Ренин-ангиотензиновая система (РАС) относится к числу важнейших регуляторных систем и принимает участие в поддержании гомеостаза организма посредством влияния практически на все аспекты его жизнедеятельности. Несмотря на то что со времени описания РАС прошло более века, до настоящего времени еще не сложилось полного представления об ее функциональной организации и механизмах реализации функционального назначения, так как основное внимание уделялось участию РАС в регуляции деятельности сердечно-сосудистой системы и поддержании водно-электролитного баланса организма.
Помимо этого, повышение активности РАС, имеющее в своей основе адаптивно-компенсаторную направленность, при чрезмерной выраженности или длительности приобретает патологический характер и приводит к развитию различных форм патологии, в частности — кардиоваскулярной. Все это обусловливает необходимость более широкого и углубленного подхода к оценке функциональной и патологической значимости РАС, постоянному уточнению и, при необходимости, пересмотру даже уже устоявшихся положений.
1.1. Основные этапы развития представлений об организации и функциональной значимости РАС
В становлении представлений о РАС, ее функциональной значимости и роли в развитии различных патологических состояний отчетливо прослеживается ряд основных этапов [197]. История открытия РАС и изучения ее роли в развитии артериальной гипертензии (АГ) берет начало в 1898 г., когда Robert Tigerstedt, профессор физиологии Каролинского института в Стокгольме, выделил и описал почечный фермент, вызывавший развитие выраженного прессорного эффекта, и дал ему название «ренин» [185, 251]. С этого времени прошло уж более 100 лет, однако функциональная значимость, принципы функционирования и регуляции активности РАС, ее роль в развитии кардиоваскулярной патологии во многих аспектах еще остаются окончательно не установленными.
Исследования, проводившиеся непосредственно после открытия и описания ренина, мало что прояснили в проблеме физиологической значимости РАС вплоть до 1934 г., когда были опубликованы результаты экспериментов Голдблатта и соавторов. В этих экспериментах была показана возможность воспроизведения стабильной выраженной АГ посредством уменьшения просвета почечных артерий, хотя авторы и не рассматривали усиленную секрецию ренина почками как причину развития АГ.
Позже было показано, что на протяжении 18-дневной инфузии ренина артериальное давление (АД) стабильно поддерживается на высоком уровне, несмотря на то что сам ренин не оказывает констрикторного действия. Установлено, что он является ферментом, а повышение АД при инфузии ренина связано с расщеплением субстрата — ангиотензиногена, находящегося в плазме крови, с образованием активного соединения, которое получило название ангиотензин (А).
Результаты дальнейших исследований свидетельствовали о том, что процесс образования активного А проходит в 2 этапа: на первом этапе из ангиотензиногена под действием ренина образуется неактивный А I, который превращается в активную форму — А II с участием ангиотензинпревращающего фермента (АПФ).
Факт преобразования неактивного А I в активный сосудосуживающий фактор А II был установлен в 1950-е годы [234], и показано, что АПФ не только определяет образование А II, но и одновременно участвует в расщеплении до неактивных фрагментов вазодилататорного вещества — брадикинина [55]. Было установлено, что АПФ продуцируется главным образом эндотелиоцитами легочных и почечных сосудов, и его угнетение сопровождается уменьшением образования А II, секреции альдостерона и деградации брадикинина.
Затем в условиях эксперимента было показано, что А II, в зависимости от дозы, способен вызывать как быстро, так и медленно развивающиеся прессорные эффекты: в высоких дозах он приводит к быстрому развитию АГ, тогда как инфузия А II в дозах, не вызывающих немедленного повышения АД, сопровождалась отсроченным развитием АГ.
В 1956 г. был впервые успешно синтезирован А II, что положило начало многочисленным экспериментальным исследованиям физиологической и патофизиологической значимости РАС. Одним из важнейших итогов этих исследований было установление зависимости между активностью РАС и интенсивностью синтеза и секреции альдостерона. Это положение значительно расширило представления о роли РАС в регуляции гомеостатического состояния организма и развитии различных патологических реакций. Прежде всего, было отмечено, что на фоне сочетанного применения минералокортикоида дезоксикортикостерона ацетата (ДОКА) и солевой нагрузки выраженность прессорного действия инфузии ренина у крыс резко увеличивалась. Это объяснили исходным снижением активности ренина в плазме крови в результате угнетения его высвобождения из клеток юкстагломерулярного аппарата.
В дальнейшем это предположение подтвердилось результатами исследований, в которых аналогичный эффект отмечен и после двусторонней нефрэктомии. Помимо этого, солевая нагрузка в сочетании с ДОКА сопровождалась уменьшением количества секреторных гранул, содержащих ренин, в мезангиальных клетках почек, тогда как ограничение потребления соли — их увеличением. При этом выявлена четкая зависимость между степенью грануляции юкстагломерулярных клеток, высвобождением ренина и секрецией альдостерона.
Было также подтверждено, что секреция ренина находится в обратной зависимости от плазменного содержания натрия, а РАС тесно и однонаправленно взаимодействует с альдостероном как в физиологических, так и в патологических условиях. Так, в исследовании, проведенном с участием здоровых добровольцев, было показано, что введение А II сопровождалось 2–10-кратным повышением экскреции альдостерона с мочой.
Однако эти данные противоречили тому, что у лиц с низким потреблением соли уровень альдостерона в плазме крови повышался непропорционально высоко в сопоставлении с уровнем ренина и А II, и отношение альдостерон/А II резко возрастало по сравнению с нормой. Это поставило под сомнение значимость А II как важнейшего фактора регуляции секреции альдостерона.
Результаты дальнейших исследований, проведенных как в эксперименте, так и на здоровых добровольцах, позволили установить, что выраженность влияния А II на образование и высвобождение альдостерона существенно изменяется в зависимости от плазменной концентрации натрия в диапазоне от значительного снижения при гипернатриемии до выраженного повышения при гипонатриемии. Наличие подобной зависимости рассматривалось как подтверждение тесной связи между РАС и альдостероном в регуляции солевого баланса организма в условиях гипонатриемии.
Этому положению противоречили данные об отсутствии способности саралазина — антагониста А II снижать уровень альдостерона в крови. Однако оказалось, что этот эффект характерен только при условии нормального солевого баланса, когда концентрация А II сохраняется на низком уровне, а при ограниченном потреблении соли и высоком плазменном уровне А II применение саралазина сочеталось со снижением уровня альдостерона до нормальных значений. Эти данные затем были подтверждены в исследованиях с участием людей, в экспериментах на бодрствующих собаках и кроликах.
Особое значение имело установление изменений активности РАС в зависимости от солевого баланса организма. Было показано, что ограничение потребления соли только на 10 ммоль/сут приводит к выраженному и прогрессирующему повышению концентрации ренина в плазме крови, и она возвращается к контрольному уровню через несколько дней после восстановления нормальной солевой диеты. Напротив, у лиц, потреблявших 400 ммоль соли ежедневно, отмечено прогрессирующее снижение уровня ренина в крови и его восстановление после перехода на нормальную солевую диету. Выявлено также, что ограничение потребления соли до 12 ммоль/сут в течение 5 дней сочеталось с достоверным повышением плазменной концентрации А II, а возврат к нормальной солевой диете сочетался с быстрой нормализацией уровня А II в крови.
Сложный характер связи между секрецией альдостерона, активностью РАС, состоянием водно-солевого обмена и уровнем АД продемонстрирован в исследованиях 1960-х годов. Установлено, что альдостерон оказывает угнетающее влияние на синтез и высвобождение ренина. В результате уровень ренина в плазме крови у лиц с адренокортикальной недостаточностью повышен более чем в 10 раз и нормализуется после замещающей терапии, тогда как при синдроме Кона и первичной гиперальдостеронемии отмечено резкое снижение плазменного уровня ренина и А II в сочетании с повышением АД. Лечение этих пациентов с применением спиронолактона — антагониста альдостерона, или посредством хирургического удаления опухоли надпочечников сопровождалось нормализацией плазменной концентрации натрия и калия, снижением АД.
Результатом существенного прогресса в понимании функциональной организации РАС была концепция локальных или тканевых РАС [55]. Эта концепция базировалась на идентификации компонентов РАС в необычных местах, прежде всего — в головном мозгу, где их наличие не могло быть объяснено с позиции эндокринного принципа организации системы. В последующем это привело к формированию новых взглядов, в соответствии с которыми функциональным назначением тканевых РАС является локальный синтез А II. Это предположение было подтверждено установлением экспрессии мРНК различных компонентов РАС во многих тканях, их гиперэкспрессия или удаление у мышей позволили детально определить функцию локальных РАС [9, 37, 38, 129 и др.].
В 1970-е годы началась эра применения специфических ингибиторов РАС, прежде всего — саралазина и аланина. Эти вещества обладают высоким сродством к рецепторам А II и слабой, но все же заметной агонистической активностью. Кратковременное применение саралазина у лиц с низкой концентрацией А II в плазме крови проявлялось умеренным, но закономерным повышением АД и плазменной концентрации альдостерона, а при длительной инфузии саралазина у крыс отмечено развитие стабильной АГ. Поэтому препараты этой группы применяли в клинической практике очень ограниченно и кратковременно.
Затем было установлено, что яд бразильской змеи Bothobs pararaca содержит пептиды, которые угнетают действие АПФ — фермента, осуществляющего образование А II из А I. Синтетические аналоги этих пептидов, прежде всего — каптоприл, получили широкое распространение в клинической практике, особенно в лечении АГ, а по антигипертензивному эффекту они оказались сопоставимыми с блокаторами β-адренорецепторов и тиазидными диуретиками.
Однако в последние годы выявлено, что длительное применение ингибиторов АПФ приводит к повышению плазменной концентрации ренина и А I с возможным развитием отрицательных эффектов. Поэтому вплоть до настоящего времени не прекращается поиск более эффективных ингибиторов РАС, которые являются специфическими блокаторами ренина и рецепторов А II, о чем и будет идти речь далее.
Особое значение в развитии представлений о физиологической и патологической значимости РАС имели исследования последних лет, в которых была установлена роль системы ренина/проренина и их рецепторов в процессах воспаления и ремоделирования сердца, сосудов и почек. Установлено, что эти эффекты в значительной степени не зависят от классического пути реализации действия РАС, который осуществляется через А II, альдостерон и их рецепторы. Однако до настоящего времени механизмы реализации этих эффектов остаются невыясненными во многих аспектах и продолжают интенсивно изучаться.
К важнейшим этапам развития представлений о регуляторной и патологической значимости РАС относится также установление ее непосредственных связей с вегетативной нервной системой, особенно симпатическим ее отделом, как на центральном, так и на периферическом уровнях, а также с иммунной системой.
Идентификация АПФ2 в 2000 г. открыла новую главу в исследованиях регуляторной роли РАС. Было установлено, что АПФ2 является начальным звеном так называемой альтернативной РАС, и его активация сопровождается уменьшением содержания в крови и тканях А ΙΙ и увеличением — А (1–7) – антиатеросклеротического пептида. Он является лигандом Mas-рецептора, сопряженного с G-белком, и интенсивно экспрессируется в сердечно-сосудистой системе. Выявлено, что А (1–7), в отличие от А II, оказывает антипролиферативное, противовоспалительное и антиоксидативное действие, замедляет развитие атеросклероза. Установлено наличие реципрокных отношений между активностью АПФ и АПФ2, а применение ингибиторов АПФ приводило к повышению экспрессии мРНК АПФ2 в сердце крысы. В настоящее время проблеме этой альтернативной РАС уделяется особое внимание как в теоретическом, так и практическом аспектах.
1.2. Современные представления о компонентах РАС и ее функции в норме и патологии
РАС имеет важнейшее значение в регуляции ряда физиологических и патофизиологических состояний в организме, определяет уровень АД, водно-солевой гомеостаз, метаболические процессы, принимает участие в развитии АГ, атеросклероза, ишемической болезни сердца (ИБС), сердечной недостаточности, влияет на характер их клинического течения.
Повышение активности РАС — адаптивный механизм, играющий защитную роль в поддержании функции сердца и центральной гемодинамики при их острых нарушениях. По мере снижения функции сердца уменьшается перфузия почек, параллельно активируется симпатоадреналовая система, что в совокупности приводит к усиленной секреции ренина. Однако длительная активация РАС сопровождается появлением отрицательных эффектов, прежде всего — развитием и прогрессированием АГ и сердечной недостаточности [258].
В настоящее время не вызывает сомнений, что повышенная активность РАС и плазменная концентрация А II как ее медиатора занимают важное место среди факторов сердечно-сосудистой патологии и прежде всего для АГ, что обусловливает широкое применение в кардиологической клинике препаратов, снижающих активность РАС. Однако, несмотря на то что ингибиторы АПФ и блокаторы рецепторов А II 1-го типа (АТ1-рецепторов) в качестве антигипертензивной терапии применяются уже более 10 лет, частота тяжелых и летальных исходов кардиоваскулярных, цереброваскулярных и почечных заболеваний остается высокой даже при эффективном контроле АД.
В соответствии с классическими представлениями, основные функции РАС — регуляция АД и поддержание водно-солевого баланса. При уменьшении объема жидкости и сопутствующем нарушении перфузии почек или развитии гипонатриемии активируются клетки юкстагломерулярного аппарата почек с секрецией ренина и последующим образованием А II.
Значимость РАС в регуляции АД особенно отчетливо проявляется у мышей с генетическим отсутствием АПФ, ангиотензиногена или АТ1-рецепторов, у которых среднее АД равно 73 мм рт. ст. по сравнению со 110 мм рт. ст. в норме. У мышей с отсутствием ангиотензиногена или АТ1-рецепторов АД сохраняется примерно на том же уровне, что и у мышей с отсутствием АПФ. Эти данные означают, что даже при наличии активности ряда систем, регулирующих АД, отсутствие функционирующей РАС неизбежно приводит к снижению АД, свидетельствуя о важности РАС в его поддержании. В то же время зависимость между активностью циркулирующей РАС и уровнем АД нечетко выражена, что указывает на относительно малое значение содержащегося в крови АПФ в регуляции АД. Так, у мышей с концентрацией АПФ в крови на уровне 75% нормальной, но с отсутствием тканевого АПФ (в легких и в почках), системное АД снижено практически до уровня, отмеченного при полном отсутствии АПФ.
Участие РАС в регуляции водно-солевого обмена имеет сложный характер, для мышей с полным отсутствием активности АПФ характерны повышенный плазменный уровень креатинина и уменьшение его клиренса, тогда как у мышей с отсутствием тканевого АПФ, но наличием АПФ в крови, эти изменения не возникают. В то же время у мышей обеих линий отмечается усиленная экскреция калия и повышенная его концентрация в моче, тогда как при отсутствии гена ангиотензиногена уровень калия в плазме крови значительно повышен на фоне снижения концентрации А II, что способствует усиленной продукции альдостерона и частично компенсирует дефицит А II [37, 38].
Роль РАС в регуляции водно-солевого обмена и АД детально изучена в условиях низкосолевой диеты. При ограниченном потреблении соли внеклеточный объем жидкости значительно уменьшается, но АД снижается только умеренно благодаря повышению активности РАС, которая предупреждает развитие гипотензии. Противоположный эффект отмечается при солевой нагрузке, при которой, несмотря на выраженное увеличение внеклеточного объема жидкости, АД повышается умеренно в результате снижения активности РАС.
На фоне применения ингибиторов АПФ и блокады РАС зависимость между АД и водно-солевым балансом приобретает пропорциональный характер, а изменение потребления соли оказывает более выраженное действие на АД [147]. Показано, что ограничение потребления соли пациентами с АГ сочетается с более значимым снижением АД и меньшим приростом активности РАС, чем у нормотензивных лиц, у которых отмечается выраженное повышение активности РАС с незначительным снижением АД. Этот же механизм объясняет наличие выраженного гипотензивного эффекта при ограничении потребления соли у пациентов с АГ, получающих терапию каптоприлом, который на 8/5 мм рт. ст. превышает эффект низкосолевой диеты у лиц с АГ, не получавших антигипертензивной терапии [147].
У здоровых собак увеличение потребления соли от 5 до 245 ммоль/сут не вызывало заметных изменений АД, и умеренно выраженный гипертензивный эффект отмечался только при суточной дозе, равной 495 ммоль. Однако при постоянной инфузии А II в низкой субпрессорной концентрации или при применении каптоприла солевая нагрузка сопровождалась развитием выраженной АГ [42].
Гиперактивность РАС сопровождается развитием ряда патологических эффектов и отмечается при многих патологических состояниях, прежде всего — поражениях сердечно-сосудистой системы. Эти эффекты развиваются в результате митогенного действия А II и альдостерона на сосудистые гладкомышечные клетки, фибробласты и кардиомиоциты, на их способности повышать провоспалительный и профибротический потенциал, индуцировать оксидативный стресс и дисфункцию эндотелия, активировать атерогенез, неоангиогенез в атеросклеротической бляшке с развитием ее нестабильности. Угнетение РАС предупреждает прогрессирование диабетической нефропатии, повышает выживаемость у пациентов с АГ, застойной сердечной недостаточностью и перенесенным инфарктом миокарда (ИМ), способствует регрессии гипертрофии левого желудочка (ЛЖ), улучшает исход у пациентов высокого кардиоваскулярного риска.
Начальным звеном в РАС является ренин, который секретируется почками и накапливается в гранулах мезангиальных клеток, входящих в состав юкстагломерулярного аппарата. В ряде исследований показано, что ренин может также синтезироваться и в других тканях, но этот вопрос остается еще малоизученным. Установлено, что у лиц с нефрэктомией активность ренина в плазме крови равна только 7% нормальной, плазменный уровень А I и А II также достигает только 6% нормального. Эти данные являются одним из доказательств доминантной значимости почек в секреции ренина и ренина в образовании пептидов А [24–26].
Почки секретируют также проренин, и его концентрация в крови превышает содержание ренина в 7–10 раз. Хотя почки являются единственным известным источником образования ренина, его предшественник — проренин — может образовываться и в ряде других тканей [26]. Биологическая значимость этого явления остается в настоящее время не полностью определенной и привлекает внимание многих исследователей.
В настоящее время идентифицированы 4 основных фактора, оказывающих регуляторное влияние на высвобождение ренина:
1 — барорецепторный аппарат в афферентных артериолах почек, который активируется снижением и угнетается повышением перфузионного давления в почечных сосудах;
2 — изменение концентрации NaCl, сенсором которой являются клетки дистальных канальцев почек (macula densa cells);
3 — активирующее действие симпатической нервной системы, которое осуществляется через β1-адренорецепторы;
4 — прямое угнетающее действие А II и альдостерона.
Секреция ренина является ключевым фактором активации РАС и сопровождается каталитическим гидролизом ангиотензиногена с высвобождением А I. Существенное угнетающее влияние на высвобождение ренина почками оказывают А II и альдостерон. Поэтому применение ингибиторов АПФ или блокаторов АТ1-рецепторов стимулирует усиленную секрецию ренина и повышение его плазменной активности.
Ангиотензиноген продуцируется главным образом в печени, но доказана его секреция и в других тканях, прежде всего — в периваскулярной жировой ткани и в фибробластоподобных клетках.
Плазменный уровень ангиотензиногена стабилен и не претерпевает острых изменений, однако его синтез и высвобождение увеличиваются под действием глюкокортикоидов, эстрогенов, половых, тиреоидных гормонов, воспалительных цитокинов (интерлейкина (ИЛ)-1, фактора некроза опухоли (ФНО)-α), А II. Концентрация ангиотензиногена в плазме крови снижается при недостаточности надпочечников, гипотиреоидизме, дефиците инсулина. Повышение уровня ангиотензиногена является фактором развития АГ и отмечается в период беременности, при воспалении, синдроме Кушинга, применении глюкокортикоидов. Однако при хроническом повышении уровня ангиотензиногена отмечается компенсаторное уменьшение секреции ренина.
В отличие от быстрых изменений плазменного содержания ренина, которые происходят в течение секунд или минут, изменения концентрации ангиотензиногена развиваются на протяжении суток и дней. Характерно, что А II, являющийся ингибитором высвобождения ренина в почках, стимулирует высвобождение ангиотензиногена в печени.
Показано, что изменения плазменного уровня ангиотензиногена не являются основной детерминантой активности РАС. В то же время, концентрация ренина в плазме крови может изменяется в 1000 раз (в 10 раз — в большинстве физиологических ситуаций), и диапазон его действия в значительной степени определяется концентрацией ангиотензиногена, который находится как в плазме крови, так и во внеклеточной жидкости.
В нормальных условиях содержание ангиотензиногена в плазме крови ниже порога его взаимодействия с ренином. Поэтому он рассматривается как фактор, лимитирующий активность РАС, ее способность контролировать АД, а введение экзогенного ангиотензиногена приводит к повышению АД. Полагают, что развитие ряда форм вторичной АГ, в частности — при приеме контрацептивов, при синдроме Кушинга, определяется повышением концентрации ангиотензиногена под действием эстрогенов или глюкокортикоидов.
Ангиотензиноген относится к числу белков острой фазы, и введение липополисахарида — индуктора воспаления — сопровождается 5-кратным повышением экспрессии мРНК ангиотензиногена в печени. Установлено, что этот эффект опосредован действием провоспалительных цитокинов: ИЛ-1, -6, ФНО-α, интерферона (ИФН)-γ. У пациентов с пневмонией, острым пиелонефритом, различными стрептококковыми инфекциями уровень ангиотензиногена повышен на 70%, что обусловливает участие РАС и А II в развитии иммунного ответа [157].
А I гидролизуется АПФ — мембраносвязанным ферментом, который также активирует катализ других пептидов, в частности — вазодилататорного пептида брадикинина, что приводит к вазоконстрикции. Хотя основным активным фактором в РАС является А II, существуют и другие метаболиты А I с выраженной биологической активностью (А III и А IV), роль которых пока недостаточно выяснена.
Идентифицированы 4 типа рецепторов А II, которые обусловливают его биологическую активность (рис. 1). А II через АТ1-рецепторы оказывает вазоконстрикторное действие и инотропное влияние на сердце, стимулирует продукцию альдостерона в клубочковой зоне надпочечников. Через рецепторы 2-го типа (АТ2-рецепторы) А II оказывает диаметрально противоположные эффекты, рецепторы 4-го типа опосредуют высвобождение ингибитора активатора плазминогена-1 (PAI-1), тогда как функция рецепторов 3-го типа остается невыясненной.

Помимо этого, результаты исследований последних лет свидетельствуют о существовании в сердце, мозгу, почках рецепторов ренина/проренина, главным образом — в субэндотелиальных гладкомышечных клетках. Предполагают, что их активация связана с усилением экспрессии митогенактивирующих протеинкиназ, посредством которых ренин и проренин могут оказывать прямой митогенный эффект и вызывать ремоделирование независимо от А II.
А II является мощным вазоактивным пептидом, вызывает сокращение сосудистой стенки и повышение АД. А II также стимулирует высвобождение альдостерона из коркового слоя надпочечников, а альдостерон активирует реабсорбцию натрия и воды в почках с увеличением объема циркулирующей крови и повышением АД. Поэтому длительное повышение активности РАС сопровождается развитием перманентной АГ. Однако патофизиологическая роль гиперактивности РАС не ограничивается ее гипертензивным эффектом и включает участие в ремоделировании сердца и сосудов, развитии сердечной недостаточности, недостаточности почек и осложнений сахарного диабета (СД).
А II оказывает мощное вазоконстрикторное действие; в почках он вызывает более выраженную констрикцию эфферентных артериол, чем афферентных, благодаря чему способствует повышению гломерулярного давления и фильтрации жидкости из крови. Однако в связи с уменьшением гидростатического и повышением онкотического давления крови в эфферентом отделе почечных клубочков этот эффект уравновешивается увеличением реабсорбции жидкости. Несмотря на уменьшение объема перфузии почек, скорость гломерулярной фильтрации поддерживается на стабильном уровне на фоне уменьшения экскреции натрия, что также способствует реабсорбции жидкости в периферических отделах нефрона.
Прямое интраренальное действие А II заключается в констрикции почечных сосудов, усиленной реабсорбции натрия, что способствует развитию АГ и застойной сердечной недостаточности. Однако при резко выраженном нарушении перфузии почек А II приводит к преимущественному сужению эфферентных артериол, тогда как афферентные более рефрактерны к его действию. Благодаря этому А II способен поддерживать гломерулярное перфузионное давление и, таким образом, скорость гломерулярной фильтрации. Усиленной реабсорбции натрия и воды способствует также альдостерон, который высвобождается из корковой зоны надпочечников под действием А II.
Центральное действие А II заключается, прежде всего, в стимуляции образования в гипоталамусе и высвобождения из задней доли гипофиза антидиуретического гормона, который обладает вазоконстрикторными свойствами, усиливает реабсорбцию натрия и вызывает ощущение жажды.
Нарушение функции эндотелия при действии А II в значительной степени определяется его способностью оказывать повреждающее действие на клетки — предшественники эндотелиоцитов. В культуре этих клеток применение А II сопровождалось развитием оксидативного стресса и последующего апоптоза. В условиях in vivo инфузия А II у мышей приводила к значительному уменьшению количества и нарушению функционального состояния предшественников эндотелиоцитов, но этот эффект не развивался у животных с генетическим отсутствием АТ1-рецепторов или на фоне применения их блокатора кандесартана. Применение кандесартана у пациентов со стабильной стенокардией в течение 3 мес сопровождалось значительным увеличением количества циркулирующих CD34/KDR-положительных клеток, которые являются предшественниками эндотелиоцитов [60].
А II является основным конечным медиатором РАС и его действие имеет решающее значение в развитии кардиоваскулярной патологии, такой как АГ, атеросклероз, ИБС, рестеноз и сердечная недостаточность.
В реализации влияния А II на сердце и сосуды принимают участие 2 типа рецепторов. Через АТ1-рецепторы опосредуются основные физиологические и патофизиологические сердечно-сосудистые эффекты А II, тогда как роль рецепторов 2-го типа остается спорной. В нормальных условиях они мало представлены в сердечно-сосудистой системе и выявляются прежде всего в сосудистом эндотелии. Однако их экспрессия значительно повышается при различных патологических состояниях, связанных с воспалением и ремоделированием сердца и сосудов: АГ, атеросклерозе, СД 2-го типа и ИМ. Полагают, что АТ2-рецепторы способны медиировать вазодилатацию, угнетать рост и пролиферацию кардиомиоцитов и сосудистых гладкомышечных клеток, стимулировать их апоптоз. Через АТ2-рецепторы осуществляется также антиангиогенное действие А II, предупреждаются гипертрофия и ремоделирование сердца и сосудистой стенки [132, 133].
К факторам, инициирующим экспрессию AT2-рецепторов, относятся различные цитокины, тогда как угнетение их экспрессии связано прежде всего с глюкокортикоидами. Через AT2-рецепторы А ІІ усиливает продукцию как брадикинина, так и NO, что вызывает вазодилатацию. Поэтому у мышей с отсутствием AT2-рецепторов отмечается значительно усиленный констрикторный ответ на А ІІ.
В ряде исследований показано, что через AT1-рецепторы реализуется способность А II вызывать развитие вазоконстрикции, стимулировать NADPH-оксидазы в сосудистых эндотелиоцитах и гладкомышечных клетках с последующей клеточной пролиферацией и стимуляцией клеточного роста, через них осуществляется способность А II оказывать проатерогенное действие, тогда как активация AT2-рецепторов сопровождается развитием противоположных эффектов, включая клеточный апоптоз. В сердце с явлениями недостаточности или в сосудистой стенке при формировании неоинтимы после повреждения отмечается активация AT2-рецепторов, которые экспрессируются пролиферирующими клетками, оказывают угнетающее действие на митогенные эффекты А II, ослабляя ремоделирование тканей. В крайней форме этот эффект находит выражение в апоптозе.
Усиление экспрессии AT2-рецепторов в кардиомиоцитах и сосудистых гладкомышечных клетках угнетает вызванные А II хронотропные и прессорные эффекты; через систему циклического гуанозинмонофосфата (цГМФ) AT2-рецепторы оказывают кардиопротекторное и вазодилататорное действие. Установлено, что защитный эффект блокаторов AT1-рецепторов осуществляется в значительной степени благодаря повышению плазменной концентрации А II с последующей активацией AT2-рецепторов [257].
Предполагают, что активация AT2-рецепторов замедляет прогрессирование атеросклероза и способствует повышению стабильности бляшки. Отмечено, что их отсутствие сочетается с повышением содержания в бляшке макрофагов, гладкомышечных клеток, накоплением коллагена, тогда как активация AT2-рецепторов сопровождается угнетением клеточной пролиферации, усилением апоптоза и предупреждает образование неоинтимы. В исследовании, проведенном на биоптатах атеросклеротических бляшек сонных артерий 14 пациентов, установлена выраженная экспрессия AT2-рецепторов, локализующаяся в зоне инфильтрации макрофагов и фибробластов [111].
Характер распределения рецепторов А II 2-го типа в сердце с явлениями ремоделирования и недостаточности остается малоизученным. При аутопсии сердца без признаков недостаточности соотношение рецепторов А II 1-го и 2-го типов составляло 59:41. При дилатационной кардиомиопатии содержание AT2-рецепторов было увеличено в 3,1 раза на фоне выраженного уменьшения экспрессии AT1-рецепторов. Рецепторы 2-го типа локализовались главным образом в интерстиции, тогда как рецепторы 1-го типа равномерно и с малой плотностью распределялись в миокарде. Эти данные позволили сделать заключение о том, что:
а) экспрессия AT2-рецепторов значительно повышается в сердце при развитии его недостаточности, ответственными за что являются фибробласты, накапливающиеся в интерстиции;
б) экспрессия рецепторов А II 1-го типа в ткани предсердий и желудочков уменьшается при сердечной недостаточности;
в) экспрессия рецепторов А II 1-го и 2-го типов в сердце при развитии недостаточности регулируется дифференцированно [257].
В последние годы особое внимание уделяется значимости полиморфизма генов, определяющих активность компонентов РАС, в развитии кардиоваскулярной патологии. Несмотря на значительное число работ, проведенных с участием большого количества добровольцев, полученные данные и их трактовка существенно варьируют.
В исследованиях, проведенных на спонтанно гипертензивных крысах, показано, что в основе развития АГ в этих условиях лежит повышение активности ренина, определяемое наличием SHR-аллели его гена. В клинических условиях установлено, что ряд форм АГ связан с мутацией генов, вовлеченных в синтез ангиотензиногена, альдостерона и реализацию их действия. Показано, что гиперэкспрессия гена ангиотензиногена сопровождается пропорциональным повышением уровня ангиотензиногена в плазме крови, что является основной детерминантой активности РАС и сочетается с повышением АД. В исследованиях, проведенных с участием американской и японской популяции, установлено, что два варианта гена ангиотензиногена (M235T и T174) сопряжены с повышенным уровнем ангиотензиногена в крови, риском развития АГ и ИБС, хотя в популяциях Англии и Германии получен отрицательный результат.
По данным ряда авторов, более чем 50% вариабельности ведущих факторов риска ИБС определяются генетическими механизмами [223]. Показано, что ген АПФ идентичен для циркулирующей и локальных РАС, и поэтому активность АПФ в крови аналогична его тканевой активности. Тем не менее, связь между I/D полиморфизмом АПФ и распространенностью ИБС остается противоречивой [23].
В ряде исследований выявлено, что D-аллель гена АПФ является независимым фактором риска коронарной патологии у лиц как с отсутствием [23], так и наличием СД 2-го типа [205]. Предполагают, что D-аллель этого полиморфизма сочетается с повышенной плазменной концентрацией АПФ [161, 262] и более высокой распространенностью ИБС в странах Европы и Азии [301].
Результаты метаанализа 46 исследований с вовлечением в целом 5215 пациентов и 4782 контрольных лиц в китайской популяции свидетельствуют о наличии четкой связи между I/D полиморфизмом гена АПФ и риском развития ИБС [296–299]. В метаанализе ряда других популяционных исследований продемонстрирована достоверная связь между полиморфизмом гена АПФ и риском развития ИБС, и при фенотипе DD риск был в 2,4 раза выше, чем фенотипе II; наличие DD-полиморфизма гена АПФ сочеталось с большей распространенностью АГ и ИБС, более высоким риском развития ИМ. Показано также, что лица с D-аллелью гена АПФ характеризуются повышенным риском развития диабетической нефропатии по сравнению с носителями I-аллели [181].
Результаты ряда других популяционных исследований также свидетельствуют о том, что более половины вариаций плазменной активности АПФ связано с полиморфизмом его гена. Данные метаанализа 15 исследований, включавших 3394 пациента с ИМ и 5479 контрольных лиц, свидетельствовали о прямой связи между DD-генотипом АПФ и риском развития ИМ [208]. При многофакторном регрессионном анализе наличие DD-генотипа сочеталось с достоверным повышением риска развития ИБС и СД 2-го типа.
Напротив, в исследовании Т. Fujimura и соавторов DD-полиморфизм гена АПФ не был детерминантой развития ИМ [76]. Не установлено также зависимости между типом полиморфизма гена АПФ и риском развития кардиоваскулярной патологии при исследовании 538 лиц с СД 2-го типа, у 220 из которых также выявлены коронарный атеросклероз и ИБС. У 40,9% исследованных отмечено наличие D-аллели, у 59,1% — I-аллели. У лиц с сочетанием СД 2-го типа и ИБС D-аллель установлена в 34,7% случаев, I-аллель — 65,3% [131]. В большинстве других исследований не установлено зависимости между полиморфизмом гена АПФ и развитием АГ, хотя наличие D-аллели более закономерно сочеталось с гипертрофией ЛЖ.
Не выявлено также четкой связи между полиморфизмом гена рецепторов А II и риском развития кардиоваскулярной патологии, хотя у женщин с СД 2-го типа 1675G/A-полиморфизм гена AT1-рецепторов отмечался параллельно с повышенным риском ИБС. A/G-полиморфизм АПФ2 сочетался с повышенным риском развития АГ в женской популяции Китая с наличием метаболического синдрома.
Относительно связи полиморфизма гена альдостеронсинтазы с кардиоваскулярной патологией также нет единого мнения. Во многих исследованиях отмечена связь между АГ и наличием Т-аллели гена альдостеронсинтазы, тогда как в исследовании с участием французской популяции не установлено подобной зависимости [176].
1.3. Механизмы физиологического и патологического действия РАС
Так как основным эффекторным звеном РАС является А II, то изучение характера ее влияния на структурно-функциональные особенности сердечно-сосудистой системы было до последнего времени сконцентрировано на эффектах А II.
А II оказывает выраженное тоническое влияние на тонус артериальных сосудов, имеющее фазный характер, и при констрикции почечной артерии АД повышается в течение нескольких минут параллельно с возрастанием концентрации ренина и А II в крови. Однако после этой начальной фазы, которая может длиться в течение 1–2 дней, уровень ренина и А II начинает снижаться, тогда как выраженность АГ увеличивается. Эти изменения сочетаются с повышением сосудистой реактивности на А II, что совпадает с данными о развитии выраженного медленного прессорного эффекта А II при его концентрации в крови, близкой к нормальной, у большинства лиц с почечной или эссенциальной АГ [74].
Показано, что быстрый прессорный эффект А II определяется его мощным вазоконстрикторным действием, тогда как природа постепенного прогрессирующего повышения АД при длительной инфузии низких субпрессорных доз А II остается менее определенной. Этот медленно развивающийся прессорный эффект А II, который установлен у людей, крыс, кроликов и собак, возникает при его концентрации в крови, не выходящей за пределы физиологической; он накладывается на быстрый ответ и усиливает его. Так, быстрый ответ, достигающий 50 мм рт. ст. в течение 2 мин, постепенно устраняется в течение 8 мин после окончания инфузии А II, тогда как медленный ответ аналогичной выраженности отмечается на 3–4-й день длительного введения А II и полностью исчезает через 4 ч после прекращения инфузии. Быстрый прессорный эффект выраженностью 50 мм рт. ст. возникает при повышении концентрации А II в крови примерно в 80 раз, тогда как медленный эффект аналогичной выраженности — только в 6–8 раз.
Длительное введение А II в высоких дозах сопровождается развитием так называемой тахифилаксии — ослабления ответа, и АД, несмотря на продолжающуюся инфузию, к концу 2-х суток возвращается к исходному значению. Предполагают, что этот эффект определяется интернализацией рецепторов А II и снижением их экспрессии на клеточной мембране. В то же время, инфузия А II в промежуточных дозах сочетается с удержанием стабильного прессорного эффекта в течение 2 мес и более, а инфузия А II в низких дозах сопровождается развитием медленно прогрессирующего прессорного эффекта. Отмечено, что для поддержания высокого уровня АД при длительной инфузии А II у здоровых лиц необходимо постепенное снижение скорости инфузии или концентрации препарата.
Предполагают, что в основе медленно развивающегося прессорного эффекта А II лежит его способность усиливать реабсорбцию натрия в почках как непосредственно, так и через высвобождение альдостерона, поскольку ограничение потребления соли значительно снижает медленно развивающийся прессорный эффект. Однако этому противоречат данные об отсутствии повышения плазменной концентрации альдостерона при медленно развивающемся прессорном ответе; не выявлена также задержка натрия в этих условиях. В то же время блокада симпатической нервной системы предупреждала развитие медленного прессорного ответа, что совпадает с данными о взаимодействии А II с вегетативной нервной системой как на центральном, так и периферическом уровнях. Однако симпатэктомия у крыс, которым осуществлялась инфузия А II в течение 6 мес, не предупреждала развития медленного прессорного ответа; ни у людей, ни у собак не отмечали также признаков гиперактивности симпатической нервной системы при развитии медленного ответа.
Наиболее реальным механизмом развития медленного прессорного ответа на инфузию низких доз А II является ремоделирование сосудистой стенки, ее выраженная гипертрофия отмечена уже в течение 10 дней инфузии препарата. Выявлено увеличение отношения стенка/просвет, что определяло развитие гиперреактивности сосуда на различные констрикторные воздействия. Эти структурные изменения отмечены даже при отсутствии повышения АД и определялись увеличением количества и размера гладкомышечных клеток, объема внеклеточного пространства в результате, по-видимому, прямого митогенного действия А II. Показано, что интенсивность клеточного митоза в конце 10-го дня инфузии увеличилась на 30%, и выраженность этого митогенного эффекта А II значительно увеличивалась при повышенной симпатической активности [75].
Помимо этого, прогипертензивное действие А II в значительной степени определяется его способностью усиливать реабсорбцию натрия и воды в дистальных канальцах почек с увеличением объема циркулирующей крови. А II оказывает также положительное инотропное действие на миокард, однако этот эффект отмечен только при супрафизиологических концентрациях и связан в значительной степени со стимуляцией симпатической нервной системы.
Известно, что длительное увеличение потока крови вызывает внешнее ремоделирование сосуда с увеличением его просвета, тогда как уменьшение потока крови сопряжено с внутренним ремоделированием и уменьшением просвета, и эти изменения направлены на нормализацию пристеночного напряжения сдвига [12]. А II участвует в развитии внутреннего ремоделирования резистивных сосудов с уменьшением просвета при хроническом уменьшении потока крови. Этот эффект не зависит от изменений АД или метаболических влияний и отмечается при АГ, ИБС, СД. Установлено, что через 1 нед после коарктации мезентериальной артерии ослабление потока крови в периферических резистивных сосудах сопровождалось уменьшением их диаметра, эндотелийзависимого расслабления и снижением экспрессии эндотелиальной синтазы оксида азота (eNOS) в сочетании с усиленной продукцией супероксидного радикала (СОР).
Тем не менее, отсутствие eNOS или угнетение продукции СОР не предупреждало ремоделирования сосудистой стенки, тогда как оно устранялось применением периндоприла или кандесартана. В соответствии с заключением, к которому пришли авторы, внутреннее ремоделирование сосудов с сокращением сосудистой стенки при уменьшении потока крови имеет активный характер и определяется действием А II. Его продукция в эндотелии в этих условиях увеличивается реципрокно к уменьшению образования оксида азота в результате снижения пристеночного напряжения сдвига [12].
Одним из наиболее сложных и спорных вопросов до настоящего времени остаются роль и механизмы участия А II в развитии гипертрофии и ремоделирования сердца при АГ. Эти эффекты практически до последнего времени рассматривались как следствие повышения АД и увеличения интрамиокардиального напряжения. Только в последние годы было установлено отсутствие корреляционной зависимости между тяжестью АГ и выраженностью гипертрофии сердца. Кроме того, в ряде случаев ремоделирование сердца предшествовало развитию АГ, а ее устранение с помощью вазодилататоров не сочеталось с нормализацией структуры сердца.
В то же время повышение плазменной концентрации А II при его длительной инфузии в субпрессорных дозах приводило к развитию гипертрофии миокарда, которое значительно опережало развитие АГ. Этот эффект определялся как прямым митогеннным действием А II посредством активации митогенактивирующей протеинкиназы, так и, в значительной степени, способностью А II стимулировать высвобождение эндотелина-1 (ЭТ-1) и трансформирующего фактора роста-β (TФР-β), которые обладают выраженным митогенным эффектом.
Гипертрофия сердца обычно сопровождается нарушением его диастолической функции в результате развития фиброза, и восстановление этой функции параллельно с регрессией гипертрофии отмечено при применении блокаторов рецепторов А II даже в дозах, не приводящих к снижению АД. Аналогичный эффект отмечен и при применении блокаторов ЭТ-1, что указывает на его участие в гипертрофическом ремоделировании сердца, вызванном А II.
В исследовании, проведенном на биоптатах сердца человека, полученных при проведении хирургического вмешательства, установлено связывание А II с высокой активностью как с фибробластами сердца, так и кардиомиоцитами. Это связывание сопровождалось активацией митогенактивирующей протеинкиназы, опосредованной через АТ1-рецепторы, тогда как параллельно активирующиеся АТ2-рецепторы ограничивали митогенное действие А II. Однако в связи с преобладанием АТ1-рецепторов длительное действие А II сопряжено с ремоделированием сердца, возникающим вследствие гипертрофии кардиомиоцитов в сочетании с усиленным синтезом коллагена и белков межклеточного матрикса в фибробластах [272, 273].
Неоднократно установлено, что основным механизмом, обусловливающим значимость А II в развитии кардиоваскулярной патологии, является его способность инициировать развитие оксидативного стресса и воспаления, которые являются следствием активации никотинамидадениндинуклеотидфосфат (NADPH)-оксидаз и усиления экспрессии ядерного фактора транскрипции (NF-κB) в гладкомышечных клетках, моноцитах, макрофагах [17].
Развитие оксидативного стресса при действии А II связано с резким увеличением продукции СОР, который образуется при участии ряда оксидаз и оксигеназ и при незавершенном окислительном фосфорилировании в митохондриях. Основным источником СОР в сосудистой стенке является фермент NADPH-оксидазы. Из известных 7 представителей семейства NADPH-оксидазы 4 (NOX1, NOX2, NOX4 и NOX5) экспрессируются в сердечно-сосудистой системе и активируются при связывании А II с рецептором 1-го типа.
В клетках сосудистой стенки представлены 4 типа NADPH-оксидазы: NOX1, NOX2, NOX4 и NOX5. NOX1 экспрессируется в эндотелиоцитах, гладкомышечных клетках и фибробластах, активируется при повреждении, при действии А II, фосфолипаз С и D, протеинкиназы С [127]. Угнетение NOX1 значительно снижает развитие АГ при введении А II [127], тогда как гиперэкспрессия NOX1 существенно повышает способность А II вызывать АГ и гипертрофию [48, 49]. В то же время, NOX1 не участвует в развитии АГ, не связанной с действием СОР, вызванной введением норэпинефрина [82] и у трансгенных по ренину животных [287].
К числу активаторов NOX1 и вызывающих его гиперэкспрессию в сосудистых гладкомышечных клетках относятся А II, окисленные липопротеины низкой плотности (ЛПНП), продукты глубокого гликозилирования белков (AGEs), что обусловливает выраженную активацию и гиперэкспрессию NOX1 в клетках сосудистой стенки животных с атеросклерозом и СД [210, 287]. В результате усиливаются миграция гладкомышечных клеток и их пролиферация под действием тромбоцитарного фактора роста, а отсутствие NOX1 угнетает эти реакции. Аторвастатин угнетает экспрессию NOX1, что может в некоторой степени определять его противовоспалительное действие.
В клетках сосудистой стенки идентифицирован также NOX2. Он определяет внутри- и внеклеточную продукцию СОР, и экспрессия NOX2 находится в обратной корреляционной связи с выраженностью эндотелийзависимого расслабления. Экспрессия NOX2 в аорте усилена у крыс со спонтанной АГ, у мышей при инфузии А II, крыс, у которых применялся альдостерон на фоне высокосолевой диеты. Антиоксидант темпол способен подавлять экспрессию NOX2 параллельно с уменьшением выраженности АГ. Удаление гена NOX2 у мышей с АГ, вызванной дезоксикортикостеронацетатом или коарктацией почечных сосудов, сочеталось со снижением прессорной реакции. Показано, что усиление экспрессии NOX2 в эндотелиоцитах, фибробластах и мигрировавших макрофагах способствует активации воспаления в сосудистой стенке и развитию атеросклероза.
Характерным отличием NOX1 и NOX2 является их активное участие в развитии АГ и повреждении органов-мишеней.
NOX4 экспрессирован во всех клетках сосудистой стенки значительно более интенсивно, чем другие NOX, и его основным активатором является ТФР-β. Эндотелиоциты экспрессируют преимущественно NOX2 и NOX4, и активность этих ферментов изменяется при СД, АГ, действии А II, что может обусловливать дисфункцию эндотелия, повышение АД, клеточную пролиферацию и развитие гипертрофии.
NOX5 является кальцийактивируемым ферментом, который конститутивно продуцирует СОР с невысокой интенсивностью. NOX5 в эндотелиоцитах способствует пролиферации и неоангиогенезу, образованию капилляроподобных трубок. Как и другие NOX, NOX5 активирует продукцию СОР и способствует связыванию оксида азота. Показано, что трансфекция гена NOX5 в эндотелий аорты мышей приводила к угнетению расслабления, вызываемого ацетилхолином, и потенцировала эффект А II, а в предварительно сокращенной аорте действие антиоксидантного фермента супероксиддисмутазы (СОД) на фоне гиперэкспрессии NOX5 сопровождалось выраженным расслаблением, что свидетельствовало о способности NOX5 активировать продукцию СОР в сосудистом эндотелии.
В то же время, в ряде других исследований установлено, что активация NOX5 сопровождалась не только усилением продукции СОР, но и увеличением высвобождения NO из эндотелиоцитов как у животных, так и у людей. Обусловливается это тем, что NOX5, как и другие формы NADPH-оксидазы, угнетают функцию эндотелия в большей степени за счет высвобождения СОР, связывания NO и снижения его бидоступности, чем посредством прямого угнетения активности фермента eNOS [292, 293]. При трансфекции гена NOX5 в эндотелиоциты быка установлено повышение активности eNOS и содержания NO во внеклеточной среде в сочетании с увеличением продукции СОР.
Ранее установлено как непосредственно в сосудистой стенке, так и в культуре эндотелиоцитов, что активация NOX5 сопровождается сочетанным увеличением продукции как СОР, так и NO с образованием пероксинитрита на фоне резкого угнетения эндотелийзависимого расслабления. Уровень нитрита в среде, содержащей культуру эндотелиоцитов, при активации NOX5 значительно повышался в результате спонтанного одновременного распада как NO, так и пероксинитрита в водной среде, и потому отражал интенсивность продукции NO [177].
Эти данные подтверждены в исследовании, проведенном на сочетанной культуре эндотелиоцитов и сосудистых гладкомышечных клеток. Гиперэкспрессия NOX5 в эндотелиоцитах на фоне применения СОД и усиленной элиминации СОР проявлялась повышением концентрации цГМФ в гладкомышечных клетках, но в отсутствие СОД продукция цГМФ резко угнеталась. Эти данные прямо свидетельствовали о том, что повышение продукции СОР при гиперэкспрессии NOX5 активирует, но не угнетает eNOS, но интегральный эффект определяется способностью СОР снижать биодоступность NO.
Высказано предположение, что стимуляция NOX в зонах мозга, расположенных возле желудочков, сопровождается повышением симпатической активности. Это приводит к активации Т-лимфоцитов в лимфатических узлах с последующей их миграцией в сосудистую стенку и в почки [98]. Т-клетки высвобождают цитокины, стимулирующие NADPH-оксидазы в этих тканях, что приводит к развитию вазоконстрикции и задержке натрия. СОР также способен самостоятельно активировать NOX-ферменты, усиливая собственную продукцию по принципу положительной обратной связи.
Высвобождение активных форм кислорода с участием NOX имеет существенное значение не только в развитии сосудистого поражения, но и в качестве сигнального пути в регуляции нормальной функции сосудов, сокращения и расслабления сосудистой стенки, регуляции выживаемости. Возможно, поэтому применение антиоксидантов в клинических условиях оказалось недостаточно эффективным в предупреждении сосудистого поражения [269], так как наряду с защитным, оно оказывало и дисрегуляторное действие.
Семейство NADPH-оксидаз — главный источник радикалов кислорода в сосудистой стенке и в почках, активные формы кислорода, продуцируемые NADPH-оксидазой, играют ведущую роль в развитии дисфункции эндотелия, воспаления, гипертрофии, апоптоза, фиброза, ангиогенеза. Поэтому они потенциально рассматриваются как возможная мишень для терапевтических воздействий при АГ и кардиоваскулярной патологии.
К другим источникам радикалов относятся также ферменты митохондриального транспорта электронов, система ксантин/ксантин-оксидаза, циклооксигеназа-2 (ЦОГ-2), липоксигеназа (ЛОГ) и разобщенная синтаза оксида азота. Активные формы кислорода являются химическими соединениями, которые включают две основные группы: свободные радикалы (СОР, гидроксильный радикал, оксид азота) и нерадикальные производные (перекись водорода, пероксинитрит, гипохлористая кислота). СОР вследствие наличия заряда не проникает через клеточные мембраны, тогда как перекись водорода свободно диффундирует через них.
СОР инактивирует оксид азота, приводя к дисфункции эндотелия и вазоконстрикции, тогда как перекись водорода действует как вазодилататор в ряде сосудистых областей, включая церебральные, коронарные и мезентериальные сосуды. Ксантин-оксидаза экспрессируется в эндотелии, и эта экспрессия усилена при АГ. Однако у крыс со спонтанной АГ длительное угнетение ксантин-оксидазы аллопуринолом не сопровождалось снижением АД, что свидетельствует о ее роли как следствия, но не причины АГ. В то же время аллопуринол значительно уменьшал выраженность гипертрофии сердца и нефросклероза, что свидетельствовало об участии ксантин-оксидазы в поражении органов-мишеней. Полагают, что этот эффект обусловлен конечным продуктом ксантин-оксидазы — мочевой кислотой.
Радикалы кислорода могут продуцироваться всеми тремя изоформами NOS при их разобщении — отсутствии субстрата L-аргинина или кофактора тетрагидробиоптерина. Этот эффект отмечался у спонтанно гипертензивных крыс и при ДOКA-солевой АГ, когда применение тетрагидробиоптерина проявлялось антигипертензивным эффектом.
Антиоксидантная защита сосудистой стенки обеспечивается тремя основными ферментными системами: СОД, каталазой и глютатионпероксидазой [178, 179].
Связь между оксидативным стрессом и повышением АД продемонстрирована во многих экспериментальных моделях АГ. Показано, что у спонтанно гипертензивных крыс увеличение продукции активных форм кислорода на фоне снижения уровня NO и антиоксидантных ферментов предшествовало развитию АГ. Помимо этого, у мышей с функционально неполноценной NADPH-оксидазой введение А II не сопровождалось усиленной продукцией СОР, развитием АГ, эндотелиальной дисфункции, ремоделирования сердца [112, 124, 125, 134]. В то же время, в другом исследовании гиперэкспрессия сосудистой NADPH-оксидазы сочеталась с развитием оксидативного стресса и сосудистой дисфункцией, но без значительных изменений АД [128].
В ряде работ применение апоцинина — ингибитора NADPH-оксидазы, при инфузии А II у мышей сопровождалось снижением сосудистой продукции СОР, предупреждением ремоделирования сердца и сосудистой стенки в сочетании с уменьшением выраженности гипертензивной реакции [195, 254]. Однако в этих исследованиях инфузия А II осуществлялась кратковременно, в течение 1–3 нед, и гипертензивная реакция носила острый характер. В то же время, при хронической АГ, связанной с трансгенной гиперэкспрессией человеческого ренина у мышей, развитие АГ не предупреждалось применением апоцинина, несмотря на уменьшение выраженности оксидативного стресса, сосудистой продукции СОР [254].
Участие NADPH-оксидазы в патогенезе АГ показано у спонтанно гипертензивных крыс, у которых отмечено значительное увеличение сосудистой, ренальной и кардиальной продукции активных форм кислорода [292–294] на фоне усиленной экспрессии NOX1 и NOX4 [2]. В ряде исследований установлено, что применение антиоксидантных витаминов, миметиков СОД типа темпола, скавенджеров свободных радикалов или тетрагидробиоптерина не только предупреждало или ослабляло поражение органов-мишеней, но и препятствовало развитию АГ или уменьшало ее выраженность [30, 31, 105, 106, 124, 125].
Результаты ряда клинических исследований также свидетельствуют об участии оксидативного стресса в развитии АГ, реноваскулярной АГ, солечувствительной АГ и АГ, связанной с применением циклоспорина [90, 130, 139, 265, 266]. У пациентов с АГ закономерно отмечали повышенный плазменный уровень H2O2, примерно 25–30% вариаций его продукции было связано с генетическим полиморфизмом. Это особенно отчетливо проявилось в исследовании, проведенном в японской популяции [164, 165].
Выявлено, что продукция СОР значительно повышена в сосудистых гладкомышечных клетках пациентов с АГ. Этот эффект был связан с гиперэкспрессией NADPH-оксидазы [254], a полиморфизм гена NADPH-оксидазы являлся маркером повреждения эндотелия, развития АГ и атеросклероза [8, 162]. АГ при ожирении также сочеталась с развитием оксидативного стресса, о чем свидетельствовала прямая зависимость между индексом ожирения, содержанием 8-изопростанов в моче и уровнем АД.
Однако, несмотря на то что гипертензивная реакция при инфузии ЭТ-1 также сопровождалась увеличением продукции СОР в результате активации NADPH-оксидазы, ее предупреждение не устраняло развития АГ [58, 59].
Одной из причин развития и повышения интенсивности оксидативного стресса является недостаточность антиоксидантной защиты, которая может иметь как первичный характер, так и возникать в результате ее истощения при хроническом стрессе. СОР метаболизируется с участием фермента СОД — супероксиддисмутазы с образованием перекиси водорода (H2O2), либо соединяется с NO, образуя пероксинитрит. Поэтому недостаточность или истощение СОД приводит к образованию пероксинитрита, обладающего мощным цитотоксическим эффектом, прежде всего — на эндотелиоциты.
Установлено, что СОР и другие активные формы кислорода продуцируются в центральной нервной и сердечно-сосудистой системе, почках и принимают участие в регуляции ряда физиологических реакций. Однако они также вовлекаются в патогенез различных процессов, в частности — в развитие ИБС, АГ, сердечной недостаточности. При этом угнетение экспрессии NOX1 значительно уменьшало выраженность прессорного действия А II наряду с резким уменьшением продукции СОР и выраженности гипертрофии гладкомышечных клеток и кардиомиоцитов, а гиперэкспрессия NOX1 потенцировала развитие гипертензивной реакции [270, 271].
В ряде исследований установлено, что гипертрофия миокарда при АГ, связанной с активацией РАС, опосредуется действием СОР, и окись углерода (CO), обладающая антиоксидантными свойствами благодаря способности угнетать окислительный потенциал СОР, уменьшала выраженность как гипертензивного действия А II, так и гипертрофию ЛЖ [119]. В исследовании, проведенном на крысах, гипертрофия миокарда, индуцированная А II, блокировалась СОД, что подтверждало патогенетическую значимость СОР в ее развитии [103].
Позже было установлено, что развитие оксидативного стресса при действии А II обусловлено усиленным образованием СОР не только в результате активации мембраносвязанной NADPH-оксидазы, но и усилением его митохондриальной продукции, которая также стимулируется цитозольными оксидазами. Поэтому применение митохондриальноориентированного антиоксиданта MitoQ10 приводило к повышению биодоступности NO в грудной аорте спонтанно гипертензивных крыс и снижению системного АД [52, 92].
В ряде исследований продемонстрирована связь между активацией NADРH-оксидазы и других источников СОР и развитием эндотелиальной дисфункции, воспаления и вазоконстрикции при действии А II. Гипертрофия стенки аорты мышей и продукция в ней СОР значительно возрастали после 2-недельной инфузии А II, тогда как при сопутствующей гиперэкспрессии в гладкомышечных клетках каталазы — одного из важнейших антиоксидантных ферментов, выраженность этих изменений значительно уменьшалась. Однако при этом АД не отличалось у животных обеих групп [292]. Эндогенный антиоксидант глютатионпероксидаза, наряду с каталазой, также способствует инактивации перекиси водорода, и у мышей, трансгенных по глютатионпероксидазе, применение А II не вызывало дисфункции эндотелия [34].
В стимуляции продукции СОР при действии А II существенную роль играет ИЛ-6, и у мышей с генетическим его отсутствием инфузия А II сопровождалась значительно менее выраженной продукцией СОР, гипертрофией стенки и дисфункцией эндотелия в сонных артериях [219]. С другой стороны, А II стимулирует продукцию ИЛ-6, и этот эффект исчезает после блокады минералокортикоидных рецепторов [147].
В сосудистой стенке А II индуцирует продукцию СОР всеми типами клеток, включая фибробласты адвентиции, гладкомышечные клетки и эндотелиоциты [102]. В результате развивается оксидативный стресс с активацией факторов транскрипции АР-1 и NF-κB, которые ответственны за регуляцию экспрессии генов, определяющих развитие воспалительного ответа. Это проявляется усиленной продукцией молекул адгезии, хемокинов типа моноцитарного хемотаксического белка-1 (МСР-1) и провоспалительных цитокинов, рекрутированием моноцитов/макрофагов в сосудистую стенку с активацией локальной РАС, так как моноциты экспрессируют ангиотензиноген, ренин, АПФ и AT1-рецепторы.
В условиях эксперимента, проведенного на нормальных крысах, показано, что 3-разовая инфузия А II в течение 2 нед сопровождалась повышением активности NADPH-оксидазы в аорте в 1,5 раза, в миокарде — 1,8 раза. Это сочеталось с развитием оксидативного стресса, ослаблением эндотелийзависимого расслабления на ацетилхолин на 70%, двукратным возрастанием экспрессии сосудисто-клеточных молекул адгезии-1 (VCAM-1).
Отмеченные изменения в значительной степени являлись отражением активации воспалительного ответа и возникали на фоне увеличения инфильтрации макрофагами и Т-клетками стенки аорты и артериальных сосудов. Развитие всех отмеченных изменений предупреждалось применением T-регуляторных клеток (Treg), которые оказывали иммуносупрессорное действие, устраняли активацию T-хелперов и развитие воспалительной реакции. На основании этих данных было сделано заключение, что при действии А ІІ как повышение АД, так и развитие воспаления и оксидативного стресса в сосудистой стенке с дисфункцией эндотелия являются в значительной степени следствием развития иммунного ответа [11].
В настоящее время большинство исследователей разделяют точку зрения о том, что повышение активности NADPH-оксидазы и интенсивности образования активных форм кислорода в почках являются важнейшим фактором развития АГ и повреждения органов-мишеней. В исследовании S. Liu и соавторов показано, что пептидный ингибитор NADPH-оксидазы снижал АД и препятствовал накоплению макрофагов в миокарде, сосудистой стенке и почках у крыс, которым вводился А II [141, 142].
Отмечено, что увеличение реабсорбции натрия при действии А II также сопряжено с его способностью активировать NADPH-оксидазу и стимулировать продукцию СОР в эпителии проксимальных канальцев, и этот эффект угнетался при действии NO. У крыс со спонтанной АГ применение антиоксиданта темпола блокировало протеинурию, нарушения функции почек, причинами которых были усиленное образование СОР и угнетение продукции NO. Эти изменения возникали в результате активации минералокортикоидных рецепторов и устранялись с помощью применения их блокаторов спиронолактона и эплеренона.
Способность А II оказывать провоспалительное действие в сосудистой стенке, миокарде и почках в значительной степени осуществляется посредством стимуляции Т-лимофцитов с усилением их инфильтрации в периваскулярную жировую ткань и продукции воспалительных цитокинов. Этому способствует истощение антиоксидантной защиты, в частности — снижение активности СОД, что свидетельствует об участии СОР в реализации активирующего действия А II на Т-клетки [143].
Результаты ряда фундаментальных исследований свидетельствуют о том, что активные формы кислорода принимают непосредственное участие в развитии гипертрофии и ремоделировании сердца и сосудистой стенки при АГ, а митогенное действие А II обусловлено усиленным синтезом СОР в гладкомышечных клетках и эндотелиоцитах. Показано, что этот эффект сопряжен с активацией в них митохондриальных ферментов, оксигеназ (ЦОГ, ЛОГ, синтазы оксида азота), перокcидаз. Генетический дефицит эндогенного антиоксиданта глютатионпероксидазы сочетался со значительным увеличением выраженности гипертрофии и дисфункции миокарда при АГ, вызванной А II, тогда как восполнение этого дефицита существенно предупреждало развитие нарушений [5]. Способность угнетать гипертрофию миокарда, вызванную А II, характерна и для моноокиси углерода, одного из продуктов гемоксигеназы, проявляющего высокую антиоксидантную активность [119].
В реализации митогенного действия А II существенную роль играют не только АТ1-, но и минералокортикоидные рецепторы, и у мышей с их селективной гиперэкспрессией в сердце значительно повышена способность А II вызывать гипертрофию миокарда с накоплением коллагена и фибронектина. Это свидетельствует о том, что митогенное действие А ІІ частично опосредовано стимуляцией секреции альдостерона [220, 221].
Участие РАС в патогенезе АГ и атеросклероза определяется в значительной степени тем, что А II — один из важнейших триггеров воспалительного ответа, механизмов его поддержания и усиления по принципу положительной обратной связи. Так, воспаление в сосудистой стенке повышает активность A II с дополнительным повышением активности NADPH-оксидазы в эндотелиоцитах и гладкомышечных клетках, усилением продукции СОР, экспрессии фактора NF-κB, продукции адгезивных молекул (ICAM-1, VCAM-1, Е-селектина) и миграции лейкоцитов в сосудистую стенку.
Развитие и генерализация воспаления при действии А II обусловлены также его способностью активировать продукцию в эндотелиоцитах и сосудистых гладкомышечных клетках МСР-1 — хемоаттрактанта моноцитов и лимфоцитов. Усиливается также секреция ИЛ-6, который оказывает дистантное действие на печень и стимулирует продукцию белков острой фазы: С-реактивного белка, сывороточного амилоида А (SАА), а также фибриногена и ангиотензиногена. Отмечено, что уровень циркулирующего ангиотензиногена в крови у пациентов с острой инфекцией повышен в 2 раза, а инициация воспаления у животных однократным введением липополисахарида сопровождалась повышением его концентрации через 8 ч в 4 раза.
Отмечено, что поражение почек при АГ связано с активацией локального воспаления, и для пациентов с АГ и хроническим поражением почек характерен высокий плазменный уровень провоспалительных цитокинов. Важнейшим медиатором повреждающего действия воспаления в этих условиях является ИЛ-6, и генетическое отсутствие его продукции у мышей сочетается со значительно уменьшенной выраженностью АГ и поражения почек при инфузии А II. Установлено также, что блокада продукции ИЛ-6 устраняет развитие АГ при холодовой пробе. Эти данные позволили сделать вывод, что ключевым медиатором прогипертензивного и провоспалительного действия А II с развитием нефросклероза является ИЛ-6. В экспериментальных исследованиях установлено, что ИЛ-6 способствует усилению экспрессии профибротических генов (проколлагена, TФР-β, PAI-1, ЭТ-1), а у лиц с хроническим поражением почек в их биоптатах выявлено повышение содержания ИЛ-6, особенно выраженное при наличии сопутствующей АГ. В исследованиях, проведенных на культуре эндотелиоцитов человека и эксплантах почек мышей, прямо продемонстрировано, что действие А II сочетается с усилением экспрессии ИЛ-6, а нефросклероз и протеинурия развиваются параллельно с повышением АД и имеют общую патогенетическую основу в виде развития воспаления.
А II стимулирует также NADPH-оксидазу в клетках почечных сосудов, приводя к их констрикции, нарушению функции канальцев, нефросклерозу и развитию почечной недостаточности. Эти эффекты предупреждались блокадой AT1-рецепторов и значительно ослаблялись антиоксидантами типа N-ацетилцистеина и α-токоферола [275–278].
Хотя положение о способности А II оказывать митогенное действие, вызывать ремоделирование и склерозирование сердца и сосудов посредством продукции радикалов кислорода в настоящее время не вызывает сомнений, роль кислородных радикалов в развитии АГ при действии А II остается противоречивой. Так, в модели низкорениновой солевой АГ угнетение NADPH-оксидазы сопровождалось уменьшением продукции СОР, нормализацией эндотелийзависимого расслабления и уменьшением выраженности воспаления, однако без существенного влияния на АД [298, 299]. В другом исследовании при хроническом действии А II антиоксидант темпол только частично уменьшал выраженность прессорного ответа на фоне выраженного антипролиферативного действия.
Помимо этого, у бурых норвежских крыс отмечены значительно повышенный плазменный уровень АПФ и А II, повышенная активность NADPH-оксидазы и увеличение продукции СОР в аорте, но без АГ [111]. У мышей с трансгенной гиперэкспрессией ренина и повышенной концентрацией А II развитие АГ и сосудистого повреждения сочеталось с усиленной продукцией СОР. Применение валсартана нормализовало все нарушения, тогда как темпол — миметик СОД, устранял оксидативный стресс, но не оказывал существенного влияния на АД [272, 273]. Эти данные дали основание предположить, что развитие АГ и сочетанных с ней поражений миокарда и сосудистой стенки при действии А II обусловлено не только развитием оксидативного стресса, но и рядом не зависимых от него механизмов.
В то же время, в ряде других исследований вазоконстрикторное и прессорное действие А II, как и активации АТ1-рецепторов в значительной степени блокировалось СОД, что свидетельствовало о ведущей роли СОР в повышении АД. Характерно, что подобное действие СОД не отмечено при АГ другой природы.
Вопрос об участии митохондриальной продукции СОР в реализации эффектов А ІІ остается спорным, и в ряде исследований ее угнетение не отражалось на сокращении сосудистой стенки и гипертензивном ответе при инфузии А II у крыс [117]. Эти данные означали, что в констрикторном и гипертензивном ответах на А II ведущую роль играет СОР, продуцируемый NADPH-оксидазой, но не митохондриями.
Проатерогенное действие А II также в значительной степени связано с развитием оксидативного стресса в результате активации NADРH-оксидазы как эндотелиоцитов и сосудистых гладкомышечных клеток, так и клеток крови. Наиболее выраженный эффект А II оказывает на NADРН-оксидазу нейтрофилов, в результате чего продукция СОР приобретает взрывной характер и на порядок превышает его продукцию в клетках другого типа.
Следствием развития оксидативного стресса и повреждающего действия СОР может быть и расслоение стенки аорты с образованием аневризм. Так, после 7 дней инфузии А II эти явления были отмечены у 23% нормальных мышей и только у 4% животных с отсутствием повышенной активности NADPH-оксидазы [214]. Развитие аневризмы аорты после 4 нед инфузии А II отмечено также у 90% мышей с отсутствием апоЕ и только у 16% подобных животных с одновременным блокированием активности NADPH-оксидазы [250].
Оксидативный стресс, развивающийся при действии А II, имеет не только локальный тканевой, но и системный характер, что сопряжено с окислением ЛПНП как непосредственно в сосудистой стенке, так и в крови. Параллельно A II индуцирует экспрессию макрофагами скавенджер-рецепторов CD36 и таким образом способствует модификации ЛПНП, их захвату макрофагами и образованию пенистых клеток [15].
Практически все патогенетические факторы атеросклероза — гиперхолестеринемия, СД, курение, сопряжены с продукцией свободных радикалов кислорода, которые способны окислять ЛПНП, повреждать эндотелий и провоцировать развитие воспалительного процесса в сосудистой стенке. Неоднократно отмечено, что основным источником активных радикалов кислорода при атеросклерозе является NADРH-оксидаза, а А II относится к числу наиболее мощных ее стимуляторов, способных провоцировать развитие воспалительной реакции. В то же время ингибиторы АПФ предупреждают активацию прооксидантных ферментных систем и уменьшают выраженность оксидативного стресса и повреждающего действия гиперэкспрессии РАС [124].
Провоспалительное действие свободных радикалов связано как с активирующим влиянием на фактор NF-κB, так и со способностью связывать NO и угнетать противовоспалительные, антиадгезивные и антипролиферативные свойства эндотелия. Так как проатерогенное действие А II осуществляется через AT1-рецепторы, то их блокада оказывает такой же противовоспалительный эффект, как и угнетение АПФ [274].
Помимо этого, участие РАС в патогенезе атеросклероза в значительной степени связано со способностью гиперхолестеринемии усиливать экспрессию AT1-рецепторов, что приводит к увеличению продукции активных форм кислорода, вазоконстрикции и клеточной пролиферации. Этот эффект предупреждается статинами, что лежит в основе их холестериннезависимого антиатерогенного действия.
Активация воспаления в сосудистой стенке при действии А II с последующей усиленной экспрессией факторов типа TФР-β и PAI-1 играет существенную роль в запуске каскада реакций, приводящих к фиброзу и ремоделированию сердца и сосудов. TФР-β стимулирует трансформацию сосудистых гладкомышечных клеток в фибробласты, способствует усиленному синтезу матриксных протеинов и интегринов, уменьшает продукцию матриксных металлопротеиназ (ММРs). В результате увеличивается продукция коллагена и уменьшается деградация внеклеточного матрикса, вызывая развитие фиброза.
В настоящее время общепризнано, что процесс ремоделирования сердца при АГ и хронической ИБС опосредован прежде всего действием А II. Он стимулирует рост фибробластов, которые являются основным источником продукции коллагена в миокарде, и этот процесс медиируется оксидативным стрессом. В изолированных фибробластах сердца крысы А II активировал фактор NF-κB, стимулировал внутриклеточное образование активных форм кислорода, увеличивал экспрессию коллагена 1-го типа — основной детерминанты жесткости миокарда, параллельно со снижением экспрессии и активности ММР-1 [30].
Известно, что трофическая функция А II связана со стимуляцией экспрессии провоспалительных цитокинов и факторов роста типа TФР-β1, PDGF-β. Эти изменения особенно выражены в клубочках почки при СД 2-го типа и приводят к быстрому развитию нефросклероза; они определяются провоспалительным действием А II, его способностью активировать продукцию ФНО-α [86, 132, 133, 154]. Выявлено, что применение блокатора рецепторов ФНО-α этанерцепта у крыс, у которых осуществлялась инфузия А II, сопровождалось значительным уменьшением выраженности протеинурии в отсутствие влияния на уровень АД [189].
А II также играет существенную роль в развитии осложненных форм течения атеросклероза, у гипертензивных мышей с гиперпродукцией А II атеросклеротические бляшки характеризовались более тонкой покрышкой, большим накоплением липидов в ядре и увеличенным содержанием макрофагов, что лежало в основе развития нестабильности бляшки. В основе этих эффектов лежит способность А II активировать продукцию ИФН-γ, который угнетает пролиферацию гладкомышечных клеток и синтез матриксных белков. В то же время, атеросклеротические бляшки у гипертензивных мышей с нормальной эндогенной продукцией А II и даже с более высоким уровнем АД относились к стабильному фенотипу [30].
Общепризнано, что один из ведущих механизмов угнетающего действия А II на функциональные возможности эндотелия заключается в снижении биодоступности NO в результате активации NADPH-оксидазы в эндотелиоцитах и гладкомышечных клетках и увеличения продукции СОР, тогда как применение ингибиторов АПФ сопровождалось устранением этих нарушений. Ранее установлено, что ослабление эндотелийзависимого расслабления у животных с АГ, атеросклерозом и гиперхолестеринемией сочеталось с повышением содержания в крови NO, однако этот эффект развивался параллельно с увеличением продукции в сосудистой стенке СОР, инактивацией NO и снижением его биодоступности. Кроме того, NO по принципу отрицательной обратной связи регулирует активность еNOS и свой собственный синтез. В связи с этим как сам NO, так и его экзогенные доноры угнетают еNOS, тогда как инактиватор NO оксигемоглобин активирует ее. Поэтому усиленный синтез и повышение концентрации NO в крови может сочетаться со снижением его биологической активности [27].
С другой стороны, NO, продуцируемый клетками эндотелия, существенно уменьшает выраженность провоспалительного действия А II, его способность индуцировать апоптоз клеток эндотелия, их быструю сменяемость, и способствовать развитию атеросклероза. В условиях культуры эндотелиоцитов пупочной вены человека А II приводил к резкому повышению интенсивности апоптоза, тогда как воздействие ламинарного напряжения сдвига, способствующее активации eNOS и усиленной продукции NO, полностью предупреждало этот эффект [50].
Результаты ряда экспериментальных исследований свидетельствуют о том, что гипертрофия миокарда, отмечаемая в условиях повышенной нагрузки на сердце, определяется усиленным высвобождением А II из кардиомиоцитов при их механическом растяжении. Полученные данные также подтвердили, что развитие гипертрофии миокарда в этих условиях связано с активацией продукции свободных радикалов и развитием оксидативного стресса, у мышей с гиперэкспрессией гена гемоксигеназы-1 (НО-1) — мощного естественного антиоксиданта, отмечено значительное угнетение гипертрофии кардиомиоцитов при действии А II [107].
НО-1 — стресс-индуцируемый фермент, который относится к семейству белков теплового шока (HSPs), вызывает деградацию гема с высвобождением ионов железа, СО и биливердина. Биливердин и продукт его метаболизма билирубин обладают антиоксидантной активностью, тогда как СО — противовоспалительной.
Одним из механизмов повреждающего действия А ІІ на кардиомиоциты и гладкомышечные клетки сосудистой стенки является способность угнетать внутриклеточное окисление свободных жирных кислот (СЖК). Выявлено, что инкубация изолированных кардиомиоцитов на протяжении 14 дней в среде, содержащей А ІІ, сопровождалась 2-кратным снижением активности ключевых ферментов, обеспечивающих утилизацию липидов. Этот эффект сочетался с гипертрофическим ответом, который проявлялся в ускоренном синтезе белков и увеличении размера клеток.
В основе развития гипертрофии лежали активация воспаления и развитие оксидативного стресса, которые реализовались через увеличение синтеза ФНО-α и полностью устранялись при его блокаде с использованием специфических антител. Аналогичный ответ зафиксирован и в отсутствие А ІІ при прямом применении ФНО-α, и эффекты как А ІІ, так и ФНО-α полностью устранялись с помощью применения ингибитора NADPH-оксидазы апоцинина или скавенджеров свободных радикалов. Стимуляция А ІІ продукции ФНО-α также устранялась апоницином и скавенджерами свободных радикалов [183].
В то же время ФНО-α по принципу отрицательной обратной связи оказывает угнетающее действие на активность локальной РАС в миокарде, у мышей с генетически рестриктированной экспрессией ФНО-α в сердце отмечены локальная активация АПФ, повышение уровня А II в миокарде с развитием гипертрофии ЛЖ. Применение у этих животных блокаторов АT1-рецепторов сопровождалось нормализацией массы ЛЖ и толщины его стенки, уменьшением содержания коллагена и количества апоптозных клеток.
Данные ряда исследований последних лет также свидетельствуют о четкой координированности между РАС и содержанием провоспалительных цитокинов, прежде всего — ФНО-α, так как они в значительной степени сдерживают по принципу отрицательной обратной связи эффект каждой из них. Особенно это значимо при воспалительных процессах в миокарде, где избыточное количество цитокинов может вызывать паралитическое расширение сосудов, истончение стенки желудочка [73].
А II оказывает активирующее действие на экспрессию тканевого фактора — основного активатора свертывающей системы крови, которое опосредовано высвобождением ФНО-α. В культуре эндотелиоцитов пупочной вены человека применение как А II, так и ФНО-α стимулировало прокоагулянтную активность, 5-кратное усиление экспрессии тканевого фактора, тогда как ингибиторы АПФ, блокаторы АТ1-рецепторов и алискирен — ингибитор ренина, угнетали этот эффект [72].
Четкое угнетающее действие на РАС с блокадой ее гипертензивного и проатерогенного действия оказывает А (1–7) — эндогенный гептапептидный фрагмент, продукт деградации А II, который во многих аспектах является его антагонистом. Пептид А (1–7) образуется из А II под действием АПФ2, который является цинксодержащей металлопротеиназой и нечувствителен к ингибиторам АПФ. Дефицит АПФ2 сочетается с повышенным уровнем тканевого и циркулирующего А II и сниженным уровнем А (1–7). Отмечено, что отсутствие гена АПФ2 приводит к активации воспаления в сосудистой стенке и ускоренному образованию атеросклеротической бляшки у мышей с генетическим отсутствием апоЕ.
Установлено также, что декапептид А I не только превращается в А II под действием АПФ и химазы, но и является субстратом для АПФ2, который катализирует образование А (1–9) [265].
Идентификация АПФ2 существенно расширила представления о регуляторной роли РАС. Установлено, что АПФ2 уменьшает содержание в крови и тканях А ΙΙ и увеличивает – А (1–7) – антиатеросклеротического пептида. АПФ2 способен трансформировать А Ι в А (1–9), а А ΙΙ – в А (1–7), причем метаболизм А ΙΙ осуществляется в 300 раз более эффективно, чем А Ι [265]. А (1–7) является лигандом АТ2-рецептора, который сопряжен с G-белком и интенсивно экспрессируется в сердечно-сосудистой системе. АПФ2/А (1–7)/АТ2-рецепторный путь оказывает антипролиферативное, противовоспалительное и антиоксидантное действие. Выявлено, что подкожное введение А (1–7) в течение 4 нед замедляло развитие атеросклероза у мышей с дефицитом как апоЕ, так и рецепторов ЛПНП, содержащихся на жировой диете. ФНО-α способствует переходу АПФ2 в растворенную форму, значимость которой пока не установлена.
Полагают, что активность циркулирующего А ΙΙ определяется АПФ, который присутствует в циркуляции, и генетический дефицит АПФ сопряжен с отсутствием А II в плазме крови. В то же время тканевой уровень А ΙΙ не определяется АПФ, и его генетическое отсутствие у мышей практически не отражалось на содержании А II в сердце, почках и легких [282]. Полагают, что тканевое образование А ΙΙ определяется главным образом химазой [153]. В то же время, АПФ2 играет важнейшую роль в регуляции активности как циркулирующей, так и тканевой РАС, и его дефицит сопровождается повышением плазменной концентрации А ΙΙ на 130%, а тканевой – на 110%. Помимо этого, дефицит АПФ2 сопровождается снижением, гиперэкспрессия — повышением тканевого уровня А (1–7).
Экспрессия АПФ2 установлена в эндотелиоцитах, гладкомышечных клетках и макрофагах в зоне атеросклеротического поражения. Его гиперэкспрессия замедляет развитие атеросклероза, а генетический дефицит сочетается с ускоренным его развитием. Помимо этого, гиперэкспрессия АПФ2 сочетается со снижением тканевой концентрации А ΙΙ и МСР-1, с угнетением экспрессии цитокинов ФНО-α, ИЛ-6, VCAM-1, MMP-2 и -9, миграции и пролиферации гладкомышечных клеток, а также с накоплением коллагена и стабилизацией бляшки.
Установлено наличие реципрокных отношений между активностью АПФ и АПФ2 в кардиальных миофибробластах, повышение уровня АПФ2 в значительной степени определяет кардио- и ангиопротекторное действие ингибиторов АПФ и блокаторов АТ1-рецепторов. У трансгенных мышей с гиперэкспрессией АПФ2 предупреждалось развитие патологической гипертрофии ЛЖ, а угнетение АПФ2 сочеталось с увеличением содержания А ΙΙ в миокарде и плазме крови с нарушением его сократимости и развитием дилатации ЛЖ. В ряде исследований, проведенных в условиях длительной плавательной нагрузки на крысах, также установлено, что физиологическая гипертрофия ЛЖ сочеталась со снижением активности АПФ и повышением активности АПФ2, экспрессии АТ1- и АТ2-рецепторов, уменьшением кардиального содержания А Ι на фоне увеличения содержания А (1–7).
Поражение почек и прогрессирующее нарушение их функции при гипертонической болезни являются следствием выраженного фиброза как клубочков и почечных сосудов, так и канальцев и интерстиция. Основная причина развития этих изменений — повышение активности локальной почечной РАС с активацией почечных фибробластов, которые экспрессируют рецепторы А ІІ, что приводит к усиленной продукции коллагена, TФР-β и развитию нефросклероза. Выявлено, что дефицит АПФ2 значительно ускоряет развитие почечной недостаточности, тогда как гиперэкспрессия АПФ2 замедляет его.
Это положение базируется на данных ряда экспериментальных исследований. Так, инфузия А ІІ в течение 4 дней мышам с отсутствием АПФ2 сопровождалась значительно более выраженным увеличением содержания А ІІ в почках и более выраженной активацией NADPH-оксидазы и экспрессией ИЛ-1β, чем инфузия нормальным мышам. Эти изменения сочетались с более выраженным усиленным повышением экспрессии TФР-β, продукции коллагена и интенсивности канальцево-интерстициального фиброза, чем у контрольных животных. Интраперитонеальное применение рекомбинантного АПФ2 у мышей, которым вводился А ІІ, сочеталось с уменьшением прессорного ответа, локального содержания А ІІ, интенсивности оксидативного стресса, Т-лимфоцитарного воспаления и фиброза почек.
Углубленный анализ позволил установить, что основным механизмом активации альтернативной РАС является изменение экспрессии специфических микро-РНК. Микро-РНК – эндогенные, мелкие и некодируемые РНК, которые функционируют как отрицательные регуляторы экспрессии генов, угнетают передачу сигнала или усиливают деградацию мРНК-мишеней. Они принимают участие в развитии ряда форм кардиоваскулярной патологии и патологической гипертрофии сердца, но играют также важную роль в развитии физиологической гипертрофии, связанной с аэробной работой. Экспрессия микро-РНК-27a и 27b является отрицательным регулятором активности АПФ (рис. 2, 3), тогда как экспрессия микро-РНК-143 оказывает угнетающее действие на активность АПФ2 [265].
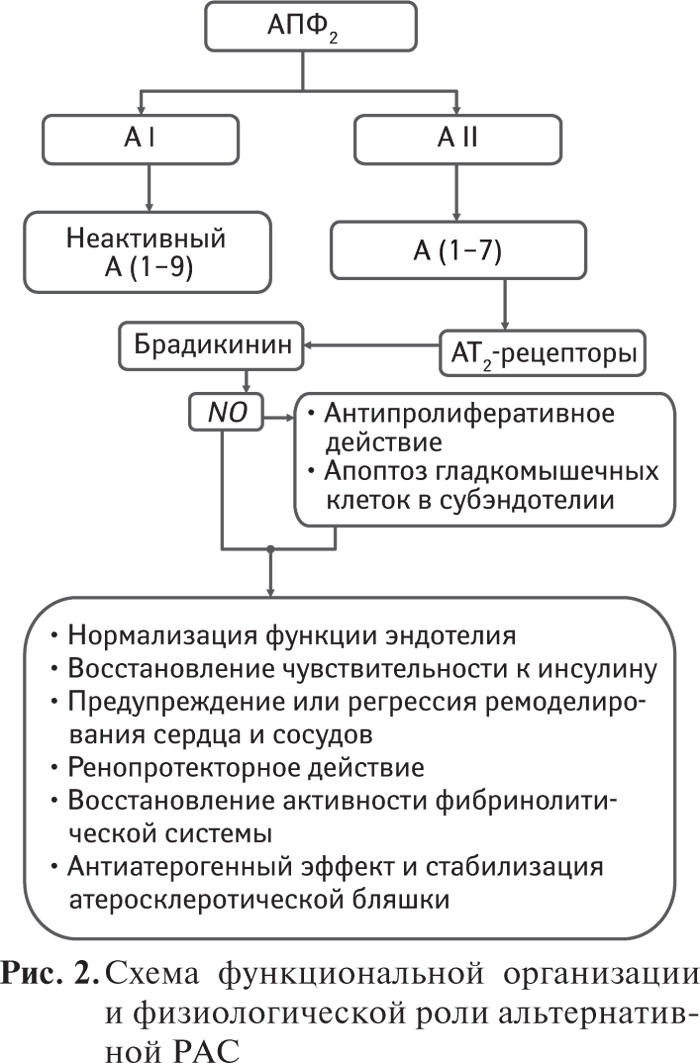
А (1–7) является эндогенным лигандом АТ2-рецепторов, которые связаны с G-белком, их стимуляция сопровождается продукцией NO и простациклина, угнетением прессорного и митогенного действия А II. Это определяет роль А (1–7) как механизма отрицательной обратной связи в отношении А II. Установлено, что антигипертензивное действие ингибиторов АПФ и блокаторов АТ1-рецепторов в значительной степени опосредовано увеличением экспрессии А (1–7).
Выявлено, что А (1–7) угнетает образование неоинтимы у крыс после имплантации стента [126], активирует eNOS и восстанавливает функцию эндотелия [68, 209]. У мышей с отсутствием апоЕ, находящихся 16 нед на атерогенной диете, применение А (1–7) в течение 4 нед сопровождалось значительным угнетением продукции СОР, повышением активности eNOS с восстановлением функции эндотелия параллельно с уменьшением отношения интима/медиа и отложения липидов в сосудистой стенке.
Экспрессия АПФ2 ограничена преимущественно кровеносными сосудами сердца и эпителием канальцев почек. Хотя отсутствие АПФ2 оказывает только минимальное действие на АД, оно резко отражается на сократимости миокарда в сочетании с повышением миокардиальной концентрации А II.
Благодаря способности АПФ2 метаболизировать А II, предотвращается его отрицательное действие на почки. У мышей с отсутствием гена АПФ2 инфузия А II сопровождалась в 2 раза более выраженным повышением его содержания в ткани почки, большим повышением локальной активности NADPH-оксидазы и более выраженной экспрессией провоспалительных цитокинов. Эти изменения сочетались с резким повышением экспрессии генов, определяющих развитие фиброза (прежде всего TФР-β), большим накоплением коллагена и более выраженными гистологическими признаками интерстициального фиброза канальцев. Применение рекомбинантного человеческого АПФ2 у нормальных мышей при инфузии А II значительно снижало прессорный эффект, интенсивность воспаления, которое медиировалось Т-клетками, и интерстициального фиброза канальцев [295].
Установлено, что защитное действие ингибиторов АПФ и блокаторов AT1-рецепторов сочетается с повышением содержания А (1–7) в крови [211]. Это позволило предположить, что А (1–7) может действовать либо как агонист AT2-, либо как блокатор AT1-рецепторов. Результаты ряда исследований свидетельствуют, что пептидные фрагменты А II и, в частности А (1–7), связывались с сайтами AT1-рецепторов и тем самым противодействовали эффектам их активации. В то же время применение блокаторов AT2-рецепторов значительно ослабляло способность А (1–7) нормализовать функцию эндотелия и уменьшать выраженность атеросклеротического поражения. Эти данные свидетельствовали о том, что в реализации защитного эффекта А (1–7) доминантную роль играют AT2-рецепторы [248].
Защитная роль AT2-рецепторов в предупреждении развития прооксидантных и провоспалительных реакций на действие А II, сосудистого и кардиального ремоделирования установлена в ряде исследований. В ряде экспериментальных исследований отмечен положительный эффект агонистов AT2-рецепторов в различных моделях поражения почек [65].
В работе, проведенной на культуре мезангиальных клеток крыс с гиперэкспрессией AT2, оценивалась их модулирующая роль в развитии профибротических изменений. Уровень фибронектина в супернатанте этих клеток, подвергшихся действию активированных макрофагов, был значительно ниже, чем в контроле. При этом не отмечено влияния гиперэкспрессии AT2-рецепторов на секрецию TФР-β и ФНО-α. Полученные данные позволили сделать вывод, что посредством AT2-рецепторов оказывается прямое антисклеротическое действие, которое реализуется в почках в значительной степени путем угнетения мезангиальных клеток, независимо от влияния на уровень АД или активность воспалительного процесса [182].
Однако в другом исследовании показано, что гиперэкспрессия AT2-рецепторов у крыс с частичной нефрэктомией оказывала защитное действие на клубочки, предотвращала накопление внеклеточного гломерулярного матрикса, клеточной пролиферации и развитие нефросклероза в сочетании с угнетением экспрессии TФР-β и тромбоцитарного фактора роста [99].
Одним из механизмов проатерогенного действия А II является способность индуцировать оксидативную модификацию ЛПНП. Недавние исследования показали, что этот эффект опосредован в значительной степени активацией секреторной фосфолипазы А2 IIА типа (сФЛА2 IIА), которая относится к белкам острой фазы и является независимым предиктором коронарных явлений как у здоровых лиц, так и у пациентов с ИБС [145]. Установлено, что между экспрессией сФЛА2 IIА в атеросклеротической бляшке у человека, тяжестью течения атеросклероза, а также риском развития рестеноза после коронарной ангиопластики существует четкая зависимость [216].
В циркуляции сФЛA2 IIA прямо гидролизует ЛПНП, что приводит к образованию их мелких проатерогенных плотных частиц с измененной конфигурацией апоВ, а также СЖК и лизофосфолипидов. Последние являются субстратом для образования провоспалительных липидных медиаторов, в частности — фактора активации тромбоцитов. Отмечено, что ингибиторы сФЛА2 IIА резко уменьшали способность A II модифицировать ЛПНП, и более всего — у лиц с АГ, для которых характерна значительно сниженная резистентность ЛПНП к оксидативной модификации [114]. Применение ирбесартана, особенно в сочетании со статинами, способствовало значительному угнетению сФЛА2 IIА, приводило к уменьшению содержания окисленных модифицированных ЛПНП и предупреждало развитие как АГ, так и атеросклероза [51].
Проатерогенное действие А ІІ в значительной степени определяется также его способностью стимулировать экспрессию в сосудистых гладкомышечных клетках протеина, связанного с рецептором ЛПНП (LRP1). Через этот белок происходит интернализация в гладкомышечные клетки эфиров холестерина из агрегатов ЛПНП, связанных с внеклеточным матриксом, что приводит к трансформации гладкомышечных клеток в пенистые клетки. Этот эффект предупреждался лозартаном — блокатором АТ1-рецепторов, что свидетельствовало о доминантной роли А II в его развитии. Инфузия A II сочеталась со значительным повышением экспрессии LRP1 и липидной инфильтрацией интимы дуги аорты у крыс даже с нормальным содержанием липидов в крови [224].
Известно, что A II, действуя через AT1-рецепторы, вызывает сосудистое воспаление, ремоделирование, дисфункцию эндотелия, угнетает действие инсулина в скелетных мышцах. В исследовании, проведенном на крысах с генетически усиленной продукцией А II, установлены значительно повышенная активность NADPH в ткани аорты, увеличенное содержание активных форм кислорода, С-реактивного белка, усиленная экспрессия ФНО-α, увеличение толщины стенки в сочетании с усиленным апоптозом. Эти изменения отражаются не только на структурно-функциональных свойствах сосудистой стенки, но и существенно уменьшают выраженность действия инсулина с нарушением метаболизма глюкозы, липидов и белков, а также активирующего действия инсулина на еNOS и продукцию NO.
Связь этих нарушений с действием А II и развитием оксидативного стресса подтверждалась тем, что они отчетливо угнетались блокатором AT1-рецепторов валсартаном и миметиком СОД темполом. Кроме этого, в ряде исследований получено прямое подтверждение способности А II через AT1-рецепторы активировать NADPH-оксидазу, стимулировать продукцию активных форм кислорода в сочетании с развитием воспаления, инсулинорезистентности, дисфункции эндотелия и клеточного апоптоза. Установлена прямая связь между выраженностью снижения чувствительности к инсулину, степенью увеличения продукции радикалов кислорода, снижения биоактивности NO, активации факторов транскрипции (NF-κB, AP-1) с усилением экспрессии провоспалительных цитокинов типа ФНО-α, MCP-1, ИЛ-6, С-реактивного белка и развитием воспаления.
Аналогичный эффект характерен и для локальных РАС. У крыс с гиперэкспрессией гена ренина в ткани аорты отмечено увеличение содержания A II с повышением активности NADPH-оксидазы и значительным увеличением продукции свободных радикалов. Эти изменения сочетались с развитием воспаления, АГ, инсулинорезистентности, микроальбуминурии, апоптоза и ремоделирования стенки с увеличением толщины интимы и адвентиции на 50%. Отмеченные изменения, за исключением АГ, примерно в равной степени устранялись применением как блокаторов AT1-рецепторов, так и ингибитора NADPH темпола. В то же время АГ значительно более выражено предотвращалась блокаторами AT1-рецепторов, что свидетельствовало о прямом констрикторном действии А ІІ на гладкомышечные клетки.
На основании этих данных был сделан вывод, что А ІІ активирует NADPH-оксидазу, что проявляется усиленной продукцией активных форм кислорода, являющихся медиатором его метаболического и провоспалительного действия, тогда как АГ в большей степени — отражение прямого вазоконстрикторного влияния А II [271, 272].
Установлено, что А II в значительной степени определяет и возможность дестабилизации атеросклеротической бляшки. У мышей с генетическим отсутствием апоЕ и реноваскулярной АГ, сочетающейся с увеличенной продукцией А ІІ, бляшки характеризовались нестабильностью, тонкой покрышкой, большим накоплением липидов в ядре и повышенным содержанием макрофагов. В то же время, у гипертензивных мышей с нормальной эндогенной продукцией А II и даже с более высоким уровнем АД бляшки относились к стабильному фенотипу. Полагают, что дестабилизация бляшки под действием А ІІ определялась его способностью стимулировать продукцию ИФН-γ, который угнетает пролиферацию гладкомышечных клеток и синтез матриксных белков, его содержание в крови у мышей с повышенным уровнем А ІІ было значительно увеличено [155].
Отрицательный эффект активации РАС на течение и исходы СД 2-го типа, связанные с поражением почек, отчетливо проявился в исследовании 3773 пациентов с различной степенью альбуминурии. На протяжении в среднем 3 лет наблюдения в группах с отсутствием альбуминурии, наличием микро- и макроальбуминурии летальность составляла 7,1; 10,8 и 21,7% соответственно, тогда как применение ингибиторов АПФ достоверно снижало летальность во всей когорте в среднем на 60%.
В соответствии с результатами ряда экспериментальных исследований, сосудистое иммунное воспаление при действии А II играет существенную роль и в развитии АГ. Показано, что инфузия А II мышам в течение 2 нед сопровождалась инфильтрацией Т-клеток в адвентицию аорты и в периаортальную жировую ткань, усиленной продукцией активных форм кислорода, дисфункцией эндотелия и АГ. У мышей с иммунодефицитом эти эффекты не возникали, тогда как трансфузия T-, но не В-клеток, их восстанавливала. Развитие АГ также предупреждалось применением антагониста ФНО-α [18]. Минералокортикоид дезоксикостеронацетат, так же, как и А II, вызывал усиленную продукцию СОР и развитие АГ посредством активации Т-лимоцитов [94].
Установлено, что способность А II усиливать миграцию макрофагов в интиму и приводить к развитию локального воспаления в сосудистой стенке с продукцией активных форм кислорода, цитокинов, хемокинов, молекул адгезии лежит и в основе его проатерогенного действия. Это подтверждено в исследованиях на мышах с генетическим отсутствием апоЕ, у которых после 2-недельного применения А II инфильтрация макрофагами зоны поражения возросла практически в 2 раза. В значительной степени это обусловлено экспрессией моноцитами рецепторов А II, благодаря чему они реагируют на него усилением продукции кислородных радикалов, окислением и захватом ЛПНП.
А II ускоряет также прогрессирование атеросклероза посредством активации продукции ММРs, разрушения эластических мембран с последующей миграцией в интиму и пролиферацией гладкомышечных клеток с продукцией внеклеточного матрикса [173]. Во многих исследованиях эти эффекты сочетались с достоверным снижением активности циркулирующей РАС, уменьшением плазменного содержания ренина и А II и, несмотря на это, с быстрым развитием АГ и атеросклероза. При этом прогрессирование атеросклероза существенно угнеталось ингибиторами АПФ или блокаторами AT1-рецепторов даже без влияния на уровень АД. Это свидетельствовало о ведущей роли локальной РАС и подтверждалось возрастанием экспрессии в аорте А II, AПФ и AT1-рецепторов.
Подобное заключение было сделано также по результатам исследования мышей с генетическим дефицитом апоЕ или рецепторов ЛПНП, у которых при сочетанном отсутствии AT1-рецепторов отмечено резкое замедление развития атеросклероза, несмотря на сохраняющиеся выраженные нарушения липидного спектра крови [267, 268].
В клинических условиях также показана способность А II приводить к дестабилизации клинического течения атеросклероза и развитию острого коронарного синдрома. Важнейшими факторами этого эффекта стали активация воспаления в сосудистой стенке и усиленная продукция сосудистого эндотелиального фактора роста (VEGF). Показано значительное повышение его экспрессии в стенке аорты и в крови при инфузии А II, тогда как блокада VEGF с помощью растворенных рецепторов сопровождалась уменьшением выраженности сосудистого ремоделирования и воспаления, уменьшением продукции МСР-1, хотя при этом не устранялись гипертрофия миокарда и АГ. На основании этих данных сделан вывод, что VEGF является важным медиатором воспаления и структурных изменений в сосудистой стенке, индуцируемых А II, тогда как ремоделирование сердца при инфузии А II определяется преимущественно наличием АГ и увеличенной гемодинамической нагрузкой.
VEGF — один из наиболее мощных ангиогенных факторов, и его применение после повреждения артерии способствует ускоренной регенерации эндотелия, нормализации эндотелийзависимого расслабления, уменьшению образования неоинтимы. Однако VEGF обладает и провоспалительным эффектом, стимулирует миграцию и активацию моноцитов посредством усиления продукции МСР-1 и экспрессии молекул адгезии [294].
Помимо этого, VEGF способствует неоваскуляризации бляшки с появлением тонкостенных сосудов, уязвимых к действию протеиназ, которые накаливаются в бляшке. В результате значительно возрастает риск развития интрамуральных геморрагий, резкого увеличения выраженности сосудистого стенозирования и разрушения бляшки.
Митогенное действие А II во многом определяет ремоделирование сердца, однако и в настоящее время остается невыясненным, в какой степени это связано с гипертрофией кардиомиоцитов, а в какой — определяется пролиферацией других клеток сердца, прежде всего клеток соединительной ткани. Известно, что до 60% клеточных элементов миокарда составляют немиоциты типа фибробластов, гладкомышечных клеток, эндотелиоцитов и макрофагов, и потому их пролиферация может существенно влиять как на структурные, так и функциональные свойства сердца.
В исследовании с длительной инфузией А II отмечены примерно двукратная пролиферация адвентициально-интерстициальных фибробластов в интрамиокардиальных артериях, усиленная пролиферация сосудистых гладкомышечных клеток в сочетании с выраженным угнетением гипертрофии кардиомиоцитов и клеток эндотелия. Несмотря на повышение внутрижелудочкового давления, клеточная пролиферация в предсердиях и желудочках была выражена в равной степени, что свидетельствовало об отсутствии связи между пролиферацией и повышением АД. Уровень альдостерона при этом практически не повысился, что означало наличие прямого гипертрофического действия А II. Лозартан угнетал пролиферацию гладкомышечных клеток и фибробластов, вызванную А II, но не оказывал влияния на угнетение пролиферации кардиомиоцитов и эндотелиоцитов. На основании этих данных был сделан вывод, что повышение уровня А II в условиях ренинзависимой АГ или сердечной недостаточности приводит к пролиферации сосудистых гладкомышечных клеток и фибробластов, опосредуемой AT1-рецепторами и не зависящей от альдостерона и уровня АД. При этом пролиферация миоэндотелиальных клеток снижается вне связи с активацией AT1-рецепторов, что является косвенным свидетельством преобладания в клетках данного типа AT2-рецепторов.
Эти данные были подтверждены в исследовании, проведенном в условиях культуры, в котором А II через AT1-рецепторы усиливал, а через AT2-рецепторы угнетал пролиферацию эндотелиоцитов коронарных сосудов. Хотя радиолигандные исследования не установили наличия AT2-рецепторов в предсердиях и желудочках сердца, прямое угнетающее действие активации этих рецепторов на пролиферацию отмечено в других тканях. В то же время, применение лозартана не оказывало влияния на пролиферацию миоэндотелиальных клеток, свидетельствуя о том, что AT1-рецепторы не задействованы в ее регуляции.
Эти данные означают, что А II оказывает дифференцированное регуляторное влияние на пролиферативные реакции клеток сердца: немиоциты пролиферируют, эта реакция опосредуется активацией AT1-рецепторов и не определяется повышением концентрации альдостерона и гемодинамической перегрузкой сердца. В то же время, угнетение пролиферации миоэндотелиоцитов, отмеченное при действии А II, сопряжено с активацией AT2-рецепторов [65, 189].
Действие А II в значительной степени связано с его способностью активировать синтез и высвобождение ЭТ-1 — мощного прессорного и митогенного фактора. В культуре сосудистых гладкомышечных клеток и эндотелиоцитов А II вызывал экспрессию гена препроэндотелина и продукцию ЭТ-1, который потенцировал гипертрофический ответ кардиомиоцитов на А II. Кроме того, показано, что ЭТ-1 оказывает многогранное влияние на РАС, угнетая синтез ренина и прямо стимулируя секрецию альдостерона в коре надпочечников. У спонтанно гипертензивных крыс раздельное применение ингибиторов АПФ и блокаторов ЭТ-1 давало только частичный эффект, тогда как сочетанное их применение практически полностью нормализовало АД. Эти данные свидетельствуют о том, что патогенез АГ включает сложное взаимодействие РАС и ЭТ-1 [201–203].
Существенное угнетающее влияние на эффекты А II оказывают агонисты рецептора активации пролиферации пероксисом-γ (PPAR-γ), в частности — пиоглитазон и другие производные тиазолидиндиона [100]. После введения крысам А II в течение 7 дней отмечено повышение системного АД со 109 до 176 мм рт. ст., тогда как при одновременном применении пиоглитазона гипертензивный ответ был ослаблен в 3,3 раза. У гипертензивных крыс отмечено развитие оксидативного стресса, сопровождающееся усиленным синтезом ДНК, пролиферацией гладкомышечных клеток с увеличением в результате отношения толщины медии к диаметру просвета сосуда, нарушение эндотелийзависимого расслабления, тогда как у крыс, получавших пиоглитазон, эти изменения отсутствовали. Усиленная экспрессия рецепторов А II 1-го типа, VCAM-1, повышенная активность NF-κB, ММРs, iNOS и увеличенная продукция ФНО-α, отмеченные у гипертензивных крыс, также в значительной мере предупреждались применением пиоглитазона в результате его ингибирующего действия на макрофаги [47]. Выявлено, что способность лигандов PPAR-γ угнетать митогенное и прессорное действие А II, а также устранять его ингибиторное действие на апоптоз гладкомышечных клеток осуществляется преимущественно вследствие снижения экспрессии АТ1-рецепторов [238].
В работе, проведенной на биоптатах атеросклеротических бляшек коронарных артерий пациентов с ИБС, установлено наличие сочетанной экспрессии А II, его рецепторов 1-го типа, АПФ, ИЛ-6 и CD68-положительных макрофагов. Эти данные означали, что в артериях, пораженных атеросклерозом, активируется АПФ с локальным образованием А II. В результате увеличивается продукция ИЛ-6 в макрофагах и гладкомышечных клетках, приводя к развитию воспаления в сосудистой стенке с высоким риском разрушения бляшки и развития острого коронарного синдрома.
В исследовании, проведенном на кроликах с гиперхолестеринемией, установлено увеличение продукции СОР в грудной аорте в 2,2 раза в сочетании с 2-кратным возрастанием способности эндотелия адгезировать моноциты. Эти эффекты почти полностью устранялись инкубацией исследуемого сегмента с антагонистом A II или антиоксидантом пирролидиндитиокарбаматом в течение 2 ч. На основании этих данных сделано заключение, что гиперхолестеринемия активирует локальную РАС с повышением адгезивности эндотелия к моноцитам и увеличением продукции ими СОР [171].
Известно, что содержание в крови ИЛ-6 значительно повышается у лиц с острыми коронарными явлениями. ИЛ-6 стимулирует экспрессию в гладкомышечных клетках белков острой фазы, вызывает миграцию и дифференциацию активированных макрофагов, усиливает продукцию в них ММРs — ферментов, разрушающих соединительнотканный матрикс, регулирует экспрессию адгезивных молекул и других цитокинов типа ИЛ-1 и ФНО-α. Эти процессы в значительной степени являются следствием активации локальной РАС, прежде всего — в макрофагах, инфильтрирующих область атеросклеротического поражения, с увеличенным образованием А II [215].
Не вызывает сомнений, что A II играет решающую роль в возникновении сосудистых повреждений при АГ, атеросклерозе и ряде почечных заболеваний. Этот эффект определяется участием как клеток сосудистой стенки, так и циркулирующих лейкоцитов, которые также экспрессируют рецепторы А II. В исследовании, проведенном на мезентериальных венулах крысы, через 60 мин после введения A II в дозе 1 нмоль/л отмечено 5-кратное повышение интенсивности прокатывания лейкоцитов по эндотелию, 13-кратное возрастание их адгезии и 20-кратное — миграции в сосудистую стенку.
Усиленное взаимодействие лейкоцитов и эндотелиоцитов под влиянием A II определяется тем, что он активирует клетки эндотелия с высвобождением Р-селектина из телец Вейбель — Паладе параллельно с увеличенной продукцией ИЛ-8. Для Р-селектина характерна способность усиливать прокатывание лейкоцитов по эндотелию, тогда как ИЛ-8 обладает хемотаксическими свойствами и способностью обеспечивать прочную адгезию лейкоцитов к эндотелию. Помимо этого, A II стимулирует высвобождение МСР-1 из артериальных эндотелиоцитов, что продемонстрировано в условиях клеточной культуры, и обусловливает посредством него хемотаксис моноцитов. Применение блокаторов рецепторов А II или антител к Р-селектину практически полностью устраняло эти реакции [180] (рис. 4).
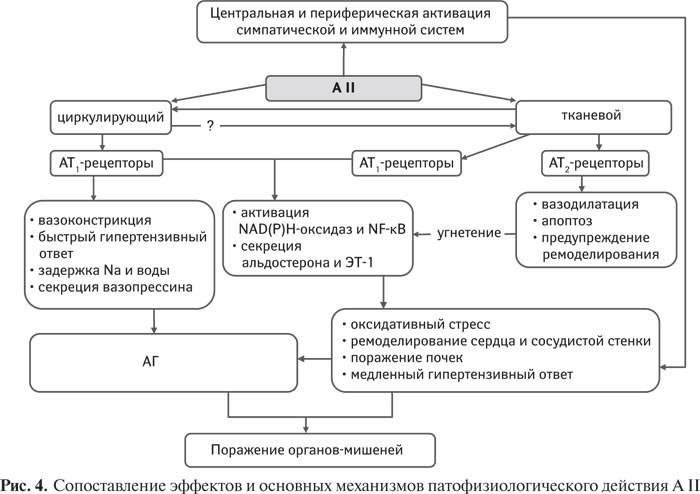
1.4. Альдостерон как компонент ренин-ангиотензиновой регуляторной системы
Множество данных свидетельствует о том, что ряд патологических процессов, особенно АГ, ожирение, СД и сердечная недостаточность сопряжены с активацией РАС и высвобождением в качестве конечного эффекторного фактора не только А II, но и альдостерона [14, 64, 166, 285–289]. Повышенный плазменный уровень альдостерона установлен у лиц с метаболическим синдромом и резистентной АГ и закономерно сочетался с гипертрофией ЛЖ и кардиофиброзом [19–22, 85–87, 104, 151, 259].
Хотя провоспалительные и фибротические изменения в сердце при недостаточности традиционно рассматриваются как следствие повышения концентрации А II, результаты исследований последних лет свидетельствуют о том, что в значительной степени они имеют опосредованный характер и связаны с действием альдостерона. В исследованиях последних лет также установлено, что повышение уровня альдостерона в плазме крови значительно чаще, чем считалось ранее, отмечается у лиц с АГ [76, 138, 139, 261], и показано, что применение спиронолактона — блокатора минералокортикоидных рецепторов, оказывает антигипертензивное действие даже на фоне применения ингибиторов АПФ и блокаторов AT1-рецепторов [205].
Альдостерон, как и А II, играет ключевую роль в гомеостатическом контроле АД посредством регуляции объема внеклеточной жидкости, сосудистого тонуса и сердечного выброса. Основное назначение альдостерона — активация реабсорбции натрия и экскреции калия в дистальных канальцах почек. Альдостерон продуцируется преимущественно в надпочечниках, но в последние годы установлено наличие локального синтеза альдостерона в жировой ткани и высказывается предположение о возможности его образования также в сердце и сосудистой стенке. Этот вопрос еще остается дискуссионным при том, что наличие рецепторов альдостерона в этих тканях не вызывает сомнений.
Помимо участия в регуляции АД посредством контроля водно-солевого баланса, альдостерон оказывает прямое действие на сосудистую стенку [280], установлено, что он может вызывать дисфункцию эндотелия и вазоконстрикцию [281].
Избыток альдостерона в крови оказывает выраженное патологическое действие, и риск развития резистентной АГ, ИМ, геморрагического инсульта, тяжелой гипертрофии ЛЖ при первичном альдостеронизме значительно выше, чем у лиц с АГ. Хотя ранее первичный альдостеронизм считался редким явлением, в последние годы установлено, что он характерен для 10–20% лиц с АГ [78], а в исследовании, результаты которого были опубликованы в 2011 г., наличие первичного альдостеронизма было установлено у 21,2% лиц с неконтролируемой АГ [113].
Так как у лиц с первичным гиперальдостеронизмом значительно увеличен объем крови, то для них характерна сниженная активность ренина плазмы крови, и отношение альдостерон/ренин превышает 30 [79, 82].
Альдостерон способен также активировать моноциты крови, индуцировать воспаление в сосудистой стенке, миокарде и почках [213], приводя к сосудистому ремоделированию, нарушению фибринолиза, развитию артериосклероза, миокардиосклероза, нефросклероза [36, 217, 218]. Системное введение альдостерона приводит к интенсивной периваскулярной лейкоцитарной инфильтрации, усиленной экспрессии остеопонтина, МСР-1, ИЛ-6 и -1β, повышает активность NADPH-оксидазы с инициацией оксидативного стресса в сердце, сосудистой стенке и почках [108], тогда как применение спиронолактона предупреждало появление этих эффектов [120, 168].
На клеточном уровне альдостерон увеличивает продукцию СОР и усиливает экспрессию NF-κB в клетках сосудистой стенки с развитием в ней воспаления, в эксперименте введение альдостерона вызывало тяжелое повреждение миокарда с периваскулярным воспалением, микроинфарктами и фиброзом [156]; аналогичные изменения отмечены и в почках.
В условиях эксперимента при хроническом применении альдостерона закономерно отмечалось развитие оксидативного стресса, повышение активности NADPH-оксидазы и содержания липидных продуктов свободнорадикального окисления, усиление экспрессии генов ICAM-1, МСР-1, ФНО-α, развитие кардиофиброза [229].
В клинических условиях также выявлено, что альдостерон способствует развитию воспаления и фиброза в миокарде, эндотелиальной дисфункции, а плазменный его уровень находится в тесной связи с выраженностью гипертрофии ЛЖ [25]. При сердечной недостаточности альдостерон обусловливает развитие водно-электролитного дисбаланса с задержкой натрия, потерей калия, увеличением внеклеточного объема жидкости.
В соответствии с данными клинических наблюдений, для пациентов с первичным альдостеронизмом характерен наиболее высокий риск кардио- и цереброваскулярных явлений. В экспериментальных условиях показано, что альдостерон стимулирует кардиофиброз, ремоделирование и гипертрофию ЛЖ, а масса миокарда у лиц с первичным альдостеронизмом значительно больше, чем у лиц с эссенциальной АГ при аналогичном уровне АД.
У лиц с первичным альдостеронизмом значительно чаще отмечается эксцентрическая гипертрофия ЛЖ, тогда как для лиц с эссенциальной АГ более характерна концентрическая гипертрофия. Эти различия могут определяться увеличением массы циркулирующей крови при объемзависимой АГ, одной из причин развития которой является повышенное содержание альдостерона в крови.
Поскольку моноциты экспрессируют на мембране минералокортикоидные рецепторы, то альдостерон приводит к их активации с усиленной продукцией TФР-β1, ФНО-α, ИЛ-6 и ИФН-γ, в результате чего развивается воспаление, активируется синтез коллагена, развивается кардиофиброз, который имеет как интерстициальный, так и периваскулярный характер [4].
Помимо этого, гиперальдостеронизм сопровождается снижением уровня в крови калия и магния, что в сочетании с кардиосклерозом, связанным с пролиферацией фибробластов и усиленной секрецией коллагена, приводит к повышению риска развития аритмий и внезапной смерти [99]. У крыс с ИМ применение спиронолактона сопровождалось уменьшением выраженности дилатации ЛЖ, интерстициального ремоделирования, улучшением наполнения ЛЖ и его систолической функции, повышением порога фибрилляции желудочков. Характерно, что способность спиронолактона восстанавливать электролитный баланс, уменьшать риск развития аритмий сердца проявлялась также на фоне применения стандартных доз ингибиторов АПФ [35].
В ряде клинических исследований установлено, что активация РАС при АГ сочеталась с гипертрофией ЛЖ и ремоделированием сердца; эти эффекты развивались на фоне усиленной секреции альдостерона [155] и предупреждались спиронолактоном [191, 192, 263].
Полагают, что способность А ІІ усиливать реабсорбцию натрия и воды в почках, вызывать увеличение объема циркулирующей крови опосредована действием альдостерона. Установлено наличие минералокортикоидных рецепторов в ряде тканей, включая сосудистую стенку, что обусловливает способность альдостерона вызывать развитие АГ, тогда как выраженность АГ и сосудистого повреждения, связанных с действием А ІІ, значительно уменьшалась при применении блокаторов минералокортикоидных рецепторов [236].
Исследования как на культуре клеток, так и in vivo на целостном организме показали, что реакции, характерные для А II, закономерно развиваются при действии альдостерона и активации минералокортикоидных рецепторов [18].
Установлено также, что альдостерон участвует в развитии сердечной недостаточности, определяет скорость ее прогрессирования и повышение риска развития внезапной кардиальной смерти. В основе развития этих эффектов лежит усиление реабсорбции натрия, развитие гипокалиемии и других отрицательных эффектов. Поэтому блокада рецепторов альдостерона устраняет часть аномалий, включая дисфункцию эндотелия, изменение барорецепторной функции, препятствует развитию сердечной и почечной недостаточности.
Одной из особенностей действия альдостерона является его способность ускорять прогрессирование сердечной недостаточности и значительно повышать риск развития фибрилляции желудочков в результате стимуляции экскреции магния. Дефицит магния в крови отмечается у 50% лиц с застойной сердечной недостаточностью, а способность спиронолактона восстанавливать и поддерживать магниевый гомеостаз определяет снижение риска внезапной кардиальной смерти у лиц с сердечной недостаточностью более чем на 30%. С другой стороны, магний участвует в регуляции секреции альдостерона, и внутривенная инфузия сульфата магния в течение нескольких часов сопровождалась снижением уровня циркулирующего альдостерона [270].
Неоднократно показано, что даже длительная и интенсивная терапия ингибиторами АПФ или блокаторами рецепторов А II сопровождается только транзиторным снижением уровня альдостерона в плазме крови и устранением связанных с этим нарушений. Этот эффект обозначается как «ускользание альдостерона» и сочетается с параллельным повышением уровня А II, тогда как дополнительное применение спиронолактона позволяет снизить риск летального исхода более чем на 30% [64, 213].
Значительное повышение активности альдостерона с отрицательными кардиальными и ренальными последствиями установлено у 1053 пациентов с СД 2-го типа, даже длительно получавших терапию ингибиторам АПФ или блокаторами АТ1-рецепторов [14]. В то же время, сочетанное применение блокаторов минералокортикоидных рецепторов существенно уменьшало выраженность поражения органов-мишеней и обеспечивало дополнительный антигипертензивный эффект у лиц с резистентной АГ [276, 277].
В исследованиях с участием пациентов с АГ и первичным альдостеронизмом неоднократно установлено повышение риска развития ИМ [21, 22], а применение блокаторов минералокортикоидных рецепторов оказывало значительное защитное действие [191]. Наличие повышенного плазменного уровня альдостерона также значительно ухудшало прогноз у лиц с перенесенным ИМ, особенно на фоне предшествовавшей АГ. Показано, что применение эплеренона на 3–14-й день после острого ИМ у лиц с предшествовавшей АГ сочеталось с выраженным снижением частоты развития конечных точек, тогда как у лиц с отсутствием АГ подобный эффект не развивался [190].
Установлено, что развитие сердечной недостаточности сочетается с резким возрастанием активности РАС в сочетании с повышением уровня альдостерона в плазме крови в 20–60 раз. Поэтому дополнительное применение блокаторов альдостерона на фоне ингибиторов АПФ или блокаторов АТ1-рецепторов оказывало выраженное защитное действие у пациентов с застойной хронической сердечной недостаточностью и с сердечной недостаточностью в постинфарктный период [191, 196]. Показано, что применение эплеренона у лиц с перенесенным ИМ оказывает значительный протекторный эффект [192].
Результаты ряда исследований, проведенных в последнее время, подтвердили, что блокада альдостерона значительно снижает летальность при сердечной недостаточности даже на фоне стандартной терапии ингибиторами АПФ [192]. Это может быть следствием того, что А II — не единственный фактор, стимулирующий высвобождение альдостерона, подтверждением чему является также «ускользание» альдостерона при длительном применении ингибиторов АПФ. Полагают, что одной из причин, определяющих эффект «ускользания», является гипергликемия, характерная для повышенного уровня альдостерона в крови.
Механизм, который определяет повреждающее действие альдостерона, заключается, главным образом, в стимуляции периваскулярного воспаления и фиброза, ремоделирования сердца и сосудистой стенки [22]. Альдостерон ответственен за развитие при сердечной недостаточности «цитокинового шторма», который проявляется резким увеличением содержания ФНО-α и ИЛ-6 в циркуляции.
В экспериментах на мышах показано, что генетическая гиперэкспрессия минералокортикоидных рецепторов сопровождается спонтанным развитием гипертрофической кардиомиопатии со всеми ее основными проявлениями: гипертрофией кардиомиоцитов, их дезориентацией и интерстициальным фиброзом. Применение спиронолактона устраняло в этих условиях фиброз, на 50% уменьшало беспорядочность ориентации кардиомиоцитов и улучшало диастолическую функцию сердца.
Кардиоваскулярное ремоделирование с усиленным развитием фиброзной ткани в миокарде и сосудистой стенке при гиперальдостеронизме, применении альдостерона и кортизона проявляется усиленной экскрецией гидроксипролина — маркера образования коллагена, и этот эффект устраняется спиронолактоном в субдепрессорных дозах. При гиперальдостеронизме отмечается также периваскулярный фиброз коронарных сосудов, которому предшествуют провоспалительные изменения в их стенке с инвазией моноцитов/макрофагов, и показано, что эти нарушения не связаны с изменениями АД [152, 199–201].
Результаты ряда исследований, проведенных в 1990-е годы, свидетельствовали о том, что в основе развития периваскулярного и интерстициального фиброза в сердце, фиброза и ремоделирования в аорте, поражения почек при действии альдостерона лежит активация факторов транскрипции АР-1 и NF-κB, гиперэкспрессия TФР-β и последующее развитие воспаления. Провоспалительное и профибротическое действие альдостерона подтверждено также на культуре гладкомышечных клеток и кардиомиоцитов.
Установлено, что блокада рецепторов альдостерона у гипертензивных крыс уменьшала выраженность АГ, интенсивность оксидативного стресса и воспаления в аорте, фиброза и гипертрофии миокарда, а у мышей с дефицитом апоЕ, содержащихся на атерогенной диете, уменьшала выраженность воспаления, экспрессии ФНО-α и МСР-1 [95]. Подобно А II, альдостерон активирует NADPH-оксидазы в гладкомышечных клетках и моноцитах с образованием активных форм кислорода и апоптозом клеток проксимальных канальцев почек, угнетением эндотелиальной NO-синтазы. Он также усиливает экспрессию ЦОГ-2 в стенке аорты с образованием целого ряда простаноидов и посредством этого индуцирует воспаление и оксидативный стресс в почках, миокарде и сосудистой стенке с дальнейшим развитием склеротических процессов [218, 219].
Помимо прямого действия на рецепторы, альдостерон активирует экспрессию АПФ и AT1-рецепторов и усиливает провоспалительное действие А II [198].
До недавнего времени не было полной ясности в вопросе о механизмах ремоделирования сердца, приводящего к развитию сердечной недостаточности. Однако в исследованиях последних лет показано, что у подобных больных закономерно возрастает содержание циркулирующего альдостерона, сопровождаясь развитием системного провоспалительного ответа с вовлечением сосудистой системы сердца [83]. Этому предшествует иммуностимуляторное состояние с активацией мононуклеаров периферической крови, которое вызывается снижением цитозольной концентрации свободного магния и последующей перегрузкой кальцием. В результате происходит инвазия моноцитов/макрофагов и CD4+-лимфоцитов в субэндотелиальное и периваскулярное пространство, развивается оксидативно-нитрозилирующий стресс с появлением пероксинитрита и его продукта 3-нитротирозина и активацией NADPH-оксидазы и NF-κB, продукцией ФНО-α, МСР-1, молекул адгезии. Активация периферических мононуклеаров в этих условиях значительно опережала развитие сосудистых изменений в виде ремоделирования стенки. Проявления провоспалительного коронарного сосудистого фенотипа и наличие 3-нитротирозина в мигрировавших воспалительных клетках в этих условиях предупреждались или устранялись увеличением содержания Mg2+ в диете, применением спиронолактона, антиоксиданта N-ацилцистеина и амлодипина, который предупреждал перегрузку моноцитов кальцием и нарушение их редокс-потенциала [1].
Увеличение содержания альдостерона в плазме крови может иметь первичный характер; в этом случае оно является следствием спонтанной избыточной секреции альдостерона надпочечниками и сопровождается развитием АГ. Вторичный гиперальдостеронизм часто развивается у лиц с СД 2-го типа и гиперинсулинемией, поскольку инсулин стимулирует секрецию альдостерона из надпочечников, а также при сердечной недостаточности вследствие активации РАС и увеличения продукции А ІІ.
Первичный альдостеронизм всегда сочетается с АГ, и в этих случаях отмечается увеличенное отношение альдостерон/ренин в плазме крови [158, 170]. При альдостеронизме распространенность резистентной АГ увеличена на 17–22% по сравнению с общей популяцией пациентов с АГ [81].
С 1990 г. существенно изменились представления о функции альдостерона, было показано, что он может синтезироваться и вне надпочечников, прежде всего — в мозгу, сосудистой ткани, миокарде [226]. Установлено, что при сердечной недостаточности синтез альдостерона в миокарде имеет даже более выраженный характер, чем в надпочечниках [230, 248]. Кроме того, минералокортикоидные рецепторы могут активироваться не только альдостероном, но и эндогенными глюкокортикоидами, А II и свободными радикалами кислорода [78].
Плазменный уровень альдостерона при циркадном ритме, АГ или сердечной недостаточности может превышать нормальный в 3–20 раз [250, 261]. Помимо этого, альдостерон может синтезироваться локально и затем высвобождаться в циркуляцию. Отмечено, что его содержание в миокарде и других тканях даже в норме превышает плазменное содержание в 20 и более раз; основными стимуляторами локальной продукции альдостерона являются А II и калий [300].
В клинических наблюдениях уровень альдостерона в значительной степени определял исход ИМ [121], а блокада рецепторов альдостерона снижала летальность у лиц с застойной сердечной недостаточностью и дисфункцией ЛЖ после ИМ [18]. По данным исследования CONSENSUS, между уровнем альдостерона и летальностью при сердечной недостаточности существует прямая зависимость. В ряде крупных многоцентровых исследований установлено, что уровень альдостерона в плазме крови у лиц с сердечной недостаточностью повышен в тесной связи с риском летального исхода.
В исследовании UK-HEART установлена прямая связь между содержанием альдостерона в плазме крови и риском внезапной сердечной смерти. Это объясняют увеличением экскреции калия и развитием гипокалиемии параллельно с повышением плазменного уровня альдостерона, что является одним из ведущих факторов развития аритмий сердца [174].
По данным исследования RALES, блокада рецепторов альдостерона спиронолактоном у пациентов с тяжелой сердечной недостаточностью сопровождалась снижением на 30% риска развития кардиальной смерти на протяжении 24 мес наблюдения [175]. В более позднем исследовании EPHESUS применение эплеренона у 6632 лиц с сердечной недостаточностью в течение 16 мес сочеталось со снижением частоты летальных исходов на 15%, внезапной кардиальной смерти — на 21%.
Результаты многих клинических наблюдений свидетельствуют о том, что альдостерон участвует в развитии АГ, застойной сердечной недостаточности, ИБС, инсульта и почечной недостаточности. Особенно сильная связь характеризует повышение плазменного уровня альдостерона, риск развития АГ и ее осложнений. В исследовании G. Rossi и соавторов среди 1125 пациентов с АГ повышенный уровень альдостерона в плазме крови отмечен у 11,2% [203], и для них было характерно значительно более выраженное поражение органов-мишеней: сердца, сосудов и почек, резкое возрастание риска развития кардиальных явлений [204].
Установлено, что повышение плазменного уровня альдостерона закономерно сочетается с развитием АГ [19, 20, 114], по средним данным, альдостерон определяет до 20% всех случаев АГ [261]. В исследовании, проведенном с участием 1688 нормотензивных лиц, было выявлено, что увеличение содержания альдостерона в крови даже в физиологических границах предрасполагало к развитию АГ [171].
Несмотря на наличие значительного числа антигипертензивных препаратов различных классов, примерно у 50% пациентов с АГ не удается добиться адекватного контроля уровня АД. Особенно это характерно для лиц с инсулинорезистентностью и СД 2-го типа, у которых АГ достоверно сочетается с увеличением содержания альдостерона в крови. Участие альдостерона в развитии АГ подтверждается выраженным антигипертензивным эффектом, уменьшением выраженности гипертрофии миокарда и альбуминурии при использовании блокаторов его рецепторов — эплеренона и спиронолактона.
Альдостерон вызывает также развитие выраженных метаболических нарушений [187–189], и в исследовании 356 пациентов с АГ плазменный уровень альдостерона положительно сочетался с уровнем глюкозы, инсулина, С-пептида и индекса HOMA-IR [39]. У больных с гиперальдостеронизмом отмечена четкая прямая связь между индексом НОМА-IR и содержанием альдостерона в крови, а адреналэктомия сопровождалась уменьшением содержания в крови глюкозы и инсулина, снижением инсулинорезистентности [85–87].
Особо выраженная экспрессия минералокортикоидных рецепторов характерна для фибробластов сердца, их активация альдостероном сопровождается интенсивным синтезом коллагена. Поэтому у лиц с АГ при избытке альдостерона гипертрофия ЛЖ выражена непропорционально степени повышения АД. Так, в исследовании с участием 21 пациента с АГ и первичным альдостеронизмом отмечено достоверно более выраженное увеличение конечно-систолического и конечно-диастолического объема ЛЖ, увеличение толщины межжелудочковой перегородки и массы ЛЖ по сравнению с лицами с АГ из группы контроля, но без признаков альдостеронизма.
Данные исследований последних лет свидетельствуют о том, что повышение плазменной концентрации альдостерона является значительно более частой причиной развития АГ и более тяжелого ее течения, чем предполагалось ранее. Первичный альдостеронизм установлен примерно у 2–13% неселективной популяции гипертензивных лиц и у 10–23% пациентов с резистентной АГ [21, 53, 78]. При исследовании 600 пациентов с АГ он диагностирован в 6,1% случаев, а у лиц с тяжелой АГ (АД выше 180/110 мм рт. ст.) — в 13% [162]. В популяционном исследовании, проведенном с участием 1392 гипертензивных пациентов, наличие первичного гиперальдостеронизма, определенного по увеличенному отношению альдостерон/ренин, отмечено в 7% случаев, при резистентной АГ — в 11,9%, АГ III стадии — в 18,3% [98].
Более того, ⅔ пациентов с рефрактерной АГ характеризуются низкой плазменной концентрацией как ренина, так и А II, и для них характерна высокая эффективность применения антагонистов альдостерона. Установлено, что для многих пациентов с АГ с увеличенным отношением концентрации в плазме альдостерона к ренину блокаторы минералокортикоидных рецепторов являются препаратами выбора даже при отсутствии первичного альдостеронизма [57].
Наиболее значимым является то, что даже у нормотензивных пациентов повышенная концентрация альдостерона в плазме крови является предиктором развития тяжелой АГ [261], особенно в сочетании со сниженной концентрацией ренина [159]. Выявлено, что наличие положительной связи между возрастанием значения отношения альдостерон/ренин в плазме и риском развития АГ [171], повышением жесткости стенки артериальных сосудов [137] и увеличением скорости распространения пульсовой волны [227] характерно даже для нормотензивных лиц молодого возраста.
Неоднократно высказывалось предположение, что низкорениновая эссенциальная АГ является следствием недиагностированного идиопатического гиперальдостеронизма [139, 140].
Примерно в 20–30% случаев АГ характеризуется резистентностью к лечению, прежде всего — среди лиц пожилого возраста, особенно, при наличии ожирения и ИМТ, превышающего 30 кг/м2. Резистентной АГ считается в тех случаях, когда не удается добиться полноценного контроля АД с помощью применения 3 антигипертензивных препаратов различных классов, одним из которых является диуретик, и для достижения целевого уровня АД необходимо применение 4 и более препаратов. Распространенность резистентной АГ достигает 20–30%, и основной стратегией терапии лиц с этой патологией является включение в схему лечения спиронолактона.
По данным ряда исследований, одной из наиболее частых причин развития резистентной АГ является первичный гиперальдостеронизм, особенно в сочетании с нормальным уровнем калия в крови, когда он определяется в 20–23% случаев [80, 162]. О первичном характере альдостеронизма судят по сочетанию сниженной плазменной активности ренина с увеличенным содержанием альдостерона в суточной моче при проведении солевой нагрузки [57]. Величина отношения альдостерон/ренин в плазме крови >20 при содержании альдостерона >15 нг/дл указывает на наличие первичного гиперальдостеронизма.
Для пациентов с резистентной АГ обычно характерна повышенная чувствительность к соли, и при содержании на низкосолевой диете АД у них на 23/9 мм рт. ст. ниже, чем при высокосолевой [186–188]. Исследования, проведенные на экспериментальных моделях с генетически повышенной активностью синтазы альдостерона, свидетельствовали о значительном повышении в этих условиях гипертензивной реакции на солевую нагрузку. Эти данные позволили высказать предположение, что наследственно повышенная активность альдостерон-синтазы может лежать в основе развития АГ при увеличенном потреблении соли, независимо от РАС и даже на фоне снижения ее активности [150].
Участие альдостерона в патогенезе резистентной АГ подтверждается высокой терапевтической эффективностью в этих условиях блокаторов минералокортикоидных рецепторов, прежде всего — спиронолактона [173]. В ряде клинических исследований его применение дополнительно к комбинации ингибитора АПФ и блокатора AT1-рецепторов или диуретика у лиц с резистентной АГ позволило снизить cистолическое АД в среднем на 25 мм рт. ст. и диастолическое — на 12 мм рт. ст. [29, 176, 260].
В одном из исследований 55 пациентов с резистентной АГ получали стандартную антигипертензивную терапию, включавшую в среднем 4 препарата, 56 других дополнительно получали спиронолактон в дозе 215 мг. Через 8 нед лечения у лиц опытной группы систолическое АД было ниже в среднем на 5,4 мм рт. ст., диастолическое — на 1,0 мм рт. ст. [260]. В крупном Скандинавском исследовании (Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm) добавление спиронолактона к стандартному лечению, состоявшему в применении 3 препаратов, приводило через 1,3 года к снижению систолического АД на 21,0 мм рт. ст., диастолического — на 9,5 мм рт. ст. [176].
Метаанализ данных 5 проспективных и одного ретроспективного исследований также свидетельствует о выраженном антигипертензивном действии блокады минералокортикоидных рецепторов спиронолактоном у пациентов с резистентной АГ. Его добавление к стандартной антигипертензивной терапии позволило добиться дополнительного снижения систолического АД в среднем на 22 мм рт. ст., диастолического — на 10 мм рт. ст. [152].
Полагают, что основную роль в терапевтическом эффекте спиронолактона у лиц с резистентной АГ играет регрессия ремоделирования сосудистой стенки с увеличением ее растяжимости, что проявляется снижением скорости распространения пульсовой волны. Этот фактор объясняет более выраженное влияние спиронолактона на систолическое АД по сравнению с диастолическим [145, 149].
У 3056 пациентов определялась зависимость между отношением плазменной концентрации альдостерон/ренин и уровнем АД. Среднее АД у всех пациентов исследованной когорты составило 141/81 мм рт. ст., отношение альдостерон/ренин — 10,2 у мужчин и 14,4 — у женщин. Средняя величина систолического АД возрастала ступенчато от 126,8 мм рт. ст. в 1-м дециле отношения альдостерон/ренин до 151,0 мм рт. ст. в 10-м дециле, средняя величина диастолического АД — от 74,3 до 86,9 мм рт. ст. с учетом пола, возраста, ИМТ, функции почек и характера лечения. При проведении регрессионного анализа отношение альдостерон/ренин было наиболее достоверным предиктором среднего как систолического, так и диастолического АД. Полученные данные свидетельствовали о том, что отношение альдостерон/ренин определяет уровень АД в широком диапазоне изменений величины этого отношения и оказывает влияние на АД при величине значительно ниже необходимой для диагностики первичного альдостеронизма.
У лиц в нижних децилях величины отношения альдостерон/ренин увеличение плазменного содержания ренина сопровождалось синхронным повышением содержания А II. Однако это происходило без увеличения содержания альдостерона, и при его содержании в 1-м дециле содержание ренина было в 10 раз выше, чем при содержании альдостерона в 10-м дециле. Эти данные означают, что при повышенном плазменном содержании ренина клетки коркового слоя надпочечников становятся менее реактивными в отношении стимуляции А II и секретируют меньше альдостерона.
С другой стороны, в исследовании с участием 3300 лиц установлено, что отношение альдостерон/ренин имеет наследственный характер, а секреция альдостерона возрастает в результате повышения чувствительности клеток коркового слоя надпочечников к А II. По мере увеличения отношения альдостерон/ренин отмечено снижение плазменного содержания не только ренина, но и А II, одним из механизмов повышения секреции альдостерона в этих условиях могло быть отмечено повышение плазменного содержания кортизола, который является одним из наиболее мощных стимуляторов синтеза альдостерона [172]. В последние годы установлено, что хроническое повышение уровня кортизола в плазме крови сочетается с гиперплазией надпочечников и повышением их чувствительности к действию калия и А II; подобная зависимость отмечается и при синдроме Кушинга [40, 41].
У лиц с синдромом Кушинга особенно отчетливо проявляется связь между повышенной активностью РАС, увеличением плазменного содержания альдостерона и резистентной АГ [21]. В основе синдрома лежит гиперсекреция кортизола, который является мощным индуктором продукции и высвобождения альдостерона. Для 70–90% лиц с синдромом Кушинга характерно наличие АГ, для 17% — тяжелой; особенностью течения АГ при синдроме Кушинга является резистентность к терапии и значительно более выраженное поражение органов-мишеней [6, 163].
Механизмы развития АГ у лиц с ожирением имеют в настоящее время еще гипотетический характер и предположительно включают избыточное потребление соли, нарушение экскреции натрия, активацию симпатической нервной системы и РАС [97]. В последние годы к числу важнейших механизмов развития резистентной АГ у лиц с ожирением относят и избыточное содержание в крови альдостерона. Установлено, что у подобных пациентов на фоне отсутствия эффективности антигипертензивной терапии уменьшение массы тела на 1 кг сочетается со снижением систолического АД на 1–2 мм рт. ст., предположительно — за счет уменьшения продукции альдостерона [171].
Частое сочетание ожирения с гиперальдостеронизмом с определяется как активацией РАС, так и способностью адипоцитов секретировать рилизинг-факторы — еще не идентифицированные соединения, активирующие образование и высвобождение альдостерона, независимо от А II [56, 63, 91, 92].
На основании результатов многочисленных исследований было сделано заключение, что гиперпродукция альдостерона с его усиленным влиянием на экскрецию калия, реабсорбцию натрия и воды, структуру и функциональные свойства сосудистой стенки является важнейшей детерминантой АД. Зависимость между АД и отношением альдостерон/ренин имеет закономерный характер уже при пограничной АГ, и это означает, что гиперальдостеронизм в различной степени имеет широкое распространение у пациентов с АГ [252, 253].
Результаты экспериментальных исследований свидетельствуют, что роль альдостерона в ремоделировании сердца (гипертрофии и склерозирования) в значительной степени модулируется потреблением соли. В частности, установлено, что содержание натрия в крови и интенсивность его экскреции с мочой находятся в положительной корреляционной связи с массой ЛЖ и толщиной межжелудочковой перегородки только у пациентов с первичным альдостеронизмом, но не в группе контроля.
Диета западного типа с высоким содержанием натрия и низким — калия предрасполагает к развитию АГ. При этом сниженное поступление калия значительно усиливает повреждающее действие избытка натрия в крови. Выявлено, что повышенная экскреция натрия с мочой, отражающая его избыточное поступление в организм с пищей, находится в прямой зависимости от уровня АД и повышения индекса массы ЛЖ.
Четкая зависимость между показателем экскреции натрия и массой сердца установлена при исследовании, включавшем 1042 практически здоровых лиц молодого возраста. Так, лица в верхнем квартиле экскреции натрия по сравнению с лицами в нижнем характеризовались достоверно большим на 11% индексом массы ЛЖ, что является предиктором кардиоваскулярной летальности, независимым от наличия АГ. Более того, у нормотензивных лиц с увеличенной массой ЛЖ достоверно повышен риск развития АГ, а у лиц с АГ ограничение потребления соли сопровождается регрессией гипертрофии ЛЖ, несмотря на повышение активности циркулирующей РАС.
Отмечено, что в основе пролиферативного и профибротического действия увеличенного плазменного и тканевого содержания натрия лежит активация локальных тканевых РАС, прежде всего, повышение экспрессии АТ1-рецепторов в миокарде, сосудистой стенке, ткани почек [45].
Эти данные и результаты ряда других исследований позволили сделать вывод, что уровень пищевого потребления соли оказывает существенное влияние на повреждение сердца посредством повышения чувствительности к альдостерону, а у лиц с гипернатриемией ограничение потребления соли может существенно снизить уровень кардиоваскулярного риска [187–189].
В исследовании, проведенном на культуре кардиомиоцитов, продемонстрировано, что альдостерон при высоком содержании соли способствует также усилению экспрессии гена АПФ [285], а в других исследованиях увеличенное потребление соли сопровождалось сочетанной активацией экспрессии рецепторов А II 1-го типа и минералокортикоидных рецепторов [10, 227].
По данным ряда исследований, основные проявления патологического действия альдостерона в виде АГ, сочетающейся с развитием оксидативного стресса и воспаления, усиленным синтезом коллагена, ремоделированием сердца и сосудов в значительной степени опосредованы участием остеопонтина. У мышей инфузия альдостерона в сочетании с солевой нагрузкой в течение 4 нед сопровождалась повышением АД и развитием альбуминурии параллельно с повышением экспрессии остеопонтина, усиленным интерстициальным фиброзом и воспалительными инфильтратами в почках на фоне повышенной экспрессии NADPH-оксидазы [245]. При генетическом отсутствии продукции остеопонтина у мышей в подобных условиях возникало аналогичное повышение АД, но поражение почек и активация NADPH были значительно менее выраженными. Эти данные свидетельствуют о том, что остеопонтин является одним из основных медиаторов воспаления, оксидативного стресса и интерстициального фиброза в почках при действии альдостерона [109].
Остеопонтин относится к регуляторам активности ММРs и посредством их активации принимает участие в ремоделировании сердца и сосудов [32]. Он также способен активировать миграцию и пролиферацию гладкомышечных клеток [286]. Установлена прямая связь между плазменным содержанием остеопонтина и жесткостью сосудистой стенки [136], кальцификацией атеромы [115]. Остеопонтин является также активатором воспаления, и его содержание положительно коррелирует с экспрессией ЦОГ-2 и провоспалительных цитокинов [119].
Концентрация остеопонтина в миокарде резко повышается при ожирении [90], атеросклерозе, СД 2-го типа, кардиомиопатиях, сердечной недостаточности. Усиленный синтез остеопонтина четко коррелирует с риском тромбоза коронарных сосудов и развития ИМ [247], тогда как при развитии ИМ у мышей с отсутствием остеопонтина отмечено резкое снижение синтеза коллагена и увеличение выраженности дилатации ЛЖ [256].
В экспериментальных условиях установлено, что содержание остеопонтина в интиме и медии артерий значительно увеличивается при действии альдостерона и А ІІ. Остеопонтин обусловливает пролиферацию гладкомышечных клеток и деградацию эластической мембраны медии артерий и является, таким образом, триггером сосудистого ремоделирования [208]. Особенно этот процесс выражен в сосудах сердца и почек и приводит к развитию кардио- и нефросклероза, сердечной и почечной недостаточности [291]. Повышенная продукция остеопонтина является фактором риска развития аневризм абдоминального отдела аорты [88, 89, 166].
По мнению ряда исследователей, остеопонтин является одним из важнейших факторов и маркеров патологического ремоделирования сердца и сосудов [246, 291], и его концентрация в плазме крови больных с сердечной недостаточностью тесно ассоциируется с тяжестью клинической картины заболевания и активностью воспаления [43].
В нормальном миокарде отсутствует экспрессия остеопонтина, ее выявляют только при развитии реакции на повреждение. При гипертрофии миокарда и развитии инфаркта миокарда остеопонтин накапливается в интерстиции после клеточной инфильтрации и локализуется преимущественно вокруг миофибробластов [232].
Остеопонтин усиленно экспрессируется в миокарде пациентов с ишемической, гипертрофической и дилатационной кардиомиопатией. Основным источником его продукции в этих условиях были непосредственно кардиомиоциты, а интенсивность синтеза остеопонтина не коррелировала с выраженностью клеточной инфильтрации интерстиция.
В то же время, хотя интенсивность секреции альдостерона прямо связана с активностью РАС и уровнем А II, она может регулироваться и независимо от них. Это особенно отчетливо проявляется при применении ингибиторов АПФ у пациентов с АГ и сердечной недостаточностью, у которых влияние терапии на уровень альдостерона в плазме крови имеет транзиторный характер на фоне стабильно сниженного уровня А II.
Неоднократно продемонстрировано, что высокий плазменный уровень альдостерона инициирует развитие миокардиального склероза и гипертрофии ЛЖ, наличие прямой зависимости между массой миокарда ЛЖ и уровнем альдостерона установлено как у здоровых лиц, так и у пациентов с АГ или первичным гиперальдостеронизмом [262]. Для предупреждения развития гипертрофии и стимуляции ее регрессии при АГ широко используются ингибиторы АПФ и блокаторы рецепторов А II. Однако они только транзиторно угнетают продукцию альдостерона, плазменное его содержание у лиц с сердечной недостаточностью увеличено на 40% даже на фоне хронического применения ингибиторов АПФ — феномен, который обозначается как «ускользание альдостерона» и значительно ограничивает терапевтический эффект ингибиторов РАС [192, 249].
В исследовании RESOLVD применение эналаприла и кандесартана у пациентов с сердечной недостаточностью сопровождалось достоверным снижением уровня альдостерона через 17 нед, но возвращением его к исходному значению через 43 нед даже при максимальной дозе применяемых препаратов. Помимо этого, блокада минералокортикоидных рецепторов эплереноном оказывала выраженное защитное действие даже на фоне эффективной терапии ингибиторами АПФ или блокаторами AT1-рецепторов.
Хотя, в соответствии с традиционными взглядами, увеличение массы сердца является следствием АГ, в ряде исследований установлено, что гипертрофия ЛЖ может предшествовать повышению АД [110, 193]. Кроме того, в большинстве наблюдений у лиц с АГ не установлено зависимости между уровнем АД и выраженностью гипертрофии миокарда [46, 71]. В то же время уровень альдостерона сочетался с массой ЛЖ независимо от АД и определял тяжесть течения патологического процесса как в экспериментальных [197, 289, 290], так и в клинических исследованиях [54, 222, 225].
Данные ряда крупных многоцентровых исследований подтвердили, что альдостерон является триггером гипертрофии ЛЖ и кардиосклероза независимо от изменений АД, а снижение его уровня может стать важным медиатором устранения или регрессии гипертрофии и ремоделирования сердца. В исследовании ALLAY (Aliskiren in Left Ventricular Hypertrophy) у 465 пациентов с избыточной массой тела, АГ и гипертрофией ЛЖ применение алискирена в течение 9 мес при сопоставлении с лозартаном сопровождалось значительно более выраженным снижением уровня альдостерона в плазме крови в сочетании с более выраженным уменьшением массы миокарда ЛЖ при аналогичном антигипертензивном эффекте [194]. Эти данные свидетельствуют о том, что плазменный уровень альдостерона и выраженность его патологического действия не всегда являются прямым отражением активности РАС и содержания А ΙΙ в крови.
Отсутствие прямой зависимости между повышением АД и гипертрофией сердца подтверждается также тем, что ремоделирование и фиброз миокарда при АГ отмечаются в обоих желудочках, хотя при этом не увеличивается нагрузка на правый желудочек. Применение спиронолактона в этих условиях значительно уменьшало выраженность ремоделирования, что свидетельствовало о доминирующем значении альдостерона в его развитии [16].
Раздельный эффект альдостерона на АД и ремоделирование сердца и коронарных сосудов выявлен у крыс, у которых экзогенное воспроизведение гиперальдостеронизма сопровождалось повышением АД на 23% параллельно с независимым от него увеличением содержания коллагена в миокарде и фибронектина в стенке коронарных артерий с повышением ее жесткости. Применение эплеренона в низких дозах не оказывало антигипертензивного действия, но устраняло, тем не менее, как кардиальные, так и сосудистые эффекты альдостерона [169]. Хотя гипертрофия сердца при АГ ограничена ЛЖ, фиброз отмечался в обоих, свидетельствуя о том, что он не обусловлен гемодинамическими влияниями [290]. Неоднократно также отмечено, что спиронолактон в низких дозах уменьшает выраженность аортального фиброза у спонтанно гипертензивных крыс без влияния на уровень АД [123].
Роль альдостерона в ремоделировании и фиброзировании сердца отчетливо проявилась в исследовании, проведенном на трех моделях АГ. В реноваскулярной модели установлено одновременное повышение уровня А II и альдостерона, при сужении аорты ниже почечных артерий уровень обоих гормонов был в пределах нормы, тогда как при хронической инфузии альдостерона и повышении его уровня уровень А II был сниженным. В двух моделях, где уровень альдостерона был повышенным, отмечались выраженная гипертрофия и фиброз в обоих желудочках. Угнетение действия альдостерона низкими дозами спиронолактона предупреждало развитие фиброза, хотя и не уменьшало выраженности АГ и гипертрофии миокарда. В то же время, в модели с пережатием аорты и нормальным уровнем альдостерона, несмотря на выраженную АГ и гипертрофию миокарда, не было отмечено признаков его фиброза [16].
Сопоставление этих данных позволило высказать предположение, что гипертрофия кардиомиоцитов связана преимущественно с гемодинамической нагрузкой, тогда как накопление коллагена и развитие фиброза определяются действием альдостерона.
Альдостерон, как и А II, оказывает повреждающий эффект двумя путями — путем действия, опосредованного активацией минералокортикоидных рецепторов, и через повышение АД. Показано, что ремоделирование сердца, фибротические изменения в миокарде, выраженный апоптоз кардиомиоцитов при действии альдостерона возникают даже без развития АГ, тогда как применение блокаторов альдостерона в дозах, не оказывающих антигипертензивного действия, значительно уменьшает выраженность повреждения органов-мишеней у крыс со спонтанной АГ, находящихся на солевой диете [199, 200]. В основе этого действия лежит способность блокаторов предупреждать повышение активности NADPH-оксидазы, зависящую от нее продукцию СОР и посредством этого поддерживать функцию эндотелия [194]. Подобный эффект отмечен также у лиц с сердечной недостаточностью, у которых спиронолактон способствовал значительной нормализации функции эндотелия на фоне угнетения продукции СОР [69, 70].
Альдостерон вызывает также развитие сосудистого воспаления, характеризующегося инфильтрацией сосудистой стенки моноцитами/макрофагами с усиленной экспрессией генов ЦОГ-2, МСР-1, остеопонтина и фокальными некротическими изменениями. Действие альдостерона на сосудистую стенку распространяется и на адвентицию с развитием периваскулярного фиброза, тогда как спиронолактон ослабляет этот эффект. Более того, этот эффект альдостерона лежит также в основе действия А II, что подтверждается способностью эплеренона предупреждать развитие сосудистого воспаления при АГ, воспроизведенной введением А II [199–201].
Характерно, что при экспериментально воспроизведенном селективном кардиоспецифическом гиперальдостеронизме поражаются прежде всего коронарные артерии с развитием периваскулярного воспаления, и этот эффект отмечен еще до развития гипертрофии и фиброза миокарда [84, 240–242, 290].
К механизмам патологического действия альдостерона относится способность стимулировать продукцию PAI-1 с угнетением фибринолиза, а также способность значительно усиливать сосудистый ответ на А II в результате повышения экспрессии АТ1-рецепторов и степени их сродства к А II [18].
Повреждающее действие альдостерона в значительной степени определяется его способностью подавлять активность парасимпатической нервной системы, что рассматривается рядом исследователей как следствие развития оксидативного стресса и снижения биодоступности NO [34]. Еще одним важнейшим патогенетическим механизмом альдостерона является способность стимулировать экскрецию калия и магния из организма, а поддержание их нормального уровня у лиц с сердечной недостаточностью способствует резкому снижению риска прогрессирования процесса и развития внезапной кардиальной смерти [7].
Помимо прямого действия, альдостерон способен усиливать профибротический эффект А II, стимулируя экспрессию его рецепторов. На культуре миокардиальных фибробластов крысы показано, что альдостерон активирует синтез коллагена в концентрациях, в которых он присутствует в крови при реноваскулярной АГ. А II также способен активировать синтез коллагена, но только в высоких концентрациях, которые могут возникать при его локальной продукции в миокарде, но не отмечаются в циркуляции.
Хотя сам факт развития кардиосклероза при повышенном уровне альдостерона в настоящее время является общепризнанным, в ряде недавно проведенных работ было установлено, что выраженность кардиосклероза при гиперальдостеронизме резко увеличивается в условиях высокосолевой диеты, так как чувствительность минералокортикоидных рецепторов к альдостерону значительно повышается при увеличенном содержании натрия в среде.
Основным активатором синтеза альдостерона является А II, однако кардиопротекторный эффект ингибитора АПФ эналаприла, значительно выраженный в первые недели применения, сильно ослабляется со временем. Это совпадает с начальным снижением содержания альдостерона в крови и постепенным восстановлением его повышенного уровня, несмотря на сохраняющийся сниженный уровень А II. Считают, что этот эффект определяется постепенным развитием гиперкалиемии, так как ионы калия являются независимым активатором синтеза альдостерона. Поэтому сочетание ингибиторов АПФ или блокаторов рецепторов А II при сердечной недостаточности со спиронолактоном приводило к значительному повышению эффективности терапии и сопровождалось уменьшением летальности 30% [191, 192]. Комбинированное применение эплеренона и эналаприла у лиц с АГ сопровождалось вдвое более выраженным антигипертензивным действием и регрессией ЛЖ, чем каждого из препаратов в виде монотерапии.
В ряде крупных клинических исследований установлено, что в качестве антигипертензивного препарата эплеренон столь же эффективен, как и лозартан. В условиях длительного применения А II эплеренон предупреждал развитие сосудистого воспаления и ремоделирование сосудистой стенки, независимо от наличия АГ и гипертрофии ЛЖ, что свидетельствует о медиаторной роли альдостерона в повреждающем действии А II.
В последние годы было неоднократно подтверждено, что альдостерон принимает прямое участие в развитии инсулинорезистентности, метаболического синдрома, резистентной АГ, независимо от активности циркулирующей РАС и плазменного содержания А II [116]. Так, при первичном альдостеронизме закономерно отмечается сниженный уровень плазменной активности ренина и А II в сочетании с повышенным уровнем глюкозы, большей распространенностью кардиометаболического синдрома и большим риском развития кардиоваскулярных явлений, чем при АГ [29, 96, 160]. У пациентов с первичным альдостеронизмом толщина стенки сонных артерий увеличена пропорционально уровню альдостерона и отношению альдостерон/активность ренина. В ряде исследований показано, что повышенный уровень альдостерона, но не активность ренина, определяет риск развития метаболического синдрома [13, 119].
В значительной части случаев альдостерон действует наряду с А II как эффекторное звено РАС. Так, развитие метаболического синдрома у лиц с абдоминальным ожирением опосредовано повышением уровня в плазме крови как ренина, так и альдостерона, и этот эффект устраняется при уменьшении массы тела [66, 67]. Установлено, что изменения, инициированные внутривенной инфузией А ІІ, в частности — повышение концентрации, в значительной степени опосредованы действием альдостерона, активацией минералокортикоидных рецепторов и блокируются спиронолактоном [146, 237]. Помимо прямого провоспалительного действия через минералокортикоидные рецепторы, альдостерон также усиливает его посредством активации экспрессии АПФ, AT1-рецепторов и повышения локальной концентрации А II [239]. В свою очередь, глюкокортикоиды активируют экспрессию рецепторов альдостерона, и потому их блокаторы снижают активность воспаления даже в условиях, когда эндогенная концентрация альдостерона не повышена.
Установлено, что многие эффекты А II типа ремоделирования, дисфункции эндотелия, гиперпродукции СОР реализуются через альдостерон [101, 264], и повышение АД, как и развитие оксидативного стресса в стенке аорты, при введении А II частично нормализуются под действием спиронолактона. Эплеренон и блокаторы рецепторов альдостерона улучшали функцию эндотелия у крыс, которым проводили инфузии А II [122, 264], и у кроликов, содержавшихся на высокожировой диете [195]. Установлено, что А II и альдостерон оказывают синергичное действие на сосуды, в частности, митогенный эффект альдостерона имеет не только прямой характер, но и определяется существенным потенцированием эффекта А II посредством повышения экспрессии его рецепторов 1-го типа, а также активности АПФ [283, 284].
В связи с сочетанными механизмами действия А ІІ и альдостерона, наиболее эффективной антиатерогенной терапией у обезьян, находящихся на атерогенной диете, было сочетание блокаторов минералокортикоидных рецепторов с ингибиторами АПФ или блокаторами AT1-рецепторов, которое приводило к уменьшению содержания МСР-1, окисленных ЛПНП, толщины интима-медиа, существенной нормализации функции эндотелия. Выявлено также снижение активности ММРs, что свидетельствовало о возможной активной роли альдостерона в дестабилизации атеросклеротической бляшки [243, 244].
Альдостерон, как и А II, оказывает двойственное действие на тонус сосудистой стенки — расслабляющее через эндотелий и прямое — констрикторное — через прямое влияние на гладкомышечные клетки [142, 143].
Для альдостерона характерно также выраженное влияние на системный метаболизм, у лиц с аденомой надпочечников примерно в 50% случаев отмечаются сниженная толерантность к глюкозе и другие метаболические аномалии, которые устраняются при хирургическом удалении опухоли [86, 87]. Поэтому наличие гиперальдостеронизма в значительной степени определяет сочетание АГ с высокой распространенностью сопутствующих метаболических аномалий, прежде всего — ожирения, инсулинорезистентности, дислипидемии и гипергликемии, то есть факторов, формирующих метаболический синдром, даже на фоне сниженной активности ренина в крови. Выявлено, что у лиц с АГ, связанной с первичным альдостеронизмом, по сравнению с лицами с АГ почти на 40% выше распространенность метаболического синдрома (41,1 по сравнению с 29,6% соответственно), на 77% — гипергликемии (27,0 и 15,2%). Эти данные объясняют большую значимость АГ при альдостеронизме как фактора кардиоваскулярного риска по сравнению с изолированной АГ [67].
В исследовании, проведенном с участием 397 лиц афроамериканской этнической группы, пациенты с АГ по сравнению с нормотензивными характеризовались большей окружностью талии, нарушенным профилем липидов, наличием инсулинорезистентности и сниженной активностью ренина в сочетании с повышенным уровнем альдостерона. АД положительно коррелировало с содержанием альдостерона и обратно — с активностью ренина, а плазменное содержание альдостерона находилось в тесной связи с окружностью талии, индексом НОМА, содержанием общего холестерина, триглицеридов и инсулина. У 17% пациентов был диагностирован метаболический синдром по критериям АТР III параллельно с повышением плазменного уровня альдостерона, но не активности ренина, плазменная концентрация которого снижалась, обратно коррелировала с АД и не коррелировала с компонентами метаболического синдрома. Кроме того, как АД, так и содержание альдостерона коррелировали с компонентами метаболического синдрома, включая инсулинорезистентность и гиперинсулинемию. Эти данные свидетельствуют о том, что альдостерон является связующим звеном между АГ, ожирением, инсулинорезистентностью и метаболическим синдромом [117].
По данным более чем 35 000 аутопсий, АГ часто сочетается как с наличием метаболического синдрома, так и с гиперплазией или аденомой надпочечников, особенно у лиц афроамериканской этнической группы [207]. Связано это с тем, что двумя важнейшими стероидами коры надпочечников являются альдостерон и глюкокортикоид кортизол, и оба они участвуют в патогенезе АГ и метаболического синдрома [142]. Избыток кортизола отмечается при синдроме Кушинга, который по всем характеристикам соответствует метаболическому синдрому и включает АГ, инсулинорезистентность и дислипидемию. В работе установлено, что у лиц с синдромом Кушинга плазменное содержание альдостерона коррелировало с АД, уровень альдостерона был повышен у лиц с АГ, несмотря на сниженный уровень активности ренина. Напротив, содержание ренина обратно коррелировало с АД и не сочеталось с компонентами метаболического синдрома.
Данные ряда исследований указывают на то, что подобная форма патогенеза АГ существует у 10% в неселективной популяции пациентов с АГ, у которых длительная терапия с применением ингибиторов АПФ не приводит к снижению уровня АД и содержания альдостерона в крови [40].
В исследовании, проведенном на пациентах с метаболическим синдромом, среди которых у 66% лиц отмечали избыточную массу тела, содержание альдостерона в крови было повышено на 20%, независимо от сочетания компонентов синдрома. В то же время, связь между активностью ренина и содержанием альдостерона была слабой, и только содержание альдостерона, но не активность ренина сочеталось с повышенным уровнем АД и наличием компонентов синдрома [13]. Во многих клинических и популяционных исследованиях установлено, что при первичном альдостеронизме метаболический синдром отмечается гораздо чаще, чем при АГ. Уменьшение массы тела сопровождается снижением уровня альдостерона и восстановлением чувствительности к инсулину у лиц как с нормальным АД, так и при АГ [231, 232].
Известно, что гиперактивность симпатической нервной системы играет важнейшую роль в развитии АГ и обменных нарушений при метаболическом синдроме, она значительно более выражена у лиц с висцеральным ожирением. Хотя длительное время основной причиной активации симпатической нервной системы при ожирении считали повышение уровня А II, в настоящее время установлено, что подобный эффект возникает и при действии альдостерона. Показано, что первичный гиперальдостеронизм и АГ сочетаются с симпатической гиперактивностью, а блокада минералокортикоидных рецепторов в центральной нервной системе устраняет ее [228]. Блокада периферических эффектов альдостерона с помощью спиронолактона также снижала симпатическую активность у лиц с АГ и метаболическим синдромом.
При уменьшении массы тела у лиц с ожирением отмечена нормализация плазменного уровня альдостерона параллельно со снижением АД. Эти данные позволили предположить, что РАС жировой ткани, наряду с классической РАС, принимает существенное участие в развитии инсулинорезистентности и АГ [61, 62, 105].
Неоднократно подтверждено наличие прямой зависимости между ожирением и АГ; ее распространенность составляет 15% среди лиц с ИМТ <25 кг/м2 и достигает 40% среди пациентов с ожирением [181]. Данные ряда исследований свидетельствуют о том, что в основе сочетания ожирения, дислипидемии, инсулинорезистентности и АГ лежит активация РАС с параллельным увеличением содержания альдостерона, который инициирует развитие оксидативного стресса и системного воспаления с последующими нарушениями структуры и функции почек, миокарда и стенки артериальных сосудов. Выявлено, что блокада минералокортикоидных рецепторов в этих условиях устраняла прогрессирование кардиоваскулярной патологии [279, 280], повышала чувствительность к инсулину в сочетании со снижением активности NADPH-оксидазы и ослаблением оксидативного стресса [237].
Особое значение в развитии АГ и метаболического синдрома имеет активация минералокортикоидных рецепторов в адипоцитах, что сопровождается усилением экспрессии провоспалительных цитокинов и оказывает адипогенное действие, тогда как их угнетение сопровождается повышением продукции адипонектина, особенно у пациентов с АГ [80].
Описано наличие как прямой, так и обратной связи между альдостероном и А II: альдостерон повышает активность АПФ и стимулирует экспрессию рецепторов А II, тогда как А II потенцирует секрецию альдостерона [134, 135]. С другой стороны, хотя А II является ведущим фактором секреции альдостерона в ответ на уменьшение объема циркулирующей крови, но продукция альдостерона увеличивается также при гиперкалиемии, под действием адренокортикотропина. В большинстве случаев альдостерон попадает в сердце из циркуляции, однако в определенных ситуациях он может синтезироваться непосредственно в миокарде, хотя экспрессия мРНК альдостеронсинтазы в миокарде примерно на 3–5 порядков ниже, чем в надпочечниках.
Выраженная экспрессия альдостеронсинтазы в сердце у людей отмечена при сердечной недостаточности [290]. У лиц с дилатационной кардиомиопатией и застойной сердечной недостаточностью установлено значительно большее содержание альдостерона в крови коронарного синуса, чем в артериальной крови, что свидетельствовало о его продукции непосредственно в миокарде [155]. В исследованиях последних лет установлено, что повышение уровня альдостерона в плазме крови значительно чаще отмечается у лиц с АГ, чем считалось ранее [77, 139, 140, 262], а применение спиронолактона — блокатора минералокортикоидных рецепторов, оказывает антигипертензивное действие даже на фоне приема ингибиторов АПФ и блокаторов AT1-рецепторов [206].
Хотя провоспалительные и фибротические изменения в сердце при недостаточности длительное время рассматривались как следствие увеличения содержания А II, в последние годы установлено, что в значительной степени они связаны с альдостероном, действие которого медиируется через систему NF-κB [212]. У спонтанно гипертензивных крыс установлена достоверно повышенная экспрессия воспалительных медиаторов в стенке, тогда как применение блокатора рецепторов альдостерона сопровождалось выраженным угнетением их продукции. В то же время, применение комбинированной антигипертензивной терапии, включающей гидралазин, гидрохлоротиазид и резерпин, сопровождалось аналогичным снижением АД, но отсутствием эффекта на экспрессию NF-κB и воспалительных медиаторов.
Установлено, что лица с АГ характеризуются наличием положительной достоверной корреляционной зависимости между плазменным содержанием альдостерона и уровнем АД, показателями инсулинорезистентности, содержанием в крови калия, кортизола и ренина [40]. Помимо этого, распространенность метаболического синдрома достоверно повышена у пациентов с АГ и высоким плазменным уровнем альдостерона по сравнению с пациентами с АГ, но с нормальным содержанием альдостерона в крови [68, 235].
Одним из факторов, определяющих связь между повышением уровня альдостерона и риском развития АГ, инсулинорезистентности и дислипидемии, является гипокалиемия как следствие гиперальдостеронизма. Это подтверждается тем, что гипокалиемия, возникающая при применении тиазидных диуретиков, также способствует развитию инсулинорезистентности, и неоднократно отмечено, что уровень калия в крови находится в обратной зависимости от содержания глюкозы [301]. Установлено также, что повышение уровня калия в крови стимулирует, а снижение — угнетает высвобождение инсулина, приводя к развитию соответствующих метаболических нарушений.
Недавно выявлено, что у лиц афроамериканской этнической группы, выходцев из Сейшельских островов, плазменное содержание альдостерона, но не ренина, сочетается с метаболическим синдромом и маркерами инсулинорезистентности, и эта зависимость имеет прямой характер, не опосредованный изменениями содержания калия в плазме крови [13, 118].
Установлено, что в основе развития инсулинорезистентности при гиперальдостеронизме лежат активация воспаления и появление оксидативного стресса, тогда как угнетение NADPH-оксидазы при введении альдостерона практически полностью угнетает эти эффекты [42].
Данные исследований последних лет свидетельствуют о том, что ожирение и АГ развиваются сочетанно с активацией РАС [14, 168, 279]. Неоднократно отмечено повышение уровня альдостерона у лиц с ожирением, метаболическим синдромом и резистентной АГ в сочетании с гипертрофией ЛЖ и кардиофиброзом [21, 87, 152], тогда как уменьшение массы тела сочеталось со снижением плазменного уровня альдостерона, А II и ренина [61, 63, 104].
Связь ожирения с гиперальдостеронизмом предположительно объясняют способностью адипоцитов секретировать минералокортикоид-рилизинг-факторы и стимулировать продукцию альдостерона в надпочечниках [56]. В ряде работ установлено, что при висцеральном ожирении содержание альдостерона в крови увеличивается независимо от содержания ренина или концентрации калия, и высказано предположение, что это связано с адипоцитарным фактором, активирующим секрецию альдостерона. Позднее было подтверждено, что жировая ткань способна секретировать пока еще не идентифицированные так называемые альдостеронстимулирующий [21, 167] или минералокортикоид-рилизинг-фактор [56], которые активируют стероидогенез.
Многочисленные клинические и экспериментальные данные свидетельствуют, что у лиц с ожирением основным источником усиленной продукции альдостерона является жировая ткань, особенно абдоминальная, в связи со способностью висцеральных адипоцитов крыс с метаболическим синдромом продуцировать факторы, активирующие высвобождение альдостерона из коркового слоя надпочечников [167].
Отмечено, что инкубация адренокортикальных клеток с продуктами секреции изолированных адипоцитов сопровождалась 7-кратным увеличением продукции альдостерона через 24 ч инкубации. Этот эффект был сопоставим с эффектом максимальной стимуляции этих клеток форсколином, при этом он не зависел от А II и не устранялся блокатором AT1-рецепторов валсартаном. Это означает, что усиление продукции альдостерона при ожирении не является прямым следствием продукции А II в жировой ткани.
В связи с усиленной продукцией альдостерона в жировой ткани, АГ при ожирении имеет объемный характер и связана преимущественно с задержкой натрия и воды. Особо значимую роль в этом процессе играет жировая ткань, локализованная в надпочечниках, что свидетельствует о паракринном действии факторов, продуцируемых в ней. Установлено, что влияние ожирения на продукцию альдостерона в надпочечниках может определяться не столько массивами жировой ткани, сколько адипоцитами, находящимися непосредственно в надпочечниках в тесном контакте с клетками, продуцирующими кортикостероиды [3].
В результате повышенного плазменного содержания альдостерона потребление соли у лиц с ожирением оказывает значительно более выраженное гипертензивное и повреждающее действие, чем у лиц без ожирения.
Закономерное развитие гиперальдостеронизма при ожирении опосредовано и рядом других механизмов. В значительной степени оно определяется способностью СЖК, высвобождаемых из жировой ткани, прямо стимулировать продукцию альдостерона [91]. Повышение концентрации в крови СЖК приводит к развитию оксидативного стресса, который, как полагают, является непосредственным фактором усиленной секреции альдостерона. Выявлено, что содержание продуктов оксидации линолевой кислоты прямо коррелирует с уровнем альдостерона и АГ у лиц с избыточной массой тела, свидетельствуя о наличии связи между оксидативным стрессом, альдостероном и АГ при ожирении [92].
В недавно опубликованных работах отмечено, что адипоциты также могут независимо продуцировать альдостерон [17].
Способность прямо стимулировать секрецию альдостерона характерна также для инсулина [184], и при ожирении неоднократно выявлено наличие прямой корреляционной зависимости между плазменным содержанием альдостерона, инсулина и маркерами инсулинорезистентности [39]. В связи с этим, гиперинсулинемия у лиц с ожирением сопровождается усиленной секрецией альдостерона и последующим развитием АГ даже при сниженном содержании ренина в крови, которое определяется задержкой натрия в результате действия альдостерона. Помимо этого, А II в большей степени усиливает секрецию альдостерона у лиц с ожирением, чем с нормальной массой тела, и у женщин с АГ и избыточной массой тела установлено значительно повышенное отношение альдостерон/активность ренина в плазме крови [141, 142].
В исследовании 2010 г. показано, что у лиц афроамериканской этнической группы с АГ и высоким уровнем потребления соли значительно усилен эффект блокады РАС. Для этой категории больных характерно интенсивное поражение органов-мишеней и повышенное содержание альдостерона, несмотря на низкорениновый статус, хотя причины, лежащие в основе этого сочетания, оставались невыясненными. В ряде других исследований была установлена независимая и прямая связь между плазменной концентрацией ангиотензиногена и альдостерона, и эта связь была особенно выраженной у лиц с избыточным потреблением натрия.
Отмечено, что лимитирующим звеном образования А I и А II является не ренин, а ангиотензиноген, и его повышенный уровень, особенно в сочетании с увеличением тканевого содержания альдостерона и натрия, способствует развитию АГ и тяжелому поражению органов-мишеней даже в отсутствие повышенной концентрации ренина.
Для многих лиц афроамериканского происхождения с АГ характерно сочетание низкого уровня ренина с непропорционально высокой концентрацией альдостерона в крови и степенью поражения органов-мишеней. Это связано с тем, что на фоне диеты с высоким содержанием натрия и низким — калия угнетается высвобождение ренина, но активируется РАС, и ее участие в повышении АД в значительной степени обусловлено повышением концентрации ангиотензиногена.
В исследовании 579 лиц африканского происхождения определялось, в какой степени ангиотензиноген в крови участвует в поддержании концентрации циркулирующего альдостерона и повышении АД при нахождении на диете с высоким Na+ и низким K+. Установлено, что в этих условиях плазменная концентрация ренина находилась в обратной связи с АД, содержанием альдостерона и потреблением соли, тогда как концентрация ангиотензиногена в плазме крови положительно коррелировала с содержанием альдостерона и системным АД. В ряде других исследований также установлено, что действие альдостерона значительно усиливается на фоне высокосолевой диеты, и это связано с повышением чувствительности коркового слоя надпочечников к действию А II в условиях гипернатриемии.
Результаты многих клинических наблюдений свидетельствуют о том, что на фоне высокосолевой диеты, вызывающей угнетение секреции ренина, ангиотензиноген становится важнейшей детерминантой активации РАС и повышения систолического АД. Это означает, что у солечувствительных лиц на фоне солевой диеты ключевым фактором повышения АД является активация РАС через ангиотензиноген, и это подтверждается наличием сильной зависимости между концентрацией ангиотензиногена и альдостерона у лиц с высоким, но не низким потреблением натрия. Предположительно это связано с тем, что на фоне высокого потребления натрия значительно повышается эффективность преобразования ангиотензиногена в А Ι под действием ренина, а также продукция альдостерона надпочечниками под действием А ΙΙ.
При уменьшении массы тела у лиц с ожирением отмечена нормализация плазменного уровня альдостерона параллельно со снижением АД. Эти данные позволили предположить, что РАС жировой ткани, наряду с классической РАС, принимает существенное участие в развитии инсулинорезистентности и АГ [61, 62, 103].
Хотя ранее считали, что А II стимулирует секрецию альдостерона в надпочечниках через AT1-рецепторы, при ожирении блокаторы этих рецепторов не оказывали влияния на плазменный уровень альдостерона.
Связь между уровнем альдостерона и риском развития метаболического синдрома подтверждена результатами Фремингемского исследования, в рамках которого у 2292 лиц среди 8 биомаркеров, отражающих гемостаз, воспаление и нейрогуморальную активность, только содержание PAI-1 и альдостерона коррелировало с риском развития синдрома, прежде всего — с систолическим АД, окружностью талии, индексом инсулинорезистентности, уровнем инсулина и нарушениями профиля липидов. У пациентов с метаболическим синдромом отмечали повышение уровня альдостерона, часто при отсутствии повышенной активности ренина и даже при его снижении [45]. Повышение плазменной активности альдостерона у лиц с ожирением и АГ на фоне сниженной активности ренина указывает на гиперчувствительность в этих условиях адренокортикоидной системы к А II [124].
Эти положения подтверждены в исследовании с участием 397 пациентов, в котором АД положительно коррелировало с уровнем альдостерона в плазме крови, тогда как последний — с окружностью талии, содержанием инсулина, индексом инсулинорезистентности и нарушениями профиля липопротеидов, а пациенты с метаболическим синдромом характеризовались повышенным уровнем альдостерона, нормальным или сниженным уровнем ренина и увеличенным отношением альдостерон/ренин [117]. В другом исследовании уровень альдостерона у лиц с метаболическим синдромом был повышен на 20%, и только он, но не плазменная активность ренина, коррелировал с АД [13, 148].
Между А II и альдостероном существуют тесная прямая и обратная взаимосвязь. Помимо прямого действия на минералокортикоидные рецепторы, альдостерон способствует повышению экспрессии АПФ и AT1-рецепторов и усиливает провоспалительный эффект А II. С другой стороны, A II обладает высоким сродством к минералокортикоидным рецепторам и способен прямо их активировать. Поэтому применение спиронолактона значительно уменьшает выраженность воспаления, повышение АД, увеличение продукции МСР-1 и F2-изопростанов, индуцированных введением А II [18].
Наличие тесной взаимосвязи между А II и альдостероном показано в исследовании, проведенном на добровольцах, находящихся в течение 5 дней на ограниченном потреблении соли (<12 ммоль/сут), у которых выраженно и прогрессирующе повышался уровень в крови как А II, так и альдостерона. Более того, уровень альдостерона повышался непропорционально высоко относительно уровня А II, что свидетельствует о повышении секреции альдостерона в ответ на А II в условиях снижения уровня натрия в крови. С другой стороны, повышение уровня альдостерона сопровождалось угнетением продукции ренина и уменьшением содержания в крови как ренина, так и А II.
Помимо этого, пролиферативное и ремоделирующее действие альдостерона на сердце и сосуды в значительной степени опосредовано усилением экспрессии ЭТ-1 в стенке мелких и крупных артерий.
Альдостерон оказывает также выраженное активирующее действие на свертывающую систему крови. После его инфузии с 2–3-кратным повышением уровня в крови отмечено развитие тромбоза венозных сосудов со значительным уменьшением периода кровотечения и повышением тромбоцитарной адгезии. Эти изменения сочетались с увеличением экспрессии PAI-1, активности NADPH-оксидазы, содержания СОР, малонового диальдегида и перекиси водорода. Применение эплеренона значительно уменьшало выраженность отмеченных изменений, свидетельствуя об их связи с активацией минералокортикоидных рецепторов [93]. На крысах со спонтанной АГ показано, что постоянное применение альдостерона сочеталось с развитием тромботических микроангиопатий, а у мышей с атеросклерозом отмечено значительное усиление тромбоза после повреждения стенки артериальных сосудов, тогда как спиронолактон блокировал эти эффекты.
Установлено, что для содержания альдостерона в крови характерен циркадный ритм, его пик отмечается в утренние часы параллельно с повышением АД и транзиторными изменениями активности свертывающей системы крови. Эти изменения у лиц с наличием клинически выраженной ИБС могут предрасполагать к развитию острых коронарных явлений.
С другой стороны, неоднократно показано, что повышение плазменного уровня калия, являющегося фактором усиленной секреции альдостерона, сочеталось с угнетением тромбообразования и снижением чувствительности тромбоцитов к индукторам агрегации.
Полагают, что одним из ведущих механизмом влияния альдостерона на систему свертывания крови является его способность усиливать синтез и экспрессию PAI-1, тогда как применение спиронолактона сопровождалось значительным угнетением экспрессии мРНК PAI-1 в ткани почек у крыс, подвергшихся радиационному воздействию [18]. В значительной степени протромбогенное действие альдостерона связано также с его способностью инициировать развитие оксидативного стресса и снижение биодоступности оксида азота.
Наличие тесных взаимосвязей между РАС и альдостероном дало основание ряду исследователей рассматривать альдостерон как один из компонентов РАС, и в настоящее время термин «ренин-ангиотензин-альдостероновая система» (РААС) отмечается в литературе так часто, как и термин РАС. Признанием этого факта является издание в США в течение ряда последних лет журнала «Journal of Renin-Angiotensin-Aldosterone System». Однако это положение, по-видимому, не всегда достаточно обосновано, поскольку повышение уровня альдостерона в плазме крови может иметь первичный характер относительно активности РАС и возникать даже на фоне ее сниженной активности.
1.5. Список использованной литературы
- Ahokas R.A., Sun Y., Bhattacharya S.K. et al. Aldosteronism and a proinflammatory vascular phenotype. Role of Mg2+, Ca2+, and H2O2 in peripheral blood mononuclear cells // Circulation. — 2005. — Vol. 111, № 1. — P. 51–57.
- Akasaki T., Ohya Y., Kuroda J. et al. Increased expression of gp91phox homologues of NAD(P)H oxidase in the aortic media during chronic hypertension: involvement of the renin-angiotensin system // Hypertens.Res. — 2006. — Vol. 29. — P. 813–820.
- Ammarguellat F.Z., Gannon P.O., Amiri F., Schiffrin E.L. Fibrosis, matrix metalloproteinases, and inflammation in the heart of DOCA-salt hypertensive rats: Role of ETA receptors // Hypertens. — 2002. — Vol. 39. — Р. 679–684.
- Anand K., Mooss A.N., Mohiuddin S.M. Aldosterone inhibition reduces the risk of sudden cardiac death in patients with heart failure // JRAAS. — 2006. — Vol. 7. — P. 15–19.
- Ardanaz N., Yang X-P., Cifuentes M.E. et al. Lack of glutathione peroxidase 1 accelerates cardiac-specific hypertrophy and dysfunction in angiotensin II hypertension // Hypertens. — 2010. — Vol. 55. — P. 116–123.
- Arnaldi G., Mancini T., Polenta B. et al. Cardiovascular risk in Cushing’s syndrome // Pituitary. — 2004. — Vol. 7. – P. 253–256.
- Ascherio A., Rimm E.B., Hernan M.A. et al. Intake of potassium, magnesium, calcium, and fiber and risk of stroke among US men // Circulation. — 1998. — Vol. 98. — P. 1198–1204.
- Auabe N., Babaev V.R., Tang Y. et al. Transiently hightened angiotensin II has distinct effects on atherosclerosis and aneurism formation in hyperlipidemic mice // Atherosclerosis. — 2006. — Vol. 184. — P. 312–321.
- Bader M., Peters J., Baltatu O. et al. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research // J. Mol. Med. — 2001. — Vol. 79. — P. 76–102.
- Barauna V.G., Magalhaes F.C., Krieger J.E. et al. AT1 receptor participates in the cardiac hypertrophy induced by resistance training in rats // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2008. — Vol. 295. — P. R381–R387.
- Barhoumi T., Kasal D.A., Li M.W. et al. T regulatory lymphocytes prevent Angiotensin II–induced hypertension and vascular injury // Hypertens. — 2011. — Vol. 57. — P. 469–476.
- Baron-Menguy C., Toutain B., Cousin M. et al. Involvement of angiotensin II in the remodeling induced by a chronic decrease in blood flow in rat mesenteric resistance arteries // Hypertens. Res. — 2010. — Vol. 33. — P. 857–866.
- Bochud M., Nussberger J., Bovet P. et al. Plasma aldosterone is independently associated with the metabolic syndrome // Hypertension. — 2006. — Vol. 48. — P. 239–247.
- Bomback A.S., Klemmer P.J. Interaction of aldosterone and extracellular volume in the pathogenesis of obesity-associated kidney disease: a narrative review. Am. J. Nephrol. — 2009. — Vol. 30. — P. 140–146.
- Brasier A.R., Recinos A., Eledrisi M.S. Vascular inflammation and the renin-angiotensin system // Arterioscler. Thromb. Vasc. Biol. — 2002. — Vol. 22. — P. 1257–1262.
- Brilla C.G., Rupp H., Maisch B. Effects of ACE inhibition versus non-ACE inhibitor antihypertensive treatment on myocardial fibrosis in patients with arterial hypertension. Retrospective analysis of 120 patients with left ventricular endomyocardial biopsies // Herz. — 2003. — Vol. 28. — Р. 744–753.
- Briones A.M., Rodríguez-Criado N., Hernanzet R. et al. Atorvastatin prevents angiotensin II–induced vascular remodeling and oxidative stress // Hypertens. — 2009. — Vol. 54. — P. 142–149.
- Brown N.J. Aldosterone and vascular inflammation // Hypertension. — 2008. — Vol. 51. — P. 161–167.
- Calhoun D.A. Aldosterone and cardiovascular disease: smoke and fire //Circulation. — 2006. — Vol. 114. — P. 572–2574.
- Calhoun D.A. Is there an unrecognized epidemic of primary aldosteronism? // Hypertens. — 2007. — Vol. 50. — P. 447–453.
- Calhoun D.A., Jones D., Textor S. et al. Resistant Hypertension: Diagnosis, Evaluation, and Treatment. A Scientific Statement From the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research // Hypertens. — 2008. — Vol. 51. — P. 1403–1419.
- Calhoun D.A., Sharma K. The role of aldosteronism in causing obesity-related cardiovascular risk // Cardiology Clinics. — 2010. — Vol. 28. – Р. 517–527.
- Cambien F., Poirier O., Lecerf L. et al. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction // Nature. — 1992. — Vol. 359. — P. 641–644.
- Campbell D.J. Critical review of prorenin and (pro)renin receptor research // Hypertens. — 2008. — Vol. 51. — P. 1259–1264.
- Campbell D.J. Angiotensin II generation in vivo: does it involve enzymes other than renin and angiotensin-converting enzyme? // Angiotensin II generation in vivo: does it involve enzymes other than renin and angiotensin-converting enzyme? // JRAAS. — 2012. — Vol. 13. — P. 314–316.
- Campbell D.J., Karam H., Menard J. et al. Prorenin contributes to angiotensin peptide formation in transgenic rats with rat prorenin expression targeted to the liver // Hypertens. — 2009. — Vol. 54. — P. 1248–1253.
- Cannon R.O. Potential mechanisms for the effect of angiotensin-converting enzyme inhibitors on endothelial dysfunction: the role of nitric oxide // Am. J. Cardiol. — 1998. — Vol. 82. — P. 8S–10S.
- Catena C., Lapenna R., Baroselli S. et al. Insulin sensitivity in patients with primary aldosteronism: a follow-up study // J. Clin. Endocrinol. Metabol. — 2006. — Vol. 91. — P. 3457–3463.
- Chapman N., Dobson J., Wilson S. et al. Anglo-Scandinavian Cardiac Outcomes Trial Investigators. Effect of spironolactone on blood pressure in subjects with resistant hypertension // Hypertens. — 2007. — Vol. 49. — P. 839–845.
- Chen K., Chen J., Li D. et al. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts. Modulation by PPAR-y ligand pioglitazone // Hypertension. — 2004. — Vol. 44. — P. 655.
- Chen H., Li D., Sawamura T. et al. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan // Biochem. Biophys. Res. Commun. — 2000. — Vol. 276. — P. 1100–1104.
- Cho H.J., Cho H.J., Kim H.S. Osteopontin: a multifunctional protein at the crossroads of inflammation, atherosclerosis, and vascular calcification // Curr. Atheroscler. Rep. — 2009. — Vol. 11. — P. 206–213.
- Chowdhary S., Townend J.N. Role of nitric oxide in the regulation of cardiovascular autonomic control // Clin. Sci. (Lond). — 1999. — Vol. 97. — P. 5–17.
- Chrissobolis S., Didion S.P., Kinzenbaw D.A. et al. Glutathione perioxidase-1 plays a major role in protecting against angiotensin II-induced vascular dysfunction // Hypertens. — 2008. — Vol. 51. — P. 872–877.
- Cittadini A., Monti M.G., Isgaard J. et al. Aldosterone receptor blockade improves left ventricular remodeling and increases ventricular fibrillation threshold in experimental heart failure // Cardiovasc.Res. — 2003. — Vol. 58. — P. 555–564.
- Cohn J.N., Colucci W. Cardiovascular effects of aldosterone and post-acute myocardial infarction pathophysiology // Am. J. Cardiol. — 2006. — Vol. 97. — P. 4F–12F.
- Cole J., Ertoy D., Bernstein K.E. Insights derived from ACE knockout mice // JRAAS. — 2000. — Vol. 1. — P. 137–141.
- Cole J.M., Xiao H., Adams J.W. et al. New approaches to genetic manipulation of mice: tissue-specific expression of ACE // Am. J. Physiol. Renal. Physiol. – 2003. — 284. — P. F599–F607.
- Colussi G., Catena C., Lapenna R. et al. Insulin resistance and hyperinsulinemia are related to plasma aldosterone levels in hypertensive patients // Diabetes Care. — 2007. — Vol. 30. — Р. 2349–2354.
- Connell J., MacKenzie S.M., Freel E.M. et al. A lifetime of aldosterone excess: long-term consequences of altered regulation of aldosterone production for cardiovascular function // Endocr. Rev. — 2008. — Vol. 29. — P. 133–154.
- Cooper S.A., Whaley-Connell A., Habibi J. et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H2009–H2023.
- DeClue J.W., Guyton A.C., Cowley A.W. et al. Subpressor angiotensin infusion, renal sodium handling, and salt-induced hypertension in the dog // Circ. Res. — 1978. — Vol. 43. — P. 503–512.
- Del Ry S., Maltini M., Poletti R. et al. Osteopontin plasma level are elevated in patients with chronic heart failure in relation to clinical severity and cytokine expression // Eur. J. Heart Failure. — 2004. — Vol. 3 (Suppl. 1). — Р. 23.
- De Paula R.B., da Silva A.A., Hall J.E. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration // Hypertens. — 2004. — Vol. 43. — P. 41–47.
- De Simone G., De Marco M. Sodium, Left Ventricular Mass, and Arterial Hypertension. Is It Time to Look for a New Paradigm? // Hypertens. — 2011. — Vol. 58. — P. 349–351.
- Devereux R.B., Dahlof B., Gerdts E. et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: The Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial // Circulation. — 2004. — Vol. 110. — Р. 1456–1462.
- Diep Q.N., Mabrouk M.E., Cohn J.S. et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats. Role of peroxisome proliferator-activated receptor-y // Circulation. — 2002. — Vol. 105. — P. 2296–2302.
- Dikalova A.E., Bikineyeva A.T., Budzyn, K. et al. Therapeutic targeting of mitochondrial superoxide in hypertension // Circ. Res. — 2010. — Vol. 107. — P. 106–116.
- Dikalova A., Clempus R., Lassegue B. et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice // Circulation. — 2005. — Vol. 112. — P. 2668–2676.
- Dimmeler S., Hermann C., Assmus B. et al. Shear stress abolishes angiotensin II-induced apoptosis of endothelial cells // Europ. Heart J. — 1998. — Vol. 19, Abstr. Suppl. — P. 381.
- Divchev D., Grothusen C., Luchtefeld M. et.al. Impact of combined treatment of angiotensin II type 1 receptor blockade and 3-hydroxy-3-methyl-CoA-reductase inhibition on secretory phospholipase A2 type — IIA and low density lipoproteins oxidation in patients with coronary artery disease // Europ. Heart J. — 2008. — Vol. 209. — P. 1956–1965.
- Doughan A.K., Harrison D.G., Dikalov S.I. Molecular mechanisms of angiotensin II mediated mitochondrial dysfunction. Linking mitochondrial oxidative damage and vascular endothelial dysfunction // Circ.Res. — 2008. — Vol. 102. — P. 488–496.
- Douma S., Petidis K., Doumas M. et al. Prevalence of primary hyperaldosteronism in resistant hypertension: a retrospective observational study // Lancet. — 2008. — Vol. 371. — P. 1921–1926.
- Duprez D.A., Bauwens F.R., De Buyzere M.L. et al. Influence of arterial pressure and aldosterone in left ventricular hypertrophy in moderate essential hypertension // Am. J. Cardiol. — 1993. — Vol. 71. — Р. 17A–20A.
- Dzau V.J. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis // Hypertens. — 2001. — Vol. 37. — Р. 1047–1052.
- Ehrhart-Bornstein M., Lamounier-Zepter V., Schraven A. et al. Human adipocytes secrete mineralocorticoid-releasing factors // JRAAS. — 2003. — Vol. 100. — P. 14211–14216.
- Eide I.K., Torjesen P.A., Drolsum A. et al. Low-renin status in therapy-resistant hypertension: a clue to efficient treatment // J. Hypertens. — 2004. — Vol. 22. — P. 2217–2226.
- Elmarakby A.A., Quigley J.E., Imig J.D. et al. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats // Am. J. Physiol. Regul. Integr. Comp. Physiol. — 2008. — Vol. 294. — Р. R76–R83.
- Elmarakby A.A., Loomis E.D., Pollock J.S. et al. NAD(P)H oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1 // Hypertens. — 2005. — Vol. 45. — P. 283–287.
- Endtmann C., Ebrahimian T., Czech T. et al. Angiotensin II Impairs Endothelial Progenitor Cell Number and Function In Vitro and In Vivo Implications for Vascular Regeneration // Hypertens. — 2011. — Vol. 58. — P. 394–403.
- Engeli S., Bohnke J., Gorzelniak K. et al. Weight loss and the renin-angiotensin-aldosterone system // Hypertension. — 2005. — Vol. 45. — P. 356–362.
- Engeli S., Negrel R., Sharma A.M. Physiology and pathophysiology of the adipose tissue renin-angiotensin system // Hypertens. — 2000. — Vol. 35. — P. 1270–1277.
- Engeli S., Schling P., Gorzelniak K. et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? // Int. J. Biochem. Cell. Biol. — 2003. — Vol. 35. — P. 807–825.
- Essick E.E., Sam F. Cardiac hypertrophy and fibrosis in the metabolic syndrome: a role for aldosterone and the mineralocorticoid meceptor // Int. J. Hypertens. — 2011. — Vol. 34. — P. 69–85.
- Esteban V., Ruperez M., Vita J.R. et al. Effect of simultaneous blockade of AT1 and AT2 receptors on the NF-κB pathway and renal inflammatory response // Kidney Int. — 2003. — Vol. 64. — Р. S33–S38.
- Fallo F., Federspil G., Veglio F. et al. The metabolic syndrome in primary aldosteronism // Cur. Hypertens.Rep. — 2007. — Vol. 9. — P. 106–111.
- Fallo F., Veglio F., Bertello C. et al. Prevalence and characteristics of the metabolic syndrome in primary aldosteronism // J. Clin. Endocrinol. Metabol. — 2006. — Vol. 91. — P. 454–459.
- Faria-Silva R., Duarte F.V., Santos R.A. Short-term angiotensin(1–7) receptor MAS stimulation improves endothelial function in normotensive rats // Hypertens. — 2005. — Vol. 46. — P. 948–952.
- Farquharson C.A., Struthers A.D. Angiotensin II receptor blockers in chronic heart failure — Not as ELITE as expected! // JRAAS. — 2000. — Vol. 1. — P. 21–22.
- Farquharson C.A., Struthers A.D. Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy // Clin. Sci. (London). — 2002. — Vol. 103. — P. 425–431.
- Ferrara L.A., de Simone G., Pisanti N. et al. Adrenergic nervous system and left ventricular mass in primary hypertension // Eur. Heart J. — 1989. — Vol. 10. — Р. 1036–1040.
- Fiorentino A.D., Cianchetti S., Celi A. et al. Aliskiren, a renin inhibitor, downregulates TNF-α-induced tissue factor expression in HUVECS // JRAAS. — 2010. — Vol. 11. — P. 243–247.
- Flesch M., Hoper A., Dell’Italia L. et al. Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor // Circulation. — 2003. — Vol. 108, № 5. — P. 598–604.
- Flever A. The fast and slowly developing pressor effect of angiotensin II // in «The Renin-Angiotensin System» ed. by J.Robertson a. M.Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 1 (part 28). — P. 28.1–28.9.
- Freel M., Connell J.M. Mechanisms of hypertension: the expanding role of aldosterone // J.Am.Soc.Nephrol. — 2004. — Vol. 15. — Р. 1993–2001.
- Fujimura T., Yokota M., Kato S. et al. Lack of association of angiotensin converting enzyme gene polymorphism or serum enzyme activity with coronary artery disease in Japanese subjects // Am. J. Hyperten. — 1997. — Vol. 10. — Р. 1384–1390.
- Funder J., Carey R., Fardella C. et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline // J. Clin. Endocrinol.Metab. — 2008. — Vol. 9. — P. 3266–3281.
- Furuhashi M., Ura N., Higashiura K. et al. Blockade of the Renin-Angiotensin System Increases Adiponectin Concentrations in Patients with Essential Hypertension // Hypertens. — 2003. — Vol. 42. — P. 76–81.
- Gallay B.J., Ahmad S., Xu L. et al. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosterone-renin ratio // Am. J. Kidney Dis. — 2001. — Vоl. 37. — P. 699–705.
- Gálvez-Prieto B., Bolbrinker J., Stucchi P. et al. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue // J. Endocrinol. — 2008. — Vol. 197. — P. 55–64.
- Garcia E.A., Lopez J.R., Meier J.L. et al. Resistant hypertension and undiagnosed primary hyperaldosteronism detected by use of a computerized database // J. Clin. Hypertens. — 2011. — Vol. 3. — P. 487–491.
- Gavazzi G., Deffert C., Trocme C. et al. NOX1 deficiency protects from aortic dissection in response to angiotensin II // Hypertens. — 2007. — Vol. 50. — P. 189–196.
- Gerling I.C., Sun Y., Ahokas R.A. et al. Aldosteronism: an immunostimulatory state precedes proinflammatory/fibrogenic cardiac phenotype // Am. J. Physiol. Heart Circ. Physiol. — 2003. — Vol. 285. — P. H813–H821.
- Giacchetti G., Ronconi V., Turchi F. et al. Aldosterone as a key mediator of the cardiometabolic syndrome in primary aldosteronism: an observational study // J. Hypertens. — 2007. — Vol. 25. — Р. 177–186.
- Giacchetti G., Sechi L.A., Rilli S. et al. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes // Trends Endocrinol. Metab. — 2005. — Vol. 16. — Р. 120–125.
- Giacchetti G., Turchi F., Boscaro M. et al. Management of primary aldosteronism: its complications and their outcomes after treatment // Cur. Vasc. Pharmacol. — 2009. — Vol. 7. — Р. 244–249.
- Golledge J., Clancy P., Jamrozik K. et al. Obesity, adipokines, and abdominal aortic aneurysm: Health in Men study // Circulation. — 2007. — Vol. 116. — P. 2275–2279.
- Golledge J., Müller J., Shephard N. et al. Association between osteopontin and human abdominal aortic aneurysm // Arterioscler. Thromb. Vasc. Biol. — 2007. — Vol. 27. – P. 655–660.
- Gomez-Ambrosi J., Catalan V., Ramirez B. et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity // J. Clin. Endocrinol. Metab. — 2007. — Vol. 92. — P. 3719–3727.
- Goodfriend T.L., Ball D.L., Egan B.M. et al. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion // Hypertens. — 2004. — Vol. 43. — Р. 358–363.
- Goodfriend T.L., Calhoun D.A. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy // Hypertens. — 2004. — Vol. 43. — Р. 518–524.
- Graham D., Huynh N.N., Hamilton C.A. et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy // Hypertens. — 2009. — Vol. 54. — P. 322–328.
- Gromotowicz A., Szemraj J., Stankiewicz A. et al. Study of the mechanisms of aldosterone prothrombotic effect in rats // JRAAS. — 2011. — Vol. 12. — P. 430–439.
- Guzik T.J., Hoch N.E., Brown K.A. et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction // J. Exp. Med. — 2007. — Vol. 204. — P. 2449–2460.
- Hall J.E. The kidney, hypertension, and obesity // Hypertens. — 2003. — Vol. 41. — Р. 625–633.
- Hannemann A., Bidlingmaier M., Friedrich N. et al. Screening for primary aldosteronism in hypertensive subjects: results from two German epidemiological studies // Eur. J. Endocrinol. — 2012. — Vol. 167. — Р. 7–15.
- Hansen P.R., Rieneck K., Bendtzen K. Spironolactone inhibits production of proinflammatory cytokines by human mononuclear cells // Immunology Letters. — 2004. — Vol. 91. — Р. 87–91.
- Harrison D.G., Guzik T.J., Lob H.E. et al. Inflammation, immunity, and hypertension // Hypertens. — 2011. — Vol. 57. — P. 132–140.
- Hashimoto N., Maeshima Y., Satoh M. et al. Overexpression of angiotensin type 2 receptor ameliorates glomerular injury in a mouse remnant kidney model // Am. J. Renal. Physiol. — 2004. — Vol. 286. — Р. F516–F525.
- He H., Yang D., Ma L. et al. Telmisartan prevents weight gain and obesity through activation of peroxisome proliferator-activated receptor-δ-dependent pathways // Hypertens. — 2010. — Vol. 55. — P. 869–879.
- Higashi Y., Sasaki S., Nakagawa K. et al. Endothelial function and oxidative stress in renovascular hypertension // N. Engl. J. Med. — 2002. — Vol. 346. — P. 1954–1962.
- Hilgers K.F., Stumpf C. Angiotensin II, the endothelium and superoxide anions // J. Hypertens. — 2002. — Vol. 20. — P. 1271–1273.
- Hingtgen S.D., Tian X., Yang J. et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy // Physiol. Genomics. — 2006. — Vol. 26. — P. 180–191.
- Ho J.T., Keogh J.B., Bornstein S.R. et al. Moderate weight loss reduces renin and aldosterone but does not influence basal or stimulated pituitary-adrenal axis function // Horm. Metab. Res. — 2007. — Vol. 39. — P. 694–699.
- Hong H.J., Hsiao G., Cheng T.H. et al. Supplementation with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats // Hypertens. — 2001. — Vol. 38. — P. 1044–1048.
- Houston M.C. Nutraceuticals, vitamins, antioxidants, and minerals in the prevention and treatment of hypertension // Prog. Cardiovasc. Dis. — 2005. — Vol. 47. — P. 396–449.
- Hu C., Chen Y., Chiang M. et al. Heme oxigenase-1 inhibits angiotensin-II -induced cardiac hypertrophy in vitro and in vivo // Circulation. — 2004. — Vol. 110, N 3. — P. 309–316.
- Irita J., Okura T., Jotoku M. et al. Osteopontin deficiency protects against aldosterone-induced inflammation, oxidative stress, and interstitial fibrosis in the kidney // AJP — Renal Physiol. — 2011. — Vol. 301. — P. F833–F844.
- Iso H., Kiyama M., Doi M. et al. Left ventricular mass and subsequent blood pressure changes among middle-aged men in rural and urban Japanese populations // Circulation. — 1994. — Vol. 89. — P. 1717–1724.
- Jalil J.E., Perez A., Ocaranza M.P. et al. Increased aortic NAPDH oxidase activity in rats with genetically high angiotensin-converting enzyme levels // Hypertens. — 2005. — Vol. 46. — P. 1362–1367.
- Johansson M.E., Fagerberg B., Bergström G. Angiotensin type 2 receptor is expressed in human atherosclerotic lesions // JRAAS. — 2008. — Vol. 9. — P. 17–21.
- Jung O., Schreiber J.G., Geiger H. et al. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension // Circulation. — 2004. — Vol. 109. — P. 1795–1801.
- Kaplan N.M. Is there an unrecognized epidemic of primary aldosteronism? // Hypertension. — 2007. — Vol. 50. — P. 454–458.
- Keidar S., Kaplan M., Shapira C. et al. Low density lipoprotein isolated from patients with essential hypertension exhibits increased propensity for oxidation and enhanced uptake by macrophages: a possible role for angiotensin II // Atherosclerosis. — 1999. — Vol. 107. — P. 71–84. Kerr P.G., Guerin A.P. Аrterial calcification and stiffness in chronic kidney disease // СЕРР. — 2007. — Vol. 34. — P. 683–687.
- Kidambi S., Kotchen J.M., Grim C.E. et al. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks // Hypertens. — 2007. — Vol. 49. — P. 704–711.
- Kimura S., Zhang G.-X., Nishiyama A. et al. Role of NADPH oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II // Hypertens. — 2005. — Vol. 5. — P. 860–866.
- Klusonova P., Rehakova L., Borchert G. et al. Chronic intermittent hypoxia induces 11beta-hydroxysteroid dehydrogenase in rat heart // Endocrinology. – 2009. – Vol. 150. — P. 4270–4277.
- Kobayashi A., Ishikawa K., Matsumato H. et al. Synergetic antioxidant and vasodilatory action of carbon monoxide in angiotensin II-mediated cardiac hypertrophy // Hypertens. — 2007. — Vol. 50. — Р. 1040–1048.
- Krug A.W., Ehrhart-Bornstein M. Aldosterone and metabolic syndrome. Is increased aldosterone in metabolic syndrome patients an additional risk factor? // Hypertension. — 2008. — Vol. 51. — P. 1252–1260.
- Lacolley P., Labat C., Pujol A. et al. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: effects of eplerenone // Circulation. — 2002. — Vol. 106. — Р. 2848–2853.
- Lacolley P., Safar M.E., Lucet B. et al. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats // J. Am. Coll. Cardiol. — 2001. — Vol. 37. — P. 662–667.
- Lamounier-Zepter V., Rotthoff T., Ansurudeen I. et al. Increased aldosterone/renin quotient in obese hypertensive women: a novel role for low-density lipoproteins? // Horm. Metab. Res. — 2006. — Vol. 38. — P. 471–475.
- Landmesser U., Dikalov S., Price S.R. et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension // J. Clin. Invest. — 2003. — Vol. 111. — P. 1201–1209.
- Landmesser U., Drexler H. Oxidative stress, the renin-angiotensin system, and atherosclerosis // Europ. Heart J. — 2003. — Vol. 5 (suppl. A). — P. A3–A7.
- Langeveld B., van Gilst W.H., Tio R.A. et al. Angiotensin-(1–7) attenuates neointimal formation after stent implantation in the rat // Hypertens. — 2005. — Vol. 45. — P. 138–141.
- Lassègue B., Griendling K.K. NADPH oxidases: functions and pathologies in the vasculature // Arterioscler.Thromb. Vasc. Biol. — 2010. — Vol. 30. — P. 653–661.
- Laude K., Cai H., Fink B. et al. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice // Am. J. Physiol. Heart. Circ. Physiol. — 2005. — Vol. 288. — P. H7–H12.
- Lavoie J.L., Bianco R.A., Sakai K. et al. Transgenic mice for studies of the renin-angiotensin system in hypertension // Acta Physiol. Scand. — 2004. — Vol. 181. — P. 571–577.
- Lee K.A., Kang M.H., Lee Y.S. et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity // Cell. Immunol. — 2008. — Vol. 251. — P. 50–55.
- Lei H.-P., Chen H.-M., Zhong S.-L. et al. Association between polymorphisms of the renin–angiotensin system and coronary artery disease in Chinese patients with type 2 diabetes // JRAAS. — 2012. — Vol. 13. — P. 305–313.
- Lemarie C.A., Paradis P., Schiffrin E.L. New insights on signalling cascades induced by cross-talk between angiotensin II and aldosterone // J. Mol. Med. — 2008. — Vol. 86. — P. 673–678.
- Lemarié C.A., Schiffrin E.L. The angiotensin II type 2 receptor in cardiovascular disease // JRAAS. — 2010. — Vol. 11. — P. 19–31.
- Li L., Fink G.D., Watts S.W. et al. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension // Circulation. — 2003. — Vol. 107. — Р. 1053–1058.
- Libby P. Fat fuels the flame: triglyceride-rich lipoproteins and arterial inflammation. Circ. Res. — 2007.— Vol. 100. — Р. 299–301.
- Lieb W., Larson M.G., Benjamin E.J. et al. Multimarker approach to evaluate correlates of vascular stiffness: the Framingham Heart Study // Circulation. — 2009. — Vol. 119. — P. 37–43.
- Lim P.O. Role of aldosterone in the pathogenesis of hypertension // Hypertens. — 2002. — Vol. 39. — Р. E14.
- Lim P.O., Dow E., Brennan G. et al. High prevalence of primary aldosteronism in the Tayside hypertension clinic population // J. Hum. Hypertens. — 2000. — Vol. 14. — Р. 311–315.
- Lim P.O., Struthers A.D., MacDonald T.M. The neurohormonal natural history of essential hypertension: towards primary or tertiary aldosteronism? // J. Hypertens. — 2002. — Vol. 20. — P. 11–15.
- Lip G.Y., Edmunds E., Nuttall S.L. et al. Oxidative stress in malignant and non-malignant phase hypertension // J. Hum. Hypertens. — 2002. — Vol. 16. — Р. 333–336.
- Liu S.L., Schmuck S., Chorazcyzewski J.Z. et al. Aldosterone regulates vascular reactivity — Short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation // Circulation. — 2003. — Vol. 108. — Р. 2400–2406.
- Liu J., Yang F., Yang X.P. еt al. NAD(P)H oxidase mediates angiotensin II–induced vascular macrophage infiltration and medial hypertrophy // Arterioscler. Thromb. Vasc. Biol. — 2003. — Vol. 23. — P. 776–782.
- Lob H.E., Marvar P.J., Guzik T.J. et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system // Hypertens. — 2010. — Vol. 55. — Р. 277–283.
- London G.M. Role of arterial wall properties in the pathogenesis of systolic hypertension // Am. J. Hypertens. — 2005. — Vol. 18. — Р. 19S–22S.
- Luchtfeld M., Bandlow N., Tietge U.J. et al. Angiotensin II typy 1-receptor antagonism prevents type IIA secretory phospholipase A(2)-dependent lipid peroxidation // Atherosclerosis. — 2006. — Vol. 25. – P. 25–32.
- Luther J.M., Gainer J.V., Murphey L.J. et al. Angiotensin II induces IL-6 in humans through a mineralocorticoid receptor-dependent mechanism // Hypertens. — 2006. — Vol. 48. — Р. 1050–1057.
- MacGregor G.A., Markandu N.D., Singer D.R.J. et al. Moderate sodium restriction with angiotensin converting enzyme inhibitor in essential hypertension: a double blind study // BMJ — 1987. — Vol. 294. — P. 531–534.
- Mahmud A., Feely J. Aldosterone-to-renin ratio, arterial stiffness, and the response to aldosterone antagonism in essential hypertension // Am. J. Hypertens. — 2005. — Vol. 18. — Р. 50– 55.
- Makhanova N., Hagaman J., Kim H.S. et al. Salt-sensitive blood pressure in mice with increased expression of aldosterone synthase // Hypertens. — 2008. — Vol. 51. — P. 134–140.
- Marcy T.R., Ripley T.L. Aldosterone antagonists in the treatment of heart failure // Am. J. Health-System Pharmacy. — 2006. — Vol. 63. — P. 49–58.
- Marrs J.C. Spironolactone Management of Resistant Hypertension // Ann. Pharmacotherap. — 2010. — Vol. 44. — Р. 1762–1769.
- Martinez D., Rocha R., Matsumura M. et al. Cardiac damage prevention by eplerenone: comparison with low sodium diet or potassium loading // Hypertens. — 2002. — Vol. 39. — P. 614–618.
- Matsumoto T., Wada A., Tsutamoto T. et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure // Circulation. — 2003. — Vol. 107. — P. 2555–2558.
- Matsumura K., Fujii K., Oniki H. et al. Role of aldosterone in left ventricular hypertrophy in hypertension. American Journal of Hypertension. — 2006. — 19(1). — Р. 13–18.
- Mazzolai L., Duchosal M.A., Korber M. et al. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in apoE-/-mice // Hypertens. — 2004. — Vol. 44. — P. 277–283.
- McEvan P.E., Gray G.A., Sherry L. et al. Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo // Circulation. — 1998. — Vol. 98. — P. 2765–2773.
- Menard J., Clauser E., Bouchnik J. et al. in «The Renin-Angiotensin System» ed. by J. Robertson a. M. Nichols. Merck Sharp a. Dohnme, GowerMedical Publishing, London, New York. — 1993. — Vol. 1 (part 8). — P. 8.1–8.10.
- Meneton P., Galan P., Bertrais S. et al. High plasma aldosterone and low renin predict blood pressure increase and hypertension in middle-aged Caucasian populations // J. Hum. Hypertens. — 2008. — Vol. 22. — Р. 550–558.
- Milliez P., Girerd X., Plouin P.F. et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism // J. Am. Coll. Cardiol. — 2005. — Vol. 45. — Р. 1243–1248.
- Moreno C., Hoffman M., Stodola T.J. et al. Creation and characterization of a renin knockout rat // Hypertens. — 2011. — Vol. 57. — P. 614–619.
- Moreno M.U., Jose G.S., Fortuno A. et al. The C242T CYBA polymorphism of NADPH oxidase is associated with essential hypertension // J. Hypertens. — 2006. — Vol.24. — P. 1299–1306.
- Mosso L.M., Carvajal C.A., Maiz A. et al. A possible association between primary aldosteronism and a lower β-cell function // J. Hypertens. — 2007. — Vol. 25. — Р. 2125–2130.
- Muiesan M.L., Lupia M., Salvetti M. et al. Left ventricular structural and functional characteristics in Cushing’s syndrome // J. Am. Coll. Cardiol. — 2003. — Vol. 41. — P. 2275–2279.
- Müller D.N. Mechanisms of hypertension-induced target organ damage // JRAAS. — 2007. — Vol. 8. — Р. 148–150.
- Müller D.N., Kleinewietfeld M., Kvakan H. Vitamin D review // J. Renin-Angiotensin-Aldosterone System. — 2011. — Vol. 12. — P. 125–128.
- Nagase M., Yoshida S., Shibata S. et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors // J. Am. Soc. Nephrol. — 2006. — Vol. 17. — P. 3438–3446.
- Nagata K., Obata K., Xu J. et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats // Hypertension. — 2006. — Vol. 47. — P. 656–664.
- Nehme J., Mercier N., Labat C. et al. Differences between cardiac and arterial fibrosis and stiffness in aldosterone-salt rats: effect of eplerenone // JRAAS. — 2006. — Vol. 7. — P. 31–39.
- Neter J.E., Stam B.E., Kok F.J. et al. Influence of weight reduction on blood pressure: a meta-analysis of randomized controlled trials // Hypertens. — 2003. — Vol. 42. — P. 878–884.
- Newton-Cheh C., Guo C.Y., Gona P. et al. Clinical and genetic correlates of alosrone-to-renin ratio and relations to blood pressure in a community sample // Hypertension. — 2007. — Vol. 49. — P. 846–856.
- Niebauer J., Tsao P.S., Lin P.S. et al. Effects of hypercholesterolemia on angiotensin II, superoxide production and monocyte binding // Europ. Heart J. — 1998. — 19. — Abstr. Suppl. — P. 379.
- Nishizaka M.K., Zaman M.A., Calhoun D.A. Efficacy of low-dose spironolactone in subjects with resistant hypertension // Am. J. Hypertens. — 2003. — Vol. 16. — P. 925–930.
- Nobuhiko A., Suganuma E., Babaev V.R. Angiotensin II amplifies macrophage-driven atherosclerosis // Arterioscler. Thromb. Vasc. Biol. — 2004. — Vol. 24. — P. 2143–2152.
- Nolan J., Batin P.D., Andrews R. et al. Prospective study of heart rate variability and mortality in chronic heart failure: results of the United Kingdom heart failure evaluation and assessment of risk trial (UK-heart) // Circulation. — 1998. — Vol. 98. — Р. 1510–1516.
- Ouzan J., Pérault C., Lincoff A.M. et al. The role of spironolactone in the treatment of patients with refractory hypertension // Am. J. Hypertens. — 2002. — Vol. 15. — Р. 333–339.
- Padmanabhan N., Padmanabhan S., Connell J.M.C. Genetic basis of cardiovascular disease — the reninangiotensin-aldosterone system as a paradigm // JRAAS. — 2000. — Vol. 1. — P. 316–324.
- Pagano P.J., Griswold M.C., Najibi S. et al. Resistance of endothelium-dependent relaxation to elevation of O(-)(2) levels in rabbit carotid artery // Am. J. Physiol. — 1999. — Vol. 277. — Р. H2109–2114.
- Paravicini T., Touyz R.M. Redox signaling in hypertension // Cardiovasc. Res. — 2006. — Vol. 71. — P. 247–258.
- Paravicini T., Touyz R.M. NADPH oxidases, reactive oxygen species, and hypertension. Clinical implications and therapeutic possibilities // Diabetes Care. — 2008. — Vol. 31 (Suppl. 2). — Р. S170–S180.
- Park J.B., Schiffrin E.L. ETA receptor antagonist prevents blood pressure elevation and vascular remodeling in aldosterone-infused rats // Hypertens. — 2001. — Vol. 37. — Р. 1444–1449.
- Parving H.H., de Zeeuw D., Cooper M.E. et al. ACE gene polymorphism and losartan treatment in type 2 diabetic patients with nephropathy // J. Am. Soc. Nephrol. — 2008. — Vol. 19. — P. 771–779.
- Pawluczyk I.Z.A., Harris K.P.G. Effect of angiotensin type 2 receptor over-expression on the rat mesangial cell fibrotic phenotype: effect of gender // JRAAS. — 2012. — Vol. 13. — Р. 221–231.
- Pellieux C., Montessuit C., Papageorgiou I. et al. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumor necrosis factor-α // Cardiovasc. Res. — 2009. — Vol. 82. — P. 341–350.
- Petrasek D., Jensen G., Tuck M. et al. In vitro effects of insulin on aldosterone production in rat zona glomerulosa cells // Life Sci. — 1992. — Vol. 50. — Р. 1781–1787.
- Phillips M.I., Schmidt-Ott K.M. The Discovery of Renin 100 Years Ago // News Physiol. Sci. — 1999. — Vol. 14. — P. 271–274.
- Pimenta E., Calhoun D.A. Aldosterone and metabolic dysfunction. An unresolved issue // Hypertens. — 2009. — Vol. 53. — P. 585–591.
- Pimenta E., Gaddam K.K., Oparil S. et al. Effects of dietary sodium restriction on blood pressure in subjects with resistant hypertension: results from a randomized trial // Hypertens. — 2009. — Vоl. 54. — Р. 444–446.
- Pimenta E., Gordon R.D., Ahmed A.H. et al. Cardiac Dimensions Are Largely Determined by Dietary Salt in Patients with Primary Aldosteronism: Results of a Case-Control Study // J. Clin. Endocrinol. Metabol. — 2011. — Vol. 96. — Р. 2813–2820.
- Piqueras L., Kubes P., Alvarez P. et al. Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT1 and AT2 receptor-mediated P-selectin upregulation // Circulation. — 2000. — Vol. 102. — P. 2118–2123.
- Pitt B., Ahmed A., Love T.E. et al. History of hypertension and eplerenone in patients with acute myocardial infarction complicated by heart failure // Hypertens. — 2008. — Vol. 52. — P. 271–278.
- Pitt B., Poole-Wilson P.A., Segal R. et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial — the Losartan Heart Failure Survival Study ELITE II // Lancet. — 2000. — Vol. 355. — P. 1582–1587.
- Post W.S., Larson M.G., Levy D. Impact of left ventricular structure on the incidence of hypertension. The Framingham Heart Study // Circulation. — 1994. — Vol. 90. — P. 179–185.
- Pouleur A-C., Uno H., Prescott M.F. et al. (for the ALLAY Investigators). Suppression of aldosterone mediates regression of left ventricular hypertrophy in patients with hypertension // JRAAS. — 2011. — Vol. 12. — P. 483–490.
- Rajagopalan S., Duquaine D., King S. et al. Mineralocorticoid receptor antagonism in experimental atherosclerosis // Circulation. — 2002. — Vol. 105. — Р. 2212–2216.
- Rey F.E., Cifuentes M.E., Kiarash A. et al Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice // Circ. Res. — 2001. — Vol. 89. — Р. 408–414.
- Robert V., Heymes C., Silvestre J.S. et al. Anigotensin AT1 receptor subtype as a cardiac target of aldosterone: role in aldosterone salt-induced fibrosis // Hypertens. — 1999. — Vol. 33. — Р. 981–986.
- Robertson J.I.S. Renin and angiotensin // in «The Renin-Angiotensin System» ed.by J.Robertson a. M. Nichols. Merck Sharp a. Dohnme, Gower Medical Publishing, London, New York. — 1993. — Vol. 1 (part 1). — P. 1.1–1.18.
- Rocha R., Martin-Berger C.L., Yang P. et al. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart // Endocrinol. — 2002. — Vol. 143. — P. 4828–4836.
- Rocha R., Rudolph A., Frierdich G. et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart // Am. J. Physiol. — 2002. — Vol. 283. — P. H1802–H1810.
- Rocha R., Stier C.T. Pathophysiological effects of aldosterone in cardiovascular tissues // Trends Endocrinol.Metab. — 2001. — Vol. 12. — Р. 308–314.
- Rossi G., Belfiore A., Bernini G. et al. Body mass index predicts plasma aldosterone concentrations in overweight-obese primary hypertensive patients // J. Clin. Endocrinol. Metab. — 2008. — 93. — Р. 2566–2571.
- Rossi G.P., Bernin G., Caliumi C. et al. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients // J. Am. Coll. Cardiol. — 2006. – Vol. 48. — P. 2293–2300.
- Rossi G.P., Boscaro M., Ronconi V. et al. Aldosterone as a cardiovascular risk factor // Trends Endocrinol.Metab. — 2005. — Vol. 16. — Р. 104–107.
- Rossing K., Schjoedt K.J., Smidt U.M. et al. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study // Diabetes Care. — 2005. — Vol. 28. — Р. 2106–2112.
- Ruiz J., Blanche H., Cohen N. et al. Insertion/deletion polymorphism of the angiotensin-converting enzyme gene is strongly associated with coronary heart disease in non-insulin-dependent diabetes mellitus // Proc.Natl. Acad. Sci. USA. — 1994. — Vol. 91. — Р. 3662–3665.
- Russell R.P., Masi A.T. The prevalence of adrenal cortical hyperplasia at autopsy and its association with hypertension // Ann. Intern. Med. — 1970. — Vol. 73. — Р. 195–205.
- Sakurabayashi-Kitade S., Aoka Y., Nagashima H. et al. Aldosterone blockade by Spironolactone improves the hypertensive vascular hypertrophy and remodeling in angiotensin II overproducing transgenic mice // Atherosclerosis. – 2009. – Vol. 206. – P. 54–60.
- Samani N.J., Thompson J.R., O’Toole L. et al. A meta-analysis of the association of the deletion allele of the angiotensinconverting enzyme gene with myocardial infarction // Circulation. — 1996. – Vol. 94. — Р. 708–712.
- Sampaio W.O., Souza dos Santos R.A., Faria-Silva R. et al. Angiotensin-(1–7) through receptor mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways // Hypertens. — 2007. — Vol. 49. — P. 185–192.
- San Martin A., Foncea R., Laurindo F.R. et al. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products // Free Radic. Biol. Med. — 2007. — Vol. 42. — Р. 1671–16791.
- Santos R.A.S., Campagnole-Santos M.J., Andrade S.P. Angiotensin-(1–7): an update // Regul. Pept. — 2000. — Vol. 91. —
P. 45–51. - Sanz-Rosa D., Cediel E., Heras N. et al. Participation of aldosterone in the vascular inflammatory response of spontaneously hypertensive rats: role of the NFkB/IkB system // Hypertension. — 2005. — Vol. 23. — P.1167–1172.
- Sato A., Saruta T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy // J. Int. Med. Res. — 2001. — Vol. 29. — Р. 13–21.
- Schappi M., Herrmann F.R., Krause K.H. NOX1 deficiency protects form aortic dissections in response to angiotensin II // Hypertens. — 2007. — Vol. 50. — Р. 189–196.
- Schieffer B., Schieffer E., Hilfiker-Kleiner D. et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques // Circulation. — 2000. — Vol. 101. — P. 1372–1378.
- Schiering A., Menschikowski M., Mueller E. et al. Analysis of secretory group II phospholipase A 2 expression in human aortic tissue in dependence on the degree of atherosclerosis // Atherosclerosis. — 1999. — Vol. 144. — P. 73–78.
- Schiffrin E.L. Vascular endothelin in hypertension // Vasc. Pharmacol. — 2005. — Vol. 43. — Р. 19–29.
- Schiffrin E.L. Effects of aldosterone on the vasculature // Hypertens. — 2006. — Vol. 47. — P. 312–318.
- Schrader L.I., Kinzenbaw D.A., Johnson A.W. et al. IL-deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy // Arteriocler. Thromb. Vasc. Biol. — 2007. — Vol. 27. — Р. 2576–2581.
- Schulman I.H., Zhou M.S., Raij L. Nitric oxide, angiotensin II, and reactive oxygen species in hypertension and atherogenesis // Curr. Hypertens. Rep. — 2005. — Vol. 7. — P. 61–67.
- Schulman I.H., Zhou M.S., Raij L. Cross-talk between angiotensin II receptor types 1 and 2: potential role in vascular remodeling in humans // Hypertens. — 2007. — Vol. 49. — Р. 270–271.
- Schunkert H., Нense H., Muscholl M. et al. Associations between circulating components of the renin-angiotensin-aldosterone system and left ventricular mass // Heart. — 1997. — Vol. 77. — Р. 24–31.
- Sekuri C., Cam F.S., Ercan E. et al. Renin-angiotensin system gene polymorphisms and premature coronary heart disease // JRAAS. — 2005. — Vol.6. — Р. 38–42.
- Sendra J., Llorente-Cortes V., Costales P. еt al. Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall: a new pro-atherogenic mechanism of hypertension // Europ. J. Heart Failure. — 2008. — Vol. 10. — P. 581–589.
- Shah N.C., Pringle S., Struthers A. Aldosterone blockade over and above ACE-inhibitors in patients with coronary artery disease but without heart failure // JRAAS. — 2006. — Vol. 7. — P. 20–30.
- Shapiro Y., Boaz M., Matas Z. et al. The association between the renin-angiotensin-aldosterone system and arterial stiffness in young healthy subjects // Clin. Endocrinol. (Oxf). — 2008. — Vol. 68. — P. 510–512.
- Shibata S., Nagase M., Yoshida S. et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease // Nat. Med. — 2008. — Vol. 14. — P. 1370–1376.
- Shieh P., Diez-Freire C., Jun J.Y. et al. Brain microglial cytokines in neurogenic hypertension // Hypertens. — 2010. — Vol. 56. — P. 297–303.
- Silvestre J.S., Robert V., Heymes C. et al. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation // J. Biol. Chem. — 1998. — Vol. 273. — P. 4883–4891.
- Singh V.P., Baker K.M., Kumar R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production // Am. J. Physiol. Heart Circ. Physiol. — 2008. — Vol. 294. — P. H1675–H1684.
- Singh V.P., Le B., Bhat V.B. et al. High glucose induced regulation of intracellular angiotensin II synthesis and nuclear redistribution in cardiac myocytes // Am. J. Physiol. Heart Circ. Physiol. — 2007. — Vol. 293. — P. H939–H948.
- Singh V.P., Le B., Khode R. et al. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis // Diabetes. — 2008. — Vol. 57. — P. 3297–3306.
- Singh R., Leehey D.J. Effect of ACE inhibitors on angiotensin II in rat mesangial cells cultured in high glucose // Biochem. Biophys. Res. Commun. — 2007. — Vol. 357. — P. 1040–1045.
- Skeggs L., Kahn J.R., Shumway N.P. The preparation and function of the hypertension converting enzyme // J. Exp. Med. — 1956. — Vol. 103. — P. 295–299.
- Sowers J.R., Whaley-Connell A., Epstein M. The emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension // Ann. Intern. Med. — 2009. — Vol. 150. — P. 776–783.
- Stas S., Whaley-Connell A., Habibi J. et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling // Endocrinology. — 2007. — Vol. 148. — Р. 3773–3780.
- Sugiyama T., Yoshimoto T., Tsuchiya K. et al. Aldosterone induces angiotensin converting enzyme gene expression via a JAK2-dependent pathway in rat endothelial cells // Endocrinol. — 2005. — Vol. 146. — Р. 3900–3906.
- Suguwara A., Takeuchi K., Uruno A.et al. Transcriptional suppression of type 1 angiotensin II receptor gene expression by PPAR-y in vascular smooth muscle cells. Endocrinology. — 2001. — Vol. 142. — P. 3125–3134.
- Sun Y. Myocardial repair/remodelling following infarction: roles of local factors // Cardiovasc.Res. — 2009. — Vol. 81. —
P. 482–490. - Sun Y., Ahokas R.A., Bhattacharya S.K. et al. Oxidative stress in aldosteronism. Cardiovasc Res. — 2006. —Vol. 71. — Р. 300–309.
- Sun Y., Zhang J., Lu L. et al. Aldosterone-induced inflammation in the rat heart : role of oxidative stress // Am. J. Pathol. — 2002. — Vol. 161. — P. 1773–1781.
- Suzuki G., Morita H., Mishima T. et al. Effects of long-term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure // Circulation. — 2002. — Vol. 106. — Р. 2967–2972.
- Suzuki Y., Ruiz-Ortega M., Lorenzo O. et al. Inflammation and angiotensin II // Int. J. Biochem. Cell.Biol. — 2003. — Vol. 35. — P. 881–900.
- Suzuki H., Shibata H., Murakami M. et al. Modulation of angiotensin II type 1 receptor mRNA expression in human blood cells: Comparison of Swedberg.
- Szalay G., Sauter M., Haberland M. et al. Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host // Circ. Res. – 2009. – Vol. 104. – P. 851–859.
- Takahashi F., Kumasaka T., Nagaoka T. Osteopontin expression in pulmonary tumor thrombotic microangiopathy caused by gastric carcinoma // Pathol. Int. – 2009. – Vol. 59. – P. 752–756.
- Takeda Y., Yoneda T., Demura M. et al. Effects of high sodium intake on cardiovascular aldosterone synthesis in stroke-prone spontaneously hypertensive rats // J. Hypertens. — 2001. — Vol. 19. — P. 635–639.
- Tesanovic S., Vinh A., Gaspari T.A. et al. Vasoprotective and atheroprotective effects of angiotensin (1–7) in apolipoprotein E-deficient mice // Arterioscler. Thromb. Vasc. Biol. — 2010. — Vol. 30. — P. 1606–1613.
- Thohan V., Torre-Amione G., Koerner M.M. Aldosterone antagonism and congestive heart failure: a new look at an old therapy // Curr. Opin. Cardiol. — 2004. — Vol. 19. — Р. 301–308.
- Thomas M., Gavrila D., McCormick M.L. et al. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice // Circulation. — 2006. — Vol. 114. — Р. 404–413.
- Tigerstedt R., Bergman P. «Niere und Kreislauf» // Scandinav. Arch. Physiol. — 1898. — Vol. 8. — P. 223–271.
- Tomaschitz A., Maerz W., Pilz S. et al. Aldosterone/Renin Ratio Determines Peripheral and Central Blood Pressure Values Over a Broad Range // JACC. — 2010. — Vol. 55. — P. 65–73.
- Tomaschitz A., Pilz S., Ritz E. et al. Associations of plasma renin with 10-year cardiovascular mortality, sudden cardiac death, and death due to heart failure // Eur. Heart J. — 2011.
- Touyz R.M. Reactive oxygen species and angiotensin II signaling in vascular cells: implications in cardiovascular disease // Braz. J. Med. Biol. Res. — 2004. — Vol. 37. — P. 1263–1273.
- Touyz R.M., Mercure C., He Y. et al. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase // Hypertens. — 2005. — Vol. 45. — P. 530–537.
- Trueblood N.A., Xie Z., Communal C. et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin // Circ. Res. – 2001. — Vol. 88. — P. 1080–1087.
- Tsutsumi Y., Matsubara H., Okhubo N. et al. Angiotensin II type 2 receptors is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major type forits expression // Circ.Res. — 1998. — Vol. 83. — P. 1035–1046.
- Unger T., Li J. The role of the renin-angiotensin-aldosterone system in heart failure // JRAAS. — 2004. —Vol. 5 (suppl.). —
P. S7–S10. - Vaclavik J., Sedlak R., Plachy M. et al. Addition of spironolactone in patients with resistant arterial hypertension (ASPIRANT): a randomized, double-blind, placebo-controlled trial // Hypertens. — 2011. — Vоl. 57. — Р. 1069–1075.
- Vasan R.S., Evans J.C., Larson M.G. et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons // N. Engl. J. Med. — 2004. — Vol. 351. — Р. 33–41.
- Velagaleti R.S., Gona P., Levy D. et al. Relations of biomarkers representing distinct biological pathways to left ventricular geometry // Circulation. — 2008. — Vol. 118. — Р. 2252–2258.
- Villard E., Tiret L., Visvikis S. Identification of new polymorphisms of the angiotensin I-converting enzyme (ACE) gene, and study of their relationship to plasma ACE levels by 2-QTL segregation-linkage analysis // Am. J. Hum. Genet. — 1996. — Vol. 58. — Р. 1268–1278.
- Virdis A., Neves M.F., Amiri F. et al. Spironolactone improves angiotensin-induced vascular changes and oxidative stress // Hypertens. — 2002. — Vol. 40. — Р. 504–510.
- Virdis A., Neves M.F., Amiri F. et al. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice // J. Hypertens. — 2004. — Vol. 22. — P. 535–542.
- Wang Y., Tikellis C., Thomas M.C. et al. Angiotensin converting enzyme 2 and atherosclerosis // Atherosclerosis. — 2013. — Vol. 226. — P. 3–8.
- Ward N.C., Croft K.D. Hypertension and oxidative stress // Clin. Exp. Pharmacol. Physiol. — 2006. — Vol. 33. — P. 872–876.
- Wassmann S., Czech T., van Eickels M. et al. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice // Circulation. — 2004. — Vol. 110. — P. 3062–3067.
- Wassmann S., Laufs U., Müller K. et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo // Arterioscler. Thromb. Vasc. Biol. — 2002. — Vol. 22. — P. 300–305.
- Weber K.T., Sun Y., Wodi L.A. et al. Toward a broader understanding of aldosterone in congestive heart failure // JRAAS. — 2003. — Vol. 4. — P. 155–163.
- Wei Y., Sowers J.R., Nistala R. et al. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells // AJP — Regu. Physiol. — 2008. — Vol. 294. — Р. R673–R680.
- Wei Y., Whaley-Connell A.T., Chen K. et al. NADPH oxidase contributes to vascular inflammation, insulin resistance and remodeling in the transgenic (mRen2) rat // Hypertens. — 2007. — Vol. 50. — Р. 384–391.
- Welch W.J. Angiotensin II–dependent superoxide effects on hypertension and vascular dysfunction // Hypertens. — 2008. — Vol. 52. — P. 51–56.
- Welch W.J., Mendonca M., Blau J. et al. Antihypertensive response to prolonged tempol in the spontaneously hypertensive rat // Kidney Int. — 2005. — Vol. 68. — Р. 179–187.
- Werner N., Nickenig G. AT1 receptors in atherosclerosis: biological effects including growth, angiogenesis, and apoptosis // Europ. Heart J. — 2003. — Vol. 5 (suppl. A). — P. A9–A13.
- Whaley-Connell A., Govindarajan G., Habibi J. et al. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic TG (mRen2) 27 Ren2 rat. // Am. J. Physiol. Endocrinol.Metab. — 2007. — Vol. 293. — Р. E355–E363.
- Whaley-Connell A.T., Habibi J., Nistala R. et al. Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment // Hypertens. — 2008. — Vol. 51. — Р. 474–480.
- Whaley-Connell A., Habibi J., Wei Y. et al. Mineralcorticoid receptor antagonism attenuates kidney renin-angiotensin-aldosterone system mediated filtration barrier remodeling in the transgenic Ren2 rat // Am. J. Physiol. Renal. Physiol. — 2009. — Vol. 296. — P. F1013–F1022.
- Whaley-Connell A., Johnson M.S., Sowers J.R. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension // Progr. in Cardiovascular. Dis. — 2010. — Vol. 52. — P. 401–409.
- Williams B. The renin angiotensin system and cardiovascular disease: hope or hype? // JRAAS. — 2000. — Vol. 1. — P. 142–146.
- Williams G.H. Cardiovascular benefits of aldosterone receptor antagonists: what about potassium? // Hypertens. — 2005. — Vol. 46. — Р. 265–266.
- Williams J.M., Pollock J.S., Pollock D.M. Arterial pressure response to the antioxidant tempol and ETB receptor blockade in rats on a high-salt diet // Hypertens. — 2004. — Vol. 44. — Р. 770–775.
- Xiao H.D., Fuchs S., Bernstein E.A. et al. Mice expressing ACE only in the heart show that increased cardiac angiotensin II is not associated with cardiac hypertrophy // Am. J. Physiol. Heart. Circ.Physiol. — 2008. — Vol. 294. — P. 659–667.
- Xiao F., Puddefoot J.R., Barker S. et al. Mechanism for aldosterone potentiation of angiotensin II — stimulated rat arterial smooth muscle cell proliferation // Hypertens. — 2004. — Vol. 44. — Р. 340–345.
- Yamamuro M., Yoshimura M., Nakayama M. et al. Direct effects of aldosterone on cardiomyocytes in the presence of normal and elevated extracellular sodium // Endocrinology. — 2006. — Vol. 147. — P. 1314–1321.
- Yan Y.P., Lang B.T., Vemuganti R. et al. Persistent migration of neuroblasts from the subventricular zone to the injured striatum mediated by osteopontin following intracerebral hemorrhage // J. Neurochem. – 2009. – Vol. 109. — P. 1624–1635.
- Yin C.C., Huang K.T. H2O2 but not O2-elevated by oxidized LDL enhances human aortic smooth muscle cell proliferation // J. Biomed. Sci. — 2007. — Vol. 14. — Р. 245–254.
- Yogi A., Mercure C., Touyz J. et al. Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension // Hypertens. — 2008. — Vol. 51. — Р. 500–506.
- Young M.J., Clyne C.D., Cole T.J. et al. Cardiac steroidogenesis in the normal and failing heart // J. Clin.Endocrinol. Metabol. — 2001. — Vol. 86. — P. 5121–5126.
- Young M.J., Moussa L., Dilley R. et al. Early inflammatory responses in experimental cardiac hypertrophy and fibrosis: effects of 11 beta-hydroxysteroid dehydrogenase inactivation // Endocrinol. — 2003. — Vol. 44. — Р. 1121–1125.
- Zahradka P. Novel role for osteopontin in cardiac fibrosis // Circ. Res. – 2008. – Vol. 102. – P. 270–272.
- Zalba G., Beaumont F.J., San Jose G. et al. Vascular NADH/NAD(P)H oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats // Hypertens. — 2000. — Vol. 35. — Р. 1055–1061.
- Zhang Y., Griendling K.K., Dikalova A. et al. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2 // Hypertens. — 2005. — Vol. 46. — Р. 732–737.
- Zhang Y., Popovic Z.B., Bibevski S. et al. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model // Circ.Heart Fail. — 2009. — Vol. 2. — P. 692–699.
- Zhao Q., Ishibashi M., Hiasa K. et al. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling // Hypertens. — 2004. — Vol. 44. — P. 264–271.
- Zhong J.C., Guo D., Chen C.B. Prevention of angiotensin II–mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2 // Hypertens. — 2011. — Vol. 57. — P. 314–322.
- Zhou M.S., Jaimes E.A., Raij L. Vascular but not cardiac remodeling is associated with superoxide production in angiotensin II hypertension // J. Hypertens. — 2005. — Vol. 23. — Р. 1737–1743.
- Zhou M.S., Schulman I.H., Pagano P.J. et al. Reduced NAD(P)H oxidase in low renin hypertension. Link among angiotensin II, atherogenesis, and blood pressure // Hypertens. — 2006. – Vol. 47. — P. 81–86.
- Zhou M.S, Schulman I.H., Raij L. Role of angiotensin II and oxidative stress in vascular insulin resistance linked to hypertension // AJP — Heart. — 2009. — Vol. 296. — Р. H833–H839.
- Zhou L., Xi B., Wei Y. et al. Meta-analysis of the association between the insertion/deletion polymorphism in ACE gene and coronary heart disease among the Chinese population // JRAAS. — 2012. — Vol. 13. — P. 296–304.
- Zillich A.J., Garg J., Basu S. et al. Thiazide diuretics, potassium, and the development of diabetes: a quantitative review // Hypertension. — 2006. — Vol. 48. — P. 219–224.
- Zintzaras E., Raman G., Kitsios G. et al. Angiotensin-converting enzyme insertion/deletion gene polymorphic variant asa marker of coronary artery disease: a meta-analysis // Arch. Intern.Med. — 2008. — Vol. 168. — Р. 1077–1089.