.
Этиология несахарного диабета различная. В 95% случаев это спорадические случаи, в 5% — а/д передающаяся селективная неполноценность вазопрессин-синтезирующих нейронов. Онкогематологи отмечают несахарный диабет при гистиоцитозе (20–50% всех больных) или при лейкозе. Несахарный диабет может развиться как исход энцефалита, туберкулеза, актиномикоза, саркоидоза, глиом зрительного нерва, краниофарингеоме, после перелома основания черепа, оперативных вмешательств вблизи гипофиза или гипотоламуса. Мы зарегистрировали случай несахарного диабета у девочки после перелома длинной трубчатой кости, что могло повлечь за собой жировую микроэмболию нейрогипофиза. Установить причину несахарного диабета при первых визитах к врачу малореально. Так, у 50% больных с опухолевой причиной несахарного диабета опухоли были выявлены только через год после начала мочеизнурения, а у 25% — через 4 года. У существенной части пациентов с так называемой идиопатической формой несахарного диабета выявлена его аутоиммунная природа: наличие антител, реагирующих с вазопрессин-синтезирующими клетками.
В противоположность пациентам с психогенной полидипсией, у больных с несахарным мочеизнурением гипотоничность мочи сохраняется и после водной паузы. Жизненно важно для пациента, чтобы масса его тела во время проведения водной паузы уменьшалась не более чем на 5%. Введение гипертонического раствора хлористого натрия этим больным на объеме мочи не отражается. При подкожной инъекции 5 ЕД вазопрессина у пациентов с истинным несахарным диабетом тут же уменьшается объем выделяемой мочи и снижается осмолярность сыворотки.
С несахарным диабетом сочетается:
- DIDMOAD синдром —- синдром Вольфрама (Diabetus Insipidus, Diabetus Mellitus, Optic Atrophy, Deafness). А/p. Проявляется сочетанием сахарного и несахарного диабета + постепенно нарастающей атрофией зрительного нерва + медленно прогрессирующей двухсторонней внутренней тугоухостью + частой эктазией мочевыводящих путей. Несахарный диабет и нарушение слуха надо выявлять целенаправленно. Манифестирует обычно до 15-летнего возраста;
- диэнцефальный синдром (астроцитома [реже — спонгиобластома или эпендимома] + кахексия + нистагм горизонтальный + диплопия + эйфория). Обычно манифестирует в возрасте от 6 мес до 3 лет;
- гипоталамический синдром.
Нефрогенный несахарный диабет (вазопрессин-резистентный несахарный диабет) развивается, как это следует из названия, при рефрактерности почечных канальцев по отношению к антидиуретическому гормону. Врожденные формы передаются сцепленно с Х-хромосомой рецессивно. Проявляются с рождения массивной полиурией и жаждой. Если заболевание диагностируется, то прогноз достаточно благоприятен. При уточнении семейного анамнеза оказывается, что в семьях нередки случаи смерти новорожденных, что свидетельствует о вероятной высокой частоте недиагностированных форм. Приобретенный несахарный диабет развивается как последствие токсического действия амфотеррицина, колхицина, винбластина и солей лития. Нефросклероз как последствие хронического пиелонефрита, рефлюкса, острого тубулярного некроза, состояния после трансплантации почек, а также гипокалиемия (в том числе синдром Кона) и хроническая гиперкальциемия (гиперпаратиреоидизм) также обусловливают развитие нефрогенного несахарного диабета. Редко его причинами являются серповидноклеточная анемия, амилоидоз, множественная миелома и болезнь Шегрена.
Прежде чем обсуждать диагностическое значение нарушений сна, врачу следует поинтересоваться профессией посетителя (сменная работа), условиями сна (шумность и уединенность комнаты, общая кровать с человеком, страдающим храпом или двигательными расстройствами, предшествующие сну кофепития, обильное употребление кока-колы, просмотр телепередач и т. п.). Среди детей самые частые причины нарушения сна — невротические реакции и желание собравшихся вечером с работы взрослых «потютюшкать» дитятю.
Первичная инсомния обычно не находит объяснений. В 90% нарушения сна обусловлены психическими заболеваниями. Чаще это затруднения засыпания, поверхностный неглубокий сон. Реактивные нарушения сна возникают у возбудимых личностей при невозможности уйти от дневных проблем, при панических атаках. В ДД необходимо учитывать органические заболевания мозга (промежуточного и ствола), гипоталамический синдром, мукополисахаридоз III типа. У новорожденных расстройства сна выявляют при синдроме отмены, то есть если мать в период беременности употребляла вещества, способные вызывать аддитивные расстройства (алкоголь, табак, барбитураты, героин, метадон и др.). Синдром проявляется дрожью новорожденного, раздражительностью, гиперактивностью, коротким поверхностным сном, пронзительным криком. Возможны судороги. Вегетативная симптоматика проявляется в виде чихания, зевоты, потливости, лихорадки.
Расстройства сна свойственны таким соматическим заболеваниям, как сердечная недостаточность с диспноэ, ортопноэ, никтурией; гипертрофия предстательной железы (поллакиурия и соответственно необходимость частого посещения туалета); патология бронхов (сильный, особенно спастического характера кашель); гипертиреоз; опухоли, сопровождающиеся выраженным болевым синдромом; рефлюкс-эзофагит с забросом пищи в пищевод в лежачем положении со жгучей загрудинной болью. Зуд различной этиологии, периодические миоклонии, карпальный синдром, мигрень и невриты также не располагают к умиротворению. Нарушения сна отмечают и при так называемом синдроме «restless-legg» — частые беспорядочные подергивания конечностей во сне, причины которых не установлены (в нашей практике мы наблюдали это состояние при передозировке галоперидола). Другие исследователи в ряде случаев указывают на связь этого синдрома с сахарным диабетом, алкоголизмом (полинейропатия), со скрытыми отеками, железодефицитными состояниями.
Частые панические пробуждения из-за апноэ отмечают при поражении диафрагмы. Само по себе часто повторяющееся апноэ невротизирует пациента, приводит к повышению АД и гипертрофии ЛЖ. Гипертрофия миокарда предрасполагает к нарушениям ритма, последнее вновь приводит к пробуждениям, и круг замыкается. Клиническим признаком апноэ является храп. Могут отмечаться беспокойство во сне, шумное дыхание, потливость, энурез, ночные страхи, утренняя разбитость, сухость или неприятный вкус во рту. Достаточно часто синдром апноэ во сне возникает у людей с избыточной массой тела. Причины этого не известны, сужение воздухоносных путей жировыми отложениями вокруг трахеи не доказано.
Обструктивные апноэ во сне могут быть обусловлены анатомическими дефектами верхних дыхательных путей, в том числе аномалиями носа, провисающим маятникообразным небом, дополнительными складками слизистой оболочки ротоглотки, гипертрофией миндалин, аденоидами, стридором, микрогнатией, аномаладой Робина, ретрогнатией, макроглоссией (синдром Видемана — Беквита). Необходимо исключить новообразования трахеи, мягких тканей шеи, диафрагмы рта. Следует помнить о поражениях межреберных мышц и диафрагмы (миозит, миастения, миопатия). Периоды апноэ возникают при новообразованиях задней черепной ямки и ствола мозга, инфарктах ствола и полушарий головного мозга, бульбарного полиомиелита.
Центрального характера апноэ возникает при грубой органической патологии головного мозга, при этом может быть полностью утеряна функция автоматической регуляции дыхания. Оно осуществляется лишь под контролем коры мозга, что отсутствует во сне. Состояние известно как «синдром проклятья Ундины» (неверный муж Ундины в результате проклятия был лишен автоматических функций, после чего и умер во сне) и определяется как первичная альвеолярная гиповентиляция. Причиной могут быть различные нарушения ствола мозга (воспаление, инфильтрация, опухоли, но иногда причину нельзя установить). Состояние проявляется склонностью к апноэ, цианозу, сомноленции, особенно в период пробуждения. Неожиданно возникают неправильные по ритму гипервентиляционные движения. Чувствительность дыхательного центра к углекислому газу снижена. Как результат гипоксемии развивается полиглобулия. Формируется застойная кардиомиопатия. В ДД синдрома необходимо исключить деструктивные заболевания ЦНС, поражения легких, сердца, грудной клетки.
Периоды апноэ возникают при глутарацидурии II типа, изолированной некетоацидотической гиперглицинемии, синдромах Юбера, Микити — Вильсона, Пиквика, фенциклидиновой фетопатии, а у детей и при коклюше как эквивалент реприз.
Жуткие ощущения падения и проваливания, после которых пациент просыпается в холодном поту, отмечаются у молодых эмоциональных людей, при аритмии, при приеме блокаторов (β-адренорецепторов. Сон нарушают прием глюкокортикоидов, антидепрессантов, амфетамина, некоторых антиастматических препаратов, как и резкая отмена снотворных средств после их длительного применения. К достаточно редким состояниям, протекающим с бессонницей, относится фатальная семейная инсомния.
Патологическая сонливость возникает при опухолях гипоталамуса и 3-го желудочка, а также у идиотов.
Необоримая сонливость в самых неожиданных местах с одновременной мышечной гипотонией, катаплексией, гипногенными галлюцинациями ха- растеризуется как нарколепсия и требует обязательной записи электроэнцефалограммы (ЭЭГ). Прежде всего необходимо исключить вторичные формы при эпидемическом энцефалите, цереброспинальном сифилисе, опухолях дна 3-го желудочка, энцефаломиелите. Первичная форма передается а/д с неполной пенетрантностью. Для состояния характерны многократно за день развивающиеся короткие необоримые припадки сонливости, галлюцинаторные переживания, гипногенные галлюцинации, существенные потери мышечного тонуса с эмоциональными всплесками (атака смеха) без потери сознания. У пациентов возникает путаница ритма дня и ночи. Возможны диффузный гипергидроз, нарушение саливации, полицитемия, склонность к гипогликемии, ожирение, нерезко выраженный несахарный диабет, гипогенитализм.
Летаргический энцефалит Экономо в современных условиях возникает достаточно редко. Но в связи с возросшей миграцией населения следует помнить о трипаносомозе и клещевом энцефалите.
Повышенная сонливость в сочетании с усиленным аппетитом типична для синдромов Пиквика (ожирение, апноэ, компенсаторная полицитемия, легочная гипертензия (ЛГ), дневная сонливость) и Клейне — Левина (периодическое непроизвольное засыпание, чередующееся с приступами гиперфагии).
С приступами летаргии протекает аргининсукцинатурия — а/p состояние, обусловленное нарушением в цикле превращений мочевой кислоты из-за дефицита аргининсукцинатлиазы с повышением аргининянтарной кислоты в крови, последнее приводит к гипераммониемии. Формы новорожденных и детская заканчиваются фатально, форма хроническая может развиваться у подростков и взрослых. Характеризуется умственной отсталостью, часто — с судорожными припадками, интермиттирующей атаксией и летаргией. Симптомы усиливаются при инфекционных заболеваниях или при повышенном употреблении белка. Аргининсукцинатемия, протеинзависимые гипераммониемия, цитрулинемия. Выраженная аргининсукцинат- и цитрулинурия. В эритроцитах определяется снижение уровня аргининсукцинатлиазы.
Сонливость развивается при гипотиреозе, приеме противоэпилептических препаратов, нейролептиков и седативных средств.
Сердечный ритм (сердцебиение) обычно не воспринимается здоровым человеком, за исключением периодов сильных волнений или физических нагрузок. Невротизированные личности воспринимают обычные сокращения сердца как усиленные или аритмичные. Эти ощущения возникают при «прислушивании» к себе, при укладывании в кровать, иногда после неосторожных высказываний врача.
В противоположность этому сердечные сокращения ощущаются пациентом при нарушениях проводимости и возбудимости миокарда, увеличении ударного объема при пролапсе клапанов, тиреотоксикозе, тяжелых анемиях, высокой лихорадке, недостаточности митрального и/или аортального клапанов. Приступообразное сердцебиение может возникать при феохромоцитомах и гипогликемии. Функциональные расстройства, такие как «солдатское сердце» и синдром Эффорта, то есть психосоматические симптомокомплексы с вегетативной дисфункцией также характеризуются аффективными приступами сердцебиения и одышки.
Кашель — защитная реакция, направленная на очищение трахеобронхиального дерева от обильного секрета или ингалированных чужеродных тел. Кашель (покашливание) иногда возникает и у здоровых людей. Но никогда у здоровых лиц кашель не длится более 3 нед. При независимом изучении с участием больших групп пациентов установлено, что 50% хронически кашляющих — курильщики или экс-курильщики. По характеру отделения секрета выделяют два вида кашля.
Продуктивным называется кашель при наличии бронхиального секрета. Именно такой вид кашля является биологически оправданным защитным механизмом, возникает при острых и хронических воспалительных заболеваниях бронхов и легких. Медикаментозное подавление кашлевого рефлекса в этих случаях нецелесообразно.
Непродуктивный кашель развивается при химическом, механическом или термическом раздражении дыхательных путей и часто полностью прекращается после устранения раздражителя. В качестве механических причин могут выступать инородные тела, давление на воздухоносные пути опухолей, метастазов, аневризматически расширенной аорты, а также подтягивание легочной паренхимы при фиброзирующих процессах (ателектаз, фиброз).
Мелкое, частое покашливание типично для раздражения плевры.
Остро развивающийся кашель типичен для вирусных, реже — бактериальных поражений верхних дыхательных путей, наличия в них инородных тел.
Хронический кашель характерен при хроническом бронхите, бронхоэктазе, БА, туберкулезе, опухолях легких. Среди взрослых у 72% хронически кашляющих выявляют аденоидные разрастания на задней стенке глотки, аллергический назофарингит или орофарингит. Поэтому при любом кашле всегда необходимо оценить прежде всего состояние верхних дыхательных путей.
Важное значение имеет характер кашлевого толчка. «Лающий» кашель патогномоничен для эпиглоттита или ларингита. Приступообразный кашель со стридорозным вдохом типичен для коклюша, ночной кашель — свидетель сердечной недостаточности. Утренний кашель характерен для курильщиков и при хроническом бронхите с бронхоэктазами. Кашель, регулярно повторяющийся во время или сразу же после еды, может свидетельствовать о грыже пищеводного отверстия диафрагмы, дивертикулах пищевода или неврогенных расстройствах. Такой же кашель, но с отхождением пенистой мокроты, у детей раннего возраста типичен для пищеводно-бронхиальных свищей. Пароксизмальный кашель может за счет повышения внутригрудного давления вести к пневмотораксу, кровоизлияниям в конъюнктиву или мозг, а за счет резкого уменьшения возврата крови в ДЖ с уменьшением, соответственно, выброса, приводить к обморочным состояниям. Дополнительные симптомы важны для уточнения причин кашля. Например, ретростернальная боль при кашле типична для вирусного трахеобронхита, слабость и уменьшение массы тела сопутствуют кашлю при опухолях и туберкулезе.
Мокрота — естественный продукт трахеобронхиального дерева. В норме за сутки бокаловидные клетки и слизистые железы образуют 100–150 мл мокроты. Этот объем дополняется физиологическим клеточным детритом и альвеолярной жидкостью. При ненарушенном цилиарном клиренсе мокрота поступает в гипофаринкс и непроизвольно проглатывается.
Избыточное или, наоборот, скудное образование мокроты выявляют при многих заболеваниях, а ее характер (объем, запах, цвет, особенно подкрепленный инструментальными и лабораторными исследованиями) позволяет в ряде случаев установить диагноз.
Мокрота ржавого цвета характерна при пневмококковых пневмониях и застойном легком. Студневидная красная мокрота по типу малинового желе отделяется при распадающемся бронхиальном раке. Зловонная мокрота типична для абсцедируюшей пневмонии, абсцесса легких, бронхоэктазов, распадающихся опухолей. Обильная пенистная белая мокрота является результатом альвеолярного рака, а обильная, но розовая мокрота возникает при отеке легкого. Обильная мокрота, отделяющаяся по утрам полным ртом и расслаивающаяся при стоянии на 3 слоя, типична для далеко зашедшей бронхоэктатической болезни. Зелено-желтая гнойная мокрота сопровождает течение бронхоэктазов, муковисцидоза, распадающихся опухолей, прогрессирующего туберкулеза легких.
Отделение обильной кровянистой мокроты или же свежей крови обозначается как гемоптиз. Массивная кровопотеря с мокротой возможна при бронхиальных карциномах, аррозиях сосудов в бронхоэктазах, туберкулезе. Правильный диагноз в 90% случаев устанавливают бронхоскопически (с возможной биопсией, цитологическими и бактериологическими исследованиями). Для ДД с кровотечениями из пищевода и желудка (гематемезис) следует учитывать, что гематемезиз типичен для пациентов с циррозом печени, раком пищевода и желудка, сопровождается рвотными движениями, pH-реакция излившейся крови кислая или нейтральная (при гемоптизе — щелочная), типичны общая анемия и наличие крови в стуле.
Зуд. Дать четкое определение этому ощущению сложно, но все знают, что это такое. Может быть обусловлен раздражением свободных нервных окончаний эпидермиса либераторами гистамина, энзимами бактерий, грибами или иметь центральный характер. Сильный и длительный зуд любой этиологии сопровождается расчесыванием кожи, инфицированием расчесов, вторичной лихенификацией. Иногда зуд объясняют изменениями нервной системы in loco: пролиферация кожных нервов, шванноподобные разрастания, дегенерация нервных окончаний. Зуд раздражает пациента, лишает его сна, изменяет психику.
Локальный зуд обычно свидетельствует о дерматологических заболеваниях. Это могут быть атопический и контактный дерматит, экзема, нейродермит, псориаз, лишаи, стрепто- и стафилодермия, вторичный сифилис, кандидоз, чесотка. Хотя понятия местного и генерализованного зуда, дерматологического и системного во многом сходны. Генерализованный зуд может быть психогенным, паразитарным (особенно на волосистой части головы), сенильным, аллергического характера с изменениями кожи или без них, уремическим, холестатическим, при железодефицитных состояниях, гипер- и гипотиреозе, подагре, сахарном диабете, карциноидном синдроме, болезни Ходжкина и неходжкинских лимфомах, истинной полицитемии, лейкозе, мастоцитозе, амилоидозе. Зуд отмечают при узелковом пруриго, пойкилодерматомиозите, молочно-щелочном синдроме.
Важно определить локализацию зуда, время его наибольшей выраженности. Например, при чесотке сильный зуд появляется преимущественно ночью или в тепле, главным образом на пальцах, запястьях, подмышках, вокруг пупка и на половых органах. Зуд обусловлен сенсибилизацией хозяина находящимися в коже мертвыми клещами, яйцами, экскрементами. Зуд продолжается до той поры, пока по мере роста кожи все эти частицы не выносятся в роговой слой и буквально стряхиваются с человека.
Действительно, при диагностике пациента с жалобами на зуд не в последнюю очередь надо исключить такие трансмиссивные состояния, как чесотка и педикулез. Хотя заболевания ассоциируются прежде всего с низким социальным статусом, никто не застрахован от них.
Чесотка — один из наиболее простых и одновременно один из самых сложных диагнозов среди всех дерматологических расстройств (рис. 2.2).
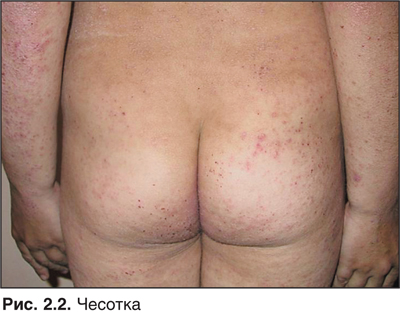
По всему миру чесоткой болеют не менее 300 млн человек. Каждый период социально-экономического спада значительно увеличивает число пораженных.
В северных широтах чесотку чаще регистрируют поздней осенью и зимой. В европейских условиях в 75% случаев в семье болен только один человек. Большинство непораженных в такой семье — люди в возрасте старше 15 лет, а 30% больных — дети школьного возраста. У взрослых чесотка весьма часто передается при половых контактах.
При обследовании квартир, где проживают пораженные, чесоточные клещи в 44% случаев были найдены на домашних вещах, из них 64% были живыми.
Чесотка передается только при тесном контакте. Сведения о вспышке зуда в семьях или у полового партнера — важнейшее свидетельство в пользу диагноза «чесотка».
Чесоточный клещ передвигается по коже со скоростью 2,5 см/мин. Самец, как правило, остается на поверхности кожи, а оплодотворенная самка внедряется в нее, где она и живет около 30 дней. Через несколько часов после вбуравливания в кожу она начинает откладывать по 2–3 яйца в сутки, каждое из которых созревает через 3–4 дня. Вышедшая из него личинка (ларва) поднимается к поверхности кожи и через стадию нимфы созревает в клеща. Незрелые формы клеща образуют так называемые ларвальные папулы, беспорядочно разбросанные по коже.
Цикл созревания завершается через 14–17 дней. Но из всех отложенных яиц только 10% созревают до взрослого клеща.
Первичное заражение в течение нескольких недель никак не проявляется. Длительность светлого промежутка без зуда зависит от числа клещей и сенсибилизации хозяина. В северном климате наибольшее число клещей выявляется на 18–150-й день после инокуляции и затем быстро снижается. При повторном заражении уже благополучно пролеченных субъектов беззудный период длится около суток, число клещей значительно меньше, чем при первичном заражении. Но число клещей на хозяине ни при первичном, ни при вторичном заражении не коррелирует с выраженностью симптомов. Абсолютного иммунитета к чесотке нет.
Клиническая картина, где ведущим и определяющим является зуд, складывается из расстройств, вызванных собственно чесоточным клещом и наличием вторичной или фоновой инфекции (табл. 2.4). Чесотка может осложнить течение любого дерматоза или дерматита, клиницисту следует помнить об этом, особенно если пациент со стабильно текущим дерматозом начинает жаловаться на сильный зуд, особенно ночной.
Таблица 2.4
Клинические проявления чесотки
| Тип чесотки и морфологический характер элементов | Локализация | Фактор риска | Среднее количество клещей | Осложнение | ДД |
| Первичные элементы: Ходы. Папулы. Пустулы. Везикулы. Узелки |
Межпальцевые промежутки; лучезапястный сустав; средняя треть живота, поясница; нижняя часть ягодиц; за ушами; под молочными железами; вокруг сосков; пенис. У детей часто — шея, голова, лицо, межягодичная складка, ладони, подошвы | Тесный физический контакт, включая сексуальный | 6 | Экземоподобные изменения; вторичная бактериальная инфекция | Контактный дерматит; атопический дерматит; себоррейный дерматит; нейродермит; медикаментозная аллергия; укусы насекомых; папулезная крапивница; вторичный зуд при системных заболеваниях; гистиоцитоз; лимфома; сифилис; хроническая пиодермия; васкулиты |
| Вторичные поражения: Экскориации. Гипо- и гиперпигментация. Пиодермия. Рубцы, шрамы | |||||
| Норвежская чесотка: Гиперкератоз. Папулы. Экскориации. Корки. Дистрофия ногтей | Подмышки, конечности, ногти, пах. Затылок, ладони, подошвы. Возможна генерализация | Иммуносупрессия. СПИД. Деменция, дебильность. Алкоголизм и плохие гигиенические условия. Спинная сухотка. Сирингомиемия. Синдром Дауна. Малегномы, Синдром Блума. Васкулиты. Сахарный диабет. Поражения сосудов головного мозга | До 2 млн | Как и при классическом варианте | Контактный дерматит; себоррейный дерматит; псориаз; болезнь Дарье; врожденная пахионихия, бленнфагическая кератодермия |
| Собачья чесотка: Папулы. Везикулы. Ходов нет | Туловище, руки, живот. Реже — лицо или половые органы | Контакт со щенками, а также с лошадьми, свиньями, козами, овцами, ламами, кошками | 1–100 | Экзема. Папулезный дерматит |
Первичное поражение чесоткой
Ходы клещей — патогномоничный признак. Выглядят как беловатые валики длиной 0,3–10 мм. Экскориации могут полностью маскировать их и другие первичные элементы. Ходы плохо видны у больных, живущих в жарком влажном климате.
Папулы. Чаще — мелкие, эритематозные, с расчесами. Их существование — результат миграции личинки или нимфы клеща в верхние слои кожи.
Везикулы — типичный элемент для чесотки детского возраста. Расположены обычно на ладонях и подошвах.
Узелки проявляются только при длительном заражении (около года) чесоткой. Чесоточные клещи внутри узелков не выявляются. Гистологически узелок — скопление лимфоидных клеток на месте укуса клеща и представляет собой, видимо, гиперергическую реакцию хозяина.
Кисти — чаще всего место, где проявляется клещевая инвазия (инфестация). При этом элементы поражения в основном располагаются в межпальцевых промежутках и на боковых поверхностях пальцев. Очень часто поражаются сгибательные поверхности лучезапястных суставов, область локтя и передняя подмышечная область, ягодицы, ягодично-бедренная складка. Для женщин типично наличие ходов и эритемы на груди и вокруг пупка, а для мужчин — на половом члене с образованием узелков.
Локализация кожных элементов при чесотке у детей отличается от таковой у взрослых. Преимущественно поражаются ладони, подошвы голова, затылок.
Норвежская чесотка (панцирная). Протекает с гиперкератозом у пациентов с иммуносупрессией, например, после пересадки почки или при СПИДе. Корки при этом варианте течения чесотки обусловлены бактериальной инфекцией у людей, у которых чесотка длительное время была не диагностирована. Корки располагаются на конечностях (особенно на ладонях и подошвах), ягодицах и ушных раковинах. Число клещей на одном хозяине может достигать 2 млн. Из-за крайне низкой степени сенсибилизации симптоматика минимальна, каждый такой пациент в высшей степени контагиозен. Описаны случаи заражения от одного больного нескольких сотен здоровых людей, в том числе и работников здравоохранения.
Собачья чесотка. Scabies sarcoptes (canis) обычно поражает собак, но может вызвать зудящие папулы у человека. Очаги располагаются на участках тела, непосредственно соприкасавшихся с больным животным. S. sarcoptes не способен завершить полный жизненный цикл в коже человека. Поэтому заболевание самоизлечивается после прекращения контакта с больным животным. Наряду с собаками человек может заразиться от кошек, верблюдов, свиней, овец, лам, коз, лис и др. Время от заражения до клинической манифестации составляет 1–10 дней.
Вторичная и сопутствующая инфекция часто осложняет чесотку. Чесотку необходимо исключать при каждом случае рецидивирующей пиодермии. Последняя обычно вызывается Streptococcus pyogenes и Staphylococcus aureus. Вторичная инфекция особенно ярко проявляется в тропиках. Описаны случаи постстрептококковых гломерулонефритов как последствия пиодермии при чесотке.
Поскольку чесотка может быть сексуально трансмиссивным заболеванием, то одновременно с диагностикой и лечением чесотки у подростков и взрослых необходимо исключить сифилис и гонорею.
Импотенция — неспособность достигнуть степени эрекции, достаточной для введения полового члена во влагалище здоровой женщины. Рассматривается как диагноз, если указанные расстройства существуют в течение 3 мес. То есть мужчина оказывается не способным оплодотворить женщину. Небольшое число пациентов сохраняют эрекцию при невозможности эякуляции. Неспособность к эякуляции при сохраненной эрекции может быть результатом приема препаратов, влияющих на симпатическую нервную систему, ретроградной эякуляцией или начальным этапом опухоли яичка, когда потенция не только сохраняется, но на время даже усиливается, а герминативный эпителий вытесняется атипичными клетками, и сперматогенез оскудевает.
Импотенция — одна из причин мужского бесплодия, но бесплодие не означает импотенцию. Например, пациент с первичной дисгинезией семиноформного эпителия или с блокадой тестикулярной экскреции может иметь полностью сохраненную эрекцию, оставаясь при этом стерильным. Полная утрата либидо влечет за собой импотенцию, но импотенция — не значит утрата либидо.
Импотенция чаще обусловлена системными процессами и достаточно редко — патологией собственно полового члена. Для клинициста важно знать, что импотенция может быть психогенной, нейрогенной, эндокринной, медикаментозной, первичной пенильной, собственно сосудистой (заболевания подвздошной артерии или нижнего отдела аорты).
1/2 всех случаев импотенции носит психогенный характер и является одной из форм соматизированной депрессии или страхов в ответ на профессиональные, семейные или другие проблемы. Органические причины импотенции — это генитальные заболевания (фимоз, индурация полового члена), повреждения нервных стволов (спинальные процессы при его травмах или заболеваниях, симпатэктомии, радикальная простатэктомия), опухоли гипофиза, пролактиномы, злокачественные опухоли и тяжелые инфекции, гипо- и гипертиреоз, гипопитуитаризм, сосудистые расстройства, синдром бифуркации аорты (слабость в ногах + перемежающаяся хромота + отсутствие пульса на ногах), синдром Nervus pelvicus (атония мочевого пузыря + недержание), синдром конского хвоста (нарушения функции мочевого пузыря + нарушения функции прямой кишки + отсутствие анального рефлекса + «рейтузная» анестезия), амилоидная полинейропатия I типа, болезнь Ламберта — Рока, первичный и вторичный ипогона- дизм. Медикаментозными причинами импотенции является прием алкоголя, наркотиков, нейролептиков, антидепрессантов, симпатолитиков, антихолинергических препаратов, гипотензивных средств, блокаторов β-адренорецепторов и эстрогенов.
Нарушения фертильности (сперматогенеза) у мужчин отмечают после травм гонад, запоздало пролеченного перекрута яичка, перенесенного вирусного эпидемического паротита, агенезии, неопущенных гонадах, при синдроме Клайнфельтера, длительном тяжелом варикоцеле. Из фармакологических средств отрицательно сказываются цитостатики, тестостерон, спиронолактон, циметидин, сульфасалазин.
Патологическая эрекция (приапизм) возникает при передозировке препаратов, применяемых иногда пациентами без совета с сексологами, в дебюте опухоли мозга, шизофрении (особенно при так называемом сатириазисе), при синдроме Клювера (гностические нарушения + оральные тенденции + гиперметаморфозы + эмоциональные нарушения + неадекватное бесстрашие), при травмах поясничного отдела спинного мозга. Мы наблюдали два случая приапизма у мальчиков, получавших пред- низолон по поводу ревматоидного артрита (РА). Но этот тип осложнений кортикостероидной терапии мы склонны считать исключительно редким. Уплотнение полового члена, ошибочно воспринимаемое как эрекция, возникает при его опухолях, при так называемом Horda venera (воспаление и фиброз при общих заболеваниях), при пластической индурации. Последнее состояние может проявиться в возрасте старше 20 лет, но чаще его выявляют в 40–60 лет. В 30% случаев сочетается с контрактурой Дюпюитрена, с рубцовой деформацией грудных желез, с образованием келоидных рубцов на других участках тела. Вначале тут же за головкой полового члена появляется гладкое эластичное образование, постепенно уплотняющееся до хрящевой твердости и распространяющееся на dorsum penis. Кожа не изменяется. Ракового перерождения не бывает. При эрекции член изгибается в сторону уплотнения, коитус становится невозможным. В итоге развивается депрессия, суицидальные мысли.
Аменореей (первичной) считается состояние отсутствия менструации (menarche) до 18 лет (по американским данным — до 16) или отсутствие менструаций (вторичная аменорея) в течение 3 мес у женщины с ранее определенными ритмическими эпизодами вагинального кровоотделения. В группу первичной аменорреи следует автоматически включать девушек в возрасте 15 лет без развитых вторичных половых признаков.
Деление аменореи на первичную и вторичную условно. Для общепрактикующего врача не менее важно помнить, что менструация — итог сложно и тонко сочетающихся психических, общесоматических, гипоталамических и гипофизарных факторов, состояния яичников, матки, ее шейки, влагалища. Нарушение в любом из этих звеньев может обусловить нарушение естественного менструального цикла. Для менструации необходима пульсативная секреция гонадотропин-рилизинг фактора, который стимулирует гипофиз к такой же волнообразной секреции лютеинизирующего и фолликулостимулирующего гормонов. Оба этих вещества приводят к стимуляции развития фолликулов в яичнике, синтезу эстрогенов и их секреции, овуляции, образованию желтого тела. Оно, в свою очередь, начинает синтез прогестерона. Эндометрий под влиянием эстрогенов и прогестерона разрастается, но когда при отсутствии имплантации оплодотворенной яйцеклетки возникает дефицит гормонов, эндометрий отторгается и исходит из матки через шейку и влагалище в виде менструального кровотечения. То есть расшифровку причин аменореи следует сводить к выявлению нарушений в одном или нескольких только что описанных звеньях.
Причинами могут быть синдромы Штейна — Левенталя, Тернера, Аддисона, адреногенитальный, Майер — Рокитанского — Кюстнера, врожденная пойкилодермия Ротмунда — Томсона (сетчатое ливедо, пойкилодермия, катаракта, гипотрихоз, алопеция, акромикрия, гипоплазия большого пальца, лучевой и локтевой костей, низкий рост, гипоплазия гонад, генитальный инфантилизм, аномалии зубов и ногтей), фенотип Свайера, опухоли надпочечников и гипофиза с вирилизацией, краниофарингеомы, гипотиреоз. Анатомический дефект в виде hymen aperforatum или нарушение естественного сообщения между маткой и шейкой или шейкой и влагалищем (мюллеровская аномалия) может быть причиной ложноположительной диагностики первичной аменорреи.
При диагностике вторичной аменореи врачу следует прежде всего исключить беременность и менопаузу.
Аменорея может развиться после применения гормональных контрацептивов (2:1000 пользовательниц), особенно при раннем по возрасту начале их приема.
Психические потрясения, истерическое желание иметь ребенка, тяжелая работа, истощающие заболевания и непродуманные курсы голодания являются причинами вторичной аменореи, как и опухоли ЦНС, яичников, поликистоз яичников, гонадотропинрезистентные яичники, дефекты эндометрия воспалительного или ятрогенного (кюретаж) характера (постравматические синехии матки — синдром Ашермана. Таким больным просто нечем менструировать).
Что же должен предпринять врач при беседе с пациенткой с аменореей? Прежде всего четко определиться с так называемой первичной и вторичной аменореей. Провести скрупулезное психосоматическое обследование. Далее, в зависимости от первичности или вторичности состояния, пути диагностического поиска несколько отличаются.
Первичная аменорея. Выделить следующие группы (если отсутствуют месячные — смотри на грудь женщины, поскольку врач всегда должен выделять наиболее яркие и просто находимые ориентиры):
- Молочные железы НЕ развиты. Матка есть. Нечастый вариант. Результат недостаточности секреции эстрогенов центральной (гипоталамо-гипо- физарная недостаточность) или периферической этиологии (дисгенезия гонад). Определить уровень гонадотропина. При низком уровне гормона исключить гипоталамо-гипофизарную недостаточность. При высоком уровне гормона исключить дисгене- зию гонад (кариотип, Y-мозаицизм).
- Молочные железы развиты. Матка НЕ выявлена или резко гипопластична. Определить уровень тестостерона. Если его уровень низок (соответствует показателям женщины), есть основания говорить о врожденной аплазии матки. Если уровень тестостерона высок (мужской вариант концентрации) определить кариотип. При наличии 46XY работать с гипотезой тестикулярной феминизации.
- Молочные железы развиты. Матка развита. Исключать варианты вторичной аменореи.
- Молочные железы НЕ развиты. Матка НЕ развита. Очень редкий вариант. Определить уровень гонадотропина и сывороточного тестостерона. При этом уровень фолликулостимулирующего гормона оказывается повышен, а тестостерон характеризуется нормальным «женским» уровнем. Изучить кариотип (выявляют — мужской). Провести поиски яичников вплоть до лапароскопии (если выявлены — удалить, поскольку высок риск развития опухоли).
Что делать при выявлении вторичной аменореи?
Провести тщательный сбор анамнеза, клинический осмотр (исключить беременность или травмы, связанные с беременностью: кюретаж; массу тела женщины; гирсутизм; галакторея; гипотиреоидизм). Определить уровень сывороточного пролактина.
При повышении уровня сывороточного пролактина выполнить КТ или ядерно-магнитный резонанс (ЯМР) черепа для исключения опухоли головного мозга.
При нормальном уровне пролактина назначить прогестерон или медроксипрогестерон (предварительно ПОВТОРНО ИСКЛЮЧИТЬ беременность!). При появлении кровотечения диагностический поиск прекращают. При отсутствии кровотечения определить уровень гонадотропина. В случае повышенной концентрации возможна первичная яичниковая недостаточность. При низком уровне гонадотропина исключить опухоль методами КТ или ЯМР. Если диагноз опухоли снят — перед вами случай гипоталамической аменорреи.
Уже при первой встрече с больным, во время беседы с ним, а затем в процессе осмотра врач отмечает особенности внешнего вида пациента, состояния тех или иных органов и систем. Внешний вид больного весьма важен для диагноза. Диагноз, по крайней мере первичный, но от этого не менее важный, определяющий набор вспомогательных методов исследования и дальнейший дифференциальный поиск, нередко строится на основании так называемого портретного метода («омнибусная диагностика»). И действительно, многие болезни делают совершенно разных людей похожими друг на друга. Но ничто не должно быть абсолютным. При оценке внешности конкретного больного необходимо помнить несколько общих правил наследования. Дети имеют сходство со своими родителями и другими членами семьи, хотя каждый индивидуум имеет уникальную персональную внешность. Поэтому необходимо оценивать внешность родителей и сиблингов. Да и характер жизни, профессия, те или иные привычки или пристрастия сильно сказываются на внешнем облике.
Существует еще групповое или расовое сходство среди индивидуумов, которое необходимо принимать в расчет. Сходство во внешности в популяционных группах, по крайней мере частично, можно объяснить географическими и расовыми группировками членов семьи и их предков. Среди физически, социально или религиозно изолированных популяций (например, амиши, евреи ашкенази) часто выявляют инбридинг и ограниченное смешение генов.
Нередко определяют заметные различия среди семей со схожими генетическими заболеваниями. Открытие того, что гемоглобин S во всех случаях серповидно-клеточной анемии получался в результате точечной мутации, приводящей к замене аминокислоты в бетаглобиновой цепи, дало возможность генетикам приписывать специфические генные заболевания изменениям отдельного гена. Но теперь мы знаем, что человеческие гены комплексны, что изменения в разных частях одного гена могут приводить к значительным вариациям клинической картины. Возможно, что индивидуумы, гомозиготные по рецессивным генным состояниям, в действительности гетерозиготны по специфическим аллелям, но оба аллеля ненормальны. Некоторые изменения являются специфическими и общими для многих семей, но различия в выраженности заболевания могут зависеть от модулирующего эффекта других компонентов гена. Другая вариабельность признака обусловлена обилием мутационных механизмов, дупликаций, делеций, сдвигов рамки считывания. Но любое состояние состоит из различных отдельных случаев (принцип гетерогенности). То есть состояние, болезнь или фенотип могут представлять общий результат аномального процесса. При генетической гетерогенности разные генетические механизмы могут вызывать развитие схожего фенотипа.
То есть наряду с синдромно значимыми внешними признаками могут быть и случайные совпадения.
В качестве примеров диагностической значимых особенностей внешнего вида можно привести несколько из бесчисленного множества состояний.
Синдром Фримена — Шелдона из-за характерной внешности пораженных лиц носит название «синдрома свистящего лица». Типичная ластовидная деформация кистей, деформация суставов заставляют думать о РА. Скованность, сутуловатость осанки, обвисшие плечи, множественные постоянные мелкоамплитудные мятникообразные движения типичны для болезни Паркинсона, сколиоз часто развивается при сирингомиелии, а выраженный поясничный лордоз — при миопатии. Конституциональный тип сложения, астенический или гиперстенический, долихо- или брахистеномелический свойственен целому ряду четко очерченных наследственных синдромов.
Один из наиболее известных синдромов с астеническим сложением — это синдром (болезнь) Марфана, выявляемый в популяции с частотой 1:10 000–1:20 000. Обусловлен изменением гена на 15-й хромосоме (15ql5–21), ответственного за синтез фибриллина, важнейшей составной части микрофибрилл эластических волокон. В одном гене описаны более 500 мутаций, что делает понятным вариабельность симптомов. Тип передачи а/д, риск рождения больного ребенка повышается у отца пожилого возраста, возраст матери значения не имеет. Семейные случаи составляют до 75% всех наблюдений, в 25% возможна новая мутация. Наиболее тяжелые формы отмечают у детей, рожденных от двух родителей-марфанов (гомозиготность). Пенетрантность гена высока, внутрисемейная вариабельность признаков широка: от неонатальных тяжелых форм до едва диагностируемых аномалий. Возможен синдром Марфана без астенического сложения (маскулинный тип взрослых — рис. 2.3), что оставляет ряд случаев не диагностированными. Есть и другие патофизиологические механизмы:
- Низкий порог протеолиза фибриллина, нарушение гомеостаза эластических волокон.
- Сниженная репаративная способность ДНК.
- Выраженная дисрегуляция активации и сигнализации трансформирующего фактора роста (ТФР)-β, приводящая к апоптозу. Выявлена мутация гена рецептора типа II, ТФР-β, обусловливающая указанную дисрегуляцию. Идентификация нарушения гена рецептора ТФР-β с локусом Зр24.2-р25 послужила основанием для выделения синдрома Марфана II типа в отличие от синдрома Марфана I типа , обусловленного мутацией гена фибриллина. Заключение о нарушении сигнализации ТФР-β радикально изменило представления о патогенезе синдрома Марфана.
- Мутации в рецепторах I и II типа ТФР-β оказались причиной синдрома Лоейес — Дитц аневризмы аорты и врожденных патологий (Online Mendelian Inheritance in Man [OMIM] 609192).

В итоге нарушения рецепторов I и II типа ТФР-β привели к выделению новой группы синдромов Марфана внутри более общего состояния генерализованной дисплазии соединительной ткани. Эта недавно выделенная группа синдромов получила название ТФР-β-сигналопатии.
Однако сложность идентификации различных вариантов синдрома Марфана, особенно в практической работе, приводит к тому, что все они объединяются лечащим врачом в одно состояние. Поэтому наши заключения о различной пенетрантности генов, вариабельности признаков объясняются, видимо, тем, что наряду с собственно множественностью мутаций в одном гене, мы в действительности сталкиваемся с мутациями в разных генах или, еще сложнее, с сочетанными мутациями в генах фибриллина и рецепторов ТФР-β.
Диагностические признаки следующие:
- Общая характеристика.
А. Лицо треугольной формы с маленьким подбородком, переносица высокая («птичье лицо»), глаза посажены близко друг к другу, глубоко, выражение грустное. Пациент напоминает персонаж с картины Эль Греко. Уши большие оттопыренные, мочки свободные («уши Будды») (рис. 2.4).
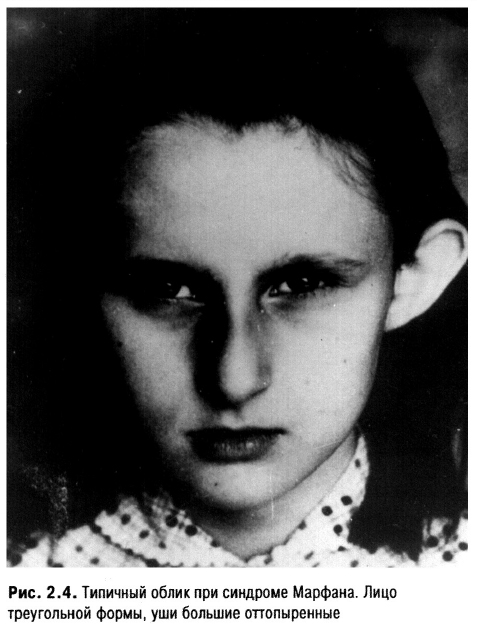
Б. Небо высокое (готическое). Неправильный рост зубов, мальокклюзия, прогнатия. Голос высокий.
В. Долихостеномелия3 (длинные тонкие конечности) — у 80% обследованных, астеничность сложения. Большой палец укладывается поперек ладони и выступает за ее ульнарный край (признак Штейнберга). Мизинцем и большим пальцем пациент свободно охватывает свое запястье (признак Мардоха) (рис. 2.5). Нижний сегмент тела больше верхнего, размах рук превышает рост.
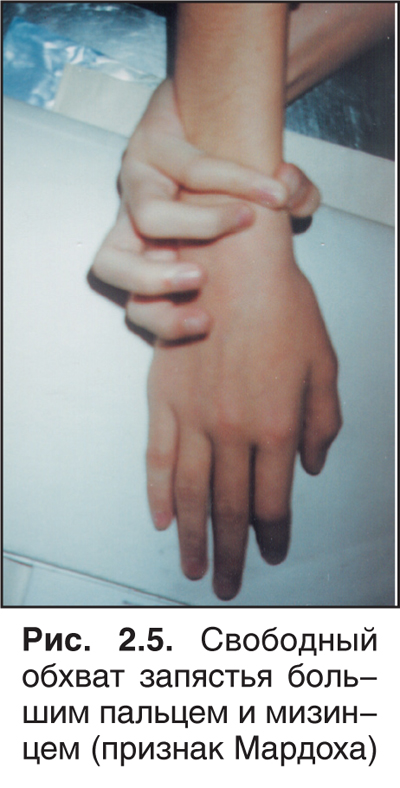
Г. Рост высокий (59%), то есть превышает средний рост здоровых родственников 1-й степени родства (не ориентироваться на популяцию!).
Д. «Куриная грудь»3 или «грудь сапожника»3 (69%), часто асимметричны.
Е. Плоская спина, кифосколиоз3 (45%).
Ж. Гиперподвижность суставов (60%).
З. Гипотрофия мышц.
И. Грыжи, часто рецидивирующие.
К. Бархатистая мягкая кожа со скудной подкожной жировой клетчаткой, стрии растяжения (20%).
Л. Кисты верхушек легких, рецидивирующий пневмоторакс (5%).
М. Эктазии твердой мозговой оболочки в люмбосакральном отделе, артериовенозные аневризмы в головном и спинном мозге. Раширение Cystema magna.
Н. Protrusio acetabuli.
- Изменения глаз.
А. Вывих/подвывих хрусталиков3, чаще двусторонний по направлению вверх и кнаружи, в 60% развивается в возрасте до 4 лет (рис. 2.6). Для своевременной диагностики важно исследование с помощью щелевой лампы на фоне мидриаза. Клинический признак дислокации хрусталиков — дрожание радужки.

Б. Миопия (у 40%), часто обусловленная шаровидным хрусталиком, мегалокорнеа, плоской роговицей.
В. Колобома радужки, глаукома, отслойка сетчатки.
Г. Узкие зрачки (недоразвитие М. dilatator pupillae), Arcus senilis.
- Сердечно-сосудистые изменения (99%).
А. Прогрессирующее расширение аорты* и/или синуса Вальсальвы*.
Б. Расслаивающая аневризма аорты и/или ее разрыв.
В. Недостаточность аортальных клапанов.
Г. Пролапс митрального клапана (95%), его недостаточность, миксоматозная дегенерация створок, обызвествление митрального кольца.
Д. Нарушения ритма сердца.
- Семейный анамнез3. Частота у родственников 1 -й степени родства — 75%, спорадические случаи — 25%.
Наиболее важным для ДД является исключение гомоцистинурии, поскольку имеется возможность ее частичной терапевтической коррекции. Тип поражения опорно-двигательного аппарата, дислокация хрусталика — общее для обоих заболеваний, однако для гомоцистинурии характерен а/p тип наследования, подвывих хрусталика чаще происходит не вверх, как при синдром Марфана, а вниз. У 30% пациентов снижен интеллект, характерны тромбозы и тромбоэмболии, остеопороз с переломами костей. В моче выявляют большое количество гомоцистина. Различают пиридоксин-чувствительную и пиридоксин-резистентную формы. Ограничение метионина в пище и применение витамина В6 при пиридоксин-зависимой форме нормализуют уровень серосодержащих аминокислот.
Синдром Элерса — Данлоса имеет много общего с синдромом Марфана. Одним из кардинальных признаков синдрома Элерса — Данлоса является гиперэластичность кожи. Она бархатистая, нежная, мягкая, с поверхностно расположенными сосудами, теплая, очень легко вытягивается и сразу возвращается в прежнее положение. Отмечается гипермобильность суставов (рис. 2.7) вплоть до симптома «телескопа», геморрагический синдром и повышенная ранимость кожи. Иногда образуются характерные рубцы типа «папиросной бумаги». Различают 11 типов синдрома Элерса — Данлоса в зависимости от клинической картины и типа наследования (см. ниже). По нашим данным у больных с синдромом Элерса — Данлоса реже, чем при синдроме Марфана возникает аневризма аорты. Общим для синдромов Сти клера и Марфана являются долихоморфное телосложение, гипопластическая мускулатура, поражение глаз (табл. 2.5), доминантный тип наследования. Но в отличие от синдрома Марфана суставы при синдроме Стиклера увеличены в объеме, иногда болезненны при движениях, отмечается покраснение кожи и местное повышение температуры над ними. Рентгенологически отмечается нарушение структуры диафизов и эпифизов, а при синдроме Марфана — остеопороз и истончение кортикального слоя. Очень характерны для синдрома Стиклера миопия высокой степени, глаукома. Лицо этих больных существенно отличается от лица больных с синдромом Марфана: оно уплощенное, возможна аномалада Робина, в ряде случаев — глухота и «волчья пасть». Много общего с синдромом Марфана имеет врожденная доброкачественная кон- трактурная арахнодактилия (синдром Билса), для которой характерны доминантный тип наследования, арахнодактилия, проксимальные и дистальные контрактуры (отличительный признак от синдрома Марфана), генерализованная остеопатия и «мятые уши». Кардиоваскулярные нарушения подобны таковым при синдроме Марфана, однако выявляются реже, могут быть разнообразные поражения глаз.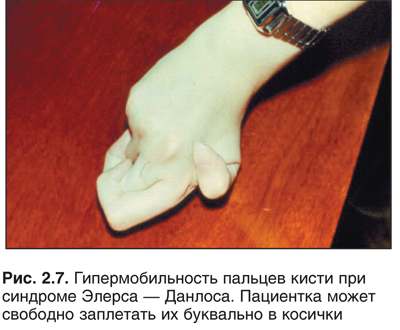
Таблица 2.5
ДД-признаки синдрома Марфана и других основных наследственных поражений соединительной ткани
| Показатель | Синдром | ||||||||||
| Марфана | Гомоцистинурия | Элерса — Данлоса VI тип | В рожденная доброкачественная контрактурная арахнодактилия | Стиклера — Маршалла | Клайнфельтера | Коудена | Множественного базальноклеточного невуса | Сохара | Ригидного позвоночника | Сиппеля | |
| Тип наследования | а/д | а/р | а/р | а/д | а/д | — | а/д | а/д | а/д | а/р | а/д |
| Время манифестации | Детство и позже | Детство и позже | Детство и позже | С рождения | С рождения и позже | Раннее детство | От 15 мес до 25 лет | Детство | Детство, снижение слуха с 10 лет | Детство | 20–60 лет |
| Марфаноидный вид | +++ | ++ | + | + | + | + | + | + | + | + | + |
| Деформация грудной клетки | ++ | ++ | + | + | + | + | Аномалии ребер | + | + | ||
| Деформации позвоночника | + | + | + | + | + | + | + | Ригидный позвоночник | + | ||
| Гипермобильность суставов | + | + | ++ | ||||||||
| Арахнодактилия | ++ | + | + | — | + | + | |||||
| Поражение глаз | Подвывих хрусталика, миопия, астигматизм | Подвывих хрусталика и др. | Миопия идр. | Миопия и др. | Миопия ++, катаракта и др. | Миопия | Миопия, катаракта, сферофакия и др. | + | |||
| Аневризма или расширение аорты | +++ | + | ++ | ||||||||
| Пролапсы клапанов сердца | +++ | + | ++ | + | + | + | + | ||||
| WPW-синдром | ++ | + | + | + | — | — | — | ||||
| Особенности | Подвывих хрусталика вверх | Подвывих хрусталика вниз, гомоцистинурия, тромбэмболии, снижение интеллекта | Повышенная ранимость глазного яблока | Контрактуры, «мятые уши» | Гипертрофия больших суставов | Кариотип XXY, гинекомастия, гипогенитализм | Кистозная гиперплазия молочных желез, кисты щитовидной железы, печени, почек | Множественные базально-клеточные эпителиомы | Прогрессирующая почечная недостаточность | Миопатический синдром, контрактуры сутавов | Феохромоцитома, карцинома щитовидной железы, полиаденоматоз |
Марфаноидный вид свойственней больным с синдромом Клайнфельтера, однако последний отличают гинекомастия, гипогонадизм, азоо- и олигоспермия, повышенная экскреция гонадотропных гормонов, снижение выделения андрогенов и задержка созревания костей. Основной признак — трисомия или даже полисомия половых хромосом. Еще раз подчеркиваем, что кариологические исследования нужны не для диагностики синдрома Марфана, а для исключения фенокопий. Близок к синдрому Клейнфельтера синдром евнухоидизма (гипогонадизм), сочетающий марфаноидное телосложение и недоразвитие вторичных половых признаков. У женщин отмечают аменорею. При гипергонадотропном гипогонадизме характерно уменьшение экскреции половых гормонов при нормальном или повышенном уровне гонадотропинов.
При гипогонадотропном гипогонадизме уменьшена экскреция гонадотропинов, андрогенов, эстрогенов.
Высокий рост, сочетающийся со снижением функции половых желез, отмечается и при гигантизме, который может возникнуть при эозинофильной аденоме гипофиза или гиперпразии эозинофильных клеток, а также после травм и инфекций. Эозинофильная аденома гипофиза в детском возрасте ведет к гигантизму, а в зрелом в связи с закрытием зон роста — к диспропорциональному росту: акромегалии. Последнее сопровождается увеличением мягких частей тела (языка, внутренних органов), носа, ушей, кистей.
Диспропорциональный рост (см. ниже) типичен для артериовенозных соустий, ангиодисплазий (синдром Клиппель — Треноне).
Следует отличать от синдрома Марфана синдром Коудена, наследуемый по доминантному типу. Общим является тип телосложения, однако рост при этом синдроме никогда не достигает показателей, характерных для синдрома Марфана. При синдроме Коудена имеются неопластические образования различной локализации: фиброаденоматоз и карциномы молочных желез, кисты яичника, кишечные полипы, папилломы кожи, липомы, менингеомы, ангиоматоз. Характерных для синдрома Марфана поражений глаз и сердечно-сосудистой системы при синдроме Коудена не описано.
Марфаноидный вид описан и при синдроме множественной невоидной базально-клеточной карциномы, наследуемой а/д. Характерны множественные пигментированные папулы на коже, опухоли ЦНС, различные поражения глаз, опухоли и кисты яичников, гипогонадизм, лейомиомы и т. д.
Аналогичное синдрому Марфана поражение опорно-двигательного аппарата отмечают при синдроме Сохара, Примерно к 10 годам развиваются тугоухость, близорукость со сферофакией, гипостенурия. Летальный исход связан с почечной недостаточностью.
Высокий рост, характерный для синдрома Марфана, отмечают также при синдроме Беквита — Видемана. Он ассоциируется с макроглоссией, пупочной грыжей, родимыми пятнами на лбу, верхнем веке, увеличением внутренних органов, умственной отсталостью, ускорением сроков костного созревания, нередко возникают опухоли различной локализации. Тип наследования не установлен. Марфаноидный вид и наличие сколиоза — общие проявления синдромов Тифри — Колера и Марфана. Наличие остеолиза и полой стопы отличают синдром Тифри — Колера от синдрома Марфана.
Синдром Маршалла — Смита (акселератные скелетные изменения) отличается кифосколиозом, аномальным лицом с выступающими глазами, расщелиной неба, деформацией моляров и умственной отсталостью. Все описанные случаи спорадические.
Вообще же «низкий», «нормальный» или «высокий» рост — понятие относительное к среднему росту популяции. В идеале необходимы данные по среднему росту каждой этнической и социальной группы. Генетические особенности родителей могут изменить стандарт среднего популяционного роста на 30%. Ожидаемый рост 95% детей этих родителей будет составлять ±8,5 см от центилей родителей. И даже в этих случаях нельзя говорить только о генетической предиспозиции роста. Дети низкорослых родителей могут «наследовать» такие семейные особенности, как плохое питание, дефицит йода, психосоциальная депривация, алкоголизм, наркомания. Средний рост человека из социально неблагополучных слоев общества и скорость его роста ниже, чем средний рост его социально благополучного сверстника. Для диагностических целей важно определение костного возраста. Костный возраст определяют по результатам рентгеновского исследования левой кисти и сравнивают со стандартными данными. Последние рассчитываются как хронологический возраст детей, для которых показатели созревания находятся на 50-й перцентили. В дальнейшем при сопоставлении индивидуальных показателей костного и хронологического возраста обследуемого ребенка или подростка можно говорить об особенностях созревания ребенка, ожидаемом конечном росте и о том, насколько фактический рост не соответствует в патологических случаях должному.
В ранний пубертатный период отмечают преимущественный рост ног как отражение увеличения секреции гормона роста. По мере развития в результате секреции тестостерона начинается усиленный рост позвоночника. Оба гормона в пубертатный период действуют синергически. В некоторых случаях этот синергизм нарушается. Так, лица с синдромом Марфана, у которых отмечают высокие концентрации гормона роста при относительно низких концентрациях тестостерона с поздним половым созреванием, растут в основном за счет увеличения длины конечностей. Соотношение длины конечностей и туловища используется в качестве одного из критериев диагноза «синдром Марфана». У людей с брахистеномелией (относительно низкие концентрации гормона роста при сравнительно высокой концентрации андрогенов) рост средний или ниже среднего при одновременном раннем половом созревании. Скорость роста подвержена значительным индивидуальным колебаниям. Но некоторые закономерности существуют. Например, мальчики, у которых объем яичек меньше 10–12 мл, никогда не достигают максимальной скорости роста. Скорость роста у детей обычно регулярная по месяцам и годам, возможные вариации зависят от сезона или тяжелых интеркурентных заболеваний. В подростковом возрасте на короткий период скорость роста взрывообразно увеличивается. Ускорение роста мальчиков начинается в возрасте 13–15,5 года. За указанный период рост увеличивается приблизительно на 20 см, из них до 10 см приходится на год максимально быстрого роста. Причина взрывного роста (catch-up — снятие щеколды) и причина его такой же резкой остановки (catch-down — набрасывание щеколды) изучены очень слабо, как и взаимодействие гормона роста и генетической детерминанты потенциала роста. Расшифровка этого феномена важна для практических и научных целей. Достаточно сказать, что дети, не пережившие скачок роста или леченные гормоном роста, никогда не достигают показателей роста своих сверстников. Таннер предположил, что в ЦНС содержится «шизостат», то есть гипотетический регулятор, предотвращающий расщепленное (shisos — расщепление), несогласованное действие гормонов. Скорость синтеза или освобождения специфической молекулы может снижаться, когда начинается созревание и другие молекулы синтезируются в пропорциях, обеспечивающих рост. Непропорциональный синтез воспринимается шизостатом и восстанавливается пропорциональный рост. В эксперименте с введением дексаметазона в проксимальную зону роста большеберцовой кости кроликов было установлено, что взрывчатый рост не только полностью зависит от ЦНС, но частично определяется и зонами роста.
Один из важнейших факторов контроля роста — гормон роста. Высокие тонкие дети выделяют больше гормона роста по сравнению с их более приземистыми и массивными сверстниками. Гормон роста состоит из 191 аминокислотного остатка и синтезируется в передней доле гипофиза. Гормон роста секретируется в двух формах. Одна фракция, тяжелая (молекулярная масса — 22 К), составляет основную часть и наиболее биологически активная, другая — менее тяжелая (масса — около 20 К) и менее активная. Соотношение фракций и способность рецепторов гормона роста реагировать на его появление в крови дает сложный спектр нарушений роста. Так, дефект гена, отвечающего за формирование рецептора гормона роста, обусловливает развитие так называемого синдрома карликовости типа Ларона. Использование человеческого гормона роста, полученного из посмертно выделенной передней доли гипофиза, в настоящее время запрещено, поскольку высок риск передачи медленных вирусов с развитием болезни Крейцфельда — Якобса. Поэтому в терапии карликовости используется рекомбинантный дериват.
Секреция гормона роста регулируется в первом приближении двумя пептидами: рилизинг-фактором (стимулирующий эффект) и соматостатином (ингибитор). Гормон роста выделяется в кровь не постоянно, а пикообразно пульсаторно преимущественно по ночам. Для нормального роста важно, чтобы в период между пиковыми значениями концентрация снижалась до неопределяемой. За ночь бывает 3–4 подъема концентрации от неопределяемой до 50–60 МЕ/л при скорости роста 10 см/год (объем тестикул 10 мл и более) и 1–2 подъема концентрации от неопределяемой до 20–30 МЕ/л при скорости роста 4,5 см/год. Наиболее массивный выброс гормона роста происходит в интервале от 22 ч до 02 ч. В случаях сдвига засыпания этот пик позднее не компенсируется. Гормон роста освобождается только в период медленного сна. Все, что нарушает естественный сон, вызывает нарушение выделения гормона роста.
Допамин-, серотонин- и норадреналинергические влияния сказываются на нейрорегуляторной системе гормона роста. Холинергические нейротрансмиттеры важны для контроля релизинг-фактора гормона роста и высвобождения соматостатина. Импульсы из гиппокампа, вероятно, и вызывающие сон, как и импульсы из миндалевидного тела, могут быть как стимуляторами, так и ингибиторами по отношению к релизинг-фактору гормона роста. Секреция соматостатина определяется через вентромедиальные ядра гипоталамуса. Ряд лекарств вызывают изменение секреции гормона роста через специфические нейротрансмиттерные патологические пути. Клонидин стимулирует выработку гормона роста через альфа-адренергический путь. Бромокриптин, пропранолол и холинергические агенты ингибируют освобождение соматостатина. Эти сведения важны при разработке и оценке стимуляционных тестов и для вероятной терапии путем стимуляции секреции гормона роста. В регуляции секреции гормона роста важны также пептиды: эндорфины, вазоактивный интестинальный пептид, нейротензин.
Определять концентрацию гормона роста в период бодрствования необходимо с нагрузочными пробами (применение аргинина, или клонидина, или леводопы, или инсулина) в динамике, так как при однократном определении без нагрузочных проб у 10–20% обследуемых можно получить ложнодефицитные состояния.
Гормон роста не ингибируется по принципу обратной связи периферическими эндокринными железами, хотя тироксин и глюкокортикоиды влияют на его секрецию. Гормон роста регулирует сам себя на гипоталамическом и гипофизарном уровне, периферическими факторами роста (инсулиноподобный фактор роста 1) и метаболическими факторами (свободные жирные кислоты и глюкоза). Сочетание этих факторов с физиологическим состоянием (сон, физические нагрузки, питание) и определяет в итоге рост ребенка. Определение инсулиноподобных факторов роста 1 и 2 могут быть малозначимыми у детей старшего возраста с классическим дефицитом гормона роста, но соотношение инсулиноподобных факторов может быть полезно при нетипичных случаях карликовости. Протеин-3 связывающий инсулиноподобный фактор роста точнее отражает дефицит гормона роста в период новорожденности, чем собственно инсулиноподобные факторы роста 1 и 2, но зависит от характера питания.
Низким считается рост в 165 см для мужчин и 155 см для женщин. Рост 154/135 рассматривается как карликовость. Отставание в росте — частая причина беспокойства подростка или его родителей. Если при этом у мальчиков выявляют пубертатное увеличение гонад и/или оволосение лобка 1-й стадии полового созревания, а у девочек — почкообразную грудь (иногда односторонне), то нет повода для беспокойства. Но если у мальчика в возрасте 13,5 года не выявляют пубертатного увеличения яичек, а у девочки нет менархе или у подростков 3-я стадия полового созревания не наступает в течение 4 лет после 2-й стадии, то можно говорить о задержке полового развития.
Следует помнить, что первичная низкорослость и дистрофия достаточно редки. Чаще эти состояния вторичны.
При низком росте или замедлении увеличения роста врач обязан провести ДЦ между следующими состояниями:
- Низкорослость при нормальной скорости роста.
- Конституционально низкий рост при родителях низкого роста.
- Нарушения роста с раннего детства или нарушения роста без внешних признаков заболеваний:
— нарушения внутриутробного развития (хронические заболевания матери и инфекции, токсины, многоплодная беременность и др.);
— врожденные пороки сердца;
— хромосомные аномалии;
— недостаточное питание, психосоциальная депривация, заболевания сердца и почек, недиагностированные синдромы мальабсорбции, клинически стертые случаи болезни Крона, неспецифического язвенного колита, целиакия.
- Эндокринные заболевания:
— пангипопитуитаризм;
— первичная или вторичная тяжелая недостаточность гормона роста (опухоли, ионизирующее облучение, гипотиреоидизм, псевдогипопаратиреоидизм, синдром Кушинга).
- Диспропорциональное сложение:
— конечности относительно короче, чем позвоночник (дисхондроплазия): ахондроплазия, гипо- хондроплазия, множественная эпифезиальная дисплазия;
— короткие конечности и позвоночник (позвоночник относительно короче): мукополисахаридоз, метатрофическая карликовость.
Низкий рост (как и высокий) — это следствие нарушения роста костей. Первичные нарушения роста костей часто связаны с хромосомными аномалиями. Среди хромосомных аномалий с низким ростом протекают синдромы Дауна, Прадера — Вилли, Нунан, прогерии. Если врач осматривает диспластично сложенную девочку ростом 150 см и ниже, он всегда должен помнить о синдроме Тернера, хромосомной болезни с моносомией X. Частота среди новорожденных девочек составляет до 1:2500. Часто возникает мозаицизм, поэтому нередки стертые формы, при которых грубые фенотопические признаки отсутствуют. Нет зависимости от возраста матери, риск повторного рождения больного ребенка в этой же семье отсутствует. У части больных в слизистой оболочке щек регистрируют половой хроматин, что может быть причиной ложноотрицательного заключения, поэтому необходимо полное исследование кариотипа. Внешний вид таких девочек достаточно типичен. Лицо «сфинкса», низкая граница оволосения на затылке; антимонголоидный разрез глаз, эпикант, птоз, страбизм; опущенные уголки рта, микрогнатия; маленькие низко посаженные, часто деформированные уши. Множественные невусы, факомы. Шея короткая, крыловидная складка (птеригиум шейный) у 50% больных. Плоская грудная клетка с гипертелоризмом сосков, первичная аменорея. Гипергонадотропный гипогонадизм. Нередки стеноз устья аорты, дефект межжелудочковой перегородки, аномалии почек. Индекс интеллектуальности изменен несущественно и не всегда, девочки привязчивы, спокойны (видимо, поэтому значительная часть пациенток состоит в браке). Некоторые дети имеют нарушения пространственного восприятия и визуального решения геометрических задач. Как следствие наличия только одной функциональной Х-хромосомы у женщин с синдромом Тернера возможно развитие сцепленных с полом состояний, характерных для мужчин (например, гемофилии А или В).
Девочкам с мозаичным содержанием клеток с Y-хромосомой необходимо удалять гонады до подросткового возраста, так как в дальнейшем высок риск развития опухолей половых желез. Некоторые женщины способны беременеть и вынашивать доношенных новорожденных (они имеют функциональные яичники). Другие с синдромом Тернера могут выносить ребенка при оплодотворении in vitro донорской яйцеклетки.
Среди генных болезней известна а/д передающаяся ахондроплазия (хондродисплазия). Вследствие нарушенного энхондрального обызвествления уменьшается длина трубчатых костей и костей основания черепа. В результате формируется непропорциональная карликовость: нормальная длина туловища при коротких конечностях.
Гормонально обусловленные задержки роста — это прежде всего гипофизарная карликовость из-за гипофизарной недостаточности вследствие гипоталамических нарушений или резистентности тканей по отношению к гормону роста. Задержка роста начинается обычно со 2-го года жизни, пациент сложен пропорционально, лицо слегка округлое, кукольное. При первичных гипофизарных нарушениях (гипо- или аплазия гипофиза, семейный изолированный дефицит гормона роста) карликовость сочетается с аномалиями развития, например, расщелиной верхней губы, челюсти и неба. Редкими причинами являются травмы и воспаления области гипофиза, объемные процессы турецкого седла. Гипоталамические нарушения, приводящие к малорослости и карликовости — это постинфекционные состояния (перенесенные менингиты), гидроцефалия (как вероятный исход постгипоксических состояний новорожденных и, особенно, недоношенных), краниофарингиомы или нейрофибромы области гипоталамуса, гис- тиоцитоз X. Если не удается установить ни одну из вышеперечисленных причин, то останавливаются на так называемой идиопатической карликовости. Хотя при тщательном сборе анамнеза во многих случаях можно с уверенностью говорить о перинатальных кровоизлияниях (ишемиях).
Опухоли надпочечников с образованием глюкокортикоидов или адренокортикотропного гормона сопровождаются задержкой роста.
Очень интересна и показательна так называемая психосоциальная карликовость (аналог материнского депривационного синдрома), возникающая при длительных тяжелых психологических конфликтах. После нормализации ситуации нормализуются и функции гипофиза, что приводит к ускорению роста ребенка.
Первичный гипотиреоз новорожденных или вторичный (в результате, например, тиреоидита Хашимото) ведут к значительной задержке роста. Характерным для первичного гипотиреоидизма является сочетание со сниженным интеллектом и тугоухостью. Среди других причин к задержке роста ведут болезни накопления, плохо корригированный сахарный диабет, витамин-В-резистентный рахит, синдром мальабсорбции, язвенный колит, болезнь Крона, недостаток цинка, железа, неполноценное белковое питание, а также такие болезни почек, как тубулярный ацидоз, болезнь Бартера, хронический гломерулонефрит. Гипоксия тканей при пороках сердца и тяжелых хронических заболеваниях легких также часто обусловливает низкорослость. О семейной низкорослости говорят в случае отсутствия всех вышеуказанных причин и при наличии низкорослых ближайших родственников. В этом случае пубертатный период наступает позже, в школьном возрасте дети отстают в росте от сверстников, но с 14–18 лет рост ускоряется и пациенты достигают нижних нормальных перцентилей.
Наряду с ростом учитывается объем движения в суставах: повышенный или пониженный.
Повышенная подвижность суставов может быть обусловлена мышечной гипотонией или дисплазией соединительной ткани. Последнее состояние известно как синдром гипермобильности и часто сочетается с повышенной эластичностью кожи, пролабирова- нием клапанов сердца, голубыми склерами, грыжей и рядом патологических состояний внутренних органов. Синдром неоднороден. К нему относятся:
- синдром «вялого ребенка (floppy-infant)»;
- MEN-синдром (гиперплазия надпочечников + аденома надпочечников + опухоли островкового аппарата поджелудочной железы + гастринома + инсулинома + гиперплазия паращитовидной желез + аденома паращитовидной железы + опухоли щитовидной железы + ганглионевромы + феохромоцитома + невриномы + марфаноидный внешний вид);
- невус Ито (гипопигментация + церебральные припадки + страбизм + мышечная гипотония + дисплазии зубов + асимметрия лица и конечностей + кифосколиоз);
- псевдоахондроплазия (низкий рост + дисплазия эпифизов и метафизов);
- SHORT-синдром (низкий рост + дисморфии лица + липодистрофии + задержка речевого развития + позднее прорезывание зубов + мезодермальная дисгенезия радужки и роговицы. Вероятно а/д наследование, хотя описанное состояние возникает при миотонической дистрофии Штайнера, при синдроме 10р- и при интерстициальной делеции 4q25-27);
- синдром Элерса — Данлоса. Это состояние является самым известным в группе синдромов гипермобильности. Представляет собой гетерогенную группу наследственных нарушений соединительной ткани с изменением кожи, суставов, связок, сосудов, глаз и внутренних органов. Классификация 11 известных в настоящее время подгрупп основывается на клинических, генетических и биохимических особенностях (табл. 2.6).
Таблица 2.6
ДД-признаки различных типов синдрома Элерса — Данлоса
| Показатель | Тип | ||||||||||
| I тяжелый |
II среднетяжелый |
III легкий, доброкачественная гипермобильность |
IV экхимотический |
V Х-сцепленный |
VI «глазной» |
VII множественный врожденный артрохолазис |
VIII «зубной» |
IX | X | XI | |
| Гиперэластичность кожи | +++ | + | ++ | + | ++ | +++ | +++ | ++ | +/- | ++ | — |
| Гипермобильность суставов | +++ | + | +++ | + | Только пальцы | +++ | +++ | + | +/- | ++ | ++ |
| Ранимость кожи | +++ | + | + | + | + | + | + | ++ | — | ++ | — |
| Кожные кровоизлияния | + | + | +/- | +++ | ++ | ++ | +/- | + | +++ | — | |
| Отслойка сетчатки | +/- | — | — | +/- | — | +++ | — | — | — | — | — |
| Генерализованный периодонтит | — | — | — | — | — | +++ | — | — | |||
| Варикозное расширение вен | ++ | + | — | — | — | — | — | +/- | — | — | |
| Образование аневризм, разрывы сосудов | ++ | +/- | — | +++ | — | — | — | — | — | + | — |
| Деформации скелета | ++ | — | — | — | — | ++ | — | — | + | — | + |
| Низкий и карликовый рост | ++ | ||||||||||
| Преждевременные роды | +++ | — | — | ■ | — | — | — | ||||
| Тип наследования | а/д | а/д | а/д | а/д | Х-сцепленный | а/р | а/д | а/д | Х-сцепленный рецессивный | а/р | а/д |
| Первичный дефект | Нарушение внеклеточного синтеза коллагена | ? | ? | Дефицит коллагена типа III | Недосточность лизилоксидазы | Недостаточность гидроксилазы | Недостаточность проколлаген-пептидазы | ? | Дефицит лизилоксидазы | Дефект фибропектина | ? |
Уменьшение объема движения в суставах может быть ложным (при парезах или параличах) и истинным. Истинная малоподвижность может быть при артрите, артрозе, кровоизлиянии в полость сустава, наличии инородных тел, миозите, миофиброзе и большой группе обменных и наследственных состояний. К ним относятся:
- диабетическая ригидность суставов (ригидность суставов + камподактилия + утолщение кожи над пораженным суставом);
- прогрессирующая ревматоидноподобная хондродисплазия (ригидность суставов + платиспондилия + дисплазия позвонков + нарушения походки + артрит);
- окуло-артро-скелетный синдром (карликовость + глаукома + катаракта + отслойка сетчатки);
- асептический остеохондроз типа Кенига;
- синдром Стиклера;
- синдесмодиспластическая карликовость.
Но наиболее разнообразной, многочисленной и сложной в ДД-плане группой состояний с ограничением подвижности суставов является так называемый артрогрипоз.
Множественный врожденный артрогрипоз известен для следующих состояний:
- Артрогрипоз с множественными птеригиями. Нельзя исключить, что представленные в литературе случаи являются новыми а/д спорадическими мутациями. Носители признака рождаются от пожилых родителей. Состояние проявляется: брахицефалия + низкая граница роста волос + выступающий нос + расщелина неба + низко посаженные уши + прогредиентная микрогения + низкий рост + малая масса тела + камцодактилия + клинодактилия + контрактуры бедер + pes equinowarus + гипоплазия мышц + позднее развитие птеригиумов (шея, подмышки) + рецидивирующие респираторные инфекции + нарастающая гипокинезия диафрагмы + прогрессирующий сколиоз + килевидная грудная клетка + постепенно увеличивающийся гипертрихоз. Гистологически: признаки немалиновой миопатии + увеличение массы жировой ткани в мышцах + увеличение диаметра мышечных волокон.
- Артрогрипоз без специфических дополнительных аномалий (табл. 2.7):
- Х-сцепленный артрогрипоз типа 1–3;
- дистальный артрогрипоз I, II (А–Е);
- синдром Незелова (см. иммунодефицита);
- синдром Пена — Шокера.
Таблица 2.7
Основные характеристики и ДД-признаки некоторых форм множественного врожденного артрогрипоза
| Синдром | Диагностический критерий | Этиология/патогенез | Пренатальная диагностика | Прогноз |
| Дистальный артрогрипоз I | Сгибательные контрактуры пальцев кисти с рождения, затем ульнарная девиация метакарпофалангеальных суставов (98%), умственное развитие не нарушено | АД, вариабельная экспрессивность/? | Нет | При лечении прогноз хороший |
| Дистальный артрогрипоз IIА — синдром Гордона | Сгибательные контрактуры, низкий рост (50%), расщепление твердого неба (50%), аномалии тазобедренного сустава (66%), птоз, эпикант, аномалии позвонков (33%), умственное развитие не нарушено | АД, вариабельная экспрессивность/? | Нет | Хороший |
| Дистальный артрогрипоз IIВ | Низкий рост, птоз, эпикант, ограничение движения глаз и гипомимия, тонкие пальцы, умственное развитие не нарушено | АД, вариабельная экспрессивность/? | Нет | Хороший |
| Дистальный артрогрипоз IIС | Сгибательные контрактуры, расщелина губы +/-, сколиз +/-, аномалии ушных раковин, умственное развитие не нарушено | АД, вариабельность высока, пенетрантность снижена/? | Нет | Сомнителен |
| Дистальный артрогрипоз IID | Сгибательные контрактуры, сколиоз, камподактилия, нистагм, асимметрия черепа, снижение умственных способностей | АД, вариабельная экспресивность/? | Нет | Сомнителен |
| Дистальный артрогрипоз IIЕ | Контрактуры в метакарпофалангеальных суставах с рождения, тризм, аномалии тазобедренных суставов, низкий рост (40%), снижение умственных способностей (35%) | АД (?), пенетрантность снижена | Нет | Хороший |
| Х-сцепленный артрогрипоз I | Сгибательные контрактуры пальцев кисти, локтей и коленей; сколиз, деформации грудной клетки, дыхательная недостаточность | Х-сцепленный рецессивный/внутриматочная спиномускулярная атрофия | По характеру движений плода при ультразвуковом обследовании беременной | Ранняя смерть |
| Х-сцепленный артрогрипоз II | Сгибательные контрактуры (преимущественно колени и бедра), аномалии гениталий, птоз, эпикант, нормальное умственное развитие | Х-сцепленный рецессивный/внутриматочная миопатия | Нет | Не прогрессирует |
| Х-сцепленный артрогиппоз III | Легкие сгибательные контрактуры, умственное развитие не нарушено | Х-сцепленный рецессивный/? | Нет | Хороший |
| Синдром Фримена — Шелдона | Сгибательные контрактуры кистей, «свистящий рот», птоз, антимонголоидный разрез глаз, нормальное умственное развитие | АД/? | Нет | Хороший |
| Синдром тризма с псевдокамтодактилией | Тризм, камптодактилия с распрямленной кистью | АД/? | Нет | Хороший |
| Котрактурная арахнодактилия | Арахнодактилия, кифосколиоз, аномалии сердечно-сосудистой системы | АД/? | Нет | Определяется состоянием сердца и магистральных сосудов |
| Камптодактилия с тугоухостью | Нейросенсорная тугоухость | АД/? | Нет | Для жизни —хороший |
| Синдром приведенного большого пальца кисти | Пальмарное сгибание большого пальца кисти, стеноз сильвиевого водопровода, умственная задержка | Х-сцепленный рецессивный/? | Нет | Сомнителен |
| Амиоплазия | Внутренняя ротация в плечевом суставе, аномалии скелета, гастросхизис | Спорадически/? | Нет | Сомнителен |
- Артрогрипоз с арахнодактилией:
- контрактурная арахнодактилия (описание см. при ДД синдрома Марфана).
- Артрогрипоз с аномалиями кожи:
- рестриктивная дерматопатия. А/p. Артрогрипоз сочетается с дисплазией кожи;
- синдром Альфи;
- синдром Лоури II (синдром Лоури — Вуда). А/p. Множественная эпифизеальная дисплазия с карликовостью и задержкой психического развития. Гиперкератоз. Контрактуры — преимущественно сгибательные в локтевых суставах. Судороги. Врожденный нистагм. Не всегда бывает миопия;
- синдром Муррей — Пуретика. Ювенильный гиалиновый фиброматоз;
- синдром ото-онихо-перонеальный. А/p? Связано с полом? Большие деформированные ушные раковины: плоский завиток с необычно высоко выступающим противозавитком и верхней ножкой. Маленькие мочки. Ногти гипопластичные или отсутствуют. Аплазия или проксимальная гипоплазия локтевой кости. Долихоцефалия. Лицо плоское. Монголоидный разрез глаз. Легкая задержка психомоторного развития;
- синдром Сентера. Артрогрипоз в сочетании с атипичной ихтиозоформной эритродермией и врожденной нейросенсорной тугоухостью.
- Артрогрипоз с преждевременным старением:
- синдром Бейтона.
- Артрогрипоз с аномалиями глаз:
- синдром блефарофимоза и шейного птеригиума. Сгибательные контрактуры пальцев кистей и стоп, остеодисплазия;
- синдром Боувен — Конради. Синдром аномалий костной и мочеполовой систем с дисплазией лица;
- синдром COFS — синдром Пена — Шокера II. Тяжелая задержка психомоторного развития, дисморфии лица, сгибательные контрактуры в больших суставах; р28-р57;
- синдром СОМ — врожденная мышечная дистрофия и дисгенезия мозга в датских семьях;
- синдром Марден — Уокера. Относится также к синдромам генерализованной дисплазии соединительной ткани.
- Артрогрипоз с аномалиями ЦНС:
- синдром Аазе — Смита;
- синдром С-тригоноцефалии;
- синдром гипертермии с менингомиелоцеле и анэнцефалией;
- синдром цереброартродигитальный. Артромиодисплазия и агенезия крестца;
- синдром отведенного большого пальца кисти;
- гелеофизическая дисплазия;
- синдром Херрика. Внутриутробная мультисистемная атрофия;
- синдром Нью — Лаксова;
- синдром ван Бирвлиета. Внутриутробная задержка роста с артрогрипозом, аномалиями мозга, лица и черепа;
- синдром Викера — Вольфа. Х-связанный. Врожденные контрактуры, окуломоторная апраксия, атрофия мышц.
- Артрогрипоз с аномалиями скелета:
- синдром Ришера — Коста. Макроцефалия, артромиодисплазия, гипопластические пальцы, аномалии ребер;
- синдром VSR — синдром Германа — Опитца II. А/д. Врожденные контрактуры локтевых суставов с гипоплазией и дислокацией головки лучевой кости, кисти, пальцев, «полая стопа». Низкий рост. Мезомелическая брахимелия верхних и ризомелическая брахимелия нижних конечностей. Тригоноцефалия. Расщелина неба. Узкие глазные щели. Ротированные кзади уши. Сколиоз. Короткая грудина, отсутствие мечевидного отростка. Крипторхизм. Необычная дерматоглифика. Умственное развитие не страдает. Склонность к инфекциям.
- Артрогрипоз с нарушениями слуха:
- синдром артрогрипоза и глухоты. Семейные аномалии кисти и сенсоневральная тугоухость;
- синдром Гордона II (см. табл. 2.7 «камподактилия с тугоухостью»).
- Артрогрипоз с поражением мышц:
- синдром Эшенна. Врожденная мышечная дистрофия со спонгиозной дисплазией белого вещества мозга;
- синдром Фукуяма. Врожденная форма прогрессирующей мышечной дистрофии;
- синдром Кузефа. А/p. Фенотип напоминает синдром Нунен. Миопатия, врожденные контрактуры, злокачественная гипертермия;
- синдром Кускоквин. Наследственная форма артрогрипоза у эскимосов Аляски.
ДД-признаки наиболее существенных для семейного врача форм артрогрипоза см. в табл. 2.7 (по Foster-Iskenius U., 1990).
Положение, которое стремится занять обследуемый, может быть достаточно патогномоничным для некоторых заболеваний. При колике пациент не находит себе место, многократно меняет положение, в то время как при перитоните скован, предпочитает лежать на спине, стремится щадить напряженный живот. Сидячее положение с опущенными плечами типично для сердечной недостаточности, горизонтальное положение сопровождается у таких больных усилением одышки. При легочной одышке горизонтальное положение не ухудшает состояние больного. Сидячее положение с поднятыми фиксированными плечами типично для БА, положение на корточках — для синих пороков типа Фалло, а на корточках с поджатыми к груди коленями — для боли при раке поджелудочной железы.
Походка короткими трясущимися шагами типична для паркинсонизма, особенно в сочетании со скованностью рук. Нарушения походки могут быть вызваны церебральными нарушениями или периферическими нейропатиями. Спотыкающаяся походка с высоким поднятием колен типична для повреждений перонеального нерва, поскольку из- за недостаточной дорсальной флексии отвисающий носок стопы цепляет почву. При ДД необходимо учитывать токсическую или диабетическую полинейропатию. При болезни Литтля и церебральных параличах вследствие высокого тонуса приводящих мышц бедра отмечается перекрещивание ног. Чем выше перекрещивание, тем тяжелее паралич. Атаксическая походка с широко расставленными ногами характерна для поражения мозжечка (рассеянный склероз, оливо-понто-церебеллярная дегенерация, аплазия мозжечка). При истерических нарушениях походки совершается такая масса вспомогательных, а иногда и вычурных движений, которые могут быть выполнены только при совершеннейшей координации.
Ожирение диагностируется при увеличении массы тела на 20% и более по сравнению с нормой. Первичное ожирение (adipositas simplex) обусловлено прежде всего неумеренным употреблением пищи, что связано с семейными факторами и невротическими реакциями. Как следствие ожирения развивается сниженная толерантность к глюкозе, повышение концентрации триглицеридов и холестерина в сыворотке крови, снижение уровня гормона роста. При ожирении возможна аменорея. Ожидаемая продолжительность жизни людей с ожирением ниже, чем с нормальной массой тела за счет высокой вероятности заболеваний сердца, АГ, сахарного диабета 2-го типа, жировой дистрофии печени, а у женщин — рака матки. При ожирении высока вероятность заболеваний суставов. Вследствие этого при добровольном страховании страховой полис для лиц с ожирением стоит больше, а людей с массой 160 кг и более многие страховые компании вообще отказываются страховать. Вторичное ожирение среди всех людей с избыточной массой тела отмечается всего в 1% случаев. Из этой сотой части наиболее распространенная причина вторичного ожирения — эндокринологические нарушения. Гипогонадизм и гипофункция яичников при синдроме Штейна — Левенталя могут быть причинами ожирения. Безусловно, синдром Кушинга с избыточной концентрацией кортикостероидов также относится к этой группе, характеризуясь типичным отложением жира на туловище и шейно-затылочной области при относительно тонких конечностях. В качестве других причин можно сослаться на инсулиномы и гипотиреоз. В последнем случае избыточная масса тела обусловлена не только ожирением, но и задержкой воды. Мягкие формы гипотиреоза возникают часто, диагностируются плохо из-за незнания симптоматики. Характерно диффузное ожирение и повышение уровня холестерина в крови.
Гипоталамические формы ожирения комбинируются с несахарным мочеизнурением или с гипогонадотропным гипогонадизмом. Причинами этого типа расстройства обмена могут быть травмы, метастазы, краниофарингеомы, энцефалиты, внутричерепная гипертензия и синдром пустого турецкого седла. Редкой причиной является адипозогенитальная дистрофия (синдром Фрелиха), характеризующаяся гипогонадотропным гипогонадизмом, резким ожирением и внутричерепной гипертензией. Заболевание необходимо дифференцировать от синдрома «раскормленного ребенка», при котором гениталии кажутся уменьшенными за счет их маскировки ожиревшими лобком и бедрами. Синдрому Прадер — Вилли присущи типичный внешний вид («мешок с мукой»), мышечная гипотония, низкий рост и сниженная толерантность к глюкозе. При болезни Альстрема наряду с ожирением развивается слепота за счет дегенерации сетчатки, прогрессирующая внутренняя тугоухость и сахарный диабет. Синдром Лоренса — Муна — Бада — Бидля наряду с ожирением проявляется слабоумием, деформациями черепа, поли- или синдактилией, экстрапирамидными нарушениями моторики, пигментным ретинитом. При КТ головного мозга могут быть выявлены нарушение дифференциации белого и серого вещества, субатрофия коры больших полушарий. Синдром Морганьи — Стюарта — Морреля диагностируют по ожирению в сочетании с гиперостозом лобной кости, гирсутизмом и в ряде случаев — отставании в умственном развитии.
Наряду с генерализованным увеличением подкожной жировой клетчатки отмечают ее локальные разрастания. Они типичны для синдрома Маделунга (синдром жирной шеи) с инфильтративно растущей липомой в области затылка и задней поверхности шеи. Характеризуется а/д типом передачи. Аналогичные разрастания возможны и при алкоголизме. Редко диагностируют генерализованный узловатый липоматоз с узлами липом по всему телу. Его сочетание с пигментными пятнами на коже цвета «кофе с молоком» позволяет отнести этот тип локального ожирения к группе нейрокутанных синдромов. Болезнь Деркума (lipomatosis dolorosa) с образованием болезненных липом на конечностях возникают у женщин в постменопаузальный период. Локальные жировые отложения известны и в местах инъекций инсулина. Подкожные некротизующиеся узлы типичны для панникулита и болезни Вебера — Кристиана, относимых в группу так называемых коллагенозов, хотя они могут сопровождать и течение панкреатита.
Недостаточная масса тела может быть просто обусловлена или являться результатом пищевых привычек. В патологических условиях недостаточную массу тела отмечают при голодании, тяжелых заболеваниях, синдромах мальабсорбции первичных и в результате заболеваний кишечника (целиакия — спру) и поджелудочной железы, при гипертиреозе и тиреотоксикозе. Трудно объяснимое и значительное уменьшение массы тела часто свидетельствует о существенных расстройствах. В подтверждение этого можно указать, что около 1/4 таких пациентов умирают в течение года с момента начала катастрофического похудания. Под похуданием понимают нежелательную потерю более 5% первоначальной массы тела за 6 мес. Уменьшение массы тела должно быть зафиксировано медиками, так как половина всех обратившихся с такой жалобой не может количественно подтвердить уменьшение массы. Более 35% всех больных с уменьшением массы тела выдвигают это состояние на первый план, хотя одновременно отмечают иные признаки: тошнота, диспепсия, лихорадка, боль различной локализации, нейропсихические проблемы и т. д.
Массу тела нельзя оценивать в отрыве от роста и окружности головы. Для детей это правило является абсолютно непреложным. Показатели при этом берут не в абсолютных величинах, а в перцентилях. Выделяют следующие группы (по Illing S., Classen М., 2000):
- Перцентиль массы тела < перцентили роста < перцентили окружности головы:
- качественно и количественно неполноценное питание: крайне строгая диета (например, при атопических состояниях), психосоциальные проблемы, anorexia nervosum, хронические инфекции, злокачественные опухоли, хроническая почечная недостаточность, сердечная недостаточность;
- дефицит калорийности при рвоте: гастроэзофагеальный рефлюкс, грыжи диафрагмы, поражения желудочно-кишечного тракта, почечная недостаточность, неврологические заболевания, гепатопатии, метаболические заболевания;
- дефицит калорийности при диарее или недостаточности пищеварения: муковисцидоз, целиакия, панкреатическая недостаточность, непереносимость белка коровьего молока, иммунологическая недостаточность, экссудативная энтеропатия, хроническая инфекция кишечника, болезнь Крона и неспецифический язвенный колит;
- усиленное потребление калорий: гипертиреоз, гиперкинезия, хронические инфекции, порок сердца.
- Уменьшение массы тела с пропорциональным уменьшением роста при нормальной окружности головы. Дистрофии нет. При ДЦ следует учитывать:
- эндокринопатии: гипотиреоз, дефицит гормона роста, гипопитуитаризм;
- семейную карликовость;
- дисплазии скелета, РА.
- Уменьшение перцентили массы тела, роста и окружности головы равномерно. Часто изменения начинаются пренатально:
- хромосомные аномалии;
- внутриматочные инфекции, плацентарная недостаточность, алкогольная и наркотическая интоксикации.
ДД-подходы расшифровки синдрома уменьшения массы тела приведены на схеме 2.3.
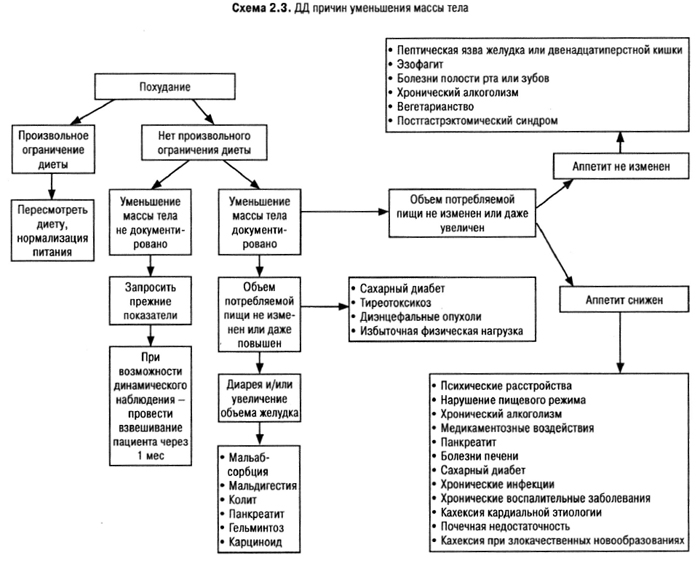
Гинекомастия — избыточное развитие молочных желез у мужчин. Объем молочной железы может увеличиваться за счет собственно железы (или протоков) или за счет стромы. При истинной гинекомастии увеличиваются оба компонента. Гинекомастия может быть асимметричной. Следует помнить, что увеличение молочных желез у мужчины может быть симулировано ожирением, при котором развивается липомастия.
В физиологических условиях гинекомастия может быть у новорожденных, юношей и стариков. Юношеская гинекомастия может возникать у подростков в пубертатный период, самостоятельно благополучно разрешается и не требует вмешательств.
Гинекомастия, развивающаяся в возрасте 20 лет и старше, требует полноценного обследования, хотя в 15–30% выяснить ее генез не удается. В остальных случаях причинами могут быть алкоголизм, опухоли и/или эндокринные расстройства. Общими механизмами являются недостаток тестостерона, повышение продукции эстрогенов или медикаментозное влияние.
Возможными причинами гинекомастий может быть рак молочной железы у мужчин, медикаментозное влияние (диазепам, трициклические антидепрессанты, кетоконазол, резерпин, спиронолактон, α-метилдопа, циметидин, гентамицин, изониазид, метронидазол, цисплатин, алкилирующие препараты, винкристин, бусульфан, героин, марихуана, метадон, D-пенициллинамин, нитромочевина, амфетамин, наперстянка, алкоголь), случаи с повышением продукции эстрогенов (опухоли половых желез, рак легкого, гермафродитизм), усиленным преобразованием андрогенов в эстрогены (опухоли надпочечников, гипертиреоз, болезни печени), снижение синтеза тестостерона (синдром Клайнфельтера, анорхия, кастрация, последствия эпидемического паротита, неврологические заболевания, гипотиреоз), тестикулярная феминизация, синдром Рейфенштейна. Последний обусловлен мутацией рецессивного гена рецепторов андрогенов на Х-хромосоме (Xq11–Xq13). Патогенез состояния заключается в том, что, в противоположность тестикулярной феминизации, развивается только частичная резистентность к андрогенам, в результате чего все-таки формируется мужской фенотип. Клиническими проявлениями являются гипоспадия различной степени, гипогонадизм с маленькими гонадами, постпубертатная атрофия гонад, азооспермия, стерильность, слабая выраженность вторичных половых признаков. В пубертатный период формируется гинекомастия различной степени выраженности, уровень тестостерона в плазме крови в норме или даже повышен, повышена концентрация эстрадиола. Андростендион — в норме (в противоположность дефициту 17-редуктазы, протекающей со схожим фенотипом).
ДД гинекомастии начинается со сбора «лекарственного анамнеза». Так, из 350 собранных нами случаев ятрогенной гинекомастии у взрослых 30% приходилось на противоязвенные препараты — циметидин, ранитидин, фамотидин, омепразол, у детей основной причиной является гентамицин. Затем проводят осмотр гонад с определением их объема, изучение функции печени и уровня гормонов, а именно: уровень 17-кетостероидов в суточной моче, концентрация эстрогенов, тестостерона и тиреотропного гормона в плазме крови. С гинекомастией протекают и состояния гипергонадотропного гипогонадизма. Причинами являются травма яичек, орхит, кастрация, лучевая и химиотерапия, далеко зашедшая миотоническая дистрофия, аплазия герминативного эпителия (сертоллиевых клеток). Клинические проявления определяются степенью дефицита андрогенов.
Примером гипергонадотропного гипогонадизма является синдром Клайнфельтера — хромосомное заболевание с дополнительными хромосомами X. Часто выявляют мозаицизм. При полноценном обследовании выявляют с частотой до 1:1000 среди всех новорожденных мальчиков и поэтому является самой частой причиной гипогонадизма с мужским фенотипом в популяции. У новорожденных мальчиков и в препубертатный период выраженные клинические признаки часто отсутствуют, в связи с чем заболевание остается не распознанным. Характеризуется высоким ростом, длинными конечностями, астеничностью или евнухоидностью сложения. Нередко возникают сколиоз, остеопороз, сахарный диабет. Гипогонадизм с гиалинизирующим склерозом семенных канальцев. Низкий уровень продукции тестостерона. Гинекомастия в части случаев односторонняя. Гистологически избыточно развитая молочная железа атипична, состоит из плотной фиброзной ткани с единичными протоками. В 60% развивается рак молочной железы. Степень снижения интеллекта, моторного и речевого развития варьируют. Больные легко внушаемы, эмоционально неустойчивы. В ДД большое значение имеет исследование кариотипа.
Природа крайне терпима к вариациям в составе половых хромосом. Для жизнеспособности необходима, по крайней мере, одна нормальная Х-хромосома, и моносомии Х-хромосомы, как при синдроме Тернера, совместимы с нормальной, но бесплодной жизнью. Другие относительно частые аномалии половых хромосом — это кариотип 47ХХХ (женщины с трисомией X) и кариотип 47XYY (мужчина с дисомией Y). Каждая аномалия возникает с частотой около 0,1%, не приводит к стерильности или к развитию уникального фенотипа. Мужчины XYY часто отличаются асоциальным поведением.
Гипогонадотропный гипогонадизм развивается при опухолях, травмах, воспалениях (в том числе васкулите) области гипоталамуса или гипофиза. Одновременно часто снижается и выработка гормона роста, что приводит к карликовости с евнухоидизмом.
Галакторея — лактация вне беременности и родов. Часто ассоциируется с повышенным уровнем пролактина в сыворотке крови. Гиперпролактинемия связана с первичным повышенным образованием гормона или с допаминобусловленной блокадой нормально сформированных лактотрофных тканей. У новорожденных галакторея появляется при избыточном поступлении материнских гормонов в молоко.
Галакторея возникает при беременности, механическом раздражении груди, повреждениях спинного мозга, стрессах, гипогликемии, гипотиреоидизме, краниофарингеомах, саркоидозе, поликистозе яичников, хронической почечной недостаточности. Галакторея может возникать при акромегалии, пролактиноме, синдроме пустого турецкого седла с микроаденомой. Галакторею могут обусловить фенотиазины, бутирофеноны, гентамицин, альфаметилдопа, резерпин, циметидин, опиаты.
При ДД галактореи абсолютно показано определение уровня пролактина в сыворотке крови и состояния щитовидной железы. Дальнейшие действия врача зависят от результатов сбора анамнеза и физикального обследования больного.
Наряду с галактореей возможны и иные выделения из соска. Жалобы на выделения из соска как повод для визита к врачу стоят на втором месте после жалоб на выявление уплотнения в молочной железе. Экссудация из тканей соска (экзема, опухоли, травмы) могут быть ошибочно приняты за выделения из молочной железы.
Выделения из молочной железы могут быть двухсторонними (обычно — системные процессы) и односторонними (более или менее локальные процессы) и обусловлены манипуляциями с молочной железой, приемом лекарственных препаратов, противозачаточных, инфекцией, травмой, фиброкистозной дисплазией, новообразованиями.
Диагностический поиск определяется анамнезом, физикальным обследованием, цитологией и маммографией. Для диагноза важно знать, являются ли выделения одно- или двусторонними, спонтанными или вызванными, персистирующими или связанными с лактацией, секрет отходит из одного или нескольких протоков, цвет, характер, связь с инфекцией, травмой, гормональными или иными препаратами, наличием узла в молочной железе, случаи рака молочной железы в семейном анамнезе. Если у пациентки выделения спонтанные из одной груди (тем более, из одного протока), возможна серьезная патология. При наличии включений в жидком отделяемом вероятность злокачественных образований составляет 35-38%, без включений — всего 9-11%.
Среди женщин с кровянистыми выделениями из соска у 25% диагностируют рак, у 20% — папилломатоз протоков. ДД-подходы при наличии отделяемого из соска приведены на схеме 2.4.
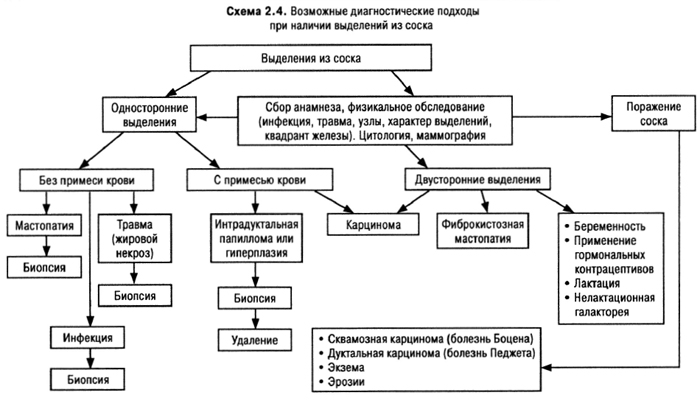
Форма и внешние особенности кисти позволяют много сказать о пациенте, его профессии и состоянии.
Типичная желто-коричневая окраска 2–3 пальцев у курильщиков, коричневатая окраска ногтей у фотографов, дюпитреновская контактура у алкоголиков и больных сахарным диабетом.
Тонкая удлиненная кисть людей с синдромом Марфана или при врожденной контрактурной арахнодактилии; маленькая кисть — при синдроме Корнеля-де-Ланге типа 1; короткая ладонь — при синдроме Дауна, трисомии 12р; большая мясистая — при акромегалии, прогрессирующей липодистрофии, синдроме Видемана — Раутенштрауха; кисть по типу тюленьих ласт, деформация межфаланговых суставов с утренней ригидностью — при РА. Штыкообразная постановка кисти — при дисхондроостеолизе Лери — Вейля. Кисть по типу «когтистой лапы» отмечают при склеродермии, хроническом артрите, боковом амиотрофическом склерозе.
Изменение концевых фаланг по типу «барабанных палочек» типично для многих заболеваний и является выражением гипертрофической остеоартропатии. Развивается при пороках сердца синего типа, бронхоэктазах, фиброзе легких, эмпиеме, муковисцидозе, мезотелиоме, раке бронхов, метастазирующих опухолях. В некоторых случаях пальцы типа барабанных палочек могут быть семейной особенностью.
Брахифалангия — при синдромах Аарского, Вейля — Маркезани, 11q, трисомии 9р, множественных дизостозов, Коэна, KBG, Рувалькаба, Рюдигера, Озеольда — Ремондини, Сетра — Хотцена, артерио- печеночной дисплазии, точечной хондродисплазии в результате Х-хромосомной делении, гелеофизической дисплазии, краниоэктодермальной дисплазии.
Трехсуставный большой палец является одним из признаков синдромов Аазе, Итона — МакКусика, Тоунеса — Брока.
Гипоплазия большого пальца характерна при наследственном сцепленном с полом стенозе Сильвиева водопровода, синдромах фацио-аурикуло-лучевом, LADD, Нагера, Робертса, пойкилодермиях типа Ротмунда — Томсона и Томсона, Холт — Орама, Юберга — Хайварда, а также при голопрозенцефалии.
Приводим описание наиболее значимых синдромов брахидактилии.
- Брахидактилия типа А1. А/д. (Именно на этом типе аномалии было доказано распространение менделевских законов наследования на человека.) Проявляется гипоплазией средней фаланги II–V пальцев кисти и основной фаланги большого пальца. При выраженном синдроме рудименты фаланг спаяны с ногтевыми (терминальный симфалангизм), что выглядит как аплазия средних фаланг. Рост пациентов значительно занижен. Различные степени укорочения фаланг отмечают в одной и той же семье, демонстрируя различную экспрессивность гена.
- Брахидактилия типа А2. Этиология гетерогенна. Синдром выявляют изолированно как а/д вариант, так и в рамках акроцефалосиндактилии типов Аперта и Пфайфера, а также при синдроме Рубинштейна — Тойби. Определяется как симметричное укорочение и нарушение формы средних фаланг двух пальцев кисти и стопы.
- Брахидактилия типа АЗ. Укорочение и деформация средней фаланги V пальца кисти. Передается а/д. Выявляют у 20% всех японцев. Но может быть при синдроме Дауна, комбинироваться с аномалиями развития эпифизов. Проявляется укорочением средней фаланги мизинца кисти с радиальной девиацией пальца.
- Брахидактилия типа В. Возможны варианты а/д наследования, но может возникать как симптомокомплекс с колобомой или при дисплазии соединительной ткани. Представляет собой гипоплазию или аплазию ногтевых фаланг II–V пальцев кисти с возможным полным отсутствием ногтей. Ногтевая фаланга большого пальца часто расширена, расщеплена или даже удвоена. Иногда — укорочение метакарпальных или метатарзальных костей, синдактилия мягких тканей. Гипоплазия отдельных пальцев стоп.
- Брахидактилия типа С. Передается а/д. Представляет собой брахимезофалангию II, III и V пальцев кисти и укорочение Os metacarpale I.
- Брахидактилия типа D. Передается а/д, выявляют как признак синдромов Рубинштейна — Тойби, Табачника и при полисиндактилии. Укорочение и расширение концевых фаланг I пальцев кистей и стоп.
- Брахидактилия типа Е. Передается изолированно а/д, но возникает также как признак генерализованной дисплазии скелета, синдрома Тернера, остеодистрофии Олбрайта, базально-клеточного невуса. Представляет собой различной степени укорочение метакарпальных костей, иногда и метатарзальных. Нередко отмечают только значительное укорочение отдельной кости. Клинически это выглядит как западение на месте укороченной метакарпальной кости при сжатом кулаке.
Список можно продолжать. Цель — не в этом. Цель — обратить внимание врача на «мелочи» и побудить к чтению.
Пальмарная эритема характерна при циррозе печени, диффузных болезнях соединительной ткани (ДБСТ), гипертиреозе, реже — при раке бронхов или терапии эстрогенами. Розоватая сухая ладонь с глубокими складками пальмарной поверхности часто сопровождает течение пептической язвы или хронического дуоденита. Пигментация всей ладони или только ее складок бывает у пациентов с болезнью Аддисона, при циррозе печени, гемохроматозе, уремии, синдроме Пейтца — Эгерса и лепре. Первичный гиперкератоз свойственней отравлению мышьяком. Плотная, деревянистая кисть формируется при склеродермии.
Местная температура ладони зависит от кровоснабжения и регуляции сосудистого тонуса. Холодная влажная кисть с легко возникающей синюхой кожи свойственна при вегетативной дистонии; холодная, бледная и сухая — гипотиреозу, сухая, синяя и холодная — сердечной недостаточности, а теплая и синяя типична для врожденных пороков сердца синего типа. Теплая рука бывает у пациентов с тиреотоксикозом, АГ. Тремор рук типичен для гипертиреоза, болезни Паркинсона, рассеянного склероза, поражения мозжечка, алкоголизма, интоксикации, старческого и семейного тремора. Гиперкератоз ладоней и подошв сопутствует тирозинемии. Вторичный гиперкератоз свойственен гипотиреозу.
Прогрессирующая слабость, часто с ночным ощущением бегания мурашек и онемением I–III пальцев с улучшением состояния после потряхивания кисти заставляют думать о туннельном синдроме. Одной из самых частых причин туннельного синдрома является минимальный отек при гипотиреозе. Кроме того, туннельный синдром возникает при амилоидозах сенильном, вторичном, типа Любарха, амилоидной полинейропатии типа II и семейной рецидивирующей политопной нейропатии.
Последнее состояние передается а/д. Обусловлено, видимо, нарушением обмена в Шванновских клетках с развитием демиелинизации, в результате чего давление и растяжение в области нервных стволов вызывает их повреждение. Проявляется на 2–3-й декадах жизни. Остро или постепенно возникают эпизоды вялых параличей, затрагивающих область иннервации одного или нескольких нервов. Начинаются безболезненно. Трофика тканей не поражена. Обратное развитие симптомов отмечают через несколько дней или недель. При рецидивах — выздоровление с дефектом. Нарушения возникают в результате неадекватно слабой травмы в области синовиальных сумок, костных выступов, туннелей. Особенно часто поражаются N. peroneus в области головки малоберцовой кости, N. radialis в области sulcus humeri, N. ulnaris в области локтя. Ликвор не изменен. Проводимость по нервам замедлена.
Достаточно редким проявлением туберкулеза являются мелкие округлые очажки разрежения костной ткани, в том числе в фалангах (туберкулез Юнглинга).
Лицо больного отражает многочисленные патологические состояния. Сардоническая улыбка при тетании за счет сочетания тризма с ограничением открывания рта и тяги мимической мускулатуры, «лицо Гиппократа» при перитоните с бледной кожей, ввалившимися глазами, опавшими щеками, заострившимся носом, цианозом губ и безучастным страдальчески остановившимся взглядом — не оставляют сомнений в диагнозе. Лицо Корвизара — при сердечной недостаточности. Лицо при базедовой болезни с блестящими расширенными глазами, потной кожей, или микседема с типичным одутловатым лицом, мешками под глазами, бледной сухой кожей, редким волосяным покровом, низкий хриплый голос и бра- дипноэ. Нефротические отеки, придающие лицу вид «надутого шарика». Яркая гиперемия лица при лихорадочных инфекционных заболеваниях, особенно при пневмококковой пневмонии, раздувание крыльев носа и герпес губ. Бледность, стонущее дыхание и пенистое отделяемое в углах рта при респираторных дистресс-синдромах. Лунообразное лицо с акне и гиперемией щек при синдроме Кушинга. Безвольное, вялое, «обвисшее» лицо кретина; лицо паркинсоника с амимичностью, блестящей сальной кожей, в тяжелых случаях со слюнотечением при сохранном интеллекте. Безвольное лицо алкоголика с паретически расширенными сосудами, конъюнктивитом, «пустым взглядом» и «водянистыми глазами». Малиновый цианоз при ЛГ, в том числе и при стенозе левого атриовентрикулярного отверстия; гиперемия лица при гипертоническом кризе и более выраженная — при истинной полицитемии. Волчаночная «бабочка» и аналогичная локальная воспалительная гиперемия, но с исходом в «минус-ткань» при туберкулезной волчанке, фиолетовые «очки» при болезни Унферрихта (дерматомиозите), параорбитальная эритема и индурация при склеродеме Бушке. Тонкая кожа, острые черты, «кисетный рот» с мелкими лучикообразными рубчиками — лицо пациента с акросклеротическим вариантом склеродермии.
В клинической генетике описывается масса мелких аномалий развития (стигм), преимущественно локализующихся на лице и являющихся патогномоничными для диагноза или специфически ассоциирующихся с изменениями некоторых внутренних органов. Например — добавочный козелок типичен для патологии почек, пылающий невус лица сочетается с ангиомой полусферы мозга этой же стороны.
Глаза больного — важный элемент для установления диагноза. Будучи построенными из нескольких типов ткани (соединительная, нервная, мышцы, пигментный эпителий, сосуды) и притом достаточно доступными для исследования, они отражают состояние многих органов и систем всего организма и являются своеобразным «микрокосмом». Изменение этого органа, характер разреза глазной щели, нарушения зрения могут быть первым проявлением системных заболеваний. Так, при массовых офтальмологических исследованиях лиц в возрасте старше 50 лет у 1,5–2,3% впервые диагностируют сахарный диабет, а при обследовании 706 пациентов со старческой катарактой гипергликемия установлена в 14,3%.
Двусторонний экзофтальм с широко раскрытой глазной щелью, видимой белочной оболочкой над радужкой (симптом Грефе, «заходящего солнца») с редким миганием (признак Штельвага) и слабость конвергенции типичны для тиреотоксикоза. Экзофтальм возможен и при эутиреоидном зобе или при болезни Хашимото. Экзофтальм с отеками лица, одышкой, цианозом и венозным застоем типичен для синдрома верхней полой вены.
Односторонний экзофтальм в случае исключения травм (ретробульбарная гематома) в 50% случаев может возникать при гиперфункции щитовидной железы. Оставшиеся 50% составляют на опухоли орбиты, придаточных пазух носа, надглоточного пространства и другие объемные процессы (гранулема при болезни Вегенера, нейрофиброматозные узлы при болезни Реклингаузена, описан и эхинококкоз орбиты); воспалительные процессы (тромбоз кавернозного синуса, флегмоны ретроорбитальные, псевдотуморозный миозит глазодвигательных мышц). Среди наследственных синдромов с экзофтальмом могут протекать цереброретинальный ангиоматоз Гиппель — Линдау, туберозный склероз (также за счет опухолей), краниофациальный или челюстнолицевой дизостозы, башенный череп.
Пульсирующий экзофтальм типичен для артерио-венозных аневризм. Состояние известно как синдром Боннета — Дешауна — Бланка. Определяется внутричерепными и ретинальными артериовенозными аневризмами с возможным их прорастанием в орбиту и на лицо.
Кроме того, экзофтальм типичен для следующих наследственных состояний:
- синдром Алерта (а/д, но, как правило, новая мутация. Преимущественно страдают дети от пожилых отцов. Типичны брахицефалия + краниосиностоз + синдактилия + ложкообразные кисти. Может сочетаться с поликистозом почек);
- синдром Коллера. Идиопатическая воспалительная неспецифическая псевдоопухоль орбиты с экзофтальмом;
- синдром Крузона. А/д. Дородовый синостоз без синдактилии. Вторичная слепота и глухота. Подвывихи в локтевом суставе.
Энофтальм возникает при тяжелом обезвоживании и при многочисленных синдромах микроцефалии и микрофтальмии, а также при наследственных синдромах Бьерсона — Форсмана — Лемана, Курца и Лове.
Парез симпатического шейного ствола при поддавливании дополнительными ребрами, опухолями, при его травмах, сирингомиелии приводят к энофтальму, миозу и птозу. Последний возникает при миастении и параличе N. oculomotorius. При отсутствии всех указанных причин после обследования родственников можно диагностировать семейный птоз, не имеющий, впрочем, прогностического значения.
Гипоплазия бровей возникает при синдроме EEC. Брови практически полностью выпадают при гипопитуитаризме. Выпадению бровей (и ресниц) способствуют блокаторы β-адренорецепторов, тиреостатики, α-интерферон, кетоконазол, депилятории, пероральные контрацептивы, нейролептики. Исчезновение латеральных частей бровей отмечают при гипотиреозе. Избыточное разрастание медиальных частей — при синдроме Видемана — Беквита. Густые сросшиеся брови характерны для людей со сниженным порогом судорожной готовности (по данным ЭЭГ), являются диагностическим критерием синдромов алкогольной эмбриофетопатии, Корнеля-де-Ланге, Оливера — МакФарлана.
Количество ресниц значительно уменьшается при рецивируюших гордеолитах (ячменях), что требует проверки на наличие сахарного диабета. Двойной ряд ресниц отмечают при синдроме Майгеля — наследственный лимфатический отек II типа (лимфатические отеки на ногах, потеря слуха, бронхоэктазы, желтые или зеленые ногти). Длинные густые ресницы типичны для лиц с туберкулезной интоксикацией и при лечении гепарином.
Воспалительные изменения век отмечают при аллергических реакциях или коллагенозах.
Блефарит может развиться при недостаточном питании, гиповитаминозе, нарушении обмена веществ, туберкулезе, хронических очагах инфекции в носоглотке, хронической интоксикации, некорригированной дальнозоркости, плохих гигиенических условиях труда и быта. Причинами могут быть поражения краев век бактериями, вирусами, клещами. Блефариты проявляются зудом, покраснением и утолщением краев век, выпадением ресниц, образованием чешуек и корок между ресницами, клейким отделяемым, слезотечением, быстрой утомляемостью глаз. Наиболее известны аллергический блефарит, язвенный (хроническая стафилококковая инфекция волосяных фолликулов ресниц), себоррейный. В последнее время появилось много публикаций по так называемому демодекозному блефариту. Вызывается клещами Demodexfolliculorum и Demodex brevis. Клещи выявлены у 29% обследованных в возрасте 0–25 лет, у 53% — в возрасте 26–50 лет и у 67% — в возрасте 51 год–90 лет. Но в 80% всех случаев отмечают носительство без клинических признаков блефарита.
Сопоставляя показатели носительства клеща с частотой клещевых блефаритов, можно (даже с поправкой на неполную диагностику) предположить, что для реализации демодекозной этиологии требуются сопутствующие факторы (сахарный диабет, синдром сухих глаз, хронические интоксикации и т. д.).
В литературе описывается синдром «красных глаз». В ДД состояния необходимо учитывать следующие причины:
- субконъюнктивальное кровоизлияние;
- острая глаукома (достаточно редкое, но прогностически неблагоприятное начало. Сопровождается болью, инъецированностью склер, головокружением, рвотой);
- ранения роговицы, кератиты. Для последних типично медленное начало, реже — острое, перилимбальная или смешанная инъецированность, язвы роговицы;
- конъюнктивиты (табл. 2.8 и 2.9), болезнь Рендю — Ослера;
- увеит (медленное начало, зрачок неправильной формы с вялой реакцией на свет).
Таблица 2.8
Клинические проявления различных видов конъюнктивита (по D. Раvan с дополнениями, 1986)
| Симптом | Бактериальный | Вирусный | Аллергический | Токсический | Хламидийный (в том числе трахомный) |
| Инъецированность | Выражена | Умеренная (при аденовирусной инфекции) | Слабо умеренная | Слабо умеренная | Умеренная |
| Геморрагии | + (очень характерны для гоноккоковой инфекции) | + | |||
| Хемоз | ++ | +/- (при аденовирусной инфекции выражен) | ++ | +/- | +/- |
| Экссудат | Гнойный или слизисто-гнойный | Скудный, водянистый | Тягучий, беловатый или водянистый | Скудный | |
| Псевдомембраны | +/- (стрептококки; коринебактерии) | +/- | — (при буллезном эпидермолизе выражены) | ||
| Фолликулез век | +/- | + | + (иногда буллы) | + | ++ |
| Бугристость конъюнктивы век | +/- | + (при весеннем катаре типично поражение) | + | ||
| Бугристость конъюнктивы глазного яблока | +
(гнойная) |
++ | — | +/- | |
| Паннус | (за исключением «весеннего») | + |
Таблица 2.9
ДД различных видов конъюнктивита (по R. Kiegman с дополнениями, 1996)
| Этиология | Клиническая симптоматика | ||||
| Одно- или двусторонний | Характер экссудата | Веки | Начало/течение | Положительный эффект от | |
| Аденовирусный | Дебют — на одном глазу, через 2–3 дня — двусторонний | Прозрачный, мукоидный | Фолликулы | Постепенное; инфекция верхних дыхательных путей; аденопатия | Симптоматической терапии |
| Герпесвирусный | Односторонний (обычно) | Прозрачный, мукоидный | Фолликулы | Постепенно; кератит; язвы | Применения ацикловира |
| Бактериальный | Одно- или двусторонний | Гнойный | Папулы, гной | Постепенно | Местного применения антибиотиков |
| Гонококковый | Односторонний (у детей — двусторонний) | Гнойный, сукровичный | Отек, воспаление | Очень остро | Системная антибиотикотерапия |
| Микоплазменный | Двусторонний, редко — односторонний | Прозрачный, мукоидный | Фолликулы | Безболезненный, персистирует; новорожденные и сексуально активные лица | Применения эритромицина, тетрациклина |
| Аллергический | Двусторонний, может быть односторонний | Водянистый | Конъюнктива век зернистая (папулы), отек век — голубоватый | Постепенно; сезонность | Применения кромонов, лекролина, сперсаллерга |
| «Весенний» | Двусторонний | Водянистый | Большие папулы, характерно поражение верхнего века | Могут быть упорные кератины, сезонность; чаще — у подростков; нередко сочетается с БА | Применения кромонов, криодиструкции булл конъюнктивы |
| Химический | Одно- или двусторонний | Водянистый | Состояние зависит от повреждающего агента | Острое | Устранить воздействие раздражителя, промыть глаза |
| Реакция на контактные линзы | Одно- или двусторонний | Водянистый | Большие папулы (крупная зернистость) | Связь с ношением контактных линз | Заменить линзы или раствор для их обработки |
Голубые склеры присущи СПИД-эмбриопатии, более чем 30 наследственным синдромам дисплазии соединительной ткани, самым известным из которых в этой группе является несовершенный остеогенез («стеклянный человек»). Заболевание развивается за счет мутации гена, ответственного за синтез проколлагена I типа. Проявляется гипермобильностью суставов, деформациями грудной клетки, тонкой эластичной кожей, пролабированием митрального клапана, множественными переломами трубчатых костей, в зрелом возрасте — тугоухостью. Возможно сочетание с медуллярной дисплазией почек. После достижения половой зрелости переломы возникают редко. Голубые склеры типичны и для остеопетроза (мраморная болезнь Альберс — Шенберга), болезни Педжета, ренальной остеодистрофии и многих других патологичексих состояний.
Желтушное окрашивание склер, как правило, является признаком поражения печени или гемолитической анемии.
Изъязвление роговой оболочки глаза характерно при эксудативной полиморфной эритеме, гиповитаминозе А.
Локальные помутнения роговицы отмечают при гиперкальциемии (гиперпаратиреоидизм, интоксикация витамином D2, саркоидозе). Диффузные помутнения выявляют при кератите (герпетическом, болезни Рейтера, Бехчета, врожденном сифилисе) или какосложнения медикаментозной терапии амиодароном, хлорохином, индометацином.
Казуистически описывают отек роговицы на фоне лечения антикоагулянтами. Но чаще отек роговицы возникают при синдромах Когана — Ризе (гетерохромия радужной оболочки + дистрофия роговицы + глаукома + эктопия зрачка), Познер — Шлосмана (глаукома + циклит + гипохромия радужки), дистрофии эпителия роговицы Фукса (помутнение роговицы и ее дистрофия).
В старости возможно образование так называемых arcus senilis или arcus lipoides, обусловленных отложением липидов.
Хрусталик теряет свою прозрачность при внутриутробных инфекциях или других повреждающих факторах в период закладки, при хроническом иридоциклите, интоксикациях динитрокрезолом, глюкокортикоидами, галоперидолом, при травмах, при дистрофической миотонии Куршмана — Штейнера, а также при нарушениях обмена (сахарный диабет, гипопаратиреоидизм, галактоземия). Катаракта в старческом возрасте обычно не вызывает диагностических затруднений.
Радужная оболочка поражается при иридоциклитах или иритах (при лептоспирозе, листериозе, токсоплазмозе, сифилисе, боррелиозе, герпесе), а также при хроническом артрите, болезнях Рейтера, Бехтерева, Бехчета, саркоидозе. Разная окраска радужной оболочки может развиваться после ее воспаления или быть врожденной, например, при синдромах «Леопарда», Когана — Риза и Ваарденбурга. Радужка изменяется при болезни Вильсона — Коновалова за счет отложения меди с формированием зеленовато- желтого кольца Кайзера — Флейшнера. Аниридия характерна при аплазии надколенника. Несмотря на большое число публикаций и массу энтузиастов, «иридодиагностика» не получила рационального объяснения и ее клиническое применение пока не оправдано.
Зрачок расширяется под действием симпатических импульсов. Раздражение парасимпатических волокон вызывает его сужение. Широкий зрачок бывает при испуге, боли, приступе глаукомы, повреждениях среднего мозга и глубокой коме. Узкий зрачок отмечают при интоксикации опием или эфирами фосфорной кислоты, применении пилокарпина, при лечении глаукомы, при ирите, повреждениях моста мозга, синдроме Горнера. Паретичный зрачок выявляют при повреждениях среднего мозга, основания черепа (энцефалит, сифилис, аневризма, опухоль), синехиях и глаукоме. Односторонний миоз характерен при раздражении шейного вегетативного ганглия.
Стекловидное тело мутнеет при ретинопатии недоношенных, травмах, амилоидозе типа III и амилоидной полинейропатии типа II, заболеваниях соединительной ткани. Стекловидное тело разжижается при синдромах Гольдмана — Фавра (витреоретинальная дегенерация + ретинопатия + ночная слепота + ретиношизис + падение зрения + отек макулы + катаракта) и Вагнера (аваскулярные области в сетчатке).
Сетчатая оболочка меняет свою окраску при пигментном ретините, синдроме Лоренса — Муна — Бидля и эластическа псевдоксантема. Диабетическая ретинопатия, коагулопатии и инфекционный хориоретинит могут быть причиной повреждений сетчатой оболочки и снижения остроты зрения.
Кандидозный эндофтальмит диагностируют у 6% больных с диссеминированным кандидозом. Фундоскопически выявляют скопления экссудата, распространяющиеся от ретины в стекловидное тело и напоминающие комки ваты. Такой тип поражения характерен при иммуносупрессии, при длительном парентеральном питании и наркомании.
Нистагм — непроизвольное ритмическое возвратно-поступательное движение глаз. Вертикальный нистагм возникает при амблиопии, альбинизме, в то время как горизонтальный нистагм связан с болезнью Меньера, рассеянным склерозом, с опухолями, интоксикациями седативными, противосудорожными препаратами и алкоголем (см. Сенсомоторные расстройства).
Синдром сухих глаз характерен при многих ревматических болезнях (синдром Шегрена, системная склеродермия), аплазии слезных желез, синдромах Уемуры, LADD, EEC, Райли — Дея, дисфункции желез в климактерический период, нарушении эпителиальной мембраны роговицы после перенесенного кератита или дистрофии роговицы, «глазном офисном и мониторном» синдроме, инстилляции блокаторов бета-адренорецепторов и кортикостероидов, реакции на контактные линзы, смог, дым, сухой кондиционированный воздух. Одной из частых причин синдрома сухих глаз являются оперативные вмешательства по поводу аномалии рефракции, широко рекламируемые в средствах массовой информации. Больные жалуются на ощущение инородного тела в конъюнктивальной полости, «жжение и резь», плохую переносимость ветра, кондиционированного воздуха, дыма, светобоязнь, колебания остроты зрения днем и ухудшение зрения к вечеру. Очень характерны болевая реакция на закапывание в конъюнктивальную полость индифферентных глазных капель, уменьшение или отсутствие у краев век слезных менисков, конъюнктивальное отделяемое в виде слизи. Выявляют локальный отек бульбарной конъюнктивы с «наползанием» на свободный край века, «вялая» гиперемия конъюнктивы, «загрязняющие» слезную пленку включения.
Самый простой диагностический тест — это пробы Ширмера. Определяют (в мм) смачивание слезой стандартной полоски фильтрованной бумаги. Также применяют пробу с флюоресцеином. Краситель не задерживается на здоровом глазу, но при ксерозе длительно сохраняется на конъюнктиве, особенно задерживаясь в местах эрозий.
Для ДД синдрома сухих глаз (ксероза) и других глазных изменений (дегенерация, воспаление) используют правило Tseng. Если подозрительные изменения локализуются исключительно в экспонируемой зоне поверхности глазного яблока (ограничена краями нормально открытых век), то их связывают с ксерозом. Если же патологические участки захватывают и «неэкспонированную» зону роговицы и конъюнктивы, то есть основание прежде всего думать о нексеротических процессах.
Следует особо выделить, что в дебюте синдрома сухих глаз при минимальных признаках заболевания компенсаторно развивается усиленное образование слезы, что приводит к ошибочной диагностике. Образование слезной жидкости усиливается на ветру, в накуренных помещениях и при воздействии других неблагоприятных факторов внешней среды.
Усиленное слезотечение характерно при атрезии или посттравматической облитерации слезно-носового протока, заболеваниях роговицы, инородных телах в глазу. Истинное усиление слезообразования отмечают при парезе лицевого нерва, врожденном дискератозе, синдроме Харлина (невралгия N. nasociliaris et Ggl. ciliare в результате воспалительных процессов по соседству с указанными структурами или закупорке внутренней сонной артерии), синдроме Тигесона (хронический поверхностный рецидивирующий кератит без конъюнктивита) и др.
Снижение остроты зрения или, тем более слепота, — крайне тяжело переносит пациент. У детей 30–35% всех случаев приобретенной слепоты приходится на травмы. При врожденном амаврозе следует исключать пороки развития, врожденную или перинатальную инфекцию, недоношенность, гипоксию. Приобретенная нетравматическая слепота или резкое снижение зрения у детей развиваются при катаракте, хориоретините, увеите, ретинобластоме, внутричерепной гипертензии, васкулите, лейкозе, демиелинизирующих заболеваниях, опухолях. Клинические проявления зависят от возраста, физических особенностей ребенка, системного заболевания, одно- или двусторонности офтальмологического процесса. Первыми признаками могут быть робость, неловкость движений, самоограничение контактов и активности в освоении пространства, снижение успеваемости, нистагм и косоглазие.
У взрослых спектр причин, обусловливающих снижение остроты зрения или даже полную его потерю, несколько иной (табл. 2.10).
Таблица 2.10
Наиболее вероятные причины потери зрения у взрослых
| Локальные
Катаракта (50–60%) Начиная с возраста 50 лет часто выявляют опалесценцию хрусталика. На зрение обычно это существенно не влияет. Частыми причинами катаракты являются травмы, хирургические вмешательства, сахарный диабет, кортикостероидная терапия Возрастная дегенерация макулы (25–30%) В 90% случаев протекает в «сухой форме» (друзы). Периферическое зрение страдает мало, дневное зрение компенсировано. У 10% больных протекает во «влажной форме» с неоваскуляризацией, кровоизлияниями и катастрофически быстрым снижением остроты зрения Глаукома (4–9%) Хроническую открытоугольную глаукому выявляют у 7% людей в возрасте 65 лет–74 года и у 8% — в возрасте 75 лет и старше. Глаукому могут обусловить системное или местное применение кортикостероидов (у генетически предрасположенных лиц), травмы, хирургическое вмешательство Отслойка сетчатки Ангиоретинопатия Окклюзия центральной вены или центральной артерии сетчатки Ишемическая нейропатия зрительного нерва Системные Транзиторные ишемические атаки (amaurosis fugax) Височный артериит Сдавление зрительного нерва |
Ушная раковина меняется при подагре (узелки как результат депонирования кристаллов мочевой кислоты), сердечной недостаточности (цианоз), охронозе (серо-голубые пятна), при холодовой аллергии (цианоз на холоде). Мягкие ушные раковины типичны для синдрома Элерса — Данлоса, обвисшие уши — для рецидивирующего полихондрита (болезнь Мейенбурга — Альтхера — Ульриха). Маленькие ушные раковины характерны при синдроме Эшера — Хирта (снижение слуха из-за нарушения проводимости в результате отстутствия связи между наковальней и стремечком). Односторонняя гипоплазия ушной раковины типична для синдрома Гольденхара. Большие оттопыренные ушные раковины свойственны при синдромах Марфана, Циммермана — Лабанда. Снижение звуковой проводимости отмечают у пациентов с поражением внешнего или среднего уха при серных пробках, ранениях барабанной перепонки, среднем отите, холеастеатоме, катаральном воспалении евстахиевой трубы, отосклерозе или анатомических дефектах. Повреждения улитки, 8-й пары черепно-мозговых нервов, центральные нарушения обусловливают нарушения звуковосприятия. Медленно прогрессирующее снижение слуха характерно при хронической акустической травме. Для взрослых это чаще профессиональная вредность. Для детей старшего возраста, подростков и юношей — это прежде всего результат посещения рок-концертов, дискотек, прослушивания музыки через наушники при интенсивности звука >110 дБ (безопасный уровень <80 дБ). Если нарушения слуха возникают остро и односторонне, можно думать о сосудистом процессе, нарушениях костной проводимости в среднем ухе, разрыве кохлеарной мембраны. Одна из причин синдрома неожиданной потери слуха — перилимфатическая фистула, которая является ни чем иным как истечением жидкости из внутреннего уха в воздушное пространство среднего. В результате возникает острое снижение слуха или колебания слуховой чувствительности. Все это может сопровождаться головокружением. Фистула возникает в наиболее слабом месте, отделяющем внутреннее ухо от среднего — овальное или круглое окно. Фистула возникает при врожденных дефектах, при усиленных физических упражнениях (особенно вращениях), резкой смене давления (подъем на самолетах, погружение с аквалангом). Лабиринтит как осложнение вирусной инфекции обусловливает одностороннее снижение слуха, в комбинации с шумом, спонтанным нистагмом и головокружением — свидетельствует о болезни Меньера. Ототоксические препараты (аминогликозиды, ванкомицин) ведут к медленному снижению слуха, как и хроническая акустическая травма. Невринома слухового нерва растет очень медленно и длительное время единственным симптомом остается одностороннее прогрессирующее снижение слуха. Опухоли ствола мозга, инфаркт, рассеянный склероз и другие изменения в височной области могут вести к снижению слуха.
Снижение слуха (тугоухость), тем более глухота, влекут за собой тяжелейшую психологическую и социальную дезадаптацию. И если эту проблему достаточно тяжело переживают взрослые, то для детей эти процессы катастрофичны, ибо к психологическим и социальным нарушениям добавляются и неизбежные нарушения развития речи, интеллекта, личности. (И действительно, весь первый год жизни ребенок слушает чужую речь и звуки окружающего мира, только затем он начинает строить свою.) Проблема усугубляется и тем, что взрослый замечает снижение слуха, а дети младшего и дошкольного возраста это состояние просто не фиксируют. Нередко врожденное снижение слуха у ребенка выявляют только в возрасте 2–3 лет. В единичных случаях тугоухость выявляли только с началом посещения школы, в 6–7 лет. То есть снижение слуха, глухота — «тихое» нарушение. И жизненно важная задача семейного врача и/или педиатра инициировать процесс диагностики тугоухости у ребенка. При оценке состояния ребенка или при расшифровке синдрома тугоухости следует учесть, что у детей, особенно младшего возраста, превалируют сенсоневральные причины снижения слуха (табл. 2.11).
Таблица 2.11
Классификация сенсоневральной тугоухости у детей
| ВРОЖДЕННЫЕ
I Генетически определенная тугоухость А. Нарушения слуха существуют с рождения
— Мишела — Мондини — Шайбе
— Александера 2. Снижение слуха сочетается с другими аномалиями
— Ваанденбурга (снижение слуха может развиться позже) — Жерваля — Ланге — Нильсена — Пендреда — Ушера
Б. Отсроченная тугоухость
Семейная прогрессирующая сенсоневральная тугоухость 2. Сочетанная
— Альпорта — Рефсума — Крузона
— Клиппеля — Фейля
II Негенетическая тугоухость А. Изолированная ототоксическая (прием матерью стрептомицина, хинина) Б. Сочетанная ототоксическая, вирусная протозойная, спирохетозная, после аноксии/гипохсии, родовой травмы, гемолитической болезни, недоношенность III Синдромы с неопределенным соотношением генетических и негенетических факторов (Вильдерванка, Гольденхара, CHARGE и др.) ПРИОБРЕТЕННЫЕ
Наряду со стандартными исследованиями слуха в соответствующие возрастные периоды необходимо активно выявлять состояния, которые могут свидетельствовать о плохом слухе:
Факторами высокого риска, обусловливающими развитие тугоухости, являются:
При анализе случаев тугоухости наиболее частыми и значимыми факторами риска были:
|
Тугоухость (глухоту) часто выявляют при следующих генетически детерминированных состояниях: врожденная краснуха, интоксикация ртутью, дефицит йода, синдромы Варденбурга, Гурлера, Гунтера, Картагенера, Киллиана/Тешлера — Николя, Коккейна, кранио-метафизальная дисплазия, синдромы Леви — Холлистера, Сентера, Стиклера, склеростеоз, трисомии 13, Тренера — Коллинза, Марото — Лами (тип мукополисахаридоза), Маршалла, Мельника — Фразера, Моркио, Моро, множественного летингенеза, Нагера, ото-палато-дигитальные I и II типа, все синдромы фацио-аурикуло-вертебрального спектра, фронто-метафизиальная дисплазия, Шпринцера, синдромокомплекс CHARGE, 1 Sq-синдром.
Реже тугоухость сопутствует следующим состояниям: акродизостоз, Баллера — Герольда, Бада — Билля, Вейля — Маркезани, дискератоза врожденного, Камурати-Энгельмана, Карпентера, кляйдо-краниального дизостоза, Крузона, де Ланге, Лангера — Гедеона, метафизальная хондродистрофия типа Янсена, Миллера, Нунен, окуло-денто-дигитальный, остеогенеза несовершенного I типа, прогерии, расщепления неба, Ригера, аномалада Робина, Фанкони анемия, Сетра — Хотцена, трисомии 8, фибродисплазии прогрессирующей оссифицирующей, церебро-костомандибулярный, Шайе, CHILD, EEC.
Шум в ушах значительно осложняет жизнь пациентов. Шум в ушах (тиннит) в тот или иной период жизни отмечает едва ли не каждый взрослый, а 6–7% всей популяции характеризуют его как «постоянный» и/или «выраженный».
Шум в ушах может быть представлен как объективный (воспринимаемый не только пациентом, но и исследователем) и субъективный.
Объективно существующий шум обусловлен региональными артерио-венозными мальформациями, передачей тонов и шумов сердца или протезов клапанов, сокращениями небных мышц, mm. tensor tympani или стремечка.
Субъективный шум возникает при серных пробках, перфорации барабанной перепонки, отитах, отосклерозе, гломусной опухоли, старческой тугоухости, в итоге физической, акустической или баротравмы, при вирусной нейропатии, поражении ототоксинами, болезни Меньера, невромах акустического нерва, опухолях мосто-мозжечкового угла, а также при рассеянном склерозе, гипо- или гипертиреозе, сахарном диабете.
Потрескивание и низкочастотные шумы больные слышат при механических повреждениях, при евстахиите. Посвистывания и высокочастотные шумы беспокоят при изменениях нерва или улитки. Прием салицилатов и хинина в высоких дозах часто обусловливает появление шума в ушах. Шум в ушах очень часто сочетается со снижением остроты слуха.
Нос уплощается при рецидивирующих хондритах, а при гранулематозе Вегенера становится седловидным. Гиперплазия сальных желез ведет к развитию ринофимы (нос по типу «цветной капусты»), а себоррейные аденомы являются признаком синдрома Прингля — Бурневилля (туберозный склероз, внутричерепные обызвествления, субунгвальные фибромы, опухоли сердца, сетчатки и почек). Крючковидный нос типичен для синдрома Рубинштейна — Тейби; грушевидный — для трихо-рино-фалангеальных синдромов типа 1 и 2; широкий плоский — для синдромов брахимелии, точечной хондродисплазии типа Шеффилда, точечной X-хромосомной доминантной хондродисплазии, Аарского, Коффина — Сириса. Клювовидный нос характерен при синдромах прогерии и микрогнатии.
Носовые кровотечения могут быть при сухости слизистой оболочки (атрофический ринит), полипах, хроническом гайморите, аномалиях сосудистого сплетения, болезни Ослера, идиопатической тромбоцитопенической пурпуре, облитерирующем аортоартериите на участке между дугой и бифуркацией. Причем в двух первых случаях кровотечения бывают, как правило, асимметричными, то есть то из одной, то из другой ноздри, а не из двух одновременно, как при коагулопатиях.
Носовые кровотечения могут быть обусловлены местными или системными причинами.
- Местные причины:
— частые:
- травмы, в том числе дигитальная (нельзя же в серьезном руководстве писать «ковыряние пальцем в носу». Но, увы, это так);
- химические повреждения;
- постоперационные;
- переломы;
- воспалительные;
— редкие:
- опухоли (в том числе ангиофибромы);
- инородные тела;
- деформация или перфорация перегородки;
- телеангиэктазии.
- Системные причины:
- АГ;
- коагулопатии наследственные и медикаментозного генеза.
Зубы изменяют цвет и формируются с дефектами эмали при рахите, гипопаратиреоидизме, целиакии, применении антибиотиков тетрациклинового ряда (рис. 2.8). Преждевременная потеря зубов характерна при витамин-D-резистентом рахите, при гипофосфатемии и лимфоме Беркита. При болезни Дауна зубы мелкие, редкие. Зубы Гетчинсона (с треугольной выемкой на резцах) — типичный признак врожденного сифилиса. Коричневая или серо-голубая окраска зубов характерна при несовершенном дентиногенезе типа II, красная — при врожденной порфирии.
Гиперплазия десен в физиологических условиях характерна при рахите, а в патологических — при лейкозе. Гипертрофия десен свойственна ювенильному фиброматозу I и II типа. Воспалительные заболевания десен типичны для болезни Дауна и сахарного диабета. Черная кайма на деснах свидетельствует о свинцовой интоксикации, серо-голубая — сопровождает серебряную или висмутовую интоксикацию.
Большой рот типичен для синдрома Ангельмана, маленький — для синдромов Фримена — Шелдона, Элерса — Данлоса, гипоплазии бедра с дисморфизмом лица. Слизистая оболочка полости рта покрывается беловатыми четко очерченными пятнами при лейкоплакии и кандидозе. Лейкоплакия на боковой поверхности языка — достаточно патогномоничный симптом СПИДа и обусловливается вирусом Эпштейна — Барр. Молочница — свидетельство чаще всего системного заболевания, возникает при ДБСТ, злокачественных опухолях, терапии кортикостероидами, иммуносупрессантами и цитостатиками. Длительное время молочница может быть единственным первым симптомом СПИДа. В физиологических условиях отмечается при беременности. Рецидивирующие афты в полости рта достаточно часто возникают при герпетических стоматитах. Они могут быть дебютом пернициозной анемии, фолиево-дефицитной анемии, недостатка железа или признаком целиакии. Изъязвления слизистой оболочки характерны для инфекционного мононуклеоза, сифилиса, экссудативной многоформной эритемы (синдром Стивена — Джонсона), опухолей слизистой оболочки, синдрома Бехчета, осложняют терапию цитостатиками.
Ксеростомия — сухость слизистой оболочки полости рта. Отмечается при воспалении слюнных желез (болезнь Бека), на фоне лечения атропином, антигистаминными и антипсихотическими препаратами, а также при сахарном диабете, гипертиреозе, синдроме Шегрена, ботулизме. Лучевая терапия также обусловливает ксеростомию. Гиперпигментация слизистой оболочки полости рта отмечается при синдроме Пейтц — Егерса, болезни Адиссона, гемохроматозе, порфирии, кровотечениях, интоксикациях свинцом, хлорохином, хлорпромазином, мышьяком, висмутом, ртутью и серебром.
Избыточное выделение слюны характерно при прорезывании зубов, при глистных инвазиях, у наркоманов и у невропатов. Истинную сиалорею отмечают у детей с последствиями внутричерепных кровоизлияний в родах. Истинную сиалорею необходимо отличать от слюнотечения при других состояниях: стоматиты, раздражающие инородные тела во рту (зубные протезы), гипертрофия миндалин и аденоидов, хроническая аспирация, спастические состояния пищевода или его стриктуры.
Макроглоссия характерна при болезни Дауна, ангионевротическом отеке, кретинизме, акромегалии, амилоидозе, синдроме Видемана — Беквита, гликогенозах, мукополисахаридозе, мышечной дистрофии Дюшена. Атрофия языка может быть одним из проявлений синдромов Джексона, Кеннеди II, Тапия, инфантильной спинальной мышечной атрофии Верднига — Гоффмана. Микроглоссия заставляет думать об оро-акральных аномалиях, узкий язык — синдром Германа (контрактуры коленных и локтевых суставов, камподактилия, лимфатические отеки, долихоцефалия), зеленый язык— болезнь SMON (subacute myelo-optico-neuropathy — результат передозировки галогенизированных гидроксихинолинов, используемых для лечения и профилактики инфекционных заболеваний кишечника и энтеропатического акродерматига).
Глоссоптоз является проявлением артрогрипоза Х-связанного типа 1, всех синдромов микрогении. Фибрилляцию языка отмечают при спинальной мышечной атрофии типа Кугельберга — Веландера, фасцикуляции — при инфантильном бульбарном параличе и болезни Махало, а постоянные ворочающиеся движения — при цервико-лингво-мастикаторном синдроме.
Обложенный язык издавна считают признаком нарушения общего состояния организма. Грубый коричневый налет по средней части языка типичен для колита и грибковых поражений. Географический язык (рис. 2.9) может быть наследственным признаком и наследуется мультифакторно или является признаком аллергии. При синдромах Дауна и Мелькерсона — Розенталя характерна так называемая lingua skrotalis. Боль в языке психогенного характера нередко отмечают у женщин в менопаузальный период. Изменение окраски языка служит диагностическим признаком скарлатины (схождение белого налета с кончика и боков с освобождением яркой поверхности с гиперплазированными сосочками — малиновый язык). Атрофия слизистой оболочки с формированием «лакированной поверхности» типична для арибофлавиноза, гунтеровского глоссита и возникает при пернициозной и фолиево-дефицитной анемии, пеллагре, сифилисе и дефиците железа.
Запах сложно описать, способность к его восприятию весьма вариабельна, однако по запаху, исходящему от больного, можно предположить наличие некоторых заболеваний. Например, появление неприятного запаха изо рта при пальпации эпигастральной области свидетельствует о гастро-эзофагеальном рефлюксе. Сладковатый гнилостный запах возможен при абсцессе легкого; сладковатый — при дифтерии, каловый — при дивертикулах пищевода, непроходимости кишечника, гастро-колитических свищах; ацетоновый — при голодании, кетоацидозе, интоксикации салицилатами; чесночный запах — при интоксикации мышьяком и фосфором; запах свежего мяса — при острой атрофии печени.
Запах пота меняется с возрастом. От грудного ребенка пахнет грудным молочком, от старика — «чем-то острым, чем всегда пахнет от стариков» (Л. Толстой «Война и мир», характеристика старого князя Болконского). Кислый запах пота отмечают при лечении салицилатами, мышиный — при фенилкетонурии, прелых яблок — при газовой гангрене, зрелого камамбера — при протеолитических инфекциях с абсцедированием.
Удушливый, резко зловонный запах моча приобретает при обильном употреблении спаржи, чеснока, хрена, каловый — при пузырно-ректальном свище, неприятный аммиачный запах — при цистите, карамельный — при болезни мочи кленового сиропа, рыбный — при тирозинемии, мышиный — при фенилкетонурии, плесневый — при лечении пенициллиновыми антибиотиками.
Гнилостный запах мокроты заставляет думать об абсцессе легкого.
Каловые массы пахнут резко гнилостно при синдроме мальабсорбции, прогорклым маслом — при шигеллезе.
Гнилостный запах вагинального секрета сопровождает вагинит, инородные тела влагалища, распадающиеся опухоли.
Спиртовый запах ликвора бывает при криптококковых менингитах.
Речь — возможность нашей связи с миром — результат сложного взаимодействия нескольких систем организма, в первую очередь ЦНС. Через нервные пути управляемое давление воздуха в фазе выдоха заставляет колебаться голосовые связки. Возникающий звук модифицируется размерами, формой и напряжением резонирующих камер. К ним относятся глотка, полости рта и носа. Дальнейшее формирование речи идет за счет движения языка, челюсти, губ и, безусловно, окрашивания ее эмоциями.
Расстройства речи объясняются нарушением моторики (дизартрией), анатомическими повреждениями звуковоспроизводящей области (афазией) или нарушениями мышления при психозах. В общей медицинской практике нарушения речи обусловлены прежде всего механическими причинами (обструкция воздухопроводящих резонирующих камер, вялость мягкого неба и т. д.). Одной из наиболее важных структур, формирующих речь, является велофарингеальный сфинктер. Он изолирует ротоглотку от атмосферного давления и носоглотку во время глотания, в этот же период открывает евстахиевы трубы и соединяет или опять разъединяет полости носа и рта во время речи. Сфинктер состоит из 6 мышц: m. levator veil palatini, т. tensor veli palatini, m. palatoglossus, m. palatopharyngeus, m. uvulae et superior m. pharyngeal constrictor. Наиболее важная мышца сфинктера — т. levator veli palatini.
Велофарингиальная недостаточность (ВФН) — это неспособность сфинктера к эффективному выполнению функции, выливающаяся в нарушение артикуляции и гнусавость. Звуки нормальной речи — звонкие или глухие. Все гласные — звонкие, согласные могут быть звонкими или глухими. Только носовые «м», «н», «р» произносятся с открытым велофарингиальным сфинктером. Все другие звуки требуют различной степени смыкания сфинктера. Если сфинктер открыт, гласные и звонкие согласные звуки резонируют в полости носа, и давление в полости рта падает. Конечный результат — выход воздуха через нос и назальная вялость, редуцированность речи. Таким образом, первичный речевой дефект ВФН — гипоназальность. Вторичный — усиленный поток воздуха из носа, искажение артикуляции, компенсаторная артикуляция и фоноторная поддержка, дисфония. Возникает гримасничанье.
Причины ВФН — органические или функциональные (табл. 2.12).
Таблица 2.12
Органические и функциональные причины ВФН
| Органические
Структурные: — а/д наследуемая диспропорция велофарингиальных структур — атрофия аденоидов в пубертатный период — нейрофиброматоз — гемифациальная микросомия — тетрада Фалло — расщелина неба — подслизистая, в том числе скрытая расщелина неба — короткое мягкое небо — большая глотка — небные перепонки — большие миндалины — токсическое действие лекарственных препаратов на плод (фенобарбитал, фенотоин) Приобретенные: — резекция неба — рубцы на небе — последствия тонзилэктомии или аденотомии Нейромышечные: Врожденные: — энцефалопатия — болезнь Мебиуса Приобретенные: — неврологические заболевания — мышечные заболевания Функциональные — имитация — низкая мотивация к правильной речи — реакция конверсии |
В 70–92% причиной ВФН является расщелина неба. После оперативной коррекции порока ВФН остается у 20–75% пациентов. Дети с полной расщелиной неба вскоре после рождения попадают к челюстно-лицевым хирургам. Это не проблемы врача общей практики, педиатра или терапевта. Проблемы терапевтические, психологические, логопедические, сексуальные4 начинаются потом. Но они ожидаемые и прогнозируемые, врач готов к ним. Однако подслизистое (в том числе скрытое) расщепление неба — явно междисциплинарная проблема.
Признаками подслизистого (в том числе скрытого) расщепления неба являются короткий апоневроз, расщепление язычка, сглаженный срединный шов, отсутствие впадинки или даже наличие выступа на заднем крае твердого неба.
Механические нарушения моторики (гнусавость при аденоидах, миастения с нарастающим в процессе речи утомлением мышц) необходимо отличать от неврологически обусловленной дизартрии. Дизартрия часто сопутствует синдромам Ангельмана, спинно-церебеллярной атаксии Герстмана — Штрейслера, атаксии Фридрейха, болезням Куру и Минаматы, метохроматическим лейкодистрофиям типов Аустина, Шольца — Гринфилда. Тихая, спотыкающаяся речь, дающаяся пациенту с неимоверными сложностями, развивается при уменьшениях коркового кровотока и носит название кортикальной дизартрии. Монотонная тихая речь типична для стволовых расстройств и характерна при паркинсонизме. Выталкивание отдельных рубленных слогов и фраз (скандированная речь) типична для мозжечковых нарушений, как это отмечают при ишемических, опухолевых, дегенеративных процессах (оливо-понто-церебеллярная дегенерация), при рассеянном склерозе. При этом часто возникает триада Шарко: скандированная речь, нистагм, тремор. При боковом амиотрофическом склерозе (бульбарная речь) с поражением области ядер продолговатого мозга речь тихая, медленная, невнятная («каша во рту»). Одновременно возникают и расстройства глотания. При афазии вследствие повреждения кортикальных центров речи нарушено ее формирование. Афазия Брока характеризуется медленной речью, требующей неимоверных усилий и построенной в телеграфном стиле, что часто сочетается с нарушениями письма. Афазия Вернике — сенсорная — речь плавная, но совершенно непонятная: названия предметов ошибочны. При утере способности давать определения (амнестическая афазия) речь изобилует описаниями предметов, а не их названиями.
Голос изменяется (дисфония, афония) при поражениях гортани (ларингиты, опухоли), а также в период полового созревания, при менструации и в менопаузальный период. Низкий, хриплый голос типичен для гипотиреоза, акромегалии, вирилизма, синдрома Вильямса — Бюрена.
Последнее состояние характеризуется в основном суправальвулярным стенозом аорты и периферическим стенозом легочной артерии (вариабельность кардиальной патологии очень велика) с другими множественными аномалиями. В их число входят врожденно низкий рост, характерное изменение черт лица по типу «лица гнома» или «лица эльфа»: полное округлое лицо, микроцефалия, нависшие щеки, выступающий лоб, гипертелоризм, сходящееся косоглазие, синофрис, нос «картошкой» с уплощенной переносицей, широкой рот, толстые сосискообразные губы, удлиненный фильтр, выступающий подбородок. Микродонтия («мышиные зубы»), дисплазия зубов, часто отсутствуют два нижних резца и/или премоляра. Умственное развитие низкое. У мальчиков — гипогенитализм (микропенис, микроскротум, крипторхизм), у девочек — преждевременное половое созревание. Дисплазия радужной оболочки и/или почек. Синдром описывается как спорадический, вероятность а/д передачи ставится под сомнение.
При гипофизарной карликовости голос высокий. Хронически хриплый голос отмечают у 2–24% детей и 1–15% взрослых. Хриплый голос может быть при усталости, ларингите, интенсивном курении, травмах, алкоголизме, а также при инородных телах, папилломах и злокачественных опухолях голосовых связок. Все, что ведет к хронической травме голосовых связок, приводит к образованию так называемых узелков на голосовых связках. Голосовые узелки ответственны за 38–78% охриплости у детей (м:д=3:1). Голосовые узелки чаще появляются с началом посещения ребенком школы, их частота значительно снижается в пре- и пубертатном возрасте. Голосовые узелки наряду с профессиональной предрасположенностью чаще бывают у агрессивных гиперактивных людей с высоким уровнем внутренней тревоги и страха. В ряде случаев существует внешняя органическая причина. Лучший пример — высокая частота голосовых узелков у пациентов с ВФН. Это — результат вторичного компенсаторного напряжения голоса и травмы голосовых связок.
Голосовые узелки обусловлены механической травмой вибрирующей слизистой оболочкой на голосовой связке. Травма — результат усиленной или длительной вибрации («сорванный командами» голос Понтия Пилата — см. «Мастер и Маргарита» М. Булгакова). Максимальное усилие приходится на середину связки. Узелки обычно двусторонние, но их размеры на правой и левой связках могут быть разными. Острая травма вызывает застойную гиперемию и отек со скоплением жидкости в подслизистом слое. Хроническая травма вызывает гиалинизацию и организацию отека. В любом случае поверхностный эпителий остается неизменным. Диагностика основывается на непрямом исследовании гортани зеркалом или гибким назофарингоскопом. Очень редко применяют прямую ларингоскопию. Во время исследований отмечают сниженную вибрационную способность и неполное смыкание связок, что и обусловливает охриплость.
ДД голосовых узелков включает полипы, кисты голосовых связок, папилломатоз, злокачественные опухоли. Полипы обычно односторонние, на ножке, возникают у лиц с непостоянной перегрузкой голоса. Папилломатоз — доброкачественное разрастание, напоминающее бородавки, с высокой частотой рецидивирования.
Из внешних причин охриплости голоса могут быть струма (или карцинома щитовидной железы), состояния после операции на щитовидной железе, опухоли средостения, аневризма аорты, опухоли и ранения в области шеи, переломы основания черепа, то есть все то, что может повредить возвратный нерв.
«Щадящая» афония возникает при первичных или возвратных формах невралгии N. glossopharingeus. Состояние известно как синдром Сикарда и может быть идиопатическим (первичным) и вторичным. Проявляется интенсивной, непереносимой, разрывающейся болью, которую пациенты сравнивают с ударами ножа. Боль длится не дольше 2 мин («болевой тик») и сменяется фазой рефрактерности к боли. Боль локализуется в одной полудуге неба, иррадиирует в язык, угол нижней челюсти, внутренне ухо, соответствующую сторону шеи. Из-за интенсивной боли пациенты боятся есть (дефицит массы тела вторичного характера). Одностороннее снижение вплоть до полной агеузии (или повышение) вкусовых ощущений (особенно по отношению к горечям по задней части языка). Во время болевых приступов возможен сухой раздражающий кашель или синкопы. Боль провоцируется глотанием твердой, горячей или холодной пищи, интенсивным жеванием, зевотой, громким разговором. Предвестниками болевого приступа являются ощущуение глухоты или истечение вязкой слюны.
Хриплый голос, слабый голос, афония могут возникать при парезе и параличе голосовых связок. Миопатический парез возникает при ларингите, дифтерии, гриппе, туберкулезе, тифе и некоторых других инфекционных заболеваниях, при голосовой травме, миопатии. Неврологические механизмы преобладают при опухолях, бульбарных нарушениях, спинной сухотке, истерии, повреждениях периферических нервов (состояние после операций на щитовидной железе, любых травмах, сдавлении).
Ложно-положительная диагностика пареза голосовых связок возможна при травмах хрящей гортани.
У новорожденных паралич голосовых связок — второе по частоте врожденное поражение гортани. Причины — изменения в ЦНС, периферической нервной системе, сердце и магистральных сосудах, дыхательных путях.
У детей более старшего возраста и взрослых односторонний парез (паралич) голосовых связок возникает в 4 раза чаше, чем двусторонний. Среди всех случаев унилатерального поражения значительно чаще поражена левая связка. При одностороннем параличе сохраняются свободное дыхание и слабый голос. Двусторонний полный паралич с нарушением дыхания — повод для неотложного вмешательства. При одностороннем параличе в случае исключения неоплазий или иных неотложных состояний самопроизвольное восстановление происходит в 55–60% случаев. Для взрослых срок выжидания составляет от 6 мес до 1 года, для детей — от 6 мес до 2–3 лет.
При всяком непонятном изменении голоса (его осиплости, хриплости, изменение высоты) необходимо проведение ларингоскопии для исключения рака гортани.
Изменения кожи отражают состояние внутренних органов. По утверждению Гебры, вся терапия произошла из дерматологии. Действительно, в свое время врачу не оставалось ничего другого, как наблюдать больного и отмечать изменение его внешнего вида. Но и сейчас описание изменений кожи весьма важно для диагноза. Окраска кожи зависит от кровоснабжения, уровня гемоглобина и от характера пигментации. Бледная кожа типична для анемии, хотя для суждения о степени анемии большее значение имеет цвет слизистой оболочки, поскольку толщина кожи и ее пигментированность в значительной степени вариабельны. При пернициозной анемии кожа голубоватая, а при гипофизарно-надпочечниковой недостаточности (синдром Симмондса — Шихана) цвет становится алебастровым. При почечной недостаточности наряду с бледностью отмечается и пастозность кожи. У лиц с гиперпигментацией кожи и представителей негроидной расы бледность и синюшность кожи проявляются как ее серость. Для микседемы типична сухая, грубая, холодная кожа.
Резкое покраснение лица, особенно в сочетании с инъецированностью конъюнктивальных сосудов, должно послужить основанием для ДД между первичной (истинной) полицитемией и вторичной полиглобулией. Вторичную полиглобулию сопровождает цианоз. Покраснение меньшей интенсивности отмечают при синдроме Кушинга, при АГ, сахарном диабете, алкоголизме.
Желтушное прокрашивание кожи типично при обильном употреблении моркови, при заболеваниях печени и гемолитической анемии, а кожа цвета «кофе с молоком» — симптом подострого бактериального эндокардита.
Пигментация кожи обусловлена присутствием меланина, который продуцируется в меланоцитах и депонируется в кератоцитах, клетках кожного эпидермиса. В зависимости от недостатка или избытка меланина говорят соответственно о гипо- или гиперпигментации. Самое частое состояние, протекающее с гипопигментацией, — витилиго. Оно возникает почти у 1 % населения и в 1/2 всех случаев начинается в возрасте до 20 лет. Заболевание обусловлено деструкцией меланоцитов. В основе лежит аутоиммунный процесс, и поражение кожи часто предшествует другим аутоиммунным заболеваниям, в том числе пернициозной анемии, тиреидиту Хашимото. У детей и подростков вероятность системных аутоиммунных расстройств (полигландулярный аутоиммунный синдром, поражение щитовидной железы, хронический слизисто-кожный кандидоз) выше, чем у взрослых.
Витилиго может быть двух вариантов: генерализированное и сегментарное.
Генерализированная форма представлена симметричными очагами поражения периорбитально, вокруг носа, рта, пупка, гениталий. Кроме того, депигментированные участки кожи образуются на ладонях, подошвах, туловище, над локтями и коленями.
Сегментарная форма витилиго чаще всего возникает у детей. При обследовании больших групп пациентов сегментарное витилиго выявлено у 19% больных детей и только у 5% взрослых.
Кроме того, депигментацию кожи вызывают следующие причины:
- хроническое белковое голодание;
- недостаточность гипофиза, заболевания щитовидной железы, очень редко — в виде локальных очагов при болезни Аддисона;
- поствоспалительные процессы на местах экземного поражения, псориаза, реже сифилиса, саркоидоза, волчанки;
- облучения, ожоги, травмы, локальная терапия кортикостероидами (феномен Кебнера);
- туберозный склероз, альбинизм, синдромы Кросса, Титце, Германски — Пудлака, Чедиака — Хигаши, мальабсорбции метионина, Ваарденбурга, ахроматический невус Ито;
- сахарный диабет.
Гиперпигментация должна послужить причиной ДД эфелид, ксантом, адрено-генитального синдрома типа 1, врожденной пойкилодермии типа Томсона, анемии Фанкони, синдрома Фелти, болезни Аддисона, кожного саркоидоза, мастоцитоза.
При последнем состоянии происходит неконтролируемая пролиферация мастоцитов, которая в случае кожных форм ограничивается кожей (и тогда говорят о так называемой пигментной крапивнице), а при системных вариантах поражаются кишечник, печень, селезенка, кости. Пигментная крапивница (рис. 2.10) чаще начинается в детстве и не прогрессирует в пубертатный период. Более того, 75% детей, заболевших мастоцитозом в возрасте 2 года, спонтанно выздоравливают в пубертатный период. Типичны коричневатые, иногда с желтоватым оттенком округлые папулезные или папуло-пустулезные высыпания, обычный диаметр которых составляет 5–6 мм. Приступы пигментной крапивницы (выброс гистамина из мастоцитов) провоцируются интенсивными физическими упражнениями, механическим раздражением кожи, горячей ванной, салицилатами, морфином, кодеином, полимиксином В, атропином, алкоголем, сырами (особенно пряными), рыбой семейства тунцовых, грецкими орехами, шоколадом. После потирания (механическое раздражение) происходит усиленное выделение гистамина из пятен, которые являются ничем иным, как скоплением мастоцитов, сопровождаемое гиперемией, зудом, местным повышением температуры и квинке-подобным отеком вплоть до образования волдырей.

Системный мастоцитоз обычно начинается в пубертатном возрасте. Так, у детей с началом мастоцитоза в возрасте до 5 лет системные формы бывают только в 3–4%, а у более старших — уже в 10–30% случаев. Заболевание характеризуется прогрессирующим течением. Кожные высыпания более яркие, красно-коричневые, обильные, нередко окружены телеангиоэктазиями. Аналогичные высыпания выявляют на слизистой оболочке рта, а при эндоскопии — и на слизистой оболочке кишечника, прежде всего в прямой кишке. Из других органов поражается ретикуло-эндотелиальная система с развитием гепато- и спленомегалии, развиваются остеопороз, остеолиз, инфильтрация костного мозга, эозинофилия, анемия, лейко- и тромбоцитопения. Для системного мастоцитоза типичны зуд и «приливы». Последние проявляются как быстро развивающаяся темно-красная экзантема на лице и верхней половине туловища. Одновременно появляются признаки массивного освобождения гистамина: рвота, понос, кишечные колики, снижение АД, возможны потрясающий озноб, гипертермия, шок. Вся клиническая картина обычно длится около 30 мин. Необходимо провести ДД с карциноидным синдромом, при котором экзантема более цианотична, а общие симптомы длятся не более 10 мин.
Из невоспалительных пигментных дерматозов самой известной группой являются нейрокутанные синдромы, при которых отмечают пятна цвета «кофе с молоком» разной степени насыщенности, количества и локализации. Нейрокутанные синдромы характерны тем, что пигментация кожи и пигментация мозга (тем более, что обе эти структуры происходят из эктодермы) определяются одним геном и происходят в одно и то же время. Поэтому нарушение пигментации кожи ассоциируется со многими нарушениями нервной системы (пример, синдром исчезающего пигмента).
Впервые термин «ретинальная факома» был употреблен в 1920 г. для описания опухолевидного образования на сетчатке при туберозном склерозе, а позднее, после включения в одну трупу нейрофиброматоза и болезни Гиппеля — Линдау, было развито понятие «факоматоз». Факоматозы рассматривают как системные дисплазии, характеризующиеся комбинированными опухолевыми пороками развития кожи (рис. 2.11), глаз, нервной системы, а факому — как врожденную группировка невусных клеток в различных органах и тканях, которая в результате пролиферации клеток имеет потенциал разрастания и образования истинных опухолей. К факоматозам в настоящее время относят следующие заболевания: нейрофиброматоз, туберозный склероз, болезнь Гиппеля — Линдау, синдром Штурге — Вебера, фиброзная дисплазия Олбрайта, атаксия-телеангиэктазия Луи — Барр, синдром базальноклеточного невуса, синдром Гарднера, синдром Клиппеля — Треноне — Вебера, синдром Маффучи, синдром множественных лентиго, синдром множественных слизистых невром, нейрокожный меланоз, синдром Рендю — Ослера — Вебера, синдром Пейтц — Егерса (как и многие кишечные семейные полипозы), миксомный синдром и др. Различие типов клеток, вовлекаемых в патологический процесс при факоматозах, указывает на то, что объединение данных заболеваний в одну группу — упрощение. С другой стороны, общими признаками этих заболеваний является вовлечение кожи и ЦНС, васкулярный компонент. Отмечено также наличие двух заболеваний этой группы у одного больного.
Туберозный склероз — наследственное заболевание с а/дтипом передачи и вариабельной экспрессивностью. Около 70% составляют спорадические случаи. Клиническая картина характеризуется поражением кожи (91%), эпилептическими припадками (88%) и умственной отсталостью (60%). Поражение кожи может быть следующим: сальные аденомы (70%), гипомеланотические макулы (57%), околоногтевые и подногтевые фибромы (18%), пятна «кофе с молоком» (16%) и/или «шагреневой кожи» (36%), кожные узелки (14%). Изменения нервной системы выражаются в наличии эпилепсии, снижении интеллекта, возникающих за счет узловатых разрастаний нейроглии в области коры мозга, базальных ганглиев, желудочков мозга. Иногда отмечают пирамидные и экстрапирамидные симптомы, гидроцефалию, внутричерепные кальцинаты. Поражения глаз (50% случаев) включают ретинальные опухоли («факомы»), эпикант, застойные соски зрительных нервов, атрофию зрительных нервов, амавроз. Могут возникать эндокринные нарушения (адипозогенитальный синдром), а также костные изменения (гиперостоз костей черепа, кистозные изменения фаланг). К другим проявлениям болезни относят кисты и опухоли (липомы, гамартомы) почек, опухоли сердца (прежде всего рабдомиомы).
Существуют два типа диагностических критериев: первичные и вторичные.
Первичные диагностические критерии позволяют установить несомненный диагноз, если присутствует по крайней мере одно из каждой из трех нижеуказанных категорий:
1. Кортикальные узлы или субэпиндемальные гамартомы (поданным КТ, ЯМР и патологоанатомических исследований).
2. Множественные ретинальные гамартомы.
3. Такие кожные поражения, как себоррейная аденома, периунгвальная или субунгвальная фиброма.
Вторичные диагностические критерии позволяют установить предварительный диагноз, если два или более признаков, представленных ниже, присутствуют при наличии только двух первичных критериев:
- Инфантильный спазм.
- Гипомеланотические макулы.
- Единичная ретинальная гамартома.
- Субэпиндемальные или кортикальные кальцинаты.
- Множественные опухоли почек.
- Кардиальная рабдомиома.
- Родственники первой степени родства с туберозным склерозом.
- Углубления на зубной эмали.
Туберозный склероз, как и нейрофиброматоз,
характеризуется вариабельностью клинических проявлений, прогрессирующим течением, возникновением новых мутаций. Могут возникать абортивные и олигосимптомные формы. Учитывая вариабельность клинических проявлений, можно сказать, что каждый пациент с эпилептическими припадками, умственной отсталостью, помимо рутинных клинических обследований, нуждается в консультациях офтальмолога, дерматолога и КТ. При подозрении на туберозный склероз, необходимо провести экскреторную пиелографию или эхоГ почек. Каждого пациента с кардиальной рабдомиомой, кистами почек или неуточненной диффузной патологией легких необходимо обследовать на туберозный склероз. ДД следует проводить с другими факоматозами, а кожные проявления необходимо отличать от множественных трихоэпителиальных сирингоцистаденом, атипичных ксантом и интрадермальных невусов. Риск рождения больного ребенка составляет 50%.
Нейрофиброматоз — а/д заболевание, характеризующееся образованием множественных нейроэктодермальных опухолей и иногда ассоциирующееся с поражением других систем. Многочисленные проявления болезни привели к подразделению ее на много типов, которые не всегда четко разграничиваются друг от друга. Выделяют следующие типы нейрофибро матоза:
- Центральный тип.
А. Менингеальный и периневральный — синдром Висхарта.
Б. Только менингеальный — синдром Штультце.
В. Только периневральный — синдром Кноблауха.
Г. Акустический тип.
- Периферический тип Реклингаузена. Классический периферический тип нейрофиброматоза характеризуется вариабельной экспрессивностью и полной пенетрантностью. Наиболее яркий клинический признак — гиперпигментированные пятна на коже. Часто опухолевидные образования на коже и нервных стволах представлены нейрофибромами или невриномами. Их количество варьирует от одного до бесчисленного множества.
- Висцеральный тип, при котором поражение локализуется преимущественно в кишечнике.
А. Е.М. Делягина описала легочный тип со множественными невриномами легких и плевры.
- Сегментарный тип с пятнами цвета«кофе с молоком» и нейрофибромами, ограничивающимися одним сегментом тела.
- Множественные пятна цвета «кофе с молоком» как а/д признак без других характерных проявлений.
- Стертые формы.
Известно семейное сочетание нейрофиброматоза с червеобразной атрофодермией, множественными эпителиальными кистами, врожденными пороками сердца, приступами брадикардии, олигофренией, болезнью Дауна, что известно как синдром Кароля — Годфрида — Праккена — Прика. Описаны также ассоциации нейрофиброматоза с болезнью Гиппель— Линдау, синдромом Марфана, наследственным дефицитом плазминогена, перонеальной мышечной атрофией, врожденной глухотой. Могут возникать нейроэктодермальные эндокринные опухоли. Диагностические признаки нейрофиброматоза:
- пятна цвета «кофе с молоком» (6 или более, больше 15 мм) или одно пятно площадью не менее 5 см2. При наличии этого признака диагноз достоверен в 100% случаев, в том числе при отсутствии другой симптоматики;
- кожные нейрофибромы:
- генерализованные;
- ареолярные (у женщин).
- фиброзные моллюски;
- другие опухоли (глиомы, менингеомы, астроцитомы, невриномы, феохромоцитомы, нейрофибросаркомы, злокачественные шванномы). У этих больных всегда много родинок (невусов), особенно выступающих над поверхностью кожи и в виде «цветной капусты».
Частота заболевания высокая. Поэтому к любому пациенту с родинками следует относиться очень внимательно с точки зрения наличия опухолей (в том числе у молодых) и болезни Реклингаузена:
- подмышечная веснушчатость;
- врожденный псевдоартроз;
- нейрофибромы любой локализации;
- кифосколиоз;
- родственники первой степени родства, по крайней мере, с двумя из вышеуказанных признаков.
Наличие 2 любых признаков достаточно, чтобы предположить этот диагноз, в то время как наличие 3 и более признаков достаточно для установления диагноза «нейрофиброматоз».
Другие признаки нейрофиброматоза неспецифичны. К ним относятся нарушения поведения, задержка речевого развития, макроцефалия, умственная отсталость, эпилептическими припадки, низкий рост, зуд.
Заболевание очень вариабельно, выраженность признаков различна не только в пределах семьи, но и у каждого больного в разное время, поскольку нейрофиброматоз имеет прогредиентное течение, а степень прогрессирования индивидуальна. При рождении наиболее часто имеются пятна цвета «кофе с молоком», могут быть диффузные васкулярные нейрофибромы и псевдоартроз. В период раннего и позднего детства могут развиться опухоль мозга, кожные нейрофибромы, глиомы зрительных нервов, эпилептические припадки, нарушения поведения.
О злокачественных дегенерациях типа нейрофибросаркомы и злокачественной шванномы следует помнить при обследовании пациентов пожилого возраста. Снижение умственных функций отмечают далеко не всегда, в то время как цереброваскулярные осложнения хорошо известны. Возможна также АГ за счет появления феохромоцитомы или вовлечения в патологический процесс сосудов почек. С нейрофибромами и пигментными пятнами сочетаются стенозы почечной артерии. Частота возникновения злокачественных опухолей при нейрофиброматозе составляет около 12–15%. При наличии пятен цвета «кофе с молоком» ДЦ следует проводить с синдромом Рассела — Сильвера, синдромом пятен цвета «кофе с молоком» с темпоральной дизритмией и болезнью Олбрайта.
Для синдрома Рассела — Сильвера характерен нанизм, гемигипертрофия.
Для болезни Олбрайта типичны пигментация кожи, преждевременное половое созревание, фиброзная остеодисплазия. Рентгенологически выявляют множественные кисты в длинных трубчатых костях, в костях черепа и таза. Болезнь чаще возникает у девочек.
В 1926 г. Линдау распознал ассоциацию болезни Гиппель (ретинального ангиоматоза) с ангиоматозными опухолями мозжечка (ангиомы мозжечка текут с эритроцитозом), других отделов ЦНС и ряда органов. Ретинальные ангиомы часто расположены по периферии, поэтому требуется особое внимание для их выявления. Часто возникают у людей в возрасте до 25 лет. Офтальмологические нарушения выражаются в наличии глаукомы, катаракты и отслойки клетчатки. Церебральные гемангиобластомы, которые могут быть кистозными или кальцинированными, также возникают поздно, на 3-й декаде жизни.
В ликворе отмечают белково-клеточную диссоциацию, во многих случаях — полицитемию. Могут появиться ангиоматозные опухоли головного и спинного мозга, лица, надпочечников, легких и печени, а также множественные кисты поджелудочной железы и почек. Ассоциированными неангиоматозными опухолями могут быть гипернефрома и феохромоцитома. ДД следует проводить с опухолями задней черепной ямки и с другими факоматозами.
Синдром Штурге — Вебера называют «четвертым факоматозом». Наиболее характерным клиническим проявлением этого заболевания является лицевой пылающий невус или плоская ангиома, расположенная чаще всего в зоне иннервации тройничного нерва. Она может прорастать хориоидальную оболочку и сопровождаться колобомами радужки. Гемангиома туловища или конечностей может сочетаться с сосудистыми мальформациями спинного мозга. Также могут возникать такие кожные поражения, как синдром Клиппеля — Треноне — Вебера. Другими характерными поражениями являются гемангиомы мягкой и паутинной мозговой оболочек мозга, особенно в темпоральной и окципитальной областях, что ведет к атрофии мозга и спастическим парезам. В глазу возможны кавернозные ангиомы сосудистой оболочки и врожденная глаукома. Кожные поражения проявляются с рождения и не прогрессируют. На первом году жизни возникают судорожные припадки, уже в это время можно выявить интракраниальные кальцификаты. У ряда пациентов отмечается умственная отсталость. Заболевание носит спорадический характер. Вариантами синдрома Штурге — Вебера являются:
- синдром Ширмера — капиллярный невус лица и ранняя глаукома;
- синдром Мильеса — капиллярный невус лица и гемангиома сосудистой оболочки глаза без глаукомы;
- синдром Кнуда — Краббе — ангиоматоз без глазных симптомов;
- нейроангиоматоз энцефалотригеминальный глазо-кожный — телеангиоэктатический невус лица, гемангиома верхней челюсти, черепно-лицевая гемигипертрофия, ангиоматоз конъюнктивы и сосудистой оболочки глаза, отслоение сетчатки;
- синдром Вебера — Димитри — ангиоматозная гемигипертрофия (Паркса — Вебера), эпилепсия, идиотизм.
ДД следует проводить с транзиторным пылающим невусом в неонатальный период, нейрофиброматозом, синдромом Вибурна — Масона (артерио-венозная аневризма среднего мозга и сосудов сетчатки, иногда — капиллярный невус лица, располагающийся в зоне иннервации тройничного нерва).
Пылающий невус лица в 90% случаев отмечают при синдроме Видемана— Беквита, однако в этом случае он располагается в области переносицы и верхнего века. Поражение сосудов и мозга при синдроме Штурге — Вебера обычно слишком обширно для традиционного хирургического лечения. В последнее время с успехом применяют интравазальную окклюзию. Противосудорожная терапия с трудом контролирует эпилептические припадки.
Гиперпигментация кожи возникает и при синдроме Кронхайта (множественные аденоматозные полипы желудка и тонкой кишки, тонкие дистрофичные волосы, дистрофия ногтей).
Темно-серые, как припудренные угольной пылью, иногда папилломатозные, с гиперкератозом высыпания в естественных складках, прежде всего в подмышках, получили название acantosis nigricans. Формируется в пубертатный период, при сахарном диабете и (самое тревожное!) может быть проявлением злокачественных опухолей, особенно часто — аденокарциномы желудка. Для сахарного диабета также типична так называемая диабетическая дермопатия, проявляющаяся коричневатыми, безболезненными атрофическими очажками с неправильными границами и располагающимися преимущественно на передней поверхности голеней. Чаще возникает у мужчин.
Одно из самых серьезных состояний, протекающих с гиперпигментацией, это меланома. Возникает преимущественно у взрослых, но 2% меланом выявляют у пациентов в возрасте моложе 20 лет. Частота развития меланом повышается с возрастом. Для представителей европеоидной расы риск развития меланомы в течение жизни составляет 1:100. У лиц монголоидной и негроидной расы это настолько редкая опухоль, что даже рассчитать ее гипотетический риск невозможно.
Группу риска составляют люди с гигантскими врожденными невусами, большим количеством родинок, синдромом семейных диспластичных невусов, меланомой в семейном анамнезе, пигментной ксеродермой, солнечными ожогами (особенно в детстве), с иммуннодефицитами, болезнью Ходжкина, после трансплантации почек. Клинические признаки меланомы: увеличение невуса в размере, изменение его цвета (25%), кровотечения из невуса (35%), зуд (18%), подкожные массы (7%), боль (5%), лимфаденопатия (7%).
Меланому характеризуют акронимом A2B2CD. Они обладают асимметричностью (Assymetry) формы и распределения пигмента. Обязательно должны быть оценены внешний вид пигментного пятна или появление нового (Appearance). Края (Borders) обычно неправильной или зубчатой формы, нередко кровоточат (Bleed). Меланома может быть (кроме типичного темного цвета) красного, голубого, розового, серого и даже белого. При любом изменении (Change) родимого пятна следует исключить меланому. Диаметр (Diameter) большинства меланом >6 мм, но и маленькое родимое пятно может быть злокачественным.
Следует подчеркнуть, что любой невус, особенно раздраженный (после травм, ожогов и т. п.) можно удалять только хирургически, решительно отказавшись от так называемых щадящих способов (лазер, криотерапия). Только биопсия с гистологическим (в том числе с иммуногистологическим) заключением позволяет с достоверностью опровергнуть диагноз «меланома» или установить его там, где ad oculus врач был убежден в наличии доброкачественного невуса.
Самая главная характеристика меланомы — ее абсолютная непредсказуемость. А если учесть, что в ранний эмбриональный период меланоциты мигрируют из нейроэктодермы кроме кожи также в глаза, кишечник, дыхательные пути, то самая непредсказуемая опухоль может появиться там, где ее совершенно не ждут.
Крапивница — четко очерченные, слегка возвышающиеся над поверхностью кожи резко зудящие красные образования. Подкожная клетчатка не изменяется. Обусловлена гиперэргической реакций пациентов с высокой концентрацией IgE или при недостатке системы комплемента (наследственный ангионевротический отек при дефиците ингибитора С1), при некротизирующем васкулите, лимфомах и сывороточной болезни.
Термин «крапивница» обозначает большую группу состояний, в том числе и ангионевротический отек. Только 10% случаев ангионевротического отека протекают изолированно от крапивницы. Крапивница чаще возникает у женщин в возрасте 20–50 лет. Один случай крапивницы в течение жизни отмечают 10–20% населения. Частота возникновения крапивницы может достигать 15–30%. Выделяют острую крапивницу (длится не более 6 нед) и хроническую (уртикарные элементы многократно повторяются более 6 нед). Если крапивница персистирует более 6 мес, то в последующие 10 лет у 40% больных сохраняются ее симптомы. Нередко причину хронического ангиневротического отека расшифровать не удается. С крапивницей протекают синдром Макла — Уэлса и ксантуренацидурия.
Синдром Макла — Уэлса — сочетание прогрессирующей внутренней глухоты и крапивницы. Передается а/д с разной степенью экспрессивности признаков. Заболевание может начаться в любом возрасте, но чаще в юношеском. Дебютирует с рецидивирующих потрясающих ознобов и крапивницы. Медленно прогрессирует тугоухость. Выявляют полую стопу, атрофию гонад, нефротический синдром, гиперхолестеринемию, гиперглобулинемию, повышение СОЭ, азотемию. В моче — протеинурия, гипераминоацидурия, возможна гиперглицинурия. Патологоанатомически — атрофия кохлео-вестибу- лярной системы, амилоидоз почек, надпочечников, селезенки. ДД проводится с синдромами Альпорта и Ольсена.
Ксантуренацидурия — редкое а/p передающееся нарушение триптофанового обмена, при котором дефицит кинурениназы после нагрузки триптофаном обусловливает усиленное выделение ксантуреновой кислоты, 3-гидроксикинуренина и кинуреновой кислоты. Кинурениназа катализирует превращение 3-ОН-кинуренина в 3-ОН-антралиновую кислоту. Блок, вероятнее всего, обусловлен низким аффинитетом апоэнзима кинуреназы к коэнзиму пиридоксаль-фосфата. Клиническая картина вариабельна. Часто возникает БА, крапивница, сахарный диабет, задержка умственного развития. Нагрузка триптофаном обусловливает усиленное выведение с мочой ксантуреновой и кинуреновой кислот, 3-ОН-кинуренина. Эта же нагрузка на фоне применения пиридоксина или пиридоксаль-фосфата не сопровождается знаками патологического метаболизма. ДД проводится с выраженным дефицитом пиридоксина.
Основные варианты крапивницы суммированы (табл. 2.13).
Таблица 2.13
Классификация крапивницы и состояния, протекающие с крапивницей
Иммунологически обусловленная
Анафилактоидная
Физическая
Другие виды
Наследственные формы
|
Пурпура — кровоизлияние в кожу. Термины петехии (мелкоточечные кровоизлияния), пятнистые (размером с монету), экхимозы (массивные, распространенные) только уточняют площадь поражения. Развиваются при капилляротоксикозе (пурпура Шенлляйн — Геноха как вариант синдрома диссеминированного внутрисосудистого свертывания — ДВС-синдрома), при синдроме Гарднера — Даймонда, менингококцемии, лептоспирозе, риккетсиозе, малярии, тифе, септическом эндокардите в результате сосудистого поражения. Кровоизляния в кожу возникают при синдроме Элерса — Данлоса, Кушинга, болезни Ослера, у стариков (сенильная пурпура), при парапротеинемиях и саркоме Капоши, анулярной телеангиэктазии (синдром Маойчи). Полиэтиологичные тромбопении (болезнь Верльгофа, опухолевые, медикаментозные, токсигенные, потребления) или нарушения функции тромбоцитов (тромбастении, синдром Бернарда — Сулье, Вискотта — Олдрича) также обусловливают кожные геморрагии.
Телеангиэктазии могут быть врожденными при болезни Рендю — Ослера, при ревматических болезнях (особенно системная склеродермия), пойкилодермии, пойкилодерматомиозите, синдроме Корнеля-де-Ланге типа И, карциноидном синдроме, склерозе легочной артерии, или приобретенными при озноблении, хронической фотоэкспозиции, локальной стероидной терапии, при беременности и алкоголизме. Частой причиной телеангиэктазий являются заболевания печени, а также инфекционные дерматозы, например, acrodermatosis atroficans при боррелиозе. Поражение больших по диаметру сосудов дает картину болезни Мондора или сосудистых гирлянд Сали. Первый вариант — это флебит поверхностных вен грудной клетки, второй — вертикально расположенные сосуды по типу бахромы на уровне VI ребра (не путать с «поясом Венеры»). Связи с портальной гипертензией не существует.
Синюшно-розоватый сосудистый рисунок на фоне бледной кожи (ливедо) отмечают у новорожденных с постгипоксической энцефалопатией, у более старших — при ДБСТ и васкулитах, пойкилодермиях.
Пустулы — образование везикул с гнойным содержимым. Гной может быть стерильным, как, например, при бактериоидном подошвенно-ладонном рецидивирующем пустулезе (синдром Эндрюса) и субкорнеальном пустулезе Снеддона — Вилкинсона. Пустулы отмечают также как побочное действие йода, брома, при инфицированных вирусных варицеллах (герпес, ветряная оспа, опоясывающий лишай), болезни Рейтера, чесотке.
Узловатые поражения кожи — это прежде всего узловатая эритема. Причина — гипериммунная реакция с образованием клеточных инфильтратов. Кожному процессу предшествуют общие неспецифические симптомы: температура, артралгия. Выглядит как синюшно-багровые узлы диаметром до 3–5 см, болезненные, не изъязвляющиеся, расположенные чаще на ногах, редко — по туловищу. В странах с широкой распространенностью туберкулеза, микобактериальная инфекция является причиной 90% всех случаев узловатой эритемы. Остальные 10% приходятся на саркоидоз, лимфогранулематоз и другие лимфомы, (3-гемолитический стрептококк (особенно у детей), иерсиниоз, хламидиоз, кампилобактериоз, грибковые и вирусные заболевания, болезнь Крона, язвенный колит, синдром Бехчета. Противозачаточные средства, антибиотики, бромиды и салицилаты также способны вызвать узловатую эритему.
Поверхностные узлы образуются при эпидермальных опухолях (меланома, пигментный невус, базалиома, карцинома, кератоакантома). Безусловно, все узловатые, папулоподобные образования кожи требуют исключения рака кожи. Из всех типов рака у человека чаше всего развивается рак кожи. У 40–50% людей в возрасте 65 лет, рак кожи возникал хотя бы один раз. Рак кожи обычно развивается у астеников с голубыми глазами, на участках кожи, особенно подвергающихся интенсивному солнечному облучению (лицо, уши, тыльная поверхность кистей). Предрасполагающими факторами являются генетическая предрасположенность, ультрафиолетовое (солнце, бактерицидные лампы и лампы для закаливания) и ионизирующее излучение, угольные и табачные смолы, ароматические полициклические углеводороды, неорганический пятивалентный мышьяк, хроматы, иприт, иммуносупрессия, пигментная ксеродерма.
Из всех видов рака кожи чаще всего развивается базально-клеточная карцинома (75%). На 2-м месте — плоскоклеточный рак кожи, который растет быстрее и может метастазировать.
Базально-клеточная карцинома презентирует в нескольких формах. Чаще это узловая форма в виде восковидного или полупрозрачного выпуклого образования, на верхушке которого видны мелкие расширенные сосуды. С ростом опухоли ее границы приподнимаются над кожей, появляется фиолетовый венчик. В центре образования появляется углубление или язвочка. Само по себе образование малочувствительно, но с образованием язвочки ее поверхность становится очень чувствительной к теплу и другим раздражителям. На нашей памяти есть весьма показательный случай диагностики фельдшером из дальнего села у самой себя базально-клеточной карциномы. Она обратила внимание, что язвочка над бровью начинала сильно болеть, когда она склонялась над печкой.
Другие варианты базально-клеточной карциномы — пигментная форма (неравномерное отложение черного, коричневого или даже голубого пигмента на поверхности), внутриэпителиальная (четко отграниченная красная бляшка), склеродермоподобная (нечетко отграниченная плотная желтая бляшка иногда с восковидной поверхностью).
ДД проводят со следующими состояниями: меланотический невус, фиброзная папула, ангиофиброма, трихоэпителиома и другие опухоли кожи.
Плоскоклеточная карцинома манифестирует в нескольких формах, представленных в табл. 2.14 по нарастанию злокачественности.
Таблица 2.14
Формы плоскоклеточного рака кожи
| Форма | Проявление | С чем дифференцировать |
| Болезнь Боуэна | Красная бляшка, покрыта чешуйками. Четко отграничена от окружающих тканей | Актинический кератоз. Выявляют на участках кожи, подвергающихся солнечному излучению |
| Эритроплазия Кейра | Ярко-красная бляшка без чешуек. Локализуется на головке полового члена | Воспалительные заболевания |
| Боэнподобный палулез | Множественные уплощенные папулы телесного или коричневого цвета, расположенные на половых органах | Воспалительные заболевания, остроконечные кондиломы, кератоакантомы |
| Инвазивный плоскоклеточный рак | Нечетко отграниченные красные папулы, покрытые чешуйками. Изъязвляются и кровоточат при травме | — |
Плоскоклеточная карцинома может метастазировать. Частота метастазирования зависит от степени злокачественности, места расположения, длительности существования и составляет от 10 до 35%.
Поверхностные мелкие узелки очень часто оказываются вульгарными бородавками, контагиозными эпителиальными опухолями, вызываемыми более чем 60 типами человеческого папилломавируса. Бородавки могут возникнуть в любом возрасте (у детей младшего возраста и стариков — редко), на любом месте, в любом количестве. Возможна аутоинокуляция. Бородавки (папилломы) могут персистировать годами, нередко спонтанно исчезают. Бородавки выглядят как четко отграниченные образования диаметром 2–10 мм, края закруглены или неправильные, поверхность шероховатая, цвет серый, желтый, коричневый или серо-черный. На лице и веках могут быть тонкими, удлиненными.
Клинические варианты папиллом (бородавок) представлены в табл. 2.15.
Таблица 2.15
Клинические варианты папиллом (бородавок)
| Клинический вариант | Тип папилломавируса | Особенность |
| Околоногтевые, ладонные, подошвенные | 1, 2, 4, 7 | Доброкачественные |
| Плоские | 3, 10 | Доброкачественные |
| Генитальные (могут также локализоваться во рту, перианально, в мочевом пузыре, в легких) | 6, 11 | У 28% инфицированных женщин отмечается дисплазия шеечного эпителия |
| 16, 18, 33 | Выявляют у 50–55% женщин с инвазивным раком шейки матки.
Тип 16 — у 80% мужчин и женщин с папулезом наружных половых органов. Может исчезнуть самостоятельно, но в дальнейшем часто развивается рак |
|
| 6 а, b, с, d, е | Гигантские кандиломы Бушке — Левенштейна могут озлокачествляться. Указанные типы вирусов выявляют при цервикальной метаплазии и опухолях гортани | |
| 34–58 | Цервикальная интраэпителиальная метаплазия | |
| «Папилломы мясника», «папилломы торговца мясом» | 7, 10 | Плоские вульгарные бородавки. Обычно доброкачественные |
| Кожные бородавки у пациентов с иммунодефицитными состояниями | 8 и др. | Часто озлокачествляются, кофактор — инсоляция |
| Злокачественная веррукозная эпидермодисплазия | 5 а, b; 8 | Часто озлокачествляются, кофакторы — инсоляция и ионизирующее излучение (прежде всего для 5) |
| Веррукозная эпидермодисплазия | 1–4, 7, 9, 10,12, 14, 17–19, 20, 23–25 | За исключением 14, 17 и 20 типов — доброкачественные |
| Папилломатоз гортани | 6, 11, 16, 30 | Может озлокачествляться и метастазировать в легкие. Может появляться у новорожденных после прохождения по родовым путям, у взрослых — после орально-генитального секса |
| Оральный папилломатоз (болезнь Хекка) | 13 | Доброкачественные |
Крупные подкожные узлы формируются при туберкулезе кожи, саркоидозе, сифилисе, ксантоматозе, метастазах опухолей (особенно рака бронхов и молочной железы), глубоком микозе, укусах клещей и при инородных телах.
Мелкие узелки паравазальные типичны для узелкового периартериита, над икроножными мышцами — для панникулита (причинами может быть болезнь Вебера — Кристиана, редко — туберкулез), в подкожной клетчатке — заставляют думать о подкожном липогранулематозе; узелки вдоль сухожилий, по фасциям характерны для артрита. Панникулиты достаточно часто изъязвляются. В качестве разрешающих факторов в первую очередь необходимо исключать хронический панкреатит и рак поджелудочной железы. Цвет, расположение, диаметр, симметричность узлов могут служить основанием для диагноза. Коричнево-красные, синюшно-красные узлы и пятна типичны для саркомы Капоши; разной степени насыщенности коричневые, безболезненные, четко очерченные в коже и подкожной клетчатке — для доброкачественного лимфаденоза Беферштедта; подкожные симметричные узлы — для семейного ангиолипоматоза; цвета кожи или желтоватые мягкие — для поверхностного липоматозного невуса Хофмана — Цурхеля; синюшные тестоватые — для синдрома голубых невусов; плотные, ливедного типа, быстро растущие — для синдрома Стюарта — Тривса (ангиосаркома с лимфэдемами),
Кальцинаты кожи и подкожной клетчатки чаще всего возникают при наследственных дисплазиях соединительной ткани (синдром Элерса — Данлоса), при склеродермии (синдром Тибьерже — Вейсенбаха), посттравматически, являются одним из проявлений эпителиомы Малерба (доброкачественная опухоль, происходящая из смещенных в дерму зачатков эпидермиса. Выявляют преимущественно у мальчиков, малигнизируется крайне редко). Возможны и другие состояния: базалиомы, наследственная остеодистрофия Олбрайта, паразитозы, интоксикация витамином D, гипопаратиреоидизм.
Изъязвления кожи возникают при застойной сердечной недостаточности, нарушениях артериального кровотока, диабетической ангиопатии, амилоидной полинейропатии I типа, сетчатом ливедо с летними изъязвлениями, наследственных чувствительных нейропатиях I (а/д) и II (а/p) типа, липоидном некробиозе, при болезни Крона, язвенном колите и злокачественных опухолях. У 25% больных с серповидно-клеточной анемией выявляют язвы на голенях с плохим естественным течением.
Микобактерии (М. ulcerans) способны вызвать «язву Бурули», по названию местности, где впервые был описан этот тип микобактериоза. Первым признаком поражения являются мелкие безболезненные узелки в коже, невидимые при осмотре, но хорошо прощупывающиеся. В последующем они разрастаются, спаиваются с подкожной клетчаткой, кожа над ними становится блестящей, синюшной. Происходит расплавление тканей, формируется глубокая безболезненная, реже — зудящая язва. Регионарные ЛУ реагируют поздно. При отсутствии вторичной инфекции общее состояние изменяется мало. Туберкулин (поздно) и гистологическая картина (рано, выявление кислотоустойчивых микобактерий) проясняют ситуацию. Туберкулез подчелюстных и шейных ЛУ с образованием вялых язв с типичным крошковидным отделяемым и исходом в звездные, втянутые рубцы даже при современной эпидемиологической ситуации остался скорее в памяти, чем наяву.
Язвы с реакцией регионарных ЛУ могут возникать при сифилисе, туляремии, чуме.
Эритемы локальные выявляют при эризопелои- де, ожогах кожи и аллергических реакциях, контактном дерматите и хронической мигрирующей эритеме. Если эритема это воспалительная гиперемия кожи, то экзантема — полиморфная воспалительная реакция с этапностью течения. Генерализованную экзантему отмечают при скарлатине, кори, краснухе, побочных эффектах лекарственных препаратов, при инфекции Коксаки и ЕСНО-вирусами, при вторичном сифилисе, тифе. Диффузная коре- или скарлатиноподобная экзантема с шелушением ладоней и подошв характерна при синдроме Кавасаки.
Везикулы (рис. 2.12), сопровождаемые сильным зудом, отмечают при экземах и паразитарных заболеваниях кожи. Болезненные везикулы на слизистой оболочке губ, гениталий, кератоконъюнктивиты типичны для герпеса, как первичного, так и рецидивирующего. Присоединение вируса простого герпеса к экземному процессу ведет к формированию герпетической экземы. При опоясывающем лишае (Herpes zoster) высыпания обычно односторонние, располагаются сегментарно, их появлению предшествует недомогание, корешковую боль, сегментарная гиперестезия. При иммунной недостаточности высыпания могут быть тотальными.
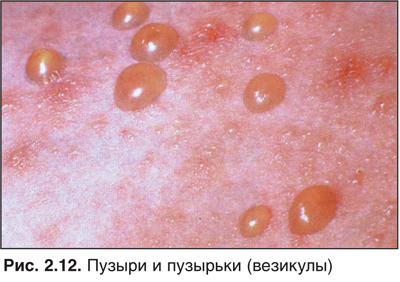
Мелкие везикулоподобные образования на коже могут оказаться контагиозным моллюском. Передается при непосредственном контакте. Представляет собой мелкие образования диаметром 2–10 мм телесного цвета с пупковидным вдавлением в центре. Содержимое — полужидкая белая масса. Гигантский моллюск может в 2–3 раза превышать обычные. До присоединения вторичной инфекции контагиозный моллюск бессимптомен. У детей младшего возраста образование может быть окружено венчиком экзематозного дерматита.
Волдыри на интактной коже типичны для пузырчатки (Pemphigus vulgaris). Наиболее часто выявляют патологию у новорожденных и лиц в возрасте 40–50 лет. Пузыри на коже легко лопаются, обнажая мокнущую поверхность. В большинстве случаев потирание кожи вызывает легкое схождение эпидермиса (симптом Никольского). Одновременно может поражаться и слизистая оболочка.
В отличие от простой пузырчатки при буллезном пемфигоиде нет акантолиза. Волдыри располагаются на гиперемированном основании. Часто поражается слизистая оболочка полости рта, пищевода, влагалища. Большинство пациентов — лица старческого возраста, прогноз в целом более благоприятен по сравнению с простой пузырчаткой, если описываемый синдром не является паранеопластическим.
С образованием везикул протекают врожденная пойкилодермия, простой буллезный эпидермолиз Огна, атрофический буллезный эпидермолиз и др. При эпидермальных пузырях рубцов не остается. При субэпидермальных пузырях возникают тяжелые поражения (рис. 2.13).
Полиморфная экссудативная эритема — заболевание кожи и слизистой оболочки с полиморфной клинической картиной. Чаще болеют молодые люди. Пусковым механизмом могут быть опухоли, коллагенозы, микоплазмы, вирусные инфекции (простого герпеса, Эпштейна — Барр, аденовирусы), а также пенициллин, сульфаниламиды и противосудорожные средства. Преимущественно на разгибательных поверхностях рук и ног, на шее появляются многочисленные экзантематозные высыпания, частично с пузырями.
Наиболее тяжелым вариантом многоформной эритемы является синдром Стивенса — Джонсона. После 1,5–2 нед продромального периода с лихорадкой, артралгией, диареей, болью в животе, кашлем развивается буллезно-десквамативное поражение кожи, слизистой оболочки, двусторонний конъюнктивит. Нередко синдром предшествует СКВ.
Если волдыри появляются после фотоэкспозиции, то логично предположить наличие порфирии или фотодерматоза. Фотосенсибилизация развивается на фоне приема тетрациклина, кислоты налидиксовой, гризеофульвина, сульфаниламидов, фенотиазина, напроксена.
Папулы — одиночные, диаметром < 1 см в плоские узелки. Полиморфная картина с папуло-везикулярными изменениями типична для герпетиформного дерматита. Хронически сохраняющиеся зудящие симметричные папулы на волосистой части головы, плечах, локтях, коленях, ягодицах могут быть проявлением асимптомной глутеновой целиакии. Папулы цвета неизмененной кожи (adenoma sebaceum) заставляют исключать болезнь Бурневиля — Прингла или амилоидоз. Коричневатые папулы отмечают при вторичном сифилисе; темно-коричневые и черные — при базалиоме, саркоме Капоши или меланоме; желтые — ксантомы и ксантелазмы при нарушениях жирового обмена. Папулы цвета кожи или слоновой кости типичны для синдрома Горштейна (полипоз толстой кишки и фибромы).
При болезни Фабри из-за дефицита α-галактозидазы-А происходит патологическое накопление тригексозилкерамида в различных органах. Болезнь наследуется Х-сцепленно. Изменения кожи при этом синдроме патогномоничны. Формируются ангиокератомы вследствие расширения мелких сосудов верхних слоев кожи с преимущественной локализацией на локтях, внутренней поверхности бедер, на мошонке, перианально, на крестце и вокруг пупка. Беспокоит резкая боль в ладонях, подошвах. Часто отмечают помутнения роговицы, извитость сосудов конъюнктивы и сетчатки. Кожа сухая, волосяной покров редкий. Депозиты тригексозилкерамида в органах ведут к вытеснению функционирующей ткани с формированием застойной сердечной недостаточности, АГ, почечной недостаточности.
Пустулы, папулы, узелки (кисты), комедоны, рубчики атрофические и гипертрофические — проявление угрей (акне). Акне — плеоморфное состояние, поражающее до 85–90% подростков. Хотя оно может проявиться в любом возрасте, в том числе и у новорожденных.
Акне — мультифакторное заболевание, обусловленное нарушениями кератинизации фолликулов, образования кожного жира, присоединяющейся размножением Propionibacterium acnes с развитием воспаления. Наиболее ранние изменения заключаются в нарушении дифференциации эпителия, в результате чего ороговевающие клетки, окружающие сальную железу, буквально передавливают ее. Образуются комедоны. Функция сальных желез во многом определяется андрогенами. Под их влиянием железа гипертрофируется и выделяет кожное сало в просвет фолликула. Внутрифолликулярные свободные жирные кислоты обладают положительным хемотаксическим эффектом по отношению к полиморфноядерным лейкоцитам и моноцитам. Жирные кислоты могут быть первичным стимулом фолликулярного гиперкератоза. Кожный жир — излюбленный субстрат для Р. аспе.
Р. аспе — анаэробный дифтероид — играет ключевую роль в развитии акне. Наряду с ним фолликулярная микрофлора состоит из Staph, epidermidis и Pityrosporum ovale. Тем не менее, только Р. аспе выделяет липазу, способную вызывать гидролиз триглицеридов кожного жира до свободных жирных кислот. Этот же возбудитель выделяет фосфатазу, гиалуронидазу, протеазу, нейроаминидазу, которые повышают проницаемость фолликулярного эпителия. Р. аспе продуцирует и низкомолекулярный фактор хемотаксиса полиморфноядерных лейкоцитов. В процессе фагоцитоза бактерий освобождается гид- ролаза, еще больше разрушающая фолликулярную стенку. В результате все содержимое — остатки кератина, липидов, волос — выдавливается в дерму. Формируется пустула. При глубоком проникновении в дерму и выраженном воспалении возникают узелки или кисты. Развивается гранулематозная реакция и реакция на инородное тело. Р. аспе далее провоцирует воспаление за счет активации С5а комплемента по классическому и альтернативному пути.
Известны несколько видов акне.
- Акне раннего грудного возраста (в первые 2–3 мес жизни). Представлены преимущественно закрытыми комедонами, хотя могут быть открытые комедоны, папулы и пустулы. Акне в этом возрасте обусловлены поступлением материнских андрогенов со стимуляцией сальных желез новорожденных. Лечения не требуется.
- Инфантильные акне. Возникают у детей в возрасте от 3 мес до 3 лет. Обычно у родителей этих детей в подростковом возрасте были множественные акне.
- Сливные акне (Аспе conglobata) характеризуются папулами, пустулами, абсцессами. Располагаются на лице, шее, туловище и верхних конечностях. Нередко выявляют у взрослых мужчин, часто сочетаются с генотипом XXY.
- Фульминантные акне присущи мальчикам- подросткам. Начинаются остро с папул на туловище.
Элементы быстро изъязвляются. Обычно отмечают системные симптомы: лихорадка, миалгия, артралгия, лейкоцитоз.
- Контактные акне. Многие вещества при нанесении на кожу способны привести к образованию комедонов у предрасположенных к акне. Контактные акне могут быть:
— мазевые акне. Отмечают у подростков, регулярно использующих мазевые препараты. На лбу и висках появляются закрытые комедоны. Предрасположены темнокожие субъекты;
— профессиональные акне. Множество веществ, используемых в промышленности (каменноугольная смола, вар, пек, битум, минеральные масла) могут вызывать акне. Характеризуются выступающими большими закрытыми комедонами, папулами, пустулами, узелками.
Волосы как производное кожи изменяются при острых (реже), хронических и наследственных заболеваниях. Их рост регулируется андрогенами, хотя существуют этнические и индивидуальные особенности. Для оценки волосяного покрова определяют густоту волос, их диаметр, твердость волосяного стержня, тип поперечного сечения (круглое, овальное, треугольное), постоянный диаметр на всем протяжении длины волоса или бамбукообразность, наличие залысин, границу роста на лбу и затылке, состояние бровей и ресниц, оволосение в подмышечных впадинах и на лобке (густота, граница роста, время появления). Так, форму волос (волнистые волосы становятся прямыми) необратимо изменяют пероральные контрацептивы. Кроме того, эти препараты ухудшают качество волос из-за снижения концентрации витамина В12 в сыворотке крови.
Выпадение волос в пожилом и преклонном возрасте является физиологическим, у некоторых мужчин — с образованием лысины. Во многом это регулируется генетическими факторами. Причинами ускоренного или ненормально массивного выпадения волос могут быть интоксикации таллием, терапия нитостатиками, реже — антикоагулянтами (производные кумарина и гепарин). Чаше всего усиленное выпадение волос происходит на фоне приема следующих препаратов: витамин А и его производные; блокаторы β-адренорецепторов (селективные и неселективные); ингибиторы ангиотензинпревращающего фермента (АПФ); антибактериальные препараты; НПВП; антикоагулянты; производные 6-аминохинолона; тиреостатические препараты; блокаторы Н2-рецепторов; нестероидные антиэстрогенные средства; препараты α-интерферона; производные имидазола; антидепрессанты; противосудорожные препараты; нейролептики; противопаркинсонические препараты; производные гидроксихлорохина; производные триазола; кетоконазол; урикозурические препараты; пероральные контрацептивы; антигельминтные препараты; препараты лития; противогерпетические препараты; бромокриптин; борная кислота.
Кроме этого выпадение волос обусловливает дефицит железа, беременность, острые массивные кровопотери, дефицит питания, прежде всего безбелковое питание, рацион с низким содержанием микроэлементов. Заболевание кожи головы (псориаз, рубцовое перерождение, экземы), инфекционные болезни, особенно протекающие с высокой лихорадкой, гипотиреоз, редко — гипертиреоз, ДБСТ — все это сопровождается значительным урежением волосяного покрова. С алопецией протекают энтеропатический акродерматит, недостаточность биотинидазы, точечная хондродисплазия, некоторые полипозы желудочно-кишечного тракта, миодистрофия. Очаговая алопеция (рис. 2.14) может быть обусловлена заболеваниями печени, рубцами, вторичным сифилисом, метастазами опухолей, трихотилломанией (самовырыванием волос). При каждой алопеции, особенно очаговой, должны быть исключены грибковые инфекции. Это касается прежде всего детей и подростков. Спектр грибковых инфекций, вызывающих алопецию, несколько изменился за последние 20–30 лет. Раньше чаще выявляли грибы рода Microsporum, сейчас — Trichophyton tousurans. Алопеция при трихофитии более диффузная, чем при микроспории. При грибковой инфекции на границе пораженного участка определяется розовофиолетовый венчик. Выявляют сломанные, поврежденные волосы. Диагноз устанавливают клинически и лабораторно. Исследование с помощью лампы Вуда с длинно-волновым ультрафиолетовым светом полезно только при микроспории, когда отмечается желто-зеленая флюоресценция. При использовании теста с КОН на соскоб на предметном стекле капают 10 или 20% раствор КОН и накрывают покровным стеклом. Препарат не следует держать при комнатной температуре более 15 мин, лучше исследовать его немедленно. Диагностическая ценность метода резко снижается при уменьшении выраженности грибкового воспаления. Поскольку диагностическая ценность двух описанных методик не всегда достаточна, необходимо инокулировать соскобы с подозрительных участков на специальные среды.
Очаговая алопеция — неожиданное и быстрое выпадение волос с четкими границами поражения. Местно нет отека, гиперемии, чешуек. Возможна диффузная форма очаговой алопеции. Тотальная алопеция — потеря всех волос на коже волосистой части головы. Потеря всех волос тела — универсальная алопеция.
При очаговой алопеции у 7–66% больных на ногтевых пластинках отмечают точечные углубления.
Очаговая алопеция — наиболее частая причина потери волос у детей, хотя возраст начала заболевания колеблется обычно от 15 до 40 лет. Связь очаговой алопеции с аутоиммунными расстройствами, витилиго, синдромом Дауна, семейные формы и наличие специфических лимфоцитарных популяций, окружающих фолликулы пораженных волос, позволяет отнести очаговую алопецию к аутоиммунным заболеваниям.
Первичная или вторичная гипофункция половых желез (синдромы Тернера, Клайнфельтера, гипофизарная карликовость, опухоли гипофиза, гипопитуитаризм, крипторхизм, анорхия) обусловливает уменьшение или даже отсутствие волос в подмышечных впадинах и на лобке, к скудной растительности на лице у мужчин.
В противоположность описанным признакам гирсутизм — избыточный рост волос у женщин на лице, груди, ягодицах, белой линии живота без признаков вирилизации. Очень часто этому сопутствуют нарушения менструального цикла. В диагностике гирсутизма всегда надо учитывать семейные и расовые факторы. У жительниц Азии скудный волосяной покров, Северной Европы — значительно меньше волос на теле и лице по сравнению с таковыми в Южной Европе и Кавказском регионе. Гирсутизм развивается при гиперплазии надпочечников из-за недостаточности 21-гидроксилазы, при синдроме Кушинга, при применении глюкокортикоидов, андрогенов.
Если при гирсутизме, развившемся в подростковом возрасте, отсутствуют признаки вирилизации и нарушения овариально-менструального цикла, то речь идет об идиопатическом гирсутизме. Причинами является избыточная продукция тестостерона или его предшественника андростендиола яичниками. Сочетание гирсутизма с низким голосом, урежением волосяного покрова на голове, угревой сыпью, гипертрофией клитора, атрофией молочных желез, олиго- и аменореей, приобретение мужских контуров тела называют вирилизмом. Вирилизация и аменорея, возникшие в пубертатный период, заставляют исключить прежде всего синдром Штейна — Левенталя. Вторичный гирсутизм выявляют при андроген продуцирующих опухолях надпочечников, при опухолях яичников (арренобластомы, лютеомы и некоторые другие).
Интенсивный очаговый рост волос на каком-либо участке тела может быть над артерио-венозным соустьем, spina bifida. Усиленный рост волос на одной ноге, как и уменьшение волосяного покрова по сравнению с противоположной ногой возможен при ишиасе.
Тотальный рост волос по типу лануго по всему телу — синдром Херцберга— Потяна— Гебауера. Объясняется гипотетическим пилоротропным фактором, циркулирующим прежде всего при метастазирующем раке органов грудной и брюшной полостей. Клинически представляет собой приобретенный ланугообразный гипертрихоз при висцеральных неоплазмах (прежде всего — карциномы с метастазами). Менее ярко синдром проявляется также при голодной дистрофии, гипертиреозе, язвенной болезни, синдроме мальабсорбции. Проявляется резким ростом ланугообразных волос вначале на лице, носу, лбу, на затылке, верхней части спины. Отдельные волосы достигают длины 4 см. Особенно выражен гипертрихоз в подмышках и паховой области. Здесь волосы достигают такой длины, что полностью скрывают гениталии. В моче — гипогонадотропинурия, гиперколтизолурия. Если синдром вызывается опухолью, прогноз крайне неблагоприятный. Ожидаемая длительность жизни не превышает 4 мес.
Цвет и некоторые другие особенности волос также могут иметь значение для диагноза. Белая прядь волос на лбу является одним из критериев диагноза «синдром Варденбурга», светлые волосы свойственны пациентам с гомоцистинурией и большинству пациентов с фенилкетонурией. Рыжие волосы вследствие большого расхода железа на их окраску ассоциируются с нарушениями иммунитета и анемией. Упорно стоящий хохолок волос на затылке сопровождает течение постгипоксической энцефалопатии у новорожденных или является признаком симпатикотонии у более старших пациентов. Твердые курчавые волосы с низкой границей оволосения на лбу бывают при синдроме Менкеса (нарушение обмена меди), тонкие мягкие — ассоциируются с дисплазией хрящей.
Осмотр волос — это и диагностика такого, увы, относительно нередкого состояния, как педикулез. Педикулез может поражать волосистую часть головы (pediculosis capitis), область лобка и брови (pediculus pubis), туловище (pediculus corporis).
Хотя педикулез выявляют прежде всего у асоциальных личностей, в семьях с полным пренебрежением к гигиене, но представитель любой социальной группы может оказаться пораженным педикулезом. В современных условиях педикулез волосистой части головы чаще выявляют у детей дошкольного и раннего школьного возраста. Его диагностируют чаще осенью после каникул, проведенных детьми в загородном лагере. Перенос вшей возможен при тесном контакте, одевании чужой одежды, использовании чужой щетки и т. д.
Лобковый педикулез выявляют обычно у людей в возрасте 15–40 лет. Это заболевание, передающимся при половых контактах. Более чем у 1/2 пациентов с лобковым педикулезом выявляют гонорею, реже — сифилис. Р. pubis паразитирует на лобке, но может селиться на других частях тела, покрытых короткими волосами: перианальная область, бедра, туловище, подмышки, борода, усы. У детей вши и гниды на веках — это практически всегда Р. pubis, вызывающие зуд, мокнутие, образование чешуек и корок. Выявление у детей Р. pubis на ресницах и бровях — очень серьезный признак насилия над ребенком.
Диагноз «педикулез» основывается на наличии зуда, расчесов (не исключена пиодермия), следов от укуса вшами, корочек, выявлении вшей и гнид. Последние плотно прикреплены ближе к корню волоса и не сдвигаются. Гниды флюоресцируют перламутровым светом в лучах лампы Вуда.
Ногти — это эпидермальная структура, производная эктодермы. Формирование ногтей плода начинается на 9-й неделе беременности и завершается иногда после 22-й недели. Скорость роста ногтей у детей и лиц молодого возраста — 0,5 мм/нед.
Для врача важно выделить наследственные, врожденные и приобретенные изменения ногтей. Наследственные изменения ногтей достаточно часто ассоциируются с эктодермальной дисплазией, аномалиями скелета и/или кожи. Врожденные изменения ногтей могут быть проявлением тератогенных синдромов (дифенилгидрантоин, варфарин, алкоголь, химиотерапевтические средства), амниотических перетяжек, врожденного гипотиреоза. Терапевт и педиатр чаще имеют дело с приобретенными изменениями ногтей.
Ногти ломкие и тонкие характерны при дефиците железа, передозировании витаминов А или D, при цитостатической терапии. У пациентов с железодефицитным состоянием — ложкообразные ногти (койлонихия). Ногти по типу «часовых стекол», выявляемые при всех тех же состояниях, что и пальцы по типу «барабанных палочек», могут быть семейной особенностью или сопровождать множественный полипоз толстой кишки, спру, первичный билиарный цирроз печени, болезнь Крона (свыше 1/3 всех пациентов) и язвенный колит. Поперечные трещины ногтей возникают при синдроме Стивенса — Джонсона, при применении цитостатиков и тетрациклина, а поперечные белые полоски часто возникают у женщин при неумелой косметической обработке ногтей или в патологических условиях — при интоксикациях таллием и мышьяком. Продольные желобки ногтевых пластинок выявляют у стариков или при синдроме Рейно.
«Свернутые», «мятые» ногтевые пластинки являются признаком псориаза, нередко этому сопутствует «изъеденность» ногтей за счет персистирующих микозов и «псориатические масляные пятна» ногтевых пластинок. Последние обусловлены просвечиванием участков псориаза на ногтевом ложе. При псориазе характерны и типичные мелкие вдавления, придающие ногтю вид наперстка (рис. 2.15).

Онихолиз (отслойка ногтей) возникает при аллергических реакциях на косметический лак, при микозе, псориазе, порфирии, применении цитостатиков, препаратов золота, тетрациклина, местном воздействии свервысоких частот (СВЧ) и кортикостероидов, при субунгвальной бактериальной инфекции.
Дистрофия ногтей может быть осложнением лечения D-пеницилламином, сопутствует буллезному эпидермолизу, прогерии, врожденному дискератозу, локальной радио- и рентгенотерапии, и Nailpatella syndrom.
Последний синдром характеризуется отсутствием одного или двух надколенников, деформациями скелета и гломерулонефритом.
Подногтевые кровоизлияния в форме продольно расположенных сосудов выявляют при эндокардите, васкулите, полицитемии истинной, циррозе печени и псориазе.
Онихофагия или обламывание, общипывание ногтей пациентом свидетельствуют о невротических реакция.
Окраска ногтей изменяется на белую при псориазе, циррозе печени, гипоальбуминемии и как семейный вариант. Желто-зеленая флюоресценция характерна при терапии хлорохином. Голубые ногти — при болезни Вильсона — Коновалова, пурпурные — при злоупотреблении пургеном (фенолфталеином), зеленые — при локальной инфекции Pseudomonas, желтые ногти — у курильщиков, при желтухе, терапии тетрациклином, при синдромах Коккейна и «желтых ногтей». Последний синдром характеризуется дополнительно лимфатическими отеками и плевральными выпотами. Серо-голубые ногти бывают при лечении препаратами серебра. Темно-коричневая окраска вплоть до черной типична при болезни Аддисона, синдромов Кушинга, Пейтц — Егерса, гипертиреоза, гемохроматоза, дефицита витамина В12.
Каждый раз, занимаясь диагностикой и ДЦ, врач должен постоянно исключать инфекцию как возможную причину расстройств. Это применимо и к заболеванию ногтей.
Грибковые поражения ногтей (рис. 2.16) выявляют у 0,1–0,2% школьников. Их частота резко нарастает в пубертатный период и затем постепенно продолжает повышаться, достигая максимума у лиц старческого возраста.
Паронихии обычно развиваются после травмы, при вросшем ногте, заусеницах. Их развитие обусловливают нарушения обмена и микроциркуляция. Возбудителями местного гнойного процесса являются стрепто- и стафилококки.
Подногтевые и околоногтевые бородавки вызываются многочисленными вирусами папилломы человека. Они могут быть болезненными и значительно труднее поддаются лечению, чем их накожные варианты. Субунгвальные и периунгвальные бородавки (папилломы) ответственны за инокуляцию вируса на другие участки тела при почесывании или на губы при обкусывании ногтя, сосании пальца.
Околоногтевая герпетическая инфекция, безусловно, может быть проявлением общей инфекции организма. У детей околоногтевой герпес может появиться вслед за первичным гингивостоматитом. По контрасту у взрослых герпетическая паронихия сопровождает генитальный герпес.
Из паразитов — чесоточный клещ способен внедряться в подногтевое ложе, вызывая гиперкератоз.
Травма ногтя может вызвать все виды его деформации и дистрофии. Острая травма с кровоизлиянием и возможным последующим отслоением ногтя не вызывает диагностических проблем. Травма же может вызвать сдвиг части матрикса из ногтевого ложа с развитием затем эктопического ногтя. Такой исход травмы вызывает проблемы только при косметических дефектах, нарушениях кровоснабжения или вторичной инфекции. Как причина поражения ногтей иногда игнорируется хроническая травма, в частности, тесная обувь, занятия спортом. Например, для теннисистов характерны хронические гематомы в подногтевом ложе большого пальца стопы. В литературе известны случаи, когда они служили основанием для ошибочной диагностики меланомы. Мы уже говорили об обкусывании и обламывании ногтей как выражении невротических реакций. В этом же ряду стоит и деформация ногтей по типу кроличьих резцов. Этот тип дистрофии обычно выявляют на больших пальцах кисти билатерально. Объясняется частой повторной абразией проксимальной части ногтя указательным пальцем этой же руки. Средняя часть ногтя истирается и вдавливается. После излечения невроза и исчезновении вредной привычки форма ногтя восстанавливается, в отличие от сохраняющейся каналообразной срединной дистрофии, причина которой пока не установлена. При диагностике патологических состояний ногтей, как и при расшифровке поражений иных структур, тканей и систем, врачу следует прежде всего рассматривать местный процесс как отражение вероятных системных заболеваний. Изолированное поражение ногтей может быть установлено только при исключении всех иных состояний.
1Три наиболее значимых фактора риска. Но у более 60% женщин в возрасте старше 50 лет с раком молочной железы нет ни одного фактора риска.
2ГИНК — гидразид изоникотиновой кислоты. Представляет собой группу препаратов: фтивазид, метазид, салюзид и т. д. ИНГА — иностранная абревиатура, соответствующая нашей ГИНК. Обе абревиатуры широко применяются в фтизиатрии.
3Диагноз «синдром Марфана» основывается на клинической картине. Необходимо наличие минимум двух основных признаков, отмеченных звездочкой (хотя бы по одному из любых четырех вышеуказанных групп) и несколько дополнительных.
4Клетки гипоталамуса, ответственные за сексуальное развитие, закладываются в области обонятельной плакоды, и только затем мигрируют собственно в мозг. Поэтому при расщеплении неба с поражением обонятельной плакоды возможен гипогонадизм.
5НПВП — нестероидный противовоспалительный препарат.