Сегодня прямая ассоциация между содержанием в плазме крови ХС ЛПНП и риском наступления кардиоваскулярных событий считается установленным фактом (рис. 14.1). В ходе многочисленных рандомизированных клинических исследований и при проведении ряда метаанализов было установлено, что редукция уровня ХС ЛПНП на 1 ммоль/л способствует 23% снижению вероятности возникновения кардиоваскулярных событий (нефатальный инфаркт миокарда и кардиоваскулярная смерть), а также 21% редукции серьезных кардиоваскулярных событий, инсульта и потребности в коронарной реваскуляризации в течение ближайших 5 лет (Hayward R.A. et al., 2006).
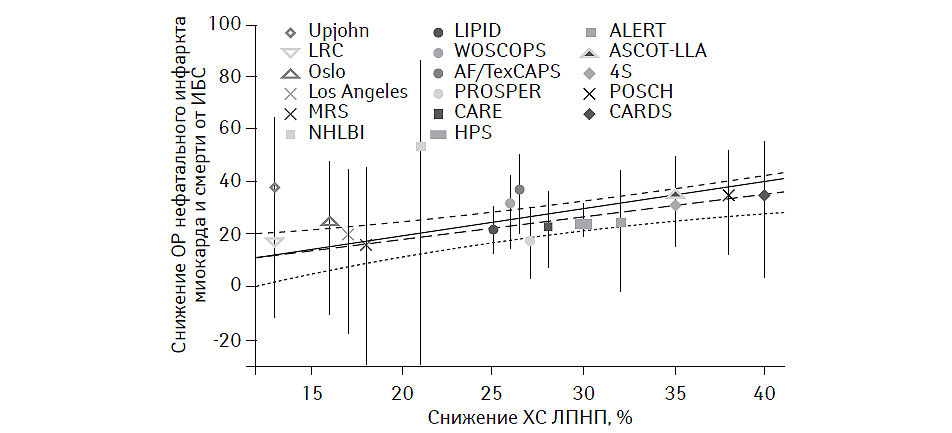
Рис. 14.1. Линейная зависимость между редукцией плазменного уровня ХС ЛПНП и величиной относительного риска возникновения серьезных кардиоваскулярных событий (модифицировано из работы Genest J., 2006).
Опыт использования различных гиполипидемических стратегий лечения способствовал возникновению представлений о том, что при сопоставимом снижении ХС ЛПНП при всех прочих равных условиях клиническая эффективность гиполипидемических лекарственных средств также не различается. С другой стороны, такое понятие, как мощность влияния препарата, невозможно отделить от популяции пациентов, тяжести гиперлипидемии и ее типа, а также суточной дозы лекарственного средства или их комбинации. Поэтому адекватное сопоставление различных гиполипидемических стратегий лечения возможно лишь при использовании методов прямого сравнения. Тем не менее, традиционно сведения о гиполипидемической эффективности различных лекарственных средств принято представлять в некотором усредненном диапазоне значений, отражающем ориентировочную эффективность достаточно широкого ранга доз. В табл. 14.1 приведена сравнительная характеристика гиполипидемической эффективности различных классов препаратов, базирующаяся именно на этих позициях.
Таблица 14.1 Сравнительная характеристика гиполипидемической эффективности лекарственных средств (модифицирована из работ The West of Scotland Coronary Prevention Study (WOSCOPS), 1998; Alessandri J.M. et al., 2004; Strandberg T.E. et al., 2004; Baigent C. et al., 2005; Hirsch M. et al., 2005; La Rosa J., 2005; Al Badarin F.J. et al., 2009)
| Основные классы препаратов | Изменения в липидном спектре плазмы крови (по сравнению с плацебо) | |||
| Общий ХС, % | ЛПНП, % | ЛПВП, % | ТГ, % | |
| Секвестранты желчных кислот | ↓ 15–20 | ↓ 15–30 | ↑ 3–5 | ↔ или ↑ |
| Эзетимиб | ↓ 12,6–14,2 | ↓ 17,4–19,7 | ↑ 5–10 | ↓ 5–10 |
| Никотиновая кислота и ее производные | ↓ 18–25 | ↓ 10–25 | ↑ 15–35 | ↓ 20–50 |
| Фибраты | ↓ 8–10 | ↓ 10–30 | ↑ 5–15 | ↓ 20–50 |
| Статины | ↓ 15–30 | ↓ 20–40 | ↑ 5–15 | ↓ 10–20 |
| ω-3-ненасыщенные жирные кислоты | ↔ | ↔ | ↔ | ↓ 20–30 |
| Пробукол* | ↓ 15–25 | ↓ 5–15 | ↑ 20–30 | ↔ |
| Торцетрапиб* | ↓ 20–25 | ↓ 15–20 | ↑ 50–60 | ↔ |
*Пробукол и торцетрапиб в настоящее время в клинической практике не используются, ↔ — отсутствие эффекта.
Основываясь на представлении о том, что ожидаемый клинический эффект является атрибутом гиполипидемического потенциала, вполне естественно предполагать, что указанные классы лекарственных средств могут оказывать несопоставимое влияние на твердые конечные точки (выживаемость, смертность, частота госпитализации и т.п.). Если фактический результат оказывался лучше ожидаемого, то его чаще всего пытались объяснить влиянием плейотропных (нелипидных) качеств лекарственных средств. Первоначально последние были описаны для статинов, а в последующем оказалось, что большинство других гиполипидемических препаратов также обладают ими. Серьезные проблемы возникают в том случае, когда фактический результат при применении гиполипидемических лекарственных средств оказывается хуже ожидаемого, особенно если целевые уровни ЛПНП или ЛПВП были достигнуты. Такие результаты были получены в ходе проведения ряда специально спланированных рандомизированных клинических исследований SANDS, ARBITER 6-HALTS, ENHANCE и ILLUMINATE (Fleg J.L. et al., 2008; Mascitelli L. et al., 2009; Ferrario C.M., 2010; Jakulj L. et al., 2010). Сложность ситуации заключалась в том, что достижение более низкого уровня ХС ЛПНП или более высокой концентрации ХС ЛПВП при использовании комбинации различных гиполипидемических лекарственных средств не отразилась на повышении вероятности выживания (то есть твердой конечной точке), ни на снижении интенсивности поражения органов-мишеней (то есть суррогатных маркеров: дисфункция эндотелия, толщина интима-медиального сегмента общей сонной артерии и т.п.). Поскольку современная концепция гиполипидемической терапии была основана на агрессивной позиции в отношении достижения более низкого уровня ХС ЛПНП (рис. 14.2), то сложность ситуации, в которой оказались эксперты, можно понять. Тем более, что при последнем пересмотре действующего клинического соглашения NCEP ATP III (2006) настаивали на необходимости хотя бы 50% редукции содержания ХС ЛПНП, если нет возможности достижения рекомендованного уровня (<70 мг/дл). В этой связи, вероятно, следует воздержаться от категоричных суждений о четком сопоставлении между клинической результативностью и гиполипидемическим потенциалом, особенно в рамках обсуждения изолированного влияния на какие-либо селективные компоненты липидного профиля плазмы крови, пусть даже такие важные для прогноза, как ХС ЛПНП или ХС ЛПВП.
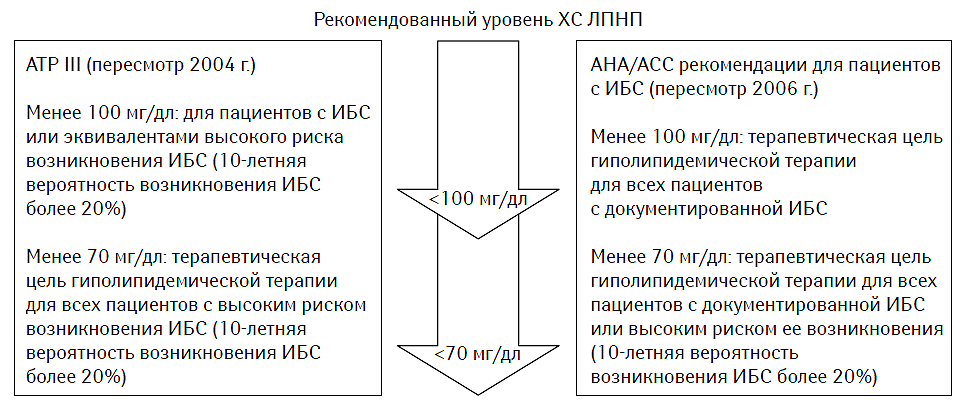
Рис. 14.2. Модификация представлений об агрессивности проведения гиполипидемических мероприятий
Таким образом, клинический аспект гиполипидемических мероприятий, вероятно, может рассматриваться как фрагмент, обусловливающий самостоятельное влияние на клинические исходы, иногда независимо от достижения липидных целей. Вместе с тем, это утверждение в некотором смысле является дискутабельным, что повышает роль врача в выборе тактики лечения, отборе пациента и формировании у последнего приверженности к терапии, желательно, конечно, на фоне достижения целевых рекомендованных уровней компонентов липидного профиля плазмы крови. Вероятно, именно поэтому столь важным является адекватный выбор инициального гиполипидемического лекарственного средства сообразно возникающей клинической ситуации (табл. 14.2). Тем более, что доказательства редукции величины кардиоваскулярной смерти представлены для статинов, фибратов, секвестрантов желчных кислот и никотиновой кислоты (и ее производных), тогда как возможность снижения риска общей смертности доказана только для статинов и производных никотиновй кислоты.
Таблица 14.2 Рекомендованные показания к применению гиполипидемических лекарственных средств основных классов
| Основные классы | Показания, основанные на подтвержденных клинических данных |
| Секвестранты желчных кислот | Первичная и вторичная гиперхолестеринемия |
| Статины | Первичная и вторичная профилактика кардиоваскулярной смертности
Лечение первичной гиперхолестеринемии и смешанной гиперлипидемии Лечение вторичной гиперлипидемии Снижение кардиоваскулярной смертности и летальности у пациентов с мягкой, умеренной и тяжелой гиперхолестеринемией и высоким кардиоваскулярным риском, в том числе с сахарным диабетом 2-го типа, а также у больных, перенесших инфаркт миокарда, ангиопластику, или у лиц с острым коронарным синдромом с нормальным или повышенным уровнем общего ХС в плазме крови Профилактика возникновения цереброваскулярных событий |
| Фибраты | Изолированная гипертриглицеридемия
Смешанная гиперлипидемия Семейная комбинированная дислипидемия, изолированная гипоальфахолестеринемия, ассоциированная с низким уровнем ХС ЛПВП Лечение дислипидемий I, IIb, III, IV, V типов и умеренной гиперхолестеринемии IIa типа у больных с ИБС при наличии противопоказаний к приему статинов Первичная и вторичная профилактика кардиоваскулярных событий |
| Никотиновая кислота и ее производные | Изолированная гипертриглицеридемия
Смешанная гиперлипидемия В комбинации со статинами при тяжелой гиперлипидемии |
| Эзетимиб | В монотерапии для лечения семейной гетерозиготной гиперхолестеринемии и первичной несемейной гиперхолестеринемии при неэффективности статинов
В комбинации со статинами для лечения семейной гомозиготной гиперхолестеринемии и смешанной гиперлипидемии |
| ω-3-ненасыщенные жирные кислоты | Изолированная тяжелая гипертриглицеридемия |
Классификация гиполипидемических лекарственных средств представлена в главе 9.
Данная глава посвящена обсуждению особенностей основных гиполипидемических лекарственных средств, обусловливающих специфику их терапевтического потенциала, уровень безопасности и переносимости, а также некоторые аспекты фармакокинетки, фармакогенетики и лекарственного взаимодействия.
Статины (ингибиторы ГМГ-КоА-редуктазы)
Статины — наиболее эффективные гиполипидемические лекарственные средства. Механизм их действия заключается в конкурентном ингибировании фермента эндоплазматического ретикулума ГМГ-КоА-редуктазы (Istvan E.S., Deisenhofer J., 2001), которая катализирует в ключевом участке биохимического каскада синтез эндогенного ХС, происходящего в гепатоците (Goldstein J.L., Brown M.S., 1990) (рис. 14.3). При этом внутриклеточное содержание ХС в гепатоцитах снижается, экспрессия мембранных рецепторов к ХС ЛПНП повышается по механизму up-regulation и, соответственно, увеличивается обратное поступление ХС ЛПНП в гепатоциты с последующим их катаболизмом.
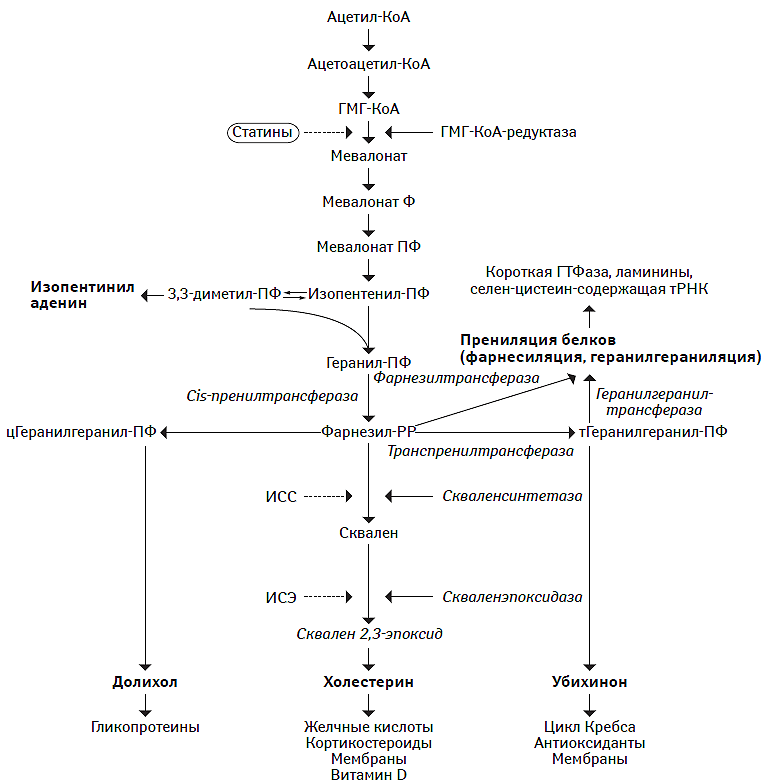
Рис. 14.3. Биохимическая мишень ингибиторов ГМГ-КоА-редуктазы
Ф — фосфат; ПФ — пирофосфат; ИСС — ингибитор скваленсинтетазы, ИСЭ — ингибитор скваленэпоксидазы.
Статины обладают доказанной эффективностью в проведении агрессивной гиполипидемической терапии в широком диапазоне доз у пациентов со многими коморбидными состояниями и различными вариантами гиперлипидемий в программах первичной и вторичной профилактики возникновения сердечно-сосудистых событий (Cannon C.P. et al., 2004). Доказано их благоприятное влияние в отношении ограничения кардиоваскулярного ремоделирования, реверсии дисфункции эндотелия, редукции провоспалительной активации и выраженности альбуминурии, снижения риска возникновения цереброваскулярных событий, повышения выживаемости при остром коронарном синдроме, после ангиопластики/стентирования, аорто-коронарного шунтирования и т.п. независимо от исходного уровня общего ХС и ХС ЛПНП (Nissen S.E. et al., 2006; Schoenhagen P. et al., 2006). Более подробно о влиянии терапии статинами на клинические исходы в селективных популяциях изложено в главе 13.
В настоящее время для клинического применения рекомендованы 6 препаратов — представителей класса статинов: правастатин, ловастатин, флувастатин, симвастатин, аторвастатин и розувастатин (Pasternak R.C. et al., 2002). Основные фармакологические характеристики статинов представлены в табл. 14.3. Питавастатин продолжают активно изучать, церивастатин отозван с рынка, а клинические исследования этого лекарственного средства прекращены ввиду его способности к повышению риска возникновения рабдомиолиза и миопатии (Law M., Rudnicka A.R., 2006; McKenney J.M. et al., 2006; Armitage J., 2007).
Таблица 14.3 Фармакологическая характеристика статинов (модифицирована из работ Williams D., Feely J., 2002; Martin P.D. et al., 2003; Mukhtar R.Y. et al., 2005)
| Название | Химический класс | Разновидность синтеза | Некоторые фармакокинетические характеристики | |||||||
| Липофильность | Собственная активность | Наличие активного метаболита | Биодоступность, % | Абсорбция с пищей % | Связь с белками плазмы крови, % | Максимальная суточная доза, мг | Период полувыведения, ч | |||
| Ловастатин | Дериват мевинивой кислоты, эфир | Производится путем ферментации Aspergillus terreus | + | – | + | 20 | 30 | 95 | 80 | 3 |
| Правастатин | Дериват нафтален-гептановой кислоты, натриевая соль | Природный статин | – | + | + | 34 | 17 | 50 | 40 | 2 |
| Симвастатин | Дериват мевинивой кислоты, эфир | Метиловый аналог ловастатина | + | – | + | 85 | 85 | 95 | 80 | 6 |
| Флувастатин | Дериват гептеновой кислоты, натриевая соль | Полностью синтетический статин | + | + | + | 98 | 24 | 98 | 40 | 2,3 |
| Аторвастатин | Дериват пиролл-гептановой кислоты, кальциевая соль | Полностью синтетическое средство | + | + | + | 95 | 86 | 98 | 80 | 14 |
| Церивастатин* | Натриевая соль пиридинилгептеновой кислоты | Полностью синтетический статин | + | + | + | 60 | 60 | 99 | 0,4 | 3 |
| Розувастатин | Кальциевая соль пиримидинил-гептеновой кислоты | Полностью синтетический статин | – | + | + | 20 | 20 | 90 | 40 | 19 |
| Питавастатин | Дериват хинолил-гептеновой кислоты | Полностью синтетический статин | ? | + | + | 80 | 80 | 96 | 4 | 11 |
*Церивастатин отозван с рынка, клинические исследования этого препарата прекращены в связи с высоким риском возникновения рабдомиолиза и миопатии (сведения представляют исторический интерес).
В качестве основных эффектов статинов принято рассматривать их способность к снижению уровня общего ХС, ХС ЛПНП, ТГ, апо-В-содержащих липопротеидов, а также повышение содержания ЛПВП, за счет чего достигается стабилизация атеромы. Необходимо отметить, что гиполипидемическая эффективность статинов может существенно варьировать и зависеть от дозы (табл. 14.4). Кроме того, линейной зависимости между удвоением суточной дозы статина и повышением его гиполипидемического потенциала обычно никогда не отмечают. Сопряженность эквивалентных доз статинов в отношении снижения общего ХС и ЛПНП представлена в табл. 14.5. Кроме липидных эффектов статинов, известны так называемые плейотропные (нелипидные) качества этих лекарственных средств (табл. 14.6).
Таблица 14.4 Гиполипидемическая эффективность основных представителей класса статинов в ранжированных дозах (по данным Strandberg T.E. et al., 2004; Hirsch M. et al., 2005; Finnish Medical Society Duodecim, 2007)
| Название статина | Суточная доза, мг/сут | Степень изменения плазменной концентрации, % | ||
| ЛПНП | ЛПВП | ТГ | ||
| Ловастатин | 20 | –24 | +3 | –5 |
| 40 | –31 | +5 | –8 | |
| Аторвастатин | 10 | –39 | +6 | –19 |
| 20 | –43 | +8 | –21 | |
| 40 | –49 | +9 | –25 | |
| 80 | –55 | +9 | –37 | |
| Симвастатин | 20 | –38 | +8 | –18 |
| 40 | –41 | +9 | –19 | |
| Правастатин | 20 | –22 | +7 | –15 |
| 40 | –34 | +12 | –24 | |
| Розувастатин | 5 | –52 | +13 | –10 |
| 10 | –52 | +14 | –35 | |
| Флувастатин | 40 | –25 | +4 | –14 |
| 80 | –36 | +7 | –19 | |
Таблица 14.5 Сопряженность эквивалентных доз статинов в отношении снижения общего ХС и ЛПНП
| Суточная доза статинов, мг/сут | Редукция плазменной концентрации, % | |||||||
| Правастатин | Флувастатин | Ловастатин | Симвастатин | Аторвастатин | Розувастатин | Питавастатин | Общий ХС | ЛПНП |
| 10 | 20 | 10 | 5 | – | 5 | 2 | 18 | 23 |
| 20 | 40 | 20 | 10 | 5 | 22 | 27 | ||
| 40 | 80 | 40 | 20 | 10 | 27 | 34 | ||
| 80 | 40 | 20 | 10 | – | 32 | 41 | ||
| 80 | 40 | 20 | 4 | 37 | 48 | |||
| – | 80 | 40 | – | 42 | 55 | |||
Таблица 14.6 Возможные плейотропные (нелипидные) эффекты статинов (по данным Hodis H.N. et al., 1996; MacMahon S. et al., 1998; Hedblad B. et al., 2001; HPS Collaborative Group, 2002; Taylor A.J. et al., 2002; Cannon C.P. et al., 2004; Crouse J.R. III et al., 2004; Schönbeck U., Libby P., 2004; Bulut D. et al., 2005; Ray K.K., Cannon C.P., 2005; LaRosa J.C. et al., 2005; Robinson J.G. et al., 2005; Fichtlscherer S. et al., 2006)
|
Плейотропные эффекты |
Название статинов |
|||||
|
|
Правастатин |
Флувастатин |
Симвастатин |
Ловастатин |
Аторвастатин |
Розувастатин |
|
Стабилизация атеромы |
+ |
+ |
+ |
+ |
+ |
+ |
|
Антипролиферативный эффект |
+ |
+ |
+ |
+ |
+ |
+ |
|
Прямое вазодилатирующее и антиишемическое действие |
+ |
+ |
+ |
+ |
+ |
+ |
|
Активация неоангиогенеза в ишемизированных тканях |
– |
– |
+ |
– |
+ |
+ |
|
Торможение миграции и ингибирование пролиферации гладкомышечных клеток |
+ |
+ |
+ |
+ |
+ |
+ |
|
Снижение синтеза тромбина |
+ |
+ |
+ |
+ |
+ |
+ |
|
Уменьшение агрегации тромбоцитов |
+ |
+ |
+ |
+ |
+ |
+ |
|
Снижение интенсивности синтеза тромбоцитов |
– |
– |
+ |
– |
– |
– |
|
Реверсия дисфункции эндотелия |
+ |
– |
+ |
+ |
+ |
+ |
|
Снижение вязкости крови |
– |
– |
+ |
– |
+ |
– |
|
Антикоагулянтный эффект |
– |
– |
+ |
– |
+ |
– |
|
Увеличение продукции оксида азота эндотелиоцитами |
– |
– |
+ |
+ |
– |
– |
|
Снижение инфильтрации сосудистой стенки макрофагами |
+ |
+ |
+ |
+ |
+ |
+ |
|
Снижение интенсивности пероксидации ЛПНП |
+ |
+ |
+ |
+ |
+ |
+ |
|
Снижение уровня СРП |
+ |
– |
+ |
+ |
+ |
+ |
|
Снижение продукции провоспалительных цитокинов |
+ |
– |
+ |
+ |
+ |
+ |
|
Потенцирование иммуносупрессивной активности циклоспорина |
+ |
– |
– |
+ |
– |
– |
|
Потенцирование антигипертензивного эффекта иАПФ и АРА |
– |
– |
+ |
– |
+ |
– |
|
Потенцирование редукции альбуминурии при применении иАПФ и АРА |
– |
– |
+ |
– |
+ |
+ |
|
Снижение риска возникновения впервые выявленного сахарного диабета |
+ |
+ |
+ |
+ |
+ |
+ |
|
Уменьшение выраженности гипертрофии ЛЖ |
– |
– |
+ |
– |
+ |
– |
|
Снижение риска возникновения эпизодов возвратной желудочковой тахикардии |
– |
– |
+ |
– |
+ |
– |
|
Уменьшение формирования депозитов β-амилоида |
– |
– |
+ |
– |
+ |
– |
|
Снижение риска возникновения васкулярной деменции |
+ |
+ |
+ |
– |
+ |
– |
|
Снижение риска возникновения болезни Альцгеймера |
– |
– |
+ |
+ |
+ |
– |
|
Уменьшение выраженности остеопороза и частоты переломов шейки бедра |
– |
– |
+ |
+ |
– |
– |
|
Снижение риска возникновения конкрементов в желчном пузыре |
– |
– |
+ |
+ |
– |
– |
|
Снижение риска возникновения онкологических заболеваний |
? (–) |
+ |
+ |
+ |
+ |
– |
|
Снижение риска возникновения и прогрессирования узураций субхондральной кости у пациентов с ревматоидным артритом |
+ |
– |
– |
+ |
+ |
– |
Правастатин
Правастатин представляет собой природный статин, выделенный из некоторых грибов. Патентная защита на оригинальный препарат Lipostat («Bristol-Myers Squibb Pharmaceuticals Ltd») закончилась в 2004 г. В настоящее время Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA; Food and Drug Administration Advisory Committee Meeting, 2010) одобрило следующие показания к применению этого лекарственного средства (табл. 14.7):
- лечение первичной гиперхолестеринемии или смешанной гиперлипидемии при неадекватности нефармакологических методов как дополнение к диетическим ограничениям;
- снижение кардиоваскулярной смертности и летальности у пациентов с умеренной и тяжелой гиперхолестеринемией и высоким кардиоваскулярным риском;
- вторичная гиперлипидемия у пациентов с ВИЧ, получающих АРВТ;
- снижение кардиоваскулярной смертности и летальности у пациентов, перенесших инфаркт миокарда, а также у лиц с нестабильной стенокардией с нормальным или повышенным уровнем общего ХС в плазме крови как дополнение к другим методам коррекции факторов риска;
- лечение посттрансплантационной гиперлипидемии у пациентов, получающих иммуносупрессивную терапию.
Таблица 14.7 Показания к применению статинов (модифицировано из работ: WHO consultation, 1998; Penzak S.R. et al., 2000; Dubé M.P. et al, 2003; Rader D.J. et al., 2003; Cheng J.W., 2004; van Wissen S. et al., 2005; Lescol [prescribing information], 2006; Crestor [prescribing information], 2007; Lipitor [prescribing information], 2007; Mevacor [prescribing information], 2007; Pravachol [prescribing information]., 2007; Zocor [package insert], 2007; Eldor R., Raz I., 2009; Macaulay T.E. et al., 2009; Minhas R. et al., 2009)
| Показания к применению | Название статинов | |||||
| Правастатин | Флувастатин | Симвастатин | Ловастатин | Аторвастатин | Розувастатин | |
| Первичная гиперхолестеринемия | + | + | + | + | + | + |
| Гомозиготная семейная гиперхолестеринемия | – | – | + | – | + | + |
| Гетерозиготная семейная гиперхолестеринемия | – | – | – | – | + | – |
| Смешанная и комбинированная гиперлипидемия | + | + | + | + | + | + |
| Гипертриглицеридемия | + | – | + | – | + | + |
| Первичная дисбеталипопротеидемия | + | – | + | – | + | + |
| Первичная профилактика возникновения ИБС | – | – | – | + | – | – |
| Ограничение прогрессирования коронарного атеросклероза у пациентов с первичной гиперхолестеринемией и документированной ИБС | – | + | – | + | + | + |
| Снижение кардиоваскулярной смертности и летальности у пациентов с умеренной и тяжелой гиперхолестеринемией и высоким кардиоваскулярным риском | + | – | + | + | + | + |
| Снижение кардиоваскулярной смертности и летальности у пациентов, перенесших инфаркт миокарда, а также у лиц с нестабильной стенокардией с нормальным или повышенным уровнем общего ХС в плазме крови как дополнение к другим методам коррекции факторов риска | + | – | + | – | + | – |
| Посттрансплантационная гиперлипидемия | + | – | + | – | – | – |
| Профилактика возникновения цереброваскулярных событий | + | + | – | – | – | |
| Вторичная профилактика возникновения кардиоваскулярных событий у пациентов, перенесших перкутанную ангиопластику | – | + | – | + | – | |
| Вторичная гиперлипидемия у пациентов с ВИЧ, получающих АРВТ | + | + | – | – | + | – |
Максимальная доза для правастатина составляет 40 мг/сут, стартовая доза — 10 мг/сут. Повышение дозы для достижения целевого уровня ХС ЛПНП обычно проводят с интервалом 4 нед.
Ловастатин
Ловастатин представляет собой полусинтетический статин, получаемый путем ферментации Aspergillus terreus. Патентная защита на оригинальный препарат Mevacor («Merck Sharp & Dohme Ltd») закончилась в 2000 г. В настоящее время FDA (Food and Drug Administration Advisory Committee Meeting, 2010) одобрила применение этого лекарственного средства по следующим показаниям (см. табл. 14.7):
- лечение первичной гиперхолестеринемии или смешанной гиперлипидемии;
- снижение кардиоваскулярной смертности и летальности у пациентов с мягкой, умеренной и тяжелой гиперхолестеринемией и высоким кардиоваскулярным риском, а также у больных, перенесших инфаркт миокарда с нормальным или повышенным уровнем общего ХС в плазме крови.
Максимальная доза для ловастатина составляет 40 мг/сут, стартовая доза — 10 мг/сут. Повышение дозы для достижения целевого уровня ХС ЛПНП обычно проводят с интервалом 4 нед.
Флувастатин
Оригинальный флувастатин Lescol («Novartis Pharmaceuticals UK Ltd») — полностью синтетический статин, выпускающийся в двух основных формах: немедленного (immediate-release — IR) в капсулах по 20 и 40 мг и медленного (extended-release — XL) высвобождения в капсулах по 80 мг. В соответствии с рекомендациями FDA (см. табл. 14.7) применение флувастатина одобрено при следующих состояниях:
- первичная гиперхолестеринемия, в том числе ассоциированная с низким уровнем ХС ЛПВП (для последней ситуации рекомендована только форма медленного высвобождения);
- смешанная гиперлипидемия;
- ограничение прогрессирования коронарного атеросклероза у пациентов с первичной гиперхолестеринемией и ИБС;
- вторичная профилактика возникновения кардиоваскулярных событий у пациентов, перенесших перкутанную ангиопластику;
- вторичная гиперлипидемия у пациентов с ВИЧ, получающих АРВТ.
Стартовая рекомендованная доза флувастатина составляет 40 мг/сут, хотя при мягкой гиперлипидемии препарат в дозе 20 мг/сут также может быть вполне адекватным. Повышение до максимально разрешенной дозы 80 мг/сут может осуществляться с интервалом 4 нед.
Симвастатин
Симвастатин является полусинтетическим статином, близким по химической структуре к ловастатину. Патентная защита на оригинальный симвастатин Zocor («Merck Sharp & Dohme Ltd») закончилась в 2003 г. В соответствии с рекомендациями FDA (см. табл. 14.7) применение симвастатина одобрено для лечения следующих состояний:
- первичная гиперхолестеринемия, а также семейная гомозиготная гиперхолестеринемия при неадекватности немедикаментозных методов лечения;
- смешанная гиперлипидемия;
- умеренная и тяжелая гиперлипидемия у пациентов с высоким кардиоваскулярным риском;
- снижение кардиоваскулярной смертности и летальности у пациентов с документированной ИБС или сахарным диабетом при нормальном или повышенном уровне общего ХС в плазме крови;
- снижение риска возникновения кардиоваскулярных событий у пациентов с умеренным суммарным 10-летним риском манифестации ИБС (10–15%).
Стартовая доза симвастатина зависит от клинической ситуации, возможно также ее повышение до максимальной (80 мг/сут) с интервалом титрования 4 нед. Максимальная суточная доза препарата 80 мг рекомендована для лечения умеренной и тяжелой гиперлипидемии у пациентов с высоким кардиоваскулярным риском.
Аторвастатин
Аторвастатин представляет собой полностью синтетический статин. Оригинальный аторвастатин Liprimar («Pfizer Ltd») утратил патентную защиту в 2005 г. Препарат рекомендован для лечения первичной, в том числе гомозиготной и гетерозиготной семейной гиперхолестеринемии, смешанной гиперлипидемии. Кроме того, препарат лицензирован для лечения вторичной гиперлипидемии у пациентов с ВИЧ, получающих АРВТ, для ограничения прогрессирования коронарного атеросклероза у пациентов с первичной гиперхолестеринемией и ИБС (см. табл. 14.7).
Обычно стартовая доза препарата составляет 10 мг/сут, которую можно постепенно повышать до максимальной (80 мг/сут) с интервалом 4 нед.
Розувастатин
Розувастатин — полностью синтетический статин. Сегодня на фармацевтическом рынке препарат представлен только оригинальной субстанцией Crestor («AstraZeneca UK Ltd»). Препарат лицензирован для лечения первичной гиперхолестеринемии (включая семейную гомозиготную гиперхолестеринемию), смешанной и комбинированной гиперлипидемии, при неэффективности немедикаментозных методов лечения, а также для ограничения прогрессирования коронарного атеросклероза у пациентов с первичной гиперхолестеринемией и ИБС (Cheng J.W., 2004).
Вторичная гиперлипидемия у пациентов с ВИЧ, получающих АРВТ, не является формальным показанием к применению розувастатина, хотя к настоящему времени уже завершены некоторые рандомизированные клинические исследования, демонстрирующие перспективность использования препарата по новому показанию (Dubé M.P. et al., 2003; Calza L. et al., 2005).
Рекомендованная стартовая суточная доза розувастатина колеблется от 5 до 10 мг/сут в зависимости от клинической ситуации (Cheng-Lai A., 2003). Максимальная суточная доза составляет 40 мг/сут и обычно используется в лечении пациентов с тяжелой гиперхолестеринемией и высоким кардиоваскулярным риском, а также резервируется для лиц с доказанной неэффективностью других медикаментозных способов лечения нарушений липидного обмена (Penzak S.R. et al., 2000). В последнем случае титрация с более низкой дозы не является необходимой. Следует отметить, что в странах Азии применение розувастатина в дозе 40 мг/сут не рекомендовано вследствие существования в этой популяции инициально высокого риска развития миопатии и рабдомиолиза (Cheng-Lai A., 2003).
Безопасность применения статинов
Популяция пациентов с показаниями к длительному приему статинов постоянно расширяется (см. табл. 14.7). Несмотря на то, что эти препараты хорошо переносятся большинством пациентов, они могут приводить к развитию серьезных побочных эффектов, сопровождающихся различными симптомами и требующими повышенного внимания (табл. 14.8). Наиболее часто отмечают гастроинтестинальные расстройства (тошнота, рвота, боль в животе, диарея, сухость во рту, изжога), головную боль, кожную сыпь, бессонницу, парестезии, головокружение, судороги, тревогу, депрессию, аллергические реакции, протеинурию, гинекомастию, нарушения функции печени и ряд других побочных эффектов (Sacks F.M. et al., 1996; LIPID Study Group, 1998; Downs J.R. et al., 2001; HPS Collaborative Group, 2002; Physician’s Desk Reference, 2005). Общая частота возникновения побочных эффектов при применении статинов колеблется в пределах 1–3%. Среди наиболее опасных побочных эффектов следует более подробно рассмотреть нарушения функции печени и миопатии.
Таблица 14.8
| Название статина | Частота возникновения побочных эффектов в группе статинов (плацебо) | |||
| Миалгия, % | Головная боль, % | Тошнота, % | Кожная сыпь, % | |
| Аторвастатин | 3,2 (1,1) | 5,4 (7,0) | 2,3 (4,1) | 3,9 (0,7) |
| Флувастатин | 1,7 (2,3) | 3,8 (3,0) | 2,0 (1,4) | 1,5 (1,6) |
| Ловастатин | 2,6 (1,7) | 2,6 (2,7) | 1,9 (2,5) | 0,8 (0,7) |
| Правастатин | 2,7 (1,0) | 6,2 (3,9) | 7,3 (7,1) | 4,0 (1,1) |
| Симвастатин | 1,2 (1,3) | 3,5 (5,1) | 1,3 (1,9) | 0,6 (0,6) |
| Розувастатин | 2,8 (1,3) | 5,5 95,0) | 3,4 (3,1) | ≥2,0 |
Нарушение функции печени обычно проявляется в виде асимптомного повышения активности печеночных трансаминаз, более чем в 3 раза превышающих референсные значения (Physician’s Desk Reference, 2005). Общая частота этого побочного эффекта обычно не превышает 0,5–2% всех негативных реакций. Так, по данным метаанализа M.R. Law и соавторов (2003) частота возникновения элевации аланинаминотрансферазы у пациентов, принимающих статины, составила 1,3%, а в группе плацебо — 1,1%. При этом повышение КФК в плазме крови было зарегистрировано у 0,17 и 0,13% пациентов, получавших соответственно статины или плацебо. Иногда повышение активности трансаминаз носит транзиторный характер, но в большинстве случаев регистрация подобного осложнения требует снижения дозы или отмены лечения. Большинство экспертов рекомендуют серьезно относиться к скринингу кандидатов для последующего лечения статинами, рассматривая коморбидные состояния как потенциальную причину для возникновения побочных эффектов (Smith C.C. et al., 2003; Libby P.J., 2005). Так, в большинстве случаев клинически значимые нарушения функции печени были зарегистрированы у пациентов с вирусным гепатитом С в анамнезе, а также у лиц, склонных к чрезмерному употреблению алкоголя (Smith C.C. et al., 2003). Кроме того, вероятность возникновения нарушений функции печени может зависеть не только от используемого препарата, но и от его суточной дозы, хотя это взаимоотношение и не носит линейного характера (рис. 14.4).
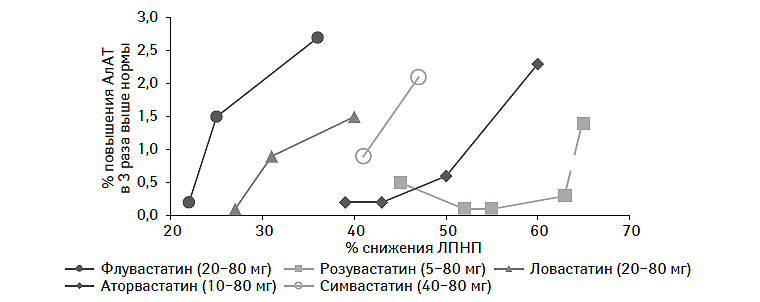
Рис. 14.4. Зависимость элевации печеночных трансаминаз от мощности гиполипидемического эффекта
При проведении метаанализа 5 крупных рандомизированных клинических исследований 4S, WOSCOPS, CARE, LIPID, AFCAPS/TexCAPS, включающих данные о клиническом статусе 30 815 пациентов, было установлено, что риск элевации печеночных трансаминаз в группе больных, получавших лечение статинами, по сравнению с пациентами, в терапии которых использовали плацебо, составил всего 1,13 (95% ДИ=0,95–1,33, р=0,17) (LaRosa J.C. et al., 1999). ОР возникновения элевации КФК в группе статинов по сравнению с группой плацебо составил 1,25 (95% ДИ=0,83–1,89, р=0,29).
Риск развития миопатии был принят во внимание в процессе отзыва с фармацевтического рынка церивастатина в августе 2001 г. после того, как была установлена ассоциация приблизительно 100 случаев смерти от рабдомиолиза с применением этого препарата (Thompson P.D. et al., 2003). Лучшее понимание молекулярных механизмов, вовлекаемых в процессы статин-индуцированной миопатии, может помочь идентифицировать группы пациентов, чувствительных к возникновению побочных эффектов статинов, и тем самым повысить их безопасность.
Дефиниция и эпидемиология статинассоциированной миопатии
Существуют весьма значительные разногласия, касающиеся использования терминологии при описании побочных эффектов. В попытке разрешить эти противоречия Американская коллегия кардиологов в сотрудничестве с Американским национальным институтом сердца, легких и крови пришла к необходимости создания ряда дефиниций (табл. 14.9) (Pasternak R.C. et al., 2002; Giannoglou G.D. et al., 2007). Частота миопатии в плацебо-контролируемых рандомизированных клинических исследованиях представлена в табл. 14.10. В этих исследованиях невозможно было пропустить случаи развития миопатии, поскольку участвовавших в них пациентов внимательно наблюдали (Law M., Rudnicka A.R., 2006).
Таблица 14.9 Дефинизирующие критерии поражений мышечной системы, ассоциированных с приемом статинов, основанные на соглашении экспертов Американской коллегии кардиологов и Американского национального института сердца, легких и крови
| Патологическое состояние | Дефиниция |
| Миопатия | Общий термин миопатия, отражающий наличие различных заболеваний мышечной системы, может иметь приобретенный или врожденный характер и манифестировать непосредственно после рождения или на протяжении всей жизни |
| Миалгия | Мышечная боль или слабость без повышения плазменной концентрации КФК |
| Миозит | Симптомы поражения мышечной системы с повышением плазменной концентрации КФК |
| Рабдомиолиз | Симптомы поражения мышечной системы с повышением плазменной концентрации КФК (более чем в 10 раз по сравнению с нормальным уровнем) и креатинина (обычно сопровождаются появлением миоглобина в моче, бурая окраска мочи) |
Таблица 14.10 Метаанализ рандомизированных плацебо-контролируемых клинических исследований, посвященных применению статинов и касающегося их побочного эффекта в виде миопатии
| Испытание | Дизайн | Доза, мг | Лечебный режим | Продолжи-тельность | Рабдомиолиз | Миопатия | Мышечная боль | Элевация КФК | |||||
| Статин | Плацебо | Статин | Плацебо | Статин | Плацебо | Статин | Плацебо | Статин | Плацебо | ||||
| Ловастатин | |||||||||||||
| AFCAPS/TexCAPS | ПП | 20–40 | 3304 | 3,301 | 5,2 | 1 | 2 | 11 | 9 | НД | – | 21 | 21 |
| CCAIT | ИБС | 36 | 165 | 166 | 2 | НД | – | НД | – | НД | НД | – | – |
| EXCEL | ПП + ИБС | 20–80 | 6582 | 1663 | 0,9 | 0 | 0 | 5 | 0 | 512 | 125 | 17 | 7 |
| ACAPS | ПП | 20–40 | 460 | 459 | 3 | 0 | 0 | НД | – | НД | – | НД | – |
| Правастатин | |||||||||||||
| CARE | ИБС | 40 | 2081 | 2078 | 5 (медиана) | 0 | 0 | 0 | 4 | НД | – | 12 | 7 |
| WOSCOPS | ИБС | 40 | 3302 (муж.) | 3293 (муж.) | 4,9 (средняя) | 0 | 0 | 20 | 19 | 97 | 102 | 3 | 1 |
| PLAC I | ИБС | 40 | 206 | 202 | 3 | НД | – | 0 | 0 | НД | – | 0 | 0 |
| PLAC II | ИБС | 10–40 | 75 | 76 | 3 | НД | – | НД | – | НД | – | НД | – |
| REGRESS | ИБС | 40 | 323 (муж.) | 330 (муж.) | 2 | 0 | 0 | 1 | 0 | НД | – | 0 | 0 |
| PREDICT | ИБС | 40 | 347 | 348 | 0,5 | НД | – | НД | – | НД | – | НД | – |
| LIPID | ИБС | 40 | 4286 | 4271 | 6 (медиана) | 0 | 0 | 8 | 10 | НД | – | НД | – |
| LCAD | ИБС | 20–40a | 70 | 56 | 2 | Д | – | НД | – | НД | – | НД | – |
| KAPS | ПП | 40 | 224 | 223 | 3 | НД | – | 0 | 0 | НД | – | НД | – |
| PRINCE | ПП | 40 | 666 | 673 | 24 нед | НД | – | НД | – | НД | – | НД | – |
| ALLHAT–L | ИБС + ПП | 40 | 5170 | 5185 | 4,8 (медиана) | НД | – | НД | – | НД | – | НД | – |
| PROSPER | ИБС + ПП | 40 | 2891 | 2913 | 3,2 | 0 | 0 | 36 | 32 | НД | – | 0 | 0 |
| FAST | ПП | 10b | 83 | 81 | 2 | НД | – | НД | – | НД | – | НД | – |
| Симвастатин | |||||||||||||
| 4S | ИБС | 10–40 | 2221 | 2223 | 5,4 (медиана) | 1 | 0 | 11 | 8 | 71 | 64 | 6 | 1 |
| CIS | ИБС | 40 | 129 | 125 | 2,3 (средняя) | НД | – | НД | – | НД | – | НД | – |
| Wenke K. et al., 1997 | ТС | 10 | 35 | 37 | 4 | 0 | – | НД | – | НД | – | 0 | 0 |
| HPS | ИБС + ТС | 40 | 10269 | 10267 | 5,3 (средняя) | 5 | 3 | 49 | 50 | 3330 | 3359 | 1 | 2 |
| MAAS | ИБС | 20 | 193 | 188 | 4 | 0 | 0 | НД | – | НД | – | НД | – |
| Флувастатин | |||||||||||||
| LCAS | ИБС | 40c | 214 | 215 | 2,5 | 0 | 0 | НД | – | НД | – | 1 | 2 |
| LiSA | ИБС | 80 | 187 | 178 | 1 | 0 | 0 | 0 | 0 | НД | – | 0 | 1 |
| FLARE | ИБС | 80 | 409 | 427 | 0,8 | 0 | 0 | 7 | 3 | НД | – | 0 | 0 |
| ALERT | ТП | 40–80 | 1045 | 1049 | 5, (средняя | 1 | 1 | 0 | 0 | 526 | 531 | 3 | 1 |
| LIPS | ИБС | 80 | 844 | 833 | 3,9 (медиана) | 0 | 0 | НД | – | НД | – | 0 | 3 |
| Аторвастатин | |||||||||||||
| AVERT | ИБС | 80 | 164 | 177 | 1,5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MIRACL | ИБС | 80 | 1538 | 1548 | 0,3 | 0 | 0 | 0 | 0 | НД | – | НД | – |
| GREACE | ИБС | 24d | 800 | 800 | 3 | 0 | 0 | 0 | 0 | НД | – | НД | – |
| ASCOT–LLA | ВР | 10 | 5168 | 5137 | 3,3 (медиана) | 1 | 3 | НД | – | НД | – | НД | – |
| SPARCL | ВП | 80 | 2365 | 2366 | 4,9 | 2 | 3 | 7 | 7 | 129 | 141 | – | – |
| Церивастатин | |||||||||||||
| ENCORE I | ИБС | 0,4 | 114 | 119 | 0,5 | НД | – | 1 | 0 | НД | 0 | 2 | 0 |
| Розувастатин | |||||||||||||
| ASTEROID | ИБС | 40b | 507 | – | 2 | 0 | – | 19 | – | НД | – | 0 | – |
| На 100 тыс. человеко-лет | – | – | – | – | – | 4,04 | 4,11 | 70 | 65 | 2090 | 1977 | 24,24 | 21 |
AFCAPS/TexCAPS — Air Force/Texas Coronary Atherosclerosis Prevention Study; CCAIT — Canadian Coronary Atherosclerosis Intervention Trial; EXCEL — EXpanded Clinical Evaluation of Lovastatin; ACAPS — Asymptomatic Carotid Artery Progression Study; CARE — Cholesterol And Recurrent Events trial; WOSCOPS — West Of Scotland COronary Prevention Study; PLAC I — Pravastatin Limitation of Atherosclerosis in the Coronary arteries; PLAC II: Pravastatin Lipids and Atherosclerosis in the Carotid arteries; REGRESS — REgression GRowth Evaluation Statin Study; PREDICT — Prevention of Restenosis by Elisor after Transluminal Coronary Angioplasty; LIPID — Long-term Intervention with Pravastatin in Ischaemic Disease study; LCAD — Lipid Coronary Artery Disease study; KAPS — Kuopio Atherosclerosis Prevention Study; PRINCE — Pravastatin INflammation/CRP Evaluation;ALLHAT–LLT — the Lipid-Lowering Trial component of the Antihypertensive and Lipid-Lowering treatment to prevent Heart ATtack trial; PROSPER — PROspective Study of Pravastatin in the Elderly at Risk; FAST — Fukuoka AtheroSclerosis Trial; 4S — Scandinavian Simvastatin Survival Study; CIS — the multicenter Coronary Intervention Study; HPS — Heart Protection Study; MAAS — Multicenter AntiAtheroma Study; LCAS — Lipoprotein Coronary Atherosclerosis Study; LiSA — Lescol In Severe Atherosclerosis; FLARE — FLuvastatin Angiographic REstenosis trial; ALERT — Assessment of LEscol in Renal Transplantation; LIPS — Lescol Intervention Prevention Study; AVERT — Atorvastatin VErsus Revascularization Treatment; MIRACL — Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering trial; GREACE — GREek Atorvastatin and Coronary heart disease Evaluation study; ASCOT-LLA — Anglo-Scandinavian Cardiac Outcomes Trial, Lipid Lowering Arm; SPARCL — Stroke Prevention by Aggressive Reduction in Cholesterol Levels; ENCORE I — Evaluation of Nifedipine and Cerivastatin On Recovery of coronary Endothelial function; ASTEROID — A STudy to Evaluate the effect of Rosuvastatin On Intravascular ultrasound-Derived coronary atheroma burden.
НД — нет данных, ПП — первичная профилактика, ВП — вторичная профилактика, ВР — высокий риск, ТП — трансплантация почек, ТС — трансплантация сердца.
aС дополнительным назначением никотиновой кислоты и колестирамина.
bРезультаты когортных исследований по первичной профилактике. Результаты когортных исследований по вторичной профилактике без плацебо-контроля.
cС применением колестирамина.
dНе является рандомизированным клиническим исследованием.
Однако некоторые группы больных, склонных к манифестации статин-индуцированной миопатии, или были исключены из рандомизированных клинических исследований как те, у которых отмечено повышение уровня КФК в плазме крови, или были недостаточно представлены в исследуемой популяции (возраст старше 75 лет с почечной и, особенно, печеночной недостаточностью) (Rosenson R.S., 2004). Кроме того, в некоторых исследованиях, таких как одно из наибольших недавно завершенных рандомизированных клинических исследований SPARCL (Stroke Prevention by Aggressive Reduction in Cholesterol Levels study) (Amarenco P. et al., 2006), пациентам разрешалось принимать статины при наличии показаний. В этих исследованиях, не посвященных статинам, частота дополнительного применения препаратов могла негативно повлиять на величину реального риска (Mukhtar R.Y., Reckless J.P., 2005). Следовательно, эти испытания могли недооценить частоту событий в том случае, когда статины применяли в обширной неселективной популяции с низким количеством накопленных данных (Thompson P.D. et al., 2003). Кроме того, эти исследования обеспечивали получение ограниченных сведений, касающихся применения статинов в высоких дозах. С другой стороны, данные, добровольно направленные в регуляторные органы, с подтверждением возникновения побочных эффектов, хотя и имели преимущества перед информацией, собранной путем опроса, могли занижать реальную частоту негативных событий (Law M., Rudnicka A.R., 2006). Общеизвестно, что о возникновении нетяжелых признаков поражения мышц, отличных от рабдомиолиза, сообщается редко, поскольку жалобы на легкую мышечную боль вследствие физической нагрузки зачастую пропускаются, а частота возникновения этих случаев может находиться в диапазоне 5% и более. Таким образом, реальную частоту возникновения статин-индуцированной миопатии точно рассчитать достаточно сложно.
Факторы, предрасполагающие к возникновению статинассоциированной миопатии
Статинассоциированная миопатия имеет дозозависимый характер и связывается с применением всех статинов. К настоящему времени уже описаны предрасполагающие условия и факторы, способствующие развитию этого состояния (Thompson P.D. et al., 2003; Antons K.A. et al., 2006; Arora R., et al., 2006) (табл. 14.11). Большинство из них приводит к формированию миопатии посредством нарушения процессов метаболизма и повышения биодоступности статинов. Кроме того, многие патологические состояния и лекарственные средства (в том числе физическое перенапряжение, употребление алкоголя, применение глюкокортикостероидов) могут рассматриваться как независимые предрасполагающие факторы возникновения миопатии, приводя к аддитивному эффекту и даже индуцированию формирования миопатии на фоне приема статинов. Кроме того, на фоне приема статинов возможна манифестация ранее латентно протекающих сопутствующих как врожденных, так и приобретенных нейромышечных (амитохондриальная миопатия) или ревматических (ревматическая полимиалгия) заболеваний.
Таблица 14.11 Факторы, предрасполагающие к возникновению статин-индуцированной миопатии
| Предрасполагающие факторы |
| Возраст |
| Женский пол |
| Почечная недостаточность |
| Печеночная недостаточность |
| Гипотиреоидное состояние |
| Полипрагмазия |
| Периоперационный период |
| Метаболические заболевания мышечной системы:
дефицит карнитин-пальминин трансферазы 2 болезнь МакАрдла дефицит миоаденилат-дезаминазы |
| Ревматическая полимиалгия |
Фармакокинетика статинов и миопатия
Существуют многочисленные сведения о наличии ассоциации между фармакологическими свойствами и фармакокинетическими характеристиками статинов с одной стороны и их миотоксическим потенциалом — с другой (табл. 14.12). Несмотря на то что точные механизмы статин-индуцированной миопатии до конца не установлены, комплексное взаимодействие процессов абсорбции, поступления в гепатоциты, растворимости, связывания с белками плазмы крови, элиминации и взаимодействия с другими лекарственными средствами, вероятно, определяют системное влияние статинов и их активных метаболитов в отношении риска возникновения миопатии. Растворимость статинов обязательно должна быть принята во внимание, как in vivo так и in vitro (Gadbut A.P. et al., 1995; Masters B.A. et al., 1995; Reijneveld J.C. et al., 1996; Hamelin B.A., Turgeon J., 1998; Nakahara K. et al., 1998). Экспериментальные исследования показали, что гидрофильные статины, такие как правастатин, гораздо реже, чем липофильные производные (ловастатин, симвастатин) способствуют развитию миопатии. Правастатин вследствие низкоэффективной пассивной диффузии обладает ограниченной способностью к проникновению в клетки различных органов и тканей (в том числе и мышечные), исключая гепатоциты. Таким образом, пропорция экстрагированного из крови в гепатоциты препарата является минимальной среди всех представителей класса статинов. В результате ожидаемая средняя плазменная концентрация правастатина почти в 10 раз выше, чем таковая ловастатина, симвастатина и флувастатина. С другой стороны, липофильность обусловливает большую экстракцию гепатоцитами препарата из венозной крови портальной системы, однако от этого зависит и способность статинов к проникновению в клетки других тканей (Schachter M., 2005). Аторвастатин, ловастатин, симвастатин и церивастатин подвергаются метаболизму с участием изофермента 3А4 системы цитохрома P450 (CYP) (см. табл. 14.12). Этот механизм отвечает за метаболизм и элиминацию широкого спектра веществ и лекарственных средств. Ожидается, что статины часто вступают во взаимодействие со многими лекарственными средствами. Так, приблизительно в 60% из сообщенных случаев рабдомиолиза последний ассоциировался с применением симвастатина, ловастатина или аторвастатина, при непосредственном лекарственном взаимодействии с препаратами, оказывающими ингибирующее влияние на систему цитохрома CYP 3A4 (Law M., Rudnicka A.R., 2006). Комбинирование статина и фибрата привлекает большое внимание в связи с широкими возможностями последнего при лечении смешанной гиперлипидемии. Почти 19% всех случаев статин-индуцированного рабдомиолиза отмечали у пациентов, получавших дополнительно фибраты, преимущественно гемфиброзил (Law M., Rudnicka A.R., 2006). Гемфиброзил ингибирует процесс глюкуронидации статинов (Prueksaritanont T. et al., 2002), который является основным механизмом, обусловливающим элиминацию их гидроксильных метаболитов (Prueksaritanont T. et al., 2002). Гемфиброзил полностью подвергается глюкуронидации при участии таких же механизмов, которые принимают участие в деградации статинов (UGT1A1 и UGT1A3 механизмы). Напротив, фенофибрат метаболизируется на иных ферментных системах (UGT1A9 и UGT2B7), что потенциально сопровождается существенно меньшим риском возникновения побочных эффектов при взаимодействии со статинами (Mukhtar R.Y., Reckless J.P., 2005). В элиминации безафибрата и ципрофибрата также используются альтернативные UGT1A1 и UGT1A3 механизмы (Mukhtar R.Y., Reckless J.P., 2005; Owczarek J. et al., 2005). Тем не менее, фармакодинамические взаимодействия между статинами и фибратами могут принимать участие в реализации миотоксического эффекта (Owczarek J. et al., 2005).
Таблица 14.12 Взаимодействие лекарственных средств и статинов
| Гиполипидемические лекарственные средства | Статины |
| Метаболизм с участием CYP 3A4 (эффект аторвастатина, ловастатина, симвастатина, церивастатина) | Фибратыа, никотиновая кислота, макролиды, противогрибковые средства, ингибиторы внутриклеточных протеаз, мифебрадилб, флуоксетин, нефазодон, верапамил, варфарин и грейпфрутовый сок |
| Метаболизм с участием CYP 2C9 (эффект флувастатина, розувастатина) | Варфарин, амиодарон, циметидин, триметоприм/сульфометоксазол, флуоксетин, флувоксамин, изониазид, метронидазол, сульфинпиразон, тиклопидин, зафирлукаст, итраконазол, кетоконазол |
При одновременном применении статинов и других лекарственных средств могут отмечать как ингибирование эффектов, так и конкуренцию в отношении путей элиминации. Врач должен знать те лекарственные средства, которые повышают элиминацию статинов (почти все барбитураты, фенитоин, карбамазепин).
aПо данным FDA (Food and DrugAdministration, США) и AERS (Adverse Events Reporting System) комбинирование статинов (но не церивастатина) с гемфиброзилом сопровождается 15-кратным повышением частоты выявляемости рабдомиолиза по сравнению с использованием сочетания статин + фенофибрат (Law M., Rudnicka A.R., 2006).
бОтмена.
Механизмы статин-индуцированной миопатии
Статины обладают более высокой аффинностью по сравнению с естественными субстратами и ГМГ-КоА в отношении способности связывать ГМГ-КоА-редуктазу (Moghadasian M.H., 1999; Istvan E.S., Deisenhofer J., 2001). Миотоксический эффект статинов может являться результатом блокады ГМГ-КоА-редуктазы, что приводит к ограничению синтеза ряда промежуточных метаболитов. На самом деле, в модели статин-индуцированной миотоксичности in vitro при использовании взвеси скелетных миофибрилл неонатальных крыс возмещение пула мевалоната (то eсть промежуточные метаболиты, продукция которых требует непосредственного участия ГМГ-КоА-редуктазы, необходимы для синтеза ГПФ и ФПФ; см. рис. 14.3) восстанавливает зависимый от концентрации субстрата синтез белка и вызывает реверсию предшествующих изменений метаболизма, вызванных статинами. Напротив, ингибирование сквален-синтетазы, которая катализирует начальный этап синтеза ХС независимо от синтеза изопрена (см. рис. 14.3), не приводит к индукции миотоксичности в культуре миоцитов крыс, клеточной линии человеческой рабдомиосаркомы и взвеси миоцитов из скелетных мышц человека (Flint O.P. et al., 1997; Nishimoto T. et al., 2003). Кроме того, блокада всех последующих этапов метаболизма посредством ингибирования скваленэпоксидазы в миоцитах крыс линии L6, причем в концентрациях, полностью блокирующих синтез ХС, не оказывает неблагоприятного влияния на выживаемость клеток (Matzno S. et al., 1997). Результаты этих исследований позволяют прийти к выводу о том, что статин-индуцированная миопатия развивается не вследствие ограничения синтеза ХС, а связана с редукцией доступности изопреноидов (ГПФ и ФПФ) для последующего вовлечения в процессы метаболизма. Это, в свою очередь, может неблагоприятно отразиться на процессах прениляции протеинов и/или способствовать активации альтернативных путей посттрансляционной модификации белков (диспрениляции). Результатами этих процессов могут стать снижение функциональной активности или полная утрата физиологической способности синтезируемых белков, а также последовательная альтерация клеточных органелл или нарушение взаимоотношений между протеинами. Более того, снижение доступности изопреноидов для метаболических процессов может негативно отразиться на биосинтезе долихола и гликации белков. Кроме того, также может пострадать и продукция убихинона. Молекулярные и клеточные эффекты статинов, лежащие в основе формирования миопатии скелетных мышц, суммированы в табл. 14.13. В исследованиях in vitro, посвященных нарушениям экспрессии генов, обусловленным применением статинов, было установлено, что миофибриллы скелетных мышц человека существенным образом более чувствительны к ингибированию синтеза ХС, чем гепатоциты (Morikawa S. et al., 2005). Эти данные только частично могут объяснить восприимчивость миофибрилл скелетных мышц к токсическому эффекту статинов генетическими причинами, поскольку эти лекарственные средства не оказывают негативного влияния в отношении кардиомиоцитов.
Табл. 14.13 Молекулярные и клеточные эффекты статинов, лежащие в основе формирования миопатии скелетных мышц
| Молекула | Эффект статинов | Молекулярные механизы | Клеточные механизмы |
| Ras-изоформа ГТФазы | Диспрениляция | Нарушение передачи сигнала | Ограничение влияния факторов роста и нарушение функционирования внутриклеточных сигнальных систем |
| Rab-изоформа ГТФазы | Диспрениляция | Нарушение внутриклеточного транспорта | Нарушение транслокации рецепторов в клеточную мембрану |
| Ламинины | Диспрениляция | Фрагментация ядра, разрушение гетерохроматина, нарушение транскрипции генов | Чувствительность к механическому стрессу и нарушение экспрессии генов |
| Селен-цистеин-содержащая РНК | Диспрениляция | Полное ограничение процессов трансляции | Продукция «дефектных» селенсодержащих протеинов |
| Долихолы | Нарушение посттрансляционной N-терминальной гликозидации | Нарушение экспрессии рецепторов на поверхности клеточных мембран и пространственной структуры протеинов | Продукция «дефектных» протеинов, нарушение или снижение экспрессии мембранных рецепторов |
| Дистрогликаны | Нарушение посттрансляционной модификации | Нарушение взаимоотношений между внеклеточным матриксом и цитоскелетом | Мышечная дистрофия (?) |
| Коэнзим Q10 | Снижение синтеза | Нарушения оксидативной фосфориляции | Нарушение энергетического метаболизма |
Влияние статинов на диспрениляцию протеинов и селен-цистеин-содержащую тРНК
Протеины, относящиеся к суперсемейству Ras-белков, такие как короткая ГТФаза, вовлекаются в процессы функционирования и дифференциации миоцитов, в отношении которых статины могут оказывать неблагоприятное влияние. Экспериментальные исследования с миобластами L6 показали, что истощение связывания Ras-протеинов с мембранами клеток при применении статинов вызывало индукцию апоптоза (Matzno S. et al., 2005). В более поздних исследованиях, проведенных на изолированных миофибриллах скелетных мышц крыс, была показана роль Rab-изоформы короткой ГТФазы в возникновении статин-индуцированной миопатии. Инактивация Rab-протеинов статинами приводит к индукции вакуолизации миофибрилл, дегенерации, а также отеку органелл и, в конечном итоге, к апоптозу клеток (Sakamoto K. et al., 2007).
Ловастатин в исследованиях in vitro блокировал процессы прениляции ламинина A и предотвращал его инкорпорирование в структуру оболочки ядра клеток (Beck L.A. et al., 1990; Lutz R.J. et al., 1992). Результаты некоторых ранних исследований свидетельствуют, что концентрация ловастатина, необходимая для ингибирования этого процесса, должна быть значительно выше, чем требуется для эффективной блокады эндогенного синтеза ХС (Sinensky M. et al., 1990). Все указанные выше наблюдения привели к появлению предположений о том, что статин-индуцированная диспрениляция ламинина действительно может лежать в основе статин-ассоциированной миопатии, особенно в тех случаях, когда статины вступают во взаимодействие с различными лекарственными средствами, что приводит к повышению плазменной концентрации первых. В качестве возможных механизмов можно рассмотреть формирование «хрупкости» (fragile) ядра вследствие нарушения скелета ядерной оболочки, что, в свою очередь, приводит к повышению его чувствительности к механическим сокращениям миофибрилл (Lammerding J. et al., 2004). Могут отмечать и нарушение устойчивости оболочки ядра, а также дефект активации процессов транскрипции генов, вовлекаемых в реализацию внутриклеточных адаптивных и протективных механизмов, активирующихся в ответ на механическую стимуляцию мышц (Lammerding J. et al., 2004). Кроме того, при врожденных ламинопатиях наряду с ядерной дисфункцией выявлены тяжелые изменения целостности структуры гетерохроматина. Все эти нарушения облегчают индукцию процессов апоптоза и, возможно, в большей или меньшей степени принимают участие в реализации статин-индуцированной миотоксичности.
G.J. Warner и соавторы (2000), изучая эффекты ловастатина in vivo, продемонстрировали роль изопентенил-аденозин-37-зависимой селен-цистеин-содержащей тРНК в процессах биосинтеза селенсодержащих протеинов. Ловастатин снижал содержание первой на 50% в клетках яичника китайских хомяков в концентрации >10 μM. Этот эффект отражал способность клеток поддерживать адекватность синтеза селенсодержащих протеинов. Все внутриклеточные селенсодержащие протеины, за исключением одного, достаточно хорошо и полно изучены.
Так, известно, что вследствие преждевременного ограничения процессов трансляции происходит накопление неадекватно собранных «обрезанных» (truncated) молекул белка, являющихся объектом для последующей деградации. В соответствии с вышеуказанными наблюдениями, G.J. Warner и соавторы (2000) показали, что при недостатке изопентенил-аденозин-37-зависимой селен-цистеин-содержащей тРНК реализуется менее эффективное негативное регулирующее влияние на активность UGA кодонов (Warner G.J. et al., 2000). Вместе с тем, значение вышеупомянутых наблюдений в отношении возникновения статин-индуцированной миопатии носит косвенный характер, в связи с чем требуется продолжение исследований в этом направлении.
Эффект статинов в отношении N-гликации белков in vitro изучался K.W. Siddals и соавторами (2004) на культуре адипоцитов. Авторы показали, что статины способствуют N-терминальной гликолизации протеинов, входящих в состав рецепторов инсулиноподобного фактора роста и рецепторов к инсулину. Установлена синергичность между деградацией кумулированных гликозилированных протеинов мембранных рецепторов и интенсивностью диспрениляции ряда метаболических интермедиатов, таких как Ras-протеины (Siddals K.W. et al., 2004). Все это приводит к нарушению влияния факторов роста и может лежать в основе возникновения статин-идуцированной миопатии. Тем не менее, экспрессия большинства клеточных мембранных рецепторов не зависит от долихол-опосредованной N-терминальной гликолизации. Этот процесс может затрагивать и другие ключевые поверхностные протеины. Одним из таких возможных кандидатов может быть дистрогликан, представляющий собой гликопротеин, входящий в состав цитоскелета, и подвергающийся существенной посттранскрипционной модификации в эндоплазматическом ретикулуме и аппарате Гольджи (Baker S.K., 2005). Дистрогликан в экстрацеллюлярном матриксе расщепляется на α-рецептор и трансмембранный дистрофинсвязывающий рецептор-b. Этот процесс обеспечивает коммуникацию между экстрацеллюлярным матриксом и цитоскелетом миофибрилл, причем недостаточная интенсивность N-терминальной гликолизации дистрогликана может нарушать эти взаимоотношения. Фактически существуют доказательства того, что подобные нарушения процессов коммуникации, лежащие в основе манифестации врожденной мышечной дистрофии, могут быть связаны с мутацией гликозилтрансферазы (Martin-Rendon E., Blake D.J., 2003). Тем не менее, непосредственный эффект статинов в отношении метаболизма дистрогликана до конца не исследован.
Роль убихинона в формировании статин-индуцированной митохондриальной миопатии
Предполагается, что статинассоциированная миопатия может быть связана с митохондриальной дисфункцией. Теоретически, статины способны ингибировать синтез коэнзима Q10 (КоQ10) в митохондриях. Вследствие этого статины могут угрожать эффективному функционированию дыхательной цепи, способствуя нарушению энергопродукции в скелетных мышцах, приводя к возникновению миопатии (Marcoff L., Thompson P.D., 2007). Несмотря на то, что статины в целом снижали уровень КоQ10 в сыворотке крови, не было зарегистрировано какого-либо негативного влияния на содержание КоQ10 в скелетных мышцах, за исключением случаев применения симвастатина в высоких дозах (Paiva H. et al., 2005; Caso G. et al., 2007; Marcoff L., Thompson P.D., 2007; Young J.M. et al., 2007; Chatzizisis Y.S. et al., 2008). Кроме того, не было выявлено прямой ассоциации между снижением внутриклеточного уровня КоQ10 и митохондриальной миопатией (Marcoff L., Thompson P.D., 2007).
Таким образом, создается впечатление, что истощение пула КоQ10 не играет существенной этиопатогенетической роли в реализации статин-индуцированной миотоксичности. Более вероятно, что содержание КоQ10 может рассматриваться в качестве предрасполагающего фактора, особенно у лиц с низким уровнем последнего в сочетании с коморбидными состояниями (Marcoff L., Thompson P.D., 2007). К таковым можно отнести пожилой возраст, повышение дозы статинов, повышение их биодоступности вследствие почечной или печеночной недостаточности, наличие наследственных метаболических синдромов, таких как семейная митохондриальная энцефалопатия, а также и другие коморбидные состояния (рак, сердечная недостаточность, сахарный диабет, семейная гиперхолестеринемия, гипофункция щитовидной железы).
Рекомендации по предотвращению возникновения статин-индуцированной миопатии
Рабочая группа по безопасности статинов национальной липидной ассоциации (США) (The National Lipid Association Statin Safety Task Force) рассмотрела данные, касающиеся доказательства безопасности статинов, и подвергла ревизии и редактированию клинические рекомендации (McKenney J.M. et al., 2006). Врачи должны сообщать пациентам о возможных побочных эффектах статинов, характере их взаимодействия с другими лекарственными средствами, факторах риска, а также о симптомах, свидетельствующих об угрозе возникновения миопатии и рабдомиолиза. Вместе с тем, измерение концентрации КФК до назначения статинов не рекомендовано в рутинной клинической практике, несмотря на то, что таковое необходимо принять во внимание при лечении пациентов с высоким риском возникновения миопатии. Также не считается рекомендованным рутинное измерение концентрации КФК на фоне лечения статинами у пациентов без симптомов миопатии (McKenney J.M. et al., 2006). При возникновении клинических признаков последней, перед принятием решения об отмене статинов необходимо предпринять попытку верификации других причин, касающихся возникновения симптомов поражения скелетных мышц. Необходимо сфокусироваться именно на дефинизирующих жалобах и клинических признаках, подтверждающих манифестацию статин-индуцированной миопатии. Измерение плазменной концентрации КФК должно быть проведено только для оценки тяжести миопатии. В клинической практике при верификации диагноза должны преобладать тяжесть клинических признаков, а не только степень элевации КФК в плазме крови. Это положение проистекает из признания того факта, что клинические признаки миопатии могут часто развиваться и при отсутствии повышения КФК в крови. Жалобы пациентов на тяжелое поражение скелетных мышц должны стать основанием для прекращения приема статинов. В тех случаях, когда жалобы больных незначительны, в первую очередь следует рассмотреть возможность снижения дозы статинов. Врачу в любом случае необходимо, прежде всего, оценивать тяжесть клинических признаков, которые могут быть основанием как для повышения дозы статинов, так и для немедленного прекращения их приема. Любое повышение плазменной концентрации КФК >10 раз выше пределов нормальных значений служит указанием для немедленного прекращения лечения статинами и должно сопровождаться мероприятиями, направленными на предотвращение манифестации рабдомиолиза (McKenney J.M. et al., 2006). Лечение обычно является успешным и разрешение ситуации в виде уменьшения тяжести клинических симптомов отмечают уже через несколько дней. После достижения полного выздоровления необходимо, отказавшись от полипрагмазии, при постоянном мониторировании клинического состояния пациента восстановить лечение статинами, начиная с низкой дозы или отдать предпочтение другому лекарственному средству — представителю этого класса (Armitage J., 2007).
Этиопатогенетическая роль КоQ10 в реализации статин-индуцированной миопатии установлена не в полной мере (Chatzizisis Y.S. et al., 2008). На основании существующих клинических данных, рутинное применение КоQ10 у всех пациентов, получающих статины, для предотвращения возникновения миотоксичности не рекомендовано. Возможно, что определенные подгруппы пациентов, предрасположенных к развитию миопатии, могут извлечь пользу от такого назначения, однако согласованное мнение по этому вопросу в литературе отсутствует. Вероятно, стоит принять решение о назначении КоQ10 у пациентов с дефицитом последнего (Marcoff L., Thompson P.D., 2007).
Таким образом, будущие экспериментальные и клинические исследования должны обеспечить более полное понимание механизмов развития статин-индуцированной миопатии, что повлечет за собой повышение профиля безопасности и расширение клинических показаний к применению статинов.
Секвестранты желчных кислот
Секвестранты желчных кислот (ионообменные смолы) рекомендованы для лечения в основном тяжелой первичной (семейной) и вторичной гиперхолестеринемии в комбинации со статинами, а также при недостаточной эффективности последних при гиперлипидемии IIа типа по классификации Фредриксона. Необходимо отметить, что отсутствие системного эффекта у этого класса лекарственных средств создает предпосылки для более широко применения последних у детей и подростков, а также у беременных и кормящих грудью с семейной гомозиготной гиперхолестеринемией (Illingworth D.R., Bacon S., 1989).
Основной механизм реализации гиполипидемического эффекта ионообменных смол состоит в супрессии нормальной рециркуляции обогащенного ХС пула желчных кислот, которые связываются в тонком кишечнике и экскретируются (Miettinen T.A., 1979; Illingworth D.R., 1984; Grundy S.M. et al., 1985). В результате снижения интенсивности экзогенного транспорта ХС развивается внутриклеточный дефицит последнего, что сопровождается повышением экспрессии мембранассоциированных рецепторов к ЛПНП, обеспечивающих активацию обратного транспорта ХС в гепатоциты (Glueck C.J., 1982). Кроме того, установлено, что ионообменные смолы при длительном применении обладают способностью стимулировать активность ГМГ-КоА-редуктазы. Мощность гиполипидемического эффекта у секвестрантов желчных кислот умеренная. Препараты способствуют снижению плазменной концентрации общего ХС и ХС ЛПНП на 15–20% и 15–30% соответственно, иногда повышая уровень ТГ. Доказана их способность к увеличению содержания ХС ЛПВП на 3–5%. Причем ионообменные смолы практически не оказывают заметного влияния на уровень хиломикронов в плазме крови (гиперлипидемия I типа по классификации Фредриксона) (Glueck C.J., 1982). Комбинированная терапия секвестрантами желчных кислот со статинами или производными никотиновой кислоты сопровождается синергичным гиполипидемическим эффектом в отношении редукции уровня общего ХС и ХС ЛПНП (в среднем на 45–60% и 20–25% соответственно), предотвращая повышение содержания ТГ (Stein E.A., Heimann K.W., 1975; Crouse J.R., Grundy S.M., 1981; LaRosa J., 1989).
Общепринятой классификации секвестрантов желчных кислот не существует. Обычно выделяют так называемые старые лекарственные средства (колестирамин и колестипол) и новые (колесевелам и колестагель). Наиболее часто в клинической практике назначают колестирамин. Рекомендованная обычная суточная доза препарата составляет 4–8 г/сут, но может быть повышена до 24 г/сут. Гиполипидемический эффект может быть зафиксирован уже на 2-й неделе лечения, достигая максимума на 4–6-й неделе постоянного приема препарата. Суточная доза колестипола составляет 5–30 г/сут. Препарат принимают в виде порошка, предварительно растворяя его в жидкости (чай, кисель). Колесевелам рекомендован в суточной дозе 3,75 мг/сут. Выпускается в таблетированной форме (625 мг в 1 таблетке препарата).
Секвестранты желчных кислот не отличаются идеальной переносимостью. В принципе, при хорошей толерантности к ним лечение можно продолжать неопределенно долго, периодически мониторируя уровень липидов, активность печеночных трансаминаз и КФК в плазме крови. Риск повышения уровня ТГ на фоне применения ионообменных смол более высок при изолированной гипертриглицеридемии (Crouse J.R., Grundy S.M., 1981; Tikkanen M.J. et al., 1987).
Прямыми противопоказаниями к назначению секвестрантов желчных кислот являются семейная гиперлипидемия III и IV типа, вторичная гиперлипидемия при билиарном циррозе печени, полная билиарная обструкция, семейная дисбеталипопротеидемия (III тип по Фредриксону), тяжелая изолированная гипертриглицеридемия (>6 ммоль/л) и обстипации (Tikkanen M.J. et al., 1987). С осторожностью можно назначать ионообменные смолы пациентам с болезнями почек, желчнокаменной болезнью, желудочно-кишечными расстройствами, синдромом мальабсорбции, язвой желудка и двенадцатиперстной кишки, геморроем, нарушением процессов коагуляции и фибринолиза, гипотиреоидизмом (Glueck C.J., 1982; LaRosa J., 1989). Данные по применению этих лекарственных средств в период беременности и кормления грудью отсутствуют (Crouse J.R., Grundy S.M., 1981; Tikkanen M.J. et al., 1987).
Ионообменные смолы нередко (в 50% случаев) вызывают запор, метеоризм и диспепсию; многие больные отказываются их принимать из-за неприятных вкусовых ощущений (Stein E.A., Heimann K.W., 1975). Клинически менее значимы и реже отмечаются такие осложнения, как диарея, отрыжка, головокружение, тошнота, абдоминальная боль и рвота. Колесевелам редко вызывает такие побочные эффекты, как миалгия, фарингит, очень редко — гриппоподобные симптомы, усиление кашля, боль в спине, ринит и синусит.
Колестирамин снижает всасывание фенилбутазона, варфарина, хлортиазида, тетрациклина, пенициллина G, фенобарбитала, тироксина, дигиталиса. Описаны негативные лекарственные взаимодействия ионообменных смол с гидрокортизоном, флувастатином, правастатином. Секвестранты желчных кислот могут оказывать отрицательное влияние на всасывание жирорастворимых витаминов (Stein E.A., Heimann K.W., 1975). На фоне приема колесевелама описано снижение концентрации формы верапамила с замедленным высвобождением лекарственного вещества. Прием ионообменных смол за час до или через 4–6 ч после приема основных лекарственных средств может существенно снизить вероятность возникновения потенциально неблагоприятного лекарственного взаимодействия (LaRosa J., 1989).
Эзетимиб
Эзетимиб в настоящее время представлен на фармацевтическом рынке оригинальной молекулой Ezetrol («Merck Sharp & Dohme Ltd» и «Schering-Plough Ltd»), срок патентной защиты на которую еще не закончился. Препарат ингибирует абсорбцию ХС в тонком кишечнике, что стимулирует интенсивность обратного транспорта ХС и снижение эндогенной продукции последнего (Bass A. et al., 2009). Предполагалось, что реализация этого эффекта будет способствовать появлению ряда протективных качеств, в том числе редукции интенсивности прогрессирования атеросклероза, что, в конечном итоге, приведет к повышению вероятности выживания пациентов и снижению кардиоваскулярного риска (Al Badarin F.J. et al., 2009; Greenberg M.E. et al. 2009). Вместе с тем, результаты ряда завершившихся недавно рандомизированных клинических исследований (SANDS, ARBITER 6-HALTS и ENHANCE) дали основания для сомнений в отношении этой гипотезы (Fleg J.L. et al., 2008; Mascitelli L. et al., 2009; Ferrario C.M., 2010; Jakulj L. et al., 2010). В то же время, многие исследователи полагают, что результаты приведенных выше рандомизированных клинических исследований в большей мере свидетельствуют о несовершенстве суррогатных критериев оценки эффективности гиполипидемических лекарственных средств (достигнутый уровень ХС ЛПНП, толщина интима-медиального сегмента общей сонной артерии и т.п.), что требует дополнительно анализа при оценке результатов специально спланированных исследований с учетом твердых клинических конечных точек (Stein E.A., 2008; Whayne T.F. Jr., 2009). Ограниченность сведений по этому вопросу пока не позволяет проводить аргументированную научную дискуссию вокруг клинической целесообразности использования эзетимиба в монотерапии и в комбинации со статином. Во всяком случае, многочисленные действующие клинические рекомендации сохраняют за эзетимибом возможность применения при рекомендованных показаниях (см. ниже) (Jackevicius C.A. et al., 2008; Minhas R. et al., 2009).
Монотерапия эзетимибом способствует редукции плазменной концентрации общего ХС на 12,6–14,2%, ХС ЛПНП — на 17,4–19,7%, ТГ — на 5–10% и повышению ХС ЛПВП на 5–10% (Drazen J.M. et al., 2008). Добавление к эзетимибу симвастатина или аторвастатина приводит к дополнительной редукции общего ХС на 10,4–15%, ХС ЛПНП на 13,9–27% (Jakulj L. et al., 2010). Таким образом, эзетимиб в сочетании со статинами проявляет отчетливый синергичный гиполипидемический эффект (Sharma M. et al., 2009). В то же время, в ходе проведения рандомизированного клинического исследования ARBITER 6-HALTS (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies) выявили, что комбинация статина и ниацина (никотиновой кислоты) более эффективна в отношении предотвращения утолщения интима-медиального сегмента общей сонной артерии, чем сочетанное использование эзетимиба и статина (Ferrario C.M., 2010). Поскольку между толщиной интима-медиального сегмента общей сонной артерии и выраженностью атеросклероза установлена прямая корреляционная взаимосвязь (Finn A.V. et al., 2010), все это ставит под сомнение существование плейотропных качеств у эзетимиба, несмотря на подтвержденное клиническими испытаниями синергичное гиполипидемическое взаимодействие со статинами (Landmesser U. et al., 2005; Kastelein J.J.P., Bots M.L., 2009; Taylor A.J. et al, 2009; Lindsay A.C. et al., 2010). С другой стороны, после завершения испытания ENHANCE достаточно сложно экстраполировать выраженность гиполипидемического эффекта на клиническую результативность лечения (Brown B.G., Taylor A.J., 2008; Whayne T.F. Jr., 2009).
В ряде рандомизированных клинических исследований эзетимиб продемонстрировал хорошую переносимость и высокий уровень безопасности при достаточно низкой вероятности развития побочных эффектов (9–18% в группе эзетимиба и 18–24% в группе плацебо). Чаще всего при монотерапии эзетимибом регистрируются головная боль (6,8%), боль в животе (3,9%) и диарея (3,2%). При применении в комбинации со статинами чаще отмечают гастроинтестинальные расстройства, головную боль, усталость, а также миопатии/миалгии, склонность к заболеванию острыми респираторными вирусными инфекциями (Sharma M. et al., 2009).
В настоящее время в качестве основного показания к применению эзетимиба рассматриваются семейная гетерозиготная гиперхолестеринемия, первичная несемейная гиперхолестеринемия, при лечении которых не удается добиться достижения целевого уровня ХС ЛПНП несмотря на применение адекватных доз статинов, а также семейная гомозиготная ситостеролемия. Кроме того, эзетимиб в комбинации со статинами или другими методами радикальной терапии (ЛПНП-аферез) рекомендован для лечения семейной гомозиготной гиперхолестеринемии (Kastelein J.J. et al., 2008). Необходимо отметить, что даже при условии одновременного применения со статинами (аторвастатин, симвастатин, правастатин) эзетимиб не рекомендован для проведения первичной или вторичной профилактики возникновения кардиоваскулярных заболеваний (National Institute for Health and Clinical Excellence, 2007; 2008).
Независимо от показания к применению и дополнительного применения статина рекомендованной дозой эзетимиба является 10 мг/сут натощак или во время еды для инициального и поддерживающего лечения. В этой же дозе препарат может быть рекомендован детям в возрасте старше 10 лет и подросткам при наличии соответствующих показаний. Опыт применения эзетимиба у детей младше 10 лет ограничен.
Сегодня существует единственная фиксированная комбинация эзетимиба с симвастатином, которая содержит 10/20 мг, 10/40 мг обоих препаратов соответственно. Основными показаниями к ее применению являются умеренная и тяжелая первичная (семейная) гомозиготная гиперхолестеринемия, а также смешанная гиперлипидемия. Обычно для достижения контроля над гиперлипидемией достаточно применения комбинации в дозах 10/20 мг/сут или 10/40 мг/сут. Иногда в когорте пациентов с тяжелой гиперхолестеринемией и очень высоким кардиоваскулярным риском целесообразно применять симвастатин в дозе 80 мг/сут и эзетимиб в дозе 10 мг/сут (Ballantyne C.M. et al., 2005). Причем использование фиксированной комбинации эзетимиба и симвастатина достоверно чаще приводит к достижению целевого уровня ХС ЛПНП (<70 мг/дл), чем при применении максимально разрешенных суточных доз аторвастатина, розувастатина или симвастатина. Комбинирование эзетимиба и аторвастатина или правастатина также возможно. Последние могут быть добавлены к 10 мг эзетимиба в широком диапазоне доз 10–80 мг/сут для аторвастатина и 10–40 мг/сут для правастатина соответственно. Эффективность и безопасность подобных нефиксированных комбинаций доказаны в ходе проведения многочисленных рандомизированных клинических исследований (Ballantyne C.M. et al., 2003; Berneis K. et al., 2010).
Основными ограничениями для использования фиксированной комбинации являются наличие гиперчувствительности к каждому из компонентов, тяжелые нарушения функции печени или почек, период беременности и кормления грудью, а также необходимость применения ингибиторов внутриклеточных протеаз у ВИЧ-инфицированных пациентов (нелфинавир, лопинавир, ритонавир), противогрибковых средств (итраконазол, кетоконазол), макролидных или кетолидных антибиотиков (кларитромицин, эритромицин, тролеандомицин), мибефрадила или нефазодона. Необходимо соблюдать осторожность при применении эзетимиба и антикоагулянтов (варфарина) в виду высокого риска кровотечений. Кроме того, дополнительное применение бозентана, карбамазепина, колестирамина, гидантоина может существенно снизить гиполипидемическую эффективность эзетимиба (Bays H. et al., 2004).
Учитывая результаты рандомизированных клинических исследований, посвященных монотерапии эзетимибом или его комбинации со статинами, можно заключить, что в настоящее время в клиническом аспекте применение сочетания статина (редуцирующего уровень ХС ЛПНП) и ниацина (повышающего уровень ХС ЛПВП) более перспективно, чем комбинации двух лекарственных средств (статин + эзетимиб), способствующих реализации более мощного влияния в отношении снижения плазменной концентрации ХС ЛПНП. После завершения двух специально спланированных рандомизированных клинических исследований AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides and Impact on Global Health Outcomes) и HPS2-THRIVE (Heart Protection Study 2: Treatment of HDL to Reduce the Incidence of Vascular Events) можно будет более детально обсудить жизнеспособность гипотезы о широком внедрении комбинации гиполипидемических лекарственных средств, оказывающих влияние на различные компоненты липидного профиля (статин + ниацин) по сравнению с сочетанием статин + эзетимиб (Kastelein J.J.P., Bots M.L., 2009; Lindsay A.C. et al., 2010).
Фибраты
Производные фиброевой кислоты (фибраты) оказывают гиполипидемический эффект в основном благодаря активации PPRA-α рецепторов, результатом чего является повышение экспрессии апо-А-I-липопротеинов и стимуляции клиренса обогащенных ТГ липопротеидов, что сопровождается редукцией плазменного уровня общего ХС на 10%, ХС ЛПНП на 10–30%, ТГ на 20–50%, а также повышению уровня ХС ЛПВП на 5–15%. Необходимо отметить, что выраженность указанного эффекта носила чрезвычайно вариабельный характер и тесно ассоциировалась с видом гиперлипидемии. Так, в когорте пациентов со смешанной гиперлипидемией использование фибратов приводило к редукции ЛПНП на 10–25%, тогда как их воздействие на уровень ТГ было более выраженным (снижение на 50–60%). С другой стороны, у больных с изолированной гипертриглицеридемией (IV тип по Фредриксону) на фоне приема фибратов уровень ХС ЛПНП мог и повышаться вследствие активации печеночной липазы, увеличивающей пул частиц ЛПНП. Избежать этого эффекта можно, добавив к терапии статин в невысокой дозе. Вместе с тем, при этой форме нарушения липидного обмена фибраты способствуют редукции плазменного содержания ТГ на 40–50%, при этом уровень ХС ЛПВП может повышаться почти на 25% (The BIP Study Group, 2000). При первичной гиперлипидемии I или V типа по классификации Фредриксона или вторичной смешанной гиперлипидемии у пациентов с сахарным диабетом 2-го типа монотерапии фибратами, как правило, бывает недостаточно. Необходимо отметить, что этот класс лекарственных средств способен весьма эффективно снижать уровень ремнантов хиломикронов и ЛПОНП, что позволяет успешно применять их при гиперлипидемии III типа.
Описаны и плейотропные эффекты фибратов. Так, последние проявляют противовоспалительный, антиоксидантный и антитромботический эффекты, а также способствуют реверсии дисфункции эндотелия и микроальбуминурии (Ansquer J.C. et al., 2005; Keech A. et al., 2005). В отличие от статинов, фибраты оказывают более выраженное влияние на реологические свойства крови и фибринолиз. В наибольшей степени изучено влияние фибратов на уровень фибриногена. В частности, известно, что фенофибрат снижает уровень фибриногена на 3% у пациентов с нормальным уровнем ХС и до 23% — у больных с различными формами гиперлипидемии, в наибольшей степени — у больных с гиперлипидемией IIb типа по классификации Фредриксона. Механизм влияния фибратов на уровень фибриногена остается невыясненным; возможно, он связан с прямым ингибированием синтеза этого протеина в гепатоцитах. Кроме того, фибраты способны улучшать пуриновый обмен и способствовать редукции уровня глюкозы натощак. Фенофибрат может уменьшать проявления жирового гепатоза, снижая плазменный уровень щелочной фосфатазы и гамма-глутарилтранспептидазы.
Большинство фибратов, за исключением гемфиброзила, метаболизируются в почках, что ограничивает их применение при почечной недостаточности. Безафибрат быстро и почти полностью абсорбируется в кишечнике. Биодоступность форм этого препарата с замедленным высвобождением действующего вещества достигает 90%. Период полувыведения безафибрата в дозе 300 мг/сут из плазмы крови самый короткий среди других фибратов — 1,5–2,0 ч. С одной стороны, это обусловливает относительно низкую гиполипидемическую эффективность, с другой — позволяет безопасно применять этот фибрат в комбинации со статинами. Период полувыведения безафибрата коррелирует с клиренсом креатинина, что обусловливает необходимость в коррекции дозы у больных с ХБП. У пожилых пациентов период полувыведения безафибрата возрастает в 3–4 раза, что также требует снижения суточной дозы. Гемфиброзил также быстро и полно абсорбируется в ЖКТ; биодоступность достигает 97%. Это лекарственное средство подвергается внутрипеченочной циркуляции, его концентрация в плазме крови очень сильно варьирует у разных пациентов. Период полувыведения гемфиброзила составляет 1,5–2,5 ч после приема стандартной дозы — 600 мг, поэтому требуется, по крайней мере, двукратный прием (до 1,2 мг/сут). Максимальная доля (97%) связывается с белками плазмы крови. Гемфиброзил метаболизируется в печени с образованием, по крайней мере, 4 основных метаболитов (конъюгаты глюкуронила). Это единственный фибрат, который с осторожностью можно назначать больным с нарушением функции почек. Фенофибрат является пролекарством, которое после абсорбции гидролизуется тканевыми и плазменными эстеразами с образованием главного метаболита — фенофиброевой кислоты. Связывание с белками плазмы крови достигает 99%. Фенофибрат в микронизированной форме обладает улучшенными фармакокинетическими свойствами по сравнению со стандартной формой выпуска, при этом его биодоступность повышается на 30%. Период полувыведения фенофибрата в микрогранулах из плазмы крови достигает 20 ч. По сравнению с другими фибратами, эффективность фенофибрата в меньшей степени зависит от приема жирной пищи. Фенофибрат метаболизируется в печени; основной путь экскреции фенофиброевой кислоты — почки, поэтому при применении данного лекарственного средства требуется контроль за уровнем креатинина в крови. По всей видимости, фенофибрат обладает наиболее сбалансированными показателями фармакокинетики среди других фибратов, что характеризуется хорошим отношением гиполипидемический эффект/переносимость. Ципрофибрат имеет наибольший период полувыведения из плазмы крови (до 80 ч), что позволяет принимать разовую суточную дозу 100 мг. Удвоение дозы, как правило, хуже переносится, особенно больными с сахарным диабетом 2-го типа.
Среди представителей этого класса чаще всего назначаются ципрофибрат, фенофибрат, безафибрат и гемфиброзил. Более подробно о принципах выбора этих лекарственных средств изложено в главе 9.
Основные показания к приему фибратов — гипертриглицеридемия, смешанная и семейная комбинированная дислипидемия, изолированная гипоальфахолестеринемия, ассоциированная с низким уровнем ХС ЛПВП, а также лечение дислипидемий I, IIb, III, IV, V типов и умеренной гиперхолестеринемии IIa типа у больных с ИБС при наличии противопоказаний к приему статинов. Гемфиброзил хорошо зарекомендовал себя в первичной и вторичной профилактике кардиоваскулярных событий. Безафибрат продемонстрировал способность предотвращать возникновение кардиоваскулярных событий, в том числе инфаркта миокарда, у пациентов с метаболическим синдромом (Tenenbaum A. et al., 2000), тогда как использование фенофибрата не привело к достоверному снижению риска возникновения нефатального инфаркта миокарда и кардиоваскулярной смерти (–11%; р=0,16), хотя и сопровождалось достоверной редукцией общего количества сердечно-сосудистых событий в когорте больных с сахарным диабетом 2-го типа (Keech A. et al., 2005). Более подробно об эффективности фибратов в селективных когортах пациентов изложено в главе 13.
Переносимость фибратов вполне приемлемая, частота возникновения серьезных побочных эффектов крайне низка. Фибраты хорошо переносятся, однако у 5–10% больных возможно развитие побочных эффектов в виде боли в животе, запора, диареи, метеоризма, а также сыпи, зуда, головной боли, бессонницы. Эти явления не носят тяжелого характера и не требуют прерывания терапии. К побочным эффектам фибратов относится и снижение потенции (более характерно для ципрофибрата). Иногда регистрируют повышение уровня печеночных ферментов, гомоцистеина и креатинина, также возможно развитие лейкопении. Миопатию, описанную на фоне клинического применения лекарственных средств этого класса, часто выявляли у пациентов с документированными нарушениями функции печени, или при использовании комбинации со статинами в высоких дозах. Кроме того, необходимо помнить и о возможности негативного взаимодействия фибратов с антикоагулянтами в виде повышения риска кровотечений. Риск возникновения конкрементов желчного пузыря обычно связывается исключительно с применением фибратов первого поколения (клофибрата, безафибрата и гемфиброзила). Из потенциально неблагоприятных плейотропных эффектов фибратов следует отметить повышение уровня гомоцистеина в плазме крови, который рассматривается в качестве дополнительного фактора риска формирования атеросклероза. Однако указанный эффект описан только для фенофибрата и безафибрата, но не характерен для гемфиброзила. Имеются сообщения о редких случаях развития панкреатита, тромбоэмболии мелких ветвей легочной артерии у больных сахарным диабетом, длительно принимавших фенофибрат.
Фибраты противопоказаны пациентам с выраженными нарушениями функции печени и почек, с врожденной галактоземией и дефицитом лактазы, детям и подросткам, в период беременности и кормления грудью.
Рекомендованные к применению дозы для гемфиброзила — 600 мг 2 раза в сутки, для безафибрата — 200 мг 2–3 раза в сутки, для ципрофибрата 100 мг 1–2 раза в сутки и для фенофибрата — 145 или 200 мг 1 раз в сутки. Препараты необходимо принимать в первой половине дня (утром и днем), поскольку синтез обогащенных ТГ липопротеидов происходит более интенсивно именно в это время.
Никотиновая кислота и ее производные
В рекомендованных дозах (3–6 г/сут) никотиновая кислота способствует снижению плазменной концентрации общего ХС на 25%, ЛПНП на 10–25%, ТГ на 20–50% и повышению уровня ЛПВП на 15–35% (McKenney J.M. et al., 2007). Механизм реализации указанного эффекта до конца не вполне понятен (McKenney J., 2004). Предполагается, что препарат обладает способностью ингибировать высвобождение ТГ из депо преимущественно за счет редукции активности липопротеинлипазы, а также ингибирует обратный захват апо-А-I-липопротеина и стимулирует интенсивность реверсивного транспорта ХС в гепатоциты (Hoeg J.M. et al., 1984; Superko H.R., 2006).
Доказана плейотропная активность ниацина, которая проявляется в виде улучшения функции эндотелия и повышения чувствительности последнего к эндогенным вазодилатирующим стимулам (McKenney J., 2004). В комбинации со статинами ниацин продемонстрировал способность к снижению риска общей и кардиоваскулярной смерти, уменьшению потребности в реваскуляризационных процедурах, а также способствовал ограничению прогрессирования коронарного атеросклероза (Brown B.G. et al., 2001; Zhao X.Q. et al., 2001; Taylor A.J., Sullenberger L.E., Lee H.J. et al., 2004; Taylor A.J. et al., 2006). Результаты рандомизированных клинических исследований COMPELL и ADVOCATE свидетельствуют, что комбинация ниацина и статина сопровождается реализацией синергичного эффекта в отношении как снижения уровня ЛПНП, так и повышения ХС ЛПВП (Kashyap M.L. et al., 2002; McKenney J.M. et al., 2007).
Никотиновая кислота не отличается хорошей переносимостью, приводя к возникновению достаточно большого количества негативных побочных эффектов, таких как эритема, кожная сыпь, зуд, гиперпигментация, многочисленные гастроинтестинальные симптомы, сердцебиение, гипотензия, которые имеют четкий дозозависимый характер (Capuzzi D.M. et al., 1998; Kashyap M.L. et al., 2002). В реализации такого побочного эффекта как эритема непосредственное участие принимают простагландины, поэтому частично реверсия симптомов возможна после приема ацетилсалициловой кислоты (Capuzzi D.M. et al., 1998; Kashyap M.L. et al., 2002). Кроме того, описаны феномен тахифилаксии, а также способность к индукции обострения подагры, нарушение толерантности к глюкозе и элевация печеночных трансаминаз (Garg A., Grundy S.M., 1990; Pandit M.K. et al., 1993). С другой стороны, многие исследователи связывают возникновение так называемых плейотропных эффектов (в основном антипролиферативных) при применении ниацина именно с его антигипертензивным потенциалом (Bays H.E., Rader D.J., 2009; Taylor A.J. et al., 2009).
В некоторой степени снижение вероятности реализации побочных эффектов достигается при применении никотиновой кислоты в новых формах выпуска — в виде форм с контролируемым высвобождением (аципимокс, никофураноза, ниацин) (Wolfe M.L. et al., 2001; McKenney J., 2004). Необходимо отметить, что результаты рандомизированных клинических исследований по изучению этих лекарственных средств продемонстрировали не только их более высокий уровень переносимости по сравнению с никотиновой кислотой, но и существенно меньшую вероятность возникновения нежелательных метаболических эффектов, таких как гипергликемия натощак (Wolfe M.L. et al., 2001; McKenney J., 2004; Taylor A.J. et al., 2004; Cefali E.A. et al., 2006). По данным рандомизированного клинического исследования IMPACT (Impact of Medical Subspecialty on Patient Compliance to Treatment) установлено, что 77% пациентов из 3245 получавших комбинацию ловастатина и ниацина в широком диапазоне доз, сохранили приверженность к лечению в течение года терапии (Rubenfire M., 2004). У 18% больных были зарегистрированы побочные эффекты, которые вызвали отказ от продолжения терапии у 6% больных. В целом, никотиновая кислота и ее производные зарекомендовали себя как достаточно действенные лекарственные средства, эффективно повышающие уровень ХС ЛПВП, обладающие синергичным со статинами гиполипидемическим потенциалом, способствующим позитивному влиянию в отношении редукции общей и кардиоваскулярной смерти, а также ограничению прогрессирования атеросклероза и возникновения атеротромботических событий (McKenney J., 2004).
ω-3 ненасыщенные жирные кислоты
В соответствии с современными представлениями эссенциальные жирные кислоты должны составлять важную часть пищевого рациона. Содержащие ω-3 ненасыщенные жирные кислоты продукты входят в состав различных диет, в том числе и в так называемую средиземноморскую, приверженность к которой достоверно снижает риск возникновения кардиоваскулярных событий (Kris-Etherton P.M. et al., 2002). Наиболее богаты ω-3 ненасыщенными жирными кислотами морепродукты, особенно сорта морских глубоководных рыб: тунец, макрель, лосось, сельдь и палтус (Mozaffarian D., Rimm E.B., 2006). В то же время, доступность таких продуктов питания достаточно ограничена, в связи с чем повышение потребления ω-3 ненасыщенных жирных кислот решается посредством применения лекарственных средств, содержащих стадартизированное количество последних (Harris W.S. et al., 1997).
Лекарственные средства, содержащие ω-3 ненасыщенные жирные кислоты, обладают доказанной способностью к снижению синтеза ЛПОНП гепатоцитами и повышению активности липопротеинлипазы за счет снижения энзиматической конверсии ацетил-КоА в СЖК (Park Y., Harris W.S., 2003; Lewis A. et al., 2004). Кроме того, ω-3 ненасыщенные жирные кислоты способны стимулировать трансфер ТГ и ТГ-обогащенных липопротеидов, таких как ЛПОНП и хиломикроны, а также способствовать ускорению клиренса ТГ, что приводит к редукции плазменного содержания ТГ (Harris W.S., Bulchandani D., 2006).
Вместе с тем, на сегодня установлено, что ω-3 ненасыщенные жирные кислоты способны приводить к умеренному снижению уровня системного артериального давления, редукции риска возникновения внезапной сердечной смерти и эпизодов нарушений ритма вентрикулярного и суправентрикулярного происхождения, частоты возникновения инфарктов миокарда, в том числе и фатальных, а также редукции общего количества кардиоваскулярных событий (Burr M.L. et al., 1989; GISSI-Prevenzione Investigators, 1999; Christensen J.H., 2003; Lee K.W. et al., 2004; Lewis A. et al., 2004; Mozaffarian D. et al., 2004; Schrepf R. et al., 2004; Frost L., Vestergaard P., 2005; Ismail H.M., 2005; Raitt M.H. et al, 2005).
В соответствии с рекомендациями FDA (Food and Drug Administration) лекарственные средства, содержащие стандартизированные комбинации ω-3 ненасыщенных жирных кислот: эйкозапентаеновую и декозагексаеновую кислоты, резервируются для лечения тяжелой изолированной гипертриглицеридемии (≥500 мг/дл [5,65 ммоль/л]). Суточная доза таких ω-3 ненасыщенных жирных кислот должна составлять 10 мг/сут. При этом необходимо отметить, что монотерапия ω-3 ненасыщенными жирными кислотами в подобной дозировке не отражается на плазменной концентрации общего ХС и в большинстве случаев у пациентов с изолированной гипертриглицеридемией может приводить к повышению уровня ХС ЛПНП.
Переносимость ω-3 ненасыщенных жирных кислот обычно хорошая, побочные эффекты (в основном гастроинтестинальные события: отрыжка, изжога и диарея) редки и чаще всего не требуют отмены терапии (Brunton S., Collins N., 2007). Данные рандомизированных клинических исследований свидетельствуют о том, что ω-3 ненасыщенные жирные кислоты не взаимодействуют с варфарином, ацетилсалициловой кислотой и другими антиагрегантами, не повышают риск кровотечений (Bender N.K. et al., 1998; Vanschoonbeek K. et al., 2004). При длительном приеме ω-3 ненасыщенных жирных кислот иногда отмечают транзиторное повышение уровня печеночных трансаминаз и глюкозы натощак (Harris W.S., 2007).
Использование ω-3 ненасыщенных жирных кислот в сочетании со статинами благоприятно отражается на липидном профиле плазмы крови и сопровождается реализацией синергичного эффекта в отношении редукции уровня ТГ, апо-В100-липопротеинов и повышения содержания ХС ЛПВП (Nordoy A. et al., 1998; Durrington P.N. et al., 2001; Chan D.C. et al., 2002). Кроме того, описано их потенцирующее влияние в отношении реверсии инсулинорезистентности у пациентов с сахарным диабетом и абдоминальным типом ожирения (Chan D.C. et al., 2002), а также многочисленные плейотропные эффекты (улучшение метаболизма хрящевой ткани, снижение интенсивности остеопороза субхондральной кости при ревматоидном артрите, снижение вариабельности пиковой скорости выдоха у пациентов с бронхиальной астмой, нефропротекторные качества при ХБП различной этиологии, вазопротекция при синдроме Рейно, снижение интенсивности демиелинизации при рассеянном склерозе, тимолептический, противотревожный эффекты и многое другое) (DiGiacomo R.A. et al., 1989; Donadio J.V. Jr., 1991; Curtis C.L. et al., 2002; Tamizi F.B., Tamizi B., 2002; Alessandri J.M. et al., 2004; Ayton A.K. et al., 2004; Facchinetti F. et al., 2005; Schwarz S., Leweling H., 2005). Несмотря на достаточно широкий клинический потенциал, ω-3 ненасыщенные жирные кислоты пока резервируются для применения в узкой нише показаний: изолированная тяжелая гипертриглицеридемия в комбинации со статинами.
Пробукол
Пробукол отличается от других представителей класса гиполипидемических лекарственных средств наличием липофильных качеств, обусловленных во многом фенольным кольцом, входящим в состав его молекулы. Необходимо отметить, что пробукол является практически единственным препаратом, способным оказывать стимулирующее влияние на нерецепторный механизм захвата ХС ЛПНП, осуществляющийся путем эндогенного пиноцитоза (Schonfeld G. et al., 1982; Dujovne C.A. et al., 1984). Длительное время с этими качествами лекарственного средства связывались надежды на осуществление адекватного лечения у пациентов с семейной гомозиготной гиперхолестеринемией с доказанной генетической мутацией гена ЛПНП (Sasaki J. et al., 1987), поскольку препарат способствовал реверсии прогрессирования атеросклероза, оказывал вазопротекторный эффект (Carew T.E. et al., 1987; Kita T. et al., 1987; Parthasarathy S. et al., 1987). Кроме того, пробукол способствовал мобилизации ХС из периферических тканей, в частности из кожи и ксантом, а также препятствовал процессам оксидации фиксированных в сосудистой стенке ЛПНП, проявляя выраженный антиоксидантный эффект (Yamamoto A. et al., 1986). Вместе с тем, не представлено доказательств существования антиатерогенного потенциала этого лекарственного средства. Более того, в последующем оказалось, что монотерапия пробуколом также малоэффективна, как и использование других гиполипидемических лекарственных средств в качестве монотерапии, по сравнению с современными методами радикальной терапии семейной гомозиготной гиперхолестеринемии (Baker S.G. et al., 1982; Witztum J.L., 1987). Все это привело к изменению представлений о месте и роли препарата в лечении первичных гиперлипидемий вообще.
Гиполипидемический потенциал пробукола расценивается как относительно умеренный. Так, препарат способствует снижению плазменного содержания общего ХС в среднем на 15%, ЛПНП на 5–15%, а также повышению ЛПВП на 20–30% соответственно. Однако для реализации данного эффекта необходим прием этого лекарственного средства на протяжении не менее 3 мес. Эти качества пробукола не позволяли рассматривать его как препарат первой линии (first-line therapy). Тем не менее, доказан синергичный гиполипидемический эффект при комбинировании пробукола с ниацином, секвестрантами желчных кислот и статинами (Schonfeld G. et al., 1982).
Несмотря на то что пробукол проявляет хороший профиль переносимости, установлена его способность к пролонгации интервала Q–T на ЭКГ и аккумуляции в жировой ткани, что приводит к сохранению достаточно высокой плазменной концентрации препарата на протяжении длительного времени (от нескольких недель до 6 мес) после его отмены. Последнее обстоятельство привело к появлению рекомендаций о прекращении лечения пробуколом как минимум за 6 мес до предполагаемой беременности. Учитывая неоптимальный профиль безопасности, отсутствие доказанного благоприятного влияния в отношении прогрессирования атеросклероза, в настоящее время пробукол практически не используется в рутинной клинической практике.
Ингибиторы конечных стадий синтеза ХС (ингибиторы сквален-синтетазы, сквален-эпоксидазы и оксидосквален-циклазы)
Установлено, что более ⅔ всей молекулы ХС синтезируется в печени. Следовательно, в качестве основного механизма блокады биосинтеза ХС рассматривается редукция синтеза ХС ЛПНП. Вместе с тем, использование альтернативных механизмов ограничения синтеза ХС может нивелировать миотоксический потенциал статинов, однако, с другой стороны, это, возможно, будет сопровождаться утратой гиполипидемической и плейотропной эффективности статинов. Ингибиторы сквален-синтетазы оказывают влияние на конечные этапы биосинтеза ХС в печени (см. рис. 14.3). Этот класс лекарственных средств также эффективно способствует снижению ХС ЛПНП, как и статины. В настоящее время ингибиторы сквален-синтетазы проходят клинические исследования, результаты которых ожидают с большим интересом (Charlton-Menys V., Durrington P.N., 2007). Кроме того, ингибиторы сквален-эпоксидазы и оксидосквален-циклазы способны воздействовать на последующие звенья в цепи синтеза ХС и находятся на различных стадиях изучения.
Торцетрапиб
Торцетрапиб представляет собой специфический ингибитор CETP, который длительное время рассматривался как перспективное гиполипидемическое лекарственное средство с уникальным механизмом действия. В ряде анимационных и клинических исследований было установлено, что препарат способствовал не только повышению плазменного уровня ЛПВП, апо-А-I-липопротеина, но и редукции ХС ЛПНП, общего ХС, апо-В100-содержащих липопротеидов, а также увеличению размеров молекул ЛПВП и ЛПНП (Nieland T.J.F. et al., 2007; Singh I.M. et al., 2007; Guerin M. et al., 2008). В ряде пилотных рандомизированных клинических исследований торцетрапиб снижал интенсивность прогрессирования атеросклероза артерий у пациентов с семейной гиперхолестеринемией (Kastelein J.J. et al., 2007). Однако в рандомизированном клиническом исследовании ILLUMINATE препарат, добавленный к аторвастатину, не оказал дополнительного влияния в отношении суррогатной точки (толщина интима-медиального сегмента общей сонной артерии), хотя уровень ХС ЛПВП достоверно возрос по сравнению с таковым при монотерапии статином (Barter P.J. et al., 2007). В последующем в рандомизированном клиническом исследовании ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation) было установлено, что комбинация аторвастатина и торцетрапиба превосходит монотерапию аторвастатином по способности повышать уровень ЛПВП (на 61% по сравнению с группой аторвастатина) и снижать содержание ХС ЛПНП (на 20% по сравнению с группой аторвастатина). Вместе с тем, у пациентов, получавших комбинацию гиполипидемических лекарственных средств, отмечалось достоверное повышение уровня системного артериального давления (на 4,6 мм рт. ст.), а способность торцетрапиба ограничивать прогрессирование атеросклероза не была подтверждена (Foody J.M., 2007). Все это привело к прекращению исследований в этом направлении и отзыва препарата с рынка.
Препараты растительного происхождения, антиоксиданты и другие лекарственные средства с гиполипидемическим потенциалом
Препараты на растительной основе, а также ряд альтернативных подходов к достижению гиполипидемического эффекта достаточно популярны не только у пациентов, но и у ряда специалистов, хотя клиническая эффективность такой стратегии не доказана (Schulz V. et al., 2000; Barnes J. et al., 2002; Chagan L. et al., 2002). Вместе с тем, станолы и стеролы, содержащиеся в растительном сырье, способны оказывать некоторое гиполипидемическое действие (Anderson J.W. et al., 2000). Так, метаанализ 41 рандомизированного клинического исследования, посвященного эффективности использования препаратов растительного происхождения, продемонстрировал возможность достижения редукции ХС ЛПНП на 10% от исходного уровня (Katan M.B. et al., 2003). В то же время, многие из них, например препараты, содержащие экстракт чеснока, не отличались по своей эффективности от плацебо (Silagy C., Neil A., 1994; Stevinson C. et al., 2000; Gardner C.D. et al., 2007).
Антиоксиданты также являются широко назначаемыми лекарственными средствами в популяции пациентов с гиперлипидемией, особенно с документированными кардиоваскулярными заболеваниями. Вместе с тем, роль большинства из них, в том числе витамина Е и КоQ10, не продемонстрировали позитивного профиля эффективности при проведении специально спланированных рандомизированных клинических исследований (Lee I.M. et al., 2005; Levy H.B., Kohlhaas H.K., 2006; Bjelakovic G. et al., 2007).
Таким образом, несмотря на то, что статины, без сомнения, занимают лидирующее положение среди других гиполипидемических лекарственных средств, существующее разнообразие других представителей препаратов этого класса предоставляет широкую возможность индивидуализировать их выбор в зависимости от потребностей сложившейся клинической ситуации и наличия доказательств их эффективности и безопасности.
Литература
- Al Badarin F.J., Kullo I.J., Kopecky S.L. et al. (2009) Impact of ezetimibe on atherosclerosis: is the jury still out? Mayo Clin. Proc., 84: 353–361.
- Alessandri J.M., Guesnet P., Vancassel S. et al. (2004) Polyunsaturated fatty acids in the central nervous system: evolution of concepts and nutritional implications throughout life. Reprod. Nutrit. Develop., 44, 509–538.
- Amarenco P., Bogousslavsky J., Callahan III A. et al. (2006) High-dose atorvastatin after stroke or transient ischemic attack. N. Engl. J. Med., 355: 549–559.
- Anderson J.W., Davidson M.H., Blonde L. et al. (2000) Cholesterol-lowering effects of psyllium intake adjunctive to diet therapy in men and women with hypercholesterolaemia: meta-analysis of 8 controlled trials. Am. J. Clin. Nut., 7: 472–479.
- Ansquer J.C., Foucher C., Rattier S. et al. (2005) Fenofibrate reduces progression to microalbuminuria over 3 years in a placebo-controlled study in type 2 diabetes: results from the Diabetes Atherosclerosis Intervention Study (DAIS). Am. J. Kidney Dis., 45: 485–493.
- Antons K.A.,Williams C.D., Baker S.K. et al. (2006) Clinical perspectives of statin-induced rhabdomyolysis. Am. J. Med., 119(5): 400–409.
- Armitage J. (2007) The safety of statins in clinical practice. Lancet, 370: 1781–1790.
- Arora R., Liebo M., Maldonado F. (2006) Statin-induced myopathy: the two faces of Janus. J. Cardiovasc. Pharmacol. Ther., 11: 105–112.
- Ayton A.K., Azaz A., Horrobin D.F. (2004) A pilot open case series of ethyl-EPA supplementation in the treatment of anorexia nervosa. Prostaglandins Leukot. Essent. Fatty Acids., 71: 205–209.
- Baigent C., Keech A., Kearney P.M. et al. (2005) Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet, 366: 1267–7128.
- Baker S.G., Joffe B.I., Mendelsohn D. et al. (1982) Treatment of homozygous familial hypercholesterolemia with probucol. S. Afr. Med. J., 62: 7–11.
- Baker S.K. (2005) Molecular clues into the pathogenesis of statin-mediated muscle toxicity. Muscle Nerve, 31: 572–580.
- Ballantyne C.M., Abate N., Yuan Z. et al. (2005) Dose-comparison study of the combination of ezetimibe and simvastatin (Vytorin) versus atorvastatin in patients with hypercholesterolemia: the Vytorin Versus Atorvastatin (VYVA) Study. Am. Heart J., 149: 464–473.
- Ballantyne C.M., Houri J., Notarbartolo A. et al. (2003) Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Circulation, 107: 2409–2415.
- Barnes J., Anderson L.A., Phillipson J.D. (Eds.) (2002) Herbal medicines. A guide for healthcare professionals. 2nd ed. Pharmaceutical Press, London, 530 p.
- Barter P.J., Caulfield M., Eriksson M. et al.; The ILLUMINATE Investigators (2007) Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med., 357: 2109–212.
- Bass A., Hinderliter A.L, Lee C.R. (2009) The impact of ezetimibe on endothelial function and other markers of cardiovascular risk. Ann. Pharmacother., 43: 2021–2030.
- Bays H., Ose L., Fraser N. et al.; The Ezetimibe Study Group (2004) A multicenter, randomized, double-blind, placebo-controlled, factorial design study to evaluate the lipid-altering efficacy and safety profile of the ezetimibe/simvastatin tablet compared with ezetimibe and simvastatin monotherapy in patients with primary hypercholesterolemia. Clin. Ther., 26: 1758–1773.
- Bays H.E., Rader D.J. (2009) Does nicotinic acid (niacin) lower blood pressure? Int. J. Clin. Pract., 63: 151–159.
- Beck L.A., Hosick T.J., Sinensky M. (1990) Isoprenylation is required for the processing of the lamin A precursor. J. Cell. Biol., 110: 1489–1499.
- Bender N.K., Kraynak M.A., Chiquette E. et al. (1998) Effects of marine fish oils on the anticoagulation status of patients receiving chronic warfarin therapy. J. Thromb. Thrombolysis, 5: 257–261.
- Berneis K., Rizzo M., Berthold H.K. et al. (2010) Ezetimibe alone or in combination with simvastatin increases small dense low-density lipoproteins in healthy men: a randomized trial. Eur. Heart. J., 31: 1366–1369.
- Bjelakovic G., Nikolova D., Gluud L.L. et al. (2007) Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA, 297: 842–857.
- Brown B.G., Taylor A.J. (2008) Does ENHANCE Diminish Confidence in Lowering LDL or in Ezetimibe? N. Engl. J. Med., 358: 1504–1507.
- Brown B.G., Zhao X.Q., Chait A. et al. (2001) Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med., 345: 1583–1592.
- Brunton S., Collins N. (2007) Differentiating prescription omega-3-acid ethyl esters (P-OM3) from dietarty-supplement omega-3-fatty acids. Curr. Med. Res. Opin., 23: 1139–1145.
- Bulut D., Hanefeld C., Bulut-Streich N. et al. (2005) Endothelial function in the forearm circulation of patients with the metabolic syndrome — effect of different lipid-lowering regimens. Cardiology, 104: 176–180.
- Burr M.L., Fehily A.M., Gilbert J.F. et al. (1989) Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet, 2: 757–761.
- Calza L., Colangeli V., Manfredi R. et al. (2005) Rosuvastatin for the treatment of hyperlipidaemia in HIV-infected patients receiving protease inhibitors: a pilot study. N. Engl. J. Med., 19: 1103–1105.
- Cannon C.P., Braunwald E., McCabe C.H. et al. (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med., 350: 1495–1504.
- Capuzzi D.M., Guyton J.R., Morgan J.M. et al. (1998) Efficacy and safety of an extended-release niacin (Niaspan): a long-term study. Am. J. Cardiol., 82: 74U–81U.
- Carew T.E., Schwenke D.C., Steinberg D. (1987) Antiatherogenic effect of probucol unrelated to its hypocholesterolemic effect: Evidence that antioxidants in vivo can selectively inhibit low density lipoprotein degradation in macrophage-rich fatty streaks and slow the progression of atherosclerosis in the Watanabe heritable hyperlipidemic rabbit. Proc. Natl. Acad. Sci. USA, 84: 7725–7729.
- Caso G., Kelly P., McNurlan M.A. et al. (2007) Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am. J. Cardiol., 99: 1409–1412.
- Cefali E.A., Simmons P.D., Stanek E.J. et al. (2006) Improved control of niacin-induced flushing using an optimized once-daily, extended-release niacin formulation. Int. J. Clin. Pharmacol. Ther., 44: 633–640.
- Chagan L., Ioselovich A., Asherova L. et al. (2002) Use of alternative pharmacotherapy in management of cardiovascular diseases. Am. J. Manag. Care, 8: 270–285.
- Chan D.C., Watts G.F., Barrett P.H. et al. (2002) Regulatory effects of HMG CoA reductase inhibitor and fish oils on apolipoprotein B-100 kinetics in insulin-resistant obese male subjects with dyslipidemia. Diabetes, 51: 2377–2386.
- Charlton-Menys V., Durrington P.N. (2007) Squalene synthase inhibitors: clinical pharmacology and cholesterol-lowering potential. Drugs, 67: 11–16.
- Chatzizisis Y.S., Vaklavas C., Giannoglou G.D. (2008) Coenzyme q10 depletion: etiopathogenic or predisposing factor in statin associated myopathy? Am. J. Cardiol., 101: 1071.
- Cheng J.W. (2004) Rosuvastatin in the management of hyperlipidemia. Clin. Ther., 26: 1368–1387.
- Cheng-Lai A. (2003) Rosuvastatin: a new HMG-CoA reductase inhibitor for the treatment of hypercholesterolemia. Heart Dis., 5: 72–78.
- Christensen J.H. (2003) n-3 fatty acids and the risk of sudden cardiac death. Emphasis on heart rate variability. Dan. Med. Bull., 50: 347–367.
- Crestor (2007) [prescribing information]. Wilmington, D.E.: AstraZeneca Pharmaceuticals LP, November.
- Crouse J.R. III, Grobbee D.E., O’Leary D.H. et al. (2004) Measuring Effects on intima media Thickness: an Evaluation Of Rosuvastatin in subclinical atherosclerosis — the rationale and methodology of the METEOR study. Cardiovasc. Drugs Ther., 18: 231–238.
- Crouse J.R., Grundy S.M. (1981) Effects of colestipol, clofibrate, and placebo on plasma lipoproteins of patients with hypercholesterolemia. Metabolism, 30: 123–8.
- Curtis C.L., Rees S.G., Cramp J. et al. (2002) Effects of n-3 fatty acids on cartilage metabolism. Proc. Nutrit. Soc., 61, 381–389.
- DiGiacomo R.A., Kremer J.M., Shah D.M. (1989) Fish-oil dietary supplementation in patients with Raynaud’s phenomenon: a double-blind, controlled, prospective study. Am. J. Med., 86: 158–164.
- Donadio J.V. (1991) Omega-3 polyunsaturated fatty acids: a potential new treatment of immune renal disease. Mayo Clin. Proc., 66: 1018–1028.
- Downs J.R. et al. (2001) Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS): Additional perspectives on tolerability of long-term treatment with lovastatin. Am. J. Cardiol., 87: 1074–1079.
- Drazen J.M., Jarcho J.A., Morrissey S. et al. (2008) Cholesterol Lowering and Ezetimibe. N. Engl. J. Med., 358: 1507–1508.
- Dubé M.P., Stein J.H., Aberg J.A. et al. (2003) Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medicine Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin. Infect. Dis., 37: 613–627.
- Dujovne C.A., Krehbiel P., Decoursey S. et al. (1984) Probucol with colestipol in the treatment of hypercholesterolemia. Ann. Intern. Med., 100: 477–482.
- Durrington P.N., Bhatnagar D., Mackness M.I. et al. (2001) An omega-3 polyunsaturated fatty acid concentrate administered for one year decreased triglycerides in simvastatin treated patients with coronary heart disease and persisting hypertriglyceridaemia. Heart, 85: 544–548.
- Eldor R., Raz I. (2009) American Diabetes Association indications for statins in diabetes: is there evidence? Diab. Care, 32: S384–S391.
- Facchinetti F., Fazzio M., Venturini P. (2005) Polyunsaturated fatty acids and risk of preterm delivery. Eur. Rev. Med. Pharmacol. Sci., 9: 41–48.
- Ferrario C.M.; ARBITER 6-HALTS (2010) Does it have the power to settle all matters? Ther Adv Cardiovasc Dis., 4: 77–81.
- Fichtlscherer S., Schmidt-Lucke C., Bojunga S. et al. (2006) Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for `pleiotropic’ functions of statin therapy. Eur. Heart J., 27: 1182–1190.
- Finn A.V., Kolodgie F.D., Virmani R. (2010) Correlation between carotid intimal/medial thickness and atherosclerosis: a point of view from pathology. Arterioscler. Thromb. Vasc. Bio., 30: 177–181.
- Finnish Medical Society Duodecim (2007) Treatment of hyperlipidaemia: aims and selection. In: EBM Guidelines. Evidence-Based Medicine [Internet]. Helsinki, Finland: Wiley Interscience. John Wiley & Sons.
- Fleg J.L., Mete M., Howard B.V. et al. (2008) Effect of statins alone versus statins plus ezetimibe on carotid atherosclerosis in type 2 diabetes: The SANDS (Stop Atherosclerosis in Native Diabetics Study) Trial. J. Am. Coll. Cardiol., 52: 2198–2205.
- Flint O.P., Masters B.A., Gregg R.E. et al. (1997) Inhibition of cholesterol synthesis by squalene synthase inhibitors does not induce myotoxicity in vitro. Toxicol. Appl. Pharmacol., 145(1): 91–98.
- Foody J.M. (2007) Torcetrapib does not limit atherosclerosis progression J. Watch. Cardiology: 2–2.
- Frost L., Vestergaard P. (2005) n-3 Fatty acids consumed from fish and risk of atrial fibrillation or flutter: the Danish Diet, Cancer, and Health Study. Am. J. Clin. Nutr., 81: 50–54.
- Gadbut A.P., Caruso A.P., Galper J.B. (1995) Differential sensitivity of C2-C12 striated muscle cells to lovastatin and pravastatin. J. Mol. Cell. Cardiol., 27: 2397–2402.
- Gardner C.D., Lawson L.D., Block E. et al. (2007) Effect of raw garlic vs. commercial garlic supplements on plasma lipid concentrations in adults with moderate hypercholesterolemia: a randomized clinical trial. Arch. Intern. Med., 167: 346–353.
- Garg A., Grundy S.M. (1990) Nicotinic acid as therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. JAMA, 264: 723–726.
- Genest J. (2006) Relationship of cholesterol lowering with statins to cardiovascular events: recent insight from meta-analyses. Expert opinions: Clinical impact. Clinical update on statin safety and efficacy. Vol. 1(9): 3–5.
- Giannoglou G.D., Chatzizisis Y.S., Misirli G. (2007) The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur. J. Int. Med., 18: 90–100.
- GISSI-Prevenzione Investigators (1999) Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet, 354: 447–455.
- Glueck C.J. (1982) Colestipol and probucol: treatment of primary and familial hypercholesterolemia and amelioration of atherosclerosis. Ann. Intern. Med., 96: 475–482.
- Goldstein J.L., Brown M.S. (1990) Regulation of the mevalonate pathway. Nature, 343: 425–430.
- Greenberg M.E., Smith J.D., Sehayek E. (2009) Moderately decreased cholesterol absorption rates are associated with a large atheroprotective effect. Arterioscler. Thromb. Vasc. Bio., 29: 1745–1750.
- Grundy S.M., Vega G.L., Bilheimer D.W. (1985) Influence of combined therapy with mevinolin and interruption of bile-acid reabsorption of low density lipoproteins in heterozygous familial hypercholesterolemia. Ann. Intern. Med., 103: 339–343.
- Guerin M., Le Goff W., Duchene E. et al. (2008) Inhibition of CETP by torcetrapib attenuates the atherogenicity of postprandial TG-rich lipoproteins in type IIB hyperlipidemia. Arterioscler. Thromb. Vasc. Bio., 28: 148–154.
- Hamelin B.A., Turgeon J. (1998) Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol. Sci., 19: 26–37.
- Harris W.S. (2007) Expert opinion: omega-3 fatty acids and bleeding-cause for concern? Am. J. Cardiol., 99: 44C–46C.
- Harris W.S., Bulchandani D. (2006) Why do omega-3 fatty acids lower serum triglycerides? Curr. Opin. Lipidol., 17: 387–393.
- Harris W.S., Ginsberg H.N., Arunakul N. et al. (1997) Safety and efficacy of Omacor in severe hypertriglyceridemia. J. Cardiovasc. Risk., 4: 385–391.
- Hayward R.A., Hofer T.P., Vijan S. (2006) Narrative review: lack of evidence for recommended low-density lipoprotein treatment targets: a solvable problem. Ann. Intern. Med., 145: 520–530.
- Hedblad B., Wikstrand J., Janzon L. et al. (2001) Low-dose metoprolol CR/XL and fluvastatin slow progression of carotid intima-media thickness: main results from the Beta-Blocker Cholesterol-Lowering Asymptomatic Plaque Study (BCAPS). Circulation, 103: 1721–1726.
- Hirsch M., O’Donnell J.C., Jones P. (2005) Rosuvastatin is cost-effective in treating patients to low-density lipoprotein-cholesterol goals compared with atorvastatin, pravastatin and simvastatin: analysis of the STELLAR trial. Eur J. Cardiovasc. Prev. Rehabil., 12: 18–28.
- Hodis H.N., Mack W.J., La Bree L. et al. (1996) Reduction in carotid arterial wall thickness using lovastatin and dietary therapy: a randomized controlled clinical trial. Ann. Intern. Med., 124: 548–556.
- Hoeg J.M., Maher M.M., Bov E. et al. (1984) Normalization of plasma lipoproteins with neomycin and niacin in type II hyperlipoproteinemia. Circulation, 70: 1004–1011.
- HPS Collaborative Group (2002) MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet, 360: 7–22.
- Illingworth D.R. (1984) Mevinolin plus cholestyramine therapy for severe heterozygous familial hypercholesterolemia. Ann. Intern. Med., 101: 598–604.
- Illingworth D.R., Bacon S. (1989) Treatment of heterozygous familial hypercholesterolemia with lipid-lowering drugs. Arteriosclerosis, 9(1 Suppl.): I121–34.
- The West of Scotland Coronary Prevention Study (WOSCOPS) (1998) Influence of pravastatin and plasma lipids on clinical events in the West of Scotland Coronary Prevention Study (WOSCOPS). Circulation, 97: 1440–1445.
- Ismail H.M. (2005) The role of omega-3 fatty acids in cardiac protection: an overview. Front. Biosci., 10: 1079–1088.
- Istvan E.S., Deisenhofer J. (2001) Structural mechanism for statin inhibition of HMG-CoA reductase. Science, 292: 1160–1164.
- Jackevicius C.A., Tu J.V., Ross J.S. et al. (2008) Use of ezetimibe in the United States and Canada. N. Engl. J. Med., 358: 1819–1828.
- Jakulj L., Vissers M.N., Groen A.K. et al. (2010) Baseline cholesterol absorption and the response to ezetimibe/simvastatin therapy: a post-hoc analysis of the ENHANCE trial. J. Lipid Res., 51: 755–762.
- Kashyap M.L., McGovern M.E., Berra K. et al. (2002) Long-term safety and efficacy of a once-daily niacin/lovastatin formulation for patients with dyslipidemia. Am. J. Cardiol., 89: 672–678.
- Kastelein J.J.P., Akdim F., Stroes E.S.G. et al. (2008) Simvastatin with or without ezetimibe in familial hypercholesterolemia. N. Engl. J. Med., 358: 1431–1443.
- Kastelein J.J.P., Bots M.L. (2009) Statin therapy with ezetimibe or niacin in high-risk patients. N. Engl. J. Med., 361: 2180–2183.
- Kastelein J.J.P., van Leuven S.I., Burgess L. et al. (2007) Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N. Engl. J. Med., 356: 1620–1630.
- Katan M.B., Grundy S.M., Jones P. et al.; Stresa Workshop Participants (2003) Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin. Proc., 78: 965–978.
- Keech A., Simes R.J., Barter P. et al. (2005) Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet, 366: 1849–1861.
- Kita T., Nagano Y., Yokode M. et al. (1987) Probucol prevents the progression of atherosclerosis in Watanabe heritable hyperlipidemic rabbit, an animal model for familial hypercholesterolemia. Proc. Natl. Acad. Sci. USA, 84: 5928–5931.
- Kris-Etherton P.M., Harris W.S., Appel L.J. (2002) Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation, 106: 2747–2757.
- Lammerding J., Schulze P.C., Takahashi T. et al. () 2004 Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest., 113: 370–378.
- Landmesser U., Bahlmann F., Mueller M. et al. (2005) Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation, 111: 2356–2363.
- LaRosa J. (1989) Review of clinical studies of bile acid sequestrants for lowering plasma lipid levels. Cardiology, 76(Suppl. 1): 55–61.
- LaRosa J. (2005) Safety and efficacy of atorvastatin induced very low LDL-C levels. A post hoc analysis of the TNT study. Circulation, 112(Suppl. II): 662–663.
- LaRosa J.C., Grundy S.M., Waters D.D. et al. (2005) Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med., 352: 1425–1435.
- LaRosa J.C., He H., Vupputuri S. (1999) Effect of statins on risk of coronary disease. A meta-analysis of randomized controlled trials. JAMA, 28: 2340–2346.
- Law M., Rudnicka A.R. (2006) Statin safety: a systematic review. Am. J. Cardiol., 97(8A): 52C–60C.
- Law M.R., Wald N.J., Rudnicka A.R. (2003) Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. BMJ, 326: 1423–1429.
- Lee I.M., Cook N.R., Baziano J.M. et al. (2005) Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized, controlled clinical trial. JAMA, 294: 56–65.
- Lee K.W., Hamaad A., MacFadyen R.J. et al. (2004) Effects of dietary fat intake in sudden death: reduction of death with omega-3 fatty acids. Curr. Cardiol. Rep., 6: 371–378.
- Lescol (2006) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation, October.
- Levy H.B., Kohlhaas H.K. (2006) Consideration for supplementing with coenzyme Q10 during statin therapy. Ann. Pharmacother., 40: 290–294.
- Lewis A., Lookinland S., Beckstrand R.L. et al. (2004) Treatment of hypertriglyceridemia with omega-3 fatty acids: a systematic review. J. Am. Acad. Nurse Pract., 16: 384–395.
- Libby P.J. (2005) The forgotten majority: unfinished business in cardiovascular risk reduction. J. Am. Coll. Cardiol., 46: 1225–1228.
- Lindsay A.C., Halcox J.P., Duivenvoorden R. et al. (2010) Niacin compared with ezetimibe. N. Engl. J. Med., 362: 1046–1047.
- LIPID Study Group (1998) Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N. Engl. J. Med., 339: 1349–1357.
- Lipitor (2007) [prescribing information]. New York, NY: Parke-Davis, November.
- Lutz R.J., Trujillo M.A., Denham K.S. et al. (1992) Nucleoplasmic localization of prelamin A: implications for prenylation-dependent lamin A assembly into the nuclear lamina. Proc. Natl. Acad. Sci. USA, 89: 3000–3004.
- Macaulay T.E., Cook A.M., Fink J.L. III et al. (2009) Pharmacists’ role in facilitating evidence-based prescribing for unlabeled use of medications. Am. J. Health Syst. Pharm., 66: 1735–1739.
- MacMahon S., Sharpe N., Gamble G. et al. (1998) Effects of lowering average of below-average cholesterol levels on the progression of carotid atherosclerosis: results of the LIPID Atherosclerosis Substudy. Circulation, 97: 1784–1790.
- Marcoff L., Thompson P.D. (2007) The role of coenzyme Q10 in statin associated myopathy: a systematic review. J. Am. Coll. Cardiol., 49: 2231–2237.
- Martin P.D., Warwick M.J., Dane A.L. et al. (2003) Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther., 25: 2822–2835.
- Martin-Rendon E., Blake D.J. (2003) Protein glycosylation in disease: new insights into the congenital muscular dystrophies. Trends Pharmacol. Sci., 24: 178–183.
- Mascitelli L., Ravnskov U., Pezzetta F. et al. (2009) After the Failure of ENHANCEd Cholesterol Lowering in Familial Hypercholesterolemia, SEAS of Problems with Ezetimibe. Angiology, 60: 127–128.
- Masters B.A., Palmoski M.J., Flint O.P. et al. (1995) In vitro myotoxicity of the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, pravastatin, lovastatin, and simvastatin, using neonatal rat skeletal myocytes. Toxicol. Appl. Pharmacol., 131: 163–174.
- Matzno S., Yamauchi T., Gohda M. et al. (1997) Inhibition of cholesterol biosynthesis by squalene epoxidase inhibitor avoids apoptotic cell death in L6 myoblasts. J. Lipid Res., 38: 1639–1648.
- Matzno S., Yasuda S., Juman S. et al. (2005) Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J. Pharm. Pharmacol., 57: 1475–1484.
- McKenney J. (2004) New perspectives on the use of niacin in the treatment of lipid disorders. Arch. Intern. Med., 164: 697–705.
- McKenney J.M., Davidson M.H., Jacobson T.A. et al. (2006) Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am. J. Cardiol., 97(8A): 89C–94C.
- McKenney J.M., Jones P.H., Bays H.E. et al. (2007) Comparative effects on lipid levels of combination therapy with a statin and extended-release niacin or ezetimibe versus a statin alone (the COMPELL study). Atherosclerosis, 192: 432–437.
- Mevacor (2007) [prescribing information]. Whitehouse Station, NJ: Merck & Co., Inc, December.
- Miettinen T.A. (1979) Effect of neomycin alone and in combination with cholestyramine on serum cholesterol and fecal steroids in hypercholesterolemic subjects. J. Clin. Invest., 64: 1485–1493.
- Minhas R., Humphries S.E, Qureshi N. et al. (2009) On behalf of the NICE Guideline Development Group. Controversies in familial hypercholesterolaemia: recommendations of the NICE Guideline Development Group for the identification and management of familial hypercholesterolaemia. Heart, 95: 584–587.
- Moghadasian M.H. (1999) Clinical pharmacology of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Life Sci., 65: 1329–1337.
- Morikawa S., Murakami T., Yamazaki H. et al. (2005) Analysis of the global RNA expression profiles of skeletal muscle cells treated with statins. J. Atheroscler. Thromb., 12: 121–131.
- Mozaffarian D., Psaty B.M., Rimm E.B. et al. (2004) Fish intake and risk of incident atrial fibrillation. Circulation, 110: 368–373.
- Mozaffarian D., Rimm E.B. (2006) Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA, 296: 1885–1899.
- Mukhtar R.Y., Reckless J.P. (2005) Statin-induced myositis: a commonly encountered or rare side effect? Curr. Opin. Lipidol., 16: 640–647.
- Mukhtar R.Y., Reid J., Reckless J.P. (2005) Pitavastatin. Int. J. Clin. Pract., 59: 239–252.
- Nakahara K., Kuriyama M., Sonoda Y. et al. (1998) Myopathy induced by HMG-CoA reductase inhibitors in rabbits: a pathological, electrophysiological, and biochemical study. Toxicol. Appl. Pharmacol., 152: 99–106.
- National Institute for Health and Clinical Excellence (2007) Ezetimibe for the treatment of primary (heterozygous-familial and non-familial) hypercholesterolaemia. Technology appraisal guidance 132 [online] (http://www.nice.org.uk/nicemedia/pdf/TA132guidance.pdf) [Accessed 23 July 2009].
- National Institute for Health and Clinical Excellence (2008) Identification and management of familial hypercholesterolaemia. Clinical guideline 71 [online] (http://www.nice.org.uk/nicemedia/pdf/CG071NICEGuideline.pdf) [Accessed 23 July 2009].
- Niacor (2003) [package insert]. Minneapolis, MN: Upsher-Smith Laboratories, Inc, June.
- Nieland T.J.F., Shaw J.T., Jaipuri F.A. et al. (2007) Influence of HDL-cholesterol-elevating drugs on the in vitro activity of the HDL receptor SR-BI. J. Lipid Res., 48: 1832–1845.
- Nikolova V., Leimena C., McMahon A.C. et al. (2004) Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest., 113: 357–369.
- Nishimoto T., Tozawa R., Amano Y. et al. (2003) Comparing myotoxic effects of squalene synthase inhibitor, T-91485, and 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors in human myocytes. Biochem. Pharmacol., 66: 2133–2139.
- Nissen S.E., Nicholls S.J., Sipahi I. et al. (2006) Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA, 295: 1556–1565.
- Nordoy A., Bonaa K.H., Nilsen H. et al. (1998) Effects of simvastatin and omega-3 fatty acids on plasma lipoproteins and lipid peroxidation in patients with combined hyperlipidaemia. J. Intern. Med., 243: 163–170.
- Owczarek J., Jasinska M., Orszulak-Michalak D. (2005) Drug-induced myopathies. An overview of the possible mechanisms. Pharmacol. Rep., 57: 23–34.
- Paiva H., Thelen K.M., van Coster R. et al. (2005) High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin. Pharmacol. Ther., 78: 60–68.
- Pandit M.K., Burke J., Gustafson A.B. et al. (1993) Drug-induced disorders of glucose tolerance. Ann. Intern. Med., 118: 529–539.
- Park Y., Harris W.S. (2003) Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J. Lipid Res., 44: 455–463.
- Parthasarathy S., Young S.G., Witztum J.L. et al. (1986) Probucol inhibits oxidative modification of low density lipoprotein. J. Clin. Invest., 77: 641–644.
- Pasternak R.C., Smith S.C. Jr., Bairey-Merz C.N. et al. (2002) ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Stroke, 33: 2337–2341.
- Penzak S.R., Chuck S.K., Stajich G.V. (2000) Safety and efficacy of HMG-CoA reductase inhibitors for treatment of hyperlipidemia in patients with HIV infection. Pharmacotherapy, 20: 1066–1071.
- Physician’s Desk Reference, 58th ed. 2005. (2007) Thomson Healthcare, Montvale, NJ.Pravachol [prescribing information]. Princeton, NJ: Bristol-Myers Squibb, March.
- Prueksaritanont T., Subramanian R., Fang X. et al. (2002) Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug. Metab. Dispos., 30: 505–512.
- Prueksaritanont T., Tang C., Qiu Y. et al. (2002) Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab. Dispos., 30: 1280–1287.
- Rader D.J., Cohen J., Hobbs H.H. (2003) Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J. Clin. Invest., 111: 1795–1803.
- Raitt M.H., Connor W.E., Morris C. et al. (2005) Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA, 293: 2884–2891.
- Ray K.K., Cannon C.P. (2005) The potential relevance of the multiple lipid-independent (pleiotropic) effects of statins in the management of acute coronary syndromes. J. Am. Coll. Cardiol., 46: 1425–433.
- Reijneveld J.C., Koot R.W., Bredman J.J. et al. (1996) Differential effects of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors on the development of myopathy in young rats. Pediatr. Res., 39: 1028–1035.
- Robinson J.G., Smith B., Maheshwari N. et al. (2005) Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J. Am. Coll. Cardiol., 46: 1855–1862.
- Rosenson R.S. (2004) Current overviewof statin-inducedmyopathy. Am. J. Med., 116: 408–416.
- Rubenfire M. (2004) Impact of Medical Subspecialty on Patient Compliance to Treatment Study Guide. Safety and compliance with once-daily niacin extended-release/lovastatin as initial therapy in the Impact of Medical Subspecialty on Patient Compliance to Treatment (IMPACT) study. Am. J. Cardiol., 94: 306–311.
- Sacks F.M., Pfeffer M.A., Moye L.A. et al. (1996) The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med., 335: 1001–1009.
- Sakamoto K., Honda T., Yokoya S. et al. (2007) Rabsmall GTPases are involved in fluvastatin and pravastatin-induced vacuolation in rat skeletal myofibers. FASEB. J., 21: 4087–4094.
- Sasaki J., Tanabe Y., Saku K. et al. (1987) Alterations in lipoprotein with discontinuation of probucol. Curr. Therap. Res., 42: 640–645.
- Schachter M. (2005) Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam. Clin. Pharmacol., 19: 117–125.
- Schoenhagen P., Tuzcu E.M., Apperson-Hansen C. et al. (2006) Determinants of arterial wall remodeling during lipid-lowering therapy: serial intravascular ultrasound observations from the Reversal of Atherosclerosis with Aggressive Lipid Lowering Therapy (REVERSAL) trial. Circulation, 113: 2826–2834.
- Schönbeck U., Libby P. (2004) Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation, 109(Suppl. II): II–18.
- Schonfeld G., Witztum J.L., Basich P. (1982) Probucol further lowers the serum cholesterol of colestipol treated patients with hypercholesterolemia. Artery, 10: 99–104.
- Schrepf R., Limmert T., Claus W.P. et al. (2004) Immediate effects of n-3 fatty acid infusion on the induction of sustained ventricular tachycardia. Lancet, 363: 1441–1442.
- Schulz V., Hansel R., Tyler V.E. (2000) Rational phytotherapy. A physicians’ guide to herbal medicine. 4th edition. Springer, Berlin.
- Schwarz S., Leweling H. (2005) Multiple sclerosis and nutrition. Multiple Sclerosis, 11: 24–23.
- Sharma M., Ansari M.T., Abou-Setta A.M. et al. (2009) Systematic review: comparative effectiveness and harms of combination therapy and monotherapy for dyslipidemia. Ann. Intern. Med., 151: 622–630.
- Siddals K.W., Marshman E., Westwood M. et al. (2004) Abrogation of insulin-like growth factor-I (IGF-I) and insulin action by mevalonic acid depletion: synergy between protein prenylation and receptor glycosylation pathways. J. Biol. Chem., 279: 38353–38359.
- Silagy C., Neil A. (1994) Garlic as a lipid lowering agent — a meta-analysis. J. R. Coll. Phys. Lond., 28: 2–8.
- Sinensky M., Beck L.A., Leonard S. et al. (1990) Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J. Biol. Chem., 265: 19937–19941.
- Singh I.M., Shishehbor M.H., Ansell B.J. (2007) High-density lipoprotein as a therapeutic target: a systematic review. JAMA, 298: 786–798.
- Smith C.C. et al. (2003) Screening for statin-related toxicity. Arch. Intern. Med., 163: 688–692.
- Stein E.A. (2008) Additional lipid lowering trials using surrogate measurements of atherosclerosis by carotid intima-media thickness: more clarity or confusion? J. Am. Coll. Cardiol., 52: 2206–2209.
- Stein E.A., Heimann K.W. (1975) Colestipol, clofibrate, cholestyramine and combination therapy in the treatment of familial hyperbetalipoproteinaemia. S. Afr. Med J., 49: 1252–1256.
- Stevinson C., Pittler M.H., Ernst E. (2000) Garlic for treating hypercholesterolaemia. A meta-analysis of randomized clinical trials. Ann. Intern. Med., 133: 420–429.
- Strandberg T.E., Feely J., Sigurdsson E.L. (2004) Twelve-week, multicenter, randomized, open-label comparison of the effects of rosuvastatin 10 mg/d and atorvastatin 10 mg/d in high-risk adults: a DISCOVERY Study. Clin. Ther., 26: 1821–1833.
- Superko H.R. (2006) The failure of LDL cholesterol reduction and the importance of reverse cholesterol transport. The role of nicotinic acid. Br. J. Cardiol., 13: 131–136.
- Tamizi F.B., Tamizi B. (2002) Treatment of chronic fatigue syndrome by dietary supplementation with omega-3 fatty acids — a good idea? Med. Hypotheses, 58: 249–250.
- Taylor A.J., Kent S.M., Flaherty P.J. et al.; ARBITER (2002) Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol: a randomized trial comparing the effects of atorvastatin and pravastatin on carotid intima medial thickness. Circulation, 106: 2055–2060.
- Taylor A.J., Lee H.J., Sullenberger L.E. (2006) The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3. Curr. Med. Res. Opin., 22: 2243–2250.
- Taylor A.J., Sullenberger L.E., Lee H.J. et al. (2004) Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation, 110: 3512–3517.
- Taylor A.J., Villines T.C., Stanek E.J. et al. (2009) Extended-release niacin or ezetimibe and carotid intima-media thickness. N. Engl. J. Med., 361: 2113–2122.
- Tenenbaum A., Motro M., Fisman E.Z et al. (2005) Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch. Intern. Med., 165: 1154–1160.
- The BIP Study Group (2000) Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation, 102: 21–27.
- Thompson P.D., Clarkson P., Karas R.H. (2003) Statin-associated myopathy. JAMA, 289: 1681–1690.
- Tikkanen M.J., Helve E., Nikkilä E.A. (1987) Treatment of familial and non-familial hypercholesterolaemia: a review of HMG-CoA reductase inhibitors and probucol. Eur. Heart J., 8(Suppl. E): 97–101.
- van Wissen S., Smilde T.J., Trip M.D. et al. (2005) Long-term safety and efficacy of high-dose atorvastatin treatment in patients with familial hypercholesterolemia. Am. J. Cardiol., 95: 264–266.
- Vanschoonbeek K., Feijge M.A., Paquay M. et al. (2004) Variable hypocoagulant effect of fish oil intake in humans: modulation of fibrinogen level and thrombin generation. Arterioscler. Thrombos. J. Vasc. Biol., 24: 1734–1740.
- Warner G.J., Berry M.J., Moustafa M.E. et al. (2000) Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem., 275: 28110–28119.
- Wenke K., Meiser B., Thiery J. et al. (1997) Simvastatin reduces graft vessel disease and mortality after heart transplantation: a four-year randomized trial. Circulation, 96: 1398–1402.
- Whayne T.F. Jr. (2009) There a Problem With Ezetimibe or Just ENHANCEd Hype? Angiology, 59: 661–663.
- Williams D., Feely J. (2002) Pharmacokinetic-pharmacodynamic drug interactions with HMG-CoA reductase inhibitors. Clin. Pharmacokinet., 41: 434–470.
- WHO consultation (1998) Familial hypercholesterolaemia (FH): report of a WHO consultation. Geneva: World Health Organization.
- Witztum J.L. (1987) Intensive drug therapy of hypercholesterolemia. Am. Heart. J., 113: 603–609.
- Wolfe M.L., Vartanian S.F., Ross J.L. et al. (2001) Safety and effectiveness of Niaspan when added sequentially to a statin for treatment of dyslipidemia. Am. J. Cardiol., 87: 476–479.
- Yamamoto A., Matsuuzawa Y., Yokoyama S. et al. (1986) Effects of probucol on xanthomata regression in familial hypercholesterolemia. Am. J. Cardiol., 27: 29H–35H.
- Young J.M., Florkowski C.M., Molyneux S.L. et al. (2007) Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am. J. Cardiol., 100: 1400–1403.
- Zhao X.Q., Morse J., Chait A. et al. (2002) Simvastatin plus niacin protect against atherosclerosis progression and clinical events in coronary artery disease patients with metabolic syndrome. J. Am. Coll. Cardiol., 39(Suppl. A): 242A; abstract 1130–1173.
- Zocor (2007) [package insert]. Whitehouse Station, NJ: Merck & Co., Inc, November.