Первый шаг при ДД двигательных нарушений и расстройств чувствительности — это тщательный сбор анамнеза.
Неожиданно возникшие очаговые неврологические изменения центрального характера при достаточно быстрой положительной динамике свидетельствуют о преходящих нарушениях мозгового кровообращения, более грубые и сохраняющиеся в динамике без существенного улучшения, могут свидетельствовать об ишемическом или геморрагическом инсульте. Медленно, в течение недель и месяцев, увеличивается выраженность очаговых изменений, заставляя думать об опухоли. Если изменения происходят медленно, затрагивают только одну функциональную систему, например экстрапирамидную, то есть основания думать о дегенеративном заболевании, например болезни Паркинсона.
Вероятность развития многих неврологических заболеваний меняется в разные возрастные периоды: болезнь Паркинсона редко развивается в возрасте до 40 лет, а рассеянный склероз чаще проявляется именно в этом возрасте.
Следующий важный этап — это очаговая диагностика. При осмотре пациента врач отвечает на вопрос, какие из выявленных неврологических симптомов относятся к одному очагу, или поражены несколько полей. Все это требует знаний анатомии и физиологии, уровней и характера рефлекторних дуг. Следующий шаг — выбор дополнительных методов исследования. Опытный врач, опираясь на анамнез, выявленную очаговую симптоматику, неврологические признаки, в большинстве случаев может установить клинический диагноз, применяя дополнительные методы исследования, если их результаты смогут повлиять на выбор тактики лечения.
Гемипарезы или гемиплегии с сенсомоторными расстройствами возникают при поражениях противоположной стороны. Двигательные и чувствительные пути в области внутренней капсулы проходят вплотную друг к другу, и в этой области достаточно часто отмечают сосудистые нарушения. Неполный паралич носит название «парез», полный известен под термином «плегия». При ходьбе пациент подволакивает ногу, движения руки ограничены. Если руки пациента поддержать в горизонтальном положении и потом освободить, то пораженная рука пронирует и сгибается в локте перед непроизвольным опусканием. Это является ранним признаком пареза, отграничивающим его от психогенного паралича, когда рука опускается в нефизиологическом положении без пронации. Собственные рефлексы повышены. Так, рефлекс с передней брюшной стенки на пораженной стороне отсутствует, рефлекс Бабинского проявляется выраженным разгибанием большого пальца с веерообразным разведением остальных. В течение ближайших дней формируется спастический гипертонус. Чувствительные расстройства касаются поверхностной и глубокой чувствительности и проявляются чуть отступя от срединной линии в пораженную сторону, поскольку всегда имеется перекрестная чувствительность. Это также позволяет отграничить истинные нарушения от психогенных. Выделяют доминирующую и недоминирующую гемисферы. У правшей доминирующей в формировании речи является левая гемисфера, она же чаще всего доминирует и у левшей. При повреждениях соответствующих областей доминирующей гемисферы появляются нарушения абстрактно-символических функций: письма, счета, чтения. Повреждения затылочных областей недоминирующей гемисферы приводят к нарушениям восприятия пространства, что затрудняет и делает невозможным понимание геометрии, фигур, чертежей. Но большая часть коры имеет ассоциативные связи, поэтому распознавание повреждений может представить значительные сложности. В частности, при нарушениях в области правой недоминирующей гемисферы пациенты воспринимают раздельно наносимые тактильные раздражения как справа, так и слева, но если раздражители применить и справа, и слева одновременно, то пациент не воспримет раздражение слева.
Острый гемипарез, развивающийся в течение от нескольких минут до нескольких часов типичен для кровоизлияния. Клинически в каждом отдельном случае нельзя с уверенностью сказать о том, является ли причиной пареза кровотечение или ишемия. ДД проводят с помощью КТ. Без применения контрастних средств очаг кровоизлияния выглядит как объемное образование повышенной плотности. Ишемический очаг может быть визуализирован только через несколько дней как образование пониженной плотности. Допплерография позволяет выявить область нарушения кровообращения, стенозы артерий, бляшки. Кровоизлияние развивается в случае аномалий сосудов или длительного предшествующего атеросклероза с артериальной гипертензией. Сосудистая мальформация может быть выявлена после рассасывания кровоизлияния методом ЯМР с контрастированием. Ишемические инфаркты развиваются при коагулопатии, эмболии, васкулите, атеросклеротических окклюзиях артерий или их преходящих спазмах.
Неотложность дополнительных обследований определяется диагностической концепцией. Ишемический инсульт у человека молодого возраста априорно объясняется иначе, чем у 70-летнего пациента с длительным «сосудистым анамнезом». Гемипарезы, развившиеся после эпилептического припадка и протекающие с быстрой благоприятной динамикой, свидетельствуют о вторичности своего происхождения и наблюдаются при узелковом периартериите, миксомах сердца, опухолях, аневризмах или сосудистых мальформациях мозга. Если КТ не выявляет объемных процессов, то это никак не исключает диагноз опухоли, что типично для ранних стадий астроцитом. Диагностические проблемы решает ЯМР с контрастированием.
Гемипарез, развивающийся медленно, в течение от нескольких дней до недель, прежде всего заставляет исключать объемные процессы мозга. Прежде всего это опухоли. Неотложным диагностическим вмешательством является КТ. Один очаг с полициклическими контурами отмечают при глиобластомах. Несколько округлых очагов с перифокальным отеком типичны для метастазирования. Чаще всего метастазируют карцинома легких, молочной железы, женских половых органов, меланома. Кроме того, следует помнить о лимфомах, субдуральной гематоме, туберкуломе или абсцессе мозга, особенно у пациентов с иммунной недостаточностью, с переломами основания черепа, с ликвореей.
Нарушения речи, фонации и артикуляции обусловлены повреждениями IX, X, XII пар черепно-мозговых нервов и их премоторных центров в коре головного мозга, стволе и мозжечке. Одностороннее повреждение этих нервов, особенно если оно развивается постепенно, хорошо компенсируется. Двусторонние повреждения почти всегда сопровождаются и нарушениями глотания. Если в течение суток выраженность этих расстройств колеблется, то необходимо исключить миастению. При дрожании кончика языка высока вероятность амиотрофического бокового склероза. Мимика (смех, улыбка, плач, движение глаз) — врожденные безусловные рефлексы, отмечаемые даже у слепых и исходящие из ствола мозга. Поэтому достаточно часто возникает ситуация, когда при кортикальном инсульте и центральном парезе лицевого нерва пациент не в состоянии произвольно поднять уголок рта, но при эмоциональных реакциях иннервация лица производит впечатление симметричной. При двустороннем нарушении кортико-бульбарных путей мимические движения совершаются непроизвольно, что отмечают при боковом амиотрофическом склерозе. Нарушения речи характерны при интактной фонации и артикуляции, но больной не способен понимать речь или генерировать ее. Так как восприятие и воспроизведение речи достаточно четко локализованы, то расстройства обусловлены соответствующей локализации инсультами или опухолями. Сенсорная афазия (афазия Вернике) развивается при поражении центра Вернике, который лежит на границе кровоснабжения средней и задней мозговыми артериями. Поэтому при резком развитии афазии Вернике причина практически всегда заключается в инсульте. Пациент путает слоги, изобретает новые, что делает невозможным восприятие. Сам пациент не воспринимает свои ошибки. Если нет сопутствующих нарушений, то необходимо лечение у психиатра. Моторная афазия (Брока) развивается при инсульте соответствующего поля и заключается в скудости речи, телеграфности, примитивности и ошибочности построения фраз. Сам больной осознает расстройство.
Нарушения движения глаз. Нейроны, управляющие движениями глаз, располагаются в стволе мозга. Поэтому при гемипарезе в результате инсульта в области внутренней капсулы движения глаз резко нарушены. Перманентные нарушения свидетельствуют о повреждениях ствола мозга. Если пациент отмечает двоение изображения и выявляется нарушение постановки глаза, то причина кроется в парезе глазодвигательной мышцы, а двойное изображение в пораженном глазу проецируется в сторону тяги паретичной мышцы. Односторонний парез глазодвигательного нерва с симметричной и нормальной реакцией зрачков развивается при повреждениях глазодвигательного нерва после его выхода из ствола мозга и до разделения на отдельные веточки в области верхушки орбиты. Наиболее частая причина — микроангиопатия, при этом парасимпатические веточки, проходящие на периферии нерва, не поражаются. Односторонний парез глазодвигательного нерва с мидриазом свидетельствует о том же уровне поражения нерва, но с вовлечением парасимпатических путей. Это отмечают при сдавлениях нерва аневризмой или при повышении внутричерепного давления и требует экстренной оценки. Аневризма обычно находится при этом в области отхождения задней мозговой артерии, где и оказывает давление на нерв, или развивается кровотечение, сопровождающееся острой головной болью. Обязательными являются КТ или ЯМР для исключения объемного процесса или свежей крови. Односторонний мидриаз без пареза внешних мышц глаза при остром развитии с головной болью или фокальными знаками свидетельствует о разрывах намета при объемных процессах или церебральних кровотечениях. Если пациент не предъявляет жалоб и мидриаз был выявлен случайно, то можно думать о состоянии после цилиарного ганглионита. Предположение можно проверить, закапав в глаза 0,1% р-р пилокарпина. В результате денервации чувствительность мидриатичного глаза высока и выявляют сужение зрачка, на здоровом глазу такой реакции получить не удается. Парез глазодвигательного нерва с контрлатеральной слабостью подъема глаза вверх развивается при поражении в области ядра, так как мотонейроны верхней прямой мышцы глаза лежат контрлатерально, а аксоны перекрещиваются в ядре. Так что при одностороннем повреждении захватываются как аксоны глаза этой же стороны, так и нейроны противоположного глаза.
Парез глазодвигательного нерва с контрлатеральной атаксией (синдром Бенедикта) или с контрлатеральным гемипарезом (синдром Вебера). Часть волокон глазодвигательного нерва интрацеребрально проходит через красное ядро, которое получает ветви и из мозжечка, и затем они пересекают кортикоспинальный тракт. Наиболее частая причина сочетания парезов глазодвигательного нерва с мышечными расстройствами — кровоизлияние или опухоль. Парез внутренней прямой мышцы глаза с сохраненной конвергенцией — межнуклеарная офтальмоплегия. Парез трохлеарного нерва не патогномоничен для какого-либо заболевания, поскольку нерв выходит из ствола мозга дорзально и протяженность его, соответственно, велика. Наиболее банальная причина — сотрясение головного мозга. Наличие пареза отводящего нерва в случае изолированности свидетельствует о патологическом процессе вне ствола мозга. Особенно часто парез отводящего нерва развивается при повышении внутричерепного давления.
Парез взгляда — неспособность содружественного движения глаз в определенном направлении. Двоения изображения нет, пациент жалуется только на легкое головокружение, но самостоятельно не регистрирует неспособность движения глаз. Парез взгляда развивается при опухолях или кровотечениях.
Нистагм — непроизвольное быстрое движение глаз вертикально, горизонтально или ротарно (Leigh R., Zee D., 1991). Нистагм можно определить как физиологический или патологический (врожденный или приобретенный).
Нистагм характеризуют по направлению, амплитуде, частоте, скорости фаз. Бистрий нистагм состоит из медленной инициальной фазы и следующей за ней быстрой «коррекционной» фазы. Маятникообразный нистагм имеет две фазы, практически равные по скорости.
Физиологический нистагм — естественная реакция, направленная на фиксацию сетчатки относительно предмета при движениях головы или самого предмета. Физиологический нистагм включает нистагм в крайних отведениях глаза и оптико-кинетический нистагм. Последний тип — реакция на быстрое движение предметов через поле зрения. Медленные сопровождающие движения глаза прерываются быстрым обратным компонентом. Повреждения теменной области (с или без гемианопсии) могут обусловить нарушение афферентных путей от зрительной коры к нижележащим зрительным центрам, и нистагм не появляется. Рефлекс может быть проверен при наблюдении вращения полосатого барабана с направлением вращения в сторону возможного поражения. При истерической слепоте оптокинетический нистагм сохранен.
В противоположность нистагму у взрослых нистагм у детей чаще всего врожденный (возникает в первом полугодии жизни) и обычно является результатом поражения сенсорного афферентного пути. Приобретенный нистагм детского возраста, подобно нистагму взрослых (особенно если ребенок в возрасте старше 2 лет), — результат патологии эфферентного двигательного звена. Врожденный нистагм характеризуется отсутствием головокружения, ему не присуще четкое разграничение быстрого и медленного компонентов. Следует сразу подчеркнуть, что у детей с кортикальной слепотой яркого, размашистого нистагма не бывает.
Врожденный нистагм — сенсорный или моторный, горизонтальный маятникообразный или толчкообразный. Характеризуется рядом особенностей, отличающих его от прогностически неблагоприятных приобретенных форм:
- быстрая фаза всегда направлена горизонтально вправо или влево (в сторону глаза — абдуктора);
- фиксация уменьшает интенсивность нистагма;
- конвергенция (аддукция) уменьшает размах нистагма;
- нет осциллопсии;
- можно найти «нулевую зону» (позиция взгляда, при которой снижается интенсивность нистагма), часто сочетающуюся с поворотом головы;
- типично начало нистагма на 8–12-й недели жизни.
Врожденный сенсорный нистагм проявляется в возрасте 8–12 нед и обычно связан с двусторонним снижением зрения:
- помутнение роговицы, катаракта, аниридия;
- аномалии зрительного нерва (колобомы, гипоплазия, атрофия);
- патология сетчатки (врожденный амавроз Лебера, альбинизм, ахроматопсия, врожденная постоянная ночная слепота).
Врожденный моторный нистагм составляет около 10% всех случаев врожденного нистагма. Как видно из названия, состояние обусловлено патологией эфферентного (моторного) нейрона. Диагноз основывается на результатах неврологического обследования и удовлетворительного зрения. Нистагм проявляется в возрасте 3–3,5 мес и смягчается с возрастом.
В практике нередко выявляют сочетание косоглазия (страбизма) и нистагма. Нистагм при этом обычно латентный и становится заметным при закрытии одного глаза. Это положение важно помнить при определении остроты зрения у детей с эзотропией (сходящееся косоглазие). Латентный нистагм — признак потери бинокулярного зрения в критические периоды развития зрительной системы, что связано с сохранением назотемпорального дисбаланса, выявляемого оптико-кинетическими пробами и исчезающего в норме к 22-й неделе жизни.
Синдром Чианчи (синдром блокады нистагма) — врожденный нистагм с вариабельной эзотропией (отклонение глазных яблок внутрь — сходящееся косоглазие). Эзотропия в этом сочетании, вероятнее всего, механизм адаптации, поскольку отклонение глаза кнутри гасит врожденный нистагм. Примечательно, что в случае патологического включения конвергенции нет аккомодации (миоза и миопии).
Моноокулярный нистагм (крайне ассиметричный нистагм) в 90% связан с так называемым кивательным спазмом (spasmus nutans). Оставшиеся 10% составляют случаи односторонней слепоты или глиомы хиазмы.
Кивательный спазм характеризуется:
- нистагмом (быстрый, мелкоразмашистый, горизонтальный или ассиметричный);
- кивательными движениями головы (низкая частота, исчезают во сне);
- кривошеей;
- началом на 6–12-м месяце жизни;
- нистагм самопроизвольно и без каких-либо последствий исчезает в возрасте 1 года–2 лет.
Если выявляют атрофию диска зрительного нерва или снижение зрения, то необходимо провести нейро-радиологические исследования для исключения глиомы.
Нейрогенный нистагм
Позиционный нистагм родственен позиционному доброкачественному нистагму взрослых и проявляется при движениях головы. Наиболее частыми причинами у детей являются лабиринтит или травма.
Вертикальный нистагм с быстрым компонентом вверх у детей обычно является результатом патологии переднего зрительного пути (катаракта, изменения сетчатки, гипоплазия зрительного нерва). Если по данным электроретинографии и нейровизуализации патологии не выявлено, то следует думать о тоническом взоре вниз или доброкачественном врожденном состоянии. У взрослых этиология может быть несколько иной (см. ниже).
Нистагм с быстрым компонентом вниз в раннем детстве свидетельствует о поражении кранио-цервикальной области (синдром Арнольда — Киари, сирингобульбия, аномалия Клиппеля — Фейля, глиома). Улучшение наступает после хирургической декомпрессии мозжечка. Более редкие причины — внутричерепная гипертензия, дегенерация мозжечка, препараты лития и противосудорожные. Описаны доброкачественные врожденные моторные формы.
Периодически альтернирующий нистагм — состояние co сменой каждые несколько минут быстрого компонента вправо на быстрый компонент влево. В промежутках смены быстрых фаз может быть вертикальный компонент. Врожденные формы могут иметь в основе неврологическую патологию, но значительно чаще они связаны с альбинизмом (Simon J., Kandel G., Krohel C., Nelsen C., 1984) (схема 11.1).
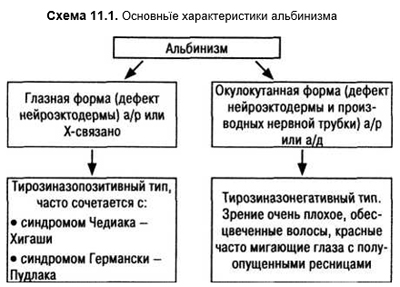
Офтальмологические проявления альбинизма:
- снижение зрения, нистагм, миопия;
- страбизм (обычно эзотропический) с амблиопией;
- повышенная прозрачность радужки co светобоязнью;
- при фундускопии:
а) сниженный фовеальный рефлекс, желтый пигмент макулы, сосуды сетчатки перекрещивают область фовеалы;
б) по периферии хорошо видна хориоидальная оболочка (снижение пигментации эпителия сетчатки);
в) аномалии зрительного нерва.
Периодически альтернирующий нистагм следует дифференцировать от других врожденных вариантов нистагма, поскольку его амплитуда снижается при фиксации взора. Приобретенные формы связаны co спиноцеребеллярной дегенерацией, опухолями или мальформациями в области задней черепной ямки, энцефалитами.
Нистагм по типу «морской волны» характеризуется движением одного глаза вверх и кнутри, а другого — вниз и наружу. Связан с большими супраселлярными опухолями (краниофарингомы, хиазмальные глиомы), сочетается с битемпоральной гемианопсией, что свидетельствует о сенсорном механизме. Нистагм может полностью исчезнуть на фоне применения клоназепама и баклофена (ингибитор высвобождения глутамата).
Флаттер глаз — быстрые маятникообразные горизонтальные движения глаз без интервалов — типичный исход вирусного энцефалита.
Опсоклонус характеризуется хаотическими мультивекторными быстрыми беспрерывными движениями глаз, свидетельствует о мозжечковой дисфункции и является тяжелым вариантом флаттера. Опсоклонус у детей может возникать при менингите, энцефалите, интоксикациях, гидроцефалии. И флаттер, и опсоклонус известны как паранеопластическая реакция на нейробластому.
Варианты нистагма, прежде всего у взрослых, и их клиническая трактовка приведены в табл. 11.1.
Таблица 11.1
Варианты нистагма и их клиническое значение
| Тип нистагма | Характеристика | Причина |
| Быстрый нистагм | Обычно горизонтальный. Редко — вертикальный или косой. Нередко с ротаторным компонентом. Быстрый компонент направлен от стороны поражения. Амплитуда увеличивается при взгляде в здоровую сторону. Сочетается с шумом в ушах, снижением слуха, головокружением, тошнотой, пошатыванием | Воспалительный или деструктивний лабиринт |
| Маятникообразный нистагм | Нарастает при взгляде вверх | Потеря центрального зрения в раннем возрасте; кивательный спазм и кривошея у детей; рассеянный склероз |
| Конвергентный нистагм | Медленное отводящее движение, быстрое приводящее | Опухоли шишковидной железы; нарушения в области покрышки моста, синдром Парино* |
| Нистагм при поражении ствола мозга | Грубый нистагм, зависимый от взгляда, горизонтальный или вертикальный. Отсутствует при закрытых глазах. Непостоянное головокружение, другие стволовые знаки | Рассеянный склероз; сосудистые нарушения в области покрышки моста (нистагм с направлением вверх); аномалия Киари; опухоль цервико-медуллярного угла (нистагм с направлением вниз); барбитураты, фенитоин |
| Нистагм с быстрым компонентом вниз | Медленный обратный компонент к средней линии | Грубые повреждения моста |
*Синдром Парино — опухоль, значительно реже — инфаркт в претектальной области, приводящие к параличу взора вверх. Сопутствующие признаки — широкие (до 6 мм) зрачки, очень плохо реагирующие на свет, лучше — на аккомодацию.
Паралич взгляда вниз обычно бывает при двустороннем повреждении среднего мозга ниже ядра III нерва. Раздражение вестибулярной системы может заставить глаза двигаться вверх или вниз по контрасту с горизонтальными движениями глаза, которые полностью прекращаются при повреждении ретикулярной формации.
Завершая описание нистагма, следует указать, что знание причин сенсомоторных расстройств в области иннервации черепно-мозговых нервов очень важно для врача общей практики: диагноз оказывается буквально написан на лице. Наиболее значимые нарушения функций черепно-мозговых нервов и их причины суммированы в табл. 11.2.
Таблица 11.2
Клинические проявления сенсомоторных расстройств при повреждении 12 пар черепно-мозговых нервов и причины нейропатий
| Клинический признак | Нерв | Функция | Состояние |
| Аносмия | 1-я пара N. olfactorius | Проводят чувствительные импульсы обоняния | Синуситы; транзиторно — после вирусных инфекций; травма; опухоли внутричерепные, полости носа и придаточных пазух |
| Преходящая моноокулярная слепота (amaurosis fugax) | 2-я пара N. opticus | Проводят чувствительные импульсы от сетчатки | Ипсилатерально — эмболия артерии сетчатки, поражение внутренней сонной артерии |
| Резкое снижение остроты зрения, боль при движении глазного яблока (неврит реторбульбарний) | Острые демиелинизирующие заболевания; сосудистые изменения n. opticus; энцефаломиелит; менингит; задний увеит; болезни Девика, Шильдера | ||
| Токсически алиментарные нейропатии | В12-дефицит; тяжелые алиментарные дистрофии (например Квашиоркор); метанол; свинец; органические соединения ртути; левомицетин; этамбутол; изониазид; НПВП; стрептомицин; сульфаниламиды; препараты наперстянки; алкалоиды спорыньи | ||
| Битемпоральная гемианопсия | Опухоль гипофиза; краниофарингиома; менингеома; аневризма кавернозного синуса | ||
| Птоз, мидриаз, невозможность взора вверх (парез) | 3-я пара N. oculomotorius | Поднимает верхнее веко; движение глаз; регулирует диаметр зрачка; изменяет кривизну хрусталика | Аневризма соединительной артерии; опухоли, абсцессы, субдуральная гематома; инфаркт среднего мозга; нейропатия (сахарный диабет; демиелинизирующие процессы) |
| Паралич, парез с невозможностью движения глаза вверх и кнаружи | 4-я пара N. trochlearis | Движение глаза вверх и кнаружи (нарушения выявляют с трудом, так как компенсируются другими мышцами и поворотом головы) | Закрытая травма головы без перелома костей (частый исход мотоциклетных аварий); пинеалома; менингеома; рассеянный склероз; ишемическая нейропатия (сахарный диабет) |
| Тройничная невралгия | 5-я пара, 1-я ветвь N. trigeminus | Проводит чувствительные импульсы от поверхности глаза, верхнего века, скальпа, лба; от слезных желез | Вероятнее всего — компрессионная нейропатия (сдавление нерва артерией или, реже, веной, при его выходе из ствола мозга) |
| 2 и 3-я ветви | Чувствительные импульсы от лица, зубов, десен, неба; движения жевательной мышцы | Шванномы; менингеомы; повреждения кавернозного синуса, орбиты; метастазы в верхнюю челюсть | |
| Внутреннее косоглазие, поворот глаза кнаружи происходит очень медленно | 6-я пара N. abducens | Движение боковой прямой мышцы | Повышение внутричерепного давления; последствия люмбальных пункций; сдавление нерва опухолью носоглотки; опухоли моста или мозжечка; инфекции менингеальной оболочки; вазопатии при диабете; энцефалопатия Вернике; аневризма; инфаркт моста; рассеянный склероз; травма. У детей без признаков внутричерепной гипертензии — после вирусной инфекции (исход благоприятный). Идиопатический — при отсутствии изменений других нервов и улучшения в ближайшие 2 мес |
| Паралич лицевого нерва | 7-я пара N. facialis | Движение мимических мышц лица; иннервация слезных и слюнных желез; чувствительные импульсы от передних 2/3 языка | Паралич Белла (идиопатический или иммунный отек при вирусных инфекциях); синдром Рамзая (герпес), а также последствия инфекции среднего уха и сосцевидного отростка; перелома пирамидной кости; саркоидоз; болезнь Лайма; лейкемическая инфильтрация нерва; акустические невриномы; опухоли и инфаркты моста; синдром Гийенна — Барра |
| Болезнь Меньера | 8-я пара N. vestibulo- cochlearis | Сенсорные импульсы слуха и равновесия | Причины обсуждаются |
| Доброкачественное позиционное пароксизмальное головокружение | Агрегация отолитов; контузия лабиринта; хирургические вмешательства на ухе; средний отит; окклюзия передней вестибулярной артерии | ||
| Вестибулярный неврит | Вирусная инфекция | ||
| Снижение слуха | Вирусная инфекция; невриномы; опухоли мосто- мозжечкового угла |
| Глоссофарингеальные невралгии и нейропатии | 9-я пара N. glosso- pharyngeus | Сенсорные импульсы от глотки, миндалин, задней части языка, сонной артерии; участвует в регуляции АД; движение мышц слюнных протоков и при глотании | Идиопатические расстройства; опухоли или аневризмы в задней черепной ямке или овальном отверстии |
| Дисфагия. Дисфония | 10-я пара N. vagus | Движения мышц при глотании, речи; передача импульсов к сердцу и гладким мышцам внутренних органов | Герпес; поражения моторного нейрона; сосудистые и опухолевые процессы продолговатого мозга; опухоли и инфекции менингеальной оболочки |
| Частичный или полный паралич mm. Sternocleidomastoideus или trapezius | 11-я пара N. accessorius | Двигательные импульсы к грудино-ключично-сосцевидным и трапециевидным мышцам | Опухоли менингеальной оболочки, мосто-мозжечкового угла, основания черепа |
| Нейропатия | Причины обсуждаются | ||
| Атрофия и фасцикуляции языка | 12-я пара N. hypoglossus | Двигательные импульсы к мышцам языка | Поражения моторного нейрона; полиомиелит; опухоли; хирургическая травма (эндартерэктомия); патология менингеальной оболочки или затылочной кости |
Острый нетравматический поперечный синдром характеризуется слабостью в ногах и, как правило, задержкой мочи, потерей контроля над дефекацией. Этих больных необходимо немедленно госпитализировать для дальнейшего ДД-объяснения неврологических нарушений. Причина инфарктов чаще всего объясняется сосудистыми нарушениями вне спинномозгового канала, поскольку спинномозговые сосуды крайне редко подвергаются атеросклеротическим нарушениям. Наиболее вероятное место сосудистых нарушений — артерия, входящая в спинномозговой канал на уровне XII грудного позвонка (ТXII) и отдающая веточки в спинной мозг на уровне X грудного позвонка (ТX), где возможно развитие аневризмы аорты с повреждением передних столбов спинного мозга. Грыжи дисков, расположенные медиально, внедряясь в спинномозговой канал, могут привести не только к сдавлению корешков, но и синдрому поперечного перерыва. Чаще всего это шейные грыжи, болевой синдром при них непостоянен. Грыжи грудного отдела обычно травматические, алюмбальные грыжи обусловливают только корешковые расстройства. При этих грыжах, сдавливающих задние корешки, типична боль, усиливающаяся при движении. Причиной поперечного синдрома могут быть и патологические переломы позвонков на фоне опухолей, особенно миеломы, или остеопороза (при хронических ревматических заболеваниях или терапии кортикостероидами). Спинальные ангиомы обусловливают кровотечения, при этом пациенты сообщают о боли в спине при нагрузке или слабости в ногах, возникших за несколько месяцев до обследования. Токсико-аллергический поперечный синдром чаще всего возникает после инъекций героина, особенно если она сделана после длительного перерыва в приеме наркотика, амфетамина, на фоне болезни Лайма, туберкулеза, грибковых или паразитарних инфекций, сифилиса, васкулита.
При наличии поперечного синдрома необходимо выполнить рентгеновское исследование позвоночника в прямой и боковой проекциях, обратив особое внимание на изменение контуров и высоты позвонков. Если по данным рентгеновского исследования выявили изменения, которые логично объясняют неврологическую симптоматику, то целесообразно выполнить КТ этой области. При вероятных сосудистых повреждениях информативны КТ и эхоГ для исключения аневризмы сосудов. Альтернативным исследованием может быть ЯМР, при невыясненном уровне поражения — миелография. Если симптоматика не успела сформироваться в целостную картину, проводят люмбальную пункцию с поддавливанием вен шеи. Если спинномозговой канал проходим, то давление спинномозговой жидкости повышается, возвращаясь к норме после восстановления проходимости вен. При частичной блокаде повышается концентрация белка в ликворе.
Подострый поперечный синдром, развивающийся в течение нескольких дней, может появиться при всех вышеуказанных причинах. Но при таком течении неврологических поражений необходимо помнить о воспалительных изменениях. Поперечный синдром может сформироваться как паранеопластическое состояние. Особенно подозрительны в этом отношении случаи с хронически-воспалительным ликвором без наличия возбудителей у лиц в возрасте старше 50 лет.
Диссеминированный энцефаломиелит — демиелинизирующее заболевание — при наличии многих очагов может дать симптоматику поперечного синдрома. Хронически прогредиентный поперечный синдром развивается у лиц пожилого возраста как цервикальная миелопатия в результате постепенного сужения спинномозгового канала при выпадениях дисков и дегенеративных изменениях. При этом состоянии рефлексы с рук отсутствуют, так как рефлекторная дуга в результате миелопатии прерывается. Рефлексы с ног и тонус спастично повышены.
Синдром Броун — Секара — следствие гетерогенного по характеру повреждения спинного мозга. В практике чаще развивается при сдавлении боковых столбов спинного мозга (обычно опухолью), реже — при колющих ранениях, у лиц молодого возраста появление синдрома Броун — Секара заставляет исключать рассеянный склероз. Для синдрома типична картина, обусловленная соотношением перекрещивающихся и неперекрещивающихся путей. Обязательное условие появления синдрома Броун — Секара — строго одностороннее поражение. Двигательный парез и нарушение глубокой чувствительности определяются на одной стороне, а нарушение поверхностной чувствительности — на противоположной. Диагностические критерии:
- Ипсилатеральное повреждение:
а) одностороннее повреждение пирамидных путей co спастическим парезом ноги, реже — дополнительно и руки с положительными пирамидными знаками;
б) нарушение восприятия тонких прикосновений, вибрации, положения тела;
в) вазомоторные нарушения;
г) гемиатаксия, чаще недиагностируемая за счет центрального пареза.
- Контрлатеральное повреждение:
а) нарушение болевой, температурной и грубой чувствительности на несколько сегментов ниже уровня повреждения.
- Тонкий соматом на уровне повреждения — ипсилатеральная анестезия.
Сирингомиелия характеризуется преимущественным нарушением температурной и болевой чувствительности. Эти изменения почти патогномоничны для заболевания, потому что афферентные волокна температурной и болевой чувствительности переключаются в заднем роге на второй нейрон, который в непосредственной близости от центрального канала переходит на противоположную сторону. Центральный канал у взрослых рудиментарный, при сирингомиелии (syrinx — полость, пещера) он расширяется и сдавливает проходящие рядом пути. Расширение происходит очень медленно, на протяжении месяцев или даже лет, в течение которых пациенты могут испытывать жгучие парестезии в участках, которые в дальнейшем характеризуются нарушенной чувствительностью. Вследствие давления на премоторные волокна и передние рога могут развиваться и локальные атрофии мышц. Чаще других поражается шейный отдел спинного мозга с распространением процесса на продолговатый мозг (сирингобульбия).
Прионные болезни нервной системы
К ним относятся трансмиссивные губчатые энцефалопатии, характеризующиеся образованием вакуолей в нейронах и глии головного мозга. Группу составляют болезни Крейцфельда — Якоба, Куру, Герстмана — Штрейсслера — Шейнкера, фатальная семейная инсомния.
При морфологическом исследовании для прионных болезней характерны уменьшение объема и массы головного мозга, атрофия его коры. При микроскопическом исследовании типичны спонгиоформная вакуолизация, атрофия и гибель нервных клеток, пролиферация астроцитов в тканях мозга, а также амилоидные бляшки, содержащие прионный белок. Указанные морфологические изменения при различных прионных заболеваниях варьируют. Возбудителем прионных болезней является инфекционный прионный белок РrРSc. Он отличается от нормального белка РrРс высокой устойчивостью к протеазе К, нерастворимостью после очищающей экстракции, способностью накапливаться во вторичных лизосомах, обогащением во время процесса выделения прионного инфекционного начала. Накопление патологической изоформы прионного белка происходит не за счет его синтеза, а путем изменения третичной или четвертичной структуры нормального прионного белка. Соединение молекул PrPSc и РrРс приводит к образованию 2 молекул PrPSc, что обеспечивает экспоненциальный рост количества молекул инфекционного прионного белка. То есть синтез PrPSc — посттрансляционный процесс. Выявлено более 20 мутаций гена прионного белка PRNP, кодированного на коротком плече 20-й хромосомы. С семейной формой болезни Крейцфельда — Якоба связаны точковые мутации в кодонах 178, 180, 183, 200, 208, 210, 323, а также восьмичленные аминокислотные повторы в кодонах от 51 до 91. Для болезни (синдрома) Герстмана — Штрейсслера — Шейнкера характерны точковые мутации в кодонах 102, 105, 117, 145, 198, 212 и 217, для фатальной семейной инсомнии — в кодоне 178.
Ранняя диагностика прионных болезней очень затруднена. Клинические признаки напоминают болезнь Альцгеймера и вначале представляется более важной ДД между ней и губчатыми энцефалопатиями, чем между различными формами этих энцефалопатий. По данным ЯМР устанавливают атрофию коры мозга. Гистологически у пациентов с болезнями Куру и Герстмана — Штрейсслера — Шейнкера превалируют амилоидные очажки, меньше спонгиоформных изменений, а при болезни Крейцфельда — Якоба и фатальной семейной инсомнии преобладают губчатые изменения. Амилоидные бляшки напоминают болезнь Альцгеймера, но при ней они не связывают анти-бета А4-протеин. Специфическая морфологическая диагностика прионных болезней осуществляется иммуноцитохимическими методами путем окрашивания патологического протеина. Достоверный диагноз прионных болезней может быть только по данным аутопсии, так как возможности биопсии ограничены в связи с небольшим объемом биоптата и необходимостью его взятия из различных участков мозга. В качестве альтернативы биопсии мозга появилась возможность выявления в спинномозговой жидкости атипичных белков (26 000 и 29 000 Да, а также 14-3-3).
Болезнь Крейцфельда — Якоба впервые описана в начале XX в. Выявляют по всему миру. Более 90% случаев — спорадическое заболевание. Остальные 10% — наследственные и ятрогенные случаи. Семейная форма передается а/д, характеризуется ранним началом. Заболеваемость 0,3–1 случай на 1 000 000. Мужчины болеют чаще, чем женщины (1,5:1). Болезнь Крейцфельда — Якоба обычно поражает взрослых в возрасте 40–65 лет. Передача от человека к человеку происходит, вероятно, при пересадке роговицы, твердой мозговой оболочки, печени, переливании крови и лечении карликовости гормоном роста, полученным из гипофиза трупа. Заболевание полностью аналогично скандально известному «бешенству коров». Обычно болезнь Крейцфельда — Якоба начинается с потери памяти, прогрессирующей в течение нескольких месяцев. У 1/5 пациентов процесс протекает буквально по дням. В этих случаях первыми симптомами являются головокружение, ухудшение зрения, диплопия. Присоединяются нарушения сна, афазия, апраксия, дисграфия. В первые 6 мес появляется миоклонус, провоцируемый сенсорными раздражителями. Выявляют признаки поражения передних рогов спинного мозга (атрофии мышц), базальных ганглиев (гипокинезия, хореоатетоз). Эпилептические припадки возникают редко и только в претерминальной стадии.
Куру — прогрессирующее неврологическое заболевание, диагностируемое исключительно на островах Новой Гвинеи. До начала 60-х годов XX в. было распространенным. Инфекция передавалась при каннибализме. Искоренение этого обычая привело к фактической элиминации куру.
Болезнь Герстмана — Штрейсслера — Шейнкера передается а/д (?) или вертикально (?). Доказана возможность заражения экспериментальных животных. Выявляют на всех материках, но в 100 раз реже, чем болезнь Крейцфельда — Якоба. Возраст начала заболевания — 40 лет (против в среднем 60 при болезни Крейцфельда — Якоба), длительность также больше (5 лет против 9 мес). Заболевание манифестирует как мозжечковая атаксия, протекает как спиноцеребеллярная дегенерация. Позже может присоединиться деменция. В некоторых семьях ведущими являются экстрапирамидные нарушения, в других — паралич взора, глухота и слепота. Характерно отсутствие сухожильных рефлексов на ногах при наличии разгибательных патологических знаков. Миоклонии редки.
Фатальная семейная инсомния — быстро прогрессирующее заболевание, передающееся а/д. Очень редкое состояние. Экспериментальным животным передается очень сложно. Средний возраст дебюта заболевания — 40 лет. Длительность течения — от 6 до 48 мес. Губчатая трансформация затрагивает прежде всего ядра таламуса, что обусловливает сбой цикла бодрствование/сон. Вначале отмечают сложности засыпания, вегетативную дисфункцию (изменения саливации, потоотделения, запор, лихорадка), затем — миоклонию, деменцию.
К демиелинизирующим заболеваниям ЦНС относят:
Наследственные миелинопатии
- Адренолейкодистрофии.
1. Прогрессирующая энцефалопатия с корковой слепотой у людей молодого возраста.
2. Прогрессирующая миелопатия у мужчин зрелого возраста.
3. Энцефалопатия в сочетании с миелопатией.
4. Злокачественная энцефалопатия у новорожденных мальчиков.
5. Преимущественно эндокринологические нарушения — болезнь Аддисона, сцепленные с Х-хромосомой и сопровождающиеся умеренными неврологическими нарушениями.
6. Миелопатия у женщин.
7. Преимущественно церебральные формы.
8. Преимущественно полиневропатические формы.
- Суданофильная лейкодистрофия Пелицеуса — Мерцбахера. Выраженное снижение содержания эфиров холестерина. Клинически — нарушения координации, спастические парезы, нарушения интеллектуальной деятельности. В зрелом возрасте течет благоприятно, сложно отличима от рассеянного склероза.
- Болезнь Александера; а/p. Спастика, нарастающая слепота, деменция, судорожные припадки. В начале — ремитирующее течение.
- Болезнь Краббе.
- Болезнь Канавана.
- Митохондриальная лейкоэнцефалопатия. Проявляется: быстрая мышечная утомляемость, офтальмоплегия, мимикрия рассеянного склероза, лактатемия, избыток лактата в спинномозговой жидкости.
Миелинокластические заболевания
- НейроСПИД.
- Прогрессирующая мультифокальная лейкоэнцефалопатия. Вызывается вирусом JC из группы паповавирусов. Развивается у больных СПИДом, СКВ, саркоидозом, лимфомой, лейкозом, иммунодефицитами, после трансплантации почки. Заболевание начинается подостро или постепенно. Появляются гемипарезы, дизартрия, афазия, гемианопсия. У 2/3 пациентов присоединяется прогрессирующее разрушение интеллекта. Судороги редки, но достаточно типичны для больных СПИДом и прогрессирующей мультифокальной лейкоэнцефалопатией.
КТ или ЯМР позволяют выявить одно- или двусторонние, одиночные или множественные очаги в белом веществе. При КТ — это очаги пониженной плотности, при ЯМР в Т1- или Т2-режимах — повышенной. Серологические исследования бесполезны: 2/3 населения имеют антитела к JC-вирусу.
Достоверный диагноз при жизни может быть поставлен с помощью стереотаксической биопсии. Но это вмешательство допустимо в исключительных случаях при нетипичных радиологических признаках для исключения опухоли.
- Тропический спинальный парапарез — HTLV-I миелопатия (Human T-Lymphotrophic Virus). Медленно прогрессирующее иммунообусловленное заболевание спинного мозга, вызванное ретровирусом и характеризующееся спастическим парезом ног. Ретровирус передается при половых контактах, внутривенных вливаниях, переливаниях крови и ребенку — через молоко матери. Распространен в тропическом и субтропическом поясе. HTLV-II, вероятно, вариант HTLV-I миелопатии.
Заболевание начинается как хроническое, медленно прогрессирует. Нарастает спастический парез ног с потерей вибрационной чувствительности. Часто отсутствуют ахилловы рефлексы, нередко — недержание мочи. В сыворотке крови выявляют антитела к вирусу, диагноз может быть подтвержден полимеразной цепной реакцией.
- Острый или подострый центральный понтинный и/или экстрапонтинный миелиноз.
- Токсическая лейкоэнцефалопатия.
- Острый рассеянный энцефаломиелит.
- Оптикомиелит Девиса (рассматривается как вариант рассеянного склероза).
- Концентрический склероз (болезнь Балло).
- Лейкоэнцефалит Шильдера.
- Болезнь Марбурга.
Двигательные парезы различаются в зависимости от уровня поражения (табл. 11.3, 11.4).
Таблица 11.3
ДД-признаки парезов в зависимости от места их возникновения
| Уровень повреждения | Центральный (1-й мотонейрон) | Периферический (2-й мотонейрон) | Двигательный синапс | Мышечный уровень |
| Тип поражения | Определенные группы мышц (например парез типа Вернике — Манна) | Корешковый или периферический тип при повреждении нервных корешков, сплетения, или периферического нерва | Чаще начинается в небольших мышцах типа век, глаз, затылка | Симметрично на конечностях, особенно выражено проксимально |
| Тонус | Вначале снижен, затем нарастает, появляется спастика и ригидность | Чаще снижен, реже — повышен (спинальная спастика) | Может быть снижен | Снижен |
| Рефлексы | Повышены, кожные брюшные — снижены, дополнительно появляются патологические | Снижены вплоть до полного угасания | Быстро истощаются | Снижены |
| Фасцикуляции | Нет | Особенно выражены при повреждении мотонейронов, чем при нарушениях периферических нервов | ||
| Атрофии | После длительного периода | Появляются через 2–3 нед | Возможны псевдо-гипертрофии | |
| Что прежде всего исключать | Резидуальные состояния (гипоксемия, кровоизлияния, инфекции), внутричерепные объемные процессы, нейрометаболические нарушения | Инфекции, травмы, спинальные объемные процессы, нейрометаболические наследственные дегенеративные заболевания | Миастения | Иммунные, наследственные обменные, постпараинфекционные (транзиторные) |
Таблица 11.4
ДД-схема топики поражения в зависимости от локализации, видов парезов (плегий) и сопутствующей патологии
| Локализация пареза (плегии), тип | Сопутствующий симптом | Локализация поражения |
| Рука (по центральному типу) | Нарушение чувствительности; дефект поля зрения; афазия (при поражении доминантного полушария) | Контрлатеральная кора |
| Нога (по центральному типу) | Ипсилатеральные сенсорные нарушения | Контрлатеральная кора |
| Нарушение болевой и температурной чувствительности на противоположной поражению стороне | Ипсилатеральное поражение спинного мозга | |
| С уровня глаза половина лица и шеи. С этой же стороны — рука и нога (по центральному типу) | Нарушение сознания; центральный парез лицевого нерва; дефект поля зрения; афазия (при поражений доминантного полушария) | Контрлатеральное полушарие |
| Сознание не нарушено; парез лицевого нерва по центральному типу | Контрлатеральная внутренняя капсула | |
| Контрлатеральный гемипарез; паралич глазодвигательного нерва; парез взора вверх | Контрлатеральное поражение среднего мозга | |
| Рука и нога гомолатерально (по центральному типу) | Дефект поля зрения; нарушение чувствительности; афазия (при поражении доминантного полушария) | Контрлатеральная кора |
| Ипсилатеральное нарушение болевой и температурной чувствительности; контрлатеральный синдром Горнера; контрлатеральный парез мышц языка и мягкого неба | Контрлатеральные отделы продолговатого мозга | |
| Потеря болевой и температурной чувствительности в контрлатеральной ноге; ипсилатеральное нарушение глубоких видов чувствительности; ипсилатеральный синдром Горнера | Ипсилатеральное поражение спинного мозга | |
| Рука и нога гомолатерально (по центральному типу), половина лица — по периферическому типу с противоположной стороны | Парез взора (больной смотрит на пораженные конечности) | Контрлатеральный мост |
| Рука и нога контрлатерально (по центральному типу) | Парез мышц и мягкого неба на стороне паретичной руки | Альтернирующий гемипарез (поражение продолговатого мозга) |
| Парапарез ног (по центральному типу) | Нарушение глубоких видов чувствительности; отсутствие контроля над функцией тазовых органов; температурная и болевая чувствительность сохранены | Двустороннее поражение коры медиальных отделов полушарий |
| Нарушение функций тазовых органов; четкий уровень нарушений чувствительности | Грудной отдел спинного мозга | |
| Парез мышц головы, шеи туловища, конечностей (полный тетрапарез) по центральному типу | Сохранено только вертикальное движение глаз | Мост |
| Тетрапарез по центральному типу с уровня плеч | Черепные нервы не поражены; нарушение дыхания (поражение СI–СIII), требующее ИВЛ; сохранение диафрагмального дыхания при поражении ниже СIV | Спинной мозг на шейном уровне |
| Движения мышц лица сохранены; нет движений мягкого неба, язычка; отсутствует речь | Продолговатый мозг | |
| Периферический паралич рук, центральный паралич ног | — | Симптомы поражения периферического мотонейрона свидетельствуют об уровне поражения. Состояние развивается при одновременном поражении двух отделов нервной системы. Могут присутствовать корешковые и проводниковые симптомы |
| Периферические парезы (одно- или двусторонние) | — |
Центрально обусловленные моторные парезы
Острая тетраплегия развивается за счет двустороннего выпадения кортикоспинальных путей при тромбозе a. basillaris. В острой стадии при локализации процесса в стволе мозга отмечают нарушения зрения, диплопию, головокружение, патологический нистагм, сомноленцию. Если ишемический очаг ограничен мостом, то развивается своеобразное коматозное состояние, при нем из произвольных движений сохраняются только вертикальные движения глазных яблок и мигание, которые регулируются мезенцефально. Ядра же мимических мышц и горизонтального движения глаз находятся в области tegmentum pontis. Эти пациенты могут жить долго, поскольку центр дыхания функционирует в продолговатом мозгу. Центральный тетрапарез чаще всего развивается в результате сосудистого поражения области моста.
При форсированной коррекции хронической гипонатриемии, прежде всего свойственной алкоголикам, развивается острая демиелинизация моста. При проведении ЯМР отмечается достаточно типичная картина — снижение плотности этой области.
Повреждения, обусловливающие тетрапарезы, всегда располагаются в области задней черепной ямки, кранио-цервикального перехода или верхних шейных позвонков. Если с помощью КТ не удается установить причину, то целесообразно провести ЯМР с сагиттальной реконструкцией, позволяющей надежно визуализировать патологию в области foramen magnum.
Спинальный шок развивается в результате контузии спинного мозга при падении на спину. Обычно это происходит при падении на лестнице, на льду, при катании на лыжах. Остро появляется парез ног, возможен и парез рук. Обычно через некоторое время парез бесследно проходит.
Боковой амиотрофический склероз — заболевание неустановленного генеза, характеризующееся прогрессивной дегенерацией кортико-спинального тракта, клеток передних рогов спинного мозга и/или бульбарных двигательных ядер. Средний возраст начала заболевания — 55 лет, чаще заболевают мужчины. 5% всех случаев — семейные с а/д типом наследования. При боковом амиотрофическом склерозе поражаются как кортико-спинальные пути, так и периферический мотонейрон с развитием хронического тетрапареза. Если не затронут язык и мышцы лица, то хронический тетрапарез, возможно, сформировался в результате объемного процесса в верхних отделах спинного мозга, например при медиальной грыже диска.
Боковой амиотрофический склероз — комбинированный центрально-периферический парез. Центральное поражение с дегенерацией кортико-спинальных путей ведет к парезу, спастике, повышению собственных рефлексов. Одновременно развивающаяся дегенерация мотонейрона обусловливает вялый парез с угасанием собственных рефлексов и атрофией мышц. Часто отмечают фасцикуляции. То есть типичным для заболевания является сочетание повышенных рефлексов, фасцикуляций и атрофии мышц. Симптоматика может быть достаточно пестрой в зависимости от преимущественно бульбарной или миелической локализации, от стадии заболевания, когда одни рефлексы могут быть угнетены, а другие повышены. Решающим для диагноза является ЗМГ, по итогам которой денервация должна быть выявлена минимум на двух конечностях. Выделяют следующие варианты бокового амиотрофического склероза:
- Прогрессирующий бульбарный паралич. Самый тяжелый вариант, завершающийся смертью в ближайшие 1–3 года. Прежде всего страдают мышцы, иннервируемые черепно-мозговыми нервами: нарушаются жевание, глотание, речь. Типичны эмоциональная лабильность, неспровоцированные и несоответствующие обстановке эмоциональные взрывы. Расстройства нарастают. Очень плохим прогностическим признаком является дисфагия.
- Прогрессирующая мышечная атрофия. В 10% случаев передается а/д. Заболевание может начаться в любом возрасте. Длительность течения — 25 лет и более. Превалируют изменения передних рогов, а кортико-спинальный путь длительное время остается интактным. Самый ранний признак — фасцикуляции. Мышечная слабость начинается в кистях, затем — в предплечьях, плечах, позже появляется и в ногах, а затем становится генерализованной.
- Первичный латеральный склероз и прогрессирующий псевдобульбарный паралич. Нарастают мышечная ригидность и слабость в периферических мышцах (латеральный склероз) и области иннервации нижних пар черепно-мозговых нервов (псевдобульбарный паралич). Фибрилляция и атрофия мышц присоединяются только через несколько лет.
Мышечные заболевания (парезы с нормальными рефлексами)
Наиболее известным заболеванием этой группы является тяжелая миастения (myasthenia gravis), при которой парез вечером выражен больше, чем утром (Newton Е., 2005; Palace J., Vincent A., Beeson D., 2001). Заболевание рассматривается как аутоиммунное. Аутоантитела класса IgG направлены против ацетилхолиновых рецепторов на поперечно-полосатых мышцах. В патологический процесс вовлечены и пресинаптические структуры: выявлены аутоантитела, направленные на ионные каналы. У пациентов молодого возраста тяжелая миастения ассоциируется с HLA В8 и DR3. Чаще диагностируют у женщин в возрасте 20–40 лет.
Пациент отмечает, что утром он чувствует себя как обычно, к обеду его беспокоит диплопия и нечеткость речи при длительных разговорах. После отдыха состояние несколько улучшается. При генерализованной форме поражаются мышцы шеи и конечностей. При длительном течении заболевания у 15–20% больных развивается мышечная атрофия. Для диагностики миастении применяют несколько проб. Общим является оценка функции мышцы до и после тонической или динамической нагрузки. Например, просят пациента смотреть вдаль и измеряют расстояния между веками. Затем в течение 20 с он должен смотреть вверх, после чего вновь переводит взгляд горизонтально вдаль, а врач тут же повторно измеряет расстояние между веками. У здорового человека оно соответствует первому измерению.
Диагностика также базируется на выявлении специфических антихолинрецепторных антител (положительно у 95% больных генерализованной формой миастении и у 58% — окулярной), на регистрации амплитуды вызванных сокращений, на улучшении сокращения мышц после инъекции блокаторов ацетилхолинэстеразы. В последнем случае пробу можно проводить только при наличии заполненного шприца с атропином (антидот при критической брадикардии) и аппарата искусственной вентиляции легких. Обязательным является КТ грудной клетки для выявления гиперплазии тимуса или тимом (с миастении дебютирует до 10% злокачественных опухолей вилочковой железы). Удаление вилочковой железы на ранних этапах улучшает прогноз из-за снижения образования антител. Если выявляется тимома, то оперативное лечение обязательно.
Миастения новорожденных — синдром генерализованной мышечной слабости, присущий 12% детей, рожденных женщинами с миастенией. Миастения новорожденных обусловлена трансплацентарной передачей антител. Симптоматика постепенно (дни–недели) исчезает вслед за падением титров антител.
Врожденная миастения, а/p. Очень редкое нарушение нейромышечной передачи, начинающееся в детстве. Антител к рецепторам ацетилхолина нет.
Симптоматические формы развиваются при назначении D-пеницилламина и аминогликозидов, способных вызывать нервно-мышечную блокаду. Миастения может сопровождать течение СКВ, РА, тиреотоксикоз, миозит. Паранеопластическая форма Итона — Ламберта развивается при мелкоклеточном раке бронхов. Развивается мышечная слабость, отличающаяся от миастенической по динамике амплитуды сокращений мышц в ответ на стимуляцию: при миастении она неуклонно снижается, а при паранеопластической форме после кратковременного снижения вновь возрастает. Периодические параличи/парезы, длящиеся иногда до нескольких суток, типичны для гипокалиемических состояний. Особенно часто это развивается у спортсменов после интенсивных физических нагрузок и последовавшей за ними обильной еды с высоким содержанием углеводов. Дыхательные мышцы не поражаются. Известны семейные формы заболевания.
Миопатии — хронические прогрессирующие парезы с повышением уровня креатинкиназы в крови, указывающей на повреждение мембран мышечных клеток. При ЗМГ регистрируют высокую электрическую активность при сниженной сократимости. Если диагноз не подтверждается семейным анамнезом, то целесообразно провести биопсию мышцы.
При ДД миопатий следует помнить о дистальной наследственной миопатии Веландера, наследственной центронуклеарной, дистальной, висцеральной и климактерической.
Миозит сопровождается парезами воспалительного генеза. Может быть изолированным или являться проявлением диффузных заболеваний соединительной ткани, при одновременных изменениях на коже можно диагностировать дерматомиозит. Парезы сочетаются с болью в пораженных мышцах. Обычно это поражение проксимальных симметричных мышечных групп с типичными изменениями ЗМГ и в биоптатах. Миопатические знаки, возникающие при нагрузках, заставляют думать о болезнях накопления, особенно если они связаны с болезненными мышечными местными судорогами (крампи). Обычно обусловлены недостаточностью ферментов гликолиза в мышцах, типично повышение креатинкиназы и других мышечных ферментов.
Парезы при эндокринопатии чаще всего развиваются при гипер- и гипофункции щитовидной железы. Наиболее частым вариантом является парез мышц глаза.
Стероидная миопатия может быть результатом как эндогенной гиперпродукции стероидов, так и результатом применения их в высоких терапевтических дозах.
Многие мышечные и нервно-мышечные поражения протекают с изменением тонуса мышц, что отражено в сводной табл. 11.5.
Таблиця 11.5
ДД мышечной гипотонии
| Показатель | Мышечный уровень | Периферический нерв | Передние рога | Мозг |
| Сила мышц | Снижена | Снижена | Снижена | Часто не изменена |
| Тонус мышц | Снижен | Снижен | Снижен | Снижен |
| Собственные рефлексы с мышц | Снижены или не определяются | Снижены или не определяются | Снижены или не определяются | Норма или повышены |
| Фасцикуляция | Нет | Возможны | Части | Нет |
| ЗМГ | Миопатический тип | Нейропатический | Нейропатический | Не изменена |
| Проводимость по нерву | Не изменена | Снижена или норма | Норма | Норма |
| Гистологические изменения в мыщцах | Миопатический тип | Нейропатическая атрофия | Нейропатическая атрофия | Норма или атрофия волокон |
| Гистологические изменения в нервах | Нет | Дегенерация аксона, миелиновых оболочек | Дегенерация | Норма |
| Креатинин в сыворотке крови | Норма или повышен | Норма | Норма | Норма |
| Вероятные заболевания | Врожденные миопатии, миодистрофии, миозит | Полиневриты, нейропатии | Спинальные миоатрофии | Нейролипидозы, эндокринопатии, повышение внутричерепного давления |
Сенсорные парезы — нарушение глубокой чувствительности (в том числе вибрационной и движения суставов — проприоцептивной) и поверхностной (температурной, болевой и тактильной) чувствительности. Сенсорные парезы чаще всего являются результатами полинейропатии. Типичным для них является дистальное нарастание симптоматики. Гемисимптоматика развивается при повреждении гипоталамуса с противоположной стороны или корковых зон. Истинно сенсорные расстройства очень редки, так как проводящие пути проходят вплотную друг к другу, и поэтому почти всегда нарушены тонкие движения. В молодом возрасте это выявляют при рассеянном энцефаломиелите. Если на первый план выдвигаются расстройства температурной чувствительности, то следует думать о патологии спинного мозга, прежде всего о сирингомиелии. Если поверхностная чувствительность нарушена на одной стороне, а глубокая — на противоположной, то есть основания для дальнейшего обследования относительно синдрома Броун — Секара.
Сенсомоторные парезы co сниженными собственными рефлексами (поражение периферического нейрона) включают парезы и плегии, снижение или полное исчезновение всех рефлексов, низкий тонус, развитие мышечных атрофий. Обусловлены генерализованными полирадикулоневритами, полинейропатиями, локальными поражениями (корешков, сплетений, стволов) наследственного, воспалительного, токсического метаболического или травматического генеза.
Острый вялый паралич. Прежде всего, неотложно и немедленно думать о полиомиелите, вызванном диким или культурным (вакцинальным) штаммом. Попутно с учетом возраста пациента и других симптомов можно и необходимо исключать иные причины, но полиомиелит, прежде всего в педиатрической практике, остается ведущим (см. также разделы по менингиту и вирусным инфекциям).
Полиомиелит — детский паралич — острый передний полиомиелит. Острая вирусная инфекция, обусловленная полиовирусом (см. главу Лихорадочные состояния), вызывающего неспецифические минимальные проявления, асептический менингит (не паралитический полиомиелит) и вялые параличи различных мышечных групп (паралитический полиомиелит).
Полиовирус очень мелкий (22–30 нм) с одноцепочечной РНК. Из трех серотипов чаще всего становится причиной эпидемии и вызывает параличи именно первый. Единственный хозяин — человек, но в сточных водах вирус полиомиелита (энтеровирус!) сохраняется до 3 мес. Вирус передается при непосредственном контакте, высоко контагиозен. Во внеэпидемические периоды выявляют редко. В период эпидемий на одного больного с клинически значимыми проявлениями (парезы) приходится 100–150 больных инаппарантной формой. В странах с плохими санитарно-гигиеническими условиями, низким уровнем иммунизации носителей вируса достаточно много, а приобретенный иммунитет формируется в первые годы жизни. Поэтому более 93% полиомиелитных параличей развиваются у детей в возрасте младше 5 лет. Но в экономически развитых странах с хорошими санитарно-гигиеническими условиями постоянно циркулирующей инфекции нет. Поэтому заболевают дети старшего возраста и взрослые. Завозные случаи полиомиелита, вызванного диким полиовирусом, были зарегистрированы в Европе в 2001 г.
Основные резервуары инфекции — Африка и Азия. Так, в Анголе регистрируют 53 случая полиомиелита в год; в Бенине — 1/8; в Конго — 12/14; в Нигерии — 5/98; в Нигере — 1/10; в Афганистане — 18/63; в Ираке — 4/67; в Сомали — 41/2; в Индии — 190/1126 (в числителе данные за 2000 г., в знаменателе — за 1999 г.). Значительная разница в количестве случаев полиомиелита за короткий период в пределах одной страны и характер жизни населения в этой стране, соотношение случаев полиомиелита и неполиомиелитного острого вялого паралича по данным регионам позволяют с большой уверенностью сказать, что истинных случаев полиомиелита в Азии и Африке значительно больше.
Наряду с полиомиелитом, вызванным «диким» штаммом вируса, существует так называемый острый паралитический полиомиелит, ассоциированный с вакцинацией. Под ним понимают острый вялый спинальный паралич, возникший не ранее 4-го и не позже 30-го дня после приема живой полиомиелитной вакцины. Случай острого вялого спинального паралича, возникший не позже 60-го дня после контакта с привитым, при котором выделен вирус полиомиелита вакцинного происхождения, классифицируется как острый паралитический полиомиелит, вызванный контактом с вакцинированным.
Выделение вируса полиомиелита вакцинного происхождения без клинических проявлений никакого клинического значения не имеет.
Частота полиомиелита, ассоциированного с вакцинацией, составляет 2 случая на 5 млн доз живой вакцины. Но каждый ребенок получает не менее 10 доз вакцины, поэтому это соотношение меняется не в лучшую сторону.
Вирус, попадая в полость рта, интенсивно размножается в лимфоидной ткани ротоглотки и желудочно-кишечного тракта (преимущественно в подвздошной кишке). Небольшое количество вируса попадает в кровь и заносится ею в ретикуло-эндотелиальную систему, где активно реплицируется.
Вторичная виремия обусловливает поражение ЦНС. Иногда вирус достигает ЦНС по нервным стволам. Вирус выявляют в лимфоидной ткани глотки и в кале весь период инкубации. После появления нервных симптомов его устанавливают в глотке еще на протяжении 1–2 нед, а в кале — 3–6 нед. Виремия длится несколько дней, пока не начнет циркулировать достаточный объем антител.
Клинически значимая патология развивается только при поражении вирусом моторных нейронов передних рогов спинного мозга, продолговатого мозга, мозжечка, двигательных зон коры (дано по убывающей вероятности). В ответ на поражение нейронов вирусом отмечается интенсивная воспалительная реакция и нейронопатия. Распространенность и степень паралича определяются только числом, локализацией и степенью поражения нейронов.
К тяжелому поражению нейронов предрасполагают:
- Возраст. Чем старше пациент, тем тяжелее течение острого переднего полиомиелита.
- Физическое истощение.
- Инокуляция вируса.
- Предшествующая тонзилэктомия.
- Беременность.
Клиническая симптоматика варьирует. Выделяют две основные формы полиомиелита — абортивную и так называемую большую (паралитическая или непаралитическая).
Абортивную выявляют у детей младшего возраста, она составляет более 90% всех случаев заболевания, проявляется умеренной лихорадкой, общим недомоганием, головной болью, першением в горле, благополучно завершается за 1–3 дня, имеет эпидемиологическое значение, но клиническое значение минимально.
Паралитическая форма полиомиелита следует за абортивной или у старших детей и взрослых начинается самостоятельно остро. Появляются лихорадка, сильная головная боль, ригидность затылочных мышц, глубокая боль в мышцах, иногда — парестезия и гиперстезия. В период активного миелита нередко возникают задержка мочи и мышечная ригидность. В зависимости от степени повреждения нейронов в спинном мозгу выявляют выпадение сухожильних рефлексов и параличи. Потеря тонуса мышц настолько полная, что ребенок напоминает тряпичную куклу-арлекина. В тяжелых случаях при высоком уровне поражения появляются гнусавость, поперхивание. Дыхательная недостаточность возникает либо из-за паралича дыхательных мышц, либо из-за поражения дыхательного центра в продолговатом мозгу.
Асимметричные вялые парезы конечностей или бульбарный паралич без нарушений чувствительности после острого лихорадочного заболевания у детей и взрослых молодого возраста — всегда позволяет клинически диагностировать полиомиелит. Другим признаком полиомиелита является стабильность параличей. Если они возникли, то остаются в пределах той группы мышц, где и возникли. Любая волнообразная динамика отсутствует. Очень редко вялые параличи или парезы, клинически неотличимые от полиомиелита, возникают после инфекции вирусами Коксаки А (чаще всего — А7) и В, ECHO-вирусами или энтеровирусом типа 71.
Поэтому каузальный генез паралича может быть установлен только лабораторно.
Окончательный диагноз полиомиелита устанавливается на оснований выделения и идентификации вируса и по серологическим исследованиям (4-кратное и более повышение в сыворотке крови титров антител к полиовирусу).
Полиомиелит весьма напоминает синдром Гийена — Барре. Но данный синдром протекает в большинстве случаев афебрильно, парезы симметричны, в 75% случаев нарушена чувствительность.
Синдром Гийена — Барре, остро начинающийся воспалительно-демиелинизирующий полирадикулоневрит (Seneviriathe U., 2000). Характеризуется симметричными и периферически возникающими парезами с угнетением собственных рефлексов. Обусловлен демиелинизацией с развитием блокады проведения импульса в области корешков. Периферическая дегенерация присоединяется значительно позже. Не исключена генетическая предрасположенность. В спинальной жидкости повышение белка при нормальном цитозе, проба Квекенштедта (с изменением давления при затруднениях венозного оттока из головы) нормальна.
Заболевание в 65% случаев начинается в ближайшие 3 нед после вирусной инфекции верхних дыхательных путей, желудочно-кишечного тракта, стресса, операции, травмы, вакцинации. Дебютирует с икроножных мышц, поднимаясь по ногам с захватом туловища и рук. Боли и нарушений функции тазовых органов обычно нет. Поражает все поперечно-полосатые мышцы, в тяжелых случаях захватывая и дыхательные. Симптоматика может нарастать по часам. Пик поражений развивается обычно на 4-й неделе, затем симптоматика может угаснуть полностью или сохраняются остаточные явления. В ДД следует учитывать полиомиелит, для которого характерны асимметричные моторные парезы, развивающиеся в течение нескольких часов–суток. Необходимо исключить острые нейропатии, например при порфирии или отравлениях тяжелыми металлами.
Полинейропатии проявляются дистально акцентуированными расстройствами чувствительности co сниженными или отсутствующими собственными рефлексами. Нейрон — единая метаболическая единица, определяющий по объему и интенсивности синтез происходит в теле клетки, откуда белок транспортируется на периферию, в аксоны. Чем длиннее аксон, тем раньше он будет поврежден при нарушениях обмена нервной клетки. Длина аксонов двигательных (моторных) клеток и поверхностной чувствительности кожи ноги достигает 1 м!
Субъективные жалобы разнообразны, чаще снижение чувствительности сочетается с неприятными парестезиями. Вначале снижается температурная чувствительность по типу носок, чулок, а позднее и перчаток. В запущенных случаях регистрируют вегетативные расстройства (при диабетических полинейропатиях они могут быть единственным проявлением в дебюте).
Клинически диагноз полинейропатии устанавливают при отсутствии рефлексов с ахилловых сухожилий или их снижении по сравнению с рефлексами с сухожилия четырехглавой мышцы бедра, в сочетании co сниженной поверхностной и глубокой чувствительностью на голенях. Дальнейшую ДД между аксональной и демиелинизирующей нейропатией проводят по итогам регистрации скорости проведения импульса.
Демиелинизирующие нейропатии — большая редкость, обусловлены циркуляцией антител, проявляются при дифтерии или как паранеопластическое состояние, при них скорость проведения замедлена. Большинство метаболических, токсических и алиментарных нейропатий протекают с повреждением аксонов. Скорость проведения импульса длительное время не изменяется.
Наследственные полинейропатии, ранее известны как болезнь Шарко — Мари, дифференцировались в настоящее время в моторно-сенсорные нейропатии I–V типа, к которым наверняка добавятся другие редкие типы. Кроме того, к наследственным нейропатиям относят нейропатию сенсорную І типа (а/д), нейропатию сенсорную II типа (а/д), наследственный семейный политопный рецидивирующий нейропатический синдром.
При каждом моторном парезе необходимо исключать порфирию, протекающую в дебюте с полинейропатиями. В этом случае возникают большие сложности в ДД co свинцовыми полинейропатиями. При уремии полинейропатии не представляют диагностических сложностей: наличие хронической почечной недостаточности объясняет симптоматику. Паранеопластические полинейропатии особенно характерны для мелкоклеточного рака бронхов, а также миеломы и лимфомы. Последние два вида опухолей могут инфильтрировать нервные стволы и вызывать локальные нейропатии с болевым синдромом.
Каждое заболевание с расстройством микроциркуляции и поражением мелких сосудов может вызвать ишемию vasa nervorum с развитием нейропатий. Особенно часто это неврологическое расстройство сопровождает узелковый периартериит и амилоидоз. Этот же механизм в сочетании с непосредственным повреждением аксонов обусловливает нейропатию при сахарном диабете. Развитие нейропатии провоцируется недостатком витаминов В1, В6, В12. В развитых странах В1-дефицитные состояния отмечаются у алкоголиков, В6 — при лечении туберкулеза изониазидом, а В12 — при синдроме мальабсорбции, исключительно вегетарианском питании на протяжении не менее 1–2 лет и при пернициозной анемии. В последнем случае развивается фуникулярный миелоз с центральными нарушениями.
Такие профессиональные вредности, как ингаляции органических растворителей, особенно гексана (известен и среди токсикоманов как оказывающий легкое эйфорическое действие), фосфорорганические соединения (инсектициды, производство пластмасс и бензиновая промышленность), мышьяк, таллий, свинец, пребывание рук в поле СВЧ и ультразвука приводят к полинейропатиям, иногда с болью по ходу нервных стволов. Ежедневный прием 70 г алкоголя мужчиной и 35 г женщиной приводят к пестрой картине поражения нервной системы, в том числе и к нейропатиям. Полинейропатии развиваются при лечении препаратами золота, хлорамфениколом, изониазидом, нитрофурантоином, винкристином.
Локальные повреждения нервной системы, как правило, протекают с болевым синдромом. Локальные повреждения чаще всего вызваны механическими причинами. Единственный вариант воспалительной локальной нейропатии — герпетическое поражение. Во всех других случаях сосудистые или воспалительные заболевания могут протекать с акцентуированным локальным повреждением нервных стволов, но и в этом случае развивается генерализованная или мультифокальная картина.
Боль в затылке, шее и руке — достаточно частые симптомы, могут развиваться при банальном статическом физическом напряжении, особенно в нефизиологическом положении, а также отражать сосудистые, воспалительные и психосоматические расстройства. При локальной «простреливающей» боли в шее с ограничением движения следует прежде всего подумать о корешковых повреждениях. Нетравматические повреждения обычно развиваются на уровне СVI–СVII.
В противоположность к поясничным грыжам дисков, медиальные грыжи в шейном отделе могут обусловить картину компрессии спинного мозга и симптомы шейной миелопатии. В связи с этим необходим тщательный сбор сведений о вегетативних функциях (импотенция, пузырные расстройства) и поиски маломанифестной параспастики: положительный рефлекс Бабинского, повышение рефлексов с ног. При электромиографии (ЗМГ) в соответствующих мышцах удается выявить признаки денервации, при рентгеновском исследовании — дегенеративные или деструктивные признаки. КТ и ЯМР выявляют пролапс диска. Если поражены многие сегменты, то необходимо исключить опухоль верхушки легкого. Резко болезненные неврологические амиотрофии плеча носят вирусный характер и распознаются по итогам ЗМГ.
Синдром сдавления периферических нервов. Запястный туннельный синдром развивается при сдавлении нерва в запястном туннеле около лучезапястного сустава с ладонной поверхности. Боль иррадирует до плеча, усиливаясь по ночам, вследствие чего заболевание до выяснения его истинной причины было известно как ночная парестезическая брахиалгия. Решающим для диагностики, как и для всех периферических нейропатий, является определение скорости проведения импульсов по нерву. При парезе локтевого нерва расстройства чувствительности в классических случаях определяются на мизинце и латеральной поверхности IV пальца. При парезе локтевого нерва расстройства чувствительности никогда не поднимаются выше лучезапястного сустава, в то время как при корешковом синдроме СVIII снижение чувствительности распространяется до середины предплечья.
Боль в области туловища, обусловленная корешковым синдромом, чаще носит травматический характер. Среди воспалительных заболеваний первое место занимает герпес. Если опоясывающая боль сочетается с очаговыми нарушениями чувствительности в этой же области, то необходимо исключить невриному. Вначале выполняют рентгеновское исследование для выявления возможных вторичных костных деформаций, а затем — КТ, ЯМР и спинномозговую пункцию.
Боль в пояснично-крестцовой области возникает при повреждениях LIV, LV, SI. Интенсивность боли, моторных или сенсорных расстройств варьирует. Обязательным является признак Лаже: пациент в положении лежа поднимает прямые ноги, что вызывает натяжение корешков и сопровождается болью. Диагноз корешковых расстройств устанавливают клинически, затем выполняют рентгеновское исследование в 2 плоскостях и только при несоответствии интенсивности боли и характера моторных и сенсорных расстройств проводят КТ, ЯМР или при наличии вопросов — миелографию.
Нарушение кровоснабжения спинного мозга редки, но проявляются патогномонично. Расстояние, которое может пройти больной, ограничивается болью в поясничном отделе позвоночника и нарастающей слабостью в ногах. После отдыха, особенно в положении сидя или при наклоне вперед, боль в спине проходит, сила мышц восстанавливается. Эта симптоматика известна как claudicatio spinalis и обусловлена преспинальными причинами. Чаще это аневризма аорты, перекрывающая артерию, питающую спинной мозг. Ангиомы спинного мозга неинвазивно выявляют только методом ЯМР. Поражение периферических нервов этой области проявляется как парестетическая мералгия и обусловлено сдавлением n. cutaneus femoris lateralis. На латеральной поверхности бедра в верхней половине появляется область нарушенной чувствительности размером с ладонь с жгучей болью.
Экстрапирамидная система выполняет моторные функции и обособлена от пирамидной системы: двигательных зон коры и кортикоспинальных путей. Она имеет связи с корой и проекции на лобные участки коры. Поэтому экстрапирамидную систему можно обследовать при условии бодрствования пациента и интактности кортикоспинального пути. Важнейшие симптомы поражения экстрапирамидной системы — ригидность, тремор и акинезия.
Акинезия: лицо пациента застывшее, которое можно принять за выражение подавленности. Каждое движение замедлено, отсутствуют движения рук при ходьбе, изменены начало ходьбы, ее прекращение, изменение направления движения требует времени и усилий.
Дискинезия или гиперкинезия — постоянное беспорядочное движение мимических мышц, мышц туловища, конечностей, усиливающиеся за счет того, что пациент пытается совершать противоположные движения для сохранения равновесия, или сдержать непроизвольные движения, контролируя их. Эти движения являются первым симптомом болезни Геттингтона, развиваются при передозировке допаминергических препаратов при болезни Паркинсона, как осложнение лечения нейролептиками или антигистаминными препаратами. В большинстве случаев дискинезия является результатом поражения базальных ганглиев.
Тик — быстрые координированные движения, отличающиеся от хорей своими повторениями. Если тик развивается в отдельных группах мимических мышц при стрессах, то их сложно отнести к патологическим реакциям, скорее это невротические реакции. Если тик сочетается с непроизвольными вскрикиваниями, то это может быть ранним признаком синдроме Жиля де ла Туретта. Синдром передается с различной пенетрантностью. Чаще выявляют у мужчин (3:1). Вокальный тик начинается как хрюкающие, придушенные звуки. Позднее они перерастают в непроизвольно выкрикиваемые слова. У 50% пациентов это проявляется копролалией (импульсивное произнесение бранных и нецензурних слов), которая в сочетании с тиком приводит к социальной дезадаптации и требует ДД с шизофренией. Тик представляет собой более сложные движения, чем миоклония, но значительно менее плавные, чем хорея.
Дистония — чаще локальные, преимущественно безболезненные судороги, например блефароспазм, писчий спазм. Редкое состояние — генерализованная дистония. Передается а/д с частичной пенетрантностью. Ген расположен на 9-й хромосоме. Болезнь начинается в детстве с инверсии и плантарной фиксации стопы во время ходьбы. В тяжелых случаях болезнь прогрессирует и больные буквально «скручиваются» в гротескных позах. Умственное развитие обычно не нарушено.
Ригидность — ощущение сопротивления при пассивном движении, если это сопротивление периодически на короткое время прерывается, то говорят о симптоме «зубчатого колеса».
Тремор — ритмичные осцилляторные движения в результате повторяющихся сокращений и расслаблений мышц. Тремор классифицируют по частоте (медленный — 3–5 Гц; быстрый — 6–12 Гц), амплитуде, ритму, времени возникновения (тремор покоя; во время мышечной активности). Причины тремора разнообразны.
Физиологический тремор переживали практически все: быстрое дрожание вытянутых пальцев. Типичен для тревоги, страха, утомления, гипогликемии, после употребления алкоголя, больших количеств кофе (кофеин, блокаторы фосфодиэстеразы). Близок к физиологическому тремору на фоне приема кортикостероидов.
Доброкачественный наследственный (эссенциальный) тремор обычно затрагивает кисти, голову и голос. В ½ случаев доказан а/д тип передачи. Тремор может быть односторонним. Он уменьшается или даже полностью прекращается в покое, усиливается при тех же условиях, что и физиологический. С возрастом усиливается, становится заметнее и ошибочно называется сенильным.
Тремор покоя мелкоамплитудный, частотой 3–5 движений в секунду, при движениях уменьшается.
Типичным для болезни Паркинсона являются движения большого пальца против указательного: «крутильщик пилюль». Болезнь Паркинсона — идиопатическое медленно прогрессирующее дегенеративное заболевание ЦНС, характеризующееся тремором покоя, медленными, оскудевающими в динамике процесса движениями, мышечной ригидностью и постуральной нестабильностью. Болезнь может начинаться у детей и подростков (ювенильный паркинсонизм), но средний возраст дебюта — 57 лет и старше. Среди людей в возрасте 40 лет эту форму нейродегенеративной болезни выявляют у 0,4% популяции, а среди лиц в возрасте 65 лет и старше — уже около 1%. В 60–80% случаев болезнь Паркинсона начинается с тремора покоя (4–8 Гц) типа «крутильщика пилюль» сначала в пальцах одной кисти. Тремор максимален в состоянии покоя, уменьшается в движении и полностью исчезает во сне, выражен в кистях, меньше — в руках, еще меньше — в ногах. По мере прогрессирования болезни, нарастании ригидности, гипо- и акинезии тремор становится менее выражен. Практически у всех больных — депрессия, а у 50% развивается деменция. У многих пациентов тремор отсутствует, в клинической картине доминирует синдром ригидности.
ДД может проводиться с эссенциальным тремором. Но эссенциальный (доброкачественный наследственный) тремор (см. выше) — это тремор движений, при неизмененной походке, а тремор при болезни Паркинсона — прежде всего тремор покоя. Значительно труднее отличить от болезни Паркинсона старческие состояния с обедненностью движений, тремором, мелкой (ревматической) походкой, умеренными депрессией и деменцией. Помогают анамнез и сведения о «естественном течении болезни».
Интенционный тремор типичен для заболеваний мозжечка, мелкий тремор вытянутых рук и пальцев характерен для тиреотоксикоза и алкоголизма, более размашистый — при заболеваниях печени, достигая амплитуды хлопающих крыльев при болезни Вильсона — Коновалова. Эти медленные неритмичные грубые движения («печеночные хлопки») вытянутых рук пациента типичны для печеночной или другой тяжелой обменной энцефалопатии. По ЗМГ-проявлениям они больше миоклонии (негативный миоклонус), а не собственно тики.
Хорея — быстрые неповторяющиеся движения, которые пациент часто стремится включить в осмысленно контролируемые. Хорея может быть изолированным симптомом (сенильная хорея), проявлением болезни Геттингтона, а у лиц молодого возраста — ревматического васкулита мозга (хорея Сиденгама).
Атетоз — медленные червеобразные движения конечностей, могут развиваться сочетанно с хореей и называются хореоатетоз.
Баллизм — движения конечностей, имитирующие разбрасывание предметов, чаще это одностороннее состояние (гемибаллизм) и развивается при повреждениях (кровоизлияниях) субталамического ядра.
Миоклония — короткие вспышкообразные сокращения мышцы или группы мышц. Миоклония в норме иногда возникает у некоторых лиц в процессе засыпания или в неглубоком сне (ночная миоклония). В сущности, икота — это миоклонические сокращения диафрагмы. Причинами патологической миоклонии могут быть тяжелые метаболические нарушения (уремия), дегенеративные заболевания нервной системы (болезнь Альцгеймера), медленные вирусные инфекции (подострый склерозирующий энцефалит и др.), тяжелые закрытые травмы мозга, его гипоксическо-ишемические повреждения.
ДД гиперкинезов сведена в табл. 11.6.
Таблица 11.6
ДД гиперкинезов
| Симптом | Характеристика | Вероятная причина | Как подтвердить |
| Гиперкинезы при нормальных движениях | Психомоторное беспокойство | Минимальная мозговая дисфункция, внутричерепная гипертензия | Психоневрологический статус |
| Фасцикуляция, фибрилляция | Непроизвольные сокращения отдельных мышечных групп, чаще лучше диагностируемые пальпаторно, чем визуально | Спинальные миоатрофии, поражение передних рогов спинного мозга | ЗМГ, биопсия мышцы |
| Миоклония | Повторные непроизвольные сокращения отдельных мышечных волокон или групп | Эквивалент судорог | ЭЭГ |
| Миоклония | Очень короткие подергивания | Пирамидальные расстройства | ЭМГ, очень короткие всплески мышечных потенциалов (<250 мс) |
| Миоклония | Мышечные подергивания длительностью до 3 с | Экстрапирамидние расстройства, подострый склерозирующий панэнцефалит, реже — наследственная энцефалопатия | ЭЭГ (отсутствует гиперсинхронная активность при неэпилептогенной миоклонии) |
| Миоклония | Односторонние или двусторонние, чаще синхронные и аритмичные с низкой частотой (20–130 мин) | Спинальные, вызванные соматосенсорными раздражителями | ЭМГ, соматосенсорные вызванные потенциалы |
| Миоклония | Миоклония, вызываемая положением тела, движением или намерением движения | Моторно индуцированные, резидуальный симптом при повреждениях мозга, дегенеративных и других редких заболеваниях мозга | Анамнез, ЭМГ, ЭЭГ |
| Миоритмия | Регулярные ритмичные синхронные подергивания мышц, особенно неба и лица, сохраняющиеся во сне | Опухоли ствола мозга, мозжечка, энцефалиты, травмы, дегенеративные заболевания | КТ, ЭЭГ |
| Хорея | Быстрые, взрывчатые, беспорядочные бесцельные непроизвольные движения меняющейся локализации | Атрофия, дегенерация полосатого тела, энцефалит, интоксикация, болезнь Вильсона | Исходя из анамнеза и общей клинической картины |
| Атетоз | Медленные, судорогообразные движения преимущественно в дистальных отделах с вычурным раздвиганием пальцев, причудливые движения в суставах, прекращаются во сне и усиливаются при аффектах и движениях | Дегенеративные заболевания мозга, поражения ствола мозга, травма, асфиксия, ядерная желтуха | ЭЭГ, далее — по анамнезу |
| Баллизм/гемибаллизм | Молниеносные движения конечностей по типу разбрасывания предметов | Энцефалит, травма, опухоль, туберкулез, менингит | КТ, далее — по анамнезу и клинической картине |
| Тик | Стереотипные короткие движения по типу морганий, поперхивания, поражаются преимущественно мышцы лица, шеи, глотки, рук | Эндогенные, экзогенные, психические нарушения, наследственные и приобретенные поражения мозга, нейролептики | По анамнезу |
| Дистония | Непроизвольные движения с атипичным положением, быстро меняющимся тонусом, контрактурами | Энцефалиты, системные неврологические заболевания, ядерная желтуха, асфиксия | ЭЭГ, ЭМГ, КТ, ЯМР, биохимический скрининг |
Прогрессирующий супрануклеарный парез (синдром Ричардсона — Стила — Ольшевского) — редкое заболевание, манифестирующее с потери возможности совершения произвольных, но не рефлекторных движений глаз, брадикинезии, мышечной ригидности с дистонией, псевдобульным синдромом (дисфалия, дизартрия с эмоциональной лабильностью), деменцией. Характеризуется паркинсоноподобной картиной, дополняющейся парезом взгляда вертикально вниз. Широко раскрытые глаза, редкие мигания, почти отсутствующие быстрые движения глаз придают пациенту специфический вид. Голова обычно находится в положении усиленного разгибания. На поздних этапах поражаются кортикоспинальные пути.
Болезнь Вильсона — Коновалова — гепатоцеребеллярная дегенерация, манифестирует обычно в подростковом возрасте, проявляется грубым высокоамплитудным тремором, дизартрией, широко открытым ртом, прогрессирующим снижением интеллекта, гепатомегалией, зеленоватым кольцом Кайзера — Флейшнера (рис. 11.1). Обусловлена нарушением обмена меди с ее депозитами во внутренних органах.
Блефароспазм — непроизвольное зажмуривание глаз. Закрывание глаз при мигании происходит нормально, но открывание доставляет пациентам массу сложностей. В современных условиях проводят инъекции ботулинистического токсина в гиперактивную мышцу.
Орофациальная дискинезия — гримасоподобные медленные движения мимических мышц (впервые описаны Мейгелем, известны и как синдром под его именем), описываются как синдром Брейгеля, по имени художника, охотно изображавшего на своих картинах кривляющиеся рожи.
Болезнь Геттингтона — симптомокомплекс хорей, атетоза и деменции — наследственно обусловленное заболевание, связанное с 4-й хромосомой. Спорадические случаи не известны. Семейный анамнез всегда положителен. Характеризуется атрофией хвостатого ядра, что хорошо выявляется при КТ и ЯМР.
Атаксии и другие мозжечковые нарушения. Мозжечок связан с сенсорно-моторными отделами коры и через спиноцеребеллярные связи проецирует сигналы в моторные зоны коры. То есть для выявления мозжечковых нарушений необходимо бодрствование пациента и сохранность кортикоспинальных путей. При гемиплегии выявить мозжечковые нарушения на пораженной стороне невозможно. Мозжечок координирует временной синергизм мышц. Некоторые симптомы мозжечковой недостаточности, такие как атаксия, могут возникнуть при нарушениях премоторных зон коры или изменениях в спинном мозгу. Атаксия — неспособность пациента совершать обычные координированные движения, особенно при отсутствии визуального контроля (проба Ромберга). Интенционный тремор — при произвольном движении оно начинается гладко, но с приближением к цели становится все более размашистым. Дисметрия — при быстрых движениях амплитуда их или повышена (гиперметрия) или снижена (гипометрия). Диадохокинез — способность совершать быстрые сменяющие друг друга движения агонистических и антагонистических мышц. В мозжечке представительство конечностей располагается с той же стороны (ипсилатерально), а туловища и глаз — в черве. Поэтому односторонняя атаксия указывает на ипси(гомо)латеральное поражение. Двусторонние поражения типичны для дегенеративных, токсических и воспалительно-паранеопластических состояний.
Наследственная атаксия Фридрайха характеризуется атаксией, снижением глубокой чувствительности, полой стопой, патологическим нистагмом и дизартрией. Обусловлена поражением афферентных мозжечковых путей. Причиной церебеллярной кортикальной атаксии является дегенерация клеток Пуркинье. Пациентов беспокоят медленно нарастающая атаксия и неуверенность походки, особенно в темноте. При ЯМР определяется атрофия коры мозжечка. Оливо-понто-церебеллярная атрофия развивается обычно в преклонном возрасте. Клиническая картина характеризуется сочетанием симптомов нарушения мозжечковых функций, проводимости по кортикоспинальным путям и деменции.
Спорадически возникающая алкогольная атаксия связана с дегенерацией червя. Паранеопластическая дегенерация развивается при опухолях женских половых органов, раке бронхов и лимфомах. В каждом случае мозжечковых расстройств без признаков воспаления или наследственных синдромов следует помнить о паранеопластических реакциях. Мозжечковая атрофия развивается у пациентов, длительное время принимавших противоэпилептический препарат фенитоин. Фокальные выпадения мозжечковых функций отмечают при метастазах опухолей, абсцессах, множественном склерозе, инсультах.
Демиелинизирующие заболевания нервной системы, хронические инфекции ЦНС, в том числе прионные, первичные и вторичные метаболические нарушения ЦНС, сосудистые и объемные процессы ведут к так называемой деменции.
Деменция — хроническое нарушение интеллектуальных, когнитивных функций с неспособностью человека обеспечить собственное выживание в стандартних бытовых условиях. Деменция может возникнуть в любом возрасте, в том числе у детей при гипоксии мозга. Но обычно это состояние лиц преклонного возраста и поражает более 15% людей старше 65 лет и более 40% — старше 80 лет.
У стариков необходимо отделять деменцию от возрастных изменений памяти. В последнем случае имеются сложности в вызывании ранее полученной информации и заучивании новой. Но в спокойной обстановке, при устранении тревоги и страхов, предоставлении дополнительного времени старики справляются с заданием и их интеллектуальные способности соответствуют полученному образованию и культурному уровню.
Диагностическими критериями деменции являются:
Развитие множественных когнитивных дефицитов:
- Дефицит памяти (затруднения в использовании уже заученной информации или запоминании новой).
- Одно (или более) из нижеследующих нарушений:
- афазия;
- апраксия;
- агнозия;
- нарушение способности планировать, организовывать, выделять приоритеты, абстрагировать.
Каждый из этих признаков возникает постепенно и развивается в динамике, принимается во внимание существенное отклонение от ранее свойственного уровня.
Для деменции при болезни Альцгеймера необходимо исключить все иные состояния, приводящие к нарушению памяти и когнитивных функций (цереброваскулярные заболевания, болезнь Паркинсона, хорею Геттингтона, субдуральную гематому, хронические инфекции ЦНС, опухоли мозга, обменные нарушения и др.).
Для диагноза деменции сосудистого генеза необходимо выявить очаговую неврологическую симптоматику (расстройство глубоких сухожильных рефлексов, псевдобульбарный паралич, нарушения походки, слабость мышц конечностей и др.), лабораторные и инструментальные подтверждения сосудистых расстройств (множественные инфаркты коры и белого вещества мозга).
Для диагноза деменции при других патологических состояниях необходимы анамнестические, физикальные, лабораторные и инструментальные подтверждения инфекционных, обменных и иных заболеваний (опухоль мозга, вирусные и прионные энцефалиты, гидроцефалия с нормальным внутричерепным давлением, ионизирующее облучение и т. д.). ДД при деменции важна для проведения оптимальных лечебных мероприятий, сохраняющих интактные области мозга.
Некоторые авторы в зависимости от причин деменции склонны выделять необратимую и обратимую деменцию. Но, как нам видится, такая градация стирает грани между деменцией и делирием.
Наиболее признанное выделение различных форм деменции — это выделение деменции по типу болезни Альцгеймера и деменции неальцгеймеровского типа.
Болезнь Альцгеймера ответственна за 68–74% всех случаев деменции у лиц старческого возраста. В 15% случаев альцгеймеровская деменция сочетается с сосудистой. Болезнь Альцгеймера, как правило, начинается в возрасте старше 60 лет, и частота ее нарастает с возрастом. Раннее начало отмечают в 2–6% случаев. Семейных случаев — 16–22%. Гены, ответственные за начало и развитие болезни Альцгеймера, расположены на 1, 14, 19 и 21-й хромосомах. 21-я хромосома ответственна за синтез протеина — предшественника амилоидного белка, откладываюшегося в мозгу людей с болезнью Альцгеймера (хотя этот же белок откладывается и при других состояниях). На 19-й хромосоме синтезируется аполипопротеин Е. Различные аллели и их комбинации могут повышать или понижать риск развития болезни Альцгеймера*. Патогенетическое значение этих факторов изучено не до конца. Например, является ли отложение бета-амилоида причиной болезни или биологической реакцией и вторичным феноменом? Все сведения о возможной провоцирующей роли снижения гормонов, интоксикации тяжелыми металлами, условий жизни, окружающей среды в независимых исследованиях не проверены.
Клинически вначале отмечается бытовая забывчивость, затруднения при подборе слов, визуальной идентификации знакомых, нарушения абстрактного мышления. Пациенты становятся раздражительными. Позднее им требуется помощь в еде, умывании, одевании, посещении туалета. Теряется ориентация в пространстве и времени. Болезнь неуклонно прогрессирует, хотя иногда отличаются своеобразные «плато». В конце концов развивается кома и больные умирают от инфекционных осложнений.
Лабораторно можно определить апо-Е-статус пациента. Но этот генетический тест не позволяет прогнозировать вероятность развития болезни Альцгеймера (у 40% лиц в возрасте 85 лет отмечают те или иные признаки деменции, связанные с апо-Е-протеином). Содержание некоторых белков в мозгу резко повышается, и они появляются в спинномозговой жидкости. Причинная роль этих субстанций не установлена, но их можно использовать как маркеры. Тау-протеин, происходящий из нейрофибрилл, высокоспецифичен, но чувствительность теста низка. Очень незначительно отличающиеся от этого варианта тау-протеины выявляют при прогрессирующем супрануклеарном параличе (см. выше). Значительно снижена активность холинацетил-трансферазы, а также содержание соматостатина, кортикотропин-рилизинг-фактора, других нейротрансмиттеров.
Неальцгеймеровские варианты деменции развиваются в исходе бактериальных эндокардитов (по механизму эмболий), туберкулезного и грибкового менингита, нейросифилиса (прогрессирующий паралич), медленных вирусных инфекций, прионных болезней.
Деменцией завершаются алкоголизм (в сочетании co свойственным ему дефицитом питательных веществ), аноксия, пеллагра, дефицит витамина В12 и, вероятно, фолиевой кислоты, гиперпаратиреоидизм с гиперкальциемией, гипотиреоидизм, гипогликемия, метаболические энцефалопатии (печеночная, уремическая, респираторная).
Другие причини деменции:
- болезнь Паркинсона;
- болезнь Вильсона — Коновалова;
- рассеянный склероз;
- опухоли мозга;
- хорея Геттингтона;
- мозжечковые дегенерации;
- облучение мозга (особенно лобных долей);
- травмы мозга, последствия оперативных вмешательств;
- деменция боксеров (последствия постоянных травм у профессиональных спортсменов). Генетически связана с 4 аллелей апо-Е;
- субдуральная хроническая гематома;
- гидроцефалия;
- сосудистые нарушения. Чаще у мужчин в возрасте старше 70 лет. Осложняющие моменты: АГ, сахарный диабет, табакокурение. Особенностью деменции является этапность прогрессирования с одновременным присоединением новой очаговой симптоматики (новые инфаркты мозга);
- деменция Бинсвангера (субкортикальная артериосклеротическая энцефалопатия). Диагностируют нечасто. Множественные инфаркты в глубоких отделах полушарий мозга в сочетании с тяжелой АГ и системными сосудистыми процессами. Клинически представляет собой вариант деменции сосудистого генеза, но выявляют у лиц молодого и зрелого возраста и имеет более тяжелое течение.
Делирий — состояние с варьирующим в динамике нарушением когнитивных функций, сменой настроения, внимания, нарушением ритма сна/бодрствования, самосохранения. Делирий возникает остро при неизмененной структуре мозга. Но чаще — при условии предшествующих изменений мозга, обычно — у людей пожилого и старческого возраста. Существенное значение имеет нарушение медиаторов (нейротрансмиттеров), разрушение клеток мозга и сопутствующие заболевания. Делирий вызывают алкоголь, антигиперзивные препараты, бензодиазепины, циметидин, дигоксин, наркотики, противорвотные, спазмолитики, миорелаксанты, паркинсонические средства, трициклические антидепрессанты, аноксия, гиперкалиемия, гиперпаратиреоидизм, тиреотоксикоз, гипотиреоз, гипогликемия, гипокалиемия, метаболический ацидоз, состояния после сотрясения, ушиба мозга, тяжелая желтуха, ишемия мозга.
Диагноз «делирия» следует верифицировать как можно скорее, поскольку это состояние прогностически очень серьезное. Среди людей пожилого возраста, доставленных с делирием в стационар, 20% погибают, а период пребывания на койке выживших в 2 раза больше, чем у лиц без делирия.
Критериями делирия являются:
- нарушения восприятия окружающего мира co снижением способности концентрации и фиксации внимания;
- дефицит памяти, дезориентация, нарушения речи;
- нарушения развиваются за короткий период (часы и дни).
Клинически для делирия характерна быстрая динамика симптомов, нарушение ориентации в пространстве и времени, самоидентификации. Внимание рассредоточено. Нередки зрительные галлюцинации. Некоторые больные возбуждены, другие могут быть апатичны, заторможены, в течение очень короткого времени демонстрировать противоположные эмоции. У ряда больных развиваются судороги.
Делирий с апатией следует отличать от депрессии, особенно у лиц старческого возраста. Ажиотация и галлюцинации при делириуме требуют исключения психозов. Но анамнестические сведения о маниакальных состояниях и шизофрении, исключение причин делирия (см. выше) позволяют установить правильный диагноз.
*Более того, у лиц разных рас апо-Е-протеин, вероятно, выполняет разную патогенетическую роль. Аллель 2 у лиц европеоидной расы снижает риск развития болезни Альцгеймера, а у лиц негроидной расы — повышает его.