В настоящее время идентификация фенотипа липидного спектра плазмы крови является важнейшим стратегическим подходом не только при диагностике тех или иных форм нарушений липидного обмена, но и в анализе риска возникновения кардиоваскулярных событий (Cupples L.A., D’Agostino R.B., 1987; Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults, 2001). Вместе с тем, существуют определенные сложности в интерпретации полученных данных, касающихся содержания липидов и их фракций у лиц, не получающих специфического лечения, и у пациентов, придерживающихся назначений врача о приеме гиполипидемических лекарственных средств (Jackson R. et al., 2005; Ridker P.M. et al., 2007). Современные системы оценки кардиоваскулярного риска не всегда корректны в отношении идентификации величины последнего у лиц с изолированной гиперлипидемией, гипер-бета-липопротеидемией, гиперхолестеринемией при высоком уровне ЛПВП и сохраненной концентрации ЛПНП и т.п (Jackson R., 2000; Conroy R.M. et al. for the SCORE Project Group, 2003; De Backer G. et al. for the Third Joint Task Force of European and Other Societies on Cardiovascular Disease Prevention in Clinical Practice, 2003; British Cardiac Society, 2005; Menotti A. et al., 2005; Hippisley-Cox J. et al., 2007; Ridker P.M. et al., 2007; Woodward M. et al. for the SIGN Group on Risk Estimation, 2007). Все это вызывает необходимость проведения более детального анализа фенотипа гиперлипидемии (особенно содержания аполипопротеинов), ассоциированного с оценкой семейного анамнеза и иногда — генетическими исследованиями (Ferrario M. et al., 2005; Zhang X.F. et al., 2005; Beswick A., Brindle P., 2006). Тем не менее, в рутинной практике принято ограничиваться стандартными процедурами, позволяющими сократить расходы на проведение исследований и увеличить число вовлекаемых в программы первичной превенции лиц из неорганизованной популяции (Brindle P. et al., 2006). Все это не умаляет значения современных технологий, направленных на идентификацию генотипа и фенотипа гиперлипидемии. Однако проведение подобных исследований в полном объеме, вероятно, является стратегическим планом на будущее.
Классификация
Одной из наиболее успешных классификаций нарушений липидного спектра плазмы крови, основанных на фенотипическом признаке, стала система Фредриксона (табл. 4.1).
Таблица 4.1 Классификация дислипидемий по Фредриксону/ВОЗ
| Тип дислипидемии | Повышение содержания липопротеидов | Повышение содержания ХС | Атерогенность |
| I | Хиломикроны | ТГ, ХС | Отсутствует |
| IIa | ЛПНП | ХС (может быть нормальным) | Резко увеличена |
| IIb | ЛПНП, ЛПОНП | ТГ, ХС | Резко увеличена |
| III | ЛПОНП, ремнанты хиломикронов | ТГ, ХС | Резко увеличена |
| IV | ЛПОНП | ТГ, ХС (может быть нормальным) | Вероятно увеличена |
| V | Хиломикроны, ЛПОНП | ТГ, ХС | Не выяснена |
Указанная концепция руководствовалась представлениями о том, что независимо от причины и формы гиперлипидемий соотношение фракций липидов и субфракций липопротеидов в плазме крови в значительной мере обусловливает атерогенность и прогностическое значение детектируемого метаболического нарушения. Более подробно о причинах формирования гиперлипидемий и их фенотипических особенностях изложено в главе 6. В то же время, идентификация фенотипических особенностей гиперлипидемии по Фредриксону требовала анализа основных фракций липидов, уровень содержания которых в плазме крови в последующем подвергался интерпретации, часто по практическим соображениям. Именно это возражение и предопределило судьбу концепции Фредриксона, которую, несмотря на немаловажные заслуги, в настоящее время рекомендуют в основном для использования при проведении научных исследований.
Основные фракции липидов и липопротеидов плазмы крови
Хиломикроны — самые крупные липопротеидные частицы. Хиломикроны обогащены ТГ, содержат апо-В48-протеин в качестве главного структурного белка и транспортируют экзогенные (пищевые) жиры и ХС из кишечника в печень и периферические ткани. Они образуются в эндоплазматическом ретикулуме кишечника, секретируются в лимфу и затем через грудной проток попадают в кровь. Период полужизни хиломикрона составляет 5–20 мин. Плазма крови здоровых людей, при взятии крови натощак, практически не содержит хиломикронов. После секреции хиломикроны получают апопротеины классов Е, С-I, C-II и C-III от ЛПВП. В кровотоке под действием фермента липопротеинлипазы, связанной с протеогликанами эндотелиальных клеток и активируемой с помощью апопротеина C-II, происходит гидролиз ТГ в составе хиломикронов. При этом последние подвергаются так называемому метаболическому ремоделированию с образованием остатков (ремнантов), которые имеют плотность, соответствующую таковой у ЛПОНП и ЛППП. Ремнанты хиломикронов, содержащие апо-В48-липопротеин и апо-Е-липопротеин, захватываются гепатоцитами с помощью рецепторов, имеющих высокое сродство к последнему.
ЛПОНП содержат около 55% ТГ, 19% ХС и 8% белка (апопротеинов В100, Е, С-I и C-II). Этот класс липопротеидов синтезируется в печени и является главной транспортной формой эндогенных ТГ и ХС. Благодаря ЛПОНП из печени выводятся ремнанты хиломикронов и ТГ, образовавшиеся из СЖК плазмы крови. Синтез ЛПОНП прямо коррелирует с повышением содержания СЖК в гепатоцитах. Это происходит при поступлении в организм больших количеств жиров с пищей, а также в случаях повышенного высвобождения адипоцитами СЖК непосредственно в кровоток, что отмечают при ряде заболеваний (ожирение, сахарный диабет, резистентный к терапии). Аполипопротеин C-II на поверхности ЛПОНП активирует эндотелиальную липопротеинлипазу, которая расщепляет ТГ до СЖК и глицерина, которые в последующем используются жировой тканью, миокардиоцитами и скелетными миоцитами в качестве энергетического субстрата. Остатки ЛПОНП превращаются в ЛППП, которые затем частично выводятся из кровотока гепатоцитами, а также могут быть трансформированы в ЛПНП.
ЛПНП — мелкие частицы, которые являются основной транспортной формой ХС. Они содержат около 6% ТГ, 50% ХС и 22% белка. Примерно две трети быстрообменивающегося пула ХС синтезируется в организме, преимущественно в печени (эндогенный ХС) и одна треть поступает в организм с пищей (экзогенный ХС). Ключевым (критическим) ферментом, определяющим скорость синтеза эндогенного ХС, является ГМГ-КоА-редуктаза. ЛПНП являются продуктом метаболизма ЛОНП и ЛППП, которые содержат наибольшее количество ХС. Около 40–60% всех ЛПНП захватываются гепатоцитами при участии аполипопротеина В и липопротеинлипазы печени. Второй путь катаболизма ЛПНП — свободнорадикальное перекисное окисление липидов, в результате которого образуются модифицированные ЛПНП. Последние захватываются макрофагами, которые трансформируются в пенистые клетки, входящие в состав атеромы. Кроме того, модифицированные ЛППП вызывают повреждение сосудистого эндотелия, формируя дисфункцию последнего, которая рассматривается как один из ключевых этапов возникновения кардиоваскулярных событий.
ЛППП — представляют собой ремнанты хиломикронов и ЛПОНП, содержащие большое количество ХС. ЛППП захватываются гепатоцитами или метаболизируются под влиянием липазы печени до ЛПНП (содержат апопротеин В).
ЛПВП — самые мелкие и плотные частицы липопротеинов. Они содержат 5% ТГ, 22% ХС, 40% аполипопротеинов А-I, A-II и С и относятся к липопротеидам, обладающим антиатерогенными свойствами. Основной функцией ЛПВП является обратный транспорт ХС из периферических органов, с поверхности хиломикронов и ЛПОНП, макрофагов и гладкомышечных клеток в печень, где происходят его утилизация и превращение в желчь. Синтез полноценных ЛПВП происходит при обязательном участии ХС, ЛПОНП и ЛПНП в энтероцитах и гепатоцитах. ЛПВП обладают потенциальным антиатерогенным действием. Последнее связывают с их активным участием в метаболизме ХС и ТГ, а также со стимуляцией простациклина, угнетением агрегации тромбоцитов, уменьшением выраженности активации эндотелиоцитов, уменьшением экспрессии на них молекул клеточной адгезии и т.п. Факторами, способствующими снижению уровня ЛПВП, являются: принадлежность к мужскому полу; ожирение; гипертриглицеридемия; высокое потребление углеводов; диабет у взрослых; курение. Высокий уровень антиатерогенных ЛПВП связывают с принадлежностью к женскому полу; высоким уровнем эстрогенов; высокой физической активностью; уменьшением массы тела; а также с умеренным потреблением алкоголя.
ЛП (а) — близок по своим физико-химическим свойствам к aпo-B100-содержащим ЛПНП, отличаясь от них наличием в оболочке дополнительного белка — aпo (а), что оказывает влияние на особенности его метаболизма и клиренса. Молекулярная масса ЛП (а) составляет 300–600 кДа, синтез ЛП (а) осуществляется в гепатоцитах путем присоединения цепочки апо-А к апо-В посредством дисульфидной связи (Kochinsky M.L., Marcovina S.M., 2004). В результате каждая молекула ЛП (а) состоит из одной молекулы апо-В100-липопротеина, одной молекулы апо-А-липопротеина, который структурно отличается от аналогичных молекул наличием углеводного остатка, придающего ему гидрофильные свойства, и содержанием четырех доменов (KIV типа 1, 3, 5 и 10), один из которых представляет собой протеазу, аналогичную плазминогену, а также липидного ядра, в состав которого входят эстерифицированный ХС и ТГ (рис. 4.1). Каждый домен состоит из 80–85 аминокислотных остатков с молекулярной массой 10 кДа, которые придают структуре ЛП (а) гетерогенность.
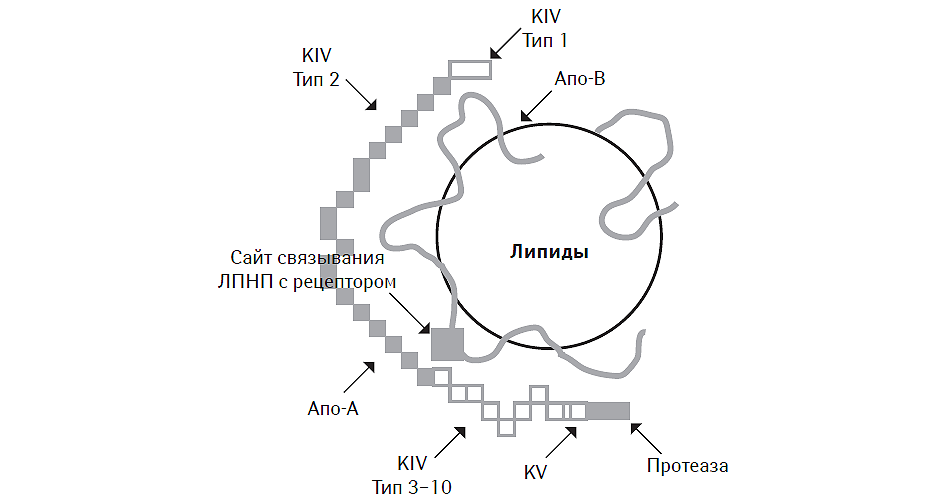
Рис. 4.1. Молекулярная структура ЛП (а)
Секреция ЛП (а) происходит немедленно после его синтеза. Циркулирующий ЛП (а) непосредственно не участвует в транспорте липидов и его метаболизм осуществляется независимо от деградации апо-В-содержащих липопротеинов.
По своим свойствам ЛП (а) близок к плазминогену и поэтому может конкурировать с ним за места связывания на фибрине, ингибируя, таким образом, фибринолитическую активность крови (Tsimikas S. et al., 2005; Kathiresan S., 2009), повышая риск возникновения тромбозов (Caplice N.M. et al., 2001). Интересно, что даже в физиологических условиях содержание ЛП (а) в плазме крови может зависеть от расовых, гендерных и возрастных различий (Marcovina S.M. et al., 1993). Так, у представителей негроидной расы уровень ЛП (а) всегда выше по сравнению с представителями белой расы, а у женщин ниже, чем у мужчин (Marcovina S.M. et al., 1996; Paultre F. et al., 2000). Это может стать отражением существования определенных генетических различий (Sandholzer C. et al., 1991; Rubin J. et al., 2002). Более того, у представителей различных рас обычно идентифицируются даже разные изоформы ЛП (а), ассоциированные с различной молекулярной массой апо-А-липопротеина и величиной ее молекулы (длинная или короткая цепь) (Parra H.J. et al., 1987) (рис. 4.2). Так, представителям негроидной расы более свойственны молекулы ЛП (а) с меньшей молекулярной массой, обладающие большей атерогенностью (Wu H.D. et al., 2004). Последняя обусловлена в основном способностью к интенсификации окисления фосфолипидов, индукции синтеза макрофагами провоспалительных цитокинов (таких как ИЛ-18), фиксации в тканях модифицированных ЛПНП и потенциации дисфункции эндотелия (Thillet J. et al., 1998; Ariyo A.A. et al., 2003; Bergmark C. et al., 2008). Кроме того, ЛП (а) способен ингибировать активность тканевого фактора роста, повышать окислительную модификацию фосфолипидов и ХС в составе ЛПНП (Grainger D.J. et al., 1994; Chasman D.I. et al., 2009).
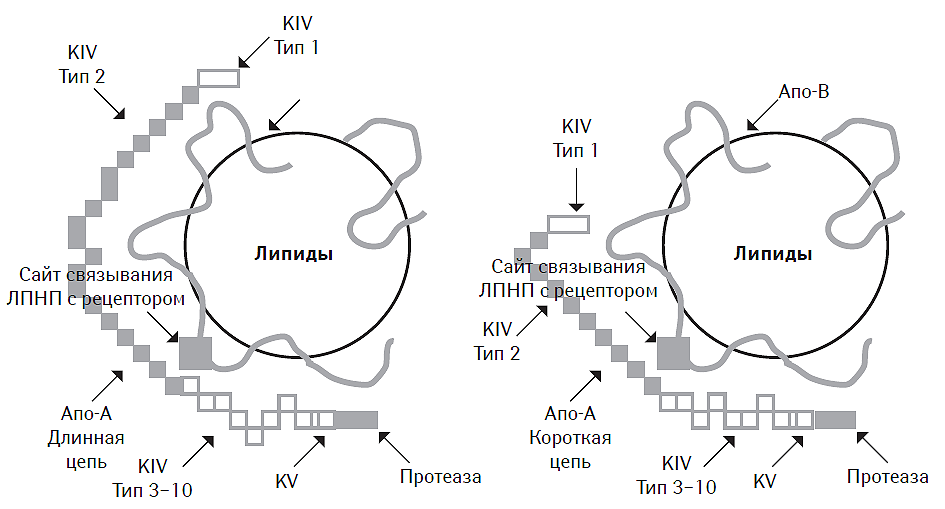
Рис. 4.2. Структура двух основных изоформ ЛП (а)
ЛП (а) относится к числу атерогенных липопротеинов, повышенный уровень которого в крови почти всегда ассоциируется с развитием атеросклероза, возникновением новых случаев ишемической болезни сердца (ИБС), инфаркта миокарда, острого коронарного синдрома, инсульта, транзиторной ишемической атаки и высоким риском иных атеротромботических осложнений, а также с некардиоваскулярной смертностью (Reuterwall C. et al., 1999; Berglund L., Ramakrishnan R., 2004; Smolders B., Lemmens R., Thijs V., 2007; Kamstrup P.R. et al., 2009; The Emerging Risk Factors Collaboration, 2009). Установлена также взаимосвязь между уровнем ЛП (a) и риском манифестации аневризмы брюшного отдела аорты (Takagi H. et al., 2009). В целом ЛП (a) рассматривается как фактор риска возникновения прежде всего ИБС, особенно у лиц с повышенным содержанием ЛПНП в плазме крови. Вместе с тем, указанная взаимосвязь имеет клиническое значение только тогда, когда содержание ЛП (a) очень высокое (Danesh J. et al., 2000). С другой стороны, в метаанализе B. Smolders и соавторов (2007), продемонстрировавшем наличие ассоциации между плазменной концентрацией ЛП (a) и риском возникновения мозгового инсульта, не было получено подтверждения об изменении этих взаимоотношений в зависимости от плазменного содержания ЛПНП. Кроме того, по данным исследования ARFY (Atherosclerosis Risk Factors in Female Youngsters), в когорте молодых женщин содержание ЛП (a) является одним из наиболее валидных предикторов высокого риска возникновения раннего атеросклероза и атеротромботических событий (Knoflach M. et al., 2009). Аналогичные данные были получены для детей и подростков с ожирением (Nascimento H. et al., 2009).
Интересно, что три региона 9-й хромосомы 6q26–27, 9p21 и 1p13 продемонстрировали сильную ассоциацию с риском возникновения новых случаев ИБС в популяции (McPherson R. et al., 2007; Broadbent H.M. et al., 2008). Причем с локусом 6q26–27 связана высокая концентрация ЛП (а), негативное влияние которой на отдаленный прогноз было продемонстрировано в ходе специально спланированного испытания PROCARDIS (Precocious Coronary Artery Disease) (PROCARDIS Consortium, 2004; Clarke R. et al., 2009).
Аполипопротеины
Все липопротеиды имеют сходную структуру и состоят из центральной части (ядра), содержащей нерастворимые в воде липиды (эфиры, ХС, ТГ, СЖК), и из оболочки, состоящей из особых белковых молекул (апопротеинов) и растворимых в воде липидов — неэстерифицированного ХС и фосфолипидов. Молекулы апопротеинов имеют неполярный гидрофобный участок, который связан с липидами, и полярный гидрофильный участок, расположенный на поверхности сферической частицы апопротеинов и обращенный к окружающей апопротеин жидкой среде (плазме крови). Гидрофильный участок апопротеина образует водорастворимые связи с молекулами воды. Такая структура липопротеидов определяет их амфифильные качества.
Характеристика основных апопротеинов и энзимов, принимающих участие в метаболизме липидов, представлены в табл. 4.2 и 4.3.
Таблица 4.2 Основные аполипопротеины, принимающие участие в метаболизме липидов
| Липопротеины | Локализация | Функция |
| Апо-А-I | Хиломикроны, ЛПВП, ЛПНП | Активация ЛХAT, активация ЛПВП-зависимого реверсивного транспорта ХС |
| Апо-А-II | Хиломикроны, ЛПВП, ЛПНП, ЛП (a) | |
| Апо-А-IV | Хиломикроны, ЛПВП, ЛПНП | |
| Апо-В100 | ЛПНП, ЛППП, ЛПОНП, ЛП (a) | Лиганд рецептора ЛПНП |
| Апо-Е | Хиломикроны, ЛПОНП, ЛПНП | Лиганд рецептора ЛПНП |
| Апо-С1 | Хиломикроны, ЛПВП, ЛПОНП, ЛПНП | Активация ЛХAT, активация ЛПВП-зависимого реверсивного транспорта ХС |
| Апо-С2 | Стимуляция липопротеинлипазы | |
| Апо-С3 | Ингибирование взаимодействия с рецепторами на поверхности гепатоцита | |
| Апо-D | Специфичный для ЛПВП представитель семейства липокаинов | Лиганд рецепторов ЛПВП, участвует в регуляции механизмов реверсивного транспорта ХС |
| Апо-F | ЛПВП, ЛПНП, ЛПОНП | Ингибирует протеин, транспортирующий ХС, регулятор активности протеина, эстерифицирующего ХС (СЕТР) |
| Апо-H, | ЛПВП, ЛПНП | Неизвестна |
| Апо-L | ЛПВП | Неизвестна |
| Апо-М | Более специфичный для ЛПВП, реже для ЛПНП, представитель семейства липокаинов | Интегральный липопротеин |
| Апо (а) | ЛП (a) | Неизвестна |
Таблица 4.3 Основные энзимы, принимающие участие в метаболизме липидов
| Энзимы | Локализация | Функция |
| АВСА1 | Внутриклеточно | Внутриклеточный транспорт ХС в направлении мембраны |
| СЕТР | ЛПНП | Модулирует перемещение эфиров ХС от ЛПНП к ЛПОНП |
| ЛХАТ | ЛПНП | Эстерификация свободного ХС для последующего транспорта и фиксации на ЛПНП |
| Хиломикроны, ЛПНП, ЛПОНП | Лиганд рецепторов ЛПНП | |
| Апо (а) | ЛП (a) | Неизвестна |
Апо-А и апо-В фенотипы
Аполипопротеины A-I и B являются структурными компонентами ЛПВП, ЛПНП и ЛПОНП. Основной функцией апо-В-содержащих липопротеидов является осуществление транспорта липидов из гепатоцитов в сайты утилизации. Напротив, апо-А1-содержащие липопротеиды осуществляют реверсивный транспорт липидов из периферических тканей в гепатоциты (Packard C.J., Shepherd J., 1997; Karpe F., 1999).
Основные направления метаболизма липопротеидов представлены на рис. 4.3. Обогащенные ТГ хиломикроны поступают в системный кровоток из воротной вены. Основными транспортными формами хиломикронов являются апо-А1- и апо-В48-липопротеины. Последний представляет собой N-терминальный фрагмент апо-B100 и подвергается быстрой метаболической деградации в ремнантные формы, транспортируемые в гепатоциты. ТГ в составе хиломикронов гидролизируются липопротеинлипазой с высвобождением СЖК. Последние затем эстерифицируются повторно с образованием нейтрального жира, который депонируется в адипоцитах. У здоровых лиц натощак своеобразным циркулирующим депо ТГ являются ЛПОНП1, которые секретируются гепатоцитами. ЛПОНП1 подвергаются быстрой конверсии с помощью липопротеинлипазы в ЛПОНП2, имеющие более мелкие размеры, а последние — в ЛППП. Конечный этап трансформации ЛППП в ЛПНП катализирует печеночная липаза (Packard C.J., Shepherd J., 1997). Уровень апо-В-липопротеинов в плазме крови в значительной мере отражает кумулятивное содержание каждого из вышеуказанных видов липопротеидов, а также тесно зависит от их синтеза и клиренса.
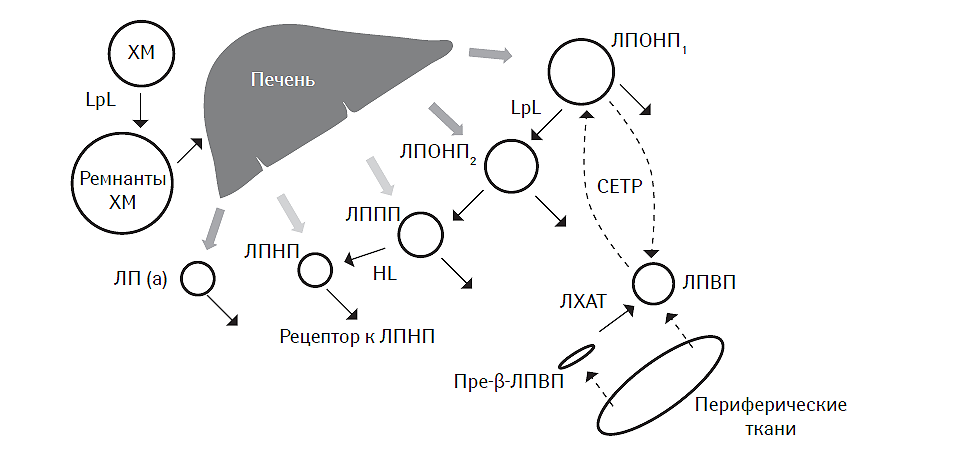
Рис. 4.3. Основные метаболические пути синтеза и деградации липопротеинов ХМ — хиломикроны, HL — печеночная липаза, LpL — липопротеинлипаза.
Апо-A-I-липопротеины являются важнейшим структурным компонентом ЛПВП, главная функция которых в основном сводится к трансферу ХС от поверхности клеточных мембран к частицам липопротеидов, а также к активации ферментных систем, эстерифицирующих ХС в плазме крови — ЛХАТ (LCAT). Обратный транспорт ХС из периферических тканей осуществляется посредством специфических рецепторов, таких как ABCA1 и ABCG1 (Rye K.A. et al., 1999; Wong N. et al., 2004; Lewis G.F., Rader D.J., 2005). Молекула ХС, включаемая в состав ЛПВП, первоначально подвергается оксидации посредством воздействия ЛХAT с образованием эстерифицированного ХС. В результате этого процесса осуществляется структурная модификация молекулы липопротеида, ассоциированная с изменением ее дисковидной пространственной конфигурации в сферическую, что способствует изоляции липопротеида в гидрофобном окружении. Эстерифицированный ХС в составе ЛПВП под действием белка-переносчика эстерифицированного ХС (CETP) трасформируется в апо-B-содержащие липопротеины (ЛПОНП или ЛПНП) и транспортируется в гепатоциты. Кроме того, возможен непосредственный трансфер эстерифицированного ХС из ЛПВП в гепатоциты посредством рецепторного механизма, осуществляемого с помощью молекулы SR-B1, экспрессируемой на мембране гепатоцита (Lewis G.F., Rader D.J., 2005).
Плазменное содержание апо-B-липопротеина модулируется достаточно большим спектром различных молекул, ферментных систем и других липопротеинов, вовлеченных в процессы транспорта, ретенции, клиренса и ресинтеза ХС и ТГ. При проведении эпидемиологических исследований было установлено, что между риском возникновения ИБС в общей популяции и уровнем апо-B-содержащих липопротеидов имеется прямая и тесная корреляционная взаимосвязь. Причем все липопротеиды проявляют больший или меньший проатерогенный эффект в зависимости от количества содержащегося в них апо-В (табл. 4.4).
Таблица 4.4 Характеристика апо-В-содержащих липопротеидов плазмы крови
| Липопротеид | Концентрация апо-В, мг/дл | Атерогенность |
| Апо-В48-содержащие липопротеины | ||
| Хиломикроны | <0,5 | ? |
| Ремнанты хиломикронов | 1,0–2,0 | ++ |
| Апо-В100-содержащие липопротеины | ||
| ЛПОНП | 5,0 | + |
| Ремнанты ЛПОНП | 2,0–5,0 | +++ |
| ЛППП | 15,0 | +++ |
| ЛПНП | 60,0 | +++ |
| ЛП (a) | 5,0 | + |
Таблица 4.5 Полиморфизм основных апопротеинов (по данным Boren J. et al., 2001; Segrest J.P. et al., 2001; Nissen S.E. et al., 2003; O’Bryan M.K. et al., 2004; Shiflett A.M. et al., 2005; Barter P.J., Rye K.A., 2006; Dahlback B., 2006)
| Тип апопротеина | Полиморфизм | Клиническое значение |
| Апо-А-I | Есть А1, А2, А3, А4; наследуется кодоминантно | Дефицит связан с ранним развитием атеросклероза, высокой частотой манифестации ИБС |
| Апо-A-II | Нет | Самостоятельное значение не установлено |
| Апо-А-III | Нет | |
| Апо-А-IV | Есть | Полиморфизм может быть связан с кардиоваскулярным риском, развитием атеросклероза |
| Апо-В100 | Есть апо-ВС, апо-ВG | Ассоциирован с ранним развитием атеросклероза, высокой частотой манифестации ИБС |
| Апо-В48 | Нет | Самостоятельное значение не установлено, описаны антиатерогенные качества |
| Апо-С-I | Нет | Высокая концентрация ассоциируется с ранним развитием ИБС в семье |
| Апо-С-II | Нет | Самостоятельное значение не установлено |
| Апо-С-III | Нет | Дефицит или отсутствие приводит к гипотриглицеридемии, самостоятельное значение не установлено |
| Апо-Е | Есть Е1, Е2, Е3, Е4; наследуется кодоминантно | Наследуемая детерминанта содержания общего ХС, ХС ЛПНП, маркер риска ИБС. Полиморфизм апо-Е (от Е1 к Е4) является независимым маркером кардиоваскулярного риска |
| Апо-D | Нет | Возможно участвует в обратном транспорте ХС |
| Апо-F | Нет | Участвует в регуляции активности протеина, транспортирующего эстерифицированный ХС |
| Апо-H | Нет | ? |
| Апо-L | Нет | Участвует в регуляции уровня ТГ и ХС |
| Апо-М | Нет | Интегральный протеин, модулирующий уровень ХС ЛПНП и ХС ЛПВП, обладает антиатерогенным потенциалом |
У здоровых лиц 70% ХС содержится в ЛПНП, преимущественно состоящих из апо-В-липопротеина. Фактически, ремнанты хиломикронов, ЛПОНП и ЛППП также содержат апо-В-липопротеин и обладают потенциальными проатерогенными качествами, как и ЛПНП (National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III), 2002). В этой связи измерение уровня апо-В-липопротеина может дать принципиальное преимущество перед оценкой расчетного показателя ЛПНП, поскольку отражает содержание всех не-ЛПВП (Pischon T. et al., 2005). Учитывая тот факт, что одна частица ЛПНП содержит только одну молекулу апо-В-липопротеина, а количество частиц последнего в большей мере отражает атерогенный потенциал плазмы крови и хорошо коррелирует с риском возникновения ИБС в популяции, то измерение содержания апо-В-липопротеина и расчет отношения апо-В-липопротеин/общий ХС являются более предпочтительными для последующей стратификации пациентов в группы высокого риска по сравнению с ЛПНП или ЛПВП (Simon A. et al., 2005; Ridker P.M. et al., 2005).
Полиморфизм аполипопротеинов
К настоящему времени описано достаточно большое количество мутаций генов, приводящих к формированию весьма специфических фенотипов аполипопротеинов (табл. 4.5). Наиболее изученными являются полиморфизм апо-А-I и апо-Е-липопротеина. Более детально полиморфизм аполипопротеинов описан в соответствующем разделе главы 6.
Интерпретация результатов измерения плазменного уровня апо-А-I-липопротеина
ЛПВП представляют собой потенциально гетерогенную метаболически активную субфракцию липопротеидов, содержащую апо-А-I-липопротеины и обладающую антиатерогенными качествами (Barter P. et al., 2003). Принято считать, что содержание ХС ЛПВП косвенно отражает интенсивность обратного транспорта ХС, хотя в этом процессе активное участие принимает рецепторный ABCA1/G1-зависимый механизм, который, в частности, является лимитирующим этапом в клиренсе ЛПВП (Eriksson M. et al., 1999; Lewis G.F., Rader D.J., 2005). Популяция ЛПВП достаточно гетерогенна и вариабельна, представлена разнообразными мелкими молекулами дисковидной формы, содержащими апо-А-I-липопротеины, а также молекулы фосфолипидов, придающие ЛПВП сферическую форму и более крупные размеры, напоминающие таковые у ЛПНП (рис. 4.4). Наиболее крупными являются ЛПВП2 и ЛПВП3. Причем апо-А-I-липопротеин представлен во всех частицах ЛПВП, независимо от их формы и размеров. Иногда в некоторых молекулах, особенно в ЛПВП3, можно выявить апо-А-ІІ-липопротеин — второй по значению структурный компонент ЛПВП. Необходимо отметить, что ЛПВП подвергаются постоянному ремоделированию, связанному с обменом ХС между другими субфракциями липопротеидов. При этом концентрация апо-А-I-липопротеина и ЛПВП является отражением некоего динамического процесса, вовлекающего липидную часть частицы ЛПВП в трансфер и обмен ХС и ТГ с другими участниками транспорта последнего. При этом необходимо помнить о том, что содержание ЛПВП может варьировать в зависимости от метаболической активности липопротеинлипазы и обогащенных ТГ липопротеидов. Так, в физиологических условиях при сохраненной активности липопротеинлипазы плазменная концентрация ЛПОНП и хиломикронов снижается очень быстро. Фосфолипиды и апо-А-I-липопротеины, высвобождаемые из последних, включаются в состав ЛПВП, повышая интенсивность их продукции (Brinton E.A. et al., 1994). Основное противоречие заключается в том, что снижение липолитической активности плазмы крови приводит к образованию мелких и плотных частиц ЛПВП, подвергающихся быстрому катаболизму (Brinton E.A. et al., 1994; Lewis G.F., Rader D.J., 2005). В то же время, ряд исследований свидетельствуют о том, что между клиренсом апо-А-I-липопротеина и размерами ЛПВП с одной стороны и уровнем ТГ — с другой отмечена обратная и прямая корреляционная взаимосвязь соответственно (Brinton E.A. et al., 1994). Так, у пациентов с повышением уровня ЛПОНП и хиломикронов содержание ЛПВП обычно низкое, а у пациентов с изолированной гипертриглицеридемией — высокое. Все это свидетельствует о том, что динамика плазменной концентрации апо-А-I-липопротеина в большей мере может зависеть именно от уровня ТГ, а не от содержания ЛПВП, что требует некоторых усилий при интерпретации результатов измерения фракций липидов и анализа атерогенности их профиля.
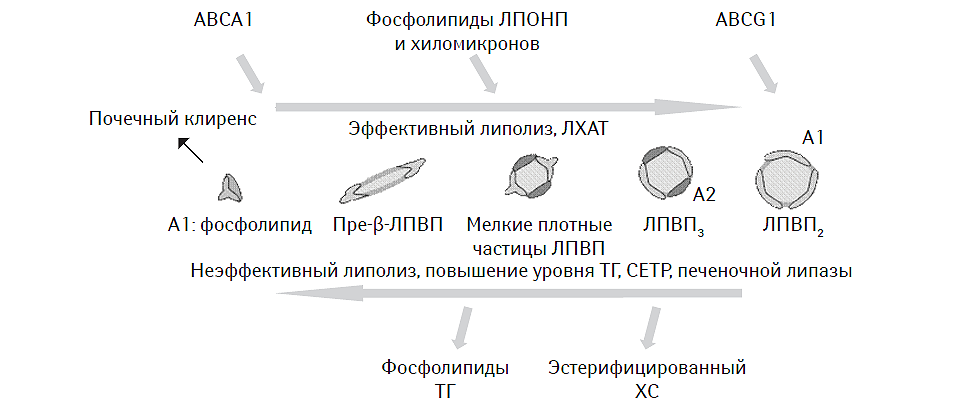
Рис. 4.4. Гетерогенность популяции субфракций ЛПВП и основные направления ее метаболического ремоделирования ABCA1, ABCG1 — молекулы-рецепторы для внутриклеточного ХС.
Формирование атерогенного фенотипа липопротеидов
Атерогенный фенотип липопротеидов характеризуется формированием особого липидного профиля или паттерна, чаще всего выявляемого у пациентов с метаболическими факторами риска возникновения кардиоваскулярных событий, таких как ожирение, метаболический синдром или сахарный диабет 2-го типа. У этих пациентов часто отмечают уменьшение плазменного содержания ЛПВП, повышение уровня ЛПОНП и превалирование мелких и плотных частиц ЛПНП (Berneis K.K., Krauss R.M., 2002). При этом содержание ТГ в плазме крови повышено (обычно >1,7 ммоль/л), а уровень ХС ЛПНП часто нормальный или умеренно повышен. Следует отметить, что формирующаяся инсулинорезистентность и сниженная активность липопротеинлипазы способствуют избыточной продукции и низкому клиренсу ЛПОНП1, повышению циркулирующего уровня ЛПНП и ремнантных форм липопротеидов. ТГ включаются в состав липидного ядра ЛПОНП и хиломикронов, что повышает их чувствительность к печеночной липопротеинлипазе и приводит к формированию мелких и плотных частиц ЛПНП (Packard C.J., Shepherd J., 1997; Berneis K.K., Krauss R.M., 2002). Последние обладают более высоким проатерогенным потенциалом, чем крупные и легкие частицы. Это связано с их способностью легко подвергаться оксидации, хорошо проникать через интиму и фиксироваться в субэндотелии благодаря своей высокой аффинности к протеогликанам, а также проявлять выраженные провоспалительные качества (Brinton E.A. et al., 1994; Packard C.J., Shepherd J., 1997; Taskinen M.R., 2003). Детекция проатерогенного профиля плазмы (сыворотки) крови базируется на анализе содержания субфракций липопротеидов методом ультрацентрифугирования, градиентного электрофореза в геле или ЯМР-спектроскопии (Berneis K.K., Krauss R.M., 2002). Вместе с тем эти методы не рекомендованы для повседневной лабораторной практики. Чаще всего используют скрининговый подход, основанный на выявлении повышенного уровня ТГ (>1,7 ммоль/л) в сочетании с увеличением значений отношения апо-B/aпо-А-I (см. главу 3).
Уровень аполипопротеинов у пациентов, которым проводят гиполипидемические мероприятия
Главная цель измерения плазменной концентрации аполипопротеинов — более точная идентификация индивидуального риска в популяции пациентов с исходным избыточным риском манифестации кардиоваскулярных событий, а также инициация решения о проведении гиполипидемических мероприятий у лиц, не удовлетворяющих критериям отбора для проведения гиполипидемической терапии по другим критериям. Кроме того, существует необходимость в мониторировании риска при проведении программ первичной и вторичной превенции кардиоваскулярных заболеваний у пациентов, уже получающих гиполипидемическую терапию. Анализ результатов некоторых рандомизированных клинических исследований, посвященных статинам (WOSCOPS, AFCAPS/TEXCAPS) и фибратам (VA-HIT) (West of Scotland Coronary Prevention Study Group, 1998; Gotto A.M. et al., 2000; Robins S.J. et al., 2001) продемонстрировал высокую вариабельность таких показателей, как содержание ЛПНП и ЛПВП в плазме крови, что ограничивает их использование как предикторов наступления неблагоприятных клинических исходов у больных в процессе лечения указанными лекарственными средствами. В то же время в некоторых рандомизированных клинических исследованиях, таких как Pravastatin Pooling Project, уровень ЛПНП оказался информативным для глобальной оценки риска, однако в то же время слабо коррелировал с частотой возникновения ишемических событий (Sacks F.M. et al., 2000). Перспективы широкого применения результатов измерения плазменного содержания аполипопротеинов базировались на их более высокой прогностической ценности по сравнению с ЛПНП и ЛПВП в когорте больных с ИБС. Вместе с тем, в рандомизированных клинических исследованиях PROSPER эти ожидания не получили подтверждения, хотя в других рандомизированных клинических исследованиях, таких как AFCAPS/TexCAPS, было установлено, что отношение апо-В/апо-А1 является более мощным предиктором наступления неблагоприятного клинического исхода, чем ЛПНП, в когорте пациентов, получающих статины (Packard C.J. et al., 2005). В этой связи применение указанного отношения может быть целесообразным для оценки риска в селективных популяциях больных, например, получающих статины или фибраты (Gotto A.M. et al., 2000).
Литература
- Ariyo A.A., Thach C., Tracy R.P. (2003) Lp(a) lipoprotein, vascular disease and mortality in the elderly. N. Engl. J. Med., 349: 2108–2115.
- Barter P., Kastelein J., Nunn A., Hobbs R.; Future Forum Editorial Board (2003) High density lipoproteins (HDL) and atherosclerosis; the unanswered questions. Atherosclerosis, 168: 195–192.
- Berglund L., Ramakrishnan R. (2004) Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler. Thromb. Vasc. Biol., 24: 2219–2226.
- Bergmark C., Dewan A., Orsoni A. et al. (2008) A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res., 49(10): 2230–2239.
- Berneis K.K., Krauss R.M. (2002) Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res., 43: 1363–1379.
- Beswick A., Brindle P. (2006) Risk scoring in the assessment of cardiovascular risk. Curr. Opin. Lipidol., 17: 375–386.
- Brindle P., Beswick A., Fahey T. et al. (2006) Accuracy and impact of risk assessment in the primary prevention of cardiovascular disease: a systematic review. Heart, 92: 1752–1759.
- Brinton E.A., Eisenberg S., Breslow J.L. (1994) Human HDL cholesterol levels are determined by apoA-I fractional catabolic rate, which correlates inversely with estimates of HDL particle size. Effects of gender, hepatic and lipoprotein lipases, triglyceride and insulin levels, and body fat distribution. Arterioscler. Thromb. Vasc. Biol., 14: 707–720.
- British Cardiac Society (2005) JBS 2: Joint British Societies’ guidelines on prevention of cardiovascular disease in clinical practice. Heart, 91: v1–v52.
- Broadbent H.M., Peden J.F., Lorkowski S. et al. (2008) Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet., 17: 806–814.
- Caplice N.M., Panetta C., Peterson T.E et al. (2001) Lipoprotein(a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood, 98: 2980–2987.
- Chasman D.I., Shiffman D., Zee R.Y. et al. (2009) Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis, 203: 371–376.
- Clarke R., Peden J.F., Hopewell J.C. et al. (2009) Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med., 361(26): 2518–2528.
- Conroy R.M., Pyorala K., Fitzgerald A.P. et al. for the SCORE Project Group (2003) Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE Project. Eur. Heart J., 24: 987–1003.
- Cupples L.A., D’Agostino R.B. (1987) Section 34: some risk factors related to the annual incidence of cardiovascular disease and death in pooled repeated biennial measurements. In: W.B. Kannel, P.A. Wolf, R.J. Garrison (Eds.) Framingham Heart Study: 30 Year Follow-Up. Bethesda, Md: US Department of Health and Human Services.
- Danesh J., Collins R., Peto R. (2000) Lipoprotein(a) and coronary heart disease: meta analysis of prospective studies. Circulation, 102: 1082–1085.
- De Backer G., Ambrosioni E., Borch-Johnsen K. et al. (2003) European guidelines on cardiovascular disease prevention in clinical practice: Third Joint Task Force of European and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of eight societies and by invited experts). Eur. Heart J., 24: 1601–1610.
- Eriksson M., Carlson L.A., Miettinen T.A. et al. (1999) Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein AI. Potential reverse cholesterol transport in humans. Circulation, 100: 594–598.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (2001) Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA, 285(19): 2486–2497.
- Ferrario M., Chiodini P., Chambless L.E. et al. (2005) Prediction of coronary events in a low incidence population: assessing accuracy of the CUORE Cohort Study prediction equation. Int. J. Epidemiol., 34: 413–421.
- Gotto A.M., Whitney E., Stein E.A. et al. (2000) Relation between baseline and on- treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation, 101: 477–484.
- Grainger D.J., Kemp P.R., Liu A.C. et al. (1994) Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature, 370: 460–462.
- Hippisley-Cox J., Coupland C., Vinogradova Y. et al. (2007) Derivation and validation of QRISK, a new cardiovascular disease risk score for the United Kingdom: prospective open cohort study. BMJ, 335: 136.
- Jackson R. (2000) Updated New Zealand cardiovascular disease risk-benefit prediction guide. BMJ, 320: 709–710.
- Jackson R., Lawes C.M., Bennett D.A. et al. (2005) Treatment with drugs to lower blood pressure and blood cholesterol based on an individual’s absolute cardiovascular risk. Lancet, 365: 434–441.
- Kamstrup P.R., Tybjaerg-Hansen A., Steffensen R. et al. (2009) Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA, 301: 2331–2339.
- Karpe F. (1999) Postprandial lipoprotein metabolism and atherosclerosis. J. Intern. Med., 246: 341–345.
- Kathiresan S. (2009) Lp(a) Lipoprotein Redux — From Curious Molecule to Causal Risk Factor. N. Engl. J. Med., 361: 2573–2574.
- Knoflach M., Kiechl S., Penz D. et al. (2009) Cardiovascular Risk Factors and Atherosclerosis in Young Women: Atherosclerosis Risk Factors in Female Youngsters (ARFY Study) Stroke, 40(4): 1063–1069.
- Kochinsky M.L., Marcovina S.M. (2004) Structure-function relationships in apolipoprotein (a) insights into lipoprotein (a) assembly and pathogenicity. Curr. Opin. Lipidol., 15: 167–174.
- Lewis G.F., Rader D.J. (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res., 96: 122–132.
- Marcovina S.M., Albers J.J., Jacobs D.R. Jr. et al. (1993) Lipoprotein(a) concentrations and apolipoprotein(a) phenotypes in Caucasians and African Americans: the CARDIA study. Arterioscler. Thromb., 13: 1037–1045.
- Marcovina S.M., Albers J.J., Wijsman E. et al. (1996) Differences in Lp(a) concentrations and apo(a) polymorphs between black and white Americans. J. Lipid Res., 37: 2569–2585.
- McPherson R., Pertsemlidis A., Kavaslar N. et al. (2007) A common allele on chromosome 9 associated with coronary heart disease. Science, 316: 1488–1491.
- Menotti A., Lanti M., Gabiti-Rosei E. et al. (2005) Riskard 2005: new tools for prediction of cardiovascular disease risk derived from Italian population studies. Nutr. Metab. Cardiovasc. Dis., 15: 426–440.
- Nascimento H., Silva L., Lourenco P. et al. (2009) Lipoprotein(a) Levels in Obese Portuguese Children and Adolescents: Contribution of the Pentanucleotide Repeat (TTTTA)n Polymorphism in the Apolipoprotein(a) Gene. Arch. Pediatr. Adolesc. Med., 163(4): 393–394.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) (2002) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation, 106(25): 3143–3421.
- Packard C.J., Ford I., Robertson M. et al. (2005) Lipoproteins and apolipoproteins as predictors of the Prospective Study of Pravastatin in the Elderly at Risk. Circulation, 112: 3058–3065.
- Packard C.J., Shepherd J. (1997) Lipoprotein heterogeneigty and apolipoprotein B metabolism. Arterioscler. Thromb. Vasc. Biol., 17: 3542–3556.
- Parra H.J., Luyeye I., Bouramoue C. et al. (1987) Black-white differences in serum Lp(a) lipoprotein levels. Clin. Chim. Acta., 167: 27–31.
- Paultre F., Pearson T.A., Weil H.F.C. et al. (2000) High levels of lipoprotein(a) carrying a small apolipoprotein(a) isoform is associated with coronary artery disease in both African American and Caucasian men. Arterioscler. Thromb. Vasc. Biol., 20: 2619–2624.
- Pischon T., Girman C.J., Sacks F.M. et al. (2005) Non-high density lipoprotein cholesterol and apolipoprotein B in the prediction of coronary heart disease in men. Circulation, 112: 3375–3383.
- PROCARDIS Consortium (2004) A trio family showing association of the lymphotoxin-alpha N26 (804A) allele with coronary artery disease. Eur. J. Hum. Genet., 12: 770–774.
- Reuterwall C., Hallqvist J., Ahlbom A. et al. (1999) Higher relative, but lower absolute risks of myocardial infarction in women than in men: analysis of some major risk factors in the SHEEP study. J. Intern. Med., 246: 161–174.
- Ridker P.M., Buring J.E., Rifai N. et al. (2007) Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds risk score. JAMA, 297: 611–619.
- Ridker P.M., Rifai N., Cook N.R. (2005) Non-HDL C, apolipoproteins AI and B100, standard lipid measures, lipid ratios and CRP as risk factors for cardiovascular disease in women. JAMA, 294: 326–333.
- Robins S.J., Collins D., Wittes J.T. et al. (2001) Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomised controlled trial. JAMA, 285: 1585–1591.
- Rubin J., Paultre F., Tuck C.H. et al. (2002) Apolipoprotein(a) genotype influences isoform dominance pattern differently in African Americans and Caucasians. J. Lipid. Res., 43: 234–244.
- Rye K.A., Clay M.A., Barter P.J. (1999) Remodelling of high density lipoproteins by plasma factors. Atherosclerosis, 145: 227–238.
- Sacks F.M., Tonkin A.M., Shepherd J. et al. (2000) Effect of pravastatin on coronary disease events in subgroups defined by coronary risk factors: the Prospective Pravastatin Pooling Project. Circulation, 102: 1893–1900.
- Sandholzer C., Hallman D.M., Saha N. et al. (1991) Effects of the apolipoprotein(a) size polymorphism on the lipoprotein(a) concentration in 7 ethnic groups. Hum. Genet., 86: 607–614.
- Simon A., Chironi G., Gariepy J. et al. (2005) Differences between markers of atherogenic lipoproteins in predicting high cardiovascular risk and subclinical atherosclerosis in asymptomatic men. Atherosclerosis, 17: 339–344.
- Smolders B., Lemmens R., Thijs V. (2007) Lipoprotein (a) and Stroke. A Meta-Analysis of Observational Studies. Stroke, 38: 1959–1969.
- Takagi H., Manabe H., Kawai N. (2009) Circulating lipoprotein(a) concentrations and abdominal aortic aneurysm presence. Interactive Cardio. Vascular. Thoracic Surgery, 9(3): 467–470.
- Taskinen M.R. (2003) Diabetic dyslipidaemia from basic research to clinical practice. Diabetologia, 46: 733–749.
- The Emerging Risk Factors Collaboration (2009) Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA, 302(4): 412–423.
- Thillet J., Doucet C., Chapman J. (1998) Elevated lipoprotein(a) levels and small apo(a) isoforms are compatible with longevity: evidence from a large population of French centenarians. Atherosclerosis, 136: 389–394.
- Tsimikas S., Brilakis E.S., Miller E.R. et al. (2005) Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N. Engl. J. Med., 353: 46–57.
- West of Scotland Coronary Prevention Study Group (1998) Influence of pravastatin and plasma lipids on clinical events in the West of Scotland Coronary prevention Study (WOSCOPS). Circulation, 97: 1440–1444.
- Wong N., Lan D., Chen W. (2004) ATP binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high density lipoproteins. Proc. Natl Acad. Sci. USA, 101: 9774–9779.
- Woodward M., Brindle P., Tunstall-Pedoe H. for the SIGN Group on Risk Estimation (2007) Adding social deprivation and family history to cardiovascular risk assessment: the ASSIGN score from the Scottish Heart Health Extended Cohort (SHHEC). Heart, 93: 172–176.
- Wu H.D., Berglund L., Dimayuga C. et al. (2004) High lipoprotein(a) levels and small apolipoprotein(a) sizes are associated with endothelial dysfunction in a multiethnic cohort. J. Am. Coll. Cardiol., 43: 1828–1833.
- Zhang X.F., Attia J., D’Este C. et al. (2005) A risk score predicted coronary heart disease and stroke in a Chinese cohort. J. Clin. Epidemiol., 58: 951–958.