В соответствии с современными представлениями РАС функционирует не только как эндокринная, но и как паракринно/аутокринная система, и включает наряду с циркулирующим и тканевой компонент. Более того, предполагают, что тканевой компонент определяет практически до 90% активности системы в целом [131, 132]. Наличие тканевой РАС установлено в почках, сосудистой стенке и миокарде, в лимфатической и пищеварительной системах, в жировой ткани. При этом локальные РАС могут функционировать как сочетанно с системной РАС, так и независимо от нее [214], повышение активности локальных систем может не сочетаться с активацией циркулирующей [26, 62, 63, 152, 153].
В связи с этим эффективность терапевтического действия препаратов, снижающих активность РАС, определяется их действием не только на циркулирующую, но и на локальные РАС. Однако определение эффективности влияния препаратов, значительно снижающих активность циркулирующей РАС, на локальном уровне остается в настоящее время проблематичным.
В отличие от циркулирующей РАС, для которой характерны эндокринная функция и регуляция общеорганизменных гомеостатических параметров — системного АД, водно-солевого обмена, системного метаболизма, для локальных РАС свойственно пара- и аутокринное влияние с местной регуляцией, направленной на поддержание локального гомеостаза. Если гиперактивность циркулирующей РАС проявляется прежде всего повышением АД, активности сердца, нарушением функции почек, то гиперактивность РАС на локальном уровне определяет как ауторегуляторные, так и патологические изменения структуры и функционального состояния органов и тканей, в которых она экспрессирована.
Установлено, что физиологическая регуляторная значимость локальных РАС, как и их значимость в развитии кардиоваскулярной патологии, во многих случаях превышает значимость циркулирующей, и активация тканевых РАС в преобладающей степени определяет ремоделирование сердца, микро- и макрососудистые поражения при СД, развитие сердечной недостаточности, ретинопатии, нефропатии. В исследовании HOPE применение ингибитора АПФ рамиприла сопровождалось выраженным снижением частоты развития конечных кардиальных точек у пациентов группы высокого риска, несмотря на незначительное угнетение активности циркулирующей РАС и умеренный гипотензивный эффект [186].
В патогенезе АГ у крыс критическую роль играет локальная продукция А II, так как в хроническую стадию процесса активность ренина и АПФ в плазме крови нормальная или даже сниженная, тогда как продукция А II в сосудистой стенке закономерно увеличивается [179]. Высказано даже предположение, что основной функцией циркулирующей РАС является доставка к тканям не А ΙΙ, а ренина и ангиотензиногена. В результате образование А Ι и А ΙΙ происходит главным образом локально, тогда как их уровень в крови отражает в основном интенсивность тканевой продукции и утечки в циркуляцию [22, 23].
Установлено также, что концентрация циркулирующего А ΙΙ остается высокой после двусторонней нефрэктомии. Это означает, что плазменное содержание А ΙΙ не определяется полностью его образованием непосредственно в крови и в значительной степени связано с оттоком в плазму крови А ΙΙ, образующегося локально.
Эти данные свидетельствуют о том, что патогенетическое и прогностическое значение повышенной активности тканевых компонентов РАС может существенно превышать значение циркулирующего А II [186]. Так, в дезоксикортикостероновой модели АГ, воспроизведенной на мышах с генетическим отсутствием апоЕ, ускоренное развитие АГ и атеросклероза происходило на фоне уменьшения плазменного содержания ренина и А II, но, тем не менее, резко угнеталось после применения ингибиторов АПФ или блокаторов АТ1-рецепторов. Эти эффекты развивались независимо от влияния на уровень АД и на фоне сохраняющихся выраженных липидных нарушений.
В другом исследовании, проведенном также на гипертензивных мышах с отсутствием апоЕ, находившихся на диете с холестерином, применение блокатора АТ1-рецепторов и блокатора кальциевых каналов сопровождалось аналогичным влиянием на уровень АД, однако только блокаторы АТ1-рецепторов замедляли развитие атеросклеротического поражения [298]. Это данные означают, что проатерогенный эффект А II, в отличие от гипертензивного, опосредован активацией не системной, а локальной РАС в сосудистой стенке, что подтверждается значительно усиленной экспрессией А II, AПФ и AT1-рецепторов в стенке аорты у гипертензивных мышей с дефицитом апоЕ.
Биохимические и иммунологические методы позволили установить наличие экспрессии и возможность синтеза ренина, как и наличие мРНК ангиотензиногена в мозгу, почках, стенке артерий и вен, сердце, надпочечниках, в культуре эндотелиоцитов и сосудистых гладкомышечных клеток. Локальное образование А II отмечено в миокарде, сосудистой стенке, почках, мозгу, надпочечниках, жировой ткани и др. При этом предположительно до 40% А II в почках образуется локально без участия АПФ и с участием химазы. Установлено, что химаза обусловливает альтернативный путь образования А ΙΙ также в сердце, коронарных артериях, атеросклеротически пораженной аорте. Поэтому активация тканевых РАС может принимать участие в развитии кардиоваскулярной патологии, независимо от циркулирующей [8].
Основным источником ренина в плазме крови и тканях являются почки, и это подтверждается практически полным исчезновением активности ренина в плазме и тканевой рениноподобной активности после нефрэктомии. Установлено, что клетки сосудистой стенки и других тканей содержат ренинсвязывающий протеин, посредством которого они осуществляют захват и депонирование ренина из крови в количестве, достаточном для локальной продукции А ΙΙ в прессорной концентрации. Поэтому при внутривенном введении ренина крысам с нефрэктомией его появление в ткани аорты имело значительно более замедленный характер по сравнению с повышением плазменной концентрации.
Эти данные послужили основой для создания новой концепции относительно принципов функционирования РАС, в соответствии с которой ее основное назначение заключается в системной доставке к тканям не А ΙΙ, а ренина и ангиотензиногена с последующим локальным образованием А Ι и А ΙΙ [185].
Установлено, что активация циркулирующей и тканевых РАС имеет различную динамику. Так, в начальную компенсированную фазу развития сердечной недостаточности после ИМ на фоне уже выраженного повышения экспрессии кардиального АПФ циркулирующая РАС еще не активировалась, не увеличивалось плазменное содержание ренина и АПФ. Активация РАС в сердце в этих условиях имела четко выраженный тканеспецифичный характер, она не отмечалась в легких, стенке аорты, почках; основным стимулом для этой активации было увеличение размера желудочка и напряжения его стенки.
Активация кардиальной РАС сочетается с повышением конечно-диастолического АД в ЛЖ, что свидетельствует о существенном влиянии А II на диастолическую функцию миокарда. Параллельно А II, образующийся локально, вызывает интенсивную констрикцию коронарных сосудов, и применение ингибиторов АПФ оказывает вазодилататорное действие, не зависящее от изменений концентрации циркулирующего А II. В то же время, активация циркулирующей РАС происходит на этапе уже выраженных гемодинамических нарушений и направлена на поддержание центральной гемодинамики.
В экспериментах на различных моделях АГ и сердечной недостаточности также показано, что при длительном применении ингибиторов АПФ изменения в сердце и сосудистой стенке отчетливо диссоциируют с активностью циркулирующей РАС, и выраженная регрессия развившихся изменений может сочетаться с высокой активностью ренина и АПФ в плазме крови. Более того, высказано предположение, что одной из основных функций локальных РАС является местное ограничение эффектов циркулирующего А II. Поэтому ни исходный уровень ренина в плазме крови, ни выраженность острого гипотензивного ответа на ингибиторы АПФ не являются предикторами эффективности лечения пациентов с АГ и сердечной недостаточностью.
Различия функционального назначения циркулирующей и локальных РАС отчетливо проявляются при применении препаратов, угнетающих ее активность. Установлено, что вазодилататорный и гипотензивный эффекты кратковременного применения ингибиторов АПФ опосредованы снижением активности циркулирующей РАС, тогда как длительный гипотензивный эффект и нормализация структуры сердца и сосудистой стенки в большей степени связаны с угнетением тканевой активности АПФ.
Показано также, что у крыс со спонтанной АГ применение каптоприла сопровождалось угнетением АПФ в сосудистой стенке значительно дольше по сравнению с плазменной фармакокинетикой препарата. Кроме того, установлено, что отдельные ингибиторы АПФ обладают разной степенью сродства к тканевым РАС, и болюсное введение каптоприла сопровождалось более длительным и выраженным угнетением кардиального АПФ, чем рамиприла и эналаприла, введенных в эквипотентных дозах и при равном гипотензивном действии.
Физиологическая роль циркулирующего и локального А ΙΙ существенно отличается, несмотря на то, что в реальных условиях как циркулирующий, так и локально продуцируемый А II оказывают аналогичное гипертрофическое влияние на кардиомиоциты и митогенное действие на фибробласты и гладкомышечные клетки. Так, применение ингибиторов АПФ сопровождалось предупреждением или устранением гипертрофии и фиброзирования миокарда и срединного слоя сосудистой стенки в различных моделях АГ. Аналогичное протективное действие ингибиторов АПФ отмечено и при повреждении сосудистой стенки, когда пролиферативный ответ определялся не повышением АД и изменениями внутрисосудистой гемодинамики, а активацией локальной РАС.
В фундаментальном исследовании, проведенном в 2000 г., выявлено, что ремоделирование сосудистой стенки и нарушение ее эластических свойств при АГ зависят от двух основных факторов — механического растяжения и активации локальной продукции А II. При воспроизведении циклического растяжения гладкомышечных клеток человека в условиях культуры отмечено значительное повышение синтеза коллагена (на 102%) и фибронектина (на 50%) в сочетании с выраженным увеличением плотности АТ1-рецепторов. А II также приводил к отчетливому увеличению синтеза как коллагена (на 92%), так и фибронектина (на 21%). Применение блокаторов АТ1-рецепторов сопровождалось значительным ослаблением эффекта как механического воздействия, так и применения А ΙΙ. Эти данные означают, что влияние циклического растяжения стенки также опосредовано участием локальной РАС, прежде всего — посредством повышения чувствительности гладкомышечных клеток к А II в результате усиленного синтеза его рецепторов [270].
Установлено, что концентрация А ΙΙ в артериальной и венозной крови примерно равна, и это означает, что интенсивный тканевой захват и метаболизм А ΙΙ балансируются эквивалентным его образованием de novo в тканях. Примерно 55% содержания как А Ι, так и А II в артериальной крови и 75% — в венозной являются следствием их локальной тканевой продукции. Результаты прямых исследований свидетельствуют о том, что локальный синтез А Ι de novo обусловливает 48% его содержания в венозной крови предплечья, 58% — конечностей, 78% — почек, 75% — печени, тогда как активность ренина в плазме крови обеспечивает только 20–30% синтеза А Ι в конечностях и почках, до 60% — в гепатомезентериальной области. В то же время, хотя сосудистая ткань проявляет рениноподобную активность, она практически полностью исчезает после нефрэктомии, и эта активность является следствием захвата циркулирующего ренина с участием ренинсвязывающего белка, который экспрессируется клетками сосудистой стенки.
В соответствии с традиционными представлениями, ренин является циркулирующим гормоном, и его реакция с ангиотензиногеном происходит непосредственно в плазме крови. Однако в ряде исследований показано сочетанное наличие ренина и А ΙΙ в клетках юкстагломерулярного аппарата почек крыс, что позволило предположить возможность внутриклеточного образования А ΙΙ.
Хотя наличие тканевого ренина длительное время оспаривалось, применение специфических антител, метода хроматографии позволило подтвердить наличие ренина у нефрэктомированых крыс в мозгу и других внепочечных тканях. Однако возможность тканевого синтеза ренина в настоящее время остается сомнительной, и большинство исследователей полагают, что наличие тканевого ренина отражает захват и депонирование циркулирующего ренина.
Эти данные позволили найти объяснение выраженному антигипертензивному действию ингибиторов АПФ и блокаторов рецепторов А ΙΙ у пациентов с нормальной и даже субнормальной активностью ренина в плазме крови. Отмечено, что эти препараты утрачивали активность через несколько часов после проведения двусторонней нефрэктомии у крыс, и это указывало на участие ренина, ранее депонированного в тканях, в локальной продукции А ΙΙ [123].
В исследованиях, проведенных на культуре клеток, в частности — на сосудистых эндотелиоцитах, получено прямое подтверждение наличия внутриклеточного синтеза А ΙΙ. Хотя А ΙΙ, синтезируемый внутриклеточно, может высвобождаться из клеток и оказывать действие через мембранные рецепторы, полагают, что основной эффект осуществляется интракринно, то есть внутриклеточно.
Хотя известно, что увеличенное потребление натрия и повышенная его плазменная концентрация приводят к снижению активности циркулирующей РАС, их влияние на активность тканевых РАС остается невыясненной. В значительном числе исследований в условиях высокосолевой диеты установлено повышение активности РАС в сердце и сосудистой стенке в отличие от активности циркулирующей РАС. Установлено, что блокаторы АТ1-рецепторов и тиаздные диуретики могут быть равноэффективными в угнетении тканевых РАС, так как диуретики уменьшают содержание натрия в тканях. Поэтому сочетание блокаторов АТ1-рецепторов и диуретиков оказалось высокоэффектиным в лечении АГ и других видов кардиальной патологии [309].
В исследованиях последнего десятилетия неоднократно показано, что А II и АПФ могут постоянно образовываться непосредственно в сердечно-сосудистой системе [60, 192]. В то же время наличие альдостерона в сердце рассматривалось как следствие его захвата из крови.
В соответствии с классическими представлениями, синтез альдостерона и кортизола из холестерина происходит главным образом в корковой зоне надпочечников. Однако в настоящее время стало очевидным, что активация тканевых РАС сопровождается локальным синтезом не только А II, но и альдостерона. Установлено, что при АГ и сердечной недостаточности синтез альдостерона осуществляется также в сосудистой стенке, а высокосолевая диета усиливает этот синтез [280, 308]. Прямыми исследованиями установлено, что экспрессия гена альдостеронсинтазы в стенке сосудов достигает 1/15 его экспрессии в надпочечниках [180, 306]. А II и альдостерон, образуемые локально, оказывают как независимое, так и сочетанное взаимоусиливающее действие на структуру и функцию сердца, сосудов, почек и мозга, оба стимулируют гипертрофию кардиомиоцитов и сосудистых гладкомышечных клеток [169].
В исследованиях последних лет неоднократно показано наличие усиленного синтеза альдостерона в сердце при АГ и сердечной недостаточности [180, 307], аналогичный эффект отмечен у крыс при гипернатриемии, связанной с получением солевой диеты [280]. Установлено, что альдостерон, особенно на фоне повышенного содержания натрия, индуцирует гипертрофию сердца, тогда как при низком тканевом содержании натрия этот эффект не развивается [309].
В миокарде также выявлено наличие альдостеронсинтазы, хотя выраженность ее экспрессии на 3 порядка ниже, чем в надпочечниках. При сердечной недостаточности усиленная экспрессия АПФ в миокарде не только сопровождается стимуляцией синтеза А II, но и сочетается с активацией альдостеронсинтазы и увеличением продукции альдостерона. Эти изменения выражены пропорционально тяжести дисфункции ЛЖ и в такой же степени они пропорциональны выраженности миокардиального фиброза [241].
Усиленный, но в меньшей степени, синтез альдостерона в миокарде установлен и у лиц с АГ. В то же время, количество альдостерона, оттекающего в плазму крови от ЛЖ, невелико, и потому физиологическое значение его локального синтеза ограничивается пара- и аутокринным действием.
Способность миокарда продуцировать и высвобождать альдостерон показана и на изолированном перфузируемом сердце крысы. Содержание альдостерона в миокарде в этих условиях достигало 16 нМ, оно было значительно меньше, чем в ткани надпочечников, где равнялось 130 мкм, но значительно больше, чем в плазме крови, где концентрация альдостерона не превышала 0,93 нМ.
Помимо этого, между продукцией в миокарде А ΙΙ и альдостерона установлено наличие прямой и обратной положительной связи: повышение концентрации А II в перфузате сопровождалось увеличением продукции альдостерона, тогда как инкубация изолированных кардиомиоцитов с альдостероном сопровождалась резким повышением экспрессии АПФ и продукции А ΙΙ.
В условиях эксперимента длительное воздействие повышенного уровня альдостерона или А II приводило к ремоделированию сердца с усиленным образованием внеклеточного матрикса и накоплением коллагена даже в отсутствие АГ, а блокатор минералокортикоидных рецепторов спиронолактон снижал АД и уменьшал выраженность гипертрофии ЛЖ у крыс с АГ [236–240]. Утверждается, что патогенез этих нарушений имеет следующий вид: активация локальной РАС приводит к усиленной секреции А II, который стимулирует продукцию альдостерона ауто- и паракринным образом. В результате развиваются гипертрофия и ремоделирование сердца, в основе которых в большей степени задействован альдостерон, так как блокаторы минералокортикоидных рецепторов более выражено угнетают эти эффекты, чем блокаторы рецепторов А ΙΙ.
2.1. Физиологическая и патологическая роль кардиоваскулярной РАС
2.1.1. Кардиальная РАС
Неоднократно отмечено, что кардиальная РАС может регулировать продукцию и содержание А II в сердце, независимо от циркулирующей РАС. В нормальном сердце экспрессируется только незначительная активность АПФ, и мыши с отсутствием кардиального гена АПФ не имеют никаких признаков кардиальной патологии. Это свидетельствует об отсутствии существенного влияния кардиальной РАС на нормальное развитие и функцию миокарда. Однако при кардиальной патологии, особенно — при сердечной недостаточности, в сердце и в сосудистом эндотелии резко повышается экспрессия компонентов РАС, прежде всего — ангиотензиногена и ренина. Аналогичный эффект отмечен также при увеличении тканевого содержания натрия.
В условиях культуры кардиомиоцитов и фибробластов сердца было подтверждено отсутствие синтеза ренина. Его наличие в миокарде имеет, по всей вероятности, внекардиальное происхождение и осуществляется как путем диффузии из крови [43, 44], так и посредством связывания циркулирующего ренина с рецепторами (ренина и проренина), которые экспрессируются на кардиомиоцитах и клетках сосудистой стенки [196, 197], При этом содержание ренина в миокарде значительно превышает таковое в плазме крови, что свидетельствует об активной секвестрации ренина из крови и накоплении его в миокарде. Отсутствие синтеза ренина в сердце подтверждается его постепенным исчезновением в миокарде при двусторонней нефрэктомии.
Установлено, что образование А II в сердце с участием циркулирующего ренина происходит непосредственно на поверхности кардиомиоцитов. Это значительно повышает эффективность действия А ΙΙ, так как позволяет ему связываться с клеточными рецептoрами с минимальной утечкой во внеклеточное пространство [240].
Наличие локальной экспрессии АПФ в сердце в настоящее время уже не вызывает сомнений и показано как в сердце крыс, так и людей [214].
Экспрессия мРНК ангиотензиногена в сердце установлена также как у животных, так и у людей, хотя синтез ангиотензиногена в миокарде значительно менее интенсивен, чем в печени [48]. В то же время, содержание ангиотензиногена в сердце значительно превышает таковое в плазме крови, что свидетельствует о преобладании локального синтеза ангиотензиногена [165]. Однако значительная часть ангиотензиногена может также захватываться из крови, что показано в экспериментах на изолированном перфузируемом сердце [48].
Роль кардиальной РАС особенно отчетливо проявляется выраженным гипотензивным эффектом ингибиторов АПФ и блокаторов АТ1-рецепторов, отмечаемым не только при высоко-, но и при низкорениновой форме АГ. Помимо этого, отмена ингибиторов АПФ при их длительном применении сопровождается развитием длительного гипертензивного ответа, несмотря на быстрый возврат активности АПФ в плазме крови к нормальным значениям. Данные ряда исследований последних лет указывают на то, что тканевые компоненты РАС могут иметь большее значение, чем циркулирующий А II, также и в атеросклеротическом поражении сосудистой системы [186].
Показано, что мощным активатором локальной РАС в сердце и сосудах является механический стресс. В культуре кардиомиоцитов — механическая стимуляция, а в целостном сердце — перегрузка давлением вызывали гиперэкспрессию генов ангиотензиногена, АТ1-рецепторов и АПФ с усиленной секрецией А II [38, 39] и последующее развитие гипертрофии ЛЖ [10, 11].
В физиологических условиях основная функция кардиальной РАС заключается в регуляции пролиферации и роста клеток сердца в зависимости от нагрузки на миокард [214], а в патологических условиях она приводит к ремоделированию сердца и сосудов в результате развития выраженной клеточной гипертрофии и пролиферации в сочетании со склерозированием.
Отмечено, что кардиальная РАС активируется при действии различных стимулов типа увеличенной пред- или постнагрузки, симпатической стимуляции, в условиях локального воспаления, а субдепрессорные дозы ингибиторов АПФ и блокаторов АТ1-рецепторов приводят к регрессии гипертрофии ЛЖ даже при неизмененном АД [287].
Данные многочисленных исследований свидетельствуют о том, что кардиальная РАС играет ведущую роль в генезе гипертрофии и ремоделирования сердца [151]. Показано, что митогенное действие А II в значительной степени определяется его способностью активировать продукцию фибробластами других факторов роста, прежде всего ЭТ-1 и TФР-β [92].
В проведенных в последнее время исследованиях получено прямое подтверждение функционирования А II не только как циркулирующего вазоконстрикторного и гипертензивного фактора, но также как пара- и аутокринного гормона, который способствует росту клеток, их апоптозу, развитию воспаления, оксидативного стресса, приводя в итоге к ремоделированию сердца, развитию сердечной недостаточности [273–275, 302]. Этому способствует значительно более высокая концентрация А II в миокарде, чем в плазме крови [194, 291].
В исследовании, проведенном на трансгенных мышах с 10–15-кратным увеличением содержания А II в миокарде при неизмененном АД и плазменной концентрации А II, в базальных условиях отмечен слабо выраженный кардиофиброз без гипертрофии. В то же время, при гемодинамической нагрузке у них развивались значительно более выраженные гипертрофия сердца и кардиофиброз, чем у контрольных животных [303–305]. Однако и в этих условиях дифференцировать действие локального продуцируемого А II от действия циркулирующего не представилось возможным.
В отличие от этого, в использованной комбинированной модели с ДOКA-солевой АГ увеличенное содержание А II в сердце сочеталось со сниженной концентрацией циркулирующего А II, что давало возможность определения прямого действия кардиального А II в условиях повышенного АД. Животные с ДOКA-солевой АГ без усиленной локальной продукции А II служили контролем.
До воспроизведения АГ АД и масса ЛЖ были аналогичными в обеих группах, свидетельствуя о том, что в отсутствие повышенной нагрузки кардиальный А II не вызывал гипертрофии сердца. Однако при воспроизведении АГ у опытных животных на фоне уменьшенного плазменного содержания ренина и А II отмечено развитие значительно более выраженной гипертрофии, кардиального и периваскулярного фиброза на фоне более выраженной инфильтрации макрофагами, интенсивности воспаления и оксидативного стресса, экспрессии NADPH-оксидазы, TФР-β1, экскреции с мочой 8-изопростанов. После введения валсартана выраженность отмеченных изменений уменьшалась только у опытных животных без значительных сопутствующих изменений функции сердца.
Анализ полученных данных позволил авторам сделать вывод, что механический стресс (сочетание увеличенной постнагрузки с объемной перегрузкой в результате ДOКA-солевой АГ) приводило к гиперэкспрессии AT1-рецепторов. В сочетании с увеличенным содержанием А II и стимулирующим действием ДОКА на минералокортикоидные рецепторы это приводило к усиленной индукции воспаления, оксидативного стресса, апоптоза и выраженному ремоделированию сердца.
Как показано в ряде клинических исследований, митогенное действие А II наиболее значимо в развитии патологических изменений, проявляющихся пролиферацией фибробластов и развитием кардиосклероза [245]. Этот процесс может иметь чисто локальную природу и развиваться после перенесенного ИМ на фоне неизмененной активности АПФ в крови, но при значительном повышении активности кардиального АПФ [116]. Поэтому применение лозартана у крыс после ИМ сопровождалось значительным уменьшением содержания коллагена за пределами зоны инфаркта по сравнению с контрольными животными [45].
Установлено, что физиологическая гипертрофия миокарда, отмечаемая у тренированных атлетов, существенно отличается от патологической, прежде всего — сохраненным характером диастолы и полноценностью диастолического наполнения желудочка, что свидетельствует об отсутствии фибротических изменений в миокарде [287].
В ряде исследований установлено, что развитие сердечной недостаточности сочетается с активацией РАС в миокарде и увеличением локальной продукции А ІІ. Это провоцирует развитие воспалительного ответа через активацию ядерного фактора kB и последующую усиленную секрецию ряда провоспалительных медиаторов, ведущую роль среди которых играет ФНО-α. С другой стороны, ФНО-α стимулирует продукцию А II и экспрессию АТ1-рецепторов в фибробластах с развитием кардиосклероза. Важнейшую медиаторную роль в генезе этих процессов играет усиленная продукция активных форм кислорода, так как применение антиоксидантов практически полностью устраняло гипертрофию и склерозирование миокарда в ответ на стимуляцию А II и ФНО-α [250].
На изолированных кардиомиоцитах также установлена способность А II через АТ1-рецепторы усиливать образование протеинкиназы С и диацилглицерола, инициировать через них воспалительный ответ. Параллельно активировался синтез ФНО-α, который усиливает экспрессию АТ1-рецепторов на фибробластах миокарда с повышением их чувствительности к митогенному действию А II и развитием в конечном итоге кардиосклероза [298].
При воспроизведении сердечной недостаточности у крыс установлено пропорциональное усиление экспрессии компонентов РАС в миокарде. Экспрессия мРНК ренина была повышена на 52% при компенсированном состоянии и на 130% — в условиях декомпенсации, аналогично повысились экспрессия мРНК и содержание белка АПФ, тогда как экспрессия мРНК AT1-рецепторов в миокарде возрастала в компенсированном состоянии и уменьшилась на 54% при декомпенсации [217].
Выявлено, что через 1–4 нед после воспроизведения ИМ в сердце крысы наибольшая плотность рецепторов А II была характерна для миофибробластов, а применение лозартана сопровождалось значительным уменьшением продукции и содержания коллагена в миокарде.
Роль кардиальной РАС в гипертрофии ЛЖ особенно отчетливо проявилась в исследовании, проведенном на крысах в условиях длительной физической нагрузки, индуцированной плаванием. В зависимости от режима нагрузок масса ЛЖ увеличилась на 20–30%, а применение эналаприла или лозартана предупреждало ее возрастание на 40–50%. Для предупреждения активации циркулирующей РАС применялась высокосолевая диета, и она не оказывала влияния на гипертрофический ответ миокарда и не приводилa к регрессии уже развившейся гипертрофии. На основании этих данных было сделано заключение, что физиологическая гипертрофия ЛЖ, связанная с повышенной физической активностью, определяется активацией кардиальной, но не циркулирующей РАС [208].
Известно, что высокосолевая диета также приводит к гипертрофии ЛЖ посредством активации кардиальной РАС даже без развития АГ, однако при этом резко возрастает плотность АТ1-рецепторов в миокарде [157, 318–320].
Локальное образование АПФ в сердце в настоящее время уже не вызывает сомнений, экспрессия мРНК АПФ установлена в сосудах сердца и эндокарде, а также непосредственно в кардиомиоцитах [214]. В то же время, наличие ренина в миокарде обусловлено, по-видимому, его внекардиальным происхождением, так как в культуре кардиомиоцитов и фибробластов сердца не установлено синтеза ренина. Отмечено также, что ренин и проренин могут оказывать гипертрофическое действие в сердце, минуя образование А II через связывание со специфическими рецепторами и активацию митогенактивирующей протеинкиназы.
Большинство исследователей разделяют точку зрения, что содержание А II в сердце в значительно большей степени определяется его локальным синтезом, чем захватом из крови [26]. В то же время, ренин и ангиотензиноген способны проходить через эндотелиальный барьер и захватываться клетками миокарда, где они участвуют в локальном образовании А ΙΙ с инотропным эффектом, стимуляцией гипертрофии миокарда и ремоделирования сердца [40, 41].
В исследованиях на изолированном сердце не установлено локальной продукции ангиотензиногена, многие авторы полагают, что основная часть его содержания в миокарде определяется захватом из плазмы крови [48]. В то же время, концентрация А I и А II в сердце выше, чем в плазме, что свидетельствует об их выраженном локальном синтезе [165]. В соответствии с результатами исследований с использованием меченых пептидов А I и А II, более 90% кардиального А I и более 75% — А II синтезируются локально [291].
Рецепторы А II в нормальном сердце локализуются преимущественно на кардиомиоцитах, их экспрессия на фибробластах отмечается только при патологических состояниях. АТ1-рецепторы являются стимулятором гипертрофии и пролиферации кардиальных клеток, АТ2-рецепторы медиируют противоположные эффекты.
Установлено, что экспрессия компонентов РАС в сердце значительно усиливается в различных патологических условиях. Так, активность АПФ не выявлена в нормальных клапанах аорты, но закономерно отмечалась в стенозированных клапанах наряду с экспрессией АТ1-рецепторов. При этом АПФ локализовалась как в макрофагах, так и внеклеточно в сочетании с апоВ и А II. В атеросклеротических бляшках коронарных артерий локализация АПФ также сочеталась с наличием апоВ.
Это сочетание АПФ с ЛПНП как в местах поражений, так и в плaзме крови свидетельствует о том, что ЛПНП могут транспортировать АПФ и обеспечивать его участие в развитии поражений как воспалительного, так и атеросклеротического характера, тогда как применение липидоснижающей терапии способно задерживать прогрессирование стенозирующего поражения аортальных клапанов [204].
Как клинические, так и экспериментальные данные свидетельствуют о выраженной активации локальной РАС в миокарде после перенесенного ИМ, результатом чего является существенное прогрессирование систолической и диастолической дисфункции ЛЖ. В этих условиях сочетанное применение ингибиторов АПФ и блокаторов АТ1-рецепторов характеризовалось менее выраженным накоплением коллагена и более полным восстановлением диастолической функции по сравнению с изолированным применением каждого из них. Этот эффект в значительной степени определялся предупреждением действия А II, который образовывался путем альтернативной продукции с участием химазы, на которую ингибиторы АПФ не оказывают влияния [189].
Значимость кардиальной РАС особенно отчетливо проявляется при таких патофизиологических состояниях, как АГ, хроническая сердечная недостаточность. В этих условиях активность циркулирующей РАС может быть даже сниженной, но в миокарде локально продуцируется А II в количестве, достаточном для развития функциональных и структурных изменений в сердце.
В ряде исследований показано, что повышенная концентрация глюкозы в крови способствует активации кардиальной РАС и увеличению продукции А II в миокарде [259, 262]. При гипергликемии у крыс отмечено также повышение экспрессии рецепторов проренина в почках с сопутствующим повышением АД и частоты сердечных сокращений [21, 263, 264, 268].
А II, образующийся в миокарде, является основным компонентом системы быстрого ответа сердца на увеличение нагрузки, и неоднократно показано, что растяжение изолированных полосок миокарда сопровождается усиленной продукцией А ΙΙ и высвобождением его в среду. Эта реакция лежит в основе ремоделирования сердца при хроническом увеличении нагрузки, которое определяется главным образом пролиферацией фибробластов под действием локально образуемого А II. Подобный эффект в значительной степени опосредован повышением экспрессии TФР-β, его угнетение траниластом значительно уменьшало выраженность фиброза без изменений АД. Другой медиатор этого процесса — остеопонтин, содержание которого значительно возрастает после применения А II.
Резкая активация кардиальной РАС показана также у пациентов с пароксизмальной или персистирующей фибрилляцией предсердий, у которых отмечено 3-кpатное повышение уровня АПФ в миокарде в сочетании с выраженным интерстициальным фиброзом. Это объясняет наличие положительного кардиопротекторного эффекта угнетения РАС у пациентов с фибрилляцией предсердий.
Хотя тканевые РАС тесно связаны с циркулирующей, они регулируются независимо, так как основной задачей циркулирующей РАС является поддержание гомеостатического состояния организма в целом, тогда как локальные непосредственно участвуют в регуляции функции и структуры органов, в которых локализуются [41, 63]. Так как эти две задачи часто не только не координированы, но находятся в противоречии, то и изменения функции циркулирующей и локальной РАС могут отличаться не только по выраженности, но и по направленности.
Это положение подтверждается данными о том, что защитное действие ингибиторов РАС в предупреждении развития гипертрофии, ремоделирования и склерозирования сердца и сосудов в значительной степени не зависит от их антигипертензивного действия. Так, применение блокатора ренина — алискирена у 465 пациентов с АГ и увеличенной толщиной межжелудочковой перегородки приводило к снижению систолического АД на 6,6 мм рт. ст., диастолического АД — на 3,6 мм рт. ст., индекса массы ЛЖ на 4,9 г/м2. Дополнительное применение лозартана сопровождалось дальнейшим уменьшением индекса массы ЛЖ на 5,8 г/м2 при том, что оно не отражалось на выраженности антигипертензивного эффекта.
Длительное время большинство исследователей рассматривали повышенный уровень циркулирующего А II в качестве основного фактора, вызывающего гипертрофию миокарда. Однако исследования последних лет свидетельствуют о том, что активация кардиальной РАС способна инициировать интенсивный локальный синтез А II, достаточный для того, чтобы вызвать первичную гипертрофию миокарда даже в отсутствие повышенной нагрузки [60]. Кроме того, даже при ее увеличении гипертрофия миокарда в значительной степени является следствием увеличения локальной продукции А II [184].
Результаты ряда исследований подтверждают, что ремоделирование сердца при АГ, гемодинамической перегрузке и ишемии определяется возрастанием экспрессии локальной РАС. Так, при хронической объемной перегрузке сердца у собак активность АПФ и содержание А II в миокарде ЛЖ прямо коррелировали с напряжением стенки [49]. В исследовании, проведенном с участием 76 пациентов с сердечной недостаточностью, образование А II в миокарде возрастало по мере прогрессирования недостаточности, и единственным его предиктором было конечно-систолическое напряжение стенки, независимо от причины развития недостаточности [251]. Действие А II было опосредовано интенсивным образованием активных форм кислорода, развитием оксидативного стресса, пролиферацией фибробластов и синтезом коллагена 1-го типа, уменьшением экспрессии ММР-1. Известно, что коллаген 1-го типа является детерминантой жесткости миокарда с развитием систолической и, особенно, диастолической сердечной недостаточности, а фибробласты относятся к числу его основных продуцентов [33, 34].
В настоящее время можно считать установленным, что как А II, так и механический стресс вызывают ремоделирование сосудистой стенки и сердца преимущественно за счет усиленного синтеза матриксных белков, а не в результате гипертрофии гладкомышечных клеток и кардиомиоцитов. При действии А II экспрессия мРНК коллагена в миокарде возрастала на 92%, фибронектина — на 20%. Механическое растяжение усиливало экспрессию мРНК коллагена на 102%, фибронектина — на 50%, и эти эффекты ослаблялись при применении блокаторов АТ1-рецепторов, несмотря на отсутствие повышения концентрации А II в крови [270].
Это положение подтверждено исследованиями на культуре фибробластов сердца крыс, инкубация которых с А II в течение 24 ч приводила к дозозависимому увеличению продукции коллагена максимально на 113%. Предварительное применение телмисартана полностью угнетало эту реакцию, тогда как применение блокаторов AT2-рецепторов в этих условиях не оказывало никакого эффекта [164].
Установлено, что характерными особенностями гипертрофии ЛЖ, которая инициируется А II, являются гиперплазия кардиальных фибробластов, стимуляция синтеза коллагена с возрастанием жесткости и нарушением функции миокарда. Показано, что баланс между гипертрофией кардиомиоцитов и гиперплазией фибробластов определяет, является ли гипертрофия ЛЖ адаптивной, как у тренированных атлетов, или патологической, сочетающейся с кардиофиброзом, как при кардиальной патологии. Помимо этого, А II способен индуцировать апоптоз кардиомиоцитов с выраженным уменьшением массы активно функционирующего миокарда [169].
Особо убедительные данные относительно роли миокардиальной РАС получены в исследовании на мышах с ее кардиоспецифичной гиперэкспрессией. У этих мышей установлено наличие первичной гипертрофии миокарда в отсутствие повышения АД. Хотя выраженность гипертрофии была умеренной и прирост массы ЛЖ составил 17%, она сочеталась с развитием электрической нестабильности миокарда, выраженными нарушениями функции сердца в виде снижения пика развиваемого АД, максимальной скорости его повышения.
Эти данные позволили сделать вывод, что хроническая локальная эндогенная гиперпродукция А II в сердце сопровождается его ремоделированием и нарушением функции, независимо от изменений АД, и этого достаточно для последующего развития сердечной недостаточности [119].
Активность кардиальной РАС модулируется различными стимулами, в частности, она повышается в сердце животных при резком увеличении потребления соли на фоне снижения активности циркулирующей РАС. Помимо этого, активация симпатической нервной системы, которая сопровождает развитие АГ и сердечной недостаточности, приводит к повышению активности миокардиальной РАС.
Детальные исследования активности миокардиальной РАС проведены на 22 пациентах с атипичной болью в области сердца путем сопоставления концентрации А I и А II в крови аорты и коронарного синуса. При нормальной солевой диете отмечено преобладание содержания А I и А II в коронарном синусе соответственно на 6,5 и 2,7 пг/мл, что свидетельствовало о выраженной их локальной продукции. При низкосолевой диете значительно повышалась активность ренина в плазме крови и содержание А I в крови аорты, но не коронарного синуса, свидетельствуя об уменьшении продукции А I в миокарде. Образования А II в миокарде в этих условиях не отмечалось. В условиях высокосолевой диеты значительно возрастала секреция миокардом А I и А II на фоне уменьшения содержания А II в крови аорты. Эти данные свидетельствовали о наличии функционально активной РАС в сердце, независимой от циркулирующей РАС [251].
2.1.2. Локальная РАС в сосудистой стенке
Концепция локальной сосудистой РАС сформировалась, когда в сосудистых клетках было установлено наличие компонентов РАС и когда стало очевидным, что А II различным образом влияет на способность этих клеток к пролиферации. Локальная продукция ренина в сосудистой стенке остается спорной, хотя она установлена у крыс в неоинтиме после баллонного повреждения стенки [125, 126]. Содержание ренина в стенке обусловлено в основном его захватом из крови через соответствующие рецепторы [195, 196]. АПФ в сосудистой стенке экспрессируется главным образом на поверхности эндотелиоцитов [321, 322] и менее выраженно — в адвентиции и в неоинтиме при ее формировании после повреждения [68], мРНК ангиотензиногена наиболее обильно представлены в периваскулярной жировой ткани [27, 28]. Локальное образование А II в сосудистой стенке крысы показано на изолированных сегментах по 50% превращению А I в А II при однократном прохождении перфузата через препарат, и этот эффект полностью блокировался ингибиторами АПФ [111].
Наличие компонентов РАС, прежде всего — ангиотензиногена и АПФ, идентифицировано как непосредственно в гладкомышечных клетках, та и в периадвентициальной жировой ткани. В то же время, рениноподобная активность сосудистой стенки предположительно связана с локальной экспрессией ренина [181], хотя большинство исследователей рассматривают ее как следствие попадания ренина и проренина в сосудистую стенку из циркуляции [60, 188]. В то же время, способность гладкомышечных клеток сосудов продуцировать ренин установлена в условиях клеточной культуры, отмечено, что содержание ренина в стенке аорты крыс со спонтанной АГ прямо коррелировало с уровнем АД.
Вопрос о возможности внепочечной секреции ренина остается в настоящее время дискуссионным, большинство исследователей полагают, что синтез ренина в сосудистой стенке в нормальных условиях если и существует, то весьма ограниченный. В ряде работ установлена экспрессия мРНК ренина непосредственно в сосудистой стенке, хотя большинство исследователей разделяют точку зрения, что даже при существовании локальной секреции ренина она недостаточна для полноценного функционирования локальной РАС.
Однако синтез ренина в сосудистой стенке может существенно возрастать в патологических условиях, кроме того, существенную роль играет захват ренина из циркуляции через ренинсвязывающий белок и рецепторы проренина на клетках эндотелия. Поэтому доминирующую роль в продукции А I и А II в сосудистых гладкомышечных клетках, клетках эндотелия и эндокарда играет захват циркулирующего ренина.
Локальное образование А II приводит в развитию вазоконстрикторного ответа как прямого характера через АТ1-рецепторы, так и посредством стимуляции высвобождения ЭТ-1 [246, 247]. Помимо этого, действие А II в сосудистой стенке опосредовано также усиленной продукцией радикалов кислорода, развитием оксидативного стресса и уменьшением биодоступности NO [248, 249].
Помимо этого, активация АТ1-рецепторов в клетках сосудистой стенки сопровождается усиленной продукцией митогенного фактора TФР-β с последующей пролиферацией гладкомышечных клеток и угнетением роста эндотелиоцитов.
Патофизиологическая значимость локальной РАС в сосудистой стенке особенно отчетливо проявляется при развитии артерио- и атеросклероза и выражается в ремоделировании сосуда с возрастанием толщины его стенки и жесткости. Эти изменения являются одним из ведущих компонентов развития АГ, сердечной и почечной недостаточности, диабетической васкулопатии.
Одним из механизмов, посредством которого А II стимулирует ремоделирование сосудистой стенки, является пролиферация гладкомышечных клеток и фибробластов. Помимо этого, СОР и перекись водорода, продукция которых активируется под действием А II, обусловливают развитие в сосудистой стенке воспаления, склерозирование в результате накопления внеклеточного матрикса, участвуют в развитии атеросклеротического поражения, а также принимают участие в неоангиогенезе [214].
Показано, что одним из факторов активации РАС в сосудистой стенке с развитием ее артерио- и атеросклеротического повреждения является хроническое угнетение продукции NO в эндотелии с реципрокным повышением активности АПФ, усилением синтеза А II и экспрессии АТ1-рецепторов.
Активация сосудистой РАС может иметь первичный характер и происходить независимо от изменений внутрисосудистого давления. Это подтверждается тем, что применение ингибиторов АПФ при АГ сопровождалось устранением миоинтимальной гиперплазии в отличие от верапамила, использованного в эквипотенциальных дозах, то есть при равном гипотензивном действии [235]. Аналогичное уменьшение миоинтимальной гиперплазии при АГ отмечено и при применении лозартана — блокатора АТ1-рецепторов, что также свидетельствовало о роли А II в гипертрофии сосудистой стенки при АГ.
АПФ в сосудистой стенке экспрессируется преимущественно в эндотелии, и эндотелиальная денудация устраняет трансформацию А I в А II. В значительно меньшей степени эта экспрессия отмечена в адвентиции, тогда как ангиотензиноген наиболее интенсивно экспрессируется в периадвентициальных жировых клетках [27, 28]. Полагают, что он диффундирует в сосудистую стенку, где вступает во взаимодействие с сосудистым ренином. Характерно, что А II через ТФР-β стимулирует рост гладкомышечных клеток, но угнетает пролиферацию эндотелиоцитов.
Сократительный ответ гладкомышечных клеток на А II медиируется преимущественно через АТ1-рецепторы и частично — через высвобождение ЭТ-1. В то же время, активация AT2-рецепторов индуцирует вазодилатацию, в основном — в микрососудистом ложе, за счет высвобождения оксида азота и, в меньшей степени — брадикинина. Локально образующийся А II через АТ1-рецепторы стимулирует также неоангиогенез.
В биоптатах стенки сонной артерии, пораженной атеросклерозом, полученных при эндартерэктомии у 21 пациента с АГ, установлена интенсивная экспрессия как мРНК, так и белка ангиотензиногена и катепсина G. Подтверждено отсутствие мРНК ренина и наличие АПФ, АТ1-рецепторов и А II в эндотелиоцитах, гладкомышечных клетках и, главным образом, макрофагах в зоне поражения. Установлено также наличие положительной достоверной корреляционной связи между экспрессией мРНК катепсина G, ангиотензиногена, АПФ и АТ1-рецепторов [158].
В экстрактах сосудистой ткани людей установлено, что образование А II на 30% обусловлено АПФ, на 70% — химазой, тогда как в экстрактах сосудов крыс образование А II определяется преимущественно действием АПФ [179]. При баллонном повреждении как артериальных, так и венозных сосудов химаза почти полностью ответственна за продукцию А ΙΙ, гипертрофию и ремоделирование сосудистой стенки. Показано, что через 28 дней после шунтирования сонной артерии безымянной веной активность химазы в ее стенке повысилась в 10 раз, тогда как экспрессия АПФ — только в 2 раза. Применение блокаторов АТ1-рецепторов сочеталось с угнетением образования неоинтимы на 26%, ингибитора химазы — на 36% [179].
Наличие АТ1-рецепторов установлено также в сосудистых гладкомышечных клетках [16], эндотелиоцитах [127], кардиомиоцитах [77], гепатоцитах и клетках почек [163]. Локально продуцируемый А II через АТ1-рецепторы, которые экспрессируются на мембране сосудистых гладкомышечных клеток, вызывает их сокращение, пролиферацию и гипертрофию [15], в миокарде — гипертрофию кардиомиоцитов [319, 320].
Показано, что в физиологических условиях поддержание нормальной активности локальной РАС в сосудистой стенке обусловливает сохранение ее структуры и функциональных свойств, тогда как уменьшение локальной экспрессии компонентов РАС устраняет возможность компенсаторного утолщения сосудистой стенки при действии повышенного гемодинамического стресса и способствует образованию аневризм. Так, в стенке аневризм церебральных артерий, полученных при проведении оперативных вмешательств у пациентов с травмой головы или глиомой, установлено значительное снижение экспрессии АПФ, A II, АТ1-рецепторов, фибробластного фактора роста (FGF), тромбоцитарного фактора роста (PDGF), тканевого ингибитора ММРs (TIMP-1) по сравнению с непораженными артериями [205].
В аорте людей, пораженной атеросклерозом, отмечено значительное повышение активности химазы и ее участие в продукции А II. В нормальной ситуации мышечные клетки, содержащие химазу, находятся в дремлющем состоянии, тогда как при повышении плазменного уровня холестерина они высвобождают химазу, что приводит к повышению локальной концентрации А II с последующим ремоделированием сердца и сосудистой стенки [290].
В экспериментальных исследованиях неоднократно подтверждено, что химазозависимая активация локальной РАС в сосудистой стенке играет существенную роль в ее атеросклеротическом поражении. Так, у кролей, находящихся на атерогенной диете, наряду с повышением активности АПФ и плотности АТ1-рецепторов, активируется и альтернативный химазный путь образования А II. Этот процесс усиливается параллельно увеличению содержания в плазме крови ЛПНП и сочетается с увеличенным отложением в стенку липидов. На этом фоне пероральное применение блокатора химазы значительно снижало активность А II в стенке аорты и отложение в нее липидов, не влияя на уровень холестерина в плазме крови [288].
Выраженная активация локальной РАС в сосудистой стенке в условиях гиперхолестеринемии установлена и в исследовании, которое было проведено на изолированных сегментах грудной аорты кролей, находящихся на атерогенной диете. Показано увеличение продукции СОР в 2,2 раза в сочетании с 2-кратным возрастанием способности эндотелия адгезировать моноциты в отсутствие повышения АД. Эти эффекты почти полностью устранялись инкубацией исследованного сегмента с антагонистом A II или антиоксидантом пирролидином в течение 2 ч [198].
Результаты ряда других исследований также свидетельствуют о том, что доминирующая роль в развитии атеросклероза в условиях АГ принадлежит не системной, а локальной РАС в сосудистой ткани. Показано, что содержание А II в сосудистой стенке может существенно превышать его уровень в крови. Помимо этого, применение каптоприла у пациентов с АГ сопровождалось выраженным увеличением диаметра плечевой и сонной артерий при том, что исходная активность АПФ плазмы крови была снижена на 95%. Характерно, что подобный эффект не отмечен у лиц с нормальным АД и нормальной активностью АПФ в плазме крови [238]. При этом жесткость сосудистой стенки коррелировала с локальной активностью РАС и не зависела от уровня циркулирующего А II [60, 238].
Хотя гипертрофия и ремоделирование сердца являются одним из важнейших последствий АГ, в ряде исследований выявлено отсутствие связи между выраженностью АГ и степенью гипертрофии ЛЖ [52]. Показано, что выраженность гипертрофии при АГ определяется преимущественно растяжимостью стенки аорты и крупных артерий и коррелирует не с диастолическим, а с систолическим и пульсовым АД. Поэтому препараты, обладающие антигипертензивным эффектом, но не влияющие на растяжимость стенки артерий, не способны уменьшать выраженность гипертрофии миокарда в отличие от препаратов, снижающих активность РАС [60].
Выраженная активация локальной РАС в сосудистой стенке с нарушением функции эндотелия отмечена также при повышении уровня СЖК в крови. Так, при внутривенной инфузии жировой эмульсии с гепарином 10 исследуемым здоровым лицам дилататорная реакция на ацетилхолин была уменьшена на 38%, и этот эффект полностью устранялся после однократного применения лозартана или периндоприла. Однако при этом не установлено каких-либо изменений активности циркулирующей РАС [299].
Возможность независимой активации локальных РАС объясняет, почему до настоящего времени остается спорным вопрос о связи между активностью циркулирующей РАС и риском развития кардиоваскулярной патологии [175, 176]. Попытка связать активность ренина в плазме крови с риском развития ИБС предпринималась в ряде исследований. В одном из первых, проведенном в 1972 г., пациенты с низкой активностью ренина характеризовались более низким риском тяжелых исходов ИБС и инсульта при той же величине АГ и гипертрофии ЛЖ [20]. В другом исследовании на большой когорте испытуемых (n=3303) подтверждено, что повышение плазменной активности ренина сочеталось с увеличением долговременной кардиоваскулярной летальности. На протяжении 10 лет наблюдения отмечено 554 случая (16,8%) кардиальной смерти, и при многофакторном регрессионном анализе лица в верхнем квартиле активности ренина по сравнению с лицами в нижнем характеризовались значительно повышенным риском ее развития (НR 1,79). На каждое стандартное отклонение активности ренина отмечено 22% повышение риска внезапной кардиальной смерти, на 23% — смерти от сердечной недостаточности. Отмеченная связь имела в значительной степени независимый характер и сохранялась после учета традиционных факторов кардиоваскулярного риска, антигипертензивной терапии, содержания в плазме крови А II и альдостерона [283].
В проспективном исследовании, проведенном с участием 1717 пациентов с АГ, также установлено повышение риска ИБС пропорционально возрастанию активности ренина в плазме крови [5]. Однако в двух других исследованиях, проведенных соответственно с участием 803 и 3532 пациентов, такой зависимости не отмечено [213]. При длительном наблюдении 3183 нелеченных пациентов с систолическим АД >160 мм рт. ст. и диастолическим АД >95 мм рт. ст. отмечено 86 кардиоваскулярных явлений, из них 35 — летальных, но риск их развития не коррелировал с активностью ренина. Напротив, была отмечена отрицательная корреляционная зависимость между плазменной активностью ренина и систолическим АД [5].
По-видимому, в этих случаях причиной развития тяжелых исходов заболевания была активация не циркулирующей, а локальной кардиоваскулярной РАС, так как в большом числе исследований продемонстрирована высокая эффективность препаратов, угнетающих РАС, даже в отсутствие повышенной активности ренина в плазме крови и увеличения содержания в ней А II.
Так как значительная часть А II в локальных РАС образуется без участия АПФ, а другими протеиназами типа химазы, то угнетение АПФ часто оказывается недостаточно эффективным. Выявлено, что при равном снижении АД перфузия почек увеличивается на 50% более выраженно при угнетении ренина, чем АПФ [73].
Установлено, что уровень А II в крови у мышей с отсутствием гена АПФ сохраняется достаточно высоким, и это подтверждает точку зрения, что не только АПФ, но и другие ферменты участвуют в образовании А II. В ряде ранних работ указывалось, что в человеческом сердце химаза является основным ферментом, ответственным за образование А II [290]. Однако это предположение противоречит тому, что применение ингибиторов АПФ сопровождается резким повышением уровня А I в плазме крови и в сердце и снижением уровня циркулирующего и кардиального А II. Помимо этого, если химаза является ответственной за продукцию А II в сердце, то блокаторы рецепторов А II были бы более действенными в угнетении РАС по сравнению с ингибиторами АПФ, тогда как в реальных условиях отмечается противоположный эффект [218–221]. Показано также, что ингибиторы химазы не приводят к снижению уровня АД и А II в плазме крови [288].
Тем не менее, в ряде работ показано наличие химазной активности в сердце и коронарных сосудах, затем оно было установлено в почках, миоцитах, мезенхимальных клетках, эндотелиоцитах.
Результаты ряда исследований свидетельствуют о том, что при оценке значимости АПФ в активности циркулирующей РАС необходимо учитывать сопутствующие изменения уровня в плазме крови ренина и А I. Так, у крыс, которым применялся периндоприл, плазменный уровень А II составил 30–40% контрольного, но уровень ренина повысился в 100 раз, уровень А I — в 30 раз, и отношение А II/А I уменьшилось на 99%. Отношение А II/А I в почках, сердце и других тканях уменьшилось примерно в такой же степени, поэтому оно значительно более точно определяло роль АПФ в образовании А II, нежели только его уровень.
Как уровень А II, так и отношение А II/А I резко снижались также в плазме крови и предсердиях у пациентов, получавших ингибиторы АПФ, что свидетельствует о его преобладающем значении в образовании А II в сердце человека [310].
Показано также, что при хроническом угнетении АПФ уровень химазы в миокардиальной интерстициальной жидкости мышей повысился в 14 раз в сочетании с повышением уровня А I в 2,4 раза, но уровень А II не изменился, а отношение А II/А I уменьшилось более чем на 50%, что также свидетельствовало о доминирующей роли АПФ в образовании А II. Об этом свидетельствуют и результаты исследований последних лет с определением содержания А II в интерстициальной жидкости миокарда [6, 94].
В ряде исследований отмечено, что химаза, обусловливающая альтернативный путь образования А II без участия АПФ, содержится в сердце у спонтанно гипертензивных крыс в секреторных гранулах тучных клеток, а уровень их экспрессии в миокарде увеличивается параллельно с выраженностью его гипертрофии и склерозирования, а также с массой коллагена [159]. У пациентов с сердечной недостаточностью количество тучных клеток в миокарде значительно возрастает, и до 75% ферментной активности, сопряженной с превращением А I в А II, определяется наличием химазы. Установлено, что содержание тучных клеток значительно увеличено в сердце пациентов с дилатационной кардиомиопатией, а их дефицит у мышей с коарктацией брюшной аорты значительно замедлял переход от компенсаторной гипертрофии к сердечной недостаточности.
Кроме того, тучные клетки содержат TФР-β, который стимулирует продукцию коллагена и дифференциацию гладкомышечных клеток в миофибробласты, выброс этого цитокина из внутриклеточных гранул происходит под действием химазы. В клинических условиях при сердечной недостаточности угнетение химазы оказывало кардиопротекторное действие посредством уменьшения продукции А II, выброса TФР-β из тучных клеток, угнетения фиброза и сохранения диастолической функции сердца [173].
Химаза — хемотрипсиноподобная сериновая протеаза, важнейшим ее эффектом является превращение А I в А II. Однако она способна также разрушать внеклеточный матрикс, активировать TФР-β, ИЛ-1, принимать участие в образовании ЭТ-1 и метаболизме липидов.
Химазозависимое образование А II установлено также в миокарде лиц с АГ на материалах аутопсий. Активность химазы регистрировалась главным образом в эпикарде, где отмечено также наличие Т-лимфоцитов и макрофагов, которые продуцируют АПФ. Хотя корреляция между активностью химазы и степенью повышения АД в большинстве исследований отсутствовала, химаза могла принимать участие в процессе тканевого ремоделирования. Поэтому применение блокаторов АТ1-рецепторов более полно устраняло ремоделирующее влияние гиперактивности РАС, чем применение ингибиторов АПФ.
А II, который продуцируется локально с участием химазы, не принимает существенного участия в регуляции АД, но в значительной степени определяет ремоделирование сосудистой стенки. Выявлено, что траниласт, ингибитор химазы, оказывал выраженное ангиопротекторное действие в экспериментальной модели атеросклероза. У собак с сердечной недостаточностью, вызванной длительной частотной стимуляцией сердца, применение ингибиторов химазы сопровождалось резким угнетением воспалительной реакции в миокарде, образования А II и развития кардиофиброза [272] в сочетании со снижением уровня А II на 18%, уменьшением накопления коллагена на 60% [173].
В физиологических условиях химазе свойственна только ограниченная роль в образовании А ІІ, на сегментах артерий желудка ответ на А I на 87% угнетался каптоприлом и только оставшиеся 13% были чувствительными к химостатину — ингибитору химазы. Однако в условиях, сопровождающихся дегрануляцией тучных клеток, химаза может существенным образом участвовать в повреждении сосудистой стенки. На фоне ее ремоделирования отмечено только умеренное повышение экспрессии АПФ и в значительно большей степени — химазы. Особенно выраженное возрастание экспрессии мРНК химазы выявлено при использовании венозного сегмента для шунтирования сонной артерии, а применение ингибиторов химазы предупреждало увеличение толщины стенки вены. Химаза является также ангиогенным фактором и принимает участие в новообразовании сосудов при диабетической ретинопатии, опухолевых процессах, ревматоидном артрите.
Если АПФ в зоне атеросклеротического поражения локализуется главным образом в макрофагах, мигрировавших в интиму, в нормальной аорте — в эндотелии, то локализация химазоположительных тучных клеток характерна для адвентиции как в норме, так и при атеросклеротическом поражении.
Повышение активности химазы в миокарде и коронарных артериях параллельно с увеличением продукции А II и ремоделированием сердца отмечено при различных видах кардиальной патологии, включая ИБС, кардиомиопатию, миокардиты [300, 301]. При сердечной недостаточности, вызванной перегрузкой сердца объемом, показано повышение кардиальной активности как АПФ, так и химазы [49]. Однако даже в зоне атеросклеротического поражения значительно чаще отмечается сочетанная локализация А II с АПФ, чем с химазой [57].
Данные последних исследований свидетельствуют о том, что увеличение плазменного содержания общего холестерина и холестерина ЛПНП сопряжено с повышением активности сосудистой химазы, тогда как у трансгенных мышей с первичной гиперактивностью сосудистой химазы отмечается ускоренное развитие атеросклероза даже с нормальным содержанием холестерина в крови. У пациентов, которым проводилось шунтирование коронарных артерий, установлена достоверная корреляция между уровнем холестерина и активностью сосудистой химазы. При содержании хомяков на атерогенной диете отмечена прямая достоверная зависимость между выраженностью гиперхолестеринемии и активностью химазы, а применение ингибитора химазы сопровождалось значительным уменьшением липидной инфильтрации аорты при отсутствии влияния на плазменный уровень липидов и холестерина.
В ряде крупных клинических исследований подтверждено значительное повышение активности химазы в миокарде, сопряженное с ремоделированием сердца, при кардиоваскулярной патологии типа ИБС, сердечной недостаточности, кардиомиопатии, вирусного миокардита. Установлено, что кардиальная химаза принимает также участие в ремоделировании сердца и развитии сердечной недостаточности после ИМ.
Характерным для альтернативного пути образования активации РАС в сосудистых гладкомышечных клетках [154], а также в жировой ткани [137] является способность химазы и катепсинов расщеплять ангиотензиноген с образованием А I, а затем — и А II без участия ренина. Хотя ренин в этом отношении в 100 000 раз более эффективен, чем химаза [97], катепсин G трансформирует А I в А II примерно с той же эффективностью, что и АПФ [279].
В атеросклеротических бляшках коронарных артерий установлено сочетанное выраженное повышение активности АПФ и содержания А II. Несмотря на это, блокада АПФ только незначительно замедляла прогрессирование атеросклероза, что подтверждало существование дополнительных путей локального образования А ІІ. Установлено, что в этих условиях А II образовывался из А I с участием катепсина D, либо прямо из ангиотензиногена, минуя этап А I, с участием катепсина G.
Отмечено, что катепсин D участвует в образовании А II в гладкомышечных клетках у крыс со спонтанной АГ, а катепсин G является компонентом ангиотензиновой системы гранулоцитов и его действие не требует участия ренина. Эти ферменты идентифицированы также в жировой ткани и участвуют в локальном образовании А II [137].
Как показано в ряде исследований с использованием гомогенатов миокарда, до 80% формирования активности А II в сердце при кардиальной патологии происходит альтернативным путем и связано с активацией химазы и только 11% обусловлено действием АПФ [290]. Наличие альтернативного пути образования А II подтверждается тем, что ингибиторы АПФ оказывают только транзиторный эффект на уровень циркулирующего А ΙΙ; через 24 ч после однократного введения препарата плазменный уровень А ΙΙ возвращается к исходному значению, несмотря на сохраняющееся угнетение АПФ. При длительном применении ингибиторов АПФ их влияние на уровень А ΙΙ также значительно ослаблено по сравнению с быстрым эффектом.
В исследованиях на крысах с применением каптоприла отмечено, что уже через 1–2 нед плазменный уровень А ΙΙ возвращался к исходному или даже превышал его, тогда как концентрация А ΙΙ в почечной ткани уменьшалась до 14% исходной. Эти данные означают, что около 86% А ΙΙ в почках образуется с участием АПФ, тогда как в сердце in vivo доминирует химазный путь образования А ΙΙ [22, 23]. Тем не менее, ингибиторы АПФ оказывают выраженное кардиопротекторное действие и эффективно предупреждают образование А II в сердце.
Однако возврат уровня А ΙΙ к исходному при длительном применении ингибиторов АПФ может объясняться и неоднократно отмеченным в этих условиях повышением концентрации ренина и А Ι. Это повышение сочетается с увеличением количества молекул ренина как в крови, так и в тканях и оказывается достаточным для того, чтобы преодолеть угнетение АПФ. Выявлено, что в этих условиях содержание А Ι в крови может возрастать более чем в 10 раз.
Наличие альтернативных путей образования А ΙΙ подтверждается тем, что на изолированных сосудистых сегментах ингибиторы АПФ только частично угнетали констрикторные реакции на А Ι и его превращение в А ΙΙ, а сохранившийся ответ устранялся ингибиторами химазы. Учитывая, что химаза, как и катепсин G, обладающий аналогичной активностью, содержатся соответственно в тучных клетках и нейтрофилах, это объясняет участие А ΙΙ в локальных сосудистых реакциях при воспалении.
2.1.3. Активность локальной почечной РАС
Почки являются не только источником компонентов РАС и регулятором ее активности, но и мишенью действия РАС. Отмечено, что повреждение почек при АГ связано не столько с повышением АД, сколько с развитием в них оксидативного стресса, воспаления и, как следствие, нефросклероза.
Эти эффекты определяются способностью А II стимулировать продукцию радикалов кислорода и высвобождение хемокинов, в частности — МСР-1, который инициирует макрофагальную инфильтрацию. Инфузия А II мышам, как нормальным, так и с отсутствием рецепторов МСР-1, сопровождалась аналогичным развитием гипертензивной реакции и гипертрофии миокарда. Однако у мышей с отсутствием рецепторов МСР-1 значительно менее выражеными были оксидативный стресс, инфильтрация макрофагов, альбуминурия и повреждение почек с сохранением более высокой скорости гломерулярной фильтрации, тогда как у контрольных мышей скорость гломерулярной фильтрации резко уменьшилась через 4 нед инфузии А II. Уровень нитротирозина, клеточная пролиферация, преимущественно — мезангиальных клеток и интерстициальных фибробластов, содержание коллагена были значительно увеличены у контрольных животных, но не изменились у мышей с отсутствием МСР-1.
Приведенные данные свидетельствуют о том, что в основе нефропатии, вызванной инфузией А II, важнейшую роль играют инфильтрация макрофагами, развитие воспаления и оксидативного стресса. В то же время эти данные не подтверждают точку зрения, что оксидативный стресс принимает участие в развитии гипертензивного ответа при инфузии А II [162]. К аналогичному выводу пришли и другие исследователи [284, 285, 317].
Предполагают, что задержка натрия, развитие интерстициального фиброза и повреждение почек при АГ являются следствием активации локальной РАС главным образом — в клетках проксимальных канальцев, которые обеспечивают до 65–70% реабсорбции натрия [315, 316]. В эпителиальных клетках этих канальцев установлена экспрессия всех компонентов РАС, включая ренин, ангиотензиноген, АПФ и рецепторы А II 1-го типа [105].
Установлено, что содержание А II в почках значительно выше, чем в плазме крови, и почки могут аккумулировать А II из плазмы [124]. Кандесартан предупреждал развитие этого эффекта [317], что свидетельствовало о доминирующей роли мембранных АТ1-рецепторов в связывании и интернализации А II.
Эти данные подтверждены результатами исследования на культуре эпителиальных клеток проксимальных канальцев, инкубация которых с А II сопровождалась 2-кратным повышением его внутриклеточной концентрации, тогда как лозартан устранял этот эффект.
Введение А II непосредственно в эпителиальные клетки проксимальных канальцев сопровождалось повышением внутриклеточной концентрации кальция. Внутриклеточное введение лозартана устраняло этот эффект, что свидетельствует о наличии внутриклеточных AT1-рецепторов [228].
Помимо участия в реабсорбции натрия и регуляции АД, А II, продуцируемый в почках, действует как провоспалительный цитокин и фактор роста, принимает участие в развитии интерстициального фиброза в проксимальных канальцах и нефросклероза. Пролиферативный эффект в значительной степени может определяться действием внутриклеточного А II на цитоплазматические и ядерные рецепторы.
Недавно выявлено, что А II способен прямо активировать факторы транскрипции NF-κB и AP-1 в почечных клетках с развитием длительных провоспалительного и митогенного эффектов и, в конечном итоге, интерстициального фиброза [321]. Полагают, что внутриклеточный синтез А II, как и усиленный захват локально продуцируемого или системного А II, особенно характерен для клеток проксимальных канальцев, что приводит к задержке натрия, усиленной пролиферации мезангиальных клеток с развитием АГ и нефросклероза [322]. В то же время, значимость внутриклеточной продукции А II, как и возможность фармакологической блокады его эффектов, остаются пока недостаточно изученными.
Установлено, что внутри- и внеклеточно образующийся А ΙΙ оказывает различное влияние как на активность натриевого насоса, так и на объем клеток почек. Так, внутриклеточное сочетанное введение ренина и антиготензиногена или А ΙΙ сопровождалось уменьшением объема клеток параллельно с активацией натриевого насоса, а внутриклеточное применение лозартана устраняло эти эффекты. Внеклеточное применение комплекса ренин/ангиотензиноген или А ΙΙ сопровождалось увеличением объема клеток и угнетением натриевого насоса. Эти данные рассматривают как свидетельство того, что активация интракринной РАС может оказывать защитное действие, в частности — на миокард при его ишемии за счет уменьшения клеточного отека [50, 51].
В значительном числе клинических наблюдений показано, что одним из важнейших патологических эффектов гипергликемии является развитие микрососудистых поражений, особенно в почках, которые отмечаются у 30–40% лиц с СД и являются ведущей причиной терминального поражения почек. В последние годы установлено, что причиной их развития является активация РАС, но не циркулирующей, как считали ранее и о чем судили по уровню А II в крови, а интраренальной РАС [292].
2.1.4. Функциональная значимость локальной РАС в центральной нервной системе
Наличие церебральной РАС подтверждено в ряде исследований по высокой локальной концентрации в ткани мозга ангиотензиногена, который продуцируется астроцитами и клетками нейроглии [178]. В нейронах мозга установлены также высокое содержание А II и выраженная экспрессия АТ1-рецепторов [182].
В настоящее время уже достаточно полно документирована роль церебральной РАС в центральном контроле кардиоваскулярного гомеостаза, а также в генезе сердечно-сосудистых нарушений, в частности — эссенциальной и реноваскулярной АГ [72]. Показано, что прямое интрацеребральное введение А II у нормальных крыс сопровождается значительным увеличением локальной продукции СОР и развитием АГ, тогда как селективное угнетение рецепторов А II в мозгу у крыс со спонтанной АГ приводит к снижению АД, а у нормальных крыс устраняет гипертензивный ответ как на локальное, так и на системное применение А II [89, 90].
В экспериментальных исследованиях неоднократно отмечено, что развитие АГ сочетается с увеличенной продукцией активных форм кислорода не только в сосудистой стенке, миокарде и почках, но и в тех отделах головного мозга, которые принимают непосредственное участие в регуляции АД. При этом ведущую роль в увеличении продукции радикалов кислорода в ткани мозга, как в периферических тканях при действии А II играет активация NADPH-оксидазы [9, 24, 201].
Установлено, что у мышей с нарушением функции NADPH-оксидазы инфузия А II или нахождение на солевой диете не сопровождались развитием АГ, тогда как применение ингибитора NADPH-оксидазы апоцинина на фоне АГ оказывало выраженное антигипертензивное действие [282, 316]. Этот эффект имел в значительной степени и центральный компонент, так как на фоне генетического отсутствия или угнетения NADPH-оксидазы в ядре солитарного тракта А ΙΙ не способен активировать локальную интрацеребральную продукцию СОР [296, 297] и вызывать развитие АГ [201].
Другим источником СОР в ткани мозга являются митохондрии, у спонтанно гипертензивных крыс установлены нарушения в системе транспорта электронов в митохондриях нейронов ядра солитарного тракта с развитием оксидативного стресса и повышением симпатической активности, тогда как введение в эти структуры коэнзима Q10, обладающего антиоксидантной активностью, сопровождалось угнетением оксидативного стресса и снижением АД [30].
Связь между активностью симпатической нервной системы, АД, продукцией СОР в регулирующих его структурах мозга и АТ1-рецепторами подтверждается результатами применения как блокаторов рецепторов ангиотензина II, так и антиоксидантов. Выявлено, что интрацеребральное введение олмесартана у гипертензивных крыс сопровождалось снижением локального оксидативного стресса, угнетением симпатической активности и выраженным антигипертензивным действием [7]. Аналогичный эффект отмечен и у аторвастатина, который также оказывал депрессорное действие, уменьшал выраженность оксидативного стресса в ядре солитарного тракта и симпатической гиперактивности у крыс со спонтанной АГ посредством повышения биодоступности оксида азота [140].
В последние годы показано, что церебральная РАС играет решающую роль в регуляции и поддержании АД, и ее компоненты, включая ангиотензиноген, ренин, АПФ и рецепторы А ΙΙ, интенсивно экспрессированы в различных ядрах, расположенных между передней частью третьего желудочка и стволом мозга. Поэтому образование А ΙΙ возможно непосредственно в ткани мозга, независимо от его уровня в циркуляции [58, 214], а гиперэкспресия АТ1-рецепторов в этих ядрах сопровождается повышением симпатического тонуса и развитием АГ [325]. Как уже указывалось выше, А ΙΙ может проникать в центральную нервную систему также из крови в субфорникальном органе, который лишен гематоэнцефалического барьера и является «окном» для поступления в центральную нервную систему мелких молекул, обусловливая наличие центрального компонента прессорной реакции на внутривенное введение А ΙΙ [108, 109, 205]. Отмечено, что разрушение area postrema значительно ослабляло прессорную реакцию на системное применение А ΙΙ, но не оказывает влияния на развитие АГ, не связанной с активацией РАС [210].
В исследованиях последних лет неоднократно показано, что гиперактивность симпатической нервной системы и циркулирующей РАС, играющая доминирующую роль в нарушениях функции ЛЖ после ИМ, определяется усиленной продукцией А II в центральной нервной системе [295–297]. Это происходит в результате локального высвобождения оубаинподобных соединений, действие которых приводит к значительному увеличению интрацеребрального содержания А ΙΙ [157, 311–314]. Развитие данного эффекта предупреждалось у крыс с селективным генетическим дефицитом содержания ангиотензиногена в ткани мозга. Помимо этого, у крыс с дисфункцией ЛЖ после ИМ блокада АТ1-рецепторов в мозгу устраняла гиперактивность симпатической нервной системы и нарушение артериальной барорефлекторной функции [54], а также снижала риск дилатации и дисфункции ЛЖ [157].
Эти данные подтверждены в исследованиях, в которых хроническое интрацеребральное введение лозартана крысам в течение 8 нед после воспроизведения ИМ сочеталось со значительным уменьшением выраженности ремоделирования ЛЖ, прежде всего — его дилатации [157]. Однако не всеми исследователями эти данные рассматривались как прямое подтверждение участия церебральной РАС в постинфарктном повреждении и ремоделировании сердца, так как лозартан мог попадать в системную циркуляцию и блокировать периферические АТ1-рецепторы.
Подобные погрешности отсутствовали у трансгенных крыс с дефицитом ангиотензиногена в мозгу. В исследовании на этих крысах получено прямое подтверждение значительной роли церебральной РАС и локальной продукции А II в регуляции симпатической активности, функции сердца и характера его повреждения в условиях развивающегося ИМ. В соответствии с полученными данными размер зоны поражения в конце 8-й недели после ИМ был аналогичным как у опытных, так и контрольных крыс. Однако плазменный уровень А II был более высоким у контрольных животных по сравнению с опытными как до, так и после воспроизведения ИМ. Только у контрольных крыс отмечены выраженные нарушения кардиогемодинамики, возрастание пика систолического АД в ЛЖ в сочетании с в 2,5 раза более выраженным повышением конечно-диастолического АД.
Максимальная скорость сокращения миокарда уменьшилась у опытных животных на 20% менее интенсивно, чем у контрольных, масса сердца и выраженность интерстициального фиброза существенно увеличились в группе контроля, но незначительно изменились у опытных крыс. У контрольных крыс был резко нарушен барорефлекторный контроль частоты сокращений сердца, повысилась активность почечных симпатических нервов, этих изменений не отмечали у животных с угнетенной активностью церебральной РАС.
Эти данные означали, что у крыс с генетической церебральной недостаточностью продукции ангиотензиногена и сниженной активностью церебральной РАС развитие ИМ сопровождалось отчетливо уменьшенной выраженностью симпатической гиперактивности, кардиальной дисфункции и ремоделирования сердца.
Длительное время считалось незыблемым, что центральная нервная система изолирована от действия циркулирующего А II гематоэнцефалическим барьером, и ведущую роль в центральной регуляции АД играет локальная церебральная РАС. Тем не менее, еще 40 лет назад было высказано предположение, что гипертензивный эффект циркулирующего А ΙΙ, особенно при длительном действии в низких и субдепрессорных дозах, может иметь и центральную нейрогенную природу и определяться способностью А ΙΙ стимулировать в центральной нервной системе зоны, ответственные за симпатическую активность.
В последнее время получены и прямые данные, свидетельствующие об участии центральной нервной системы в развитии АГ в этих условиях [12]. Показано, что ингибиторы АПФ и блокаторы АТ1-рецепторов оказывали кардио- и вазопротекторное действие у пациентов с АГ даже в отсутствие повышения активности циркулирующей РАС и при концентрации А II в крови, не оказывающей прямого гипертензивного эффекта, что в значительной степени определялось центральным действием А II. Помимо этого, применение симпатоингибиторов центрального действия [55], ганглиоблокаторов [89, 90], симпатической денервации [215] устраняло или уменьшало выраженность развития медленного прессорного ответа на введение А ΙΙ в низких дозах. Еще более убедительное подтверждение было получено в исследовании, в котором удаление субфорникального органа у крыс предупреждало повышение АД после инфузии А ΙΙ [108, 109].
Результаты ряда исследований свидетельствовали о том, что активация эндокринной РАС с увеличением концентрации циркулирующего А ΙΙ сопровождалась стимуляцией АТ1-рецепторов в центральной нервной системе с повышением симпатической активности и была одной из ведущих причин развития АГ [206]. Зависимость между выраженностью оксидативного стресса в стволе мозга, активностью симпатической нервной системы и уровнем АД показана у крыс как со спонтанной, так и реноваскулярной АГ [30, 143]. Эти данные подтверждают способность циркулирующего А II индуцировать развитие оксидативного стресса как центрального, так и периферического характера [207, 208].
Установлено, что развитие гипертензивного ответа на системное введение А II в значительной степени связано с усилением экспрессии ядерного фактора транскрипции kB и развитием локального воспаления в важнейшем кардиоваскулярном регуляторном центре, локализованном в гипоталамическом паравентрикулярном ядре. Помимо этого, интрацеребральная блокада рецепторов ФНО-α в этих условиях существенно снижала гипертензивную реакцию. Выявлено, что крысы с интрацеребральным угнетением экспрессии NF-κB характеризовались сниженным АД, уменьшенным содержанием в крови провоспалительных цитокинов, прежде всего — ФНО-α, радикалов кислорода, а также сниженной активностью АПФ. Помимо этого, блокада NF-κB в центральной нервной системе сопровождалась повышением активности антигипертензивных компонентов альтернативной РАС, прежде всего А (1–7) [25].
В ряде исследований показано, что при хронической системной инфузии А II в дозах, не оказывающих вазоконстрикторного действия и рассматривающихся как «субпрессорные» или «медленно прессорные», происходит постепенное градуальное повышение АД. Длительное время этот эффект рассматривали как следствие захвата А II из крови и накопления в сосудистой стенке. Однако в последние годы установлено, что разрушение area postrema [72] или латерального парабрахиального ядра [90] в центральной нервной системе, применение центральных симпатоблокаторов [215], ганглиоблокаторов и неселективных блокаторов α-адренорецепторов [167], денервация почек [121, 122] устраняли гипертензивное действие длительной инфузии субпрессорных доз А II. Участие церебральной РАС в развитии АГ проявляется также значительно меньшим повышением АД при инфузии А II на фоне ее угнетения [19].
Роль церебральной РАС в АГ, развивающейся при применении субпрессорных доз А II, достаточно полно определена также у трансгенных крыс со специфическим угнетением мРНК ангиотензиногена в центральной нервной системе. У этих животных отмечено уменьшенное на 90% содержание ангиотензиногена в ткани мозга, закономерно сниженное по сравнению с нормой АД (122,5 против 128,9 мм рт. ст.), уменьшенное содержание в плазме вазопрессина в сочетании со значительно менее выраженным возрастанием систолического АД на 7-й день непрерывной инфузии А II в субпрессорной дозе (на 28,9 по сравнению с 46,3 мм рт. ст.). У нормальных крыс длительная инфузия А II приводила к достоверному увеличению массы тела (на 11,1%), эта реакция отсутствовала у крыс с угнетенной церебральной РАС. В то же время, экспрессия мРНК натрийуретического пептида и коллагена в миокарде ЛЖ после инфузии А II в обеих группах животных была выражена в равной степени, свидетельствуя о том, что она прямо стимулировалась А II.
Эти данные, как и результаты ряда других исследований, свидетельствовали о том, что градуальное развитие гипертензивного ответа на системное введение А II требует участия церебральной РАС. Определяется это способностью А II проникать в головной мозг через определенные «окна» в гематоэнцефалическом барьере и активировать центральные механизмы развития АГ [67, 129, 174].
В ряде исследований установлено, что увеличение продукции СОР при действии циркулирующего А ΙΙ преимущественно ограничено субфорникальным органом, и его разрушение, как и нейтрализация свободных радикалов в этой области локальным введением антиоксидантов, резко ограничивало гипертензивную реакцию, вызванную системным действием А ΙΙ [122].
В настоящее время положение о том, что активность церебральной РАС оказывает существенное влияние на центральную регуляцию симпатического тонуса и функцию сердца и сосудов, получило широкое признание [54].
Одним из важнейших прямых подтверждений положения о центральном механизме гипертензивного действия А ΙΙ стали результаты исследования, которое было проведено на культуре нейронов, выделенных из гипоталамуса и ствола мозга. Установлено, что добавление А II в среду сопровождалось значительным повышением нейрональной активности, опосредованным усиленной продукцией СОР NADPH-оксидазой. Блокатор АТ1-рецепторов лозартан, как и апоцинин — пептидный ингибитор NAD(P)H-оксидазы, и темпол, миметик СОД, уменьшали как выраженность продукции СОР, так и частоту нейрональных разрядов [42].
В другом исследовании повышение АД, учащение сердечного ритма и повышение активности почечных симпатических нервов при кратковременной системной инфузии А II сочетались с усилением экспрессии NADPH-оксидазы в вентролатеральном отделе продолговатого мозга и в области ствола мозга, то есть в тех отделах, которые регулируют симпатический вазомоторный тонус.
Помимо этого, у солечувствительных гипертензивных крыс линии Dahl показано наличие прямой корреляционной зависимости между активностью гипоталамической NADPH-оксидазы и активностью почечных симпатических нервов, секрецией норадреналина и развитием АГ, тогда как локальное применение темпола или специфических ингибиторов NADPH-оксидазы сопровождалось нормализацией АД.
Установлено также, что развитие АГ при системном введении А II сочеталось с интрацеребральной инфильтрацией Т-клеток и лейкоцитов и увеличением локального содержания СОР; параллельно отмечено возрастание симпатической модуляции сердечного ритма и содержания СОР в сосудистой стенке [84].
Результаты исследований, проведенных в последние годы, позволили установить, что не только системное, но и прямое интрацеребральное введение А ΙΙ сопровождается усиленной локальной продукцией СОР в стенке сосудов, и этот эффект является одним из важнейших факторов развития транзиторной системной прессорной реакции [323, 324].
Подтверждено также, что даже при системном введении А ΙΙ развитие АГ имеет в значительной степени центральный характер и определяется активацией нейронов в ряде специализированных областей мозга, именуемых circumventricular organs — структур, окружающих третий и четвертый желудочки мозга[258]. Эти структуры, осуществляющие контроль АД, характеризуются недостаточно сформированным гематоэнцефалическим барьером и подвержены действию циркулирующего А ΙΙ. Они включают organum vasculosum laminae terminalis, area postrema, субфорникальный орган и антровентрикулярную область третьего желудочка.
В гипоталамическом субфорникальном органе — одном из основных регуляторов симпатической активности, установлена выраженная экспрессия АТ1-рецепторов и высокая активность СОД. Отмечено, что разрушение этих зон значительно ослабляло центральный компонент прессорного ответа на интрацеребральное введение А ΙΙ, включая активацию симпатической нервной системы, стимуляцию высвобождения вазопрессина, а также выраженность АГ, вызываемой циркулирующим А ΙΙ [108, 109].
NOX2 — изоформа NАDPH-оксидазы, — основной источник СОР в процессе нейрональной активации, вызванной А ΙΙ. В культуре нейронов гипоталамуса и ствола мозга лозартан, пептидный ингибитор, ингибитор NOX2 и темпол — миметик СОД, значительно уменьшали продукцию СОР при действии А ΙΙ и частоту нейрональных импульсов. Установлено, что при развитии АГ у солечувствительных крыс гипоталамический NOX2 задействован в регуляции секреции норадреналина и активности почечных симпатических нервов. Отмеченная у этих животных усиленная экспрессия NOX2 угнеталась при применении темпола наряду с устранением АГ.
Выявлено также, что при системной инфузии А ΙΙ развитие АГ сочеталось с усиленной продукцией СОР в субфорникальном органе — основном сенсоре А ΙΙ крови в центральной нервной системе. На фоне селективного угнетения СОД в субфорникальном органе системное введение А II сочеталось со значительным усилением лейкоцитарной, лимфоцитарной инфильтрации и продукции СОР в сосудистой стенке в сочетании с резко выраженным повышением частоты сердечных сокращений и АД. Эти данные свидетельствовали о том, что активные формы кислорода в центральной нервной системе в значительной степени определяют характер участия периферических органов в развитии АГ [42].
Усиленная продукция активных форм кислорода в ядре солитарного тракта также сопровождалась развитием АГ, а топическое введение в него СОД приводило к снижению АД, частоты сердечных сокращений и экскреции норадреналина с мочой. При кратковременной системной инфузии А ΙΙ отмечено повышение АД, активности почечных симпатических нервов, частоты сокращений сердца параллельно с усилением экспрессии NOX2 в области ствола мозга, которая регулирует симпатический вазомоторный тонус. Помимо этого, у крыс как нормальных, так и со спонтанной АГ, при введении А ΙΙ в этой области отмечена дисфункция митохондрий с усиленной продукцией радикалов кислорода, а применение коэнзима Q10 восстанавливало нормальный транспорт электронов по дыхательной цепи параллельно со снижением АД и активности симпатических нервов.
В ряде работ последних лет также установлено, что активные формы кислорода участвуют в центральном нейрогенном контроле АД и определяют развитие АГ, сопряженной с гиперпродукцией кислородных радикалов, при действии А ΙΙ и альдостерона. Отмечено, что интрацеребровентрикулярное введение А ΙΙ приводило к развитию транзиторного прессорного ответа в сочетании с тахикардией [43], и эти эффекты устранялись при помощи генетической гиперэкспрессии антиоксидантного фермента СОД в субфорникальном органе. Помимо этого, в условиях культуры нейронов, выделенных из субфорникального органа, А ΙΙ стимулировал усиленную продукцию СОР, которая устранялась применением как лозартана, так и СОД. Все эти данные свидетельствуют о высокой функциональной значимости активных форм кислорода в этой зоне мозга и зависимости их продукции как от эндокринной, так и локальной церебральной РАС [324].
Активация РАС, особенно в сочетании с активацией симпатической нервной системы, имеет критическое значение в патогенезе АГ от начальных этапов до повреждения органов-мишеней и развития сердечной и почечной недостаточности, инсульта. Более чем у 50% всех пациентов с АГ установлено повышение активности симпатической нервной системы, особое значение при этом имеют центры, локализованные в стволе мозга [65].
Как в экспериментальных, так и в клинических исследованиях показано, что фармакологическая блокада синтазы оксида азота сопровождается резким повышением АД, которое в значительной степени определяется повышением активности симпатической нервной системы [237]. Однако в последние годы установлено, что оксид азота играет важную роль в регуляции АД не только на периферии, но и центрально посредством модуляции активности автономной нервной системы. Установлено, что NO — один из важнейших медиаторов центральной нервной системы, принимающий непосредственное участие в регуляции активности системы кровообращения. В ряде исследований выявлено, что оксид азота способен угнетать, а свободные кислородные радикалы — активировать симпатическую нервную систему. Поэтому нарушение баланса между образованием кислородных радикалов и антиоксидантной активностью с уменьшением биодоступности оксида азота в центральной нервной системе, особенно в стволе мозга, приводит к активации симпатической нервной системы и является одним из важнейших механизмов развития АГ нейрогенной природы [113–115].
У нормальных крыс и крыс с сердечной недостаточностью, воспроизведенной перевязкой коронарной артерии, исследовались наличие и функциональная значимость экспрессии еNOS в гипоталамическом паравентрикулярном и супраоптическом ядрах. Полученные результаты свидетельствовали о выраженной экспрессии еNOS, связанной преимущественно с астроглиальными клетками и эндотелиоцитами, в исследованной области у нормальных крыс и значительном угнетении этой экспрессии у крыс с сердечной недостаточностью, сочетающимся с выраженным повышением симпатической и угнетением парасимпатической активности [14].
Хорошо известно, что хроническое применение у крыс l-NAME — ингибитора эндотелиальной NOS, приводит к повышению АД. Однако этот эффект имеет не только периферический характер в связи с угнетением продукции оксида азота и нарушением функции эндотелия. Введение кандесартана или проведение ганглиоблокады в этих условиях сопровождались значительно более выраженным снижением АД, чем в контроле, что свидетельствовало о повышении активности как РАС, так и симпатической нервной системы. Аналогичный эффект отмечен при введении А II в ядро солитарного тракта. Эти данные означают, что хроническое угнетение синтазы оксида азота в ядре солитарного тракта, как и активация локальной интрацеребральной РАС со стимуляцией АТ1-рецепторов, приводят к значительному повышению симпатической активности и, как следствие, развитию АГ [64].
Исследования на крысах с гиперэкспрессией гена eNOS и хронически усиленной продукцией NO в ядре солитарного тракта также свидетельствовали о том, что оксид азота в центральной нервной системе оказывает угнетающее действие на активность симпатической нервной системы с развитием гипотензии и брадикардии [236]. У животных с различными экспериментальными моделями АГ усиленный локальный синтез или повышенная доставка как NO, так и его предшественника L-аргинина в ствол мозга сопровождались снижением АД.
В то же время, центральная гиперэкспрессия индуцируемой NOS (iNOS) сопровождалась развитием локального оксидативного стресса с последующей активацией симпатической нервной системы и развитием АГ. Связано это с тем, что при гиперэкспрессии iNOS продукция NO возрастает в 2 раза больше, чем при гиперэкспрессии eNOS, что сопряжено с истощением субстрата L-аргинина и кофактора тетрагидробиоптерина. В результате этого происходит разобщение iNOS с продукцией не оксида азота, а СОР с образованием мощного оксиданта пероксинитрита [138, 139].
Данные значительного числа исследований свидетельствуют о том, что развитие оксидативного стресса в стволе мозга в результате усиленной продукции СОР и гидроксильных радикалов приводит к активации нейрогенных механизмов АГ и может быть одним из основных факторов ее развития [114, 211, 212]. Этому способствует также истощение локальной антиоксидантной защиты. Установлено, что АГ, развивающаяся при длительном введении низких доз А II, как и физиологические и патологические реакции, связанные с продукцией А II в центральной нервной системе, связаны с усиленной продукцией СОР в ядрах ствола мозга [234]. Способность А ΙΙ усиливать продукцию СОР в ткани мозга подтверждена также прямыми исследованиями, в которых установлено значительное возрастание флуоресценции в субфорникальном органе через 8 дней подкожного введения А ΙΙ, через 16 дней инфузии интенсивность флуоресценции удваивалась.
На этом основании локальная продукция СОР в центральной нервной системе и ее зависимость от активности локальной РАС и стимуляции АТ1-рецепторов рассматриваются как ведущий фактор в нейрогенной регуляции АД [116].
В исследовании, проведенном на мышах, длительная инфузия А ΙΙ в субпрессорной дозе вызывала постепенное развитие гипертензивной реакции, которая регистрировалась радиотелеметрически. Среднее АД возрастало через 24 ч от 105 до 128 мм рт. ст., а пик реакции (145 мм рт. ст.) отмечался на 5–6-й день инфузии. Селективная гиперэкспрессия СОД в ткани мозга с помощью аденовируса, как и локальное применение лозартана, предупреждали развитие гипертензивной реакции. В то же время, гиперэкспрессия СОД в ткани мозга не оказывала влияния на гипертрофию и ремоделирование сердца, которые развивались в данных условиях. Эти данные означали, что гипертрофия и ремоделирование сердца были преимущественно следствием прямого кардиотропного эффекта А ΙΙ, а не отражением его центрального действия.
Неоднократно отмечено, что в генезе АГ и кардиоваскулярной патологии ведущую роль играют как РАС и автономная нервная, так и иммунная системы, однако их роль в большинстве исследований оценивали раздельно. Только в последние годы было установлено, что А II и симпатическая нервная система могут модулировать иммунный статус, инициировать и активировать врожденный и адаптивный ответы иммунной системы и посредством этого участвовать в развитии и прогрессировании кардиоваскулярной патологии.
В настоящее время уже не вызывает сомнений, что способность А II вызывать развитие иммунного ответа относится к числу важнейших факторов, посредством которых РАС участвует в патогенезе АГ и повреждении органов-мишеней. Однако этот эффект имеет не прямой характер, а в значительной степени опосредованный участием центральной нервной системы. Так, в исследовании, проведенном на мышах, было продемонстрировано, что разрушение передней области третьего желудочка устраняло способность А ΙΙ не только активировать симпатическую систему и оказывать гипертензивное действие, но и приводить к сопутствующей активации циркулирующих Т-лимфоцитов и инфильтрации лейкоцитов в сосудистую стенку [172].
Эти данные означают, что способность А ΙΙ индуцировать оксидативный стресс и воспаление на периферии также имеют в значительной степени центральную природу и связаны с активацией симпатической нервной системы [156]. В то же время, разрушение центральных структур не устраняло АГ, лимфоцитарную активацию и лейкоцитарную инфильтрацию при периферическом действии норадреналина. Эти данные позволили сделать вывод, что прессорный эффект А ΙΙ центральной природы является также фактором, детерминирующим активацию Т-лимфоцитов и развитие сосудистого воспаления. Благодаря этому умеренное повышение АД при хроническом воздействии субпрессорных доз А ΙΙ приводит к активации Т-лимфоцитов с развитием воспаления и дальнейшим повышением АД до тяжелой АГ.
В ряде работ подтверждено, что гипертензивное действие А ΙΙ сочетается с его способностью увеличивать продукцию и плазменное содержание ИЛ-1β, -6, ФНО-α, МСР-1 и клеточных молекул адгезии. Более того, эта реакция в значительной степени определяет развитие АГ, мыши с генетическим отсутствием ИЛ-6 резистентны к развитию АГ, индуцированной А ΙΙ [18]. Причем в связи с этими факторами воспаление может играть первичную роль и определять активацию РАС с последующим действием А ΙΙ на центральные нейроны в субфорникальном органе мозга, инициацией и поддержанием симпатической гиперактивности [95].
Бесспорным является наличие прямой симпатической и парасимпатической регуляции активности лимфатических узлов и клеток иммунной системы, а также возможность иммуномодуляции агонистами холино-, адренорецепторов и АТ1-рецепторов. Тем не менее, концепция, связывающая долговременные эффекты автономной нервной системы, прежде всего — развитие АГ, с ее влиянием на иммунную систему, пока не приобрела достаточного признания [190, 242].
Наличие автономной регуляции иммунного ответа подтверждается тем, что топическая инфузия А ΙΙ в область третьего желудочка мозга сопровождалась развитием АГ с параллельной активацией как симпатической нервной системы, так и иммунного ответа и увеличением плазменного содержания ИЛ-1β, -2, -5, -16, TФР-β [114]. Активация вагуса оказывала угнетающее действие на иммунный статус, и через 4 нед после субдиафрагмальной вагусотомии в стенке толстой кишки, для которой характерно значительное накопление лимфоидной ткани, отмечено выраженное увеличение содержания ФНО-α [209]. В другом исследовании ваготомия сопровождалась достоверным возрастанием продукции провоспалительных цитокинов, включая ИФН-γ, ФНО-α и ИЛ-6 в Т-лимфоцитах селезенки мыши [136].
Третьим доказательством противовоспалительного действия парасимпатической нервной системы стала ее способность уменьшать тяжесть септического шока и сердечной недостаточности. У собак с сердечной недостаточностью, индуцированной частотной стимуляцией, активация вагуса сопровождалась увеличением фракции выброса, уменьшением размеров сердца и повышением барорефлекторной чувствительности параллельно с уменьшением плазменного содержания медиаторов и маркеров воспаления, прежде всего — ФНО-α и С-реактивного белка [311, 314].
Стимуляция вагуса у мышей также оказывала защитное действие при сепсисе, индуцированном липополисахаридом, и сочеталась с угнетением продукции ФНО-α. Этот эффект был обозначен как холинергический воспалительный рефлекс, основой которого является наличие отрицательной обратной регуляции высвобождения цитокинов. Предполагают, что вагусные афференты, широко представленные в большинстве внутренних органов, включая сердце и кишечник, активируются цитокинами, которые высвобождаются при локальном воспалении. Рефлекс медиируется центрально через ядро солитарного тракта и дорзальное двигательное ядро вагуса с активацией вагусных эфферентных волокон, угнетающих высвобождение цитокинов из макрофагов селезенки и кишечника [295–297].
В 2009 г. в мемориальной лекции, посвященной Walter B. Cannon, называвшейся «В поисках автономного баланса: хорошее, плохое и ужасное», F.M. Abboud рассматривал повышение активности парасимпатической нервной системы при интенсивном воспалительном процессе как защитный механизм, повышение симпатической активности — как отрицательный, повреждающий, а возрастание продукции кислородных радикалов в результате повышения активности РАС — как катастрофический. Если симпатическая активность превалирует над активностью парасимпатической, то вектор приобретает «смертельную» направленность, когда доминирует парасимпатическая активность, то образуется «вектор выживания» в результате угнетения воспалительной реакции [1, 2].
Аналогичная зависимость отмечена и в клинических условиях. Еще в 1984 г. сообщалось, что выживаемость пациентов с застойной сердечной недостаточностью в течение 2 лет была менее 10% при плазменном содержании норадреналина >800 пг/мл и >50% в течение 4 лет, если это содержание составляло <400 пг/мл. При этом возрастание вариабельности сердечного ритма высокой частоты, отражающее превалирование парасимпатических влияний, сочеталось с более благоприятным исходом, тогда как возрастание низкочастотного компонента, отражающего повышенную активность симпатической нервной системы, сочеталось с повышением летальности [155, 227].
Установлено также, что электрическая стимуляция вагуса у крыс с дилатационной кардиомиопатией сочеталась с 90% выживаемостью через 4 мес после перевязки коронарной артерии, тогда как в контроле выживаемость составляла только 50% [160]. Стимуляция вагуса при ишемии миокарда снижала активность ММРs, а блокада β-адренорецепторов в этих условиях угнетала апоптоз кардиомиоцитов [289].
Эти данные означают, что развитие воспаления при АГ и других видах кардиоваскулярной патологии может быть связано с потерей ингибиторного парасимпатического или усилением активирующего симпатического влияния на иммунный статус, а также с аномальной чувствительностью стимулирующих или тормозящих рецепторов на иммунных клетках.
Взаимосвязь между нервной и иммунной системами осуществляется как на центральном, так и периферическом уровнях. В центральной нервной системе циркулирующие цитокины и цитокины, которые высвобождаются локально астроцитами и клетками микроглии, активируют специфические центры в гипоталамусе и стволе мозга, приводя к симпатической активации и развитию АГ. Кроме того, миграция в центральную нервную систему клеток врожденного иммунного ответа типа макрофагов и моноцитов, а также Т-регуляторных или цитотоксических лимфоцитов может индуцировать локальный воспалительный ответ и аналогичную нейрональную активацию.
Второй уровень взаимодействия между автономной нервной и иммунной системами реализуется посредством экспрессии клетками врожденного иммунного ответа адренергических, холинергических и ангиотензиновых рецепторов. Их активация существенно изменяет характер иммунной реакции и высвобождения цитокинов при активации toll-like рецепторов (TLR) при ряде патологических состояний, включая АГ [106]. Эти изменения сочетаются с модуляцией активности цитотоксических Т-лимфоцитов, которые определяют поражение органов-мишеней: сердца, сосудов, почек [101, 103, 104].
Показано также существование в селезенке популяции Т-клеток, способных высвобождать ацетилхолин и являющихся компонентом периферического нейрогенного контроля врожденного иммунитета [229].
Связь между АГ и иммунной системой подтверждена в исследовании, проведенном на крысах со спонтанной АГ, у которых установлено значительное усиление врожденного иммунного ответа и увеличенное высвобождение цитокинов из спленоцитов под действием А II. Это способствует активации симпатической нервной системы посредством центрального действия циркулирующих цитокинов. В то же время, у нормальных крыс активация холинергических рецепторов оказывала угнетающее действие на клетки иммунной системы [106].
Миграция Т-лимфоцитов в почки, миокард и сосудистую стенку обусловливает развитие в них воспаления и патологических процессов, приводящих к ремоделированию. Эти данные подтверждают точку зрения, что АГ является в значительной степени иммунопатологическим процессом, который находится под тесным контролем автономной нервной системы. Высказано положение, что ИЛ-17 — цитотоксический цитокин, является основным специфическим повреждающим фактором в почечной и сосудистой ткани и высвобождается CD8+ Т-клетками, тогда как хелперные CD4+ Т-лимфоциты играют защитную роль [102, 103].
Ранее выявлено, что симпатические волокна плотно иннервируют лимфоидные ткани, а Т-лимфоциты интенсивно экспрессируют адренергические рецепторы. Поэтому повышение активности симпатических волокон, иннервирующих селезенку, при введении А ΙΙ в латеральный желудочек мозга сопровождалось увеличением продукции различных провоспалительных цитокинов в спленоцитах. Симпатическая денервация селезенки устраняла эти ответы, что позволило связать центральные эффекты А ΙΙ и последующую активацию симпатической нервной системы с активацией иммунного ответа и усиленной продукцией цитокинов [144].
И хотя в ряде других исследований разрушение передней области третьего желудочка также полностью устраняло иммунный ответ на центральное действие А ΙΙ, авторы не установили прямой связи между этими эффектами. Это суждение основано на том факте, что иммунный ответ в виде активации лимфоцитов и инфильтрации сосудистой стенки нейтрофилами при центральном действии А ΙΙ устранялся снижением АД гидралазином, хотя гидралазин не обладает способностью прямо угнетать активность иммунных клеток. Эти данные, по мнению авторов, позволили исключить участие симпатической активации в развитии иммунного ответа и свидетельствовали о том, что его непосредственной причиной является АГ [172].
2.1.5. РАС в скелетных мышцах, воспалительных клетках и жировой ткани
Существует достаточно подтверждений наличия РАС в скелетных мышцах, которые в различной степени экспрессируют ее компоненты, включая ангиотензиноген, АПФ, А I, А II и его рецепторы. Не определено в скелетных мышцах экспрессии ренина, но рениноподобный фермент катепсин D выявлен в супернатанте культуры пролиферирующих миобластов. Механический стресс в виде растяжения мышцы индуцировал усиление продукции ангиотензиногена, А I и А II [130].
Показана существенная роль тканевой РАС в скелетных мышцах как в обеспечении адекватного их кровоснабжения и доставки энергетических субстратов, прежде всего — глюкозы, так и поддержании центральной гемодинамики в условиях повышенной физической активности. В этом процессе существенную роль играет АПФ — основной фермент, ответственный за деградацию брадикинина в скелетных мышцах при повышении их функциональной активности. Установлено, что снижение экспрессии АПФ при выполнении физической работы лежит в основе взаимодействия РАС и тканевой калликреин-кининовой системы в многофакторной регуляции биоэнергетики скелетных мышц, поддержании гомеостаза АД и глюкозы.
Сокращающиеся мышцы высвобождают гуморальные факторы типа аденозина и кининов, которые обеспечивают расслабление сосудистой стенки с адекватным увеличением кровотока и доставки глюкозы. Кинины активируют еNOS, ЦОГ-2, цитохром Р450-зависимую монооксигеназу, приводя к развитию вазодилатации и, помимо этого, усиливают транслокацию переносчика глюкозы GLUT4 на клеточную мембрану. Регуляция активности РАС скелетных мышц, локализованной в нутритивных сосудах и в паренхиме миофибрилл, в значительной степени связана с участием симпатической нервной системы.
Неоднократно указывалось на существование единых патогенетических механизмов инсулинорезистентности, АГ и атеросклероза [46, 278]. Полагают, что к числу этих механизмов относится гиперактивность РАС в скелетных мышцах, что приводит к нарушению гомеостаза глюкозы, АД, способствует развитию локальных структурных нарушений в сосудистой стенке.
При физических нагрузках снижается активность АПФ в скелетных мышцах, уменьшаются образование А II и деградация кининов, что способствует увеличению кровоснабжения мышц, захвата и утилизации ими глюкозы. Однако эти реакции при избыточной выраженности могут приводить к перераспределению потока крови к скелетным мышцам, снижению АД и развитию системной гипогликемии. Сохранение гомеостаза организма и поддержание центральной гемодинамики в этих условиях обеспечивается симпатической нервной системой, которая способствует повышению активности сердца, увеличению его производительности, усиливает высвобождение ренина почками, а в скелетных мышцах функционирует сочетанно с РАС и способствует повышению ее активности.
Этот эффект достигается посредством, прежде всего, локальной усиленной продукции А II непосредственно в скелетных мышцах, что противодействует опасному для организма снижению АД, ограничивает экспрессию переносчика глюкозы GLUT4 и захват глюкозы. Помимо этого, повышение концентрации ренина в плазме крови сопровождается усиленным высвобождением антидиуретического гормона и альдостерона с увеличением объема циркулирующей крови. Однако этот механизм ограничивается по принципу отрицательной обратной связи действием А (1–7) — продукта деградации А II, который активирует высвобождение NO и простагландинов, усиливает в результате вазодилатацию и доставку энергетических субстратов [56].
Установлено, что для лиц с факторами риска типа избыточной массы тела и ожирения характерна гиперактивность РАС как циркулирующей, так и локализованной в скелетных мышцах, и она сочетается с выраженными гипертензивными реакциями, сниженным захватом глюкозы. Если эти нарушения сохраняются длительное время, то компенсаторные реакции на избыточную активность АПФ типа гиперсекреции инсулина и повышения растяжимости сосудистой стенки неизбежно истощаются, приводя к развитию АГ и СД 2-го типа. Этот механизм в значительной степени определяет эффективность применения ингибиторов АПФ и блокаторов рецепторов А II в лечении АГ и инсулинорезистентности.
При выполнении физической работы мышечный кровоток может возрастать от 1 до 20 л/мин, потребление глюкозы мышцами – от 1 до 20 г/ч, что составляет около 80% всей продукции глюкозы печенью. Этот эффект достигается благодаря активации симпатической нервной системы, что способствует увеличению сердечного выброса, уменьшению диаметра сосудов за пределами работающих мышц, ускорению глюконеогенеза и гликогенолиза, повышению скорости липолиза. При этом значительно ограничивается потребление глюкозы в неработающих мышцах за счет снижения экспрессии переносчика глюкозы GLUT4, и в хроническом состоянии это является предпосылкой для развития инсулинорезистентности скелетных мышц [56].
Недостаточность функции РАС в скелетных мышах приводит не только к нарушению их кислородного и энергетического обеспечения при физических нагрузках, но и контроля в этих условиях центральной гемодинамики. Так как масса скелетных мышц превосходит суммарную массу всех остальных тканей, то значительное увеличение ее кровоснабжения может приводить к критическому снижению АД, и этот эффект предупреждается активацией симпатической нервной системы и сопряженной с ней РАС в скелетных мышцах.
В нормальных условиях благодаря активации АПФ осуществляется деградация брадикинина и поддерживается баланс между обеспечением адекватного кровоснабжения скелетных мышц и возможностями центральной гемодинамики. Однако при гиперактивности АПФ, характерной для лиц с факторами риска, усиливается компенсаторный прирост тонических влияний на сосуды и ограничивается потребление мышцами кислорода и глюкозы. Если эта гиперактивность имеет хронический характер, то она приводит к развитию компенсаторной АГ и гипергликемии. Напротив, при сниженной активности РАС в скелетных мышцах повышение их активности может сочетаться с нарушениями центральной гемодинамики и снижением АД.
Адаптация центральной гемодинамики к условиям физической нагрузки включает также активацию циркулирующей РАС с увеличением плазменного содержания ренина и последующим высвобождением антидиуретического гормона из гипофиза и альдостерона из надпочечников, что приводит к усиленной реабсорбции натрия и воды и увеличению объема циркулирующей крови.
Наличие мРНК ренина, как и рениноподобной иммунореактивности, установлено также в циркулирующих и резидентных воспалительных клетках, в перитонеальных макрофагах крыс. В другом исследовании выявлено наличие белка ренина как в циркулирующих, так и в альвеолярных моноцитах и макрофагах крыс и мышей. Это свидетельствует о том, что активированные в ряде патологических ситуаций макрофаги могут становиться источником ренина [53, 126]. Наличие А I, еще более выраженное — А II, а также АТ1-рецепторов отмечено также в мононуклеарных лейкоцитах человека [141].
Установлено, что нейтрофилы человека способны трансформировать А I в А II с помощью лизосомальной сериновой протеазы катепсина G. Более того, катепсин G способен продуцировать А II непосредственно из ангиотензиногена без участия ренина и АПФ, минуя этап А I.
Наличие АПФ установлено в моноцитах, А II и его рецепторов — в нейтрофилах у людей, а макрофаги и моноциты мышей и крыс содержат А I и А II. Выявлено, что А II индуцирует высвобождение из эндотелиоцитов хемоаттрактанта для нейтрофилов, что способствует локальной регуляции воспалительного ответа.
Лейкоциты экспрессируют на мембране АТ1-рецепторы, что обусловливает их участие в развитии оксидативного стресса у больных АГ [61], тогда как эпросартан устраняет этот эффект [149, 150].
Экспрессия компонентов РАС установлена также как в белой жировой ткани, где они участвуют в развитии инсулинорезистентности, так и в бурой, прилежащей к адвентиции сосудов, благодаря чему они способны взаимодействовать с сосудистой РАС. Наличие ренина отмечено в жировой ткани лиц с ожирением; ренин локализовался преимущественно в адипоцитах бурой жировой ткани, его содержание не уменьшалось после двусторонней нефрэктомии, что свидетельствовало о локальной продукции [255].
Наличие ренина и всех других компонентов РАС установлено в культуре преадипоцитов и адипоцитов человека, в адипоцитах подкожной и экстраперитонеальной жировой ткани, в адиапоцитах периартериальной и периаортальной бурой жировой ткани [27, 28]. В жировой ткани лиц с ожирением, особенно в адипоцитах, установлена также экспрессия АПФ. Рецепторы А II генерализованно экспрессированы в белой, бурой, подкожной, ретроперитонеальной и мезентериальной жировой ткани, в клетках сальника.
Основной функцией РАС в жировой ткани является регуляция термогенеза, снижение температуры тела сопровождается локальной продукцией А II с повышением активности симпатической нервной системы и активацией двух основных липогенных ферментов: синтазы жирных кислот и глицерол-3-фосфатдегидрогеназы с увеличением содержания триглицеридов. Это приводит к увеличению термопродукции без повышения концентрации циркулирующего А II и без развития побочных реакций [27].
РАС принимает участие в развитии ожирения, установлено, что масса тела у людей положительно коррелирует с содержанием ангиотензиногена в жировой ткани [79–81, 291]. Показано, что у мышей с отсутствием АТ1-рецепторов существенно замедлено развитие ожирения при высоколипидной диете и значительно увеличенных энергозатратах.
Наличие и функциональная значимость РАС в жировой ткани в последнее время привлекают особое внимание [214]. А II, образующийся в жировой ткани, регулирует рост, дифференциацию и метаболизм адипоцитов, а усиленная локальная продукция А II, установленная у гипертензивных пациентов с избыточной массой тела [17], рассматривается как связующее звено между АГ и ожирением [82, 83, 91].
Исследованиями последних лет установлено, что адипоциты относятся к числу важнейших источников провоспалительных медиаторов, а основным регулятором этого процесса является А II, синтезируемый в жировой ткани. В условиях ожирения А II, продуцируемый локально, в 3–4 раза усиливал продукцию в адипоцитах провоспалительных медиаторов, прежде всего ИЛ-6 и -8, приводя к развитию как локального, так и системного воспаления. В изолированных адипоцитах действие А II в концентрации 1•10-5–1•10-6 М сопровождалось увеличением продукции ИЛ-6 и -8 в 3–4 раза, этот эффект полностью блокировался ингибитором NF-κB и кандесартаном [266].
Помимо этого, жировая ткань является важнейшим продуцентом ангиотензиногена, также относящегося к числу белков острой фазы. В ряде исследований установлено наличие четкой корреляции между ИМТ и уровнем ангиотензиногена в плазме крови, что свидетельствует о прямой связи между ожирением и АГ. Отмечено, что у мышей, содержащихся на высокожировой диете, экспрессия гена ангиотензиногена в висцеральных депо повышалась в 6–7 раз в ткани сальника, в 4–5 раз — в околопочечной жировой ткани, но не изменялась в подкожной. Уровень ангиотензиногена в крови в этих условиях повышался параллельно с возрастанием АД, а блокада АТ1-рецепторов предупреждала развитие как ожирения, так и АГ [223].
Активация РАС в жировой ткани сопряжена также с резким увеличением локальной продукции альдостерона [230–233], что имеет особое значение в развитии инсулинорезистентности при ожирении. В исследовании на мышах генетических линий ob/ob и db/db с врожденным ожирением, связанным с отсутствием продукции лептина или его рецепторов, применение в течение 3 нед эплеренона — блокатора минералокортикоидных рецепторов, сопровождалось угнетением инфильтрации макрофагов в жировую ткань, образования в ней СОР и, в результате, значительным уменьшением выраженности воспаления и инсулинорезистентности [112]. В изолированных адипоцитах альдостерон приводил к усиленной секреции СОР, что является ключевым механизмом образования цитокинов в жировой ткани, развития в ней воспаления и инсулинорезистентности [78].
Адипоциты способны высвобождать различные факторы, включая компоненты РАС, блокаторы АТ1-рецепторов при ожирении оказывают защитное действие, предупреждают развитие АГ [17], повышение кардиоваскулярного риска [107] и повышают выживаемость [128].
Помимо этого, адипоциты продуцируют факторы, прямо стимулирующие секрецию альдостерона адренокортикальными клетками с последующим повреждением органов-мишеней. При действии на культуру гладкомышечных клеток среды, в которой культивировались дифференцированные адипоциты, установлены фосфорилирование и активация ряда протеинкиназ, включая митогенактивирующую киназу, с повышением экспрессии провоспалительных и пролиферативных маркеров и медиаторов. Эплеренон — блокатор минералокортикоидных рецепторов, и кандесартан — блокатор АТ1-рецепторов, угнетали эти эффекты [29].
Установлено, что мРНК ангиотензиногена, АІ и А ІІ экспрессируются не так интенсивно непосредственно в сосудистой стенке, как в периваскулярной и интраперикардиальной жировой ткани. Предполагают, что ангиотензиноген может секретироваться в ней и затем диффундировать в сосудистую стенку и миокард, где он вступает во взаимодействие с ренином.
В последние годы отмечается, что развитие атеросклероза не ограничивается интимой сосуда, а в нем принимают активное участие также клетки адвентиции [59, 170], в частности — периваскулярные адипоциты, которые могут оказывать паракринное влияние на сосудистую стенку. Выявлено, что при развитии атеросклероза происходит выраженное увеличение массы жировых клеток в периаортальной жировой ткани с резко усиленной продукцией хемокинов и цитокинов, что может быть причиной инициации воспаления в стенке аорты [88, 110]. При содержании мышей на жировой диете в течение 2 нед установлено значительное угнетение экспрессии в периаортальной и периваскулярной жировой ткани адипонектина, PPARγ и значительное повышение экспрессии лептина и провоспалительного белка MIP-1α. Отмечено также повышение экспрессии CD3, но не CD68, что свидетельствовало об усиленной инфильтрации Т-клеток, но не макрофагов.
К особенностям периваскулярной жировой ткани относятся значительно уменьшенная продукция адипонектина и значительно увеличенная — ИЛ-1, -6, МСР-1. При содержании на жировой диете выраженность этих особенностей периваскулярной жировой ткани увеличивается даже в отсутствие усиленного рекрутирования макрофагов [31].
Среди 1205 пациентов из когорты лиц, исследованных в рамках Framingham Heart Study Offspring, было выявлено, что лица с избыточной массой периаортальной жировой ткани имеют повышенный риск развития периферического сосудистого поражения и сниженный коленно-плечевой индекс. В построенной регрессионной модели увеличение массы периаортальной жировой ткани на 1 SD сочеталось с повышением риска до 1,52, этот показатель повышался в еще большей степени после учета ИМТ (1,69) и висцеральной абдоминальной жировой ткани (1,67). При этом не отмечено никакой связи общего или абдоминального ожирения с риском развития периферического сосудистого поражения [74].
Данные экспериментальных исследований свидетельствуют о том, что периваскулярная жировая ткань способна высвобождать провоспалительные и ростовые факторы, обусловливающие пролиферацию гладкомышечных клеток и развитие воспаления непосредственно в сосудистой стенке [13, 31].
При избыточной гипертрофии адипоциты периваскулярной жировой ткани теряют способность секретировать адипонектин и утрачивают противовоспалительное действие [93].
Практически все артерии, включая аорту, окружены значительным количеством жировой ткани, которая способна высвобождать провоспалительные цитокины. Эти цитокины диффундируют в сосудистую стенку, приводя к инфильтрации макрофагов, развитию локального воспаления и разрушению внеклеточного матрикса. Выявлено, что ожирение способствует резкому увеличению массы периаортальной жировой ткани, секретирующей большое количество А II и МСР-1 с миграцией макрофагов в стенку аорты и развитием аневризмы.
По функциональным характеристиками периаортальная жировая ткань является бурой, экспрессирующей разобщающий белок-1 (UCP-1), и характеризуется меньшим размером адипоцитов. В жировой ткани, как периваскулярной, так и мезентериальной, установлено наличие всех компонентов РАС за исключением ренина с примерно равной экспрессией ангиотензиногена, АПФ и АПФ2, тогда как экспрессия рецепторов (про)ренина в периваскулярной жировой ткани была в 5 раз выше, а экспрессия химазы, АТ1- и AT2-рецепторов — достоверно ниже, чем в белой жировой ткани. В культуре адипоцитов бурой жировой ткани установлена значительно более выраженная экспрессия ангиотензиногена, и она повышалась под действием СЖК, глюкокортикоидов, ФНО-α.
Отличительной особенностью РАС в периваскулярной жировой ткани является определяющая роль химазы в продукции А ΙΙ, и потому применение ингибиторов АПФ существенно не угнетало его локальное образование. В последнее время в жировой ткани, особенно в бурой периаортальной, установлено также наличие мРНК рецепторов (про)ренина [3, 36–41].
Популяционные исследования свидетельствуют о том, что риск развития аневризмы брюшной аорты четко коррелирует с ИМТ [86, 87]. При содержании мышей на жировой диете в течение 4 мес и последующей инфузии А II в течение 28 дней частота развития аневризмы аорты составила 18% через 1, 36% — через 2 и 60% — через 4 мес. Инфузия А II мышам с генетическим ожирением повышала частоту развития аневризмы от 32% в контроле до 76% параллельно с усилением миграции макрофагов в периаортальную жировую ткань.
При длительной инфузии A II развитие аневризмы брюшной аорты у мышей было отмечено более чем в 80% случаев [28] параллельно с накоплением макрофагов в зоне поражения [238]. При этом риск развития аневризмы коррелиpовал с ИМТ, но не с инсулинорезистентностью. Это свидетельствовало о его зависимости от интенсивности воспаления, а не метаболических нарушений, и даже 10-кратное повышение уровня холестерина при содержании на атерогенной диете дополнительно не потенцировало этот эффект (рис. 5).
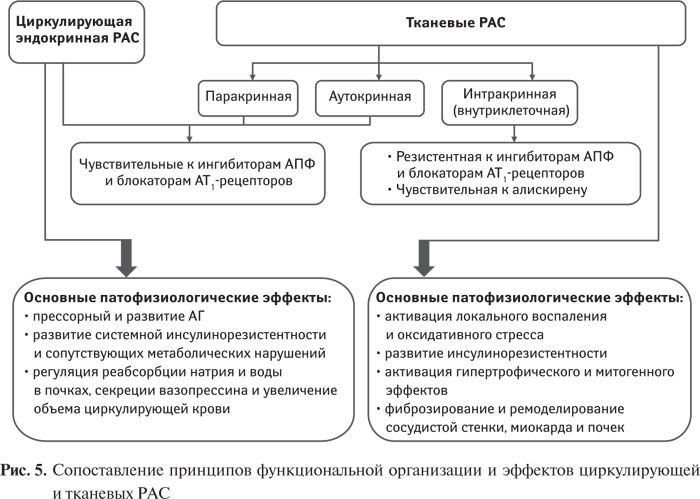
2.1.6. Функциональная значимость интракринной РАС
Практически до настоящего времени представления о локальных РАС ограничивались возможностью локализации их компонентов на плазматической мембране. Однако в последние годы появились сведения о существовании внутриклеточной РАС, о возможности внутриклеточного происхождения и внутриклеточных, так называемых интракринных эффектах А II, не связанных с активацией мембранных АТ1-рецепторов.
Наличие внутриклеточной, интракринной РАС с внутриклеточным синтезом А II, эффекты которого резистентны к действию блокаторов его рецепторов [261, 262], установлено в кардиомиоцитах, сосудистых гладкомышечных клетках, фибробластах, почечных мезангиальных клетках [146, 147]. Установлено, что внутриклеточное содержание А II в кардиомиоцитах у лиц с СД 2-го типа увеличено в 3,4 раза и еще в 2 раза — при сочетании СД с АГ [36–41, 239, 240], тогда как интернализация (про)ренина может сопровождаться развитием клеточных реакций даже в отсутствие А II в среде [76].
Помимо этого, прямое введение А II в гладкомышечные клетки и в кардиомиоциты сочеталось с увеличением цитозольной концентрации кальция, сокращением и гипертрофией клеток, что свидетельствовало о наличии внутриклеточных рецепторов А ΙΙ. Эти эффекты были практически нечувствительными к современным ингибиторам РАС в результате, по-видимому, их неспособности проходить через клеточную мембрану.
Исследования в этой области имеют еще только ориентировочный характер, однако уже не вызывает сомнений, что, помимо традиционного рецепторопосредованного действия, А II в комплексе с рецептором может поглощаться клетками и поступать во внутриклеточные компартменты. Однако пока остается невыясненным, в какой степени этот поглощенный А II может оказывать интракринное действие, активируя цитоплазматические или ядерные рецепторы.
Ранее уже неоднократно установлена способность кардиомиоцитов секретировать А II при действии механического стресса, однако это рассматривалось как следствие высвобождения А II, предварительно захваченного и депонированного в комплексе с рецепторами. Так, отмечено, что через 60 мин после введения А II в плазму крови он накапливается в сердце, почках и надпочечниках. Однако захват внеклеточного А II предупреждается применением блокаторов АТ1-рецепторов, которые не оказывают эффекта на внутриклеточный синтез А II.
Результаты ряда недавних исследований подтверждают положение о том, что А II может функционировать в качестве интракринного пептида как интернализированного из крови, так и синтезированного внутриклеточно [50, 51, 71, 152, 153, 224].
Возможность захвата циркулирующего меченого А Ι показана по его наличию в ядрах гладкомышечных клеток и кардиомиоцитов при системном введении. Однако в последние годы на культуре кардиомиоцитов установлено, что синтез А II может осуществляться внутриклеточно и вызывать как ауто-, так и интракринные эффекты. Выявлено, что микроинъекция А II в сосудистые гладкомышечные клетки крыс приводила к увеличению содержания свободного внутриклеточного кальция, и этот эффект устранялся внутриклеточным введением блокаторов АТ1-рецепторов [98, 99], что свидетельствовало о вовлечении внутриклеточных рецепторов А II в реализацию его аутокриного действия.
Характерно, что внутриклеточный синтез А ΙΙ не связан с активацией АПФ; он был опосредован значительным повышением внутриклеточной активности химазы и предупреждается путем применения алискирена, тогда как ингибитор АПФ беназеприл не оказывал заметного эффекта [259–261].
Клиническое значение внутриклеточной РАС в кардиомиоцитах особенно значимо в том отношении, что как блокаторы АТ1-рецепторов, так и ингибиторы АПФ способны блокировать только циркулирующую и аутопаракринную РАС, но практически не оказывают влияния на интракринную. Гипертрофия кардиомиоцитов при внутриклеточном введении А II не устранялась блокатором АТ1-рецепторов лозартаном [10, 11]. Кроме того, внутриклеточное образование А II связано с преимущественным действием химазы, ингибиторы АПФ при этом также неэффективны, перспективными в отношении блокады внутриклеточной РАС являются ингибиторы ренина типа алискирена [267, 268].
Внутриклеточный синтез А II и его интракринный эффект особенно выражены при СД. В условиях гипергликемии в кардиомиоцитах отмечаются отчетливо повышенная активность химазы и ренина, увеличенное содержание ангиотензиногена и образование А II. В связи с этим антигипертензивная терапия с применением лозартана и ингибиторов АПФ у гипертензивных пациентов с СД сочеталась со значительно меньшей регрессией гипертрофии сердца, большей летальностью, чем у лиц с изолированной АГ [10, 11].
В соответствии с данными ряда исследований последних лет, глюкоза в высокой концентрации является мощным стимулом для внутриклеточного синтеза А II. При экспериментальном воспроизведении СД отмечено увеличение содержания А II в изолированных кардиомиоцитах в 9,4 раза параллельно с увеличением содержания ангиотензиногена и ренина, несмотря на отсутствие изменений уровня циркулирующего А II. Нормализация содержания глюкозы в крови с помощью инсулина, как и применение алискирена, полностью устраняли повышение внутриклеточного содержания А II. В то же время, применение кандесартана только частично уменьшало содержание А II в кардиомиоцитах, что подтверждало внутриклеточную природу его образования.
Так как синтез A II в условиях гипергликемии обеспечивается преимущественно химазой, а не АПФ [35, 145, 154, 261, 262], то применение ингибиторов АПФ у пациентов с СД не устраняет увеличенного содержания А II в кардиомиоцитах [76].
Отмечены существенные отличия в эффектах вне- и внутриклеточного А II. Эти отличия в значительной степени связаны с тем, что внеклеточный А II приводит к активации мембранных рецепторов с усилением трансмембранного транспорта кальция в клетку и высвобождением кальция из внутриклеточных депо, тогда как эффекты внутриклеточного А II связаны только с мобилизацией внеклеточного кальция и потому более чувствительны к блокаторам медленных кальциевых каналов. Другим принципиальным отличием внутриклеточной РАС является низкая чувствительность к применяемым ингибиторам АПФ и блокаторам АТ1-рецепторов в связи с резко ограниченным их проникновением в цитозоль клетки через мембрану.
Наличие эффекта внутриклеточного А II означает наличие и внутриклеточных его рецепторов. Ими могут быть как интернализированные мембранные рецепторы, прежде всего — АТ1-рецепторы, так и рецепторы, синтезированные de novo внутриклеточно.
Эти данные позволили существенно уточнить понятие «локальной РАС» как независимой от «циркулирующей РАС» и разделить его на «интерстициальную РАС», локализованную на плазматических клеточных мембранах, и «внутриклеточную РАС» [71].
2.2. Основные механизмы регуляции активности тканевых РАС и их повреждающего действия
Как уже упоминалось, в нормальных условиях покоя активность РАС как циркулирующей, так и тканевых незначительна, и А II, АПФ, альдостерон находятся в циркуляции и продуцируются в миокарде, почках, жировой ткани и сосудистой стенке только в следовых количествах [60, 161, 192]. В то же время, эта продукция существенно увеличивается при АГ, сердечной недостаточности, СД 2-го типа и других патологических состояниях [180, 306, 307].
Одним из важнейших факторов регуляции активности тканевых РАС является характер солевой диеты, показано, что компоненты РАС в сердце и сосудистой стенке, в отличие от циркулирующей РАС, активируются при избыточном потреблении соли. Помимо этого, высокосолевая диета индуцирует и синтез альдостерона в сердце у крыс последующим развитием его гипертрофии [280]. Напротив, уменьшение содержания натрия в диете, как и применение диуретиков, стимулирующих его экскрецию, способно угнетать тканевые РАС, однако в сочетании с одновременным повышением активности циркулирующей РАC. Параллельно усиливается экспрессия в миокарде минералокортикоидных рецепторов, что в комплексе способствует развитию кардиофиброза и сердечной недостаточности [256].
Эти данные означают, что тканевые РАС могут быть активированы даже в отсутствие активации циркулирующей РАС и, более того, на фоне снижения ее активности [308].
Низкосолевая диета приобрела в настоящее время значение ключевой стратегии в лечении пациентов с АГ, так как способность альдостерона вызывать гипертрофию миокарда устраняется при низком содержании натрия [308]. Аналогичный эффект, как уже упоминалось, оказывают диуретики, способствующие усиленной экскреции натрия и уменьшению его содержания как в крови, так и в тканях.
Блокаторы АТ1-рецепторов эффективны в высокоренинном состоянии, то есть, при активации циркулирующей РАС, тиазидные диуретики — в низкоренинном, тогда как сочетанное их применение оказывает аддитивное действие. Определяется это тем, что тиазидные диуретики на фоне высокого потребления соли снижают АД посредством усиленной экскреции натрия и снижения активности тканевых РАС, но с одновременным повышением активности циркулирующей РАС, тогда как блокаторы АТ1-рецепторов угнетают активность как тканевых, так и циркулирующей РАС. Потому сочетание свойств блокаторов АТ1-рецепторов и тиазидных диуретиков при одновременном применении способствует синергичному угнетению тканевых и циркулирующей РАС.
Результаты клинических наблюдений свидетельствуют о том, что около 50% всех гипертензивных лиц являются солечувствительными. АГ у них традиционно рассматривается как «объемзависимая», при которой роль РАС и А ІІ считается незначительной в связи с низким содержанием ренина в плазме крови.
Однако исследования, проведенные на солечувствительных крысах линии DS, свидетельствовали о том, что при содержании на солевой диете у них происходила активация тканевых РАС, прежде всего — сосудистой и почечной, с усиленной локальной продукцией А II, развитием АГ и повышением среднего АД до 168 мм рт. ст. Это сочеталось с нарушением эндотелийзависимого расслабления и увеличением продукции СОР в аорте на фоне низкой активности ренина в плазме крови и низкой плазменной концентрации А II [142, 199, 200]. При блокаде NADPH-оксидазы уменьшалась продукции СОР и нормализовалась функция эндотелия, несмотря на недостоверное снижение системного АД (до 160 мм рт. ст.) [317].
Результаты исследований последних лет свидетельствуют о том, что повышение АД не является ведущей причиной поражения органов-мишеней при АГ, ключевую роль в этом процессе играет усиленная локальная продукция А II с развитием воспаления и иммунного ответа [100, 277]. Выявлено, что отсутствие Т-клеток почти полностью устраняло гипертензивный ответ на инфузию А II, гипертрофию и склерозирование миокарда, тогда как последующее восстановление популяции Т-клеток сочеталось с развитием этих эффектов. Помимо этого, применение у мышей иммуносупрессорных Т-регуляторных клеток (Treg) на фоне инфузии А II значительно снижало инфильтрацию миокарда макрофагами, CD4+ и CD8+ Т-клетками. Несмотря на сохраняющуюся АГ, существенно уменьшались выраженность гипертрофии и фиброза миокарда, а также нарушения его функциональных и электрофизиологических свойств [96].
Значимость локального воспаления в органах мишенях как ведущего фактора их повреждения при активации РАС показана в экспериментах на мышах с гиперэкспрессией генов ренина и ангиотензиногена. У них отмечено развитие гипертрофии и склерозирования сердца, нефросклероза на фоне резкой активации воспаления, тогда как блокада РАС предупреждала эти нарушения, независимо от уровня АД [252]. В другом исследовании длительное применение А II у мышей сочеталось с инфильтрацией Т-клеток в адвентицию сосудистой стенки и периваскулярную жировую ткань, повышением концентрации в крови маркеров их активации на фоне увеличения продукции Т-клетками ФНО-α, эти эффекты не развивались у животных с дефицитом NAD(P)H-оксидазы. Этанерцепт — блокатор рецепторов ФНО-α, также предупреждал увеличение продукции СОР, ремоделирование сосудистой стенки и сердца, склерозирование почек, вызванные A II, и в меньшей степени — развитие АГ.
Способность РАС индуцировать воспалительный и иммунный ответы в значительной степени связана с экспрессией рецепторов А ІІ на Т-лимфоцитах, макрофагах и дендритных клетках. Однако длительная инфузия А II мышам сопровождалась увеличением количества и активацией Т-клеток как за счет прямого рецепторопосредованного действия, так и через NF-κB — главный фактор транскрипции, который медиирует воспаление и врожденный иммунный ответ. Это положение подтверждалось тем, что блокада NF-κB уменьшала выраженность гипертрофии миокарда и альбуминурии у мышей с гиперэкспрессией генов ренина и ангиотензиногена.
В ранее проведенном исследовании применение ацетилсалициловой кислоты на фоне инфузии А ІІ также угнетало как экспрессию NF-κB, так и гипертрофию миокарда, хотя АД сохранялось на уровне, превышающем 170 мм рт. ст. [186].
Ведущая роль активации Т-клеток в повреждении органов-мишеней, вызванном А II, была подтверждена результатами применения иммуносупрессивной терапии. Так, длительная инфузия А II у мышей сочеталась с увеличением количества циркулирующих Т-клеток, интенсивности инфильтрации миокарда макрофагами, CD4+ и CD8+ T-клетками, экспрессии ФНО-α, выраженность этих изменений значительно уменьшалась после трансфера Т-регуляторных клеток. Масса сердца увеличилась после инфузии А ΙΙ с 3,36 до 4,57 мг/кг массы тела, тогда как применение Treg-клеток приводило к уменьшению ее прироста до 3,87 мг/кг. Толщина межжелудочковой перегородки возросла соответственно с 1,57 до 1,96 мм в контроле и до 1,73 мм — при сопутствующем применении Treg-клеток. Выраженность фиброза миокарда увеличилась после инфузии А ΙΙ практически вдвое и уменьшилась ниже контрольного значения при проведении иммуносупрессивной терапии. В то же время, АД повысилось после инфузии А ІІ со 110 до 151 мм рт. ст. и сохранялось на этом уровне после применения Treg-клеток. Эти данные свидетельствовали о том, что как гипертрофия, так и склерозирование миокарда были прямым следствием активации кардиальной РАС, но не определялись повышением АД [148], а участие иммунной системы при повышении активности РАС обусловливает главным образом повреждение органов-мишеней и в значительно меньшей степени — развитие гипертензивного ответа [294].
В многочисленных клинических исследованиях установлено частое сочетание СД 2-го типа с АГ и резко выраженным повышением активности тканевых РАС. В условиях эксперимента неоднократно установлено, что одним из важнейших факторов, определяющих наличие этой зависимости, является хроническая гиперинсулинемия [117]. Так, при подкожной инфузии инсулина крысам в течение 6 нед его уровень в крови повысился в 2–2,5 раза без изменений уровня глюкозы, систолическое АД — со 134 до 157 мм рт. ст., частота сердечных сокращений — с 380 до 423 уд./ мин, значительно увеличилось плазменное содержание норадреналина. Сочетанное с инфузией применение фозиноприла или лозартана предупреждало повышение систолического АД и частоты сердечных сокращений, практически нормализовало содержание норадреналина в плазме крови [66]. Эти данные свидетельствовали о том, что гиперинсулинемия приводит к активации РАС, усиленной продукции А ІІ с последующим развитием АГ как непосредственно, так и через активацию симпатической нервной системы.
Одним из важнейших факторов, регулирующих активность РАС, является витамин D. Полагают, что он действует как ингибитор синтеза ренина, активация рецепторов витамина D снижала активность РАС, уменьшала выраженность гипертрофии миокарда, угнетала воспаление и иммунный ответ. Установлено, что рецепторы витамина D экспрессируются всеми клетками иммунной системы, особенно моноцитами и макрофагами, дефицит этой экспрессии сочетается с повышением риска развития аутоиммунных заболеваний типа множественного склероза и СД 1-го типа.
Результаты экспериментальных исследований свидетельствуют о наличии связи между витамином D, механизмами развития АГ и поражения органов-мишеней. Показано, что применение витамина D при АГ стимулировало усиленную секрецию противовоспалительного цитокина ИЛ-10 Т-клетками, сопровождалось уменьшением объема воспалительного инфильтрата и интенсивности фиброза миокарда, несмотря на сохраняющуюся АГ. У мышей с генетическим отсутствием рецепторов витамина D отмечено значительное повышение плазменного уровня ренина и А II с развитием АГ и гипертрофии ЛЖ [186].
Установлено, что витамин D является не только регулятором кальциевого обмена, его рецепторы экспрессируются во многих тканях. Отмечено, что дефицит витамина D сопряжен с аутоиммунной и кардиоваскулярной патологией [193], доказано наличие обратной связи между плазменной концентрацией витамина D и уровнем АД у людей, независимой от плазменного уровня Ca2+ и паратиреоидного гормона. По данным Фремингемского исследования, риск развития кардиоваскулярных явлений на протяжении 6 лет удваивался у лиц с низкой концентрацией витамина D [295–297], тогда как его применение у лиц с СД 2-го типа и низким его уровнем в крови способствовало нормализации функции эндотелия и снижению АД [271].
Для крыс с дефицитом витамина D уже в раннем возрасте характерно развитие АГ, сочетающейся с нарушением продукции в стенке артерий двух дилататорных факторов — NO и эндотелийзависимого фактора гиперполяризации, а также снижением чувствительности сосудистых гладкомышечных клеток к оксиду азота. При снижении концентрации витамина D в крови примерно в 10 раз уровень АД был повышен на 11–20 мм рт. ст., частота сокращений сердца была увеличена на 20–40 уд./мин при неизмененной плазменной концентрации кальция. Миогенный тонус мезентериальной артерии был повышен примерно вдвое, тогда как эндотелийзависимое расслабление было уменьшено в 2 раза, а дилатация, связанная с действием эндотелийзависимого фактора гиперполяризации, полностью отсутствовала на фоне выраженного угнетения эндотелийнезависимого расслабления, связанного с действием нитроглицерина [281].
Существенное значение в модуляции как гипертензивного, так и митогенного действия А II имеет сосудистый эндотелий. Повышение активности eNOS с увеличением синтеза NO при активации AT2-рецепторов существенно ограничивает констрикторный эффект А II, а нарушение функции эндотелия потенцирует его прессорное действие. Кроме того, на фоне нормальной функции эндотелия применение А II только умеренно увеличивало экспрессию PDGF в сосудистой стенке, тогда как в отсутствие эндотелия или при резком нарушении его функционального состояния эта экспрессия возрастала в 10 раз. В результате толщина интимы увеличивалась при сохраненном эндотелии только на 25%, тогда как в деэндотелизированном сосуде — на 200%. Поэтому повреждение эндотелия при хронических воспалительных процессах, действии окисленных ЛПНП способствует резкому усилению эффекта А II, приобретающего патологическую направленность.
2.2.1. Связь между симпатической нервной системой и РАС
РАС и симпатическая нервная система — две основных системы регуляции кровообращения, для их функционирования характерно наличие взаимных влияний. О существовании тесной связи между активностью симпатической нервной системы и РАС свидетельствует богатая постганглионарная симпатическая иннервация юкстагломерулярных клеток почек, а стимуляция симпатических нервов сопровождалась высвобождением ренина даже в отсутствие изменений почечного кровотока или содержания натрия в крови. Блокада β-адренорецепторов устраняла этот эффект и обусловливала снижение активности ренина в плазме крови.
Раздражение определенных областей головного мозга, прежде всего — гипоталамуса, сосудодвигательного центра продолговатого мозга, сопровождалось высвобождением ренина из почек, и эта реакция устранялась при денервации почки или на фоне применения пропранолола. Значительное увеличение продукции ренина выявлено при умственных и физических нагрузках, при ортостатической пробе.
Наличие прямой связи между действием А II и симпатической нервной системой проявлялось резким повышением симпатического тонуса и АД у крыс при введении АII в спинномозговой канал, тогда как применение резерпина или симпатэктомия почки в этих условиях предупреждали развитие АГ [276].
Одним из факторов угнетения симпатической активности со снижением активности почечных нервов и сопутствующим уменьшением высвобождения ренина является растяжение предсердия. Однако этот эффект существенно угнетается у лиц с длительным течением АГ, особенно на фоне развивающейся сердечной недостаточности. В этих условиях он выражен примерно на 70% слабее по сравнению с нормой в сочетании с пропорционально менее выраженным уменьшением содержания ренина в плазме крови. Однако после проведенной антигипертензивной терапии и регрессии гипертрофии миокарда эти реакции восстанавливались.
Роль симпатической нервной системы в регуляции активности РАС проявляется и в том, что у животных с двусторонней денервацией почек при ожирении не развиваются АГ, задержка натрия и увеличение объема циркулирующей крови [253, 254].
С другой стороны, РАС способна потенцировать действие симпатической нервной системы на сердце и сосуды по принципу положительной обратной связи. Установлено, что действие А II сопровождается значительным увеличением высвобождения норадреналина из симпатических терминалей посредством активации специфических пресинаптических рецепторов, а также повышением чувствительности адренергических рецепторов к норадреналину [44].
Показано, что внутривенная инфузия А II в субпрессорных дозах нормальным лицам сочеталась со значительным увеличением выраженности реакций сердца на стимуляцию изопреналином, и эта зависимость становилась еще более отчетливой в патологических ситуациях. Так, при погружении в воду пациентов с тяжелым поражением коронарных артерий отмечено резкое повышение АД, коронарного сосудистого сопротивления и уменьшение коронарного кровотока. После перорального приема 25 мг каптоприла погружение сочеталось с нормальными реакциями — уменьшением коронарного сопротивления и увеличением коронарного кровотока.
Благодаря этим взаимным влияниям эффект сочетанной активации симпатических влияний и РАС прогрессирует независимо от того, какая система была первично активирована. Эта связь имеет существенное физиологическое значение, предполагают, что у лиц, постоянно принимающих ингибиторы АПФ, постепенноe восстановление нормальной реакции на ортостатическую пробу связано с компенсаторной активацией симпатической нервной системы. Однако возможно, что этот эффект определяется и другими факторами, прежде всего «ускользанием» продукции А II от угнетения АПФ [171].
Эффект блокады симпатической нервной системы на РАС и содержание ренина в плазме крови в большей степени характерен для физиологических условий, тогда как у лиц с АГ блокаторы β-адренорецепторов даже в высоких дозах значительно слабее влияют на активность ренина. Характерно, что селективные блокаторы β-адренорецепторов менее эффективно снижают плазменную активность ренина по сравнению с неселективными, что свидетельствует о реализации влияния симпатической нервной системы преимущественно через β1-, а не β2-адренорецепторы.
Связь между симпатической нервной системой и РАС существует также на тканевом уровне. Показано, что стимуляция препаратов мезентериальной артерии β-адреномиметиками приводила к высвобождению А II, и этот эффект блокировался пропранололом. На культуре кардиомиоцитов также продемонстрировано, что применение β-адреностимулятора изопреналина сопровождалось усилением как вне-, так и внутриклеточного синтеза А II [262].
В то же время, отношения между симпатической нервной системой и РАС в регуляции АД имеют сложный характер. У пациентов с АГ и высокой исходной активностью ренина в плазме крови применение блокаторов β-адренорецепторов сопровождается значительным снижением АД, тогда как у лиц с нормальной или сниженной активностью ренина гипотензивное действие блокаторов β-адренорецепторов выражено значительно слабее. Это позволило сделать вывод, что антигипертензивное действие блокаторов β-адренорецепторов опосредовано не только уменьшением сердечного выброса, но и уменьшением продукции ренина и снижением его плазменной активности.
Сложный характер отношений между симпатической нервной системой и РАС в регуляции АД проявляется также в особенностях реакции на применение блокаторов β-адренорецепторов у лиц с АГ. В течение первого часа отмечается выраженное уменьшение сердечного выброса, частоты сердечных сокращений и плазменной активности ренина, но АД не изменяется. Это связано с тем, что параллельно рефлекторно повышается симпатический тонус, так как барорецепторные зоны адаптированы к повышенному АД. После денервации этих зон латентный период между первой дозой блокаторов β-адренорецепторов и снижением АД значительно уменьшается.
Хотя компенсаторные механизмы в виде гипертрофии и ремоделирования стенки желудочка и сосудистой стенки при АГ, опосредованные через активацию локальной РАС, позволяют длительное время поддерживать нормальный сердечный выброс и нормализовать внутристеночное напряжение, в конечном итоге они приводят к развитию патологических изменений. Так, А II является стимулом для развития периваскулярного фиброза, кардио- и артериосклероза и в конечном итоге приводит к резкому уменьшению коронарного расширительного резерва и развитию сердечной недостаточности. Во многих клинических исследованиях отмечено, что гипертензивные пациенты с выраженной гипертрофией ЛЖ характеризуются значительно худшим прогнозом.
Данные метаанализа более 105 исследований свидетельствуют, что как ингибиторы АПФ, так и блокаторы β-адренорецепторов способны вызывать регрессию гипертрофии ЛЖ. Однако ингибиторы АПФ более эффективны в этом отношении, независимо от выраженности антигипертензивного действия и длительности лечения, хотя блокаторы β-адренорецепторов также выраженно уменьшали толщину стенки желудочка и межжелудочковой перегородки. В определенной степени различие в действии препаратов этих классов связано с тем, что блокаторы β-адренорецепторов, в отличие от ингибиторов АПФ, меньше восстанавливали растяжимость стенки артериальных сосудов и снижали постнагрузку, хотя локальное содержание ренина и А II в стенке артерий также уменьшалось при применении пропранолола.
Характер взаимодействия РАС и параcимпатической нервной системы изучен значительно меньше. Тем не менее, установлено наличие АТ1-рецепторов также на терминалях парасимпатических преганглионарных нейронов и показано, что А II угнетает высвобождение из них ацетилхолина у интактных собак и в изолированном предсердии морской свинки в отличие от увеличения высвобождения норадреналина из симпатических терминалей [222].
2.2.2. Система ренина, проренина и рецепторов ренина/проренина; ее значимость в регуляции активности циркулирующей и тканевых РАС
Исследования последних двух десятилетий показали, что начальным компонентом РАС является не ренин, а его предшественник проренин. Установлено также, что проренин, помимо значимости как субстрата для образования ренина, играет самостоятельную физиологическую роль, которая как по характеру, так и по механизмам реализации во многом остается еще невыясненной и в настоящее время.
К открытию проренина привели результаты исследования, в котором повышение кислотности плазмы крови или добавление к ней протеолитических ферментов сопровождалось 7-кратным повышением активности ренина [168, 257]. Последующие детальные исследования показали, что значительная часть ренина находится in vivo в плазме крови в неактивной форме и переходит в активную в кислой среде или под действием протеаз. Позже эта неактивная форма ренина получала название проренина.
Проренин, как и ренин, продуцируется главным образом в почках и секретируется в кровь. В отличие от ренина, источником проренина является не юкстагломерулярный аппарат, а собирательные протоки (tubulus renalis colligens) в почках. Характерно, что А ΙΙ угнетает образование ренина в юкстагломерулярном аппарате, но стимулирует продукцию проренина в собирательных протоках параллельно с активацией экспрессии рецепторов проренина [134, 135].
Выявлено, что циркулирующий проренин связывается с внутренней поверхностью сосудистой стенки, и в условиях активации протеаз плазмы крови, в частности — при превращении прекалликреина в калликреин или плазминогена в плазмин, он трансформируется в активный ренин. Аналогичный эффект отмечен при проведении фибринолитической терапии, когда концентрация ренина в плазме крови повышается в 2 раза. У лиц с генетической недостаточностью активности прекалликреина отмечено уменьшение отношения ренин/проренин на фоне повышенного уровня проренина в плазме крови.
Высокое содержание ферментов, способных конвертировать проренин в ренин, отмечено в нейтрофилах, благодаря чему образование ренина, а затем и А ΙΙ резко увеличивается в областях повреждения и воспаления.
В последние годы установлено, что почки — не единственный источник проренина. О возможности продукции проренина в других тканях свидетельствуют данные о том, что его концентрация в плазме крови нефрэктомированных животных уменьшалась, но оставалась достаточно высокой, хотя плазма практически не содержала ренина [244]. В частности, у крыс отмечено наличие прямой экспрессии и высвобождения проренина в циркуляцию из печени, в результате чего его плазменная концентрация может возрастать более чем в 400 раз. Характерно, что у этих животных не возникает АГ, но развиваются выраженные сосудистые поражения и поражение почек. Возможность образования проренина вне почечной ткани подтверждается тем, что его уровень в крови при нефрэктомии достигает 66% нормы и определяет наличие остаточной активности ренина.
У мышей и крыс с отсутствием генов ренина и проренина практически полностью отсутствует А I в крови. Это происходит, несмотря на повышение уровня ангиотензиногена в этих условиях в 2–4 раза, что подтверждает доминирующую роль ренина и проренина в образовании А II [94, 183]. Еще одним доказательством является резкое снижение уровня А I и А II в плазме крови при применении ингибитора ренина алискирена [202, 203]. Хотя сериновые протеазы — химаза и катепсины, участвуют в образовании пептидов А в местах воспаления, эти альтернативные пути образования А II не могут эффективно компенсировать дефицит или угнетение ренина [22].
Впервые роль повышенного уровня циркулирующего проренина в развитии кардиального и почечного поражения при отсутствии АГ была установлена у трансгенных мышей в 1996 г. [293]. Однако между концентрацией проренина в крови и выраженностью поражения почек не было отмечено прямой зависимости, при тяжелой диабетической нефропатии уровень циркулирующего проренина был повышен только на 50–200% [166]. В то же время, в дальнейших исследованиях при повышении уровня циркулирующего проренина у мышей в 30–60 [177], в 200 [216] и даже в 1000 раз [22, 23] было отмечено выраженное повышение АД, которое было связано с увеличением концентрации А ΙΙ и устранялось ингибиторами АПФ, но при отсутствии поражения почек и сердца.
До настоящего времени эти различия не получили четкого объяснения, но высказано предположение, что наличие проренина в крови в высокой концентрации приводит к развитию АГ посредством стимуляции системной продукции А ΙΙ, тогда как поражение органов-мишеней связано в большей степени с тканевым содержанием проренина.
Одним из важнейших эффектов проренина и механизмом его повреждающего действия является активация транскрипционного фактора NF-κB с развитием воспаления и оксидативного стресса, и эти эффекты устранялись лозартаном, но не кандесартаном. Эти данные означают, что экспрессия гена проренина и последующая его продукция регулируются внутриклеточным А II посредством стимуляции АТ1-рецепторов, локализующихся в цитоплазме. Лозартан способен проникать в клетку и блокировать эти рецепторы, тогда как другой блокатор АТ1-рецепторов — кандесартан — не проникает в клетку. Он связывается только с рецепторами, экспрессированными на поверхности клетки, блокирует их, препятствует образованию и интернализации комплекса «А ΙΙ — АТ1-рецептор». Поэтому кандесартан в отличие от лозартана не способен блокировать внутриклеточные рецепторы А II и угнетать его интракринное действие [216].
Традиционно проренин длительное время рассматривали как неактивный предшественник ренина. Однако данные недавних исследований свидетельствуют, что в определенных условиях, особенно патологических, проренин имеет самостоятельное значение, а тканевое образование А ΙΙ может в большей степени быть связано с проренином, чем с ренином. Выявлено, что концентрация проренина в крови пациентов с СД 2-го типа, сердечной недостаточностью, поражением почек резко повышена и достигает в этих условиях 95% общего содержания ренина.
Внимание к функциональной значимости проренина было привлечено двумя установленными фактами. Во-первых, у трансгенных мышей с усиленной экстраренальной продукцией проренина показана связь между его уровнем в крови и риском развития тяжелой АГ [187]. Во-вторых, отмечено, что у больных СД микроциркуляторные поражения развиваются в тесной связи с увеличением плазменной концентрации проренина, независимо от наличия АГ [75]. В обоих случаях развитие указанных эффектов сочеталось со снижением плазменной активности ренина.
В исследовании, проведенном на приматах, при экзогенном введении рекомбинантного проренина в количестве, достаточном для увеличения его плазменной концентрации в 100 раз, не отмечено развития прессорного или вазоконстрикторного ответа. Напротив, у трансгенных мышей с повышенным плазменным содержанием эндогенно продуцируемого проренина отмечено транзиторное развитие АГ на фоне снижения концентрации ренина как в почках, так и в плазме крови. Эти данные стали основанием для предположения, что физиологическое действие проренина определяется его тканевой концентрацией в месте продукции, но не содержанием в плазме крови.
Полагают, что высокая концентрация проренина в плазме крови имеет не самостоятельное значение, а только отражает повышенную интенсивность его синтеза в тканях, хотя источники синтеза проренина в подобных случаях пока не определены. Однако наличие экстраренального синтеза проренина не вызывает сомнений, так как после двусторонней нефрэктомии содержание проренина в плазме крови уменьшается только на 50%, а затем стабилизируется на этом уровне [265].
Установлено, что в физиологических условиях превращение проренина в ренин происходит в почках. Поэтому инфузия проренина не сопровождается его трансформацией в ренин и повышением плазменной активности ренина, а плазма лиц с нефрэктомией содержит только проренин и не содержит ренина. В то же время, полученные ранее данные свидетельствовали о том, что в почках, как и в сердце крыс со спонтанной АГ блокаторы рецепторов проренина не оказывали влияния на содержание ренина.
В ряде исследований выявлено, что проренин оказывает самостоятельное действие, не связанное с его трансформацией в ренин. Он так же, как и ренин, способен активировать митогенактивирующие протеинкиназы и стимулировать высвобождение TФР-β1 в мезангиальных клетках. Это сопровождается развитием фиброзных изменений, так как TФР-β1 активирует PAI-1 и приводит к синтезу фибронектина и коллагена. Связь этих эффектов с действием проренина подтверждается тем, что они сохраняются при наличии ингибиторов ренина и АПФ, блокаторов АТ1-рецепторов [195–197, 293]. Проренин оказывает и самостоятельное гипертензивное действие, что подтверждается наличием прямой зависимости между его концентрацией в крови и уровнем АД у гипертензивных мышей с генетическим отсутствием рецепторов А ΙΙ [121].
Результаты исследований in vivo свидетельствовали о том, что высокое АД крови у гипертензивных животных сочеталось с повышением уровня тканевого проренина параллельно с повышением экспрессии его рецепторов и угнетением трансформации проренина в ренин в юкстагломерулярном аппарате. Более того, отмечено резкое угнетение этого эффекта в сочетании с нормализацией тканевого уровня А Ι и А ΙΙ при специфической блокаде рецепторов проренина [122] и отсутствии изменений содержания или активности других компонентов РАС. Это означало прямое действие проренина, приводящее к активации тканевой РАС и увеличению локальной продукции А ΙΙ, без предшествующей его протеолитической трансформации в ренин.
В патологических условиях, особенно при СД 2-го типа, уровень циркулирующего проренина возрастает многократно, а кардиомиоциты способны его захватывать и использовать в синтезе А II [216]. Эти данные подтверждены в исследовании, проведенном на крысах с СД, воспроизведенным применением стрептозотоцина. Было установлено значительное увеличение содержания A II, ангиотензиногена и ренина в кардиомиоцитах, сопровождающееся усиленной продукцией СОР, развитием апоптоза и миокардиального фиброза. Отмеченные эффекты не устранялись кандесартаном или эзенаприлом, ингибитором АПФ, что означало активацию внутриклеточного синтеза А II, а не его интернализацию через АТ1-рецепторы. В то же время, внутриклеточный синтез А II и его эффекты устранялись алискиреном — ингибитором ренина.
Концепция, в которой постулируется способность проренина принимать прямое участие в развитии кардиоваскулярной патологии, базируется на 4 основных фактах [225, 226]. Во-первых, выявлено, что повышение уровня циркулирующего проренина связано с наличием диабетических микро- и макроваскулярных поражений, а часто и предшествует их развитию [75]. Во-вторых, у трансгенных мышей, у которых резко увеличивалась эндогенная продукция проренина и отмечалось 400-кратное повышение его концентрации в крови, отмечено тяжелое поражение миокарда и почек при отсутствии АГ [293]. Это свидетельствовало о том, что повреждающее действие проренина не ограничивается его способностью активировать образование А ΙΙ.
Третьим моментом явилась идентификация проренинового рецептора, который способен активировать как непротеолитическую трансформацию проренина в ренин с последующим образованием А ΙΙ, так и независимо активировать митогенактивирующие протеинкиназы [118, 197, 239, 240, 243]. Этот эффект сохранялся на фоне ингибиторов РАС и, таким образом, не ограничивался способностью проренина активировать образование А ΙΙ. Помимо этого, доказательством способности рецептора проренина приводить к непротеолитической активации проренина является угнетение гипертрофии сердца и кардиофибpоза без снижения АД при использовании блокатора рецептора проренина.
Эта концепция получила существенное подтверждение в результатах исследований последних лет, в которых было установлено, что активация РАС включает образование новых компонентов типа проренинового рецептора, экспрессированного на клеточных мембранах и обладающего высоким сродством к ренину и проренину. Связывание проренина и ренина с этим рецептором в значительной степени определяло активацию тканевых РАС, усиленную локальную продукцию А II в тканях и, в результате, его тканевой уровень, превышающий уровень в плазме крови.
Специфичные рецепторы проренина идентифицированы с высокой экспрессией в сердце, мозгу, с меньшей — в почках и печени. Результаты ряда исследований последних лет прямо указывали, что функция проренина и ренина не ограничивается образованием А II, и их связывание со специфическим рецептором сопровождается прямой активацией митогенактивирующих протеинкиназ, активацией РАI-1 развитием гипертрофического ответа, кардио- и нефросклероза даже без повышения АД. Неоднократно показано, что на фоне снижения уровня ренина и А II в крови при СД 2-го типа уровень проренина повышается в несколько раз [134, 135].
В ряде исследований прямо установлено, что проренин обладает способностью инициировать тканевое образование А I и А II, но оказывает также прямые эффекты, независимые от А II, которые медиируются взаимодействием проренина с соответствующим рецептором [196].
Описание проренина и механизмов его действия позволило существенно уточнить принципы связи между активностью РАС и развитием АГ. Известно, что при многих патогенетических вариантах АГ активность ренина в плазме крови не повышена или даже угнетена. Предположительно, ведущей причиной развития АГ, а также фактором, определяющим тяжесть ее течения и исходов, является активность тканевых РАС в органах-мишенях, а одна из детерминант этой активации и усиленной локальной тканевой продукции А Ι и А ΙΙ — действие проренина. Проренин способен оказывать действие как в результате собственной протеолитической активации с образованием ренина, так и посредcтвом связывания с прорениновым рецептором. Рецепторы проренина экспрессируются в ряде органов, прежде всего — в сердце и почках; они не выявлены в плазме крови, и потому проренин не способен приводить к активации циркулирующей РАС. Повышенная активность проренина отмечена у крыс с генетической АГ и показано, что она сопровождается активацией тканевых РАС и повреждением органов-мишеней, независимо от активности циркулирующей РАС [197].
Результаты ряда исследований подтвердили, что проренин оказывает двойственное действие, как через образование А I, так и через активацию селективного рецепторопосредованного сигнального пути [262]. Это объясняет, почему при СД, который сочетается с резким усилением экспрессии как проренина, так и его рецепторов, прежде всего — в почках, эффективность ингибиторов АПФ, блокаторов рецепторов А II существенно снижена [70, 286].
В экспериментальных условиях неоднократно установлено, что при увеличенном содержании проренина в крови АД может не повышаться, но при этом развиваются выраженные сосудистые поражения. У людей повышение уровня проренина в крови также отмечено при СД 2-го типа, которое сочеталось с выраженными структурными и функциональными нарушениями в микроваскулярном русле как при наличии АГ, так и в ее отсутствие [191].
Поскольку проренин, как и ренин, оказывает биологический эффект посредством связывания с рецептором проренина [196], он также должен рассматриваться как гормон. Рецептор проренина был идентифицирован в 2002 г., однако его роль в развитии АГ и органного повреждения пока не отражена ни в одном руководстве.
Значимость проренина в регуляции активности РАС показана в исследованиях на экспериментальных моделях, в которых применение блокатора рецепторов проренина у крыс с СД сопровождалось значительным уменьшением тканевого содержания А Ι, А ΙΙ, нормализацией почечного кровотока, препятствовало развитию нефропатии [121], а также угнетало возникновение кардиосклероза у спонтанно гипертензивных крыс [122]. У трансгенных крыс с селективной гиперэкспрессией рецепторов проренина человека выявлено закономерное повышение уровня АД, развитие тахикардии [21], протеинурии и гломерулонефроза [133]. У пациентов с СД 2-го типа отмечено сочетание снижения плазменной активности ренина с повышением его интраренальной активности и показано, что оба эти эффекта возникали в результате повышения экспрессии проренина и его рецепторов в почках.
Неоднократно установлено, что СД 2-го типа характеризуется значительно повышенным уровнем проренина в плазме крови, а выраженность вазодилататорного эффекта каптоприла на этом фоне находилась в четкой связи с плазменным уровнем проренина [269].
Характерно, что у лиц с СД 2-го типа активность ренина в плазме крови закономерно снижена. Однако это сочетается с повышением активности интраренальной РАС и увеличением локальной продукции А II в почках, что сопровождается угнетением высвобождения ренина и приводит к снижению его уровня в плазме крови.
Роль проренина в активации локальных РАС установлена и в культуре сосудистых гладкомышечных клеток человека, инкубация которых с ангиотензиногеном и проренином сопровождалась выраженной продукцией А I. Этот эффект практически полностью угнетался алискиреном. Установлено также, что терапевтическое действие препаратов, угнетающих РАС, в значительной степени определяется их влиянием на уровень проренина, и у 35 пациентов с СД 2-го типа отмечена прямая зависимость между вазодилататорным, антигипертензивным эффектами каптоприла и изменениями содержания проренина в плазме крови [269].
Существование рецептора проренина, обладающего сродством как к проренину, так и ренину, было подтверджено только в последние годы, а в 2002 г. рецептор проренина впервые был клонирован. Выявлено, что его блокада у крыс с СД сопровождалась значительным снижением уровня А I и А II и предупреждала развитие нефропатии [120]. У крыс со спонтанной АГ блокада рецептора проренина приводила к снижению уровня А II и уменьшению выраженности кардиофиброза [121]. Добавление алискирена в среду, в которой инкубировались гладкомышечные клетки, стимулированные ангиотензиногеном и проренином, приводило к дозозависимому угнетению экспрессии мРНК рецептора проренина. Это сопровождалось угнетением экспрессии мРНК, TФР-β, PAI-1, коллагена 1-го типа, то есть факторов, обусловливающих ремоделирование сосудистой стенки. Экспрессия рецептора проренина на клеточной мембране угнеталась также алискиреном [69].
Экспрессия мРНК рецепторов проренина/ренина установлена в центральной нервной системе, сердце, печени и поджелудочной железе [197]. Активация этих рецепторов стимулирует митогенактивирующие протеинкиназы с повышением экспрессии профибротических факторов: TФР-β, PAI-1, усилением продукции коллагена и фибронектина [118, 313, 314]. У гипертензивных диабетических крыс резко усилена экспрессия мРНК рецепторов проренина в клетках клубочков и канальцев почек с развитием прогрессирующей микроальбуминурии, а алискирен почти полностью предупреждал эти эффекты.
Аналогичное действие алискирен оказывал также на сосудистые гладкомышечные клетки, что определяет снижение их реактивности к действию проренина. Эти данные означают, что алискирен угнетает активность РАС не только посредством связывания с рецепторами проренина, но и через угнетение их экспрессии.
Показано, что экспрессия проренинового рецептора значительно усилена в диабетической почке и обусловливает гиперпродукцию профибротических молекул мезангиальными клетками, вызванную глюкозой в высокой концентрации.
Однако механизм, определяющий усиление экспрессии гена проренинового рецептора под действием глюкозы, до последнего времени оставался неустановленным. В исследовании, результаты которого были опубликованы в 2012 г., установлено, что стимуляция культуры мезангиальных клеток человека А II при высокой концентрации глюкозы в среде сопровождалась усиленной экспрессией фибронектина и рецепторов проренина. Данный эффект блокировался лозартаном, но не кандесартаном. Это означает, что выраженность экспрессии гена рецептора проренина контролируется внутриклеточно образующимся А II. Поскольку глюкоза в высокой концентрации усиливает внутриклеточный синтез А II, то это приводит к последующему усилению экспрессии рецепторов проренина, что сопряжено с гиперпродукцией фибронектина и развитием гломерулярного склероза.
Помимо этого, установлено, что стимуляция рецепторов проренина как ренином, так и проренином, усиливает экспрессию А II не только внутриклеточно, но и на клеточной мембране. Однако в этих условиях не увеличивается содержание А II в среде и, таким образом, его образование в данных условиях имеет внутриклеточную природу [195].
Неоднократно отмечено, что повышение внутриклеточной концентрации А II в кардиомиоцитах, фибробластах, мезангиальных клетках при действии глюкозы в высоких концентрациях определяется преимущественно усиленным внутриклеточным синтезом А II, а не его интернализацией из внеклеточного пространства через связывание с АТ1-рецепторами. Это подтверждается данными о том, что стимуляция клеток внеклеточным А II не сопровождалась усилением экспрессии рецепторов проренина, а также о преимущественной локализации рецепторов проренина в цитоплазме [261].
В ряде исследований продемонстрировано, что усиление экспрессии рецепторов проренина способствует его неферментативной активации, то есть без превращения в ренин, с последующим повышением тканевой концентрации А Ι и А ΙΙ и развитием фиброза. Эти изменения особенно характерны для гипертензивного сердца и диабетической почки.
У нормальных животных применение блокаторов рецептора проренина не оказывало влияния на содержание А Ι и А ΙΙ в миокарде, но резко уменьшало его у гипертензивных крыс без изменений их уровня в циркуляции. Эти данные означают, что система проренина и его рецепторов не принимает участия в регуляции АД в норме и активируется в патологических условиях, а непротеолитическая активация проренина и его действие реализуются только в тканях, но не в плазме крови. Поэтому блокаторы рецепторов проренина у гипертензивных крыс и крыс с СД значительно уменьшали содержание в сердце А Ι и А ΙΙ посредством угнетения непротеолитической активации проренина, не оказывая влияния на уровень А Ι и А ΙΙ в крови.
Хотя рецепторы проренина способны взаимодействовать и с ренином, проренин связывается с рецепторами в 3 раза более эффективно, чем ренин, и после способен катаболизировать ангиотензиноген с такой же кинетикой, как и ренин [195].
Установлено, что неконтролируемая активация рецепторов проренина, эффект которой осуществляется независимо от образования А II, играет существенную роль в патогенезе кардиоваскулярной и ренальной дисфункции, в повреждении сердца и почек. Так, трансгенная гиперэкспрессия рецепторов проренина человека у мышей сочеталась с повышением АД, плазменного уровня альдостерона, экспрессии ЦОГ-2 в корковом слое почек. Эти нарушения сопровождались отсутствием изменений плазменной активности ренина и тканевого содержания А ΙΙ, что означает независимую от А ΙΙ природу их действия. В то же время, проренин способствует также и образованию А ΙΙ в тканях, что особенно значимо у лиц с СД, у которых отмечено значительное увеличение содержания проренина в плазме крови [21, 133].
Значимость активации системы проренина/проренинового рецептора в поражении сердца при АГ была определена в исследовании, проведенном на генетически гипертензивных крысах. При их нахождении на солевой диете АД начинало повышаться через 8 нед параллельно с усилением экспрессии рецепторов проренина, тогда как через 12 нед уже было отмечено развитие кардиофиброза. При этом в сердце, но не в плазме крови животных иммуногистохимически установлены повышение непротеолитической активации проренина и активация тканевой РАС, которые устранялись блокаторами рецепторов проренина. В результате предупреждалось развитие кардиофиброза без существенного влияния на циркулирующую РАС и уровень АД.
В соответствии с высказанным недавно предположением, система проренина/проренинового рецептора играет доминирующую роль в развитии нефропатии при СД. Определяется это способностью глюкозы в высокой концентрации вызывать гиперэкспрессию рецепторов проренина, локализующихся преимущественно в цитоплазме мезангиальных клеткок, наряду с усиленной продукцией проренина и повышением его концентрации в крови. Ранее выявлено, что у лиц с СД и гипергликемией повышение концентрации проренина в плазме крови сочеталась с развитием такой патологии, как диабетическая микроангиопатия [47, 191], тогда как блокаторы рецепторов проренина уменьшали выраженность диабетического гломерулосклероза и протеинурии у крыс и мышей даже на фоне блокады рецепторов А ΙΙ 1-го типа [120].
В почке экспрессия рецептора проренина установлена как в мезангиальных клетках [197], так и в собирательных канальцах [4]. Применение блокатора рецептора проренина у спонтанно гипертензивных крыс сопровождалось уменьшением содержания креатинина, выраженности гипертрофии ЛЖ, фиброза миокарда и нарушений функциональных свойств сердца без изменений АД [121]. Эти данные позволили рассматривать повышение плазменной концентрации проренина при СД как предиктор развития микроциркуляторных нарушений, по значимости равный микроальбуминурии.
На основании этих данных была сформулирована гипотеза, в соответствии с которой ренин и проренин приводят к ремоделированию сердца, поражению почек, микроваскулярным повреждениям и ретинальной неоваскуляризации не только посредством активации локальной РАС, но и через взаимодействие со специфическим прорениновым рецептором, активирующим сигнал, не связанный с образованием А ΙΙ. Это открыло возможность для предупреждения поражения органов-мишеней при АГ и СД посредством блокирования взаимодействия ренина и проренина с соответствующим прорениновым рецептором.
Таким образом, в соответствии с современными представлениями, система проренина/проренинового рецептора, особенно в условиях усиленной экспрессии рецепторов проренина с последующей его непротеолитической активацией, оказывает критическое влияние на активность тканевых РАС и должна рассматриваться как важная мишень для методов, направленных на предупреждение повреждения сердца при АГ.
Высокая значимость системы проренина/проренинового рецептора была подтверждена в ряде исследований последних лет. Установлено, что высокое АД у гипертензивных животных сочеталось как с повышением уровня тканевого проренина, так и повышением экспрессии его тканевых рецепторов и угнетением трансформации проренина в ренин в юкстагломерулярном аппарате. Этот эффект резко угнетался специфической блокадой рецепторов проренина в сочетании с нормализацией тканевого уровня А Ι и А ΙΙ. При этом не отмечено изменений содержания или активности других компонентов РАС, что означало ведущую роль непротеолитической активации проренина в активации тканевой РАС.
Ранее полученные данные свидетельствовали о том, что в почках, как и в сердце у крыс со спонтанной АГ, блокаторы рецепторов проренина не оказывали влияния на содержание ренина и уменьшали продукцию А Ι и А ΙΙ посредством угнетения непротеолитической активации проренина без изменения уровня А Ι и А ΙΙ в циркуляции (рис. 6) [122].
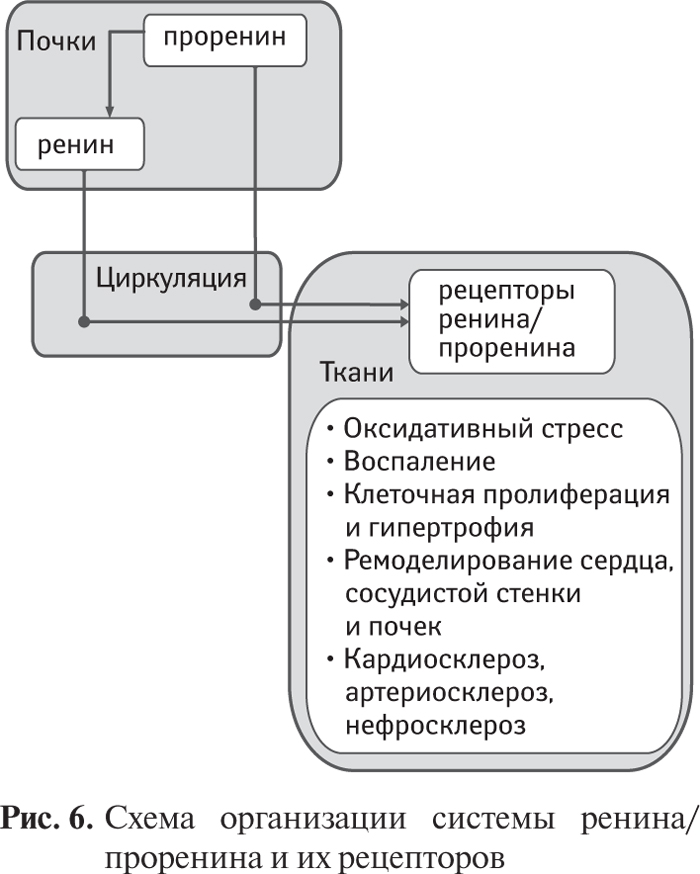
2.3. Список использованной литературы
- Abboud F.M. The Walter B. Cannon Memorial Award Lecture, 2009. Physiology in perspective: the wisdom of the body – in search of autonomic balance: the good, the bad, and the ugly // Am. J. Physiol. Regul. Integr.Comp. Physiol. — 2010. — Vol. 298. — Р. R1449–R146.
- Abboud F., Harwani S., Chapleau M. Autonomic Neural Regulation of the Immune System Implications for Hypertension and Cardiovascular Disease // Hypertens. — 2012. — Vol. 59. — P. 763–768.
- Achard V., Boullu-Ciocca S., Desbriere R. Renin receptor expression in human adipose tissue // Am. J. Physiol. Regul. Integr. Compar. Physiol. — 2007. — Vol. 292. — P. 274–282.
- Advani A., Kelly D.J., Cox A.J. et al. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney // Hypertens. — 2009. — Vol. 54. — P. 261–269.
- Alderman M.H., Madhavan S., Ooi W.L. et al. Association of the renin-sodium profile with the risk of myocardial infarction in patients with essential hypertension // N. Engl. J. Med. — 1991. — Vol. 324. — P. 1098–1104.
- Alexiou T., Boon W.M., Denton D.A. et al. Angiotensinogenand angiotensin converting enzyme gene copy numberand angiotensin and bradykinin peptide levels in mice // J. Hypertens. — 2005. — Vol. 23. — P. 945–954.
- Araki S., Hirooka Y., Kishi T. et al. Olmesartan reduces oxidative stress in the brain of stroke-prone spontaneously hypertensive rats assessed by an in vivo ESR method // Hypertens. Res. — 2009. — Vol. 32. — P. 1091–1096.
- Atlas S.A. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition // J. Manag. Care. Pharm. — 2007. — Vol. 13, (suppl. S-b). — P. S9–S20.
- Bai Y., Jabbari B., Ye S. et al. Regional expression of NAD(P)H oxidase and superoxide dismutase in the brain of rats with neurogenic hypertension // Am. J. Nephrol. — 2009. — Vol. 29. — P. 483–492.
- Baker K.M., Chernin M.I., Schreiber T. et al. Evidence of a novel intracrine mechanism in angiotensin II-induced cardiac hypertrophy // Regul. Pept. — 2004. — Vol. 120. — P. 5–13.
- Baker K.M., Kumar R. Intracellular angiotensin II induces cell proliferation independent of AT1 receptor // Am. J. Physiol. Cell. Physiol. — 2006. — Vol. 291. — P. C995–C1001.
- Baltatu O., Silva J.A., GantenD. et al. The Brain Renin-Angiotensin System Modulates Angiotensin II–Induced Hypertension and Cardiac Hypertrophy // Hypertens. — 2000. — Vol. 35. — P. 409–412.
- Barandier C., Montani J.P., Yang Z. Mature adipocytes and perivascular adipose tissue stimulate vascular smooth muscle cell proliferation: effects of aging and obesity // Am. J. Physiol. Heart Circ. Physiol. — 2005. — Vol. 289. — P. H1807–H1813.
- Biancardi V.C., Son S.J., Sonner P.M. et al. Contribution of Central Nervous System Endothelial Nitric Oxide Synthase to Neurohumoral Activation in Heart Failure Rats // Hypertens. — 2011. — Vol. 58. — P. 454–463.
- Billet S., Aguilar F., Baudry C. et al. Role of angiotensin II AT1 receptor activation in cardiovascular diseases // Kidney Int. — 2008. — Vol. 74. — Р. 1379–1384.
- Bkaily G., Sleiman S., Stephan J. et al. Angiotensin II AT1 receptor internalization, translocation and de novo synthesis modulate cytosolic and nuclear calcium in human vascular smooth muscle cells // Can. J. Physiol.Pharmacol. — 2003. — Vol. 81. — Р. 274–287.
- Boustany C.M., Brown D.R., Randall D.C. et al. AT1-receptor antagonism reverses the blood pressure elevation associated with diet-induced obesity // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2005. — Vol. 289. — P. R181–R186.
- Brands M.W., Banes-Berceli A.K., Inscho E.W. et al. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation // Hypertens. — 2010. — Vol. 56. — P. 879–884.
- Brooks V.L. Interactions between angiotensin II and the sympathetic nervous system in the long-term control of arterial pressure // Clin. Exp. Pharmacol. Physiol. —1997. — Vol. 24. — P. 83–90.
- Brunner H.R., Laragh J.H., Baer L. et al. Essential hypertension: renin and aldosterone, heart attack and stroke // N. Engl. J. Med. — 1972. — Vol. 286. — Р. 441–449.
- Burcklé C.A., Danser J.A.H., Muller D.N. et al. Elevated blood pressure and heart rate in human renin receptor transgenic rats // Hypertens. — 2006. — Vol. 47. — P. 552–556.
- Campbell D. Angiotensin II generation in vivo: does it involve enzymes other than renin and angiotensin-converting enzyme? // Angiotensin II generation in vivo: does it involve enzymes other than renin and angiotensin-converting enzyme? // JRAAS. — 2012. — Vol. 13. — P. 314–316.
- Campbell D., Karam H., Menard J. et al. Prorenin contributes to angiotensin peptide formation in transgenic rats with rat prorenin expression targeted to the liver // Hypertens. — 2009. — Vol. 54. — P. 1248–1253.
- Campese V.M., Sindhu R.K., Ye S. et al. Regional expression of NO synthase, NAD(P)H oxidase and superoxide dismutase in the rat brain // Brain Res. — 2007. — Vol.1134. — P. 27–32.
- Cardinale J.P., Sriramula S., Mariappan N. et al. Angiotensin II–Induced Hypertension Is Modulated by Nuclear Factor-κB in the Paraventricular Nucleus // Hypertens. — Vol. — 2012. — Vol.59. — P. 113–121.
- Carey R.M., Siragy H.M. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation // Endocr. Rev. — 2003. — Vol. 24. — P. 261–271.
- Cassis L.A., Fettinger M.J., Roe A.L. et al. Characterization and regulation of angiotensin II receptors in rat adipose tissue. Angiotensin receptors in adipose tissue // Adv. Exp. Med. Biol. — 1996. — Vol. 396. — P. 39–47.
- Cassis L.A., Rateri D.L., Lu H. et al. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms // Arterioscler. Thromb. Vasc. Biol. — 2007. — Vol.27. — P. 380–386.
- Cat A.N.D., Briones A.M., Callera G.E. et al. Adipocyte-Derived Factors Regulate Vascular Smooth Muscle Cells Through Mineralocorticoid and Glucocorticoid Receptors // Hypertens. — 2011. — Vol. 58. — Р. 479–488.
- Chan S.H., Wu K.L., Chang A.Y. et al. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension // Hypertens. — 2009. — Vol. 53. — P. 217–227.
- Chatterjee T.K., Stoll L.L., Denning G.M. et al. Proinflammatory рhenotype of рerivascular аdipocytes. Influence of high-fat feeding // Circ. Res. — 2009. — Vol. 104. — P. 541–549.
- Chen K., Chen J., Li D. et al. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts. Modulation by PPAR-y ligand pioglitazone // Hypertens. — 2004. — Vol. 44. — P. 655.
- Chen H., Li D., Sawamura T. et al. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan // Biochem. Biophys. Res. Commun. — 2000. — Vol. 276. — P. 1100–1104.
- Chen X., Touyz R.M., Park J.B. et al. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR // Hypertens. — 2001. — Vol. 38. — P. 606–611.
- Cristovam P.C., Arnoni C.P., de Andrade M.C. et al. ACE- and chymase-dependent angiotensin II generation in normal and glucose-stimulated human mesangial cells. // Exp. Biol. Med. — 2008. — Vol. 233. — P. 1035–1043.
- Danser A.H. Cardiac angiotensin II: does it have a function? // AJP — Heart. — 2010. — Vol. 299. — P. H1304–H1306.
- Danser A.H., Batenburg W.W., van Esch J.H.M. Prorenin and the (pro)renin receptor — an update // Nephrol. Dial. Transplant. — 2007. — Vol. 22. — P. 1288–1292.
- Danser A.H., Deinum J. Renin, proreinin and the putative (pro)renin receptor // Hypertens. — 2005. — Vol. 46. — P. 1069–1076.
- Danser A.H., Saris J.J. Prorenin uptake in the heart: a prerequisite for local angiotensin generation? // J. Mol. Cell. Cardiol. — 2002. — Vol. 34. — P. 1463–1472.
- Danser A.H., Saris J.J., Schuijt M.P., van Kats J.P. Is there a local renin–angiotensin system in the heart? // Cardiovasc. Res. — 1999. — Vol. 44. — Р. 252–265.
- Danser A.H., van Kats J.P., Admiraal P.J. et al. Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis // Hypertens. — 1994. — Vol. 24. — P. 37–48.
- Datla S.R., Griendling K.K. Reactive oxygen species, NADPH oxidases, and hypertension // Hypertens. — 2010. — Vol. 56. — P. 325–330.
- Davisson R.L., Oliverio M.I., Coffman T.M. et al. Divergent functions of angiotensin II receptor isoforms in the brain // J. Clin. Invest. — 2000. — Vol. 106. — P. 103–106.
- Day M.D., Owen D.A. The interaction between angiotensin and sympathetically-induced vasoconstriction in the isolated perfused central ear artery of the rabbit.
- De Carvalho F., Sun Y., Weber K.T. Angiotensin II receptor blockade and myocardial fibrosis of the infarcted rat heart // J. Lab. Clin. Med. — 1997. — Vol. 129. — P. 439–446.
- DeFronzo R.A., Ferranini E. Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardio-vascular disease // Diabetes Care. — 1991. — Vol. 14. — P. 173–194.
- Deinum J., Ronn B., Mathiesen E. et al. Increase in serum prorenin precedes onset of microalbuminuria in patients with insulin-dependent diabetes mellitus // Diabetologia. — 1999. — Vol. 42. — P. 1006–1011.
- De Lannoy L.M., Danser A.H., van Kats J.P. et al. Renin-angiotensin system components in the interstitial fluid of the isolated perfused rat heart: local production of angiotensin I // Hypertens. — 1997. — Vol. 29. — P. 1111–1117.
- Dell’Italia L.J., Meng Q.C., Balcells E. et al. Increased ACE and chymase-like activity in cardiac tissue of dogs with chronic mitral regurgitation // Am. J. Physiol. — 1995. — Vol. 269. — P. H2065–H2073.
- De Mello W.C. Intracellular and extracellular renin have opposite effects on the regulation of heart cell volume. Implications for myocardial ischaemia // JRAAS. — 2008. — Vol. 9. — Р. 112–118.
- De Mello W.C., Danser A.H. Angiotensin II and the heart: on the intracrine renin-angiotensin system // Hypertens. — 2000. — Vol. 35. — P. 1183–1188.
- Devereux R.B., Dahlof B., Gerdts E. et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: The Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial // Circulation. — 2004. — Vol. 110. — Р. 1456–1462.
- Dezso B., Nielsen A.H., Poulsen K. Identification of renin in resident alveolar macrophages and monocytes: HPLC and immunohistochemical study // J. Cell. Sci. — 1988. — Vol. 91. — P. 155–159.
- DiBona G.F. Central sympathoexcitatory actions of angiotensin II: role of type 1 angiotensin II receptors // J. Am. Soc. Nephrol. — 1999. — Vol. 10. — P. S90–S94.
- Dickinson C.J., Lawrence J.R. A slowly developing pressor response to small concentrations of angiotensin. Its bearing on the pathogenesis of chronic renal hypertension // Lancet. — 1963. — Vol. 1. — P. 1354–1356.
- Dietze G.J., Henriksen E.J.Angiotensin-converting enzyme in skeletal muscle: sentinel of blood pressure control and glucose homeostasis // JRAAS. — 2008. — Vol. 9. — Р. 75–88.
- Doggrel S.A., Wanstall J.C. Vascular chimase: pathophysiological role and theraputic potential of inhibition // Cardiovasc. Res. — 2004. — Vol. 61, № 4. — P. 653–662.
- Doobay M.F., Talman L.S., Obr T.D. et al. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2007. — Vol. 292. — P. R373–R381.
- Doran A.C., Meller N., McNamara C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis // Arterioscler. Thromb. Vasc. Biol. — 2008. — Vol. 28. — P. 812–819.
- Dzau V.J. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis // Hypertens. — 2001. — Vol. 37. — Р. 1047–1052.
- El Bekay R., Alvarez M., Monteseirin J. et al. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NK-kB // Blood. — 2003. — Vol. 102. — P. 662–671.
- Engeli S., Bohnke J., Gorzelniak K. et al. Weight loss and the renin-angiotensin-aldosterone system // Hypertens. — 2005. — Vol. 45. — P. 356–362.
- Engeli S., Negrel R., Sharma A.M. Physiology and pathophysiology of the adipose tissue renin-angiotensin system // Hypertens. — 2000. — Vol. 35. — P. 1270–1277.
- Eshima K., Hirooka Y., Shigematsu H. et al. Angiotensin in the nucleus tractus solitarii contributes to neurogenic hypertension caused by chronic nitric oxide synthase inhibition // Hypertens. — 2000. — Vol. 36. — P. 259–263.
- Esler M. Sympathetic nervous activation in essential hypertension: commonly neglected as a therapeutic target, usually ignored as a drug side effect // Hypertens. — 2010. — Vol. 55. — Р. 1090–1091.
- Fang T.C., Huang W.C. Role of angiotensin II in hyperinsulinemia-induced hypertension in rats // J. Hypertens. — 1998. — Vol. 16. — P. 1767–1774.
- Ferguson A.V., Bains J.S. Actions of angiotensin in the subfornical organ and area postrema: implications for long term control of autonomic output // Clin. Exp. Pharmacol. Physiol. — 1997. — Vol. 24. — P. 96–101.
- Fernandez-Alfonso M., Martorana P., Licka I. et al. Early induction of angiotensin-I-converting enzyme in rat carotid artery after balloon injury // Hypertens. — 1997. — Vol. 30. — P. 272–277.
- Ferri N., Greco C.M., Maiocchi G. et al. Aliskiren reduces prorenin receptor expression and activity in cultured human aortic smooth muscle cells // JRAAS. — 2011. — Vol. 12. — Р. 469–474.
- Ficociello L.H., Perkins B.A., Silva K.H. et al. Determinants of progression from microalbuminuria to proteinuria in patients who have type 1 diabetes and are treated with angiotensin-converting enzyme inhibitors // Clin. J. Am. Soc. Nephrol. — 2007. — Vol. 2. — P. 461–469.
- Filipeanu C.M., Henning R.H., Nelemans A. et al. Intracellular angiotensin II: from myth to reality? // J. Renin-Angiotensin-Aldosterone System. — 2001. — Vol. 2. — Р. 219–226.
- Fink G.D., Watts S., Chen A. et al. Superoxide anion contributes to hypertension during chronic inhibition of nitric oxide synthesis // Hypertens. — 2002. — Vol. 40. — P. 380–385.
- Fisher N.D., Hurwitz S., Ferri C. et al. Altered adrenal sensitivity to angiotensin II in low-renin essential hypertension // Hypertens. — 1999. — Vol. 34. — P. 388–394.
- Fox C.S., Massaro J.M., Schlett C.L. et al. Periaortic fat deposition is associated with peripheral arterial disease. The Framingham Heart Study // Circulation: Cardiovascular Imaging. — 2010. — Vol. 3. — P. 515–519.
- Franken A., Derkx F., Man in’t Veld A. et al. High plasma prorenin in diabetes mellitus and its correlaions with some complications // J. Clin. Endocrinol. Metabol. — 1990. — Vol. 71. — P. 1008–1015.
- Frustaci A., Kajstura J., Chimenti C. et al. Myocardial cell death in human diabetes // Circ. Res. — 2000. — Vol. 87. — P. 1123–1132.
- Fu M.L., Schulze W., Wallukat G. et al. Immunohistochemical localization of angiotensin II receptors (AT1) in the heart with anti-peptide antibodies showing a positive chronotropic effect // Receptors Channels. — 1998. — Vol. 6. — Р. 99–111.
- Furukawa S., Fujita T., Shimabukuro M. et al. Increased oxidative stress in obesity and its impact on metabolic syndrome // J. Clin. Invest. — 2004. — Vol. 114. — Р. 1752–1761.
- Giacchetti G., Faloia E., Sardu C. et al. Gene expression of angiotensinogen in adipose tissue of obese patients // Int. J. Obes. Relat. Metab. Disord. — 2000. — Vol. 24, (Suppl 2). — P. S142–S143.
- Giacchetti G., Faloia E., Mariniello B. et al. Overexpression of the renin–angiotensin system in human visceral adipose tissue in normal and overweight subjects // Am. J. Hypertens. — 2002. — Vol. 15. — P. 381–388.
- Giacchetti G., Ronconi V., Turchi F. et al. Aldosterone as a key mediator of the cardiometabolic syndrome in primary aldosteronism: an observational study // J. Hypertens. — 2007. — Vol. 25. — Р. 177–186.
- Giacchetti G., Sechi L.A., Rilli S. et al. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes // Trends Endocrinol. Metab. — 2005. — Vol. 16. — Р. 120–125.
- Giacchetti G., Turchi F., Boscaro M. et al. Management of primary aldosteronism: its complications and their outcomes after treatment // Cur. Vasc. Pharmacol. — 2009. — Vol. 7. — Р. 244–249.
- Gill P., Wilcox C. NADPH oxidases in the kidney // Antioxid. Redox Signal. — 2006. — Vol. 8. — P. 1597–1607.
- Gojanovic B., Feihl F., Liaudet L. et al. Concomitant calcium entry blockade and inhibition of the renin-angiotensin system: a rational and effective means for treating hypertension // JRAAS. — 2008. — Vol. 9. — Р. 1–9.
- Golledge J., Clancy P., Jamrozik K. et al. Obesity, adipokines, and abdominal aortic aneurysm: Health in Men study // Circulation. — 2007. — Vol. 116. — P. 2275–2279.
- Golledge J., Müller J., Shephard N. et al. Association between osteopontin and human abdominal aortic aneurysm // Arterioscler. Thromb. Vasc. Biol. – 2007. – Vol. 27. – P. 655–660.
- Gomez-Ambrosi J., Catalan V., Ramirez B. et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity // J. Clin. Endocrinol. Metab. – 2007. – Vol. 92. – P. 3719–3727.
- Gorbea O.V., Fink G.D. Clonidine reverses the slowly developing hypertension produced by low doses of angiotensin II // Hypertens. — 1994. — Vol. 23. — P. 844–847.
- Gorbea O.V., Fink G.D. Cerebroventricular injection of angiotensin II antagonist: effects on blood pressure responses to central and systemic angiotensin II // J. Pharmacol. Exp. Ther. — 1995. — Vol. 273. — P. 611–616.
- Gorzelniak K., Engeli S., Janke J. et al. Hormonal regulation of the human adipose-tissue renin–angiotensin system: relationship to obesity and hypertension // J. Hypertens. — 2002. — Vol. 20. — P. 965–973.
- Gray M.O., Long C.S., Kalinyak J.E. et al. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts // Cardiovasc. Res. — 1998. — Vol. 40. — P. 352–363.
- Greenstein A.S., Khavandi K., Withers S.B. et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese pаtients // Circulation. — 2009. — Vol. 119. — P. 1661–1670.
- Gribouval O., Moriniere V., Pawtowski A. et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis // Hum. Mutat. — 2012. — Vol. 33. — P. 316–326.
- Guyenet P.G. The sympathetic control of blood pressure // Nat. Rev. Neurosci. — 2006. — Vol. 7. — P. 335–346.
- Guzik T.J., Hoch N.E., Brown K.A. et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction // J. Exp. Med. — 2007. — Vol. 204. — P. 2449–2460.
- Hackenthal E., Hackenthal R., Hilgenfeldt U. Isorenin, pseudorenin, cathepsin D and renin. A comparative enzymatic study of angiotensin-forming enzymes // Biochim. Biophys. Acta. —1978. — Vol. 522. — P. 574–588.
- Haller H., Lindschau C., Erdmann B.L. et al. Effects of intracellular angiotensin II in vascular smooth muscle cells // Circ. Res. — 1996. — Vol. 79. — P. 765–772.
- Haller H., Lindschau C., Quass P. et al. Intracellular actions of angiotensin II in vascular smooth muscle cells // J. Am. Soc. Nephrol. — 1999. — Vol. 10, (suppl. 11). — P. S75–S83.
- Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease // N. Engl. J. Med. — 2005. — Vol. 352. — P. 1685–1695.
- Harrison D.G., Cai H., Landmesser U. et al. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease // JRAAS. — 2003. — Vol. 4. — P. 51–61.
- Harrison D.G., Gongora M.C. Oxidative stress and hypertension // Med. Clin. N. Am. — 2009. — Vol. 93. — P. 621–635.
- Harrison D.G., Guzik T.J., Lob H.E. et al. Inflammation, immunity, and hypertension // Hypertens. — 2011. — Vol. 57. — P. 132–140.
- Harrison D.G., Vinh A., Lob H. et al. Role of the adaptive immune system in hypertension // Curr. Opin. Pharmacol. — 2010. — Vol. 10. — P. 203–207.
- Harrison-Bernard L.M., Zhuo J., Kobori H. et al. Intrarenal AT(1) receptor and ACE binding in ANG II induced hypertensive rats // Am. J. Physiol. Renal. Physiol. — 2002. — Vol. 282. — P. F19–F25.
- Harwani S.C., Chapleau M.W., Legge K. Autonomic dysregulation of innate immunity in genetic hypertension [abstract] // Hypertens. — 2011. — Vol. 58. — Р. е50.
- He H., Yang D., Ma L. et al. Telmisartan prevents weight gain and obesity through activation of peroxisome proliferator-activated receptor-δ-dependent pathways // Hypertens. — 2010. — Vol. 55. — P. 869–879.
- Hendel M.D., Collister J.P. Role of the subfornical organ in chronic Ang II induced hypertension // FASEB J. — 2004. — Vol. 18. — Р. A672.
- Hendel M.D., Collister J.P. Contribution of the subfornical organ to angiotensin II-induced hypertension // Am. J. Physiol. Heart Circ. Physiol. — 2005. — Vol. 288. — H680–H685.
- Henrichot E., Juge-Aubry C.E., Pernin A. et al. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? // Arterioscler. Thromb. Vasc. Biol. — 2005. — Vol. 25. — P. 2594–2599.
- Hilgers K.F., Stumpf C. Angiotensin II, the endothelium and superoxide anions // J. Hypertens. — 2002. — Vol. 20. — P. 1271–1273.
- Hirata A., Maeda N., Hiuge A. et al. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice // Cardiovasc. Res. — 2009. — Vol. 84. — P. 164–172.
- Hirooka Y., Kishi T., Sakai K. et al. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension // AJP — Regul. Physiol. — 2011. — Vol. 300. — Р. R818–R826.
- Hirooka Y., Peterson J.R., Sharma R.V. et al. Reactive oxygen species in the neuropathogenesis of hypertension // Curr. Hypertens. Rep. — 2006. — Vol. 8. — Р. 232–241.
- Hirooka Y., Sagara Y., Kishi T. et al. Oxidative stress and central cardiovascular regulation: pathogenesis of hypertension and therapeutic aspects // Circ. J. — 2010. — Vol. 74. — Р. 827–835.
- Hirsch A.T., Talsness C.E., Schunkert H. et al. Tissue-specific activation of cardiac angiotensin converting enzyme in experimental heart failure // Circ. Res. — 1991. — Vol. 69. — P. 475–482.
- Hsieh P.S., Huang W.C. Chemical sympathectomy attenuates hyperinsulinemia-induced hypertension in conscious rats // Nutr. Metab. Cardiovasc. Dis. — 1993. — Vol. 3. — P. 173–178.
- Huang Y., Wongamorntham S., Kasting J. et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms // Kidney Int. — 2006. — Vol. 69. — P. 105–113.
- Huggins C.E., Domenighetti A.A, Pedrazzini T. et al. Elevated intracardiac angiotensin II leads to cardiac hypertrophy and mechanical dysfunction in normotensive mice // JRAAS. — 2003. — Vol. 4. — P. 186–190.
- Ichihara A., Hayashi M., Kaneshiro Y. et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the ‘handle’ region for nonproteolytic activation of prorenin // J. Clin. Invest. — 2004. — Vol. 114. — P. 1128–1135.
- Ichihara A., Kaneshiro Y., Takemitsu T. et al. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension // Hypertens. — 2006. — Vol. 47. — P. 894–900.
- Ichihara A., Suzuki F., Nakagawa T. et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice // J. Am. Soc.Nephrol. — 2006. — Vol. 17. — P. 1950–1961.
- Inagami T. Intracellular renin: validation and function // in «The Renin-Angiotensin System» ed. by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993, Vol. 1, (part 30). — P. 30.1–30.8.
- Ingert C., Grima M., Coquard C. et al. Contribution of angiotensin II internalization to intrarenal angiotensin II levels in rats // Am. J. Physiol. Renal. Physiol. — 2002. — Vol. 283. — P. F1003–F1010.
- Iwai N., Inagami T., Ohmichi N. et al. Renin is expressed in rat macrophage/ monocyte cells // Hypertens. — 1996. — Vol. 27. — P. 399–403.
- Iwai N., Izumi M., Inagami T. et al. Induction of renin in medial smooth muscle cells by balloon injury // Hypertens. — 1997. — Vol. 29. — P. 1044–1050.
- Jacques D., Malak A.N.A., Sader S. et al. Angiotensin II and its receptors in human endocardial endothelial cells: role in modulating intracellular calcium // Can. J. Physiol. Pharmacol. — 2003. — Vol. 81. — Р. 259–266.
- Janiak P., Bidouard J.P., Cadrouvele C. et al. Long-term blockade of angiotensin AT1 receptors increases survival of obese Zucker rats // Eur. J. Pharmacol. — 2006. — Vol. 534. — P. 271–279.
- Johnson A.K., Cunningham J.T., Thunhorst R.L. Integrative role of the lamina terminalis in the regulation of cardiovascular and body fluid homeostasis // Clin. Exp. Pharmacol. Physiol. — 1996. — Vol. 23. — P. 183–191.
- Johnston A.P., Baker J., De Lisio M. et al. Skeletal muscle myoblasts possess a stretch-responsive local angiotensin signalling system // J. Renin-Angiotensin-Aldosterone System. — 2011. — Vol. 12. — Р. 75–84.
- Jorde U.P., Vittorio T., Katz S.D. et al. Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme in chronic heart failure. Circulation. — 2002. — Vol. 106. —Р. 1055–1057.
- Jorde U.P., Vittorio T.J., Dimayuga C.A. et al. Comparison of suppression of the circulating and vascular renin-angiotensin system by enalapril versus trandolapril in chronic heart failure // Am. J. Cardiol. —2004. — Vol. 94. — P. 1501–1505.
- Kaneshiro Y., Ichihara A., Sakoda M. et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats // J. Am. Soc. Nephrol. — 2007. — Vol. 18. — P. 1789–1795.
- Kang Y.S., Park Y.G., Kim B.K. et al. Angiotensin II stimulates the synthesis of vascular endothelial growth factor through the p38 mitogen activated protein kinase pathway in cultured mouse podocytes // J. Mol. Endocrinol. — 2006. — Vol. 36. — P. 377–88.
- Kang J.J., Toma I., Sipos A. et al. The collecting duct is the major source of prorenin in diabetes // Hypertension. — 2008. — Vol. 51. — P. 1597–1604.
- Karimi K., Bienenstock J., Wang L. et al. The vagus nerve modulates CD4+ T cell activity // Brain Behav. Immun. — 2010. — Vol. 24. — P. 316–323.
- Karlsson C., Lindell K., Ottosson M. et al. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II // J. Clin. Endocrinol. Metab. — 1998. — Vol. 83. — Р. 3925–3929.
- Kimura Y., Hirooka Y., Sagara Y. et al. Overexpression of inducible nitric oxide synthase in rostral ventrolateral medulla causes hypertension and sympathoexcitation via an increase in oxidative stress // Circ. Res. — 2005. — Vol. 96. — P. 252–260.
- Kimura S., Zhang G.-X., Nishiyama A. et al. Role of NADPH oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II // Hypertens. — 2005. — Vol. 5. — P. 860–866.
- Kishi T., Hirooka Y., Mukai Y. et al. Atorvastatin causes depressor and sympatho-inhibitory effect with upregulation of nitric oxide synthases in stroke-prone hypertensive rats // J. Hypertens. — 2003. — Vol. 21. — P. 379–386.
- Kitazono T., Padgett R.C., Armstrong M.L. et al. Evidence that angiotensin II is present in human monocytes // Circulation. — 1995. — Vol. 91. — P. 1129–1134.
- Kobori H., Nishiyama A., Abe Y. et al. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet // Hypertens. — 2003. — Vol. 41. — P. 592–597.
- Koga Y., Hirooka Y., Araki S. et al. High salt intake enhances blood pressure increase during development of hypertension via oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats // Hypertens. Res. — 2008. — Vol. 31. — P. 2075–2083.
- Kohm A.P., Sanders V.M. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo // Pharmacol. Rev. — 2001. — Vol. 53. — Р. 487–525.
- Koka V., Wang W., Huang X.R. et al. Advanced glycation end products activate a chymase-dependent angiotensin II-generating pathway in diabetic complications. // Circulation. — 2006. — Vol. 113. — P. 1353–1360.
- Kumar R., Singh V.P., Baker K.M. The intracellular renin-angiotensin system: a new paradigm // Trends Endocrinol. Metab. — 2007. — Vol. 8. — P. 208–214.
- Kumar R., Singh V.P., Baker K.M. The intracellular renin-angiotensin system: implications in cardiovascular remodelling // Curr. Opin. Nephrol. Hypertens. — 2008. — Vol. 7. — P. 168–173.
- Кvakan H., Kleinewietfeld M., Qadri F. et al. Regulatory T cells аmeliorate angiotensin II–induced cardiac damage // Circulation. — 2009. — Vol. 119. — P. 2904–2912.
- Labiós M., Martínez M., Gabriel F. et al. Effects of eprosartan on mitochondrial membrane potential and hydrogen peroxide production in leucocytes in hypertension // J. Human Hypertens. — 2008. — Vol. 22. — P. 493–500.
- Labiós M., Martínez M., Gabrie F.et al. Superoxide dismutase and catalase anti-oxidant activity in leucocyte lysates from hypertensive patients: effects of eprosartan treatment // JRAAS. — 2009. — Vol. 10. — Р. 24–30.
- Lal A., Veinot J.P., Leenen F.H. Prevention of high salt diet induced cardiac hypertrophy and fibrosis by spironolactone // Am. J. Hypertens. — 2003. — Vol. 16. — P. 319–323.
- Lavoie J.L., Bianco R.A., Sakai K. et al. Transgenic mice for studies of the renin-angiotensin system in hypertension // Acta Physiol. Scand. — 2004. — Vol. 181. — P. 571–577.
- Lavoie J.L., Liu X., Bianco R.A. et al. Evidence supporting a functional role for intracellular renin in the brain // Hypertens. — 2006. — Vol. 47. — P. 461–466.
- Lavrentyev E.N., Estes A.M., Malik K.U. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells // Circ. Res. — 2007. — Vol. 101. — Р. 455–464.
- Lawrence I.G., Weston P.J., Bennett M.A. et al. Is impaired baroreflex sensitivity a predictor or cause of sudden death in insulin-dependent diabetes mellitus? // Diabet Med. — 1997. — Vol. 14. — P. 82–85.
- Lazartigues E. Inflammation and Neurogenic Hypertension. A New Role for the Circumventricular Organs? // Circ.Res. — 2010. — Vol. 107. — Р. 166–167.
- Leenen F.H., Zhang W., Huang B.S. et al. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction // Am. J. Physiol. — 1999. — Vol. 276. — P. H1608–H1615.
- Legends L., Randon J., Sessa C. et al. Cathepsin G is associated with atheroma formation in human carоtid artery // J. Hypertens. — 2004. — Vol. 22. — P. 157–166.
- Levick S.P., McLarty J.L., Murray D.B. et al. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart // Hypertens. — 2009. — Vol. 53. — P. 1041–1047.
- Li M., Zheng C., Sato T. et al. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats // Circulation. — 2004. — Vol. 109. — P. 120–124.
- Liang B., Leenen F.H. Prevention of salt induced hypertension and fibrosis by angiotensin converting enzyme inhibitors in Dahl S rats // Br. J. Pharmacol. — 2007. — Vol. 152. — P. 903–914.
- Liao T-D., Yang X-P., Liu Y-H. et al. Role of inflammation in the development of renal damage and dysfunction in angiotensin II–induced hypertension // Hypertens. — 2008. — Vol. 52. — P. 256–263.
- Licea H., Walters M.R., Navar L.G. Renal nuclear angiotensin II receptors in normal and hypertensive rats // Acta Physiol. Hung. — 2002. — Vol. 89. — Р. 427–438.
- Lijnen P.J., Petrov V.V., Fagard R.H. Angiotensin II-induced stimulation of collagen secretion and production in cardiac fibroblasts is mediated via angiotensin II subtype 1 receptors // JRAAS. — 2000. — Vol. 2. — P. 117–122.
- Lindpaintner K., Jin M., Wilhelm M.J. et al. Intracardiac generation of angiotensin and its physiologic role // Circulation. — 1988. — Vol. 77, (Suppl 1). — P. I-18–I-23.
- Luetscher J.A., Kraemer F.B., Wilson D.M. et al. Increased plasma inactive renin in diabetes mellitus: a marker of microvascular complications // N. Engl. J. Med. — 1985. — Vol. 312. — P. 1412–1417.
- Luft F.C., Wilcox C.S., Unger T. et al. Angiotensin-induced hypertension in the rat: sympathetic nerve activity and prostaglandins // Hypertens. — 1989. — Vol. 14. — P. 396–403.
- Lumbers E.R. Activation of renin in human amniotic fluid by low pH // Enzymologia. — 1971. — Vol. 40. — P. 329–336.
- MacKenzie S.M., Fraser R., Connell J.M.C. et al. Local renin-angiotensin systems and their interactions with extra-adrenal corticosteroid production // J. Renin-Angiotensin-Aldosterone System. — 2002. — Vol. 3. — P. 214–221.
- Maiellaro K., Taylor W.R. The role of the adventitia in vascular inflammation // Cardiovasc. Res. — 2007. — Vol. 75. — Р. 640–648.
- Mancia G., De Backer G., Dominiczak A. et al. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension and of the European Society of Cardiology // J. Hypertens. — 2007. — Vol. 25. — Р. 1105–1187.
- Marvar P.J., Thabet S.R., Guzik T.J. et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension // Circ.Res. — 2010. — Vol. 107. — Р. 263–270.
- Matsumoto T., Wada A., Tsutamoto T. et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure // Circulation. — 2003. — Vol. 107. — P. 2555–2558.
- McKinley M.J., Pennington G.L., Oldfield B.J. Anteroventral wall of the third ventricle and dorsal lamina terminalis: headquarters for control of body fluid homeostasis? // Clin. Exp. Pharmacol. Physiol. — 1996. — Vol. 23. — P. 271–281.
- Meade T. Plasma renin and the incidence of cardiovascular disease // JRАAS. — 2010. — Vol. 11. — P. 91–98.
- Meade T., Imeson J.D., Gordon D., Peart W.S. The epidemiology of plasma renin // Clin. Sci. — 1983. — Vol. 64. — P. 273–280.
- Mercure C., Prescott G., Lacombe M.J. et al. Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies // Hypertens. — 2009. — Vol. 53. — P. 1062–1069.
- Milsted A., Barna B.P., Ransohoff R.M. et al. Astrocyte cultures derived from human brain tissue express angiotensinogen mRNA // Proc. Natl. Acad. Sci. USA. — 1990. — Vol. 87. — P. 5720–5723.
- Miyazaki M., Takai S. Role of chymase on vascular proliferation // JRAAS. — 2000. — Vol. 1. — P. 23–26.
- Mizuno Y., Yoshimura M., Yasue H. et al. Aldosterone production is activated in failing ventricle in humans // Circulation. — 2001. — Vol. 103. — P. 72–77.
- Molteni A, Dzau VJ, Fallon JT, Haber E: Monoclonal antibodies as probes of renin gene expression // Circulation. — 1984. — Vol. 70, (suppl II). — Р. 11–16.
- Monti J., Schinke M., Bohm M. et al. Glial angiotensinogen regulates brain angiotensin II receptors in transgenic rats TGR(ASrAOGEN) // Am. J. Physiol. — 2001. — Vol. 280. — P. R233–R240.
- Moreno C., Hoffman M., Stodola T.J. et al. Creation and characterization of a renin knockout rat // Hypertens. — 2011. — Vol. 57. — P. 614–619.
- Morgan T., Aubert J.F., Brunner H. Interaction between sodium intake, angiotensin II, and blood pressure as a cause of cardiac hypertrophy // Am. J. Hypertens. — 2001. — Vol. 14. — Р. 914–920.
- Morton J.J. Biochemical aspects of angiotensins // in «The Renin-Angiotensin System» ed. by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993, Vol. 1, (part 9). — P. 9.1–9.12.
- Mueller C.F.H., Nickenig G. Angiotensin II. One driving force behind atherogenesis // Hypertension. — 2008. — Vol. 51. — P. 175–181.
- Mullins J.J., Peters J.J., Ganten D. Fulminant hypertension in transgenic rats harboring the mouse Ren-2 gene // Nature. — 1990. — Vol. 344. — P. 541–544.
- Naftilan A.J., Ryan T.J., Pratt R.E. et al. Angiotensinogen gene expression in the vascular wall is abnormal in the genetic hypertensive rat // Clin. Res. — 1988. — Vol. 36. — Р. 430A.
- Nakamura Y., Yoshiyama M., Omura T. et al. Beneficial effects of combination of ACE inhibitor and angiotensin II type 1 receptor blocker on cardiac remodelling in rat myocardial infarction // Cardiovasc. Res. — 2003. — Vol. 57. — P. 48–54.
- Nance D.M., Sanders V.M. Autonomic innervation and regulation of the immune system // Brain Behav. Immun. — 2007. — Vol. 21. — P. 736–745.
- Naruse M., Wasada T., Naruse K. et al. Pathophysiological significance of plasma total renin and prorenin in patients with diabetes mellitus // Endocr. J. — 1995. — Vol. 42. — P. 225–233.
- Navar L.G., Mitchell K.D., Harrison-Bernard L.M. et al. Intrarenal angiotensin II levels in normal and hypertensive states // J. Renin-Angiotensin-Aldosterone System. — 2001. — Vol. 2. — P. S176–84.
- Nemerovski C.W., Dorsch M.P., Simpson R.U. et al. Vitamin D and cardiovascular disease // Pharmacotherapy. — 2009. — Vol. 29. — P. 691–708.
- Neri Serneri G.G., Boddi M., Cecioni I. et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function // Circ. Res. — 2001. — Vol. 88. — P. 961–968.
- Nguyen G., Burckle C., Sraer J.D. The renin receptor: the facts, the promise and the hope // Curr. Opin. Nephrol. Hypertens. — 2003. — Vol. 12. — Р. 51–55.
- Nguyen G., Burckle C.A., Sraer J.D. Renin/prorenin-receptor biochemistry and functional significance // Curr. Hypertens. Rep. — 2004. — Vol. 6. — P. 129–132.
- Nguyen G., Delarue F., Burcklé C. et al. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin // J. Clin. Invest. — 2002. — Vol. 109. — P. 1417–1427.
- Niebauer J., Tsao P.S., Lin P.S. et al. Effects of hypercholesterolemia on angiotensin II, superoxide production and monocyte binding // Europ. Heart J. — 1998. — 19, Abstr. Suppl. — P. 379.
- Nishiyama A., Fukui T., Fujisawa Y. et al. Systemic and regional hemodynamic responses to tempol in angiotensin II-infused hypertensive rats // Hypertens. — 2001. — Vol. 37. — P. 77–83.
- Nishiyama A., Yoshizumi M., Rahman M. et al. Effects of AT1 receptor blockade on renal injury and mitogen-activated protein activity in Dahl salt-sensitive rats // Kidney Int. — 2004. — Vol. 65. — P. 972–981.
- Nozoe M., Hirooka Y., Koga Y. et al. Inhibition o f Rac-1 derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats // Hypertens. — 2007. — Vol. 50. — Р. 62–68.
- Nussberger J., Aubert J.F., Bouzourene K. et al. Renin inhibition by aliskiren prevents atherosclerosis progression: comparison with irbesartan, atenolol, and amlodipine // Hypertens. — 2008. — Vol. 51. — P. 1306–1311.
- Nussberger J., Wuerzner G., Jensen C. et al. Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100): comparison with enalapril // Hypertens. — 2002. — Vol. 39. — Р. e1–e8.
- O’Brian K.D., Shavelle D.M., Caulfield M.T. et al. Association of angiotensin // Circulation. — 2002. — Vol. 106. — P. 2224–2230.
- Ohkuma H., Suzuki S., Fujita S. et al. Role of a decreased expression of the local renin-angiotensin system in the etiology of cerebral anevrism // Circulation. — 2003. — Vol. 108, № 7. — P. 785–787.
- Oliveira E.B., Dugaich A.P., Carillo B.A. et al. Oxidative stress contributes to renovascular hypertension // Am. J. Hypertens. — 2008. — Vol. 21. — P. 98–104.
- Oliveira E.B., Nishi E.E., Carillo B.A. et al. AT1 receptor expression levels in the RVLM and the paraventricular nucleus of the hypothalamus are enhanced in this rat renovascular model of hypertension // Am. J. Hypertens. — 2009. — Vol. 22. — Р. 484–492.
- Oliveira E.M., Sasaki M.S., Cerêncio M. et al. Local renin-angiotensin system regulates left ventricular hypertrophy induced by swimming training independent of circulating renin: a pharmacological study // JRAAS. — 2009. — Vol. 10. — Р. 15–23.
- O’Mahony C., van der Kleij H., Bienenstock J. et al. Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2009. — Vol. 297. — P. R1118–R1126.
- Otsuka A., Barnes K.L., Ferrario C.M. Contribution of area postrema to pressor actions of angiotensin II in dog // Am. J. Physiol. Heart Circ. Physiol. — 1986. — Vol. 251. — P. H538–H546.
- Paravicini T., Touyz R.M. Redox signaling in hypertension // Cardiovasc. Res. — 2006. — Vol. 71. — P. 247–258.
- Paravicini T., Touyz R.M. NADPH oxidases, reactive oxygen species, and hypertension. Clinical implications and therapeutic possibilities // Diabetes Care. — 2008. — Vol. 31, (Suppl.2). — Р. S170–S180.
- Parikh N.I., Gona P., Larson M.G. et al. Plasma renin and risk of cardiovascular disease and mortality: the Framingham Heart Study // Eur. Heart J. — 2007. — Vol. 28. — Р. 2644–2652.
- Paul M., Mehr A.P., Kreutz R. Physiology of local renin–angiotensin systems // Physiol. Rev. — 2006. — Vol. 86. — Р. 747–803.
- Pawloski C.M., Kanagy N.L., Fink G.D. Contribution of increased neurogenic vascular tone to angiotensin II-induced hypertension // FASEB J. — 1989. — Vol. 3. — Abstract 551.
- Peters J., Farrenkopf R., Clausmeyer S. et al. Functional significance of prorenin internalization in the rat heart // Circ. Res. — 2002. — Vol. 90. — P. 1135–1141.
- Pieruzzi F., Abassi Z.A., Keiser H.R. Expression of renin-angiotensin system components in the heart, kidneys, and lungs of rats with experimental heart failure // Circulation. — 1995. — Vol. 92. — P. 3105–3112.
- Pitt B., Ahmed A., Love T.E. et al. History of hypertension and eplerenone in patients with acute myocardial infarction complicated by heart failure // Hypertens. — 2008. — Vol. 52. — P. 271–278.
- Pitt B., Poole-Wilson P.A., Segal R. et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial — the Losartan Heart Failure Survival Study ELITE II // Lancet. — 2000. — Vol. 355. — P. 1582–1587.
- Pitt B., Remme W., Zannad F. et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction // N. Engl. J. Med. — 2003. — Vol. 348. — Р. 1309–1321.
- Pool J.L., Schmieder R.E., Azizi M. et al. Aliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartan // Am. J. Hypertens. — 2007. — Vol. 20. — P. 11–20.
- Potter E.K. Angiotensin inhibits action of vagus nerve at the heart // Br. J. Pharmacol. — 1982. — Vol. 75. — Р. 9–11.
- Rahmouni K., Mark A.L., Haynes W.G. et al. Adipose depot-specific modulation of angiotensinogen gene expression in diet-induced obesity // Am. J. Physiol. Endocrinol. Metab. — 2004. — Vol. 286. —
P. E891–E895. - Re R.N. Intracellular renin and the nature of intracrine enzymes // Hypertens. — 2003. — Vol. 42. — Р. 117–122.
- Reudelhuber T. Prorenin, renin, and their receptor. Moving Targets // Hypertens. — 2010. — Vol. 55. — P. 1071–1074.
- Reudelhuber T., Bernstein K., Delafontaine E. Is angiotensin II a director mediator of left ventricular hypertrophy? Time for another look // Hypertens. — 2000. — Vol. 49. — P. 1196–1201.
- Robinson T.G., Dawson S.L., Eames P.J. et al. Cardiac baroreceptor sensitivity predicts long-term outcome after acute ischemic stroke // Stroke. — 2003. — Vol. 34. — P. 705–712.
- Romero M.F., Hopfer U. et al. Angiotensin actions in the rabbit proximal tubule // Ren. Physiol. Biochem. — 2003. — Vol. 14. — Р. 199–207.
- Rosas-Ballina M., Olofsson P.S., Ochani M. et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit // Science. — 2011. — Vol. 334. — P. 98–101.
- Rossi G., Bernin G., Caliumi C. et al. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients // J. Am. Coll. Cardiol. — 2006. — Vol. 48. — P. 2293–2300.
- Rossi G, Belfiore A., Bernini G. et al. Body mass index predicts plasma aldosterone concentrations in overweight-obese primary hypertensive patients // J. Clin. Endocrinol. Metab. — 2008. — Vol. 93. — P. 2566–2571.
- Rossi G., Boscaro M., Ronconi V. et al. Aldosterone as a cardiovascular risk factor // Trends Endocrinol. Metab. — 2005. — Vol. 16. — Р. 104–107.
- Rossi G., Di Bello V., Ganzaroli P. et al. Excess aldosterone is associated with alterations of myocardial texture in primary aldosteronism // Hypertens. — 2002. — Vol. 4. — P. 23–27.
- Rossi G., Pessina A.C., Heagerthy A.M. Primary aldosteronism an update on screening diagnosis and treatment // J. Hypertens. — 2008. — Vol. 26. — P. 613–621.
- Safar M.E., Laurent S.L., Bouthier J.D. et al. Effect of converting enzyme inhibitors on hypertensive large arteries in humans // J. Hypertens. — 1986. — Vol. 4, (suppl 5). — P. S285.
- Sakai K., Hirooka Y., Matsuo I. Overexpression of eNOS in NTS causes hypotension and bradycardia in vivo // Hypertens. — 2000. — Vol. 36. — P. 1023–1028.
- Sander M., Chavoshan B., Victor R.G. A large blood pressure-raising effect of nitric oxide synthase inhibition in humans // Hypertens. — 1999. — Vol. 33. — Р. 937–942.
- Saraff K., Babamusta F., Cassis L.A., Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice // Arterioscler. Thromb. Vasc. Biol. — 2003. — Vol. 23. — P. 1621–1626.
- Saris J.J., Hoen P.A., Garrelds I.M. et al. Prorenin induces intracellular signalling in cardiomyocytes independently of angiotensin II // Hypertens. — 2006. — Vol. 48. — P. 564–567.
- Saris J.J., van den Eijnden M.M., Lamers J.M. et al. Prorenin-induced myocyte proliferation: no role for intracellular angiotensin II // Hypertens. — 2002. — Vol. 39. — P. 573–577.
- Satoh M., Nakamura M., Saitoh H. et al. Aldosterone synthase (CYP11B2) expression and myocardial fibrosis in the failing human heart // Clin. Sci. — 2002. — Vol. 102. — P. 381–386.
- Schauenstein K., Felsner P., Rinner I. et al. In vivo immunomodulation by peripheral adrenergic and cholinergic agonists/antagonists in rat and mouse models // Ann. N Y Acad. Sci. — 2000. — Vol. 917. — P. 618–627.
- Schefe J.H., Menk M., Reinemund J. et al. A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein // Circ. Res. — 2006. — Vol. 99. — P. 1355–1366.
- Schelekamp M., Derkx F. Biohemistry of prorenin // in «The Renin-Angiotensin System» ed. by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993, Vol. 1, (part 6). — P. 6.1–6.13.
- Schelling P., Fischer H., Ganten D. Angiotensin and cell growth: a link to cardiovascular hypertrophy? // J. Hypertens. — 1991. — Vol. 9. — P. 3–15, 1991.
- Schiffrin E.L. Vascular endothelin in hypertension // Vasc. Pharmacol. — 2005. — Vol. 43. — Р. 19–29.
- Schiffrin E.L. Effects of aldosterone on the vasculature // Hypertens. — 2006. — Vol. 47. — P. 312–318.
- Schulman I.H., Zhou M.S., Raij L. Nitric oxide, angiotensin II, and reactive oxygen species in hypertension and atherogenesis // Curr. Hypertens. Rep. — 2005. — Vol. 7. — P. 61–67.
- Schulman I.H., Zhou M-S., Raij L. Cross-talk between angiotensin II receptor types 1 and 2: potential role in vascular remodeling in humans // Hypertens. — 2007. — Vol. 49. — Р. 270–271.
- Sekiguchi K., Li X., Coker M. et al. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure // Cardiovasc.Res. — 2004. — Vol. 63. — P. 433–442.
- Serneri G.G., Boddi M., Cecioni I. et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function // Circ. Res. — 2001. — Vol. 88. — P. 861–863.
- Shagdarsuren E., Wellner M., Braesen J.H. et al. Complement activation in angiotensin II-induced organ damage // Circ. Res. — 2005. — Vol. 97. — P. 716–724.
- Sharma N., Okere I.C., Barrows B.R. et al. High sugar diets increase cardiac dysfunction and mortality in hypertension compared to low carbohydrate or high starch diets // J. Hypertens.2008. — Vol. 26. — P. 1402–1410.
- Sharma N., Okere I.C., Duda M.K. et al. High fructose diet increases mortality in hypertensive rats compared to a complex carbohydrate or high fat diet // Am. J. Hypertens. — 2007. — Vol. 20. — P. 403–409.
- Shenoy U., Cassis L. Characterization of renin activity in brown adipose tissue // Am. J. Physiol. Cell. Physiol. — 1997. — Vol. 272. — P. C989–C999.
- Shibata S., Nagase M., Yoshida S. et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease // Nat. Med. — 2008. — Vol. 14. — P. 1370–1376.
- Shulkes A.A., Gibson R.R., Skinner S.I. The nature of inactive rennin in plasma and amniotic fluid // Clin. Science. — `1978. — Vol. 55. — P. 41–50.
- Simpson J.B. The circumventricular organs and the central actions of angiotensin // Neuroendocrinology. — 1981. — Vol. 32. — Р. 248–256.
- Singh V.P., Baker K.M., Kumar R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production // Am. J. Physiol. Heart Circ. Physiol. — 2008. — Vol. 294. — P. H1675–H1684.
- Singh V.P., Le B., Bhat V.B. et al. High glucose induced regulation of intracellular angiotensin II synthesis and nuclear redistribution in cardiac myocytes // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H939–H948.
- Singh V.P., Le B., Khode R. et al. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis // Diabetes. — 2008. — Vol. 57. — P. 3297–3306.
- Singh R., Leehey D.J. Effect of ACE inhibitors on angiotensin II in rat mesangial cells cultured in high glucose. // Biochem. Biophys. Res. Commun. — 2007. — Vol. 357. — P. 1040–1045.
- Siragy H.M. Renin inhibition: a new modality for hypertension management // Curr. Hypertens. Rep. — 2007. — Vol. 9. — P. 291–294.
- Siragy H.M., Huang J. Renal (pro)renin receptor upregulation in diabetic rats through enhanced angiotensin AT1-receptor and NADPH oxidase activity // Exp.Physiol. — 2008. — Vol. 93. — P. 709–714.
- Skinner S. The pathophysiology of prorenin // in «The Renin-Angiotensin System» ed. by J.Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993, Vol. 1, (part 7). — P. 7.1–7.14.
- Skurk T., Harmelen V., Hauner H. Angiotensin II stimulates the release of interleukin-6 and interleukin-8 from cultured human adipocytes by activation of NF-kB // Arterioscler. Thromb. Vasc. Biol. — 2004. — Vol. 24. — P. 1199–1206.
- Solomon S.D., Appelbaum E., Manning W.J. et al. Effect of the direct renin inhibitor aliskiren, the angiotensin receptor blocker Losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy // Circulation. — 2009. — Vol. 119. — P. 530–537.
- Solomon S.D., Shin S.H., Shah A. et al. Effect of the direct rennin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction // Eur. Heart J. — 2011. — Vol. 32. — Р. 1227–1234.
- Stankovic A.., Fisher N.D., Hollenberg N.K. Prorenin and angiotensin-dependent renal vasoconstriction in type 1 and type 2 diabetes // J. Am. Soc. Nephrol. — 2006. — Vol. 17. — Р. 293–299.
- Stanley A.G., Patel H., Knight A.L et al. Mechanical strain-induced human vascular matrix synthesis: The role of angiotensin II // JRAAS. — 2000. — Vol. 1. — P. 32–35.
- Sugden J.A., Davies J.I., Witham M.D. Vitamin D improves endothelial function in patients with Type 2 diabetes mellitus and low vitamin D levels // Diabet Med. — 2008. — Vol. 25. — P. 320–325.
- Sukenaga Y., Kamoshita K.,Takai S. et al. Development of the chymase inhibitor as an anti-tissue remodelling drug in myocardial infarction and some other possibilities // Jpn. J. Pharmacol. — 2002. — Vol. 90. — P. 218–222.
- Sun Y. Myocardial repair/remodelling following infarction: roles of local factors // Cardiovasc. Res. — 2009. — Vol. 81. — P. 482–490.
- Sun Y, Ahokas RA, Bhattacharya SK. et al. Oxidative stress in aldosteronism // Cardiovasc. Res. — 2006. — Vol. 71. — Р. 300–309.
- Sun Y., Zhang J., Lu L. et al. Aldosterone-induced inflammation in the rat heart: role of oxidative stress // Am. J. Pathol. — 2002. — Vol. 161. — P. 1773–1781.
- Suter C., Coote J.H. Intrathecally administered angiotensin II increases sympathetic activity in the rat // J. Auton. Nerv. Syst. —1987. — Vol. 19. — Р. 31–37.
- Suzuki J., Iwai M., Mogi M. et al. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation // Arterioscler. Thromb. Vasc. Biol. — 2006. — Vol. 26. — Р 917–921.
- Taegtmeyer H. Insulin resistance and atherosclerosis. Common roots for two common diseases? // Circulation. — 1996. — Vol. 93. — P. 1777–1779.
- Takai S., Shiota N., Yamamoto D. et al. Purification and characterization of angiotensin II-generating chymase from hamster cheek pouch // Life Sci. — 1996. — Vol. 58. — P. 591–597.
- Takeda Y., Yoneda T., Demura M. et al. Effects of high sodium intake on cardiovascular aldosterone synthesis in stroke-prone spontaneously hypertensive rats // J. Hypertens. — 2001. — Vol. 19. — P. 635–639.
- Tare M., Emmett S.J., Coleman H.A. et al. Vitamin D insufficiency is associated with impaired vascular endothelial and smooth muscle function and hypertension in young rats // J. Physiology. — 2011. — Vol. 589. — Р. 4777–4786.
- Tian N., Moore R.S., Phillips W.E. et al. NADPH oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2008. — Vol. 295. — P. R1858–R1865.
- Tomaschitz A., Maerz W., Pilz S. et al. Aldosterone/Renin Ratio Determines Peripheral and Central Blood Pressure Values Over a Broad Range // JACC. — 2010. — Vol. 55. — P. 65–73.
- Touyz R.M., Mercure C., He Y. et al. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase // Hypertens. — 2005. — Vol. 45. — P. 530–537.
- Touyz R.M., Yao G., Quinn M.T. et al. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II // Arterioscler. Thromb. Vasc. Biol. — 2005. — Vol. 25. — P. 512–518.
- Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials // Lancet. — 2003. — Vol. 362. — P. 1527–1535.
- Turner A.J., Hooper N.M. The angiotensin-converting enzyme gene family: genomics and pharmacology // Trends Pharmacol. Sci. — 2002. — Vol. 23. — P. 177–183.
- Uehara Y., Urata H., Ideishi M.et al. Chymase inhibition supresses high-cholesterol diet-induced lipid accumulation in the hamster aorta // Cardiovasc. Res. — 2002. — Vol. 55, № 1. — P. 870–876.
- Uemura L.M., Tsutsumi T., Yamazaki T. et al. Efferent vagal nerve stimulation induces tissue inhibitor of metalloproteinase-1 in myocardial ischemia-reperfusion injury in rabbit // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H2254–H2261.
- Urata H. Pathological involvement of chymase-dependent angiotensin II formation in the development of cardiovascular disease // JRAAS. — 2000. — Vol. 1, (suppl.). — P. S35–S37.
- van Kats J.P., Methot D., Paradis P. et al. Use of a biological peptide pump to study chronic peptide hormone action in transgenic mice. Direct and indirect effects of angiotensin II on the heart // J. Biol. Chem. — 2001. — Vol. 276. — P. 44012–44017.
- Velez J.C. The importance of the intrarenal renin-angiotensin system // Nat. Clin. Pract. Neph. — 2009. — Vol. 5. — Р. 89–100.
- Véniant M., Ménard J., Bruneval P. et al. Vascular damage without hypertension in transgenic rats expressing prorenin exclusively in the liver // J. Clin. Invest. — 1996. — Vol. 98. — Р. 1966–1970.
- Verlohren S., Muller D.N., Luft F.C. Immunology in hypertension, preeclampsia, and target-organ damage // Hypertens. — 2009. — Vol. 54. — P. 439–446.
- Wang G., Anrather J., Glass M.J. et al. Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in nucleus tractus solitarius // Hypertens. — 2006. — Vol. 48. — P. 482–489.
- Wang H., Huang B.S., Ganten D. et al. Prevention of Sympathetic and Cardiac Dysfunction After Myocardial Infarction in Transgenic Rats Deficient in Brain Angiotensinogen // Circ. Res. — 2004. — Vol. 94. — P. 843–849.
- Wang T.J., Pencina M.J., Booth S.L. et al. Vitamin D deficiency and risk of cardiovascular disease // Circulation. — 2008. — Vol. 117. — P. 503–511.
- Wassmann S., Czech T., van Eickels M.et al. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice // Circulation. — 2004. — Vol. 110. — P. 3062–3067.
- Watanabe S., Tagawa T., Yamakawa K. et al. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans // Arterioscler. Thromb. Vasc. Biol. — 2005. — Vol. 25. — P. 2376–2383.
- Wei Y., Sowers J.R., Nistala R. et al. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells // AJP — Regu. Physiol. — 2008. — Vol. 294. — Р. R673–R680.
- Wei Y., Whaley-Connell A.T., Chen K. et al. NADPH oxidase contributes to vascular inflammation, insulin resistance and remodeling in the transgenic (mRen2) rat // Hypertens. — 2007. — Vol. 50. — Р. 384–391.
- Whaley-Connell A., Govindarajan G., Habibi J. et al. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic TG (mRen2) 27 Ren2 rat. // Am. J. Physiol. Endocrinol. Metab. — 2007. — Vol. 293. — Р. E355–E363.
- Xu J., Carretero O.A., Cavasin M.A. et al. Role of cardiac overexpression of angiotensin ii in the regulation of cardiac function and remodeling post-myocardial infarction // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H1900–H1907.
- Xu J., Carretero O.A., LiaoT-D. et al. Local angiotensin II aggravates cardiac remodeling in hypertension // AJP — Heart. — 2010. — Vol. 299. — Р. H1328–H1338.
- Xu H., Fink G.D., Chen A. et al. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats // Am. J. Physiol. Heart Circ. Physiol. — 2001. — Vol. 281. — P. H975–H980.
- Yamamoto M., Yang G., Hong C. et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy // J. Clin. Invest. — 2003. — Vol. 112. — P. 1395–1406.
- Yamamoto N., Yasue H., Mizuno Y. et al. Aldosterone is produced from ventricles in patients with essential hypertension // Hypertens. — 2002. — Vol. 39. — P. 958–962.
- Yamamuro M., Yoshimura M., Nakayama M. et al. Direct effects of aldosterone on cardiomyocytes in the presence of normal and elevated extracellular sodium // Endocrinology. — 2006. — Vol. 147. — P. 1314–1321.
- Yoshimura M., Kawai M. Synergistic inhibitory effect of angiotensin II receptor blocker and thiazide diuretic on the tissue renin-angiotensin-aldosterone system //JRAAS. — 2010. — Vol. 11. — Р. 124–12.
- Zeitz C.J., Campbell D.J., Horowitz J.D. Myocardial uptake and biochemical and hemodynamic effects of ACE inhibitors in humans // Hypertens. — 2003. — Vol. 41. — P. 482–487.
- Zhang Y., Jang R., Mori T.A. et al. The antioxidant tempol reverses and partially prevents adrenocorticotrophic hormone-induced hypertension in the rat // J. Hypertens. — 2003. — Vol. 21. — P. 1513–1518.
- Zhang G.X., Kimura S., Nishiyama A. et al. ROS during the acute phase of Ang II hypertension participates in cardiovascular MAPK activation but not vasoconstriction // Hypertens. — 2004. — Vol. 43. — Р. 117–124.
- Zhang Q., Malik P., Pandey D. et al. Paradoxical аctivation of endothelial nitric oxide synthase by NADPH oxidase // Аrterioscler. Thromb. Vasc. Biol. — 2008. — Vol. 28. — P. 1627–1633.
- Zhang J., Noble N.A., Border W.A. et al.Receptor-dependent prorenin activation and induction of PAI-1 expression in vascular smooth muscle cells // Am. J. Physiol. Endocrinol. Metab. — 2008. — Vol. 295. — P. E810–E819.
- Zhou M.., Schulman I.H., Pagano P.J. et al. Reduced NAD(P)H oxidase in low renin hypertension. Link among angiotensin II, atherogenesis, and blood pressure // Hypertens. — 2006. — Vol. 47. — P. 81–86.
- Zhou M., Schulman I.H., Raij L. Role of angiotensin II and oxidative stress in vascular insulin resistance linked to hypertension // AJP — Heart. — 2009. — Vol. 296. — Р. H833–H839.
- Zhou L., Xi B., Wei Y. et al. Meta-analysis of the association between the insertion/deletion polymorphism in ACE gene and coronary heart disease among the Chinese population // JRAAS. — 2012. — Vol. 13. — P. 296–304.
- Zhu J., Gao D. Losartan reduces collagen content and intimal thickening of iliac arteries after balloon injury in rabbits // JRAAS. — 2000. — Vol. 1. — P. 278–282.
- Zhu Y.C., Zhu Y.Z., Lu N. et al. Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and cardiac remodelling // Clin. Exp. Pharmacol. Physiol. — 2003. — Vol. 30. — Р. 911–918.
- Zhu Z., Zhu S., Wu Z. et al. Effect of sodium on blood pressure, cardiac hypertrophy, and angiotensin receptor expression in rats // Am. J. Hypertens. — 2004. — Vol. 17. — Р. 21–24.
- Zhuo J.L, Imig J.D., Hammond T.G. et al. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT(1) receptor // Hypertens. — 2002. — Vol. 39. — Р. 116–121.
- Zhuo J.L., Li X.C. Novel roles of intracrine angiotensin II and signalling mechanisms in kidney cells // JRAAS. — 2007. — Vol. 8. — P. 23–33.
- Zimmerman M.C., Lazartigues E., Lang J.A. et al. Superoxide mediates the actions of angiotensin II in the central nervous system // Circ. Res. — 2002. — Vol. 91. — P. 1038–1045.
- Zimmerman M.C., Lazartigues E., Sharma R.V. et al. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system // Circ. Res. — 2004. — Vol. 95. — Р. 210–216.
- Zucker I.H., Schultz H.D., Patel K.P. et al. The regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure // Am. J. Physiol. Heart Circ. Physiol. — 2009. — Vol. 297. — P. H1557–H1566.