Аритмогенная кардиомиопатия ПЖ — заболевание мышцы сердца, характеризующееся частичным или полным прогрессирующим фиброзно-жировым замещением миокарда ПЖ, позднее — вовлечением в процесс ЛЖ с относительной интактностью перегородки.
Эпидемиология
Заболевание недавно идентифицировано и трудно диагностируется, поэтому его распространенность точно неизвестна, но считается, что может варьировать в пределах от 1:3000 до 1:10 000, соотношение мужчины : женщины составляет 2,5:1. Первые клинические проявления могут возникнуть в юношеском возрасте, редко — старше 40 лет.
Этиология
Точная причина заболевания неизвестна, однако в некоторых семьях существуют несомненные доказательства его наследования. В большинстве семей, где более одного заболевшего, наиболее вероятным типом наследования является аутосомно-доминантный. Описан также по крайней мере один хорошо известный вариант аритмогенной кардиомиопатии ПЖ, который наследуется по аутосомно-рецессивному типу.
Генетическими исследованиями идентифицировано по крайне мере 7 локусов генов, ответственных за развитие заболевания. С аритмогенной кардиомиопатией ПЖ также ассоциируются мутации генов, кодирующих белки вставочных дисков (десмоплакин, плакоглобин со специфическим фенотипом, плакофилин, десмоглеин, десмоколлин). Признаки заболевания могут варьировать даже у членов одной семьи, и патология может проявиться через поколения. Считается, что занятия спортом не могут вызывать аритмогенную кардиомиопатию ПЖ, тем не менее заболевание чаще регистрируют среди спортсменов. Мутации генов рианодиновых рецепторов сердца (RyR2) ассоциируются с полиморфной желудочковой тахикардией, вызванной физическими нагрузками и ювенильной внезапной смертью.
Патологическая анатомия
При морфологическом исследовании сердца вовлеченным оказывается чаще ПЖ, который имеет пятнистый вид: измененные участки могут быть окружены нормальными тканями. Вовлечение ПЖ может быть регионарным (20%) или диффузным (80%). Миокард ПЖ прогрессивно редуцируется, замещаясь жировой и фиброзной тканью, которая отличается от нефиброзной жировой инфильтрации, возникающей в ПЖ с возрастом. На ранних стадиях заболевания стенки правых отделов сердца утолщаются, но в дальнейшем из-за накопления жировой ткани и появления участков дилатации они становятся более тонкими (рис. 12.1а, б).
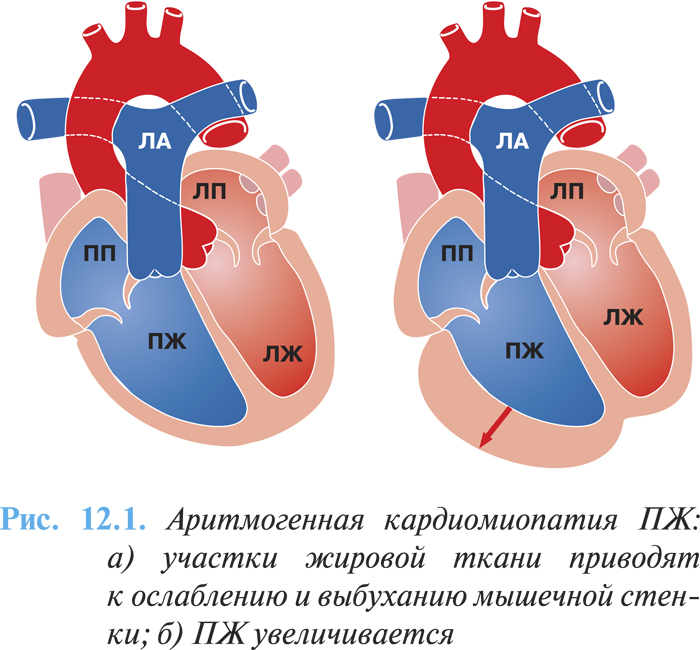
Жировое перерождение миокарда распространяется чаще от эпикардиальных слоев к эндокарду. Миокард поражается преимущественно в области выносящего тракта, верхушки и субтрикуспидальной зоны, которые рассматриваются в качестве «треугольника дисплазии».
При аритмогенной кардиомиопатии ПЖ липоматоз сопровождается преимущественно дилатацией выносящего тракта ПЖ или генерализованной дилатацией. Фибролипоматоз характеризуется наличием фокальной аневризмы ПЖ и выпячиванием в области верхушки, нижней стенки, субтрикуспидальной и инфундибулярной зоны.
По мере прогрессирования фиброзно-жировая дистрофия поражает также ЛЖ и предсердия.
Патогенез
Среди молекулярных механизмов аритмогенной кардиомиопатии ПЖ рассматриваются генетически детерминируемые мутации в десмосомальных протеинах, а также ингибирование сигнальных путей. Стресс-индуцируемый разрыв десмосомальных связей клеток может запускать процесс апоптоза, вызывать атрофию миокарда и замещение его жировой тканью.
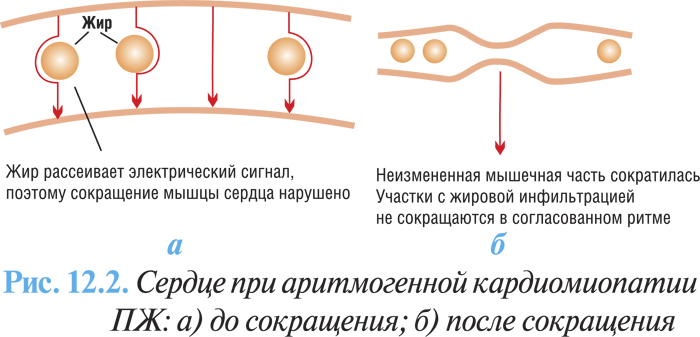
Очаги жирового перерождения и интерстициального фиброза при аритмогенной кардиомиопатии ПЖ не проводят электрические импульсы, вследствие чего дезорганизованная структура сердца обусловливает возникновение беспорядочной электрической активности, электрические импульсы могут становиться рассеянными, вследствие чего, помимо нарушений ритма сердца, могут возникать нарушения его сократимости (рис. 12.2а, б).
Клиническая картина
Основными клиническими симптомами аритмогенной кардиомиопатии ПЖ являются:
- ощущение сердцебиения, перебоев в работе сердца, приступы желудочковой тахикардии;
- повышенная утомляемость, головокружение, обмороки;
- симптомы СН;
- внезапная остановка кровообращения.
Описаны 4 клинические стадии заболевания:
- субклиническая, незначительные желудочковые аритмии могут отмечаться или отсутствовать;
- стадия явных электрических нарушений, правожелудочковые аритмии и риск остановки сердца связаны с морфофункциональными изменениями ПЖ;
- стадия правожелудочковой недостаточности с прогрессирующим вовлечением ПЖ и последующей его глобальной систолической дисфункцией;
- стадия конечной бивентрикулярной СН.
Диагностика
На ЭКГ определяются:
- спонтанные желудочковые тахикардии с изменением комплекса QRS по типу блокады левой ножки пучка Гиса;
- отрицательные зубцы Т в отведениях V1–4 на фоне синусового ритма;
- уширение комплекса QRS;
- неполная блокада правой ножки пучка Гиса;
- эктопические тяжелые аритмии: желудочковая экстрасистолия, фибрилляция желудочков, предсердная тахикардия, фибрилляция предсердий.
Приблизительно у ⅓ пациентов регистрируется характерная эпсилон-волна и ППЖ.
Методом холтеровского мониторирования можно диагностировать эпизоды желудочковой тахиаритмии. Для оценки прогрессирования заболевания важно проводить регистрацию ЭКГ в динамике.
При эхоКГ-исследовании выявляются:
- дилатация ПЖ и нарушение его сократимости (асинергия, диффузная гипокинезия, снижение ФВ);
- локальная аневризма ПЖ;
- повышенная трабекулярность;
- трикуспидальная регургитация;
- эмболия ЛА;
- увеличение правого предсердия;
- левые отделы сердца чаще не изменены.
С помощью допплер-эхоКГ определяется нарушение диастолической функции ПЖ и трикуспидальная регургитация. Для более точной визуализации ПЖ применяют контрастную эхоКГ миокарда.
Методом МРТ визуализируются участки замещения миокарда жировой тканью, фокальное истончение стенки и локальные аневризмы. Продемонстрирована хорошая корреляция между результатами этого метода и результатами морфологического исследования миокарда.
Для подтверждения диагноза используют рентгенконтрастную вентрикулографию, при которой выявляют дилатацию ПЖ с сегментарными нарушениями его сокращения, выпячивания контура в области дисплазии и повышение трабекулярности.
При эндомиокардиальной биопсии определяют фиброзно-жировую инфильтрацию миокарда ПЖ.
Из-за трудностей и риска проведения биопсии для подтверждения диагноза «аритмогенная кардиомиопатия ПЖ», а также неточностей в оценке структуры и функции ПЖ с помощью неинвазивных тестов Европейским кардиологическим обществом и Международным обществом и кардиологической федерацией разработаны критерии, согласно которым диагноз устанавливают при наличии 2 больших или 1 большого + 2 малых или 4 малых диагностических критериев (Corrado D. et al., 2000).
Большие диагностические критерии:
- семейный характер заболевания, подтвержденный данными аутопсии или при хирургическом вмешательстве;
- эпсилон-волна или локализованное уширение комплекса QRS (>110 мс) в правых грудных отведениях (V1–V3);
- фибролипоматозное замещение миокарда по данным эндомиокардиальной биопсии;
- значительная дилатация и снижение ФВ ПЖ при отсутствии или минимальном вовлечении ЛЖ;
- локализованная аневризма ПЖ;
- выраженная сегментарная дилатация ПЖ.
Малые диагностические критерии:
- наличие в семейном анамнезе случаев преждевременной внезапной смерти (у лиц в возрасте моложе 35 лет) вследствие предполагаемой аритмогенной кардиомиопатии ПЖ;
- ППЖ на усредненной ЭКГ;
- инвертированный зубец Т в правых грудных отведениях у лиц в возрасте старше 12 лет при отсутствии блокады правой ножки пучка Гиса;
- желудочковая тахикардия с признаками блокады левой ножки пучка Гиса, документированная по ЭКГ или результатам холтеровского мониторирования или во время нагрузочного теста;
- частые желудочковые экстрасистолы (>1000/24 ч при холтеровском мониторировании ЭКГ);
- умеренная глобальная дилатация или снижение ФВ ПЖ при неизмененном ЛЖ;
- умеренная сегментарная дилатация ПЖ;
- регионарная гипокинезия ПЖ.
Лечение
Для выбора антиаритмической терапии необходимо проведение инвазивного ЭФИ и проб с дозированной физической нагрузкой.
Среди антиаритмических препаратов эффективны амиодарон и соталол. Дигоксин применяют при тахисистолической форме фибрилляции предсердий для замедления ЧСС. Для восстановления синусового ритма проводят кардиоверсию.
Диуретики применяют при СН у больных с задержкой жидкости.
Из хирургических методов лечения применяют абляцию, если источник нарушенной электрической активности идентифицирован с помощью электрофизиологических тестов. В случаях если аритмии не контролируются с помощью лекарственных средств или абляции (обширное поражение или наличие множественных аритмогенных очагов), вшивают имплантируемый кардиовертерный дефибриллятор, в некоторых случаях требуется имплантация водителя ритма. Трансплантацию сердца применяют редко, если контроль ритма невозможен другими методами.
Прогноз
Результаты недавно проведенного исследования, включавшего 130 пациентов с аритмогенной кардиомиопатией ПЖ, показали, что сердечно-сосудистая смертность составила 16% (n=24), наиболее частой причиной была внезапная смерть (29%) и СН (59%). Анализ факторов риска выявил самые неблагоприятные — наличие дисфункции ПЖ или ЛЖ и желудочковую тахикардию (Hulot J.S. et al., 2004).
ЛИТЕРАТУРА
- Коваленко В.Н., Несукай Е.Г. (2001) Некоронарогенные болезни сердца. Практ. руководство. Морион, Киев, 480 с.
- Пархоменко А.Н. (1998) Аритмогенная кардиомиопатия правого желудочка: диагностика, лечение, прогноз. Кардиология, 2: 59-64.
- Angelini A., Thiene G., Boffa G.M. et al. (1993) Endomyocardial biopsy in right ventricular cardiomyopathy. Int. J. Cardiol., 40: 274-282.
- Auffermann W., Wichter T., Breithardt G. et al. (1993) Arrhythmogenic right ventricular disease: MR imaging versus angiography. Amer. J. Radiol., 161: 549-555.
- Beffagna G., Occhi G., Nava А. et al. (2005) Regulatory mutations in transforming growth fаctоr-β3 gene cause агrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res., 65: 366-373.
- Blake L.M., Scheinman M.M., Higgins C.B. (1994) MR features of arrhythmogenic right ventricular dysplasia. Amer. J. Radiol., 162: 809-812.
- Bomma C., Rutberg J., Tandri H. et al. (2004) Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Cardiovasc. Electrophysiol., 15: 300-306.
- Breithardt G., Wichter T., Haverkamp W. et al. (1994) Implantable cardioverter defibrillator therapy in patients with arrhythmogenic right ventricular cardiomyopathy? Long QT syndrome or no structural heart disease. Amer. Heart J., 127: 1151-1158.
- Castillo E., Tandri H., Rodriguez E.R. et al. (2004) Arrhythmogenic right ventricular dysplasia: ex vivo and in vitro fat detection with black-blood MR imaging. Radiology, 232: 38-48.
- Cooper L.T., Baughman K.L., Feldman A.M. et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease. Eur. Heart J., 28: 3076-3093.
- Corrado D., Fontaine G., Marcus F.I. et al. (2000) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation, 101: E101-E106.
- Danieli G.A., Rampazzo A. (2002) Genetics of arrhythmogenic right ventricular cardiomyopathy. Curr. Opin. Cardiol.,17: 218-221.
- Elliott P., Andersson B., Arbustini E. et al. (2008) Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J., 29: 270-276.
- Galderisi M., Mondillo S. (2007) Echocardiography in clinical practice. One Way S.r.l., 120 p.
- Haverkamp W., Rolf S., Osterziel K. et al. (2005) Die arrhythmogene rechts-ventriculare kardiomyopathie. Herz., 30: 565-570.
- Hodgkinson К.А., Parfrey P.S., Bassett A.S. et al. (2005) The impact of implantabIe cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J. Amer. Coll. Cardiol., 45: 400-408.
- Hulot J.S., Jouven X., Empana J.P. et al. (2004) Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation, 110: 1879-1884.
- Kinoshita O., Fontaine G., Rosas F. et al (1995) Time- and frequency domain analysis of the signal-averaged ECG in patients with arrhythmogenic right ventricular dysplasia. Circulation., 91: 715-721.
- Leclercq J.F., Coumel P.(1993). Late potentials in arrhythmogenic right ventricular dysplasia. Europ.Heart J.,14, (H):67-93.
- Mallat Z., Tedgui A., Fontaliran F. et al. (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. New Engl. J. Med., 335: 1224-1226.
- Maron B.J., Towbin J.A., Thiene G. et al. (2006) Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation., 113: 1807-1816.
- Paul М., Schulze-Bahr Е., Breithardt G. et al. (2003) Genetics of arrhythmogenic right ventricular cardiomyopathy — status quo and future perspectives. Z. Kardiol. 92: 128-136.
- Peters S., Trummel М. (2003) Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: value of standard ECG revisited. Аnn. of Noninvasive Electrocardiol., 8: 238-245.
- Piccini J.P., Nasir K., Bomma C. et al.(2005) Electrocardiographic findings over time in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Amer. J. Cardiol. 96:122-126.
- Sen-Chowdhry S., Syrris P., McKenna W.J. (2005) Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol., 16: 927-935.
- Tansey D.K., Aly Z., Sheppard M.N. (2005) Fat in the right ventricle of the normal heart. Histopathology, 46: 98-104.
- Topol E.J. (Ed.) (2007) Textbook of cardiovascular medicine. 3th ed. Lippincott Williams&Wilkins, 1628 p.
- Turrini Р., Corrado О., Basso С. et al. (2003) Noninvasive risk stratification in arrhythmogenic right ventricular cardiomyopathy. Ann. Noninvasive Electrocar-diol., 8:161-169.
- Wichter Т., Paul М., Wollmann С. et al. (2004) lmplantabIe cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single center ехреrience of long-term follow-up and complications in 60 patients. Circulation., 109: 1503-1508.
- Yoerger D.M., Marcus F., Sherill D. et al. (2005) Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insight from multidisciplinary study of right ventricular dysplasia. J. Amer. Coll. Cardiol., 45: 860-865.