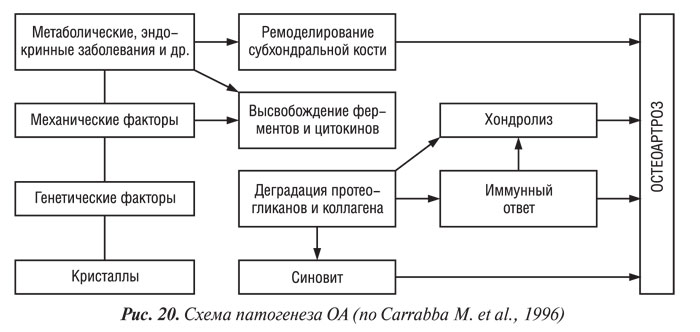
Роль биомеханических факторов в патогенезе и прогрессировании ОА
Результаты целого ряда эпидемиологических исследований, описанных в главе 1, показали, что профессии, связанные с длительным повторяющимся использованием определенных групп суставов, сопряжены с высоким риском развития ОА. Однако очень часто трудно или даже невозможно разделить долю механического фактора в патогенезе ОА и влияние возраста, генетических, гормональных и других факторов, которые могут способствовать возникновению и прогрессированию болезни. Так, профессии фермера (Thelin A., 1990; Croft P. et al., 1992; Axmacher B., Lindberg H., 1993), балерины (Andersson S. et al., 1989), строителя (Partridge R.E.H., Duthie O.R., 1968; Andersson J.A.D., 1984; Lindberg H., Montgomery F., 1987; Felson D.T. et al., 1991; Vingard E. et al., 1991), а также профессиональное занятие футболом (Klunder K.B. et al., 1980; Lindberg H. et al., 1993; Vingard E. et al., 1993), лыжным спортом, теннисом (Marti B. et al., 1989; Vingard E. et al., 1993; Kujala U. et al., 1994; Lequesne M.G. et al., 1997) ассоциированы с развитием ОА. Возникает вопрос, насколько точно можно связать это заболевание с первичной дегенерацией суставного хряща, а не со вторичными его изменениями после перенесенных неизбежных при этих видах деятельности травм других тканей сустава (менисков, связок, капсулы)? Травма или разрыв менисков, а также разрыв передних крестовидных связок коленного сустава относительно часто сопровождают профессиональных игроков в футбол (Roos H., 1994). Исследования кинетики высвобождения протеогликанов суставного хряща в синовиальную жидкость у профессиональных футболистов показало, что их концентрация была значительно повышена в течение нескольких часов после травмы и, хотя со временем их уровень снижался, он оставался повышенным на протяжении нескольких лет (Lohmander L.S. et al., 1989). Рентгенологические признаки ОА у этой категории лиц появлялись по прошествии не менее 15 лет после травмы (Roos H., 1994). На мениски коленного сустава воздействует масса тела человека, они играют важную механическую роль в нормальной функции сустава (Shrive N.G. et al., 1978; Seedhom B.B., Hargreaves D.J., 1979; Kurosawa H. et al., 1980), поэтому их травмирование приводит к тому, что суставные поверхности несут значительно бо′льшую нагрузку, чем в норме, ускоряя дегенерацию хряща и развитие ОА (De Haven K.E., 1985).
Влияние физических упражнений на суставной хрящ. Популярность джоггинга среди населения многих стран мира в последнее время привлекла внимание к бегу на длинные дистанции как к фактору риска развития ОА. Ретроспективные и проспективные исследования показали, что клинические и рентгенологические критерии ОА у бегунов на средние дистанции и марафонцев обнаруживают не чаще, чем у людей, не занимающихся бегом (Puranen J. et al., 1975; Lane N.E. et al., 1986; 1987; Panush R.S. et al., 1986; Sohn R.S., Lyle M.J., 1987; Konradsen L. et al., 1990; Lane N.E., Buckwalter A.J., 1993; Lane N.E. et al., 1993; Lequesne M.G. et al., 1997). Однако в связи с тем, что дизайн большинства этих исследований имеет ряд недостатков (некорректный статистический анализ, некорректные методы диагностики или оценки ОА и др.) их результаты вызывают сомнения (Lequesne M.G. et al., 1997). N.E. Lane и соавторы (1986, 1987, 1993) попытались исправить ошибки предыдущих исследователей. В течение 9 лет они изучали рентгенологические признаки ОА у бегунов-любителей пожилого возраста (средний возраст 65 лет). Обнаружено, что у этой категории лиц заболеваемость ОА (рентгенологически подтвержденным) не превышала таковую в группе лиц того же возраста, не увлекающихся бегом. Хотя в группе бегунов-любителей у женщин чаще регистрировали субхондральный склероз, а у лиц обоего пола — чаще обнаруживали остеофиты на рентгеновских снимках, тем не менее авторы заключили, что непрофессиональное занятие легкой атлетикой не является фактором риска ОА. Таким образом, приведенные данные указывают на то, что у лиц со «здоровыми» суставами бег на длинные дистанции не является причиной дегенерации хряща и развития ОА.
Исследования биомеханики ОА на моделях у животных подтверждают вышеприведенное заключение. P.M. Newton и соавторы (1997) исследовали гончих, которых тренировали бегом со скоростью 3,3 км/ч по 75 мин в сутки в течение 5 дней в неделю. Каждая собака несла дополнительную «экзогенную» нагрузку 11,5 кг (130% массы тела). Контрольную группу составили взрослые гончие, которых не тренировали и не применяли дополнительную нагрузку. Через 52 нед после начала тренировок было проведено гистологическое исследование суставного хряща, менисков и связок. Оказалось, что примененный уровень нагрузки не вызвал у собак дегенеративных изменений в суставных тканях. Никакой разницы между биомеханическими свойствами хряща у тренированных и нетренированных собак не выявлено.
В другом исследовании молодых гончих (с незрелым скелетом) тренировали по программе средней сложности (4 км/ч на тредмиле с наклоном 15°) в течение 15 нед (Kiviranta E. et al., 1987; 1988). Авторы обнаружили утолщение хряща и усиление синтеза протеогликанов по сравнению с контрольной (нетренированной) группой животных. Однако бо′льшая часть протеогликанов в хряще тренированных животных утратила способность к агрегации с гиалуроновой кислотой и содержала большее количество хондроитин-6-сульфатов (Kiviranta E. et al., 1988). Авторы исследования предположили, что такой уровень нагрузки ускоряет созревание матриксных депозитов в суставном хряще животных.
В исследовании, проведенном с участием молодых гончих, программа тренировки была несколько усложнена: 20 км в сутки на протяжении 15 нед (Kiviranta E. et al., 1992). Такая нагрузка вызвала снижение концентрации коллагена, повышение содержания воды, уменьшение соотношения хондроитин-6- и хондроитин-4-сульфатов в суставном хряще латеральных мыщелков бедренных костей. Увеличение дистанции до 40 км в день и длительности тренировки до 52 нед сопровождалось снижением содержания протеогликанов в ВКМ хряща (Arokoski J. et al., 1993; 1994). Наиболее выраженную потерю гликозаминогликанов отмечали на верхушках мыщелков бедренных костей, особенно в поверхностной зоне хряща (Helminen H.J. et al., 1992; Arokoski J. et al., 1994).
C. Little и соавторы (1997) продемонстрировали, что длительные интенсивные тренировки могут индуцировать изменения метаболизма протеогликанов в суставах запястья у лошадей. В рамках этого исследования авторы изучали влияние умеренных или высоких тренировочных нагрузок на синтез и деградацию крупных агрегированных протеогликанов (аггрекана) и двух мелких дерматансульфат-содержащих протеогликанов (декорин и бигликан). Эксплантаты суставного хряща были взяты из трех участков третей запястной кости, на которые приходится наибольшая нагрузка и которые чаще всего травмируются у спортивных лошадей. Двенадцать лошадей в возрасте от 3 до 5 лет без клинических или рентгенологических признаков патологии среднего запястного сустава были включены в исследование. Программа тренировок включала в себя бег со скоростью 6 м/с 2000 м 3 дня в неделю с увеличением дистанции до 4000 м к концу 8-й недели исследования. Затем всех животных разделили на две группы — животные группы А продолжали тренировки в прежнем режиме, а у животных группы В режим тренировок был усилен (бег со скоростью 8 м/с на дистанцию 4000 м 4 дня в неделю в течение 17 нед). Через 16 нед после окончания тренировок был произведен забор материала из определенных участков третьей запястной кости с обеих сторон.
В гистологическом исследовании хряща животных обеих групп обнаружена депрессия поверхностных его участков и разрушение кальцифицированного хряща и «волнистой границы» только в области дорзального радиального мыщелка третьей запястной кости. Существенной разницы обнаруженых гистологических изменений между группами А и В не выявлено. В культуре эксплантатов суставного хряща у животных группы В высвобождалось большее количество протеогликанов из хряща дорзального радиального мыщелка в среду, чем у животных группы А, что свидетельствует о более высоком уровне катаболизма в группе В. Включение 35S в протеогликаны было менее выражено в эксплантатах, полученных от животных группы В; в то же время у животных этой группы наблюдали усиление биосинтеза декорина, изменений интенсивности биосинтеза бигликана не выявлено. Таким образом, полученные результаты свидетельствуют о том, что длительные интенсивные тренировки лошадей индуцируют угнетение синтеза аггрекана и усиление синтеза дерматан сульфатсодержащих протеогликанов.
Функциональная роль декорина в соединительной ткани вообще и в хрящевой, в частности, по-прежнему остается предметом для исследований (см. гл. 3). Предполагают, что декорин играет центральную роль в организации коллагеновых макромолекул (Brown D.C., Vogel K.G., 1989), пролиферации клеток (Kresse H. et al., 1993) и модулировании активности факторов роста (например, ТФР-β) (Yamagouchi Y. et al., 1990). Добавление декорина к коллагеновому гелю вызвало отложение более однородных тонких коллагеновых фибрилл, чем в его отсутствие (Vogel K.G., Trotter J.A., 1987). В ткани шейки матки после родов разрушение коллагеновой сети коррелировало с повышенным уровнем декорина (Rechberger T., Woessner J.F.Jr., 1993). Таким образом, декорин, вероятнее всего, играет роль «дирижера» процессов репарации и ремоделирования соединительной ткани.
Повышение синтеза декорина хондроцитами суставного хряща лошадей на фоне высоких динамических нагрузок можно интерпретировать следующим образом: высвобождающийся из поврежденных хондроцитов в ответ на механическую перегрузку декорин выполняет роль мессенджера. Эта гипотеза подтверждена исследованиями in vitro и in vivo, которые продемонстрировали повышенную продукцию декорина хондроцитами, подвергшимися надфизиологической механической нагрузке. T.H.V. Korver и соавторы (1992) сообщили, что циклическая нагрузка, in vitro, применяемая в течение 7 дней, увеличивает в 3 раза синтез декорина в эксплантатах суставного хряща. Похожие результаты были получены N.A. Vissen и соавторами (1994), которые использовали эксплантаты зрелого и незрелого суставного хряща. В модели раннего (гипертрофического) ОА, индуцированного у собак путем пересечения передних крестовидных связок, G.S. Dourado и соавторы (1996) наблюдали повышение уровня мРНК бигликана, декорина и фибромодулина в хряще дестабилизированных суставов.
Влияние менискэктомии на суставной хрящ. Как указывалось ранее, суставные мениски играют важную роль в нормальной функции суставов. Мениски — структуры, которые увеличивают конгруэнтность суставных поверхностей бедренной и большеберцовой кости, повышают латеральную стабильность и улучшают распределение синовиальной жидкости, а также обмен питательными веществами с суставным хрящом (Seedhom B.B., Hargreaves D.J., 1979; Kurosawa H. et al., 1980). Тотальная или парциальная менискэктомия ведет к изменению направления нагрузки на суставную поверхность большеберцовой кости (рис. 11), вследствие чего развивается дегенерация суставного хряща (Helfet A.J., 1959; Tapper E.M., Hoover N.W., 1969; Appel H., 1970; Johnson R.J. et al., 1974; Cox J.S., Cordell L.D., 1977; Colombo C. et al., 1983).
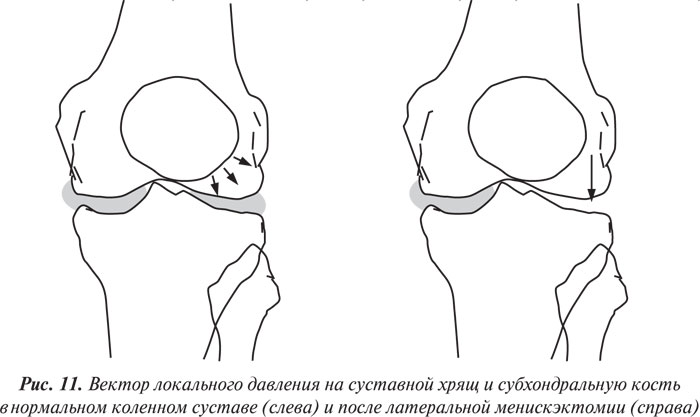
Изучению влияния менискэктомии на биомеханику сустава, а также индукции дегенеративных процессов в суставном хряще и субхондральной кости у животных (обычно собак и овец) посвящено много исследований. Вначале исследователи производили эктомию медиального мениска коленного сустава (Ghosh P. et al., 1983; 1990; 1993; Armstrong S. et al., 1994), однако впоследcтвии оказалось, что эктомия латерального мениска ведет к более быстрому развитию ОА (Ghosh P. et al., 1993; 1997; Little C.B. et al., 1996; 1997).
Используя латеральную менискэктомию у овец, C. Little и соавторы (1997) исследовали изменения суставного хряща и субхондральной кости из нескольких участков коленного сустава. Типичными гистологическими находками, иллюстрирующими индуцированные изменения в суставном хряще через 6 мес после операции, были разволокнение хряща, снижение концентрации протеогликанов, уменьшение количества хондроцитов. Под областями измененного хряща в субхондральной кости отмечено прорастание капилляров в зону кальцифицированного хряща, смещение кнаружи «волнистой границы» и утолщение губчатого вещества субхондральной кости.
В исследовании P. Ghosh и соавторов (1998) показано, что через 9 мес после латеральной менискэктомии у овец появляются признаки ремоделирования субхондральной кости и повышение ее минеральной плотности вторично по отношению к дегенерации суставного хряща. В зонах, претерпевающих аномально высокую механическую нагрузку вследствие удаления латерального мениска (латеральный мыщелок бедренной кости и латеральная пластинка большеберцовой кости), обнаружен повышенный синтез дерматан сульфатсодержащих протеогликанов (Little C.B. et al., 1996), хотя в хряще медиальной пластинки также обнаружено повышение синтеза протеогликанов того же вида. Оказалось, что дерматан сульфатсодержащие протеогликаны представлены главным образом декорином (Little C.B. et al., 1996; Ghosh P. et al., 1997). Наивысшая его концентрация обнаружена в средних и глубоких зонах суставного хряща (Poole A.R. et al., 1996; Ghosh P. et al., 1997).
Одновременно с повышением синтеза дерматан сульфатсодержащих протеогликанов в зонах хряща, несущих высокую нагрузку вследствие удаления латерального мениска, выявлен повышенный катаболизм аггрекана, о чем свидетельствует высвобождение его фрагментов в питательную среду из эксплантатов хряща (Little C.B. et al., 1996), а также высокая активность ММП и аггреканаз (Ghosh P. et al., 1998). Так как воспалительная активность в данной модели ОА была минимальной, авторы предположили, что источником ферментов были хондроциты.
Несмотря на то, что еще остается много нерешенных вопросов, в описанных исследованиях раскрывается возможная роль биомеханических факторов в патогенезе ОА. Ясно, что хондроциты способны «чувствовать» механические свойства своего окружения, реагируя на их изменения синтезом ВКМ, способного переносить бо′льшую нагрузку и таким образом предотвращать повреждение хряща. У молодых животных умеренные физические упражнения вызывали синтез богатого аггреканом ВКМ. Эта гипертрофическая (или адаптивная) фаза ответа хондроцитов может длиться несколько лет, обеспечивая стабильный уровень механической нагрузки на суставной хрящ. Однако нарушение этого баланса в результате повышения интенсивности или длительности нагрузки, или изменения нормальной биомеханики сустава после травмы или хирургического вмешательства, или снижения способности хондроцитов усиливать синтез ВКМ в ответ на увеличение нагрузки (при старении), действие эндокринных факторов влечет за собой значительные изменения на клеточном и матриксном уровне: угнетается синтез протегликанов и коллагена ІІ типа, стимулируется синтез декорина и коллагенов І, ІІІ и Х типов. Одновременно с изменением биосинтеза повышается катаболизм ВКМ, а также уровень ММП и аггреканаз. Не известно, как механическая нагрузка способствует резорбции хондроцитами окружающего их ВКМ, возможно, этот процесс опосредуется простаноидами, цитокинами (такими, как ИЛ-1β или ФНО-α, свободными кислородными радикалами). Здесь необходимо упомянуть о роли синовита при ОА, так как наиболее вероятным источником вышеназванных медиаторов катаболизма могут выступать макрофагоподобные синовициты и лейкоциты, инфильтрирующие синовиальную оболочку сустава.
В исследовании O.D. Chrisman и соавторов (1981) показано, что травматическое повреждение сустава стимулирует продукцию предшественника простагландинов — арахидоновой кислоты. Источником арахидоновой кислоты считают мембраны поврежденных хондроцитов. Хорошо известно, что арахидоновая кислота быстро конвертируется в простагландины с помощью фермента циклооксигеназы (ЦОГ). Было продемонстрировано, что простагландины, в частности ПГЕ2, взаимодействуют с рецепторами хондроцитов, изменяя эспрессию их генов. Однако остается неясным, стимулирует или угнетает арахидоновая кислота продукцию протеиназ и аггреканаз. Более ранние исследования показали, что ПГЕ2 увеличивает продукцию ММП и вызывает деградацию суставного хряща. По результатам других исследований ПГЕ2 обладает анаболическим влиянием на ВКМ, а также способствует целостности ВКМ, угнетая продукцию цитокинов хондроцитами. Возможно, противоположные данные этих исследований обусловлены различными концентрациями ПГЕ2, которые в них использовались.
Небольшое количество ИЛ-1β (основной цитокин, стимулирующий синтез и высвобождение ММП, а также угнетающий активность их естественных ингибиторов) может образоваться в ответ на повреждение суставного хряща, что ведет к дальнейшей деградации ткани.
Таким образом, описанные в данном разделе исследования показали, что поддержание подпороговой динамической нагрузки на сустав вызывает размножение хондроцитов, способных переносить новые механические условия, что означает наступление гипертрофической стадии ОА (рис. 12). Гипертрофированные хондроциты — клетки, находящиеся в последней стадии дифференцировки, а значит, экспрессия генов основных элементов матрикса в них изменена. Поэтому синтез аггрекановых протеогликанов и коллагена ІІ типа угнетен, а синтез декорина, коллагенов І, ІІІ и Х типов увеличен.

Снижение содержания аггрекана и коллагена ІІ типа в ВКМ, связанное с нарушением баланса между процессами синтеза и деградации, сообщает суставному хрящу свойство неадекватно реагировать на механическую нагрузку. Как следствие хондроциты становятся незащищенными, процесс переходит в третью, катаболическую, стадию (см. рис. 12), характеризующуюся избыточной протеолитической активностью и секрецией аутокринных и паракринных факторов регуляции. Морфологически эта стадия характеризуется деструкцией ВКМ суставного хряща, клинически она соответствует манифестному ОА. Эта гипотеза, безусловно, представляет собой упрощенное видение всех сложных процессов, протекающих при ОА, однако она обобщает современную концепцию патобиологии ОА.
Генетические и метаболические аспекты патогенеза ОА
Роль механических факторов в патогенезе ОА несомненна, однако существуют убедительные данные о том, что некоторые формы ОА наследуются по законам Менделя. Наследственные остеоартропатии можно подразделить на:
- первичный генерализованный ОА (ПГОА);
- кристаллассоциированные артропатии;
- преждевременный ОА вследствие наследственной остеохондродисплазии (Williams C.J., Jimenez S.A., 1999).
В 1803 г. W. Heberden описал «слегка плотные узлы, размером с мелкую горошину» на тыльной поверхности дистальных межфаланговых суставов кистей. Этот признак, по мнению автора, отличает ОА от других заболеваний суставов, включая подагру. J. Hayagarth (1805) расширил клиническое описание узлов Гебердена, отметив их нередкую ассоциацию с артрозом других локализаций. В дальнейшем Bouchard описал аналогичные узлы на тыльной поверхности проксимальных межфаланговых суставов кистей. Используя термин «узлы Гебердена и Бушара», W. Osler разделил «гипертрофический артрит» и «деформирующий артрит» (1909). В 1953 г. R.M. Stecher и H. Hersh обнаружили распространение узлов Гебердена среди членов семьи и пришли к выводу, что они наследуются по аутосомно-доминантному типу. Последовавшие за открытием R.M. Stecher и H. Hersh исследования выявили ассоциацию узлов Гебердена и Бушара с дегенеративным поражением других суставов (Kellgren J.H., Moore R., 1952; Stecher R.M. et al., 1953; Allison A.C., Blumberg B.S., 1958; Crain D.C., 1961; Kellgren J.H. et al., 1963; Buchanan W.W., Park W.M., 1983; Nuki G., 1983). Основываясь на данных клинического обследования и HLA-типирования, J.S. Lawrence (1977), J.S. Lawrence и соавторы (1983) предположили наличие полигенного наследования, а не дефекта единичного гена.
Фенотипический спектр наследственного ОА широко варьирует от легких форм, которые проявляются клинически лишь по достижении позднего взрослого возраста, до очень тяжелых форм, манифестирующих в детском возрасте. Традиционно все эти формы классифицировали как вторичный ОА. В настоящее время известно, что причиной некоторых из этих фенотипов является мутация генов, кодирующих макромолекулы ВКМ суставного хряща, что нарушает целостность хрящевого матрикса, а также регуляцию пролиферации хондроцитов и экспрессии генов. Данные наследственные заболевания представляют собой определенную подгруппу ОА, отличающуюся от вторичного ОА (табл. 11).

Хондродисплазии/остеохондродисплазии — группа клинически гетерогенных болезней, характеризующихся аномалиями роста и развития суставного хряща и ростовой пластинки. Некоторые ХД/ОХД приводят к раннему развитию ОА, клинически характеризующегося тяжелым течением. Среди них можно выделить следующие заболевания (табл. 12):
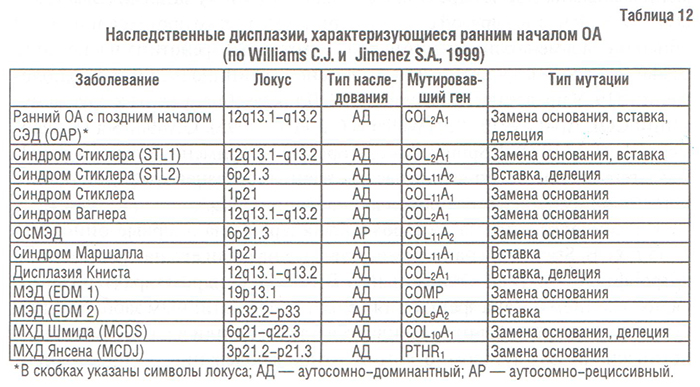
- спондилоэпифизиальные дисплазии (СЭД),
- синдром Стиклера,
- дисплазия Книста,
- множественные эпифизиальные дисплазии (МЭД),
- метафизиальные хондродисплазии (МХД),
- некоторые отоспондилометаэпифизиальные дисплазии (ОСМЭД).
Спондилоэпифизиальные дисплазии (СЭД) включают гетерогенную группу болезней с аутосомно-доминантным типом наследования, характеризующихся аномальным развитием осевого скелета и тяжелыми изменениями эпифизов длинных трубчатых костей, часто вызывающими карликовость. Нередко СЭД клинически тяжело протекают, сопровождаются укорочением тела и в меньшей степени конечностей (Spranger J., 1976; Rimoni D.L., Lachman R.S., 1990; Horton W.A., Hecht J.T., 1993; Pyeritz R.E., 1993; Byers P.H., 1994).
При формах СЭД, манифестирующих в более позднем возрасте, фенотип часто мало изменен и может клинически не проявляться до подросткового возраста, когда развивается тяжелый ОА. Деформация поясничного отдела позвоночника может проявляться сужением межпозвоночных дисков, платиспондилией и незначительным кифосколиозом. Также обнаруживают аномалии эпифизов в периферических суставах и ранние дегенеративные изменения в них. Наиболее постоянным признаком поражения периферических суставов является уплощение суставных поверхностей голеностопных и коленных суставов, а также уплощение межмыщелковой борозды бедренной кости. Нередко обнаруживают аномалии головки и шейки бедренной кости с развитием ОА тазобедренного сустава, манифестирующего в подростковом возрасте.
В связи с тем что коллаген ІІ типа — главный компонент ВКМ гиалинового хряща, высказано предположение, что причиной СЭД является кодирующий его ген COL2A1. Первое описание генетической связи между фенотипом раннего ОА, связанного с поздно манифестирующим СЭД, и геном проколлагена ІІ типа COL2A1 относится к 1989 (Palotie A. et al.) и 1990 г. (Knowlton R.G. et al.). Первое сообщение о мутации COL2A1 у родственников с ранним ОА, связанным с поздно манифестирующим СЭД, касалось замены основания Arg519>Cys (Ala-Kokko L. et al., 1990). К настоящему времени выявлены еще четыре семьи с аналогичными мутациями (Bleasel J.F. et al., 1998). У членов другой семьи с ранним ОА и легко протекающим СЭД обнаружена замена основания Arg75>Cys, хотя СЭД-фенотип у членов этой семьи не похож на фенотип семьи с заменой аргинина на цистеин в положении 519. У представителей семей с СЭД также обнаружены и другие мутации COL2A1—Gly976>Ser (Williams C.J. et al., 1995), Gly493>Ser (Katzenstein P.L. et al., 1992). J. Spranger и соавторы (1994) применили термин «тип ІІ коллагенопатии» для описания наследственных болезней хрящевой ткани с первичной мутацией гена проколлагена II типа COL2A1.
Классическая форма синдрома Стиклера была впервые описана в 1965 г. G.B. Stickler и соавторами, которые назвали его наследственной артроофтальмопатией. Описанный G.B. Stickler синдром характеризовался поражением органа зрения и тяжелым дегенеративным заболеванием суставов, которое обычно развивается в третьем или четвертом десятилетии жизни. Это аутосомно-доминантное заболевание, распространенность которого составляет приблизительно 1 на 10 тыс. новорожденных. Клиническая картина болезни включает в себя миопию, прогрессирующую глухоту, расщелину твердого нёба, гипоплазию нижней челюсти (аномалия Пьера — Робена) и гипоплазию эпифизов. В неонатальный период на рентгенограммах больных с синдромом Стиклера обнаруживают увеличенные эпифизы, главным образом проксимальный бедренной и дистальный большеберцовой костей. В процессе роста развивается дисплазия эпифизов, которая проявляется нерегулярностью оссификации эпифизов и последующими дегенеративными изменениями.
Так как COL2A1 экспрессируется в суставном хряще и стекловидном теле глазного яблока, с патологией этого гена связывали возникновение синдрома Стиклера. Однако обследование нескольких семей с синдромом Стиклера показало, что не во всех семьях заболевание связано с COL2A1 (Francomano C.A. et al., 1987; Knowlton R.G. et al., 1989; Bonaventure J. et al., 1992). Такую форму болезни называют І типом синдрома Стиклера (символ локуса STL1).
Спектр клинических проявлений синдрома Стиклера широко варьирует, в настоящее время выделено несколько фенотипов. Среди них — синдром Вагнера, который характеризуется преобладанием поражения глазного яблока; ОА при синдроме Вагнера фактически никогда не развивается, хотя у больных выявлена мутация именно гена COL2A1 (замена основания Gly67>Asp) (Korkko J. et al., 1993). Остается неясным, почему такая мутация COL2A1 компрометирует только функцию стекловидного тела и не влияет на гиалиновый хрящ.
Еще одной формой синдрома Стиклера является так называемый голландский вариант; он характеризуется всеми классическими проявлениями синдрома за исключением поражения органа зрения. H.G. Brunner и соавторы (1994) показали, что голландский фенотип синдрома Стиклера связан с мутацией гена COL11A2: доминирующей мутацией является делеция 54 пары оснований с последующей делецией экзона (Vikkula M. et al., 1995). M. Sirko-Osadsa и соавторы (1998) сообщили о другой семье, не связанной с описанной предыдущими авторами, с аналогичным фенотипом и мутацией гена COL11A2 (делеция 27 пары оснований), что подтверждает данные H.G. Brunner и соавторов (1994). Этот вариант называют ІІ типом синдрома Стиклера (символ локуса STL2).
Недавно был определен третий локус синдрома Стиклера у членов семьи с патологией стекловидного тела и сетчатки глаза, которые фенотипически значительно отличаются от изменений, наблюдаемых при «классическом» варианте синдрома. У представителей этой семьи обнаружена мутация гена COL11A1 (замена оснований Gly97>Val) (Richards A.J. et al., 1996). Разумеется, для подтверждения находки A.J. Richards и соавторов необходимы новые описания случаев такого фено- и генотипа синдрома Стиклера.
Долгое время обсуждался вопрос о нозологической связи синдрома Маршалла и классического варианта синдрома Стиклера. Сейчас синдром Маршалла классифицируют как отдельный фенотип главным образом благодаря более выраженной деформации лицевого скелета (Williams C.J., Jimenez S.A., 1999), хотя поражение периферических суставов схоже с таковым при І типе синдрома Стиклера. При синдроме Маршалла ОА коленных суставов и пояснично-крестцового отдела позвоночника начинается после 30 лет. Причиной синдрома является мутация гена коллагена ІХ типа COL11A1 (Griffith A.J. et al., 1998).
ОСМЭД. Этот фенотип был описан в голландской семье, у членов которой дегенеративные изменения в суставах, напоминающие ОА, появлялись в подростковом возрасте и поражали, главным образом, тазобедренные, коленные, локтевые и плечевые суставы; также обнаружены своеобразные черты лица, усиление поясничного лордоза, увеличение межфаланговых суставов, тугоухость, однако не выявлено никаких аномалий органа зрения (Vikkula M. et al., 1995). Исследователи обнаружили мутацию гена, кодирующего α2-цепь коллагена ІІ типа COL11A2.
Дисплазия Книста характеризуется укорочением туловища и конечностей, уплощением лица и спинки носа, экзофтальмом и тяжелой аномалией суставов (Maroteaux P., Spranger J., 1973; Kniest W., Lieber B., 1977). У больных с синдромом Книста суставы, обычно крупные от рождения, продолжают увеличиваться в детстве и раннем подростковом возрасте. У них также часто можно обнаружить миопию, тугоухость, расщелину твердого нёба, косолапость; у большинства больных рано развиваются тяжелые дегенеративные изменения, особенно выраженные в коленных и тазобедренных суставах. На рентгенограммах позвоночника обнаруживают уплощение и значительное удлинение тел позвонков, платиспондилию. Длинные трубчатые кости деформированы по типу гантели, оссификация эпифизов замедлена. В суставах кистей уплощены эпифизы и сужены суставные щели. Суставной хрящ мягкий, его эластичность снижена; гистологически в нем обнаруживают крупные кисты (симптом «швейцарского сыра»). Причиной синдрома Книста является мутация гена проколлагена ІІ типа COL2A1.
Множественные эпифизиальные дисплазии (МЭД) — гетерогенная группа болезней, характеризующихся аномалией развития ростовых пластинок длинных трубчатых костей, а также ранним (манифестирующим в детском возрасте) тяжелым ОА, поражающим как осевые, так и периферические суставы (чаще всего коленные, тазобедренные, плечевые и суставы кистей). Клинически МЭД проявляются болью и скованностью в суставах, изменением походки. У больных с МЭД обнаруживают также минимальные изменения со стороны позвоночного столба (различной степени уплощение тел позвонков), иногда позвоночник интактный. Также характерен низкий рост больных, хотя карликовость развивается редко. Орган зрения не поражается. МЭД включают несколько вариантов, например фенотип Фербэнкса (Fairbanks T., 1947) и Риббинга (Ribbing S., 1937).
МЭД наследуются по аутосомно-доминантному типу с различной степенью пенетрантности. Так как отличительной чертой МЭД является аномалия ростовой пластинки эпифизов, было высказано предположение, что причиной этих дисплазий является дефект генов, кодирующих макромолекулы хряща ростовой пластинки. Оказалось, что по крайней мере три локуса связаны с фенотипом МЭД. Исследования E.J. Weaver и соавторов (1993), J.T. Hecht и соавторов (1992) исключили из списка «виновников» МЭД гены коллагенов ІІ и VI типов, стержневого белка протеогликанов и соединительного белка хряща. J.T. Hecht и соавторы (1993), R. Oehelmann и соавторы (1994) обнаружили связь между МЭД, а также клинически близким к нему синдромом псевдоахондроплазии, и перицентромерным регионом 19-й хромосомы. Последующие исследования идентифицировали мутацию гена, кодирующего олигомерный матриксный протеин хряща (ОМПХ) у трех больных с МЭД (символ локуса EDM1) (Briggs M.D. et al., 1995). Так как все три мутации произошли в участке гена, кодирующем кальцийсвязывающий домен ОМПХ, вероятно, именно кальцийсвязывающая функция этого белка является неотъемлемой для нормального развития хряща ростовой пластинки.
M.D. Briggs и соавторы (1994) сообщили о семье из Голландии, МЭД-фенотип которой был связан с участком 1-й хромосомы, содержащей один из генов коллагена ІХ типа COL9A2 (символ локуса EDM2). Примечательно, что обнаруженная мутация оказалась первым доказательством роли коллагена ІХ типа, локализующегося на поверхности фибрилл коллагена ІІ, в поддержании целостности гиалинового хряща. M. Deere и соавторы (1995) показали, что фенотип Фербэнкса генетически не связан ни с локусом EDM1, ни с локусом EDM2, что подтвердило гетерогенность МЭД.
Метафизиальные хондродисплазии (МХД) — гетерогенная (описано более 150 типов) группа наследственных заболеваний гиалинового хряща, которые клинически проявляются ранним ОА (Sutcliffe J., Stanley P., 1973; Kozlowski K., 1976; Lachman R.S. et al., 1988). МХД характеризуются изменениями метафизов костей. Клинически они проявляются низким ростом, укорочением конечностей, искривлением голеней, «утиной» походкой. Также у больных с МХД обнаруживают признаки поражения других систем (например, иммунной и пищеварительной). Наблюдают дезорганизацию хряща ростовой пластинки, что гистологически проявляется скоплениями пролиферированных и гипертрофированных хондроцитов, окруженных утолщенными перегородками и дезорганизованным матриксом, а также проникновением некальцифицированного хряща в субхондральную кость.
Синдромы Янсена, Шмида и МакКузика — наиболее хорошо изученные МХД. Они сходны по особенностям аномалий скелета, но различаются по степени тяжести (синдром Янсена>синдром МакКузика> синдром Шмида). Наиболее часто встречается синдром Шмида (символ локуса MCDS), который наследуется по аутосомно-доминантному типу. Рентгенологически синдром проявляется coxa vara, укорочением и искривлением трубчатых костей, чашеобразной деформацией метафизов (более выражена в проксимальном, чем в дистальном отделе бедренной кости). Наиболее выраженные изменения наблюдают в ростовых пластинках длинных трубчатых костей.
По меньшей мере 17 различных видов мутаций гена коллагена Х типа описаны у больных с синдромом Шмида. Коллаген Х типа экспрессируется в гипертрофированных хондроцитах ростовых пластинок и, возможно, участвует в процессах оссификации (Jacenko O. et al., 1993). Таким образом, мутация кодирующего коллаген Х типа гена COL10A1 — наиболее вероятная причина синдрома Шмида (Kuivaniemi H. et al., 1997).
У детей с синдромом Янсена обнаруживают гиперкальциемию, а также повышенный уровень фосфатов в моче, снижение уровня паратгормона (ПГ) и ПГ-связанного пептида (Lenz W., 1969). С аномалией последнего, вероятно, связано возникновение синдрома Янсена. В 1994 г. A.C. Karaplis и соавторы опубликовали результаты оригинального исследования. После разрушения гена, кодирующего ПГ-связанный пептид в стволовых клетках эмбрионов мышей, мыши с дефицитом по этому аллелю умирали сразу после рождения. У них были обнаружены аномалия развития субхондральной кости, нарушение роста хряща и снижение пролиферации хондроцитов. В 1995 г. E. Schipani и соавторы сообщили о гетерозиготной мутации гена рецептора ПГ-связанного пептида у пациента с синдромом Янсена. Мутация заключалась в замене основания Gys223>Arg, что приводило к накоплению цАМФ; это означает, что аминокислота гистидин в положении 223 играет решающую роль в передаче сигнала. Позже E. Schipani и соавторы (1996) сообщили о трех других больных синдромом Янсена, у двух из которых обнаружена аналогичная мутация, а у третьего — замена Try410>Pro.
Наиболее часто встречающейся наследственной формой ОА является первичный генерализованный ОА (ПГОА), который был впервые описан как отдельная нозология J.H. Kellgren и R. Moore в 1952 г. Клинически для ПГОА характерно появление узлов Бушара и Гебердена, полиартикулярное поражение. ПГОА характеризуется ранним началом манифестации ОА и быстрым его прогрессированием. Рентгенологически ПГОА не отличается от ненаследственного ОА. Несмотря на то, что вопрос об этиопатогенезе ПГОА все еще дискутируется, проведенные исследования демонстрируют важную роль наследственной предрасположенности в возникновении и прогрессировании ПГОА (Stecher R.M. et al., 1953; Allison A.C., Blumberg B.S., 1958; Kellgren J.H. et al., 1963; Harper P., Nuki G., 1980; Nuki G., 1983).Так, J.H. Kellgren и соавторы (1963) обнаружили узлы Бушара и Гебердена у 36% родственников мужского пола и у 49% родственников женского пола, тогда как в общей популяции эти цифры составили соответственно 17 и 26%. У лиц с ПГОА чаще обнаруживают HLA A1B8 гаплотип (Lawrence J.S. et al., 1983; Pattrick M. et al., 1989) и MZ-изоформу α1-антитрипсина (Pattrick M. et al., 1989). В классическом исследовании с участием близнецов T.D. Spector и соавторы (1996) выполнили рентгенографию коленных суставов и суставов кистей у 130 одно- и 120 двуяйцовых близнецов женского пола на предмет наличия изменений, характерных для ОА. Оказалось, что конкордантность рентгенологических признаков ОА всех локализаций была в 2 раза выше у однояйцовых близнецов по сравнению с двуяйцовыми, а вклад генетических факторов колебался от 40 до 70%. В исследовании узелкового ОА, проведенном G.D. Wright и соавторами (1997), продемонстрированы раннее начало болезни, высокая степень тяжести и отрицательная корреляционная связь между возрастом начала болезни у пациентов и возрастом их зачатия родителями.
Среди кристаллассоциированных артропатий отложение кристаллов мочевой кислоты и кальцийсодержащих кристаллов в полости сустава имеют семейную предрасположенность (табл. 13).

В 1958 г. D. Zintan и S. Sitaj представили клинические описания патологии, которую они назвали «хондрокальцинозом» у 27 больных. Большинство пациентов принадлежали к пяти семьям, что указывало на наследственный компонент в этиопатогенезе болезни. Позже D. McCarty и J.L. Hollander (1961) сообщили о двух больных, у которых предполагали подагру с отложением неуратных кристаллов в полости суставов. Рентгенологическое исследование выявило аномальную кальцификацию гиалинового хряща многих суставов.
Рентгенологически болезнь отложения кристаллов пирофосфата кальция дигидрата, или пирофосфатная артропатия, напоминает спорадический ОА, однако она чаще поражает суставы, не типичные для обычных форм ОА (например, пястно-фаланговые, ладьевидно-лучевой, пателло-феморальный отдел коленного сустава). При пирофосфатной артропатии чаще формируются кисты субхондральной кости. Хотя в большинстве случаев хондрокальциноз возникает раньше манифестации вторичного ОА, у некоторых лиц заболевание может начинаться как идиопатический ОА, который сопровождается расстройствами обмена веществ (гемохроматоз, гиперпаратиреоидизм, гипомагнеземия и др.) (Williams C.J., Jimenez S.A., 1999).
Вероятнее всего, структурные изменения ВКМ суставного хряща индуцируют отложение кристаллов пирофосфата кальция дигидрата. A.O. Bjelle (1972, 1981) обнаружил в средней зоне матрикса суставного хряща членов семьи из Швеции с пирофосфатной артропатией снижение содержания коллагена и фрагментацию коллагеновых волокон. Так как эти участки не содержали кристаллов, авторы предположили, что описанная аномалия матрикса может предрасполагать к их отложению и развитию дегенеративных изменений в суставах. На основании изучения спорадических случаев пирофосфатной артропатии K. Ishikawa и соавторы (1989), I. Masuda и соавторы (1991) пришли к выводу, что причиной хондрокальциноза является мутация генов, кодирующих протеины ВКМ. C.J. Williams и соавторы (1993), A.J. Reginato и соавторы (1994) обнаружили гетерозиготную мутацию COL2A1 (замена оснований Arg75>Cys) у членов большой семьи с клиническим фенотипом тяжелого раннего ОА с анкилозированием, поздним развитием спондилоэпифизиальной дисплазии и хондрокальцинозом гиалинового и волокнистого хрящей. Однако оказалось, что у членов этой семьи хондрокальциноз носил вторичный характер по отношению к ОА.
Также было высказано предположение о том, что образованию кристаллов способствуют неорганические компоненты ВКМ. Например, гипомагнеземия вызывает развитие хондрокальциноза путем угнетения фермента пирофосфатазы, что в свою очередь снижает растворение кристаллов (Bennett R.M. et al., 1975). В синовиальной жидкости больных с пирофосфатной артропатией обнаружено повышенное содержание неорганических фосфатов (Silcox D.C., McCarty D., 1974). Это и другие наблюдения позволили высказать предположение, что у больных с пирофосфатной артропатией возникает локальное нарушение метаболизма пирофосфатов (Altman R.D. et al., 1973; Silcox D.C., McCarty D., 1974). Описан фермент нуклеозид-трифосфат-пирофосфогидролаза, который, возможно, участвует в образовании кристаллов пирофосфата в зоне их отложения в ВКМ (Howell D.S. et al., 1984; Muniz O. et al., 1984; Ryan L.M. et al., 1984; 1985). В спорадических случаях пирофосфатной артропатии обнаружено повышенное содержание этого фермента, однако в семейных формах болезни такую аномалию не наблюдали (Ryan L.M. et al., 1986). Тем не менее при культивировании фибробластов и лимфобластов пациентов с семейной пирофосфатной артропатией обнаружено повышение содержания неорганических фосфатов (Lust G. et al., 1981), что также подтверждает предположение о роли нарушений локального метаболизма пирофосфатов в патогенезе заболевания.
В последние годы были предприняты попытки определения генов, «виновных» в возникновении семейных случаев пирофосфатной артропатии. Так, анализ генетического материала, полученного от членов большой семьи с пирофосфатной артропатией (штат Мэн, США), при которой хондрокальциноз развивался вторично по отношению к тяжелому быстро прогрессирующему недиспластическому ОА, исключил связь заболевания с локусом COL2A1 (Baldwin C.T. et al., 1995). Однако авторы данного исследования обнаружили связь между изучаемым фенотипом пирофосфатной артропатии и локусом, расположенным на длинном плече 8-й хромосомы (символ локуса CCAL2). A.G. Hughes и соавторы (1995) обнаружили связь между фенотипом первичного хондрокальциноза в семье из Великобритании и локусом CCAL1, который локализуется на коротком плече 5-й хромосомы в регионе 5р15. По данным C.J. Williams и соавторов (1996), локус CCAL1 у членов семьи из Аргентины с пирофосфатной артропатией локализовался несколько проксимальнее, чем в предыдущем случае, — в регионе 5р15.1. Аналогичный генотип был обнаружен у членов семьи из Франции (Gaucher A. et al., 1977).
Таким образом, данные описанных исследований свидетельствуют о том, что семейная форма пирофосфатной артропатии представляет собой клинически и генетически гетерогенное заболевание, причиной которого могут служить мутации по крайней мере трех различных генов.
Роль изменений в субхондральной кости в патогенезе ОА
Наряду с дегенерацией суставного хряща, в патологический процесс при ОА вовлекается и подлежащая костная ткань. Предполагают, что утолщение субхондральной пластинки способствует прогрессированию ОА (Radin E.L. et al., 1970; Radin E.L., Rose R.M., 1986). По мере прогрессирования ОА суставной хрящ, который является объектом для механического и химического стрессов, медленно эрозируется благодаря дисбалансу процессов катаболизма и репарации хряща. В частности, механический стресс в отношении «несущих» массу тела суставов способствует образованию большого количества микропереломов в субхондральной пластинке и хряще. По мере эрозирования суставного хряща прогрессирует склероз субхондральной кости, повышается жесткость костной ткани, которая в свою очередь способствует дальнейшему нарушению структуры суставного хряща. Однако вопрос о первичности или вторичности изменений субхондральной кости при ОА остается нерешенным.
До недавнего времени считали, что определяемые рентгенологически изменения в губчатом веществе субхондральной кости, такие, как склероз или образование кист, у больных ОА носят вторичный характер. Однако результаты клинических и экспериментальных исследований свидетельствуют о возможной инициирующей роли субхондральной кости в патогенезе ОА. Одним из возможных механизмов является резкое повышение градиента жесткости субхондральной кости (Radin E.L. et al., 1970; Radin E.L., Rose R.M., 1986) в связи с тем, что целостность надлежащей хрящевой ткани зависит от механических свойств ее костного «ложа». Исследования у приматов показали, что изменения в субхондральной кости могут предшествовать изменениям в суставном хряще (Carlson C.S. et al., 1994; 1996). Появившиеся в результате проведенных исследований на моделях ОА у животных (Brandt K.D. et al., 1991; Dedrick D.K. et al., 1993; Armstrong S. et al., 1994) и клинических исследований (Chai B.F., 1991; Grynpas M.D. et al., 1991; Hulth A., 1993; Shimizu M. et al., 1993) свидетельства в пользу этой гипотезы и против нее только обострили дискуссию. Утолщение трабекул в субхондральной кости не всегда сопровождается повышением минерализации костной ткани, а вернее, увеличением объема остеоида (Grynpas M.D. et al., 1991). Этот признак аномальной минерализации (Puzas J.E., 1993) свидетельствует о том, что нарушение регуляции ремоделирования костной ткани является неотъемлемой частью ОА, а также свидетельствует в пользу концепции о дефекте клеток костной ткани при ОА. Группа J. Dequeker (1989) рассматривают последний как «генерализованную метаболическую болезнь костной ткани».
Костная ткань постоянно обновляется. Этот динамичный процесс, который называется ремоделированием костной ткани, представляет собой сложную последовательность процессов резорбции и минерализации. Остеокласты резорбируют костную ткань, а остеобласты секретируют белки, формирующие основной органический компонент для минерализации (Raisz L.G., 1988). Образование и резорбция кости неслучайно происходят по всему скелету, это — запрограммированный процесс, происходящий в различных участках скелета, называемый единицами костного ремоделирования (Parfitt A.M., 1979). В начале цикла остеокласты появляются на неактивной поверхности; в течение 2 нед они образуют туннель в кортикальном слое кости или лакуну на поверхности трабекулярной кости. Частота активации новых единиц костного ремоделирования определяет степень обновления костной ткани. У здорового молодого человека процессы формирования и резорбции костной ткани уравновешены, поддерживается нормальная масса костной ткани. В гормональной регуляции резорбции костной ткани, по крайней мере ПТГ и ПГЕ2, принимают участие не только остеокласты, но и остеобласты, так как под действием этих гормонов высвобождаются факторы, стимулирующие резорбцию кости остеокластами. В настоящее время известно более 12 локальных и системных регуляторов роста костной ткани, влияющих на ее ремоделирование, в частности ПТГ, 1,25(ОН)2D3, кальцитонин, гормон роста, глюкокортикоиды, гормоны щитовидной железы, инсулин, ИФР (1 и 2), эстрогены, ПГЕ2, андрогены (Simpson E., 1984; Centrella M., Canalis E., 1985).
Костные клетки высвобождают ряд белков и цитокинов, которые осуществляют эндокринную регуляцию и передачу сигнала. Вырабатываемые остеобластами белки включают белки костного матрикса, такие, как коллаген, остеопонтин, остеокальцин, костные сиалопротеины (Whitson S.W. et al., 1984; Price P.A., 1985; Kream B.E. et al., 1986; Lo Y.Y.C. et al., 1996). Кроме того, эти клетки высвобождают протеазы как в активной, так и в латентной форме, которые принимают участие в процесее ремоделирования костной ткани — ММП (Heath J.K. et al., 1984; Otsuka K. et al., 1984; Meikle M.C. et al., 1994), компоненты системы активатор плазминогена (АП)/плазмин (Allan E.H. et al., 1991; Hoekman K. et al., 1991; Fawthrop F.W. et al., 1992). Высвобождаемые остеобластами цитокины могут действовать как посредством аутокринных механизмов, так и паракринным путем на местные клетки (другие остеобласты, остеокласты) (Horowitz M.C., Jilka R.L., 1992; Lowik C.W.G.M., 1992).
Пока неизвестно, каким путем регулируются эти сигналы — механическим стрессом или другими химическими сигналами, индуцированными механическим стрессом. Однако известно, что повторяющийся механический стресс вызывает локальную пролиферацию костных клеток и/или белков. В условиях in vivo механическая нагрузка способна активировать остеобласты (Pead M.J. et al., 1988), повышать уровень циклических нуклеотидов (Somjen D. et al., 1980; Shimshoni Z. et al., 1984), продукцию простагландинов (Somjen D. et al., 1980; Yeh C.K., Rodan G.A., 1984), а также вызывать морфологические изменения, ассоциированные с ремоделированием костной ткани (Pead M.J. et al., 1988). В условиях in vitro механический стресс вызывает пролиферацию культуры остеобластов (Cheng M.Z. et al., 1997), экспрессию мРНК костных белков, принимающих участие в образовании остеоида и в процессе минерализации (Harter L.V. et al., 1995; Cheng M.Z. et al., 1997), высвобождение локальных факторов роста, таких, как ИФР-1 и ИФР-2 (Zaman G. et al., 1997) и молекул адгезии (Keles A.O. et al., 1994). Передача сигнала механического стресса может осуществляться посредством механочувствительных ионных каналов (Yamagouchi D.T. et al., 1987; Moreau R. et al., 1996; 1997).
Существуют косвенные доказательства нарушения функции остеобластов при ОА. G. Gevers и J. Dequeker (1987) продемонстрировали повышение уровня сывороточного остеокальцина у женщин с ОА суставов кистей, а также в эксплантатах кортикальной зоны кости, что свидетельствует о том, что патология костной ткани может выступать частью ОА. При аутопсии обнаружено не только утолщение субхондральной кости, но и аномально низкую минерализацию головки бедренной кости (Grynpas M.D. et al., 1991). У морских свинок с хирургически индуцированным ОА с помощью компьютерной томографии обнаружено значительное утолщение костной фракции в субхондральной зоне (Dedrick D.K. et al., 1991). Дисбаланс между коллагеновыми и неколлагеновыми (остеокальцин и др.) белками может привести к увеличению объема костной ткани, но не влияет на ее минеральную плотность (Hannan M.T. et al., 1993; Li B., Aspden R.M., 1997). По данным M. Shimizu и соавторов (1993), прогрессирование дегенеративных изменений суставного хряща ассоциируется с более интенсивным ремоделированием субхондральной кости и повышением ее жесткости, что также указывает на дефект клеток костной ткани при ОА. Согласно предложенной B. Lee и M. Aspden (1997) гипотезе, пролиферация дефектных костных клеток может привести к повышению жесткости костной ткани, но не вызывает повышения ее минеральной плотности.
C.I. Westacott и соавторы (1997) выдвинули гипотезу о том, что аномальные остеобласты непосредственно влияют на метаболизм хрящевой ткани. Культивируя остеобласты пациентов с ОА с хондроцитами людей, у которых не было болезней суставов, авторы наблюдали значительное изменение высвобождения гликозаминогликанов нормальной хрящевой тканью in vitro, однако уровень высвобождения цитокинов оставался неизмененным. G. Hilal и соавторы (1998) показали, что культура остеобластов субхондральной кости больных с ОА in vitro имеет измененный метаболизм — активность системы АП/плазмин и уровень ИФР-1 в этих клетках повышены. Наблюдение C.I. Westacott и соавторов (1997) можно объяснить повышением активности протеаз клетками субхондральной кости.
Остается неизвестным, инициируют ли ОА изменения в субхондральной кости или способствуют его прогрессированию? D.K. Dedrick и соавторы (1993) продемонстрировали, что у собак с хирургически индуцированным ОА утолщение субхондральной кости не является необходимым условием для развития ОА-подобных изменений суставного хряща, однако способствует прогрессированию дегенеративных процессов в хряще. Результаты исследования А. Saїеd и соавторов (1997) противоречат данным предыдущего исследования. Используя 50 МГц эхографию для оценки начальных морфологических изменений и их прогрессирования в суставном хряще и кости при экспериментальном ОА, индуцированном инъекциями монойодуксусной кислоты в коленный сустав крыс, авторы продемонстрировали одновременный процесс изменений в кости и хряще в течение первых трех дней после инъекции.
Остеобласты секретируют факторы роста и цитокины, принимающие участие в локальном ремоделировании костной ткани, что может способствовать ремоделированию надлежащей хрящевой ткани в «несущих массу тела» суставах после их проникновения через микротрещины в кальцифицированном слое суставного хряща (Sokoloff L., 1993). Более того, продукты секреции костных клеток обнаруживают в синовиальной жидкости (Sharif M. et al., 1995). Наиболее вероятными продуктами, выделяемыми аномальными остеобластами, способными запускать процесс локального ремоделирования хрящевой ткани, являются ТФР-β и костные морфометрические протеины (КМП). Оба представителя семейства ТФР выделяются и хондроцитами и остеобластами и оба способны модифицировать ремоделирование как костной, так и хрящевой ткани (Erickson D.M. et al., 1997; Lietman S.A. et al., 1997; Ripamonti U. et al., 1997). J. Martel Pelletier и соавторы (1997) наблюдали повышение уровня ТФР-β в эксплантатах субхондральной кости больных ОА по сравнению со здоровыми людьми, что свидетельствует о вероятной роли этого фактора роста в патогенезе ОА. ИФР также продуцируются остеобластами. В культуре остеобластоподобных клеток, полученных от больных с ОА, обнаружено повышение уровня ИФР (Hilal G. et al., 1998), которые изменяют метаболизм хряща (Hilal G. et al., 1998).
ТФР-β, ИФР, КМП и цитокины, продуцируемые остеобластами в субхондральной кости, могут влиять на продукцию коллагеназы и других протеолитических ферментов в хряще, что в свою очередь, может способствовать ремоделированию/деградации хрящевого матрикса (Martel-Pelletier J. et al., 1999). Остается неясным, вырабатывают ли остеобласты при ОА меньше макрофагального колониестимулирующего фактора (М-КСФ — стимулятор костной резорбции), чем нормальные клетки. Результаты исследований A.G. Uitterlinden и соавторов (1997) показали, что определенную роль в образовании остеофитов могут играть рецепторы витамина D, которые экспрессируются остеобластами и регулируют экспрессию ряда факторов, синтезируемых этими клетками, что частично объясняет роль остеобластов в патогенезе этого заболевания.
Учитывая результаты вышеприведенных исследований, G. Hilal и соавторы (1998), J. Martel-Pelletier и соавторы (1997) предложили следующую рабочую гипотезу взаимоотношений ремоделирования субхондральной кости и надлежащего суставного хряща при ОА (рис. 13). На ранней или развернутой стадии патогенеза ОА интенсифицируется процесс ремоделирования костной ткани в субхондральной кости. Одновременно повторяющаяся нагрузка ведет к локальным микропереломам и/или появлению дисбаланса системы ИФР/ИФР-связывающий белок (ИФРСБ) вследствие аномального ответа остеобластов субхондральной кости, что способствует ее склерозу. Последнее в свою очередь может способствовать появлению микропереломов надлежащего хряща и повреждению его матрикса.
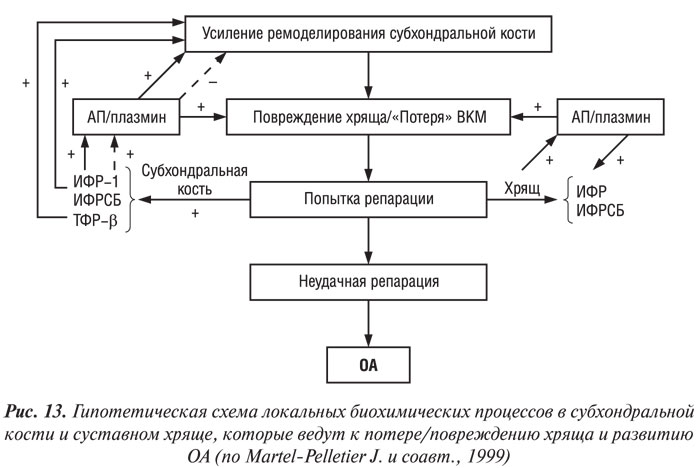
В нормальных условиях это повреждение устраняется путем локального синтеза и высвобождения ИФР-1 и ИФР-связывающего белка, которые стимулируют образование ВКМ суставного хряща. В то же время ИФР-система способствует росту клеток субхондральной кости и формированию костного матрикса. Анаболическая активность ИФР-системы повышена в субхондральной кости больных ОА, тогда как локальная активация системы АП/плазмин (местный регулятор ИФР-системы) в суставном хряще обусловливает его локальные изменения (Martel-Pelletier J. et al., 1991). В остеобластах при ОА ИФР-1 нарушает регуляцию АП плазмином по типу положительной обратной связи, следовательно, может сдерживать ремоделирование в костной ткани, что в итоге приводит к субхондральному склерозу (Hilal G. et al., 1998). Таким образом, в костной и хрящевой ткани локальная индукция ИФР-1 и протеаз ведет, с одной стороны, к повреждению хряща, с другой — к утолщению субхондральной кости, последнее в свою очередь способствует дальнейшему повреждению хряща. Дисбаланс между повреждением хряща, связанным с субхондральным склерозом, и его репаративными способностями ведет к прогрессирующему изменению ВКМ хряща и к развитию ОА. По мнению авторов, эта гипотеза также объясняет медленное прогрессирование болезни.
Роль ферментов и цитокинов в патогенезе ОА
В последние годы большое внимание исследователей фокусируется на идентификации протеаз, ответственных за деградацию ВКМ суставного хряща при ОА (рис. 14). Согласно современным представлениям, важную роль в патогенезе ОА играют матриксные металлопротеазы (ММП) (Dean D.D., 1991; Pelletier J.P. et al., 1997). У больных с ОА обнаруживают повышенный уровень трех представителей ММП — коллагеназ, стромелизинов и желатиназ. Коллагеназа ответственна за деградацию нативного коллагена, стромелизин — коллагена IV типа, протеогликанов и ламинина (Fini M. et al., 1987), а желатиназа — за деградацию желатина, коллагенов IV, V и XI типов, эластина (Mohtai M. et al., 1993). Кроме того, предполагают наличие еще одного фермента — аггреканазы, который обладает свойствами ММП и отвечает за протеолиз хрящевых протеогликановых агрегатов (Sandy J.D. et al., 1992).
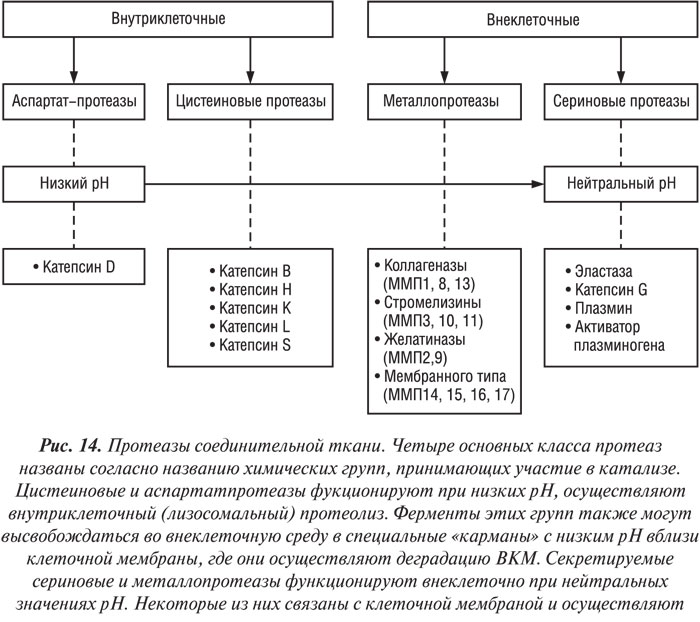
В суставном хряще человека идентифицированы три типа коллагеназ, уровень которых значительно повышен у больных ОА, — коллагеназа-1 (ММП-1), коллагеназа-2 (ММП-8) и коллагеназа-3 (ММП-13). Сосуществование трех разных типов коллагеназ в суставном хряще свидетельствует о том, что каждая из них играет свою специфическую роль (Martel-Pelletier J., Pelletier J.P., 1996). Действительно, коллагеназы-1 и -2 локализуются главным образом в поверхностной и верхней части промежуточной зоны суставного хряща, тогда как коллагеназу-3 обнаруживают в нижней части промежуточной и в глубокой зонах (Nguyen Q. et al., 1992; Cole A.A. et al., 1996; Martel-Pelletier J., Pelletier J.P., 1996; Moldovan F. et al., 1997; Fernandes J.C. et al., 1998). Более того, результаты иммуногистохимического исследования продемонстрировали, что в процессе прогрессирования ОА уровень коллагеназы-3 достигает плато и даже снижается, тогда как уровень коллагеназы-1 постепенно повышается (Fernandes J.C. et al., 1998). Имеются данные о том, что при ОА коллагеназа-1 главным образом участвует в воспалительном процессе в суставном хряще, тогда как коллагеназа-3 — в ремоделировании ткани (Martel-Pelletier J. et al., 1999). Экспрессируемая в хряще больных с ОА коллагеназа-3 осуществляет деградацию коллагена II типа более интенсивно, чем коллагеназа-1 (Mitchell P. et al., 1996).
Из представителей второй группы металлопротеаз, стромелизинов у человека идентифицированы также три — стромелизин-1 (ММП-3), стромелизин-2 (ММП-10) и стромелизин-3 (ММП-11). Сегодня известно, что только стромелизин-1 вовлечен в патологический процесс при ОА (Sirum K.L., Brinckerhoff C.E., 1989; Okada Y. et al., 1992; Hembry R.M. et al., 1995). В синовиальной мембране больных с ОА не определяется стромелизин-2, однако он обнаружен в очень малом количестве в синовиальных фибробластах больных с ревматоидным артритом (Sirum K.L., Brinckerhoff C.E., 1989; Hembry R.M. et al., 1995). Стромелизин-3 также обнаружен в синовиальной оболочке больных с ревматоидным артритом вблизи фибробластов, особенно в зонах фиброза (Sirum K.L., Brinckerhoff C.E., 1989).
В группе желатиназ в хрящевой ткани человека идентифицировано только две — желатиназа 92 кД (желатиназа В, или ММП-9) и желатиназа 72 кД (желатиназа А, или ММП-2); у больных с ОА определяют повышение уровня желатиназы 92 кД (Mohtai M. et al., 1993).
Не так давно была идентифицирована еще одна группа ММП, которые локализуются на поверхности клеточных мембран и называются ММП мембранного типа (ММП-МТ). К этой группе принадлежат четыре фермента — ММП-МТ1 — ММП-МТ-4 (Martel-Pelletier J. et al., 1999). Экспрессия ММП-МТ обнаружена в суставном хряще человека (Buttner F.H. et al., 1997). Хотя ММП-МТ-1 обладает свойствами коллагеназы (Ohuchi E. et al., 1997), оба фермента ММП-МТ-1 и ММП-МТ-2 способны активировать желатиназу-72 кД и коллагеназу-3 (Takino T. et al., 1995; Imai K. et al., 1996; Buttner F.H. et al., 1997). Роль этой группы ММП в патогенезе ОА требует уточнений.
Протеиназы секретируются в форме зимогена, который активируется другими протеиназами или органическими соединениями ртути. Каталитическая активность ММП зависит от наличия цинка в активной зоне фермента.
Биологическая активность ММП контролируется специфическими ТИМП (рис. 15). К настоящему времени идентифицированы три типа ТИМП, которые обнаруживают в суставных тканях у человека, — ТИМП-1 — ТИМП-3 (Dean D.D. et al., 1989; Apte S.S. et al., 1994; Martel-Pelletier J. et al., 1994). Четвертый тип ТИМП идентифицирован и клонирован, однако он до сих пор не был обнаружен в суставных тканях человека (Greene J. et al., 1996). Эти молекулы специфически связываются с активным центром ММП, хотя некоторые из них способны связывать активный центр прожелатиназы 72 кД (ТИМП-2, -3, -4) и прожелатиназы 92 кД (ТИМП-1 и -3) (Will H. et al., 1996; Bigg H.F. et al., 1997). Данные свидетельствуют о том, что при ОА в суставном хряще существует дисбаланс между ММП и ТИМП, результатом которого является относительный дефицит ингибиторов (Dean D.D. et al., 1989; Pelletier J.P. et al., 1990), что, возможно, частично связано с повышением уровня активных ММП в ткани. ТИМП-1 и -2 обнаруживают в суставном хряще, они синтезируются хондроцитами (Dean D.D. et al., 1989; Wolfe G.C. et al., 1993; Martel-Pelletier J. et al., 1994). При ОА в синовиальной оболочке и синовиальной жидкости обнаружен только первый тип ТИМП (Clarck I.M. et al., 1993; Lohmander L.S. et al., 1993; Hembry R.M. et al, 1995). ТИМП-3 обнаруживают исключительно в ВКМ (Gomez D.E. et al., 1997). ТИМП-4 почти на 50% имеет идентичную аминокислотную последовательность с ТИМП-2 и -3 и на 38% — с ТИМП-1 (Leco K.J. et al., 1997). В других клетках-мишенях ТИМП-4 ответственен за модуляцию активации прожелатиназы 72 кД на поверхности клеток, что свидетельствует о важной роли в качестве тканеспецифического регулятора ремоделирования ВКМ (Bigg H.F. et al., 1997).
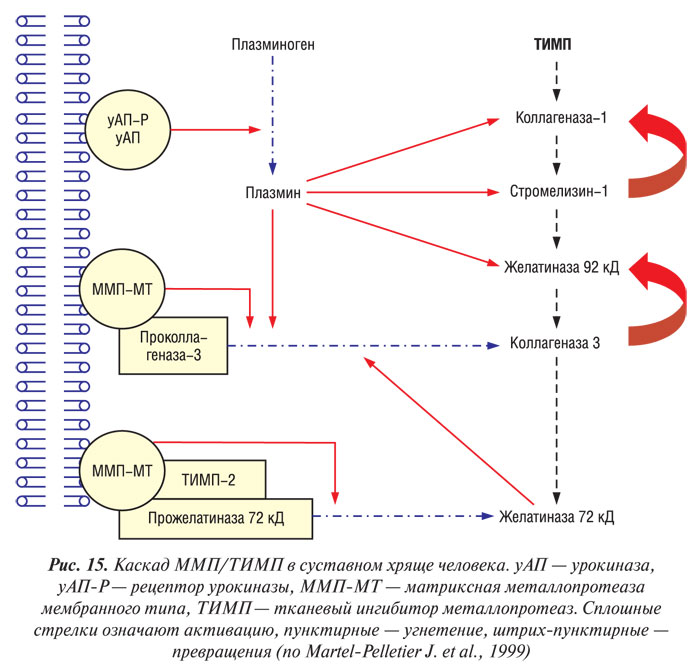
Другим механизмом контролирования биологической активности ММП является их физиологическая активация. Существует мнение, что ферменты из семейства сериновых и цистеиновых протеаз, таких, как АП/плазмин и катепсин В соответственно, и являются физиологическими активаторами ММП (Eeckhout Y., Vaes G., 1977; Nagase H. et al., 1990). В суставном хряще больных с ОА обнаружен повышенный уровень урокиназы (уАП) и плазмина (Martel-Pelletier J. et al., 1991).
Несмотря на то, что в тканях сустава обнаруживают несколько типов катепсинов, наиболее вероятным активатором ММП в хряще считают катепсин-В (Martel-Pelletier J. et al., 1990; Buttle D.J. et al., 1993). В тканях сустава человека обнаружены физиологические ингибиторы сериновых и цистеиновых протеаз. Активность ингибитора АП-1 (иАП-1), а также цистеиновых протеаз снижена у больных с ОА (Martel-Pelletier J. et al., 1990; 1991). Аналогично ММП/ТИМП — именно дисбалансом между сериновыми и цистеиновыми протеазами и их ингибиторами можно объяснить повышенную активность ММП в суставном хряще больных с ОА. Кроме того, ММП способны активировать друг друга. Например, стромелизин-1 активирует коллагеназу-1, коллагеназу-3 и желатиназу 92 кД; коллагеназа-3 активирует желатиназу 92 кД; ММП-МТ активирует коллагеназу-3, а желатиназа-72 кД потенциирует эту активацию; ММП-МТ также активирует желатиназу 72 кД (Murphy G. et al., 1987; Ogata Y. et al., 1992; Atkinson S.J. et al., 1995; Knauper V. et al., 1996; 1997; d’Ortho M.P. et al., 1997).
Цитокины можно разделить на три группы — деструктивные (провоспалительные), регуляторные (в том числе противовоспалительные) и анаболические (факторы роста).
Деструктивные цитокины, в частности ИЛ-1, индуцируют увеличение высвобождения протеаз и угнетают синтез протеогликанов и коллагенов хондроцитами. Регуляторные цитокины, в частности ИЛ-4 и -10, угнетают продукцию ИЛ-1, увеличивают продукцию антагониста рецептора ИЛ-1 (ИЛ-1РА) и снижают уровень и NО-синтазы в хондроцитах. Таким образом, ИЛ-4 противодействует ИЛ-1 по трем направлениям: 1) снижает продукцию, препятствует его эффектам, 2) увеличивает продукцию основного «скавенджера» ИЛ-1РА и 3) снижает продукцию основного вторичного «мессенджера» NO. Кроме того, ИЛ-4 снижает ферментативную деградацию ткани. В условиях in vivo оптимальный терапевтический эффект достигается при комбинации ИЛ-4 и ИЛ-10. Анаболические факторы, такие, как ТФР-β и ИФР-1, реально не препятствуют продукции или действию ИЛ-1, а проявляют противоположную активность, например, стимулируют синтез протеогликанов и коллагена, подавляют активность протеаз, а ТФР-β еще и угнетает высвобождение ферментов и стимулирует их ингибиторы.
Провоспалительные цитокины отвечают за повышенный синтез и экспрессию ММП в суставных тканях. Они синтезируются в синовиальной оболочке, а затем диффундируют в суставной хрящ через синовиальную жидкость. Провоспалительные цитокины активируют хондроциты, которые в свою очередь также способны вырабатывать провоспалительные цитокины. В пораженных ОА суставах роль эффектора воспаления играют главным образом клетки синовиальной мембраны. Именно синовициты макрофагального типа секретируют протеазы и медиаторы воспаления. Среди них в патогенезе ОА в наибольшей мере «задействованы» ИЛ-1β, ФНО-α, ИЛ-6, лейкемический ингибирующий фактор (ЛИФ) и ИЛ-17.
Данные литературы свидетельствуют, что ИЛ-1β и, возможно ФНО-α, — главные медиаторы деструкции суставных тканей при ОА (Pelletier J.P. et al., 1997). Однако до сих пор не известно, действуют ли они независимо друг от друга или между ними существует функциональная иерархия. На моделях ОА у животных показано, что блокада ИЛ-1 эффективно предотвращает деструкцию суставного хряща (van de Loo F.A. et al., 1995; Caron J.P. et al., 1996), тогда как блокада ФНО-α приводит лишь к ослаблению воспаления в тканях сустава (van de Loo F.A. et al., 1995; Plows D. et al., 1995). В синовиальной мембране, синовиальной жидкости и хряще больных обнаружены повышенные концентрации обоих цитокинов (Wood D.D. et al., 1983; Pelletier J.P. et al., 1989; 1993; 1997; Chikanza I.C. et al., 1993; Farahat M.N. et al., 1993). В хондроцитах они способны увеличивать синтез не только протеаз (главным образом ММП и АП), но и минорных коллагенов, например І и ІІІ типов, а также уменьшать синтез коллагенов ІІ и ІХ типов и протеогликанов (Martel-Pelletier J. et al., 1999). Эти цитокины также стимулируют активные формы кислорода и такие медиаторы воспаления, как ПГЕ2. Результатом таких макромолекулярных изменений в суставном хряще при ОА является неэффективность репаративных процессов, что приводит к дальнейшей деградации хряща.
Вышеназванные провоспалительные цитокины модулируют процессы угнетения/активации ММП при ОА. Например, дисбаланс между уровнями ТИМП-1 и ММП в хряще при ОА может опосредоваться ИЛ-1β, так как исследование in vitro продемонстрировало, что повышение концентрации ИЛ-1β приводит к снижению концентрации ТИМП-1 и увеличению синтеза ММП хондроцитами (Martel-Pelletier J. et al., 1994). Синтез АП также модулируется ИЛ-1β. Стимуляция in vitro хондроцитов суставного хряща с использованием ИЛ-1 вызывет дозозависимое увеличение синтеза АП и резкое снижение синтеза иАП-1 (Martel-Pelletier J. et al., 1991). Способность ИЛ-1 уменьшать синтез иАП-1 и стимулировать синтез АП является мощным механизмом генерации плазмина и активации ММП. Кроме того, плазмин является не только ферментом, активизирующим другие ферменты, он также принимает участие в процессе деградации хряща путем прямого протеолиза.
ИЛ-1β синтезируется в виде неактивного предшественника с массой 31 кД (пре-ИЛ-1β), а затем, после отщепления сигнального пептида, превращается в активный цитокин с массой 17,5 кД (Mosley B. et al., 1987; Siders W.M. et al., 1993). В тканях суставов, включая синовиальную мембрану, синовиальную жидкость и суставной хрящ, ИЛ-1β обнаруживают в активной форме, а в исследованиях in vivo продемонстрирована способность синовиальной мембраны при ОА секретировать этот цитокин (Pelletier J.P. et al., 1995). Некоторые сериновые протеазы способны превращать пре-ИЛ-1β в его биоактивную форму (Black R.A. et al., 1988). У млекопитающих такие свойства обнаружены лишь у одной протеазы, которая относится к семье цистеиновых аспартатспецифических ферментов и называется ИЛ-1β-конвертирующий фермент (ИКФ, или каспаза-1). Этот фермент способен специфически превращать пре-ИЛ-1β в биологически активный «зрелый» ИЛ-1β с массой 17,5 кД (Black R.A. et al., 1988; Kronheim S.R. et al., 1992). ИКФ — это профермент с молекулярной массой 45 кД (р45) (Black R.A. et al., 1988; Kronheim S.R. et al., 1992), который локализуется в клеточной мембране. После протеолитического расщепления проэнзима р45 с образованием двух субъединиц, известных как р10 и р20, которым свойственна ферментативная активность (Wilson K.P. et al., 1994).
ФНО-α также синтезируется в виде мембранно-связанного предшественника с массой 26 кД; путем протеолитического отщепления он высвобождается из клетки в виде активной растворимой формы с массой 17 кД (Aggrawal B.B. et al., 1985; Gearing A.J. et al., 1994). Протеолитическое отщепление осуществляется ФНО-α-конвертирующим ферментом (ФНО-КФ), который относится к семье адамализинов (Black R.A. et al., 1997). A.R. Amin и соавторы (1997) обнаружили повышенную экспрессию мРНК ФНО-КФ в суставном хряще больных с ОА.
Биологическая активация хондроцитов и синовицитов ИЛ-1 и ФНО-α опосредуется связыванием со специфическими рецепторами на поверхности клеток — ИЛ-Р и ФНО-Р. Для каждого цитокина идентифицировано два типа рецепторов — ИЛ-1Р І и ІІ типов (Slack J. et al., 1993) и ФНО-Р І (р55) и ІІ (р75) типов (Tartaglia L.A., Goeddel D.V., 1992). За передачу сигналов в клетках тканей суставов отвечают ИЛ-1РІ и р55 (Martel-Pelletier J. et al., 1992; Arend W.P., 1993; Westacott C.I. et al., 1994; Sadouck M. et al., 1995; Alaaeddine N. et al., 1997). ИЛ-1Р І типа обладает несколько большей аффинностью к ИЛ-1β, чем к ИЛ-1α; ИЛ-1Р ІІ типа — наоборот, имеет большее сродство к ИЛ-1α, чем к ИЛ-1β. До сих пор остается неясным, может ли ИЛ-1Р ІІ типа опосредовать сигналы ИЛ-1 или он служит только для конкурентного ингибирования связи ИЛ-1 с ИЛ-1Р І типа (Martel-Pelletier J. et al., 1999). В хондроцитах и синовиальных фибробластах больных с ОА обнаруживают большое количество ИЛ-1РІ и р55 (Martel-Pelletier J. et al., 1992; Westacott C.I. et al., 1994; Sadouck M. et al., 1995; Alaaeddine N. et al., 1997), что в свою очередь объясняет высокую чувствительность этих клеток к стимуляции соответствующими цитокинами (Martel-Pelletier J. et al., 1992). Этот процесс приводит как к повышению секреции протеолитических ферментов, так и к деструкции суставного хряща.
Не исключается участие ИЛ-6 в патологическом процессе при ОА. В основе этого предположения лежат следующие наблюдения:
- ИЛ-6 увеличивает количество клеток воспаления в синовиальной мембране (Guerne P.A. et al., 1989),
- ИЛ-6 стимулирует пролиферацию хондроцитов,
- ИЛ-6 усиливает эффекты ИЛ-1 в отношении повышения синтеза ММП и угнетения синтеза протеогликанов (Carroll G.J., Bell M.C., 1993).
Однако ИЛ-6 способен индуцировать продукцию ТИМП (Lotz M., Guerne P.A., 1991), но не влияет на продукцию ММП, поэтому считают, что именно этот цитокин принимает участие в процессе сдерживания протеолитической деградации суставного хряща, который осуществляется по механизму обратной связи.
Еще одним представителем семьи ИЛ-6 является ЛИФ — цитокин, который вырабатывается хондроцитами, полученными от больных с ОА, в ответ на стимуляцию провоспалительными цитокинами ИЛ-1β и ФНО-α (Lotz M. et al., 1992; Campbell I.K. et al., 1993; Hamilton J.A. et al., 1993). ЛИФ стимулирует резорбцию протеогликанов хряща, а также синтез ММП и продукцию NO (Carroll G.J., Bell M.C., 1993). Роль этого цитокина при ОА окончательно не выяснена.
ИЛ-17 — гомодимер массой 20–30 кД (Yao Z. et al., 1995), обладающий ИЛ-1-подобным действием, однако значительно менее выраженным. ИЛ-17 стимулирует синтез и выделение ряда провоспалительных цитокинов, в их числе ИЛ-1β, ФНО-α, ИЛ-6, а также ММП в клетках-мишенях, например в макрофагах человека (Jovanovic D. et al., 1998). Кроме того, ИЛ-17 стимулирует продукцию NO хондроцитами (Attur M.G. et al., 1997). Подобно ЛИФ, роль ИЛ-17 в патогенезе ОА мало изучена.
Неорганический свободный радикал NO играет важную роль в деградации суставного хряща при ОА. Хондроциты, полученные от больных с ОА, вырабатывают большее количество NO как спонатанно, так и после стимуляции провоспалительными цитокинами в сравнении с нормальными клетками (Pelletier J.P. et al., 1996). Высокое содержание NO обнаружено в синовиальной жидкости и сыворотке крови больных с ОА (Farrell A.J. et al., 1992; McInnes I.B. et al., 1996) — это результат увеличения экспрессии и синтеза индуцированной NO-синтазы (иNOС) — фермента, ответственного за продукцию NO (McInnes I.B. et al., 1996 ; Grabowski P.S. et al., 1997). Недавно была клонирована ДНК хондроцитспецифической иNOС, определена аминокислотная последовательность фермента (Charles I.G. et al., 1993). Аминокислотная последовательность указывает на 50% идентичность и 70% сходство с иNOС, специфичной для эндотелия и нервной ткани.
NO угнетает синтез макромолекул ВКМ суставного хряща и стимулирует синтез ММП. Более того, увеличение продукции NO сопровождается снижением синтеза антагониста ИЛ-1Р (ИЛ-1РА) хондроцитами (Pelletier J.P. et al., 1996). Таким образом, повышение уровня ИЛ-1 и снижение — ИЛ-1РА приводит к гиперстимуляции NO хондроцитов, что в свою очередь ведет к усилению деградации хрящевого матрикса. Имеются сообщения о терапевтическом эффекте in vivo селективного ингибитора иNOС в отношении прогрессирования экспериментального ОА (Stefanovic-Racic M. et al., 1994; Connor J.R. et al., 1995; Pelletier J.P. et al., 1998).
Естественные ингибиторы цитокинов способны непосредственно препятствовать связыванию цитокинов с рецепторами клеточных мембран, снижая их провоспалительную активность. Естественные ингибиторы цитокинов можно разделить на три класса по способу их действия.
К первому классу ингибиторов относят антагонисты рецепторов, которые препятствуют связыванию лиганда с его рецептором путем конкуренции за связывающий центр. К настоящему времени такой ингибитор найден только для ИЛ-1 — это вышеупомянутый конкурентный ингибитор системы ИЛ-1/ИЛ1Р ИЛ-1РА (Arend W.P., 1993; Dinarello C.A., 1996). ИЛ-1РА блокирует многие эффекты, которые наблюдаются в тканях суставов при ОА, включая синтез простагландинов синовиальными клетками, продукцию коллагеназы хондроцитами и деградацию ВКМ суставного хряща.
ИЛ-1РА обнаруживают в различных формах — одной растворимой (рИЛ-1РА) и двух межклеточных (мкИЛ-1РАІ и мкИЛ-1РАІІ) (Arend W.P., 1993). Аффинность растворимой формы ИЛ-1РА в 5 раз превышает таковую у межклеточных форм (Martel-Pelletier J. et al., 1999). Несмотря на интенсивный научный поиск, функция последних остается неизвестной. Экспериметы in vitro показали, что для угнетения активности ИЛ-1β необходима концентрация ИЛ-1РА, в 10–100 раз превышающая норму, в условиях in vivo требуется тысячекратное повышение концентрации ИЛ-1РА (Pelletier J.P. et al., 1995; Campion G.V. et al., 1996; Joosten L.A. et al., 1996; Bakker A.C. et al., 1997). Этот факт может частично объяснить относительный дефицит ИЛ-1РА и избыток ИЛ-1 в синовии больных с ОА.
Второй класс естественных ингибиторов цитокинов представлен растворимыми рецепторами цитокинов. Примером таких ингибиторов у человека, имеющих отношение к патогенезу ОА, являются рИЛ-1Р и рр55 (Giri J.G. et al., 1990; Lantz M. et al., 1990). Растворимые рецепторы цитокинов представляют собой укороченные формы нормальных рецепторов, связываясь с цитокинами, они препятствуют их связыванию с мембранно-ассоциированными рецепторами клеток-мишеней, действуя по механизму конкурентного антагонизма.
Основным предшественником растворимых рецепторов являются мембранно-связанные ИЛ-1РІІ. Аффинность рИЛ-1Р по отношению к ИЛ-1 и ИЛ-1РА различна. Так, рИЛ-1РІІ обладает бo′льшим сродством к ИЛ-1β, чем к ИЛ-1РА, а рИЛ-1РІ — проявляет бo′льшую аффинность к ИЛ-1РА, чем к ИЛ-1β (Arend W.P., 1993; Svenson M. et al., 1993; Dinarello C.A., 1996).
Для ФНО также существуют два типа растворимых рецепторов — рр55 и рр75, как и растворимые рецепторы ИЛ-1, они образуются путем «шеддинга» (сброса). В условиях in vivo оба рецептора обнаруживают в тканях пораженных суставов (Cope A.P. et al., 1992; Chikanza I.C. et al., 1993; Roux-Lombard P. et al., 1993). Роль растворимых рецепторов ФНО в патогенезе ОА дискутируется. Предполагают, что в низких концентрациях они стабилизируют трехмерную структуру ФНО и повышают период полужизни биоактивного цитокина (Arderka D. et al., 1992), тогда как высокие концентрации рр55 и рр75 могут снижать активность ФНО путем конкурентного антагонизма (Higuchi M., Aggrawal B.B., 1992). Вероятно, рр75 может выступать как переносчик ФНО, облегчая его связывание с мембранно-ассоциированным рецептором (Tartaglia L.A. et al., 1993).
Третий класс естественных ингибиторов цитокинов представлен группой противовоспалительных цитокинов, к которым относят ТФР-β, ИЛ-4, ИЛ-10 и ИЛ-13. Противовоспалительные цитокины снижают продукцию провоспалительных, а также некоторых протеаз, стимулируют продукцию ИЛ-1РА и ТИМП (Hart P.H. et al., 1989; Lacraz S. et al., 1992; de Waal Malefyt R. et al., 1993; Jenkins J.K. et al., 1994; Jovanovic D. et al., 1998).
Репарация суставного хряща и факторы роста в патогенезе ОА
Благодаря прогрессу биотехнологии, в частности технологии клонирования, в последнее время интенсивно пополняется перечень факторов роста, которые, являясь анаболическими факторами, играют важную, но не до конца понятную роль в патогенезе ОА (табл. 14).
Первой группой факторов роста, о которых пойдет речь ниже, являются ИФР. Они в большом количестве содержатся в сыворотке крови, имеют ряд общих свойств с инсулином. ИФР-2 — более характерен для эмбриональной стадии развития, тогда как ИФР-1 — доминирующий представитель группы у взрослого человека. Оба представителя этой группы действуют путем соединения с рецепторами ИФР І типа. Если функция ИФР-2 остается неизвестной, значение ИФР-1 уже определено — он способен стимулирвовать синтез протеогликанов хондроцитами и значительно угнетать катаболические процессы в суставном хряще. ИФР-1 — главный анаболический стимул для синтеза протеогликанов хондроцитами, присутствующий в сыворотке крови и синовиальной жидкости (Schalkwijk J. et al., 1989). ИФР-1 — важный фактор для культивирования хондроцитов в экспериментальных моделях ОА in vitro (Schalkwijk J. et al., 1989; Tyler J.A., 1989; Tesch G.H. et al., 1992; van Beuningen H.M. et al., 1993). Предполагают, что ИФР-1 попадает в синовиальную жидкость из плазмы крови. Кроме того, нормальные хондроциты продуцируют оба фактора — экспрессия ИФР-1 и ИФР-2 обнаружена в синовиальной оболочке и хряще больных ОА (Middlton J.F., Tyler J.A., 1992; Keyszer G.M. et al., 1995). В нормальном хряще ИФР-1 не обладает митогенными свойствами, однако способен стимулировать пролиферацию клеток в поврежденном матриксе, что свидетельствует об участии в репаративных процессах (van den Berg W.B. et al., 1999).
Действия ИФР-1 и ИФР-2 контролируются различными ИФР-связывающими белками (ИФР-СБ), которые также вырабатываются хондроцитами (Martel-Pelletier J. et al., 1998). ИФР-СБ могут выполнять функцию переносчика, а также обладают блокирующей ИФР-активностью. Изолированные из суставного хряща больных с ОА клетки вырабатывают избыточное количество ИФР-СБ, что свидетельствует о блокировании ними эффектов ИФР (Dore S. et al., 1994). J. Martel-Pelletier и соавторы (1998) показали, что хотя синтез ИФР-1 в хряще при ОА возрастает, хондроциты слабо реагируют на стимуляцию ИФР-1. Оказалось, что этот феномен связан (по меньшей мере частично) с повышением уровня ИФР-СБ. ИФР-СБ имеет высокое сродство к ИФР и является важным биомодулятором его активности. К настоящему времени изучено семь типов ИФР-СБ, нарушение регуляции ИФР-СБ-3 и ИФР-СБ-4 играет важную роль при ОА (Marlet-Pelletier J. et al., 1998).
Еще одна категория факторов роста, проявляющих различное действие в отношении хондроцитов, включает фактор роста, полученный из тромбоцитов (ФРПТ), ФРФ и ТФР-β. Эти факторы вырабатываются не только хондроцитами, но и активированными синовицитами (Remmers E.F. et al., 1991; Nalashima M. et al., 1994). ФРФ обладает как анаболическими, так и катаболическими свойствами в зависимости от концентрации и состояния суставного хряща (Sah R.L. et al., 1994). ФРПТ принимает участие в поддержании гомеостаза ВКМ суставного хряща, не обладая явными митогенными свойствами. Для этого фактора роста известна способность усиливать синтез протеогликанов и уменьшать их деградацию (Schafer S.J. et al., 1993).
ТФР-β представляет особый интерес в плане изучения его роли в патогенезе ОА. Он является членом большого ТФР-суперсемейства, имеет общие фунциональные и сигнальные свойства с недавно открытыми факторами роста КМБ (костных морфогенетических белков).
ТФР-β — плейотропный фактор: c одной стороны, он обладает иммуносупрессивными свойствами, с другой — это хемотаксический фактор и мощный стимулятор пролиферации фибробластов (van den Berg W.B. et al., 1999). Уникальными свойствами ТФР-β являются способность угнетать высвобождение ферментов из разных клеток и значительно увеличивать продукцию ингибиторов ферментов (например, ТИМП) (Wright J.K. et al., 1991; Gunter M. et al., 1994). ТФР-β считают важным регулятором повреждения ткани вследствие воспаления. Так, в ткани суставного хряща ТФР-β значительно стимулирует продукцию матрикса хондроцитами (van der Kraan P.M. et al., 1992; Morales T.I., 1994; van Beuningen H.M. et al., 1994), особенно после преэкспозиции этим фактором. Нормальный хрящ нечувствителен к ТФР-β. У больных с ОА ТФР-β стимулирует продукцию аггрекана и малых протеогликанов в суставном хряще (Roughley P.J. et al., 1994; CS-Szabo G. et al., 1995).
ТФР-β вырабатывается многими клетками, в частности хондроцитами. Он высвобождается в латентной форме, связанной со специальным белком, который называется «белок, ассоциированный с латентностью» (БАЛ). Диссоциация с этим белком осуществляется протеазами, которые в большом количестве вырабатываются в воспаленных тканях. Не считая ТФР-β, который продуцируется активированными клетками, запасы латентной формы этого фактора являются важным элементом реактивности ТФР-β в ткани после локального повреждения. ТФР-β в значительном количестве содержится в синовиальной жидкости, синовиальной мембране и хряще сустава, пораженного ОА (Fava R. et al., 1989; Schlaak J.F. et al., 1996; Chambers M.G. et al., 1997). В участках поврежденной ткани, где имеются воспалительные инфильтраты, обнаруживают коэкспрессию ФНО и ИЛ-1, тогда как в участках с явлениями фиброза обнаруживают исключительно экспрессию ТФР-β (Chu C.Q. et al., 1991).
Инкубация культуры хондроцитов, полученных от больных с ОА, с ТФР-β вызывает значительное увеличение синтеза протеогликанов этими клетками. Стимуляция ТФР-β нормальных хондроцитов вызывает увеличение синтеза протеогликанов только после многих дней инкубации. Возможно, это время необходимо для изменения фенотипа клеток под влиянием ТФР-β (например, для изменения так называемой компартментализации протеогликанов: вновь созданные протеогликаны локализуются только вокруг хондроцитов).
Известно, что активация синтеза факторов роста, в частности ТФР-β, — важное звено в патогенезе фиброза почек и печени, формировании рубца при заживлении раны (Border W.A., Ruoslahti E., 1992). Увеличение нагрузки на хондроциты in vitro приводит к гиперпродукции ТФР-β, тогда как уменьшение синтеза протеогликанов после иммобилизации конечности может быть нивелировано ТФР-β (Zerath E. et al., 1997). ТФР-β индуцирует формирование остеофитов в краевой зоне суставов как механизм приспособления к изменению нагрузки. ИЛ-1, вызывая умеренный воспалительный процесс в синовии в ответ на повреждение сустава, способствует образованию хондроцитов с измененным фенотипом, которые вырабатывают избыточное количество (van den Berg W.B. et al., 1999).
Повторные локальные инъекции рекомбинантного ТФР-β в высоких концентрациях привела к развитию ОА у мышей линии C57Bl — образованию остеофитов, что характерно для ОА человека, и значительной потере протеогликанов в зоне «волнистой границы» (van den Berg W.B. et al., 1999).
Для понимания того, как избыток ТФР-β вызывает известные изменения хряща, необходимо заметить, что экспозиция ТФР-β индуцирует характерный фенотип хондроцита с изменением подкласса синтезируемых протеогликанов и нарушением нормальной интеграции элементов ВКМ. И ИФР-1, и ТФР-β стимулируют синтез протеогликанов хондроцитами, культивируемыми в алгинате, однако последний еще и индуцирует так называемую компартментализацию протеогликанов (van Osch G.J.V.M. et al., 1998). Более того, обнаружено, что ТФР-β повышает уровень коллагеназы-3 (ММП-13) в активированных хондроцитах, что расходится с общим представлением о ТФР-β, как факторе, который, наоборот, уменьшает высвобождение деструктивных протеаз (Moldovan F. et al., 1997). Хотя не известно, участвует ли ТФР-β-индуцированный синтез ММП-13 в патогенезе ОА. ТФР-β не только стимулирует синтез протеогликанов, но и способствует их отложению в связках и сухожилиях, увеличивая скованность и уменьшая объем движений в суставах (Kawaguchi H. et al., 1992; Hoshi K. et al., 1997).
КМП — члены суперсемейства ТФР-β. Некоторые из них (КМП-2, КМП-7 и КМП-9) обладают свойством стимулировать синтез протеогликанов хондроцитами (Luyten F.P. et al., 1992; Lietman S.A. et al., 1997; van Beuningen H.M. et al., 1998). КМП осуществляют свои эффекты путем связывания со специфическими рецепторами на поверхности клеток; сигнальные пути ТФР-β и КМП несколько отличаются (Yamashita H. et al., 1996; Attisano L., Wrana J.L., 1998). Подобно ТФР-β, передача сигнала от КМП осуществляется через комплекс рецепторов сериновой/треониновой киназы І и ІІ типа. В этом комплексе рецептор ІІ типа трансфосфорилируется и активирует рецептор І типа, который передает сигнал сигнальным молекулам, называемым Smad (Attisano L., Wrana J.L., 1998). После получения сигнала Smad быстро фосфорилируются. В настоящее время известно, что в сигнальном пути КМП фосфорилируются Smad-1, -5 и -8, а в сигнальном пути ТФР-β — Smad-2 и Smad-3. Затем названные Smad ассоциируются с Smad-4, который является общим для сигнальных путей всех представителей суперсемейства ТФР-β. Этот факт объясняет наличие перекрестных функций у членов суперсемейства ТФР-β, а также явление взаимного угнетения сигнальных путей ТФР-β и КМП путем конкуренции за общие компоненты. Не так давно идентифицирован еще один класс Smad-белков, который представлен Smad-6 и -7. Эти молекулы выступают в качестве регуляторов сигнальных путей ТФР-β и КМП (Hayashi H. et al., 1997).
Несмотря на то, что стимулирующее влияние КМП на синтез протеогликанов известно давно, их роль в регуляции функции суставного хряща остается спорной в связи с известной способностью КМП вызывать дедифференцировку клеток, стимулировать кальцификацию и образование костной ткани (van den Berg W.B. et al., 1999). M. Enomoto-Iwamoto и соавторы (1998) показали, что взаимодействие КМП с КМП-рецептором ІІ типа необходимо для поддержания дифференцированного фенотипа хондроцитов, а также контроля их пролиферации и гипертрофии (Enomoto-Iwamoto M. et al., 1998). По данным L.Z. Sailor и соавторов (1996), КМП-2 поддерживает фенотип хондроцитов в культуре на протяжении 4 нед, не вызывая их гипертрофирования. КМП-7 (идентичен остеогенному протеину-1) длительно поддерживает фенотип зрелых хондроцитов суставного хряща, культивируемых в алгинате (Flechtenmacher J. et al., 1996).
Введение КМП-2 и -9 в коленные суставы мышей на 300% увеличило синтез протеогликанов, причем значительно больше, чем ТФР-β (van Beuningen H.M. et al., 1998). Однако стимулирующий эффект оказался временным, и через несколько дней уровень синтеза вернулся к исходному. ТФР-β вызвал более длительную стимуляцию синтеза протеогликанов (рис. 16), что, вероятно, связано с аутоиндукцией ТФР-β и сенсибилизацией хондроцитов к этому фактору.
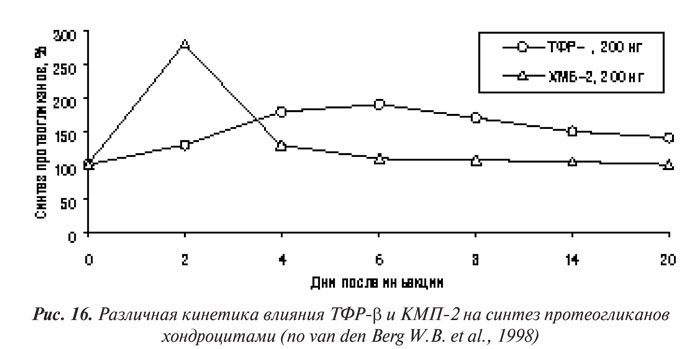
ТФР-β отвечает за образование хондрофитов, что можно считать нежелательным эффектом его действия, КМП-2 также способствует образованию хондрофитов, но в другом участке суставного края (главным образом, в области ростовой пластинки) (van Beuningen H.M. et al., 1998).
Хрящевые морфогенетические протеины (ХМП-1 и -2) — еще одни представители суперсемейства ТФР-β, необходимые для образования хрящевой ткани в период развития конечностей (Luyten F.P., 1997). Мутация гена ХМП-1 вызывает хондродисплазию (Thomas J.T. et al., 1996). Возможно, у ХМП более селективный, направленный на хрящ, профиль. Несмотря на то, что ТФР-β и КМП способны стимулировать хондроциты, они могут действовать и на многие другие клетки, поэтому их использование для репарации хряща может сопровождаться побочными эффектами. ХМП обоих типов обнаруживают в хряще здоровых и пораженных ОА суставов, они способствуют репарации ВКМ суставного хряща после ферментативной деградации, поддерживая нормальный фенотип (Erlacher L. et al., 1998).
Синергизм факторов роста. Один фактор роста способен индуцировать сам себя, а также другие факторы роста, это взаимодействие тонко регулируется. Так, например, ФРФ вместе с другими факторами роста обеспечивает более эффективную репарацию суставного хряща после травматического дефекта (Frenz D.A. et al., 1994; Otsuka Y. et al., 1997). ИФР-1 вместе с ТФР-β в значительно большей степени индуцируют нормальный фенотип хондроцитов при культивировании их in vitro (Tsukazaki T. et al., 1994; Yaeger P.C. et al., 1997). Было продемонстрировано, что ТФР-β препятствует продукции ИФР-1 и ИФР-СБ, а также дефосфорилирует рецептор ИФР-1, стимулирует связывание ИФР-1 (van den Berg W.B. et al., 1999). В интактном хряще мышей обнаружено явление синергизма ИФР-1 со многими факторами роста (Verschure P.J. et al., 1994). Однако слабую реакцию хондроцитов на ИФР-1 нельзя нивелировать использованием в комбинации с другими факторами роста (Verschure P.J. et al., 1994).
Взаимодействие анаболических и деструктивных цитокинов. Факторы роста демонстрируют сложное взаимодействие с ИЛ-1. Так, например, преэкспозиция хондроцитов ФРФ увеличивает высвобождение протеаз после экспозиции ИЛ-1; возможно, это связано с увеличением экспрессии рецепторов ИЛ-1 (Chandrasekhar S., Harvey A.K., 1989). ФРПТ также стимулирует ИЛ-1-зависимое высвобождение протеаз, однако он снижает ИЛ-1-опосредованное угнетение синтеза протеогликанов (Smith R.J. et al., 1991). Это может означать, что некоторые факторы роста могут одновременно стимулировать процесс репарации хряща и способствовать его разрушению. Другие факторы роста, например ИФР-1 и ТФР-β, стимулируют синтез суставного матрикса и угнетают ИЛ-1-опосредованное разрушение суставного хряща, т.е. их активность связана только с репарацией ткани. Такое взаимодействие не зависит от преэкспозиции хондроцитов ИЛ-1. Интересно, что кинетика эффектов ИЛ-1 и ТФР-β может быть различной: способность ТФР-β угнетать деградацию суставного хряща ослабляется его медленным действием на мРНК ТИМП (Lum Z.P. et al., 1996). С другой стороны, отмечается повышение уровня иNOС и NO в отсутствие ТФР-β (Vodovotz Y. et al., 1996). Учитывая NO-зависимость супрессорного эффекта ИЛ-1 в отношении синтеза протеогликанов хондроцитами (van de Loo F.A.J. et al., 1998), можно объяснить, почему мы наблюдаем значительно более сильное противодействие ТФР-β ИЛ-1-зависимому угнетению синтеза протегликанов в сравнении с разрушением протеогликанов in vivo.
В исследовании на мышах, которым внутрисуставно вводили ИЛ-1 и факторы роста, было продемонстрировано, что ТФР-β значительно противодействует ИЛ-1-опосредованному угнетению синтеза протеогликанов суставного хряща, тогда как КМП-2 не способен на такое противодействие: его стимуляторный потенциал полностью угнетался ИЛ-1 даже при условии высокой концентрации КМП-2 (van Beuningen H.M. et al., 1994; Glansbeek H.L. et al., 1997). Примечательно, что в отсутствие ИЛ-1 КМП-2 значительно интенсивнее стимулировал синтез протеогликанов, чем ТФР-β (рис. 17).
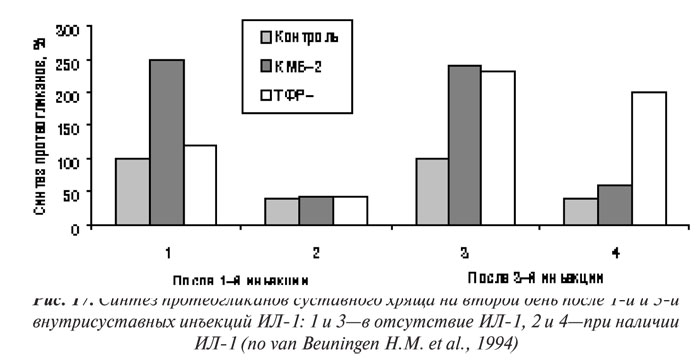
Кроме влияния на синтез протеогликанов, ТФР-β также значительно влияет на ИЛ-1-индуцированное снижение содержания протеогликанов в хряще. Возможно, в зависимости от относительной концентрации ИЛ-1 и ТФР-β содержание протеогликанов снижается или повышается. Интересно, что описанное выше противодействие ИЛ-1 и ТФР-β наблюдали в толще хряща, однако вблизи хондрофитов по краям суставных поверхностей такого явления не было. Образование хондрофитов индуцируется ТФР-β, который влияет на хондрогенные клетки в периосте, вызывая развитие хондробластов и отложение протеогликанов (van Beuningen H.M. et al., 1994; 1998). По-видимому, эти хондробласты не чувствительны к ИЛ-1.
H.L. Glansbeek и соавторы (1998) изучали способность ТФР-β и КМП-2 противодействовать угнетению синтеза протеогликанов в суставах мышей с зимозаниндуцированным артритом (т.е. в модели «чистого» ИЛ-1-индуцированного воспаления). Внутрисуставное введение ТФР-β значительно противодействовало угнетению синтеза протеогликанов, вызванного воспалением, в то время как КМП-2 был практически не способен противодействовать этому ИЛ-1-зависимому процессу. Повторные инъекции ТФР-β в коленный сустав исследуемых животных значительно стимулировали синтез протеогликанов хондроцитами, способствовали сохранению существующих протеогликанов истощенного воспалением хряща, но не угнетали воспалительный процесс.
При изучениии протеогликансинтезирующей функции хондроцитов с использованием экспериментальных моделей ОА у животных всегда отмечали повышение содержания и стимуляцию синтеза протеогликанов (Adams M.E., Brandt K.D.,1991; van der Kraan P.M. et al., 1992; Gaffen J.D. et al., 1995) на ранних стадиях ОА в отличие от воспалительных моделей, в которых наблюдается значительное угнетение синтеза (ИЛ-1-зависимый процесс) (van de Loo A.A.J. et al., 1995; 1998). Повышение активности анаболических факторов, в частности факторов роста, что наблюдается при ОА, нивелирует действие таких супрессорных цитокинов, как ИЛ-1. Среди факторов роста наибольшее значение имеет ТФР-β; КМП-2 вряд ли играет значительную роль в этом процессе. Хотя ИФР-1 способен стимулировать синтез протеогликанов in vitro, в условиях in vivo такое свойство не наблюдают при локальном применении ИФР-1. Возможно, это связано с тем, что эндогенный уровень этого фактора роста является оптимальным. На более поздних стадиях ОА появляются признаки угнетения синтеза протеогликанов, что, вероятно, связано с доминирующим действием ИЛ-1 и неспособностью факторов роста противодействовать ему вследствие снижения активности.
Анализ экспрессии факторов роста у мышей линии STR/ORT со спонтанным ОА продемонстрировал повышение уровня мРНК ТФР-β и ИЛ-1 в поврежденном хряще (Chambers M.G. et al., 1997). Необходимо заметить, что активация ТФР-β из латентной формы является важным элементом репарации ткани. Понимание роли ТФР-β осложняет результаты исследования экспрессии рецепторов ТФР-β II типа у кроликов линии ACL (Boumediene K. et al., 1998). Сразу после индукции ОА обнаружен сниженный уровень этих рецепторов, что свидетельствует о недостаточной сигнальной функции ТФР-β. Интересно, что у дефицитных по рецептору ТФР-β II типа мышей обнаружены признаки спонтанного ОА, что также свидетельствует о важной роли сигнальной функции ТФР-β в ухудшении репарации хряща и развитии ОА (Serra R. et al., 1997).
Абсолютное содержание факторов роста в суставах больных с ревматоидным артритом или ОА могут свидетельствовать об их возможной роли в патогенезе этих заболеваний. Однако несмотря на то, что в суставах при ОА и ревматоидном артрите обнаруживают высокие концентрации факторов роста, характер процессов деградации и репарации при обоих заболеваниях совершенно различен. Вероятно, существуют другие, еще не идентифицированные факторы, играющие основную роль в патогенезе этих болезней, или иные аспекты изучаемых явлений определяют течение процессов деградации и репарации в тканях суставов (например, экспрессия определенных рецепторов на поверхности хондроцитов, растворимые рецепторы, связывающие белки, или дисбаланс анаболических и деструктивных факторов).
Роль отложения кристаллов в патогенезе ОА
У 30–60% больных с ОА обнаруживают кристаллы основного фосфата кальция (ОФК) в суставной жидкости (Dieppe P.A. et al., 1972; Gibilisco P.A. et al., 1985). По мнению A. Swan и соавторов (1994), кальцийсодержащие кристаллы находятся в суставной жидкости у гораздо большего количества больных с ОА, однако из-за чрезмерно мелких размеров кристаллов или их малого количества они не идентифицируются с помощью общепринятых методик. Наличие кристаллов ОФК в суставной жидкости коррелирует с рентгенологическими признаками дегенерации суставного хряща (Halverson P.B., McCarty D.J., 1986) и ассоциируется с большим объемом выпота по сравнению с выпотом в коленных суставах без кристаллов (Carroll G.J. et al., 1991). Изучение факторов, влияющих на рентгенологическое прогрессирование гонартроза, показало, что отложение кристаллов пирофосфата кальция дигидрата (ПФКД) является предиктором неблагоприятного клинического и рентгенологического исхода (Ledingham J. et al., 1995). При исследовании пациентов пожилого возраста обнаружили, что ОА ассоциируется с хондрокальцинозом, особенно в латеральном тибиофеморальном отделе коленного сустава и в первых трех пястно-фаланговых суставах (Sanmarti R. et al., 1996). Нередко у больных с ОА обнаруживают оба типа кристаллов — ОФК и ПФКД (Dieppe P. et al., 1978).
Клинически дегенерация суставного хряща, обусловленная отложением кальцийсодержащих кристаллов, отличается от таковой при первичном ОА (Ryan L., McCarty G.M., 1997). Если бы кристаллы были простым эпифеноменом дегенерации хряща, их обнаруживали бы в суставах, которые чаще всего поражаются первичным ОА, т.е. в коленных, тазобедренных, мелких суставах кистей. Напротив, болезни отложения кристаллов чаще поражают нетипичные для первичного ОА суставы — плечевые, лучезапястные, локтевые. Наличие кристаллов в суставной (выпотной) жидкости ассоциируется с более тяжелой дегенерацией суставного хряща. Обсуждается вопрос о том, что является причиной, а что следствием — отложение кристаллов или дегенерация хряща (Bjelle A., 1981). Промежуточную позицию занимает следующее предположение: первичная аномалия метаболизма хряща ведет к его дегенерации, а вторичное отложение кристаллов ускоряет его деградацию (так называемая теория амплификационной петли) (Doherty M. et al., 1982).
Точный механизм повреждения суставного хряща кальцийсодержащими кристаллами не известен, его отдельные элементы приведены ниже. Теоретически кальцийсодержащие кристаллы могут непосредственно повреждать хондроциты. Однако при гистологическом исследовании кристаллы редко локализуются вблизи хондроцитов, еще реже поглощаются ними (Schumacher H., 1988). Наиболее вероятным является фагоцитоз кристаллов клетками синовиальной выстилки с последующим выделением ими протеолитических ферментов или секрецией цитокинов, стимулирующих выделение ферментов хондроцитами. Эта концепция подтверждается исследованием роли ПФКД-индуцированного синовита в развитии быстро прогрессирующего ОА при пирофосфатной артропатии (Fam A.G. et al., 1995). В ходе этого исследования кроликам с ОА, индуцированным частичной латеральной менискэктомией, в правый коленный сустав вводили кристаллы ПФКД (1 или 10 мг) 1 раз в неделю. Оказалось, что после 8 инъекций в правом коленном суставе были значительно более серьезные изменения по сравнению с левым. Интенсивность синовиального воспаления коррелировала с внутрисуставными инъекциями кристаллов ПФКД и их дозой. Несмотря на то, что использованные в данном исследовании дозы кристаллов ПФКД превышают таковые in vivo, результаты свидетельствуют о роли ПФКД-индуцированного воспаления в прогрессировании ОА при пирофосфатной артропатии.
Потенциальные механизмы индуцирования кальцийсодержащими кристаллами повреждения суставного хряща связаны с их митогенными свойствами, способностью индуцировать ММП и стимулировать синтез простагландинов.
Митогенный эффект кальцийсодержащих кристаллов. При кристаллассоциированных артропатиях часто обнаруживают пролиферацию клеток синовиальной выстилки, причем сами кристаллы лишь частично ответственны за этот процесс (Garancis J.C. et al., 1981). Увеличение количества синовиальных клеток сопровождается усилением секреции цитокинов, которые способствуют хондролизу и вызывают секрецию протеолитических ферментов. Кристаллы ОФК в концентрациях, обнаруживаемых при патологии суставов у человека, дозозависимо стимулируют митогенез культуры покоящихся фибробластов кожи, синовиальных фибробластов собак и мышей (Barnes D., Colowick S., 1977; Cheung H.S. et al., 1984). Кристаллы ПФКД, урата, сульфата, карбоната и дифосфата кальция стимулируют рост клеток (Barnes D., Colowick S., 1977; Cheung H., McCarty G.M., 1985). Начало и пик включения (3Н)-тимидина, индуцируемые этими кристаллами, смещены на 3 ч по сравнению со стимуляцией клеток сывороткой крови. Возможно, этот отрезок времени необходим для фагоцитоза и растворения кристаллов. Добавление контрольных кристаллов того же размера (например, бриллиантовой пыли или частичек латекса) не стимулировало митогенез. Кристаллы урата натрия моногидрата обладали слабыми митогенными свойствами и значительно уступали таковым урата кальция, что свидетельствует о важном значении в митогенезе содержания кальция в кристаллах. Синтетические кристаллы ОФК обладали такими же митогенными свойствами, как и кристаллы, полученные от пациентов с хондрокальцинозом (Cheung H. et al., 1984). Митогенный эффект кальцийсодержащих кристаллов не был результатом повышения содержания кальция в окружающей клетку питательной среде in vitro, так как при растворении кристаллов ОФК в питательной среде не стимулировало включение (3Н)-тимидина фибробластами (Cheung H., McCarty G.M., 1985).
Одним из предполагаемых механизмов ОФК-индуцированного митогенеза является следующий: аномальная пролиферация синовиальных клеток может быть связана (по крайней мере частично) с эндоцитозом и внутриклеточным растворением кристаллов, что приводит к повышению концентрации Са2+ в цитоплазме клеток и к активации кальцийзависимого пути, ведущего к митогенезу. В поддержку этой концепции выступает необходимость прямого контакта клетка — кристалл для стимуляции митогенеза, так как экспозиция культуры клеток кристаллам вызывала рост клеток, а экспозиция клеток, лишенных возможности такого контакта, не вызвала их роста (Bowen-Pope D., Rubin H., 1983). Для изучения необходимости фагоцитоза кристаллов, следующего за взаимодействием клетка — кристалл, клетки культивировали с 45Са-ОФК и (3Н)-тимидином. Оказалось, что содержащие 45Са-ОФК клетки включали гораздо большее количество (3Н)-тимидина, чем клетки без меченых ОФК (Borkowf A. et al., 1987). В культуре макрофагов угнетение цитохалазином эндоцитоза кристаллов вызывало ингибицию растворения кристаллов, что также подчеркивает необходимость фагоцитоза (Owens J. et al., 1986).
Кальцийсодержащие кристаллы растворимы в кислоте. После фагоцитоза кристаллы растворяются в кислой среде фаголизосом макрофагов. Хлорохин, аммония хлорид, бафиломицин А1 и все лизосомотрофные агенты, повышающие лизосомальный рН, дозозависимо угнетают внутриклеточное растворение кристаллов и поглощение (3Н)-тимидина в фибробластах, культивируемых с кристаллами ОФК (McCarty G.M. et al., 1998).
Добавление ОФК-кристаллов к монослойной культуре фибробластов вызвало немедленное десятикратное повышение содержания внутриклеточного кальция, которое вернулось к исходному уровню через 8 мин. Источником кальция преимущественно был внеклеточный ион, так как кристаллы ОФК были добавлены в бескальциевую питательную среду. Следующее повышение концентрации внутриклеточного кальция наблюдалось через 60 мин и продолжалось не менее 3 ч. Здесь источником кальция были фагоцитированные кристаллы, растворенные в фаголизосомах (Halverson P. et al., 1998).
Установлено, что митогенный эффект ОФК-кристаллов похож на таковой у ФРПТ в качестве фактора роста; подобно последнему, ОФК-кристаллы проявляют синергизм по отношению к ИФР-1 и плазме крови. Блокада ИФР-1 снижает митогенез клеток в ответ на ОФК (Cheung H. et al., 1986). P.G. Mitchell и соавторы (1989) показали, что индукция ОФК-кристаллами митогенеза фибробластов Balb/c-3T3 требует наличия сериновой/треониновой протеинкиназы С (ПКС) — одного из главных медиаторов сигналов, генерированных при внешней стимуляции клеток гормонами, нейротрансмиттерами и факторами роста. Снижение активности ПКС в клетках Balb/c-3T3 угнетает ОФК-опосредованную индукцию протоонкогенов c-fos и c-myc, но не влияет на стимуляцию этих онкогенов, опосредованную ФРПТ (Mitchell P.G. et al., 1989).
Повышение содержания внутриклеточного кальция после растворения фагоцитированных кристаллов — не единственный сигнальный путь для митогенеза. Когда факторы роста, такие, как ФРПТ, связываются со своим мембранным рецептором, стимулируется фосфолипаза С (фосфодиэстераза), которая гидролизирует фосфатидилинозитол 4,5-бисфосфат с образованием внутриклеточных мессенджеров — инозитол-3-фосфата и диацилглицерола (Berridge M., 1987). Первый высвобождает кальций из эндоплазматической сети, модулируя активность кальцийзависимых и кальций/кальмодулинзависимых ферментов, таких, как протеинкиназы и протеазы (Rasmussen H., 1986).
R. Rothenberg и H. Cheung (1988) сообщили о повышенной деградации фосфолипазой С фосфатидилинозитола 4,5-бифосфата в синовиальных клетках кроликов в ответ на стимуляцию ОФК-кристаллами. Последние значительно повышают содержание инозитол-1-фосфата в клетках с меченым (3Н)-инозитолом; пик достигался в течение 1 мин и сохранялся около 1 ч.
Диацил-глицерол — потенциальный активатор ПКС (Kikkawa U., Nishizuka Y., 1986). Так как ОФК-кристаллы повышают активность фосфолипазы С, что ведет к аккумуляции диацилглицерола, следовательно, можно ожидать повышения активации ПКС. P.G. Mitchell и соавторы (1989) сравнивали влияние ОФК-кристаллов и ФРПТ на синтез ДНК фибробластами Balb/c-3T3. В культуре клеток ПКС была инактивирована инкубацией клеток с туморподдерживающим форболовым диэфиром (ТФД), аналогом диацилглицерола. Длительная стимуляция низкими дозами ТФД снижает активность ПКС, тогда как однократная стимуляция высокой дозой — активирует. Стимуляция ОФК-кристаллами синтеза ДНК была угнетена после инактивации ПКС, что свидетельствует о важности этого фермента в ОФК-индуцированном митогенезе. Ранее G.M. McCarthy и соавторы (1987) продемонстрировали связь митогенного ответа фибробластов человека на ОФК-кристаллы с активацией ПКС. Однако ОФК-кристаллы не активируют фосфатидилинозитол-3-киназу или тирозинкиназы, подтверждая тот факт, что механизм активации клеток кристаллами ОФК селективен (McCarty G.M. et al., 1987).
Пролиферацию клеток контролирует группа генов, называемых протоонкогенами. Белки foc и myc, продукты протоонкогенов c-fos и c-myc локализованы в клеточном ядре и связаны со специфическими последовательностями ДНК (Kelly K. et al., 1983; Cochran B. et al., 1984). Стимуляция 3Т3-фибробластов ОФК-кристаллами приводит к экспрессии c-fos в течение нескольких минут, которая достигает максимума через 30 мин после стимуляции. Индукция транскрипции c-myc ОФК-кристаллами или ФРПТ происходит в течение 1 ч и достигает максимума через 3 ч после стимуляции. Не менее 5 ч клетки поддерживают повышенный уровень транскрипции c-fos и c-myc (Cheung H.S. et al., 1989). В клетках с инактивированной ПКС стимуляция c-fos и c-myc кристаллами ОФК или ТФД значительно угнетена, в то время как индукция этих генов ФРПТ не изменяется.
Представители семейства митогенактивированных протеинкиназ (МАПК) — ключевые регуляторы различных внутриклеточных сигнальных каскадов. Один из подклассов данного семейства — р42/р44 — регулирует пролиферацию клеток с помощью механизма, включающего активацию протоонкогенов с-fos и c-jun. ОФК- и ПФКД-кристаллы активируют протеинкиназный путь передачи сигнала, в котором участвуют оба р42 и р44, что свидетельствует о роли этого пути в митогенезе, индуцированном кальций-содержащими кристаллами (Nair D. et al., 1997).
Наконец, в ОФК-индуцированном митогенезе участвует транскрипционный ядерный фактор κВ (NF-κВ), который впервые был описан как ген легкой цепи иммуноглобулина κ (Igκ). Это индуцированный транскрипционный фактор, важный для многих сигнальных путей, поскольку он регулирует экспрессию различных генов. Индукция NF-κВ обычно сопряжена с высвобождением из цитоплазмы ингибиторных белков, называемых IκВ. Вслед за индуцией NF-κВ происходит транслокация активного транскрипционного фактора в ядро. Кристаллы ОФК индуцируют NF-κВ в Balb/c-3T3 фибробластах и фибробластах кожи человека (McCarty G.M. et al., 1987).
Несколько путей могут быть вовлечены в передачу сигнала после активации NF-κВ, однако все они включают протеинкиназы, которые фосфорилируют (и таким образом деградируют) ІκВ (Barnes P., Karin M., 1997). Основываясь на результатах исследований in vitro, ранее предполагали, что ІκВ служит субстратом для киназ (например, ПКС и протеинкиназа А) (Baeuerle P.A., Baltimore D., 1988; Shirakawa F., Mizel S.B., 1989). Однако недавно был идентифицирован ІκВ-киназный комплекс большой молекулярной массы (Mercurio F. et al., 1997). Эти киназы специфически фосфорилируют сериновые остатки ІκВ. Активация NF-κВ ФНО-α и ИЛ-1 требует эффективного действия NF-κВ-индуцирующей киназы (НИК) и IκВ-киназы (Woronicz J.D. et al., 1997). Молекулярный механизм активации НИК в настоящее время не известен. Несмотря на то, что кристаллы ОФК активируют и ПКС и NF-κВ, неизвестно, в какой степени эти два процесса могут быть связаны. Так как модификация IκВ-киназы осуществляется путем фосфорилирования, не исключается роль ПКС в индукции кристаллами ОФК NF-κВ путем фосфорилирования и активации IκВ-киназы (DiDonato J.A. et al., 1997). В поддержку этой концепции может служить угнетение ингибитором ПКС стауроспорином ОФК-кристаллиндуцированного митогенеза и экспрессии NF-κВ (McCarty G.M. et al., 1987). Аналогично стауроспорин может угнетать IκВ-киназу, а значит, угнетает протеинкиназу А и другие протеинкиназы (Tamaoki T., 1991).
Таким образом, механизм ОФК-кристаллиндуцированного митогенеза в фибробластах включает не менее двух различных процессов (рис. 18):
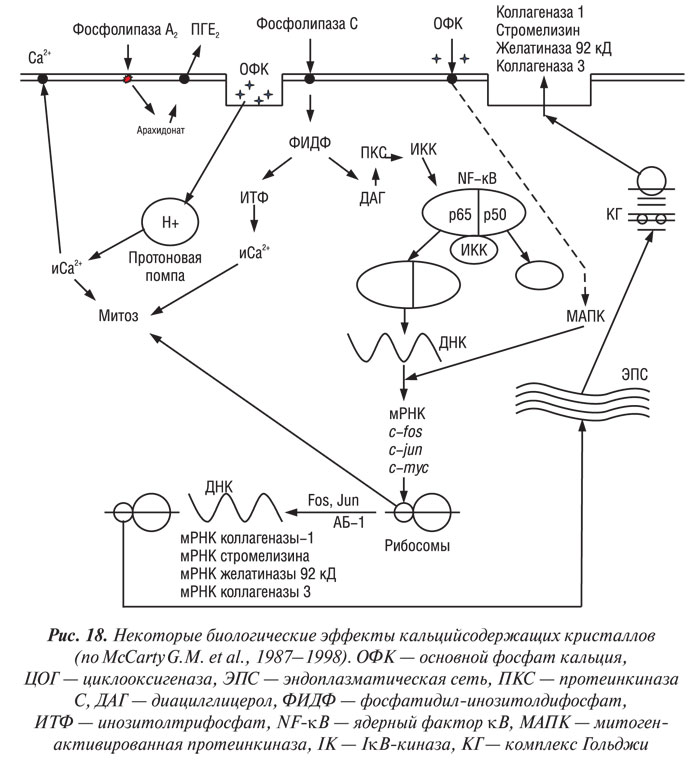
1) быстрое мембранно-связанное событие, которое приводит к активации ПКС и МАПК, индукции NF-κВ и протоонкогенов,
2) более медленное внутриклеточное растворение кристаллов, что ведет к повышению внутриклеточного содержания Са2+, а затем к активации ряда кальцийзависимых процессов, стимулирующих митогенез.
Индукция ММП кальцийсодержащими кристаллами. Медиаторами повреждения ткани кальцийсодержащими кристаллами являются ММП — коллагеназа-1, стромелизин, желатиназа 92 кД и коллагеназа-3.
С учетом связи между содержанием кристаллов ОФК и деструкции тканей суставов была высказана гипотеза, согласно которой кристаллы ОФК и, возможно, некоторые коллагены фагоцитируются синовиальными клетками. Стимулированные синовициты пролиферируют и секретируют протеазы. Эта гипотеза была проверена in vitro путем добавления естественных или синтетических кристаллов ОФК, ПФКД и других в культуру синовицитов человека или собаки. Уровень активности нейтральных протеаз и коллагеназ дозозависимо возрос и был приблизительно в 5–8 раз выше по сравнению с контрольной культурой клеток, которые культивировали без кристаллов (Cheung H. et al., 1981).
В клетках, культивируемых в кристаллсодержащей среде, обнаружена коиндукция мРНК коллагеназы-1, стромелизина и желатиназы-92 кД с последующей секрецией ферментов в среду (McCarty G.M. et al., 1992; 1998). Кристаллы ОФК также вызывали аккумуляцию мРНК коллагеназы-1 и коллагеназы-2 в зрелых хондроцитах свиньи с последующей секрецией ферментов в среду (Mitchell P.G. et al., 1992; McCarty G.M. et al., 1997).
G.M. McCarty и соавторы (1998) изучали роль внутриклеточного растворения кристаллов в кристаллиндуцированной продукции ММП. Повышение лизосомального рН с помощью бафиломицина А1 угнетало внутриклеточное растворение кристаллов, а также ослабляло пролиферативный ответ фибробластов человека на кристаллы ОФК, но не угнетало синтез и секрецию ММП (McCarty G.M. et al., 1998).
Ни кристаллы ОФК, ни ПФКД не индуцировали продукцию ИЛ-1 in vitro, в отличие от кристаллов урата натрия (Malawista S. et al., 1985). Современные данные четко свидетельствуют о прямой стимуляции продукции ММП фибробластами и хондроцитами при контакте с кальцийсодержащими кристаллами (McCarty G.M. et al., 1999).
Клиническая картина ОА свидетельствует о значительной роли ММП в прогрессировании болезни. Наличие кальцийсодержащих кристаллов усиливает дегенерацию тканей пораженных суставов.
Стимуляция синтеза простагландинов. Наряду со стимуляцией роста клеток, секрецией ферментов кальцийсодержащие кристаллы вызывают высвобождение простагландинов из культур клеток млекопитающих, особенно ПГЕ2 (Cheung H. et al., 1981; McCarty G.M., Cheung H., 1985; Dayer J.M. et al., 1987). Высвобождение ПГЕ2 во всех случаях происходит в течение первого часа после экспозиции клеток кристаллам. R. Rothenberg (1987) определил, что основными источниками арахидоновой кислоты для синтеза ПГЕ2 являются фосфатидилхолин и фосфатидилэтаноламин, а также подтвердил, что фосфолипаза А2 и ЦОГ — доминирующие пути продукции ПГЕ2.
В ответ на действие кристаллов ОФК также может выделяться ПГЕ1 (Rashad S. et al., 1989). G.M. McCarty и соавторы (1993, 1994) изучали влияние ПГЕ2, ПГЕ1 и его аналога мизопростола на митогенный ответ фибробластов человека на кристаллы ОФК. Все три агента угнетали митогенный ответ дозозависимым путем, причем ПГЕ1 и мизопростал проявляли более выраженную ингибиторную активность. ПГЕ1 и мизопростол, но не ПГЕ2 угнетали аккумуляцию мРНК коллагеназы в ответ на действие кристаллов ОФК (McCarty G.M. et al., 1993; 1994).
M.G. McCarty и H . Cheung (1994) исследовали механизм ОФК-опосредованной активации клеток ПГЕ. Авторы показали, что ПГЕ1 — более мощный индуктор внутриклеточного цАМФ, чем ПГЕ2, и ПГЕ1 угнетает ОФК-индуцированный митогенез и продукцию ММП через цАМФ-зависимый путь передачи сигнала. Возможно, повышение продукции ПГЕ, индуцированное кристаллами ОФК, ослабляет их другие биологические эффекты (митогенез и продукцию ММП) по механизму обратной связи.
Воспаление, индуцированное кристаллами. Кальцийсодержащие кристаллы часто обнаруживают в синовиальной жидкости у больных с ОА (McCarty G.M. et al., 1999), однако эпизоды острого воспаления с лейкоцитозом встречаются редко как при ОА, так и при кристаллассоциированных артропатиях (например, синдроме «плечевой сустав Милуоки») (McCarty G.M. et al., 1999). Флогистический потенциал кристаллов может быть модифицирован рядом ингибиторных факторов. R. Terkeltaub и соавторы (1988) продемонстрировали способность сыворотки и плазмы крови значительно угнетать ответ нейтрофильных гранулоцитовов на кристаллы ОФК. Факторами, которые вызывают такое угнетение, являются кристаллсвязывающие белки. Исследование одного из таких белков — α2-HS гликопротеина (АHSГ) — показало, что АHSГ является наиболее мощным и специфическим ингибитором ответа нейтрофильных гранулоцитов на кристаллы ОФК. АHSГ — сывороточный протеин печеночного происхождения; известно, что по сравнению с другими белками сыворотки крови он в относительно высоких концентрациях содержится в костной и минерализирующейся ткани (Triffit J., 1978). Кроме того, АHSГ присутствует в «невоспаленной» синовиальной жидкости, а также обнаружен на кристаллах ОФК в нативной синовиальной жидкости (Terkeltaub R. et al., 1988). Таким образом, не исключается вероятность модулирования АHSГ флогогенного потенциала кристаллов ОФК в условиях in vivo.
Резюмируя все вышесказанное, приведем две схемы патогенеза ОА, предложенные W.B. van den Berg и соавторами (1999) (рис. 19) и M. Carrabba и соавторами (1996) (рис. 20), которые объединяют механические, генетические и биохимические факторы.