ВИДЫ И ПРИЧИНЫ НАРУШЕНИЙ КРОВООБРАЩЕНИЯ
Расстройства кровообращения связаны с изменениями функции сердца, артерий, микрососудов, вен, нарушениями реологических свойств текущей по ним крови, проницаемости гистогематического барьера или нейрогуморальной регуляции. Отдельные звенья сердечно-сосудистой системы функционируют в тесной взаимосвязи, поэтому изменения, возникающие в одном из них, влияют на функцию всех остальных. При этом возможны изменения состава тканевого метаболизма вплоть до повреждения и гибели клеток и стромальных структур. Расстройства кровообращения могут иметь общий характер, когда изменяется функция всей кровеносной системы, или же относится к отдельным участкам сосудистого русла. В морфологической картине повреждений, обусловленных расстройствами крово- и лимфообращения, сочетаются признаки как общие, так и определяемые структурно-функциональными особенностями данной ткани.
Общие и местные нарушения кровообращения, отмечаемые при многих заболеваниях, осложняют их течение. В настоящее время различают следующие виды нарушений: артериальное и венозное полнокровие (гиперемия), ишемия, стаз крови, тромбоз, ДВС-синдром, эмболия, инфаркт, кровотечение и кровоизлияние.
Артериальная гиперемия
Артериальная гиперемия — повышенное кровенаполнение ткани, обусловленное избыточным притоком артериальной крови, может возникать в нормальных и патологических условиях, иметь общий или местный характер, развиваться в здоровом организме или в условиях патологии.
Общая артериальная гиперемия наблюдается при эритремии (повышение числа эритроцитов в крови) либо при увеличении всего ОЦК (плетора). Кожные покровы таких больных приобретают отчетливый красноватый оттенок, а уровень АД повышается. Однако значительно чаще артериальная гиперемия ограничивается каким-либо конкретным регионом. В физиологических условиях она связана с функцией сосудосуживающих и сосудорасширяющих нервов либо с действием физических факторов. Интенсивность кровотока в микрососудах возрастает в результате увеличения просвета периферических артерий, что способствует поддержанию метаболических процессов в питаемой ими ткани. Гиперемия, связанная с усилением функции органов, обозначается как функциональная, или рабочая.
Механизмы развития артериальной гиперемии в условиях патологии значительно разнообразнее. В соответствии с причиной различают ангионевротическую (нейропаралитическую), коллатеральную, постанемическую, вакатную, возникающую на почве артериовенозного свища, и воспалительную артериальную гиперемию.
Ангионевротическая гиперемия связана с нарушением соотношения процессов возбуждения и торможения в отделах вегетативной нервной системы, регулирующих тонус соответствующих артерий. Доминирование парасимпатических воздействий над симпатическими ведет к дилатации артерий и преобладанию объема притока крови над ее оттоком по венозным сосудам. Этот вид вазомоторной гиперемии может возникать рефлекторно, например при психомоторных аффектах, повреждении нервных проводников или сплетений, некоторых инфекционных заболеваниях (грипп, сыпной тиф), протекающих с поражением узлов симпатической нервной системы. Органы, кожные покровы, слизистая оболочка при артериальной гиперемии покрасневшие, иногда припухшие, температура их повышена. Ощущение пульсации гиперемированного поля свидетельствует о распространении пульсовой волны в периферическую сеть микрососудов. Обычно этот вид артериальной гиперемии имеет быстропроходящий, транзиторный характер и не оставляет следов.
Коллатеральная гиперемия развивается при ограничении проходимости магистрального артериального ствола, например при его тромбозе, сдавливании извне, пороке развития сосуда. В результате притекающая кровь устремляется в ветви, отходящие до препятствия, переполняет и расширяет их. Формирование обходных путей-коллатералей определяется темпами обтурации сосуда, количеством предсуществовавших анастомозов и степенью развития чувствительной иннервации по ходу сосудов, которая обеспечивает более или менее полное и быстрое раскрытие коллатеральных путей. Образование коллатералей стимулируется метаболитами, поступающими из ишемизированной ткани и оказывающими вазодилататорный эффект. Сосудистая сеть, оказавшаяся в зоне коллатеральной гиперемии, подвергается приспособительной перестройке. Отмечается усиление эластического и аргирофильного каркаса, гипертрофия и гиперплазия гладкомышечных клеток стенки входящих в нее сосудов. Коллатеральная гиперемия является важным компенсаторным механизмом, благодаря которому поддерживается кровоснабжение ткани при закрытии основного артериального ствола.
Вакатная (от лат. vacuus — пустой) гиперемия является следствием градиента барометрического давления между каким-либо участком тела или всем организмом и окружающей средой. Примером местного артериального полнокровия такой природы является гиперемия кожи, вызванная с помощью медицинской банки. Общую вакатную гиперемию отмечают при резком снижении давления в окружающей среде вследствие быстрого подъема водолазов или кессонных работах, разгерметизации высотных летательных аппаратов. Резкое расширение сосудов в этих условиях может привести к полнокровию слизистых оболочек, кровотечению из носа и ушей, кровоизлияниям с тяжелыми повреждениями внутренних органов.
Постанемическая гиперемия развивается в тех случаях, когда восстанавливается проходимость артерий, ранее частично или полностью выключенных из кровотока. Такая ситуация бывает при снятии хирургической лигатуры, удалении опухоли или жидкости, сдавливавших сосуд, канализации тромбированной или стенозировании артерии. Это может приводить к ишемизации других органов, в частности головного мозга, с развитием обморочного состояния (коллапса).
Гиперемия на почве артериовенозного свища чаще всего является следствием огнестрельного ранения с повреждением артерии и вены и образованием соустья между ними. Артериальная кровь, устремляющаяся по новому пути в вену, повышает температуру ткани, изменяя обменные процессы и способствуя ускоренному росту волос на коже.
Воспалительная гиперемия — обязательный компонент воспалительного процесса. В ее основе лежит полнокровие периферических сосудов, связанное с нарушением обменных процессов и парезом сосудов. Усиленный приток крови к воспаленному участку повышает локальную температуру, а при поражении кожи или слизистых оболочек окрашивает их в красный цвет. Гиперемия более выражена в острой фазе процесса и проходит вместе с его окончанием.
Венозное полнокровие
Венозная (застойная, пассивная) гиперемия — патологическое изменение кровообращения, обусловленное затруднением оттока венозной крови при сохраненной доставке ее в ткани по соответствующим артериям. Венозное полнокровие может быть общим и местным, острым и хроническим.
Острая общая венозная гиперемия возникает при быстро развивающейся СН, которая может быть следствием ИМ, токсико-инфекционных поражений сердечной мышцы, утрачивающей способность эффективно поддерживать общую гемодинамику или деструкции митрального или аортального клапанов. Это приводит к переполнению кровью емкостных сосудов. Скорость кровотока в венах и капиллярах снижается, а давление в них значительно повышается. Просветы всех микрососудов расширяются, а ранее не фунционировавшие раскрываются. В этих условиях максимально дилатируются мелкие вены, венулы и венулярные отделы капилляров вследствие относительно большего повышения давления в дистальных отделах сосудистой сети. Так как редуцированный гемоглобин венозной крови беден кислородом, то ткани испытывают гипоксию. Проницаемость стенок микрососудов и растяжимость опорной соединительной ткани повышаются, а объем тканевой жидкости существенно увеличивается.
Признаками венозной гиперемии являются понижение температуры, синюшность (цианоз) кожи и слизистых оболочек, что связано с заполнением поверхностно расположенных сосудов кровью, содержащей восстановленный гемоглобин. Цианотическая окраска более заметна в периферических частях тела — на губах, носу (акроцианоз), кончиках пальцев. В выраженных случаях цианоз сочетается с отеком жировой клетчатки и водянкой полостей. В тканях отмечают плазматическое пропитывание (плазморрагия), отек стромы, замедление и остановку кровотока в микрососудах, множественные кровоизлияния диапедезного характера. Так как при острой венозной гиперемии нарушается большинство основных функций крови (транспортная, дыхательная, трофическая, экскреторная, поддержание водного баланса ткани), в паренхиматозных органах возникают дистрофические изменения и рассеянные мелкоочаговые некрозы. Быстро нарастающий застой крови в бассейне магистральной вены ведет к застойному геморрагическому инфаркту — обширному некрозу, формирующемуся на фоне выраженного цианоза, отека и пропитывания ткани кровью. Такие инфаркты отмечают при закрытии селезеночной, почечной, брыжеечных вен, тромбозе синусов мозговой оболочки или коронарного синуса.
Венозное полнокровие проявляется неодинаково в различных органах в зависимости от архитектоники сосудов. В легких выявляют дилатацию и переполнение кровью капилляров, пропитывание фильтратом крови межальвеолярных перегородок и заполнение им значительной части альвеол (интерстициальный и альвеолярный отек), диапедезный выход эритроцитов. Острая венозная гиперемия печени характеризуется увеличением ее объема. На разрезе ткань органа приобретает темно-красный цвет, становится влажной со стертым рисунком.
Остро возникающая регургитация венозной крови из сердца при дисфункции сопровождается не только полнокровием тканей, но и кровоизлияниями в центральных зонах печеночных долек. Почки при остром венозном полнокровии увеличиваются в объеме, их масса возрастает от 250–300 до 400–500 г. Наиболее полнокровно мозговое вещество пирамидок, что связано с внутриорганным перераспределением кровотока. Дистрофические изменения, развивающиеся на фоне отека ткани почек, максимально выражены в наиболее чувствительных к гипоксии проксимальных отделах канальцевой системы нефрона. Объем селезенки в условиях острого венозного застоя увеличивается в 2,5 раза, что сопровождается резким напряжением ее капсулы, а масса достигает >300 г. При разрезе с темно-красной поверхности ткани селезенки обильно стекает кровь. В сердечной мышце отмечаются избыточное кровенаполнение интрамуральных вен и микрососудов, капилляростазы, отечность интерстиция, мелкоочаговые повреждения мышечных волокон.
Иногда венозная гиперемия имеет активный характер. Это выявляют в зонах коллатерального венозного полнокровия, например при обструкции крупного венозного ствола, циррозе печени или шоке. В последнем случае в печени, селезенке, подкожной жировой клетчатке, венах ЖКТ, таза депонируется до 40% всей крови, что приводит к дефициту наполнения полостей сердца и фибрилляции желудочков. В целом, достаточно быстро ликвидируемая острая венозная гиперемия оставляет незначительные последствия.
Хроническую общую венозную гиперемию отмечают значительно чаще, чем острую. Она является следствием ХСН, осложняет многие длительно текущие заболевания, которые приводят к прогрессирующему нарушению функции сердца: врожденные и приобретенные пороки сердца, кардиомиопатия, хроническая алкогольная интоксикация, миокардиты различного генеза, амилоидоз сердца, некоторые системные поражения соединительной ткани, перикардит, хронические воспалительные процессы в легких диффузного характера и др.
Основными причинами нарушения насосной функции сердца являются:
- Слабость поврежденного миокарда, сокращения которого не могут обеспечить уровня внутрижелудочкового давления, необходимого для перемещения притекающей к сердцу крови в артерии большого и малого кругов кровообращения.
- Декомпенсированные пороки сердца, когда энергия сердечного сокращения оказывается недостаточной для продвижения требуемой массы крови через суженные сердечные отверстия либо когда неполное смыкание сердечных клапанов приводит к возврату части крови в ретроградно расположенную камеру сердца и венозные сосуды.
- Ограничение емкости полостей сердца вследствие массивного субэндокардиального тромбоза (эндокардит Леффлера), фиброэластоза эндокарда, резкого утолщения стенки желудочков и межжелудочковой перегородки с субаортальным стенозом (ГКМП).
- Невозможность достаточного расширения полостей сердца во время диастолы из-за скопления значительного объема жидкости в полости перикарда или ее облитерации и петрификации.
- Уменьшение ОЦК и, как следствие, недостаточный возврат крови к сердцу.
Длительный венозный застой поддерживает состояние тканевой гипоксии, которая вместе с гемодинамическими факторами нарушает проницаемость гистогематического барьера и приводит к повторяющимся плазмо- и геморрагиям, стойкому отеку, дистрофии и постоянно возникающим микронекрозам. Вследствие этого хроническая венозная гиперемия ведет к атрофии паренхиматозных органов и прогрессирующим склеротическим изменениям. Склерозирование ткани обусловлено стимулирующим влиянием хронической гипоксии на пластические процессы в соединительной ткани. Новообразование гликозаминогликанов и коллагеновых волокон приводит к постепенному наращиванию массы и огрубению стромы органов, вытесняющей паренхиматозные структуры, которые атрофируются в условиях гипоксии. В фокусах микронекроза происходит формирование звездчатых рубчиков. Вследствие прогрессирующего характера таких диффузных и мелкоочаговых склеротических изменений развивается цианотическая индурация (застойное уплотнение) органов и тканей. Органы становятся плотными, увеличиваются в размерах, приобретают характерный синюшно-темно-красный цвет.
Венозный застой более выражен в нижних частях тела, чаще всего в нижних конечностях, а при длительном соблюдении постельного режима — в нижних отделах легких. Это явление называют гипостазом. Гипостаз в нижних конечностях связан также с расширением просвета вен и недостаточностью их клапанов. Длительно удерживающаяся венозная гиперемия сопровождается гипертрофией мышечной оболочки вен.
Переполненные кровью неравномерно расширенные вены часто становятся извилистыми и обретают узловатые выпячивания — варикозные узлы. В коже выявляют цианоз, отек дермы и подкожной клетчатки, дополняющиеся разрастанием соединительной ткани, атрофическими изменениями дермы и эпидермиса.
В печени хроническая венозная гиперемия сопровождается сложной структурно-функциональной перестройкой гистоархитектоники органа. Наряду с увеличением размеров печени, закруглением краев и напряжением капсулы отмечается уплотнение ткани, приобретающей на разрезе рисунок, напоминающий мускатный орех вследствие чередования мелких зон темно-красного и серовато-желтого цвета. Темно-красные участки соответствуют срединным зонам печеночных долек, где на месте центральных вен, прилежащих к ним отделов синусоид и лежащих между ними печеночных клеток образуются кровяные озера, располагаются элементы пролиферирующей соединительной ткани.
На периферии печеночных долек цитоплазма гепатоцитов приобретает зернистый вид и содержит множество липидных капелек, придающих ткани желтоватый оттенок. Такая неравномерность окраски ткани объясняется особенностями кровоснабжения органа и строением печеночных долек. В печень поступает кровь из 2 источников: воротной вены и печеночной артерии. Оба сосуда, последовательно делясь, в конечном итоге разветвляются на капилляры, которые, внедряясь между балками из паренхиматозных клеток, проникают в печеночные дольки. Здесь капилляры, приносящие артериальную и венозную кровь, сливаются, образуя синусоидальные капилляры портальной системы, которые впадают в центральную вену печеночной дольки. Центральная вена является дренажной системой, несущей кровь в нижнюю полую вену.
Внутридольковые кровеносные капилляры — синусоиды — на всем своем протяжении выстланы уплощенными эндотелиоцитами с незамкнутыми межклеточными щелями и лишены базальной мембраны. Обращенная к ним поверхность гепатоцитов образует множество микроворсинок, увеличивающих всасывающую поверхность клеток. Особенности строения стенок синусоидов и обращенной к ним плазмолеммы гепатоцитов обеспечивает свободный обмен между печеночной паренхимой и кровью. Гепатоциты и стенку микрососуда разделяет щель — пространство Диссе, которое заполнено плазмой и в которой располагаются звездчатые ретикулоэндотелиоциты. Повышение давления в венозной системе ретроградно распространяется от магистральных сосудов на печеночные, собирательные и центральные вены, а затем и на синусоиды, вызывая их расширение. Однако последние дилатируются только в центральных и срединных отделах дольки, так как ближе к ее периферии возросшее венозное давление уравновешивается артериальной кровью, поступающей в синусоиды из капилляров печеночной артерии.
Расширение микрососудов в центре печеночной дольки сопровождается кровоизлияниями, тогда как в зоне слияния синусоидов и артериальных капилляров отмечают только отек. В этих условиях происходит активирование адвентициальных фибробластов центральных и собирательных вен, пролиферация клеток синусоидов, приобретающих способность активно продуцировать склеропротеины. Застой крови при хронической венозной гиперемии нарушает циркуляцию в лимфатической системе, в сосудах и капиллярах которой скапливается богатая белками и метаболитами лимфа. Затруднение лимфооттока и, следовательно, освобождения внутритканевой среды от избытка жидкости, содержащей продукты тканевого обмена, потенцирует гипоксию и способствует новообразованию соединительной ткани в печени и других органах.
По мере прогрессирования процесса в печени разрастается соединительная ткань, что ведет к склерозу центральных и собирательных вен, портальных трактов, образованию сплошной базальной мембраны у синусоидов, узелковой гиперплазии сохранившихся печеночных клеток. Такая глубокая перестройка сосудистого русла и стромально-паренхиматозных соотношений, сопровождающаяся деформацией органа, обозначается как мускатный (сердечный) цирроз печени.
В легких хроническая венозная гиперемия приводит к так называемой бурой индурации. Легкие увеличиваюттся в размере, их ткань уплотняется, приобретая на поверхности разреза буроватую окраску. Усиливается рисунок междольковых и межсегментарных прослоек фиброзной стромы, не содержащей капилляры, что связано со склерозом, обусловленным застоем лимфы. Склерозированные межсегментарные прослойки определяются на рентгенограмме (линии Керли). В терминальной стадии процесса, когда развивается интерстициальный и альвеолярный отек, с поверхности разреза легочной ткани обильно стекает пенистая жидкость, смешанная с темной венозной кровью. Бурая окраска легких связана с явлениями гемосидероза — накоплением буро-коричневого пигмента гемосидерина, образующегося в макрофагах при множественных диапедезных кровоизлияниях, а уплотнение ткани представляет собой следствие прогрессирования фиброза легочной ткани.
Бурая индурация легких отмечается при митральном пороке сердца, особенно при сужении левого венозного отверстия, а также при обширном кардиосклерозе. Ей предшествует ряд компенсаторно-приспособительных реакций со стороны артериального и венозного русла легких, направленных на предупреждение переполнения кровью капилляров и легочных альвеол и нарушения функции аэрогематического барьера. На начальных этапах процесса это достигается сокращением сфинктеров легочных вен, предупреждающим обратный заброс крови из правого предсердия в легочные вены, а также заполнением депо большого круга кровообращения, что уменьшает массу крови, притекающей к ПЖ сердца. Затем возникает спазмирование мелких легочных вен. Параллельно через барорецепторы растянутых кровью устьев легочных вен и левого предсердия передается импульс сокращения на артериолы и мелкие ветви ЛА (веноартериальный рефлекс Ф.Я. Китаева). Адаптивная веноартериальная реакция ведет к гипертрофии мышечной оболочки легочных вен и ЛА, которые перестраиваются по типу замыкающих артерий.
Со временем гипертрофия гладкомышечных элементов стенки сокращенных сосудов дополняется явлениями ангиосклероза. Миоэластофиброз снижает реактивность артерий, артериол и мелких вен, ограничивая их способность предохранять капилляры легкого от резкого переполнения кровью. Наступает декомпенсация легочного кровообращения, и капилляры межальвеолярных перегородок переполняются кровью. Стенки растянутых капилляров выбухают в альвеолы, их проницаемость повышается. Плазма и эритроциты выходят в просветы альвеол и в межальвеолярный интерстиций, эритроциты разрушаются и поглощаются макрофагами, превращающими гемоглобин в гемосидерин, который после распада макрофагов оказывается свободно лежащим в ткани.
Белки плазмы крови, продукты клеточного распада, гемосидерин поступают в лимфатическое русло легких, нарушая его дренажную функцию. В результате к расстройствам кровообращения постепенно присоединяется лимфостаз и лимфатический отек межуточной ткани легких. Нарастающая гипоксия ткани активирует фиброциты, которые в большом количестве присутствуют в межальвеолярных перегородках. Фиброциты трансформируются в фибробласты, длинные уплощенные отростки которых проникают в заполненные отечной жидкостью щели между кровеносными капиллярами и стенкой альвеолы. Около тел отростков фибробластов из секретируемых ими склеропротеинов формируются коллагеновые волокна. Склерозирование стенок кровеносных сосудов, капилляров и межальвеолярных перегородок вместе с отеком еще более ухудшает условия газообмена. В зависимости от степени и длительности венозного застоя явления склероза и гемосидероза в легких приобретают генерализованный характер. Скопления свободно лежащих пигментов гемосидерина и ферритина и нагруженных ими сидерофагов выявляют не только в межальвеолярных перегородках и альвеолах, но и по ходу лимфатических сосудов и в лимфоузлах. Сидерофаги, появляющиеся в мокроте, обозначают как «клетки сердечного порока».
Бурая индурация легких приводит к прогрессирующему ухудшению гемодинамики в малом круге, усугубляя течение основного заболевания.
Почки при хроническом общем венозном застое не только увеличиваются в размерах, но и уплотняются, становятся цианотичными. Нарушение кровообращения в большом круге приводит к спазму почечных артериол, что ограничивает клубочковую фильтрацию. В результате способность почек экскретировать натрий и воду снижается, а объем плазмы крови и тканевой жидкости в организме увеличивается, что еще более ухудшает кровообращение и тканевый обмен. На фоне застойного полнокровия и расширения почечных вен развивается лимфостаз и отек стромы. Ее набухание наиболее выражено в мозговом слое, где основное вещество межуточной ткани особенно богато гликозаминогликанами. Клубочки также несколько увеличены и полнокровны. Внеклеточные пространства в области тубулярных элементов нефрона расширены вследствие отека и вторичного нарушения реабсорбции жидкости. В условиях нарастающей гипоксии возникает дистрофия эпителия канальцев. На этой стадии процесса возможно огрубение базальной мембраны и их склероз, приводящие к альбуминурии.
Индурация селезенки при венозной гиперемии связана с застоем крови, отеком и склерозированием стромы, а также с относительной неподатливостью фиброзной капсулы органа. При хронической венозной гиперемии капсула селезенки напряжена, утолщена, пульпа ее уплотнена, поверхность среза ткани темно-коричнево-красного цвета. Микроскопически выявляют расширение щелей пульпы и синусов, обусловленное полнокровием, увеличением массы и огрубением волокнистых структур стромы.
В других органах (ЦНС, ЖКТ, эндокринные железы) венозное полнокровие индуцирует описанный выше стереотипный комплекс изменений: переполнение вен, цианоз, нарушение лимфооттока и отек ткани, плазмо- и геморрагии, дистрофические изменения и повреждение клеток паренхимы, прогрессирующее склерозирование стромы.
Местное венозное полнокровие возникает при затруднении оттока крови от какой-либо части тела или органа. К наиболее частым причинам местного полнокровия относятся сужение и обтурация просвета вены при тромбозе, хроническом воспалительном процессе в стенке сосуда, сдавливании вены извне разрастающейся опухолевой или рубцовой тканью, а также наложение повязки, гипса, лигатуры. Как и при общем венозном застое, вены дистальнее препятствия расширены, застойные участки тела синюшны, их температура понижена. В основе патологических изменений, развивающихся непосредственно в области регионального венозного застоя, лежат принципиально те же закономерности и механизмы, которые описаны выше. Так, мускатная печень и ее цирроз могут быть следствием облитерирующего тромбофлебита печеночных вен (синдром Бадда — Киари), а цианотическая индурация почек или селезенки — тромбоза основной магистральной вены соответствующего органа. Иногда местное венозное полнокровие возникает вследствие формирования венозных коллатералей, что требует значительного времени и возможно только при постепенном ограничении оттока крови по вене. В тех ситуациях, когда блокада венозного ствола развивается быстро, возможно подключение только предсуществовавших путей оттока крови.
Основными последствиями венозной гиперемии является развитие циркуляторной гипоксии, накопление в интерстиции белков и плазмы, продуктов нарушенного обмена и деструкции тканевых структур, которые активируют процессы фибриллогенеза и приводят к развитию органосклероза. Вторичные осложнения, связанные с общей и местной венозной гиперемией, — снижение функциональных возможностей органов, предрасположенность к хроническим воспалительным процессам. При компенсаторном формировании коллатералей переполненные кровью вены резко расширяются, стенка их истончается. В условиях хронического течения процесса сочетание атрофии и гипертрофии гладкомышечных элементов венозной стенки в совокупности с перестройкой ее соединительнотканного каркаса приводит к недостаточности венозных клапанов, появлению варикозных узлов, а иногда и к опасным кровотечениям. В нижних конечностях, в венах таза подобные изменения способствуют тромбообразованию (флеботромбоз) и воспалению (флебит), которые часто сочетаются (тромбофлебит). Примером компенсаторного венозного полнокровия являются коллатерали, образующиеся при застое крови в воротной вене вследствие цирроза печени.
Ишемия
Ишемией называют патологическое состояние, обусловленное ограничением притока артериальной крови. Несмотря на многообразие непосредственных причин, вызывающих ишемию, принято различать следующие ее виды: ангиоспастическую, компрессионную, связанную с перераспределением крови.
Ангиоспастическая (рефлекторная) ишемия обусловливается возбуждением вазомоторных центров и сосудосуживающих нервов, а также действием гуморальных факторов, циркулирующих в крови или образующихся в сосудистой стенке. Большое значение в развитии ангиоспастической ишемии имеет состояние сосудистого эндотелия, который продуцирует факторы как констрикции, так и расслабления сосудистой стенки, а повреждение эндотелиального монослоя извращает сосудистую реакцию на гуморальные воздействия. Неадекватное кровоснабжение ткани может быть также связано с нарушением расслабления гладких мышц артерий, что ведет к их длительной констрикции.
Обтурационная ишемия — следствие сужения или закрытия просвета артерии при воспалительном процессе (обтурирующий эндартериит), эмболизации, образовании массивной атеросклеротической бляшки, тромба. В механизмах обтурационной ишемии способствующим моментом является увеличение вязкости крови вследствие усиленной агрегации ее форменных элементов или повышения свертываемости.
Компрессионная ишемия развивается при рубцевании ткани, сдавлении артерии жгутом, лигатурой, скоплением жидкости, разрастающейся опухолью.
Ишемия в результате перераспределения крови возникает под влиянием факторов, которые вызывают гиперемию в других участках тела. Такова, например, причина сонливости после приема пищи или обморочного состояния вследствие быстрого извлечения значительного объема жидкости или массивной опухоли из брюшной полости. В этих случаях к ранее сдавленным тканям и органам устремляется большая масса крови с одновременным уменьшением кровотока в сосудах головного мозга, кожи и других областей тела.
Морфологические изменения в органах при ишемии возникают вследствие нарушения микроциркуляции и развития гипоксии. Макроскопически органы уменьшены в размерах, гипотермичны, дряблые, со сморщенной капсулой. Ткань на разрезе в зоне ишемии тусклая, более светлая, чем участки, получающие артериальную кровь.
В зависимости от интенсивности ишемизации ткани, ее чувствительности к гипоксии, длительности последней изменения могут развиваться либо на уровне микроструктур, либо в виде грубых повреждений вплоть до формирования инфаркта. Макроскопически выявляют компенсаторную дилатацию микрососудов, нарушение кровотока в них, повышение сосудистой проницаемости, признаки расстройства обменных процессов в паренхиматозных клетках, деструктивные изменения различных элементов ткани. В строме органов отмечают отек, накопление и перераспределение гликозаминогликанов, набухание, гомогенизацию и разрушение аргирофильных и коллагеновых волокон.
Ишемия вследствие спазма артерий обычно непродолжительна и ликвидируется без существенных последствий. В тех случаях, когда ангиоспазм становится затяжным, а тканевой метаболизм характеризуется высокой потребностью в кислороде (как, например, в сердечной мышце или головном мозгу), развивается инфаркт.
Наиболее часто к тяжелым последствиям приводит остро наступающая обтурационная ишемия. Ее последствия зависят и от регионарных особенностей ткани. Если соответствующая область богато васкуляризирована и ее кровоснабжение осуществляется не одним магистральным сосудом, а несколькими артериальными ветвями, то предпосылки для развития ишемии уменьшаются. Кроме того, чем быстрее развивается ишемия, тем меньше возможностей для реализации компенсаторных механизмов, в частности открытие уже имеющихся коллатералей и формирование новых. При постепенном уменьшении проходимости сосуда возможности для восстановления кровоснабжения ткани по коллатералям существенно возрастают и последствия такой ишемии могут быть незначительны. Длительно сохраняющаяся ишемия в выраженности, не приводящей к необратимым некротическим изменениям ткани, постепенно ведет к атрофии паренхимы и склерозу.
Инфаркт
Инфаркт — очаг некроза, развившегося вследствие нарушения кровообращения. Инфаркт называют также циркуляторным, или ангиогенным некрозом. Термин «инфаркт» (от лат. нафаршировать) был предложен Вирховым для формы некроза, при которой омертвевший участок ткани пропитывается кровью. Размеры и морфологические особенности инфаркта определяются калибром обтурированного сосуда, наличием других нарушений кровообращения, на фоне которых он развивается. При магистральном типе ветвления артерии инфаркт по своим очертаниям напоминает конус, узкая часть которого (вершина) обращена к воротам органа, а основание ориентировано на периферию, к зоне терминального разветвления внутриорганных артерий. Инфаркты такой формы обычно выявляют в селезенке, почке, легких. В органах с преобладанием рассеянного типа ветвления артерии, например в головном мозгу, кишечнике, сердце, кровоснабжаемая ею территория не образует конусовидных контуров и инфаркты не имеют определенной формы.
Виды инфаркта
Зона инфаркта может занимать весь орган или большую его часть (тотальный и субтотальный инфаркт) или выявляться только под микроскопом (микроинфаркт). По макроскопическим признакам различают 3 вида инфаркта: белый, белый с геморрагическим венчиком и красный.
Белый (ишемический) инфаркт формируется при непроходимости магистрального артериального ствола и запустевании всего сосудистого русла в его бассейне вследствие недостаточного развития сосудистых анастомозов и коллатералей. Чаще всего выявляют в селезенке, иногда в головном мозгу, печени. Зона некроза хорошо видна при макроскопическом обследовании примерно через 24 ч после нарушения кровоснабжения. Под микроскопом ткань уплотнена, бледно-желтой окраски, структура ткани неразличима, а образующие ее элементы сливаются в гомогенную массу. По периферии зона инфаркта отграничивается воспалительным демаркационным валом.
Белый инфаркт с геморрагическим венчиком выглядит как участок белесовато-желтого цвета, окруженный темно-красной зоной кровоизлияний. Такой инфаркт развивается в случаях, когда компенсаторному включению коллатералей и реактивной артериальной гиперемии сосудов периферической зоны предшествует ангиоспазм, сменяющийся паралитическим расширением. В результате резкое полнокровие сосудов сопровождается явлениями стаза крови и диапедезными кровоизлияниями в некротизированную ткань. Белый инфаркт с геморрагическим венчиком развивается в сердце, селезенке, иногда в почках.
Красный (геморрагический) инфаркт обычно выявляют в легких, что связано с особенностями их кровоснабжения. Иногда геморрагический инфаркт возникает на фоне выраженной гиперемии и в других органах: кишечнике, головном мозгу, почках. При красном инфаркте зона ишемии пропитывается кровью, приобретает темно-красный цвет и четкие границы. Этот эффект возникает, если вслед за закупоркой артерии периферические сосуды омертвевшей ткани переполняются кровью, поступившей по коллатералям. При венозном застое ретроградное поступление крови из вен в зону ишемии также ведет к пропитыванию некротизированной ткани кровью.
Геморрагический инфаркт может развиваться также в результате выраженного венозного застоя при быстром прекращении оттока крови по крупным венозным стволам или одновременном выключении из кровотока большого количества мелких вен. Венозные застойные инфаркты выявляют в селезенке при тромбозе вены, отводящей от нее кровь, в головном мозгу — при нарушении проходимости синусов твердой мозговой оболочки или яремных вен, в сердце — при обтурации венечного синуса тромботическими массами, в тканях нижних конечностей — при перевязке бедренной вены. Микроскопически в очаге геморрагического инфаркта отмечают массы гемолизированных эритроцитов, инфильтрирующих некротизированную ткань.
Общие закономерности формирования и заживления инфаркта
Стадия ишемии и некроза. Развитию инфаркта предшествует ишемия. Первые сдвиги, обусловленные нарушением кровоснабжения, определяются угнетением тканевого дыхания, компенсаторной активацией анаэробного гликолиза, быстрым накоплением метаболитов в клетках в токсических концентрациях. Недостаточное воспроизводство энергии и гистотоксический эффект ишемии нарушают электролитный гомеостаз клеток и подавляют пластические процессы, что приводит к прогрессирующей диссоциации цитомембран, закислению внутриклеточной среды, денатурации белков, гибели и разрушению клеток.
Электронно-микроскопически при ишемизации выявляют внутриклеточный отек или, напротив, дегидратацию цитоплазматического матрикса. Органеллы клеток набухают, их мембраны подвергаются гомогенизации и фрагментации, гранулы лабильного гликогена исчезают, отмечают накопление липидов в виде капель вследствие их высвобождения из диссоциирующих фосфолипидов цитомембран и нарушения липидного обмена. В лизосомах накапливаются продукты внутриклеточного распада. Происходят перераспределение, конденсация либо вымывание ядерного хроматина и разрушение ядрышек, расплавление цитоплазматических рибосом и органелл немембранной структуры.
Гистохимически и биохимически в ишемизированной ткани определяется снижение уровня макроэргических фосфатов, активности окислительно-восстановительных ферментов, накопление недоокисленных метаболитов, нарушение обмена электролитов, уменьшение содержания гликогена, РНК и ДНК, а со временем — накопление продуктов распада стромальных структур. На некротической стадии инфаркта при микроскопическом исследовании ядра клеток не окрашиваются, все структурные элементы ткани сливаются в однородную массу.
Стадия репаративных изменений наступает вслед за формированием некроза. По периферии инфаркта всегда существует зона дистрофических изменений и реактивного воспаления — так называемый демаркационный вал. Микроскопически воспалительная реакция отмечается уже через несколько часов, а максимум ее развития приходится на 3-и–5-е сутки. Воспаление в зоне демаркационного вала сопровождается выходом форменных элементов крови из капилляров. Некротические массы постепенно частично расплавляются под действием протеолитических ферментов, выходящих из нейтрофильных лейкоцитов, частично подвергаются фагоцитозу или резорбируются лимфатической сетью и выводятся по ее сосудам.
Организация зоны некроза — замещение некротических масс соединительной тканью, которая врастает со стороны демаркационного вала и к 7–10-м суткам трансформируется в грануляционную (юную) соединительную ткань, а со временем созревает в рубцовую.
Особенности развития инфаркта в различных органах
Морфология инфаркта во многом зависит от органной архитектоники сосудистой системы. В клинической практике наиболее часто отмечают инфаркт сердца (миокарда), головного мозга, кишечника, легких, почек и селезенки. Время, необходимое для развития инфаркта в различных органах, неодинаково и зависит от функциональных энергозатрат и филогенетически сложившегося метаболизма, что определяет потребность ткани в обеспечении кислородом.
Для развития ИМ достаточно полного прекращения его кровоснабжения на 20–25 мин, однако ишемия длительностью 5 мин уже ведет к гибели отдельных мышечных клеток. В реальной жизни формирование инфаркта сердечной мышцы требует несколько большего промежутка времени, так как в зоне ишемии всегда частично сохраняется кровоток по сосудистым анастомозам и коллатералям. Он недостаточен для того, чтобы полностью предотвратить некроз, но несколько увеличивает срок его развития и ограничивает размеры.
Инфаркт обычно локализуется в ЛЖ, чаще всего в передней стенке. По типу это белый инфаркт с геморрагическим венчиком, имеющий неправильную форму. В зависимости от объема и локализации пораженной ткани миокарда различают мелко- и крупноочаговый, субэпикардиальный, интрамуральный, субэндокардиальный и трансмуральный ИМ, охватывающий все слои сердечной стенки. В зоне перехода инфаркта на эпикард или эндокард развивается реактивное воспаление, в первом случае приводящее к фиброзному перикардиту (выпот в полость перикарда плазмы крови, обогащенной фибрином, и образование фибринозных наслоений на эпикарде), во втором — к тромбоэндокардиту (пристеночный тромбоз соответственно зоне инфаркта).
Формирование ИМ начинается с ишемической стадии. Наряду с прогрессирующим нарушением метаболизма и дезинтеграцией клеточных мембран отмечают фрагментацию, растяжение и дезинтеграцию миофибрилл кардиомиоцитов. В результате снижается активность внутриклеточных энзимов, изменяется характер окрашивания клеток при использовании основных или кислотных гистологических красителей, нарушается способность клеток к лучепреломлению в поляризованном свете и люминесцентно-микроскопические свойства. Эти явления используются для ранней диагностики метаболических и ишемических повреждений сердца.
Гистологические признаки гибели клеток — сморщивание, набухание и разрушение клеточного ядра, исчезновение продольной и поперечной исчерченности, гомогенизирование саркоплазмы выявляют через 12 ч (рис. 2.1).
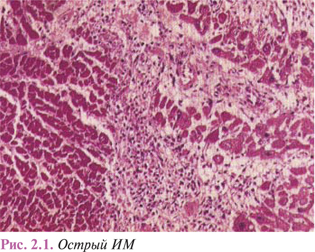
Параллельно с деструктивными изменениями рабочих клеток миокарда происходит сосудисто-тканевая реакция, характеризующаяся спазмированием и паретической дилатацией интрамуральных артерий и артериол, плазматическим пропитыванием и повышением проницаемости их стенок, а также нарушением микроциркуляции с внутрисосудистой агрегацией эритроцитов, отеком интерстиция.
При развитии некроза кровоток в некротической зоне прекращается, а в периинфарктной увеличивается. Наряду с диапедезными кровоизлияниями в ней происходит экстравазация лейкоцитов и формируется лейкоцитарный вал. В толще некротической зоны вокруг сохранившихся сосудов иногда выявляют островки жизнеспособной ткани, по периферии которых отмечают такие же явления, как и в окружающей инфаркт зоне.
В течение первых 18–24 ч от начала патологического процесса миокард в бассейне пораженной артерии отличается бледностью на фоне подчеркнуто неравномерного кровенаполнения остальной ткани. В конце 1-х суток участок некроза становится различимым макроскопически. В связи с непрерывной деятельностью сердца, высокой активностью ферментов, выделяющихся из лейкоцитов, на 3-и–5-е сутки начинается размягчение (миомаляция) погибшей ткани. Постепенное рассасывание (резорбция) некротизированной массы осуществляется при активном участии микрофагальных клеток, которые появляются на 4-й день кнаружи от лейкоцитарного вала.
Фибропластическая реакция интерстиция также возникает на 4–5-е сутки, а первые волокнистые элементы новообразованной соединительной ткани в зоне инфаркта появляются еще через 3 сут. В течение последующей недели зона некроза представлена распадающимися мышечными волокнами, пропитана отечной жидкостью и инфильтрирована распадающимися лейкоцитами. По ее периферии и вокруг периваскулярных островков сохранившегося миокарда происходит новообразование соединительной ткани.
Процесс организации продолжается 2–2,5 мес. В дальнейшем соединительная ткань, образовавшаяся на месте некротических масс, уплотняется, ее сосуды запустевают и облитерируются, на месте некроза образуется рубец (рис. 2.2).
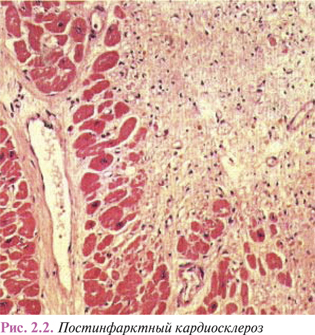
Проводящая система сердца более устойчива к гипоксии по сравнению с рабочим миокардом и способна дольше сохраняться в зонах ишемии, что важно для восстановления ритмичной работы сердца после экстренной инвазивной антиишемической терапии.
В почках обычно развивается белый инфаркт с геморрагическим венчиком. Вследствие хорошего развития сосудистых анастомозов и коллатералей инфаркт возникает только при нарушениях проходимости сосудов большего калибра, чем дольковая артерия. Характерное расположение инфаркта — передняя латеральная поверхность органа, так как в этой зоне почечные артерии ветвятся не по магистральному, а по рассеянному типу, при котором межсосудистые коллатерали выражены значительно слабее. Обычно инфаркт почки напоминает по форме конус, обращенный основанием к капсуле, верхушкой к почечной лоханке. Однако иногда процесс ограничивается только корой, не затрагивая пирамидки, и поражение приближается по форме к квадрату.
Инфаркт почки часто сопровождается гематурией вследствие попадания крови в мочевые канальцы при разрыве мелких сосудов. Ишемическая стадия инфаркта почек развивается по общим закономерностям. Некроз всех структур почечной паренхимы наступает через 24 ч, однако повреждение эпителия почечных канальцев возникает значительно раньше. Так, уже через 6 ч отмечается гибель эпителия извитых, а через 12 ч — прямых канальцев нефрона. К этому же времени по периферии инфаркта развивается реактивное воспаление, достигающее своего максимума примерно к 3-му дню процесса.
Формирование демаркационной зоны сопровождается нарушениями кровотока в микрососудах, явлениями отека, плазморрагиями и диапедезными кровоизлияниями, активной миграцией лейкоцитов. Это приводит спустя сутки к образованию периферической геморрагической зоны инфаркта и лейкоцитарного вала. Примерно с этого же времени появляются макрофаги и начинается процесс резорбции некротических масс. На 7-е сутки деструктивно-резорбтивные процессы сочетаются с отчетливо выраженными явлениями организации, которая через несколько недель завершается образованием плотного соединительнотканного рубца, реже — кисты.
В селезенке обычный морфологический тип инфаркта — белый (ишемический). В условиях выраженного венозного застоя инфаркт селезенки может быть геморрагическим, приобретающим в течение нескольких суток серую или белую окраску. Ишемический инфаркт селезенки конической формы, бледно-желтой окраски. На поверхности капсулы органа в области широкой части этого конуса, а также на границе зоны инфаркта развиваются реактивное воспаление, процессы лизиса, резорбции и организации некротических масс. Непосредственно в зоне некроза вначале разрушается красная пульпа, затем фолликулы и трабекулы. Организация инфаркта осуществляется по общим закономерностям. Созревание постинфарктного рубца сопровождается деформированием селезенки.
Инфаркт головного мозга в 85–90% случаев является белым, в остальных — красным или смешанного характера. Белый инфаркт может поражать любые отделы мозга. Первоначально это нечетко отграниченный участок дряблой или крошащейся консистенции, красновато-серого цвета, со стертым естественным рисунком базальных узлов или коры головного мозга. Геморрагические инфаркты в виде небольших очагов красного цвета локализуются преимущественно в пределах скоплений серого вещества, чаще всего в коре. Смешанные инфаркты состоят из белых и красных участков, причем последние располагаются в сером веществе.
Топография различных морфологических типов инфарктов мозга предопределена особенностями кровоснабжения его различных областей. Наиболее часто они возникают в бассейне средней мозговой артерии, реже — позвоночных и базиллярных артерий. Геморрагические инфаркты формируются в хорошо васкуляризированных зонах — скоплениях серого вещества или в коре головного мозга.
Развитие инфаркта головного мозга включает ишемическую и некротическую стадии. Ишемическая стадия характеризуется дистрофическими изменениями нервной ткани, кровоизлияниями и деструкцией клеточных мембран с необратимой дезорганизацией обменных процессов и электролитного гомеостаза нервных клеток. При микроскопическом исследовании отмечают лизис глыбок базофильного вещества, просветление цитоплазмы, гиперхроматоз и деформацию ядра. В результате нервные клетки и их ядра приобретают угловатую форму, а цитоплазма гомогенизируется, утрачивает базофильные включения и просветляется. Нарушение циркуляции крови в микрососудах сочетается с перицеллюлярным отеком — появлением светлого промежутка между капиллярной стенкой или телом нейрона и окружающей тканью. Вокруг капилляров отмечают отек и набухание отростков окружающих их глиальных клеток.
Некротическая стадия инфаркта — стадия нарастающего аутолиза ишемизированной ткани мозга. Гибели нейронов предшествует их резкое просветление либо уплотнение и превращение в пикноморфные (уплотненные дегидратированные) клетки, а затем и в гомогенную бесструктурную массу. Вместе с нейроцитами в деструктивные изменения вовлекаются и клетки глии. Из мелких сосудов происходят диапедезные кровоизлияния, небольшие и единичные в очагах белого инфаркта, множественные и сливающиеся между собой при геморрагическом инфаркте.
К началу 2-х суток начинается резорбция некротизированной нервной ткани. На границе с очагом ишемического поражения скапливаются лейкоциты. Вместе с ними в зону некроза внедряются многочисленные активированные астроциты и появляются зернистые шары с липидными включениями. Часть астроцитов утрачивает цитоплазматические отростки, в их цитоплазме выявляют многочисленные фибриллы, приобретающие способность к образованию волокнистых структур. Вокруг очага некроза начинается новообразование сосудов, капилляров и сосудистых петель.
В организации некротических масс участвуют как глиальные, так и соединительнотканные клетки — фибробласты. Однако в конечной стадии процесса при небольших размерах инфаркта продукты мезодермальной пролиферации полностью вытесняются глиоволокнистыми структурами, образующими рубец. В крупных очагах срединная зона организовавшегося инфаркта остается соединительнотканной, а в центре сформировавшегося рубца образуется одна или несколько полостей, снаружи окруженных разрастаниями глии.
Инфаркт легких, как правило, имеет геморрагический характер, причиной чего являются двойное кровоснабжение легких и венозный застой. Кровь попадает в легкие как по бронхиальным артериям, входящим в систему большого круга кровообращения, так и по артериям малого круга кровообращения. Между ЛА и бронхиальными артериями существуют многочисленные анастомозы, которые имеют строение артерий замыкающего типа и в обычных условиях не функционируют. При обтурации достаточно крупной ветви ЛА в ее бассейн под большим давлением устремляется кровь из бронхиальных артерий по рефлекторно открывшимся анастомозам. Переполняющиеся кровью легочные капилляры резко дилатируются, их стенки разрываются, кровь изливается в интерстиций альвеолярных перегородок и в полости альвеол, имбибируя соответствующий участок ткани. Благодаря автономному артериальному кровоснабжению бронхи в зоне инфаркта сохраняют жизнеспособность. Нередко геморрагический инфаркт в легком развивается на фоне хронической венозной гиперемии, так как повышение давления в крупных венах способствует ретроградному поступлению крови в зону инфаркта.
Инфаркт чаще всего развивается в периферических зонах средних и мелких отделов легких. При этом макроскопически выявляют очаги более плотной консистенции, чем окружающая ткань, конусовидной формы, основанием обращенные к плевре, которая покрывается фибринозным налетом и гиперемируется вследствие реактивного воспаления. На разрезе некротизированная ткань темно-красного цвета, слегка зернистая, выбухает над поверхностью.
В 1-е сутки в зоне инфаркта микроскопически определяются отек и кровоизлияния в виде скоплений частично гемолизированных эритроцитов в интерстициальной ткани, в просветах альвеол и мелких бронхов, что сопровождается кровохарканьем. Затем присоединяются признаки некроза стенок альвеол, накапливаются сидерофаги. На 3-и–4-е сутки инфаркт представляет собой гомогенизированную массу из разрушившихся эритроцитов, на фоне которых видны следы некротизированных альвеолярных перегородок. Расплавление некротизированной ткани и излившейся крови, их резорбция и организация начинаются с периферии и из сохранившихся периваскулярных и перибронхиальных зон. Через 2–8 мес на месте инфаркта остается рубец или киста.
Белый инфаркт в легком выявляют редко. Возникает при нарушении кровотока в бронхиальных артериях на фоне затруднения капиллярного кровотока, например вследствие сдавления внутриальвеолярным экссудатом или при уплотнении (гепатизации) легочной ткани, обусловленного пневмонией.
В кишечнике инфаркт развивается по типу геморрагического. Наиболее характерная локализация — бассейн верхней брыжеечной артерии, которая в связи с большой протяженностью чаще подвергается обструкции. Макроскопически инфаркт кишки имеет вид темно-красного участка, который достаточно ясно отграничен от непораженного кишечника. Серозная оболочка в области инфаркта кишки становится тусклой, на ней появляются фибринозные наложения. Стенка кишки утолщена, слизистая оболочка синюшная.
Некротические и реактивные изменения в ишемизированном сегменте кишечника развиваются быстро. Через 15–20 мин после прекращения кровоснабжения в его стенке выявляют выраженные микроциркуляторные нарушения: тотальный отек ткани, замедление и прекращение движения крови в резко полнокровных капиллярах и венулах, множественные кровоизлияния. Через 30 мин в отечной строме слизистой оболочки кишки появляются лейкоциты, лимфоциты, развивается макрофагальная реакция. В течение 1–1,5 ч стенка кишки подвергается некрозу, который начинается с изъязвления ее слизистой оболочки.
В сетчатке глаза инфаркт имеет характер белого, который в условиях венозного застоя трансформируется в геморрагический. Участок пораженной ткани в виде конуса обращен вершиной к зрительному диску, обычно локализуется в височном сегменте. Микроскопически выявляют деструкцию внутренних слоев сетчатки, ганглиозных клеток и нервных волокон на фоне нарушения микроциркуляции, отека и кровоизлияний. Очень редко отмечают инфаркты в печени, мышцах, костях.
Последствия инфаркта чрезвычайно существенны для организма. Так, поражение при ИМ >30% ткани ЛЖ сопровождается развитием ОСН с остановкой сердца. Повреждение проводящей системы сердца при формировании некроза влечет тяжелые нарушения ритма. При обширном трансмуральном инфаркте иногда происходит выбухание некротизированного участка сердечной стенки и его истончение — развивается острая аневризма сердца. В некоторых случаях десинхронизация процессов миомаляции, резорбции некротических масс и организации зоны инфаркта приводит к разрыву аневризмы, заполнению полости перикарда кровью с летальным исходом. В результате ИМ могут возникать разрывы межжелудочковой перегородки, отрыв папиллярных мышц, что также ведет к серьезным последствиям. В более отдаленные сроки обширная рубцовая зона, изменяя геометрию сокращения сердца и внутрисердечную гемодинамику, способствует развитию ХСН и общей венозной гиперемии.
Инфаркт головного мозга сопровождается его отеком, расстройством микроциркуляции и метаболическими нарушениями как в непосредственной близости от очага поражения, так и в отдаленных участках. Исход инфаркта определяется его размерами, локализацией и темпами развития патологического процесса. Смерть таких больных может быть обусловлена как самим очагом поражения в головном мозгу, так и причинами, непосредственно с ним не связанными. Нередко при медленном формировании инфаркта больные погибают не от деструктивных изменений, затрагивающих жизненно важные центры головного мозга, а вследствие СН, пневмонии и другой присоединившейся патологии, осложнившей течение инфаркта. Серьезным осложнением инфаркта головного мозга является кровоизлияние в размягченную ткань. Как и отек мозга, так и увеличение его объема вследствие восстановления кровотока по сосудам в зоне ишемии могут вызывать дислокацию и ущемление ствола мозга. При благоприятном исходе на месте инфаркта формируется рубец или киста с более или менее значительными нарушениями функции ЦНС.
Инфаркт кишечника требует обязательного хирургического вмешательства, так как конечной фазой его развития является гангрена с прободением кишечной стенки. Попадание содержимого кишечника в брюшную полость влечет за собой развитие перитонита. Причиной перитонита может стать также инфаркт селезенки, обычно заканчивающийся формированием грубого рубца, деформирующего орган.
Инфаркт легкого обычно не несет непосредственной угрозы жизни больному. Однако его течение может осложниться постинфарктной пневмонией, нагноением и распространением воспалительного процесса на плевру с развитием пневмоторакса и гангрены легкого. Одной из наиболее характерных причин нагноения инфаркта является попадание в сосуд гнойного эмбола. Это вызывает гнойное расплавление ткани легкого и образование абсцесса на месте инфаркта.
При инфаркте почек, обычно заживающем посредством рубцевания соответствующего участка, угрожающие жизни осложнения возникают при нагноении либо при обширных поражениях, особенно при симметричных некрозах коркового слоя, следствием которых может быть ОПН.
Стаз крови
Этиология и патогенез. Стаз (от лат. остановка) — местное прекращение тока крови в микроциркуляторном русле, главным образом в капиллярах, посткапиллярах и венулах. Полной остановке кровотока в микрососудах предшествует его резкое замедление, обозначаемое как предстаз. Механизмами стаза крови являются уменьшение разности давления между проксимальными и дистальными отделами микрососудов и повышение сопротивления в микрососудах. В зависимости от причин различают ишемический, застойный и истинный капиллярный стаз крови.
Уменьшение артериолярно-венулярного градиента давления крови приводит к замедлению кровотока в микроциркуляторном русле и развитию ишемического стаза. В основе уменьшения артериолярно-венулярного градиента давления могут лежать также венозная гиперемия и повышение давления в венулярном сегменте микрогемоциркуляторного русла. Истинный капиллярный стаз связан с первичным повышением сопротивления кровотоку в микрососудах вследствие нарушения реологических свойств крови и появления препятствий в их просветах.
Реологические свойства крови — вязкость и текучесть в микрососудах определяются главным образом агрегационной способностью ее форменных элементов, прежде всего эритроцитов, способных слипаться с образованием «монетных столбиков» и полиморфных конгломератов (рис. 2.3).
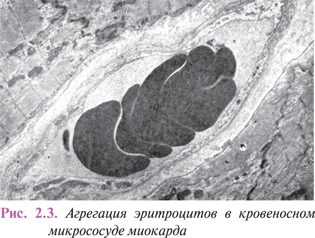
Агрегации эритроцитов способствуют факторы, повышающие проницаемость сосудистой стенки. Выход в ткань жидкости и мелкодисперсных альбуминов повышает концентрацию глобулинов и фибриногена в плазме крови, а также облегчает поступление в венулярные отделы продуктов нарушенного метаболизма и биологически активных веществ из тканей. Агрегация эритроцитов усиливается при снижении поверхностного потенциала их цитолеммы и при замедлении скорости движения крови в капиллярах.
Морфологическая картина. Гистологически стаз проявляется наличием расширенных микрососудов, выполненных однородной массой из слившихся форменных элементов крови. Однако на электронограммах эритроциты еще некоторое время сохраняют четкие контуры (см. рис. 2.3), а со временем границы между ними стираются вследствие гемолиза и свертывания крови. Это объясняет обратимость изменений при непродолжительной остановке крови в микрососудах, тогда как стойкий стаз приводит к образованию гиалиновых тромбов.
Исходы. Стаз крови — явление неспецифическое и возникает под воздействием причин общего и местного характера — при патологических процессах, сопровождающихся интоксикацией и расстройством кровообращения, при физических (термических, лучевых, вибрационных), химических (кислоты, щелочи), биологических (переливание несовместимой группы крови) воздействиях. Преходящие стазы, особенно в тканях, малочувствительных к гипоксии, могут разрешаться без каких-либо существенных последствий. При отсроченном разрешении стаза, когда приток артериальной крови восстанавливается, но еще сохраняется гипоксия тканей и сосудов, возникают множественные кровоизлияния путем диапедеза и разрыва микрососудов. Исходом стаза крови, длительно сохраняющегося в тканях с высоким потреблением кислорода, являются некробиоз и некроз.
Тромбоз
Тромбозом (от греческого trombosis — свертывание) называют прижизненное нарушение естественного состояния крови в просветах сосудов или в полостях сердца с образованием сгустка, называемого тромбом. В основе тромбоза лежит физиологическая способность крови к свертыванию (гемостазу) при повреждении сосудистой стенки, которая является важнейшей защитной реакцией организма, останавливающей кровотечение. При внутрисосудистом свертывании лимфы также формируются тромбы, однако закономерности лимфотромбоза существенно отличаются. Сохранение жидкостного состояния крови обеспечивается антигемостатическими свойствами интактного эндотелия сосудов, а также функциональной сбалансированностью систем, одна из которых осуществляет свертывание крови, другая препятствует этому, третья способствует растворению образовавшегося тромба. Благодаря взаимодействию этих систем, постоянно координируемому нервной и эндокринной системами, условия для образования тромба в норме отсутствуют.
Сосудистая стенка и гемостаз. Интактный эндотелиальный монослой выполняет роль атромбогенного барьера между стенкой сосуда и циркулирующей кровью, препятствует свертыванию крови и тромбообразованию. Он синтезирует и катаболизирует метаболиты, регулирующие взаимодействие форменных элементов крови и факторов гемостаза, содержащихся в плазме и сосудистой стенке. Атромботические свойства эндотелия обеспечиваются прежде всего его гликокаликсом — пристеночным слоем гликопротеидов, насыщенных гликозаминогликанами и сиаловыми кислотами. Вместе с полярными фосфолипидами плазмолеммы эндотелиоцитов они сообщают внутренней поверхности сосудистой стенки отрицательный потенциал, такой же, как и у форменных элементов крови. Атромбогенность эндотелия усиливается способностью кумулировать на поверхности комплекс биологически активных веществ, поступающих из ткани и элиминируемых из крови.
Тромборезистентность эндотелия определяется рядом факторов. Одним из них является связывание и активация антитромбина III, который ингибирует тромбин и другие факторы свертывания, к другим относятся гепаринсульфаты, присутствующие в гликокаликсе эндотелиоцитов, и белок тромбомодулин, который ингибирует тромбин и другие факторы коагуляционного каскада. К факторам тромборезистентности эндотелия относится активация комплексом тромбин — тромбомодулин системы С-протеина, мощного антикоагулянтного комплекса, который ингибирует циркулирующие в крови факторы свертывания V–VIII. При этом белок С блокирует ингибитор тканевого активатора плазминогена, что усиливает фибринолиз. Эндотелиоциты осуществляют также секрецию активаторов плазминогена тканевого и сывороточного (урокиназного) типов, синтез и выделение простациклина и оксида азота (NO) — высокоэффективных антиагрегантов тромбоцитов и вазодилататоров.
Прокоагулянтные свойства клеток эндотелия связаны с высвобождением фактора Виллебранда — макромолекулярного белка, синтезируемого и запасаемого в специфических органеллах (тельца Вейбела — Палладе). Фактор Виллебранда связывает и переносит регуляторный белок — плазменный фактор VII, а также служит в качестве рецептора для гликопротеинов поверхности тромбоцитов. Кроме того, эндотелиоциты выделяют тканевой тромбопластический фактор (фактор III), стимуляторы агрегации тромбоцитов и высвобождения ими биологически активных веществ.
При повреждении и отторжении эндотелиоцитов обнажается субэндотелий сосудистой стенки, который активно связывает белки плазмы и тромбоциты, провоцируя тромбообразование. В структуру субэндотелия входят различные типы коллагена, эластин, гликопротеины и гликозаминогликаны, фибронектин, ламинин, тромбоспондин, ассоциирующиеся с фибриногеном и способствующие адгезии тромбоцитов.
Наиболее мощным стимулятором тромбоцитов является фибриллярный коллаген, который осуществляет также контактную активацию факторов так называемой внутреннего пути свертывания крови. Тромбоспондин способен ассоциироваться с волокнами фибрина и полимеризоваться подобно фибриногену. Усиливает клеточное взаимодействие, превращая обратимую агрегацию тромбоцитов в необратимую, специфически связывается с моноцитами и служит молекулярным мостиком между ними и активированными тромбоцитами в участках повреждения сосудистой стенки. Фиброкинетин, основной компонент соединительнотканного матрикса, образует ковалентные связи с фибрином и осуществляет рецептор-опосредованное осаждение активированных тромбоцитов.
Тромбоцитарное звено является важнейшим в системе гемостаза. Участие тромбоцитов в гемостазе обусловлено их способностью к адгезии и агрегации, содержанием собственных и адсорбированных факторов свертывания крови, физиологически активных веществ. Поверхность тромбоцитов, как и клеток эндотелия, покрыта гликокаликсом. Реактивность тромбоцитов зависит от величины отрицательного заряда, обусловленного полианионными свойствами гликокаликса и фосфатными группами плазмолеммы. Плазмолемма тромбоцитов имеет обычное для клеточной мембраны строение, образует множественные инвагинации (поверхностно-связанную систему каналов), многократно увеличивающие ее площадь. На тромбоцитах адсорбируются факторы свертывания, иммуноглобулины. Помимо того, тромбоциты являются источником факторов агрегации и дезагрегации форменных элементов крови, в частности фосфолипидов, тромбоксана А2 — стимулятора агрегации и вазоконстрикции, ряда простагландинов. С ними ассоциированы рецепторные и регуляторные белки, в том числе аденилатциклаза и фосфофолипаза А2, адениннуклеотиды, комплекс ферментов, катализирующих образование и трансформацию арахидоновой кислоты в эндопероксиды и конечные продукты их метаболизма.
Любые агенты, изменяющие физикохимическое состояние гликокаликса и проницаемость плазмолеммы, активируют тромбоциты, повышая их агрегационную способность и провоцируя реакцию высвобождения — секрецию в окружающую среду содержимого тромбоцитарных гранул, являющихся депо биологически активных веществ и адгезивных белков. Тромбоциты содержат 2 основных их типа — α-гранулы и плотные тельца. α-Гранулы депонируют фибриноген, фибронектин, фактор Виллебранда, тромбоспондин, а также фактор роста, стимулирующий миграцию и пролиферацию гладкомышечных клеток сосудистой стенки, тромбоцитарный фактор IV (антигепарин), тромбоцитоспецифические глобулины. Плотные тельца богаты АДФ и ионизированным кальцием, содержат гистамин, эпинефрин, серотонин.
Реакции тромбоцитов на действие активирующих агентов опосредуются повышением концентрации в цитоплазме ионов кальция, которые депонированы в плазмолемме и тромбоцитарных гранулах, в плотной тубулярной системе, расположенной в субмембранной зоне рядом с элементами цитолеммы. Кальций поступает в тромбоциты также из среды в виде трансмембранного тока. Обязательным условием агрегации тромбоцитов является присутствие фибриногена. Фосфолипиды плазмолеммы тромбоцитов служат катализатором для тканевых и плазменных тромбопластов, предшественников тромбина. Поэтому участие тромбоцитов в гемостазе определяется их способностью адсорбировать на своей поверхности плазменные факторы коагуляции, секретировать комплекс биологически активных веществ и адгезивных белков, поставлять в окружающую среду комплексы, активирующие прокоагулянты, а также прочно ассоциироваться с сосудистой стенкой и друг с другом. Роль в гемостазе других форменных элементов, эритроцитов и лейкоцитов обусловлена содержанием в них большинства факторов свертывания крови, которые вовлекаются в процесс образования фибрина при повреждении сосудистой стенки.
Общие закономерности гемостаза. Факторы свертывания крови в норме находятся в неактивном состоянии, в форме предшественников. Плазменные факторы свертывания крови и их функции представлены в табл. 2.1.
Таблица 2.1
Плазменные факторы свертывания крови
| Фактор | Название | Активная форма, функция |
| I | Фибриноген | Образует полимер фибрин |
| II | Протромбин | Тромбин, фермент. Полимеризуется в фибриноген, активирует факторы V, VIII, XIII, стимулирует противосвертывающую систему |
| III | Тромбопластин (фермент) | Кофактор фактора VII |
| IV | Ионы кальция | Участвует в активировании и агрегации тромбоцитов, полимеризации фибриногена, стабилизации фибрина. Связывает факторы протромбинного комплекса с фосфолипидами |
| V | Проакцелерин (плазменный Ас-глобулин) | Регуляторный белок, активирует фактор Х |
| VI | Исключен из классификации | |
| VII | Проконвертин | Активирует фактор Х, ускоряет превращение протромбина в тромбин |
| VIII | Антигемофильный глобулин | Кофактор фактора Х |
| IX | Плазменный компонент тромбопластина (Кристмас-фактор) | Участвует в качестве катализатора, активирует фактор Х в комплексе с фактором VIII и IV |
| X | Фактор Стюарта — Пауэра | Участвует в образовании протромбиназы, превращающей протромбин в тромбин |
| XI | Предшественник плазменного тромбопластина | Участвует в активировании факторов VIII и IX |
| XII | Фактор Хагемана | Участвует в активировании фактора XI, превращении прекалликреина в калликреин |
| XIII | Фибриназа | Стабилизирует фибрин, участвует в формировании плотного сгустка |
Активирование факторов свертывания крови происходит последовательно, причем фермент, являющийся продуктом соответствующей реакции, действует на свой специфический субстрат, вызывая появление другого фермента, который начинает следующий этап в цепи этого каскадного процесса, завершающегося превращением растворимого фибриногена в нерастворимый фибрин. Каждый такой этап представляет комплекс реакций, в которых участвуют активированный коагуляционный фактор — фермент, субстрат — проэнзимная форма сопряженного коагуляционного фактора и кофактор — ускоритель реакции. Все компоненты этих реакций собираются на фосфолипидах и удерживаются вместе ионами кальция. Такой белково-липидной матрицей, на которой собираются и активируются ферментные и другие факторы свертывания, является поверхность тромбоцитов.
В механизме свертывания крови можно условно выделить внешний и внутренний пути, тесно связанные между собой. Внешний путь запускается при повреждении сосудистой стенки и тканей и высвобождении в кровь тканевого фактора свертывания (фактор III, тромбопластин). Тромбопластин представляет липопротеидный комплекс, белковая часть которого работает как кофактор фактора VII свертывания крови, а фосфолипидная служит матрицей для активной формы последнего и его субстрата — фактора X.
Внутренний путь свертывания формируется факторами, содержащимися в крови, активируется при контакте плазмы с субэндотелием, измененными клеточными мембранами, с заряженной поверхностью либо под влиянием биогенных аминов и протеаз. Сопряжен с калликреин-кининовой системой, системой комплемента и другими ферментными системами крови. Калликреин участвует во взаимодействии факторов XII и XI, связывая внутренний и внешний пути свертывания крови. Исходным пунктом внутреннего пути является активация фактора Хагемана, за которым последовательно активируются факторы VII, IX, XI. Вместе с кальцием они образуют на поверхности активированных тромбоцитов или поврежденной сосудистой стенки комплекс, активирующий фактор X, на уровне которого объединяются внешний и внутренний пути гемостаза.
Между механизмами обоих путей свертывания крови существуют сложные взаимоотношения. Небольшое количество тромбина, образующегося при активации внешнего пути, стимулирует агрегацию тромбоцитов и реакцию высвобождения тромбоцитарных факторов, но оно недостаточно для образования фибрина. При этом активируется фактор V, являющийся рецептором фактора X, который активируется при фиксации на поверхности тромбоцитов. Основная масса фактора X трансформируется в активное состояние посредством более сложного и эффективного внутреннего пути гемостаза.
Схема дальнейшего этапа, общего для обоих путей свертывания крови после активации фактора X, включает стадии образования тромбина из протромбина и свертывания фибриногена. Каждая из них осуществляется при участии соответствующих активированных комплексов, состоящих из высокомолекулярного неферментного белка, активной протеиназы и кальция. Они фиксированы на фосфолипидной или другой отрицательно заряженной подложке, образуемой поверхностью клеток крови или стенкой сосудов. Жесткая связь таких комплексов с фосфолипидами обеспечивает их оптимальную защиту от ингибиторов, выход в окружающую среду только конечного фермента в цепи превращений тромбина и локализацию процесса свертывания в поврежденном участке. При этом ферментные факторы запускают аутокаталитический процесс гемостаза, а неферментные компоненты реакции ускоряют их и обеспечивают специфичность действия на субстраты.
Общий путь внешнего и внутреннего путей свертывания крови начинается активацией фактора X и завершается поляризацией фибриногена. Субстратом фактора X служит протромбин, синтезируемый в печени, от которого последовательно отщепляются 2 фрагмента и образуется тромбин — сериновая протеиназа. Основные функции тромбина: ограниченный протеолиз фибриногена с последующей полимеризацией образовавшихся фибрин-мономеров в фибрин; стимуляция тромбоцитов и эндотелия; стимуляция синтеза простагландинов; освобождение адгезивных белков; активирование регуляторных белков — факторов свертывания крови, а также фибринстабилизирующего фактора XIII. Между новообразованными полимерами фибрина устанавливаются дополнительные перекрестные связи, что повышает их эластичность и резистентность к действию фибринолитических агентов.
При активировании гемостаза в 1 мл крови может образоваться примерно 150 ед. тромбина — количество, достаточное для свертывания нескольких ее литров. Однако в организме жидкое состояние крови сохраняется даже при массивных травмах. Это обеспечивается сложной системой, предотвращающей цепную реакцию, которая могла бы привести к свертыванию всей массы крови в сердце и сосудах. Тромбообразованию препятствует антикоагулянтная система, которая включает факторы как образующиеся непосредственно при активации гемостаза, так и существующие независимо от него. Она функционально сопряжена с системой фибринолиза, растворяющей образовавшиеся тромбы.
Антигемостатическая система крови включает следующие механизмы:
1. Снижение локальной концентрации факторов свертывания посредством вымывания и разведения в кровотоке.
2. Истощение остающейся в фокусе повреждения части факторов свертывания за счет их утилизации.
3. Освобождение крови от активированных факторов свертывания вследствие их элиминации и катаболизма гепатоцитами и мононуклеарной системой. Этот механизм может быть эффективен только при сохранении циркуляции в зоне повреждения.
4. Ингибирование активных факторов и кофакторов крови физиологической противосвертывающей системой, регулирующей уровень тромбина.
В крови циркулирует сложный набор протеаз и других биохимических ингибиторов, взаимодействующих с одним или несколькими факторами коагуляции. К их числу относится основной плазменный ингибитор ферментов — антитромбин III, который в присутствии гепарина инактивирует тромбин, факторы свертывания XII, XI, X, IX и кининоген. Протеин С, приобретающий под действием тромбина способность к протеолизу, инактивирует факторы свертывания V, VIII, XI, XII. Скорость инактивации возрастает при связывании факторов с тромбомодулином на поверхности эндотелиоцитов в присутствии ионов кальция и фосфолипидов. Кроме того, протеин С блокирует активацию комплемента, нейтрализует тканевый ингибитор плазминогена, что ускоряет его превращение в плазмин, лизирующий сгустки фибрина, и т.д. Таким образом, система биохимической регуляции гемостаза функционально объединяет механизмы, направленные как на активацию факторов свертывания крови, так и на блокирование их активных форм.
5. Лизис фибрина противосвертывающей системой, осуществляющей ферментативный и неферментативный фибринолиз. Эта система активируется при избыточном накоплении тромбина, ее эффекторным звеном является выброс в кровь гепарина и активаторов фибринолиза из тканевых источников и клеток крови. У фибринолиза внутренний и внешний механизмы активации, первый обеспечивается лейкоцитарными протеазами и плазминогеном, который превращается в плазмин при участии фактора XII и калликреина. Внутренний ферментативный механизм фибринолиза запускается тканевыми кининами, которые синтезируются главным образом эндотелием и активируются при образовании комплексов с фибрином.
Неферментативный фибринолиз инициируется посредством выброса в кровоток гепарина, который связывается с тромбином, фибриногеном и другими тромбогенными протеинами, с катехоламинами. Образующиеся комплексы обладают противосвертывающей активностью, расщепляют нестабилизированный фибрин, блокируют полимеризацию его мономеров, а также являются антагонистами фактора XIII, стабилизирующего свежепреципитированный фибрин. Продукты ферментативного и неферментативного лизиса фибрина приобретают свойства дезагрегантов и антикоагулянтов.
В зависимости от масштабов повреждения и степени участия отдельных компонентов системы свертывания крови различают сосудисто-тромбоцитарный и коагуляционный механизмы, тесное взаимодействие которых обеспечивает надежность гемостаза. Сосудисто-тромбоцитарный механизм гемостаза останавливает кровотечение из периферических сосудов небольшого калибра при ограниченном участии второго механизма. При этом отмечают быстро преходящий спазм травмированных сосудов вследствие рефлекторного выброса в кровоток катехоламинов и повышения тонуса вегетативной нервной системы. Вслед за этим происходит накопление тромбоцитов в зоне повреждения, их адгезия к раневой поверхности с последовательным развитием всех фаз активирования — формированием псевдоподий, распластыванием и реакцией высвобождения.
Накопление необратимо агрегированных тромбоцитов, которые в течение 1–3 с адгезируют к поврежденным эндотелиальным клеткам или обнажившемуся субэндотелию, обеспечивает формирование гемостатического тромба. Это сочетается со вторичным спазмом поврежденных сосудов, обусловленным выделением из тромбоцитов целого ряда биологически активных веществ, запуском процессов преципитации фибриногена и формирования волокон фибрина, активирование антикоагулянтных и фибринолитических механизмов, координирующих процесс гемостаза.
Коагуляционный механизм гемостаза, который реализуется при повреждении крупных сосудов, в общих чертах аналогичен описанному выше. Также начинается рефлекторной реакцией сосудистой стенки, опосредуемой нейрогуморальной системой регуляции, и осаждением тромбоцитов в зоне повреждения. Выделение сосудисто-тканевого и коагуляционного механизмов гемостаза достаточно условно, так как они функционально сопряжены и связующим звеном являются тромбоциты, представляющие собой центр формирования тромба.
Морфология и виды тромбов. По морфологическим особенностям различают тромбы белые (агглютинационные), смешанные (слоистые) и гиалиновые.
Белый тромб возникает в отделах сосудистой системы с быстрым током крови, например в полостях сердца и на створках его клапанов, в аорте и коронарных артериях. Образуется при снижении атромбогенных свойств эндотелия и накопления в крови факторов, стимулирующих тромбоциты, представляет собой суховатую светло-серую массу с тусклой гофрированной поверхностью плотной консистенции, спаян со стенкой сосуда, легко крошится при попытке отделения. Основу белого тромба составляют тромбоциты, склеившиеся с сосудистой стенкой и между собой. Тромбоцитарные конгломераты формируют коралловидные фигуры, ориентированные перпендикулярно току крови, пространства между которыми выполнены сетью фибрилл со скоплениями нейтрофильных лейкоцитов.
Отложения тромбоцитов слоистого характера. Это обусловлено чередованием фаз тромбообразования с преобладанием адгезии и агглютинации тромбоцитов и полимеризации мономеров фибрина на их поверхности, играющей роль матрицы. Во время реакции высвобождения, сопровождающей активирование и агглютинацию тромбоцитов, из них вместе с адгезивными протеинами и биологически активными веществами выделяется фермент ретрактозим. Фермент вызывает сокращение гладкомышечных клеток сосудистой стенки и уплотняет трехмерную сеть, образуемую волокнами фибрина, обеспечивая тем самым консолидацию всех его элементов. Тромб теряет часть жидкости, местами отделяясь от сосудистой стенки, возникшие в нем щели облегчают тромболизис и процесс организации.
Красный тромб образуется вследствие повышения потенциала гемокоагуляционных механизмов при относительно невысокой активности тромбоцитов и снижения антиагрегационных свойств сосудистой стенки. Наиболее частая локализация красных тромбов — емкостные сосуды с относительно низкой скоростью кровотока. Вследствие высоких темпов образования и меньшего содержания тромбоцитов красный тромб легче отделяется от сосудистой стенки. Он рыхлый с гладкой влажной, лишь местами гофрированной поверхностью, что придает ему сходство с посмертным сгустком крови. Новообразованные тромбы этого типа темно-красной окраски, со временем приобретают бурый оттенок; их поверхность утрачивает блеск. Структурную основу красного тромба составляет трехмерная сеть волокон фибрина различной толщины, петли которой заполнены агглютинированными и в различной степени выщелоченными эритроцитами с незначительной примесью лейкоцитов и небольшими скоплениями тромбоцитов. Однако коралловидные фигуры, образуемые ими в белых тромбах, отсутствуют.
Смешанный тромб включает участки, по своей структуре соответствующие белому или красному тромбу. Чем медленнее тромбообразование, тем лучше выражена скелетная часть тромба, образуемая коралловидно-ветвящимися агрегациями тромбоцитов и характерная для белого тромба, и тем меньше зоны коагуляции крови, представленные сетью полимеризованного фибрина, ячейки которого заполнены осевшими эритроцитами с вкраплением других форменных элементов. Присутствие в смешанных тромбах светлых и темных участков придает им пестрый слоистый вид как на поверхности, так и на разрезах. Такие тромбы чаще всего выявляют в артериях различного калибра, крупных венах, аневризмах сердца и артерий. Так же, как и красные тромбы, они имеют в сосудах удлиненную форму. Макроскопически в них различают головку, обычно конической или уплощенной формы, плотно соединенную со стенкой сосуда, соответствующую по своему строению белому тромбу. Головка тромба переходит в тело (собственно смешанный тромб), продолжающееся в рыхло связанный с ним свободно расположенный в просвете сосуда хвост, который представляет собой красный тромб.
Связь смешанного тромба с сосудистой стенкой и описанные выше особенности строения отличают его от посмертного сгустка крови. Наибольших размеров смешанные тромбы достигают в крупных венах, где, как правило, располагаются по току крови. Такой тромб может начинаться в бедренной вене, где его головка плотно прикреплена к сосудистой стенке, тело (смешанный тромб) продолжается в наружную подвздошную вену, переходя в рыхлый темно-красный хвост, иногда достигающий нижней полой вены.
Гиалиновый тромб представляет собой однородную гиалиноподобную массу, образующуюся при агглютинации и деструкции эритроцитов, лейкоцитов и преципитированных белков плазмы крови в мелких периферических сосудах. Содержание фибрина в гиалиновых тромбах сравнительно невелико, а присутствие его непостоянно. Образованию гиалинового тромба часто предшествует стаз крови в микрососудах.
Тромбы классифицируются также в зависимости от их локализации, отношения к просвету сосуда, в котором они сформировались, и этиологических факторов, способствовавших тромбообразованию. Тромбы, только частично ограничивающие сосудистый просвет, называют пристеночными, полностью закрывающие его — обтурирующими. Для последних характерно развитие как в дистальном, так и в проксимальном направлении по току крови. В тех случаях, когда такой тромб имеет строение слоистого или смешанного, определение места, где началось его образование и соответственно расположена головка, представляет большие трудности.
Пристеночные тромбы обычно выявляют в просветах крупных сосудов, в камерах сердца и на клапанах при атеросклерозе и воспалительных процессах (тромбартериит, тромбоэндокардит, тромбофлебит), при венозной гиперемии, сопровождающейся замедлением кровотока (марантические тромбы). Патологическая дилатация артерий или камер сердца (аневризмы), варикозное расширение вен также способствуют тромбообразованию (дилатационные тромбы). Обтурирующие тромбы наиболее характерны для мелких сосудов. Нередко при росте пристеночного тромба посредством наслоения вновь образующихся тромботических масс возможна закупорка магистральных сосудов — коронарных артерий сердца или кишечника, крупных артерий головного мозга, печеночных, бедренных и других вен. Такой тромбоз называют прогрессирующим.
Промежуточное положение между пристеночным и обтурирующим тромбами по влиянию на кровоток занимают так называемые аксиальные тромбы, которые, прикрепляясь свободной частью к сосудистой стенке только в области головки и частично тела, существенно ограничивают проходимость сосуда. В предсердии крупный растущий тромб, оторвавшись от стенки, может оставаться в его полости во взвешенном состоянии, приобретая под действием кровотока шаровидную форму (шаровидные тромбы). Фактором, провоцирующим тромбоз, может стать разрастание опухоли, проникающей в просвет вены и образующей поверхность, на которой инициируется тромбообразование (опухолевые тромбы).
Факторы развития тромбоза. Инициирование тромбоза определяется общими и местными предпосылками, при сочетании которых нарушается равновесие процессов про-, антикоагуляции и фибринолиза. Наиболее существенными факторами общего характера, предрасполагающими к тромбообразованию, являются нарушение гемодинамики при СН, изменения состава крови при заболеваниях системы крови, инфекционно-аллергических процессах, патологических нейрогуморальных реакциях (хронический стресс) и нарушениях кровообращения с наклонностью к ангиоспастическим явлениям.
Из местных факторов, способствующих тромбозу, следует назвать прежде всего изменения сосудистой стенки и локальные нарушения гемодинамики. Изменения сосудистой стенки, оказывающие тромбогенный эффект, имеют различную природу, однако во всех случаях происходит повреждение сосудистого эндотелия, приводящее к утрате его антигемостатических свойств. Непосредственными причинами этого может стать механическое повреждение или воспаление, запускающее сосудисто-тромбоцитарный механизм гемостаза, к которому присоединяются гемокоагуляционные процессы. Таковы же последствия распада атеросклеротической бляшки, ангиоспазма, резкого повышения уровня АД и сосудистой проницаемости с последующей отслойкой и десквамацией эндотелиоцитов, обнажающей субэндотелий. Тромбозу способствует также появление завихрений в потоке крови, травмирующих эндотелиальный монослой и тромбоциты.
Замедление скорости кровотока создает благоприятные условия для агрегации тромбоцитов к сосудистой стенке и ограничивает вымывание выделяемых ими факторов. О важном значении этих изменений для развития тромбоза свидетельствуют в 5 раз более частая локализация тромбов в местах ветвлений сосудов или атеросклеротических бляшек, деформирующих их стенку, более частое тромбирование вен, чем артерий, с типичной локализацией в нижних конечностях, синусах венозных клапанов, варикозных расширениях и аневризмах сосудов и сердца.
Однако большинство из названных предпосылок не имеет абсолютного значения для тромбоза, и только их сочетание с острым или хроническим нарушением свертывающей и противосвертывающей систем становится достаточным условием для его развития.
Исходы тромбоза, как и его непосредственные причины или строение тромбов, неодинаковы. При неосложненном развитии тромба в нем отмечают асептическое расплавление (аутолиз), наступающее как под влиянием литических ферментов (катепсинов, гидролаз, пептидаз), высвобождающихся из полиморфно-ядерных лейкоцитов и тромбоцитов, так и вследствие фибринолиза, обусловленного действием плазмина и пептидаз плазмы крови.
Расплавление тромбов начинается со срединной зоны, где скапливается наибольшее количество энзимов. Образующийся кашицеобразный детрит и полужидкие массы в белом тромбе желтоватого оттенка, а в красном приобретают красно-коричневую окраску в результате изобилия эритроцитов. Иногда продукты аутолиза попадают в кровоток и уносятся током крови. Мелкие тромбы могут аутолизироваться полностью.
Параллельно с аутолизом к концу 1-х суток начинается организация тромба, в которой участвует сосудистая стенка. В тех участках тромба, которые позже других вовлекаются в асептический аутолиз, в первые 4 дня происходят распад и гомогенизация форменных элементов крови и нитей фибрина со слиянием детрита в гиалиноподобную массу.
На 2-е сутки отмечают пролиферацию эндотелиоцитов сосудистой стенки, которые как бы наползают на поверхность тромба, постепенно покрывая ее. Наряду с этим отмечают размножение клеток интимы, накопление активированных макрофагов, некротические изменения еще сохранившихся лейкоцитов и проникновение фибропластических элементов в тромб. В последующие дни явления лизиса детрита и выраженная макрофагальная реакция сочетаются с врастанием в тромб тяжей от пролиферирующих эндотелиоцитов, из которых затем образуются кровеносные капилляры. В организации тромба вместе с фибробластами и макрофагами активно участвуют недифференцированные гладкомышечные клетки сосудистой стенки, продуцирующие гликопротеины и коллаген.
Организация тромба начинается с его головки, распространяясь потом на тело. Новообразованные сосуды соединяются с vasa vasorum или с просветом тромбированного сосуда. По мере созревания соединительной ткани в тромбе появляются щели и каналы, выстланные эндотелием (канализация тромбов), а с 5-й недели выявляют дифференцированные сосуды (васкуляризация тромба), из которых иногда формируются сосудистые полости (кавернозная трансформация тромба). Канализация и васкуляризация тромба частично восстанавливают проходимость сосуда. Эволюция тромба завершается созреванием новообразованной соединительной ткани в рубцовую и последующим формированием фиброзномышечной бляшки, стенозирующей просвет сосуда. При нарушении процесса организации в гиалинизированные участки тромба выпадают соли кальция, что приводит к обызвествлению тромботических масс. В венах этот процесс иногда завершается петрификацией — образованием камней (флеболитов).
Значение тромбоза для организма неоднозначно. Тромбы, образующиеся при повреждениях сосудов, защищают организм от фатальной кровопотери, организация тромботических масс в аневризмах сердца и сосудов предупреждает разрывы их стенки. Однако в большинстве случаев, когда тромбоз развивается как патологический процесс, существует угроза возникновения его более или менее опасных осложнений. Это определяется локализацией и скоростью образования тромба, степенью ограничения просвета сосуда, наличием или отсутствием коллатералей, а также последующей эволюцией образовавшегося тромба. Наиболее опасные осложнения тромбоза обусловлены:
- Локальными нарушениями кровотока вследствие ограничения проходимости просвета тромбированного сосуда.
- Способностью тромба или его части отделяться от стенки сосуда и переноситься потоком крови на значительные расстояния (тромбоэмболия) при вялом развитии процессов организации либо вследствие аутолиза.
- Инфицированием тромба и переходом асептического аутолиза в септический.
Обтурация тромбом магистрального сосуда при недостаточном развитии коллатералей вызывает ишемию или венозную гиперемию с возможными неблагоприятными последствиями. В то же время постепенное растянутое во времени формирование пристеночного тромба даже в крупных артериальных стволах не обязательно приводит к тяжелым последствиям, например к развитию инфаркта, так как в этих случаях кровоток успевает частично восстановиться за счет коллатералей. Опасность осложнений при тромбозе резко возрастает при его прогрессирующем развитии, что свидетельствует о существенных общих нарушениях регуляции гемостаза и кровообращения. Последствиями этого могут быть рост и превращение тромбов из пристеночного или аксиального в обтурирующий либо быстрое увеличение хвоста, рыхло связанного с телом, возникновение в различных сосудах множественных тромбов, слабо фиксированных к сосудистой стенке. Отрыв от нее всего или части такого тромба превращает его в тромбоэмбол, свободно мигрирующий с током крови. Развитие тромбоэмболии возможно при любой локализации тромбов, однако наиболее часто это отмечают при флеботромбозе, тромбофлебите или тромбозе полостей и особенно ушек сердца.
Аутолиз тромба бывает не только асептическим. Попадание в него гноеродных бактерий обусловливает септическое расплавление тромботических масс с последующим распространением образующихся инфицированных продуктов распада по организму, вызывающим тромбобактериальную эмболию сосудов и образование очагов гнойного воспаления в различных органах и тканях.
В патологоанатомической практике нередко возникает необходимость дифференцировать тромбы от посмертных сгустков крови, которые также бывают белыми или смешанными и иногда имеют весьма значительное сходство с тромбами. Такое сходство определяется подобием механизмов, обусловливающих посмертное свертывание крови. Считается, что до окончательной остановки метаболических процессов, протекающих в сосудистой стенке, в ней происходит накопление и диффузия в просвет сосуда АДФ с последующей активацией тромбоцитов и запуском внутреннего пути свертывания крови. Вместе с тем отличие условий, в которых это происходит, от процесса тромбообразования в живом организме находит отражение в морфологии посмертных сгустков и тромбов.
ДВС-синдром
ДВС-синдром — патологическая ситуация, непосредственно связанная с дисфункцией системы гемостаза организма. Основное различие локального тромбоза и ДВС-синдрома заключается в относительно ограниченном характере этих изменений в первом случае и их генерализацией с преимущественной локализацией в микроциркуляторном русле — во втором. Тромбоз более крупных сосудов может развиваться как компонент или финал данного синдрома. В соответствии с этиопатогенетическими особенностями синдрома преобладают нарушения прокоагулянтного или сосудисто-тромбоцитарного звена гемостаза либо оба они активируются в равной степени.
ДВС-синдром неспецифичен и развивается при многочисленных заболеваниях и патологических состояниях: сердечно-сосудистых (крупноочаговый ИМ, врожденные «синие» пороки сердца, СН и др.), всех видах шока, включая кардиогенный. ДВС-синдром входит в число осложнений трансфузии несовместимой крови, травматологической, инфекционно-септической, онкологической, акушерской, ятрогенной, аутоиммунной и иммунокомплексной патологии, аллергических реакций, отравлений гемокоагулянтами и гемолитическими ядами, обширных оперативных вмешательств, в том числе с применением АИК.
Патогенез ДВС-синдрома определяется активацией факторов свертывания крови с последующим их истощением, избыточной стимуляцией фибринолиза, что сопровождаются массивными, крайне трудно купируемыми кровотечениями и кровоизлияниями. Непосредственные причины развития ДВС-синдрома неоднозначны. Чаще всего его факторами становятся продукты гемолиза, амниотическая жидкость, эндотоксины, прогрессирующий ацидоз, протеолитические энзимы, эллаговая кислота, избыток АДФ, некоторые липидные фракции плазмы и циркулирующие иммунные комплексы, гиперадреналинемия, общие гемодинамические нарушения и др. В основе активации этими факторами системы коагуляции и снижения эффективности процессов, сдерживающих свертывание крови, лежат 2 главных механизма: высвобождение в кровоток тканевых факторов и альтерация эндотелиального монослоя.
Многочисленные факторы, инициирующие ДВС-синдром, часто тесно взаимосвязаны. Даже ограниченное повреждение эндотелия стимулирует прокоагулянтную активность посредством экспрессии тканевого фактора. В числе причин, способных провоцировать повреждение эндотелия, особое значение имеют гипоксия, ацидоз и шок как наиболее характерные для кардиологической патологии. Продукция тканевого тромбопластина резко возрастает при расширении зоны альтерации эндотелия с обнажением субэндотелиальных структур, активированием тромбоцитов и внутреннего пути свертывания крови через контактный XII фактор Хагемана, который действует на калликреин-кининовую систему, фибринолиз, систему комплемента. Это сопровождается блокадой фагоцитарной функции мононуклеаров, которые в физиологических условиях поддерживают равновесие процессов гемостаза, элиминируя из крови растворимые комплексы фибрина.
Инициация ДВС-синдрома непосредственно связана с действием как тромбопластина и тромбина, так и эндотелиотропных медиаторов. В результате этого коагуляция и тромбогенез оказываются первичными процессами с последующей активацией и агрегацией тромбоцитов, которые высвобождают биологически активные соединения в сочетании с интенсивным потреблением факторов свертывания крови. При этом компенсаторная активация тромбином противосвертывающей системы, в норме обеспечивающей адекватное повышение антикоагулянтного и фибринолитического фона, оказывается недостаточной. Распространенный микротромбогенез сопровождается ростом активности системы фибринолиза с появлением в крови плазмина, который гидролизирует фибрин, инактивирует факторы V, VIII, IX, XI и снижает их концентрацию в крови.
Протеиназы тромбин и плазмин обусловливают преципитацию фибрина. В то же время они расщепляют его и фибриноген с образованием ранних и поздних продуктов деградации, которые препятствуют полимеризации фибрин-мономера и вызывают дисфункцию тромбоцитов. Вместе с тем некоторая часть фибрин-мономеров полимеризуется в микрососудах и, захватывая форменные элементы крови, провоцирует реакцию фибрин — эритроциты и микроангиопатогенную гемолитическую анемию с высвобождением в кровь фосфолипидов и АТФ, которые являются индукторами ДВС. Часть тромбоцитов, предварительно активированных различными индукторами, включая тромбин и коллаген, связывается в этих микротромботических комплексах, высвобождая тромбоспондин, фибропластин и другие адгезивные белки, что также способствует истощению защитных механизмов противосвертывающей системы. Избыточное потребление факторов свертывания, высокий уровень растворимых комплексов фибрина, тромбоцитопения, дисфункция противосвертывающих механизмов приводят к реализации вторичных процессов гиперкоагуляции, недостаточности гемостаза, кровотечениям и кровоизлияниям.
В развитии ДВС-синдрома различают 4 стадии, которые некоторыми исследователями рассматриваются скорее как формы этого патологического процесса. Первая стадия морфологически характеризуется массированным микротромбозом с блокированием микрососудов, обусловленным гиперкоагуляцией и внутрисосудистой агрегацией форменных элементов крови на фоне активации плазменных систем гемостаза. Морфологическими эквивалентами этой стадии ДВС-синдрома являются фибриновые, гиалиновые, глобулярные, тромбоцитарные, лейкоцитарные, эритроцитарные (красные) микротромбы, состав и строение которых не соответствует структуре тромбов в макрососудах. Фибриновые микротромбы, которым при морфологической диагностике ДВС-синдрома отводится решающая роль, как и гиалиновые, состоят преимущественно из фибрина с более или менее значительной примесью фибриногена (рис. 2.4).
Каркасом глобулярных микротромбов служат агрегированные эритроциты с явлениями гемолиза, на выщелоченных оболочках которых откладываются фибриновые массы. Тромбоцитарные или пластинчатые микротромбы, наряду с компактно расположенными кровяными пластинками, включают единичные эритроциты, лейкоциты и нити фибрина. Предрасполагающим фактором для образования таких микротромбов являются альтеративные изменения эндотелиоцитов.
Белые или лейкоцитарные тромбы чаще образуются при ДВС-синдроме инфекционной этиологии, располагаясь преимущественно в дистальных отделах микрогемоциркуляторного русла. Красные тромбы, главным компонентом которых являются выщелоченные сладжированные эритроциты и преципитаты фибрина, выявляют на всех участках микрогемоциркуляторного русла.
Вторая стадия ДВС-синдрома, коагулопатия потребления, определяется тромбоцитопенией, внутриваскулярной преципитацией фибрина со снижением в крови содержания фибриногена и других плазменных факторов свертывания. Проявляется как гипер-, так и гипокоагуляцией в виде кровотечений и признаков геморрагического диатеза. Коагулопатия потребления является следствием распространенного микротромбоза с повышенным использованием факторов свертывания крови и удаления коагулятов фибрина из кровотока фагоцитами, а также клетками печени и селезенки.
Активация фибринолиза в третьей стадии ДВС-синдрома обеспечивает восстановление гемоперфузии в микрогемоциркуляторном русле, освобождая просветы микрососудов от тромботических масс. Вместе с тем появление в крови плазмина — высокоактивной протеазы, расщепляющей фибриноген и фибрин, способствует вторичному формированию так называемых гиалиновых микротромбов.
Четвертая, восстановительная, стадия сводится к остаточным проявлениям предшествовавшей блокады микрососудов в виде дистрофических и некротических изменений наиболее пострадавших тканей, завершается выздоровлением либо при неблагоприятном течении процесса развитием ОПН, острой печеночной, надпочечниковой, легочной или другой органной недостаточности.
Клинико-морфологическая картина ДВС-синдрома определяется характером этиологических факторов, их интенсивностью и длительностью действия, адекватностью и эффективностью лечебных мероприятий. Несмотря на генерализованный характер патологического процесса, в общей картине часто доминируют региональные нарушения с преимущественным поражением легких (68%), почек (66%), селезенки и печени (соответственно 52 и 50%).
Частое повреждение легких объясняется их функцией своеобразного сосудистого фильтра. В кровеносных капиллярах легких задерживаются продукты альтерации и инородные частицы, являясь триггером для запуска ДВС-синдрома, провоцируя внутрисосудистую коагуляцию, агрегацию, сладж и агглютинацию форменных элементов крови с образованием всех вариантов микротромбов. Множественные полиморфные по составу микротромбы вызывают явления дистрофии в паренхиматозных органах, а при пролонгированном течении процесса — некробиотические и некротические изменения. Микрогемодинамические нарушения при значительной распространенности и продолжительности способны приводить к органной недостаточности, резко усугубляющей клиническую картину. Интенсивное распространенное микротромбообразование иногда осложняется тромботической окклюзией предрасположенных к этому артерий мышечного и мышечно-эластического типа, например при атеросклерозе или системных поражениях соединительной ткани.
Облигатным компонентом клинико-морфологической картины крови при ДВС является тромбогеморрагический синдром, который почти в 40% случаев протекает с выраженной кровопотерей, множественными точечными или обширными кровоизлияниями в различных органах. Чаще всего это легочные альвеолы, толща надпочечников, паренхима печени, селезенки, почек, субэндо- и субэпикардиальные зоны в сердце. Отмечают также появление мелко- и крупнопятнистой геморрагической сыпи на коже, множественные кровоизлияния в местах инъекций и операционных разрезов.
В ЦНС микронекрозы, чаще всего вызываемые фибриновыми тромбами, сочетаются с внутритканевыми и интраоболочечными кровоизлияниями. В ЖКТ распространенный микротромбоз и кровоизлияния в слизистую оболочку приводят к острым язвам желудка, эрозивному гастриту, энтероколиту. Моно- или полиорганная недостаточность, гипофибриногенемия и тромбогеморрагический синдром часто дополняются постгеморрагической анемией и выраженной гипотензией, нарушениями сердечного ритма и мозговой симптоматикой.
В зависимости от темпов развития и особенностей течения принято различать острую, подострую и хроническую формы ДВС-синдрома. Генерализованный характер острой формы вследствие быстрого массированного поступления тромбопластинового компонента в кровоток в период от нескольких часов до суток приводит к шоковому состоянию: затемнению сознания, гипотензии, острой полиорганной недостаточности, часто с явлениями очагового панкреонекроза и эрозивно-язвенного энтероколита.
Подострая форма развивается в течение нескольких суток, а иногда и 1 нед. Мозаичность сопутствующей симптоматики свидетельствует о полиорганном процессе, однако в клинической картине чаще всего доминируют признаки преимущественного повреждения какого-либо одного органа или системы. В целом умеренно выраженные признаки тромбогеморрагического синдрома могут резко усиливаться, приобретая выраженный генерализованный характер, при присоединении даже небольшого экзо- или эндогенного стимула, например при онкологической, ятрогенной патологии, затяжных коронарных кризах.
Хронический ДВС-синдром, сопутствующий воспалительным и аутоиммунным процессам (гепатиту, панкреатиту, пневмониям, системным поражениям соединительной ткани, онкопатологии, ИБС), может продолжаться месяцами. Ослабляясь на период ремиссии и усиливаясь при обострении, ДВС-синдром может оказывать существенное влияние на клиническую картину основного заболевания.
Исход ДВС-синдрома определяется причиной, степенью выраженности и характером развития. При наиболее тяжелой, острой форме почти в 50% случаев угрожающей летальным исходом, решающее значение имеет своевременность диагностики и сбалансированность лечебных мероприятий, ориентированных на оптимизацию процессов гемокоагуляции. При менее тяжелых пролонгированных формах ДВС-синдрома акцент смещается на адекватную терапию провоцирующих его заболеваний и процессов.
Эмболия
Эмболией (от греч. — вторжение, вставка) называют патологический процесс перемещения в потоке крови субстратов (эмболов), которые отсутствуют в нормальных условиях и способны обтурировать сосуды, вызывая острые регионарные нарушения кровообращения. Для классификации эмболий используется ряд признаков: характер и происхождение эмболов, их объемы, пути миграции в сосудистой системе, а также частоту повторения эмболии у данного больного.
В зависимости от места возникновения эмболы перемещаются:
- Из полости левого предсердия, ЛЖ или магистральных сосудов в периферические отделы большого круга кровообращения. Таковы же пути миграции эмболов из легочных вен, попадающих в левые отделы сердца (ортоградная эмболия).
- Из сосудов различного калибра венозной системы большого круга в правое предсердие, ПЖ и далее по току крови в артерии малого круга кровообращения.
- Из ветвей портальной системы в воротную вену печени.
- Против тока крови в венозных сосудах значительного калибра (ретроградная эмболия). Это отмечают в случаях, когда удельный вес тромба позволяет ему преодолеть движущую силу потока крови, в котором он находится. Через нижнюю полую вену такой эмбол может опускаться в почечную, подвздошную и даже бедренные вены, обтурируя их.Из вен большого круга в его артерии, минуя легкие, что становится возможным при наличии врожденных либо приобретенных дефектов в межпредсердной или межжелудочковой перегородках, а также при небольших размерах эмболов, способных проходить через артериовенозные анастомозы (парадоксальная эмболия).
Источниками эмболии могут быть тромбы и продукты их разрушения; содержимое вскрывшейся опухоли или костного мозга; жиры, высвобождающиеся при повреждении жировой клетчатки или костей; частицы тканей, колонии микроорганизмов, содержимое околоплодных вод, инородные тела, пузырьки газа и др. В соответствии с природой эмболов различают тромбоэмболию, жировую, тканевую, бактериальную, воздушную, газовую эмболии и эмболию инородными телами.
Тромбоэмболия — наиболее распространенный вид эмболии. Непрочно фиксированный тромб или его часть могут отделяться от места прикрепления и превращаться в эмбол. Этому способствуют внезапное повышение АД, изменение ритма сердечных сокращений, резко возросшая физическая нагрузка, колебание внутрибрюшного или внутригрудного давления (при кашле, дефекации). Иногда причиной мобилизации тромба становится его распад при аутолизе. Свободно движущийся эмбол заносится током крови в сосуд, просвет которого меньше размеров эмбола, и фиксируется в нем вследствие ангиоспазма.
Чаще всего тромбоэмболию отмечают в емкостных сосудах большого круга кровообращения, в первую очередь в венах нижних конечностей и малого таза (венозная тромбоэмболия). Образовавшиеся здесь тромбоэмболы обычно заносятся током крови в систему ЛА. Артериальную эмболию в большом круге кровообращения выявляют в 8 раз реже. Ее основными источниками являются тромбы, локализованные в ушке левого предсердия, между трабекулами ЛЖ, на створках клапанов, образовавшиеся в зоне инфаркта, аневризме сердца, аорты или ее крупных ветвей, сформировавшиеся на атеросклеротической бляшке.
Состояние, при котором отмечается повышенная склонность к внутрисосудистому тромбообразованию и повторным тромбоэмболиям, определяется как тромбоэмболический синдром. Данный синдром развивается при сочетанном нарушении механизмов, контролирующих процессы гемостаза и поддерживающих текучесть крови, с другими общими и местными факторами, способствующими тромбообразованию. Это отмечают при тяжелых оперативных вмешательствах, онкологической патологии, заболеваниях сердечно-сосудистой системы.
Жировая эмболия возникает вследствие попадания в кровь капелек собственного или инородного нейтрального жира. Причинами этого являются травма скелета (закрытые переломы или огнестрельные ранения длинных трубчатых костей, множественные переломы ребер, костей таза), обширные повреждения мягких тканей с размозжением подкожной жировой клетчатки, тяжелые ожоги, интоксикации или электротравмы, жировая дистрофия печени, закрытый массаж сердца, некоторые виды наркоза. Жировая эмболия может возникать также при введении больному лечебных или диагностических препаратов на масляной основе. Жировые капли обычно попадают в легкие и задерживаются в мелких сосудах и капиллярах. Часть жировых капелек проникает через артериовенозные анастомозы в большой круг кровообращения и разносится кровью в головной мозг, почки и другие органы, блокируя их капилляры. При этом макроскопические изменения в органах отсутствуют. Однако целенаправленное исследование гистологических препаратов с использованием выявляющих жир красителей позволяет диагностировать жировую эмболию в большинстве подобных ситуаций.
Тканевую (клеточную) эмболию отмечают при попадании в кровоток частиц ткани, продуктов их распада или отдельных клеток, которые становятся эмболами. Тканевая эмболия возникает при травмах, прорастании злокачественных опухолей в просветы сосудов, язвенном эндокардите. Эмболами могут стать комплексы костномозговых клеток и мегакариоциты, обрывки дермы, мышечной ткани, частицы печени, мозга, продукты деструкции створок клапанов сердца или комплексы опухолевых клеток. Возможна также эмболия околоплодными водами, содержащими роговые чешуйки и попадающими в капилляры легких; при неполном отслоении плаценты, когда эмболами становятся ворсинки хориона, оказавшиеся в венах матки. Угроза тканевой эмболии существует также в случаях, когда нарушается методика проведения пункционных биопсий внутренних органов, неправильно выполняется катетеризация крупных вен.
Бактериальная (микробная) эмболия осложняет воспалительные процессы, вызванные пиогенной или грибковой микрофлорой, отмечается при заражении простейшими или животными паразитами. Попавшие в кровоток колонии микроорганизмов, друзы грибов, патогенные амебы задерживаются в легких или обтурируют периферические сосуды большого круга кровообращения, питающие ткань почек, печени, сердца, головного мозга и других органов. В новом месте возможно развитие патологического процесса, подобного тому, который явился источником эмболии.
Воздушная эмболия возникает при попадании в кровь пузырьков воздуха, которые мигрируют в сосудистом русле, задерживаются в местах ветвления мелких сосудов и капилляров и обтурируют просвет сосуда. В выраженных случаях возможны блокада более крупных сосудистых ветвей и даже скопление пены, образуемой воздухом и кровью в полости правого сердца. В связи с этим при подозрении на воздушную эмболию вскрытие полостей сердца производят без извлечения его из грудной клетки, под водой, заполнив ею раскрытую полость перикарда.
Причиной воздушной эмболии является повреждение вен, в которые воздух засасывается вследствие отрицательного давления крови. Наиболее часто это отмечают при ранении яремных или подключичных вен, открытой травме синусов твердой мозговой оболочки, баротравме легких. Воздух может поступать в зияющие после родов вены внутренней поверхности матки. Угроза воздушной эмболии существует при операциях на сердце с использованием АИК, во время вскрытия грудной клетки либо наложении диагностического или лечебного пневмоперитонеума, а также при неосторожном внутривенном введении лекарственных средств.
Газовая эмболия при определенном сходстве с воздушной имеет несколько иные механизмы развития. В основе ее лежат изменения растворимости газов в жидкости пропорционально давлению в среде. Так, при быстром подъеме водолазов, находившихся на значительной глубине, при скоростном подъеме в разгерметизированном высотном летательном аппарате газы воздуха либо специальной дыхательной смеси, растворенные в крови, высвобождаются (эффект «газированной воды») и, свободно циркулируя в ней, становятся источником эмболии. В связи с более значительным объемом крови, находящейся в большом круге, чем в малом, изменения в его бассейне выражены сильнее.
Эмболия инородными телами возможна вследствие проникновения их в сосудистое русло при огнестрельных ранениях (осколки, дробь, пули), иногда при попадании в сосуды обломков катетеров. Значительно чаще источником этого вида эмболии оказываются известь, кристаллы ХС, содержащиеся в атероматозной массе, попадающей в кровоток при разрушении и изъявлении атеросклеротических бляшек. Эмболия инородными телами, имеющими большой удельный вес, может быть ретроградной. Такие эмболы способны перемещаться при изменении положения тела.
Значение для организма и исходы эмболии определяются размерами и количеством эмболов, маршрутами миграции в сосудистой системе и характером образующего их материала. В зависимости от величины эмболов различают эмболию крупных сосудов и микроциркуляторного русла (ДВС-синдром). Все эмболии, за исключением воздушной и газовой, являются осложнениями других заболеваний, течение которых они отягощают вплоть до летального исхода. Наиболее часто выявляют венозную тромбоэмболию, при которой эмболы, в зависимости от своего размера, задерживаются в периферических ветвях ЛА, вызывая геморрагические инфаркты легких, либо закрывают их просвет уже в начальных отделах, что ведет к внезапной смерти. Однако нередки случаи, когда тромбоэмболы при сравнительно небольшом диаметре, но значительной протяженности под действием потока крови обтурируют сосуды значительно большего калибра, чем их собственный, либо задерживаются в месте разветвления общего легочного ствола.
Основным патогенетическим фактором, определяющим клинику ТЭЛА и ее крупных ветвей, является резкое повышение сопротивления кровотоку в малом круге кровообращения. Внезапное отключение сосудов сопровождается местной рефлекторной вазоконстрикцией, иногда распространяющейся на всю артериальную систему легкого. Эта реакция усугубляется массивным выбросом катехоламинов и повышением вязкости крови вследствие стресса. Подъем давления в ЛА может стать причиной резкой перегрузки правого сердца с дилатацией его полостей и развитием острого легочного сердца. Спазм артерий при их внезапной механической обтурации отмечают не только в легких. Так, причиной остановки сердца при закупорке ЛА являются и острое легочное сердце, и рефлекторный спазм коронарных артерий сердца. При заносе эмболов в небольшие ветви ЛА, не сопряженном непосредственно с летальным исходом, на ЭКГ отмечают изменения, характерные для острой коронарной недостаточности.
Выраженный ангиоспазм повреждает сосудистый эндотелий, усиливает адгезию и агрегацию тромбоцитов, включает каскадный процесс гемостаза и обусловливает трансформацию эмбола в растущий тромб (эмболотромбоз). Следствием тромбоэмболии артерий большого круга кровообращения является ишемизация соответствующих органов и тканей с последующим развитием инфарктов. Инфицирование тромбов существенно осложняет последствия тромбоэмболии, источниками которой они становятся, так как в местах фиксации таких эмболов к изменениям, связанным с нарушением кровообращения, присоединяется гнойное воспаление (тромбобактериальная эмболия).
Последствия жировой эмболии определяются объемом попавшего в кровь жира и зависят от исходных физико-химических свойств крови, состояния липидного обмена и системы гемостаза. При травмах значительная часть жировых капель формируется из липидов крови, что сопровождается резким повышением ее коагуляционной активности. В связи с этим жировую эмболию нередко рассматривают как вариант травматической коагулопатии. Летальный исход при жировой эмболии может явиться также следствием закупорки капилляров и циркуляторной гипоксии головного мозга. При небольшом объеме обтурированных микрососудов жировая эмболия протекает без существенной клинической симптоматики. Жир, попавший в легкие, частично расщепляется либо омыляется макрофагами и выводится через дыхательные пути. В более тяжелых случаях возможно присоединение пневмонии, а при выключении ⅔ легочных капилляров развивается острая легочная недостаточность с угрозой остановки сердца.
Тканевую (клеточную) эмболию отмечают в сосудах большого круга кровообращения чаще, чем в малом. Наибольшее практическое значение имеет эмболия клетками злокачественной опухоли, лежащая в основе гематогенной диссеминации опухолевого процесса. В результате опухолевые клетки могут заноситься током крови практически в любой регион и давать начало новому очагу разрастания опухоли. Это явление получило название метастазирования, а очаги опухолевого роста, возникшие в результате, — метастатических. При распространении подобных клеток с током лимфы говорят о лимфогенном метастазировании. Аналогичный механизм лежит в основе гематогенного распространения патогенной микрофлоры с появлением фокусов инфекции в любом пункте большого или малого кругов кровообращения, куда заносятся бактериальные эмболы, — в легких, почках, селезенке, головном мозгу, сердечной мышце. Бактериальные эмболии часто сопровождаются клиническими и биохимическими признаками ДВС-синдрома.
Последствия газовой эмболии могут варьировать от стертых форм кессонной болезни до тяжелых и даже приводящих к летальному исходу нарушений в системе кровообращения и внутренних органах, главным образом в головном мозгу. Кессонная болезнь развивается при пассивном выделении газа, растворенного в крови, в результате резкого снижения давления окружающей среды. Образующиеся пузырьки газа — эмболы попадают в микрососуды ЦНС и внутренних органов, скелетных мышц, кожи, слизистых оболочек, нарушая их кровоснабжение и вызывая кессонную болезнь. В дальнейшем могут появиться множественные мелкие кровоизлияния и мелкие некрозы в различных органах, в частности в головном мозгу и сердце, следствием чего являются параличи и нарушения сердечной деятельности.
Скопление значительного объема выделившегося из крови газа в камерах сердца способно стать причиной блокады кровотока и летального исхода, так как сжимающийся и расширяющийся воздушный пузырь делает невозможным перекачивание сердцем крови. Газовая эмболия может осложнять газовую гангрену, возникающую при инфицировании раны анаэробной инфекцией, когда скапливающиеся в пораженной ткани газы прорываются в кровоток.
Кровотечение
Кровотечение — выход крови из просветов сосудов или полостей сердца при нарушении целостности их стенки или повышения ее проницаемости. Варианты этого процесса разнообразны, их дифференцируют в зависимости от причины, механизмов и места возникновения, от того, куда изливается покидающая сосудистое русло кровь, от временных характеристик процесса. Так, если кровь изливается вовне, говорят о наружном кровотечении, если в полость организма — о внутреннем.
К наружным кровотечениям относят также кровохарканье, кровотечение из носа, рвоту кровью, выделение крови с калом, из полости матки. Примерами внутреннего кровотечения являются поступление крови в брюшную полость (гемоперитонеум), в полость перикарда (гемоперикардиум) или плевры (гемоторакс).
Накопление крови в результате внутреннего кровотечения непосредственно в тканях называют кровоизлиянием. При этом в зависимости от места кровотечения, его объема и скорости возможно пропитывание ткани кровью, раздвигающей ее клеточные и стромальные элементы, заполняющей периваскулярные зоны (геморрагическая инфильтрация), либо образование полости, выполненной кровью, сопровождающееся деструкцией тканевых структур (гематома). Плоскостные кровоизлияния в коже или слизистых оболочках называют кровоподтеками, а мелкие точечные — петехиями или экхимозами.
Гематомы образуются в результате повреждения вен или артерий. Чем выше давление крови в поврежденном сосуде и меньше сопротивление окружающей ткани, тем больше вероятность образования гематомы, а не геморрагической инфильтрации. В мягких тканях бедра, в области ягодиц гематома может вмещать 100–200 мл и более, в забрюшинной клетчатке — 1–2 л крови. Иногда между поврежденной артерией и возникшей в тканях полостью сохраняется прямая связь, пульсовая волна передается на содержащуюся в ней кровь. Гематома в таком случае называется пульсирующей.
В основе кровотечений лежат следующие механизмы: разрыв, разъедание либо резкое повышение проницаемости сосудистой стенки при отсутствии ее видимых повреждений.
Кровотечение путем разрыва стенки сосуда или сердца происходит в результате травмы либо патологического процесса, снижающего ее прочность. Ими могут быть заболевания воспалительного или аутоаллергического характера, при которых поражаются и некротизируются гладкомышечные и волокнистые структуры сосудистой стенки (сифилис, туберкулез, медионекроз, ревматизм, атеросклероз, инфаркт и др.). Разрыву способствует внезапное повышение давления, часто оказывающееся пусковым фактором при наличии других предрасполагающих изменений. Таков механизм спонтанных надклапанных разрывов аорты при медионекрозе, разрывов врожденных и приобретенных аневризм, различных сосудов, когда стенки их изменены и истончены вследствие деструкции эластического каркаса и рубцевания. Разрывы подобного характера наиболее часты при ИМ, в артериях эластического типа, менее адаптированных к резким перепадам давления, чем артерии с хорошо развитым мышечным слоем, в венах, которые расположены под слизистыми оболочками или на поверхности, выбухающей в просвет полых органов, например пищевода или прямой кишки.
Иногда тяжелое внутреннее кровотечение возникает при разрыве капсулы и ткани органа вследствие повышения внутриорганного давления либо патологической перестройки его структуры. Таковы последствия разрывов печени и ее капсулы при родовой травме, метастазах рака, стенки и сосудов матки при осложненных родах, селезенки и ее капсулы при портальной гипертензии, кровяных инфекциях (малярия, возвратный тиф).
Кровотечение в результате разъедания стенки сосудов (аррозивное кровотечение) возникает при вторичном ее вовлечении в патологический процесс. Это отмечают при инфильтративном росте злокачественной опухоли, разрушающей сосудистую стенку, под действием протеолитических ферментов в очаге нагноения, в дне язвы желудка под влиянием желудочного сока, в процессе распада некротизированной ткани опухолевого узла или при специфическом воспалении (казеозный некроз и распад стенки туберкулезной каверны), вследствие разрушения стенки и сосудов фаллопиевой трубы ворсинками хориона при внематочной беременности, в кишечнике, как осложнение брюшного тифа или дизентерии.
Кровотечение путем диапедеза представляет собой просачивание крови через сосудистую стенку при повышении ее проницаемости. Эритроциты и плазма крови выходят из сосудов через расширенные межэндотелиальные стыки и временные трансэндотелиальные канальцы, образуемые при слиянии микропиноцитозных пузырьков. После прохождения эритроцита, как бы «перетекающего» в периваскулярную зону, межклеточная щель восстанавливается до исходного состояния. Диапедезные геморрагии возникают, как правило, из артериол, венул, капилляров и проявляются в виде мелких кровоизлияний с геморрагической инфильтрацией, иногда принимающих системный характер (геморрагический синдром).
Причины диапедезных кровоизлияний многочисленны и разнообразны. Основными факторами их возникновения являются гипоксия, интоксикация и аутоаллергические процессы, нарушение нейрососудистой регуляции, реологических свойств крови и системы гемостаза, повышение внутрисосудистого давления, авитаминозы, радиационные воздействия. В связи с этим диапедезные кровоизлияния сопутствуют различным заболеваниям сердечнососудистой системы, поражениям головного мозга, гипертонической болезни и симптоматическим гипертензиям. Также отмечаются при коллагеновых и ряде тяжелых инфекционных заболеваний, при воспалительных процессах, вызванных высокотоксическими веществами или микроорганизмами, при болезнях системы кроветворения, коагулопатиях, острой форме лучевой болезни.
Диапедезные кровоизлияния лежат в основе развития петехий и экхимозов. При атеросклерозе возможны диапедезные кровоизлияния в атеросклеротические бляшки. Распространенные диапедезные кровоизлияния спонтанного характера, отмечаемые при целом ряде заболеваний, являются одним из их ведущих проявлений и обозначаются как геморрагический диатез.
Повышенная кровоточивость мелких сосудов связана с комплексом изменений, включающим повышение проницаемости сосудистой стенки, нарушения состава и свойств крови, снижающие ее свертываемость. Так, при острой лучевой болезни происходит включение всех этих патогенетических звеньев. Геморрагические диатезы при наследственной патологии (недостаток или отсутствие тех или иных факторов свертывания крови) имеют первичный характер, тогда как при общих нарушениях в организме, например при болезнях системы кроветворения, С-авитаминозе, некоторых инфекциях, отравлениях мышьяком, фосфором, грибами, укусах змей, введении в организм избыточных доз антикоагулянтов, лучевой болезни и т.д., они являются осложнениями основного заболевания.
В зависимости от источника различают сердечное, артериальное, венозное, артериально-венозное и капиллярное кровотечения. По срокам они могут быть первичными и вторичными, ранними, поздними, рецидивирующими или повторными.
Первичные кровотечения имеют место при любом проникающем повреждении сосудистой стенки и по механизму своего возникновения относятся к геморрагиям. Вторичные кровотечения происходят из сосудов, травмированных в момент повреждения ткани или подвергшихся деструкции при дальнейшем течении патологического процесса, например вследствие разъедания при нагноении. Опасное вторичное кровотечение отмечают также при септическом расплавлении тромба, закрывшего дефект, который образовался в сосудистой стенке.
Исход и значение кровотечения для организма определяются видом, особенностями источника, продолжительностью, интенсивностью и объемом кровопотери. Иногда кровотечение имеет физиологический (менструальная геморрагия) или компенсаторный характер (викарные носовые кровотечения при нарушениях менструального цикла). Однако в большинстве случаев оно является патологическим процессом, вред которого для организма пропорционален объему кровопотери.
Наиболее опасны кровотечения из полостей сердца или крупных сосудов, которые, как правило, приводят к внезапной смерти. Опасность быстрой массивной кровопотери состоит не столько в потере эритроцитарной массы, сколько в резком уменьшении ОЦК. Это приводит к глубоким гемодинамическим нарушениям, когда защитные механизмы организма оказываются недостаточными для предупреждения геморрагического шока. В результате смерть от острого кровотечения наступает в течение нескольких минут без признаков общей анемии, до того как успевает мобилизироваться кровь из депо и периферических сосудов.
Повреждение сосудов сравнительно небольшого калибра при снижении гемостатических свойств крови или отсутствии квалифицированной помощи приводит к длительному кровотечению, продолжающемуся в течение нескольких суток, с потерей значительного суммарного объема крови. Смерть от наступающего острого малокровия обусловлена тканевой гипоксией. Так, обескровливание головного мозга вначале приводит к коллапсу, а снижение САД до 70 мм рт. ст. и потеря 40% гемоглобина вызывают острую анемию головного мозга с угрозой гибели. Ткани при таком кровотечении становятся бледными, тусклыми, селезенка сморщивается, ее пульпа становится дряблой. В почках резкое полнокровие мозгового вещества сочетается с обескровливанием коркового слоя вследствие рефлекторного спазма междольковых артерий и сброса крови по расположенным на границе артериовенозным анастомозам.
Незначительное по разовому объему, но длительно (в течение месяцев и лет) рецидивирующее кровотечение, например при пептической язве желудка, нарушении менструального цикла, неспецифическом язвенном колите, геморрое, существенно не нарушает общую гемодинамику, но приводит к развитию хронической постгеморрагической анемии.
При кровоизлияниях, наряду с объемом кровопотери, большое значение приобретает его локализация. Так, даже сравнительно небольшое кровоизлияние в ЦНС при АГ, разрыве аневризмы мозговых артерий приводит, как правило, к летальному исходу. Вместе с тем массивные кровоизлияния в клетчатку или мышцы часто не представляют прямой опасности для организма.
Кровь, инфильтрирующая ткань, резорбируется макрофагами и удаляется лимфатическими путями; образующая гематому — сворачивается. Гемоглобин высвобождается из эритроцитов и трансформируется вначале внутриклеточно, в макрофагах, в гемосидерин, а спустя 7–8 сут — в расположенный вне клеток гематоидин, придающий содержимому гематомы желто-коричневый цвет. В гематоме гемосидерин расположен по периферии, а гематоидин — в центре. При обширных кровоизлияниях отмечают так называемую гемолитическую желтуху, которая возникает в результате превращения гематоидина в билирубин.
Свернувшаяся кровь подвергается организации, начинающейся на 2–4-е сутки с периферии образовавшейся полости. При больших ее размерах формируется фиброзная сумка, заключающая детрит, который постепенно разжижается и резорбируется (кистозная гематома). Кровь, излившаяся в полости организма или образовавшая гематому, утрачивает свои бактерицидные свойства и становится питательной средой для микрофлоры. В результате возникает серьезная угроза нагноения, что может резко ухудшить состояние больного. Рассасывание крови, попавшей в брюшную, плевральную или перикардиальную полость, сопровождается образованием большого количества спаек.
Множественные кровоизлияния в ткани при геморрагических диатезах приводят ко вторичным изменениям. На поверхности кожных покровов и слизистых оболочек, непосредственно соприкасающихся с воздухом, иногда возникают эрозии и изъязвления, в головном мозгу — множественные петехии или геморрагические инсульты, в легких возможно образование ателектазов.
ЛИТЕРАТУРА
- Струков А.І., Сєров В.В. (2004) Патологічна анатомiя. Підручник (Пер. з рос.). 4-те вид., стереотипне. Факт, Харків. 864 с.
- Пальцев М.А., Аничков Н.М. (2001) Патологическая анатомия. Учебник. В 2 т. Т. 2, ч. 1. Медицина, Москва. 736 с.
- Зербино Д.Д., Лукасевич Л.Л. (1989) Диссеминированное внутрисосудистое свертывание крови: факты и концепции. Медицина, Москва. 256 с.
- Общая патология человека: Руководство для врачей (1990) Под ред. A.И. Струкова, В.В. Серова, Д.С. Саркисова. В 2 т. Т. 1. 2-е изд., перераб. и доп. АМН СССР. Медицина, Москва. 448 с.
- Патологическая анатомия. Курс лекций. Учебное пособие (1998) Под ред. B.В. Серова, М.А. Пальцева. Медицина, Москва. 640 с.
- Шлопов В.Г. (2004) Патологічна анатомія. Нова книга, Вінниця. 758 с.