2.1. Кальцийрегулирующие и системные гормоны
2.1.1. Паратиреоидный гормон
Регуляция движения кальция и фосфатов в организме, поддержание постоянной концентрации Са2+ в крови осуществляется ПТГ, кальцитонином, витамином D. Они образуют единый гомеостатический механизм регуляции метаболизма Са2+ и фосфора. (Витамин D по структуре относится к группе стероидных гормонов, а по функции — к гормонам, регулирующим обмен Са2+ и фосфатов.) ПТГ выделен из паращитовидных желез крупного рогатого скота в 1970 г. Его молекула состоит из одной полипептидной цепи, в состав которой входит 84 аминокислотных остатка. Синтезируется из препропаратиреоидного гормона (или препроПТГ), состоящего из 115 аминокислотных остатков. Удаление из него 25 аминокислотных остатков приводит к образованию пропаратиреоидного гормона (или проПТГ), а затем еще 6 — к ПТГ. Биологическая активность молекулы ПТГ связана с N-терминальной 1-34-аминокислотной последовательностью. ПТГ секретируется паращитовидными железами в ответ на снижение содержания кальция в крови. Гиперкальциемия подавляет секрецию ПТГ, а кальцитриол — его синтез (Маршалл В.Дж., 2002). Повышение содержания кальция в сыворотке крови является результатом действия ПТГ на кишечник, костную ткань, почки (система регуляции по принципу обратной связи). ПТГ усиливает высвобождение кальция из костной ткани и обратное всасывание кальция в почечных канальцах. В результате повышается концентрация кальция в плазме крови, наблюдается гиперкальциурия. Действие ПТГ на кость сопровождается (Уайт А. и соавт., 1981б) рядом эффектов: торможением синтеза коллагена в активных остеобластах, усилением остеолиза остеоцитами и остеокластами, повышением скорости созревания клеток-предшественников, дифференцирующихся в остеобласты и остеокласты. Эти эффекты также обнаруживаются in vitro. ПТГ повышает активность коллагеназы остеокластов, растворимость гидроксиапатитов. Усиливает поступление в кровь из костной ткани кальция и фосфора. Стимулирующее влияние ПТГ на костную ткань проявляется в присутствии остеобластов и остеобластных факторов — остеокластстимулирующего и остеокластингибирующего. ПТГ ингибирует реабсорбцию фосфора в почечных канальцах (снижается его содержание в плазме крови). Но изменение концентрации фосфатов непосредственно не влияет на секрецию ПТГ. Секреция ПТГ является магнийзависимым процессом: умеренная гипомагниемия стимулирует, а выраженная подавляет высвобождение ПТГ. Введение ПТГ обусловливает деполиминерализацию протеогликанов, постепенное исчезновение кристаллической структуры и матрикса, появление в плазме крови кислых гликопротеидов. При этом наблюдается повышение всасывания кальция и фосфора из пищеварительного тракта клетками ворсинок слизистой оболочки кишечника. ПТГ снижает 1α-гидроксилирование 25-гидроксихолекальциферола в почках. Снижение реабсорбции бикарбонатов в почках приводит к развитию ацидоза. Основные эффекты действия ПТГ приведены в табл. 2.1.
Поражение костей, развивающееся при гиперпаратиреоидизме, неравномерно, в них образуются полости (фиброзный остит). Концентрация кальция в сыворотке крови здорового человека поддерживается на уровне 2,22,6Гммоль/л при суточных колебаниях не выше 35%. При недостатке кальция в рационе, нарушенном всасывании его в кишечнике усиливается синтез ПТГ. В результате этого интенсивно вымываются цитратные и фосфатные соли кальция из костей, снижается реабсорбция фосфатов в дистальных канальцах, повышается канальцевая реабсорбция кальция. После инъекции ПТГ увеличивается отдача цитрата клетками костной ткани. Цитрат доставляет к другим органам с током крови кальций в форме анионного комплекса (кальций/цитрат), проходящего через ультрафильтры. В этих органах часть цитрата может подвергаться окислительному расщеплению. Действие ПТГ на клетки костной ткани и почек опосредуется через систему аденилатциклаза — цАМФ. Плазматические мембраны преостеобластов, остеобластов, остеоцитов имеют рецепторы, через которые действует ПТГ. Посредством системы аденилатциклаза — цАМФ ПТГ влияет на клеточную пролиферацию, дифференциацию, повышает продукцию ИПФР-1, ТФР-в.
ПТГ проявляет следующие эффекты:
- повышает митотическую активность остеобластов, увеличивает поглощение этими клетками неорганического фосфора;
- влияет на синтез остеобластами ЩФ (эффект зависит от дозы гормона, видовой специфичности);
- ингибирует синтез остеобластами коллагена, остеокальцина;
- повышает остеолитическую активность остеобластов и остеоцитов (активирует остеоцитарный остеолиз);
- оказывает стимулирующее влияние на дифференциацию, формирование клеток остеокластной линии (остеокластогенез);
- влияет на остеокласты опосредованно через остеобласты и вырабатываемые ими ФР, цитокины (в частности ГМ-КСФ, активизирующий остеокластическую резорбцию кости);
- активизация резорбтивной активности остеокластов под влиянием ПТГ сопровождается изменением морфологии этих клеток;
- ПТГ стимулирует движение остеокластов к месту резорбции и их активность;
- действие ПТГ при экзогенном введении зависит от его дозы: в низких дозах (и при прерывистом введении) он стимулирует костеобразование (усиливает синтез остеобластами ЩФ, остеокальцина, концевого пептида проколлагена), в высоких — усиливает резорбцию кости, ингибирует формирование кости в результате торможения синтеза коллагена и костного матрикса. Это действие опосредовано через продукцию цитокинов (ИЛ-6 и ГМ-КСФ стимулируют остеокластогенез, резорбцию кости, а ИПФР-1 и -2, ТФР-β усиливают костеобразование).
Результаты исследования Н.В. Дедух и С.В. Малышкиной (2002) свидетельствуют, что длительное (в течение 1 мес) введение крысам ПТГ в высокой дозе (0,5 МЕ/г массы тела) сопровождается увеличением количества остеокластов, площадей лакун резорбции в губчатой и компактной костной ткани тел позвонков с преобладанием процесса в компактном слое кости. В этом слое определяются расширенные центральные каналы остеонов с неравномерными границами, прободающие каналы в виде полостей, расширенные лакуны резорбции на эндостальной костной поверхности. В губчатой костной ткани снижается плотность поперечных костных трабекул, появляются микротрещины, переломы вертикальных трабекул, увеличивается количество остеокластов, расширяется слой остеоида, нарушается процесс минерализации. В итоге меняются анатомические параметры тел позвонков (особенно в грудном отделе), уменьшается ширина их тел.
Болезнь Реклингхаузена (гиперпаратиреоз при новообразованиях или гиперплазии паращитовидных желез) сопровождается высоким содержанием кальция, ЩФ, низким содержанием фосфатов в крови, что требует применения кальция, витамина D.
Концентрация ПТГ в крови составляет около 1 мкг/л. Его образуется тем больше, чем ниже концентрация Са2+ в крови (Мусил Я., 1985). Период полураспада интактного ПТГ в крови составляет 34 мин: он быстро метаболизируется в печени, почках. В крови присутствуют продукты расщепления: N-терминальный фрагмент (период полураспада — 34 мин), С-терминальный (период полураспада 23 ч) и другие. Современные иммунологические методы позволяют определять только интактный ПТГ, который адекватно характеризует паратиреоидный статус пациента. Влияние различных факторов на функциональную активность остеокластов представлено в табл. 2.2.
2.1.2. Кальцитонин
Это полипептидный гормон, включающий 32 аминокислотных остатка, который преимущественно секретируют парафолликулярные (С-) клетки щитовидной железы. Он не образуется в виде прогормона, а сразу синтезируется его активная форма. Кальцитонин также вырабатывается в вилочковой железе, паращитовидных железах, надпочечниках, легких, головном мозге. Действие осуществляется через цАМФ и протеинкиназы. Поскольку действие ПТГ и кальцитонина направлено на одни и те же клетки и связано с цАМФ, Я. Мусил (1985) предполагает наличие разных специфичных рецепторных белков, например протеинкиназ, способных изменять (повышать и понижать) внутриклеточную концентрацию цАМФ. В настоящее время используется в качестве лечебного средства для профилактики и лечения остеопороза. Уровень секреции кальцитонина определяется содержанием Са2+ (особенно ионизированной формы) в крови: при повышении содержания Са2+ секреция кальцитонина усиливается, при снижении — угнетается. Основной функцией кальцитонина является угнетение костной резорбции. Этот процесс сопровождается гипокальциемией, гипофосфатемией. Поступлению кальция и фосфора в костную ткань под действием кальцитонина способствует образование лабильной формы фосфата кальция в межклеточной жидкости. Он ингибирует остеолиз, стимулированный ПТГ, витамином D, способствует репарации костей после переломов. Анаболическое действие кальцитонина на костную ткань (при гиперпаратиреозе, метастазах злокачественных опухолей в костях, болезни Педжета) сопровождается снижением активности ЩФ и выделения оксипролина с мочой (при болезни Педжета повышена активность ЩФ и уровень гидроксипролина в моче). На гипокальциемический эффект кальцитонина влияет уровень потребления кальция и фосфора: гипокальциемический эффект выше у пациентов с низким содержанием кальция в рационе. Кальцитонин является функциональным антагонистом ПТГ: стимулирует поступление кальция и фосфора из крови в кости, ускоряет отложение кальция, ингибирует его выход из костей (таким образом способствует формированию органического матрикса, процессу его минерализации, повышает прочность костей). В клетках почечных канальцев кальцитонин вызывает повышенный клиренс и выведение Са2+, фосфатов, Mg2+, К+ и Nа+. На выведение фосфатов кальцитонин и ПТГ действуют как синергисты. Вместе с ПТГ кальцитонин стимулирует превращение в почках неактивной формы витамина D3 в активную — 1,25-(ОН)2D3 (кальцитриол). Кальцитонин может быть использован в период подготовки больных к операции на паращитовидных железах. Он замедляет снижение МПКТ, прежде всего в позвонках поясничного отдела позвоночника. Кальцитонин угнетает распад коллагена, его введение сопровождается уменьшением выведения с мочой оксипролина (Рассказов Л.В., Савченко Е.А., 1982), увеличением диуреза, экскреции кальция, фосфора, натрия, калия. Ингибирует пролиферацию и дифференциацию предшественников остеокластов, уменьшает количество остеокластов, угнетает их активность (Azria M., 1989) и тем самым тормозит костную резорбцию, обусловливает гипокальциемию, гипофосфатемию. Остеобласты и остеокласты имеют рецепторы к кальцитонину (Martin T.J., Dempster D.W., 1998). Полипептид замедляет (блокирует) остеолиз, стимулированный ПТГ, витамином D, цАМФ, а также положительно влияет на костеобразование, тормозит распад гликогена (Azria M., 1989), уменьшает выход минералов из костного матрикса. Повышает способность остеобластов, остеоцитов к синтетическим процессам, стимулирует синтез костного матрикса, отложение кальция в костях, стимулирует поглощение костями фосфора, снижает содержание фосфатов в крови. Влияние кальцитонина опосредуется почками: он ускоряет превращение витамина D в 1,25-дигидроксихолекальциферол, снижает реабсорбцию кальция в почках. Кальцитонин в невысоких дозах снижает абсорбцию кальция в кишечнике, стимулированную витамином D (Рожинская Л.Я., Марова Е.И., 1996). Предполагают (Уайт А. и соавт., 1981б), что кальцитонин оказывает двоякое влияние на костные клетки: с одной стороны, активирует кальциевый насос, что способствует выходу кальция из клетки, с другой — стимулирует его поглощение митохондриями. Этот процесс энергозависимый и зависит от соотношения Na+/К+. Свое действие на клетки (в том числе костной ткани, почек) кальцитонин реализует через аденилциклазу, цАМФ. Результатом такого действия является снижение содержания кальция в цитоплазме клеток. При резком замедлении костной резорбции проявляется гипокальциемический эффект кальцитонина. Кальцитонин вместе с ПТГ обеспечивают постоянный уровень кальция в крови. Уровень кальцитонина у мужчин в несколько раз выше, чем у женщин (его уровень возрастает в период беременности и лактации). По своей структуре, биологической активности кальцитонины лососевых рыб, быка, свиньи, овцы мало отличаются.
В.Дж. Маршалл (2002) считает, что физиологическое значение снижения активности остеокластов под действием кальцитонина не установлено. Свою точку зрения он аргументирует тем, что у больных с тотальной тиреоидэктомией не формируется клинический синдром, который можно было бы обьяснить недостаточностью кальцитонина. Гомеостаз кальция поддерживается у пациентов с медуллярной карциномой щитовидной железы, клетки которой секретируют большое количество кальцитонина. Кальцитонин может блокировать действие кальцитриола на костную ткань, приводить к увеличению поступления кальция из кишечника без его потери из кости. Кальцитонин обнаружен во многих тканях, в том числе кишечника, центральной нервной системы (предполагается, что он выполняет роль нейротрансмиттера). В некоторых тканях мРНК кальцитонина транслируется в пептиды, родственные по гену кальцитонину, функция которых не изучена.
Кальцитонин обладает центральным анальгетическим эффектом, сопоставимым с действием опиоидных анальгетиков. Обезболивающее действие кальцитонина связывают также с угнетением синтеза ПГЕ, тромбоксана, стимуляцией выделения в-эндорфина (агониста опиоидных рецепторов). Сочетание антирезорбтивного и анальгетического действия придает большую практическую ценность препаратам кальцитонина. Обезболивающий эффект кальцитонина подтвержден при РА, остеопорозе, переломах костей, болезни Педжета, метастазах в кости, мигрени, фантомной боли, поражении периферических артерий. Кальцитонин также обладает противовоспалительным и антигистаминным эффектом.
На образование кальцитонина влияют половые гормоны: тестостерон и эстрогены повышают его уровень в крови. Он повышается в период беременности и кормления грудью. Уровень кальцитонина в крови снижается по мере старения человека (Скоромец А.А. и соавт., 1995), это наиболее выражено в постменопаузальный период (Deftos L. et al., 1980). Кальцитонин тормозит желудочную и панкреатическую секрецию. Установлена связь между секрецией кальцитонина и гастрина: снижение содержания Са2+ в пищеварительной системе способствует усилению секреции гастрина, что приводит к усилению продукции и секреции кальцитонина. Кроме гастрина, секрецию кальцитонина стимулируют глюкагон и соматостатин. Секреция кальцитонина увеличивается при действии стрессорных факторов (Држевецкая И.А. и соавт., 1981): его уровень повышается при иммобилизации, алкогольной интоксикации, инсулиновой гипогликемии, интенсивной мышечной работе и др. Гипокальциемия, возникающая под действием кальцитонина, обусловливает торможение активности гипоталамо-гипофизарно-надпочечниковой системы, ограничение катаболического эффекта стрессовой реакции (Држевецкая И.А. и соавт., 1983). Кальцитонин замедляет секрецию инсулина при нагрузочной пробе глюкозой, снижает тормозящее действие глюкозы на секрецию глюкагона, нарушает толерантность к глюкозе (Хасанова Э.Р., 1989). Причиной этого автор считает перераспределение кальция в островковых клетках. У больных, длительно принимающих кальцитонин, диабетогенного действия не выявлено. Дефицит кальцитонина клинически не выявляется. Его гиперпродукцию (чаще без гипокальциемии) отмечают при медуллярной карциноме щитовидной железы.
У здоровых людей уровень кальцитонина в крови колеблется в пределах 50–450 нг/л и зависит от возраста обследованных, физиологического состояния организма (период интенсивного роста, период беременности и лактации, менопауза), циркадных и сезонных ритмов, сопутствующей патологии. С целью определения кальцитонина в тканях и биологических жидкостях используют биологические и радиоиммунологические методы исследования.
2.1.3. Витамин D, кальцитриол
Витамин D является прогормоном. Вместе с двумя другими гормонами — ПТГ (или паратирином) и кальцитонином — витамин D в виде кальцитриола принимает участие в регуляции метаболизма кальция.
К витаминам D относятся эргокальциферол (D2), холекальциферол (D3), дигидроэргокальциферол (D4), D5, D6, D7. Витамин D2 образуется из эргостерина — растительного предшественника, D3 — из 7-дегидрохолестерина, который образуется в коже после облучения ультрафиолетовым светом. У лиц в возрасте старше 70 лет способность кожи синтезировать витамин D3 снижается более чем в 2 раза. Биологически более активен витамин D3. По своему действию витамин D3 — гормон, обмен которого связан с митохондриями клеток почек. Активные формы эргокальциферола и холекальциферола образуются в процессе метаболизма. Вместе с метаболитами являются регуляторами кальциево-фосфорного обмена, поддерживают гомеостаз кальция, участвуют в минерализации костной ткани. Витамин поступает в пищеварительный тракт в виде эргокальциферола (D2) и под влиянием 25-гидроксилазы в печени превращается в транспортную форму — гидроксихолекальциферол (25-ОН-D3). Под влиянием 1α-гидроксилазы в эпителии проксимальных почечных канальцев образуется кальцитриол, то есть 1,25-дигидроксикальциферол (1,25-(ОН)2D3), при воздействии 24-гидроксилазы — 24,25-(ОН)2D3. Гидроксилирование в печени не подчинено контролю по механизму обратной связи, а в почках процесс строго регулируется. После синтеза в коже холекальциферол транспортируется кровью в печень, где в митохондриях превращается в 25-ОН-D3. Этот промежуточный продукт превращается в 25-(ОН)2D3 или в 24,25-(ОН)2D3. 1,25-(ОН)2D3 образуется, как было отмечено, в митохондриях клеток почек под действием 1α-гидроксилазы. Активность 1α-гидроксилазы регулирует ПТГ (без него кальцитриол не влияет на транспорт кальция и фосфора). Как отмечает Я. Мусил (1985), активность этого фермента регулируется двумя путями: в минутных интервалах — изменением концентрации Са2+ в митохондриях (недостаток Са2+ повышает активность); в часовых интервалах — действием ПТГ, который ингибирует синтез 1α-гидроксилазы de novo. Избыток кальцитриола ингибирует синтез и секрецию ПТГ: в цитозоле клеток паращитовидных желез имеются рецепторы для него (действует механизм обратной связи). Синтез и высвобождение ПТГ ингибирует и избыток кальция, вызванный избытком 1,25-(ОН)2D3 (вторая обратная связь). Избыток 1,25-(ОН)2D3 индуцирует синтез 24-гидроксилазы, которая катализирует синтез 24,25-(ОН)2D3, являющегося функциональным антагонистом 1,25-(ОН)2D3. Механизм действия 1,25-(ОН)2D3 аналогичен механизму действия стероидных гормонов. Физиологическая роль 24,25-дигидроксихолекальциферола полностью не изучена. Указанные метаболиты стимулируют кишечную абсорбцию кальция, фосфора, усиливают экскрецию кальция с мочой, активируют костный обмен. В остеоцитах повышается мобилизация Са2+ и фосфатов. К эффектам метаболитов витамина D относят:
- стимуляцию остеокластической резорбции кости и остеоцитарного остеолиза;
- усиление дифференциации клеток — предшественников остеокластов;
- усиление реабсорбции кальция в дистальных почечных канальцах;
- увеличение пула кальция в организме.
Кальцитриол (1,25-дигидроксихолекальциферол) способствует всасыванию кальция в кишечнике, усиливает его поступление в ткани, регулирует образование и минерализацию костного матрикса. Из почек (места синтеза) он поступает с кровью в кишечник, где в клетках слизистой оболочки стимулирует синтез специфического гликопротеида (кальцийсвязывающего протеина), который, связывая кальций, облегчает его транспорт внутрь клеток слизистой оболочки. Посредством действия Са2+-Nа+-АТФазы через базальный полюс клетки кальций попадает в кровоток. Рецепторы кальцитриола обнаружены на остеобластах, их клетках-предшественниках, хондробластах, хондроцитах. Связывание кальцитриола остеобластами обусловливает увеличение продукции ЩФ и кальцийсвязывающего белка, остеокальцина, функция которых не полностью изучена.
T.J. Martin, D.W. Dempster (1998) считают, что кальцитриол стимулирует остеокласты, способствует образованию из клеток — предшественников остеокластов и остеобластов. Кальцитриол в высоких концентрациях стимулирует резорбцию костной ткани остеокластами, в результате чего кальций и фосфаты поступают во внеклеточную жидкость. В почках кальцитриол ингибирует свой синтез (по механизму обратной связи). Действуя вместе с ПТГ, он оказывает незначительный стимулирующий эффект на реабсорбцию кальция. Кальцитриол действует медленно: его эффект наблюдается через несколько часов и даже дней.
Наличие рецепторов к кальцитриолу во многих тканях дает основание предполагать, что, кроме роли в гомеостазе кальция, он выполняет дополнительные функции. Он влияет на клеточную дифференцировку в нормальных и опухолевых тканях, стимулирует выработку некоторых цитокинов и, возможно, участвует в иммуномодуляции.
Если ультрафиолетовое облучение кожи недостаточно или витамин D отсутствует в пищевых продуктах, развивается витаминная недостаточность, следствием которой является рахит, остеомаляция.
2.1.4. Адреналин
Под влиянием адреналина повышается секреция лизосомальных гидролаз в результате дестабилизации лизосомальных мембран (Loegering D. et al., 1975) и внеклеточная концентрация остеокластных гидролаз, равновесие между их ингибиторами и ферментами меняется в сторону последних. В результате нарушается структура костной ткани вне зоны резорбции. Адреналин может оказывать и опосредованное влияние на состояние костного метаболизма при стрессовых состояниях (в том числе при хроническом стрессе). Концентрация адреналина в крови в норме составляет около 0,06 мкг/л или 10-10 ммоль/л. В стрессовом состоянии она может вырасти почти в 1000 раз за секунды или минуты. Опосредованное влияние этого гормона на метаболизм костей осуществляется через повышение экскреции глюкокортикостероидов (ГКС), развитие ацидоза (в результате усиления анаэробного распада гликогена до молочной кислоты), сопровождающихся усилением костной резорбции, развитием остеопении. Адреналин также усиливает свободнорадикальные процессы (свободные радикалы повышают функцию остеокластов), окисление полиненасыщенных жирных кислот, синтез эйкозаноидов, среди которых негативное влияние на состояние костной ткани оказывают ПГЕ1 и ПГЕ2. Механизмы развития остеопении при возникновении общего неспецифического адаптационного синдрома представлены на схеме 2.1.
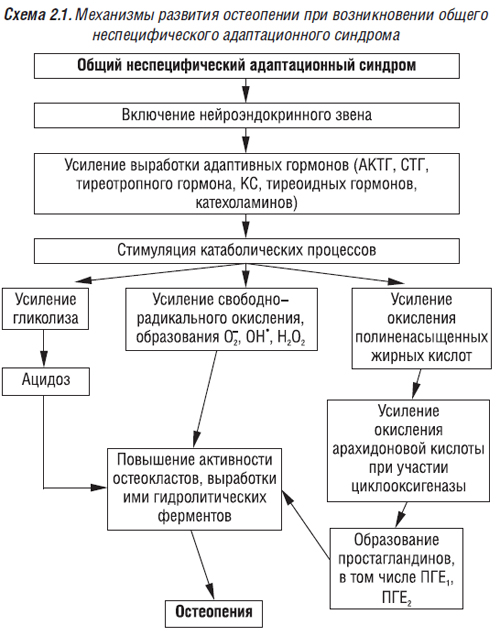
Запасы минеральных веществ в костной ткани, по мнению Е.П. Подрушняка (1987), позволяют организму быстро реагировать на стрессовые ситуации, выбрасывая их в кровь и сохраняя жизнь человека. В результате при повторных стрессовых ситуациях, хроническом стрессе понижаются все основные функции костной системы. С возрастом повышается напряженность минерально-органического гомеостаза, особенно при нагрузках, стрессовых ситуациях.
2.1.5. Глюкокортикоиды
Кортизол (гидрокортизон) принимает участие в регуляции белкового и углеводного обмена, стимулирует распад белков и превращение аминокислот в глюкозу, повышает уровень глюкозы в крови. Синтетические ГКС оказывают противовоспалительное и иммунодепрессивное действие. ГК ингибируют всасывание кальция и фосфора в кишечнике, снижают реабсорбцию кальция в почечных канальцах и тем самым повышают его экскрецию с мочой. На цитоплазматических мембранах остеобластов и остеокластов имеются рецепторы к ГК. Они угнетают пролиферацию остеобластов, синтез ими коллагена, неколлагеновых белков, выделение интегринов. При длительном воздействии ГК на культуру костных клеток снижается синтез ИПФР-1, белков, коллагена, РНК остеобластами, уменьшается образование клеток — предшественников остеобластов. Эффект действия ГК на костные клетки зависит от их дозы: ГК в физиологических дозах стимулируют синтез остеобластами коллагена I типа (вследствие усиления связывания ИПФР с его рецепторами), ЩФ, ДНК; при повышении дозы уменьшается площадь остеобластов, увеличивается количество остеокластов. ГК в высоких (терапевтических) дозах снижают синтез РНК, белка, в том числе коллагена, ингибируют синтез протеогликанов, снижают содержание кальция в кости, что обусловливает деминерализацию костей (Gallagher J.A. et al., 1984) и повышение содержания кальция и дезоксипиридинолина в моче. При продолжительном (до 12 нед) введении ГК в высоких дозах уменьшается объем трабекулярной кости, трабекулы истончаются, расширяются межтрабекулярные пространства, снижается минерализация. Аналогичный эффект (снижение костеобразования, минерализации, усиление резорбции, уменьшение объема кости) вызывают и ГК в низких дозах при длительном введении. Лечение ГКС способствует снижению динамики кальцификации остеоида, уменьшению его толщины, увеличению остеоидных пространств, повышению показателей резорбции костной ткани (Meunier P.J. et al., 1984). Изучение (Дедух Н.В., Малышкина С.В., 2002) длительного влияния дексаметазона (1,2 мг/кг массы тела) на фоне дефицита кальция выявило выраженные деструктивные изменения в губчатой и компактной костной ткани тел позвонков, изменение их формы. Авторы обнаружили резкое истончение костных трабекул, преобладание вертикальной ориентации трабекул, расширение межтрабекулярных пространств, спонгизацию компактной кости. Плотность остеоцитов в костных трабекулах была сниженной, среди них преобладали клетки с пикнотическими ядрами. Обнаруживались «запустевшие» лакуны остеоцитов. Резко уменьшилось количество остеобластов на эндостальной поверхности кости, в них снизилась плотность цитоплазматических структур, рецепторов.
Побочное действие терапии КС проявляется повышенным выделением кальция, калия, задержкой натрия, остеопорозом вследствие сниженного синтеза основного вещества костей.
2.1.6. Тироксин (тетрайодтиронин, Т4)
Образуется из аминокислоты тирозина и содержит атомы йода. Повышает скорость метаболизма и стимулирует развитие эмбриона. Активирует костный обмен, оказывает стимулирующее влияние на остеобласты и остеокласты кортикальной и трабекулярной костной ткани, а также на образование хряща. На остеобластах обнаружены (Martin T.J., Dempster D.W., 1998) рецепторы к тироксину, воздействие на которые стимулирует образование ИПФР-1. При гипертиреозе повышены как новообразование кости, так и костная резорбция (она преобладает), при гипотиреозе костная перестройка замедлена. Тиреотоксикоз сопровождается гиперкальциемией, прием препаратов щитовидной железы увеличивает выраженность остеопении у пациенток в период менопаузы. Тироксин в высоких дозах усиливает явления недостаточности жирорастворимых витаминов — эргокальциферола, токоферола, витамина А (некоторые из этих витаминов тормозят функцию щитовидной железы). Гормоны щитовидной железы в высоких дозах задерживают процессы регенерации.
Тиреоидные гормоны способствуют пролиферации костных клеток, стимулируют активность остеобластов, биосинтез компонентов матрикса. Влияя на эпифизарный хрящ, способствуют росту костей. При гипертиреоидизме отмечают усиление костной резорбции (ее стимулирует тироксин), снижение МПКТ, гиперкальциемию (развивается тиреогенная костная дистрофия). В постменопаузальный период у многих женщин выявляют снижение функции щитовидной железы, что обусловливает большую выраженность остеопороза. Снижение выработки йодированных L-тироксинов сопровождается подавлением активности остеобластов, замедлением роста длинных костей, запаздыванием образования вторичных центров окостенения, замедлением образования костной мозоли после переломов костей. Применение тиреоидных гормонов предотвращает эти процессы. Тиреоглобулин может предотвратить развитие кариеса у экспериментальных животных. Влияние различных гормонов на скорость образования и резорбции кости представлено в табл. 2.3.
2.1.7. Эстрогены
- Важнейшим представителем эстрогенов является эстрадиол, который синтезируется в яичниках, а в период беременности — в плаценте. Он отвечает за развитие вторичных женских половых признаков, регулирует менструальный цикл, стимулирует пролиферацию клеток слизистой оболочки матки. Установлено наличие рецепторов к эстрогенам на остеобластах, остеокластах и остеоцитах (Eriksen E.F. et al., 1988; Komm B.S. et al., 1988). Эстрогены регулируют транскрипцию специфических генов, отвечающих за биосинтез ядерных рецепторов для эстрогенов.
- Эстрогены являются регуляторами костной резорбции, предотвращают резорбирующий эффект ПТГ (Cosman F. et al., 1993). При действии эстрогенов отмечается снижение синтеза ПТГ, увеличивается продукция кальцитонина, снижается резорбционное действие кальцитриола.
- Эстрогены ингибируют резорбцию костной ткани, формирование остеокластоподобных клеток путем прямого воздействия на гемопоэтические бластные клетки и непрямого — на остеобласты. Это действие реализуется через цАМФ-протеинкиназу. Они регулируют секрецию остеокластами катепсина-L, β-глюкуронидазы, лизоцима, катепсина В, ЩФ (Judd J. et al., 1995).
- Увеличивают количество остеобластов, их размеры, стимулируют выработку остеобластами фактора, который ингибирует резорбтивную активность остеокластов (эстрогены ингибируют активность остеокластстимулирующего фактора остеобластов, повышают активность остеокластингибирующего фактора).
- Эстрогены усиливают разрушение остеокластов.
- В репродуктивный период эстрогены поддерживают костный баланс, минеральный гомеостаз (в том числе у лиц мужского пола). Стимулируют процессы костеобразования, биосинтез ДНК, индуцируют экспрессию иРНК для ЩФ и остеокальцина.
- Эти гормоны подавляют резорбцию костной ткани путем ингибирования синтеза остеобластами ИЛ-1, -6, ФНО-α, ГМ-КСФ, стимулируют синтез ТФР-β. 17-β-эстрадиол активирует синтез ИПФР-1 и -2, ТФР-β (Slater M. et al., 1994).
- Снижение их концентрации в крови сопровождается увеличением количества остеокластов, усилением продукции перечисленных факторов, развитием остеопороза (Lindsay R., 1998).
- Эстрогены влияют на обмен белков, пуринов, углеводов, активируют ДНКзу.
У женщин в период менопаузы дефицит эстрогенов является ведущим фактором развития остеопороза. Дефицит эстрогенов, возникающий после овариоэктомии, сопровождается увеличением продукции моноцитами, другими клетками костного мозга цитокинов, регулирующих остеокластогенез, увеличивающих количество остеокластов. Он сопровождается повышением секреции ИЛ-6, уменьшением массы костных трабекул, объема губчатой кости, снижением минеральной плотности (МП). Из эстрогенных гормонов (эстрон, эстрадиол, эстриол) по отношению к метаболизму костной ткани наиболее активен эстрадиол. У детей и подростков эстрогены обеспечивают развитие хрящей, окостенение эпифизов трубчатых костей, формирование «женского» скелета, стимулируют синтез белка (обладают анаболическим действием), обеспечивают синтез коллагена, отложение кальция и фосфора в эпифизах костей. Их дефицит до периода полового развития сопровождается задержкой окостенения эпифизарных хрящей, потерей кальция и фосфора, отрицательным азотистым балансом. После введения радиоактивного эстрона (меченого 14С) его обнаруживают в метафизе костей. Позднее половое созревание приводит к нарушению образования матрицы кости. Остеопенический синдром, возникающий на фоне дефицита эстрогенов, связан с увеличением остеокластической резорбции губчатой костной ткани, снижением процессов костеобразования. То, что у тучных женщин с избыточной массой тела реже отмечают остеопороз, объясняют образованием в жировой ткани эстрогенов из андростендиола коры надпочечников.
2.1.8. Андрогены
Наиболее важным представителем андрогенов (мужских половых гормонов) является тестостерон, который синтезируется клетками Лейдига и контролирует развитие и функцию половых желез. Андрогены синтезируются не только в семенниках, но и в коре надпочечников, яичниках (аналогично: эстрогены образуются не только в яичниках, но и в коре надпочечников и семенниках). Развитие вторичных мужских половых признаков определяет гормон (точнее, они определяются соотношением секретируемых андрогенов и эстрогенов). Механизм воздействия андрогенов на костную ткань окончательно не установлен. Как у мужчин, так и у женщин (в одинаковом количестве) имеются рецепторы к андрогенам на остеобластоподобных клетках (Vanderschueren D., Bouillon R., 1995), остеобластах (Abu E.O. et al., 1997), остеоцитах, хондроцитах, мононуклеарах, эндотелиальных клетках сосудов костного мозга. Андрогены оказывают прямое влияние на метаболизм костной ткани: усиливают пролиферацию остеобластов, выработку ими ЩФ, синтез коллагена III типа. Они усиливают продукцию СТГ, ИПФР-1. Не исключается, что андрогены могут воздействовать на костную ткань и через эстрогенные рецепторы: в жировой ткани андрогены метаболизируются в эстрон. Лечение андрогенами приостанавливает развитие остеопении у мужчин с гипогонадизмом (сенильный остеопороз у них является результатом недостатка андрогенов). J.E. Dixon и соавторы (1989) отмечали гиперандрогению (гирсутизм) у женщин с нормальным уровнем костной массы. Андрогены оказывают более сильный, чем эстрогены, анаболический эффект: усиливают биосинтез белков в тканях, в том числе в костях (стимулируют синтез костного матрикса). Они способствуют развитию и минерализации эпифизарных зон роста костей (отложению фосфорно-кальциевых солей), формированию скелета. При нарушении андрогенной функции процессы окостенения эпифизов костей запаздывают. Препараты тестостерона, анаболические стероиды улучшают сращивание костей при переломах.
Анаболические половые гормоны, находясь в равновесии с гормонами коры надпочечников с антианаболическим действием, оказывают регулирующее влияние на строение основного вещества кости. У лиц пожилого возраста антианаболическое действие кортикостероидов становится преобладающим. Влияние кортикостероидов и половых гормонов на костный метаболизм представлено на схеме 2.2.

При сенильном остеопорозе кортизол способствует усиленному выделению кальция из организма.
2.1.9. Соматотропный гормон (гормон роста)
Гормон роста, образующийся в эозинофильных α1-клетках передней доли гипофиза, оказывает прямое регулирующее влияние на различные ферменты белкового, жирового и углеводного обмена. Он стимулирует синтез белка, ДНК, хондроитинсерной кислоты в растущих костях, повышает содержание РНК в клетках. Соматотропин и адренокортикотропный гормон (АКТГ) в большинстве случаев действуют как антагонисты (схема 2.3).
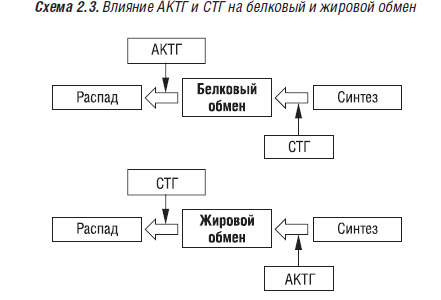
На углеводный обмен СТГ и АКТГ оказывают одинаковое действие. Избыточная секреция СТГ проявляется гигантским ростом. До окостенения эпифизарного хряща отмечается рост костей в длину и ширину, после окончания окостенения усиливается рост периостальной ткани и внутренних органов. Гормон роста стимулирует клетки-предшественники в эпифизарном хряще. Установлено, что этот гормон сохраняет свое регулирующее влияние на метаболические процессы (в том числе, в костной ткани) в организме взрослого человека, а также на процессы ремоделирования. Гормон роста оказывает как прямое, так и непрямое воздействие на остеобласты человека. На этих клетках выявлены рецепторы к СТГ (Barnard R. et al., 1991). Он способствует продукции остеобластами ТФР-β, КМГБ, стимулирует синтез ИПФР-1 и в печени, регулирует образование ИПФР-связывающих белков. В свою очередь, ИПФР-1 влияет на скелет подобно гормону роста. ИПФР играют ведущую роль в остеобластическом ответе на гормон роста. Остеобласты имеют рецепторы к ИПФР-1. Уровень ИПФР-1 в плазме крови снижается с возрастом, после менопаузы (лечение эстрогенами нормализует его уровень). Гормон роста усиливает синтез остеобластами коллагена I типа, ЩФ, костного Gla-протеина (Kassern M. et al., 1993). Он усиливает пролиферацию хондроцитов ростовой пластинки, рост костей в длину, что позволяет лечить низкорослость у детей. СТГ активирует 1α-гидроксилазу почек, способствует трансформации транспортной формы витамина D в кальцитриол (Bengtsson M.B., 1994) и, таким образом, усилению абсорбции кальция в кишечнике, фосфатов — в кишечнике и почках.
2.1.10. Инсулин
Обладает стимулирующим действием на костную ткань (синтез костного матрикса, образование хряща). Способствует дифференциации остеобластов, увеличению количества клеток, продуцирующих коллаген (McCarty M.F., 1995), минерализации костной ткани, не влияет на процессы резорбции. Влияние инсулина на костные клетки осуществляется прямым влиянием на их рецепторы, а также опосредованно через продукцию ИПФР в печени. Дефицит инсулина сказывается на метаболизме костной ткани: нарушается транспорт кальция в клетки, угнетается белковый синтез, повышается распад белков, нарушается рост детей.
К вторичным эффектам инсулина относят снижение содержания калия в крови и фосфатов в крови и моче.
2.1.11. Глюкагон
Осуществляет стимулирующее влияние на остеогенез опосредованно через кальцитонин. В культуре ткани он угнетает резорбцию кости.
2.2. Цитокины, ростовые факторы
Цитокины представляют собой низкомолекулярные пептиды (менее 80 кДа), секретируемые клетками воспаления и иммунного ответа. Они контролируют активность и рост этих клеток. Некоторые из цитокинов оказывают эндокринный (дистанционный) эффект, но большинство их функций локального характера. В.Дж. Маршалл (2002) выделяет четыре класса цитокинов:
- ИЛ (регулируют процессы воспаления);
- ИФН (антивирусные вещества, оказывающие ингибирующий эффект на рост клеток);
- колониестимулирующие факторы (КСФ) (стимулируют рост макрофагов и лейкоцитов);
- ФНО (стимулируют пролиферацию многих клеток, в том числе цитолитических Т-клеток).
Цитокины имеют множество свойств, взаимодействуют между собой (эффект одного цитокина зависит от присутствия других), влияют на секрецию других факторов.
Как отмечает В.Дж. Маршалл (2002), цитокины проявляют функциональное сходство с пептидными ФР, которые влияют на рост неимунных клеток. Цитокины и ФР, продуцируемые костными клетками, представлены в табл. 2.4.
Наиболее изучены ЭФР, ТрФР, ТФР и ИПФР. На состояние костного метаболизма активно влияют ФР (табл. 2.5), которые продуцируются в костной и других тканях и циркулируют в крови (Рожинская Л.Я., 2000; Дедух Н.В., Малышкина С.В., 2002). В процессе ремоделирования костей принимают участие следующие из них:
- ИПФР-1, -2;
- ТФР-α;
- ТФР-β;
- МГБ;
- ЭФР;
- ФРФ;
- ТрФР.
Большинство ФР циркулируют в кровотоке. В значительных количествах в костной ткани представлены ИПФР-2 и ТФР-β. ФР имеют рецепторы на остеобластах и влияют на их дифференциацию, формирование органического матрикса (ИПФР-1, ТФР-β, МГБ, ФРФ), а также резорбцию костной ткани (ЭФР, ТФР-α, ТрФР) (Martin T.J., Dempster D.W., 1998). Так, ТФР-β посредством рецепторов костных клеток усиливает репликацию остеобластов, синтез этими клетками коллагена, остеопонтина, остеонектина, фибронектина, протеогликанов, стимулирует активность ЩФ, ингибирует резорбирующую активность остеокластов, стимулирует костеобразование, активирует онкоген c-fos, активно участвующий в морфогенезе кости. Он оказывает модулирующее действие на синтез полипептидных факторов остеогенеза и ПГ. Синтез ТФР-в усиливают кальцитриол, половые гормоны. ИПФР-1 (в 7 раз активнее ИПФР-2) усиливает репликацию остеобластов, синтез коллагена, образование белков костного матрикса, участвует в пролиферации, дифференцировке остеокластов и их предшественников. Синтез ИПФР-1 регулирует соматотропин, усиливает ПТГ в физиологических концентрациях, а угнетают ГК. ИПФР-2 активирует остеокластоподобные многоядерные клетки, стимулирует пролиферацию преостеобластов, дифференцировку остеобластов. Ген ИПФР-2 локализован в 11-й хромосоме рядом с геном инсулина. Тропность ИПФР к клеткам-мишеням увеличивают ИПФР-связывающие белки и прежде всего белок III типа. Эти белки обладают активностью и могут стимулировать пролиферацию остеобластов. ФРФ (обнаруживается в костном матриксе) стимулирует синтез остеобластами коллагена I типа, протеогликанов. Он оказывает митогенное действие на преостеобласты, увеличивает пул дифференцированных остеобластных клеток. ТрФР является регулятором репликации костных клеток, костной резорбции. Он оказывает митогенное действие на предшественников остеобластов. Влияние ТФР осуществляется с участием ИПФР-1. М-КСФ регулирует остеокластогенез, костную резорбцию.
В процессах ремоделирования костной ткани принимают участие ИЛ, ФНО-α и -β, ГМ-КСФ (табл. 2.6). В развитии остеокластов участвуют ИЛ-1, -3, -6, -11, ФНО-α, -β, ГМ-КСФ. ИЛ-6 стимулирует гемопоэз, остеокластогенез, способствует костной резорбции (Насонов Е.Л. c соавт., 1997). В развитии остеопороза участвуют ИЛ-1 (стимулирует костную резорбцию), ФНО-α (Mundy G.R., 1994).
Установлена тесная связь между остеогенезом, гемопоэзом и иммунной системой. На активацию остеокластов, нарушение баланса между остеобластами и остеокластами, резорбцию кости оказывают влияние местные факторы микроокружения, клетки иммунной системы (Бутенко Г.М., 2002). Клетки селезенки продуцируют фактор, способствующий резорбции кости путем активации остеокластов (Abe E. et al., 1986). Этот фактор представлен группой провоспалительных цитокинов (ИЛ-1, ФНО-α, -β, колониестимулирующими факторами КСФ-1, M-КСФ), ПГ. На состояние костной ткани оказывают влияние цитокины, выделяемые лимфоцитами и моноцитами. Лимфоциты секретируют ФНО-β, который стимулирует костную резорбцию. Патогенез ряда заболеваний костной ткани, для которых характерно преобладание скорости костеобразования над резорбцией, связывают с нарушением секреции ИФН. Патологический остеогенез наблюдается при нарушенной функции естественных киллеров. ИФН-γ, секретируемый Т-лимфоцитами, подавляет костную резорбцию, индуцированную ИЛ-1, ФНО-α. Лимфоциты имеют рецепторы к ПТГ, потенцирующему костную резорбцию. Взаимодействие моноцитов с костным матриксом сопровождается локальной продукцией ИЛ-1. У пациентов с остеопорозом с высоким уровнем костного обмена отмечают повышенное выделение ИЛ-1 в моноцитах. ФНО-α моноцитов стимулирует костную резорбцию, ингибирует костное формирование, индуцирует гиперкальциемию. Наиболее важной функцией моноцитов и макрофагов в отношении кости является продукция ФНО-α, ИЛ-1α и -1β, ГМ-КСФ. На функцию костных клеток оказывают влияние молекулы белков и гликопротеинов органического матрикса (некоторые связывающие полипептидные факторы остеогенеза белки, протеогликаны декорин, бигликан). Другой компонент внеклеточного матрикса — гликопротеин теасцин (его секретируют клетки остеобластной линии) — модулирует взаимодействие предшественников остеобластов, остеокластов и фибронектина.
Локальные факторы полипептидной природы стимулируют митогенетическую активность клеток-мишеней. Они синтезируются различными клетками и тканями, в том числе костной, и действуют локально, через мембранные рецепторы клеток или через межклеточные контакты. В локальной регуляции катаболической фазы ремоделирования принимают участие ПГ, цитокины (остеокластактивирующий фактор, ИЛ-1β, ФНО-α). В анаболической фазе ремоделирования участвуют гликопротеиды и неколлагеновые белки — фибронектин, остеонектин, остеопонтин, остеокальцин и др.
Итак, процессы остеогенеза и резорбции кости находятся под контролем не только системных, но и локальных факторов регуляции, которые играют важную роль в патогенезе развития остеопороза. Предполагается возможное использование их в будущем в лечебных целях. Механизмы системной и локальной регуляции ремоделирования, их взаимодействие изучены недостаточно, имеющаяся информация противоречива. Очевидно, что действие локальных факторов корректируется системными.
2.3. Простагландины
Имеется связь между функционированием костной ткани и обменом незаменимых полиненасыщенных жирных кислот — линолевой, линоленовой и арахидоновой. При отсутствии незаменимых полиненасыщенных жирных кислот в рационе экспериментальных животных установлено замедление их роста, нарушение работы почек. Большое значение полиненасыщенных жирных кислот отражает высокая физиологическая активность, образующихся из них ПГ. Источником арахидоновой кислоты является линолевая: она последовательно превращается в γ-линоленовую, гомо-γ-линоленовую, затем в арахидоновую. Полиненасыщенные жирные кислоты могут окисляться в митохондриях путем модификации β-окисления, но они не являются значительным источником энергии. Большое биологическое значение имеет их использование для синтеза эйкозаноидов (ПГ, простациклинов, тромбоксанов, метаболитов ПГ, гидроперекисей, лейкотриенов), а также участие в свободнорадикальных процессах. Эйкозаноиды — большая группа медиаторов с широким спектром биологической активности, образующихся почти во всех клетках организма.
Первичными источниками ПГ являются арахидоновая и гомо-γ-линоленовая кислоты. ПГ имеются во всех тканях, оказывают разнообразные и многочисленные действия. Концентрация этих веществ в тканях и жидкостях организма очень низкая (1010 ммоль/л). Эйкозаноиды инактивируются в течение нескольких секунд в результате восстановления двойных связей и окисления гидроксигрупп. Поэтому дальность их действия ограничена. Внутривенное введение натриевой соли арахидоновой кислоты экспериментальным животным обусловливает быстрое проявление биологического действия ПГ, например ускорение агрегации тромбоцитов, образование внутрисосудистых тромбов, усиление желудочно-кишечной моторики (Уайт А. и соавт., 1981б). Конечные продукты биосинтеза ПГ отличаются в различных органах. Реакции организма на ПГ подобны реакциям, обычно связанным с цАМФ. В зависимости от природы ткани ПГ повышают или снижают синтез цАМФ аденилатциклазой или его расщепление фосфодиэстеразой. Эндокринные железы (паращитовидные, щитовидная, кора надпочечников) на введение ПГ реагируют повышением секреции гормонов. Сами ПГ посредством аденилатциклазы имитируют гормональное действие. То, что ПГ влияют опосредованно через стимуляцию аденилатциклазы, затрудняет определить, какие эффекты обусловлены только ими. In vivo ПГ стимулируют образование стероидных гормонов в надпочечниках, освобождение инсулина посредством цАМФ и оказывают прямое действие на образование и секрецию гормонов щитовидной железы, гормонов желтого тела. Разнонаправленность действия различных видов ПГ объясняют стимуляцией разных циклических нуклеотидов — цАМФ или цГМФ. Таким образом, влияя на синтез системных гормонов, ПГ могут изменить метаболизм костной ткани.
Высвобождение арахидоновой кислоты из фосфолипидов клеточных мембран происходит под действием фосфолипазы А2 (схема 2.4). Активность этого фермента контролируется гормонами и другими биорегуляторами, сопряженными с G-белками. Свободная арахидоновая кислота также является биологически активным соединением. Дальнейшее превращение свободной арахидоновой кислоты может проходить по пути, который инициируется ПГ-синтазой (имеет свойства циклооксигеназы и пероксидазы) и/или по липоксигеназному пути с образованием нестабильных промежуточных соединений. ПГ-синтаза (циклооксигеназа-пероксидаза) катализирует процесс только в присутствии кислорода. (В настоящее время различают циклооксигеназу-1, -2 и -3). Продуктом реакции является пероксикислота в виде радикала, который подвергается циклизации с одновременным воздействием молекулы кислорода в положении С-15, в результате чего образуется эндоперекись (ПГG) (Мусил Я., 1985). При восстановлении ПГG с образованием ОН-группы образуются ПГH, дальнейшее превращение которых происходит по двум путям с образованием первичных ПГЕ и ПГF. Другие (вторичные) ПГ (относящиеся к типу А и В) синтезируются в ходе дополнительных реакций (цифры 1, 2, 3 указывают на количество двойных связей в боковых цепях). В легких, тромбоцитах PGH превращается в тромбоксан А, который далее превращается в тромбоксан В. Итак, в результате метаболизма по этому пути (с участием ПГ-синтазы) образуются ПГ, простациклины и тромбоксаны. Окисление полиненасыщенных жирных кислот при участии липоксигеназы обусловливает образование гидроперокси- и гидроксипроизводных жирных кислот, из которых путем дегидратации образуются лейкотриены. Эйкозаноиды служат вторичными мессенджерами гидрофильных гормонов, обладают разносторонней физиологической активностью, в том числе по отношению к метаболизму костной ткани. Эйкозаноиды действуют как локальные биорегуляторы через мембранные рецепторы в непосредственной близости от места их синтеза как на синтезирующие их клетки (аутокринное действие), так и на соседние клетки (паракринное действие) (Кольман Я., Рем К. ., 2000). Биосинтез ПГ осуществляется в микросомах. В сутки образуется около 100 мкг ПГ, их содержание в тканях составляет доли микрограмма на 1 г массы тела. Катаболизм ПГ совершается под действием 15-гидроксипростагландиндегидрогеназы, ПГ-редуктазы. ПГ чрезвычайно активны: одинаковый эффект ПГЕ, гистамина, ацетилхолина отмечается при соотношении их концентрации 1:12,5:25. Наиболее высокая активность ферментов распада ПГ характерна для легочной ткани: ПГ, попадающие в кровяное русло, исчезают из крови после однократного прохождения через легкие. Высвобождаясь из одного органа, они действуют на другой или окружающие ткани. Но основной эффект ПГ проявляется в той клетке, где они синтезируются.

ПГ принимают участие в ремоделировании костной ткани. Так, ПГЕ2 влияет на формирование и активность остеокластов, процессы резорбции костной ткани. ПГЕ1 и ПГЕ2 являются стимуляторами формирования остеокластоподобных клеток in vitro. Их влияние сходно с ПТГ и, вероятно, опосредовано цАМФ. Усиление костной резорбции при иммобилизации, воспалении, злокачественных процессах E. Canalis (1993) объясняет действием ПГ. Влияние ПГЕ2 на костное формирование зависит от концентрации: в концентрации 10-9–10-7 ммоль/л он повышает костный синтез коллагена, при концентрации 10-6 ммоль/л — замедляет. ПГЕ1 ускоряет выход кальция из кости (Уайт А. и соавт., 1981б). Как и ПГТ, ПГЕ1 стимулирует высвобождение из клеток костной ткани лизосомальных ферментов (Мецлер Д., 1980). Ацетилсалициловая кислота, ингибируя биосинтез ПГ, устраняет эти эффекты. Установлена взаимосвязь между ПГ и системными гормонами: эстрогены инактивируют ПГЕ2 в костной ткани, ГКС — ингибируют их, а паратиреодный гормон — стимулирует (Raiz L.G., 1988). У остеобластов имеются рецепторы к ПГЕ.
2.4. Влияние избыточного образования лактата, ацидоза
В костных клетках осуществляется гликолиз, в них образуется молочная кислота. При введении ПТГ образование лактата повышается. Вследствие снижения рН может возникать локальная деминерализация кости, сопровождающаяся поступлением в плазму крови цитрата. Образование лактата ингибируют эстрогены. При ацидозе усиливается резорбция костной ткани остеокластами. Особенно наглядна роль ацидоза при тубулопатиях — генетически обусловленных (врожденных) заболеваниях почек. Резорбирующий эффект ПТГ в определенной мере обусловлен ацидозом, возникающим в результате нарушения реабсорбции бикарбонатов. На состоянии костной ткани сказывается хроническая гипоксия, ацидоз, сопровождающие различные заболевания и, прежде всего, заболевания легких, сердечно-сосудистую патологию.
2.5. Регуляция процесса ремоделирования кости
Обновление костной ткани, которое происходит в течение всей жизни человека, является результатом осуществления двух одновременных динамических процессов — образования и разрушения кости. Ремоделирование — это замена костных структур новыми, то есть процесс регенерации, результат двух разнонаправленных процессов — резорбции и костеобразования.
Резорбция и новообразование кости происходят в участках костной ткани, так называемых КРЕ (БМЕ). В состав этих участков размером 0,051 мм3 входят остеобласты, остеокласты, активные мезенхимальные клетки, капиллярные петли. В организме человека одновременно функционирует от 100 тыс. до 1 млн БМЕ. От их количества зависит абсолютный объем кости. Метаболизм кости постоянно в динамике. При ремоделировании у детей новообразованная кость формируется в ином положении, чем разрушенная, меняется форма скелета. В процессе ремоделирования у взрослых форма кости не меняется (резорбция и формирование происходят в одном и том же месте), старая кость заменяется новой. Количество вновь сформированной кости равно разрушенной (нулевой баланс). Механизм взаимодействия («сцепления») процессов резорбции и формирования кости не изучен. Процесс ремоделирования регулируется системными и локальными факторами, механическими воздействиями. Системное воздействие оказывают гормоны: гормон роста, ПТГ, тироксин, трийодтиронин, кальцитонин, инсулин, половые гормоны, кальцитриол. В гомеостазе кальция участвуют ПТГ, кальцитонин, половые гормоны, ГК, кальцитриол. Действие системных факторов на костную ткань опосредуется через локальные (цитокины, ФР, ПГ). Факторы местной (локальной) регуляции процесса резорбции кости представлены в табл. 2.7.
Выделяют остеоцитарное ремоделирование (ограничено прелакунарной областью), остеобластно-остеокластное (обеспечивает изменение структуры костного органа, формы, размеров). А.С. Аврунин и соавторы (1998) отмечают несколько этапов остеобластно-остеокластного ремоделирования:
1) формирование участков активного ремоделирования;
2) резорбция костного матрикса;
3) формирование органического матрикса;
4) формирование минерального матрикса.
В цикле ремоделирования также выделяют фазы активации, резорбции, реверсии, формирования и покоя (Frost H.M., 1973; Baron R., 1977). Возникновение зоны активного ремоделирования начинается с приема сигнала локальной группой выстилающих клеток (происходят от остеобластов) или остеоцитов, в результате чего начинается резорбция костной ткани остеокластами, продолжающаяся от 2 до 4 нед. Для активации остеокластов необходимо присутствие остеобластов. После резорбции остеобласты в течение 34 мес образуют новую костную ткань. Как свидетельствуют приведенные А.С. Авруниным и соавторами (1998) сведения, резорбция костного матрикса происходит в три этапа.
На первом этапе происходит дифференцировка мононуклеаров гемопоэтического происхождения (попадающих сюда через сосуды) в остеокласты после контакта наружной мембраны моноцитов с костным матриксом. Циркулирующие мононуклеарные клетки образуют скопления, сливаются друг с другом и формируют остеокласты (Tran Van P. et al., 1982). Миграцией управляют хемотаксические факторы: ИПФР-1, макрофагальный воспалительный белок-1α. В регулировании остеокластной резорбции костной ткани главную роль играют клетки остеобластной линии. Остеокласты выделяют различные семейства интегринов — β1, β2, α2, α5, αV. Контакт остеокластов с костным матриксом обеспечивают интегрины αV, β3, его адгезию — связи с остеопонтином. Адгезию регулируют Са2+, ПГ, ИЛ-4, -13, перекись водорода и др. ПГ могут стимулировать и угнетать резорбцию. Так, ПГЕ2 усиливает дифференцировку остеокластов, резорбцию посредством цАМФ и угнетает активность остеокластов также через цАМФ. Перекись водорода регулирует формирование и резорбтивную функцию остеокластов. Стимуляция резорбции перекисью водорода дозозависима. ИЛ-4 и -13 угнетают резорбцию, ингибируя синтез ПГ в остеобластах. После контакта с костной тканью и процесса дифференцировки остеокласты выделяют лизосомальные ферменты, которые разрушают основное вещество и фибриллы коллагена (кислая фосфатаза и ЩФ, коллагеназа I и IV типа, катепсины В, С, Д, L, К, арилсульфатаза, β-глюкуронидаза, β-глицерофосфатаза, стромализин). В физиологических условиях синтез ферментов ограничивают их ингибиторы. Они регулируют активность металлопротеиназ — коллагеназы, стромализина, желатиназы. В зоне резорбции накапливается углекислота, способствующая растворению минералов. При участии карбоангидразы Н+ взаимодействует с СО2. Высокая активность Н+-АТФазы в щеточной каемке мембран остеокластов обеспечивает достаточное количество Н+ для резорбции костной ткани. В растворении минералов участвует имеющаяся в зоне резорбции лимонная кислота. На процесс резорбции влияют продукты распада костного матрикса (детрит органического матрикса, коллагена I типа) и уровень внеклеточного кальция: его высокий уровень предотвращает прилипание остеокласта. В активации остеокласта в начале резорбции участвуют свободные радикалы кислорода, вырабатываемые остеокластами (Hall T. еt al., 1995). В процессе сложной регуляции различные факторы (табл. 2.8) дополняют и дублируют друг друга. Образующиеся фрагменты костного матрикса фагоцитируются остеокластами и перевариваются.
На стадии резорбции в губчатой кости появляются углубления до 40 мкм (Eriksen E.F. et al., 1985; 1990) , в кортикальной кости — конусовидные пустоты 2,5 мм длиной и 150 мкм в диаметре (Agerbaek M.O. et al., 1991). Средняя продолжительность фазы резорбции в губчатой части подвздошной кости составляет 42 дня, в кортикальном слое — 27 дней (Agerbaek M.O. et al., 1991; Eriksen E.F., 1986). В заключительной фазе резорбции появляются преостеобласты, начинает формироваться костная ткань. Эта фаза реверсии (или переходной период) составляет 9 дней для губчатой кости и 4 дня для кортикальной (Baron R., 1977). Механизмы фазы реверсии не изучены. Вначале резорбция осуществляется многоядерными остеокластами, а завершается мононуклеарными клетками.
Из мигрирующих в полость резорбции клеток-предшественников дифференцируются остеобласты, которые синтезируют компоненты органического матрикса (коллаген, протеогликаны, неколлагеновые белки — ФР, костные морфогенные белки и др.). В результате биосинтеза и экскреции коллагенового белка, протеингликанов, агрегации молекул формируется волокнистая основа костной ткани (примерный путь агрегации молекул коллагена: молекулы коллагена > протофибриллы > микрофибриллы > коллагеновые фибриллы > волокна). Параметры коллагеновых волокон зависят от биомеханических функций костного органа, химического состава окружающей среды, свойств компонентов. Как отмечают А.С. Аврунин и соавторы (1998), условия, в которых образуется коллагеновый матрикс, записываются в его структуре. Секреция белков костного матрикса достигает максимума к периоду полового созревания. К четвертому десятилетию жизни их секреция снижается и изменения в составе матрикса являются одним из факторов, способствующих склонности к переломам, усиливающейся с возрастом. Образовавшийся органический (коллагеновый) матрикс подвергается дальнейшей минерализации. Процесс минерализации остеоида начинается через 25 дней в губчатой и через 35 — в кортикальной кости (Parfitt A.M., 1990). Доставка неорганического фосфата через плазматические мембраны регулируется ПТГ, ПТГ-связывающим белком, ИПФР-1, ТрФР. Накапливание фосфатов в матриксных пузырьках обусловливает индукцию минерализации. Гормональные и другие факторы (в том числе ионы кальция, неорганического фосфата) могут приводить к индукции минерализации посредством цАМФ. Матриксные пузырьки продуцируются остеобластами и содержат липиды, кальций, пирофосфатазу, ЩФ. Пирофосфатаза и ЩФ разрушают ингибиторы кальцификации и гидролизуют фосфорные эфиры с освобождением свободных фосфатов. Из кальция и фосфата образуются аморфные фосфатно-кальциевые комплексы, которые трансформируются в гидроксиапатит. Рост кристаллов приводит к разрыву мембран матриксных пузырьков. Период между формированием органического матрикса и его минерализацией составляет 8–12 дней. Процесс появления минеральных структур подавляют протеогликаны, ингибирующая активность которых определяется степенью их сульфатирования. Минерализация органического матрикса происходит после гидролиза протеогликанов. Образование ядер кристаллизации регулирует сиалопротеин (располагается в межфибриллярных промежутках), формирование правого кристалла — остеопонтин, размеры и скорость образования кристаллов — остеокальцин и остеонектин. На основании приведенных выше сведений А.С. Аврунин и соавторы (1998) отмечают, что строение минерального матрикса определяется структурой органического.
Ремоделирование кости начинается у плода и продолжается всю жизнь. Оно контролируется геномом. Размеры и массивность скелета определяют:
- реализованная в течение периода роста скелета генетическая программа;
- гормональный статус (прежде всего продукция половых гормонов);
- питание;
- механическая нагрузка.
Отложение в субпериосте остеоида приводит к расширению кости, удлинение обеспечивается за счет площадки ростового хряща. Особенно интенсивно рост и обновление костей (ремоделирование) происходят в детском возрасте. В каждом цикле ремоделирования костный баланс является положительным в период роста скелета, в молодом возрасте, как в губчатой, так и кортикальной кости. Объем губчатой кости в гребне подвздошной кости мужчин и женщин достигает пика во второй декаде жизни. Максимальная величина костной массы (пиковая костная масса) в результате нормального роста достигается в третьей декаде жизни. Она имеет резервное значение и в последующем определяет устойчивость или склонность к переломам. Губчатая костная ткань метаболически более активна и быстрее ремоделируется, чем компактная (ежегодно обновляется 45% компактной и 25% губчатой кости). С возрастом (после 35 лет) темпы ремоделирования костей снижаются, исчезает поверхность ростового хряща, увеличивается наружный и внутренний диаметр кости. По мере старения (особенно у женщин в постменопаузальный период) уменьшается количество матрикса, синтезируемого остеобластами, происходит потеря трабекул. После 35 лет трабекулы истончаются в среднем на 0,6% (1 мкм) в год (Weinstein R.S. et al., 1987; Mellish R.W.E. et al., 1989; Parfitt A.M., 1992). Баланс ремоделирования кости становится отрицательным. Потеря костной массы ускоряется после менопаузы, происходит полное разрушение трабекулярных пластин, нарушение структуры трабекулярной решетки. С возрастом происходит потеря и кортикальной кости с ускорением процесса в постменопаузальный период. Кортикальный слой истончается, увеличивается его порозность. В пожилом и старческом возрасте развиваются атрофические процессы в костной ткани. Эндостальный диаметр кости увеличивается с большей скоростью, чем периостальный, уменьшается толщина кортикального слоя. Ширина кортикального слоя считается достоверным прогностическим показателем МП относительно переломов проксимального отдела бедренной кости, поясничных позвонков. Уменьшение костной массы сопровождается нарушением микроархитектоники кости.
При остеопорозе дисбаланс ремоделирования возникает раньше всего (и преимущественно) в губчатой кости. С возрастом человека время, необходимое для завершения ремоделирования КРЕ, увеличивается. Резорбция начинает превалировать над процессом образования кости, уменьшается костная масса. К 85 годам у женщин костная масса кортикальной костной ткани в среднем уменьшается на 30%, трабекулярной — на 50%, у мужчин соответственно на 20 и 35% (Fransis R.M. et al., 1998). Темп потери костной ткани более выражен у женщин: в возрасте старше 35 лет они теряют 0,75–2,5% костной массы в год, после наступления менопаузы — 34%. У мужчин в возрасте старше 50 лет костная масса ежегодно уменьшается на 0,4–1,2%. Эстрогены и тестостерон снижают продукцию ИЛ-6. При овариоэктомии, орхиэктомии, менопаузе повышается продукция ИЛ-6 и резорбция кости. При использовании антирезорбентов (эстрогены, кальцитонин, бисфосфонаты) происходит нестойкое общее увеличение на 34% скелетного кальция, МПКТ при высоком темпе ремоделирования. Если угнетение резорбции прекратить, костная масса возвращается к исходному уровню. При условии перестройки кости с низкой скоростью увеличение костной массы будет продолжаться. Характер влияния различных факторов на скорость ремоделирования костной ткани представлен в табл. 2.9.
Строение живой кости меняется в зависимости от механической нагрузки (Бэссет А., 1971). В ответ на механическую нагрузку изменяется ориентация структурных элементов кости и масса составляющего ее вещества (матрикс, минеральные компоненты перестраиваются по линиям механического напряжения). Согласно закону Ю. Вольфа «при данной форме кости ее элементы перестраиваются в зависимости от направления функциональной нагрузки, а увеличение или уменьшение их массы отражает величину этой нагрузки». Способность костей к подобным изменениям объясняют пьезоэлектрическими свойствами: при механической нагрузке в них возникает электрический ток. В основе явления, описываемого законом Вольфа, лежит принцип обратной связи. Кость обладает выраженными свойствами кристалла: при ее механической деформации возникает электрический ток. На поверхности кристаллов существует электрический двойной слой (Гельмгольца), который, связывая воду, образует поляризованный монослой (Ньюман У., Ньюман М., 1961). При контакте клеточных мембран с кристаллами меняются их функция и клетки в целом (Финеан Дж. и соавт., 1977). По мнению А. Бэссета (1971), теоретически электричество в кости может возникнуть в результате любого из трех способов (или всех трех):
1) напряжения или изгиба коллагеновых волокон;
2) изгиба молекул мукополисахаридов;
3) напряжения на поверхности соприкосновения коллагена и гидроксилапатита.
Данные этого исследования свидетельствуют, что источники электричества — множество точек соприкосновения кристаллов гидроксиапатита и коллагена. Возникающий электрический заряд избирательно притягивает, отталкивает или ориентирует в определенном порядке другие заряженные молекулы и ионы, находящиеся в непосредственной близости. Характер электрических импульсов, регистрируемых в кости, варьирует в зависимости от частоты, мощности и длительности деформирующего воздействия. Кроме пьезоэлектрических потенциалов, механизмы трансформации механического усилия в биохимическую стимуляцию кости включают высвобождение ПГ, увеличение притока крови в кости, механизмы, опосредованные гормонами. При увеличении физической нагрузки остеоциты повышают синтез бета-актина, остеокальцина, ИПФР-1 и других факторов (D. Mason et al., 1996), также перестраивается матрикс, окружающий остеоцит. Существует несколько видов нормальной физической нагрузки на скелет: гидростатическое давление крови, сокращение всего сердца, сила тяжести, попеременное сокращение мышц, ходьба. Имеется корреляция плотности кости с мышечной массой, силой, максимальным потреблением кислорода.
Таким образом, для нормального развития и функционирования костной ткани, кроме регулирующего влияния системных и локальных факторов, большое значение имеет физическая нагрузка. С помощью физических упражнений можно снизить скорость потери костной ткани. Существует зависимость между физической активностью и возрастными изменениями МПКТ. Повышенная нагрузка различного типа сопровождается повышением МПКТ. В зрелом возрасте физическая активность уменьшает потерю костной массы, обусловленную возрастом. Обратный процесс отмечается при снижении нагрузки. Физические перегрузки вызывают локальные расстройства кровообращения, нарушения циркуляции жидкости в костной ткани. Уменьшение костной массы при избыточной физической нагрузке у женщин объясняют нарушениями эндокринной регуляции (снижением функции яичников), дефицитом массы тела. В период роста значение генетической программы больше, чем эффект физической нагрузки. Формирование пика костной массы генетически детерминировано (Slemenda W. et al., Kanis J.A., 1994). Генетический фактор определяет 50–80% плотности костной массы. Для пиковой костной массы важное значение имеет масса тела, от которой зависит величина нагрузки на скелет. В постменопаузальный период избыток массы тела частично компенсирует дефицит эстрогенов. Дефицит костной массы особенно значителен при сочетании дефицита массы тела и дефицита эстрогенов. При анорексии уменьшение костной массы связано с продолжительностью дефицита массы тела.
Костные структуры отличаются метаболической индивидуальностью (Аврунин А.С. и соавт., 1998). Распределение и количество неколлагеновых белков в костном матриксе зависит от типа кости (Bonucci E., Silvestrini G., 1996). По мнению указанных исследователей, функциональные особенности костных органов, характер метаболизма определяют их структуру и форму. Каждая кость имеет свои фенотипические особенности метаболизма и на одни и те же регуляторы кости реагируют по-разному. Условия, в которых протекает ремоделирование кости, определяют ее структуру. При нарушении регуляции ремоделирования устойчивость структуры костного матрикса нарушается.