На сегодня установлено, что кардиоваскулярные заболевания имеют мультифакториальную этиологическую природу, ассоциированную с воздействием многочисленных факторов риска (Brunzell J.D. et al., 2008). Среди этих факторов гиперлипопротеидемии занимают одно из важнейших мест (Peyser P.A., 1997). Появление доказательств, касающихся наличия прямой взаимосвязи между плазменным уровнем общего ХС, ХС ЛПНП и ЛПВП с величиной общей и кардиоваскулярной смертности в популяции привело к появлению программ профилактики возникновения неблагоприятных клинических исходов (Cui Y. et al., 2001; Alexander C.M. et al., 2003). При этом наибольшее внимание уделялось вопросам диетических ограничений (Hegsted D.M. et al., 1993; Cobb M.M., Teitlebaum H., 1994; Lichtenstein A.H. et al., 2006), расширения физической активности (Leon A.S., Sanchez O.A., 2001) и медикаментозным методам лечения гипердислипидемий (Jacobs D.R. et al., 1983; McNamara D.J. et al., 1987; Schaefer E.J. et al., 1997).
В последующем оказалось, что среди больных можно идентифицировать пациентов с достаточно высоким и неадекватным ответом на проводимое лечение (hyper- and hyporesponders) (Katan M.B. et al., 1986; Beynen A.C. et al., 1987). Более того, по данным обсервационных исследований, плазменное содержание ХС более чем в половине случаев непосредственно обусловлено именно генетическими причинами (Genest J.J. Jr. et al., 1992), а не особенностями питания или сопутствующими заболеваниями (Jacobs D.R. et al., 1983; McNamara D.J. et al., 1987).
Все это поддерживало представление о существовании генетической предрасположенности к формированию гиперлипопротеидемии. Действительно, развитие последней может быть обусловлено не только сопутствующими заболеваниями, такими как сахарный диабет, ожирение, нефропатия, гипотиреоз (вторичные гиперлипопротеидемии), но и генетическими причинами (первичные гиперлипопротеидемии) (Brunzell J.D., Deeb S.S., 2000). Причем фенотипические проявления подобных нарушений тесно ассоциированы с воздействиями факторов внешней среды (Brunzell J.D., Failor R.A., 2006).
В соответствии с NCEP (The National Cholesterol Education Program) ATP (Adult Treatment Panel) III пересмотра гиперлипидемия рассматривается как состояние, при котором плазменные концентрации основных компонентов липидного спектра выходят за пределы установленного интервала референсных значений, а именно: для общего ХС — >240 мг/дл (6,21 ммоль/л), для ХС ЛПНП — >130 мг/дл (3,36 ммоль/л), для ТГ — >200 мг/дл (2,26 ммоль/л) и для ХС ЛПВП — <35 мг/дл (0,91 ммоль/л).
Существуют различные классификационные подходы к систематизации гиперлипидемий. Одним из наиболее популярных является ставший уже классическим классификационный подход Фредриксона, базирующийся на биохимических фенотипических признаках гиперлипидемий (табл. 6.1). Последний рекомендован ВОЗ, однако при его клиническом применении возникает ряд трудностей в основном онтодологического характера, в связи с чем классификацию Фредриксона чаще всего используют в научных исследованиях. Вместе с тем, подход Фредриксона позволяет оценить потенциальную атерогенность гиперлипидемии независимо от ее этиологии.
Таблица 6.1 Биохимическая фенотипическая классификация гиперлипидемий по Фредриксону, рекомендованная ВОЗ
| Тип гиперлипидемии | Содержание общего ХС | Содержание ТГ | Характеристики сыворотки | Клиническое значение |
| I | Нормальное | ↑↑ | Кремовая, хиломикроны | Дефицит липопротеинлипазы, дефицит апо-С2-липопротеина (тип Ib первичной гиперлипидемии) |
| IIa | ↑↑ | Нормальное | Прозрачная, ЛПНП | СГХ, СКГЛ |
| IIb | ↑↑ | ↑ | Прозрачная, ЛПНП и ЛПОНП | СКГЛ |
| III | ↑ | ↑ | Мутная, ЛППП | Дисбеталипопротеидемия |
| IV | ↑ или нормальное | ↑↑ | Мутная, ЛПОНП | Семейная гипертриглицеридемия, СКГЛ |
| V | ↑ | ↑↑ | Кремовая с мутным осадком, хиломикроны, ЛПОНП | Мутация гена APOA5, приводящая к исключению A-V-аполипопротеина |
СГХ — семейная гиперхолестеринемия, СКГЛ — семейная комбинированная гиперлипидемия.
Кроме того, гиперлипидемии классифицируют в зависимости от биохимической структуры липидов, основных видов патофизиологических нарушений, приводящих к их возникновению (табл. 6.2), а также особенностей программ первичной и вторичной профилактики (табл. 6.3).
Первичные гиперлипидемии
Таблица 6.2 Этиология основных форм гиперлипопротеидемий
| Тип гиперлипидемии по Фредриксону | Первичные причины | Вторичные причины |
| I | Дефицит липопротеинлипазы | Системная красная волчанка |
| Дефицит апо-С-II | ||
| IIa | Семейная гиперхолестеринемия | Гипотиреоз |
| IIb | Семейная комбинированная гиперлипидемия | Нефротический синдром, сахарный диабет, нервная анорексия |
| III | Тип III семейной гиперлипопротеидемии | Гипотиреоз, сахарный диабет, абдоминальное ожирение |
| IV | Семейная комбинированная гиперлипидемия | Хроническая болезнь почек, сахарный диабет, употребление алкоголя |
| Семейная гипертриглицеридемия | ||
| V | Семейная гипертриглицеридемия | Диуретики, блокаторы β- адренорецепторов, пероральные контрацептивы |
| Дефицит апо-С-II |
Таблица 6.3 Характеристика основных типов гиперлипидемий
| Характер гиперлипидемии | Вид гиперлипидемии | Фенотипические проявления и их клиническое значение | Диагностические критерии |
| Первичная гиперлипидемия | |||
| С высоким уровнем ХС и нормальным содержанием ТГ | Полигенная гиперхолестеринемия | Полигенный тип наследования, истинная частота неизвестна, отмечают как во взрослой, так и в детской популяциях | Диагноз устанавливается на основании клинических признаков, семейного анамнеза и повышения общего ХС >7,5 ммоль/л у взрослых и >6,7 ммоль/л у детей до 16 лет |
| Моногенная семейная гиперхолистеринемия | Верифицировано более 400 видов мутаций рецептора к ЛПНП
Гетерозиготы (распространенность 1:500) с 5% риском инфаркта миокарда в возрасте <60 лет |
Общий ХС >7,5 ммоль/л у взрослых и >6,7 ммоль/л у детей до 16 лет
ХС ЛПНП >4,9 ммоль/л у взрослых. Ксантомы сухожилий у пробанда и родственников 1-й и 2-й степени родства |
|
| Семейный дефицит апо-В | Клинически идентичен гетерозиготной форме моногенной семейной гиперхолестеринемии | Общий ХС >7,5 ммоль/л у взрослых и >6,7 ммоль/л у детей до 16 лет
ХС ЛПНП >4,9 ммоль/л у взрослых |
|
| С высоким содержанием ХС и ТГ | Семейная комбинированная гиперлипидемия | 1:100–200 в популяции; повышение содержания частиц ЛПОНП; фенотипические проявления варьируют в зависимости от соотношения содержания общего ХС и ТГ, характерен ксантоматоз, случаи раннего возникновения ИБС в семье, часто отмечают различные факторы кардиоваскулярного риска: ожирение, сниженную физическую активность | Общий ХС >8 ммоль/л, ТГ >5 ммоль/л |
| Ремнантная гиперлипидемия | 1:10 000 населения; апо-Е2/Е2-генотип, содержание ЛППП повышено, характерен пальмарный ксантоматоз, отмечают различные факторы кардиоваскулярного риска, в частности сахарный диабет | Общий ХС >11 ммоль/л, ТГ >13 ммоль/л | |
| Тяжелая гипертриглицеридемия | Врожденный дефицит липопротеинлипазы или апо-С2 (известен как тип Ib первичной гиперлипидемии), ассоциирована с высоким риском панкреатита, риск ИБС не повышен
Функциональная недостаточность липопротеинлипазы, связанная с избыточной продукцией ЛПОНП (тип V гиперлипопротеидемии), распространенность 1:10 000 населения, ассоциируется с различными вторичными причинами возникновения панкреатита, особенно при уровне ТГ >20 ммоль/л |
Общий ХС >15 ммоль/л, ТГ >60 ммоль/л | |
| С высоким содержанием ХС и ТГ | Тяжелая хиломикронемия с/без повышения ЛПОНП | Клинически идентична тяжелой гипертриглицеридемии | Общий ХС >15 ммоль/л, ТГ >60 ммоль/л |
| С нормальным содержанием ХС и высоким уровнем ТГ | Умеренная гипертриглицеридемия | Ассоциированная с инсулинорезистентностью: часто — с подагрой, сахарным диабетом, ожирением, метаболическим синдромом, нарушением содержания глюкозы натощак, сниженной физической активностью
Семейная гипертриглицеридемия Гипергликемия Высокое содержание липидов в пище |
Нормальный (<5,2 ммоль/л) или пограничный (5,2–6,2 ммоль/л) уровень общего ХС, ХС ЛПВП <1,0 ммоль/л, ТГ >5 ммоль/л |
К настоящему времени описано достаточно большое количество наследственных форм нарушений липидного обмена в виде гиперлипопротеидемий, среди которых наиболее распространенными являются семейная дисбеталипопротеидемия, семейная гиперхиломикронемия, семейная гиперхолестеринемия, полигенная гиперхолестеринемия, семейная комбинированная гиперлипидемия, семейная гипертриглицеридемия (Brunzell J.D. et al., 1983; Brunzell J.D., Failor R.A., 2006). В то же время, точный биохимический дефект, лежащий в основе их манифестации, идентифицирован только для первых двух (Goldstein J.L. et al., 1973; Brunzell J.D., Failor R.A., 2006). Более того, до сих пор не существует согласованного мнения о критериях диагностики большинства наследственных форм гиперлипопротеидемий (Ceveira F., 2004; Brunzell J.D., Failor R.A., 2006; Brunzell J.D. et al., 2008).
Первичная гипертриглицеридемия
Согласно NCEP ATP III гипертриглицеридемия определяется как повышение плазменной концентрации ТГ >150 мг/дл (National Cholesterol Education Program, 2001). При использовании этого критерия в NHANES (The Third National Health and Nutrition Examination Survey) выявлено, что в США избыточный уровень ТГ отмечают у >35% мужчин и 25% женщин в возрасте старше 20 лет. Традиционно гипертриглицеридемии разделяют на первичные (ассоциированные с генетическими причинами) и вторичные (связанные с другими причинами) (табл. 6.4). И хотя этот подход довольно условен, подобное деление прижилось и широко используется. Так, элевация плазменного пула ТГ может быть ассоциирована с рядом генетических дефектов и рассматривается в рамках следующих патологических состояний: семейной комбинированной гиперлипидемии с аутосомно-доминантным типом наследования, резидуальной гиперлипидемии у пациентов с контролируемым сахарным диабетом 2-го типа, семейной гипоальфалипопротеидемией, моногенной наследственной гипертриглицеридемией. Первые три типа нарушений липидного обмена верифицируются при метаболическом синдроме и выступают в качестве непосредственной причины возникновения ИБС (Carr M.C., Brunzell J.D., 2004). Хотя подобные семейно-ассоциированные гиперлипидемии составляют всего 1% среди популяции, а резидуальную гиперлипидемию при сахарном диабете 2-го типа отмечают почти в 5 раз чаще (Hokanson J.E., Austin M.A., 1996; Hopkins P.N. et al., 2003; Watanabe H. et al., 2006), именно с ними связывают почти 50% всех случаев ИБС в когорте пациентов с метаболическими нарушениями (Zambon A. et al., 2006). Необходимо отметить, что риск развития ИБС у пациентов с метаболическим синдромом или документированным сахарным диабетом значительно выше, чем в популяции. Вместе с тем, точно не установлено, какую роль в этом играют семейно-ассоциированные нарушения обмена ТГ (Alexander C.M. et al., 2003; Després J.P., Lemieux I., 2006). Во всяком случае, если вклад инсулинорезистентности, повышение уровня эндогенного инсулина и гипергликемии, а также других дефинизирующих компонентов метаболического синдрома на клинический исход достаточно четко определен (Manninen V. et al., 1992), то изолированное влияние гипертриглицеридемии на прогноз вне влияния сопутствующих нарушений липидного обмена (таких как снижение пула апо-А, увеличение уровня ХС ЛПНП, ЛПОНП, апо-В) оценить очень сложно (Lamarche B. et al., 1997; Lemieux I. et al., 2000; Jacobson T.A. et al., 2007; Nordestgaard B.G. et al., 2007). С другой стороны, распространенность изолированных наследственных гипертриглицеридемий, включающих семейную гипертриглицеридемию с аутосомно-доминантным типом наследования и моногенную наследственную гипертриглицеридемию, не превышает 1% в популяции. В то же время, роль последней как фактора риска ИБС не установлена (Brunzell J.D. et al., 1983; Austin M.A., 1989). Исследователи полагают, что моногенная наследственная гипертриглицеридемия не требует медикаментозного лечения в отличие от семейной гипертриглицеридемии с аутосомно-доминантным типом наследования, после верификации которой должна проводиться агрессивная гиполипидемическая терапия (Austin M.A. et al., 2000; Zambon A. et al., 2006; Jacobson T.A. et al., 2007).
Таблица 6.4 Основные причины формирования гипертриглицеридемии
| Причины гипертриглицеридемии | Типы нарушений | Характеристика | Заболевания, при которых чаще всего отмечают нарушение обмена ТГ |
| Генетические причины | I тип гиперлипопротеидемии | Дефицит липопротеинлипазы, а также ее кофактора или апо-С-II с повышением ТГ, ХС ЛПНП и ЛПОНП, нарушением обмена хиломикронов | Сахарный диабет 1-го и 2-го типа |
| Семейная комбинированная гиперлипидемия с аутосомно-доминантным типом наследования | Ассоциированная с элевацией ТГ и (или) ХС ЛПНП | ИБС с семейно-отягощенным анамнезом | |
| Семейная гипертриглицеридемия с аутосомно-доминантным типом наследования | Изолированная гипертриглицеридемия | ИБС с семейно-отягощенным анамнезом | |
| Семейная гипоальфалипопротеидемия | Снижение плазменной концентрации апо-А, ХС ЛПВП и увеличение ТГ | ИБС с семейно-отягощенным анамнезом | |
| Моногенная наследственная гипертриглицеридемия | Изолированная гипертриглицеридемия | Низкий риск развития ИБС | |
| Резидуальная гиперлипидемия у пациентов с контролируемым сахарным диабетом 2-го типа | Снижение плазменной концентрации апо-А, ХС ЛПВП и увеличение апо-В100 и ТГ | Сахарный диабет 2-го типа | |
| Метаболические причины | Первично изолированная гипертриглицеридемия | Дефицит инсулина и липопротеинлипазы, ее кофактора | Сахарный диабет 1-го типа |
| Гипертриглицеридемия в сочетании с повышением ХС ЛПОНП и ЛППП, дисбеталипопротеидемией | Дефицит липопротеинлипазы, неэффективный метаболизм ЛПОНП с повышением концентрации последних и формированием дисбеталипопротеидемии | Сахарный диабет 2-го типа | |
| Мягкое или умеренное изолированное повышение ТГ | Дефицит липопротеинлипазы, ассоциированный с повышением продукции ЛПОНП | Ожирение | |
| Мягкое или умеренное изолированное повышение ТГ в сочетании с повышением ХС ЛПНП или формирование смешанной гиперлипидемии | Снижение активности печеночноклеточной липазы, снижение катаболизма ЛПОНП. У пациентов с гомозиготной апо-Е может быть причиной гипербеталипопротеидемии | Гипотиреоидное состояние | |
| Изолированное повышение уровня ТГ или формирование смешанной гиперлипидемии | Снижение синтеза и деградации ЛПОПН, ЛПНП | Нефротический синдром | |
| Использование лекарственных средств | Изолированное повышение уровня ТГ или формирование смешанной гиперлипидемии | Снижение активности печеночноклеточной и (или) плазменной липазы, снижение катаболизма ЛПОНП, ЛПНП, нарушение транспорта ЛПВП, повышение апо-В100 | Высокие дозы тиазидных диуретиков и β-адреноблокаторов без внутренней симпатомиметической активности, тамоксифен, пероральные эстрогенсодержащие контрацептивы, глюкокортикостероиды, АРВП, атипические антипсихотические средства |
| Другие причины | Изолированное повышение уровня триглицеридов или формирование смешанной гиперлипидемии | Снижение активности печеночноклеточной и (или) плазменной липазы, снижение катаболизма ЛПОНП, ЛПНП, нарушение транспорта ЛПВП иногда вследствие билиарной обструкции | Высоко калорийная углеводная диета, злоупотребление алкоголем, беременность, острый панкреатит |
В настоящее время в группу первичных гипертриглицеридемий принято включать семейную гипертриглицеридемию, семейный дефицит липопротеинлипазы, семейный дефицит апо-С-II, семейный дефицит печеночной липазы и семейный тип ингибирования липопротеинлипазы. Все перечисленные типы гипертриглицеридемий объединяют первичное преимущественное повышение плазменного пула хиломикронов натощак, или ХС ЛПОНП, или обоих компонентов липидного спектра с формированием фенотипа I, IV, V по классификации Фредриксона. Доказательством наследственного характера этих нарушений обычно является выявляемый у родственников пробанда 1-й степени родства аналогичный метаболический дефект.
Семейная гипертриглицеридемия
Семейная гипертриглицеридемия представляет собой нарушение липидного обмена с аутосомно-доминантным типом наследования, при котором у пробанда и его ближайших родственников выявляют значительное повышение плазменного уровня ТГ (преимущественно в составе хиломикронов и ЛПОНП) при нормальной концентрации ХС ЛПНП (табл. 6.5). Причем снижение уровня ХС ЛПВП регистрируется не всегда (Hopkins P.N. et al., 2003). Таким образом, с формальной точки зрения, у таких пациентов чаще всего отмечают гиперлипопротеидемию I, IV и V типов по классификации Фредриксона (см. табл. 6.1). Наиболее часто заболевание манифестирует в возрасте 30 лет и старше (Hopkins P.N. et al., 2003). Распространенность в общей популяции широко варьирует от 1% (в когорте молодых мужчин) до 8% (у пациентов с ИБС пожилого и старческого возраста). В последнее время отмечается ренессанс интереса исследователей к различным формам гипертриглицеридемий (Lemieux I. et al., 2000; Jacobson T.A. et al., 2007), поскольку ранее их самостоятельное значение в модуляции кардиоваскулярного риска ставилось под серьезное сомнение (Jap T.S. et al., 2003; Oh R. et al., 2008). На данный момент установлено, что манифестация семейной гипертриглицеридемии может быть ассоциирована с рядом биохимических нарушений, таких как дефицит липопротеинлипазы или печеночной липазы, а также наличием ингибитора липопротеинлипазы (Veerkamp M.J. et al., 2002). Однако генетический дефект, лежащий в основе манифестации семейной гипертриглицеридемии, пока не удалось идентифицировать. Наиболее приемлемым с клинической точки зрения биохимическим маркером, позволяющим проводить скрининг, является повышение отношения ТГ/апо-В. Тем не менее, диагноз семейной гипертриглицеридемии часто остается предположительным, даже после детального обследования родственников пробанда 1-й степени родства (Oh R. et al., 2008).
В качестве основы для проведения гиполипидемических мероприятий рассматриваются диетические ограничения, физическая нагрузка, модификация образа жизни и медикаментозная терапия (фибраты, препараты никотиновой кислоты, ω-3-ненасыщенные жирные кислоты) (Rader D.J., Rosas S., 2000; National Cholesterol Education Program, 2001; Hooper L. et al., 2006).
Семейный дефицит липопротеинлипазы
Семейный дефицит липопротеинлипазы (СДЛПЛ) является достаточно редким дефектом (1:1 млн населения в неорганизованной популяции), известным как I тип семейной гиперлипопротеидемии, верифицирующимся уже в детском возрасте. Основными признаками этого нарушения являются гипертриглицеридемия и хиломикронемия, связанные со снижением клиренса хиломикронов вследствие дефицита циркулирующей липопротеинлипазы. Результатом является повышение плазменного содержания хиломикронов. Характер наследования — аутосомно-рециссивный, не сопряженный с полом (Nagasaka H. et al., 2003). При этом заболевании снижение употребления ТГ с пищей способствует уменьшению продукции ХС ЛПОНП гепатоцитами, несмотря на то, что уровень ЛПОНП в плазме крови остается в пределах нормальных значений. Тем не менее, содержание ХС ЛППП и ХС ЛПНП в плазме крови снижается (McKenney J., 2005).
Основными клиническими признаками СДЛПЛ являются эпизоды достаточно выраженной боли в животе, иногда носящей опоясывающий характер и часто напоминающей таковую при остром панкреатите (Yuan G. et al., 2007). Кроме того, отмечаются ксантомы сухожилий и гепатоспленомегалия, ассоциированные с повышением плазменного содержания ТГ в пределах 50–100 ммоль/л. Необходимо отметить, что аккумуляция хиломикронов в плазме крови больных с СДЛПЛ часто приводит к липидной инфильтрации сетчатки и впоследствии — к офтальмопатии. Избыточный уровень хиломикронов способствует также увеличению плазменного содержания ТГ, при котором соотношение ТГ/общий ХС превышает 9:1. Уровень ХС ЛПОПН обычно нормальный или сниженный, тогда как ХС ЛПНП и ХС ЛПВП практически всегда выявляются в очень низких концентрациях.
Диагноз СДЛПЛ преимущественно базируется на анализе характера липидного спектра плазмы крови, а также измерении активности липопротеинлипазы после внутривенного введения 5000 ЕД гепарина. Диагноз считается достоверным при выявлении дефицита липопротеинлипазы (10% и менее от нормальных значений) после теста с гепарином. Необходимо отметить, что иногда отмечают ложноотрицательные результаты указанного выше теста. Это обстоятельство объясняется тем, что плазменная активность других циркулирующих липаз, таких как печеночная липаза, фосфолипаза, моноглицеридлипаза, часто оказывается выше нормальных значений после внутривенного введения гепарина, а перекрестные реакции между ними и липопротеинлипазой не являются редкостью. Таким образом, при наличии клинических признаков и липидного спектра, характерного для СДЛПЛ, необходимо принимать во внимание вероятность получения ложноотрицательного результата при попытках окончательной верификации диагноза. В этих случаях рекомендовано использование альтернативных методов определения липопротеинлипазы, таких как ингибирование активности последней с помощью натрия хлорида или протаминсульфата, а также хроматографию на бумаге или разделение в колонке с гепарин-сахарозой. Кроме того, в некоторых случаях у родственников пробанда 1-й степени родства отмечают снижение более чем на 50% активности липопротеинлипазы не только в плазме крови, но и в адипоцитах, что рассматривается как подтверждение гетерозиготности мутантного гена, определяющего формирование СДЛПЛ.
Главной целью лечения пациентов с СДЛПЛ является минимизация продукции хиломикронов преимущественно путем снижения поступления длинноцепочечных ТГ с пищей (<50 г/сут). Это приводит к редукции вероятности манифестации субклинического атерослероза и риска возникновения острого панкреатита. Необходимо отметить, что в последнем случае попытки измерения активности липопротеинлипазы часто оказываются малоперспективными, поскольку последняя интерферирует с циркулирующей амилазой. В этих случаях рекомендовано повторное определение активности липопротеинлипазы после 10-кратного разведения сыворотки крови. У больных с документированным СДЛПЛ риск возникновения острого панкреатита считается минимальным, если концентрация ТГ в плазме крови удерживается на уровне <20 ммоль/л.
Семейный тип ингибирования липопротеинлипазы
Этот редкий дефект был описан в трех последовательных поколениях одной семьи и представляет собой I тип гиперлипопротеидемии, возникшей вследствие синтеза циркулирующего ингибитора липопротеинлипазы. Появление последнего в плазме крови сопровождается выраженной хиломикронемией и гипертриглицеридемией. Клиническое значение этого феномена до сих пор не установлено.
Семейный дефицит апо-С-II
Это нарушение связано с аутосомно-рецессивным наследованием мутантного гена, который у гомозигот способствует исчезновению в плазме крови нормального апо-С-II, что ассоциируется с редукцией липолитической активности плазмы крови, повышением уровня ТГ в пределах 17–107 ммоль/л, формируя I или V тип фенотипа по классификации Фредриксона и высокий риск развития панкреатита. К настоящему времени уже описаны как минимум 4 функционально неактивных варианта апо-С-II. В результате существования последних липопротеинлипаза, являясь функционально активной, не способна к адекватному гидролизу хиломикронов или ЛПОНП в отсутствие нормального апо-С-II. У гетерозигот уровень апо-С-II обычно снижен на 30–50% от нормального, но липолитическая активность плазмы крови при этом не изменяется (Heath B. et al., 2003). Диагноз устанавливается на основании идентификации повышенного уровня ТГ, хиломикронов и/или ЛПОНП в сочетании с подтверждением дефицита апо-С-II. Ранние случаи возникновения кардиоваскулярных заболеваний в семьях больных с дефицитом апо-С-II обычно не выявляют (Nauck M.S. et al., 1998).
Семейный дефицит печеночной липазы
Впервые в 1974 г. у пациента с гипертриглицеридемией была описана новая форма дислипидемии, получившая название гиперальфатриглицеридемия, поскольку основным проявлением указанного метаболического дефекта стала аккумуляция ТГ в ЛПВП или α-фракции липопротеина. В последующем было установлено, что у этого пациента и его родного брата была снижена активность печеночной липазы, хотя активность липопротеинлипазы сохранялась на нормальном уровне. У обоих пациентов было верифицировано повышение плазменной концентрации общего ХС и ТГ в пределах 4,5–93 ммоль/л в зависимости от характера диеты. В клинической картине заболевания обращали на себя внимание наличие corneal arcus, ксантоматоза, пальмарных стрий, а также клинические признаки стенокардии напряжения и ишемические изменения на ЭКГ в покое. Попытки лечения пациентов фибратами оказались безрезультатными. Сегодня принято считать, что дефицит печеночной липазы способствует формированию ненормально крупных частиц ЛПВП, обогащенных ТГ. Причем ТГ преимущественно содержатся в частицах ЛПВП2, тогда как частицы ЛПВП3 фактически не определяются. Выявленный метаболический дефект негативно отражается на чувствительности первых к гидролизу. Более того, дефицит печеночной липазы ответственен за накопление именно ЛПВП2, поскольку с его помощью не образуются ЛПВП3. С другой стороны, нарушения аккумуляции ЛПОНП и ЛППП приводят к изменению соотношения ХС и ТГ в ЛПОНП. Несмотря на то что липопротеинлипаза гидролизует ТГ, содержащиеся в ЛПОПН, печеночная липаза не способна оказывать влияние на ремнантные формы ЛПОНП и ЛППП. Таким образом, дефицит печеночной липазы способствует аккумуляции ремнантов ЛПОНП в плазме крови. Интересно, что обогащенные ХС ремнанты ЛПОНП, формируя III тип гиперлипопротеидемии, обычно выявляются у пациентов, гомозиготных по апо-Е2 и апо-Е3, причем последние тесно связаны с дефицитом печеночной липазы и часто выявляются в семьях больных с дислипидемией, проживающих в Канаде и Швеции. Интересно, что фенотипически пациенты этих стран существенно различаются между собой: в Канаде дефицит печеночной липазы часто ассоциируется с ожирением. А лица, проживающие в Швеции, обычно имеют нормальную массу тела. По-видимому, эти наследственные формы нарушений липидного обмена ассоциируются с некоторыми демографическими и, возможно, расовыми отличиями.
Первичная гиперхолестеринемия
К первичным гиперхолестеринемиям относят семейную гиперхолестеринемию (гетеро- и гомозиготные формы), семейную дисаполипопротеинемию (гетеро- и гомозиготные формы), полигенную гиперхолестеринемию, семейную гиперальфалипопротеидемию и болезнь накопления эстерифицированного ХС.
Семейная гиперхолестеринемия
Семейная гиперхолестеринемия ассоциируется с кодоминантно наследуемым дефектом синтеза рецепторов к ЛПНП, что приводит к редукции деградации мелких и плотных гранул ЛПНП и существенно повышает уровень ХС в плазме крови (World Health Organisation, Human Genetics Programme, 1997). В популяции превалируют гетерозиготные формы семейной гиперхолестеринемии в соотношении 1:500, но в некоторых селективных популяциях (христиане Ливана) могут составлять 1:50 и иногда даже выше. К настоящему времени уже идентифицировано более 1000 мутаций рецепторов к ЛПНП. Пациенты с семейной гиперхолестеринемией отличаются склонностью к раннему развитию атеросклероза и клинически манифестных форм кардиоваскулярных заболеваний, в том числе ИБС (Marks D. et al., 2003). При отсутствии лечения семейная гиперхолестеринемия способствует 100-кратному повышению риска ИБС у молодых мужчин. В женской популяции семейная гиперхолестеринемия также рассматривается как независимый маркер высокого кардиовасулярного риска, но менее значимый по сравнению с таковым в мужской популяции. Так, по данным D. Marks и соавторов (2003) ИБС в женской популяции с семейной гиперхолестеринемией развивается в среднем на 10 лет позднее, чем в мужской. В настоящее время во всем мире насчитывается около 10 млн пациентов с семейной гиперхолестеринемией, из которых ежегодно 200 тыс. умирает преждевременно преимущественно вследствие коронарных причин (World Health Organisation, Human Genetics Programme, 1997).
Молекулярный дефект, определяющий формирование семейной гиперхолестеринемии, впервые описан J.L. Goldstein и соавторами (1973). Основной причиной гиперхолестеринемии принято считать мутацию рецептора к ЛПНП, что приводит к избыточному накоплению мелких и плотных гранул последних и повышению плазменной концентрации ХС. Установлено, что степень редукции экспрессии рецепторов к ЛПНП устойчиво коррелирует с уровнем ХС ЛПНП, тогда как в отношении фенотипических проявлений заболевания такой взаимосвязи не получено (Goldstein J.L., Brown M.S., 2001). На сегодня описано более 100 вариантов мутаций гена рецептора к ЛПНП, которые ассоциированы с полным отсутствием рецептора на поверхности клеточной мембраны, либо нарушением транспорта специфического протеина-переносчика к поверхности клетки, либо препятствуют связыванию ЛПНП на поверхности клеточных мембран, либо ограничивают интернационализацию рецептора после фиксации на нем молекулы ЛПНП (Goldstein J.L., Brown M.S., 2001) (табл. 6.6).
На рис. 6.1 представлены классические представления J.L. Goldstein, M.S. Brown (2001) о локализации выявляемых метаболических нарушений при основных классах мутаций гена рецептора ЛПНП.
Таблица 6.6 Основные классы мутаций гена рецептора ЛПНП
| Класс мутации | Синтез | Транспорт из СПР в комплекс Гольджи | Связывание с ЛПНП | Кластеризация в экзосомы |
| 1 | × | – | – | – |
| 2 | – | × | – | – |
| 3 | – | – | × | – |
| 4 | – | – | – | × |
СПР — саркоплазматический ретикулум.
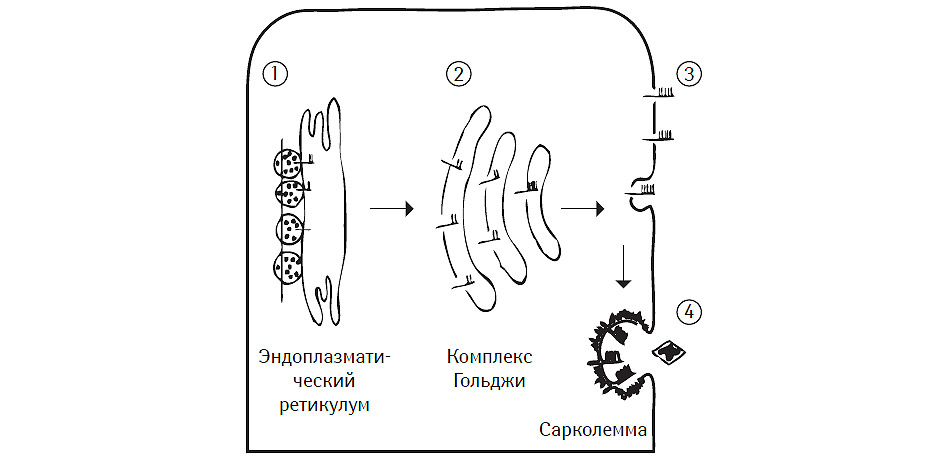
Рис. 6.1. Локализация выявляемых метаболических нарушений при основных классах мутаций гена рецептора ЛПНП. Модифицировано с изменениями из работы J. Boren и соавторов (2001)
Большую роль в нарушении связывания молекулы ЛПНП с рецептором могут играть функциональные и наследственные изменения его лиганда — апопротеина В100 (апо-В100). Частота распространенности наследственного дефекта апо-В100 в популяции сопоставима с таковой для семейной гиперхолестеринемии (Motulsky A.G., 1989).
Клинические признаки семейной гиперхолестеринемии очень вариабельны, особенно это справедливо для гетерозиготных форм заболевания (табл. 6.7). Вместе с тем, при гомо- и гетерозиготных формах семейной гиперхолестеринемии плазменное содержание общего ХС может существенным образом различаться и составлять 18,4–20,3 ммоль/л и 8,9–10,8 ммоль/л соответственно. Не получено доказательств наличия прямой взаимосвязи между генетическими и фенотипическими проявлениями у пациентов с различными формами семейной гиперхолестеринемии. Во всех случаях, чаще всего, выявляют высокий уровень общего ХС, ХС ЛПНП, наличие ксантом кожи и сухожилий, склонность к раннему возникновению атеросклероза любой локализации и ИБС. При этом у родственников пробанда определяют такую же форму гиперлипопротеидемии (IIA или IIB) (Umans-Eckenhausen M.A. et al., 2001). Вместе с тем, необходимо отметить, что у пациентов с гетерозиготной формой семейной гиперхолестеринемии ксантомы сухожилий хоть и являются патогномоничным признаком, однако верифицируются не всегда, а частота их выявляемости может зависеть от целого ряда причин. Среди последних наиболее важное значение имеет возраст пациентов (см. табл. 6.7).
Таблица 6.7 Факторы, ассоциированные с возникновением ксантом сухожилий и ИБС у пациентов с гетерозиготной формой семейной гиперхолестеринемии
| Характеристики | Ксантомы сухожилий | ИБС |
| Возраст | ++ | ++ |
| Мужской пол | – | ++ |
| Повышение уровня ХС ЛПНП | + | + |
| Снижение уровня ХС ЛПВП | – | + |
| Увеличение содержания ТГ | – | ± |
К настоящему времени предложены различные критерии (WHO, Dutch Lipid Clinic Network Criteria, Simon Broome Register Group) для скрининга и последующей детальной диагностики семейной гиперхолестеринемии (World Health Organisation, Human Genetics Programme, 1997). Практически все они основаны на оценке уровня ХС ЛПНП, наличия кожных и сухожильных ксантом у пациента и его родственников, а также идентификации типа гиперлипопротеидемии в семье (Ceveira F., International Panel on Management of Familial Hypercholesterolemia 2004). При этом не только пробанд, но и его родственники 1-й степени родства должны быть подвергнуты детальному обследованию не только с целью верификации заболевания, но и для оценки кардиоваскулярного риска (EUROASPIRE II Study Group, 2001; Pearson T.A. 2007).
Наиболеее часто используют диагностические критерии Simon Broome Register Group, которые базируются на биохимических и клинических признаках. Диагноз достоверной семейной гиперхолестеринемии устанавливается при наличии следующих критериев:
- плазменное содержание общего ХС превышает 7,5 ммоль/л (290 мг/дл) у взрослых или 6,7 ммоль/л (260 мг/дл) у детей младше 16 лет или ХС ЛПНП превышает 4,9 ммоль/л (190 мг/дл) у взрослых (4,0 ммоль/л у детей);
в сочетании со следующим признаком:
- ксантомы сухожилий у родственников 1-й и 2-й степени родства.
Диагноз вероятной семейной гиперхолестеринемии устанавливается при наличии одного из следующих признаков:
- инфаркт миокарда у родственников 2-й степени родства в возрасте моложе 50 лет или у родственников 1-й степени родства моложе 60 лет;
- повышение уровня общего ХС в плазме крови >7,5 ммоль/л (290 мг/дл) у родственников 1-й или 2-й степени родства.
Установлено, что именно так называемый каскадный скрининг (cascade screening) обладает наивысшим соотношением эффективность/стоимость в популяции пациентов из группы риска (Marks D. et al., 2002). При этом идентификация наибольшего количества случаев заболеваний в семье является основой успешности в верификации диагноза, поскольку специфических биохимических маркеров семейной гиперхолестеринемии до сих пор не предложено (EUROASPIRE II Study Group, 2001; Pearson T.A. 2007). Анализ частоты выявляемости мутаций гена рецептора к ЛПНП в популяции показал, что более чем в 30% случаев диагноз семейной гиперхолестеринемии, основанной на фенотипических проявлениях, вообще не верифицируется (Leren T.P., 2004). С другой стороны, специфические мутации гена рецептора для ЛПНП обычно выявляют не более чем у 70% пациентов с фенотипическими признаками семейной гиперхолестеринемии и у 20% лиц, у которых этот диагноз является вероятным или предположительным. С этими обстоятельствами связан отказ от необходимости проведения исследований ДНК с целью верификации изменений, специфичных для семейной гиперхолестеринемии (ten Asbroek A.H. et al., 2001).
Таким образом, неэтично исключать из группы скрининга лиц тех пациентов с гиперлипопротеидемией и отягощенным семейным анамнезом, у которых не было выявлено мутации гена рецептора ЛПНП (Marks D. et al., 2003; Leren T.P., 2004).
Необходимо отметить, что генетический скрининг может быть менее эффективным в гетерогенной популяции поскольку достаточно большое количество мутаций гена рецептора ЛПНП может определять широкое разнообразие клинических проявлений семейной гиперхолестеринемии (Leren T.P., 2004). В этой связи генетический скрининг можно не проводить, если диагноз семейной гиперхолестеринемии установлен на основании клинических проявлений и данных семейного анамнеза (Marks D. et al., 2003; Leren T.P., 2004). Однако многие эксперты указывают на тот факт, что зачастую сбор семейного анамнеза может быть затруднен вследствие не только медицинских причин, но и в связи с культурными, религиозными, традиционными и иными причинами (Marks D. et al., 2003), что создает серьезные трудности для проведения эффективной дифференциальной диагностики семейной гиперхолестеринемии (van Maarle M.C. et al., 2003; Agard A. et al., 2005). Наиболее сложной задачей считается скрининг семейной гиперхолестеринемии у детей и подростков (Rodenburg J. et al., 2004; Kavey R.E. et al., 2006). Вместе с тем, в этой возрастной популяции семейная гиперхолестеринемия сохраняет свою ассоциацию с высоким кардиоваскулярным риском и преждевременным возникновением ИБС, что делает необходимым настойчивое проведение скрининга несмотря на все возникающие затруднения (Rodenburg J. et al., 2004). Кроме того, установлено, что модификация образа жизни в когорте детей и подростков с установленной семейной гиперхолестеринемией позволяет добиться редукции плазменного уровня ХС более чем на 12% (Rodenburg J. et al., 2004; Kavey R.E. et al., 2006). Это достаточно важный факт, поскольку безопасность длительного применения статинов в этой возрастной группе пациентов с семейной гиперхолестеринемией до сих пор четко не установлена (Avis H.J. et al., 2007), несмотря на существование доказательств ее высокой гиполипидемической эффективности (Rodenburg J. et al., 2004). Сегодня принято считать, что при определении соотношения риск/польза статины при семейной гиперхолестеринемии можно назначать мальчикам старше 10 лет и девочкам после установившихся месячных (Kavey R.E. et al., 2006; Kwiterovich P.O. Jr., 2008). Кроме того, у детей и подростков с семейной гиперхолестеринемией необходимо принять решение о профилактике и возможном лечении артериальной гипертензии, ожирения, сахарного диабета, метаболического синдрома (Marks D. et al., 2003; Tonstad S., Thompson G.R., 2004; Pearson T.A., 2007). Скрининговые мероприятия в отношении семейной гиперхолестеринемии в детской популяции принято проводить не ранее 2-го года жизни ребенка, исключение составляет только гомозиготная форма семейной гиперхолестеринемии (Kwiterovich J.R. et al., 1973; Ceveira F.; International Panel on Management of Familial Hypercholesterolemia, 2004; Rodenburg J. et al., 2004).
В целом профилактические и лечебные мероприятия при семейной гиперхолестеринемии являются высокозатратными, но принципиально эффективными (Marks D. et al., 2002). Принято считать, что система лечения семейной гиперхолестеринемии является образцом для создания других моделей лечения наследственных заболеваний (Scientific Steering Committee on behalf of the Simon Broome Register Group, 1999). Так, модификация образа жизни, включая диетические ограничения, гиполипидемическая терапия, особенно использование статинов, позволяют существенным образом снизить кардиоваскулярный риск в когорте пациентов с семейной гиперхолестеринемией независимо от возраста и гендерной принадлежности (Hoogerbrugge N., 1998; Marks D. et al., 2003; Grundy S.M. et al., 2004). Однако, несмотря на то, что современное понимание патофизиологии семейной гиперхолестеринемии достаточно четко определено, в более чем 80% случаев эта патология не диагностируется и, соответственно, программы профилактики не проводятся (Ceveira F.; International Panel on Management of Familial Hypercholesterolemia, 2004).
Семейная дисаполипопротеинемия
Среди молекулярных дефектов, ассоциированных с дефицитом аполипопротеинов, выделяют гомо- и гетерозиготные формы (табл. 6.8). В большинстве случаев наибольшее влияние на композицию и концентрацию всех главных классов липопротеидов оказывают дефекты апо-А-I (Genest J.J. et al., 1992). Установлено, что гены, кодирующие апо-А-I, апо-С-III, апо-А-IV, находятся на одной хромосоме, что обусловливает частое вовлечение целого генного комплекса в процесс делеции. Большинство исследователей считают, что именно последнее обстоятельство лежит в основе манифестации такого тяжелого дефекта, как отсутствие в плазме крови ХС ЛПВП, апо-А-I, апо-С-III, апо-А-IV, ассоциированных с ранним развитием атеросклероза и высоким кардиоваскулярным риском (Genest J.J. Jr. et al., 1992; Hopkins P.N. et al., 2003).
Таблица 6.8 Молекулярные дефекты, ассоциированные с апопротеинами
| Характер дефекта | Форма | Генетический маркер | Значение |
| Дефицит апо-А-I | |||
| Апо-А-I Milano | Гетерозиготная | Замена цистеина аргинином в позиции 173 | Оказывает влияние на композицию и концентрацию всех классов апо-В-содержащих липопротеидов |
| Апо-А-I Iowa | Замена глицина аргинином в позиции 26 | ||
| Апо-А-I Seattle | Делеция в позиции 146-160 апопротеина | ||
| Дефект апо-А-I |
Гетерозиготная | Замена пролина аргинином в позиции 165 | |
| Дефект апо-А-I, апо-С-III, апо-А-IV |
Гомозиготная | Делеция генного комплекса апо-А-I, апо-С-III, апо-А-IV | Отсутствие в плазме крови ХС ЛПВП, апо-А-I, апо-С-III, апо-А-IV |
| Гетерозиготная | Низкий или субнормальный уровень ХС ЛПВП, апо-А-I. Апо-С-III и апо-А-IV не выявляют или уровень низкий | ||
| Дефект апо-А-II |
Гетерозиготная | Точечная мутация гена апо-А-II | Не оказывает существенного влияния на уровень ЛПВП, самостоятельное значение не установлено |
| Дефицит апо-А-II |
Гетерозиготная | ||
| Дефект апо-В100 | Гомозиготная | Замена глутамина аргинином в позиции 3500 | Идентифицирован у лиц с семейной гиперхолестеринемией и ассоциирован с высоким риском возникновения ИБС |
| Гетерозиготная | |||
| Дефект апо-В100-Hopkins | Гомозиготная | Точечная мутация, влияющая на сродство апо-В100 к рецептору, а также модифицирующая размеры и состав апопротеина | Ассоциирован с гиперлипопротеидемией |
| Гетерозиготная | |||
| Дефект апо-В | Гомозиготная | Точечная мутация кодирующей части гена, ведущая к появлению апо-В37 | Ассоциирован с гиполипопротеидемией |
| Гетерозиготная | |||
| Гомозиготная | Точечная мутация кодирующей части гена, ведущая к появлению апо-В90 | Ассоциирован со снижением апо-В и ХС ЛПНП | |
| Гетерозиготная | |||
| Гомозиготная | Точечная мутация кодирующей части гена, ведущая к появлению апо-В40 | ||
| Гетерозиготная | |||
| Избыток апо-С-I | Гомозиготная | Мутация гена апо-С-I | Ассоциирован с увеличением распространенности ИБС в популяции |
| Гетерозиготная | |||
| Дефект апо-С-II |
Гомозиготная | Мутация гена апо-С-II | Самостоятельное значение не установлено |
| Гетерозиготная | |||
| Дефект апо-С-III |
Гомозиготная | Мутация гена апо-С-III | |
| Гетерозиготная | |||
Необходимо отметить, что не все апопротеины могут оказывать самостоятельное влияние в отношении кардиоваскулярного риска, особенно ассоциированного с преждевременным развитием атеросклероза в семье (табл. 6.9). Большинство дисапопротеинемий, имеющих реальное клиническое значение, касаются нарушения синтеза и дефекта апо-В, апо-А и апо-Е протеинов. Достаточно часто весьма сложно установить, какой именно дефект играет определяющую роль в повышении атерогенности липидного профиля плазмы крови: дисапопротеинемия или гиперхолестеринемия. Так, апо-В-содержащие липопротеиды широко представлены в различных липопротеидах, даже в тех из них, которые традиционно не рассматриваются как атерогенные и не ассоциируются с элевацией кардиоваскулярного риска (хиломикроны, ЛПОНП) (табл. 6.10). Тем не менее, гипер-апо-В-протеинемия, характеризующаяся повышением концентрации апо-В содержащих ЛПНП >120 мг/дл при нормальном уровне ХС ЛПНП (<5,0 ммоль/л), ассоциируется с избыточным кардиоваскулярным риском и преждевременным развитием атеросклероза любой локализации (Hopkins P.N. et al., 2003). Интересно, что в семьях пациентов с гипер-апо-В-протеинемией выявляют ксантомы сухожилий без значительного повышения плазменного уровня общего ХС и ХС ЛПНП (Schneeman O. et al., 1993).
Таблица 6.9 Влияние основных апопротеинов на липидный профиль (по данным Boren J. et al., 2001; Segrest J.P. et al., 2001; Nissen S.E. et al., 2003; O’Bryan M.K. et al., 2004; Shiflett A.M., 2005; Barter P.J., Rye K.A., 2006; Dahlback B., 2006)
| Тип апопротеина | Полиморфизм | Предполагаемая функция | Клиническое значение |
| Апо-А-I | Имеется: А1, А2, А3, А4, наследуется кодоминантно | Активация ЛХАТ, акцепция ХС из клеток, структурный компонент ЛПВП | Дефицит связан с ранним развитием атеросклероза, высокой частотой манифестации ИБС |
| Апо-A-II | Нет | Модификация состава ЛПНП, структурный компонент ЛПВП | Самостоятельное значение не установлено |
| Апо-А-III | Нет | ||
| Апо-А-IV | Имеется | Активация ЛХАТ, акцепция ХС из клеток, модификация состава ЛПНП, ЛПВП, ЛПОНП, модуляция уровня ТГ | Полиморфизм может быть связан с кардиоваскулярным риском, развитием атеросклероза |
| Апо-В100 | Имеется: апо-Вс, апо-Вg | Модификация состава ЛПНП, транспорт ХС, структурный компонент ЛПНП, ЛПОНП, ЛППП | Ассоциирован с ранним развитием атеросклероза, высокой частотой манифестации ИБС |
| Апо-В48 | Нет | Структурный компонент хиломикронов | Самостоятельное значение не установлено, описаны антиатерогенные качества |
| Апо-С-I | Нет | Активирует ЛХАТ, структурный компонент ЛПВП и обогащенных ТГ липопротеидов | Высокая концентрация ассоциируется с ранним развитием ИБС в семье |
| Апо-С-II | Нет | Активирует ЛХАТ, структурный компонент ЛПВП и обогащенных ТГ липопротеидов | Самостоятельное значение не установлено |
| Апо-С-III | Нет | Ингибирование ЛХАТ, структурный компонент ЛПВП и обогащенных ТГ липопротеидов | Дефицит или отсутствие приводит к гипотриглицеридемии, самостоятельное значение не установлено |
| Апо-Е | Имеется: Е1, Е2, Е3, Е4; наследуется кодоминантно | Модулирует катаболизм ТГ, ЛПОНП, лишенных ремнантов хиломикронов, активирует ЛХАТ, является лигандом для ЛПНП и рецептором для ремнантов хиломикронов, структурный компонент ЛПВП и обогащенных ТГ липопротеидов | Наследуемая детерминанта содержания общего ХС, ХС ЛПНП, маркер риска ИБС. Полиморфизм апо-Е (от Е1 к Е4) является независимым маркером кардиоваскулярного риска |
| Апо-D | Нет | Представитель семейства липокалинов, содержит сайт связывания с короткими гидрофобными лигандами | Возможно участвует в обратном транспорте ХС |
| Апо-F | Нет | Ингибирует транспорт липидов | Участвует в регуляции активности протеина, транспортирующего эстерифицированный ХС |
| Апо-H | Нет | Четко не установлена | ? |
| Апо-L | Нет | Структурный компонент преимущественно ЛПВП | Участвует в регуляции уровня ТГ и ХС |
| Апо-М | Нет | Структурный компонент преимущественно ЛПВП | Интегральный протеин, модулирующий уровень ХС ЛПНП и ЛПВП, обладает антиатерогенным потенциалом |
Таблица 6.10 Плазменная концентрация и атерогенность апо-В-содержащих липопротеидов (модифицирована из работы S. Marcovina, C.J. Packard, 2006)
| Липопротеид | Концентрация апо-В (мг/дл) | Атерогенность |
| Хиломикроны | <0,5 | ? |
| Ремнанты хиломикронов | 1–2 | ++ |
| ЛПОНП | 5,0 | + |
| Ремнанты ЛПОНП | 2,0–5,0 | +++ |
| ЛППП | 15,0 | +++ |
| ЛПНП | 60,0 | +++ |
| ЛП (а) | 5,0 | + |
В программах лечения семейной дисапопротеинемии наибольшее значение имеют модификация образа жизни (диета, физическая нагрузка, отказ от употребления алкоголя и курения) и медикаментозная терапия (статины, ω-3 ненасыщенные жирные кислоты) (Grundy S.M. et al., 2004; Hooper L. et al., 2006).
Полигенная гиперхолестеринемия
Полигенная гиперхолестеринемия является одним из наиболее часто отмечаемых наследственных нарушений липидного обмена, для которых до сих пор не установлен соответствующий биохимический маркер. Для этого заболевания характерны полигенный тип наследования, отсутствие специфических клинических признаков, ассоциация со спецификой питания, уровнем физической активности и наличием сопутствующих заболеваний (см. табл. 6.5).
Распространенность полигенной гиперхолестеринемии в популяции и среди пациентов с документированной ИБС может достигать 14% и более. Возможно, что частота выявляемости полигенной гиперхолестеринемии среди пациентов с семейной гиперлипопротеидемией существенно выше, чем считалось ранее. Диагноз достаточно сложен и в основном основан на клинических признаках и исключении других форм семейных гиперлипопротеидемий, особенно ассоциированных с бимодальным распределением содержания ХС у пробанда и его ближайших родственников (табл. 6.11). Необходимо отметить, что полигенная гиперхолестеринемия также рассматривается как одна из составляющих высокого кардиоваскулярного риска и ассоциируется с ранним развитием атеросклероза.
Таблица 6.11 Полиморфизм аполипопротеина Е, ассоциированный с III типом гиперлипопротеидемии*
| Тип полиморфизма | Изменения апо-Е3 у родителей пробанда | Молекулярный дефект | Способность рецептора к связыванию апо-Е3, % |
| Е1 Harrisburg | Нет у обоих | Lys146 → Glu | Дефектная |
| Е2 | Нет у одного | Arg158 → Cys | <2 |
| Е2 | Нет у одного | Arg145 → Cys | 45 |
| Е2 | Нет у одного | Arg146 → Gln | 40 |
| Е2 Christchurch | Нет у одного | Arg136 → Ser | 41 |
| Е3 A | 0 | Cys112 → Arg, Arg142 → Cys | <5 |
| Е3 Leiden | 0 | Дуплификация в положении 121–127 | 25 |
| Е Bethesda | Нет у обоих | – | – |
| E Deficiency | – | – | – |
*Таблица модифицирована из работы G.R. Thompson (1989).
Наиболее эффективными гиполипидемическими вмешательствами в этой когорте пациентов, кроме традиционных немедикаментозных способов лечения, является использование статинов. Последние зарекомендовали себя как достаточно эффективное и безопасное средство для адекватного контроля кардиоваскулярного риска и улучшения ближайшего и отдаленного прогноза (National Cholesterol Education Program, 2001).
Семейная гиперальфалипопротеидемия
Семейная гиперальфалипопротеидемия представляет собой гетерогенную группу гиперлипидемий, основным признаком которых являются преимущественно изолированное повышение уровня ХС ЛПВП, аутосомно-доминантный характер наследования, манифестация у лиц европеоидной расы, ассоциация с высоким риском возникновения ранних случаев ИБС в семье. Точный механизм возникновения этого дефекта не выяснен, хотя существуют данные о том, что повышение уровня ХС ЛПВП может быть результатом редукции активности белка-переносчика эстерифицированного ХС. Это приводит к уменьшению размеров частиц ЛПНП и увеличению частиц ЛПВП, обогащенных апо-А липопротеином. Клиническая картина заболевания сводится иногда к появлению arcus senilis и модулированию кардиоваскулярного риска.
Болезнь накопления эстерифицированного ХС
Одной из редких причин формирования первичной гиперхолестеринемии является наследственный дефицит гидролазы эстерифицированного ХС, который обусловливает манифестацию так называемой болезни накопления эстерифицированного ХС (cholesterol ester storage disease). Для липидного профиля при этом заболевании характерно не только увеличение плазменного пула общего ХС и ХС ЛПНП, но и редукция ХС ЛПВП, что отражает его атерогенный характер и соответствует IIa типу по классификации Фредриксона. Клиническая картина заболевания достаточно размыта, одним из наиболее характерных признаков является гепатоспленомегалия. Ксантоматоз обычно не верифицируется. Диагноз устанавливается на основании идентификации избыточного накопления эстерифицированного ХС в гепатоцитах, полученных при проведении прижизненной пункционной биопсии печени, а также при подтверждении факта сниженной активности гидролазы эстерифицированного ХС в культуре фибробластов. Клиническое значение болезни накопления эстерифицированного ХС состоит исключительно в формировании высокого риска возникновения и прогрессирования системного атеросклероза и потенцирования наступления атеротромботических событий.
Первичные смешанные гиперлипидемии
К первичным смешанным гиперлипидемиям обычно относят семейную комбинированную гиперлипидемию, семейную гиперхиломикронемию, III тип гиперлипопротеидемии, болезнь «рыбьих глаз» («fish-eye disease») и семейный дефицит ЛХАТ.
Семейная комбинированная гиперлипидемия
Термин «семейная комбинированная гиперлипидемия» (familial combined hyperlipidemia) был предложен J.L. Goldstein и соавторами (1973) для идентификации наследственного заболевания с аутосомно-доминантным характером наследования, отличительной особенностью которого является наличие различных типов гиперлипидемий, в основном IIA, IIB и IV, у пробанда и его родственников 1-й степени родства (см. табл. 6.5). Предполагают, что у около 15% пациентов с документированной ИБС в возрасте <60 лет отмечают семейную комбинированную гиперлипидемию (Veerkamp M.J.et al., 2002; Suviolahti E. et al., 2006).
Попытки выявления топического и молекулярного дефектов, приводящих к манифестации заболевания, оказались не вполне успешными, хотя удалось идентифицировать молекулярный дефект, находящийся в локусе 1q21-q23 11-й хромосомы. Тем не менее, не выявлено доказательств того, что основной причиной возникновения семейной комбинированной гиперлипидемии могут быть нарушения синтеза апо-В, дефицит липопротеинлипазы, мутации комплекса генов апо-А-I/апо-С-III/апо-А-IV (Zambon A. et al., 2006; Zechner R., Goldberg I.J., 2007). Также не увенчались успехом исследования, направленные на идентификацию биохимического дефекта, позволяющего проводить дифференциальную диагностику этой формы нарушений липидного обмена с другими наследственными и приобретенными гиперлипопротеидемиями (Zechner R., Goldberg I.J., 2007). Таким образом, согласно современным представлениям семейная комбинированная гиперлипидемия является генетически гетерогенным заболеванием.
Обычно первые признаки заболевания выявляют в возрасте старше 20 лет, хотя в некоторых случаях они могут быть идентифицированы и в более раннем возрасте (Cortner J. et al., 1990). Частота выявления в популяции семейная комбинированная гиперлипидемия составляет 0,5–5%, а среди пациентов с документированной ИБС в пожилом и старческом возрасте этот показатель достигает даже 20% (Kwiterovich P., 1993).
При семейной комбинированной гиперлипидемии выявляют существенное увеличение содержания апо-В-100 в плазме крови, а также в ЛПНП и ЛНОНП, что связано с селективным повышением синтеза этого апопротеина (Zechner R., Goldberg I.J., 2007). Необходимо отметить, что при этой форме наследственной гиперлипидемии наблюдается повышение содержания в плазме крови обогащенных ТГ ЛПОНП, но в несколько меньшей степени, чем это обычно выражено при семейной гипертриглицеридемии. (Vakkilainen J. et al., 2002) Кроме того, при семейной комбинированной гиперлипидемии пропорция ЛПОНП, конвертирующихся в ЛПНП, обычно имеет нормальные значения, тогда как для семейной гипертриглицеридемии характерен субнормальный уровень этого отношения (Davignon J., Genest J. Jr., 1998). При этом молекулы ЛПНП и ЛПОНП, в которых преобладают мелкие тяжелые гранулы липопротеидов, в когорте пациентов с семейной комбинированной гиперлипидемией содержат достоверно меньше ХС и фосфолипидов, чем у здоровых лиц. В этой связи у больных с документированной семейной комбинированной гиперлипидемией часто фиксируют сохранение соотношения ТГ/апо-В в ЛПОНП и ХС/апо-В в ЛПНП (Garcia-Otin A.L. et al., 1999).
Традиционно используемые диетические ограничения и модификация образа жизни при семейной комбинированной гиперлипидемии обычно демонстрируют ограниченный гиполипидемический потенциал. Применение монотерапии фибратами и производными никотиновой кислоты дополнительно к программам модификации образа жизни оказывают достаточно мощное влияние в отношении редукции плазменного уровня ТГ, не влияя на концентрацию ХС ЛПНП и апо-В-100. В этой связи наиболее предпочтительной терапевтической стратегией в настоящее время принято считать комбинацию статина и фибрата при постоянном мониторинге соотношения риск/польза.
Семейная гиперхиломикронемия
Семейная гиперхиломикронемия ассоциирована с аккумуляцией в плазме крови хиломикронов и ремнантов ЛПОНП (см. табл. 6.5). При этом молекулы ЛПНП содержат преимущественно апо-Е3 или апо-Е4 в различных соотношениях. Генетическим дефектом, опосредующим появление подобного фенотипа, является замена аргинина на цистеин в аминокислотной позиции 112-го гена, кодирующего синтез апо-Е3. У большинства пациентов отмечают гомозиготную форму, определяющую преимущественный синтез апо-Е3, или гетерозигоные формы, ассоциированные с фенотипом апо-Е2/апо-Е3 или апо-Е3/апо-Е4. При этой форме наследственной гиперлипопротеидемии выявляют также сниженный клиренс ремнантов ЛПОНП и хиломикронов. Биохимическим маркером для дифференциальной диагностики является повышение плазменной концентрации общего ХС преимущественно за счет хиломикронов приблизительно до 10 ммоль/л. Иногда у пробанда и родственников, преиущественно при гомозиготных формах, можно выявить кожные ксантомы и arcus senilis. Несмотря на то, что семейная гиперхиломикронемия ассоциируется с повышением плазменной концентрации потенциально не- или малоатерогенных фракций липопротеидов, в этой когорте пациентов кардиоваскулярный риск обычно несколько выше, чем в общей популяции. Вместе с тем, риск возникновения ИБС меньше такового при семейной гиперхолестеринемии, семейной комбинированной гиперлипидемии, полигенной гиперхолестеринемии и выше, чем при семейной гипертриглицеридемии. В качестве лечебных мероприятий часто рассматривают модификацию образа жизни и прием секвестрантов желчных кислот (National Cholesterol Education Program, 2001).
Таблица 6.5 Диагностические критерии основных форм наследственных гиперлипопротеидемий
| Диагностические признаки | Популяция | Формы гиперлипопротеидемии | |||||
| Семейная гиперхиломикронемия | Семейная дисбеталипопротеидемия | Семейная гиперхолестеринемия | Полигенная гиперхолестеринемия | Семейная комбинированная гиперлипидемия | Семейная гипертриглицеридемия | ||
| Тип гиперлипопротеидемии по классификации Фредриксона | Пробанд | I | III | IIA, IIB | IIA | IIA, IIB,IV | IV, V |
| Родственники | I | III | IIA, IIB | IIA | IIA, IIB, IV | IV, V | |
| Наличие кожных ксантом | Пробанд | Нет | Нет | Присутствуют хотя бы у одного родственника | Могут быть | Могут быть | Могут быть |
| Родственники | Нет | Нет | Присутствуют хотя бы у одного родственника | Нет | Нет | Нет | |
| Наличие сухожильных ксантом | Пробанд | Нет | Нет | Присутствуют | Могут быть | Могут быть | Нет |
| Родственники | Нет | Нет | Присутствуют | Нет | Нет | Нет | |
| IV, V типы гиперлипидемии в семье | Пробанд | Нет | Нет | Нет | Нет | Могут быть | Присутствуют |
| Родственники | Нет | Нет | Нет | Нет | Могут быть | Присутствуют | |
| Тип наследования | – | Аутосомно-доминантный | Аутосомно-доминантный | Аутосомно-доминантный | Полигенный | Аутосомно-доминантный | Аутосомно-доминантный |
| Характерная популяция пациентов и их возраст | Пробанд | Дети и взрослые | Дети и взрослые | Обычно после 20 лет | Дети и взрослые | После 20 лет, иногда в детском возрасте | После 30 лет |
| Генетический дефект | Пробанд | Неизвестен | Неизвестен | Дефект ЛПНП-рецептора | Неизвестен | Неизвестен | Неизвестен |
| Биохимический маркер | Пробанд | Повышение плазменной концентрации общего ХС преимущественно за счет хиломикронов приблизительно до 10 ммоль/л | Увеличение концентрации апо-В содержащих ЛПНП более 120 мг/дл при нормальном уровне ХС ЛПНП (менее 5,0 ммоль/л) | Высокий уровень общего ХС (более 220 мг/дл) и ЛПНП (155 мг/дл) | Неизвестен | Не установлен | Повышение отношения ТГ/апо-В |
| Критерии дифференциальной диагностики | Пробанд | Клинические признаки, липидный профиль, семейный анамнез | Клинические признаки, липидный профиль, семейный анамнез | Уровень ХС ЛПНП больше 95% интервала в соответствии с полом и возрастом
Наличие ксантом сухожилий у пробанда или его родственников |
Клинические признаки, семейный анамнез | Клинические признаки, семейный анамнез, сохранение отношения ТГ/апо-В в ЛПОНП и ХС/апо-В в ЛПНП | Клинические признаки, семейный анамнез, повышение плазменного уровня ТГ (преимущественно в составе хиломикронов и ЛПОНП) при нормальной концентрации ХС ЛПНП |
Таким образом, в заключение необходимо отметить, что наследственные нарушения липидного обмена — не редкость в современной клинической практике и требуют настойчивой верификации. Вместе с тем, ограниченные возможности для скрининга преимущественно обусловлены отсутствием высокоспецифичного биохимического маркера для большинства наследственных форм гиперлипопротеидемий. С другой стороны, большинство последних могут быть хотя бы заподозрены, а иногда и четко верифицированы при адекватном анализе семейного анамнеза и клинической картины. Возможно, в будущем практикующий врач будет иметь больше возможностей для качественной диагностики наследственных гиперлипопротеидемий.
III тип гиперлипопротеидемии (семейная дисбеталипопротеидемия)
Это нарушение липидного обмена характеризуется накоплением в плазме крови хиломикронов и ремнантных форм ЛПОНП вследствие преимущественно нарушения их клиренса при нормальной экспрессии на поверхности мембран гепатоцитов рецепторов к липопротеидам (Smelt A.H., de B.F., 2004). В исследованиях in vitro было установлено, что ЛПНП или апо-В/апо-Е-рецепторы способны связывать с высокой степенью аффинности частицы, содержащие апо-Е3 или апо-Е4. Молекула последнего отличается от апо-Е3 заменой аргинина на цистеин в положении 112 аминоксилотной последовательности. Частицы липопротеида, содержащие апо-Е2, в которых отмечают замену цистеина на аргинин в положении 158, не способны связываться с апо-В/апо-Е-рецепторами (см. табл. 6.11).
Большинство пациентов с III типом гиперлипопротеидемии являются гомозиготами по апо-Е2. Вместе с тем, некоторые гетерозиготные по этому признаку больные имеют полиморфизм апо-Е в виде апо-Е2/апо-Е3 или апо-Е2/апо-Е4. У некоторых пациентов выявлено нарушение способности рецептора ЛПНП к связыванию нормального апо-Е3 (см. табл. 6.11). Аналогичные нарушения описаны также в некоторых семьях с абсолютным дефицитом апо-Е (Smelt A.H., de B.F., 2004).
Многочисленные исследования показали, что врожденный дефект апо-Е обычно недостаточен для манифестации клиинчески значимой семейной дисбеталипопротеидемии. Так, апо-Е2/апо-Е2 фенотип определяют в популяции с частотой 1:100, тогда как сама семейная дисбеталипопротеидемия верифицируется с гораздо меньшей частотой: 1:5000 в популяции. Таким образом, очевидно, что необходимы иные метаболические нарушения, способствующие реализации клинической значимости изменений фенотипа апо-Е. По мнению большинства исследователей, «критическим» потенциалом могут обладать традиционные факторы риска кардиоваскулярных событий: ожирение, артериальная гипертензия, сахарный диабет, гипотиреоидизм, отягощенный семейный анамнез. Возможно, что именно комбинация этих факторов, включающая нарушение клиренса ремнантных форм липопротеидов, снижение экспрессии апо-В/апо-Е-рецепторов на поверхности клеточных мембран, повышение синтеза ЛПОНП, суммирует негативное влияние семейных вариантов смешанной гиперлипидемии и традиционные факторы кардиоваскулярного риска в отношении вероятности возникновения атеросклероза и атеротромботических событий. Интересно, что III тип гиперлипопротеидемии редко манифестируется у мужчин до наступления пубертатного периода, а у женщин — до периода менопаузы, что подтверждает существование гормонального контроля за семейным нарушением лиидного обмена.
Клинические признаки III типа гиперлипопротеидемии включают corneal arcus, ксантаматоз вокруг коленных и локтевых суставов, ксантелазмы и пальмарные стрии, которые считаются патогномоничным признаком. Плазменное содержание общего ХС обычно превышает 10 ммоль/л, при проведении электрофореза на бумаге проявляется широкая полоска ремнантного β-липопротеида. При ультрацентрифугировании фракция с удельной плотностью <1,006 содержит обогащенные ХС ремнанты β-ЛПОНП. При этом отношение масс ХС/ТГ превышает 0,42 единицы. Содержание ХС ЛПНП обычно снижено, поскольку при этом заболевании выявляется нарушение конвертации ЛППП в ЛПНП. Несмотря на это, III тип гиперлипопротеидемии рассматривается как атерогенная гиперлипидемия, поскольку ремнанты β-ЛПОНП участвуют в процессе активации макрофагов и инфильтрации субинтимы. Кардиоваскулярные заболевания обычно верифицируются почти у 50% пациентов, причем достаточно часто идентифицируются атеросклеротические поражения не только коронарных, но и периферических, в том числе мозговых артерий.
Диагноз подтверждается при идентификации апо-Е-фенотипа (см. табл. 6.11). Нарушения толерантности к глюкозе в виде повышения ее уровня натощак, а также гиперурикемия являются частым компонентом этой формы заболевания. Возможны также случаи острого панкреатита.
Лечение III типа гиперлипопротеидемии в основном направлено на коррекцию сопутствующих факторов риска, таких как артериальная гипертензия, ожирение, сахарный диабет или метаболический синдром. Для достижения контроля за уровнем ТГ наряду с диетой рекомендованы производные фиброевой кислоты (бензафибрат, гемфиброзил) как в монотерапии, так и в сочетании с никотиновой кислотой. Это позволяет не только снизить риск возникновения ИБС, но и способствует реверсии кожных поражений. Ионно-обменные смолы и секвестранты желчных кислот могут усугубить гипертриглицеридемию и в этой связи не рекомендованы для лечения пациентов с III типом гиперлипопротеидемии. Статины могут быть иногда полезными, особенно при сочетании этого заболевания с семейной гиперхолестеринемией.
Семейный дефицит ЛХАТ
Семейный дефицит ЛХАТ является редким заболеванием, характеризующимся диффузным помутнением роговицы, гемолитической анемией, протеинурией, а также гипертриглицеридемией. Этот наследственный дефект был впервые описан в 1967 г. у 26 представителей одной из шведских семей. Выявленный метаболический дефект был связан с точечной генной мутацией, опосредующей неспособность эстерифицировать свободный ХС в плазме крови, что приводило к повышению содержания практически во всех фракциях липопротеидов свободного ХС и лецитина при снижении пула лизолецитина (Thompson G.R., 1989). Накопление неэстерифицированного ХС в мембранах эритроцитов объясняло появление нормохромной анемии. Содержание общего ХС в крови часто было нормальным, однако ХС ЛПВП значительно снижался. Уровень ТГ натощак обычно незначительно превышал нормальные значения, отражая повышение содержания обогащенных ТГ ЛПОПН. Полагают, что помутнение роговицы и инфильтрация паренхимы почек неэстерифицированными хиломикронами, приводящая в последующем к нефропатии и почечной недостаточности, может быть опосредована как повышением уровня ЛПНП, так и дефицитом ЛПВП, возникающим при нарушении транспорта свободного ХС.
Диагноз подтверждается при выявлении абсолютного дефицита активности ЛХАТ в плазме крови гомозиготного пробанда и снижении активности последней в гетерозиготных случаях (Thompson G.R., 1989).
В лечении семейного дефицита ЛХАТ используют диеты со сниженным содержанием жира и ТГ, на стадии почечной недостаточности рекомендуется заместительная терапия (диализ, трасплантация почки). Медикаментозные подходы к лечению указанного дефекта не разработаны.
Болезнь «рыбьих глаз» («fish-eye disease»)
Болезнь «рыбьих глаз» («fish-eye disease») была впервые описана в 1979 г. в Швеции и характеризовалась появлением помутнения роговицы, подобно той, которая верифицируется при семейном дефиците ЛХАТ (Carlson I.A., Holmquist I., 1985). Однако в отличие от последнего в клинической картине отсутствовали анемия и нефропатия. У больных обычно выявляют умеренное повышение плазменной концентрации ТГ натощак, а также ЛППП и ЛПОНП. При этом уровень общего ХС и активность ЛХАТ не отличается от нормальных значений, тогда как существенно снижается содержание ЛПВП, апо-А и эстерифицированного ХС. Эти данные позволили I.A. Carlson и I. Holmquist (1985) предположить, что существует как минимум две формы ЛХАТ. Первая, так называемая αЛХАТ, проявляет активность только в отношении ЛПВП, тогда как вторая — βЛХАТ, оказывает влияние на ЛПОНП и ЛПНП. В дальнейшем оказалось, что манифестация болезни «рыбьих глаз» является следствием дефицита активности αЛХАТ, а семейный дефицит ЛХАТ связан с дефектом обеих изоформ (Thompson G.R., 1989). Предполагается также, что синтез обеих изоформ этого фермента опосредуется двумя различными генами. Подходы к профилактике и лечению болезни «рыбьих глаз» пока не совсем ясны.
Вторичные гиперлипидемии
Вторичные гиперлипидемии чаще всего носят смешанный характер или представляют собой изолированные гипертриглицеридемии. Другие формы вторичных гиперлипидемий выявляют реже.
Вторичные гиперхолестеринемии
Гиперхолестеринемия является документированным фактором риска возникновения атеросклероза, повышая вероятность манифестации ИБС и атеротромботических событий иной локализации в 2–4 раза. В этой связи коррекция причин, приводящих к нарушениям липидного обмена, а также гиполипидемические мероприятия в этой популяции пациентов позволяют оказывать благоприятное влияние на ближайший и отдаленный прогноз. В то же время, согласно современным представлениям, повышение содержания плазменной концентрации общего ХС и ХС ЛПНП обычно связано с генетическими причинами, а основным разрешающим фактором, особенно для гетерозиготных форм, являются нарушения характера питания, метаболизма, включая сахарный диабет, метаболический синдром, идиопатическую и вторичную подагру; нефропатии с протеинурией/нефротическим синдромом; дисфункция щитовидной железы и т.п. Таким образом, дифференциация вторичных гиперлипидемий на гиперхолестеринемии и гипертриглицеридемии достаточно условна, и, вероятно, отражает существование патогенетических этапов в развитии дислипидемии, причем изначальным моментом для большинства из них является формирование гипертриглицеридемии, ассоциированной с повышенным синтезом ЛПОНП.
Вторичные гипертриглицеридемии
Вторичные гипертриглицеридемии обычно формируются у пациентов с нелеченным или неконтролируемым сахарным диабетом, а также у лиц, получающих ряд лекарственных средств (высокие дозы тиазидных диуретиков и блокаторов β-адренорецепторов без внутренней симпатомиметической активности, тамоксифен, пероральные эстрогенсодержащие контрацептивы, глюкокортикостероиды, антиретровирусные препараты (АРВП), атипические антипсихотические средства) и пищевых продуктов (алкоголь, высокоуглеводная диета [более 60% от общего количества калорий]) (табл. 6.12). Кроме того, вторичная гипертриглицеридемия может явиться также следствием гипотиреоидного состояния, ХБП, нефротического синдрома, острого панкреатита, а также результатом приема АРВП, особенно при лечении ВИЧ (Sanderson S.L. , 1991; Bainton D. et al., 1992; Fortson M.R. et al., 1995; Hsia S.H. et al., 1995; Rader D.J., Rosas S., 2000; Athyros V.G. et al., 2002; Bamba V., Rader D.J., 2007).
Таблица 6.12 Основные причины, приводящие к формированию гипертриглицеридемий
| Первичные гипертриглицеридемии | Вторичные гипертриглицеридемии |
| Семейная гипертриглицеридемия | Адипозопатия |
| Семейная комбинированная гиперлипидемия | Сахарный диабет |
| Семейный дефицит липопротеинлипазы | Гипотиреоидизм |
| Дефицит апо-С-II | Нефротический синдром |
| Семейная дисбеталипопротеидемия | Ятрогенные (использование медикаментов: АРВП, фенотиазины, секвестранты желчных кислот, неселективные блокаторы β-адренорецепторов, тиазидные диуретики, циклофосфамид, тамоксифен, глюкокортикостероиды, пероральные эстрогены, изотретиноин) |
| Употребление алкоголя |
Вторичные гипертриглицеридемии, как и первичные формы, могут быть градуированы по классической классификации Фредриксона (Fredrickson D.S., Lees R.S., 1965). Вместе с тем, исключая тип IIa гиперлипидемий, при всех остальных видах нарушения обмена липидов отмечают повышение уровня ТГ в плазме крови (Jacobson T.A. et al., 2007). С другой стороны, абсолютный уровень элевации ТГ при различных формах гипертриглицеридемий неодинаков. Полагают, что пациенты с плазменной концентрацией ТГ >2000 мг/дл (22,6 ммоль/л) практически всегда имеют генетические причины для элевации ТГ (Brunzell J.D., Deeb S.S., 2000). Подобный дефект выявляют приблизительно в 1,8 случаев на 100 тыс. населения. Более тяжелые формы гипертриглицеридемии, сопровождающиеся повышением плазменного пула ТГ >2000 мг/дл (22,6 ммоль/л), наблюдаются не чаще 1 случая на 1 млн населения или даже реже (Fortson M.R. et al., 1995). Потенциально опасным для жизни осложнением гипертриглицеридемии является острый панкреатит (Gan S.I. et al., 2006), риск возникновения которого считается минимально приемлемым при концентрации ТГ в плазме крови <500 мг/дл (5,65 ммоль/л) (Athyros V.G. et al., 2002; Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults, 2002; Yadav D., Pitchumoni C.S., 2003). Особенно высоким риск панкреатита становится при повышении ТГ в плазме крови >1000 мг/дл (11,3 ммоль/л) (Kyriakidis A.V., et al., 2006). Полагают, что избыточный уровень ТГ способствует повышению включения последних в молекулы ЛПОНП, которые у здоровых лиц практически на 90% состоят из ТГ (Heath B. et al., 2003). После приема пищи в плазме крови возрастает содержание ТГ-обогащенных ЛПОНП и хиломикронов (Schneeman O. et al., 1993). Гипотетически постпрандиальная гиперхиломикронемия способна вызывать нарушение кровообращения в зоне микроциркуляции паренхимы поджелудочной железы, что и лежит в основе отека, ишемии и асептического панкреонекроза при тяжелых формах гипертриглицеридемии (Monga A. et al., 2003; Gan S.I. et al., 2006).
Большинство экспертов полагают, что изолированные вторичные гипертриглицеридемии не могут рассматриваться в рамках потенциально атерогенных нарушений липидного обмена. Действительно, тесной ассоциации между величиной кардиоваскулярного риска и содержанием в плазме крови ТГ не установлено. Тем не менее, многие вторичные гипертриглицеридемии с большим допущением можно градуировать как изолированные формы. В большинстве случаев определяется смешанная гиперлипидемия, вклад которой в формирование кардиоваскулярного риска достаточно хорошо установлен (см. главу 8).
Таким образом, вторичные гипертриглицеридемии представляют собой достаточно гетерогенную группу нарушений липидного обмена, имеющих сходный патофизиологический механизм и различающихся в основном инициальным этиологическим фактором. В этой связи вторичные гипертриглицеридемии, как и другие вторичные гиперлипидемии, принято рассматривать в непосредственной ассоциации с основной причиной их возникновения.
Гиперлипидемии, связанные с гормональными причинами
Беременность
Беременность в большинстве случаев ассоциируется с умеренным повышением в плазме крови содержания общего ХС, ХС ЛПВП, ХС ЛПНП, ХС ЛПОНП и ТГ, которые регрессируют к нормальному уровню в первые дни после рождения ребенка и связаны с эволюцией эстрогенного фона. Особенно значительно уровень общего ХС повышается у беременных с семейными формами гиперхолестеринемии, однако в этом случае отмечают снижение ХС ЛПНП и даже регресс ксантом (Gursoy A. et al., 2006; Oh R.C., Lanier J.B., 2007). Во время беременности обычно ухудшается течение гипертриглицеридемий, особенно в том случае, когда последние связаны с дефицитом липопротеинлипазы (Loo C.C., Tan J.Y., 2002).
Гормонзаместительная терапия
В ряде предшествующих исследований было установлено, что у женщин моложе 45 лет, принимающих пероральные контрацептивы, отмечается достоверное повышение содержания общего ХС, ХС ЛПОНП, ХС ЛПНП и ТГ в сыворотке крови по сравнению с теми, кто не использовал эти лекарственные средства. Вместе с тем, у пациенток старше 45 лет в период менопаузы, принимающих гормонзаместительную терапию на основе эстрогенов, не были зарегистрированы указанные выше изменения, напротив в этой когорте женщин чаще отмечали изолированное повышение ХС ЛПВП. Более того, при сопоставлении двух популяций женщин в возрасте старше 45 лет, получающих и не получающих эстрогены, элевация ХС ЛПВП верифицировалась только в первой из них. Исследователи пришли к заключению о том, что эстрогены способствуют редукции проатерогенных фракций липопротеидов и повышению ХС ЛПВП, что отражается в более низком кардиоваскулярном риске у пациентов с сохраненной фертильностью вне приема сбалансированных контрацептивов (Gardner C.D. et al., 2007).
Тем не менее, существуют наблюдения, свидетельствующие о том, что использование эстрогенов как трансдермальных контрацептивов или в качестве гормонзаместительной терапии, а также в случаях их применения в составе комбинированной терапии злокачественных новообразований простаты возможна индукция достаточно выраженной гипертриглицеридемии, иногда сопровождающейся возникновением острого панкреатита. В качестве механизма, объясняющего возникновение эстроген-индуцированной гипертриглицеридемии, рассматривают редукцию липолиза и/или повышение синтеза ЛПОНП. Снижение ХС ЛПВП при проведении гормонзаместительной терапии является результатом влияния гестагенов, входящих в состав последней. Причем включение в фиксированную комбинацию эстроген-гестагенных контрацептивов норэтистерона или левоноргэстрела способствует реализации различных по тяжести проатерогенных дислипидемий.
Результаты завершенных к настоящему времени рандомизированных клинических исследований свидетельствуют, что у женщин с высоким риском возникновения кардиоваскулярных событий необходимо взвешенно подойти к использованию даже низкодозовых фиксированных комбинаций эстрогенов и гестагенов, поскольку подобное решение может негативно отразиться на вероятности манифестации атеротромботических событий. В то же время, изолированное применение эстрогенов с гормонзаместительной целью, с одной стороны, не способствует ожидаемой редукции кардиоваскулярного риска, а с другой — приводит к повышению риска рака молочной железы и новообразований эндометрия.
Аналоги тестостерона в прошлом применяли для лечения V типа гиперлипидемии у мужчин, поскольку они способствовали повышению активности печеночной липазы. Однако в последующем оказалось, что метилтестостерон оказывает негативное влияние на липидный профиль плазмы крови, способствуя повышению уровня ХС ЛПНП и снижению ХС ЛПВП, а также повышает риск возникновения кардиоваскулярных событий, в связи с чем использование этого класса лекарственных средств имеет исключительно исторический интерес (Shils M.E. et al., 2007).
Анаболические стероиды достоверно ухудшают липидный профиль за счет редукции содержания антиатерогенных фракций липопротеидов, таких как ЛПВП.
Гипотиреоидное состояние
Субклинический и манифестный гипотиреоз часто ассоциируется с персистирующей гиперлипидемией, обычно имеющей обратимый характер (Yuan G. et al., 2007). В этой популяции пациентов обычно детектируется гиперхолетеринемия IIa и IIb фенотипа по классификации Фредриксона, но иногда отмечают и другие формы, такие как III или IV фенотип. В липидном профиле выявляют одновременное повышение ХС ЛПНП и ХС ЛПВП, вероятно, обусловленное потенциально обратимым снижением их клиренса, подобно таковому у пациентов с семейными формами гиперхолестеринемии. При проведении популяционных исследований было установлено, что у около 8% всех пациентов с плазменным содержанием общего ХС, превышающим 8 ммоль/л, отмечали субклинический гипотиреоз (McKenney J., 2005). Большинство из этих больных — женщины в возрасте старше 40 лет.
Поскольку нарушения липидного профиля плазмы крови при гипотиреозе имеют хотя и обратимый, но потенциально атерогенный характер, действующие клинические рекомендации обращают внимание специалистов на необходимость проведения скринирующих мероприятий в популяции пациентов с гиперхолестеринемией, особенно женского пола, на наличие сопутствующего субклинического гипотиреоза путем мониторирования плазменного пула тиреотропного гормона и Т4.
Гиперлипидемии вследствие метаболических нарушений
Сахарный диабет 2-го типа
Среди факторов кардиоваскулярного риска гипер- и дислипидемия у пациентов с сахарным диабетом занимает особое место. Если при сахарном диабете 1-го типа достижение адекватного контроля за гликемией натощак способствует редукции избыточного содержания ТГ, то при сахарном диабете 2-го типа такого подхода недостаточно (Ginsberg H.N. et al., 2005). При отсутствии диетических ограничений, модификации образа жизни и настойчивой гиполипидемической стратегии лечения смертность от кардиоваскулярных причин в течение первых 5 лет после возникновения заболевания у пациентов этой категории может достигать 65%.
Наиболее характерными и общими признаками гипер- и дислипидемии у пациентов с сахарным диабетом 2-го типа являются преимущественное повышение уровня ТГ и ЛПОНП в плазме крови, ассоциированное с редукцией концентрации ЛПВП (табл. 6.13). Предполагают, что инициальным триггером, запускающим каскад последующих патогенетических механизмов, является инсулинорезистентность (Marcoux C. et al., 2000). Вместе с тем, установлено, что дислипидемия может быть верифицирована у пациентов уже на ранней стадии заболевания, как, например, на этапе преддиабета — нарушение содержания глюкозы натощак, или как дефинизирующий компонент в составе метаболического синдрома (Cohn J.S. et al., 1999). Кроме того, гипердислипидемия в этой популяции пациентов может быть опосредована и иными факторами, такими как нарушение характера питания, связанного с употреблением высококалорийной пищи с повышенным содержанием углеводов и/или жиров, а также с рядом наследственных причин (Wilson D.E. et al., 1993). К последним относят мутацию экзона гена, кодирующего синтез циркулирующей липопротеинлипазы (Wilson D.E. et al., 1993; Jap T.S. et al., 2003), а также точечные генные мутации, затрагивающие процессы синтеза рецепторов для апопротеинов (Ayyobi A.F., Brunzell J.D., 2003) (табл. 6.14).
Таблица 6.13 Характеристика основных нарушений липидного обмена у пациентов с сахарным диабетом 1-го и 2-го типов
| Характеристика популяции | Основные классы липопротеидов | |||
| Общий ХС/ ХС ЛПНП |
ТГ | ХС ЛПВП | Не-ЛПНП | |
| Адекватно контролируемый сахарный диабет | ||||
| 1-й тип | Нормальный или ↓ | Нормальный или ↓ | Нормальный или ↑ | Нормальный или ↑ |
| 2-й тип | Нормальный | ↑ | ↓ | ↓ |
| Неадекватно контролируемый сахарный диабет | ||||
| 1-й тип | ↑ | ↑ | Нормальный | Нормальный или ↓ |
| 2-й тип | ↑ | ↑ или ↑↑ | ↓ | ↓ |
| Диабетическая нефропатия | ↑ или ↑ | ↑ или ↑↑ | ↓ | ↓ |
Таблица 6.14 Первичные и вторичные причины гиперлипидемии у пациентов с сахарным диабетом
| Клинические состояния | Плазменное содержание липидов | ||
| Общий ХС | ТГ | ХС ЛПВП | |
| Первичные причины | |||
| Семейная гиперхолестеринемия | ↑↑ | Нормальный | ↓ |
| Семейная комбинированная гиперхолестеринемия | ↑↑ или ↑↑↑ | Нормальный или ↑ | ↓ или ↓↓ |
| Вторичные причины | |||
| Гипотиреоидизм | ↑ | Нормальный | Нормальный или ↓ |
| Ятрогенные причины | |||
| Алкоголь | Нормальный | ↑ | ↑ |
| Эстрогены | Нормальный | ↑ | ↑ |
| Тиазидные диуретики | ↑ | ↑ | Нормальный или ↓ |
| Блокаторы β-адренорецепторов | Нормальный или ↑ | ↑ | ↓ |
Предполагают, что инсулинорезистентность приводит к усилению липолиза и высвобождению большого количества СЖК из адипоцитов, что в сочетании с гипергликемией создает условия для ресинтеза ТГ в гепатоциты по глицерофосфатному пути (Bays H. et al., 2005). В результате в плазме крови выявляют избыточное содержание ЛПОНП, обогащенных ТГ. Кроме того, возникающая гипертриглицеридемия может быть связана со снижением катаболизма ЛПОНП, непосредственно обусловленным редукцией активности внепеченочной липопротеинлипазы (Julius U., 2003). Результатом двух описанных процессов является уменьшение содержания в плазме крови ЛПВП (Bays H. et al., 2004).
Большое значение в формировании и прогрессировании атеросклероза у пациентов с сахарным диабетом 2-го типа имеют качественные и количественные изменения структуры липопротеидов и апопротеинов. Среди последних условно можно выделить следующие: наличие малых плотных ЛПНП, которые формируются из ЛПНП, обогащенных ТГ, а также неферментативная модификация структуры ЛПНП, ЛПВП и апо-А/В вследствие гликозилирования. Последний процесс непосредственно ассоциируется с выраженностью гликемии и хорошо коррелирует с абсолютным уровнем гликолизилированного гемоглобина. Модифицированные ЛПНП характеризуются повышенной доступностью для последующей пероксидации макрофагами. Этот процесс рассматривается как ключевой механизм образования пенистых клеток, инфильтрации интимы липидами и ускорения развития атеросклероза. Качественные изменения отмечают также и в структуре ЛПВП в виде гликозилирования апопротеинов, что приводит к повышению содержания в них ТГ и количества малых плотных ЛПВП. Эти изменения негативно отражаются на функциональной способности ЛПВП к транспортировке ХС.
Метаболический синдром
Гиперлипидемия при метаболическом синдроме отличается преимущественным повышением плазменного уровня ТГ и редукцией ХС ЛПВП, в частности ЛПВП2, что напоминает в целом липидный профиль при сахарном диабете 2-го типа (Yuan G. et al., 2007). Фактически, являясь дефинизирующим компонентом метаболического синдрома, сахарный диабет в значительной мере определяет характер сопутствующих нарушений липидного обмена (Ginsberg H.N. et al., 2005). Однако, выраженность последних при метаболическом синдроме без верифицированного сахарного диабета по сравнению с когортой пациентов с сахарным диабетом отличается. К настоящему времени установлено, что между количеством компонентов метаболического синдрома и тяжестью гиперлипидемии есть прямая корреляционная взаимосвязь (Ayyobi A.F. et al., 2003). Вместе с тем, риск возникновения кардиоваскулярных событий максимальный у тех пациентов, в состав метаболического синдрома которых входят не только инсулинорезистетность и/или андроидный тип ожирения, но и сахарный диабет 2-го типа. С другой стороны, количественный анализ состава липидного профиля плазмы крови показывает, что концентрации ЛПНП у больных с метаболическим синдромом с и без сахарного диабета не различаются (Cohn J.S. et al., 1999; Marcoux C. et al., 2000; Sacks F.M., 2002). Однако в плазме крови больных с сахарным диабетом аполипопротеины представлены в основном мелкими и плотными частицами, тогда как популяции больных без диабета — крупными частицами (Zambon A. et al., 2006). Известно, что атерогенный потенциал апо-В липопротеинов, представленных виде мелких и плотных частиц, более высок, чем у крупных апо-А-содержащих липопротеидов (Austin M.A. et al., 1990; Lamarche B. et al., 1997; Sacks F.M., 2002).
Таким образом, в количественном отношении липидный спектр плазмы крови у пациентов с метаболическим синдромом напоминает таковой у пациентов с сахарным диабетом, а в качественном выражении может существенным образом отличаться. Это в какой-то мере объясняет существующие различия в расчетной величине кардиоваскулярного риска между этими двумя популяциями пациентов (Smith S.C. Jr. et al., 2006).
Ожирение
Традиционно абдоминальное ожирение (ИМТ >30 кг/м2, окружность талии >102 см у мужчин и >88 см у женщин) рассматривают как один из наиболее значимых факторов кардио- и цереброваскулярного риска, включая риск общей смерти (Manson J.E. et al., 1995; Williams M.A. et al., 2002; Fontaine K.R. et al., 2003), ассоциированного с гиперлипидемией, инсулинорезистентностью, нарушением уровня глюкозы натощак, а также включают в качестве дефинизирующего компонента в состав метаболического синдрома (Chan D.C. et al., 2004).
Распространенность ожирения прогрессивно возрастает не только в развитых странах мира. Так, в США и странах Европы у около 30% людей отмечают ожирение, а избыточную массу тела регистрируют почти у 63% мужчин и 55% женщин в общей популяции (Mokdad A.H. et al., 2001; Flegal K.M. et al., 2002; Weil E. et al., 2002).
Полагают, что негативное влияние ожирения в отношении клинических исходов опосредуется не только гиперлипидемией, но и другими факторами кардиоваскулярного риска, такими как артериальная гипертензия, снижение физической активности, инсулинорезистентность, сахарный диабет. В то же время, необходимо отметить, что многие из них потенцируют друг друга (Mann G.V., 1974; Turcato E. et al., 2000). Так, постпрандиальная гипертриглицеридемия тесно коррелирует с уровнем физической активности и выраженностью ожирения (Weil E. et al., 2002; Nishijima H. et al., 2007; Zhang J.Q. et al., 2007), а риск возникновения инсульта ассоциируется с тяжестью артериальной гипертензии (Abbott R.D. et al., 1994; Rexrode K.M. et al., 1997). Вместе с тем, согласно результатам популяционного исследования Physicians’ Health Study в мужской популяции увеличение массы тела может рассматриваться как самостоятельный фактор риска ишемического инсульта независимо от наличия артериальной гипертензии, сахарного диабета или гиперлипидемии (Kurth T. et al., 2002). Напротив, в женской популяции подобных результатов верифицировано не было (DiPietro L. et al., 1994; Rexrode K.M. et al., 1997; Dey D.K. et al., 2002; Ford E.S. et al., 2003; Suk S.H. et al., 2003). Вместе с тем, в мужской популяции негативное влияние ожирения в отношении риска возникновения цереброваскулярных событий сохранялось для представителей различных рас и этнических групп, участвующих в Northern Manhattan Study (Suk S.H. et al., 2003). Необходимо отметить, что, в отличие от кардиоваскулярных заболеваний, частота воникновения цереброваскулярных событий, в том числе транзиторной ишемической атаки, ишемического инсульта, интракраниальных кровоизлияний, не снижается при уменьшении массы тела, хотя при этом и наблюдается регресс уровня артериального давления, улучшение липидного профиля плазмы крови, снижение уровня глюкозы натощак (Anderson J.W., Konz E.C., 2001). Более того, приверженность к средиземноморской диете способствует улучшению липидного профиля плазмы крови, снижению уровня артериального давления и редукции риска возникновения кардиоваскулярных событий, включая случаи внезапной сердечной смерти, тогда как риск мозгового ишемического инсульта при этом обычно не редуцируется (Renaud S. et al., 1995; Singh R.B. et al., 2002; Krauss R.M. et al., 2000).
Таким образом, необходимо заключить, что гипердислипидемию не следует рассматривать как определяющий фактор риска, обусловливающий влияние на ближайший и отдаленный прогноз у пациентов с избыточной массой тела или ожирением.
Подагра
Одним из наиболее часто диагностируемых нарушений липидного обмена у пациентов с идиопатической подагрой является гипертриглицеридемия. Последняя идентифицируется приблизительно у 5% больных с сохраненной почечной функцией и без подагрической нефропатии. Плазменное содержание ТГ натощак колеблется от 2 до 14 ммоль/л в зависимости от диетических предпочтений пациента. Обычно в этой популяции больных выявляют V или реже IV фенотип гиперлипидемии по Фредриксону (McKenney J., 2005). Причем между выраженностью гиперурикемии и повышением уровня ТГ в плазме крови прямой ассоциации не выявлено. Рутинное применение аллопуринола не способствует улучшению липидного профиля плазмы крови, несмотря на достижение адекватного контроля над концентрацией мочевой кислоты в крови (Fung A., Frohlich J.J., 2002). Предполагают, что характерные для идиопатической подагры сопутствующие факторы кардиоваскулярного риска, такие как ожирение и употребление алкоголя, а также, возможно, использование тиазидных диуретиков способны оказывать неблагоприятное влияние на формирование нарушений липидного обмена независимо от выраженности гиперурикемии (Dunbar R.L., Rader D.J., 2005). С другой стороны, установлено, что у пациентов с первичным IV типом гиперлипидемии часто отмечают сопутствующую гиперурикемию, а использование фибратов приводит к редукции не только плазменной концентрации ТГ, но и мочевой кислоты. Напротив, в этой популяции пациентов производные никотиновой кислоты, способствуя уменьшению выраженности гипетриглицеридемии, индуцируют ухудшение гиперурикемии (Bays H., Stein E.A., 2003; Harper C.R., Jacobson T.A., 2006). Таким образом, гиперлипидемия в когорте пациентов с идиопатической подагрой включает различные состояния, многие из которых могут быть рассмотрены как ятрогенные или первичные.
Прогрессирующая частичная липодистрофия
Прогрессирующая частичная липодистрофия представляет собой достаточно редкое заболевание, иногда ассоциированное с семейным характером наследования, характеризующееся прогрессирующей редукцией объема подкожной клетчатки верхней половины тела, а также перераспределением аккумуляции жировой ткани преимущественно на нижних конечностях. В клинический картине доминируют нарушение толерантности к углеводам с повышением концентрации глюкозы натощак с последующим высоким риском возникновения сахарного диабета, гломерулонефрит, гепатоцеллюлярная недостаточность и тяжелая гипертриглицеридемия. Этиологическая природа заболевания не установлена, а лечение и профилактика не разработаны.
Болезни накопления
Болезнь Гоше, как и другие болезни накопления гликогена, сопровождается формированием гипертриглицеридемии, значение которой в формировании кардиоваскулярного риска в этой популяции пациентов точно не установлено. Известно, что формирование порто-кавального анастомоза способствует реверсии гипертриглицеридемии.
Гиперлипидемии у пациентов с нефропатиями
ХБП и нефротический синдром
Общее содержание циркулирующих липидов у больных с нефротическим синдромом, ассоциированным с ХБП, существенно повышено (приблизительно в 8–10 раз по сравнению с нормальными значениями). Для этой формы вторичной гиперлипидемии характерно повышение концентрации всех основных липидных фракций: ТГ, общего ХС и фосфолипидов, НЭЖК. Обычно у пациентов идентифицируют элевацию ЛПНП, ЛПОНП, хиломикронов при нормальных или сниженных концентрациях ЛПВП. Формирующийся тип нарушений липидного профиля соответствует IIb и IV типу по классификации Фредриксона. Механизм развития этой формы гиперлипидемии окончательно не выяснен. Тем не менее, полагают, что возникновение последней может быть связано с задержкой липопротеидов как высокомолекулярных веществ в сосудистом русле, с усиленным синтезом ХС в печени, снижением активности липолитических ферментов, возможно, с нарушением метаболической функции почек. Кроме того, обсуждается роль гипоальбуминемии, снижения онкотического давления, повышения вязкости крови, агрегации тромбоцитов и выделения ими тромбоцитарного фактора роста, дисбаланса циклооксигеназных метаболитов (тромбоксана и простагландина I2), снижения активности печеночной липазы, повышения активности процессов свободно-радикального и перекисного окисления липидов и белков клеточных мембран и т.д. Кроме того, при хроническом гломерулонефрите нарушается утилизация почками мевалоновой кислоты, основного промежуточного продукта цепи образования ХС из ацетил-КоА.
Вместе с тем, проатерогенный потенциал плазмы крови у этих пациентов может оказывать негативное влияние на риск манифестации атеротромбоза. Однако некоторые исследователи подвергают сомнению выше указанное предположение, поскольку формирующаяся при нефротическом синдроме гипопротеинемия и необычно крупные размеры частиц, обогащенных ТГ, прежде всего ЛПНП, некоторым образом ограничивают способность последних к инфильтрации субинтимы. Напротив, гиперлипидемия при терминальной почечной дисфункции рассматривается как одна из основных причин возникновения кардиоваскулярных осложнений, в том числе и атеротромботических событий, таких как инфаркт миокарда и ишемический инсульт. Кроме того, накопление липидов в мезангии и интерстиции канальцевого аппарата нефрона способствует прогрессированию заболевания и укорачивает додиализный период.
Терминальная почечная дисфункция, требующая проведения заместительной терапии
Гиперлипидемия при терминальной почечной дисфункции представлена преимущественно гипертриглицеридемией, которая в основном обусловлена метаболическими нарушениями. Дополнительное поступление в организм больного глюкозы при лечении гемодиализом с раствором, содержащим глюкозу, усугубляет гипертриглицеридемию. Концентрация общего ХС и СЖК в плазме крови больных с терминальной почечной дисфункцией обычно нормальная, тогда как уровень ЛПНП и ЛПОНП обычно повышен, а ЛПВП нормален или снижен. Перечисленные сдвиги в липидном спектре крови у этой категории больных соответствуют IV типу гиперлипидемии по классификации Фредриксона, который является потенциально атерогенным.
Гиперлипидемия развивается также у больных с почечным трансплантатом, несмотря на его удовлетворительную функцию. Она значительно резче выражена в первый год после процедуры трансплантации почки и характеризуется повышением содержания в плазме крови как ТГ, так и общего ХС. В большинстве случаев гиперлипидемия относится к IIb и IV, реже — к IIa типу. В ее генезе важное значение имеет иммуносупрессивная терапия, носящая наиболее интенсивный характер в первый год после трансплантации (см. ниже).
Гиперлипидемии при первичном билиарном циррозе и синдроме холестаза
Длительный холестаз, в том числе и при первичном билиарном циррозе, сопровождается формированием выраженной гиперлипидемии вследствие повышения содержания липопротеида-Х (Lp-X). В результате проведения клинических исследований было установлено, что прогрессирование холестаза при первичном билиарном циррозе может приводить к дефициту ЛХАТ вследствие ретенции лецитина из желчи в плазму крови. В результате взаимодействия между свободным ХС, подвергающимся эстерификации ЛХАТ, альбумином, апо-С формируется Lp-X. Диагноз верифицируется при выявлении последнего при проведении электрофореза на бумаге.
Кожные ксантомы и нейропатия тесно ассоциированы с тяжестью гиперлипидемии и плазменным содержанием Lp-X. Более того, установлено, что риск возникновения и прогрессирования атеросклероза в этой популяции пациентов хорошо коррелирует с содержанием Lp-X.
Проведение гиполипидемических мероприятий у пациентов с первичным билиарным циррозом может представлять значительные трудности. Так, фибраты обычно усугубляют гиперхолестеринемию, секвестранты желчных кислот в этих случаях малоэффективны, а применение статинов ограничивается печеночно-клеточной недостаточностью (Havel R.J., Rapaport E., 1995).
Гиперлипидемии, связанные с употреблением алкоголя или токсических веществ
Алкоголь
Употребление этанола обычно рассматривается как одна из наиболее частых причин формирования в популяции вторичной гипертриглицеридемии, которая градуируется как IV или V фенотип по классификации Фредриксона. Даже умеренное употребление легких алкогольных напитков (менее 2 унций этанола в сутки) способствует возникновению клинически значимой гипертриглицеридемии, причем последняя может быть более выражена у лиц с предшествующей первичной гиперлипопротеидемией IV типа. Кроме того, для этих пациентов характерно повышение плазменного содержание ЛПВП2 и ЛПВП3 вследствие блокады активности циркулирующей липопротеинлипазы.
Один из возможных механизмов, объясняющих возникновение гипертриглицеридемии при употреблении алкоголя, состоит в конкурентной блокаде окисления СЖК в гепатоцитах, вследствие чего последние включаются в избыточный синтез ТГ. Отказ от дальнейшего употребления алкоголя обычно приводит к редукции или даже полной реверсии гипертриглицеридемии.
Иногда длительный прием алкоголя, особенно крепких напитков, способствует возникновению так называемого синдрома Циве, который характеризуется тяжелой гиперлипидемией, гемолизом и желтухой. Плазменное содержание ТГ обычно колеблется в пределах 3–327 ммоль/л, а детектируемая гиперлипидемия обычно градуируется как V фенотип по классификации Фредриксона.
Хлорсодержащие соединения
Отравление дихлордифенилтрихлорметилметаном (ДДТ) и близкими к нему по структуре инсектицидами обычно сопровождается селективным повышением плазменного уровня ХС ЛПВП. Существующие к настоящему времени наблюдения за популяцией подобных пациентов свидетельствуют о потенциально обратимом характере выявленных нарушений липидного обмена. Значение последних как факторов кардиоваскулярного риска до конца не установлено.
Диоксин
Отравление диоксином, который рассматривается как своеобразный индустриальный токсин, сопровождается появлением гирсутизма, угревой сыпью, рассеянной неврологической симптоматикой и гиперхолестеринемией. Возможно, последний симптом обусловлен диоксин-индуцированным гипотиреозом, который может длительно оставаться субклиническим (см. Гиперлипидемии, связанные с гормональными причинами).
Гиперлипидемии вследствие ятрогенных причин
Среди ятрогенных причин, способствующих формированию гиперлипидемии, наибольшее значение имеют некоторые антигипертензивные лекарственные средства (блокаторы β-адренорецепторов, тиазидные диуретики), иммуносупрессанты, включая глюкокортикостероиды, а также АРВП.
Гиперлипидемия, развивающаяся при применении антигипертензивных лекарственных средств
Доказанным негативным влиянием в отношении липидного обмена обладают тиазидные диуретики и блокаторы β-адренорецепторов. Так, длительное применение гидрохлоротиазида и хлорталидона способствует повышению плазменной концентрации общего ХС, ТГ, а также снижению уровня ЛПОНП и ЛПНП. Интересно, что при этом концентрация ремнентных форм ЛПВП не претерпевает существенных изменений. Необходимо отметить, что чаще всего гиперлипидемия не детектируется как изолированный побочный эффект этих лекарственных средств. В большинстве случаев удается выявить и повышение уровня глюкозы натощак в сочетании с умеренной гиперурикемией. Указанные нарушения особенно часто идентифицируются у женщин в период постменопаузы и могут быть в значительной мере минимизированы с помощью диетических ограничений. В то же время для индапамида, являющегося тиазидоподобным диуретиком, не было получено доказательств существования клинически значимых изменений липидного профиля плазмы крови даже при длительном применении в различных популяциях пациентов с артериальной гипертензией.
Традиционно блокаторы β-адренорецепторов без внутренней симпатомиметической активности рассматриваются как лекарственные средства, особенно часто индуцирующие вторичную гиперлипидемию в виде гипертриглицеридемии или снижения содержания ЛПВП. В то же время, согласно результатам завершенных рандомизированных клинических исследований, элевация ТГ регистрируется у 15–30% пациентов, получавших лекарственные средства этого класса, а редукция ЛПВП — у 6–8% больных. Напротив, широкое применение блокаторов β-адренорецепторов с внутренней симпатомиметической активностью или с дополнительным α-блокирующим эффектом показало, что эти средства не обладают клинически значимым негативным влиянием в отношении липидного обмена. Однако, принимая во внимание вероятность возникновения подобных побочных эффектов, во многих клинических рекомендациях по лечению артериальной гипертензии блокаторы β-адренорецепторов исключены из списка препаратов первой линии. В то же время они сохраняют свои позиции в популяции пациентов с так называемыми принудительными показаниями: клиническими состояниями, при которых позитивное влияние блокаторов β-адренорецепторов в отношении клинических исходов доказано и польза от их применения превышает ожидаемый риск (National Institute of Clinical Excellence, 2004; Williams B. et al., 2004; Mancia G. et al., 2007; Zanchetti A. et al., 2009).
Основными механизмами возникновения дислипидемии при применении антигипертензивных лекарственных средств являются снижение активности циркулирующей липопротеинлипазы и ингибирование аденилатциклазы в адипоцитах, что приводит к повышению уровня ТГ и нарушению конвертации ЛОПНП в ЛПВП. В результате этого уровень ЛПВП снижается. В то же время нельзя исключить и тот факт, что указанные нарушения обмена липидов могут возникать у предрасположенных к этому лиц, например, с гетерозиготной формой семейной гипертриглицеридемии. В этом случае применение блокаторов β-адренорецепторов или тиазидных диуретиков, супрессирующих активность липопротеинлипазы, является своеобразным разрешающим фактором. Тем не менее, большинство клинических рекомендаций, посвященных лечению и профилактике артериальной гипертензии, обращают внимание врача на необходимость постоянного мониторинга безопасности приема блокаторов β-адренорецепторов и тиазидных диуретиков (National Institute of Clinical Excellence, 2004; Williams B. et al., 2004; Mancia G. et al., 2007).
Посттрансплантационная гиперлипидемия
Гиперлипидемию достаточно часто выявляют у реципиентов трансплантированных органов, определяя вероятность манифестации атеросклероза и кардиоваскулярных событий, ассоциированных с атеротромбозом. Причем последние в значительной мере определяют выживаемость больных в посттрансплантационный период (Oliver M.F., 1995). Мониторирование липидного профиля плазмы крови может иметь определяющее значение для выбора тактики лечения и стратегии минимизирования кардиоваскулярного риска в этой популяции пациентов (Oliver M.F., 1995). В многочисленных клинических рандомизированных и популяционных исследованиях убедительно подтверждена высокая эффективность гиполипидемических мероприятий, базирующихся на диетических ограничениях и различных медикаментозных режимах (статины, фибраты, секвестранты желчных кислот), в программах первичной и вторичной превенции кардиоваскулярных событий (Scandinavian Simvastatin Survival Study Group, 1994; Havel R.J., Rapaport E., 1995; Shepherd J. et al., 1995). Полученные данные привели к формированию мнения о возможности экстраполяции успехов, достигнутых в общей популяции, на когорту пациентов, перенесших трансплантацию органов с целью улучшения ближайшего и отдаленного исходов за счет редукции кардиоваскулярного риска. Необходимо отметить, что ряд метаболических нарушений (метаболический синдром, повышение уровня глюкозы в плазме крови натощак, гиперурикемия, гипергомоцистеинемия), а также гиперлипидемия не только детектируются у потенциальных реципиентов перед процедурой трансплантации, но и манифестируются впервые после ее проведения (McDiarmid S.V. et al., 1992). Чаще всего в качестве основной причины, способствующей их появлению, рассматривают агрессивную иммуносупрессорную терапию. Хотя нельзя исключать реализацию традиционных факторов риска, таких как курение, употребление алкоголя, артериальная гипертензия, нарушение диетического рациона, а также мужской пол и отягощенный семейный анамнез, как вероятных индукторов метаболических нарушений и атеросклероза независимо от возраста реципиента (Gokal R. et al., 1979; Raine A.E.G. et al., 1987).
Иммуносупрессорные стратегии лечения, включающие глюкокортикостероиды, циклоспорин и такролимус (tacrolimus), обладают доказанной потенциально высокой способностью к индукции возникновения гиперлипидемии и новых случаев сахарного диабета (Morrisett J.D. et al., 1991; Dunn S.P. et al., 1994; Jindal R.M., 1994). Частота посттрансплантационной гиперлипидемии (ПТГЛ) при применении различных иммуносупрессоров варьирует от 22 до 54% в популяции реципиентов донорских органов. Так, при сопоставлении двух различных протоколов иммуносупрессорной терапии в когорте больных, перенесших трансплантацию почки, оказалось, что ПТГЛ детектируется у 42,2% пациентов, получавших комбинацию азатиоприна и преднизолона, и у 26,3% — при лечении циклоспорином и преднизолоном. У больных с ортотопической трансплантацией сердца плазменное содержание общего ХС и ХС ЛПНП было выше при использовании в качестве иммуносупрессорной стратегии лечения комбинации циклоспорина и преднизолона по сравнению с комбинацией циклоспорина и азатиоприна (Taylor D.O. et al., 1989; Vatsala A. et al., 1989). По данным R.M. Jindal и соавторов (1996) ПТГЛ верифицируется у 58% после ортотопической трансплантации печени. Кроме того, в этой популяции у 37% больных отмечали посттрансплантационный сахарный диабет. Схожие данные были получены S.J. Munoz и соавторами (1991). В целом, многие эксперты склоняются к мнению о том, что гиперлипидемия у реципиентов донорских органов неоднородна по своему составу и включает различные виды нарушений липидного обмена, как первичные (особенно у детей, подростков и молодых лиц с семейно-отягощенным анамнезом), так и вторичные, в том числе связанные с проведением иммуносупрессорной терапии. В любой ситуации ПТГЛ рассматривается как фактор высокого кардиоваскулярного риска и требует проведения гиполипидемических мероприятий. Исключая ситуации с использованием иммуносупрессорных лекарственных средств, подход к лечению гиперлипидемии у реципиентов донорских органов не отличается от такового у лиц общей популяции. В этой связи более детально ПТГЛ, индуцированная агрессивной иммуносупрессорной терапией, рассматривается в отдельном разделе, который приведен ниже.
Гиперлипидемия, связанная с применением иммуносупрессантов
Глюкокортикостероиды
Существуют доказательства негативного влияния длительной терапии глюкокортикостероидами на состояние липидного обмена (Bierman E.L., 1992). Глюкокортикостероиды также индуцируют формирование инсулинорезистентности и способствуют избыточной продукции гепатоцитами ЛПОНП и снижению активности липопротеинлипазы, что, в свою очередь, ассоциируется с повышением синтеза ТГ (Gilman A.G., Goodman L.S., 1990; Havel R.J., 1990). В целом, терапия глюкокортикостероидами ассоциируется с повышением риска развития ПТГЛ, инсулинорезистентности, сахарного диабета 2-го типа и атеросклероза (Garg A., Grundy S.M., 1990; Reaven G.M., Laws A., 1994; Stem M.P., 1996). Кроме того, развивающаяся диабетическая нефропатия не только ухудшает ближайшие клинические исходы, но и является самостоятельным фактором риска возникновения кардиоваскулярных событий, оказывающих негативное влияние на отдаленный прогноз (The Diabetes Control and Complications Trial Research Group., 1993; Bucala R. et al., 1994; Defronzo R.A., Goodman A.M., 1995; Savage P.J., 1996 ).
Циклоспорин, такролимус, микофеноловая кислота, имуран, сиролимус
Многочисленные исследования показали, что циклоспорин способствует повышению плазменного содержания общего ХС и ТГ с последующей аккумуляцией липидов в кардиомиоцитах, паренхиме трансплантата, почек и печени. Механизм этого процесса является мультифакторным. Так, установлено, что циклоспорин блокирует синтез желчных кислот посредством ингибирования 26-гидроксилазы (ключевого фермента первого этапа синтеза желчных кислот), ограничивая деградацию ХС. Это, в свою очередь, приводит к снижению обратного захвата ХС ЛПНП и экспрессии соответствующих рецепторов на поверхности мембран гепатоцитов по механизму down-regulation (deGroen P., 1988). В экспериментальных исследованиях установлено, что циклоспорин стимулирует синтез ХС посредством повышения активности ключевого фермента ГМГ-КoA-редуктазы. Кроме того, препарат оказывает негативное влияние на активность циркулирующей липопротеинлипазы, что снижает клиренс ЛПОНП и хиломикронов, приводя к гипертриглицеридемии (Superko H. et al., 1990). В качестве еще одного механизма, благодаря которому циклоспорин индуцирует гиперхолестеринемию, рассматривается нарушение секреции фосфолипидов с желчью. Существует вероятность того, что и генетические факторы способны принимать участие в проатерогенном эффекте циклоспорина А (Ballantyne C.M. et al., 1989).
Такролимус (FK506) способен индуцировать процесс окисления ЛПНП, являющегося важным этапом в формировании и прогрессировании атеросклероза (Palumbo J.D. et al., 1987; Apanay D.C. et al., 1994). В ряде рандомизированных клинических исследований удалось установить, что при сопоставимом терапевтическом потенциале профиль переносимости у такролимуса по сравнению с циклоспорином значительно лучше (The US Multicenter FK506 Liver Study Group, 1994), а способность индуцировать ПТГЛ существенно ниже (Jindal R.M. et al., 1994). Причем такролимус превосходил циклоспорин и по способности оказывать меньшее негативное влияние в отношении углеводного обмена и индукции инсулинорезистентности (Jindal R.M. et al., 1994). Сходные данные были получены и другими исследователями (Krentz A.J. et al., 1993; Munoz S.J., 1995).
Микофеноловая кислота и азатиоприн обычно не оказывают неблагоприятного влияния на липидный или углеводный обмен (Halloran P. et al., 1997). Тем не менее, такая возможность не исключается, особенно при комбинированном применении с глюкокортикостероидами. В ряде исследований установлен факт реверсии ПТГЛ после отмены циклоспорина и назначения микофеноловой кислоты или азатиоприна (Sandbom W.J. et al., 1994).
Сиролимус по химической структуре является макролидом, подобным такролимусу, и демонстрирует достаточно близкий к последнему фармакокинетический и фармакодинамический профиль (Murgia M. et al., 1996). Установлено, что у пациентов, перенесших трансплантацию почки, препарат часто проявляет способность повышать плазменный уровень липидов, особенно общего ХС, ХС ЛПНП, тогда как содержание ТГ обычно не изменяется (Murgia M. et al., 1996). Механизм этого явления пока точно не выявлен.
Установлено, что глюкокортикостероиды и цитостатики при одновременном применении могут оказывать синергичное влияние в отношении вероятности возникновения ПТГЛ. Так, оказалось, что кумулятивная доза глюкокортикостероидов и циклоспорина хорошо коррелирует с величиной общего ХС плазмы крови (European FK506 Multicenter Study Group, 1994).
Гиперхолестеринемия как фактор риска реакции отторжения трансплантата
К настоящему времени установлен факт взаимосвязи между интенсивностью процессов реакции отторжения трансплантата и выраженностью системного атеросклероза (Perez R.V., Alexander J.W., 1988). В экспериментальных исследованиях удалось продемонстрировать, что гиперхолестеринемия существенным образом повышает интенсивность пролиферативных процессов в интиме артерий, а также индуцирует повреждение эндотелиоцитов и формирование дисфункции эндотелия в сосудах графта, что, в частности, играет негативную роль при остром и хроническом отторжении трансплантата (Dimeny E. et al., 1995). Существующие объяснения этого феномена в основном эксплуатируют гипотезу о возможном повышении экспрессии рецепторов антигенов II класса по механизму up-regulation за счет ЛПНП. Не исключено, что последние могут оказывать хемотаксическое влияние в отношении моноцитов и NK-клеток (Dimeny E. et al., 1995). Более того, ЛПНП способны оказывать непосредственное повреждающее воздействие в отношении эндотелиоцитов, способствовать инфильтрации субинтимы липидами через привлечение в субинтиму активированных макрофагов, трансформирующихся впоследствии в пенистые клетки (Erickson K.L. et al., 1983; Hwang D., 1989).
Статины способны сохранять свое значение в качестве важнейших гиполипидемических лекарственных средств, оказывающих благоприятное влияние на ближайший и отдаленный прогноз, даже в популяции пациентов с ПТГЛ (Gerson E.J. et al., 1989; Kobashigawa J.A. et al., 1994). Причем существуют доказательства позитивного влияния статинов на течение хронической реакции отторжения трансплантата у больных с ортотопической трансплантацией сердца (Schror K. et al., 1989). В то же время, необходимо сдержанное и осторожное отношение к статинам как вероятным адъювантным средствам у больных с реакцией отторжения трансплантата, поскольку в специально спланированных рандомизированных клинических исследованиях не получено отчетливой корреляционной зависимости между выживаемостью графта и выраженностью ПТГЛ (Bumgardner G.L. et al., 1995). Таким образом, иммуносупрессия остается единственным методом контроля за выживаемостью этих пациентов с доказанным уровнем эффективности (Katznelson S., Wilkinson A.H., 1995).
Гиперлипидемия у пациентов с ВИЧ-1, получающих АРВП
В настоящее время благодаря широкому применению антиретровирусной терапии (АРВТ), основанной на использовании ингибиторов протеаз и нуклеозидных ингибиторов обратной транскриптазы, прогноз пациентов, инфицированных ВИЧ-1, значительно улучшился. Вместе с тем, указанные лекарственные средства отличаются достаточно высокой токсичностью, относительно низкой переносимостью и широким спектром негативных побочных эффектов, среди которых метаболические осложнения занимают особое место. Так, липодистрофии, связанные с различными нарушениями распределения и аккумуляции липидов, особенно ярко выраженные на лице, шее, груди, животе и бедрах, а также гиперлипидемия, инсулинорезистентность, сопутствующие нарушения уровня глюкозы в крови натощак рассматриваются как дополнительные факторы кардиоваскулярного риска, способные в значительной мере оказывать негативное влияние на прогноз в когорте пациентов, получающих АРВТ (Thiebaut R. et al., 2000; Penzak S.R., Chuck S.K., 2000; Carr A. et al., 1998; Carr A. et al., 1999). Классификация морфологических и метаболических нарушений, возникающих в результате проведения АРВТ, рекомендованная Antiretroviral-associated Lipodystrophy European Comparative Study (ALECS) Group, приведена в табл. 6.15. При этом выраженность метаболических нарушений, особенно часто детектируемых в этой когорте пациентов, также градуируется в зависимости от тяжести (табл. 6.16).
Таблица 6.15 Классификация морфологических и метаболических нарушений, возникающих в результате проведения АРВТ, рекомендованная Antiretroviral-associated Lipodystrophy European Comparative Study (ALECS) Group
| Тип | Основные морфологические характеристики | Клинические признаки |
| I | Липоатрофия | (a) Без редукции тел Биша |
| (b) С редукцией тел Биша | ||
| II | Аккумуляция липидов (липогипертрофия) | (с) Локальная аккумуляция (1 участок) |
| (d) Локальная аккумуляция (2 и более участков) | ||
| (e) Липоматоз | ||
| III | Комбинированная форма | a/b + c/d/e |
| IV | Изолированные метаболические нарушения | – |
Таблица 6.16 Классификация тяжести метаболических нарушений, ассоциированных с АРВТ
| Степень тяжести | Характеристика метаболических нарушений | |||
| Плазменное содержание общего ХС натощак, мг/дл | Плазменное содержание ХС ЛПНП натощак, мг/дл | Плазменное содержание ТГ натощак, мг/дл | Плазменное содержание глюкозы в крови натощак, мг/дл | |
| 0 (норма) | <200 | <160 | <200 | <110 |
| 1 (легкая) | 200–239 | 160–189 | 200–399 | 110–125 (интермиттирующее) |
| 2 (средняя) | 240–300 | 190–220 | 400–1000 | 110–125 (персистирующее) |
| 3 (тяжелая) | >300 | >220 | >1000 | >125 |
Необходимо отметить, что нарушения липидного обмена различной степени выраженности выявляли у пациентов, инфицированных ВИЧ-1, и до широкого внедрения в клиническую практику АРВТ, детальное изучение их природы и клинических последствий было начато позднее (Sellmeyer D.E., Grunfeld C., 1996; Penzak S.R., Chuck S.K., 2000). Так, в ранних исследованиях установлено, что гиперлипидемия, неассоциированная с АРВТ, отличается инициальным снижением в плазме крови общего ХС, ХС ЛПНП, ХС ЛПВП и повышением уровня ТГ. Таким образом, на ранних стадиях инфицированя ВИЧ-1 характерным признаком нарушений липидного обмена является изолированная гипертриглицеридемия. Отмеченные нарушения не являются специфическими для пациентов, инфицированных ВИЧ-1, посколько могут быть выявлены и у больных с различными бактериальными, вирусными или паразитарными инфекциями длительного течения. Полагают, что одной из важнейших причин, приводящих к повышению плазменного уровня ТГ, является цитокиновая провоспалительная активация, преимущественно за счет избыточной продукции интерферона α (ИФ-α). В когорте пациентов с ВИЧ-1 циркулирующий уровень последнего существенно повышается на поздних стадиях заболевания и устойчиво коррелирует с синтезом и клиренсом ТГ (позитивно и негативно соответственно). Несмотря на то что уровень ИФ-α является достаточно точным маркером нарушений липидного обмена в этой когорте пациентов, точный механизм его влияния на содержание ТГ до сих пор не установлен (Grunfeld C. et al., 1991; Grunfeld C. et al., 1992; Hellerstein M.K., Grunfeld C., Wu K. et al., 1993; Penzak S.R., Chuck S.K., 2000; Thiebaut R. et al., 2000). Полагают, что фактор некроза опухоли (ФНО)-α может вовлекаться в процесс индукции дислипидемии у пациентов с ВИЧ. В то же время, у пациентов, инфицированных ВИЧ-1, при отсутствии оппортунистических инфекций плазменное содержание ФНО-α обычно не превышает нормальных значений. В этой связи многие исследователи полагают, что редукция плазменного содержания общего ХС, ХС ЛПНП и ХС ЛПВП является ответом на возникновение инфекционных осложнений и, возможно, опосредовано повышением содержания интерлейкина-1 (ИЛ-1). С другой стороны, избыток циркулирующего ИЛ-1 у пациентов, инфицированных ВИЧ-1, обычно вообще не детектируется, а реальный плазменный пул не ассоциируется с содержанием общего ХС, ХС ЛПНП и ХС ЛПОВП (Shor-Posner G. et al., 1993; Constans J. et al., 1994; Kotler D.P., 2000). По мнению ряда исследователей, гиперхолестеринемия и гипертриглицеридемия могут косвенно отражать эволюцию ВИЧ-инфекции и зависеть от стадии заболевания, поскольку частота верификации и тяжесть гиперлипидемии негативно ассоциируются не только со снижением концентрации CD4-лимфоцитов, но и с риском возникновения оппортунистических инфекций (Constans J. et al., 1994; Mirete G. et al., 1998). Кроме того, необходимо отметить, что разнообразные эндокринные дисфункции, особенно гипотиреоз, обычно развиваются как побочный эффект при применении АРВТ, что необходимо принимать во внимание при оценке эволюции гиперлипидемии и ее клинического значения (Dobs A.S. et al., 1988; Sammalkorpi K. et al., 1988; Raffi F. et al., 1999).
Факторы риска гиперлипидемии, ассоциированной с АРВТ
К настоящему времени установлено, что АРВТ приводит к достаточно широкому спектру разнообразных нарушений липидного обмена (Behrens G. et al., 1999; Roberts A.D. et al, 1999; Mulligan K. et al., 2000). Предполагают, что характер терапии может иметь определяющее значения для формирования специфических форм метаболических нарушений. Так, для зидовудина, ламивудина, ставудина и ненуклеозидных ингибиторов обратной транскриптазы наиболее характерным является повышение уровня ТГ и общего ХС на фоне снижения ХС ЛПВП (Manfredi R., 2000; Hadigan C. et al., 2001). При этом часто отмечают формирование инсулинорезистентности, увеличение плазменного содержания С-реактивного протеина (СРП), манифестация сахарного диабета 2-го типа и синдром липодистрофии (Saint-Marc T., Touraine J.L., 1998; Walli R. et al., 1998; Save’s M. et al., 2002). В популяции пациентов, получающих ингибиторы внутриклеточных протеаз в качестве основного режима терапии, гиперлипидемия верифицируется особенно часто (в 28–80% случаев). Нарушения липидного профиля плазмы крови в основном связаны с гипертриглицеридемией (40–80%), гиперхолестеринемией (10–50%) и гипергликемией (5–30%) на фоне нарушений распределения подкожно-жировой клетчатки (10–80% случаев) (Shaw A.J. et al., 1998; Dong K.L. et al., 1999; Save’s M. et al., 2002). Именно в этой категории пациентов липодистрофия наиболее часто ассоциируется с гипер- и дислипидемией (Vigouroux C. et al., 1999; Save’s M. et al., 2002), особенно с содержанием ТГ, а также с СРП и выраженностью инсулинорезистентности (De Luca A. et al., 1998; Yanovski J.A. et al., 1999; Christeff N. et al., 1999).
В этой связи возникло предположение о том, что подобный широкий спектр метаболических нарушений, возникающих на фоне применения АРВТ, может стать результатом ассоциации особенностей фармакодинамики и фармакокинетики молекулы лекарственного средства с предшествующими факторами риска, такими как возраст, пол, увеличение массы тела, артериальная гипертензия. Действительно, в ряде клинических исследований такая взаимосвязь была установлена (Foster C.J. et al., 1987; Silva M. et al., 1998; Stricker R.B., Goldberg B., 1998). Тем не менее, существуют клинические исследования, в которых не было выявлено корреляционной взаимосвязи между половой принадлежностью больных, уровнем липидов в крови натощак, стадией СПИДа и массой тела до начала использования АРВТ (Tsiodras S. et al., 2000).
Вместе с тем, принято считать, что предрасполагающими факторами в отношении гиперлипидемии у пациентов, инфицированных ВИЧ-1, является не только применение АРВТ, но и ряд дополнительных характеристик, таких как уровень липидов и CD4-лимфоцитов, величина вирусной нагрузки и масса тела до лечения, а также возраст больных (Penzak S.R., Chuck S.K., 2000). Кроме того, многие исследователи подчеркивают, что содержание общего ХС в плазме крови пациентов наиболее тесно коррелирует с их возрастом, массой тела и пулом CD4-лимфоцитов (Tsiodras S. et al., 2000; Mooser V., Carr A., 2001). Тем не менее, многие данные, касающиеся механизмов возникновения гиперлипидемии, индуцированной различными ингибиторами протеаз, остаются во многом противоречивыми. Так, максимальную элевацию ТГ в плазме крови отмечают у пациентов, получающих ритонавир, а также его комбинацию с саквинавиром или индинавиром, при этом плазменное содержание ТГ может достигать 1000 мг/дл (Danner S.A. et al., 1995; Sullivan A.K., Nelson M.R., 1997; Di Perri G. et al., 1998; Sullivan A.K. et al., 1998; Moyle G.J., Baldwin C., 1999; Benson C.A. et al., 2002; Yeni P.G. et al., 2002). В то же время умеренное повышение уровня общего ХС в плазме крови чаще отмечают у больных, получающих в качестве АРВТ ритонавир и нелфинавир (Periard D. et al., 1999; Currier J.S., 2000). При проведении ретроспективного анализа было установлено, что использование индинавира ассоциируется с наиболее низким по сравнению с другими представителями этого класса риском индукции гиперхолестеринемии и гипертриглицеридемии (Penzak S.R., Chuck S.K., 2000). Таким образом, гиперлипидемия, развивающаяся вследствие АРВТ, может тесно зависеть от характеристик молекулы применяемого лекарственного средства, а также, возможно, и от вида используемой комбинации препаратов.
Манифестацию гиперлипидемии обычно отмечают между 3-м и 12-м месяцем после начала лечения. Ритонавир в монотерапии, а также его комбинация с саквинавиром или лопинавиром приводит к наиболее раннему появлению гиперлипидемии по сравнению с другими представителями АРВТ (Currier J.S., 2000). В ряде завершенных рандомизированных клинических исследований было установлено, что тяжесть гиперлипидемии также зависит от продолжительности применения ставудина, хотя характер влияния ингибиторов обратной транскриптазы в отношении нарушений липидного обмена продолжает дискутироваться. Так, некоторые исследователи полагают, что элевация ТГ в плазме крови больных, получающих АРВТ, может быть признаком метаболического синдрома или сахарного диабета и непосредственно не связана с эффектом лекарственных средств (Paparizos V.A. et al., 2000; Petit J.M. et al., 2000). В когорте детей и подростков, инфицированных ВИЧ-1 и получающих АРВТ, гиперлипидемия регистрируется в 70% случаев при приеме ритонавира и более чем в 50% — при применении нелфинавира (Fiore P. et al., 2000).
Патофизиологические механизмы формирования липоатрофического синдрома и гиперлипидемии у пациентов, инфицированных ВИЧ-1
Установлено, что наиболее вероятным механизмом, вовлекаемым в процесс формирования гиперлипидемии у пациентов, инфицированных ВИЧ-1, является существование структурной гомологичности между каталитическим участком протеазы ВИЧ-1 с одной стороны и двумя ключевыми для биосинтеза липидов протеинами — цитоплазматическим ретиноидным кислотосвязанным протеином 1-го типа (cytoplasmic retinoic acid-binding protein type 1, CRABP-1) и рецепторзависимым ЛПНП (low-density lipoprotein-receptor-related protein, LRP). CRABP-1 ответственен за процесс связывания и презентации ретинола для системы цитохрома P450 (CYP) 3A, которая конвертирует последний в cis-9-ретиноловую кислоту. Эта молекула связывается со специфическими гетеродимерами Х-рецептора для ретинола (RXR) и рецептором активации пролиферации пероксисом типа g (peroxisome proliferator-activated receptor gamma, PPARg) в ядре адипоцитов. Гетеродимер RXR-/PPARg, ассоциированный с cis-9-ретиноловой кислотой, ингибирует апоптоз адипоцитов, а также стимулирует пролиферацию и дифференцирование последних. Возможно, ингибиторы протеаз связываются с CRABP-1, который гомологичен вирусной протеазе, и способствуют ингибированию образования cis-9-ретиноловой кислоты, приводя к редукции активности RXR-PPARg. Это, в свою очередь, усиливает апоптоз адипоцитов и снижает степень их дифференцирования. Полагают, что этот механизм является ключевым для формирования липоатрофического синдрома у пациентов, инфицированных ВИЧ-1 (Carr A. et al., 1998; Penzak S.R., Chuck S.K., 2000).
Другим возможным механизмом возникновения метаболических нарушений является способность некоторых ингибиторов протеаз (ритонавир) подавлять активность комплекса цитохрома CYP 3A, ответственного за образование cis-9-ретиноловой кислоты и экспрессию энзимов системы RXR-PPARg (Kakuda T.N. et al., 1998). Последние, в частности, модулируют активность циркулирующей липопротеинлипазы, которая ответственна за аккумуляцию ТГ в хиломикронах, ЛПОНП и адипоцитах. Предполагают, что ингибиторы протеаз ингибируют активность липопротеинлипазы, что приводит к элевации плазменной концентрации ТГ и обогащенных ими ЛПОНП и ЛПНП (Arpadi S.M. et al., 1999; Berthold H.K. et al., 1999; Martinez E., Gatell J.M., 1999; Penzak S.R., Chuck S.K., 2000). Кроме того, ингибиторы протеаз способны непосредственно участвовать в регулировании синтеза ТГ через стимуляцию экспрессии соответствующей мРНК (Graham N.M., 2000; Lenhard J.M. et al., 2000; Distler O. et al., 2001).
Таким образом, гиперлипидемия у пациентов, инфицированных ВИЧ-1, является результатом взаимодействия как минимум трех основных механизмов: непосредственного влияния вируса в отношении регуляторных систем синтеза ТГ, ЛПОНП и ЛПНП; модуляции активности основных ферментных систем, ответственных за продукцию липидов гепатоцитами, АРВП; а также влияния традиционных факторов риска (Gallet B. et al., 1998; Henry K. et al., 1998; Klein D. et al., 1999). Результатом этих процессов является индукция раннего возникновения нарушений липидного и углеводного обмена, манифестация атеросклероза и атеротромботических осложнений (Flynn T.E., Bricker L.A., 1999; Friedl A.C. et al., 2000; Hadigan C. et al., 2000; Maggi P. et al., 2000; Passalaris J.D. et al., 2000; Depairon M. et al., 2001; Meng Q., et al., 2002).
Литература
- Abbott R.D., Behrens G.R., Sharp D.S. et al. (1994) Body mass index and thromboembolic stroke in nonsmoking men in older middle age: the Honolulu Heart Program. Stroke, 25: 2370–2376.
- Agard A., Bolmsjö I.A., Hermerén G. et al. (2005) Familial hypercholesterolemia: ethical, practical and psychological problems from the perspective of patients. Patient. Educ. Couns., 57: 162–167.
- Alexander C.M., Landsman P.B., Teutsch S.M. et al. (2003) NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes, 52: 1210–1214.
- Anderson J.W., Konz E.C. (2001) Obesity and disease management: effects of weight loss on comorbid conditions. Obes. Res., 9(Suppl. 4): 326S–334S.
- Apanay D.C., Neyland J.F., Ragab M.S. et al. (1994) Cyclosporine increases the oxidizability of low-density lipoproteins in renal transplant recipients. Transplantation, 58(6): 663–669.
- Arpadi S.M., Cuff P.A., Horlick M. et al. (1999) Visceral obesity, hypertriglyceridemia and hypercortisolism in a boy with perinatally acquired HIV infection receiving protease inhibitor-containing antiviral treatment. AIDS, 13: 2312–2313.
- Athyros V.G., Giouleme O.I., Nikolaidis N.L. et al. (2002) Long-term follow-up of patients with acute hypertriglyceridemia-induced pancreatitis. J. Clin. Gastroenterol., 34: 472–475.
- Austin M.A. (1989) Plasma triglyceride as a risk factor for coronary heart disease. The epidemiologic evidence and beyond. Am. J. Epidemiol., 129: 249–259.
- Austin M.A., King M.C., Vranizan K.M. et al. (1990) Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation, 82: 495–506.
- Austin M.A., McKnight B., Edwards K.L. et al. (2000) Cardiovascular disease mortality in familial forms of hypertriglyceridemia: A 20-year prospective study. Circulation, 101: 2777–2782.
- Avis H.J., Vissers M.N., Stein E.A. et al. (2007) A systematic review and meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler., Thromb., Vasc. Biol., 27: 1803–1810.
- Ayyobi A.F., Brunzell J.D. (2003) Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. Am. J. Cardiol., 92: 27J–33J.
- Bainton D., Miller N.E., Bolton C.H. et al. (1992) Plasma triglyceride and high density lipoprotein cholesterol as predictors of ischaemic heart disease in British men. The Caerphilly and Speedwell Collaborative Heart Disease Studies. Br. Heart J., 68: 60–66.
- Ballantyne C.M., Podet E.J., Patsch W.P. et al. (1989) Effects of cyclosporine therapy on plasma lipoprotein levels. JAMA, 262: 53–56.
- Bamba V., Rader D.J. (2007) Obesity and atherogenic dyslipidemia. Gastroenterology, 132: 2181–2190.
- Barter P.J., Rye K.A. (2006) The rationale for using apoA-I as a clinical marker of cardiovascular risk. J. Intern. Med., 259: 447–454.
- Bays H., Abate N., Chandalia M. (2005) Adiposopathy: sick fat causes high blood sugar, high blood pressure and dyslipedmia. Future Cardiol., 1: 39–59.
- Bays H., Mandarino L., DeFronzo R.A. (2004) Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of Type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab., 89: 463–447.
- Bays H., Stein E.A. (2003) Pharmacotherapy for dyslipidaemia — current therapies and future agents. Expert Opin. Pharmacother., 4: 1901–1938.
- Behrens G., Dejam A., Schmidt H. et al. (1999) Impaired glucose tolerance, beta cell function and lipid metabolism in HIV patients under treatment with protease inhibitors. AIDS, 13: 63–70.
- Benson C.A., Deeks S.G., Brun S.C. et al. (2002) Safety and antiviral activity at 48 weeks of lopinavir/ritonavir plus nevirapine and two nucleoside reverse-transcriptase inhibitors in human immunodeficiency virus type 1-infected protease inhibitor-experienced patients. J. Infect. Dis., 185: 599–607.
- Berthold H.K., Parhofer K.G., Ritter M.M. et al. (1999) Influence of protease inhibitor therapy on lipoprotein metabolism. J. Intern. Med., 246: 567–575.
- Beynen A.C., Katan M.B., van Zutphen L.F.M. (1987) Hypo- and hyperresponders: individual differences in the response of serum cholesterol concentration to changes in diet. Adv. Lipid Res., 22: 115–171.
- Bierman E.L. (1992) Atherogenesis in diabetes. Arterioscler Thromb, 12: 647–656.
- Boren J., Ekstrom U., Agren B. et al. (2001) The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J. Biol. Chem., 276: 9214–9218.
- Brunzell J.D., Albers J.J., Chait A. et al. (1983) Plasma lipoproteins in familial combined hyperlipidemia and monogenic familial hypertriglyceridemia. J. Lipid Res., 24: 147–155.
- Brunzell J.D., Davidson M., Furberg C.D. et al. (2008) Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the american diabetes association and the american college of cardiology foundation. J. Am. Coll. Cardiol., 51: 1512–1524.
- Brunzell J.D., Deeb S.S. (2000) Familial lipoprotein lipase deficiency, apo CII deficiency and hepatic lipase deficiency. In: C.R. Scriver, A.I. Beaudet, W.D. Sly, D. Valle (Eds). The metabolic and molecular bases of inherited disease. 8th ed. Vol. 3. McGraw-Hill, New York, pp. 2789–2816.
- Brunzell J.D., Failor R.A. (2006) Diagnosis and treatment of dyslipidemia. In: D.C. Dale, D.D. Federman (Eds.). ACP medicine, section 9, chap. 11. WebMD, New York.
- Bucala R., Makita Z., Vega G. et al. (1994) Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc. Natl. Acad. Sci. USA, 91: 9441–9445.
- Bumgardner G.L., Wilson G.A., Tso G.A. et al. (1995) Impact of serum lipids on long-term graft and patient survival after renal transplantation. Transplantation, 60: 1418–1424.
- Carlson I.A., Holmquist I. (1985) Evidence for deficience of high density lipoprotein lecitine: cholesterol acyltransferase activity (α-LCAT) in fish eye disease. Acta Med. Scand., 218: 189–196.
- Carr A., Samaras K., Burton S. et al. (1998) A syndrome of peripheral lipodystrophy, hyperlipidemia and insulin resistance due to HIV protease inhibitors. AIDS, 12: F51–58.
- Carr A., Samaras K., Thorisdottir A. et al. (1999) Diagnosis, prediction, and natural course of HIV-1 protease inhibitor-associated lipodystrophy, hyperlipidaemia, and diabetes mellitus: a cohort study. Lancet, 353: 2093–2099.
- Carr M.C., Brunzell J.D. (2004) Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J. Clin. Endocrinol. Metab., 89: 2601–2607.
- Ceveira F.; International Panel on Management of Familial Hypercholesterolemia (2004) Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis, 173: 55–68.
- Chan D.C., Barrett H.P., Watts G.F. (2004) Dyslipidemia in visceral obesity: mechanisms, implications, and therapy. Am. J. Cardiovasc. Drugs, 4: 227–246.
- Christeff N., Melchior J.C., De Truchis P. et al. (1999) Lipodystrophy defined by a clinical score in HIV infected men on highly active antiretroviral therapy: correlation between dyslipidemia and steroid hormone alterations. AIDS, 13: 2251–2260.
- Cobb M.M., Teitlebaum H. (1994) Determinants of plasma cholesterol responsiveness to diet. Br. J. Nutr., 71: 271–282.
- Cohn J.S., Marcoux C., Davignon J. (1999) Detection, quantification, and characterization of potentially atherogenic triglyceride-rich remnant lipoproteins. Arterioscler. Thromb. Vasc. Biol., 19: 2474–2486.
- Constans J., Pellegrin J.L., Peuchant E. et al. (1994) Plasma lipids in HIV-infected patients: a prospective study in 95 patients. Eur. J. Clin. Invest., 24: 416–420.
- Cortner J., Coates P., Gallacher P. (1990) Prevalence and expression of familial combined hyperlipidemia in childhood. J. Pediatr., 116: 514–519.
- Cui Y., Blumenthal R.S., Flaws J.A. et al. (2001) Non-high-density lipoprotein cholesterol level as a predictor of cardiovascular disease mortality. Arch. Intern. Med., 161: 1413–1419.
- Currier J.S. (2000) How to manage metabolic complications of HIV therapy: what to do while we wait for answers. AIDS Read, 10: 162–169, 171–174.
- Dahlback B. (2006) Isolation and characterization of human apolipoprotein M-containing lipoproteins. J. Lipid Res., 47: 1833–1843.
- Danner S.A., Carr A., Leonard J.M. et al. (1995) A short-term study on the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. N. Engl. J. Med., 333: 1528–1533.
- Davignon J., Genest J. Jr. (1998) Genetics of lipoprotein disorders. Endocrinol. Metab. Clin. North Am., 27: 521–550.
- De Luca A., Murri R., Damiano F. et al. (1998) «Buffalo hump» in HIV-1 infection. Lancet, 352: 320.
- Defronzo R.A., Goodman A.M., The Multicenter Metformin Study Group (1995) Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. N. Engl. J. Med., 333: 541–549.
- deGroen P. (1988) Cyclosporine, low-density lipoprotein, and cholesterol. Mayo Clin. Proc., 262: 53–56.
- Depairon M., Chessex S., Sudre P. et al. (2001) Premature atherosclerosis in HIV-infected individuals/focus on protease inhibitor therapy. AIDS, 15: 329–334.
- Després J.P., Lemieux I. (2006) Abdominal obesity and metabolic syndrome. Nature, 444: 881–887.
- Dey D.K., Rothenberg E., Sundh V. et al. (2002) Waist circumference, body mass index, and risk for stroke in older people: a 15 year longitudinal population study of 70- year-olds. J. Am. Geriatr. Soc., 50: 1510–1518.
- Di Perri G., Del Bravo P., Concia E. (1998) HIV-protease inhibitors. N. Engl. J. Med., 339: 773–774.
- Dimeny E., Fellstrom B., Larsson E. et al. (1995) Hyperlipoproteinemia in renal transplant recipients: is there a linkage with chronic vascular rejection. Transplant. Proc., 25: 2065–2066.
- DiPietro L., Ostfeld A.M., Rosner G.L. (1994) Adiposity and stroke among older adults of low socioeconomic status: the Chicago Stroke Study. Am. J. Public. Health, 84: 14–19.
- Distler O., Cooper D.A., Deckelbaum R.J. et al. (2001) Hyperlipidemia and inhibitors of HIV protease. Curr.Opin. Clin. Nutr. Metab. Care, 4: 99–103.
- Dobs A.S., Dempsey M.A., Ladenson P.W. et al. (1988) Endocrine disorders in men infected with human immunodeficiency virus. Am. J. Med., 84: 611–616.
- Dong K.L., Bausserman L.L., Flynn M.M. et al. (1999) Changes in body habits and serum lipid abnormalities in HIV-positive women on highly active antiretroviral therapy (HAART). J. Acquir. Immune Defic. Syndr., 21: 107–113.
- Dunbar R.L., Rader D.J. (2005) Demystifying triglycerides: a practical approach for the clinician. Cleve. Clin. J. Med., 72: 661–680.
- Dunn S.P., Falkenstein K., Lawrence J.P. et al. (1994) Monotherapy with cyclosporine for chronic immunosuppression in pediatric liver transplant recipients. Transplantation, 57: 544.
- Erickson K.L., Adams D.A., McNeill C.J. (1983) Dietary lipid modulation of immune responsiveness. Lipids., 18: 468–474.
- EUROASPIRE II Study Group (2001) Lifestyle and risk factor management and use of drug therapies in coronary patients from 15 countries: principal results from EUROASPIRE II Euro Heart Survey Programme. Eur. Heart J., 22: 554–572.
- European FK506 Multicenter Study Group (1994) Randomized trial comparing tacrolimus (FK506) and cyclosporine in prevention of liver allograft rejection. Lancet, 344: 423–428.
- Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (2002) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation, 106: 3143–3421.
- Fiore P., Donelli E., Boni S. et al. (2000) Nutritional status changes in HIV-infected children receiving combined antiretroviral therapy including protease inhibitors. Int. J. Antimicrob. Agents, 16: 365–369.
- Flegal K.M., Carroll M.D., Ogden C.L. et al. (2002) Prevalence and trends in obesity among US adults, 1999–2000. JAMA, 288: 1723–1727.
- Flynn T.E., Bricker L.A. (1999) Myocardial infarction in HIV-infected men receiving protease inhibitors. Ann. Intern. Med., 131: 548.
- Fontaine K.R., Redden D.T., Wang C. et al. (2003) Years of life lost due to obesity. JAMA, 289: 187–193.
- Ford E.S., Mokdad A.H., Giles W.H. (2003) Trends in waist circumference among U.S. adults. Obes. Res., 11: 1223–1231.
- Fortson M.R., Freedman S.N., Webster P.D. 3rd (1995) Clinical assessment of hyperlipidemic pancreatitis. Am. J. Gastroenterol., 90: 2134–2139.
- Foster C.J., Weinsier R.L., Birch R. et al. (1987) Obesity and serum lipids: an evaluation of the relative contribution of body fat and fat distribution to lipid levels. Int. J. Obes., 11: 151–161.
- Friedl A.C., Attenhofer Jost C.H., Schalcher C. et al. (2000) Acceleration of confirmed coronary artery disease among HIV-infected patients on potent antiretroviral therapy. AIDS, 14: 2790–2792.
- Fredrickson D.S., Lees R.S. (1965) A system for phenotyping hyperlipoproteinemia. Circulation, 31: 321–327.
- Fung A., Frohlich J.J. (2002) Common problems in the management of hypertriglyceridemia. CMAJ, 167: 1261–1266.
- Gallet B., Pulik M., Genet P. (1998) Vascular complications associated with use of HIV protease inhibitors. Lancet, 351: 1958–1959.
- Gan S.I., Edwards A.L., Symonds C.J. et al. (2006) Hypertriglyceridemia-induced pancreatitis: a case-based review. World J. Gastroenterol., 12: 7197–7202.
- Garcia-Otin A.L., Civeira F., Peinado-Onsurbe J. (1999) Acquired lipoprotein lipase deficiency associated with chronic urticaria. A new etiology for type I hyperlipoproteinemia. Eur. J. Endocrinol., 141: 502–505.
- Gardner C.D., Kiazand A., Alhassan S. et al. (2007) Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA, 297: 969–977.
- Garg A., Grundy S.M. (1990) Management of dyslipidemia in NIDDM. Diabetes Care, 13: 153–71.
- Genest J.J. Jr., Martin-Munley S.S., McNamara J.R. et al. (1992) Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation, 85: 2025–2033.
- Gerson E.J., MacDonald J.S., Alberts A.W. et al. (1989) Animal safety and toxicology of simvastatin and related hydroxy-methylglutaryl-coenzyme A reductase inhibitors. Am. J. Med., 87: 28–38.
- Gilman A.G., Goodman L.S. (1990) The pharmacological basis of therapeutics. Pergamon, New York.
- Ginsberg H.N., Zhang Y.L., Hernandez-Ono A. (2005) Regulation of plasma triglycerides in insulin resistance and diabetes. Arch. Med. Res., 36: 232–240.
- Gokal R., Mann J.I., Moore R.A. et al. (1979) Hyperlipidemia following renal transplantation. Q. J. Med., 192: 507–517.
- Goldstein J.L., Brown M.S. (2001) Familial hypercholesterolemia. In: C.R. Scriver, B. Childs (Eds.). The metabolic and molecular basis of inherited disease. 8th ed. McGraw-Hill, Medical Publishing Division, New York.
- Goldstein J.L., Schrott H.G., Hazzard W.R. (1973) Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Invest., 52: 1544–1568.
- Graham N.M. (2000) Metabolic disorders among HIV-infected patients treated with protease inhibitors: a review. J. Acquir. Immune Defic. Syndr., 25(Suppl. 1): S4–S11.
- Grundy S.M., Cleeman J.I., Merz C.N.B. et al. (2004) Implication of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation, 110: 227–239.
- Grunfeld C., Kotler D.P., Shigenaga J.K. et al. (1991) Circulating interferon-alpha levels and hypertriglyceridaemia in the acquired immunodeficiency syndrome. Am. J. Med., 90: 154–162.
- Grunfeld C., Pang M., Doerrler W. (1992) Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J. Clin. Endocrinol. Metab., 74: 1045–1052.
- Gursoy A., Kulaksizoglu M., Sahin M. et al. (2006) Severe hypertriglyceridemia-induced pancreatitis during pregnancy. J. Natl. Med. Assoc., 98: 655–657.
- Hadigan C., Corcoran C., Basgoz N. (2000) Use of metformin for the treatment of the HIV lipodystrophy syndrome: a randomized controlled trial. J. Am. Med. Assoc., 284: 472–477.
- Hadigan C., Meigs J.B., Corcoran C. et al. (2001) Metabolic abnormalities and cardiovascular disease risk factors in adults with human immunodeficiency virus infection and lipodystrophy. Clin. Infect. Dis., 32: 130–139.
- Halloran P., Mathew T., Tomlanovich S. et al. (1997) Mycophenolate mofetil in renal allograft recipients: a pooled efficacy analysis of three randomized, double-blind, clinical studies in prevention of rejection. Transplantation, 63: 39–47.
- Harper C.R., Jacobson T.A. (2006) An evidence-based approach to the use of combination drug therapy for mixed dyslipidemia. JCOM, 13: 57.
- Havel R.J. (1990) Role of triglyceride-rich lipoproteins in progression of atherosclerosis. Circulation, 81: 694–696.
- Havel R.J., Rapaport E. (1995) Management of primary hyperlipidemia. N. Engl. J. Med., 332: 1491–1498.
- Heath B., Karpe F., Milne R.W. (2003) Selective partitioning of dietary fatty acids into the VLDL TG pool in the early postprandial period. J. Lipid Res., 44: 2065–2072.
- Hegsted D.M., Ausman L.M., Johnson J.A. et al. (1993) Dietary fat and serum lipids: an evaluation of the experimental data. Am. J. Clin. Nutr., 57: 875–83.
- Hellerstein M.K., Grunfeld C., Wu K. et al. (1993) Increased de novo hepatic lipogenesis in human immunodeficiency virus infection. J. Clin. Endocrinol. Metab., 76: 559–565.
- Henry K., Melroe H., Huebsch J. et al. (1998) Severe premature coronary artery disease with protease inhibitors. Lancet, 351: 1328.
- Hokanson J.E., Austin M.A. (1996) Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J. Cardiovasc. Risk., 3: 213–219.
- Hoogerbrugge N. (1998) Effects of atorvastatin on serum lipids of patients with familial hypercholesterolaemia. J. Intern. Med., 244: 143–147.
- Hooper L., Thompson R.L., Harrison R.A. et al. (2006) Risks and benefits of omega 3 fats for mortality, cardiovascular disease, and cancer: systematic review. BMJ, 332: 752–760.
- Hopkins P.N., Heiss G., Ellison R.C. et al. (2003) Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation,108: 519–523.
- Hsia S.H., Connelly P.W., Hegele R.A. (1995) Successful outcome in severe pregnancy-associated hyperlipemia: a case report and literature review. Am. J. Med. Sci., 309: 213–218.
- Hwang D. (1989) Essential fatty acids and immune response. FASEB J., 3: 2052–2061.
- Jacobs D.R., Anderson J.T., Hannan P. et al. (1983) Variability in individual serum cholesterol response to change in diet. Arteriosclerosis, 3: 349–356.
- Jacobson T.A., Miller M., Schaefer E.J. (2007) Hypertriglyceridemia and cardiovascular risk reduction. Clin. Ther., 29: 763–777.
- Jap T.S., Jenq S.F., Wu Y.C. et al. (2003) Mutations in the lipoprotein lipase gene as a cause of hypertriglyceridemia and pancreatitis in Taiwan. Pancreas, 27: 122–126.
- Jindal R.M. (1994) Posttransplant diabetes mellitus — a review. Transplantation, 58: 1289–1298.
- Jindal R.M., Popescu I., Emre S. et al. (1994) Serum lipid changes in liver transplant recipients in a prospective trial of cyclosporine versus FK506. Transplantation, 57: 1395–1398.
- Jindal R.M., Sidner R.A., Hughes D. et al. (1996) Metabolic problems in recipients of liver transplants. Clin. Transplant., 10: 213–217.
- Julius U. (2003) Influence of plasma free fatty acids on lipoprotein synthesis and diabetic dyslipidemia. Exp. Clin. Endocrinol. Diabetes., 111: 246–250.
- Kakuda T.N., Struble K.A., Piscitelli S.C. (1998) Protease inhibitors for the treatment of human immunodeficiency virus infection. Am. J. Health Syst. Pharm., 55: 233–254.
- Katan M.B., Beynen A.C., de Vries J.H. et al. (1986) Existence of consistent hypo- and hyperresponders to dietary cholesterol in man. Am. J. Epidemiol., 123: 221–234.
- Katznelson S., Wilkinson A.H. (1995) HMGCoA reductase inhibitors and omega-3 fatty acids as adjunctive agents in maintenance immunosuppression after solid organ transplantation. Clin. Transplant., 9: 197–200.
- Kavey R.E., Allada V., Daniels S.R. et al. (2006) Cardiovascular risk reduction in high risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation, 114: 2710–2738.
- Klein D., Hurley L., Sidney S. (1999) Do protease inhibitors increase the risk for coronary heart disease among HIV positive patients? In: Sixth Conference on Retroviruses and Opportunistic Infections, Chicago [Abstract 657].
- Kobashigawa J.A., Gleeson M.P., Stevenson L.W. et al. (1994) Prevention of severe cardiac allograft rejection with pravastatin: a randomized trial. J. Am. Coll. Cardiol., 23: 483A.
- Kotler D.P. (2000) Nutritional alterations associated with HIV infection. J. Acquir Immune Defic. Syndr., 25(Suppl. 1): S81–87.
- Krauss R.M., Eckel R.H., Howard B. et al. (2000) AHA dietary guidelines: revision 2000: a statement for healthcare professionals from the Nutrition Committee of the American Heart Association. Circulation, 102: 2284–2299.
- Krentz A.J., Dousset B., Mayer D. et al. (1993) Metabolic effects of cyclosporine A and FK506 in liver transplant recipients. Diabetes, 42: 1753–1759.
- Kurth T., Gaziano J.M., Berger K. et al. (2002) Body mass index and the risk of stroke in men. Arch. Intern. Med., 162: 2557–2562.
- Kwiterovich J.R., Levy R., Fredrickson D. (1973) Neonatal diagnosis of familial type-II hyperlipoproteinemia. Lancet, 301: 118–122.
- Kwiterovich P. (1993) Genetics and molecular biology of familial combined hyperlipidemia. Curr. Opin. Lipid., 4: 133–143.
- Kwiterovich P.O. Jr. (2008) Recognition and management of dyslipidemia in children and adolescents. J. Clin. Endocrinol. Metab., 93: 4200–4209.
- Kyriakidis A.V., Raitsiou B., Sakagianni A. et al. (2006) Management of acute severe hyperlipidemic pancreatitis. Digestion, 73: 259–264.
- Lamarche B., Tchernof A., Moorjani S. et al. (1997) Small, dense low-density lipoprotein particles as a predictor of the risk of ischemic heart disease in men. Prospective results from the Quebec Cardiovascular Study. Circulation, 95: 69–75.
- Lemieux I., Pascot A., Couillard C. et al. (2000) Hypertriglyceridemic waist: a marker of the atherogenic metabolic triad (hyperinsulinemia; hyperapolipoprotein B; small, dense LDL) in men? Circulation, 102: 179–184.
- Lenhard J.M., Croom D.K., Weiel J.E. et al. (2000) HIV protease inhibitors stimulate hepatic triglyceride synthesis. Arterioscler. Thromb. Vasc. Biol., 20: 2625–2629.
- Leon A.S., Sanchez O.A. (2001) Response of blood lipids to exercise training alone or combined with dietary intervention. Med. Sci. Sports Exerc., 33(Suppl.): S502–S515.
- Leren T.P. (2004) Cascade genetic screening for familial hypercholesterolemia. Clin. Genet., 66: 483–487.
- Lichtenstein A.H., Appel L.J., Brands M. et al. (2006) Summary of American Heart Association Diet and Lifestyle Recommendations revision 2006. Arterioscler. Thromb. Vasc. Biol., 26: 2186–2191.
- Loo C.C., Tan J.Y. (2002) Decreasing the plasma triglyceride level in hypertriglycerideia-induced pancreatitis in pregnancy: a case report. Am. J. Obstet. Gynecol., 187: 241–242.
- Maggi P., Serio G., Epifani G. et al. (2000) Premature lesions of the carotid vessels in HIV-1-infected patients treated with protease inhibitors. AIDS, 14: F123–128.
- Mancia G., De Backer G., Dominiczak A. et al. (2007) Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens., 25: 1105–1187.
- Manfredi R. (2000) Management of dyslipidemia in patients with HIV disease. Clin. Microbiol. Infect., 6: 579–584.
- Mann G.V. (1974) The influence of obesity on health (second of two parts). N. Engl. J. Med., 291: 226–232.
- Manninen V., Tenkanen L., Koskinen P. et al. (1992) Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation, 85: 37–45.
- Manson J.E., Willett W.C., Stampfer M.J. et al. (1995) Body weight and mortality among women. N. Engl. J. Med., 333: 677–685.
- Marcoux C., Hopkins P.N., Wang T. et al. (2000) Remnant-like particle cholesterol and triglyceride levels of hypertriglyceridemic patients in the fed and fasted state. J. Lipid Res., 41: 1428–1436.
- Marcovina S., Packard C.J. (2006) Measurement and meaning of apolipoprotein AI and apolipoprotein B plasma levels. J. Int. Med., 259: 437–446.
- Marks D., Thorogood M., Neil H.A. et al. (2003) A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis, 168: 1–14.
- Marks D., Wonderling D., Thorogood M. et al. (2002) Cost-effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ, 324: 1303–1308.
- Martinez E., Gatell J.M. (1999) Metabolic abnormalities and body fat redistribution in HIV-1 infected patients: the lipodystrophy syndrome. Curr. Opin. Infect. Dis., 12: 13–19.
- McDiarmid S.V., Gombein J.A., Fortunat M. et al. (1992) Serum lipid abnormalities in pediatric liver transplant patients. Transplantation, 53: 109–115.
- McKenney J. (2005) Dyslipidemias, atherosclerosis, and coronary heart disease. In: M.A. Koda-Kimble, L.Y. Young, W.A. Kradjan et al. (Eds). Applied therapeutics: the clinical use of drugs (8th Edition). Lipincott, Williams & Wilkins, Philadelphia.
- McNamara D.J., Kolb R., Parker T.S. et al. (1987) Heterogeneity of cholesterol homeostasis in man: response to changes in dietary fat quality and cholesterol quantity. J. Clin. Invest., 79: 1729–1739.
- Meng Q., Lima J.A., Lai H. et al. (2002) Coronary artery calcification, atherogenic lipid changes, and increased erythrocyte volume in black injection drug users infected with human immunodeficiency virus-1 treated with protease inhibitors. Am. Heart J., 144: 642–648.
- Mirete G., Masia M., Gutierrez F. (1998) Acute pancreatitis as a complication of ritonavir therapy in a patient with AIDS. Eur. J. Clin. Microbiol. Infect. Dis., 17: 810–811.
- Mokdad A.H., Ford E.S., Bowman B.A. et al. (2003) Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA, 289: 76–79.
- Monga A., Arora A., Makkar R.P. et al. (2003) Hypertriglyceridemia-induced acute pancreatitis — treatment with heparin and insulin. Indian J. Gastroenterol., 22: 102–103.
- Mooser V., Carr A. (2001) Antiretroviral therapy-associated hyperlipidaemia in HIV disease. Curr. Opin. Lipidol., 12: 313–319.
- Morrisett J.D., Northrup S.R., Gotto A.M. et al. (1991) Effect of FK506 and cyclosporine on plasma cholesterol levels. Transplant. Proc., 23: 3185.
- Motulsky A.G. (1989) Genetic aspects of familial hypercholesterolemia and its diagnosis. Arteriosclerosis, 9(Suppl. 1): I3–7.
- Moyle G.J., Baldwin C. (1999) Lipid abnormalities during saquinavir soft-gel-based highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr., 15: 423–424.
- Mulligan K., Grunfeld C., Tai V.W. et al. (2000) Hyperlipidemia and insulin resistance are induced by protease inhibitors independent of changes in body composition in patients with HIV infection. J. Acquir. Immune Defic. Syndr., 23: 35–43.
- Munoz S.J. (1995) Hyperlipidemia and other coronary risk factors after orthotopic liver transplantation: pathogenesis, diagnosis, and management. Liver Transplant. Surg., 1(Suppl. 1): 29–38.
- Munoz S.J., Deems R.O., Moritz M.J. et al. (1991) Hyperlipidemia and obesity after orthotopic liver transplantation. Transplant. Proc., 23: 1480—1483.
- Murgia M., Jordan S., Kahan B.D. (1996) The side effect profile of sirolimus: a phase I study in quiescent cyclosporin-prednisolone-treated renal transplant patients. Kidney Int., 49: 209–216.
- Nagasaka H., Kikuta H., Chiba H. et al. (2003) Two cases with transient lipoprotein lipase (LPL) activity impairment: evidence for the possible involvement of an LPL inhibitor. Eur. J. Pediatr., 162: 132–138.
- National Cholesterol Education Program (2001) Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA, 285: 2486–2497.
- National Institute of Clinical Excellence (NICE) (2004) Clinical Guideline 18 — Management of hypertension in adults in primary care. London: NICE, www.nice.org.uk/CG018NICEguideline.
- Nauck M.S., Nissen H., Hoffmann M.M. et al. (1998) Detection of mutations in the apolipoprotein CII gene by denaturing gradient gel electrophoresis. Identification of the splice site variant apolipoprotein CII-Hamburg in a patient with severe hypertriglyceridemia. Clin. Chem., 44: 1388–1396.
- Nishijima H., Satake K., Igarashi K. et al. (2007) Effects of exercise in overweight Japanese with multiple cardiovascular risk factors. Med. Sci. Sports Exerc., 39: 926–933.
- Nissen S.E., Tsunoda T., Tuzcu E.M. et al. (2003) Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA, 290: 2292–2300.
- Nordestgaard B.G., Benn M., Schnohr P. et al. (2007) Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA, 298: 299–308.
- O’Bryan M.K., Foulds L.M., Cannon J.F. et al. (2004) Identification of a novel apolipoprotein, ApoN, in ovarian follicular fluid. Endocrinology, 145: 5231–5242.
- Oh R., Malani A., Ammar H. et al. (2008) Hypertriglyceridemia. N. Engl. J. Med., 358: 310–310.
- Oh R.C., Lanier J.B. (2007) Management of hypertriglyceridemia. Am. Fam. Physician., 75: 1365–1371.
- Oliver M.F. (1995) Statins prevent coronary heart disease. Lancet, 346: 1378–1379.
- Palumbo J.D., Lopes S.M., Zeisel S.H. et al. (1987) Effectiveness of orthotopic liver transplantation on the restoration of cholesterol metabolism in patients with end-stage liver disease. Gastroenterology, 93: 1170–1177.
- Paparizos V.A., Kyriakys K.P., Botsis C. et al. (2000) Protease inhibitor therapyassociated lipodystrophy, hypertriglyceridemia and diabetes mellitus. AIDS, 14: 903–905.
- Passalaris J.D., Sepkowitz K.A., Glesby M.J. (2000) Coronary artery disease and human immunodeficiency virus infection. Clin. Infect. Dis., 31: 787–797.
- Pearson T.A. (2007) The prevention of cardiovascular disease: have we really made progress? Health Aff. (Millwood), 26: 49–60.
- Penzak S.R., Chuck S.K. (2000) Hyperlipidemia associated with HIV protease inhibitor use: pathophysiology, prevalence, risk factors and treatment. Scand. J. Infect. Dis., 32: 111–123.
- Perez R.V., Alexander J.W. (1988) Immune regulation by lipids. Transplant. Proc., 20: 1162–1165.
- Periard D., Telenti A., Sudre P. et al. (1999) Atherogenic dyslipidemia in HIV-infected individuals treated with protease inhibitors. The Swiss HIV Cohort Study. Circulation, 100: 700–705.
- Petit J.M., Duong M., Duvillard L. et al. (2000) HIV-1 protease inhibitors induce an increase of triglyceride level in HIV-infected men without modification of insulin sensitivity: a longitudinal study. Horm. Metab. Res., 32: 367–372.
- Peyser P.A. (1997) Genetic epidemiology of coronary artery disease. Epidemiol. Rev., 19: 80–90.
- Rader D.J., Rosas S. (2000) Management of selected lipid abnormalities. Hypertriglyceridemia, low HDL cholesterol, lipoprotein(a), in thyroid and renal diseases, and post-transplantation. Med. Clin. North. Am., 84: 43–61.
- Raffi F., Brisseau J.M., Planchon B. et al. (1999) Endocrine function in 98 HIV-infected patients: a prospective study. AIDS, 5: 729–733.
- Raine A.E.G., Carter R., Mann J.I. et al. (1987) Increased plasma LDL cholesterol after renal transplantation associated with cyclosporine immunosuppression. Transplant. Proc., 21: 1820–1821.
- Reaven G.M., Laws A. (1994) Insulin resistance, compensatory hyperinsulinaemia, and coronary heart disease. Diabetologia, 37: 948–52.
- Reaven G.M., Lawsa A. (1994) Insulin resistance, compensatory hyperinsulineamia, and coronary heart disease. Diabetologia, 37: 948–52.
- Renaud S., de Lorgeril M., Delaye J. et al. (1995) Cretan Mediterranean diet for prevention of coronary heart disease. Am. J. Clin. Nutr., 61(Suppl.): 1360S–1367S.
- Rexrode K.M., Hennekens C.H., Willett W.C. et al. (1997) A prospective study of body mass index, weight change, and risk of stroke in women. JAMA, 277: 1539–1545.
- Roberts A.D., Muesing R.A., Parenti D.M. et al. (1999) Alterations in serum lipid levels of lipids and lipoproteins with indinavir therapy for human immunodeficiency virus-infected patients. Clin. Infect. Dis., 29: 441–443.
- Rodenburg J., Vissers M.N., Wiegman A. et al. (2004) Familial hypercholesterolemia in children. Curr. Opin. Lipidol., 15: 405–411.
- Sacks F.M., The Expert Group on HDL Cholesterol: (2002) The role of high-density lipoprotein (HDL) cholesterol in the prevention and treatment of coronary heart disease: Expert Group recommendations. Am. J. Cardiol., 90: 139–143.
- Saint-Marc T., Touraine J.L. (1998) «Buffalo hump» in HIV-1 infection. Lancet, 352: 319–320.
- Sammalkorpi K., Valtonen V., Kerttula Y. et al. (1988) Changes in serum lipoprotein pattern induced by acute infections. Metabolism, 37: 859–865.
- Sandbom W.J., Hay J.E., Porayko M.K. et al. (1994) Cyclosporine withdrawal for nephrotoxicity in liver transplant recipients does not result in sustained improvement in kidney function and causes cellular and ductopenic rejection. Hepatology, 19: 925–932.
- Sanderson S.L., Iverius P.H., Wilson D.E. (1991) Successful hyperlipemic pregnancy. JAMA, 265(14): 1858–1860.
- Savage P.J. (1996) Cardiovascular complications of diabetes mellitus: what we known and what we need to know about their prevention. Ann. Intern. Med., 124: 123–126.
- Save’s M., Raffi F., Capeau J. et al. (2002) Factors related to lipodystrophy and metabolic alterations in patients with human immunodeficiency virus infection receiving highly active antiretroviral therapy. Clin. Infect. Dis., 34: 1396–1405.
- Scandinavian Simvastatin Survival Study Group (1994) Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet, 344: 1383–1384.
- Schaefer E.J., Lamon-Fava S., Ausman L.M. et al. (1997) Individual variability in lipoprotein cholesterol response to National Cholesterol Education Program Step 2 diets. Am. J. Clin. Nutr., 65: 823–830.
- Schneeman O., Kotite L., Todd K.M. et al. (1993) Relationships between the responses of triglyceride-rich lipoproteins in blood plasma containing apolipoproteins B-48 and B-100 to a fat-containing meal in normolipidemic humans. Proc. Natl Acad. Sci. USA, 90: 2069–2073.
- Schror K., Lopel P., Steinhagen-Thiessen E. (1989) Simvastatin reduces platelet thromboxane formation and restores normal platelet sensitivity against prostacyclin in type IIa hypercholesterolemia. Eicosanoids, 2: 39–45.
- Scientific Steering Committee, The Simon Broome Register Group (1999) Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Atherosclerosis, 142: 105–112.
- Segrest J.P., Jones M.K., De Loof H. et al. (2001) Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res., 42: 1346–1367.
- Sellmeyer D.E., Grunfeld C. (1996) Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immune deficiency syndrome. Endocr. Rev., 17: 518–532.
- Shaw A.J., McLean K.A., Evans B.A. (1998) Disorders of fat distribution in HIV infection. Int. J. STD. AIDS, 9: 595–599.
- Shepherd J., Cobbe S.M., Ford I. et al. (1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med., 333: 1301–1307.
- Shiflett A.M., Bishop J.R., Pahwa A. et al. (2005) Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J. Biol. Chem., 280: 32578—32585.
- Shils M.E., Shike M., Ross A.C. et al. (2007) Lipids, sterols, and their metabolites. In: M.E. Shils, M. Shike, A.C. Ross et al. (Eds). Modern Nutrition in Health and Disease (10th Edition). Lippincott, Williams & Wilkins.
- Shor-Posner G., Basit A., Lu Y. et al. (1993) Hypocholesterolemia is associated with immune dysfunction in early human immunodeficiency virus-1 infection. Am. J. Med., 94: 515–519.
- Silva M., Skolnik P.R., Gorbach S.L. et al. (1998) The effect of protease inhibitors on weight and body composition in HIV-infected patients. AIDS, 12: 1645–1651.
- Singh R.B., Dubnov G., Niaz M.A. (2002) Effect of an indo-mediterranean diet on progression of coronary artery disease in high risk patients (Indo-Mediterranean Diet Heart Study): a randomised single-blind trial. Lancet, 360: 1455–1461.
- Smelt A.H., de Beer F. (2004) Apolipoprotein E and familial dysbetalipoproteinemia: clinical, biochemical, and genetic aspects. Sem. Vasc. Med., 4: 249–257.
- Smith S.C. Jr., Allen J., Blair S.N. et al.; AHA/ACC; National Heart, Lung and Blood Institute (2006) AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update: endorsed by the National Heart, Lung, and Blood Institute. Circulation, 113: 2363–2372.
- Stem M.P. (1996) Do non-insulin-dependent diabetes mellitus and cardiovascular disease share common antecedents? Ann. Intern. Med., 124: 110–116.
- Stricker R.B., Goldberg B. (1998) Weight gain associated with protease inhibitor therapy in HIV-infected patients. Res. Virol.149: 123–126.
- Suk S.H., Sacco R.L., Boden-Albala B. et al., The Northern Manhattan Stroke Study. (2003) Abdominal obesity and risk of ischemic stroke: the Northern Manhattan Stroke Study. Stroke, 34: 1586–1592.
- Sullivan A.K., Feher M.D., Nelson M.R. et al. (1998) Marked hypertriglyceridemia associated with ritonavir therapy. AIDS, 12: 1393–1394.
- Sullivan A.K., Nelson M.R. (1997) Marked hyperlipidemia on ritonavir therapy. AIDS, 11: 938–939.
- Superko H., Haskell W., DiRiccio C. (1990) Lipoprotein and hepatic lipase activity and high-density lipoprotein sub-classes after cardiac transplantation. Am. J. Cardiol., 66: 1131–1134.
- Suviolahti E., Lilja H.E., Pajukanta P. (2006) Unraveling the complex genetics of familial combined hyperlipidemia. Ann. Med., 38: 337–335.
- Taylor D.O., Thompson J.A., Hastillo A. et al. (1989) Hyperlipidemia after clinical heart transplantation. J. Heart Transplant., 8: 209–213.
- ten Asbroek A.H., de Mheen P.J., Defesche J.C. et al. (2001) Results from a family and DNA based active identification programme for familial hypercholesterolaemia. J. Epidemiol. Community Health, 55: 500–502.
- The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med., 329: 977–986.
- The US Multicenter FK506 Liver Study Group (1994) A comparison of tacrolimus (FK506) and cyclosporin for immunosuppression in liver transplantation. N. Engl. J. Med. 331: 1110–1115.
- Thiebaut R., Dabis F., Malvy D. et al. (2000) Serum triglycerides, HIV infection, and highly active antiretroviral therapy, Aquitaine Cohort, France, 1996 to 1998. J. Acquir. Immune Defic. Syndr., 23: 261–265.
- Thompson G.R. (1989) Handbook of hyperlipidaemia. London, Current Science, 236 p.
- Tonstad S., Thompson G.R. (2004) Management of Hyperlipidemia in the Pediatric Population. Curr. Treat. Options Cardiovasc. Med., 6: 431–437.
- Tsiodras S., Mantzoros C., Hammer S. et al. (2000) Effects of protease inhibitors on hyperglycemia, hyperlipidemia, and lipodystrophy: a 5-year cohort study. Arch. Intern Med., 160: 2060–2066.
- Turcato E., Bosello O., Di Francesco V. et al. (2000) Waist circumference and abdominal sagittal diameter as surrogates of body fat distribution in the elderly: their relation with cardiovascular risk factors. Int. J. Obes. Relat. Metab. Disord., 24: 1005–1010.
- Umans-Eckenhausen M.A., Defesche J.C., Sijbrands E.J. (2001) Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet, 357: 165–168.
- Vakkilainen J., Jauhiainen M., Ylitalo K. et al. (2002) LDL particle size in familial combined hyperlipidemia: effects of serum lipids, lipoprotein-modifying enzymes, and lipid transfer proteins. J. Lipid Res., 43: 598–603.
- van Maarle M.C., Stouthard M.E., Bonsel G.J. (2003) Quality of life in a family based genetic cascade screening programme for familial hypercholesterolaemia: a longitudinal study among participants. J. Med. Genet., 40: e3–e8.
- Vatsala A., Weinberg R.B., Schoenberg L. et al. (1989) Lipid abnormalities in cyclosporin-prednisonetreated renal transplant recipients. Transplantation, 48: 37–43.
- Veerkamp M.J., de Graaf J., Bredie S.J.H. (2002) Diagnosis of familial combined hyperlipidemia based on lipid phenotype expression in 32 families: results of a 5-year follow-up study. Arterioscler. Thromb. Vasc. Biol., 22: 274–282.
- Vigouroux C., Gharakhanian S., Salhy Y. et al. (1999) Diabetes, insulin resistance and dyslipidaemia in lipodystrophic HIV-infected patients on highly active antiretroviral therapy (HAART). Diabetes. Metab., 25: 225–232.
- Watanabe H., Söderlund S., Soro-Paavonen A. et al. (2006) Decreased high-density lipoprotein (HDL) particle size, prebeta-, and large HDL subspecies concentration in Finnish low-HDL families: relationship with intima-media thickness. Arterioscler. Thromb. Vasc. Biol., 26: 897–902.
- Walli R., Herfort O., Michl G.M. et al. (1998) Treatment with protease inhibitors associated with peripheral insulin resistance and impaired oral glucose tolerance in HIV-1-infected patients. AIDS,12: F167–173.
- Weil E., Wachterman M., McCarthy E.P. et al. (2002) Obesity among adults with disabling conditions. JAMA, 288: 1265–1268.
- Williams B., Poulter N.R., Brown M.J. et al. (2004) British Hypertension Society Guidelines for hypertension management 2004 (BHS-IV): summary. BMJ, 328: 634-640.
- Williams M.A., Fleg J.L., Ades P.A. et al.; American Heart Association Council on Clinical Cardiology Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention (2002) Secondary prevention of coronary heart disease in the elderly (with emphasis on patients 75 years of age): an American Heart Association scientific statement from the Council on Clinical Cardiology Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Circulation, 105: 1735–1743.
- Wilson D.E., Hata A., Kwong L.K. et al. (1993) Mutations in exon 3 of the lipoprotein lipase gene segregating in a family with hypertriglyceridemia, pancreatitis, and non-insulin-dependent diabetes. J. Clin. Invest., 92: 203–211.
- World Health Organisation, Human Genetics Programm (1997) Familial Hypercholesterolemia (FH): report of a WHO Consultation, Paris, 3 October. Geneva, Switzerland.
- Yadav D., Pitchumoni C.S. (2003) Issues in hyperlipidemic pancreatitis. J. Clin. Gastroenterol., 36: 54–62.
- Yanovski J.A., Miller K.D., Kino T. et al. (1999) Endocrine and metabolic evaluation of human immunodeficiency virus-infected patients with evidence of protease inhibitor-associated lipodystrophy. J. Clin. Endocrinol. Metab., 84: 1925–1931.
- Yeni P.G., Hammer S.M., Carpenter C.C. et al. (2002) Antiretroviral treatment for adult HIV infection in 2002. Updated recommendations of the International AIDS Society/USA Panel. J. Am. Med. Assoc., 288: 222–235.
- Yuan G., Al-Shali K.Z., Hegele R.A. (2007) Hypertriglyceridemia: its etiology, effects and treatment. CMAJ, 176: 1113–1120.
- Zambon A., Brown B.G., Deeb S.S. et al. (2006) Genetics of apolipoprotein B and apolipoprotein AI and premature coronary artery disease. J. Intern. Med., 259: 473–480.
- Zanchetti A., Grassi G., Mancia G. (2009) When should antihypertensive drug treatment be initiated and to what levels should systolic blood pressure be lowered? A critical reappraisal. J. Hypertens., 27: 923–934.
- Zechner R., Goldberg I.J. (2007) Session: Lipid metabolism. Program and abstracts of the XVI International Symposium on Drugs Affecting Lipid Metabolism; October 4–7, New York, NY.
- Zhang J.Q., Ji L.L., Fogt D.L. et al. (2007) Effect of exercise duration on postprandial hypertriglyceridemia in men with metabolic syndrome. J. Appl. Physiol., 103: 1339–1345.