Результаты клинических и экспериментальных работ последних лет не оставляют сомнений в том, что повышение активности РАС в значительной степени определяет развитие важнейших форм кардиоваскулярной патологии, прежде всего — ИБС, атеросклероза, являющегося ее патогенетической основой, АГ и метаболического синдрома, которые относятся к числу важнейших факторов риска ИБС [26, 216, 337]. Данные более чем столетних исследований свидетельствуют о доминирующей роли РАС, А ΙΙ и альдостерона в развитии и прогрессировании поражений почек, сердца и сосудов. Отмечено, что повышение плазменной активности ренина на 2 ед. сопровождается повышением риска развития ИМ на 25% [5], тогда как применение антагонистов РАС оказывает четко выраженное кардио- и ангиопротекторное действие у лиц с АГ, ИМ, сердечной недостаточностью, диабетической нефропатией даже при нормальной и сниженной активности циркулирующей РАС.
Особое значение в развитии кардиоваскулярной патологии имеют тканевые РАС, локализованные в сердце, сосудах и почках. Их активация сопровождается возникновением оксидативного стресса и воспаления с последующим развитием структурных, функциональных и регуляторых изменений, ремоделирования и нарушения реактивности миокарда и сосудистой стенки.
Участие оксидативного стресса в развитии АГ, почечного и кардиоваскулярного поражения интенсивно изучается с начала 90-х годов ХХ в. Установлено, что он участвует в повреждении эндотелия, развитии атеросклероза [168], почечной [280] и кардиальной патологии [297], тогда как применение в этих условиях антиоксидантов типа темпола — миметика супероксиддисмутазы или N-ацилцистеина оказывает защитное действие [280].
Особое внимание уделяется роли оксидативного стресса в ремоделировании сердца и сосудов, их гипертрофии и склерозировании, которые возникают в различных патологических условиях: при АГ, атеросклерозе, ожирении, метаболическом синдроме, СД, сердечной недостаточности [4]. Однако до настоящего времени остается спорным положение, в какой степени эти нарушения являются следствием оксидативного стресса, а в какой — причиной его развития. Одним из основных эффекторных звеньев РАС, который обусловливает как физиологическое, так и патологическое ее действие, является А II — многофункциональный пептид. Он активирует сокращение сердца, вазоконстрикцию, вызывает развитие воспаления и оксидативного стресса, а также ремоделирование миокарда и сосудистой стенки. Это ремоделирование включает как гипертрофию кардиомиоцитов, так и пролиферацию сосудистых гладкомышечных клеток, фибробластов, усиленный синтез ими коллагена, эластина и гликозоаминогликанов с увеличением объема внеклеточного матрикса [60].
На экспериментальных моделях АГ показана способность А II стимулировать интерстициальное накопление коллагена, тогда как применение ингибиторов АПФ сопровождалось регрессией гипертрофии и склероза миокарда даже при их применении в дозах, не вызывающих гипотензивного действия.
Гипертрофия кардиомиоцитов и стенок сердца является основным компенсаторным ответом на увеличение нагрузочного режима и позволяет нормализовать интрамиокардиальное напряжение и поддерживать адекватную сократительную и насосную функцию сердца. Выполнение физической работы в аэробных условиях приводит к физиологической гипертрофии ЛЖ, которая реализуется посредством увеличения как длины, так и толщины сакромеров. В то же время, патологическая гипертрофия сочетается с фиброзом миокарда и уменьшением его функционального потенциала.
Установлено, что в генезе патологического ремоделирования желудочка, которое отмечается при АГ, ИМ и сердечной недостаточности, ведущую роль играет кардиальная РАС с увеличением локального содержания ангиотензиногена, АПФ и А II [202], тогда как механизмы, определяющие физиологическую гипертрофию сердца, остаются в значительной степени дискуссионными. Полагают, что кардиальный А II, продуцируемый непосредственно в миокарде, участвует в развитии кардиосклероза, но не требуется для гипертрофии кардиомиоцитов. Тем не менее, кардиальная РАС принимает участие и в физиологической гипертрофии ЛЖ, так как блокада АТ1-рецепторов предупреждает ее развитие при увеличении нагрузки сопротивлением [16] и при аэробной физической работе [235, 236], независимо от активности циркулирующей РАС.
Выявлено, что оксидативный стресс и воспаление играют ведущую роль в ремоделировании и склерозировании сердца при активации кардиальной РАС. Это проявляется значительным увеличением выраженности повреждения на фоне угнетения антиоксидантной защиты при СД 2-го типа, сердечной недостаточности, старении даже при неизмененном уровне АД. Об этом свидетельствует также способность антиоксидантов и липопротеинов высокой плотности (ЛПВП), обладающих антиоксидантными свойствами, угнетать гипертрофию сердца. Установлена также четкая связь между выраженностью эффекта А ΙΙ, экспрессией NADPH-оксидазы и усиленной продукцией СОР, а повышенный уровень С-реактивного белка, фактора и маркера воспаления, был предвестником активации кардиальной РАС, повышения тканевой концентрации А II и развития гипертрофии сердца.
Помимо этого, выраженность гипертрофии, индуцированной А ΙΙ, существенно уменьшается при действии адипонектина, оказывающего противовоспалительное и антиоксидантное действие. Повышение уровня адипонектина в крови при применении пиоглитазона значительно уменьшает выраженность ремоделирования сердца, связанного с АГ [179].
Способность А ΙΙ вызывать склеротические изменения продемонстрирована также в исследовании, проведенном с участием 50 пациентов с гепатитом В по сравнению с 20 лицами группы контроля. Среднее плазменное содержание АПФ составило по группам 48,4 и 26,2 Ед./л соответственно. У 22 пациентов опытной группы отмечен выраженный фиброз печени, у 28 — умеренный. У лиц с выраженным фиброзом установлен более высокий уровень АПФ в плазме крови (60,3 по сравнению с 52,5 еЕд./л), разделительным было значение 52,5 Ед./л. При этом АД достоверно не отличалось у больных обеих групп [247].
Механизмы развития физиологической гипертрофии миокарда детально изучены на крысах в условиях длительной плавательной нагрузки. При ее умеренной выраженности прирост массы ЛЖ составил 13%, при значительной — 27%, масса правого желудочка увеличилась соответственно на 15 и 35%. Диаметр кардиомиоцитов ЛЖ увеличился на 13,2 и 14,4 мкм, при этом не изменилась экспрессия гена натрийуретического фактора как показателя патологической гипертрофии.
Развитие физиологической гипертрофии сердца сочеталось со снижением активности кардиальной РАС и уменьшением локального содержания ангиотензиногена при двух режимах нагрузки на 26 и 44%, содержания в миокарде А I на 26 и 44%, А ΙΙ — на 23 и 20%, снижением активности кардиального АПФ на 22 и 32%. В то же время, экспрессия АТ1-рецепторов в миокарде была повышена в группе животных с умеренной нагрузкой на 300%, нагрузкой средней выраженности — на 332% [226].
Одной из наиболее характерных черт физиологической гипертрофии сердца стало отсутствие увеличенной продукции активных форм кислорода и оксидативного стресса.
Помимо этого, нагрузка сопровождалась повышением активности так называемой альтернативной РАС. Эта система включает АПФ2, который играет значительную роль в регуляции функции сердца и в образовании А (1–7). А (1–7) является агонистом АТ2-рецепторов, принимает участие в вазодилатации и угнетает развитие фибротических изменений в миокарде, сосудистой стенке и почках [71, 140, 249].
Углубленный анализ позволил установить, что основным механизмом активации альтернативной РАС явилось изменение экспрессии специфических микро-РНК. Микро-РНК — эндогенные, мелкие и некодируемые РНК, которые функционируют как отрицательные регуляторы экспрессии генов, угнетают передачу сигнала или усиливают деградацию мРНК-мишеней. Они принимают участие в развитии ряда форм кардиоваскулярной патологии и патологической гипертрофии сердца, но играют также важную роль в развитии физиологической гипертрофии, связанной с аэробной работой. Экспрессия микро-РНК-27a и 27b является отрицательным регулятором активности АПФ, тогда как экспрессия микро-РНК-143 оказывает угнетающее действие на активность АПФ2 [24].
В проведенном исследовании экспрессия микро-РНК-27а повысилась у крыс после плавательной нагрузки в двух режимах соответственно на 26 и 83%, микро-РНК-27b — на 27 и 45%, экспрессия микро-РНК-143, регулирующей АПФ2, недостоверно снизилась в группе с мягкой плавательной нагрузкой и уменьшилась на 35% у крыс с умеренной нагрузкой. Соответственно этому активность АПФ2 в миокарде ЛЖ в двух группах крыс повысилась после нагрузки соответственно на 68 и 91% параллельно с усилением экспрессии АТ2-рецепторов в 1,6 и 2,2 раза и увеличением содержания А (1–7) на 160 и 120%. Отношение А (1–7)/А II увеличилось на 180 и 160%, свидетельствуя об увеличении образования А (1–7) из А II с участием АПФ2.
Полученные данные позволили авторам сделать вывод, что аэробная нагрузка плаванием у крыс приводила к гипертрофии ЛЖ, имеющей физиологический характер, не коррелировала с маркерами патологической гипертрофии, но сочеталась с повышением экспрессии АТ1- и АТ2-рецепторов, снижением активности кардиального АПФ и уровня А II, повышением уровня АПФ2 и А (1–7).
Эти данные означают, что физиологическая гипертрофия ЛЖ в результате увеличенной аэробной работы происходит с участием микро-РНК и повышением экспрессии АТ1-рецепторов без участия А II. Параллельно с этим повышается экспрессия АПФ2, А (1–7), АТ2-рецепторов. Эти данные означают, что неклассическая (альтернативная) РАС обусловливает физиологическую гипертрофию миокарда и противодействует активации классической, которая сопровождается развитием патологической гипертрофии с наличием фибротического компонента [248, 253, 363].
Данное положение подтверждается результатами ряда других исследований, в которых выявлено, что выраженность гипертрофии ЛЖ при нагрузке плаванием не определяется активацией кардиальной РАС, не коррелирует с активностью кардиального АПФ [84] и происходит без участия кардиального А ΙΙ [247]. Помимо этого, в других исследованиях установлено, что увеличение локальной продукции А ΙΙ в сердце прямо не сопровождается развитием гипертрофии за исключением случаев, когда избыток кардиального А ΙΙ попадает в циркуляцию и вызывает развитие АГ [254, 345].
В то же время, применение лозартана и блокирование активации АТ1-рецепторов предупреждало развитие гипертрофии ЛЖ при обоих типах нагрузки. Предположительно это определяется тем, что АТ1-рецепторы являются сенсором механической нагрузки, они способны прямо активироваться при растяжении миокарда и инициировать развитие гипертрофии ЛЖ. В ряде исследований показана усиленная экспрессия и активация АТ1-рецепторов в миокарде при нагрузке давлением и повышении интрамиокардиального напряжения [16, 84].
В ряде фундаментальных исследований отмечено, что способность АТ1-рецепторов инициировать гипертрофию кардиомиоцитов опосредована активацией рецепторов эпидермального фактора роста, их блокада устраняла у крыс развитие гипертрофии ЛЖ при АГ, связанной с действием А ΙΙ [149]. Отсутствие способности А ΙΙ вызывать гипертрофию миокарда даже при резком повышении его концентрации в нормальном сердце определяется незначительной выраженностью экспрессии АТ1-рецепторов в миокарде при отсутствии механического стресса.
Аэробная работа усиливала также экспрессию АТ2-рецепторов, специфичных по отношению к А (1–7), активность которых сочетается с угнетением митогенактивирующей протеинкиназы, а также усилением продукции оксида азота с развитием вазодилатации. Показано, что гиперэкспрессия АТ2-рецепторов у крыс способствует поддержанию функции ЛЖ в условиях ИМ [354].
Неоднократно отмечено, что при тренировке аэробными нагрузками значительно повышается защитная функция альтернативной РАС в виде усиления экспрессии АПФ2. Его отличительным свойством является способность вызывать деградацию А Ι с образованием неактивного продукта А (1–9), но еще в 500 раз более выраженную деградацию А ΙΙ с образованием А (1–7), проявляющего вазодилататорную и антипролиферативную активность.
Установлено, что повышение уровня АПФ2 в значительной степени определяет кардио- и ангиопротекторное действие ингибиторов АПФ и блокаторов АТ1-рецепторов [43, 90, 91]. У трансгенных мышей с гиперэкспрессией АПФ2 предупреждалось развитие патологической гипертрофии ЛЖ, а угнетение АПФ2 сочеталось с увеличением содержания А ΙΙ в миокарде и плазме крови с нарушением его сократимости и развитием дилатации ЛЖ. В ряде исследований, проведенных в условиях длительной плавательной нагрузки на крысах, также показано, что физиологическая гипертрофия ЛЖ сочеталась со снижением активности АПФ и повышением активности АПФ2, экспрессии АТ1- и АТ2-рецепторов, уменьшением кардиального содержания А Ι на фоне увеличения содержания А (1–7) [88].
Приведенные данные позволяют понять, почему аэробная физическая нагрузка рекомендуется как средство предупреждения развития АГ, сопровождающееся нормализацией АД.
Однако длительно удерживающаяся и прогрессирующая гипертрофия сердца в результате хронической перегрузки приобретает патологические черты, приводит к развитию сердечной недостаточности, является одной из основных причин внезапной смерти [100]. Ведущим механизмом, обусловливающим переход компенсаторной гипертрофии в патологическую, являются развитие стресса оксидативного и нитрозилирующего типов [111, 129, 215].
Оксидативный стресс развивается в тех случаях, когда продукция активных форм кислорода превышает антиоксидантный потенциал, а нитрозилирующий стресс является следствием взаимодействия оксида азота с СОР, образования пероксинитрита и ряда других реактивных соединений. В нормальных условиях еNOS секретирует оксид азота, оказывающий угнетающее влияние на развитие гипертрофии и фиброза миокарда, ремоделирование сердца. Однако кислородные радикалы связывают оксид азота, снижают его биодоступность, способствуют разобщению еNOS и продукции ею СОР. СОР и оксид азота в комплексе с пероксинитритом обусловливают развитие нитрозилирующего стресса.
Кислородные радикалы — нормальный компонент окислительного фосфорилирования, играющий существенную роль в функционировании физиологических сигнальных систем в качестве медиатора. Однако избыточная продукция СОР приводит к дисфункции клеток, пероксидации липидов, необратимому повреждению клеток и их гибели.
К активным формам кислорода относятся СОР, гидроксильный радикал и компоненты типа перекиси водорода, которые могут переходить в радикальную форму. СОР образуется как в результате активации ферментов типа NADPH-оксидазы и ксантиноксидазы, так и вследствие утечки из митохондрий при нарушении транспорта электронов по дыхательной цепи. Перекись водорода является субстратом образования гидроксильного радикала в реакции Фентона при ряде патологических ситуаций.
Активные формы кислорода стимулируют митогенактивирующие протеинкиназы с развитием патологической гипертрофии миокарда, ремоделирования матрикса и клеточной дисфункции.
В ряде фундаментальных исследований определено, что пролиферативное действие А II и ЭТ-1 в значительной степени обусловлено их способностью усиливать продукцию кислородных радикалов. Радикалы кислорода оказывают мощное влияние на образование внеклеточного матрикса, стимулируя пролиферацию кардиальных фибробластов и активируя MMPs, которые высвобождаются в неактивной форме и приобретают активность под действием активных форм кислорода. Они также усиливают экспрессию факторов транскрипции — κB, АР-1 и вызывают развитие воспалительного ответа, тогда как перекись водорода выступает в качестве медиатора клеточного повреждения и апоптоза. Наконец, радикалы кислорода прямо угнетают сократительную функцию миокарда, оказывая угнетающее влияние на белки, определяющие сопряжение возбуждения с сокращением.
Известно, что А II приводит к развитию гипертрофии сердца, которая длительное время рассматривается как следствие АГ. Однако применение А II в субпрессорных дозах также сопровождается увеличением массы миокарда, и прямое кардиотропное действие А II установлено также в культуре кардиомиоцитов. Независимость кардиотропного эффекта А II от влияния повышенного давления показана на крысах, получавших инфузию А II на фоне применения гидралазина, у которых сохранялся нормальный уровень АД, но развивалась выраженная гипертрофия миокарда.
В работах последних лет также неоднократно отмечено, что в основе кардиальных, сосудистых и почечных нарушений при длительной инфузии А II лежит развитие оксидативного стресса, независимо от изменений уровня АД.
В исследованиях, проведенных на культуре кардиомиоцитов, установлено также выраженное митогенное действие повышенного уровня ионов натрия в среде с развитием гипертрофии и увеличенным внутриклеточным содержанием белка [122]. Помимо этого, увеличенное потребление соли приводило к увеличению массы сердца in vivo у нормотензивных крыс и увеличивало выраженность гипертрофии у гипертензивных. У людей установлено наличие четкой зависимости между потреблением соли и индексом массы ЛЖ как при наличии, так и отсутствии АГ, а низкосолевая диета сопровождалась уменьшением выраженности гипертрофии миокарда, независимо от влияния на уровень АД [211].
Установлено, что в основе кардиотропного и ремоделирующего действия как А II, так и повышенной концентрации натрия, которое в значительной степени не зависит от влияния АД, лежит их способность инициировать развитие оксидативного стресса. Так, у крыс с непрерывной инфузией А II в течение 10 дней развивалась АГ, продукция СОР с миокарде увеличилась на 50%, продукция H2O2 — на 60–70% в сочетании с выраженным поражением сердца и почек [360]. Сопутствующее применение низкосолевой диеты не оказывало выраженного антигипертензивного действия, но полностью нормализовало системную продукцию СОР и перекиси водорода, уменьшало выраженность альбуминурии и гипертрофии сердца. Эти данные позволили сделать вывод, что защитное действие низкосолевой диеты на фоне повышенного уровня А II в крови опосредовано антиоксидантным эффектом в отсутствие существенного влияния на уровень АД, а длительное применение низкосолевой диеты может предупреждать поражение сердца и почек у лиц как с наличием, так и отсутствием АГ [260].
Результаты ряда исследований подтвердили, что кислородные радикалы, которые продуцируются NADPH-оксидазой, играют ключевую роль в гипертрофии и ремоделировании сердца при действии А II. Так, у нормальных мышей субпрессорные дозы А II вызывали гипертрофию сердца, которая не развивалась у мышей с генетическим отсутствием NOX2-изоформы NADPH-оксидазы [19].
Характерно, что устранение повышенной активности NADPH-оксидазы в условиях перегрузки сердца давлением предупреждало пролиферацию фибробластов, усиленную продукцию внеклеточного матрикса и развитие кардиофиброза, но не предотвращало гипертрофию кардиомиоцитов [120, 200]. Эти данные означают, что оксидативный стресс является ведущим механизмом развития только патологической, но не физиологической гипертрофии сердца.
Существенную роль в развитии оксидативного стресса и ремоделировании сердца при хронической сердечной недостаточности играет система ксантин/ксантиноксидазы. Повышенная ее экспрессия в сердце выявлена у пациентов с тяжелым течением сердечной недостаточности и у собак с сердечной недостаточностью, вызванной длительным навязыванием сокращений высокой частоты. Помимо этого, применение аллопуринола — ингибитора ксантиноксидазы, предупреждало ремоделирование сердца у мышей после ИМ и у мышей с дилатационной кардиомиопатией [308].
Усиленная митохондриальная продукция СОР также принимает участие в развитии миокардиального оксидативного стресса и последующей сердечной недостаточности. Показано, что гиперэкспрессия митохондриального антиоксиданта пероксиредоксина-3 значительно уменьшала выраженность гипертрофии ЛЖ у мышей после ИМ. Однако в развитии гипертрофии миокарда, вызванной повышенной физической нарузкой, не установлена роль как митохондриальной продукции СОР, так и в целом оксидативного стресса [199].
Существенное модулирующее влияние на гипертрофическое ремоделирование сердца играют антиоксидантные ферменты. Так, угнетение тиоредоксина-1 сопровождалось значительным увеличением выраженности оксидативного стресса и гипертрофии миокарда при перегрузке сердца объемом, тогда как гиперэкспрессия этого фермента сочеталась с уменьшением выраженности стресса и ремоделирования сердца в этих условиях [352, 353].
По данным ряда исследований, нахождение гипертензивных крыс на диете с увеличенным содержанием сахарозы или фруктозы приводит к значительному повышению летальности и степени дисфункции ЛЖ, что связано с развитием резко выраженного оксидативного стресса [279]. В этих условиях значительное снижение концентрации СОД в миокарде сочеталось с пропорционально выраженным усилением оксидантного поражения.
В исследовании, проведенном на мышах, перегрузка сердца давлением в контрольных условиях сопровождалась увеличением его массы на 22%, тогда как у мышей, содержащихся на диете с добавлением фруктозы в течение 8 нед, отмечена значительно более выраженная гипертрофия миокарда, и масса сердца увеличилась на 55%. При сочетании перегрузки сердца с обогащением диеты фруктозой и применением антиоксиданта темпола прирост массы сердца уменьшился до 18%. Выраженность систолического укорочения волокон ЛЖ у мышей, находившихся на диете с фруктозой, уменьшилась с 38% в контроле до 22%, на фоне темпола — до 33%. Изменения показателей пероксидации липидов также заметно уменьшились на фоне применения темпола [297]. В других исследованиях показано, что наличие фруктозы в диете у крыс сопровождалось развитием оксидативного стресса и воспаления в стенке аорты, значительным увеличением плотности АТ1-рецепторов в сосудистой стенке с нарушениями ее реактивности и развитием АГ [4, 222].
Еще несколько лет назад дoминировало положение, в соответствии с которым А ΙΙ оказывает прямое трофическое действие на кардиомиоциты и фибробласты сердца при АГ, являясь непосредственной причиной гипертрофии и ремоделирования сердца [254]. И в настоящее время ряд исследователей продолжают рассматривать А ΙΙ как прямой медиатор гипертрофии и фиброза миокарда, базирующийся на действии А ΙΙ, установленном на культуре кардиомиоцитов. Однако в ряде работ последних лет отмечено, что А ΙΙ определяет ремоделирование сердца только в условиях, когда он сочетается с другими гуморальными, механическими или патологическими стимулами. Поэтому вопрос о том, в какой степени защитное действие ингибиторов АПФ и блокаторов АТ1-рецепторов является прямым эффектом, а в какой определяется их гипотензивным действием, остается еще открытым.
При АГ отмечается гипертрофия ЛЖ, которая начально является компенсаторным механизмом и включает как гипертрофию кардиомиоцитов, так и периваскулярный и интерстициальный фиброз. Однако, в соответствии с данными ряда исследований последних лет, А II не является фактором, непосредственно инициирующим ремоделирование сердца; он только усиливает ремоделирование, связанное с увеличенной постнагрузкой, в результате развития в сердце воспаления низкой градации [349].
В то же время, данные других исследований свидетельствуют о том, что А ΙΙ прямо инициирует ремоделирование сердца при АГ, независимо от уровня АД, хотя отделить прямое действие А ΙΙ от эффекта, связанного с повышением АД, не всегда представляется возможным.
Для решения этого вопроса была воспроизведена модель АГ у крыс с низким уровнем циркулирующего А ΙΙ и высоким — кардиального. В этой модели значительно увеличенное интрамиокардиальное содержание А II не сопровождалось влиянием на структуру сердца в условиях нормального АД, но значительно усиливало гипертрофию и ремоделирование ЛЖ при наличии сопутствующей АГ. Это проявлялось выраженным апоптозом кардиомиоцитов, макрофагальной инфильтрацией, усиленной экспрессией NADPH-оксидазы и TФР-β1 [347, 348].
Выраженность ремоделирующего действия А II существенно увеличивалась также при АГ, сочетающейся с выраженной вариабельностью АД. В этих условиях отмечена более интенсивная активация кардиальной РАС, усиленная экспрессия TФР-β1, ангиотензиногена и АТ1-рецепторов. Параллельно происходила активация локального воспаления в результате увеличенной секреции МСР-1, макрофагальной инфильтрации, а сочетание этих факторов приводило к увеличению выраженности гипертрофии миокарда и кардиофиброза [162].
Значительная гипертрофия сердца с увеличением индекса массы ЛЖ у лиц без АГ отмечена также при действии альдостерона. Этот эффект был особенно выражен на фоне повышенного уровня натрия в плазме крови и не отмечался у лиц в нижнем тертиле его содержания. Показано также, что уменьшение массы сердца при применении ингибиторов АПФ и блокаторов АТ1-рецепторов сочеталось и коррелировало не столько со снижением АД, сколько со снижением плазменной концентрации натрия и альдостерона [166].
Тем не менее, вопрос о том, в какой степени ремоделирование сердца является следствием локальной продукции А ΙΙ в сердце, не решен и в настоящее время. Многие исследователи полагают, что способность ингибиторов РАС угнетать ремоделирование сердца и вызывать его регрессию в значительной степени связана с блокадой прямого действия локально продуцируемого А ΙΙ на миокард. Однако отмеченные эффекты возникали на фоне выраженного гипотензивного эффекта препаратов и в определенной степени могли быть его следствием. Несмотря на то что в условиях клеточной культуры продемонстрирована способность А II индуцировать гипертрофию кардиомиоцитов, стимуляция миокардиальной продукции А ΙΙ с увеличением его концентрации даже в 20–50 раз не сопровождалась развитием гипертрофии сердца, и в большинстве случаев отмечено только развитие легкого фиброза [311].
В большинстве исследований закономерное и выраженное ремоделирующее действие локально продуцируемого А ΙΙ проявлялось только в сочетании с другими факторами при АГ, СД 2-го типа, при активации минералокортикоидных рецепторов, но не возникало изолировано. С другой стороны, повышение АД при ДОКА-солевой АГ без увеличенной кардиальной продукции А ΙΙ и на фоне угнетения циркулирующей РАС также не сочеталось с выраженным ремоделированием сердца [60, 61].
В соответствии с результатом анализа и трактовки данных многих клинических наблюдений решающее значение в способности ингибиторов РАС оказывать влияние на долговременные исходы имеет их антигипертензивный эффект. Хотя в ряде крупных исследований установлено, что кардиопротекторное действие ингибиторов РАС возникает и на фоне незначительного снижения АД, полагают, что при этом не учитывается возможность существенно более выраженного уменьшения ночного АД (на 17/8 мм рт. ст.) при снижении амбулаторного АД только на 3,3 мм рт. ст. [295].
Значимость антигипертензивного действия ингибиторов АПФ как фактора, лежащего в основе их кардиопротекторного действия, показана в исследовании эффекта 4 групп антигипертензивных препаратов, которые включали диуретики, блокаторы β-адренорецепторов, блокаторы медленных кальциевых каналов и ингибиторы РАС. Отмечен примерно равный эффект терапии на кардиоваскулярный риск, включая ИМ и общую смертность [17]. В исследовании VALUE (Valsartan Antihypertensive Long-term Use Evaluation) также не установлено различий в уменьшении тяжести течения и смертности при применении ингибиторов АПФ и блокаторов кальциевых каналов у пациентов с АГ на протяжении 4,2 года наблюдения [147]. Более того, у лиц с СД 2-го типа отмечено менее выраженное влияние ингибиторов АПФ на уровень АД и снижение риска развития кардиоваскулярных исходов по сравнению с другими классами препаратов.
Тем не менее, этот вопрос остается еще дискуссионным и, в противоположность приведенным данным, в исследовании LIFE (Losartan Intervention For Endpoint reduction in hypertension) установлено значительное превосходство ингибиторов РАС в уменьшении гипертрофии ЛЖ и смертности у пациентов с АГ по сравнению с блокаторами β-адренорецепторов [336]. С другой стороны, в исследовании Second Australian National Blood Pressure Trial, проведенном с участием пациентов пожилого возраста с АГ, установлено только сомнительное превосходство ингибиторов АПФ по сравнению с диуретиками [265]. По мнению авторов, эти данные ставят под сомнение наличие прямого влияния РАС на сердце, не связанного с развитием АГ.
Большинство данных, свидетельствующих о прямом трофическом действии А ΙΙ на кардиомиоциты, получены в условиях культуры и при их анализе не учитывалось сопутствующее влияние А ΙΙ на сосудистый тонус, секрецию альдостерона, реабсорбцию натрия, объем циркулирующей крови, уровень АД, хотя эти факторы в целом могут в значительной степени определять конечный эффект А ΙΙ. Полагают, что данные исследований с применением субпрессорных доз А ΙΙ также не являются абсолютным доказательством прямого кардиотропного действия А II, так как нельзя исключить, что эти субпрессорные дозы вызывают повышение АД в ночное время.
Практически во всех исследованиях со стимуляцией кардиоспецифической продукции А ΙΙ не установлено его прямого влияния на гипертрофическое ремоделирование сердца, хотя это не исключает участия кардиальной РАС в ремоделировании помимо А ΙΙ, прежде всего — через усиленную экспрессию АТ1-рецепторов.
В интактном сердце не выявлено экспрессии АТ1-рецепторов кардиомиоцитами, тогда как их гиперэкспрессия у новорожденных мышей при использовании промоутера сопровождалась резким увеличением размера предсердий и смертью от блокады сердца даже в отсутствие увеличенного миокардиального содержания А II. Показано, что у трансгенных мышей с усиленной в 200–400 раз экспрессией АТ1-рецепторов развивалась выраженная гипертрофия ЛЖ с увеличением размера кардиомиоцитов, гипертрофия быстро прогрессировала с развитием сердечной недостаточности и летального исхода [232]. Выраженная экспрессия АТ1-рецепторов в миокарде у крыс через 1 нед после ИМ установлена и в областях, окружающих зону инфаркта. Эти данные означают, что характер экспрессии АТ1-рецепторов может быть фактором, определяющим действие РАС на сердце.
Хотя в соответствии с рядом данных гиперэкспрессия АТ1-рецепторов в миокарде более значима для развития гипертрофии сердца, чем локальная концентрация А ΙΙ, другие данные противоречат этому положению. Так, у мышей с селективным отсутствием АТ1-рецепторов в почках при инфузии А ΙΙ в течение 4 нед не отмечено повышения АД и не развивалась гипертрофия сердца, несмотря на наличие АТ1-рецепторов в сердце и значительное повышение концентрации циркулирующего А ΙΙ.
На основании ряда подобных данных многие исследователи продолжают рассматривать гипертрофию сердца под влиянием А ΙΙ преимущественно как следствие его гипертензивного действия. При этом не исключают и прямого эффекта А ΙΙ, но только в сочетании с другими механическими, гуморальными или патологическими стимулами, особенно усиливающими экспрессию миокардиальных АТ1-рецепторов типа гемодинамической перегрузки сердца, развития воспаления и оксидативного стресса. На этом основании остается доминирующим положение, что ведущим принципом лечения лиц с АГ для предупреждения прогрессирования и индукции регрессии гипертрофии сердца должен быть эффективный контроль АД [253].
С целью отделения кардиотропного действия А II от влияния гемодинамических факторов использовались трансгенные мыши с гиперэкспрессией белка, стимулирующего высвобождение А II в миокарде, на фоне воспроизведения ДОКА-солевой АГ для подавления активности циркулирующей РАС и уменьшения плазменного содержания А ΙΙ. На этой модели определялось, в какой степени А II, продуцируемый интракардиально, способен индуцировать воспаление, оксидативный стресс, апоптоз, гипертрофию сердца и кардиофиброз посредством связывания с АТ1-рецепторами.
Как у опытных, так и контрольных животных с АГ, но без увеличенной кардиальной продукции А II, отмечены аналогичные по выраженности повышение АД и изменение функции сердца. Однако у опытных животных с усиленной локальной продукцией А ΙΙ развивались более выраженные гипертрофия миокарда, интерстициальный и периваскулярный фиброз, инфильтрация макрофагов, экспрессия NADPH-оксидазы и TФР-β1. Валсартан частично устранял эти эффекты у опытных животных, но не у контрольных. Эти данные позволили сделать вывод, что у животных с неизмененной гемодинамической нагрузкой кардиальный А II не оказывает влияния на размеры и функцию сердца. Однако у животных с АГ А ΙΙ, продуцируемый локально, через АТ1-рецепторы усиливает воспаление, оксидативный стресс и гибель клеток, вызывая гипертрофию сердца и развитие кардиофиброза [348].
Данные многих современных исследований свидетельствуют о том, что радикалы кислорода играют критическую роль в повреждении сердца, развитии интерстициального фиброза при действии как А II, так и альдостерона и ДОКА — агонистов минералокортикоидных рецепторов, а основным источником кислородных радикалов является NADPH-оксидаза. Однако в проведенном исследовании в базальных условиях А II не оказывал влияния на NADPH-оксидазу, не приводил к развитию оксидативного стресса, и эти эффекты развивались только при сочетании кардиальной гиперэкспрессии А II с ДОКА-солевой АГ даже при угнетении циркулирующей РАС.
Известно, что повышение нагрузки на сердце и увеличение интрамиокардиального напряжения сочетаются с резким повышением экспрессии миокардиальных АТ1-рецепторов, которая практически не отмечается при отсутствии механического стресса. По-видимому, именно это сочетание создает условия для реализации возможности А II инициировать воспаление и оксидативный стресс, приводя к развитию интерстициального фиброза, гипертрофии и ремоделирования сердца.
3.1. Роль РАС в развитии АГ
На протяжении многих десятилетий значительное внимание уделялось и продолжает уделяться в настоящее время определению роли РАС в патогенезе АГ, однако и в этой проблеме еще сохраняется множество нерешенных вопросов. По многим аспектам роль РАС в развитии АГ остается противоречивой, и прежде всего в связи с тем, что у большинства лиц с АГ не выявлено повышенного уровня ренина в плазме крови, а нарушения почечного кровотока при эссенциальной АГ имеют вторичный характер и не являются причиной повышения АД. Это положение доказывается тем, что увеличение сопротивления почечных сосудов, их структурные изменения и уменьшение почечного кровотока являются признаками уже устоявшейся, но не развивающейся АГ.
В исследовании, проведенном в 1972 г. в Нью-Йорке с участием 1717 лиц с АГ, установлена прямая связь между активностью ренина в плазме крови и риском развития ИБС. За 8,3 года наблюдения зарегистрировано 107 летальных исходов, из них 70 — кардиоваскулярной природы, среди которых 27 — в результате ИМ. Риск его развития составил 14,7; 5,6 и 2,8 на 100 пациенто-лет у лиц соответственно с высокой, нормальной и сниженной активностью ренина в плазме крови. При этом не отмечено связи между активностью ренина и частотой развития инсульта, некардиальной смерти и смерти от всех причин. Отсутствие подобной зависимости отмечено также у лиц с АГ, принимающих блокаторы β-адренорецепторов, хотя у них выявлено повышение риска развития ИМ [204].
Однако наличие зависимости между плазменной активностью ренина и кардиоваскулярными исходами не было подтверждено в популяционном исследовании, проведенном в Лондоне с участием 863 лиц. По данным Фремингемского исследования, которое включало 3532 участника, активность ренина статистически также не отличалась у лиц с различным уровнем кардиоваскулярного риска при том, что было отмечено наличие независимой связи между АД и риском развития кардиоваскулярных явлений [233].
В исследовании, результаты которого были опубликованы в 1997 г., активность ренина прямо коррелировала с частотой развития ИМ, но не цереброваскулярных проявлений [9], тогда как в другом исследовании отмечено наличие обратной зависимости между активностью ренина в плазме крови и уровнем АД и отсутствие достоверной зависимости между уровнем АД, активностью ренина с одной стороны и риском развития коронарных явлений — с другой [113]. В исследовании NPHS кардиоваскулярный риск у лиц с активностью ренина в плазме крови в верхнем тертиле по отношению к нижнему составил 1,26, но различие не достигало статистической значимости [204].
На основании результатов многочисленных клинических наблюдений предположение о ведущей роли увеличенного высвобождения ренина и повышения активности циркулирующей РАС в патогенезе АГ и ее исходов было оспорено многими авторами. В ряде популяционных исследований установлено наличие обратной зависимости между уровнем систолического АД (в меньшей степени — диастолического) и активностью ренина в плазме крови, которая снижалась на 8,4% на каждое стандартное отклонение систолического АД. Однако эта зависимость была слабой, и изменения АД определяли не более чем 1% вариаций плазменной активности ренина.
В исследовании 89 пациентов с АГ активность ренина была сниженной относительно группы контроля, сопоставимой по возрасту и полу, а в целом по популяции пациентов с АГ установлено умеренное снижение активности ренина в плазме крови параллельно с уменьшением выраженности ее угнетения при применении мочегонных препаратов или при солевой нагрузке.
Нет также единого мнения относительно влияния повышенной активности РАС на риск развития конечных кардиоваскулярных точек. В соответствии с данными ряда исследований, низкорениновая форма сочетается со значительно меньшим риском кардио- и цереброваскулярных осложнений по сравнению с высокорениновой. Так, при наблюдении 219 пациентов с АГ в течение 10 лет ни у одного из 59 лиц с низкой активностью ренина в плазме крови не было отмечено кардио- и цереброваскулярных явлений, тогда как у лиц с нормальной и высокой активностью ренина они выявлены соответственно в 11 и 14% случаев. В то же время, в популяционном исследовании, проведенном в 1983 г., у пациентов с низкорениновой формой АГ отмечена более высокая частота развития кардиальных и церебральных явлений, что авторы объясняли более высоким уровнем АД [204].
У лиц с АГ активность циркулирующей РАС резко варьирует; примерно у ⅓ пациентов с АГ отмечается низкое содержание ренина, сочетающееся с повышенным уровнем альдостерона. Повышенный плазменный уровень ренина выявлен примерно у 15% пациентов, а у большинства (50–60%) он остается в пределах нормы.
Однако эта норма характерна для нормотензивных пациентов, тогда как при АГ уровень ренина в плазме крови даже в пределах нормальных значений может рассматриваться как повышенный, и в этом случае активация циркулирующей РАС у лиц с АГ будет характерна для более чем 70% пациентов. Помимо этого, даже у оставшихся 30% может происходить активация локальных РАС, что особенно характерно для почечной РАС. Поэтому применение у таких пациентов ингибиторов РАС не только оказывает антигипертензивное действие, но и значительно задерживает развитие нефросклероза.
Тем не менее, несмотря на сохраняющиеся в настоящее время противоречия и спорные положения, участие РАС в патогенезе АГ в настоящее время уже не может вызывать сомнений. Повышение плазменного уровня А II и альдостерона в сочетании с гипертрофией ЛЖ и кардиофиброзом неоднократно установлено у лиц с метаболическим синдромом и резистентной АГ [34–36, 110].
Помимо этого, воспаление и явления фиброза в сердце при развитии его недостаточности большинство исследователей рассматривают как следствие активации миокардиальной РАС с повышением локальной концентрации А II и альдостерона даже при нормальном или сниженном уровне в циркуляции ренина и А II. Поэтому выраженный клинический эффект в значительно большей степени проявляется при фармакотерапии, которая не только оказывает антигипертензивное действие, но и предупреждает или ослабляет ремоделирование сердца и сосудистой стенки.
Установлено, что лечение АГ сопровождается снижением риска развития цереброваскулярных поражений на 48%. В то же время, снижение риска развития ИБС выглядит менее впечатляющим и не превышает 16%, и даже достижение уровня АД 120/80 мм рт. ст. у лиц с АГ не устраняет полностью риска развития ИБС. Исследования показали, что изменения структуры и функциональных свойств артериальных сосудов у многих лиц с АГ отмечают еще до повышения АД, и ведущей их причиной является повышение активности РАС, в меньшей степени циркулирующей и в большей — тканевых.
Ингибиторы АПФ и блокаторы рецепторов А II значительно более эффективны, чем другие антигипертензивные препараты, в нормализации сопротивления почечных сосудов, восстановлении ауторегуляторных свойств сосудов мозга, несмотря на сопоставимое снижение АД и независимо от изменений активности ренина в плазме крови. Результаты недавно проведенных исследований подтвердили, что антигипертензивные препараты, не влияющие на сосудистое и кардиальное ремоделирование, не способны длительно поддерживать терапевтический эффект после отмены, который определяется прежде всего нормализацией структурных свойств сердца и сосудистой стенки.
В ряде крупных исследований подтверждено наличие зависимости между классом применяемых антигипертензивных препаратов и выраженностью регрессии гипертрофии миокарда и сосудистой стенки, которая в большей степени характерна для препаратов, угнетающих РАС, чем для диуретиков, и особенно вазодилататоров прямого действия. Показано, что частота развития конечных кардиоваскулярных точек и уменьшение массы ЛЖ у лиц с АГ в течение 1 года наблюдения значительно более выражены при применении ингибиторов АПФ и блокаторов рецепторов А II, чем блокатора β-адренорецепторов атенолола, тогда как АД снижалось примерно в равной степени. Это означает, что угнетение РАС при АГ оказывает и прямое протекторное действие, независимое от изменений АД [67].
В других исследованиях также подтверждено, что угнетение активности РАС при проведении антигипертензивной терапии сочетается со значительно более выраженным защитным действием, чем аналогичное снижение АД, но достигаемое путем применения антигипертензивных препаратов других классов. Показано, что при лечении диабетической АГ риск ИМ был вдвое ниже при применении ингибиторов АПФ, чем блокаторов медленных кальциевых каналов.
Результаты ряда крупных клинических исследований также свидетельствует о выраженном защитном эффекте антигипертензивной терапии даже при отсутствии существенного влияния на уровень АД. Так, в исследовании RENAAL у 1513 пациентов с СД 2-го типа и нефропатией применение лозартана оказывало выраженное протекторное действие при минимальном влиянии на AД. Применение ирбесартана у 590 гипертензивных пациентов с СД 2-го типа приводило к снижению риска развития клинической нефропатии на 40–70% (в зависимости от дозы) также при незначительном влиянии на уровень АД [322]. Защитный эффект угнетения РАС и повышение выживаемости были отчетливо выражены у 3773 пациентов с СД 2-го типа, у которых летальность на протяжении 3 лет наблюдения снизилась в среднем на 60% [286].
В соответствии с современными представлениями, в патогенезе АГ существенную, а порой и доминирующую роль играет повышение активности тканевых РАС, часто не сочетающееся с активацией циркулирующей РАС [296]. Поэтому вещества, блокирующие действие РАС, остаются препаратами выбора в терапии АГ, независимо от ее формы и выраженности.
При детальном анализе результатов, установленных на больших массивах пациентов, установлено, что снижение уровня ренина у многих лиц как с наличием, так и отсутствием АГ имеет вторичный характер и происходит в результате высокого потребления соли. Это положение подтверждено при исследовании здоровых добровольцев, у которых высокое потребление соли (100–200 ммоль/сут) сопровождалось снижением уровня ренина в плазме крови до минимально определяемого уровня.
На основании этих данных было высказано предположение, что при низкорениновой АГ преобладает объемный фактор, связанный с повышенной активностью минералокортикоидов, задержкой натрия и жидкости, при высокорениновой — вазоконстрикторный, связанный с активацией симпатической нервной системы и сопутствующей усиленной секрецией ренина почками.
Это предположение было подтверждено результатами исследования, проведенного в 1970 г., в котором было выявлено, что антигипертензивный эффект блокаторов β-адренорецепторов в значительной степени определяется угнетением секреции ренина и снижением его уровня в плазме крови примерно на 70% [171]. Параллельно уменьшалась секреция альдостерона, что затем было подтверждено во многих других исследованиях [23, 290]. Эти данные стали основанием для разработки дифференцированных схем лечения лиц с АГ — при низкорениновой форме с применением диуретиков, при высокорениновой — блокаторов β-адренорецепторов.
Однако применение этих схем в клинических условиях не дало достаточно убедительного подтверждения данной гипотезы. В частности, было установлено, что антигипертензивное действие блокаторов β-адренорецепторов при высокорениновой АГ сочеталось с незначительным влиянием на плазменный уровень ренина, а пропранолол в высоких дозах оказывал антигипертензивное действие даже при низкорениновой форме.
Помимо этого, в последующих исследованиях не отмечено наличия у лиц с низкорениновой АГ закономерно повышенной минералокортикоидной активности. Уровень альдостерона в плазме крови не превышал в большинстве случаев нормальных значений, не отмечалось снижения плазменного уровня калия, хотя эти случаи характеризовались отсутствием угнетения секреции альдостерона при солевой нагрузке. В соответствии с другой гипотезой объемная АГ определялась централизацией кровообращения в результате снижения растяжимости стенки венозных сосудов.
Во многих крупных клинических исследованиях последних лет было показано, что у пациентов с низкорениновой формой АГ более эффективна антигипертензивная терапия с применением диуретиков, с высокорениновой — ингибиторов РАС и блокаторов β-адренорецепторов [171]. Высокорениновая форма АГ характеризуется увеличенным сердечным выбросом, что соответствует представлениям о повышенной симпатической активности. В подобных случаях высокая антигипертензивная эффективность присуща как ингибиторам АПФ, так и блокаторам β-адренорецепторов.
Трактовка значимости ренина как показателя активности РАС в патогенезе АГ существенно затруднена в связи с тем, что изменения содержания ренина в плазме крови могут быть как причиной развития АГ, так и следствием повышения АД или, чаще всего, сочетания обоих факторов. Особенно затруднена оценка значимости активности ренина в плазме крови как причины развития АГ у лиц пожилого возраста. Для них характерен, как правило, повышенный риск дистрофических изменений в почках типа мягкого нефросклероза с уменьшением продукции ренина, а также в стенке сонных артерий с ослаблением афферентной активности каротидных барорецепторов. Сочетание этих причин может существенно способствовать снижению активности ренина в плазме крови и развитию низкорениновой АГ нейрогенной природы.
К важнейшим факторам регуляции секреции ренина относится А II, который угнетает высвобождение ренина по принципу отрицательной обратной связи. Поэтому снижение уровня ренина в плазме крови не всегда является отражением низкой активности РАС, особенно в острых ситуациях.
Существенное значение в зависимости между активностью РАС и развитием АГ имеют изменения реактивности сосудов, прежде всего — сосудов почек, на действие А II, которая существенно повышается при высокосолевой диете и гипернатриемии. Изменение выраженности ответа сосудов этих тканей на А II обозначается как модуляция, и она отсутствует примерно у 45% пациентов с нормальной и высокорениновой АГ.
Роль отсутствия феномена модуляции в патогенезе АГ остается еще недостаточно изученной. Полагают, что в основе этой патологии лежат генетические факторы; у подобных лиц АГ часто имеет семейный характер, даже у нормотензивных потомков с отсутствием модуляции отмечается значительно ослабленная продукция альдостерона в ответ на инфузию А II. Коррекция этих нарушений с помощью АПФ свидетельствует о том, что в ее основе лежит усиленная продукция А II. С другой стороны, выраженность гипертензивного действия А II в этих условиях значительно уменьшается в связи с ослаблением способности А II индуцировать секрецию альдостерона надпочечниками при гипонатриемии.
Согласно клиническим данным примерно 50% лиц с АГ могут рассматриваться как солечувствительные; для них характерны высокая степень дисфункции эндотелия, инсулинорезистентность и микроальбуминурия. Длительное время эта форма АГ рассматривалась как независимая от А II в связи с нормальной или даже сниженной плазменной активностью ренина. Однако в настоящее время установлено, что она сочетается со значительным повышением активности локальных РАС в миокарде и сосудистой стенке и увеличение тканевой продукции А II [159, 220].
Показано, что одним из важнейших факторов патогенеза низкорениновой объемной формы АГ является повышение активности сосудистой и миокардиальной РАС, которое в значительной степени является следствием повышенного содержания натрия в крови. Гипернатриемия является также ведущей причиной увеличения ее объема, но одновременно приводит к снижению активности циркулирующей РАС и плазменной концентрации ренина [262]. Поэтому в терапии низкорениновой формы АГ наиболее эффективным оказалось сочетание ингибиторов АПФ и диуретиков, которые уменьшают как объем циркулирующей крови, так и содержание в ней натрия и, посредством этого, дополнительно угнетают РАС в сосудистой стенке и миокарде. В то же время, при высокорениновой форме предпочтительно сочетание ингибиторов АПФ с блокаторами кальциевых каналов (амлодипином), которые блокируют вазоконстрикторное и кардиотропное действие А II в результате угнетения входящего кальциевого тока.
Результаты многих работ последних лет свидетельствуют о том, что повреждающий эффект АГ не ограничивается действием повышенного АД. В одном из ранних крупных ретроспективных клинических исследований отмечено, что для пациентов с низкорениновой формой АГ характерен значительно меньший риск развития ИМ и инсульта, независимо от выраженности АГ [33]. Позднее это было подтверждено в ряде проспективных исследований, в одном из которых частота развития ИМ составила 13 на 1000 пациенто-лет у лиц с высокой, 5,3 — нормальной и 3,3 — низкой активностью ренина в плазме крови. Эта закономерность сохранялась после учета уровня АД, холестерина, курения, возраста, содержания глюкозы в крови [5].
Известно, что важнейшим патологическим следствием АГ является гипертрофия миокарда и сосудистой стенки как ответ на увеличение нагрузки и растяжение. В результате развивается клеточная пролиферативная реакция, и чем выше АД и больше длительность АГ, тем больше выраженность гипертрофии. При стабильной тяжелой АГ гипертрофия ЛЖ выявляется более чем у 90% пациентов, у 15% масса сердца не увеличивается, но существенно увеличивается толщина стенки относительно диаметра полости ЛЖ, что обозначается как концентрическое ремоделирование сердца.
Гипертрофия ЛЖ при длительной АГ является в значительной степени адаптивным процессом, и в то же время она оказывает независимое отрицательное влияние на клиническое течение АГ и ее исходы. Установлено, что предиктором высокого риска летального исхода у лиц с АГ является не уровень АД, а наличие гипертрофии ЛЖ. В 10-летнем наблюдении 280 пациентов с исходно неосложненным течением АГ примерно у 20% масса ЛЖ превышала 125 г/м2, и риск летального исхода у них был примерно в 4 раза выше, чем у лиц без гипертрофии миокарда.
В других исследованиях, в частности — во Фремингемском, также показана значимость массы ЛЖ как предиктора кардиальной, цереброваскулярной и общей смертности, независимо от уровня АД и других факторов риска как в общей популяции, так и у лиц с различными формами кардиоваскулярной патологии, включая АГ.
Во многих случаях не выявлено прямой зависимости между выраженностью АГ и степенью ремоделирования сосудистой стенки и ЛЖ. Так, в эксперименте хроническая инфузия как катехоламинов, так и А II в субпрессорных дозах сопровождалась гипертрофией ЛЖ, которая устранялась применением соответственно симпатолитиков или ингибиторов РАС на фоне неизмененного АД. Помимо этого, у потомков лиц с АГ уже в детском возрасте закономерно отмечена увеличенная масса ЛЖ, выраженность которой является прогностическим фактором последующего развития АГ.
Показано, что сочетание повышенного АД с выраженными структурными изменениями сердца и сосудов характерно для патогенеза АГ, начиная с самых ранних этапов развития заболевания. Более того, эти изменения могут даже предшествовать развитию АГ, и у крыс со спонтанной АГ гипертрофия и уменьшение растяжимости ЛЖ отмечаются в ранний возрастной период еще на фоне нормального АД. Последующее применение вазодилататоров не оказывало нормализующего влияния на изменения структуры сосудистой стенки, но они полностью устранялись при применении ингибиторов АПФ.
Эти и многие другие наблюдения послужили основанием для предположения, что гипертрофия миокарда может развиваться первично, независимо от повышения АД, а развитие АГ при этом имеет вторичный характер. Так, в проспективном исследовании 15 лиц, которые на протяжении 5 лет оставались нормотензивными, и 15 лиц из той же популяции, у которых развилась пограничная АГ, наиболее достоверным предиктором ее развития была исходно увеличенная масса ЛЖ. В последующем эти наблюдения были подтверждены результатами многих исследований, что послужило основанием для гипотезы, в соответствии с которой гипертрофия ЛЖ более тесно связана с патогенезом АГ, чем повышенное АД.
Тем не менее, не может вызывать сомнения, что увеличение механической нагрузки на сердце является одним из важнейших стимулов для развития его гипертрофии. Однако ведущим фактором в этой реакции является активация локальной РАС в сердце и сосудистой стенке на фоне возможного отсутствия изменений активности циркулирующей РАС, содержания А II и активности ренина в плазме крови. Так, у 52 пациентов с АГ не установлено зависимости между величиной общего сосудистого сопротивления и массой ЛЖ, выявлена более высокая распространенность гипертрофии миокарда у лиц с нормальным, но не увеличенным сосудистым сопротивлением (50% по сравнению с 27%). В другом исследовании у 29 пациентов с АГ масса ЛЖ не коррелировала с сопротивлением сосудов предплечья, его уменьшение после проведенного лечения не сопровождалось регрессией гипертрофии сердца.
Во всех классических руководствах по кардиологии гипертрофия и ремоделирование сердца и сосудистой стенки при АГ рассматриваются как основное патологическое следствие заболевания. Морфологически они проявляются клеточной гипертрофией (увеличением размера клеток) и увеличением объема внеклеточного матрикса в результате возрастания продукции коллагена, эластина, гликозаминогликанов; для мелких резистивных сосудов более характерным является сочетание накопления матрикса с клеточной гиперплазией, то есть увеличением количества клеток, прежде всего — фибробластов. Отмечена также инвазия воспалительных клеток, которые инициируют процесс ремоделирования посредством секреции факторов роста, протеаз, цитокинов. Эндотелиоциты сосудов при АГ теряют вазодилататорную активность, способность угнетать агрегацию тромбоцитов и адгезию лейкоцитов. Активация ММРs вызывает разрушение внутренней эластической мембраны, миграцию гладкомышечных клеток в субинтимальное пространство, их пролиферацию и усиленную продукцию матриксных белков, что способствует утолщению интимального слоя [351].
Подобные структурные изменения в стенке церебральных сосудов у пациентов с АГ предрасполагают к образованию микроаневризм, следствием которых является повышение риска развития геморрагических инсультов. В миокарде эти изменения сопровождаются уменьшением вазодилататорного резерва, что в сочетании с гипертрофией миокарда провоцирует развитие ишемии, особенно в субэндокардиальных слоях. Ремоделирование почечных сосудов обусловливает повышение сосудистого сопротивления, нарушение экскреторной функции почек, усиленную секрецию ренина в связи со снижением перфузионного давления в артериолах.
В этих условиях ингибиторы АПФ значительно более эффективно предупреждают поражение сосудов и органов-мишеней по сравнению с блокаторами β-адренорецепторов, независимо от изменений активности ренина в плазме крови и при сопоставимом антигипертензивном действии [207].
Участие локально продуцируемых гуморальных факторов при АГ особенно значимо в развитии гипертрофии миокарда; в кардиомиоцитах значительно увеличивается содержание белков, особенно сократительных. Эти изменения не подвергаются регрессии при нормализации АД в результате применения вазодилататоров и устраняются только блокаторами РАС.
Эти данные означают, что процесс сосудистого ремоделирования и поражения органов-мишеней определяется не только механическим фактором, то есть повышением АД, но и активацией ауто- и паракринного действия локальных РАС. Однако при этом необходимо учитывать, что механическая перегрузка миокарда и сосудистой стенки является одним из факторов активации локальных РАС.
Подобные представления подтверждаются данными, полученными в исследовании на крысах со спонтанной АГ, у которых только ингибиторы АПФ и блокаторы рецепторов А II сохраняли функцию эндотелия, предупреждали гипертрофию и ремоделирование миокарда и стенки сосудов. В то же время, блокаторы β-адренорецепторов атенолол и карведилол не оказывали защитного действия при сопоставимом гипотензивном эффекте [314].
В другом длительном наблюдении у крыс со спонтанной АГ на фоне тяжелой АГ отмечено быстрое и закономерное развитие гипертрофии и недостаточности сердца с инфильтрацией миокарда макрофагами, нарушением функции ЛЖ, высокой частотой летальных исходов. Однако эти изменения не были следствием повышенного АД и значительно ослаблялись в результате применения эпросартана в течение 28 нед в низких дозах на фоне только умеренного (на 16%) снижения АД [12, 13].
Результаты многих клинических наблюдений свидетельствуют о том, что антигипертензивная терапия с применением блокаторов β-адренорецепторов снижает риск цереброваскулярных проявлений, но относительно малоэффективна в предупреждении поражения органов-мишеней, развития ИБС, снижении общей и кардиоваскулярной смертности у лиц с АГ. Позже ряд авторов пришел к заключению, что «…пришло время заявить, что блокаторы β-адренорецепторов не могут рассматриваться как терапия первого ряда при неосложенной АГ» [217].
Несмотря на то что развитие оксидативного стресса при действии А ΙΙ и его участие в патогенезе АГ являются несомненными, результаты исследований с определением терапевтической эффективности антиоксидантов в предупреждении развития, прогрессирования АГ и ее осложнений оказались неубедительными. В ряде небольших работ отмечен их умеренный защитный эффект при АГ, особенно в комбинации, однако в большинстве крупных многоцентровых исследований не установлено выраженного антигипертензивного действия даже длительной антиоксидантной терапии. Разноречивый характер имеют также результаты применения витамина Е у пациентов с АГ [209, 326]. Более того, в одном из исследований 6-недельное применение витамина С и полифенолов из косточек винограда сочеталось с повышением АД у пациентов с АГ [330].
Полагают, что отсутствие выраженного защитного действия антиоксидантной терапии в значительной степени может быть связано с неадекватной доставкой применяемых антиоксидантов в те области клетки, где происходит образование СОР, прежде всего — в митохондрии, являющиеся одним из ведущих источников активных форм кислорода при АГ [72].
Установлено, что при нормальной функции митохондрий только незначительный процент электронов, перемещающихся по дыхательной цепи, восстанавливает кислород до СОР. Однако при старении, ряде патологических процессов, включая атеросклероз, ишемию/реперфузию, нарушается функция митохондрий и значительно увеличиваются утечка электронов и образование СОР [192].
Значимость митохондрильного образования СОР в поражении сердца продемонстрирована в экспериментальных исследованиях. Так, мыши с отсутствием в митохондриях специфической формы СОД (СОД2), активность которой критична в отношении защиты от избытка СОР, погибают уже через 10 дней после рождения от кардиомиопатии, а у гетерозиготных мышей с отсутствием одной аллели СОД2 (СОД2±) при старении или содержании на солевой диете быстро развивается тяжелая АГ [257].
Продукция СОР в митохондриях активируется также А II, применение которого приводит к снижению мембранного потенциала митохондрий в результате активации NADPH-оксидазы и развития внутриклеточного оксидативного стресса [76]. Эта связь подтверждается тем фактом, что мыши с гиперэкспрессией митохондриального антиоксидантного фермента тиреодоксина резистентны по отношению к развитию АГ и дисфункции эндотелия при введении А II [338].
В ряде исследований, проведенных на нормальных мышах, показано, что как А II, так и содержание на солевой диете стимулировали продукцию СОР в митохондриях эндотелиоцитов. Применение в этих условиях специфичного для митохондрий антиоксиданта mitoТЕМРО после стабилизации АГ сопровождалось угнетением митохондриальной и общей продукции СОР, снижением активности NADPH-оксидазы, восстанавлением биодоступности NO в сочетании со снижением АД на 30 мм рт. ст.
Аналогичные эффекты отмечены и при гиперэкспрессии митохондриальной СОД2, тогда как применение мощного антиоксиданта темпола, не проникающего в митохондрии, было неэффективным. В другом исследовании темпол предупреждал развитие АГ и дисфункции эндотелия и сосудов почек в ряде моделей АГ, однако в дозе, превышающей дозу mitoTEMPO на 3 порядка [67, 303]. Значительное уменьшение выраженности АГ и ее повреждающего действия в органах-мишенях установлено у крыс со спонтанной АГ и при применении специфического митохондриального антиоксиданта mQ10 [114].
Повышение сосудистого сопротивления, отмечаемое у пациентов с АГ, включает как активный компонент, вызванный сокращением гладкомышечных клеток, так и структурный, который определяется ремоделированием стенки главным образом резистивных сосудов — мелких артерий и артериол. Отмечается значительное утолщение стенки наряду с уменьшением просвета, пропорционально этому повышается реактивность сосуда на различные констрикторные влияния.
До настоящего времени остается нерешенным вопрос, в какой степени ремоделирование сосудистой стенки определяет повышение АД, а в какой является его следствием, однако в любом случае оно приводит к поддержанию высокого АД и прогрессированию АГ. Эти изменения отмечаются также и у потомков лиц с АГ на фоне нормального АД, что свидетельствует об их возможной первичной природе. Кроме того, значительно легче добиться нормализации АД, чем восстановления нормальной структуры стенки резистивных артерий, и в одном из исследований полное устранение увеличенного сопротивления сосудов предплечья отмечено только через 6 лет после нормализации АД в результате антигипертензивной терапии.
В последние годы неоспоримо доказано, что повышенное АД при АГ не является единственной и даже основной причиной развития кардиоваскулярных, цереброваскулярных и ренальных осложнений. В то же время, роль активации РАС в их развитии еще не может считаться бесспорно установленной, хотя показано, что высокие дозы А ΙΙ как экзо-, так и эндогенного происхождения у крыс приводят к развитию некроза в стенке артериол, аналогично тому, что наблюдается при тяжелой АГ. Внутривенное введение А ΙΙ кроликам сопровождалось развитием также мультифокального миокардиального некроза, выраженность которого коррелировала с плазменной концентрацией А ΙΙ. Параллельно развивались некроз почечных канальцев и почечная недостаточность, а выраженность поражения значительно увеличивалась в условиях гипернатриемии в результате солевой нагрузки или применения кортикостероидов. Аналогичные изменения отмечены и при первичном альдостеронизме на фоне резко сниженного плазменного уровня ренина и А ΙΙ [108].
Мозг, сердце и почки являются органами-мишенями при АГ, в значительной степени в связи с тем, что их сосуды характеризуются наиболее высокой чувствительностью как к констрикторному действию А ΙΙ, так и дилататорному — брадикинина. При этом локальная концентрация А II в условиях АГ повышается, тогда как брадикинина — снижается в результате активации АПФ. Высокая чувствительность церебральных сосудов к А ΙΙ лежит в основе повышения риска развития ишемического инсульта у пациентов с АГ. В то же время, антигипертензивная терапия, основанная на длительном применении антагонистов А ΙΙ, сопровождалась повышением риска развития церебральных геморрагий, так как устраняла защитное действие вазоконстрикции и гипертрофического ремоделирования стенки церебральных сосудов, которые медиируются А ΙΙ [31, 32].
Не вызывает сомнений, что существенную роль в повреждающем действии А ΙΙ на миокард играет локальная РАС. Однако большое значение в этом эффекте имеет и циркулирующая РАС, так как в условиях двусторонней нефрэктомии и отсутствия ренина в плазме крови частота кардиальных эпизодов на фоне АГ значительно уменьшается. Полагают, что это свидетельствует об участии циркулирующего А ΙΙ в их развитии, однако при этом не учитывается, что нефрэктомия сопровождается угнетением и кардиальной РАС в связи с уменьшением плазменного содержания ренина и возможности его захвата миокардом.
Хотя этот вопрос остается еще в сфере дебатов, положение о преимущественном значении кардиальной РАС в ремоделировании сердца при АГ подтверждается тем, что применение ингибиторов АПФ в дозах, не оказывающих гипотензивного действия, предупреждает прогрессирование гипертрофии ЛЖ, тогда как даже снижение АД с помощью вазодилататоров не оказывает подобного эффекта.
Активация РАС играет важнейшую роль в развитии не только АГ, но и осложенных форм ее течения, сочетающихся с аритмиями сердца. Установлено, что у пациентов с АГ риск развития фибрилляции предсердий в 1,42 раза выше, чем у нормотензивных лиц. В ряде исследований выявлено, что фибрилляция предсердий является одним из осложнений неконтролируемой АГ, и эффективный контроль АД значительно снижает риск ее развития.
Длительное время этот эффект рассматривался как следствие ремоделирования левого предсердия в результате активации кардиальной РАС, усиленной экспрессии в предсердии АПФ и действия А ІІ в качестве ключевого фактора прогрессирующего фиброза миокарда. Полагали, что А ІІ способствует усиленной локальной продукции альдостерона, сопровождающейся стимуляцией пролиферации фибробластов, повышением содержания коллагена на фоне снижения активности коллагеназ, наряду с увеличением размера предсердия. Однако полученные в последний годы данные свидетельствуют о том, что при равном влиянии карведилола и телмисартана на размеры предсердия и уровень АД, способность угнетать фибрилляцию предсердий значительно более выражена у телмисартана. Так, с участием 132 пациентов с умеренной АГ (систолическое АД 140–159, диастолическое АД 90–99 мм рт. ст.) с синусовым ритмом, у которых на протяжении предшествовавших 6 мес документировано наличие пароксизмов фибрилляции предсердий, проведено сравнительное исследование терапевтического эффекта телмисартана и карведилола. У лиц, получавших телмисартан, на протяжении 1 года отмечено достоверно более редкое развитие эпизодов фибрилляции предсердий (14,3% по сравнению с 37,1%) при том, что диаметр левого предсердия (3,4 и 3,6 см) и средние величины АД как до, так и после лечения были аналогичными у пациентов обеих групп. Эти данные позволили авторам заключить, что телмисартан значительно более эффективен в предупреждении фибрилляции предсердий по сравнению с карведилолом, несмотря на аналогичное антигипертензивное действие и равную способность устранять ремоделирование левого предсердия [107].
В исследовании Val-HeFT (Valsartan Heart Failure Trial) применение блокаторов АТ1-рецепторов дополнительно к стандартной терапии сердечной недостаточности (включая в 92,5% и ингибиторы АПФ) сопровождалось снижением частоты развития фибрилляции предсердий на 37%. Это сочеталось не только с предупреждением или устранением ремоделирования сердца, но и повышением активности парасимпатической нервной системы, восстановлением баланса между симпатической и вагусной активностью, уменьшением гетерогенности желудочковой реполяризации. Установлено, что в этих условиях телмисартан не только выраженно нормализовал скорость проведения импульса по миокарду предсердий, но и ослаблял дисперсию Р-волны даже по сравнению с рамиприлом.
3.1.1. Роль тканевых РАС в развитии локального воспаления и оксидативного стресса в миокарде, сосудистой стенке и почках как факторов их поражения и патогенеза АГ
Увеличение продукции активных форм кислорода является компонентом патогенеза различных форм кардиоваскулярной патологии, включая АГ, хроническую сердечную недостаточность, ИБС и СД 2-го типа. Большинство исследований, посвященных изучению механизмов развития АГ при введении А ΙΙ, были сконцентрированы на сосудистой стенке, прежде всего — на увеличении в ней продукции СОР с развитием оксидативного стресса и воспаления, результатом чего является гипертрофическое ремоделирование стенки [130, 131, 184, 185].
В ряде работ показано, что АГ, индуцированная введением А ΙΙ, сочетается с развитием оксидативного стресса в сердце, периферических сосудах и почках [328]. Еще в 1994 г. было установлено, что А ΙΙ активирует NADPH-оксидазу — основной источник СОР в сосудистых гладкомышечных клетках и эндотелиоцитах [119], тогда как применение липосомальной формы СОД эффективно предупреждало развитие АГ [303, 304]. Помимо этого, генетическая недостаточность NADPH-оксидазы существенно ограничивала как продукцию СОР в сосудистой стенке, так и гипертензивную реакцию при введении А ΙΙ [170]. Установлено также, что развитию оксидативного стресса может способствовать не только прямое усиление продукции СОР, но и первичное ослабление антиоксидантной защиты [42].
Неоднократно отмечено, что в основе развития АГ, связанной с активацией РАС, и сопутствующего хронического поражения почек лежит активация воспаления. Так, у лиц с хроническим поражением почек значительно повышена экспрессия ИЛ-6, особенно при наличии сопутствующей АГ, а при инфузии А II крысам с генетическим отсутствием ИЛ-6 отмечается значительно меньшая выраженность АГ и поражения почек. В условиях блокады продукции ИЛ-6 устраняется развитие АГ при холодовой пробе, а у лиц с АГ и выраженной протеинурией в биоптатах почек отмечена усиленная экспрессия ИЛ-6. При этом развитие АГ предшествовало появлению протеинурии и являлось, таким образом, причиной поражения почек и развития нефросклероза.
На основании этих данных был сделан вывод, что ИЛ-6 является ключевым медиатором прогипертензивного и провоспалительного действия А II с развитием нефросклероза. В основе этих эффектов лежит прежде всего способность ИЛ-6 активировать экспрессию профибротических генов (проколлагена, TФР-β, PAI-1, ЭТ-1).
Известно, что далеко не у всех больных антигипертензивная терапия позволяет добиться целевого уровня АД. Помимо этого, во многих клинических исследованиях не установлено связи между выраженностью снижения АД и уменьшением риска развития ИБС, а высокая кардиоренальная летальность остается даже у лиц с эффективно контролируемым АД. При этом повышенный риск развития тяжелых кардиоваскулярных, цереброваскулярных и почечных исходов при АГ определяется в значительной степени сопутствующими метаболическими нарушениями, активацией свободнорадикальных реакций, ремоделированием сердца и сосудов, не связанных непосредственно с наличием АГ и определяемых активацией тканевых РАС. Поэтому задачами антигипертензивной терапии должны быть не только нормализация АД, но и устранение воспаления и оксидативного стресса, обусловливающих поражение органов-мишеней, нормализация метаболического статуса.
Продукция активных форм кислорода относится к числу компонентов врожденного иммунного ответа; образование свободных радикалов кислорода является следствием респираторного взрыва прежде всего нейтрофилов, моноцитов и обусловливает хемотаксис воспалительных клеток в зону поражения.
К оксидантам относятся как свободные радикалы, так и нерадикальные формы (пероксинитрит, гипохлористая кислота, перекись водорода, миелопероксидаза нейтрофилов и моноцитов), которые обладают высоким окислительным потенциалом. Пероксинитрит, помимо прямого повреждающего действия, может окислять тетрагидробиоптерин — кофактор еNOS, приводя к разобщению eNOS и продукции СОР взамен оксида азота [131].
Основной источник aктивных форм кислорода в сосудистой ткани — NADPH-оксидаза, А II является ее прямым активатором. В ряде исследований отмечено, что активность NADPH-оксидазы значительно возрастает при действии гипертензивных стимулов, а у мышей со сниженной ее экспрессией отмечается уменьшение выраженности АГ, вызванной введением А II или содержанием на солевой диете с применением ДОКА [170, 198]. Показано также, что введение СОД или ее миметиков типа темпола крысам с экспериментальными моделями АГ приводило к снижению АД и уменьшению сопротивления сосудов почек [2, 269].
Результаты многочисленных исследований послужили основанием для утверждения, что оксидативный стресс является непосредственным индуктором АГ [315, 316], особенно возникающей при действии А II [62], хотя защитное действие антиоксидантов в этих условиях не носило закономерного характера [10, 309].
Значимость оксидативного стресса как причины, а не следствия развития АГ при применении А II, подтверждается тем, что АГ, вызываемая введением норэпинефрина, не сочетается с наличием оксидативного стресса [173].
Установлено, что развитие оксидативного стресса у крыс при длительном содержании на высоколипидной диете сопровождалось дисфункцией эндотелия и развитием АГ, и эти нарушения устранялись при переходе на нормальную диету [256]. Закономерное наличие сосудистого оксидативного стресса в результате повышения активности NADPH-оксидазы с увеличенным образованием СОР отмечено в экспериментальных условиях у крыс со спонтанной и ДОКА-солевой АГ, АГ, вызванной введением А II, ингибиторов eNOS, а также АГ, сочетающейся с ожирением.
В соответствии с традиционными представлениями, которых и в настоящее время продолжают придерживаться ряд исследователей, в основе развития АГ лежит дисфункция эндотелия со снижением его вазодилататорной активности и последующей вазоконстрикцией. Полагают, что этот эффект обусловлен усиленной продукцией СОР и развитием оксидативного стресса, результатом чего является уменьшение продукции NO или его инактивация со снижением биодоступности [154, 156].
Это предположение основывается на многих данных экспериментальных исследований. В частности, применение L-аргинина, субстрата для образования оксида азота, способствовало увеличению его продукции, восстановлению функции эндотелия и предупреждало или устраняло гипертензивный ответ на А ΙΙ или солевую нагрузку. Показано, что инфузия крысам А ΙΙ в течение 9 дней в дозе 20 нг/кг/мин сопровождалась резким нарушением функции эндотелия, повышением АД со 124 до 199 мм рт. ст. в сочетании с выраженным повышением индекса гломерулярного повреждения. Однако при сопутствующем введении L-аргинина полностью предупреждалось уменьшение содержания нитритов в плазме крови и повреждение клубочков почек, прирост АД был уменьшен на 45%. Аналогичный эффект с полным или частичным устранением АГ в ряде ее экспериментальных моделей отмечен при применении антиоксидантов [315].
NO рассматривается в настоящее время как главный регулятор сосудистого тонуса, нарушение его продукции или биодоступности считается одним из факторов развития АГ [20]. Поэтому применение субстрата его образования L-аргинина у пациентов с мягкой и умеренной АГ также оказывало выраженное антигипертензивное действие и способствовало нормализации функции эндотелия [284].
Хотя гипотеза о дисфункции эндотелия как основной причины развития АГ на первый взгляд представляется убедительной, однако это нарушение отмечается при ряде патологических состояний (атеросклероз, СД, ревматоидный артрит и др.), которые не сопровождаются закономерным развитием АГ.
В то же время, усиленная продукция активных форм кислорода и оксидативный стресс закономерно приводят к развитию воспаления с трансцитозом воспалительных клеток, гипертрофическим процессам, усиленной продукции коллагена и ремоделированию сосудистой стенки. Ранее отмечено, что применение антиоксидантов нормализует метаболизм NO и уменьшает выраженность АГ у крыс с хронической почечной недостаточностью [316] и спонтанной АГ [269].
Однако результаты других исследований с применением антиоксидантов как в экспериментальных, так и клинических условиях не всегда подтверждали эту точку зрения. Так, воздействия с антиоксидантной направленностью типа диеты, обогащенной овощами и фруктами, особенно в сочетании с уменьшением потребления жиров и увеличенным — белков, приводили к снижению АД у лиц с АГ. Этот эффект отмечался уже через 2 нед, и в результате у большинства пациентов был достигнут целевой уровень АД [9]. Гипотензивное действие сочеталось с повышением антиоксидантной активности плазмы крови и уменьшением плазменного содержания конечных продуктов пероксидации липидов.
На фоне выраженного антигипертензивного действия пищевых антиоксидантов применение фармакологических препаратов с антиоксидантным действием в ряде исследований было неэффективным или даже оказывало отрицательное влияние [139].
В одном из крупных многоцентровых исследований применение комбинации аскорбиновой кислоты, витамина Е и β-каротена на протяжении 5 лет у лиц с АГ не сопровождалось антигипертензивным эффектом и не оказывало влияния как на кардиоваскулярную, так и общую летальность [295]. В другом исследовании сочетанное применение комбинации цинка, аскорбиновой кислоты, α-токоферола и β-каротена сопровождалось только умеренным снижением систолического АД у лиц как с наличием, так и отсутствием АГ при недостоверном изменении диастолического АД [106].
При оценке значимости оксидативного стресса в развитии гипертензивной реакции следует учитывать, что антиоксиданты способствуют удалению активных форм кислорода, но не оказывают влияния на их продукцию. Кроме того, антиоксиданты не оказывают влияния на H2O2, которая имеет не меньшее значение в патогенезе АГ, чем СОР. Поэтому вмешательства, основанные на угнетении продукции активных форм кислорода, могут оказать более выраженный терапевтический и профилактический эффект, чем скавенджеры свободных радикалов.
К числу возможных причин отсутствия закономерного антигипертензивного действия антиоксидантов является то, что они не устраняют действия такого фактора повышения АД в условиях оксидативного стресса, как пероксинитрит. Пероксинитрит образуется при взаимодействии оксида азота с СОР и приводит к пероксидации липидов плазмы крови с образованием конечных продуктов 8-изопростанов, оказывающих мощное вазоконстрикторное действие. Недостаточная выраженность защитного действия антиоксидантов может обусловливаться также тем, что они связывают активные формы кислорода, обладающие как повреждающим эффектом, так и являющиеся медиатором сигнальных систем или оказывающие защитное действие, в частности — оксид азота.
Хотя АГ закономерно сочетается с развитием оксидативного стресса, до настоящего время остается спорным вопрос о том, является он причиной или следствием АГ. С одной стороны, оксидативный стресс приводит к пролиферации и гипертрофии сосудистых гладкомышечных клеток, отложению коллагена, что сопровождается утолщением стенки и уменьшением просвета сосуда. Это сочетается с повреждением эндотелия, нарушением его вазомоторной активности и усилением вазоконстрикторных реакций, что в совокупности может лежать в основе развития АГ. В то же время, клинические исследования не показали способности антиоксидантов оказывать антигипертензивное действие, хотя антигипертензивная терапия сопровождалась уменьшением выраженности оксидативного стресса, что свидетельствовало в пользу предположения о его вторичном характере [121].
В соответствии с результатами ряда исследований, вазоконстрикторные соединения типа ЭТ-1 и А II приводят к развитию оксидативного стресса в сосудистой стенке, однако он не относился к числу факторов, определяющих повышение АД. Выявлено, что у крыс с хронической инфузией ЭТ-1 два антиоксиданта — темпол и апоцинин, предупреждали повышение плазменного уровня 8-изопростанов и увеличение продукции СОР в стенке аорты, но не оказывали влияния на повышенное АД [78, 79, 271].
Хотя в ряде исследований, проведенных на крысах со спонтанной АГ и на моделях АГ, индуцированной введением А II, содержанием на высокосолевой диете, применение антиоксидантов сопровождалось снижением АД [101, 323, 324], подобные результаты не были получены в других исследованиях [79, 362, 363].
Основными источниками СОР в сосудистой стенке являются как NADPH-оксидаза, так и эндотелиальная синтаза оксида азота. При ее разобщении на фоне истощения субстрата eNOS L-аргинина или кофактора тетрагидробиоптерина при воспалении или действии проатерогенных факторов, прежде всего — окисленных липопротеинов, eNOS становится продуцентом СОР. В результате взаимодействия СОР с NO образуется пероксинирит — мощный оксидант, оказывающий повреждающее действие на клеточные и плазменные белки, липиды и лежащий в основе развития нитрозилирующего стресса [92].
Хотя роль оксидативного стресса в развитии АГ и кардиоваскулярной патологии изучается с 1990-х годов, механизмы этой связи еще далеко не ясны. В соответствии с данными исследований последних лет, связующим звеном между оксидативным стрессом и развитием АГ является активация тканевых РАС в сердце, сосудистой стенке, почках [52]. Показано, что А II стимулирует продукцию СОР в мезангиальных клетках, а угнетение РАС уменьшает выраженность оксидативного стресса в ряде патологических ситуаций [63].
Кроме того, экспрессия ядерного фактора транскрипции-κB — ключевого звена в развитии воспаления, усиливается как радикалами кислорода, так и при активации AT1-рецепторов, что подтверждает наличие связи между оксидативным стрессом, воспалением и РАС [208]. У трансгенных мышей с гиперэкспрессией IκBα, соответственно сниженной экспрессией NF-κB и предупреждением воспалительной реакции, развитие АГ при инфузии А II сочеталось со значительно менее выраженным поражением сосудов, канальцев и гломерул почек при сохранении выраженности АГ [133].
Вопрос о том, являются ли медиаторы воспаления и активные формы кислорода непосредственным фактором повышения АД, остается спорным и в настоящее время [85].
Прямым подтверждением патогенетической связи между оксидативным стрессом и развитием АГ стали результаты исследований с хроническим введением крысам мощного оксиданта BSO, ингибитора синтеза восстановленного глутатиона. Это воздействие вызывало развитие оксидативного стресса, нарушение как эндотелийзависимого, так и эндотелийнезависимого расслабления и умеренное, а в сочетании с солевой диетой — значительное повышение АД. Темпол уменьшал выраженность оксидативного стресса, нормализовал АД, восстанавливал функцию эндотелия [12, 13].
В другом исследовании применение BSO у крыс в течение 2 нед сопровождалось 3-кратным уменьшением тканевого содержания восстановленного глутатиона, выраженным повышением АД и сниженной экскрецией с мочой нитратов и нитритов, что свидетельствовало о снижении биодоступности NO. Это сочеталось с тканевым накоплением нитротирозина, отражающее усиленную инактивацию NO активными формами кислорода. Сочетанное применение витаминов Е и С уменьшало выраженность АГ, восстанавливало почечную экскрецию нитритов и нитратов, уменьшало накопление нитротирозина, несмотря на сохраняющееся истощение восстановленного глутатиона. Полученные данные рассматривались как прямое подтверждение доминирующей роли оксидативного стресса в патогенезе АГ в данной модели.
При применении BSO у крыс в течение 3 нед с питьевой водой отмечено развитие оксидативного стресса, о котором судили по повышению уровня малонового диальдегида в стенке проксимальных канальцев и 8-изопростанов в моче, параллельно с развитием АГ с повышением АД на 27 мм рт. ст. Эти эффекты сопровождались значительным повышением экспрессии AT1-рецепторов в клетках проксимальных канальцев почек, повышением в результате чувствительности к А ΙΙ и увеличением реабсорбции в почках натрия и воды, что существенно усиливало влияние оксидативного стресса на АД [12, 13].
Отмеченные изменения блокировались применением как антиоксиданта темпола, так и блокатора AT1-рецепторов кандесартана, что свидетельствовало о связи между активацией РАС, развитием оксидативного стресса и АГ [14]. На этом основании сформировалось представление об антиоксидантах как препаратах с возможным антигипертензивным действием, которое реализуется посредством инактивации кислородных радикалов и восстановления биодоступности оксида азота [348].
Однако применение антиоксидантной терапии паралельно с BSO не приводило к полной нормализации АД, несмотря на устранение оксидативного стресса. Это противоречие рассматривалось как следствие того, что восстановленный глутатион является важнейшим компонентом системы антиоксидантной защиты, и его истощение не может восполняться применением других антиоксидантов [316].
В значительном числе клинических исследований связь между развитием оксидативного стресса и АГ не имела закономерного характера [328]. Так, в одном из исследований у лиц с АГ не установлено зависимости между активностью СОД и уровнем АД, а также показано, что усиленная продукция СОР и перекиси водорода имела вторичный характер и нормализовалась после снижения АД [167, 238]. Помимо этого, концентрация в моче 8-изопростанов — продуктов и маркеров оксидативного стресса, как и выраженность антиоксидантной защиты были аналогичными у нормальных лиц и нелеченных пациентов с АГ [329].
Теоретически генез АГ, возникающей при остром введении А ΙΙ, едва ли можно связать только с генерализованным нарушением функции эндотелия и системной вазоконстрикцией, так как эти эффекты должны неизбежно приводить к повышению давления в артериолах почек. В связи с тем, что сосудистая система почек относится к открытому типу, в результате должны увеличиться скорость гломерулярной фильтрации, уменьшиться объем циркулирующей крови, что устранило бы повышение АД. Поэтому, в соответствии с высказыванием всемирно известного американского патофизиолога А. Гайтона, повреждение почек является обязательным компонентом патогенеза АГ, и это подтверждено в ряде исследований значительным повышением индекса гломерулярого повреждения при введении А ΙΙ и развитии АГ [249].
Одним из основных доказательств патогенетической роли оксидативного стресса в развитии АГ считается способность антиоксиданта темпола — миметика СОД, угнетать оксидативный стресс и восстанавливать функцию эндотелия у крыс с различными моделями АГ параллельно со снижением АД. В исследованиях на генетически гипертензивных крысах и крысах с АГ, вызванной инфузией А II, темпол также оказывал гипотензивное действие [206, 346, 347], и оно не было отмечено у нормотензивных животных с отсутствием гиперпродукции СОР [72].
Выраженный антигипертензивный эффект однократного введения темпола отмечен также у крыс с длительной инфузией А ΙΙ [220] и в других моделях АГ, сочетающихся с увеличением продукции ЭТ-1, включая ДОКА-солевую АГ и АГ у солечувствительных крыс [206]. В ряде проведенных в последнее время экспериментальных исследований длительное введение темпола или апоцинина предупреждало развитие АГ, индуцированной А II и ЭТ-1 [228, 272], а антигипертензивное действие темпола у крыс с АГ, вызванной введением А II, блокировалось угнетением eNOS [220]. Темпол также значительно, хотя и транзиторно, уменьшал выраженность АГ при длительной инфузии ЭТ-1 [340].
В то же время, другие исследователи отмечали отсутствие закономерного антигипертензивного действия темпола, несмотря на выраженный антиоксидантный эффект. Полагают, что в определенной степени это может быть связано с выраженным и прогрессирующим увеличением продукции в почках перекиси водорода, отмеченным при хроническом применении темпола, что может оказывать самостоятельное гипертензивное действие [193].
Наиболее отчетливо прогипертензивное действие оксидативного стресса проявилось при хроническом применении А ΙΙ у крыс в субпрессорных дозах, которое сопровождалось развитием так называемого медленного прессорного ответа. Повышение АД отмечалось только через несколько дней, несмотря на то, что плазменная концентрация А ΙΙ не достигала вазоконстрикторного и прессорного уровней [115]. Динамика развития АГ в этих условиях коррелировала с содержанием маркеров оксидативного стресса как в плазме крови, так и в почках [251], а системное применение антиоксидантов типа витамина Е и темпола значительно ослабляло гипертензивный ответ [155].
Помимо этого, данные многих экспериментальных исследований свидетельствуют о том, что угнетение NAD(P)H-оксидазы с помощью апоцинина при инфузии А ΙΙ предупреждало продукцию СОР и сопровождалось не только регрессией сосудистого ремоделирования, улучшением функции эндотелия, но и оказывало антигипертензивное действие [172, 255, 323, 324]. Установлено также, что антигипертензивная эффективность ингибиторов АПФ и блокаторов рецепторов А II в значительной степени обусловлена их способностью предупреждать развитие оксидативного стресса.
В исследованиях на крысах, содержащихся на высокосолевой диете, были получены отличные от этого результаты. Отмечено развитие АГ с повышением среднего АД до 168 мм рт. ст. в сочетании с увеличением продукции СОР и пероксинитрита в стенке аорты (соответственно на 115 и 157%), усилением экспрессии эндотелиоцитами лектиноподобного рецептора LOX- 1 на 130% и МСР-1 — на 166%, гипертрофией ЛЖ, протеинурией, нарушением эндотелийзависимого расслабления [210]. Угнетение NADPH-оксидазы в этих условиях сопровождалось полной нормализацией продукции радикалов кислорода, функции эндотелия, экспрессии LOX-1 и MCP-1 при незначительном влиянии на выраженность гипертрофии ЛЖ и протеинурии. Не выявлено и выраженного антигипертензивного эффекта, а среднее АД было снижено только до 159 мм рт. ст. Эти данные рассматривают как подтверждение того, что механизмы гипертензивного, ремоделирующего и повреждающего действия А II не ограничиваются активацией NADPH-оксидазы, только частично обусловлены развитием оксидативного стресса и включают ряд других факторов [367, 368].
Результаты исследований последних лет существенно углубили представление о механизмах развития АГ при действии А II в субпрессорных дозах. Было подтверждено, что постепенное развитие гипертензивной реакции у крыс в этих условиях закономерно сочеталось с прогрессированием оксидативного стресса, однако оно предупреждалось селективной блокадой рецепторов ЭТ-1 с помощью бозентана при сохранении высокой выраженности стресса [228]. Аналогичные данные получены при инфузии А II крысам в дозе 5 нг/кг/мин в течение 2 нед, что приводило к повышению АД в среднем от 132 до 151 мм рт. ст. параллельно с почти 2-кратным повышением уровня в плазме крови и моче маркеров оксидативного стресса — малонового диальдегида и изопростанов, но в сочетании с повышением уровня ЭТ-1. Эти реакции предупреждались или значительно ослаблялись с помощью применения антиоксидантной терапии темполом или витамином Е. Однако блокада АТ1-рецепторов и рецепторов ЭТ-1 в этих условиях также устраняла развитие АГ, несмотря на то, что оксидативный стресс в этих условиях сохранялся в полном объеме и устранялся только под действием лозартана. Эти данные послужили основанием для предположения, что оксидативный стресс возникал вследствие действия А ΙΙ, но не был непосредственной причиной развития АГ, которое было опосредовано усиленной секрецией мощного прессорного и митогенного фактора ЭТ-1. Поэтому блокада его рецепторов при хронической инфузии А ΙΙ полностью устраняла гипертензивную реакцию и дисфункцию эндотелия, несмотря на сохраняющийся интенсивный оксидативный стресс [229].
Основным источником секреции ЭТ-1 являются эндотелиоциты, хотя он может секретироваться и другими клетками. ЭТ-1 через соответствующие рецепторы, которые экспрессируются сосудистыми гладкомышечными клетками, вызывает их сокращение, гипертрофию и пролиферацию, приводя к ремоделированию сосудистой стенки. Поэтому в последние годы особое внимание уделяется способности А ΙΙ активировать синтез и секрецию ЭТ-1 наряду с активными формами кислорода [6, 263, 298].
Еще более усложнили представления о связи между А ΙΙ, оксидативным стрессом, секрецией ЭТ-1 и АГ результаты исследований, в которых было установлено, что антиоксидант темпол не оказывал влияния на продукцию ЭТ-1 и развитие прессорного ответа в условиях оксидативного стресса, хотя в культуре эндотелиоцитов и гладкомышечных клеток радикалы кислорода стимулировали образование ЭТ-1 [148]. Более того, показано, что ЭТ-1 способен индуцировать продукцию активных форм кислорода в изолированных препаратах аорты, и блокатор рецепторов ЭТ-1 бозентан устранял развитие оксидативного стресса в этих условиях.
В исследованиях М. Ortiz и соавторов также показано, что медленный прессорный ответ на системное введение А ΙΙ сочетался как с развитием оксидативного стресса, так и с увеличением продукции ЭТ-1 [228]. Оба эти эффекта устранялись бозентаном, что свидетельствовало о доминирущей роли продукции ЭТ-1 как причины развития оксидативного стресса. Однако в этом же исследовании применение антиоксидантной терапии в виде темпола или сочетания витаминов Е и С ослабляло экспрессию ЭТ-1, связанную с действием А ΙΙ, что противоречило заключению о первичной роли ЭТ-1 в индуцировании оксидативного стресса [195, 229].
Отсутствие прямой связи между оксидативным стрессом и развитием АГ отмечено также при инфузии ЭТ-1 крысам в течение 12 дней на фоне высокосолевой диеты. Выявлено развитие АГ с повышением АД от 114 до 132 мм рт. ст. на фоне увеличения плазменного содержания 8-изопростанов в 2,5 раза, продукции СОР — более чем на 120% по сравнению с контрольными животными, находившимися только на высокосолевой диете. Сопутствующее применение темпола сопровождалось уменьшением прироста плазменного содержания 8-изопростанов на 80%, применение апоцинина — в 2 раза в сочетании с выраженным уменьшением продукции СОР. Однако ни апоцинин, ни темпол не оказывали достоверного влияния на выраженность гипертензивного эффекта [78, 79].
Существенно отличающиеся результаты получены в исследовании на крысах, которым осуществлялась системная инфузия ЭТ-1 в течение 9 дней. У них отмечено повышение среднего АД от 125 до 141 мм рт. ст., возрастание сопротивления почечных сосудов на 50% и уменьшение почечного кровотока более чем на 30%. Эти изменения сопровождались развитием оксидативного стресса и значительным увеличением содержания его маркеров: малонового диальдегида в почках и 8-изопростанов в моче. Повышение АД полностью устранялось применением темпола, который также значительно уменьшал прирост сопротивления почечных сосудов, уменьшал выраженность оксидативного стресса. Полученные данные трактовались как свидетельство доминирующей роли активных форм кислорода в гипертензивном и вазоконстрикторном действии ЭТ-1 [272]. Результаты ряда других исследований также свидетельствовали о том, что вазоконстрикция и АГ при введении ЭТ-1 являются следствием усиленной продукции СОР и развития оксидативного стресса [105].
Данные исследований in vitro на культуре гладкомышечных клеток стали прямым доказательством способности ЭТ-1 приводить к усиленному образованию СОР и развитию оксидативного стресса, что позволило авторам рассматривать его как основу гипертензивного эффекта ЭТ-1.
В других исследованиях получено подтверждение, что способность А II и альдостерона приводить к ремоделированию сосудистой стенки в значительной степени медиируется ЭТ-1, и блокаторы его рецепторов предупреждают как повышение АД, так и развитие сосудистого повреждения при длительной инфузии А II [243]. При хронической АГ, вызванной введением альдостерона, блокада рецепторов ЭТ-1 также сочеталась с уменьшением выраженности как оксидативного стресса и активности NADPH-оксидазы, так и АГ и ремоделирования сосудов [181].
Однако последующие исследования показали, что увеличение продукции ЭТ-1 при действии альдостерона имеет также опосредованный характер, и медиаторная роль в этом процессе принадлежит вазопрессину. Блокада его рецепторов угнетала продукцию ЭТ-1 и СОР [180], предупреждала развитие оксидативного стресса и повышение АД у крыс с ДОКА-солевой моделью АГ [246].
В то же время, результаты других наблюдений хотя и свидетельствовали о способности ЭТ-1 приводить к развитию оксидативного стресса, однако не подтверждали положения о доминирующей роли стресса в развитии АГ [272]. В исследовании, проведенном на солечувствительных крысах, выраженное антигипертензивное действие темпола также не сочеталось с угнетением оксидативного стресса и нормализацией функции эндотелия [206]. Кроме того, гипотензивное действие темпола установлено и у нормотензивных крыс без признаков оксидативного стресса [282, 348, 349].
В ряде исследований отмечено, что основой антигипертензивного действия темпола являются не антиоксидантные свойства, а способность прямо угнетать симпатическую активность. Так, нормализация АД у гипертензивных крыс при системном введении темпола сочеталась со снижением активности почечных симпатических нервов и уменьшением плазменного содержания норадреналина [51, 134, 282, 283, 361, 362].
Аналогичные данные получены при применении темпола у крыс с ДОКА-солевой АГ, которое сопровождалось снижением среднего АД от 140 до 80 мм рт. ст., симпатической активности — на 50%, уменьшением частоты сокращения сердца от 435 до 390 уд./мин. Однако эти изменения не сочетались с угнетением продукции СОР в стенке аорты и полой вены. Дополнительное применение апоцинина — ингибитора NADPH-оксидазы сопровождалось уменьшением выраженности оксидативного стресса, но не приводило к дальнейшему угнетению симпатической активности, не сопровождалось урежением сердечных сокращений и не потенцировало антигипертензивного действия темпола.
В других работах антигипертензивный эффект темпола также не сочетался с угнетением продукции СОР в сосудистой стенке и не зависел от снижения биодоступности NO, но развивался на фоне выраженного снижения активности симпатической нервной системы [93].
Выраженное симпатоингибиторное действие темпола отмечено и у нормальных животных, его введение приводило к развитию отчетливой гипотензии и брадикардии. Однако, по мнению авторов, эти эффекты не отрицают антиоксидативного характера гипотензивного действия темпола, которое может определяться влиянием темпола на высшие нервные центры, обеспечивающие физиологическую регуляцию АД и в функционировании которых задействованы радикалы кислорода (см. раздел 2.1.4) [282, 346].
Неоднозначные данные о роли воспаления в развитии АГ получены в ряде клинических и экспериментальных работ, в которых продемонстрировано как наличие [18, 285, 299], так и отсутствие антигипертензивного действия статинов [124, 350, 359]. В то же время статины существенно предупреждали ремоделирование сосудистой стенки, что связывают с их способностью угнетать продукцию активных форм кислорода, уменьшать выраженность оксидативного стресса и воспаления в сосудистой стенке [65, 331, 365–368].
В исследовании, проведенном на крысах, инфузия А II в течение 2 нед сопровождалась развитием АГ на фоне оксидативного стресса и повышения уровня малонового диальдегида в плазме крови. В мезентериальных резистивных артериях отмечено увеличение сосудистой продукции СОР, снижение активности еNOS и СОД в сочетании с выраженным ремоделированием стенки. Это проявлялось уменьшением диаметра просвета, повышением соотношения толщины стенки к просвету, увеличением количества клеточных элементов (фибробластов, гладкомышечных клеток и эндотелиоцитов) и массы коллагена, уменьшением количества фенестр во внутренней эластической мембране, повышением жесткости стенки.
Применение на этом фоне аторвастатина не оказывало влияния на уровень АД, но устраняло развитие оксидативного стресса, нормализовало активность еNOS и СОД, снижало экспрессию NAD(P)H-оксидазы и предупреждало изменение структуры стенки. На основании этих данных был сделан вывод, что статины обладают выраженными антиоксидантными свойствами, оказывают выраженный защитный эффект в условиях ремоделирования сосудистой стенки, связанного с действием А II, но не оказывают гипотензивного действия [29].
Однако в данном исследовании применялись низкие дозы аторвастатина, тогда как в других исследованиях отмечено, что в более высоких дозах аторвастатин способен снижать АД в различных моделях АГ [65, 359].
Представлениям о прямом гипертензивном эффекте оксидативного стресса противоречат также результаты исследования, проведенного на крысах с ДОКА-солевой АГ, у которых длительная инфузия СОД сопровождалась резким уменьшением выраженности оксидативного стресса, но не приводила к снижению АД, урежению сердечного ритма или угнетению симпатической нервной системы. Введение антиоксидантов типа аскорбиновой кислоты в этих условиях также не оказывало антигипертензивного эффекта [347], как и применение у лиц с АГ препарата тирона с мощным антиоксидантным действием. Не было отмечено снижения АД, хотя при этом выявлено угнетение хемилюминесценции в большой подкожной вене и внутренней грудной артерии более чем на 70%.
Значительные различия в участии активных форм кислорода в развитии АГ отмечены при однократном и длительном применении А ΙΙ. При острой инфузии А ΙΙ темпол не оказывал антигипертензивного действия, хотя угнетал продукцию ЭТ-1 в сочетании со снижением активации митогенактивирующей протеинкиназы, сосудистого ремоделирования и уменьшения выраженности поражения клубочков почек. В условиях длительной инфузии А ΙΙ темпол, как и бозентан и спиронолактон, устраняли эти эффекты параллельно с предупреждением развития АГ.
Это различие в эффектах антиоксидантной терапии при различных режимах действия А ΙΙ позволило высказать предположение, что основным стимулом для развития острой АГ является прямое действие А II, тогда как развитие оксидативного стресса и усиление гипертензивной реакции являются следствием хронического повышения концентрации циркулирующих минералокортикоидов, прежде всего — альдостерона с усиленной продукцией ЭТ-1 [187].
Результатом этих многочисленных, но противоречивых данных стала достаточно компромиссная позиция, в соответствии с которой оксидативный стресс рассматривают как один из ведущих механизмов развития АГ, а антиоксиданты — как препараты с мощным терапевтическим потенциалом, применение которых является одним из путей предупреждения развития и прогресссирования АГ и сопутствующих осложнений. В качестве основного доказательства данной концепции рассматривалась способность темпола оказывать антигипертензивное действие параллельно с угнетением продукции СОР и уменьшением выраженности оксидативного стресса, несмотря на то, что антигипертензивное действие нехарактерно для других антиоксидантов (апоцинин, СОД), которые также угнетают оксидативный стресс.
Одним из объяснений этого противоречия является достаточно гипотетическое предположение, что гипертензивный эффект СОР может проявляться даже в отсутствие значительного повышения его уровня, а антиоксидантное действие СОД, апоцинина, витаминов Е и С может быть недостаточно выраженным, чтобы оказать антигипертензивный эффект. Помимо этого, полагают, что антиоксиданты следует применять непрерывно, в высокой дозе и длительно [349].
Однако СОР не только участвует в развитии ряда сердечно-сосудистых патологических процессов типа АГ, рестеноза, воспаления, атеросклероза и СД, он задействован также в реализации большого числа клеточных функций, включая дифференциацию, рост, пролиферацию, апоптоз, миграцию и сокращение. Поэтому длительное применение антиоксидантов может оказывать не только защитное, но и патологическое действие.
В анализе причин отсутствия закономерных антигипертензивных эффектов антиоксидантов необходимо также учитывать, что терапевтическое их действие сопряжено с определенными ограничениями. Установлено, что после захвата и связывания свободных радикалов антиоксиданты должны восстанавливаться, иначе они приобретают прооксидантные свойства, теряют способность оказывать защитное действие или даже оказывают повреждающее влияние [42].
Помимо этого, необходимо учитывать, что при воспалении и оксидативном стрессе могут активироваться не только про-, но и антигипертензивные факторы. При воспалении отмечается усиленное высвобождение кининов, активируется индуцибельная синтаза оксида азота (iNOS) в сочетании с повышением активности ЦОГ-2, что приводит к резкому повышению тканевой и плазменной концентрации соединений, обладающих вазодилататорным эффектом — NO, простациклина, простагландина Е2. В крайней форме это проявляется паралитическим расширением сосудов с резким снижением АД, что отмечается при септическом шоке и часто приводит к летальному исходу.
В оценке значимости оксидативного стресса в развитии АГ необходимо также принимать во внимание наличие центральных механизмов в гипертензивном действии А II и альдостерона. Установлено, что АГ, развивающаяся при их инфузии и в ряде других экспериментальных моделей, устраняется разрушением области мозга в окружении третьего желудочка. В последние несколько лет установлено, что активность этих центров мозга, принимающих участие в регуляции АД, повышается при усиленной локальной продукции активных форм кислорода с повышением симпатической активности и развитием АГ [30]. Подтверждается это тем, что топическое применение аденовируса, кодирующего СОД, или апоцинина значительно уменьшало выраженность АГ, вызванной А II, при его локальной и системной инфузии [369].
При определении роли РАС как ведущего фактора патогенеза АГ недостаточно внимания уделяется анализу изменений активности тканевых РАС, прежде всего — интрацеребральной [30]. В значительной степени это связано с отсутствием адекватных методов определения активности тканевых РАС, которое доступно почти исключительно в условиях эксперимента и имеет очень сложный характер. Тем не менее, в ряде исследований показано наличие центрального компонента патогенеза АГ, который определяется как существованием интрацеребральной РАС, так и способностью циркулирующего А ІІ воздействовать на АТ1-рецепторы, локализованные в определенных зонах головного мозга [151].
Поскольку А II представляет собой крупный полипептид, он не способен проходить через гематоэнцефалический барьер и осуществляет центральное действие в пределах циркумвентрикулярных органов, где этот барьер отсутствует или недостаточно развит. Сигналы от этих органов активируют нейроны в различных кардиорегуляторных центрах в гипоталамусе и стволе мозга, включая паравентрикулярное ядро. Активность этих центров находится также под влиянием локальной интрацеребральной РАС, и совокупность сигналов, внешних и внутренних по отношению к этим центрам, интегрально определяет их активность и характер модуляции симпатических посылок на периферию.
Влияние А ІІ на активность высших центров регуляции кардиоваскулярной активности, также как и его действие на периферии, обусловлено в значительной степени способностью вызывать развитие оксидативного стресса и усиление продукции провоспалительных цитокинов. Недавно выявлено, что активные формы кислорода, особенно СОР, являются важнейшим интрацеребральным сигнальным путем, посредством которого А II и цитокины вызывают активацию симпатической нервной системы и развитие нейрогенной АГ. В ряде исследований у крыс с интрацеребровентрикулярным введением антиоксиданта темпола на фоне системной инфузии А II установлено значительное снижение АД в сочетании со снижением активности почечных симпатических нервов.
Установлено, что паравентрикулярное ядро гипоталамуса является важнейшим кардиорегуляторным центром головного мозга и рассматривается как зона центральной координации солевого аппетита и симпатического оттока. Отмечено, что при развитии АГ в результате усиления влияния как циркулирующей, так и локальной РАС в паравентрикулярном ядре повышается концентрация провоспалительных цитокинов, активных форм кислорода с повышением экспрессии АТ1-рецепторов и последующим угнетением нейрональной синтазы оксида азота (nNOS). Следствием этого являются активация высших центров регуляции вегетативной активности, увеличение симпатического оттока и плазменного содержания норадреналина с развитием АГ [39, 225].
В ряде экспериментальных исследований показано, что экспрессия nNOS в структурах ствола мозга обратно связана с интенсивностью симпатического оттока. Локальная блокада NO в этих условиях сочеталась с повышением активности симпатических волокон, увеличением плазменной концентрации норадреналина, и эти эффекты устранялись билатеральным угнетением NF-κB в паравентрикулярном ядре.
Неоднократно подтверждено, что активация NF-κB в паравентрикулярном ядре гипоталамуса играет важнейшую роль в развитии системного гипертензивного ответа на повышение концентрации А ІІ в системной циркуляции. Помимо этого, активация интрацеребральной РАС в пределах паравентрикулярного ядра также сопровождается усиленной продукцией провоспалительных цитокинов и активных форм кислорода, повышением активности симпатической нервной системы, развитием и прогрессированием АГ. Установлено, что активные формы кислорода, особенно СОР, являются важнейшим интрацеребральным сигнальным путем, через который А II и цитокины вызывают активацию симпатической нервной системы, повышение плазменной концентрации норадреналина и развитие нейрогенной АГ.
Развитие выраженного оксидативного стресса в центральной нервной системе с последующим повышением АД отмечено также у крыс с поражением почек, вызванным длительной солевой нагрузкой. В его основе лежала активация интрацеребральной РАС с увеличением содержания альдостерона, а топическая блокада альдостерон-синтазы или минералокортикоидных рецепторов в этих условиях препятствовала развитию АГ [138, 139, 143]. Аналогичные результаты получены также при исследовании крыс с острым поражением почек, воспроизведенным фенолом [278]. Активация интрацеребральной РАС отмечена также у крыс с хронической почечной недостаточностью [38, 219, 370].
Установлено, что ключевым регулятором продукции свободных радикалов кислорода, провоспалительных цитокинов и развития воспалительного ответа, которые отмечаются при АГ как в периферических тканях, так и в центральной нервной системе, является NF-κB. Его специфическая блокада в паравентрикулярном ядре гипоталамуса при АГ, вызванной действием А ІІ, как циркулирующего, так и продуцируемого интрацеребрально, значительно уменьшала локальную продукцию провоспалительных цитокинов и активных форм кислорода в сочетании с частичной нормализацией АД. Эти данные стали убедительным подтверждением центральной роли NF-κB, локализованного в паравентрикулярном узле, в регуляции нейрогенного компонента АГ как при системном действии А ІІ, так и при активации локальной интрацеребральной РАС [39].
Это положение получило дополнительное подтверждение в исследовании, проведенном на 12-недельных крысах линии Sprague-Dawley. При подкожной инфузии А ІІ в течение 2 нед развитие АГ у них сочеталось со значительным усилением экспрессии в паравентрикулярном ядре гипоталамуса мРНК ФНО-α, ИЛ-1β, -6 и МСР-1 в сочетании с увеличением продукции СОР и пероксинитрита, экспрессии АТ1-рецепторов и АПФ, ослаблением экспрессии АПФ2, угнетением nNOS, являющейся непрямым индикатором нейрональной активности.
Однако сочетание подкожной инфузии А II с микроинъекцией ингибитора NF-κB в паравентрикулярное ядро сопровождалось значительно меньшей активацией симпатической нервной системы, меньшим приростом АД и менее выраженным образованием кислородных радикалов и провоспалительных цитокинов, чем в контрольных экспериментах с изолированной инфузией А ІІ. Помимо этого, блокада NF-κB в паравентрикулярном ядре уменьшала экспрессию АПФ и АТ1-рецепторов, ослабляла способность А II поддерживать повышенный уровень АД, обусловливала повышение экспрессии АПФ2 и реализацию его антигипертензивного действия [53].
На основании этих данных был сделан вывод, что специфическая блокада NF-κB в паравентрикулярном узле значительно уменьшает интрацеребральную продукцию провоспалительных цитокинов и активных форм кислорода при АГ, вызванной введением А II, что свидетельствует о центральной роли NF-κB, локализованного в паравентрикулярном узле, в регуляции нейрогенного компонента АГ при активации как циркулирующей, так и интрацеребральной РАС.
В ряде исследований, проведенных в 2000–2003 гг., установлено, что эмоционально-болевой стресс у кроликов сопровождается генерализованным повышением симпатической активности и развитием АГ. В то же время, микроинъекция темпола в дорсомедиальную область гипоталамуса или ростальную вентролатеральную область мозжечка значительно уменьшала выраженность этих изменений. Помимо этого, хроническая интрацеребральная антиоксидантная терапия с применением темпола сопровождалась нормализацией АД и уменьшением поражения почек, несмотря на продолжающееся эмоционально-болевое воздействие, что свидетельствовало в этих условиях об участии оксидативного стресса центрального характера в повышении симпатической активности, развитии АГ и нарушении функции почек.
Известно, что АГ является частым проявлением поражения почек и способствует его прогрессированию. Однако и в этих условиях в патогенезе АГ существенную роль играет активация церебральной РАС с повышением симпатической активности и плазменной концентрации катехоламинов. Выявлено, что применение центральных симпатоингибиторов типа клонидина или моксонидина у крыс с почечной АГ, как и денервация почек у лиц с резистентной АГ, сопровождались снижением симпатической активности, АД и резким уменьшением выраженности альбуминурии.
Отмечено, что развитие интрацеребрального оксидативного стресса лежит в основе активации симпатической нервной системы и развития АГ также у крыс, содержащихся на высокосолевой диете, несмотря на снижение активности циркулирующей РАС. Так, проведение солевой нагрузки у крыс линии Sprague-Dawley с односторонней нефрэктомией сопровождалось развитием АГ, протеинурии и глобального гломерулосклероза на фоне повышения симпатической активности. В основе развития этих эффектов лежало наличие оксидативного стресса в гипоталамусе, который регистрировался по выраженности локальной хемилюминесценции. При хроническом введении в латеральный желудочек мозга темпола — антиоксиданта, способного проникать через мембрану, значительно нормализовалась активность почечных симпатических волокон, уменьшалась выраженность АГ и сопутствующих нарушений функции почек. Аналогичный эффект отмечен и при пероральном применении моксонидина — симпатолитика центрального действия [102].
В последние годы установлено, что гипотензивный эффект денервации почек у лиц с резистентной АГ определяется устранением не только эфферентных, но и афферентных волокон, также принимающих участие в центральной симпатической активации и повышении АД при первичном поражении почек [73]. Выявлено, что афферентная денервация предупреждает передачу сигнала в области мозга, регулирующие АД, предупреждает развитие АГ у 5/6 крыс с двусторонней нефрэктомией, и этот эффект в значительной степени определяет антигипертензивное действие денервации почек, которую в последние годы начинают использовать в лечении больных с резистентной АГ [80].
В нормальных физиологических условиях афферентные волокна в почечных нервах играют важнейшую роль в адаптивном ответе на пероральную перегрузку натрием, а в патологических условиях рено-ренальный рефлекс индуцирует повышение АД. Установлено, что солевая нагрузка приводит к повышению АД в значительной степени посредством возбуждения симпатической системы центральной природы, одним из механизмов которого является гиперпродукция в центральной нервной системе активных форм кислорода. Продемонстрирована также роль центральной продукции радикалов кислорода в симпатической активации и развитии АГ у крыс с повреждением почек, вызванным фенолом, при АГ, вызванной пережатием почечных артерий.
У крыс с первичным поражением почек афферентная активность проецируется в области мозга, регулирующие АД, с развитием локального оксидативного стресса и повышением симпатической активности. Антигипертензивная эффективность антиоксидантов отмечена при их интрацеребровентрикулярном введении в области, принимающие непосредственное участие в регуляции активности кардиоваскулярной системы — в субфорникальный орган, паравентрикулярное гипоталамическое ядро.
Ранее установлено, что развитие оксидативного стресса в этих зонах мозга характерно для интрацеребрального введения А ІІ, тогда как у крыс с хронической почечной недостаточностью выявлена активации локальной церебральной РАС. Установлено также, что при солевой нагрузке увеличивается интрацеребровентрикулярное содержание альдостерона, и у солечувствительных крыс локальное введение ингибитора альдостерон-синтазы, как и блокатора минералокортикоидных рецепторов, препятствует развитию АГ при солевой нагрузке. Эти данные свидетельствуют о том, что увеличение интрацеребральной продукции кислородных радикалов с центральной активацией симпатической нервной системы является характеристикой и механизмом развития не только нейрогенной АГ, но и АГ, связанной с поражением почек и их дисфункцией.
Значительно более однородные данные получены при исследовании роли воспаления и оксидативного стресса в ремоделировании сердца и сосудистой стенки при АГ. В ряде фундаментальных исследований установлено, что это ремоделирование имеет активный характер и реализуется посредством активации NADPH-оксидазы, усиленной продукции активных форм кислорода и закономерно предупреждается или ослабляется применением пептидного ингибитора NADPH-оксидазы апоцинина и антиоксидантов типа темпола.
Аналогичные по характеру и выраженности изменения оксидативного стресса и воспаления, а также их сочетание с характерными изменениями структуры отмечены в миокарде и сосудистой стенке как при механическом растяжении, так и при инфузии А II. Эти данные стали прямым подтверждением активации локальной РАС с усиленной продукцией А II и последующим ремоделированием сосудов и сердца в условиях повышенного АД [131].
Данные ряда экспериментальных исследований также свидетельствуют о том, что способность А II вызывать ремоделирование сосудистой стенки связана не только с повышением АД, но и определяется развитием локального оксидативного стресса в результате активации NADPH-оксидазы и образования большего количества радикалов кислорода. Так, повышение систолического АД у мышей, вызванное инфузией А II в течение 14 дней, ослаблялось как апоцинином — ингибитором NADPH-оксидазы, так и гидралазином. Однако развитие оксидативного стресса, снижение эндотелийзависимого расслабления, увеличение толщины стенки мезентериальных артерий и содержания в ней коллагена устранялись только апоцинином. В то же время, несмотря на полное угнетение NADPH-оксидазы в этих условиях, гипертензивный эффект инфузии А II снижался только частично, что свидетельствовало о наличии его редокснезависимого компонента [323, 324].
Показано также, что угнетение NF-κB при АГ сопровождается уменьшением выраженности протеинурии, хотя и не оказывает влияния на выраженность АГ, что подтверждает отсутствие прямой зависимости между поражением почек и повышением уровня АД, а также между воспалением и развитием АГ [134]. В другом исследовании угнетение оксидативного стресса у крыс со спонтанной АГ приводило к значительному уменьшению поражения органов-мишеней, выраженности фиброзирования миокарда, его инфильтрации макрофагами, экспрессии ФНО-α, ИЛ-6, TФР-1β, но только к частичному устранению АГ. При этом у нелеченных животных летальность к 8-й неделе составила 100%, у леченных — уменьшалась до 10% [214].
3.1.2. Роль иммунного компонента в патогенезе АГ
В последние годы представление о патогенезе АГ значительно усложнилось, было отмечено, что он включает иммунный компонент с инфильтрацией сердца, почек и сосудистой стенки иммунокомпетентными клетками, тогда как оксидативный стресс — только одно из проявлений воспалительного процесса.
Вопрос об участии иммунного воспаления в развитии АГ широко дебатируется с начала 1990-х годов. Многие факторы, причастные к повышению АД (А II, альдостерон, цитокины, растяжение, напряжение сдвига) стимулируют источники активных форм кислорода, которые являются эффекторным звеном врожденного иммунного ответа и, по данным многих исследований, участвуют в развитии АГ [130]. В центральной нервной системе активные формы кислорода способствуют повышению симпатической активности, в сосудах индуцируют констрикцию, в почках вызывают задержку натрия и воды, и каждый из этих факторов оказывает прогипертензивное действие.
На начальных этапах развития представлений о роли иммунной системы в развитии АГ доминировало представление, что ведущую роль в повышении АД и повреждении органов-мишеней в этих условиях играет активация врожденного иммунного ответа. Это проявлялось усилением экспрессии NF-κB в эндотелиоцитах и активацией воспаления в сосудистой стенке, тогда как роль адаптивного иммунитета оставалась спорной. Даже в настоящее представления о значимости адаптивного иммунного ответа в патогенезе АГ еще не получили существенного развития. В настоящее время значимость и природа участия адаптивного иммунитета, в частности — лимфоцитов, в развитии АГ остаются еще недостаточно изученными. Тем не менее, развитие стабильной АГ при применении А ΙΙ, стимуляции минералокортикоидных рецепторов, введении соли и ДОКА отмечено только у крыс с наличием нормально функционирующих Т-лимфоцитов [227]. Помимо этого, применение иммунодепрессанта циклофосфамида у спонтанно гипертензивных крыс сопровождалось выраженным, хотя и транзиторным, снижением АД [77], а угнетение иммунного ответа у генетически гипертензивных крыс сопровождалось снижением АД в сочетании с уменьшением продукции радикалов кислорода [234].
В соответствии с классическими положениями иммунологии, врожденный иммунный ответ — первая линия защиты от патогенов, включает клетки эпителия, фагоциты (нейтрофилы, макрофаги), систему комплемента и систему распознающих рецепторов, среди которых важнейшими являются toll-like-рецепторы (TLR). LOX, важнейшее звено патогенеза атеросклероза, аналогичны мембранным компонентам бактерий и могут активировать TLR4, которые экспрессированы на фагоцитах, приводя к развитию воспалительного ответа. Фундаментальным компонентом и эффекторным звеном врожденного иммунитета являются активные формы кислорода и азота.
При развитии адаптивного ответа происходит презентация антигена в сочетании с главным комплексом гистосовместимости. CD4+-лимфоциты, экспрессирующие CD28 и ответственные за развитие гуморального иммунитета, активируются антигенпрезентирующими клетками, в роли которых выступают дендритные клетки, макрофаги, В лимфоциты и эндотелиоциты, которые экспрессируют CD80 и CD86. Эти клетки презентируют антиген в сочетании с главным комплексом гистосовместимости 2-го типа. Однако для полной активации лимфоцитов в этих условиях необходима костимуляция, и к числу костимулирующих молекул относится ФНО-α.
В отличие от CD4+-клеток, CD8+-клетки (цитотоксические Т-лимфоциты) активируются антигенными пептидами в комплексе с главным комплексом гистосовместимости 1-го типа, который экспрессируется не только типичными антигенпрезентирующими, но и всеми ядросодержащими клетками.
Практически все клетки иммунной системы, относящиеся как к врожденному, так и адаптивному иммунному ответу: моноциты, резидентные макрофаги, нейтрофилы, Т-лимфоциты, экспрессируют рецепторы к А ΙΙ. Поэтому повышение концентрации А ΙΙ приводит к их активации, усиленному высвобождению как СОР и других оксидантов, так и провоспалительных цитокинов, что обусловливает неразрывную связь между действием А ΙΙ, развитием воспаления и оксидативного стресса.
В исследовании на крысах показано, что инфузия А II сопровождается развитием иммунного ответа преимущественно по клеточному типу, что проявлялось увеличением содержания в крови ИФН-γ — цитокина Т-хелперов 1 и уменьшением содержания ИЛ-4 — цитокина Т-хелперов 2 [276]. Проведение в этих условиях антигипертензивной терапии с применением блокаторов АТ1-рецепторов, как и применение иммуносупрессорных Treg-клеток устраняли дисбаланс между подфракциями Th-лимфоцитов в сочетании с предупреждением гипертрофии и склерозирования миокарда. В то же время, снижение АД с помощью гидралазина не оказывало подобного действия [319], что свидетельствовало о независимости иммунного ответа от наличия АГ.
В последние годы в ряде работ подтверждено, что способность А II вызывать развитие иммунного ответа, как врожденного, так и приобретенного, является важнейшим фактором, посредством которого РАС участвует патогенезе АГ и в повреждении органов-мишеней [27, 68, 130–132, 275]. Тем не менее, в этих условиях также сохраняется отчетливое различие в факторах, приводящих к повышению АД и к ремоделированию сердца и сосудов. При том, что участие врожденного иммунного ответа в повреждении органов-мишеней при АГ установлено бесспорно и не вызывает сомнений, его роль в развитии гипертензивного ответа остается сомнительной. Так, иммуносупрессия параллельно с инфузией А II с применением дексаметазона или этанерцепта — блокатора ФНО-α, устраняла инфильтрацию тканей иммунокомпетентными клетками при том, что практически не оказывала влияния на выраженность гипертензивной реакции [213].
В то же время, инфузия А II у мышей с иммунодефицитом и недостаточностью клеток Лангерганса, дендритных клеток и натуральных киллеров не сопровождалась развитием АГ при том, что у них отмечалась нормальная констрикторная реакция на норэпинефрин, а изолированные сосуды сохраняли реактивность по отношению к А II. Выраженная резистентность к развитию как АГ, вызванной А ΙΙ, так и ДОКА-солевой АГ, отсутствие у них воспаления и оксидативного стресса в условиях инфузии А ΙΙ отмечены у мышей с генетическим дефицитом Т-лимфоцитов [123].
В ряде исследований, проведенных на мышах с тяжелым генетическим иммунодефицитом и отсутствием В- и Т-клеток, при длительной инфузии А ΙΙ отмечено развитие значительно менее выраженных гипертензивного ответа, гипертрофии ЛЖ, фиброза миокарда, альбуминурии, нарушения функции эндотелия, чем у контрольных животных [55, 56]. Восстановление в этих условиях популяции Т-, но не В-клеток сопровождалось развитием гипертензивного ответа, усиленной продукцией СОР и уменьшением выраженности эндотелийзависимого расслабления, что свидетельствовало о преимущественной значимости нарушений в этих условиях клеточного иммунитета по сравнению с гуморальным [123]. Показано также, что угнетение иммунного ответа у генетически гипертензивных крыс сопровождалось снижением АД в сочетании с уменьшением продукции радикалов кислорода [258].
Выявлено, что Т-клетки играют решающую роль в развитии АГ не только при введении А ΙΙ, но и при повышенном потреблении соли, применении ДОКА и норэпинефрина [197].
Неоднократно установлено, что способность РАС вызывать АГ, оксидативный стресс и сосудистое воспаление в значительной степени медиируется альдостероном с последующим вовлечением макрофагов, Т-лимфоцитов и развитием иммуновоспалительного ответа. Установлено, что применение Tregs (Т-регуляторных CD4+CD25+ клеток) предотвращает или значительно уменьшает выраженность эффектов как А ІІ, так и альдостерона. Так, инфузия альдостерона 13 мышам в течение 14 дней сопровождалась умеренным, но стабильным повышением АД (в среднем на 14 мм рт. ст.), уменьшением выраженности эндотелийзависимого расслабления на 66%, увеличением толщины медии бедренной артерии на 60%, двукратным повышением активности NAD(P)H-оксидазы и продукции СОР в стенке аорты, миокарде и почках в сочетании с инфильтрацией макрофагов и Т-клеток в почки и уменьшением содержания в них Tregs. Внутривенное введение CD4+-,CD25+-клеток с последующей инфузией альдостерона устраняло все сосудистые и почечные эффекты альдостерона. Выявлено, что одним из важнейших механизмов повреждающего действия А ІІ является способность вызывать апоптоз T-регуляторных клеток с уменьшением их количества и активацией в результате врожденного и приобретенного иммунного ответа.
В Т-лимфоцитах значительно преобладает экспрессия рецепторов А ІІ, тогда как макрофаги экспрессируют как АТ1-, так и минералокортикоидные рецепторы. Поэтому участие Т-лимфоцитов в воспалительном ответе, вызванном введением А ІІ, эффективно устраняется применением блокаторов АТ1-рецепторов, тогда как полная блокада продукции макрофагами СОР достигается только сочетанным применением блокаторов АТ1- и минералокортикоидных рецепторов.
3.1.3. Роль РАС в атерогенезе, патогенезе ИБС и сердечной недостаточности
Результаты исследований последних лет неопровержимо свидетельствуют, что А II, как содержащийся в крови, так и продуцируемый локально и действующий пара- и аутокринно, принимает существенное участие в атерогенезе. В атероматозных бляшках отмечено наличие практически всех компонентов РАС, включая А ΙΙ и его рецепторы, в макрофагах и гладкомышечных клетках пораженной сосудистой стенки установлено значительное увеличение содержания А II и экспрессии АПФ. Интенсивная экспрессия АПФ характерна также для стенки vasa vasorum, локализованных в атероматозной бляшке.
В исследовании, проведенном на биоптатах коронарных артерий человека, установлена выраженная активация локальной РАС с продукцией А II, усиленным синтезом ИЛ-6 в макрофагах и гладкомышечных клетках. В бляшках отмечена сочетанная экспрессия А II, его рецепторов 1-го типа, АПФ, ИЛ-6 и CD68-положительных макрофагов. Ранее отмечалось, что увеличенное содержание ИЛ-6 в крови и сосудистой стенке способствует развитию в ней локального воспаления, синтеза в макрофагах, инфильтрирующих область атеросклеротического поражения, MCP-1, MMP — ферментов, разрушающих соединительнотканный матрикс, экспрессии адгезивных молекул и цитокинов типа ИЛ-1 и ФНО-α с дестабилизацией бляшки и развитием острого коронарного синдрома [266].
Вовлечение сосудистой РАС в развитие атеросклеротической бляшки обусловлено в значительной степени тем, что повышение плазменной концентрации ЛПНП сопровождается усиленной сосудистой экспрессией АПФ, AT1-рецепторов и продукцией А II.
Дисфункция эндотелия, снижение биодоступности NO и развитие воспаления в сосудистой стенке, которые закономерно отмечаются при АГ и в значительной степени определяются активацией сосудистой РАС, рассматриваются как предшественники атеросклероза и механизмы его развития [172, 270, 292, 298, 365–368]. В ряде экспериментальных исследований установлено, что ключевую роль в этом процессе играет А ΙΙ с развитием проатерогенных нарушений как следствия повышения активности NADPH-оксидазы с продукцией активных форм кислорода и развитием воспаления.
Установлено, что как ингибиторы АПФ, так и блокаторы AT1-рецепторов значительно уменьшали выраженность накопления липидов в сосудистой стенке у животных с генетической гиперхолестеринемией и с гиперхолестеринемией, возникающей при нахождении на атерогенной диете. Этот эффект развивался даже в отсутствие влияния терапии на содержание в крови как холестерина, так и триглицеридов.
Аналогичные данные получены также на обезьянах, содержащихся на атерогенной диете, у которых образование липидных полосок значительно угнеталось имидаприлом, а инфузия лозартана в течение 6 нед уменьшала выраженность липидной инфильтрации аорты, сонных и коронарных артерий более чем на 50%. Эти сдвиги не зависели от изменений АД или содержания липидов в плазме крови, но сочетались со снижением плазменного уровня МСР-1 и экспрессии CD11b на моноцитах. Лозартан значительно угнетал также продукцию МСР-1 в изолированных моноцитах, стимулированных А II [294].
Одним из наиболее важных патофизиологических эффектов А II и механизмов его проатерогенного действия является способность индуцировать воспаление. Выявлено, что повышенная экспрессия А II в сосудистой стенке сочеталась с ускоренным развитием ее атеросклеротического поражения в значительной степени посредством инициации локального воспаления низкой градации. Это показано in vitro при инкубации эндотелиоцитов и сосудистых гладкомышечных клеток человека в течение 24 ч в среде с А II по значительному повышению экспрессии моноцитарных хемоаттрактантов МСР-1 и -2. MCР-1 является хемоаттрактантом и активатором макрофагов, тогда как МСР-2 активирует различные воспалительные клетки, включая моноциты/макрофаги, Т-клетки, базофилы и эозинофилы. В результате усиливалась инфильтрация стенки моноцитами, которые экспрессируют все компоненты РАС, и эта экспрессия значительно повышалась при дифференциации моноцитов в макрофаги. Все отмеченные реакции, прежде всего — усиленная продукция МСР-1 и -2, устранялись блокатором АT1-рецепторов.
Помимо этого, А II оказывал прямое усиливающее действие на экспрессию в макрофагах, эндотелиоцитах и гладкомышечных клетках 19 генов, прежде всего — генов, ответственных за МСР-1 и -2, и этот эффект был опосредован АT1-рецепторами и устранялся при их блокаде [302].
Инфильтрация моноцитов в сосудистый субэндотелий является ключевым моментом атерогенеза. Она осуществляется с участием моноцитарного хемоаттрактанта MCР-1, экспрессии на поверхности моноцитов адгезивных молекул CD11b/CD18, которые являются лигандами для межклеточных молекул адгезии-1 (ICAM-1) и сосудисто-клеточных молекул адгезии (VCAM-1), экспрессируемых клетками эндотелия. Эта экспрессия значительно усиливается под действием А IΙ и предшествует субэндотелиальному накоплению моноцитов.
А II — важный стимул не только для рекрутирования моноцитов в сосудистую стенку, но и для их транформации в пенистые клетки. Отмечено, что миграция моноцитов в стенку мезентериальных артерий и их трансформация в макрофаги и пенистые клетки значительно усиливались при действии А II, независимо от развития прессорного эффекта.
Гладкомышечные клетки и фибробласты под действием А II секретируют коллаген I и III типов, что приводит к увеличению массы внеклеточного матрикса, ремоделированию сосудистой стенки и предрасполагает к фиксации в ней липопротеинов крови с последующим развитием атеросклеротической бляшки.
С другой стороны, ММРs, продуцируемые резидентными макрофагами, вызывают деградацию матрикса, способствуя разрушению бляшки. Этот процесс угнетался лозартаном, что свидетельствует об участии в нем А II и АТ1-рецепторов. Помимо этого, А II играет также существенную роль в активации иммунного воспалительного ответа, и чувствительность циркулирующих моноцитов к активации липополисахаридом значительно снижается на фоне применения валсартана.
Одним из механизмов проатерогенного действия А II является его способность индуцировать экспрессию на эндотелиоцитах LOX-1 — скавенджер-рецепторов, окисленных ЛПНП. Эти рецепторы слабо экспрессированы в норме, но их экспрессия значительно возрастает при действии модифицированных окисленных ЛПНП, провоспалительных цитокинов, А II и ЭТ-1 [220]. Связывание окисленных ЛПНП с LOX-1 сопровождается увеличением продукции в эндотелиоцитах СОР и нарушением их функции.
У мышей с отсутствием рецепторов ЛПНП в сосудистой стенке отмечено значительное увеличение выраженности проатерогенных факторов: NF-κB, митогенактивирующих протеинкиназ и маркера воспаления CD68, который экспрессируется активированными макрофагами. Это сочетается с угнетением экспрессии противовоспалительного цитокина ИЛ-10, активности СОД и eNOS, и все отмеченные эффекты не возникали у мышей с генетическим отсутствием LOX-1. При содержании мышей с одновременным отсутствием рецепторов ЛПНП и LOX-1 на атерогенной диете в течение 18 нед площадь поражения аорты была уменьшена по сравнению с контролем почти вдвое (с 61 до 36%).
Захват окисленных ЛПНП эндотелиоцитами через LOX-1 приводит к каскаду событий, непосредственно связанных с атерогенезом, прежде всего — к нарушению функции эндотелия. При действии окисленных ЛПНП как генетическое отсутствие LOX-1, так и применение специфических антител к ним способствовало сохранению функции эндотелия, накоплению в стенке CD68-положительных макрофагов, ослабляло экспрессию NF-κB, фосфорилирование и активацию митогенактивирующих протеинкиназ [205].
Экспрессия LOX-1 не ограничивается эндотелием и установлена в меньшей степени в гладкомышечных клетках и макрофагах, способствуя их пролиферации и образованию пенистых клеток. Зависимость этих эффектов от активности сосудистой РАС подтверждалась тем, что как повышение экспрессии LOX-1, так и утолщение интимы сосудов у кроликов, содержащихся на атерогенной диете, значительно ослабевали при применении блокаторов АТ1-рецепторов [45].
Скавенджер-рецепторы классов А (SRA), В (CD36) и Е (LOX-1) обеспечивают захват около 90% всех окисленных ЛПНП, и элиминация этих рецепторов замедляет образование атеросклеротических поражений у мышей с отсутствием апоЕ или рецепторов ЛПНП. Установлено, что макрофаги жировой ткани экспрессируют скавенджер-рецепторы типа SRA и LOX-1. Экспрессия LOX-1 угнеталась адипонектином, нейтрализация которого сопровождалась повышением этой экспрессии в 2 раза.
Дисфункция эндотелия, снижение биодоступности NO и развитие воспаления в сосудистой стенке закономерно отмечаются при АГ и рассматриваются как предшественники атеросклероза [270, 292, 368]. При содержании крыс на высокосолевой диете у них выявляют развитие АГ (систолическое АД 168 мм рт. ст.), гипертрофии ЛЖ, протеинурии, нарушение функции эндотелия, увеличение продукции СОР и пероксинитрита в аорте (соответственно на 115 и 157%), усиление экспрессии LOX-1 (на 130%) и МСР-1 (на 166%). Блокада АТ1-рецепторов, как и угнетение NAD(P)H-оксидазы в этих условиях сопровождались нормализацией продукции радикалов кислорода, функции эндотелия, экспрессии LOX-1 и MCP-1 при незначительном влиянии на систолическое АД (159 мм рт. ст.), выраженность гипертрофии ЛЖ и протеинурии. Эти данные свидетельствуют о наличии помимо активации NAD(P)H-оксидазы и других факторов, которые медиируют повреждающее действие А II [152, 365, 366].
Участие РАС в развитии атеросклероза в настоящее время представляется настолько значимым, что применение ингибиторов АПФ предложено рассматривать как стандартную терапию у всех пациентов с атеросклерозом за исключением случаев низкого АД. Особенно эта терапия показана у лиц с сочетанием АГ с инсулинорезистентностью или СД 2-го типа, которое является практически закономерным и при котором значительно повышается чувствительность сосудов к действию А II и альдостерона. Об этом свидетельствует высокая распространенность АГ у лиц с инсулинорезистентностью и частое развитие гипертрофии ЛЖ и диффузного утолщения интимы артериальных сосудов при СД 2-го типа даже у лиц с отсутствием АГ.
Установлено, что длительное применение ингибиторов АПФ у лиц с СД 2-го типа и АГ сопровождается уменьшением числа кардиальных явлений на 50% более выраженно, чем терапия блокаторами кальциевых каналов, несмотря на аналогичное снижение АД. В значительной степени эффект ингибиторов АПФ может быть связан с их способностью предупреждать истощение NO, которое возникает в результате повышенной активности А II. Это обеспечивает противовоспалительный эффект, сохранение нормальной функции эндотелия, устраняет ремоделирование сосудистой стенки и способствует поддержанию высокой чувствительности к инсулину [223].
Одним из важнейших механизмов проатерогенного действия A II является его способность активировать экспрессию в сосудистых гладкомышечных клетках белка, связанного с рецептором ЛПНП (LRP1). Через этот белок осуществляется интернализация в гладкомышечных клетках эфиров холестерина из агрегатов ЛПНП, связанных с внеклеточным матриксом. Применение A II у мышей с генетическим дефицитом апоЕ сопровождалось атеросклеротической инфильтрацией липидов в аорте при отсутствии изменений уровня липидов в крови. Даже у крыс с нормальным содержанием липидов в крови инфузия A II сочеталась со значительным повышением экспрессии LRP1 и липидной инфильтрацией интимы дуги аорты [11].
В исследовании, проведенном на культуре гладкомышечных клеток, установлено повышение экспрессии LRP1 под действием А II в через 24 ч в 4,1 раза. Этот эффект сочетался с 2-кратным усилением захвата эфиров холестерина из агрегатов ЛПНП и предупреждался блокатором LRP1 лактоферрином. Способность A II вызывать усиленную экспрессию LRP1 осуществлялась через АТ1-рецепторы и устранялась лозартаном, но не блокаторами AT2-рецепторов.
Стимулирующее действие А II на экспрессию LRP1 в культуре гладкомышечных клеток отмечено даже при отсутствии в среде ЛПНП, что свидетельствует о прямом характере этого эффекта. Влияние A II на экспрессию LRP1 по выраженности аналогично эффекту агрегированных ЛПНП — одного из наиболее мощных путей модификации ЛПНП в сосудистой стенке. Помимо этого, в условиях in vivo эффект A II на экспрессию LRP1 достоверно коррелировал с повышением систолического АД, что подтверждало значимость РАС как общего звена в патогенезе как АГ, так и атеросклероза [273].
3.1.4. Роль РАС в развитии метаболического синдрома, СД и их кардиоваскулярных проявлений
Активация локальных РАС имеет особое значение в сочетанном развитии АГ с сопутствующими метаболическими нарушениями, инсулинорезистентностью [70, 177], тяжелым поражением органов-мишеней [259, 332]. В этих условиях в сердце, сосудистой стенке и почках закономерно отмечают повышение активности NAD(P)H-оксидазы, увеличение содержания активных форм кислорода, перекисей липидов, экспрессию ФНО-α и С-реактивного белка, интенсивный апоптоз. Эти изменения сочетаются и лежат в основе нарушений функции эндотелия, развития инсулинорезистентности, воспаления, пролиферации и фиброза [49]. В то же время, блокада рецепторов А II и минералокортикоидных рецепторов предотвращала развитие оксидативного стресса, инсулинорезистентности, ремоделирования сосудистой стенки и сердца даже при сохранении повышенного АД [333, 337].
Связь между активностью РАС и чувствительностью к инсулину имеет как прямой, так и обратный характер, и интенсивность секреции А II находится под прямым контролем инсулина. Установлено, что инсулин оказывает двойственный эффект на продукцию А II в сосудистой стенке. Он прямо стимулирует секрецию ангиотензиногена в гладкомышечных клетках, однако параллельно активирует еNOS в клетках эндотелия с усиленным высвобождением оксида азота, который оказывает угнетающее действие на локальную РАС. При инкубации культуры эндотелиоцитов с инсулином в дозе 1000 мкЕД/мл в течение 48 ч отмечено уменьшение содержания в них ангиотензиногена в сочетании с повышением активности АПФ в 16,7 раза, тогда как в культуре гладкомышечных клеток инсулин оказывал митогенное действие в дозе свыше 100 мкЕд/мл. В сочетанной культуре клеток эндотелия и гладкомышечных клеток инсулин в дозе до 100 мкЕД/мл не оказывал влияния на рост гладкомышечных клеток, но приводил к их гипертрофии в дозе 1000 мкЕд/мл [150].
Данные ряда последних клинических исследований свидетельствуют о том, что основой антидиабетического действия блокаторов АПФ является не снижение АД, а прямая блокада метаболического эффекта А II. Это подтверждалось тем, что применение ингибиторов АПФ и блокаторов рецепторов А II приводило к значительному снижению риска развития СД 2-го типа или его прогрессирования, независимо от наличия АГ или выраженности антигипертензивного эффекта. Так, у пациентов с АГ при применении каптоприла частота развития СД была на 30% меньше по сравнению с лицами контрольной группы, которые находились на лечении блокаторами β-адренорецепторов и диуретиками, при аналогичной выраженности антигипертензивного эффекта [144]. В исследовании RESOLVD на протяжении 2,9 года наблюдения среди лиц с дисфункцией ЛЖ в группе эналаприла частота развития СД 2-го типа составила 5,9% по сравнению с 22,4% — в группе плацебо [201].
Неоднократно установлено, что блокаторы рецепторов А II оказывают такое же выраженное антигипертензивное действие, как и ингибиторы АПФ, однако данные об их влиянии на чувствительность к инсулину остаются противоречивыми [179–181, 227]. После 6-месячного курса применения эналаприла и лозартана у лиц с АГ отмечены сопоставимый антигипертензивный эффект, примерно равное восстановление функции эндотелия и угнетение свободнорадикального окисления, но только эналаприл способствовал нормализации чувствительности к инсулину [356].
Предполагают, что различие в способности препаратов этих классов влиять на чувствительность к инсулину определяется особенностями их действия на брадикинин, который оказывает существенное влияние на метаболизм глюкозы, усиливая приток крови и, соответственно, доставку глюкозы и инсулина к тканям [301]. Известно, что ингибиторы АПФ обладают двойной функцией — блокируют образование А II и предупреждают разрушение брадикинина, и оба эти механизма участвуют в кардиопротекторном и антидиабетическом действии ингибиторов АПФ.
Участие РАС в развитии инсулинорезистентности в значительной степени определяется ее способностью потенцировать развитие ожирения. Выявлено, что А II, локально продуцируемый в жировой ткани, преимущественно — висцеральной, является одним из регуляторов ее роста и развития. Он принимает участие в процессе дифференциации преадипоцитов, гипертрофии и гиперплазии зрелых адипоцитов. У крыс с врожденным ожирением в жировой ткани установлено значительное повышение экспрессии мРНК и белка ангиотензиногена, прогрессирующее с возрастом, эти изменения не отмечались в клетках печени [125].
Существенная роль А II в развитии ожирения подтверждается данными о том, что применение антагонистов АТ1-рецепторов позволяет добиться уменьшения размера адипоцитов, массы жировой ткани и предупреждает развитие и прогрессирование инсулинорезистентности [54].
В клинических и экспериментальных исследованиях неоднократно показано, что РАС принимает активное участие в развитии СД 2-го типа и атеросклероза, а ее блокада оказывает антидиабетическое и антиатерогенное действие. При исследовании 21 пациента с СД 2-го типа и клиническими проявлениями атеросклероза установлено возрастание в 2–5 раз экспрессии мРНК и белка ангиотензиногена, АПФ и AT1-рецепторов как в атероме, так и в гладкомышечных клетках макроскопически интактных участков стенки коронарных сосудов. Помимо этого, введение инсулина сопровождалось дальнейшим повышением экспрессии мРНК ангиотензиногена, АПФ и AT1-рецепторов. Эти данные подтвердили предположение о гиперактивации РАС в стенке артерий при развитии атеросклероза у лиц с СД 2-го типа, одним из механизмов которой является действие гиперинсулинемии [135].
Неопровержимо установлено, что РАС играет центральную роль в микро- и макроваскулярных поражениях при СД. У лиц с СД 2-го типа применение ингибиторов АПФ дополнительно к пероральным гипогликемизирующим препаратам сопровождалось снижением общей смертности от 17,1% в контрольной группе до 8,6%, кардиальной летальности от 5,2 до 3,4% при продолжительности наблюдения в среднем 3,6 года [83].
Однако при монотерапии ингибиторами АПФ начальное уменьшение содержания альдостерона в крови сопровождалось возвратом его уровня к исходному или даже превышением его в результате, как предполагают, «ускользания». Поэтому применение антагонистов альдостерона, особенно селективных (эплеренон), способствует устранению отрицательного действия ингибиторов АПФ, и сочетание антагонистов альдостерона с ингибиторами АПФ или блокаторами AT1-рецепторов позволяет существенно уменьшить выраженность микроальбуминурии при СД 2-го типа [163, 164].
Помимо этого, активация РАС и усиленная продукция А II лежат также в основе связи между воспалением, СД 2-го типа и атеросклерозом. При этом гипергликемия является одним из важнейших факторов активации РАС с последующим увеличением продукции ФНО-α и экспрессии RAGE — рецепторов конечных продуктов гликозилирования белково-липидно-углеводных комплексов (AGE), характерных для гипергликемии. Взаимодействие AGE с RAGE сопровождается активацией ядерного фактора транскрипции-κB и продукцией провоспалительных цитокинов типа IСАМ-1, VCAM-1, Р-селектина. Поэтому угнетение АПФ и блокада AT1-рецепторов приводит к снижению функциональной активности системы «ФНО-α, RAGE, NF-κB», оказывая, в результате, противовоспалительное, антиатеросклеротическое и антидиабетическое действие [101].
Одно из важнейших осложнений СД — развитие почечной патологии с поражением клубочков, прогрессирующей экспансией мезангиального матрикса и пролиферацией мезангиальных клеток. Эти эффекты связаны в значительной степени с локальной продукцией А II, стимуляцией экспрессии провоспалительных цитокинов и факторов роста. На крысах с СД применение блокатора АТ1-рецепторов ирбесартана в течение 6 мес сопровождалось значительным уменьшением выраженности воспаления в почках, их поражения, протеинурии, продукции цитокинов: TФР-β1, PDGF-B, TNF-α и VEGF. В изолированных клубочках почек крыс ирбесартан также значительно уменьшал экспрессию цитокинов. Аналогичные результаты получены в исследовании пациентов с СД 2-го типа и нефропатией [178]. Эти данные свидетельствуют о том, что даже при системном введении ирбесартана защитное действие на почки обусловлено в значительной степени блокадой почечной РАС [321].
В настоящее время большинство исследователей и клиницистов разделяют точку зрения, что патогенез метаболического синдрома и СД 2-го типа, которые являются важнейшими факторами риска ИБС, непосредственно связан с РАС, так как ее блокада позволяет нормализовать метаболизм липидов и углеводов, предупредить развитие коронарного атеросклероза [126]. РАС принимает непосредственное участие в патогенезе инсулинорезистентности и гепатостеатоза — неалкогольного жирового перерождения печени, которые отмечаются у 70–80% больных с избыточной массой тела и ожирением. Спиронолактон также оказывал выраженное защитное действие у мышей с моделью этих состояний, свидетельствуя о роли альдостерона как одного из эффекторных путей их развития [244].
В ряде исследований установлено, что блокада РАС у лиц с метаболическим синдромом уменьшает выраженность гиперинсулинемии, ожирения, гипертрофии миокарда [230], способствует повышению уровня адипонектина, возрастанию чувствительности к инсулину, сохранению функции эндотелия, угнетению пролиферации сосудистых гладкомышечных клеток и образованию пенистых клеток, существенно снижает риск развития атеросклероза и ИБС [104, 174, 242].
Неоднократно продемонстрировано, что пациенты с АГ характеризуются нарушенной толерантностью к глюкозе, которая в значительной степени обусловлена действием А II. Поэтому у пациентов с АГ ингибиторы АПФ способствовали повышению чувствительности к инсулину и на 11–25% увеличивали элиминацию глюкозы из крови, стимулированную инсулином, как при наличии, так и отсутствии СД. В то же время, антигипертензивный эффект сам по себе не приводил к восстановлению чувствительности к инсулину, а антигипертензивная терапия блокаторами β-адренорецепторов и диуретиками сопровождалась даже ухудшением метаболического контроля с частым развитием новых случаев СД [118].
Связь между АГ и инсулинорезистентностью в значительной степени обусловлена и генетическими факторами, отмечено, что даже для нормотензивных потомков лиц с АГ характерен аномальный метаболизм глюкозы [289].
В настоящее время является бесспорным то, что активация РАС — наиболее характерный признак и патогенетическая основа кардиометаболического синдрома с увеличением продукции А II и альдостерона, развитием оксидативного стресса, дисфункцией эндотелия и угнетением чувствительности к инсулину [37, 109, 158]. Выявлено, что у крыс с гиперэкспрессией гена ренина на фоне значительного повышения активности РАС, содержания А II и альдостерона в плазме крови отмечено сочетанное развитие АГ, инсулинорезистентности и гепатостеатоза [21].
На изолированных миоцитах установлено, что А II приводит к развитию инсулинорезистентности посредством активации AT1-рецепторов в результате повышения активности NAD(P)H-оксидазы, образования большого количества СОР и развития оксидативного стресса. Это приводит к угнетению фосфорилирования тирозина в инсулинзависимом субстрате-1 (IRS-1) и нарушению перемещения переносчика глюкозы GLUT4 на клеточную мембрану [172]. При воспроизведении инсулинорезистентности у крыс путем содержания на диете с фруктозой в течение 8 нед отмечалось повышение АД, 2-кратное усиление продукции СОР в гомогенате аорты, значительно усиленная экспрессия АТ1-рецепторов. Предварительное применение лозаратана или ингибиторов NAD(P)H-оксидазы (апоницин) значительно уменьшало продукцию СОР, нарушение функции эндотелия и реактивности полосок аорты на А ІІ. Эти изменения отсутствовали при содержании на фруктозной диете крыс с генетическим отсутствием АТ1-рецепторов, что свидетельствовало о доминирующей роли А ІІ в их развитии [281].
Участие А II в сочетанном развитии АГ и инсулинорезистентности продемонстрировано и у мышей линии Zucker с генетическим ожирением, которые характеризовались выраженным снижением чувствительности к инсулину, развитием гипергликемии, в 6 раз повышенным уровнем триглицеридов в крови. Применение блокатора AT1-рецепторов олмесартана сопровождалось значительным повышением чувствительности к инсулину, снижением плазменного уровня СЖК и уменьшением более чем вдвое скорости продукции и содержания триглицеридов в печени при умеренном антигипертензивном эффекте [250].
Сопряженность инсулинорезистентности и АГ отмечена также у крыс со спонтанным СД [46] и крыс с ожирением [291], тогда как при вторичной АГ подобная зависимость не отмечалась. Это означает, что инсулинорезистентность и гиперинсулинемия не являются следствием АГ, а наличие связи между ними обусловлено общими патогенетическими механизмами, прежде всего — активацией РАС, повышением плазменной концентрации А ІІ и развитием оксидативного стресса. В экспериментальных исследованиях получено прямое подтверждение того, что нарушение передачи в клетку сигнала инсулина при действии А ІІ опосредовано усиленной продукцией СОР, снижением биодоступности NO и устраняется при сопутствующем введении СОД [289].
Результаты исследования, проведенного на крысах с генетической гиперэкспрессией РАС, свидетельствовали о сочетанном развитии у них АГ и инсулинорезистентности. Изменения в сердце включали метаболические нарушения, снижение чувствительности к инсулину, периваскулярный фиброз, увеличение содержания митохондрий, активацию NADPH-оксидазы и развитие оксидативного стресса, ремоделирование миокарда с утолщением межжелудочковой перегородки и нарушением систолической функции.
Применение в этих условиях блокаторов AT1-рецепторов уменьшало выраженность как АГ, так и оксидативного стресса, тогда как антиоксидант темпол не оказывал выраженного гипотензивного действия [336]. Эти данные указывают на существенные различия в механизмах гипертензивного действия А II и его способности вызывать ремоделирование сердца и метаболические нарушения в миокарде. Если гипертензивный эффект определяется прямым действием А II на гладкомышечные клетки сосудистой стенки, то ремоделирование и метаболические эффекты связаны с развитием оксидативного стресса.
Данные клинических исследований последних лет также свидетельствуют о закономерном сочетании АГ с инсулинорезистентными состояниями, в основе которых лежит активация РАС [317]. У нелеченных пациентов с АГ уровень инсулина в крови повышен как натощак, так и после приема пищи, отмечается прямая корреляция между уровнем АД и содержанием инсулина [289]. В этих случаях ингибиторы АПФ и блокаторы АТ1-рецепторов снижали риск развития кардиоваскулярных явлений примерно на 50% больше, чем можно было ожидать только от снижения АД, что главным образом связано с устранением инсулинорезистентности и сочетанных метаболических нарушений [44].
В исследовании с участием 484 лиц было установлено, что как масса тела, так и АД независимо связаны с нарушением толерантности к глюкозе, и риск ее нарушений был повышен при ожирении и АГ соответственно в 1,8 и 2,2 раза, тогда как при сочетанном наличии обоих факторов риск возрастал в 4 раза. Индекс НОМА был увеличен на 18% при АГ, на 60% — при ожирении и на 90% — при их сочетании [123].
В длительном 30-летнем исследовании было показано, что распространенность АГ значительно повышена у пациентов с СД 2-го типа или нарушенной толерантностью к глюкозе, и это различие было отчетливо выражено как в исходном состоянии, так и через 5, 20 и 30 лет. С другой стороны, у лиц с наличием АГ был отмечен значительно более высокий риск развития СД 2-го типа [28].
Неоднократно отмечалось, что А ΙΙ оказывает прямое угнетающее влияние на чувствительность к инсулину с сопутствующими метаболическими нарушениями, устранением вазодилататорного, противовоспалительного и антимитогенного действия инсулина. В результате развивается гиперинсулинемия, которая сопровождается усилением экспрессии ангиотензиногена, А ΙΙ и его рецепторов 1-го типа. Применение препаратов, угнетающих РАС, приводило к восстановлению чувствительности к инсулину и, по данным исследования HOPE, снижало на 34% риск развития СД и на 16% — его осложнений [136, 358]. Блокада AT1-рецепторов также способствовала восстановлению чувствительности к инсулину, и в исследовании LIFE применение лозартана у лиц с АГ сочеталось с 25% снижением риска развития СД 2-го типа по сравнению с эффектом атенолола [182, 327]. Значительно более выраженным по сравнению с действием атенолола было также снижение риска развития конечных кардиальных точек при равном антигипертензивном эффекте [59].
Неоднократно показано, что применение блокаторов AT1-рецепторов у лиц с метаболическим синдромом сочеталось с увеличением содержания в крови адипонектина [105] параллельно со снижением уровня лептина, восстановлением чувствительности к инсулину и снижением риска развития ИБС [242].
Отличительной особенностью состояния РАС при СД 2-го типа является увеличение секреции и концентрации проренина в плазме крови как в абсолютном значении, так и по сравнению с концентрацией ренина. Этот эффект предопределяет поражение почек, развитие микроальбуминурии и является прогностическим признаком нефро-, ретино- и нейропатии [61]. Выявлено, что риск развития микроциркуляторных поражений при СД 2-го типа резко повышен у лиц с высоким уровнем проренина в плазме крови. Однако у лиц с сочетанием СД 2-го типа и АГ прогностическая значимость повышенного плазменного уровня проренина значительно уменьшена, и по мере повышения АД уровень ренина и проренина в плазме крови снижается параллельно с повышением риска развития микроальбуминурии и нефропатии.
В условиях СД значительно возрастает сосудистая реактивность по отношению к А II, и его доза, необходимая для повышения диастолического АД на 20 мм рт. ст., значительно уменьшена по сравнению с нормой. Этот эффект находится в обратной зависимости от исходной активности ренина в плазме крови и определяется, как полагают, ремоделированием сосудистой стенки и гипернатриемией. Применение тиазидных диуретиков восстанавливает реактивность сосудистой стенки параллельно со снижением плазменной активности ренина. Восстановление сосудистой реактивности отмечено также при применении блокаторов кальциевых каналов, что свидетельствует о роли изменений внутриклеточного кальциевого ответа в ее нарушении.
Ремоделирование сосудистой стенки при СД 2-го типа определяется в значительной степени гиперинсулинемией, так как инсулин стимулирует пролиферацию сосудистых гладкомышечных клеток, фиброзирование и повышение жесткости стенки, возрастание сопротивления потоку крови. Следствием гиперинсулинемии является также повышение активности симпатической нервной системы с возрастанием сосудистого тонуса и сократимости миокарда. Есть данные о том, что при гиперинсулинемии повышается экспрессия АТ1-рецепторов, прежде всего — в эфферентных артериолах клубочков, что способствует усиленной реабсорбции натрия [335].
Связь АГ с инсулинорезистентностью и механизмы этой связи изучались в исследовании, проведенном на крысах, находящихся на высокосолевой диете в течение 6 нед. Развитие АГ в этих условиях (145 против 102 мм рт. ст.) сочеталось с повышением экспрессии AT1-рецепторов в аорте на 100%, продукции СОР — на 104%, уменьшением эндотелийзависимого расслабления на 25%. Эти изменения сопровождались снижением чувствительности к инсулину и на 60% — его способности вызывать эндотелийзависимое расслабление.
Применение блокатора AT1-рецепторов и антиоксиданта темпола в этих условиях сопровождалось нормализацией продукции СОР, функции эндотелия и чувствительности к инсулину, хотя при этом АД снизилось только на 25%. Эти данные свидетельствовали о том, что активация РАС в условиях гипернатриемии сопровождается снижением чувствительности к инсулину, которое обусловлено активацией оксидативного стресса и в значительной степени независимо от сопутствующего развития АГ [367].
Закономерные данные были установлены при исследовании роли РАС в динамике постинфарктного периода. Впервые активация РАС при ИМ была установлена непрямым образом по высокому уровню альдостерона в плазме крови и моче, особенно выраженным этот ответ был у лиц с развивающейся сердечной недостаточностью. Позднее в прямых исследованиях было установлено повышение уровня ренина и А II в плазме крови на 3-й день после ИМ, и эти изменения коррелировали с развитием осложнений типа сердечной недостаточности, кардиогенного шока, фибрилляции желудочков. В другом исследовании в острую фазу ИМ уровень ренина и А II в плазме крови был повышен только у лиц с признаками сердечной недостаточности, тогда как в последующие 3–10 дней он повышался у всех пациентов, хотя более выраженно — у лиц с прогрессирующей недостаточностью сократительной функции сердца. В значительной степени этот эффект определялся повышением симпатической активности и коррелировал с плазменным содержанием катехоламинов. Кроме того, одной из причин увеличения продукции ренина могло быть нарушение внутрипочечной гемодинамики со стимуляцией почечных барорецепторов.
Хотя активация РАС при ИМ имеет компенсаторную направленность, так как способствует поддержанию центральной гемодинамики, она сопровождается развитием ряда отрицательных эффектов типа констрикции венечных сосудов, возрастания работы сердца, его потребности в кислородном обеспечении и кровоснабжении и, в результате, увеличения тяжести ишемии. Помимо этого, электролитные нарушения, прежде всего — гипокалиемия в результате развития вторичного гиперальдостеронизма, создают условия для развития аритмий.
Следствием активации РАС в постинфарктный период является ремоделирование сердца, прежде всего — его гипертрофия, начальные проявления которой отмечают уже на 3-й день ИМ. Этот эффект развивается в результате как возрастания постнагрузки, так и прямого митогенного действия А II. А II вызывает также увеличение преднагрузки в связи с увеличением притока крови к сердцу, что является причиной дилатации правого желудочка и развития его недостаточности. Указанные осложнения в значительной степени предупреждались применением каптоприла, что подтверждает вовлечение РАС в ремоделирование сердца в условиях ИМ.
Изменения активности РАС у нелеченных лиц с сердечной недостаточностью, по вполне понятным причинам, определялись только в очень ограниченном количестве работ, и данные, отражающие эти изменения, получены преимущественно в экспериментальных исследованиях. Однако и в клинических наблюдениях у пациентов с сердечной недостаточностью установлено закономерное и значительное повышение активности ренина в плазме крови как при низком, так и нормальном сердечном выбросе. Часто это сочеталось с гиперплазией и гипергрануляцией клеток юкстагломерулярного аппарата, увеличением продукции и высвобождения ренина, что впоследствии было подтверждено на собаках с сердечной недостаточностью, вызванной наложением аортокавальной фистулы.
Однако повышение активности ренина в плазме крови характерно главным образом для начального ответа РАС, независимо от того, возникала сердечная недостаточность вследствие коарктации аорты или легочной артерии, недостаточности трехстворчатого клапана или навязывания высокой частоты сокращений сердца. Этот ранний прирост активности ренина сочетался с параллельным повышением концентрации циркулирующего альдостерона и выраженной задержкой натрия. Повышенная активность ренина сохранялась весь период ухудшающейся гемодинамической ситуации, сниженного АД, тогда как при стабилизации центральной гемодинамики плазменная активность ренина выходила на плато или даже восстанавливалась, иногда до исходного уровня [218].
Менее закономерный характер имели изменения, установленные в условиях клиники. Так, из 21 пациента с застойной сердечной недостаточностью плазменная концентрация ренина была субнормальной у 3, нормальной — у 12 и высокой — у 6 [31]. Близкие результаты получены в ряде других исследований, и у многих бессимптомных пациентов с дисфункцией ЛЖ, не получавших лечения, уровень циркулирующего ренина оставался в пределах нормы, а его повышение, сочетающееся с задержкой натрия, отмечено только у лиц с острой или тяжелой сердечной недостаточностью. Отмечено, что уровень ренина повышался у лиц с сердечным индексом <2,51 л/мин/м2, но мало изменялся у лиц с более высоким его значением.
Повышение плазменной концентрации ренина у животных с сердечной недостаточностью в значительной степени определялось активацией симпатической нервной системы, а острая денервация почек в этих условиях сопровождалась снижением уровня ренина на 50%. Кроме того, у собак с сердечной недостаточностью, вызванной навязыванием высокой частоты сердечных сокращений, активность ренина в плазме крови коррелировала с содержанием норадреналина, тогда как зависимость между продукцией ренина и АД имела обратный характер.
Существенную роль в изменениях активности ренина имеет активация блуждающего нерва, блокада которой приводила к 2-кратному повышению уровня циркулирующего ренина. Значительная роль в регуляции плазменной активности ренина принадлежит также барорецепторам почечных сосудов, и интраренальное введение папаверина у собак с сердечной недостаточностью сопровождалось снижением секреции ренина на 50%. Отмечено также значительное стимулирующее действие интраренально продуцируемых простагландинов, в результате чего применение индометацина у собак с коарктацией нижней полой вены сопровождалось снижением уровня ренина в циркуляции на 43%, хотя подобный ответ не был отмечен у собак с сердечным выбросом, находящимся в пределах нормальных значений.
Отрицательный эффект повышения активности РАС при сердечной недостаточности обусловлен возрастанием сопротивления сосудов сердца, почек и мозга, и кровоток в этих органах значительно увеличивается при применении ингибиторов АПФ или блокаторов AT1-рецепторов. В то же время, кровоток в скелетных мышцах в этих условиях мало изменяется при кратковременном применении блокаторов РАС и возрастает только при длительном ее угнетении. Помимо этого, активация РАС сопряжена с положительным инотропным эффектом, однако он в значительной степени сочетается с повышением пред- и постнагрузки на сердце под действием А II.
Установлено, что А II принимает существенное участие в регуляции функции почек при сердечной недостаточности, и в исследовании 34 пациентов установлена статистически достоверная связь между плазменной активностью ренина и скоростью клубочковой фильтрации. Установлено, что при умеренном повышении плазменной концентрации ренина и А II угнетение АПФ сопровождалось снижением фильтрационной функции почек и повышением концентрации мочевины и креатинина в плазме крови, тогда как при тяжелой сердечной и почечной недостаточности ингибиторы АПФ улучшали почечную функцию.
Отягощающее действие А II при сердечной недостаточности в значительной степени связано со стимуляцией секреции альдостерона, что проявляется наличием обратной зависимости между плазменной концентрацией ренина и калия.
3.2. Значимость и принципы фармакологической коррекции активности РАС в терапии кардиоваскулярной патологии
В настоящее время достигнуты существенные успехи в изучении фундаментальных и клинических аспектов роли РАС в развитии кардиоваскулярной патологии. Однако, несмотря на это, сохраняются значительные расхождения в оценке терапевтического потенциала препаратов, корригирующих состояние РАС, и показаний для их применения в кардиологической практике.
Данные ряда клинических и экспериментальных исследований показывают, что РАС играет центральную роль в развитии и прогрессировании АГ и связанного с ней поражения органов-мишеней. РАС принимает также непосредственное участие в патогенезе ИБС на всех этапах развития, и ее активация сопряжена с серьезными патофизиологическими последствиями. Показано, что ингибиторы АПФ оказывают существенное кардиопротекторное действие, однако у 10–40% пациентов развиваются резистентность к терапии или ее непереносимость.
Отличительной чертой блокаторов РАС является их способность снижать АД без сопутствующей активации симпатической нервной системы, увеличения частоты сердечных сокращений и плазменной концентрации норадреналина на фоне повышения тонуса блуждающего нерва.
Результаты сравнительных исследований свидетельствуют о том, что применение блокаторов ингибиторов РАС приводит к значительному улучшению исходов кардиоваскулярной патологии, особенно у пациентов с застойной сердечной недостаточностью.
Общепризнано, что активация РАС сочетается с поражением кардиоваскулярной системы в значительной степени независимо от изменений АД. Метаанализ ряда исследований свидетельствует, что блокаторы β-адренорецепторов при выраженном гипотензивном действии значительно менее эффективны по сравнению с другими классами антигипертензивных препаратов, прежде всего — ингибиторами РАС, в снижении частоты возникновения инсульта и других кардиоваскулярных исходов [169].
3.2.1. Особенности терапевтического действия ингибиторов АПФ, блокаторов АТ1-рецепторов и ренина
Класс антигипертензивных препаратов — ингибиторов АПФ, оказался высокоэффективным в снижении АД у больных АГ и способствовал нормализации гемодинамики у пациентов с сердечной недостаточностью. В ряде рандомизированных контролируемых исследований установлено, что применение ингибиторов АПФ у лиц с застойной сердечной недостаточностью, систолической дисфункцией после острого ИМ улучшает клиническое течение и увеличивает продолжительность жизни, и уже более десятилетия назад они стали основой терапии при этой патологии [261].
Ингибиторы АПФ широко и эффективно применяются в лечении пациентов с АГ, СД, сердечной недостаточностью и ИМ, выявлено, что наибольшая эффективность ингибиторов АПФ характерна для лиц с высоким кардиоваскулярным риском [312]. В ряде крупных исследований отмечено, что ингибиторы АПФ позволяют снизить риск развития ИМ и других конечных точек у лиц с тяжелым течением ИБС, АГ, СД 2-го типа примерно на 8% на каждый год лечения [96, 97, 306, 342, 358].
Ингибиторы АПФ способствуют также нормализации перфузионного давления в почечных сосудах, экскреции натрия и воды, предупреждают развитие и прогрессирование протеинурии. Частично эти эффекты связаны с предупреждением деградации брадикинина, которое сопровождается увеличением продукции оксида азота, простациклина и простагландина Е2, что в значительной степени определяет вазодилататорное, антитромботическое, антиатерогенное и антипролиферативное действие ингибиторов АПФ [188].
Их применение в виде монотерапии позволяет добиться целевого уровня АД у 40–60% лиц с мягкой и умеренной АГ в сочетании с регрессией гипертрофии миокарда [141]. У пациентов с сердечной недостаточностью ингибиторы АПФ сбалансированно снижают пред- и постнагрузку, уменьшают давление в правом предсердии, капиллярное заклинивающее давление и способствуют увеличению сердечного выброса на фоне уменьшения давления наполнения ЛЖ [187, 188]. В метаанализе результатов исследований SAVE, AIRE и TRACE дополнительное включение ингибиторов АПФ в стандартную терапию сопровождалось снижением риска летального исхода у лиц с ИМ, а также снижением риска развития повторного ИМ на 20%, значительно уменьшало риск развития сердечной недостаточности после ИМ.
Однако данные о применении ингибиторов РАС в терапии хронической ИБС характеризуются неоднородностью. Противоречивые результаты получены в трех крупных многоцентровых исследованиях: HOPE, 2000 [136]; FOX, 2003 [96]; PEACE, 2004 [237], в которых определялось влияние ингибиторов АПФ на течение стабильной ИБС.
В исследовании HOPE, в которое были включены 9297 пациентов с наличием в анамнезе инсульта, коронарного атеросклероза, поражения периферических сосудов, СД 2-го типа и других факторов риска (АГ, дислипидемии, микроальбуминурии), применение рамиприла (10 мг/сут) на протяжении 4,5 года сопровождалось снижением частоты развития конечных точек в виде ИМ, кардиальной смерти и инсульта на 22% по сравнению с плацебо. Значительно снизился также риск развития новых случаев СД 2-го типа, острой сердечной смерти, необходимости проведения реваскуляризации.
Несмотря на то что снижение АД было в пределах 2–3 мм рт. ст., кардиоваскулярная летальность уменьшилась на 25%, риск развития инсульта — на 32%, сердечной недостаточности — на 22%, ИМ — на 20%, частота развития первичной композитной конечной точки — на 22%. Применение ингибиторов АПФ у лиц с АГ и ИБС сочеталось также со снижением частоты развития новых случаев СД 2-го типа [237].
В исследовании EUROPA, проведенном с участием 12 218 пациентов с манифестной ИБС, применение периндоприла (8 мг) на протяжении 4,2 года также приводило к снижению частоты развития композитной конечной точки на 20%. В то же время, в исследовании PEACE с участием 8290 пациентов с коронарным атеросклерозом применение трандолаприла (4 мг) в течение 4,8 года не сопровождалось наличием достоверного эффекта относительно тех же конечных точек.
Тем не менее, метаанализ результатов исследований с применением ингибиторов АПФ у пациентов с коронарным атеросклерозом и хроническим течением ИБС свидетельствовал о положительном, хотя и умеренно выраженном эффекте терапии [7].
В ряде рандомизированных контролируемых исследований показана способность ингибиторов АПФ снижать летальность у лиц с сердечной недостаточностью. Этот эффект особенно выражен у лиц с наличием систолической дисфункции ЛЖ, и уже более десятилетия ингибиторы АПФ являются основой терапии у лиц с этой тяжелой патологией [58].
В исследовании STOP-Hypertension продемонстрировано значительное преимущество ингибиторов АПФ относительно блокаторов кальциевых каналов в предупреждении ИМ и сердечной недостаточности. Аналогичные данные получены в исследовании Glasgow Blood Pressure Clinic.
Выявлено, что кардио-, ангио- и ренопротекторное действие ингибиторов АПФ не связано с их антигипертензивным действием [358] и в значительной степени определяется предупреждением или регрессией ремоделирования сердца, сосудистой стенки и развития нефросклероза [59].
В исследовании, проведенном на крысах со спонтанной АГ, установлено также, что ингибиторы АПФ и блокаторы кальциевых каналов, как изолированно, так и сочетанно, предупреждали или устраняли нефросклероз, вызванный применением L-NAME, ингибитора еNOS, тогда как диуретик гидрохлоротиазид усиливал его при равном антигипертензивном действии.
Значительное число рандомизированных исследований свидетельствует о том, что ингибиторы АПФ значительно улучшают исход ИБС у различных групп пациентов, то есть при первичной и вторичной профилактике, с наличием и отсутствием дисфункции ЛЖ, у лиц с АГ и без нее. Выраженное терапевтическое действие ингибиторов АПФ определено у лиц с ИМ, осложненным сердечной недостаточностью или дисфункцией ЛЖ, и этот эффект проявлялся снижением частоты летальных исходов на 27% и риска повторного развития ИМ — на 25% [94].
В то же время, применение различных ингибиторов АПФ существенно отличалось по выраженности эффекта, что остается пока необъяснимым. Так, наибольшее снижение частоты первичных конечных точек, включая летальность (на 20–22%), отмечено при применении периндоприла и рамиприла, тогда как трандолаприл и квинаприл не оказывали достоверного эффекта на выживаемость и повторное развитие кардиальных явлений [75].
В метаанализе, опубликованном в 2003 г. и включившим результаты 5 основных исследований (HOPE, EUROPA, INVEST, ACTION и CAMELOT), отмечено, что ингибиторы АПФ оказывали более выраженное действие по сравнению с блокаторами кальциевых каналов в предупреждении ИБС, но противоположный вывод был сделан относительно риска развития инсульта [191, 318]. В исследовании CAPPP (Captopril Prevention Project) каптоприл по эффективности не превосходил традиционную терапию. Отсутствие выраженного эффекта объясняли тем, что в условиях применения ингибиторов АПФ активируются альтернативные пути образования ренина, А I и А II, тогда как блокаторы AT1-рецепторов более полно предотвращают развитие побочных эффектов РАС [128].
Полагают, что в определенной степени эти различия в результатах исследований связаны с особенностями трактовки полученных данных. Так, многие исследователи считали антигипертензивный эффект около 1–3 мм рт. ст. незначительным, тогда как в соответствии с результатами последних исследований подобный эффект, сохраняющийся на протяжении многих лет, может оказать существенное влияние на исходы. В частности, в исследовании FEVER (Felodipine Event Reduction) фелодипин в низких дозах вызывал устойчивое снижение АД только на 4/2 мм рт. ст., но этого было достаточно, чтобы снизить частоту развития кардиальных явлений на 35% [184, 185].
Показано, что даже незначительное на первый взгляд антигипертензивное действие терапии существенно отражается на риске развития осложнений. В одном из исследований, проведенных в Великобритании, при агрессивном лечении АГ и строгом контроле АД (144/82 мм рт. ст.) риск летального исхода при СД был ниже на 32%, риск инсульта — на 44% по сравнению с менее агрессивной терапией, хотя различие в величине АД было в пределах 10/5 мм рт. ст.
Помимо этого, эффективность и исходы лечения в значительной степени определяются количеством и выраженностью факторов риска, а отсутствие выраженного эффекта применения трандолаприла в исследовании PEACE объясняли более низким уровнем риска в исследованной популяции [327]. Однако это объяснение не представлялось достаточно убедительным, так как исходные характеристики пациентов в исследовании PEACE были сопоставимыми с теми, которые отмечались в исследовании EUROPA.
Помимо этого, кардиопротекторные свойства ингибиторов АПФ не ограничиваются их антигипертензивным действием. В исследовании HOPE защитный эффект рамиприла был значительно более выражен, чем можно было ожидать от отмеченного только умеренного гипотензивного эффекта, и между выраженностью уменьшения систолического и диастолического АД и снижением кардиоваскулярного риска не отмечено достоверной зависимости. В последнее время показано, что уменьшение АД у лиц со стабильной стенокардией не является предпосылкой снижения частоты кардиальных явлений, и применение блокаторов кальциевых каналов сопровождалось значительно менее выраженным кардиопротекторным действием по сравнению с ингибиторами АПФ при равном антигипертензивном действии [184].
В многоцентровых исследованиях CONSENSUS, SOLVD, V-HeFT-II доказана способность ингибиторов АПФ существенно увеличивать продолжительность жизни и снижать летальность у лиц с сердечной недостаточностью, а в исследованиях SAVE, AIRE, TRACE применение ингибиторов АПФ у лиц с ИМ сопровождалось снижением риска развития сердечной недостаточности, предупреждало ремоделирование сердца, уменьшало летальность и увеличивало продолжительность жизни [358]. В этих исследованиях более половины пациентов с высоким кардиоваскулярным риском не были гипертензивными, применение ингибиторов АПФ сопровождалось достоверным снижением смерти от кардиоваскулярных причин (ИМ, инсульта). Аналогичные результаты были получены в крупном исследовании, проведенном в 2007 г. [344]. Эти данные означают, что кардиопротекторное действие ингибиторов АПФ не ограничивается их антигипертензивным эффектом.
Установлено, что блокаторы РАС эффективны в ренопротекции при СД 2-го типа, что также не может быть связано в полном объеме с антигипертензивным действием препаратов [179]. Показано, что в развитии нефропатии и морфофункциональных изменений клубочков в виде экспансии мезангиального матрикса и гипертрофии мезангиальных клеток ведущую роль играет гиперактивация ренальной РАС с усиленным ауто- и паракринным действием А ΙΙ, который продуцируется локально.
Однако, несмотря на многократно установленную способность ингибиторов АПФ снижать концентрацию циркулирующего А II, секрецию вазопрессина и альдостерона, активность симпатических нервов, эффективность их влияния на тканевые РАС и в настоящее время остается спорной.
В экспериментальных исследованиях показано, что при хронической сердечной недостаточности ингибиторы АПФ с различным сродством к тканевым РАС оказывают различный эффект на уровень ее активности, и на крысах ингибиторы АПФ с высоким тканевым сродством в большей степени предупреждали гипертрофию миокарда [363, 364].
Однако эти данные не получили достаточного подтверждения в клинических наблюдениях. Хотя в исследовании BANFF только ингибиторы АПФ с высоким тканевым сродством значительно улучшали эндотелийзависимое расслабление у лиц с ИБС [8], в исследовании 33 пациентов с хронической сердечной недостаточностью и фракцией выброса менее 40% применение в течение 8 нед эналаприла (40 мг/сут) — препарата с низким и трандолаприла (4 мг) — с высоким тканевым сродством сопровождалось аналогичным отсутствием влияния на уровень альдостерона (12,6 vs 13,3 г/дл и 12,5 vs 14,5 нг/дл). В других исследованиях также не установлено выраженных различий в эффективности ингибиторов АПФ с различным тканевым сродством в угнетении РАС у лиц с хронической сердечной недостаточностью [145].
Одним из ингибиторов АПФ, наиболее широко применяемых в настоящее время в кардиологической клинике, является периндоприл. Периндоприл — ингибитор АПФ третьей генерации, был синтезирован в 1982 г., в 1988 г. был рекомендован для лечения лиц с АГ, с 1991 г. применяется в клинике сердечной недостаточности. В 2005 г. периндоприл рекомендован для вторичной профилактики кардиальных явлений у лиц с ИБС, основанием для чего стали данные о его способности восстанавливать функциональные свойства эндотелия [40].
Отличительным свойством периндоприла является повышенная липофильность, что увеличивает продолжительность действия, обеспечивает возможность проникновения в ткани и воздействия на тканевые РАС [90, 91]. Выявлено, что применение периндоприла у лиц с ИБС уменьшает содержание А ІІ на 70% в эндотелии и адвентиции, на 57% — в плазме крови [368].
Благодаря повышенному сродству к тканевым РАС приндоприл эффективно предупреждает прогрессирование и способствует регрессии ремоделирования сосудистой стенки, гипертрофии миокарда и склеротического поражения почек [81, 82]. Этот эффект особенно выражен при наличии СД 2-го типа и в значительной степени независим от антигипертензивного действия терапии [132]. Установлено, что применение периндоприла у лиц с АГ приводит к регрессии сосудистого ремоделирования с увеличением диаметра мелких артерий, уменьшению отношения толщина медиа/сосудистый просвет. Снижение жесткости стенки периферических артерий сопровождается нормализацией перфузии периферических органов, а также уменьшением отраженной пульсовой волны и снижением центрального АД, которое является более значимым фактором постнагрузки и развития гипертрофии ЛЖ, чем системное АД [146].
К особенностям действия периндоприла относится также его повышенная способность предупреждать деградацию брадикинина, что является одним из важнейших факторов защитного действия ингибиторов АПФ и проявляется восстановлением функции эндотелия и чувствительности к инсулину.
Для периндоприла в большей степени, чем для других ингибиторов АПФ, характерна повышенная способность уменьшать плазменное содержание фибриногена, экспрессию PAI-1 и восстанавливать фибринолитический потенциал крови, сниженный в условиях активации РАС [95].
К особенностям кратковременного применения ингибиторов АПФ относится снижение концентрации А II и альдостерона в крови, однако при длительном их применении этот эффект в значительной части случаев имел транзиторный характер. Примерно у ⅓ пациентов плазменный уровень А II после начального снижения повышался в сочетании восстановлением клинической симптоматики, что обозначается как «ускользание АПФ» [127]. Полагают, что отсутствие длительного эффекта препаратов этого класса может быть в значительной степени связано как с недостаточной дозировкой и неполной блокадой АПФ, так и с активацией альтернативных путей образования А II.
Определение величины прессорной реакции на введение А I позволило установить, что даже максимальные рекомендованные дозы препаратов не подавляют полностью активность АПФ, и для этого требуется повышение дозы, по меньшей мере, в 2 раза. Эти данные послужили основанием для разработки нового подхода к угнетению РАС, основанного на блокаде рецепторов А II. Установлено, что основным преимуществом блокаторов AT1-рецепторов является более полное угнетение активности РАС, помимо этого, их применение лучше переносится больными [241].
Однако отсутствие ожидаемого эффекта терапии с применением различных ингибиторов АПФ могло быть связано не только с недостаточным влиянием на активность локальных РАС, но и с феноменом «ускользания». Известно, что он в значительной степени определяется альтернативным путем образования А ΙΙ с участием химазы, на который ингибиторы АПФ не оказывают влияния [325].
Ингибиторы РАС как, практически, и все фармакологические препараты, имеют наряду с корригирующими и побочные эффекты. Установлено, что важнейшей отрицательной стороной действия ингибиторов АПФ является резкое, примерно 15-кратное повышение активности ренина в плазме крови с последующим увеличением продукции А Ι и постепенной активацией альтеративного пути образования А ΙΙ. Связано это, прежде всего, с уменьшением плазменного содержания А ΙΙ, который является мощным ингибитором секреции ренина.
Этих отрицательных качеств лишены блокаторы рецепторов А ΙΙ и прямые ингибиторы ренина, эффективность одного из которых — алискирена — в настоящее время широко исследуется в клинической практике [7, 246].
Так как повреждающее действие А II реализуется главным образом через рецепторы 1-го типа, то было высказано предположение, что блокада этих рецепторов должна более селективно предупреждать отрицательные последствия активации РАС, независимо от наличия и активации альтернативных путей образования А II.
Эпоха блокаторов AT1-рецепторов началась в 1990 г. с разработки и испытания лозартана. Уже на первых этапах исследования было показано, что применение блокаторов AT1-рецепторов сопровождается повышением концентрации А II, что обусловливало усиленную активацию рецепторов 2-го типа и развитие защитного эффекта через систему брадикинин/оксид азота.
Определение кардиопротекторной эффективности блокаторов АТ1-рецепторов было проведено в многоцентровом исследовании VALUE, в которое было включено 15 245 гипертензивных пациентов в возрасте старше 50 лет с высоким риском развития кардиоваскулярных событий. Все испытуемые рандомизированно распределялись на группы, находившиеся на лечении амлодипином или валсартаном. Длительность лечения составила в среднем 4,2 года.
Отмечен антигипертензивный эффект при обоих режимах лечения, несколько более выраженный при применении амлодипина (на 4,0/2,1 мм рт. ст. после 1-го месяца, 1,5/1,3 мм рт. ст. — в конце года наблюдения). Первичная композитная конечная точка, включавшая кардиоваскулярную и общую смертность, отмечена у 810 пациентов в группе валсартана (10,6%) и у 789 — в группе амлодипина (10,4%). Помимо этого, в подгруппе валсартана риск развития ИМ был на 79% выше, особенно в первые 6 мес наблюдения. В более отдаленный период, когда антигипертензивная эффективность терапии в обеих группах была сопоставимой, это различие сглаживалось. Характерно, что в обеих группах при аналогично выраженном снижении АД риск развития инсульта снижался в значительно большей степени, чем риск ИМ, поэтому амлодипин оказывал более выраженное влияние в предупреждении инсульта, чем валсартан. Таким образом, сделан вывод об отсутствии существенного различия в эффективности обоих режимов лечения в плане предупреждения конечных точек, хотя отсутствие преимущественного эффекта валсартана могло быть следствием его менее выраженного антигипертензивного действия [146].
В настоящее время блокаторы АТ1-рецепторов, в частности — валсартан, являются альтернативной стратегией лечения АГ, которая направлена на ряд факторов ИБС, помимо АД обусловливающих ремоделирование сердца и сосудов, повреждение почек. В виде монотерапии блокаторы AT1-рецепторов позволяли добиться антигипертензивного эффекта у 40–60% пациентов с мягкой и умеренной АГ в сочетании с нормализацией жесткости стенки артериальных сосудов [190, 277].
Применение блокаторов AT1-рецепторов в клинике АГ оказалось более эффективным и характеризовалось меньшей частотой побочных эффектов, чем ингибиторов АПФ. В ряде крупных клинических исследований установлена высокая эффективность блокаторов рецепторов А II в лечении больных с сердечной недостаточностью и ИМ [241]. У больных с хирургической васкуляризацией сердца применение кандесартана в течение 24 мес сопровождалось достоверным уменьшением частоты развития композитной кардиальной конечной точки, независимо от влияния на АД [161].
Однако при стабильном течении ИБС блокаторы рецепторов А II оказались менее эффективными, чем ингибиторы АПФ, что большинство исследователей связывают со способностью последних блокировать деградацию брадикинина [59].
В исследовании LIFE (Losartan Intervention For Endpoint reduction in hypertension) применение блокатора AT1-рецепторов валсартана сопровождалось выраженным снижением АД в первый год и менее выраженным — во второй. Однако масса ЛЖ уменьшалась и во второй год применения препарата непропорционально выраженно относительно АД [157]. Параллельно уменьшалась выраженность альбуминурии, свидетельствуя о восстановлении функции почек [213].
Вопрос о сравнительной эффективности ингибиторов АПФ и блокаторов АТ1-рецепторов в клинике сердечной недостаточности остается дискуссионным до настоящего времени [264]. В исследовании ELITE I, в которое было включено 722 пациента с хронической сердечной недостаточностью II–IV классов по NYHA, среди 352 пациентов, получавших лозартан (50 мг/сут), отмечен более низкий на 56% риск смерти от всех причин по сравнению с 370 пациентами, получавшими каптоприл (по 50 мг 3 раза в сутки). Это различие определялось снижением риска развития внезапной сердечной смерти на 64% и риска развития смерти от некардиальных причин на 26%.
В то же время, при сопоставлении эффектов каптоприла и лозартана в значительно более крупном исследовании ELITE II с участием 3152 аналогичных пациентов не было установлено статистически значимого различия, а со временем проявилась достоверно большая летальность у лиц, получавших лозартан (280 по сравнению с 250 случаями).
Тем не менее, полагают, что полная блокада АТ1-рецепторов примерно на 20% более эффективна по сравнению с угнетением АПФ, несмотря на то, что сочетается с 4–5-кратным повышением плазменной концентрации А II. Однако подобный эффект не отмечен при сочетанном применении при сердечной недостаточности блокаторов АТ1-рецепторов и ингибиторов АПФ, и комбинированное лечение сопровождалось уменьшением летальности более чем на 20% по сравнению с применением ингибиторов АПФ в виде монотерапии [128, 203].
Теоретически ингибиторы АПФ только частично предупреждают образование А II из А I, которое может осуществляться альтернативным путем при участии химазы, катепсинов, и потому блокада AT1-рецепторов должна оказывать более выраженный эффект. Однако блокаторы AT1-рецепторов предупреждают накопление брадикинина, который может оказывать положительный эффект при сердечной недостаточности, хотя, с другой стороны, брадикинин стимулирует высвобождение норадреналина из симпатических окончаний в ишемизированном миокарде с отрицательным эффектом. Тем не менее, у лиц с сердечной недостаточностью и непереносимостью АПФ блокаторы AT1-рецепторов являются препаратами выбора [86].
Блокаторы АТ1-рецепторов концептуально более эффективны в снижении АД и кардиопротекции в высокорениновом состоянии, тогда как тиазидные диуретики — в низкорениновом. Выявлено, что при содержании крыс на высокосолевой диете не только увеличивается объем циркулирующей крови, но и активируются тканевые РАС. Определяется это тем, что низкосолевая диета, как и прием диуретиков, сопровождаются угнетением локальных РАС в результате уменьшения тканевого содержания натрия. Однако это может сочетаться с развитием гипонатриемии и активацией циркулирующей РАС, тогда как блокаторы AT1-рецепторов угнетают действие преимущественно циркулирующей, но не локальных РАС. При этом отмечается повышение чувствительности к соли, склонности к развитию АГ, что обусловливает значительное повышение эффективности диуретиков. Поэтому сочетанное применение диуретиков и блокаторов АТ1-рецепторов позволяет устранить побочные эффекты, характерные для применения препаратов этих классов в виде монотерапии [357].
Также установлено, что на фоне высокосолевой диеты, вызывающей угнетение секреции ренина, важнейшей детерминантой активации РАС и повышения систолического АД становится ангиотензиноген, что подтверждается наличием сильной зависимости между концентрацией ангиотензиногена и альдостерона у лиц с высоким потреблением натрия. Это может быть связано с тем, что на фоне повышенного тканевого содержания натрия значительно повышается эффективность преобразования ангиотензиногена в А І под действием ренина, а также продукции альдостерона надпочечниками под действием А ІІ [99]. Поэтому комбинированная терапия может оказывать выраженный эффект посредством уменьшения продукции альдостерона как в надпочечниках, так и на тканевом уровне, а также в связи с уменьшением выраженности действия альдостерона при сниженном под действием мочегонных препаратов тканевом содержании натрия.
Исходя из этих данных, было высказано предположение, что сочетанное применение блокаторов рецепторов А ІІ и диазидных диуретиков может приводить к синергичному угнетению как циркулирующей, так и тканевой РАС [15, 146, 174, 183, 231]. Отмечено, что сочетанное применение блокаторов АТ1-рецепторов и диуретика гидрохлоротиазида способствует значительному улучшению контроля АД без проявления отрицательных эффектов [217]. Для действия гипотиазида характерен гипокалиемический эффект, он также угнетался валсартаном, по-видимому, в результате снижения уровня альдостерона. В то же время, ингибиторы АПФ уменьшали продукцию альдостерона более выраженно, чем блокаторы АТ1-рецепторов, поэтому применение ингибиторов АПФ в клинике часто сопровождается развитием гиперкалиемии.
Установлено, что хроническое применение лозартана у нормотензивных крыс с нормальным потреблением соли и нормальной активностью ренина сопровождалось стабильным снижением АД на 25–30 мм рт. ст., однако этот эффект не возникал при высокосолевой диете в связи со снижением активности циркулирующей РАС. При постоянном применении лозартана уровень ренина повышался независимо от характера потребления соли, и переход от высокого ее потребления к нормальному и наоборот не отражался на уровне АД. Эти результаты означают, что РАС принимает существенное участие в поддержании АД и в нормальных условиях, а гипотензивное действие лозартана определяется устранением эффектов преимущественно циркулирующего А II и не связано с натрийурезом [49].
Ряд данных свидетельствует о том, что длительное антигипертензивное действие блокаторов АТ1-рецепторов определяется в значительной степени пептидом А (1–7), так как применение его антагониста значительно уменьшало выраженность действия лозартана [50]. В то же время, эффект лозартана ослаблялся и даже полностью блокировался у крыс, находящихся на высокосолевой диете и характеризующихся низким уровнем активности ренина в плазме крови. Эти данные также означают, что реализация антигипертензивного действия лозартана определяется прежде всего угнетением активности циркулирующей РАС.
В ряде работ показано, что блокаторы АТ1-рецепторов способны подавлять активность и тканевых РАС. В исследовании, проведенном на крысах с СД, установлено, что значительная часть эффекта ирбесартана связана с прямой блокадой клубочковой РАС и предупреждением усиленной экспрессии в почках провоспалительных медиаторов и факторов роста. Применение у этих крыс ирбесартана в течение 8 мес сопровождалось достоверным уменьшением поражения почек и выраженности протеинурии, значительно угнеталась и практически не отличалась от нормальной выраженность гломерулярной цитокинной экспрессии. Эти данные подтверждены в исследовании, в котором инкубация изолированных почечных клубочков в течение 24 ч в среде, содержащей ирбесартан, сопровождалась достоверным снижением экспрессии цитокинов [321].
У лиц с атеросклерозом применение блокаторов AT1-рецепторов приводило к снижению активности воспаления и нормализации функции эндотелия [160], оказывало также органосберегающее действие, способствовало регрессии ремоделирования сердца и сосудистой стенки, уменьшало частоту развития конечных точек и новых случаев СД 2-го типа [59].
Хотя в кардиологичeской практике ингибиторы АПФ применяются уже длительное время, их терапевтический потенциал ограничен тем, что они устраняют не только отрицательные эффекты активации рецепторов А ΙΙ 1-го типа, но и защитное действие активации рецепторов ΙΙ типа. В то же время, селективные блокаторы AT1-рецепторов сохраняют и даже усиливают положительные эффекты активации AT2-рецепторов, так как приводят к увеличению секреции А ΙΙ.
Установлено, что отличительной особенностью активации АТ2-рецепторов является их нормализующее действие на метаболизм, в частности — на чувствительность к инсулину, а также отсутствие активации симпатической нервной системы. Эти эффекты сочетаются с выраженной кардиопротекцией, регрессией ремоделирования сердца и сосудов, уменьшением жесткости стенки артерий и повышения ее растяжимости, устранением дисфункции эндотелия [1].
Результаты последних исследований свидетельствуют о том, что ряд блокаторов АТ1-рецепторов (телмисартан, ирбесартан) обладают также частичной агонистичной активностью в отношении PPARγ адипоцитов, приводя к повышению экспрессии генов-мишеней PPARγ, прежде всего — гена адипонектина, что может оказывать инсулинсенситизирующее и противоспалительное действие [90, 91].
Поэтому, по мнению многих исследователей, телмисартан — представитель 5-го поколения блокаторов AT1-рецепторов, соответствует всем требованиям, которые предъявляются к «идеальному антигипертензивному средству», так как оказывает нормализующее действие не только на АД, но и на метаболический статус и осуществляет защиту органов-мишеней [194, 195, 300].
На этом фоне неожиданными стали результаты исследования с применением блокаторов АТ1-рецепторов у пациентов с ИБС, в котором было установлено повышение риска развития ИМ [293], и этот парадокс в настоящее время пока еще не получил объяснения.
Применение лозартана у крыс с нормальной активностью РАС сопровождалось снижением АД на 25–30 мм рт. ст., что свидетельствует о выраженной тонической активности РАС в состоянии покоя. Этот эффект не связан с блокадой периферического вазоконстрикторного действия А II, так как снижение АД происходило значительно позже по сравнению с прямым влиянием А ΙΙ на сосуды. Ряд данных свидетельствует о том, что в основе этого гипотензивного эффекта лежит блокада центрального действия А II, показано, что разрушение зон вблизи желудочков мозга (area postrema и organum subfornicale) значительно уменьшало выраженность гипотензивного эффекта лозартана [48].
В эксперименте установлено, что в развитие и прогрессирование нарушений структуры сердца и сосудов при АГ вовлечены нейрогенные механизмы. Отмечено, что прогрессирование гипертрофии ЛЖ, как и развитие проатерогенного метаболического профиля, зависят не только от уровня АД, но и от повышения кардиальной симпатической активности, повышения в результате уровня СЖК в крови и развития инсулинорезистентности [116, 117, 146]. Применение телмисартана, в отличие от блокаторов медленных кальциевых каналов амлодипина и нифедипина, не сочеталось с повышением активности адренергических нервов и, более того, приводило к повышению активности вагуса, улучшению метаболического профиля, устранению гипергликемии.
У крыс со спонтанной АГ и крыс с длительной инфузией А II телмисартан предупреждал ремоделирование сердца [252, 305], повышение синтеза коллагена и развитие кардиофиброза даже в дозе, не оказывающей антигипертензивного действия [25]. Способность вызывать регрессию гипертрофии ЛЖ сочеталась с уменьшением размеров левого предсердия и риска развития фибрилляции предсердий [41], с уменьшением скорости распространения пульсовой волны [212, 307, 341].
В исследовании, проведенном с участием 17 пациентов с АГ, применение блокатора AT1-рецепторов в течение 1 года сочеталось с нормализацией структуры стенки мелких артерий, тогда как блокатор β-адренорецепторов атенолол не вызывал аналогичного эффекта при равном гипотензивном действии. После применения лозартана отношение медиа/просвет уменьшалось с 8,4 до 6,7%, тогда как при действии атенолола оно практически не изменялось. Аналогичные эффекты были отмечены при определении жесткости сосудистой стенки [234].
Способность лозартана устранять ремоделирование сосудистой стенки выявлена также у крыс со спонтанной АГ, у которых его применение сопровождалось уменьшением выраженности миграции, пролиферации гладкомышечных клеток и синтеза ими матриксных белков. Аналогичный эффект применения лозартана отмечен в стенке аорты гипертензивных крыс через 2 дня после ее баллонного повреждения.
Предупреждение ремоделирования и регрессия гипертрофии сосудистой стенки при применении лозартана в значительной степени связаны с его способностью стимулировать апоптоз гладкомышечных клеток, мигрировавших в субэндотелий. Этот эффект объясняют участием AT2-рецепторов, так как блокада рецепторов А I типа сочетается с повышением концентрации А II, усиленной активацией рецепторов ΙΙ типа, которые способны стимулировать апоптоз и экспрессия которых в сосудистой стенке значительно повышается после повреждения. Так, на 5-й и 10-й дни применения лозартана у крыс отмечено уменьшение толщины неоинтимы соответственно на 50 и 64%, повышение интенсивности апоптоза гладкомышечных клеток на 61 и 68% и, в результате, уменьшение их количества в неоинтиме на 10-й день на 56% без влияния на размер срединного мышечного слоя. Ранее установлено, что аналогичное усиление апоптоза после повреждения может вызываться применением L-аргинина, который стимулирует продукцию оксида азота [176].
Применение у крыс со спонтанной АГ с блокадой еNOS и тяжелым нефросклерозом валсартана в дозе, не вызывающей антигипертензивного эффекта, как изолированно, так и в сочетании с эналаприлом, сопровождалось возрастанием выживаемости с 37 до 67–85%, тогда как выживаемость при изолированном применении эналаприла составляла 55%. Несмотря на повышение выживаемости, негипотензивные дозы валсартана не предупреждали нарушения структуры и функции почек, развитие и персистирование протеинурии. В то же время, применение в этих условиях валсартана в антигипертензивной дозе сопровождалось дополнительным повышением выживаемости, которая достигала 95%. Это данные означали, что уменьшение почечного перфузионного давления является одним из существенных терапевтических эффектов валсартана в высоких дозах [64].
Существенное отличие действия валсартана от эналаприла в этих условиях заключалось в выраженном повышении плазменного уровня натрия, который не изменялся при применении эналаприла, и сохранении уровня калия в крови в пределах нормальных величин. В то же время, валсартан предупреждал повышение концентрации креатинина, отмеченный в контроле и у животных, получавших эналаприл, oднако структурные нарушения в почках и протеинурия предупреждались только валсартаном в высоких дозах, применение которого сопровождалось антигипертензивным действием.
Плазменный уровень альдостерона был резко повышен в исходе и снижался при всех видах терапии на 50 и 90% — при применении валсартана в высокой дозе. Нефросклероз и кардиосклероз не подвергались существенной регрессии при всех видах терапии.
Эти данные означают, что сочетание ингибиторов АПФ и блокаторов AT1-рецепторов у генетически гипертензивных крыс сопровождается увеличением выраженности эффекта за счет более полной блокады РАС даже без изменений АД и в отсутствие достоверного улучшения функции почек. Предполагают, что этот эффект может определяться увеличением продукции брадикинина и, соответственно, NO. Улучшение функции эндотелия при применении валсартана отмечено и в ряде других исследований, в частности, у мышей с отсутствием апоЕ, несмотря на сохраняющуюся гиперхолестеринемию.
Помимо этого, условия механической перегрузки, воспаления и повышения концентрации натрия приводят к усилению экспрессии AT1-рецепторов и активации минералокортикоидных рецепторов [16]. Поэтому комбинированная терапия может оказывать выраженный эффект посредством уменьшения продукции альдостерона как в надпочечниках, так и на тканевом уровне, а также в связи с уменьшением выраженности действия альдостерона при низком тканевом содержании натрия.
Исходя из этих данных, было высказано предположение, что сочетанное применение блокаторов рецепторов А ΙΙ и тиазидных диуретиков может приводить к синергичному угнетению как циркулирующей, так и тканевой РАС [15, 55, 130, 156, 165, 209].
Установлено, что сочетание блокаторов АТ1-рецепторов с диуретиком гидрохлоротиазидом способствует значительному улучшению контроля АД без появления отрицательных эффектов [218]. Для действия гипотиазида характерен гипокалиемический эффект, и он также угнетался валсартаном в результате, по-видимому, снижения уровня альдостерона. В то же время, ингибиторы АПФ уменьшали продукцию альдостерона более выраженно, чем блокаторы АТ1-рецепторов, поэтому применение ингибиторов АПФ в клинике часто сопровождается развитием гиперкалиемии.
Достижение целевого уровня АД при антигипертензивной терапии отмечается только у 64–84% пациентов, у лиц с изолированной систолической АГ — не более чем в 50% случаев и еще меньше — у пациентов сопутствующими СД 2-го типа или нефропатией, у которых целевым является уровень АД в пределах 130/80 мм рт. ст. [116, 117]. Еще более сложной является задача длительной стабилизации АД на целевом уровне. В то же время, антигипертензивный эффект телмисартана отмечается уже через 1–2 нед, достигает плато через 8–10 нед и стабилизируется на нем.
В руководстве ESC/ESH 2007 г. указывается, что задачей антигипертензивной терапии является не только нормализация АД, но ее сочетание с устранением метаболических нарушений, сопутствующих факторов кардиоваскулярного риска и повреждения органов-мишеней [195, 196]. Блокаторы AT1-рецепторов, в частности телмисартан, характеризуются выраженными кардио- и ангиопротекторными свойствами, способностью нормализовывать функцию эндотелия, структурные свойства сосудистой стенки, прежде всего — ее растяжимость, чувствительность к инсулину, и нейтральным действием в отношении симпатической нервной системы [103].
Одним из отличий в действии препаратов, модулирующих активность РАС, являются особенности их влияния на фибринолитическую систему. Установлено, что на фоне ее угнетения у лиц с АГ с уменьшением экспрессии t-PA и повышением плазменной концентрации PAI-1 применение ингибиторов АПФ сопровождается ее снижением и усилением экспрессии t-PA [32]. Для блокаторов АТ1-рецепторов, напротив, характерна способность угнетать фибринолитическую систему и повышать свертываемость крови. Однако дополнительное применение блокаторов АТ1-рецепторов у 24 пациентов с неконтролированной АГ на фоне недостаточно эффективной монотерапии ингибиторами АПФ сопровождалось достоверным снижением систолического АД (со 155 до 139 мм рт. ст.) и диастолического АД (с 91 до 81 мм рт. ст.) без отрицательного влияния на активность фибринолитической системы и С-реактивный белок. Эти данные позволили авторам сделать вывод, что сочетание ингибиторов АПФ и блокаторов рецепоров А II позволяет лучше контролировать АД без отрицательного влияния на активность воспаления и системы фибринолиза [3].
Установлено, что у лиц с сочетанием АГ и СД 2-го типа при любом виде терапии регрессия гипертрофии ЛЖ имеет менее выраженный характер. Так, в исследовании LIFE применение лозартана у лиц с АГ и без СД 2-го типа сопровождалось более выраженным уменьшением массы ЛЖ, чем применение атенолола, но у лиц с наличием СД отмечена обратная зависимость. Объясняют это тем, что у лиц с СД 2-го типа лозартан в большей степени способствует повышению активности ренина в плазме крови [67].
Однако, несмотря на патогенетический характер действия, как монотерапия ингибиторами АПФ и блокаторами AT1-рецепторов, так и их сочетание у части пациентов не позволяет добиться полной нормализации АД, и их комбинация с блокаторами кальциевых каналов обеспечивает дополнительную антигипертензивную эффективность и максимальную защиту от развития кардиоваскулярных и почечных осложнений [112].
Учитывая отсутствие альтернативных путей образования ренина, его блокаторы концептуально должны обладать более выраженным терапевтическим эффектом, чем ингибиторы АПФ и блокаторы АТ1-рецепторов. Это особенно связано с тем, что уровень ренина в плазме крови значительно повышен у лиц с высоким риском развития ИМ, инсульта и летального исхода и в еще большей степени может повышаться под действием блокаторов РАС [33]. Однако разработка эффективных и безопасных ингибиторов ренина растянулась на долгие годы, и только в настоящее время один из них — алискирен нашел применение в клинической практике.
Алискирен — первый представитель препаратов класса прямых ингибиторов ренина с доказанной эффективностью в отношении АГ [113, 343], экспериментального атеросклероза [143, 189, 221], диабетической нефропатии [47, 87]. Алискирен синтезирован около 30 лет назад и установлено, что его прием сочетается со снижением активности ренина в плазме крови, хотя содержание ренина возрастает параллельно за счет угнетения продукции А II. Однако математический анализ показал, что при применении алискирена в терапевтических дозах его концентрация в плазме крови достаточна для того, чтобы связать увеличенное даже в несколько раз количество молекул ренина.
Этот эффект алискирена подтвержден в исследовании, в котором его применение в виде монотерапии приводило к снижению активности ренина в плазме крови на 66%, несмотря на выраженное повышение его плазменной концентрации [320]. Эти изменения сочетались со снижением плазменного уровня А I, А II и альдостерона, а антигипертензивное действие алискирена было сопоставимым с действием лозартана [268].
В ряде исследований подтверждено, что применение прямых ингибиторов ренина сопровождается повышением плазменного содержания проренина и ренина в результате снижения плазменного уровня А II и устранения его угнетающего действия на продукцию ренина. Этот эффект может иметь самостоятельное повреждающее действие, так как ренин и проренин активируют (про)рениновые рецепторы и индуцируют развитие фиброза [153]. Однако параллельно ингибиторы ренина угнетают профибротические явления, что может предупреждать развитие отрицательных эффектов повышения плазменной концентрации ренина и проренина.
К числу наиболее значимых механизмов защитного действия алискирена при АГ, сердечной недостаточности и СД 2-го типа относится его способность уменьшать продукцию СОР и выраженность оксидативного стресса, воспаления, а также угнетать секрецию альдостерона [74, 142, 310].
В двойном слепом пилотном исследовании, проведенном с участием пациентов с хроническим недиабетическим почечным поражением, применение алискирена в течение 12 нед сопровождалось повышением плазменной концентрации ренина на 1131%, тогда как на фоне применения периндоприла содержание ренина в плазме крови увеличилось на 626%, плазменное содержание проренина, соответственно, нa 100 и 52,3%. В то же время, экскреция с мочой профибротического TФР-β достоверно снизилась по сравнению с плацебо и в равной степени после применения как алискирена, так и периндоприла [98]. Эти данные свидетельствовали о том, что как алискирен, так и периндоприл уменьшают выраженность профибротических явлений в почках, несмотря на значительное увеличение плазменного содержания ренина и проренина [186]. Антифибротическое и нефропротекторное действие алискирена отмечено и в ряде других исследований.
В двойном слепом рандомизированном исследовании 694 пациента с АГ получали алискирен, атенолол или их комбинацию в течение 6 нед. Снижение систолического АД к концу исследования составляло по группам соответственно 14,3; 14,3 и 17,3 мм рт. ст., диастолического — 11,3; 13,7 и 14,1 мм рт. ст. Снижение активности ренина в плазме крови в конце лечения достигло соответственно 65; 52 и 61% [69].
Установлено, что гипотензивное действие алискирена при монотерапии превосходило эффект гидрохлоротиазида и рамиприла и было аналогичным эффекту валсартана, лозартана и лизиноприла. В исследовании, проведенном с участием 14 тыс. пациентов с АГ, выявлено, что алискирен обладает высокой антигипертензивной активностью даже на фоне ингибиторов АПФ или блокаторов АТ1-рецепторов. При сочетанном применении алискирен значительно повышал эффективность других антигипертензивных препаратов [245, 274], а дополнительное его применение позволило в большинстве случаев добиться снижения АД до целевого уровня [357].
Результаты исследований ALOFT, AVOID и ALLAY свидетельствуют о том, что алискирен оказывает выраженный положительный эффект у лиц с СД 2-го типа и нефропатией, предупреждает кардиоваскулярные и почечные осложнения, особенно развитие гипертрофии ЛЖ, сердечной и почечной недостаточности [59, 358]. В двойном слепом рандомизированном исследовании гипотензивный эффект алискирена у больных АГ превосходил эффект гидрохлоротиазида на 12-й неделе (снижение систолического/диастолического АД на 17,4/12,2 по сравнению с 14,7/10,3 мм рт. ст.), 26-й (на 20,3/14,2 по сравнению с 18,6/13,0 мм рт. ст.) и на 52-й неделе (на 22,1/16,0 по сравнению с 21,2/15,0 мм рт. ст.).
Добавление алискирена к стандартной терапии у пациентов с СД 2-го типа сопровождалось снижением систолического АД у лиц в нижнем тертиле содержания HbA1c на 0,5 мм рт. ст., в среднем — на 0,6 мм рт. ст., в верхнем — на 1,1 мм рт. ст. без достоверного влияния на уровень диастолического АД [239].
В исследовании AVOID (Aliskiren in the EValuation of PrOteinuria In Diabetes) 599 испытуемых с СД 2-го типа, АГ и нефропатией были распределены на подгруппы соответственно тертилям содержания HbA1c (<7,1%, 7,1–8,4% и >8,4%). Часть пациентов получали алискирен дополнительно к лозартану и стандартной антигипертензивной терапии в течение 6 мес. Полученные данные свидетельствовали о способности алискирена уменьшать выраженность протеинурии практически у всех больных, но наибольший эффект с максимальным уменьшением отношения альбумин/креатинин в моче был получен в группе лиц с наиболее высоким содержанием HbA1c (>8,4%) [240].
В исследовании ALLAY (Aliskiren in Left Ventricular Hypertrophy), включавшем 465 гипертензивных пациентов, определялась эффективность угнетения РАС в отношении регрессии гипертрофии ЛЖ. Длительность лечения составляла 36 нед. Данные показали, что терапия с комбинацией лозартана и алискирена характеризовалась значительно более выраженным уменьшением гипертрофии ЛЖ у лиц с АГ, сочетающейся с СД 2-го типа, по сравнению с лицами без СД. Этот эффект сопровождался и в значительной степени был следствием более выраженного снижения плазменной концентрации альдостерона. В то же время, у пациентов без СД 2-го типа оба принципа терапии в одинаковой степени влияли как на выраженность гипертрофии ЛЖ, так и на уровень альдостерона [313].
Ранее неоднократно отмечено, что пациенты с сочетанием АГ и СД 2-го типа более подвержены развитию микро- и макроваскулярных поражений, которым предшествует появление гипертрофии ЛЖ [224]. Это подтверждено в исследовании AVOID с участием пациентов с СД 2-го типа, АГ и протеинурией, которые получали стандартную антигипертензивную и ренопротекторную терапию лозартаном. Установлено, что добавление к этой терапии алискирена достоверно уменьшало выраженность протеинурии, и этот эффект не был связан со снижением АД. В другом исследовании применение алискирена дополнительно к ирбесартану уменьшало выраженность альбуминурии на 31% [240].
В исследовании ASPIRE (Aliskiren Study in Post-MI Patients to Reduce Remodeling) применение алискирена на фоне ингибиторов АПФ или блокаторов рецепторов А II у постинфарктных пациентов с СД сопровождалось достоверно более выраженным восстановлением функции ЛЖ и уменьшением его конечно-систолического объема [288]. Помимо этого, при комбинированной терапии отмечено более выраженное снижение активности ренина в плазме крови и плазменного содержания альдостерона.
Однако применение алискирена сопровождалось многократным повышением плазменной концентрации ренина, поскольку устраняло угнетающее действие А II на его секрецию. Тем не менее, концентрация алискирена в крови при его длительном применении оказывается достаточной для того, чтобы блокировать действие ренина даже при резком повышением его уровня в плазме крови.
Алискирен оказывал также значительно менее выраженное побочное действие по сравнению с мочегонными средствами, и частота развития гипокалиемии составила соответственно 0,9 и 17,9%. Поэтому ингибиторы ренина концептуально рассматриваются многими исследователями как перспективное оптимальное средство для угнетения РАС в кардиологической практике.
Однако окончательное практическое решение этого вопроса еще остается в отдаленной перспективе. Ранее установленное достоверное повышение ренопротекторной эффективности у лиц с диабетическим поражением почек при добавлении алискирена к блокаторам АТ1-рецепторов позволило высказать предположение, что сочетание традиционных ингибиторов РАС с алискиреном может повысить эффективность устранения отрицательных эффектов гиперактивности РАС [235]. Проверка этого предположения проведена в двойном слепом исследовании, в которое был включен 8561 пациент с СД 2-го типа; часть больных, получавших сочетанное лечение ингибиторами АПФ и блокаторами АТ1-рецепторов, дополнительно принимали алискирен. Эффективность терапии оценивалась по первичной конечной точке (время до развития кардиальной смерти или остановки сердца с реанимацией, нефатального ИМ или инсульта, терминальной стадии поражения почек). Исследование было прекращено преждевременно через 32,9 мес, так как конечная точка была отмечена в 18,3% случаев у пациентов группы сочетанной терапии по сравнению с 17,0% в группе контроля (HR 1,08). Достоверно повысился риск развития инсульта (HR 1,34), существенное различие отмечено и в эффективности влияния терапии на функцию почек. Частота развития гиперкалиемии у лиц, получавших дополнительно алискирен, повысилась на 30%, что стало одной из основных причин преждевременного прекращения исследования [236]. Эти данные свидетельствовали, что дополнение стандартной терапии ингибиторами РАС алискиреном у пациентов высокого кардиоваскулярного риска с СД 2-го типа не только не снижало частоту развития кардиоваскулярных и ренальных явлений, но приводило даже к ее повышению в сочетании с увеличением выраженности гиперкалиемии, ренальной дисфункции (рис. 7).
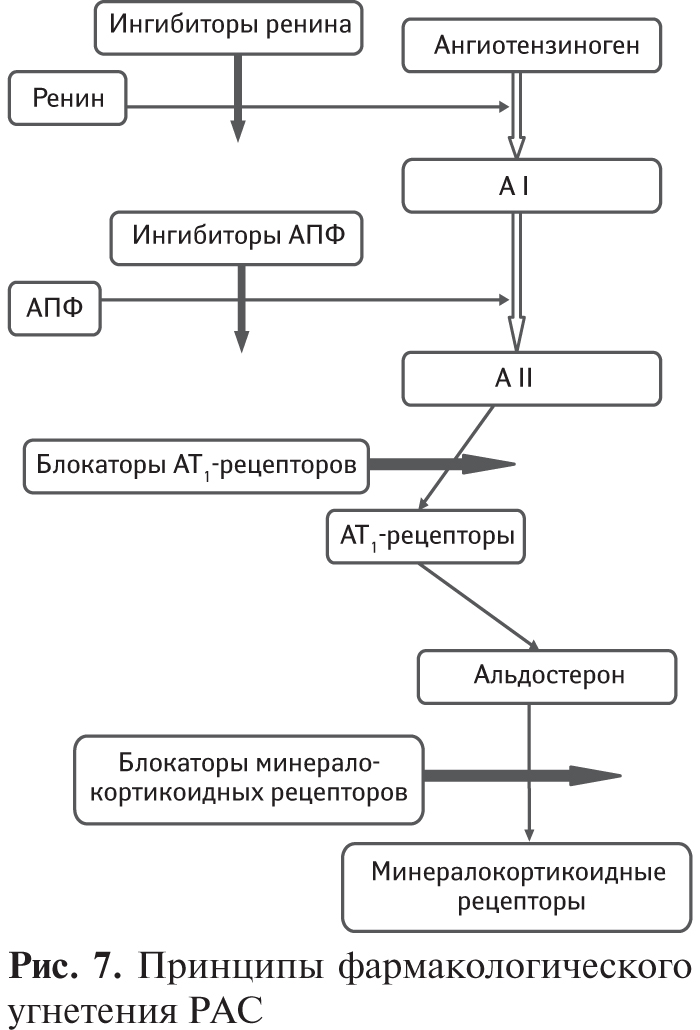
Тем не менее, вопрос об эффективности применения различных стратегий в лечении лиц с АГ остается открытым и в настоящее время. Ни в одном из крупных метаанализов результатов применения различных стратегий контроля АД у пациентов с АГ не получено убедительных данных о преимуществах какого-то определенного режима фармакотерапии в предупреждении инсульта, ИМ и других конечных кардиоваскулярных точек. Расчетным путем определено, что эффективность современных стратегий, основанных на применении ингибиторов АПФ или блокаторов АТ1-рецепторов, должна превысить эффективность применения блокаторов β-адренорецепторов и диуретиков на 25%, но ни в одном из проведенных исследований это предположение не подтвердилось.
В исследовании ALLHAT [15] не установлено достоверных преимуществ применения ингибиторов РАС у лиц с АГ и даже более частое развитие сердечной недостаточности по сравнению с тиазидными диуретиками. Не установлено преимуществ ингибиторов АПФ даже у лиц с сочетанием АГ и СД 2-го типа. Хотя у пациентов, получавших диуретики, отмечена большая частота развития СД 2-го типа, это не отразилось на частоте развития конечных точек в связи с относительно небольшой длительностью наблюдения. На основании полученных данных авторы пришли к выводу, что тиазидные диуретики остаются препаратами первого ряда в лечении АГ.
Однако сделанные выводы оспариваются многими авторами в связи с рядом погрешностей в протоколе исследования [274].
Положение о преобладании защитного эффекта ингибиторов АПФ при АГ остается в настоящее время неоднозначным, и в исследовании PEACE не было установлено эффекта трандолаприла на первичные конечные точки [237]. В крупном метаанализе, включившем 16 исследований и более 70 000 пациентов, не выявлено различий в эффективности предупреждения инсульта и кардиоваскулярных конечных точек между ингибиторами АПФ, диуретиками и блокаторами β-адренорецепторов [22]. В данное время широко исследуется эффективность сочетанного применения блокаторов АТ1-рецепторов и ингибиторов АПФ (исследование ASCOT).
До настоящего времени остается спорным вопрос и о том, в какой степени защитный эффект блокаторов РАС определяется их антигипертензивным действием, а в какой связан со способностью уменьшать выраженность ремоделирования сердца и сосудов.
Исследование НОРЕ было проведено в 19 странах с включением 9297 пациентов в возрасте старше 55 лет с наличием кардиоваскулярной патологии и, как минимум, одного из факторов риска (АГ, гиперхолестеринемия, низкий уровень холестерина ЛПВП, микроальбуминурия). У 40% пациентов выявлен СД 2-го типа, у 50% — АГ. Из 651 пациентов, отобранных в группу рамиприла, 14% достигли композитной конечной точки по сравнению с 17,8% в группе плацебо, что означало снижение риска на 22%. Кардиоваскулярная летальность уменьшилась на 26%, риск ИМ — на 20%, инсульта — на 32%, смерти от всех причин — на 16%. Эффект был достоверным, независимо от наличия СД, АГ и микроальбуминурии, а также от возраста. Авторы исследования пришли к выводу о кардиопротекторном действии рамиприла, не связанном с антигипертензивным действием, так как его влияние на АД было крайне незначительным (уменьшением систолического/диастолического АД на 3/2 мм рт. ст. в среднем и 2,2/1,4 мм рт. ст. — у пациентов с СД). Часть пациентов получали витамин Е с отрицательным результатом.
Однако ранее в исследовании HOT (Hypertension Optimal Treatment) продемонстрировано, что даже небольшое снижение АД в физиологическом диапазоне (снижение диастолического АД с 90 до 80 мм рт. ст.) сочеталось с выраженным уменьшением кардиоваскулярной летальности, особенно у пациентов с СД 2-го типа. В других исследованиях у пациентов с АД, равным в среднем 175/85 мм рт. ст., его снижение на 8,6/3,9 мм рт. ст. сочеталось со снижением риска развития конечных точек на 69%, кардиоваскулярной смерти — на 76%, общей летальности — на 55%. Даже в исследовании HOPE снижение риска первичной конечной точки составило 6% на 1 мм рт. ст. снижения систолического АД и 9% — на 1 мм диастолического АД. Это данные трактуются как подтверждение того, что почти весь эффект применения рамиприла в исследовании HOPE определялся его антигипертензивным действием [339].
В большинстве случаев кардиоваскулярной патологии комбинация ингибиторов АПФ с блокаторами АТ1-рецепторов сочеталась с умеренным, но достоверным повышением эффективности лечения. Однако в терапии пациентов после перенесенного ИМ и лиц с высоким кардиальным риском сочетанное применение ингибиторов АПФ с блокаторами АТ1-рецепторов не оказывало дополнительного эффекта.
В то же время, в ряде крупных клинических исследований применение как ингибиторов АПФ, так и блокаторов AT1-рецепторов у пациентов с АГ во многих случаях не позволило добиться полного контроля АД, оставались высокими частота госпитализации и летальность у лиц с застойной сердечной недостаточностью, диабетической нефропатией.
Отсутствие закономерной эффективности действия ингибиторов РАС в клинической практике может быть в значительной степени связано со сложностью организации и недостаточным пониманием принципов функционирования РАС, несовершенством современной фармакотерапии в снижении ее активности. Большинство современных препаратов не вызывает полной блокады РАС, их применение сопровождается компенсаторным увеличением плазменного содержания ренина, А ΙΙ и альдостерона [364]. Установлено, что применение блокаторов АТ1-рецепторов приводит к повышению концентрации А ΙΙ в крови и развитию прямых эффектов, не связанных с его действием рецепторным путем.
Основными эффектами применения ингибиторов АПФ являются снижение концентрации в крови А II и уменьшение выраженности его действия. Однако А II является мощным естественным ингибитором продукции ренина в юкстагломерулярном аппарате почек, и снижение его уровня в крови сопровождается повышением плазменного содержания и активности проренина и ренина, которые могут оказывать самостоятельное повреждающее действие. В прямых исследованиях подтверждено, что применение как ингибиторов АПФ, так и блокаторов А II сочетается с повышением активности ренина в плазме крови, что является независимым предиктором возрастания риска тяжелых клинических исходов. Поэтому полагают, что применение прямых блокаторов ренина может устранить этот эффект и предупредить увеличение продукции А I и А II [287, 288].
В настоящее время не вызывает сомнений, что значимость АГ как фактора кардиоваскулярного риска определяется не столько повышением уровня АД, сколько сочетанием АГ с метаболическими нарушениями, развитием воспаления и оксидативного стресса. Установлено, что пациенты с этим сочетанием характеризуются значительно более выраженным поражением органов-мишеней, чем пациенты в наличием изолированной АГ [57, 63, 267]. Поэтому и в настоящее время не прекращаются дебаты относительно того, связан ли кардиопротекторный эффект ингибиторов АПФ с их антигипертензивным действием, как повторно заявляют авторы исследования HOPE, или же он обусловлен угнетением других механизмов РАС.
Помимо того, что развитие АГ сочетается с прямым поражением органов-мишеней, в последние 20 лет неоднократно отмечено наличие зависимости между повышенным АД и развитием инсулинорезистентности. У пациентов с АГ закономерно выявляют значительно более высокий уровень инсулина, С-пептида и индекса инсулинорезистентности (НОМА-IR) по сравнению с нормотензивными лицами [89].
В соответствии с современными представлениями, АГ в большинстве случаев развивается в сочетании с нарушением толерантности к глюкозе и является одним из компонентов кардиометаболического синдрома [196, 290]. Поэтому в лечении больных с АГ оптимальным является только тот тип фармакотерапии, который сочетает антигипертензивное действие с устранением или уменьшением выраженности сопутствующих метаболических нарушений даже при отсутствии достоверных признаков метболического синдрома или СД 2-го типа.
3.3. Список использованной литературы
- Abramov D., Carson P.E. The role of angiotensin receptor blockers in reducing the risk оf cardiovascular disease // JRAAS. — 2012. — Vol. 13. — P. 317–327.
- Adeagbo A.S., Zhang X., Patel D. et al. Cyclo-oxygenase-2, endothelium and aortic reactivity during deoxycorticosterone acetate salt-induced hypertension // J. Hypertens. — 2005. — Vol. 23. — P. 1025–1036.
- Agirbasli M., Cincin A., Baykan O.A. Short-term effects of angiotensin receptor blockers on blood pressure control, and plasma inflammatory and fibrinolytic parameters in patients taking angiotensin-converting enzyme inhibitors // JRAAS. — 2008. — Vol. 9. — P. 22–26.
- Al Awwadi N.A., Bornet A., Azay J. et al. Red wine polyphenols alone or in association with ethanol prevent hypertension, cardiac hypertrophy, and production of reactive oxygen species in the insulin-resistant fructose-fed rat //J. Agric. Food Chem. — 2004. — Vol. 52. — P. 5593–5597.
- Alderman M.H., Madhavan S., Ooi W.L. et al. Association of the renin-sodium profile with the risk of myocardial infarction in patients with essential hypertension // N. Engl. J. Med. — 1991. — Vol. 324. — P. 1098–1104.
- Alexander B.T., Cockrell K.L., Rinewalt A.N. et al. Enhanced renal expression of preproendothelin mRNA during chronic angiotensin II hypertension // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2001. — Vol. 280. — P. R1388–R1392.
- Al-Mallah M., Tleyjeh I., Abdel-Latif A. et al. Angiotensin-converting enzyme inhibitors in coronary artery disease and preserved left ventricular systolic function // J.Am.Coll.Cardiol. — 2006. — Vol. 47. — P. 1576–1583.
- Anderson T.J., Elstein E., Haber H. et al. Comparative study of ACE-inhibition, angiotensin II antagonism, and calcium channel blockade on flow-mediated vasodilation in patients with coronary disease (BANFF study) // J. Am. Coll. Cardiol. — 2000. — Vol. 35. — P. 60–66.
- Appel L.J., Moore T.J., Obarzanek E. et al. A clinical trial of the effects of dietary patterns on blood pressure: DASH Collaborative Research Group // N. Engl. J. Med. — 1997. — Vol. 336. — P. 1117–1124.
- Atarashi K., Ishiyama A., Takagi M. et al. Vitamin E ameliorates the renal injury of Dahl salt-sensitive rats // Am. J. Hypertens. — 1997. — Vol. 10. — P. 116S–119S.
- Auabe N., Babaev V.R., Tang Y. et al. Transiently hightened angiotensin II has distinct effects on atherosclerosis and aneurism formation in hyperlipidemic mice // Atherosclerosis. — 2006. — Vol. 184. — P. 312–321.
- Banday A.A., Lokhandwala M.F. Oxidative stress-induced renal angiotensin AT1 receptor upregulation causes increased stimulation of sodium transporters and hypertension // Am. J. Physiol. Renal. Physiol. — 2008. — Vol. 295. — P. F698–F706.
- Banday A.A., Lokhandwala M.F. Oxidative stress causes renal angiotensin II type 1 receptor upregulation, Na+/H+ еxchanger 3 overstimulation, and hypertension // Hypertens. — 2011. — Vol. 57. — P. 452–459.
- Banday A.A., Muhammad A.B., Fazili F.R. et al. Mechanisms of oxidative stress-induced increase in salt sensitivity and development of hypertension in sprague-dawley rats // Hypertens. — 2007. — Vol. 49. — P. 664–671.
- Bangalore S., Sawhney S., Messerli F.H. Relation of beta-blocker–induced heart rate lowering and cardioprotection in hypertension // JACC. — 2008. — Vol. 52. — P. 1482–1489
- Barauna V.G., Magalhaes F.C., Krieger J.E. et al. AT1 receptor participates in the cardiac hypertrophy induced by resistance training in rats // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2008. — Vol. 295. — P. R381-R387.
- Barzilay J.I., Davis B.R., Cutler J.A. et al. Fasting glucose levels and incident diabetes mellitus in older nondiabetic adults randomized to receive 3 different classes of antihypertensive treatment: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) // Arch. Intern. Med. — 2006. — Vol. 166. — P. 2191–2201.
- Bayorh M.A., Ganafa A.A., Eatman D. et al. Simvastatin and losartan enhance nitric oxide and reduce oxidative stress in salt-induced hypertension // Am. J. Hypertens. — 2005. — Vol. 18. — P. 1496–1502.
- Bendall J.K., Cave A.C., Heymes C. et al. Pivotal role of a gp91(phox)-containing NAD(P)H oxidase in angiotensin II-induced cardiac hypertrophy in mice // Circulation. — 2002. — Vol. 105. — P. 293–296.
- Berry C., Brosnan M.J., Fennell J. et al. Oxidative stress and vascular damage in hypertension // Curr. Opin. Nephrol. Hypertens. — 2001. — Vol. 10. — P. 247–255.
- Blendea M.C., Jacobs D., Stump C.S. et al. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression // Am. J.Physiol. Endocrinol. Metab. — 2005. — Vol. 288. — Р. E353–E359.
- Blood Pressure Lowering Treatment Trialists Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs; results of prospectively designed overviews of randomised trials // Lancet. — 2000. — Vol. 355. — Р. 1955–1964.
- Blumenfeld J.D., Sealey J.E., Mann S.J. et al. β-Adrenergic receptor blockade as a therapeutic approach for suppressing the renin-angiotensinaldosterone system in normotensive and hypertensive subjects // Am. J. Hypertens. — 1999. — Vol. 12. — Р. 451–459.
- Boetger T., Beetz N., Kostin S. et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster // J. Clin. Invest. — 2009. — Vol. 119. — P. 2634–2640.
- Bohm M., Lee M., Kreutz R. et al. Angiotensin II receptor blockade in TGR(mREN2)27: effects of renin-angiotensin system gene expression and cardiovascular functions // J. Hypertens. — 1995. — Vol. 13. — P. 891–899.
- Bomback A.S., Klemmer P.J. Interaction of aldosterone and extracellular volume in the pathogenesis of obesity-associated kidney disease: a narrative review // American Journal of Nephrology. — 2009. — Vol. 30(2). — Р. 140–146.
- Bondeva T., Roger T., Wolf G. Differential regulation of Toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU1 transcriptional sites // Am. J. Nephrol. — 2007. — Vol. 27. — P. 308–314.
- Brancati F.L., Kao W.H., Folsom A.R. et al. Incident type 2 diabetes mellitus in African American and white adults: the Atherosclerosis Risk in Communities Study // JAMA. — 2000. — Vol. 283. — Р. 2253–2259.
- Briones A.M., Rodríguez-Criado N., Hernanzet R. et al. Atorvastatin prevents angiotensin II–induced vascular remodeling and oxidative stress // Hypertens. — 2009. — Vol. 54. — P. 142–149.
- Brody M.J. Central nervous system and mechanisms of hypertension // Clin. Physiol. Biochem. — 1988. — Vol.6. — P. 230–239.
- Brown N.J. Aldosterone and vascular inflammation // Hypertension. — 2008. — Vol. 51. — P. 161–167.
- Brown N.J., Kim K.S., Chen Y.Q. et al. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production // J. Clin. Endocrinol. Metab. — 2000. — Vol. 85. — P. 336–344.
- Brunner H.R., Laragh J.H., Baer L. et al. Essential hypertension: renin and aldosterone, heart attack and stroke // N. Engl. J. Med. — 1972. — Vol. 286. — Р. 441–449.
- Calhoun D.A. Is there an unre-cognized epidemic of primary aldosteronism? //Hypertens. — 2007. — Vol. 50. — P. 447–453.
- Calhoun D.A., Jones D., Textor S. et al. Resistant Hypertension: Diagnosis, Evaluation, and Treatment. A Scientific Statement From the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research // Hypertens. — 2008. — Vol. 51. — P. 1403–1419.
- Calhoun D.A., Sharma K. The role of aldosteronism in causing obesity-related cardiovascular risk // Cardiology Clinics. — 2010. — Vol. 28. — Р. 517–527.
- Callera G.E., Touyz R.M., Tostes R.C. et al. Aldosterone activates vascular p38MAP kinase and NAD(P)H oxidase via c-Src // Hypertens. — 2005. — Vol. 45. — Р. 773–779.
- Campese V.M., Shaohua Y., Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity // Hypertens. — 2005. — Vol. 46. — P. 533–539.
- Cardinale J.P., Sriramula S., Mariappan N. et al. Angiotensin II-induced hypertension is modulated by nuclear factor-κB in the paraventricular nucleus // Hypertens. — 2012. — Vol. 59. — P. 113–121.
- Ceconi C., Fox K.M., Remme W.J. et al. ACE inhibition with perindopril and endothelial dysfunction. Results of a sustudy of the EUROPA study PERTINENT // Cardiovasc. Res. — 2007. — Vol. 73. — P. 237–246.
- Celik T., Iyisoy A., Kursaklioglu H. et al. The comparative effects of telmisartan and ramipril on P-wave dispersion in hypertensive patients: a randomized clinical study // Clin. Cardiol. — 2005. — Vol. 28. — P. 298–302.
- Ceriello A. Possible role of oxidatіve stress in the pathogenesis of hypertension // Diabetes Care. — 2008. — Vol. 31, (Suppl.2). — P. S181–S184.
- Chapell M.C. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1–7)-Mas receptor axis: more than regulation of blood pressure // Hypertens. — 2007. — Vol. 50. — P. 596–599.
- Cheetham C., Collis J., O’Driscoll G. et al. Losartan, an angiotensin type1 receptor antagonist, improves endothelial function in non-insulin-dependent diabetes // J. Am. Coll. Cardiol. — 2000. — Vol. 36. — P. 1461–1466.
- Chen K., Chen J., Li D.et al. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts. Modulation by PPAR-y ligand pioglitazone // Hypertension. — 2004. — Vol. 44. — P. 655.
- Cheng Z.J., Vaskonen T., Tikkanen I. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats // Hypertens. — 2001. — Vol. 37. — Р.433–438.
- Cherney D.Z., Lai V., Scholey J.W. et al. Effect of direct renin inhibition on renal hemodynamic function, arterial stiffness, and endothelial function in humans with uncomplicated type 1 diabetes: a pilot study // Diabetes Care. — 2007. — Vol. 33. — P. 361–365.
- Collister J.P., Hendel M.D. Role of the subfornical organ in the chronic hypotensive response to losartan in normal rats // Hypertens. — 2003. — Vol. 41. — P. 576–582.
- Collister J.P., Hendel M.D. The role of Ang (1–7) in mediating the chronic hypotensive effects of losartan in normal rats // JRAAS. — 2003. — Vol. 4. — P. 176–179.
- Collister J.P., Nahey D.B. Changing dietary sodium alters the chronic cardiovascular effects of losartan in rats // JRAAS. — 2008. — Vol. 9. — P. 10–16.
- Comporti M., Signorini C., Arezzini B. et al. F2-isoprostanes are not just markers of oxidative stress // Free Radic. Biol. Med. — 2008. — Vol. 44. — P. 247–256.
- Cooper S.A., Whaley-Connell A., Habibi J. et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H2009–H2023.
- Crackower M.A., Sarao R., Oudit G.Y. et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function // Nature. — 2002. — Vol. 417. — P. 822–828.
- Crandall D.L., Herzlinger H.E., Saunders B.D. et al. Developmental aspects of the adipose tissue renin-angiotensin system: therapeutic implications // Drug Dev. Res. — 1994. — Vol. 32. — P. 117–125.
- Crowley S.D., Song Y.S., Lin E.E. et al. Lymphocyte responses exacerbate angiotensin II-dependent hypertension // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2010. — Vol. 298. — P. R1089–R1097.
- Crowley S.D., Vasievich M.P., Ruiz P. et al. Glomerular type 1 angiotensin receptors augment kidney injury and inflammation in murine autoimmune nephritis // J. Clin. Invest. — 2009. — Vol. 119. — P. 943–953.
- Cuspidi C., Meani S., Fusi V. et al. Metabolic syndrome and target organ damage in Dab H., Hachani R., Dhaouadi N. et al. Physiological regulation of MMPs and tPA/PAI in untreated essential hypertensives // J. Hypertens. — 2004. — Vol. 22. — Р. 1991–1998.
- Dagenais G.R., Pogue G., Fox K. et al. Angiotensin-converting enzyme inhibitors in stabe vascular disease without left ventriculasr systolic dysfunction or heart failure: a combined analysis of these trails // Lancet. — 2006. — Vol. 368. — P. 581–588.
- Dahlof B., Devereux R.B., Kjeldsen S.E. et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol // Lancet. — 2002. — Vol. 359. — P. 995–1003.
- Danser A.H. Cardiac angiotensin II: does it have a function? // AJP — Heart. — 2010. — Vol. 299. — P. H1304–H1306.
- Danser A.H., Batenburg W.W., van Esch J.H.M. Prorenin and the (pro)renin receptor — an update // Nephrol. Dial. Transplant. — 2007. — Vol. 22. — P. 1288–1292.
- Datla S.R., Griendling K.K. Reactive oxygen species, NAD(P)H oxidases, and hypertension // Hypertens. — 2010. — Vol. 56. — P. 325–330.
- De Cavanagh E.M., Inserra F., Ferder M. et al. From mitochondria to disease: role of the renin-angiotensin system // Am.J.Nephrol. — 2007. — Vol. 27. — Р. 545–553.
- De Gasparo M., Hess P., Nuesslein-Hildesheim B. et al. Combination of non-hypotensive doses of valsartan and enalapril improves survival of spontaneously hypertensive rats with endothelial dysfunction // JRAAS. — 2000. — Vol. 1. — P. 151–158.
- Delbosc S., Cristol J.P., Descomps B. et al. Simvastatin prevents angiotensin II-induced cardiac alteration and oxidative stress // Hypertens. — 2002. — Vol. 40. — P. 142–147.
- Demers C., McMurray J.J., Swedberg K. et al. Impact of candesartan on nonfatal myocardial infarction and cardiovascular death in patients with heart failure // JAMA. — 2005. — Vol. 2941. — P. 1794–1798.
- Devereux R.B., Dahlof B., Gerdts E. et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: The Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial // Circulation. — 2004. — Vol. 110. — Р. 1456–1462.
- Diep Q.N., Mabrouk M.E., Cohn J.S. et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats. Role of peroxisome proliferator-activated receptor-y // Circulation. — 2002. — Vol.105. — P.2296–2302.
- Dietz R., Dechend R., Yu C-M. Effects of the direct renin inhibitor aliskiren and atenolol alone or in combination in patients with hypertension // JRAAS. — 2008. — Vol. 9. — P. 163–175.
- Dietze G.J., Henriksen E.J. Angiotensin-converting enzyme in skeletal muscle: sentinel of blood pressure control and glucose homeostasis // JRAAS. — 2008. — Vol. 9. — Р. 75–88.
- Díez-Freire C., Vasquez J., Correa de Adjounian M. et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR // Physiol. Genomics. — 2006. — Vol. 27. — Р. 12–19.
- Dikalova A.E., Bikineyeva A.T., Budzyn K. et al. Therapeutic targeting of mitochondrial superoxide in hypertension // Circ. Res. — 2010. — Vol. 107. — P. 106–116.
- Ditting T., Tiegs G., Veelken R. Autonomous innervation in renal inflammatory disease-innocent bystander or active modulator? // J. Mol. Med. — 2009. — Vol. 87. — P. 865–870.
- Dong Y.F., Liu L., Kataoka K. et al. Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes // Diabetologia. — 2010. — Vol. 53. — Р. 180–191.
- Donnelly R., Manning G. Angiotensin-converting enzyme inhibitors and coronary heart disease prevention // JRAAS. — 2007. — Vol. 8. — P. 13–22.
- Doughan A.K., Harrison D.G., Dikalov S.I. Molecular mechanisms of angiotensin II mediated mitochondrial dysfunction. Linking mitochondrial oxidative damage and vascular endothelial dysfunction // Circ. Res. — 2008. — Vol. 102. — P. 488–496.
- Dzielak D.J. Immune mechanisms in experimental and essential hypertension // Am. J. Physiol. — 1991. — Vol. 260. — P. R459–R467.
- Elmarakby A.A., Loomis E.D., Pollock J.S. et al. NAD(P)H oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1 // Hypertens. — 2005. — Vol. 45. — P. 283–287.
- Elmarakby A.A., Quigley J.E., Imig J.D. et al. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2008. — Vol. 294. — Р. R76–R83.
- Esler M.D., Krum H., Sobotka P.A. et al. Renal sympathetic denervation in patients with treatment-resistant hypertension (the Symplicity HTN-2 Trial): a randomised controlled trial // Lancet. — 2010. — Vol. 376. — P. 1903–1909.
- Eurich D.T., Majumdar S.R., Tsuyuki R.T. et al. Reduced mortality associated with the use of ACE inhibitors in patients with type 2 diabetes // Diabetes Care. — 2004. — Vol. 27. — P. 1330–1334.
- Evangelist F., Krieger J. Small gene effect and exercise training-induced cardiac hypertrophy in mice: an Ace gene dosage study // Physiol. Genomics. — 2006. — Vol. 27. — P. 231–236.
- Fanelli C., Zatz R. Linking oxidative stress, the renin-angiotensin system, and hypertension // Hypertens. — 2011. —
Vol. 57. — P. 373–374. - Farquharson C.A., Struthers A.D. Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy // Clin.Sci. (London). — 2002. — Vol. 103. — P. 425–431.
- Feldman D.L., Jin L., Xuan H. et al. Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats // Hypertens. — 2008. — Vol. 52. — P. 130–136.
- Fernandes T., Hashimoto N.Y., Magalhаes F.C. et al. Aerobic Exercise Training–Induced Left Ventricular Hypertrophy Involves Regulatory MicroRNAs, Decreased Angiotensin-Converting Enzyme-Angiotensin II, and Synergistic Regulation of Angiotensin-Converting Enzyme 2-Angiotensin (1–7) // Hypertension. — 2011. — Vol. 58. — P. 182–189.
- Ferrannini E., Buzzigoli G., Bonadonna R. et al. Insulin resistance in essential hypertension // N. Engl. J. Med. — 1987. — Vol. 317. — Р. 350–357.
- Ferrari R. Angiotensin-converting enzyme inhibition in car diovascular disease: evidence with perindopril // Expert. Rev. Cardiovasc. Ther. — 2005. — Vol. 3.— P. 15–29.
- Ferrari R., Fox K.M. The mode of action of perindopril // in: Perindopril: a major contribution to the prevention and reatment of cardiovascular diseases, ed. R. Ferrari a. K.M. Fox. — 2008. — P. 3–14.
- Ferrario C.M. Role of angiotensin II in cardiovascular disease –therapeutic implications of more than a century of research // JRAAS. — 2006. — Vol. 7. — P. 3–14.
- Ferrario C.M. Angiotensin-converting enzyme 2 and angiotensin-(1–7): an evolving story in cardiovascular regulation // Hypertens. — 2006. — Vol. 47. — P. 515–521.
- Ferroni P., Basili S., Falco A. et al. Oxidant stress and platelet activation in hypercholesterolemia // Antioxid.Redox Signal. — 2004. — Vol. 6. — P. 747–756.
- Fink G.D., Watts S., Chen A. et al. Superoxide anion contributes to hypertension during chronic inhibition of nitric oxide synthesis // Hypertens. — 2002. — Vol. 40. — P. 380–385.
- Flather M.D., Yusuf S., Kober L. et al. ACE-Inhibitor Myocardial Infarction Collaborative Group. Long-term ACEinhibitor therapy in patients with heart failure or left ventricular dysfunction: a systematic overview of data from individual patients // Lancet. — 2000. — Vol. 355. — P. 1575–1581.
- Fogart R., Mugellini A., Zoppi A. et al. Losartan and perindopril effects on plasma plasminogen activator inhibitor-1 and fibrinogen in hypertensive type 2 diabetic patients // Am. J. Hypertens. — 2002. — Vol. 15. — P. 316–320.
- Fox K.M. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study) // Lancet. — 2003. — Vol. 362. — P. 782–788.
- Fox C.S., Massaro J.M., Schlett C.L. et al. Periaortic fat deposition is associated with peripheral arterial disease. The Framingham Heart Study // Circulation: Cardiovascular Imaging. — 2010. — Vol. 3. — P. 515–519.
- Fraser D., Wakefield L., Phillips A. Independent regulation of transforming growth factor beta 1 transcription and translation by glucose and platelet derived growth factor // Am. J. Pathol. — 2002. — Vol. 161. — P. 1039–1049.
- Frederic S. Michel, Gavin R. Norton, Olebogeng H.I. Majane et al. Contribution of Circulating Angiotensinogen Concentrations to Variations in Aldosterone and Blood Pressure in a Group of African Ancestry Depends on Salt Intake // Hypertens. — 2012. — Vol. 59. — P. 62–69.
- Frey N., Olson E.N. Cardiac hypertrophy: the good, the bad, and the ugly // Annu. Rev. Physiol. — 2003. — Vol. 65. — Р. 45–79.
- Fujii S., Zhang L., Igarashi J. et al. L-arginine reverses p47phox and gp91phox expression induced by high salt in Dahl rats // Hypertens. — 2003. — Vol. 42. — P. 1014–1020.
- Fujita M., Ando K., Kawarazaki H. et al. Sympathoexcitation by Brain Oxidative Stress Mediates Arterial Pressure Elevation in Salt-Induced Chronic Kidney Disease // Hypertens. — 2012. — Vol. 59. — P. 105–112.
- Fujita M., Okuda H., Tsukamoto O. et al. Blockade of angiotensin II receptors reduces the expression of receptors for advanced glycation end products in human endothelial cells // Arterioscler. Thromb. Vasc.Biol. — 2006. — Vol. 26. — P. e138–e140.
- Furuhashi M., Ura N., Higashiura K. et al. Blockade of the Renin-Angiotensin System Increases Adiponectin Concentrations in Patients with Essential Hypertension // Hypertens. — 2003. — Vol. 42. — P. 76–81.
- Galle J., Lehmann-Bodem C., Hubner U. et al. CyA and OxLDL cause endothelial dysfunction in isolated arteries through endothelin-mediated stimulation of O2− formation // Nephrol. Dial. Transplant. — 2000. — Vol. 15. — P. 339–346.
- Galley H.F., Thornton J., Howdle P.D. et al. Combination oral antioxidant supplementation reduces blood pressure // Clin. Sci. — 1997. — Vol. 92. — P. 361–365.
- Galzerano D., Di Michele S., Paolisso G. et al. A multicentre, randomized study of telmisartan versus carvedilol for prevention of atrial fibrillation recurrence in hypertensive patients // JÀAS. — 2012. — Vol. 13. — P. 496–503.
- Garvas I., Garvas H. Angiotensin II — possible adverse effects on arteries, heart, brain and kidney: experimental, clinical, and epidemiological evidence // in «The Renin-Angiotensin System» ed. by J.Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 1, (part 40). — P. 40.1.–40.11.
- Giacchetti G., Ronconi V., Turchi F. et al. Aldosterone as a key mediator of the cardiometabolic syndrome in primary aldosteronism: an observational study // J. Hypertens. — 2007. — Vol. 25. — Р. 177–186.
- Giacchetti G., Turchi F., Boscaro M. et al. Management of primary aldosteronism: its complications and their outcomes after treatment // Cur. Vasc. Pharmacol. — 2009. — Vol. 7. — Р. 244–249.
- Giordano F.J. Oxygen, oxidative stress, hypoxia, and heart failure // J. Clin. Invest. — 2005. — Vol. 115. — Р. 500–508.
- Gojanovic B., Feihl F., Liaudet L. et al. Concomitant calcium entry blockade and inhibition of the renin-angiotensin system: a rational and effective means for treating hypertension // JRAAS. — 2008. — Vol. 9. — Р. 1–9.
- Gradman A.H., Kad R. Renin inhibition in hypertension // J. Am. Coll. Cardiol. — 2008. — Vol. 51. — P. 519–528.
- Graham D., Huynh N.N., Hamilton C.A. et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy // Hypertens. — 2009. — Vol. 54. — P. 322–328.
- Granger J.P., Schnackenberg C.G. Renal mechanisms of angiotensin II-induced hypertension // Sem.Nephrol. — 2000. —
Vol. 20. — P. 417–425. - Grassi G., Mancia G. Neurogenic hypertension: is the enigma of its origin near the solution? // Нypertens. — 2004. — Vol. 43. — P. 154–155.
- Grassi G., Quarti-Trevano F., Mancia G. Cardioprotective effects of telmisartan in uncomplicated and complicated hypertension // JRAAS. — 2008. — Vol. 9. — P. 66–74.
- Gress T.W., Nieto F.J., Shahar E. et al. Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus/ Atherosclerotic Risk in Community Study // New Engl. J. Med. — 2000. — Vol. 342. — P. 905–912.
- Griendling K.K., Minieri C.A., Ollerenshaw J.D. et al. Angiotensin II stimulates NADH and NAD(P)H oxidase activity in cultured vascular smooth muscle cells // Circ. Res. — 1994. — Vol. 74. — P. 1141–1148.
- Grieve D.J., Byrne J.A., Siva A. et al. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload // J. Am. Coll. Cardiol. — 2006. — Vol. 47. — P. 817–826.
- Grossman E. Does increased oxidative stress cause hypertension? // Diabetes Care. — 2008. — Vol. 31, (Suppl.2). — P. S185–S189.
- Gu J.W., Anand V., Shek E.W. et al. Sodium induces hypertrophy of cultured myocardial myoblasts and vascular smooth muscles cells // Hypertens. — 1998. — Vol. 31. — P. 1083–1087.
- Guzik T.J., Hoch N.E., Brown K.A. et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction // J. Exp. Med. — 2007. — Vol. 204. — P. 2449–2460.
- Habibi J., Whaley-Connell A., Qazi M.A. et al. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats // Endocrinology. — 2007. — Vol. 148. — P. 2181–2188.
- Hainault I., Nebout G., Turban S. et al. Adipose tissue-specific increase in angiotensinogen expression and secretion in the obese (fa/fa) Zucker rat // Am. J. Physiol. Endocrinol. Metab. — 2002. — Vol. 282. — P. E59-E66.
- Hammoud R.A., Vaccari C.S., Nagamia S.H. et al. Regulation of the renin–angiotensin system in coronary atherosclerosis: A review of the literature // Vasc. Health Risk Manag. — 2007. — Vol. 3. — P. 937–945.
- Hanonv S., Vijayaraman P., Sonnenblick E. et al. Persistent formation of angiotensin II despite treatment with maximally recommended doses of angiotensin converting enzyme inhibitors in patients with chronic heart failure // JRAAS. — 2000. — Vol. 1. — P. 147–50.
- Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease // N. Engl. J. Med. — 2005. — Vol. 352. — P. 1685–1695.
- Hare J.M., Stamler J.S. NO/redox disequilibrium in the failing heart and cardiovascular system // J.Clin.Invest. — 2005. — Vol. 15. — Р. 509–517.
- Harrison D.G., Guzik T.J., Lob H.E. et al. Inflammation, immunity, and hypertension // Hypertens. — 2011. — Vol. 57. — P. 132–140.
- Harrison D.G., Vinh A., Lob H. et al. Role of the adaptive immune system in hypertension // Curr.Opin.Pharmacol. — 2010. — Vol. 10. — P. 203–207.
- Heeneman S., Slumer J.C., Deamen M.J. Angiotensin-converting enzyme and vascular remodelling // Circ. Res. — 2007. — Vol. 101. — P. 441–454.
- Henke N., Schmidt-Ullrich R., Dechend R. et al. Vascular endothelial cell-specific NF-kappaB suppression attenuates hypertension-induced renal damage // Circ. Res. — 2007. — Vol. 101. — P. 268–276.
- Hoagland K.M., Maier K.G., Roman R.J. Contributions of 20-HETE to the antihypertensive effects of tempol in Dahl salt-sensitive rats // Hypertens. — 2003. — Vol. 41. — P. 697–702.
- Hodroj W., Legedz L., Foudi N. et al. Increased insulin-stimulated expression of arterial angiotensinogen and angiotensin type 1 receptor in patients with type 2 diabetes mellitus and atheroma // Arterioscler. Thromb. Vasc. Biol. — 2007. — Vol. 27. — P. 525–532.
- [HOPE] The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients // N. Engl. J. Med. — 2000. — Vol. 342. — P. 145–153.
- Huan Y., DeLoach S., Keith S. W. et al. High blood pressure and obesity increase the risk of abnormal glucose tolerance in young adult african Americans // J. Clin. Hypertens. — 2011. — Vol. 13. — P. 397–403.
- Huang B.S., White R.A., Jeng A.Y. et al. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in dahl salt-sensitive rats // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2009. — Vol. 296.— Р. R994–R1000.
- Huang Y., Wongamorntham S., Kasting J. et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms // Kidney Int. — 2006. — Vol. 69. — P. 105–113.
- Huentelman M.J., Grobe J., Vasquez J.et al. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats // Exp. Physiol. — 2005. — Vol. 90. — Р. 783–790.
- Ibrahim M.M. RAS inhibition in hypertension // J. Hum. Hypertens. — 2006. — Vol. 20. — P. 101–108.
- Ino J., Kojima C., Osaka M. et al. Dynamic observation of mechanically injured mouse femoral artery reveals an antiinflammatory effect of renin inhibitor // Arterioscler. Thromb. Vasc. Biol. — 2009. — Vol. 29. — P. 1858–1863.
- Ito K., Hirooka Y., Sunagawa K. Blockade of mineralocorticoid receptors improves salt-induced left-ventricular systolic dysfunction through attenuation of enhanced sympathetic drive in mice with pressure overload // J. Hypertens. — 2010. — Vol. 28. — P. 1449–1458.
- Jandeleit-Dahm K.A.M., Tikellis C., Reid K.M. et al. Why blockade of the renin-angiotensin system reduces the incidence of new-onset diabetes // Hypertension. — 2005. — Vol. 23. — P. 463–473.
- Jorde U.P., Vittorio T.J., Dimayuga C.A. et al. Comparison of suppression of the circulating and vascular renin-angiotensin system by enalapril versus trandolapril in chronic heart failure // Am. J. Cardiol. — 2004. — Vol. 94. — P. 1501–1505.
- Julius S. Effectiveness of perindopril in hypertension // in: Perindopril: a major contribution to the prevention and reatment of cardiovascular diseases, ed. R. Ferrari a. K.M. Fox. — 2008.— P. 15–30.
- Julius S., Kjeldsen S.E., Weber M. et al. VALUE Trial Group. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial // Lancet. — 2004. — Vol. 363. — P. 2022–2031.
- Kaehler J., Sill B., Koester R. et al. Endothelin-1 mRNA and protein in vascular wall cells is increased by reactive oxygen species // Clin. Sci. — 2002. — Vol. 103. — P. 176S–178S.
- Kagiyama S.,Qian K., Kagiyama T. et al. Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy //Hypertens. — 2003. — Vol. 41. — P. 824–829.
- Kamide K., Rakugi H., Nagai M. et al. Insulin-mediated regulation of the еndothelial renin-angiotensin system and vascular cell growth // J. Hypertens. — 2004. — Vol. 22. — P. 121–127.
- Kang Y.M., Ma Y., Zheng J.P. et al. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension // Cardiovasc. Res. — 2009. — Vol. 82. — P. 503–512.
- Kang Y.S., Park Y.G., Kim B.K et al. Angiotensin II stimulates the synthesis of vascular endothelial growth factor through the p38 mitogen activated protein kinase pathway in cultured mouse podocytes // J. Mol. Endocrinol. — 2006. — Vol.3 6. — P. 377–388.
- Kang J.J., Toma I., Sipos A. et al. The collecting duct is the major source of prorenin in diabetes // Hypertension. — 2008. — Vol. 51. — P. 1597 –1604.
- Katusic Z.S. Superoxide anion and endothelial regulation of arterial tone // Free Rad. Bio. Med. — 1996. — Vol. 20. — P. 443–444.
- Kawada N., Imai E., Karber A. et al. A mouse model of angiotensin II slow-pressor response: role of oxidative stress // J. Am. Soc. Nephrol. — 2002. — Vol. 13. — P. 2860–2868.
- Kitamoto S., Egashira K., Kataoka C. et al. Chronic inhibition of nitric oxide synthesis in rats increases aortic superoxide anion production via the action of angiotensin II // J. Hypertens. — 2000. — Vol. 18. — P. 1795–1800.
- Kjeldsen S., Dahlof B., Devereux R.B. et al. Lowering of blood pressure and predictors of response in patients with left ventricular hypertrophy: the LIFE study. Losartan Intervention for Endpoint // Am. J. Hypertens. — 2000. — Vol. 8. — Р .899–906.
- Kobayashi N., Yoshida K., Nakano S. et al. Cardioprotective mechanisms of eplerenone on cardiac performance and remodeling in failing rat hearts // Hypertension. — 2006. — Vol. 47. — P. 671–679.
- Kobori H., Nishiyama A., Abe Y. et al. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet // Hypertens. — 2003. — Vol. 41. — P. 592–597.
- Koh K.K., Ahn J.Y., Han S.H. et al. Pleiotropic effects of angiotensin II receptor blocker in hypertensive patients // J. Am. Coll. Cardiol. — 2003. — Vol. 42. — P. 905–910.
- Kondo J., Sone T., Suboi H. et al. Effects of low-dose angiotensin II receptor blocker candesartan on cardiovascular events in patients with coronary artery disease // Am. Heart J. — 2003. — Vol. 146. — P. 1022–1027.
- Kudo H., Kai H., Kajimoto H. et al. Exaggerated blood pressure variability superimposed on hypertension aggravates cardiac remodeling in rats via angiotensin II system-mediated chronic inflammation // Hypertens. — 2009. — Vol. 54. — Р. 832–838.
- Kumar R., Singh V.P., Baker K.M. The intracellular renin-angiotensin system: a new paradigm // Trends Endocrinol. Metab. — 2007. — Vol. 8. — P. 208–214.
- Kumar R., Singh V.P., Baker K.M. The intracellular renin-angiotensin system: implications in cardiovascular remodelling // Curr. Opin. Nephrol. Hypertens. — 2008. — Vol. 7. — P. 168–173.
- Kumar R., Winocour P.H. Dual blockade of the renin angiotensin system in diabetes — rationale and risks // Br. J. Diabetes Vasc. Dis. — 2005. — Vol. 5, № 5. — P. 266–271.
- Kurdi M., Booz G.W. New Take on the Role of Angiotensin II in Cardiac Hypertrophy and Fibrosis // Hypertens. — 2011. — Vol. 57. — Р. 1034–1038.
- Lacy F., Kailasam M.T., O’Connor D.T. et al. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity // Hypertens. — 2000. — Vol. 36. — P. 878–884.
- Lahera V., Goicoechea M., de Vinuesa S.G. et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins // Curr. Med. Chem. — 2007. — Vol. 14. — P. 243–248.
- Lakkis J., Lu W.X., Weir M.R. RAAS escape: a real clinical entity that may be important in the progression of cardiovascular and renal disease // Curr.Hypertens.Rep. — 2003. — Vol. — P. 408–417.
- Landmesser U., Drexler H. Oxidative stress, the renin-angiotensin system, and atherosclerosis // Europ. Heart J. — 2003. — Vol. 5 (suppl.A). — P. A3–A7.
- Laragh J. Laragh’s lessons in pathophysiology and clinical pearls for treating hypertension // Am. J. Hypertens. — 2001. — Vol. 14. — P. 296–304.
- Lassègue B., Griendling K.K. NAD(P)H oxidases: functions and pathologies in the vasculature // Arterioscler. Thromb. Vasc. Biol. — 2010. — Vol. 30. — P. 653–661.
- Laursen J.B., Rajagopalan S., Galis Z. et al. Role of superoxide in angiotensin II–induced but not catecholamine-induced hypertension // Circulation. — 1997. — Vol. 95. — P. 588–593.
- Law M.R., Wald N.J., Morris J.K. et al. Value of low dose combination treatment with blood pressure lowering drugs: analysis of 354 randomised trials // BMJ. — 2003. — Vol. 326. — P. 1427–1434.
- Lee K.A., Kang M.H., Lee Y.S. et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity // Cell Immunol. — 2008. — Vol. 251. — P. 50–55.
- Lemay J., Hamet P., deBlois D. Losartan-induced apoptosis as a novel mechanism for the prevention of vascular lesion formation after injury // JRAAS. — 2000. — Vol. 1. — P. 46–50.
- Leoncini G., Ratto E., Viazzi F. et al. Metabolic syndrome is associated with early signs of organ damage in nondiabetic, hypertensive patients // J. Int. Med. — 2005. — Vol. 257. — Р. 454–460.
- Lewis E.J., Hunsicker L.G., Clarke W.R. et al. Collaborative Study Group: Renoprotective effect of the angiotensin receptor antagonist Irbesartan in patients with nephropathy due to type 2 diabetes // New Engl. J. Med. — 2001. — Vol. 345. — Р. 851–860.
- Li L., Fink G.D., Watts S.W. et al. Endothelin-1 increases vascular superoxide via endothelin(A)-NAD(P)H oxidase pathway in low-renin hypertension // Circulation. — 2003. — Vol. 107. — Р. 1053–1058.
- Li J.S., Lariviere R., Schiffrin E.L. Effect of a nonselective endothelin antagonist on vascular remodeling in deoxycorticosterone acetate-salt hypertensive rats. Evidence for a role of endothelin in vascular hypertrophy // Hypertens. — 1994. — Vol. 24. — Р. 183–188.
- Li J.M., Wheatcroft S., Fan L.M. et al. Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2-production, vascular tone, and mitogen-activated protein kinase activation // Circulation. — 2004. — Vol. 109. — P. 1307–1313.
- Lindholm L.H., Ibsen H., Dahlöf B. et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention for Endpoint Reduction in Hypertension Study (LIFE): a randomized trial against atenolol // Lancet. — 2002. — Vol. 359. — P. 1004–1010.
- Lithell H., Hansson L., Skoog I. et al. SCOPE Study Group. The study on cognition and prognosis in the elderly (SCOPE): principal results of a randomised double-blind intervention trial // J.Hypertens. — 2003. — Vol. 21. — Р. 875–886.
- Liu S.L., Schmuck S., Chorazcyzewski J.Z. et al. Aldosterone regulates vascular reactivity — Short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation // Circulation. — 2003. — Vol. 108. — Р. 2400–2406.
- Liu J., Yang F., Yang X.P. еt al. NAD(P)H oxidase mediates angiotensin II–induced vascular macrophage infiltration and medial hypertrophy // Arterioscler. Thromb. Vasc. Biol. — 2003. — Vol. 23. — P. 776–782.
- Lizakowski S., Tylicki L., Renke M. et al. Aliskiren and perindopril reduce the levels of transforming growth factor-β in patients with non-diabetic kidney disease // Am. J. Hypertens. — 2012.— Vol. 25. — P. 636–639.
- Lopez B., Salom M.G., Arregui B. et al. Role of superoxide in modulating the renal effects of angiotensin II // Hypertens. — 2003. — Vol. 42. — P. 1150–1156.
- Lopez-Sendon J., Swedberg K., McMurray J. et al.; for the Task Force on ACE-inhibitors of the European Society of Cardiology. Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease // Eur. Heart J. 2004. — Vol. 25. — P. 1454–1470.
- Lu H., Rateri D.L., Feldman D.L. et al. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice // J. Clin. Invest. — 2008. — Vol. 118. — P. 984–993.
- Ma C., Cao J., Lu X-C. et al. Cardiovascular and cerebrovascular outcomes in elderly hypertensive patients treated with either ARB or ACEI // J. Geriatric. Cardiol. — 2012. — Vol. 9. — P. 252–257.
- Mackay J.A. Blocking the renin-angiotensin-aldosterone system or lowering the blood pressure: does EUROPA help? // J. Renin-Angiotensin-Aldosterone System. — 2003. — Vol. 4. — Р. 205–206.
- Madamanchi N.R., Runge M.S. Mitochondrial dysfunction in atherosclerosis // Circ. Res. — 2007. — Vol. 100. — Р. 460–473.
- Makino A., Skelton M.M., Zou A.P. et al. Increased renal medullary H2O2 leads to hypertension // Hypertens. — 2003. — Vol. 42. — Р. 25–30.
- Mancia G. Total cardiovascular risk: a new treatment concept // J. Hypertens. — 2006. — Vol. 24, (suppl 2). — Р. S17–S23.
- Mancia G., De Backer G., Dominiczak A. et al. Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension and of the European Society of Cardiology // J. Hypertens. — 2007. — Vol. 25. — Р. 1105–1187.
- Manrique C., Lastra G., Whaley-Connell A. et al. Hypertension and the cardiometabolic syndrome // J. Clin. Hypertens. — 2005. — Vol. 7. — Р. 471–476.
- Marvar P.J., Thabet S.R., Guzik T.J. et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension // Circ. Res. — 2010. — Vol. 107. — P. 263–270.
- Matsuno K., Yamada H., Iwata K. et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice // Circulation. — 2005. — Vol. 112. — P. 2677–2685.
- Matsushima S., Ide T., Yamato M. et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice // Circulation. — 2006. — Vol. 113. — P. 779–1786.
- Maytin M., Siwik D.A., Ito M. et al. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox // Circulation. — 2004. — Vol. 109. — P. 1168–1171.
- McKelvie R.S., Yusuf S., Pericak D. et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators // Circulation. — 1999. — Vol. 100. — P. 1056–1064.
- McMullen J.R., Jennings G.L. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure // Clin. Exp. Pharmacol. Physiol. — 2007. — Vol. 34. — P. 255–262.
- McMurray J.J., Ostergren J., Swedberg K. et al. Effects of candesartan in patients with chronic heart failure and reduced left ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial // Lancet. — 2003. — Vol. 363. — P. 767–771.
- Meade T. Plasma renin and the incidence of cardiovascular disease // JRAAS. — 2010. — Vol. 11. — P. 91–98.
- Mehta J.L., Sanada N., Hu C.P. et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet // Circ. Res. — 2007. — Vol. 100. — P. 1634–1642.
- Meng S., Cason G.W., Gannon A.W. et al. Oxidative stress in Dahl salt-sensitive hypertension // Hypertens. — 2003. — Vol. 41. — P. 1346–1352.
- Messerli F.H., Beevers G., Franklin S.S. et al. Beta-blockers in hypertension—the emperor has no clothes: an open letter to present and prospective drafters of new guidelines for the treatment of hypertension // Am. J. Hypertens. — 2003. — Vol. 16. — Р. 870–873.
- Mezzano S.A., Ruiz-Ortega M., Egido J. Angiotensin II and renal fibrosis // Hypertens. — 2001. — Vol. 38. — P. 635–638.
- Mishra G.D., Malik N.S., Paul A.A. et al. Childhood and adult dietary vitamin E intake and cardiovascular risk factors in mid-life in the 1946 British Birth Cohort // Eur. J. Clin. Nutr. — 2003. — Vol. 57. — P. 1418–1425.
- Morawietz H. LOX-1 and аtherosclerosis рroof of сoncept in LOX-1–knockout mice // Circ. Res. — 2007. — Vol. 100. — P. 1534–1536.
- Morgan T., Aubert J.F., Brunner H. Interaction between sodium intake, angiotensin II, and blood pressure as a cause of cardiac hypertrophy //Am. J. Hypertens. — 2001. — Vol. 14. — Р. 914–920.
- Morimoto S., Yano Y., Maki K. et al. Renal and vascular protective effects of telmisartan in patients with essential hypertension // Hypertens.Res. — 2006. — Vol. 29. — P. 567–572.
- Muirhead N., Feagan B.F., Mahon J. The effects of valsartan and capropril on reducing microalbuminuria in patients with type 2 diabetes mellitus: a placebo-controlled trial // Cur. Therap.Res. — 2000. —
Vol. 60. — P. 650–660. - Müller D.N. Mechanisms of hypertension-induced target organ damage // JRAAS. — 2007. — Vol. 8. — Р. 148–150.
- Murdoch C.E., Zhang M., Cave A.C. et al. NAD(P)H oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure // Cardiovasc.Res. — 2006. — Vol. 71. — Р. 208–215.
- Nagase M., Yoshida S., Shibata S. et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors // J. Am. Soc.Nephrol. — 2006. — Vol. 17. — P. 3438–3446.
- Neutel J.M. Hypertension and its management: a problem in need of new treatment strategies // JRAAS. — 2000. — Vol. 1, (suppl.). — P. S10–S13.
- Nicholis M.G., Riegger A.J. Renin in cardiac failure // in: «The Renin -Angiotensin System» ed. by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 2, (part 76). — P. 76.1–76.76.21.
- Nishimura M., Takahashi H., Yoshimura M. Upregulation of the brain renin-angiotensin system in rats with chronic renal failure // Acta Physiol. (Oxf). — 2007. — Vol. 189. — P. 369–377.
- Nishiyama A., Yoshizumi M., Rahman M. et al. Effects of AT1 receptor blockade on renal injury and mitogen-activated protein activity in Dahl salt-sensitive rats // Kidney Int. — 2004. — Vol. 65. — P. 972–981.
- Nussberger J., Aubert J.F., Bouzourene K. et al. Renin inhibition by aliskiren prevents atherosclerosis progression: comparison with irbesartan, atenolol, and amlodipine // Hypertens. — 2008. — Vol. 51. — P. 1306–1311.
- Nyby M.D., Abedi K., Smutko V. et al. Vascular angiotensin type 1 receptor expression is associated with vascular dysfunction, oxidative stress and inflammation in fructose-fed rats // Hypertens. Res. — 2007. — Vol. 30. — P. 451–457.
- O’Keefe J.H., Wetzel M., Moe R.R. et al. Should an angiotensin-converting enzyme inhibitor be standard therapy for patients with atherosclerotic disease // JACC. — 2001. — Vol. 37, № 1. — Р. 1–8.
- Okin P.M., Wachtell K., Devereux R.B. et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension // JAMA. — 2006. — Vol. 296. — P. 1242–1248.
- Oliveira E.B., Nishi E.E., Carillo B.A. et al. AT1 receptor expression levels in the RVLM and the paraventricular nucleus of the hypothalamus are enhanced in this rat renovascular model of hypertension // Am. J. Hypertens. — 2009. — Vol. 22. — Р. 484–492.
- Oliveira E.M., Sasaki M.S., Cerêncio M. et al. Local renin-angiotensin system regulates left ventricular hypertrophy induced by swimming training independent of circulating renin: a pharmacological study // JRAAS. — 2009. — Vol. 10. — Р. 15–23.
- Olsen M.H., Andersen U.B., Wachtell K. et al. A possible link between endothelial dysfunction and insulin resistance in hypertension. A LIFE sub study. Losartan Intervention for Endpoint-Reduction in Hypertension // Blood Press. — 2000. — Vol. 9. — P. 132–139.
- Ortiz M.C., Manriquez M.C., Romero J.C. et al. Antioxidants block angiotensin II–induced increases in blood pressure and endothelin // Hypertens. — 2001. — Vol. 38. — P. 655–659.
- Ortiz M.C., Sanabria E., Manriquez M.C. et al. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II // Hypertens. — 2001. — Vol. 37. — P. 505–510.
- Ortlepp J.R., Breuer J., Eitner. et al. Inhibition of the renin-angiotensin system ameliorates genetically determined hyperinsulinemia // Eur. J. Pharmacol. — 2002. — Vol. 43. — P. 145–50.
- Palatini P., Malacco E., Di S.S. et al. Trough peak ratio and smoothness index in the evaluation of 24-h blood pressure control in hypertension: a comparative study between valsartan/hydrochlorothiazide combination and amlodipine // Eur. J. Clin. Pharmacol. — 2002. — Vol. 57. — Р. 765–770.
- Paradis P., Dali-Youcef N., Paradis F.W. et al. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling // Proc. Natl. Acad. Sci. USA. — 2000. — Vol. 97. — P. 931–936.
- Parikh N.I., Gona P., Larson M.G. et al. Plasma renin and risk of cardiovascular disease and mortality: the Framingham Heart Study // Eur. Heart J. — 2007. — Vol. 28. — Р. 2644–2652.
- Park J.B., Schiffrin E.L. ETA receptor antagonist prevents blood pressure elevation and vascular remodeling in aldosterone-infused rats // Hypertens. — 2001. — Vol. 37. — Р. 1444–1449.
- Parving H., Brenner B.M., McMurray J.J.V. et al. Cardiorenal End Points in a Trial of Aliskiren for Type 2 Diabetes // New Engl. J. Med. — 2012. — Vol. 367. — P. 2204–2213.
- Parving H-H., Persson F., Lewis J.B. et al. Aliskiren combined with losartan in type 2 diabetes and nephropathy // N. Engl. J. Med.— 2008. — Vol. 358. — P. 2433–2466.
- [PEACE] The PEACE Trial Investigators. Angiotensin-converting-enzyme inhibition in stable coronary artery disease // N. Engl. J.Med. — 2004. — Vol. 35. — P. 2058–2068.
- Pedro-Botet J., Covas M.I., Martin S. et al. Decreased endogenous antioxidant enzymatic status in essential hypertension // J. Hum. Hypertens. — 2000. — Vol. 14. — Р. 343–345.
- Persson F., Lewis J.B., Lewis E.J. et al. Impact of glycaemic control on the effect of direct renin inhibition in the AVOID study // JRAAS. — 2012. — Vol. 3. — P. 250–253.
- Persson F., Rossing P., Reinhard H. et al. Renal effects of aliskiren compared with and in combination with irbesartan in patients with type 2 diabetes, hypertension, and albuminuria // Diabetes Care. — 2009. — Vol. 32. — Р. 1873–1879.
- Pfeffer M., McMurray J., Velazquez E. et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both // N. Engl. J. Med. — 2003. — Vol. 349. — P. 1893–906.
- Pischon T., Girman C.J., Hotamisligil G.S. et al. Plasma adiponectin levels and risk of myocardial infarction in men // JAMA. — 2004. — Vol. 291. — P. 1730–1737.
- Pollock D.M. Endothelin, Angiotensin, and Oxidative Stress in Hypertension // Hypertens. — 2005. — Vol. 45. — Р. 477–480.
- Polyzos S.A., Kountouras J., Zafeiriadou E. et al. Effect of spironolactone and vitamin E on serum metabolic parameters and insulin resistance in patients with nonalcoholic fatty liver disease // RAAS. — 2011. — Vol. 12. — Р. 498–503.
- Pool J.L., Schmieder R.E., Azizi M., et al. Aliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartan // Am. J. Hypertens. — 2007. — Vol. 20. — P. 11–20.
- Pu Q., Neves M.F., Virdis A. et al. Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling // Hypertens. — 2003. — Vol. 42. — Р. 49–55.
- Purnak T., Beyazit Y., Oztas E. et al. Serum angiotensin-converting enzyme level as a marker of fibrosis in patients with chronic hepatitis B // JRAAS. — 2012. — Vol. 13. — P. 244–249.
- Raizada M.K., Ferreira A.J. ACE2: a new target for cardiovascular disease therapeutics // J. Cardiovasc. Pharmacol. — 2007. — Vol. 50. — Р. 112–119.
- Rajapakse N.W., De Miguel C., Das S. et al. Exogenous l-Arginine Ameliorates Angiotensin II-Induced Hypertension and Renal Damage in Rats // Hypertens. — 2008. — Vol. 52. — P. 1084–1090.
- Ran J., Hirano T., Adachi M. Angiotensin II type 1 receptor blocker ameliorates overproduction and accumulation of triglyceride in the liver of Zucker fatty rats // Am. J. Physiol. Endocrinol. Metab. — 2004. — Vol.287. — P. E227-E232.
- Reckelhoff J.F., Zhang H., Srivastava K. et al. Subpressor doses of angiotensin II increase plasma F(2)-isoprostanes in rats // Hypertens. — 2000. — Vol. 35. — P. 476–479.
- Remuzzi A., Remuzzi G. Potential protective effects of telmisartan on renal function deterioration // JRAAS. — 2006. — Vol. 7. — Р. 185–191.
- Reudelhuber T. Prorenin, renin, and their receptor. Moving Targets // Hypertens. — 2010. — Vol. 55. — P. 1071–1074.
- Reudelhuber T., Bernstein K., Delafontaine E. Is angiotensin II a director mediator of left ventricular hypertrophy? Time for another look //Hypertens. — 2000. — Vol. 49. — P. 1196–1201.
- Rey F.E., Cifuentes M.E., Kiarash A. et al. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice // Circ. Res. — 2001. — Vol. 89. — Р. 408–414.
- Roberts C.K., Vaziri N.D., Wang X.Q. et al. Enhanced NO inactivation and hypertension induced by a high-fat, refined-carbohydrate diet // Hypertens. — 2000. — Vol. 36. — P. 423–429.
- Rodriguez C.J., Bibbins-Domingo K., Jin Z.et al. Association of Sodium and Potassium Intake With Left Ventricular Mass Coronary Artery Risk Development in Young Adults // Hypertens. — 2011. — Vol. 58. — P. 410–416.
- Rodriguez-Iturbe B., Quiroz Y., Nava M. et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats // Am. J. Physiol. Renal. Physiol. — 2002. — Vol. 282. — P. F191–F201.
- Rosen E.D., Spiegelman B.M. Adipocytes as regulators of energy balance and glucose homeostasis // Nature. — 2006. — Vol. 444. — P. 847–853.
- Rugale C., Delbosc S., Cristol J-P. et al. Sodium restriction prevents cardiac hypertrophy and oxidative stress in angiotensin II hypertension // AJP — Heart. — 2003. — Vol. 284. — Р. H1744–H1750.
- Ruschitzka F., Taddei S. Angiotensin-converting enzyme inhibitors: first-line agents in cardiovascular protection? // Eur. Heart J. — 2012. — Vol. 33. — P. 1996–1998.
- Samani N.J., Thompson J.R., O’Toole L. et al. A meta-analysis of the association of the deletion allele of the angiotensinconverting enzyme gene with myocardial infarction // Circulation. — 1996. — Vol. 94. — Р. 708–712.
- Sasser J.M., Pollock J.S., Pollock D.M. Renal endothelin in chronic angiotensin II hypertension // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2002. — Vol. 283. — P. R243–R248.
- Savarese G., Costanzo P., Cleland J.G.F. et al. A Meta-Analysis Reporting Effects of Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers in Patients Without Heart Failure // J. Am. Coll. Cardiol. — 2013. — Vol. 61. — P. 131–142.
- Sawicki P.T., McGauran N. Have ALLHAT, ANBP2, ASCOT-BPLA, and so forth improved our knowledge about better hypertension care? // Hypertens. — 2006. — Vol. 48. — P. 1–7.
- Schieffer B., Schieffer E., Hilfiker-Kleiner D. et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques // Circulation. — 2000. — Vol. 101. — P. 1372–1378.
- Schillaci G., Pirro M., Vaudo G. et al. Prognostic value of the metabolic syndrome in hypertension // J. Am.Coll. Cardiol. — 2004. — Vol. 43. — Р. 1817–1822.
- Schmieder R.E., Philipp T., Guerediaga J. et al. Long-term antihypertensive efficacy and safety of the oral direct renin inhibitor aliskiren. A 12-month randomized, double-blind comparator trial with hydrochlorothiazide // Circulation. — 2009. — Vol. 119. — P. 417–425.
- Schnackenberg C.G., Wilcox C.S. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin F2α // Hypertens. — 1999. — Vol. 33. — Р. 424–428.
- Schulman I.H., Zhou M-S., Raij L. Cross-talk between angiotensin II receptor types 1 and 2: potential role in vascular remodeling in humans // Hypertens. — 2007. — Vol. 49. — Р. 270–271.
- Sedeek M.H., Elmarakby A.A., Loomis E.D. et al. NAD(P)H oxidase inhibition attenuates oxidative stress but not the hypertension produced by chronic ET-1 // Hypertens. — 2005. — Vol. 45. — Р. 283–287.
- Sedeek M.H., Llinas M.T., Drummond H. et al. Role of reactive oxygen species in endothelin-induced hypertension // Hypertens. — 2003. — Vol. 42. — Р. 806–810.
- Sendra J., Llorente-Cortes V., Costales P. еt al. Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall : a new pro-atherogenic mechanism of hypertension // Europ. J. Heart Failure. — 2008. — Vol. 10. — P. 581–589.
- Sever P.S., Gradman A.H., Azizi M. Managing cardiovascular and renal risk: the potential of direct renin inhibition // J. Renin-Angiotensin-Aldosterone System. — 2009. — Vol. 10. — P. 65–76.
- Shagdarsuren E., Wellner M., Braesen J.H. et al. Complement activation in angiotensin II-induced organ damage // Circ. Res. — 2005. — Vol. 97. — P. 716–724.
- Shao J., Nangaku M., Miyata T., Inagi R. et al. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury // Hypertension. — 2003. — Vol. 42. — Р. 31–38.
- Shargorodsky M., Leibovitz E., Lubimov L. et al. Prolonged treatment with the AT1 receptor blocker, valsartan, increases small and large artery compliance in uncomplicated essential hypertension // Am. J. Hypertens. — 2002. — Vol. 15. — P. 1087–1091.
- Shaohua Y., Zhong H., Yanamadala S. et al. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension // Hypertens. — 2006. — Vol. 48.— P. 309–315.
- Sharma N., Okere I.C., Barrows B.R. et al. High sugar diets increase cardiac dysfunction and mortality in hypertension compared to low carbohydrate or high starch diets // J. Hypertens. — 2008. — Vol. 26. — P. 1402–1410.
- Shimizu M.H., Coimbra T.M., de Araujo M. et al. N-Acetylcysteine attenuates the progression of chronic renal failure // Kidney Int. — 2005. — Vol. 68. — P. 2208–2217.
- Shinozaki K., Ayajiki K., Nishio Y. et al. Evidence for a causal role of the renin-angiotensin system in vascular dysfunction associated with insulin resistance // Hypertens. — 2004. — Vol. 43. — P. 255–262.
- Shokoji T., Fujisawa Y., Kimura S. et al. Effects of local administration of tempol and diethyldithio-carbamic on peripheral nerve activity // Hypertens. — 2004. — Vol. 44. — P. 236–243.
- Shokoji T., Nishiyama A., Fujisawa Y. et al. Renal sympathetic nerve responses to tempol in spontaneously hypertensive rats // Hypertens. — 2003. — Vol. 41. — P. 266–273.
- Siani A., Pagano E., Iacone R. et al. Blood pressure and metabolic changes during dietary L-arginine supplementation in humans // Am. J. Hypertens. — 2000. — Vol. 13. — P. 547–551.
- Sicard P., Delemasure S., Korandji C. et al. Anti-hypertensive effects of Rosuvastatin are associated with decreased inflammation and oxidative stress markers in hypertensive rats // Free Radic.Res. — 2008. — Vol. 42. — P. 226–236.
- So W.Y., Ozaki R., Chan N.N. et al. Effect of angiotensin-converting enzyme inhibition on survival in 3773 chinese type 2 diabetic patients // Hypertension. — 2004. — Vol. 44. — P. 294–301.
- Solomon S.D., Appelbaum E., Manning W.J. et al. Effect of the direct renin inhibitor aliskiren, the angiotensin receptor blocker Losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy // Circulation. — 2009. — Vol. 119. — P.530–537.
- Solomon S.D., Shin S.H., Shah A. et al. Effect of the direct rennin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction // Eur. Heart J. — 2011. — Vol. 32. — Р. 1227–1234.
- Sowers J.R. Insulin resistance and hypertension // Am. J. Physiol. Heart Circ. Physiol. — 2004. — Vol. 286. — Р. H1597–H1602.
- Staessen J.A., Li Y., Richart T. Oral renin inhibitors // Lancet. — 2006. — Vol. 368. — P. 1449–1456.
- Standley P.R., Rose K., Sowers J.R. Increased basal arterial smooth muscle glucose transport in the Zucker rat // Am. J. Hypertens. — 1995. — Vol. 8. — Р. 48–52.
- Stocker R., Keaney J.F.Jr. Role of oxidative modifications in atherosclerosis // Physiol Rev. — 2004. — Vol. 84. — P. 1381–1478.
- Strauss M.H., Hall A.S. Angiotensin receptor blockers may increase risk of myocardial infarction: unravelling he ARB-MI paradox // Circulation. — 2006. — Vol. 114. — P. 838–854.
- Strawn W.B., Dean R.H., Ferrario C.M. et al. Novel mechanisms linking angiotensin II and early atherogenesis // JRAAS. — 2000. — Vol. 1. — P. 11–17.
- Svensson P., de Faire U., Sleight P. et al. Comparative effects of ramipril on ambulatory and office blood pressures: a HOPE Substudy // Hypertens. — 2001. — Vol. 38. —Р. E28–E32.
- Swales J.D. The renin-angiotensin system in essential hypertension // in «The Renin-Angiotensin System» ed.by J.Robertson a. M.Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 1 (part 62). — P. 62.1–62.12.
- Takimoto E., Kass D.A. Role of oxidative stress in cardiac hypertrophy and remodeling // Hypertens. — 2007. — Vol. 49. — P. 241–248.
- Taniyama Y., Griendling K.K. Reactive oxygen species in the vasculature: molecular and cellular mechanisms // Hypertens. — 2003. — Vol. 42. — P. 1075–1081.
- Terzoli L., Mircoli L., Raco R. et al. Lowering of elevated ambulatory blood pressure by HMG-CoA reductase inhibitors // J. Cardiovasc. Pharmacol. — 2005. — Vol. 46. — P. 310–331.
- The ONTARGET investigators. Telmisartan, ramipril, or both in patients at high risk of vascular events // New Engl. J. Med. — 2008. — Vol. — 358. — P. 1547–1559.
- Tomiyama H., Kushiro T., Abeta H. et al. Kinins contribute to the improvement of insulin sensitivity during treatment with angiotensin converting enzyme inhibitor // Hypertens. — 1994. — Vol. 23. — P. 450–455.
- Tone A., Shikata K., Ogawa D. et al. Changes of gene expression profiles in macrophages stimulated by angiotensin II — Angiotensin II induces MCP-2 through AT1-receptor // JRAAS. — 2007. — Vol. 8. — P. 45–50.
- Touyz R.M. Reactive oxygen species and angiotensin II signaling in vascular cells: implications in cardiovascular disease // Braz. J.Med. Biol. Res. — 2004. — Vol. 37. — P. 1263–1273.
- Touyz R.M., Mercure C., He Y. et al. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NAD(P)H oxidase // Hypertens. — 2005. — Vol.45. — P.530–537.
- Tsunenari I., Ohmura T., Seidler R. et al. Renoprotective effects of telmisartan in the 5/6 nephrectomised rats // JRAAS. — 2007. — Vol. 8. — P. 93–100.
- Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials // Lancet. — 2003. — Vol. 362. — P. 1527–1535.
- Uchida H., Nakamura Y., Kaihara M. et al. Practical efficacy of telmisartan for decreasing morning home blood pressure and pulse wave velocity in patients with mild-to-moderate hypertension // Hypertens.Res. — 2004. — Vol. 27. — P. 545–550.
- Ukai T., Cheng C.P., Tachibana H. et al. Allopurinol enhances the contractile response to dobutamine and exercise in dogs with pacing-induced heart failure // Circulation. — 2001. — Vol. 103. — P. 750–755.
- Umegaki K., Ichikawa T. Vitamin E did not prevent platelet activation, but prevented increase of lipid peroxides and renal dysfunction in DOCA-salt hypertensive rats // J. Nutr. Sci. Vitaminol. — 1993. — Vol. 39. — P. 437–449.
- Uresin Y., Taylor A.A., Kilo C. et al. Efficacy and safety of the direct renin inhibitor aliskiren and ramipril alone or in combination in patients with diabetes and hypertension // JRAAS. — 2007. — Vol. 8. — P. 190–200.
- Van Kats J.P., Methot D., Paradis P. et al. Use of a biological peptide pump to study chronic peptide hormone action in transgenic mice. Direct and indirect effects of angiotensin II on the heart // J. Biol. Chem. — 2001. — Vol. 276. — P. 44012–44017.
- van Vark L.C., Bertrand M., Akkerhius M. et al. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosteron system inhibitors involving 159 998 patients // Eur. Heart J. — 2012. — Vol. 33. — P. 2088–2097.
- Vardeny О., Pouleur A-C., Takeuchi M. et al. Influence of diabetes on efficacy of aliskiren, losartan or both on left ventricular mass regression // JRAAS. — 2012. — Vol. 13. — P. 265–272.
- Vasan R.S., Evans J.C., Larson M.G. et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons // N. Engl. J. Med. — 2004. — Vol. 351. — Р. 33–41.
- Vasdev S., Ford C.A., Longerich L. et al. Aldehyde induced hypertension in rats: prevention by N-acetyl cysteine // Artery. — 1998. — Vol. 23. — P. 10–36.
- Vaziri N.D., Wang X.Q., Oveisi F. et al. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats // Hypertens. — 2000. — Vol. 36. — P. 142–146.
- Vecchione C., Colella S., Fratta L. Impaired insulin-like growth factor-1 vasorelaxant effects in hypertension // Hypertens. — 2001. — Vol. 37. — P. 1480–1485.
- Verdecchia P., Beboldi G., Angeli F. Angiotensin-converting enzyme inhibitors and calcium channel blockers for coronaryheart disease and stroke prevention // Hypertens. — 2005. — Vol. 46. — Р. 386–92.
- Verlohren S., Muller D.N., Luft F.C. Immunology in hypertension, preeclampsia, and target-organ damage // Hypertens. — 2009. — Vol. 54. — P. 439–446.
- Verma S., Gupta M., Holmes D.T. et al. Plasma renin activity predicts cardiovascular mortality in the Heart Outcomes Prevention Evaluation (HOPE) study // Europ. Heart J. — 2011. — Vol. 32. — P. 2135–2142.
- Vieitez P., Gómez O., Uceda E.R. et al. Systemic and local effects of angiotensin II blockade in experimental diabetic nephropathy // JRAAS. — 2008. — Vol. 9. — P. 96–102.
- Vijan S., Hayward R.A. Treatment of hypertension in type 2 diabetes mellitus: blood pressure goals, choice of agents, and setting priorities in diabetes care // Ann. Int. Med. — 2003. — Vol. 138. — P. 593–602.
- Virdis A., Neves M.F., Amiri F. et al. Spironolactone improves angiotensin-induced vascular changes and oxidative stress // Hypertens. — 2002. — Vol. 40. — Р. 504–510.
- Virdis A., Neves M.F., Amiri F. et al. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice // J. Hypertens. — 2004. — Vol. 22. — P. 535–542.
- Vittorio T.J., Ahuja K., Kasper M. et al. Comparison of high- versus low-tissue affinity ACE-inhibitor treatment on circulating aldosterone levels in patients with chronic heart failure // JRAAS. — 2007. — Vol. 8. — P. 200–204.
- Vivekananthan D.P., Penn M.S., Sapp S.K. et al. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials // Lancet. — 2003. — Vol. 361. — P. 2017–2023.
- Wachtell K., Lehto M., Gerdts E. et al. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: the Losartan Intervention For End Point Reduction in Hypertension (LIFE) study // J. Am. Coll. Cardiol. — 2005. — Vol. 45. — P. 712–719.
- Ward N.C., Croft K.D. Hypertension and oxidative stress // Clin. Exp. Pharmacol. Physiol. — 2006. — Vol. 33. — P. 872–876.
- Ward N.C., Hodgson J.M., Croft K.D. еt al. The combination of vitamin C and grape seed polyphenols increases blood pressure: a randomized, double-blind, placebo-controlled trial // J. Hypertens. — 2005. — Vol. 23. — P. 427–434.
- Ward N.C., Hodgson J.M., Puddey I.B. et al. Oxidative stress in human hypertension: association with antihypertensive treatment, gender, nutrition, and lifestyle // Free Radic.Biol.Med. — 2004. — Vol. 36. — P. 226–232.
- Wassmann S., Czech T., van Eickels M. et al. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice // Circulation. — 2004. — Vol. 110. — P. 3062–3067.
- Wei Y., Sowers J.R., Nistala R. et al. Angiotensin II-induced NAD(P)H oxidase activation impairs insulin signaling in skeletal muscle cells // AJP — Regu. Physiol. — 2008. — Vol. 294. — Р. R673–R680.
- Wei Y., Whaley-Connell A.T., Chen K. et al. NAD(P)H oxidase contributes to vascular inflammation, insulin resistance and remodeling in the transgenic (mRen2) rat // Hypertens. — 2007. — Vol. 50. — Р. 384–391.
- Welch W.J. Angiotensin II–dependent superoxide effects on hypertension and vascular dysfunction // Hypertens. — 2008. — Vol. 52. — P. 51–56.
- Weuidmann P., Ferrari P., Shaw S.G. Renin in diabetes mellitus // in «The Renin-Angiotensin System» ed.by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 2, (part 75). — P. 75.1–75.26.
- Whaley-Connell A., Habibi J., Wei Y. et al. Mineralcorticoid receptor antagonism attenuates kidney renin-angiotensin-aldosterone system mediated filtration barrier remodeling in the transgenic Ren2 rat // Am. J. Physiol. Renal. Physiol. — 2009. — Vol. 296. — P. F1013–F1022.
- Whaley-Connell A., Johnson M.S., Sowers J.R. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension // Progr. in Cardiovascular. Dis. — 2010. — Vol. 52. — P. 401–409.
- Widder J.D., Fraccarollo D., Galuppo P. et al. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of thioredoxin 2 // Hypertens. — 2009. — Vol. 54. — Р. 338–344.
- Williams G.H. Cardiovascular benefits of aldosterone receptor antagonists: what about potassium? // Hypertens. — 2005. — Vol. 46. — Р. 265–266.
- Williams J.M., Pollock J.S., Pollock D.M. Arterial pressure response to the antioxidant tempol and ETB receptor blockade in rats on a high-salt diet // Hypertens. — 2004. — Vol. 44. — Р. 770–775.
- Willum-Hansen T. Staessen J.A. et al. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population // Circulation. — 2006. — Vol. 113. — Р. 664–670.
- Wing L.M., Reid C.M., Ryan P. et al. A comparison of outcomes with angiotensin-converting enzyme inhibitors and diuretics for hypertension in the elderly // N. Engl. J. Med. — 2003. — Vol. 348. — Р. 583–592.
- Wood J.M., Maibaum J., Rahuel J. et al. Structure-based design of aliskiren, a novel orally effective renin inhibitor // Biochem. Biophys. Res. Commun. — 2003. — Vol. 308. — P. 698–700.
- Woodward A.C., Chalmers J., Zanchetti AmacMahon S. Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system // J. Hypertens. — 2007. — Vol. 25. — P. 951–958.
- Xiao H.D., Fuchs S., Bernstein E.A. et al. Mice expressing ACE only in the heart show that increased cardiac angiotensin II is not associated with cardiac hypertrophy // Am. J. Physiol. Heart. Circ. Physiol. — 2008. — Vol. 294. — P. 659–667.
- Xu J., Carretero O.A., Cavasin M.A. et al. Role of cardiac overexpression of angiotensin ii in the regulation of cardiac function and remodeling post-myocardial infarction // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H1900–H1907.
- Xu J., Carretero O.A., LiaoT-D. et al. Local angiotensin II aggravates cardiac remodeling in hypertension // AJP — Heart. — 2010. — Vol. 299. — Р. H1328–H1338.
- Xu H., Fink G.D., Chen A. et al. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats // Am. J. Physiol. Heart Circ. Physiol. — 2001. — Vol. 281. — P. H975–H980.
- Xu H., Fink G.D., Galligan J.J. Tempol lowers blood pressure and sympathetic nerve activity but not vascular O2- in DOCA-salt rats // Hypertens. — 2004. — Vol. 43. — P. 329–334.
- Yagi S., Aihara K., Ikeda Y. et al. Pitavastatin, an HMG-CoA reductase inhibitor, exerts eNOS-independent protective actions against angiotensin II induced cardiovascular remodeling and renal insufficiency // Circ. Res. — 2008. — Vol. 102. — Р. 68–76.
- Yamabe H., Osawa H., Kaizuka M. et al. Platelet derived growth factor, basic fibroblast growth factor, and interferon gamma increase type IV collagen production in human fetal mesangial cells via a transforming growth factor beta dependent mechanism // Nephrol. Dial. Transplant. — 2000. — Vol. 15. — Р. 872–876.
- Yamamoto M., Yang G., Hong C. et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy // J. Clin. Invest. — 2003. — Vol. 112. — P. 1395–1406.
- Yamamoto N., Yasue H., Mizuno Y. et al. Aldosterone is produced from ventricles in patients with essential hypertension // Hypertens. — 2002. — Vol. 39. — P. 958–962.
- Yang Z., Bove C.M., French B.A. et al. Angiotensin II type 2 receptor overexpression preserves left ventricular function after myocardial infarction // Circulation. — 2002. — Vol. 106. — P. 106–111.
- Yasuda N., Miura S., Akazawa H. et al. Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation // EMBO Rep. — 2008. — Vol. 9. — P. 179–186.
- Yavuz D., Koç M., Toprak A. et al. Effects of ACE inhibition and AT1-receptor antagonism on endothelial function and insulin sensitivity in essential hypertensive patients // JRAAS. — 2003. — Vol. 4. — P. 197–203.
- Yoshimura M., Kawai M. Synergistic inhibitory effect of angiotensin II receptor blocker and thiazide diuretic on the tissue renin-angiotensin-aldosterone system //JRAAS. — 2010. — Vol. 11. — Р. 124–12.
- Yusuf S., Sleight P., Pogue J. et al.; for The Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients // N. Engl. J. Med. — 2000. — Vol. 342. — Р. 145–153.
- Zhai Y., Gao X., Wu Q. et al. Fluvastatin decreases cardiac fibrosis possibly through regulation of TGF-beta(1)/Smad 7 expression in the spontaneously hypertensive rats // Eur. J. Pharmacol. — 2008. — Vol. 587. — Р. 196–203.
- Zhang Y., Griendling K.K., Dikalova A. et al. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2 // Hypertens. — 2005. — Vol. 46. — Р. 732–737.
- Zhang J., Noble N.A., Border W.A. et al.Receptor-dependent prorenin activation and induction of PAI-1 expression in vascular smooth muscle cells // Am. J. Physiol. Endocrinol. Metab. — 2008. — Vol. 295. — P. E810–E819.
- Zhang Y., Popovic Z.B., Bibevski S. et al. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model // Circ.Heart Fail. — 2009. — Vol. 2. — P. 692–699.
- Zhang C., Zhao Y.X., Zhang Y.H. et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells // Proc. Natl. Acad. Sci. USA. — 2010. — Vol. 107. — P. 15886–15891.
- Zheng Z., Shi H., Jia J., Dong Li et al. A systematic review and meta-analysis of aliskiren and angiotension receptor blockers in the management of essential hypertension // JRAAS. — 2011. — Vol. 12. — P. 102–112.
- Zhou M.., Schulman I.H., Pagano P.J. et al. Reduced NAD(P)H oxidase in low renin hypertension. Link among angiotensin II, atherogenesis, and blood pressure // Hypertens. — 2006. — Vol. 47. — P. 81–86.
- Zhou M., Schulman I.H., Raij L. Role of angiotensin II and oxidative stress in vascular insulin resistance linked to hypertension // AJP — Heart. — 2009. — Vol. 296. — Р. H833–H839.
- Zhou L., Xi B., Wei Y. et al. Meta-analysis of the association between the insertion/deletion polymorphism in ACE gene and coronary heart disease among the Chinese population // JRAAS. — 2012. — Vol. 13. — P. 296–304.
- Zhuo J.L., Mendelson F.A., Ohishi M. Perindopril alter vascular angiotensin-converting enzyme, AT1 receptor, and nitric oxide synthase expression in patients with coronary heart disease // Hypertens. — 2002. — Vol. 39. — P. 634–638.
- Zimmerman M.C., Lazartigues E., Lang J.A. et al. Superoxide mediates the actions of angiotensin II in the central nervous system // Circ. Res. — 2002. — Vol. 91. — P. 1038–1045.
- Zimmerman M.C., Lazartigues E., Sharma R.V. et al. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system // Circ. Res. — 2004. — Vol. 95. — P. 210–216.