Дилатационная кардиомиопатия (ДКМП) — заболевание сердечной мышцы, характеризующееся дилатацией и систолической дисфункцией ЛЖ при отсутствии нарушений наполнения (гипертензия, клапанные пороки) или ИБС, способных вызвать глобальное ухудшение систолической дисфункции. Может также присутствовать дилатация и дисфункция ПЖ, однако это не обязательно для постановки диагноза.
Эпидемиология
Об истинной распространенности ДКМП судить трудно, поскольку частота ее выявления в различных регионах неодинакова. Идиопатическую ДКМП отмечают в 0,4 случая на 1 тыс. населения, ежегодно выявляют 0,08 нового случая на 1 тыс. населения, что составляет приблизительно 25% всех случаев кардиомиопатии и является причиной ежегодной смерти 10 тыс. больных. Мужчины болеют в среднем в 3 раза чаще, чем женщины.
Этиология
Этиология ДКМП окончательно не установлена. Многие исследователи придерживаются полиэтиологической гипотезы происхождения заболевания — описано достаточно случаев развития ДКМП, являющейся конечным результатом различных патологических процессов. Выделяют идиопатическую, семейную (или генетическую), вирусную (и/или иммунную) и ассоциированную с известными сердечно-сосудистыми заболеваниями ДКМП.
Идиопатическая ДКМП, которая является первичным заболеванием миокарда неизвестной причины, развивается у 40% больных. Семейная ДКМП главным образом ассоциируется с мутациями в генах цитоскелета и экстрацеллюлярного матрикса.
В последнее время достигнут значительный прогресс в области молекулярной генетики ДКМП. У 20% больных заболевание наследуется или имеются указания в семейном анамнезе. При наследственных формах установлен аутосомно-доминантный, аутосомно-рецессивный, а также связанный с Х-хромосомой гена или митохондриальной трансмиссией тип наследования. Мутации выявлены в генах кардиомиоцитов, которые кодируют контрактильные белки или их регулирующие элементы, включая компоненты саркомера, цитоскелета, а также различные механизмы, обеспечивающие сопряжение процессов возбуждения — сокращения, бета-адренергические пути проведения и процессы, приводящие к дефициту энергетических механизмов, в том числе мутации митохондрий, метаболизм гликогена, обмен кальция, а также транскрипторную регуляцию.
Возникновение ДКМП связывают с вариантами мутаций гена актина, важное значение также отводится патологии гена белка дистрофина, входящего в состав комплекса, связывающего мышечный цитоскелет кардиомиоцита с экстрацеллюлярным матриксом.
Результаты исследования CARDIGENE (1999) свидетельствуют, что при ДКМП имеют значение генетические нарушения путей эндотелина и полиморфизм гена эндотелиновых рецепторов типа А — первый идентифицированный генетический фактор риска развития заболевания.
Существует вирусно-иммунологическая теория возникновения ДКМП. При ДКМП выявлен ряд нарушений иммунной регуляции, включая гуморальную и клеточную аутоиммунную реактивность по отношению к миоцитам, снижение содержания и клеточной активности естественных киллеров и отклонения в деятельности супрессорных клеток. Наличие нарушений иммунной регуляции и множества антимиокардиальных антител при ДКМП согласуется с этой гипотезой: при иммунологическом исследовании повышение титров антител к вирусу Коксаки В3 выявляли у 40% больных с ДКМП и только у 2% — в группе здоровых лиц, при этом в эндомиокардиальных биоптатах не определяли признаков миокардита. У 50% больных ДКМП выявляют антитела к миокарду, у 49% — антиинтерфибриллярные антитела, у 30% — сенсибилизацию к сердечному антигену. Активация аутоиммунных процессов приводит к образованию антител к миозину и β1-адренорецепторам.
Причиной угнетения активности естественных киллеров может служить первичное нарушение их созревания, детерминированное антигенами системы HLA, у больных с ДКМП чаще всего выявляют гаплотипы HLA B27, HLA A2, HLA DQ4 и HLA DR4, что указывает на наследственную предрасположенность к заболеванию и свидетельствует о возможной его иммунной основе. Несмотря на выявление нарушений гуморального иммунитета, в целом вирусно-инфекционно-аутоиммунная гипотеза на сегодня остается недоказанной.
Патогенез
Патогенез ДКМП следует прежде всего рассматривать на молекулярном уровне, поскольку нарушается экспрессия генов, приводящая к изменению фенотипа. Выделяют несколько механизмов, лежащих в основе развития заболевания: одиночный генный дефект; полиморфные изменения генов-модификаторов (в гене АПФ и других компонентах РААС и β2-адренергических рецепторах); нарушение экспрессии нормальных генов, кодирующих белки, которые регулируют сократительную функцию сердца или формируют структуру его полостей.
Мутации генов, кодирующих белки внеклеточного матрикса, могут служить причиной ослабления механической взаимосвязи между последним и кардиомиоцитами, возможным следствием чего является прогрессирующая дилатация сердца. В оставшихся кардиомиоцитах происходит повреждение внутриклеточных органелл, в том числе и отвечающих за энергетику клетки. Вследствие этого быстро расщепляется АТФ, что ведет не только к нарушению процесса сокращения, но и к контрактуре миокарда в результате нехватки энергии и кальциевой перегрузки.
Определенная роль в развитии фиброза отводится деградации нормального коллагенового матрикса металлопротеиназами, активация которых происходит вследствие активации провоспалительных цитокинов и экспозиции свободных кислородных радикалов (оксидантного стресса).
При повреждении миокарда и дилатации сердца происходит активация различных компенсаторных механизмов, направленных на нормализацию сердечной деятельности (схема 9.1).
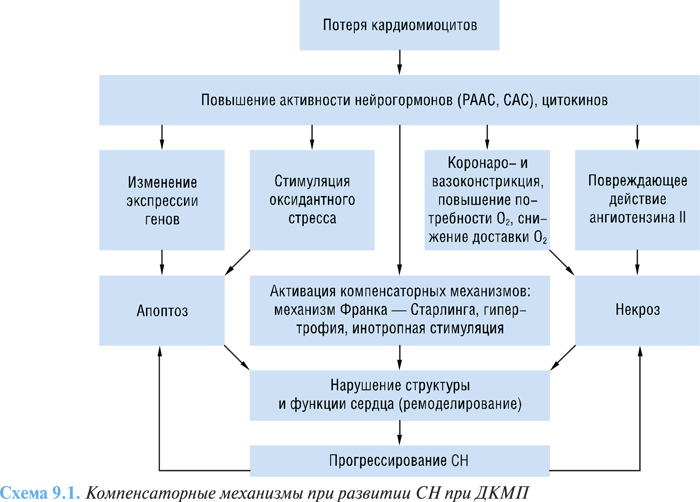
Долговременные компенсаторные механизмы включают развитие гипертрофии оставшихся жизнеспособных кардиомиоцитов и изменение геометрии камер сердца, что составляет суть ремоделирования ЛЖ.
Гипертрофия миокарда не достигает адекватной степени и не соответствует нуждам дилатированного сердца, поскольку сократительная активность гипертрофированного миокарда на единицу массы ниже, чем в здоровом сердце. Постепенно происходит изменение геометрии желудочка от нормальной эллипсоидной до сферической формы (ремоделирование) и постепенное преобладание дилатации над гипертрофией его стенки. Сердечный выброс поддерживается тахикардией и большим объемом диастолического наполнения, который в свою очередь повышает внутримиокардиальное напряжение как в систолу, так и диастолу и увеличивает потребность миокарда в кислороде. Эти процессы являются пусковым механизмом активации РААС и стимулируют выделение норадреналина симпатическими терминалями, а также секрецию НУП.
Длительная чрезмерная активация РААС оказывает повреждающее действие, следствием чего является потенцирование активности других нейрогормональных систем, коронарная и системная вазоконстрикция, непосредственное токсическое повреждающее действие ангиотензина II на кардиомиоциты, что приводит к их дисфункции и гибели.
Длительная активация САС оказывает ряд негативных эффектов на сердечно-сосудистую систему, включая повышение потребности в кислороде, уменьшение силы сокращения (вследствие повышения ЧСС), повышение концентрации катехоламинов в плазме крови, снижение чувствительности β-адренорецепторов к катехоламинам, угнетение функции митохондрий, нарушение окислительно-восстановительного равновесия, что опосредуется через β-рецепторы сердца и цАМФ.
В результате нарушений со стороны β-адренергических путей проведения, в значительной мере модулирующих функцию сердца на рецепторном и клеточном уровнях, происходит значительное уменьшение количества и плотности β1-рецепторов в пораженном сердце, тогда как плотность β2-рецепторов остается практически без изменений. В совокупности с повышением концентрации блокирующих G-протеинов и усилением процессов β-рецепторного фосфорилирования эти изменения усугубляют нарушения сократительной функции пораженного миокарда.
Активация нейрогормонов, цитокинов, перестройка биомеханики миокарда обусловливают изменение генной экспрессии, гибель кардиомиоцитов, клеточное ремоделирование, что в свою очередь является причиной дисфункции миокарда и его ремоделирования. Возможно, определенный генотип изначально приводит к активации нейрогормонов и цитокинов. Большую роль играют эндотелин-1 и ФНО-α.
Нарушение насосной функции сердца при ДКМП может быть обусловлено уменьшением количества самих кардиомиоцитов. Активация процессов, связанных с механическим перерастяжением стенок пораженного сердца, с эффектами системы нейрогуморальной регуляции и цитокинами отражается в прогрессирующей потере контрактильных элементов путем апоптоза и некроза.
Патологическая анатомия
Заболевание характеризуется резкой дилатацией всех полостей сердца, масса сердца может достигать 1000 г и более.
В полостях предсердий и желудочков часто (при вскрытии >50%) выявляют пристеночные тромбы. AV-отверстие значительно растянуто, створки клапана слегка белесоваты, удлинены, как и сухожильные нити. Сосочковые мышцы гипертрофированы, часто резко склерозированы.
Микроскопически специфических изменений нет, определяют неравномерную гипертрофию кардиомиоцитов с большими неправильной формы ядрами, также заметна очаговая жировая дистрофия мышечных волокон, дезорганизация миофибрилл, потеря миофиламентов, миоцитолиз, коллапс стромы, интерстициальный фиброз. Вновь образуемые коллагеновые структуры характеризуются извращенным соотношением между типами коллагена и нарушением архитектоники взаиморасположения волокон. При ДКМП выявляют преобладание коллагенов I+III типа, умеренное содержание коллагена VI типа и сниженное содержание коллагена IV типа.
Отмечают выраженные дегенеративные и некробиотические изменения кардиомиоцитов (исчезновение поперечной исчерченности, вакуолизация ядер, некроз, умеренное содержание гликогена). Нередко определяют воспалительные инфильтраты, при гистологическом исследовании биоптатов миокарда в интерстициальной ткани миокарда насчитывают от 5 до 10 лимфоцитов при увеличении в 400 или 200 раз соответственно.
Клиническая картина
Доминирующим клиническим симптомом является нарастающая СН. Как правило, ранние стадии заболевания выявляют случайно при профилактическом рентгенологическом или эхоКГ-исследовании. Период от появления первых симптомов до возникновения развернутой клинической картины составляет обычно 2 года.
Клинические симптомы обусловлены дисфункцией левого, правого или обоих желудочков. У ⅓ больных возможна боль за грудиной, нередко — различные нарушения ритма, характерными являются эпизоды тромбоэмболии сосудов большого и малого круга кровообращения.
При прогрессировании заболевания и появлении декомпенсации симптомы в общем характерны для СН.
Диагностика
На ЭКГ специфичных признаков нет, могут выявляться нарушения внутрижелудочковой проводимости, блокада левой ножки пучка Гиса, снижение вольтажа зубца R, возможны нарушения ритма сердца, в том числе экстрасистолическая аритмия, пароксизмальная тахикардия, фибрилляция предсердий. По данным суточного мониторирования ЭКГ часто выявляют тяжелые желудочковые нарушения ритма, не определяемые при обычной регистрации ЭКГ.
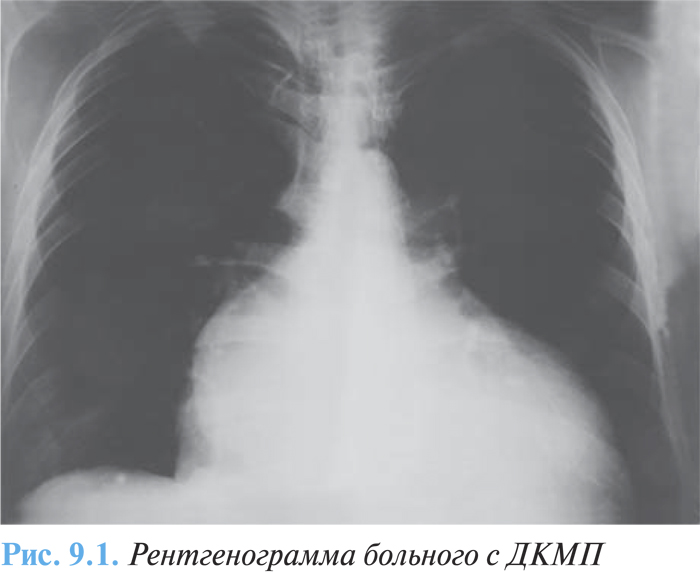
При рентгенологическом исследовании выявляют кардиомегалию, застойные изменения в малом круге кровообращения (рис. 9.1).
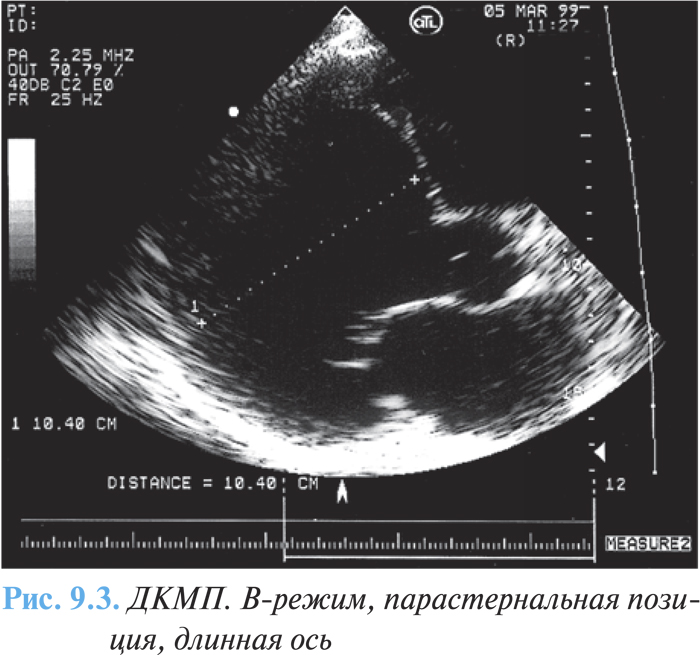
ЭхоКГ является основным методом диагностики, с помощью которой определяют размеры ЛЖ, степень повышения конечного диастолического давления, нарушения его систолической и диастолической функции. У больных с ДКМП отмечают выраженную дилатацию ЛЖ (рис. 9.2) и левого предсердия (рис. 9.3), толщина стенок нормальная или уменьшена. Важным является также определение наличия дилатации ПЖ, измерение систолического давления в ПЖ по степени трикуспидальной регургитации с помощью допплеровского исследования. Часто выявляют внутриполостные тромбы.

Ремоделирование миокарда ассоциируется с нарушением гемодинамики и уменьшением ФВ, ударного объема, увеличением объемов и повышением давления в камерах сердца. Нередко выявляют дилатацию кольца митрального и трехстворчатого клапанов и признаки клапанной регургитации.
Данные иммунологических исследований свидетельствуют о достоверном снижении активности естественных киллеров, повышении уровня ФНО-α по сравнению со здоровыми лицами, а также о наличии специфических циркулирующих антител, которые являются важными маркерами ДКМП: антимиозиновые антитела (к тяжелым цепям α- и β-миозина), антимитохондриальные антитела (к М7, к адениннуклеотидному транслокатору — ферменту внутренней мембраны митохондрий сердца, осуществляющему перенос АТФ и АДФ между цитоплазмой кардиомиоцитов и матриксом этих органелл), антитела к β-адренорецепторам.
При проведении прижизненной биопсии миокарда выявляют неспецифические дегенеративные изменения различной степени, миоцитолиз, очаги некроза.
Характерные морфологические признаки ДКМП при гистологическом исследовании отсутствуют. Как правило, выявляют множественные очаги фиброзного замещения миокарда, иногда воспалительные инфильтраты.
При радионуклидной вентрикулографии определяют расширение полостей сердца, нарушение локальной сократимости на фоне диффузного снижения сократимости миокарда, значительное снижение ФВ ЛЖ и ПЖ.
При сцинтиграфии миокарда с 201Tl выявляют диффузные и очаговые дефекты накопления изотопа. При сцинтиграфии миокарда с 67Gl изотоп накапливается в воспалительных очагах при миокардите и не накапливается при ДКМП.
Катетеризацию полостей сердца проводят, если диагноз остается под сомнением после неинвазивного исследования. Сердечный выброс может быть нормальным или сниженным, ФВ снижена, на ангиограмме отмечается диффузный гипокинез. Повышение конечно-диастолического давления в ЛЖ отмечают в поздних стадиях заболевания. Во время катетеризации можно произвести биопсию миокарда из каждого желудочка.
Диагностические критерии ДКМП включают подтверждающие и исключающие признаки. К подтверждающим признакам относят прогрессирующую недостаточность кровообращения, резистентную к лечению; кардиомегалию с наличием относительной недостаточности митрального и трехстворчатого клапанов; тромбоэмболический синдром; нарушения ритма и проводимости; относительно молодой возраст; отсутствие признаков воспалительного процесса; отсутствие связи заболевания с инфекционным или каким-либо другим этиологическим фактором.
К исключающим признакам относят нормальные размеры сердца, наличие ИБС (обструкция >50% просвета основных коронарных артерий), документированную АГ (>160/100 мм рт. ст.), врожденные пороки сердца и приобретенные изменения клапанного аппарата сердца, указания в анамнезе на хроническое употребление алкоголя (>40 г в день для женщин и >80 г в день для мужчин в течение свыше 5 лет) с ремиссией алкогольной кардиомиопатии после 6 мес абстиненции, системные заболевания, поражения перикарда.
Лечение
Должно быть направлено на решение следующих основных задач:
- предотвращение прогрессирования СН, дальнейшего ремоделирования сердечно-сосудистой системы (снижение нейроэндокринной, клеточной активации);
- уменьшение выраженности симптоматики (улучшение систолической функции сердца, периферического кровообращения);
- улучшение качества жизни (улучшение переносимости физических нагрузок);
- увеличение продолжительности жизни (улучшение прогноза);
- уменьшение количества случаев госпитализации;
- уменьшение количества осложнений.
Лечение включает немедикаментозные подходы (диета, ограничения приема жидкости и поваренной соли, физические нагрузки), применение лекарственных препаратов (диуретиков, сердечных гликозидов, ингибиторов АПФ, блокаторов β-адренорецепторов, блокаторов рецепторов ангиотензина II, инотропных агентов, антикоагулянтов, антиаритмических средств), применение вспомогательных устройств для поддержки функции ЛЖ, а также современные хирургические вмешательства (кардиомиопластика, восстановление митрального и трехстворчатого клапана, частичная вентрикулоэктомия ЛЖ и трансплантация сердца).
Важное значение в лечении больных с СН имеет снижение постнагрузки путем нормализации уровня АД, а также коррекция анемии для повышения транспорта кислорода тканям.
За последние годы кардинально изменились взгляды на значение физической активности: установлено, что продолжительная малоподвижность может приводить к атрофии скелетных мышц, дальнейшему ухудшению переносимости физических нагрузок, венозному тромбозу, легочной эмболии и прогрессированию симптомов СН.
Рабочая группа по кардиологической реабилитации совместно с Рабочей группой по СН Европейского общества кардиологов в 2001 году опубликовали рекомендации по проведению физических тренировок у больных с ХСН.
К относительным противопоказаниям к использованию физической нагрузки у больных со стабильной СН отнесены:
- Увеличение массы тела на 1,8 кг и более за предшествующие 1–3 дня;
- Сопутствующая постоянная или периодическая терапия добутамином;
- Снижение САД при нагрузках;
- IV функциональный класс по NYHA;
- Сложные нарушения ритма в покое или появляющиеся при нагрузке;
- Исходная ЧСО 100 уд./мин.
К абсолютным противопоказаниям отнесены:
- Прогрессивное ухудшение переносимости физической нагрузки или одышка в покое или при нагрузке в течение 3–5 предшествующих дней;
- Значительная ишемия при небольших нагрузках (50 Вт);
- Неконтролируемый сахарный диабет;
- Острое системное заболевание или лихорадка;
- Предшествующие эмболии;
- Тромбофлебит;
- Активный перикардит или миокардит;
- Регургитация на клапанах, требующая хирургического вмешательства;
- Вновь возникшая фибрилляция предсердий.
Относительные критерии, при которых рекомендуется начинать нагрузочные тренировки:
- компенсированная СН в течение по крайней мере 3 нед;
- способность разговаривать без одышки (с частотой дыхания менее 30 в 1 мин);
- ЧСС в покое не более 100 уд./мин;
- незначительная усталость;
- сердечный индекс не менее 2,1 мин/м2 (для инвазивно мониторируемых больных);
- уровень ЦВД <12 мм рт. ст. (для инвазивно мониторируемых больных).
Относительные критерии, при которых необходимо изменить или прекратить тренировки:
- выраженная одышка или усталость;
- частота дыхания более 40 в 1 мин во время упражнений;
- появление III тона или хрипов в легких;
- усиление хрипов в легких;
- усиление второго компонента II тона;
- недостаточное пульсовое давление (<10 мм рт. ст. между САД и ДАД);
- снижение АД (>10 мм рт. ст.) во время увеличивающихся нагрузок;
- увеличение числа суправентрикулярных или желудочковых экстрасистол во время нагрузок;
- профузное поотделение, бледность, спутанность сознания.
Медикаментозное лечение ДКМП по сути является лечением СН.
Диуретики остаются средствами первого ряда, поскольку это единственная группа препаратов, которые могут адекватно контролировать задержку жидкости при СН. При умеренной СН лечение обычно начинают с тиазидных диуретиков, при более выраженной применяют петлевые диуретики (торасемид, фуросемид или этакриновую кислоту). Комбинация диуретиков, действующих на различные участки нефрона, позволяет предупредить развитие резистентности, а также возникновение побочных эффектов.
Ингибиторы АПФ играют ключевую роль в долговременной терапии больных с ДКМП и систолической дисфункцией независимо от степени ее выраженности.
Ингибиторы АПФ следует применять с осторожностью у больных с очень низким системным АД (САД <80 мм рт. ст.), значительно повышенным уровнем креатинина в сыворотке крови (>3 мг/дл), повышенным уровнем калия в сыворотке крови (>5,5 ммоль/л). Начинать лечение ингибиторами АПФ следует с низкой дозы под контролем уровня АД, удваивая дозу каждые 3–7 дней при хорошей переносимости и титруя ее до целевой.
Наиболее частыми побочными эффектами ингибиторов АПФ, которые потенциально могут ограничивать их применение, являются артериальная гипотензия, ухудшение функции почек, сухой кашель, ангионевротический отек. Возможна замена ингибиторов АПФ антагонистами рецепторов ангиотензина II.
Всем пациентам с СН вследствие систолической дисфункции ЛЖ необходимо назначать блокаторы β-адренорецепторов (метопролол, бизопролол, карведилол, небиволол), кроме случаев, когда они им противопоказаны или плохо переносятся.
Лечение блокаторами β-адренорецепторов начинают с низких доз — с ⅛ средней терапевтической дозы, удваивая дозу каждые 2–3 нед до оптимальной. Терапию у больных с ДКМП следует начинать в стационаре под наблюдением врача. Начало лечения блокаторами β-адренорецепторов может сопровождаться клиническим ухудшением у 10–20% больных, что проявляется снижением переносимости физической нагрузки, усугублением застоя в легких, усилением утомляемости, увеличением периферических отеков и, как следствие, ухудшением качества жизни. Однако при дальнейшем лечении признаки кратковременного гемодинамического ухудшения исчезают.
У больных с выраженной СН используют антагонисты альдостерона (спиронолактон) как средства дополнительного влияния на нейрогуморальные механизмы развития СН, улучшающие прогноз.
Дигоксин показан больным с ДКМП с явлениями декомпенсации для уменьшения выраженности симптомов СН. У пациентов с тахисистолической формой фибрилляции предсердий применение сердечных гликозидов позволяет снизить частоту сокращений желудочков. Отмена сердечных гликозидов у больных с ДКМП как с синусовым ритмом, так и фибрилляцией предсердий и низкой ФВ приводит к нарастанию симптомов декомпенсации сердечной деятельности.
Непрямые антикоагулянты (варфарин) показаны пациентам с постоянной формой фибрилляции предсердий, тромбоэмболическими осложнениями в анамнезе, при наличии тромбов в полостях сердца.
Имплантация кардиовертера-дефибриллятора при наличии рецидивирующей фибрилляции желудочков или стойкой желудочковой тахикардии и резистентности к антиаритмическим препаратам, а также имплантация трехкамерного электрокардиостимулятора в режиме DDDR у больных со значительными нарушениями внутрижелудочковой проводимости и десинхронизацией сокращения желудочков при тяжелой СН, рефрактерной к медикаментозной терапии, входит в перечень дополнительного ассортимента медицинских услуг у больных с ДКМП (приказ МЗ Украины № 436 от 03.07.2006 г.).
Патогенетическая терапия не разработана. Применение иммунодепрессантов не оправдано.
Поскольку прогноз при ДКМП неблагоприятный и любая терапия не предотвращает летального исхода, эти больные представляют наибольшую группу реципиентов для трансплантации сердца, которая позволяет увеличить продолжительность жизни. На благоприятный исход оперативного вмешательства можно рассчитывать у больных без сопутствующих системных заболеваний, нарушений психики, высокого необратимого сопротивления легочных сосудов, у пациентов в возрасте моложе 60 лет.
ЛИТЕРАТУРА
- Амосова Е.Н. (1999) Кардиомиопатии. Книга плюс, Киев, 422 с.
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практическое руководство. Морион, Киев, 480 с.
- Мравян С.Р., Канвар С., Голухова Е.З. (1997) Клинико-инструментальные показатели в оценке прогноза миокардита и дилатационной кардиомиопатии. Кардиология, 7: 67-72.
- Наказ № 436 Міністерства охорони здоров’я України від 03.07.2006 р. «Про затвердження протоколів надання медичної допомоги за спеціальністю «Кардіологія» (2006) Укр. кардіол. журн., 6; 89-115.
- Оганов Р.Г., Фомина И.Г. (ред.) (2006) Болезни сердца: руководство для врачей. Литтерра, Москва, 1328 с.
- Ardehali H., Kasper E.K., Baughman K.L. (2005) Diagnostic approach to the patient with cardiomyopathy: whom to biopsy. Amer. Heart J., 149: 7-12.
- Ardehali H., Qasim A., Cappola T. et al. (2004) Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Amer. Heart J., 147: 919-923.
- Aukrust P., Ueland T., Lien E. et al. (1999) Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Amer. J. Cardiol., 83: 376-382.
- Burkett E.L., Hershberger R.E. (2005) Clinical and genetic issues in familial dilated cardiomyopathy. J. Amer. Coll. Cardiol., 45: 969-981.
- Charron Ph., Tesson F., Poirier O. et al. (1999) Identification of a genetic risk factor for idiopathic dilated cardiomyopathy. Europ. Heart J., 20: 1587-1591.
- Chojnowska L., Ruzyllo W. (2000) Rodzinna kardiomiopatia przerostowa. Kardiol. Pol., 53(III): 88-95.
- Elliot P. (2000) Cardiomyopathy. Diagnosis and management of dilated cardiomyopathy. Heart, 84: 106-112.
- Elliot P., Anderson B., Arlustini E. et al. (2008). Classification of the cardiomyopathy; a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J., 29: 270-276.
- Galderisi M., Mondillo S. (2007) Echocardiography in clinical practice. One Way S.r.l., 120.
- Giordano F.J. (2005) Oxygen, oxidative stress, hypoxia and heart failure. J. Clin. Invest., 115: 500-508.
- Graham R.M., Owens W.A. (1999) Pathogenesis of inherited forms of dilated cardiomyopathy. N. Engl. J. Med., 341: 1759-1762.
- Gustafson A.B., Gottlieb R.A. (2004) Mechanisms of apoptosis in the heart. J. Clin. Immunol., 23: 447-459.
- Hannuksela J., Leppilampi M., Peuhkurinen K. et al. (2005) Hereditary hemochromatosis gene (HFE) mutations C282Y, H63D and S65C in patients with idiopathic dilated cardiomyopathy. Eur. J. Heart Fail., 7: 103-108.
- Kamisago M., Sharma S.D., DePalma S.R. et al. (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med., 34: 1688-1696.
- Keller D.I., Carrier i., Schwartz K. (2002) Genetics of familial cardiomyopathies and arrhythmias. Swiss. Med. Weekly, 132: 401-407.
- Mahon N.G., Murphy R.T., MacRae C.A. et al. (2005) Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann. Intern. Med., 143: 108-115.
- Manolio T.A., Baughman K.L., Rodeheffer R. et al. (1992) Prevalence and etiology of idiopathic dilated cardiomyopathy. Amer. J. Cardiol., 69: 1458-1466.
- Maron B.J., Towbin J.A., Thiene G. et al. (2006) Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation, 113: 1807-1816.
- Mogensen J., Murphy R.T., Shaw T. et al. (2004) Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Amer. Coll. Cardiol., 44: 2033-2040.
- Morita H., Seidman J., Seidman C.E. (2005) Genetic causes of human heart failure. J. Clin. Invest., 115: 518-526.
- Muller J., Wallukat G., Dandel M. et al. (2000) Immunoglobulin absorption in patients with idiopathic dilated cardiomyopathy. Circulation, 101: 385-391.
- Nelson G.S., Berger R.D., Febics B.J. et al. (2000) Left ventricular or biventricular pacing improves cardiac function at diminished energy cost in patients with dilated cardiomyopathy and left bundle-branch block. Circulation, 102: 3053-3059.
- Report of the WHO/ISFC task force on the definition and classification of cardiomyopaties (1996) Circulation, 93: 841-842.
- Tesson F., Charron P., Peuchmaurd M. et al. (1999) Characterization of a unique genetic variant in the beta-1 adrenoceptor gene and evaluation of its role in idiopathic dilated cardiomyopathy. J. Mol. Cell Cardiol., 31: 1025-1032.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Williams&Wilkins, Lippincott, 1628 p.