Саксенда® (Saxenda®) (557707) - инструкция по применению ATC-классификация
Саксенда инструкция по применению
Фармакологические свойства
фармакодинамика.
Механизм действия
Лираглутид является ацилированным аналогом ГПП-1 с последовательностью аминокислот на 97% гомологичной эндогенной человеческой ГПП-1, которая связывается с ГПП-1-рецепторами и активирует их.
ГПП-1 является физраствором аппетита и потребления пищи, но точный механизм его действия полностью не установлен. В исследованиях на животных периферическое введение лираглутида привело к его накоплению в специфических участках мозга, отвечающих за регуляцию аппетита, где лираглутид благодаря специфической активации рецептора ГПП-1Р повышал ощущение насыщения и снижал сигналы голода, что способствовало снижению массы тела.
Рецепторы ГПП-1 также экспрессируются в определенных участках сердца, сосудах, иммунной системе и почках. При моделировании атеросклероза у мышей лираглутид предотвращал прогрессирование аортальной бляшки и снижал воспаление в бляшке. Кроме того, лираглутид оказывал положительное влияние на липиды плазмы крови. Лираглутид не уменьшал размер уже имеющихся бляшек.
Фармакодинамические эффекты
Снижение массы тела происходит благодаря преимущественной потере висцерального жира по сравнению с подкожным. Лираглутид регулирует аппетит, усиливая чувство сытости и наполненности желудка, снижая при этом чувство голода и приводит к снижению потребления пищи. Лираглутид не увеличивает энергозатраты по сравнению с плацебо.
Лираглутид стимулирует секрецию инсулина и уменьшает чрезмерно высокую секрецию глюкагона в зависимости от уровня глюкозы, что приводит к снижению глюкозы натощак и после еды.
У пациентов с преддиабетом и сахарным диабетом эффект снижения уровня глюкозы более выражен по сравнению с пациентами с нормогликемией. Клинические испытания свидетельствуют, что лираглутид улучшает и поддерживает функцию бета-клеток в соответствии с НОМА-В и соотношение проинсулин/инсулин.
Клиническая эффективность и безопасность
Клиническая эффективность и безопасность лираглутида при применении для снижения массы тела как дополнение к диете с пониженной калорийностью и увеличенной физической активностью были изучены в четырех рандомизированных двойных слепых плацебо-контролируемых исследованиях фазы 3 с участием 5358 пациентов.
Исследование 1 (SCALE Ожирение и преддиабет – 1839):
Всего 3731 пациент с ожирением (индекс массы тела (ИМТ) ≥30 кг/м2) или с избыточной массой тела (ИМТ ≥27 кг/м2), с дислипидемией и/или АГ, были стратифицированы с помощью скрининга в соответствии к статусу преддиабета и начального уровня ИМТ (≥30 кг/м2 или ≤30кг/м2). Все пациенты (3731) были рандомизированы по продолжительности лечения 56 недель, а 2254 пациента с преддиабетом, при скрининге были рандомизированы на 160 недель лечения. Оба периода лечения сопровождались 12-недельным периодом наблюдения за группами препарат/плацебо. Коррекция образа жизни посредством диеты с пониженной калорийностью и увеличенной физической активностью была базовой терапией для всех пациентов.
56-недельное исследование 1 показало потерю массы тела у всех (3731) рандомизированных пациентов (2590 пациентов завершили курс лечения). 160-недельное исследование 1 показало время до развития сахарного диабета 2 типа у 2254 рандомизированных пациентов, больных преддиабетом (1128 пациентов завершили курс лечения).
Исследование 2 (SCALE Диабет – 1922):
Исследование продолжительностью 56 недель, при котором оценивали потерю массы тела у 846 рандомизированных пациентов с ожирением и избыточной массой тела (628 пациентов завершили курс лечения), которые имели недостаточно контролируемый сахарный диабет 2 типа (HbA1c в диапазоне 7-10%). Основным методом лечения в начале исследования была либо диета, либо увеличенная физическая активность, либо применение отдельных лекарственных средств, таких как метформин, сульфонилмочевина и глитазон, или их комбинаций.
Исследование 3 (SCALE Апноэ во время сна – 3970):
Исследование продолжительностью 32 недели, в котором оценивали степень тяжести апноэ во время сна и снижение массы тела у 359 рандомизированных пациентов (276 пациентов завершили курс лечения), страдавших ожирением и средней или тяжелой степенью обструктивного апноэ во время сна.
Исследование 4 (SCALE Поддерживающее лечение – 1923):
Исследование продолжительностью 56 недель, в котором оценивали поддерживающее лечение после потери массы тела ≥5%, вызванной диетой с пониженной калорийностью у 422 рандомизированных пациентов с ожирением и избыточной массой тела (305 пациентов завершили курс лечения) с АГ или дислипидемией.
Масса тела
При применении лираглутида было достигнуто снижение массы тела по сравнению с плацебо у пациентов с ожирением и избыточной массой тела во всех исследуемых группах. У большинства пациентов потеря массы тела достигала ≥5% и >10% при применении лираглутида по сравнению с плацебо. В течение 160 недель исследования 1 подавляющая потеря массы тела произошла в первый год терапии и продолжалась в течение всех 160 недель.
• В исследовании 1 средняя потеря массы тела на 56-й неделе составила 8,0% (8,4 кг) при применении лираглутида и 2,6% (2,8 кг) при применении плацебо (расчетная разница при лечении (РРЛ) (средняя потеря у процентах): -5,4 [95% ДИ -5,8; -5,0], р<0,0001, РРЛ (середня потеря в килограммах): -5,6 [95% ДИ -6, 0; -5,1], p<0,0001). Часть пациентов, потерявших 5% и 10% массы тела на 56-й неделе, составляла 63,5% и 32,8% соответственно при применении лираглутида против 26,6% и 10,1% соответственно при применении плацебо (коэффициент вероятности (потери ≥5% массы тела): 4,8 [95% ДИ 4,1; 5,6], р <0,0001, коэффициент вероятности (потери >10% массы тела): 4,3 [95% ДИ 3,5; 5,3] р <0,0001).
• В исследовании 1 средняя потеря массы тела на 160 неделе составила 6,2% (6,5 кг) при применении лираглутида и 1,8% (2,0 кг) при применении плацебо РРЛ (средняя потеря в процентах): -4,3 [95 % ДИ –4,9; -3,7], р< 0,0001), РРЛ (средняя потеря в килограммах): -4,6 [95% ДИ -5,3; -3,9], p<0,0001). Часть пациентов, потерявших 5% и 10% массы тела на 160-й неделе, составляла 49,6% и 24,4% соответственно при применении лираглутида против 23,4% и 9,5% соответственно при применении плацебо (коэффициент вероятности ( потери ≥5% массы тела): 3,2 [95% ДИ 2,6; 3,9], р<0,0001, коэффициент вероятности (потери >10% массы тела): 3,1 3;4,1] р<0,0001).
• В исследовании 2 средняя потеря массы тела на 56-й неделе составила 5,9% (6,2 кг) при применении лираглутида и 2,0% (2,2 кг) при применении плацебо РРЛ (средняя потеря в процентах): -4 ,0 [95% ДИ –4,8; -3,1], р <0,0001), РРЛ (средняя потеря в килограммах): -4,1 [95% ДИ -5,0; -3,1], p<0,0001). Часть пациентов, потерявших 5% и 10% массы тела на 56-й неделе, составляла 49,8% и 22,9% соответственно при применении лираглутида против 13,5% и 4,2% соответственно при применении плацебо (коэффициент вероятности ( потери ≥5% массы тела): 6,4 [95% ДИ 4,1; 10,0], р <0,0001, коэффициент вероятности (потери >10% массы тела): 6,8 4;13,8] г <0,0001).
• В исследовании 3 средняя потеря массы тела на 32 неделе составила 5,7% (6,8 кг) при применении лираглутида и 1,6% (1,8 кг) при применении плацебо РРЛ (средняя потеря в процентах): -4 ,2 [95% ДИ –5,2; -3,1], р <0,0001), РРЛ (средняя потеря в килограммах): -4,9 (95% ДИ -6,2; -3,7], p<0,0001). Часть пациентов, потерявших 5% массы тела на 32-й неделе, составляла 46,4% при применении лираглутида против 18,1% соответственно при применении плацебо (оцененное соотношение шансов: 3,9 [95% ДИ 2,4; 6,4], <0,0001)
• В исследовании 4 большинство пациентов поддерживали массу тела, достигнутую в начале лечения лираглутидом по сравнению с плацебо (81,4% и 48,9% соответственно). Средняя потеря массы тела на 56-й неделе составила 6,3% (6,0 кг) при применении лираглутида и 0,2% (0,2 кг) при применении плацебо (РРЛ (средняя потеря в процентах)): -6,1 [95% ДИ -7,5; -4,6], р 0,0001), РРЛ (середня втрата у кілограмах): -5,9 [95% ДИ -7,3; -4,4], p<0,0001). Часть пациентов, потерявших 5% и 10% массы тела на 56-й неделе, составляла 50,7% и 27,4% соответственно при применении лираглутида против 21,3% и 6,8% соответственно при применении плацебо (коэффициент вероятности (потеря ≥5% массы тела): 3,8 [95% ДИ 2,4;6,0], р 0,0001, коефіцієнт вірогідності (втрати >10% маси тіла): 5,1 [95% ДИ 2,7;9,7] p <0,0001).
Данные по потере массы тела, срока лечения и кумулятивного распределения изменения массы тела (%) представлены на рисунках 1, 2 и 3.
Потеря массы тела через 12 недель при лечении лираглутидом (3,0 мг)
После 12 нед применения лираглутида зарегистрирована потеря массы тела ≥5% (4 недели — эскалация дозы и 12 недель — лечебная доза) у 67,5% пациентов в исследовании 1 продолжительностью 56 недель. В исследовании 2 50,4% пациентов достигли потери массы тела ≥5%, через 12 недель. После применения лираглутида в течение года у 86,2% пациентов снижение массы тела составило ≥5%, а у 51% пациентов — ≥10%. Средняя потеря массы тела у пациентов, применявших лираглутид в течение 1 года, составила 11,2% от их исходной массы тела (9,7% для мужчин и 11,6% для женщин). Доля пациентов, достигших потери массы тела после 12 недель терапии <5% и после года применения лираглутида не достигших потери массы тела ≥10%, составила 93,4%.
Контроль гликемии
Лечение лираглутидом значительно улучшило показатели гликемии у пациентов с нормогликемией, преддиабетом и сахарным диабетом 2 типа. В 56-недельной части исследования 1 сахарный диабет 2 типа развился у меньшего количества пациентов, получавших лираглутид, по сравнению с пациентами, получавшими плацебо (0,2% против 1,1% соответственно). У большинства пациентов с преддиабетом в начале лечения наблюдалось обратное развитие данного заболевания после применения лираглутида по сравнению с плацебо (69,2% против 32,7% соответственно). По сравнению с начальным значением HbA 1c у 5,6% пациентов, применявших лираглутид, наблюдалось снижение среднего значения HbA1c на 56-й неделе на -0,3% против -0,1% пациентов, применявших плацебо (РРЛ: - 0,23 [95 % ДИ -0,25; -0,21], р<0,0001). По сравнению с начальным уровнем глюкозы в плазме крови натощак (ГПН) 5,3 ммоль/л у пациентов, применявших лираглутид, наблюдалось снижение ОПН в среднем на -0,4 ммоль/л против -0,01 ммоль/л у пациентов, применявших плацебо на 56-й неделе (РРЛ: -0,38 [95% ДИ -0,42; -0,35], р<0,0001).
Показателем первичной эффективности 160-недельной части исследования 1 являлось отношение количества пациентов, у которых возник сахарный диабет 2 типа, к периоду проявления. На неделе 160 исследования, 3% пациентов, получавших Саксенда® и 11% пациентов, получавших плацебо, были диагностированы с диабетом 2 -го типа. Приблизительный период развития сахарного диабета 2 типа у пациентов, получавших лираглутид 3 мг, был в 2,7 раза длиннее (95% ДИ [1,9; 3,9]), а коэффициент риска развития сахарного диабета 2 типа составил 0,2 для лираглутида по сравнению с плацебо. По сравнению с начальным значением HbA1c у 5,8% пациентов, применявших лираглутид, и у 5,7% пациентов, применявших плацебо, наблюдалось снижение значения HbA1c на 160-й неделе в среднем на 0,4% и 0,1% соответственно (РРЛ: -0,21 [95% ДИ -0,24; -0,18], р<0,0001). По сравнению с начальным значением ГПН (5,5 ммоль/л) у пациентов, применявших лираглутид, наблюдалось снижение ОПН в среднем на 0,4 ммоль/л против 0,04 ммоль/л у пациентов, применявших плацебо на 160-м. недели (РРЛ: -0,4 [95% ДИ -0,5; -0,4], р<0,0001). В исследовании 2, по сравнению с начальным значением HbA1c, у 7,9% пациентов, применявших лираглутид, наблюдалось снижение среднего значения HbA1c на 56-й неделе на 1,3% против 0,4% пациентов, применявших плацебо (РРЛ): -0,9 [95% ДИ -1,1;-0,8], р<0,0001). По сравнению с начальным значением ГПН 8,8 ммоль/л для пациентов, применявших лираглутид, и 8,6 ммоль/л для пациентов, применявших плацебо, у пациентов, применявших лираглутид, наблюдалось снижение ГПН в среднем на 1,9 ммоль/ л против 0,1 ммоль/л у пациентов, применявших плацебо на 56-й неделе (РРЛ: -1,8 [95% ДИ -2,1; -1,4], р<0,0001).
Кардиометаболические факторы риска
Лечение лираглутидом значительно улучшило показатели систолического АД, также наблюдалось уменьшение окружности талии по сравнению с плацебо. В исследовании 1 первоначальное значение систолического АД составляло 123,00 мм рт. ст., на 56-й неделе наблюдалось снижение систолического АД в среднем на 4,3 мм рт. ст., и на 1,5 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -2,8 [95% ДИ -3,6; -2,1], р<0,0001). Начальное значение диастолического АД составляло 78,7 мм рт. ст. у пациентов, применявших лираглутид, и 78,9 мм рт. ст. у пациентов, применявших плацебо; на 56-й неделе наблюдалось снижение диастолического АД на -2,7 мм рт. ст. и 1,8 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -0,9 [95% ДИ -1,4; -0,4], р<0,05). Первоначальное значение окружности талии составляло 115,0 см для пациентов, применявших лираглутид, и 114,5 см для пациентов, применявших плацебо; на 56-й неделе наблюдалось уменьшение окружности талии на -8,2 см и -4,0 см у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -4,2 [95% ДИ -4,7; -3, 7], р<0,0001). В исследовании 1 первоначальное значение систолического АД составляло 124,8 мм рт. ст. для пациентов, применявших лираглутид, и 125,0 мм рт. ст. для пациентов, применявших плацебо; на 160-й неделе наблюдалось снижение систолического АД на -3,2 мм рт. ст., и на -0,4 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -2,8 [95% ДИ -3,8; -1,8], р<0,0001). Начальное значение диастолического АД составляло 79,4 мм рт. ст. для пациентов, применявших лираглутид, и 79,8 мм рт. ст. для пациентов, применявших плацебо; на 160-й неделе наблюдалось снижение диастолического АД на 2,4 мм рт. ст. и –1,7 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -0,6 [95% ДИ -1,3; -0,1]). Первоначальное значение окружности талии составляло 116,6 см для пациентов, применявших лираглутид, и 116,7 см для пациентов, применявших плацебо; на 160-й неделе наблюдалось уменьшение окружности талии на 6,9 см и на 3,4 см у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -3,5 [95% ДИ -4,2; -2,8] , р<0,0001). В исследовании 2 первоначальное значение систолического АД составляло 128,9 мм рт. ст. у пациентов, применявших лираглутид, и 129,2 мм рт. ст. у пациентов, применявших плацебо; на 56-й неделе наблюдалось снижение систолического АД на 3,0 мм рт. ст. и 0,4 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -2,6 [95% ДИ -4,6; -0,6], р<0,0001). Начальное значение диастолического АД составляло 79 мм рт. ст. для пациентов, применявших лираглутид, и 79,3 мм рт. ст. для пациентов, применявших плацебо; на 56-й неделе наблюдалось снижение диастолического АД на 1,0 мм рт. ст. и 0,6 мм рт. ст. у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -0,4 [95% ДИ -1,7; -1,0], р=0,5918). Первоначальное значение окружности талии составляло 118,1 см для пациентов, применявших лираглутид, и 117,3 см для пациентов, применявших плацебо; на 56-й неделе наблюдалось уменьшение окружности талии на 6,0 см и на 2,8 см у пациентов, применявших лираглутид и плацебо соответственно (РРЛ: -3,2 [95% ДИ -4,2; -2,2] , р<0,0001).
Индекс апноэ-гипное (ИАГ)
При применении лираглутида наблюдалось значительное снижение тяжести обструктивного апноэ во время сна по сравнению с плацебо, которое было оценено с помощью уменьшения по сравнению с плацебо (12,2 события/час для лираглутида против 6,1 события/час для плацебо (РРЛ: -6,1 [95% ДИ -11,0; -1,2], р<0,05).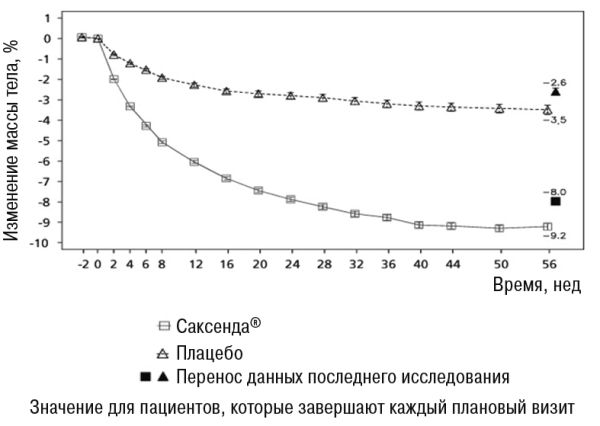
Рисунок 1. Изменение массы тела (%) от первоначального показателя по времени в исследовании 1 (0-56 недель)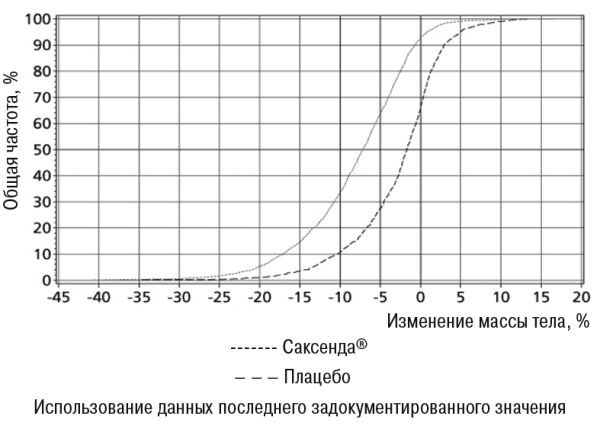
Рисунок 2. Кумулятивное распределение изменения массы тела (%) после 56 недель лечения в исследовании.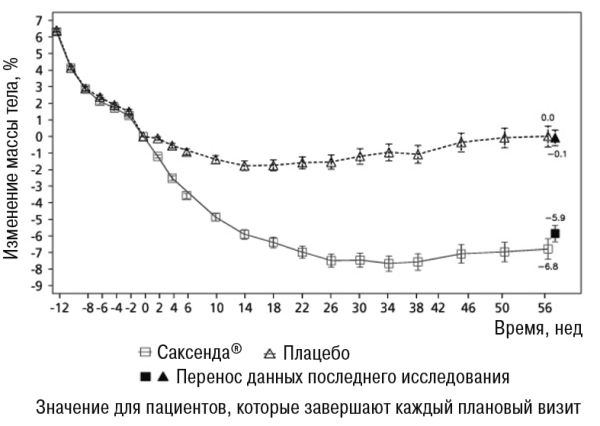
Рисунок 3. Изменение рандомизации (неделя 0) в массе тела (%) по времени в исследовании 4
До недели 0 пациенты находились только на диете с пониженной калорийностью, также для них была увеличена физическая активность. На неделе 0, пациенты были рандомизированы для получения препарата Саксенда® или плацебо.
Иммуногенность
Учитывая потенциальные иммуногенные свойства лекарственных средств, содержащих белки или пептиды, можно прогнозировать, что пациенты могут вырабатывать антитела к лираглутиду после применения препарата Саксенда®. Во время клинических исследований у 2,5% пациентов, получавших лираглутид, образовывались антитела к лираглутиду. Образование антител не приводило к понижению эффективности лираглутида.
Воздействие на сердечно-сосудистую систему
Основные серьезные неблагоприятные сердечно-сосудистые явления (МАСЕ), определенные внешней независимой экспертной группой: нелетальный инфаркт миокарда, нелетальный инсульт, летальный случай из-за сердечно-сосудистой патологии. Во всех длительных клинических испытаниях препарата Саксенда® сообщалось о 6 MACE у пациентов, получавших лираглутид, и 10 MACE у пациентов, получавших плацебо. Соотношение риска и ДИ 95% при сравнении Саксенда® и плацебо было 0,33 [0,12; 0,90].
В клинических исследованиях 3-й фазы наблюдалось незначительное повышение ЧСС на 2,5 уд/мин (от 1,6 до 3,6 уд./мин в отдельных исследованиях). Максимальное увеличение ЧСС наблюдалось приблизительно после 6 недель терапии. Длительное клиническое влияние увеличения ЧСС не установлено. Это увеличение было обратимым и исчезало после прекращения терапии лираглутидом (см. ОСОБЫЕ УКАЗАНИЯ).
В исследовании LEADER® участвовало 9340 пациентов с недостаточно контролируемым сахарным диабетом 2 типа. Подавляющее число из них страдали сердечно-сосудистыми заболеваниями. Пациентов рандомизированно распределяли для применения лираглутида в суточной дозе до 1,8 мг (4668) и плацебо (4672) для предоставления стандартной медицинской помощи в обеих группах рандомизации.
Продолжительность терапии составляла от 3,5 до 5 лет. Средний возраст пациентов составил 64 года, средний ИМТ — 32,5 кг/м2. Среднее значение начального уровня HbA1c составило 8,7 и улучшилось через 3 года на 1,2% у пациентов, которым был назначен лираглутид, и на 0,8% у пациентов, которым было назначено плацебо. Первичной конечной точкой эффективности было время от рандомизации до первого возникновения любых основных MACE: нелетального инфаркта миокарда, нелетального инсульта, летального случая из-за сердечно-сосудистой патологии. Лираглутид значительно снизил частоту возникновения основных неблагоприятных сердечно-сосудистых явлений (события первичной конечной точки, MACE) по сравнению с плацебо (3,41 против 3,90 на 100 пациенто-лет в группах лираглутида и плацебо соответственно), уменьшив риск на 13%, HR 0,87, [0,78; 0,97] [95% ДИ]) (р = 0,005) (см. рисунок 4).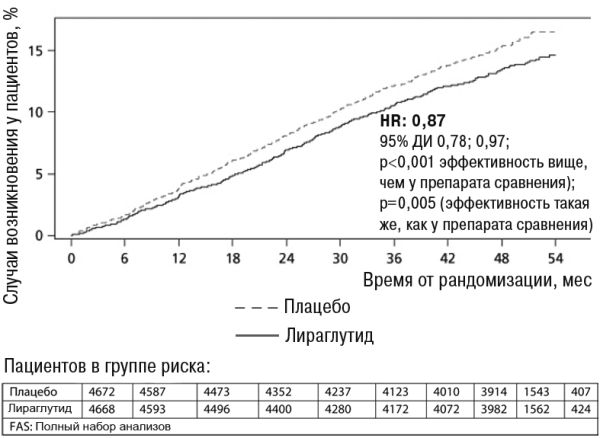
Рисунок 4. График времени Каплана-Майера в начале исследования MACE – FAS популяции
Дети Двойно слепое исследование эффективности и безопасности применения лекарственного средства Саксенда по сравнению с плацебо для снижения массы тела у пациентов детского возраста от 12 лет с ожирением показало, что лекарственное средство Саксенда является более эффективным, чем плацебо, для снижения массы тела (оценивалось по шкале) стандартного отклонения ИМТ или Z-score) через 56 недель лечения (табл. 1). Среди пациентов, принимавших лираглутид, больший процент пациентов достиг снижения ИМТ на ≥5% и ≥10%, чем среди пациентов, принимавших плацебо. В среднем снижение ИМТ и массы тела также было больше в группе пациентов, принимавших лираглутид (табл. 1). После 26 недель периода последующего наблюдения без применения исследуемого лекарственного средства наблюдалось восстановление массы тела у пациентов, принимавших лираглутид, по сравнению с принимающими плацебо (табл. 1) Таблица 1 Исследование 4180: изменения от исходного уровня массы тела и ИМТ на 56-й неделе и изменение по шкале стандартного отклонения ИМТ (BMI SDS) от 56-й недели до 82-й недели.
| Саксенда (N=125) | Плацебо (N=126) | Саксенда по сравнению с плацебо | |
| BMI SDS | |||
| Исходный показатель, BMI SDS (СВ) | 3,14 (0,65) | 3,20 (0,77) | |
| Среднее изменение на 56-й неделе (95% ДИ) | -0,23 | -0,00 | -0,22* (-0,37; -0,08) |
| 56-я неделя, BMI SDS (СВ) | 2,88 (0,94) | 3,14 (0,98) | |
| Среднее изменение от 56-й недели до 82-й недели, BMI SDS (95% ДИ) | 0,22 | 0,07 | 0,15** (0,07; 0,23) |
| Масса тела | |||
| Исходный показатель, кг (СВ) | 99,3 (19,7) | 102,2 (21,6) | - |
| Среднее изменение на 56-й неделе, % (95% ДИ) | -2,65 | 2,37 | -5,01** (-7,63; -2,39) |
| Среднее изменение на 56-й неделе, кг (95% ДИ) | -2,26 | 2,25 | -4,50** (-7,17; -1,84) |
| ИМТ | |||
| Исходный показатель, кг/м2 (СВ) | 35,3 (5,1) | 35,8 (5,7) | - |
| Среднее изменение на 56-й неделе, кг/м2 (95% ДИ) | -1,39 | 0,19 | -1,58** (-2,47; -0,69) |
| Процент пациентов со снижением на ≥5% исходного ИМТ на 56 неделе, % (95% ДИ) | 43,25 | 18,73 | 3,31** (1,78; 6,16) |
| Процент пациентов со снижением на ≥10% исходного ИМТ на 56 неделе, % (95% ДИ) | 26,08 | 8,11 | 4,00** (1,81; 8,83) |
Полная выборка пациентов. Для BMI SDS, массы тела и ИМТ исходные показатели являются средними величинами, изменения от исходного уровня на 56 неделе являются рассчитанными средними величинами (методом наименьших квадратов), а различия в лечении на 56 неделе — это рассчитанные разницы между методами лечения. Для BMI SDS показатели на 56-й неделе являются средними величинами, изменения от 56-й недели до 82-й недели являются рассчитанными величинами (методом наименьших квадратов), а отличия в лечении на 82-й неделе — это рассчитанные разницы между методами лечения. Для доли пациентов, потерявших ≥5%/≥10% исходного ИМТ, также приведено рассчитанное соотношение шансов. Пропущенные наблюдения интерполированы из группы пациентов, принимавших плацебо, на основе перехода к референционному подходу множественной (×100) подстановке данных. *p<0,01, **p<0,001. ДИ – доверительный интервал. СВ — стандартное отклонение. Согласно переносимости лекарственного средства 103 пациента (82,4%) увеличили дозу и продолжали принимать дозу 3,0 мг, 11 пациентов (8,8%) увеличили дозу и продолжали принимать дозу 2,4 мг, 4 пациента (3,2%). ) увеличили дозу и продолжали принимать дозу 1,8 мг, 4 пациента (3,2%) увеличили дозу и продолжали принимать дозу 1,2 мг и 3 пациента (2,4%) продолжали принимать дозу 0,6 мг. Через 56 недель лечения не было зарегистрировано никакого влияния на рост или половое созревание пациентов. Исследование с 16-недельным двойно слепым периодом и 36-недельным открытым периодом было проведено для оценки эффективности и безопасности применения лекарственного средства Саксенда педиатрическим пациентам с синдромом Прадера — Вилли и ожирением. Исследование включало 32 пациента в возрасте от 12 до 18 лет (часть А) и 24 пациента в возрасте от 6 до 12 лет (часть В). Пациенты были рандомизированы в соотношении 2:1 для приема лекарственного средства Саксенда или плацебо. Пациенты с массой тела менее 45 кг начали увеличивать дозу на меньшую величину (0,3 мг вместо 0,6 мг) и увеличивали до максимальной дозы 2,4 мг. Рассчитана разница в лечении по средним значениям ИМТ SDS на 16 неделе (часть A: -0,20 против -0,13, часть B: -0,50 против -0,44) и на 52 неделе (часть A: -0,31 против -0,17, часть B: -0,73 против -0,67) была сходной при применении лекарственного средства Саксенда и плацебо. Никаких дополнительных проблем безопасности при исследовании не было замечено. Европейское агентство по лекарственным средствам отложило обязательство подавать результаты исследований применения лекарственного средства Саксенда в одной или нескольких подгруппах детского населения при лечении ожирения и при лечении синдрома Прадера-Вилли (см. ПРИМЕНЕНИЕ).
Фармакокинетика.
Абсорбция
Абсорбция лираглутида после п/к введения происходит медленно, максимальная концентрация достигается через 11 ч после введения. У пациентов, страдающих ожирением (ИМТ 30-40 кг/м2), после введения 3 мг лираглутида его средняя равновесная концентрация (AUCt/24 ) достигала примерно 31 нмоль/л. Экспозиция лираглутида увеличивалась пропорционально дозе. Абсолютная биодоступность лираглутида после п/к введения составляет около 55%.
Распределение
Средний видимый объем распределения после подкожного введения составляет 20-25 л (для человека с массой тела примерно 100 кг). Лираглутид связывается преимущественно с белками плазмы крови (98%).
Метаболизм
В течение 24 ч после введения разовой дозы [3H]-лираглутида здоровым добровольцам основным компонентом в плазме крови был неизмененный лираглутид. В плазме крови было обнаружено в незначительном количестве два метаболита (≤9% и ≤5% от общего уровня радиоактивности в плазме крови).
Выведение
Лираглутид эндогенно метаболизируется, как все крупные белки, без участия специфического органа как основного пути элиминации. После введения дозы [3H]-лираглутида в моче и кали не было обнаружено неизмененного лираглутида. Только небольшая часть введенной радиоактивности в виде метаболитов лираглутида выводилась почками и через кишечник (6% и 5% соответственно). Радиоактивные вещества выводятся почками или через кишечник в основном в течение первых 6-8 суток в виде 3-х метаболитов. После однократного п/к введения лираглутида среднее значение клиренса составляет примерно 0,9-1,4 л/ч, Т½ — около 13 ч.
Особые группы пациентов
Пациенты пожилого возраста
На основании данных фармакокинетического анализа группы пациентов в возрасте от 18 до 82 лет с избыточной массой тела или ожирением был сделан вывод, что возраст не оказывает клинически значимого влияния на фармакокинетику лираглутида. Поэтому нет необходимости в коррекции дозы относительно возраста.
Пол
Данные фармакокинетического анализа показали, что у женщин наблюдается на 24% более низкий клиренс лираглутида по сравнению с мужчинами. На основании этих данных можно заключить, что коррекция дозы относительно пола не требуется.
Этническое происхождение
На основании данных фармакокинетического анализа группы пациентов европеоидной, монголоидной, латиноамериканской и негроидной рас с избыточной массой тела или ожирением был сделан вывод, что этническое происхождение не оказывает какого-либо существенного клинического влияния на фармакокинетику лираглутида.
Масса тела
Экспозиция лираглутида уменьшается при увеличении начальной массы тела. Как показали исследования, суточная дозировка лираглутида 3,0 мг обеспечивает нормальное системное влияние на организм пациента с массой тела 60-234 кг. Экспозиция лираглутида у пациентов с массой тела более 234 кг не изучалась.
Нарушение функции печени
Фармакокинетика лираглутида исследована у пациентов с разной степенью нарушений функции печени в процессе исследования с применением однократной дозы (0,75 мг). Было показано, что у пациентов с легкими и умеренными нарушениями функции печени экспозиция лираглутида снижалась на 13-23% по сравнению со здоровыми добровольцами. У пациентов с тяжелыми нарушениями функции печени (9 баллов по классификации Чайлда – Пью) экспозиция была значительно ниже (на 44%).
Нарушение функции почек
Экспозиция лираглутида была снижена у пациентов с нарушением функции почек по сравнению с лицами с нормальной почечной функцией в процессе исследования с применением однократной дозы (0,75 мг). У пациентов с легкими нарушениями (клиренс креатинина 50-80 мл/мин) экспозиция снижалась на 33%, с нарушениями умеренной тяжести (клиренс креатинина 30 — 50 мл/мин) – на 14%, с тяжелыми нарушениями (клиренс креатинина <30 мл) мин) – на 27%, а на конечных стадиях заболеваний почек, требующих проведения диализа – на 26%.
Дети
Фармакокинетические свойства лираглутида в дозе 3,0 мг были оценены в ходе клинических исследований с участием пациентов детского возраста ≥12 лет с ожирением (134 пациента с массой тела 62–178 кг). Экспозиция лираглутида у детей ≥12 лет была похожа на экспозицию у взрослых с ожирением. Фармакокинетические свойства также оценивались в ходе клинико-фармакологического исследования с участием пациентов детского возраста 7–11 лет с ожирением (13 пациентов, масса тела 54–87 кг). Была обнаружена сравнительная экспозиция при введении 3,0 мг лираглутида у взрослых, детей ≥12 лет и детей в возрасте 7-11 лет после коррекции массы тела. Доклинические данные по безопасности Доклинические данные, основанные на исследованиях по фармакологической безопасности, токсичности повторных доз и генотоксичности, не выявили риска для человека. В процессе двухлетних исследований канцерогенности у крыс и мышей были обнаружены опухоли С-клеток щитовидной железы, не приводившие к летальному исходу. Нетоксичная доза (NOAEL) у крыс не была установлена. У обезьян, получавших лечение в течение 20 мес, таких опухолей не обнаружено. Опухоли у грызунов обусловлены негенотоксическим специфическим ГПП-1-рецептор-опосредованным механизмом, к которому частично чувствительны грызуны. Значимость этого механизма у людей достаточно низкая, но не может быть полностью исключена. Развития других опухолей не было обнаружено. В процессе исследований на животных не было выявлено прямого вредного воздействия на фертильность, однако при введении высоких доз отмечалось незначительное повышение ранней эмбриональной летальности. Введение лираглутида в период середины беременности приводило к снижению массы тела самки, замедлению роста плода с невыясненным влиянием на развитие ребер у крыс и скелета у кроликов. При введении лираглутида отмечено замедление роста новорожденных крыс, сохраняющееся в период отлучения от кормления молоком в группе приема высокой дозы. Неизвестно, замедление ли роста новорожденных крыс обусловлено снижением потребления ими молока в результате прямого воздействия ГПП-1, или уменьшением молока у матери, что обусловлено снижением калорийности потребляемой пищи. У ювенильных крыс применение лираглутида приводило к задержке полового созревания как самцов, так и самок при клинически значимых концентрациях препарата в плазме крови. Такие задержки не влияли на фертильность и репродуктивную способность самок и самцов или способность самок вынашивать беременность.
Показания Саксенда
лекарственное средство Саксенда применяют для уменьшения массы тела как дополнение к диете с пониженной калорийностью и увеличенной физической активностью у взрослых пациентов с начальным индексом массы тела (ИМТ) более 30 кг/м2 (ожирение) или от 27 до 30 кг/м2 (чрезмерная масса тела) при наличии хотя бы одного сопутствующего заболевания, связанного с массой тела, такого как дисгликемия (преддиабет или сахарный диабет 2 типа), гипертензия, дислипидемия или обструктивное апноэ сна. Если через 12 нед после приема суточной дозы 3,0 мг пациент не потерял по меньшей мере 5% от начальной массы тела, применение лекарственного средства Саксенда следует прекратить. Дети ≥12 лет Лекарственное средство Саксенда можно применять в качестве дополнения к здоровому питанию и увеличенной физической активности для коррекции массы тела у пациентов детского возраста от 12 лет. • ожирением (ИМТ ≥30 кг/м2 для взрослых пациентов по международным предельно допустимым значениям)* и • массой тела более 60 кг. Лечение препаратом Саксенда следует прекратить и пересмотреть, если пациент не потерял по меньшей мере 4% своего ИМТ или ИМТ по оценке Z-score через 12 нед применения препарата в дозе 3,0 мг/сут или в максимальной переносимой дозе. * Предельно допустимые значения ИМТ, установленные Международной рабочей группой по изучению ожирения, для пациентов 12–18 лет с ожирением в разбивке по полу (см. табл. 2). Таблица 2 Предельно допустимые значения ИМТ, установленные Международной рабочей группой по изучению ожирения, для пациентов с ожирением в возрасте 12–18 лет в разбивке по полу.
| Возраст (лет) | ИМТ, который соответствует 30 кг/м2 для взрослых пациентов по международным предельно допустимым значениям | |
| Мальчики | Девочки | |
| 12 | 26,02 | 26,67 |
| 12,5 | 26,43 | 27,24 |
| 13 | 26,84 | 27,76 |
| 13,5 | 27,25 | 28,20 |
| 14 | 27,63 | 28,57 |
| 14,5 | 27,98 | 28,87 |
| 15 | 28,30 | 29,11 |
| 15,5 | 28,60 | 29,29 |
| 16 | 28,88 | 29,43 |
| 16,5 | 29,14 | 29,56 |
| 17 | 29,41 | 29,69 |
| 17,5 | 29,70 | 29,84 |
| 18 | 30,00 | 30,00 |
Применение Саксенда
дозировка
Взрослые
Начальная доза составляет 0,6 мг/сут. Для улучшения переносимости со стороны желудочно-кишечного тракта дозу следует повышать еженедельно на 0,6 мг до достижения суточной дозы 3,0 мг (см. таблицу 3). При плохой переносимости последующей повышенной дозы в течение двух последовательных недель следует рассмотреть вопрос о прекращении лечения. Суточная дозировка выше 3,0 мг не рекомендуется.
Таблица 3
График увеличения дозы
| Доза, мг | Недели | |
| Увеличение дозы в течение 4 недели | 0,6 | 1 |
| 1,2 | 1 | |
| 1,8 | 1 | |
| 2,4 | 1 | |
| Поддерживающая доза | 3,0 мг | |
Дети ≥12 лет Для детей от 12 лет следует применять такую же схему повышения дозы, как и для взрослых (см. таблицу 3). Дозу следует увеличивать до достижения 3,0 мг (поддерживающая доза) или максимальной переносимой дозы. Суточные дозы выше 3 мг не рекомендуются. Пропущенная доза Если инъекция пропущена в течение 12 ч с момента ее обычного введения, пациент должен принять дозу как можно скорее. Если до следующего введения остается менее 12 ч, пациент не должен принимать пропущенную дозу, а продолжать режим приема 1 раз в сутки — принять следующую запланированную дозу препарата. Не следует принимать дополнительную дозу или увеличивать дозу для компенсации пропущенной инъекции. Пациенты с сахарным диабетом 2 типа Лекарственное средство Саксенда не следует применять в комбинации с другими агонистами рецепторов ГПП-1. Для снижения риска развития гипогликемии в начале применения лекарственного средства Саксенда следует рассмотреть возможность уменьшения дозы одновременно применяемых инсулина или стимуляторов секреции инсулина (например, сульфонилмочевины). Необходим самоконтроль уровня глюкозы в крови для коррекции дозы инсулина или стимуляторов секреции инсулина (см. ОСОБЫЕ УКАЗАНИЯ). Особые группы пациентов Пациенты пожилого возраста (≥65 лет) Коррекция дозы в связи с возрастом не требуется. Опыт применения препарата пациентам ≥75 лет ограничен, поэтому не рекомендуется применять его данной категории пациентов (см. Фармакокинетика и ОСОБЫЕ УКАЗАНИЯ). Пациенты с нарушениями функции почек Коррекция дозы не требуется у пациентов с легкой или средней степенью нарушения функции почек (клиренс креатинина ≥30 мл/мин). Не рекомендуется применять Саксенда пациентам с тяжелым нарушением функции почек (клиренс креатинина 30 мл/мин), включая пациентов с терминальной стадией нарушения функции почек (см. Фармакокинетика, ОСОБЫЕ УКАЗАНИЯ и ПОБОЧНЫЕ ЭФФЕКТЫ). Пациенты с нарушениями функции печени Не рекомендуется корректировать дозу пациентам с легкой или средней степенью нарушения функции печени. Применение лекарственного средства Саксенда не рекомендуется пациентам с тяжелым нарушением функции печени, а пациентам с легким или умеренным нарушением функции печени препарат следует применять с осторожностью (см. Фармакокинетика и ОСОБЫЕ УКАЗАНИЯ).
Способ введения
Лекарственное средство Саксенда® предназначено только для подкожного введения. Его нельзя вводить в/в или в/м. Препарат вводят п/к 1 раз в сутки в любое время вне зависимости от еды. Его можно вводить п/к в участок передней брюшной стенки, бедра или плеча. Место и время введения можно изменять без коррекции дозы, однако желательно вводить примерно в одно и то же наиболее удобное время. Дополнительные сведения о вводе см. в разделе. См. раздел Руководство по использованию шприц-ручки.
Инструкция по использованию шприц-ручки Саксенда®, 6 мг/мл, раствор для инъекций в предварительно заполненной шприц-ручке
Необходимо внимательно прочитать инструкцию перед использованием шприц-ручки Саксенда®.
Не использовать шприц-ручку без получения соответствующей информации по ее использованию от врача или медсестры.
Применение препарата следует начинать с тестом шприца пера, чтобы убедиться, что она содержит лекарственное средство Саксенда®, 6 мг/мл, то вам нужно посмотреть на фотографиях ниже, чтобы узнать о различных частях шприца ручки и иголки.
Если у пациента плохое зрение или он не видит вообще, можно применять шприц-ручку без посторонней помощи. Помогать необходимо человеку с хорошим зрением, который может увидеть счетчик дозы на шприц-ручке Саксенда® и умеющий пользоваться им.
Шприц-ручка Саксенда® предварительно заполнена. Она содержит 18 мг лираглутида, что позволяет вводить дозы 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг и 3,0 мг. Шприц-ручка Саксенда® предназначена для использования с одноразовыми иглами НовоФайн® или НовоТвист длиной до 8 мм и толщиной 32 G. Иглы не входят в комплект.
Важная информация
Необходимо обратить особое внимание на эту отметку, поскольку она важна для безопасного пользования шприц-ручкой.
Саксенда® шприц ручка и игла (пример)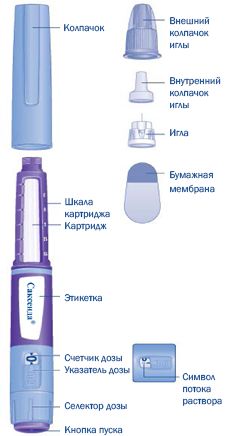
| 1. Подготовка шприц-ручки с новой иглой для использования • Проверьте название и цветную этикетку Вашей шприц-ручки, чтобы убедиться, что она содержит лекарственное средство Саксенда®. Это особенно важно в том случае, если вы применяете различные инъекционные лекарственные средства. Применение неправильного лекарственного средства может оказаться вредным для Вашего здоровья. • Снимите колпачок шприц-ручки. |  |
| Убедитесь, что раствор в шприце-ручке прозрачный и бесцветный. Посмотрите в окно шкалы картриджа. Если препарат мутный, использовать шприц-ручку запрещено. |  |
| • Возьмите новую одноразовую иглу и удалите защитную мембрану. |  |
| • Навинтите иглу на шприц-ручку и поверните ее, чтобы игла плотно держалась на шприц-ручке. |  |
| • Снимите внешний колпачок и сохраните его. Он пригодится после завершения инъекции для безопасного снятия иглы. |  |
| Снимите внутренний колпачок иглы. Если Вы попытаетесь надеть внутренний колпачок снова на иглу, то можете пораниться. На конце иглы может появиться капля раствора. Это нормальное явление, однако необходимо проверить поступление препарата при использовании новой шприц-ручки впервые. Не подсоединяйте новую иглу до тех пор, пока не будете готовы сделать инъекцию. Для каждой инъекции всегда используйте новую иглу. Это снизит риск закупорки иглы, заражения, попадания инфекции и введения неправильной дозы препарата. Никогда не используйте иглу, если она погнута или повреждена. |  |
| 2 Проверка работы шприц-ручки • Перед первым введением каждой новой шприц-ручки проверяйте поток. Если вы уже использовали эту шприц-ручку, перейдите к пункту 3. • Поверните селектор дозы, пока счетчик дозы не укажет символ проверки потока |  |
| • Держите шприц-ручку иглой вверх. Нажмите и удерживайте кнопку дозы до тех пор , пока счетчик дозы не вернется к 0. Значение 0 должно соответствовать показателю дозы. На кончике иглы должна появиться капля раствора. На кончике иглы может остаться маленькая капля, но она не будет вводиться. Если ни одна капля не появилась , повторите шаг 2 «Проверка работы шприц-ручки» до 6 раз. Если капли еще нет, измените иглу и повторите шаг 2 «Проверка работы шприц-ручки» еще раз. Если капля все-таки не появилась, утилизируйте ручку и используйте новую. • Необходимо всегда убедиться, что капля появляется на кончике иглы, прежде чем впервые использовать новую ручку. Это гарантирует, что раствор будет введен. Если капля не появится, не используйте шприц-ручку, даже если счетчик дозы изменяет значение. Это может указывать на заблокированную или поврежденную иглу. Если вы не проверите поток перед первым введением новой ручки, вы можете не получить необходимую дозу раствора для обеспечения желаемого эффекта препарата. |  |
| 3. Выставление дозы Возвращайте селектор дозы до тех пор, пока счетчик дозы не покажет необходимую дозу (0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг или 3,0 мг). Если выбрана неправильная доза, можно вернуть селектор дозы вперед или назад к правильной дозе. Шприц-ручка содержит максимум 3,0 мг препарата. С помощью селектора можно изменить дозу. Только счетчик дозы и показатель дозы покажут, какое количество миллиграмм Вы выбираете для инъекции. Если шприц-ручка содержит менее 3,0 мг, счетчик дозы останавливается до 3,0 мг. При вращении селектора вперед или назад слышен разный щелчок. Не считайте щелчки. Всегда используйте счетчик дозы и показатель дозы, чтобы увидеть, сколько миллиграмм Вы выбрали перед введением этого препарата. Не используйте картридж. Она показывает примерное количество раствора, оставшееся в шприц-ручке. Должны быть выставлены дозы 0,6 мг, 1,2 мг, 1,8 мг, 2,4 мг или 3,0 мг. Цифры на дисплее должны совпадать с показателем дозы, чтобы обеспечить правильную дозу для введения. |  |
| Остаток раствора в шприц-ручке • Шкала шприц-ручки показывает примерно, сколько раствора осталось в шприц-ручке. |  |
| • Чтобы точно увидеть, сколько раствора осталось , воспользуйтесь счетчиком дозы: Поверните селектор дозы влево, пока счетчик дозы не остановится. Если он указывает 3,0 мг, в шприц-ручке осталось не менее 3,0 мг. Если счетчик дозы останавливается до 3,0 мг, значит, в шприц-ручке не хватает раствора для полной дозы 3,0 мг. Если вам нужна более высокая доза, чем та, оставленной в пера. Вы можете разделить свою дозу между текущей шприц-ручкой и новой шприц-ручкой только при условии, что Вас проинструктировал врач или медсестра. Будьте очень осторожны, чтобы правильно рассчитать дозу. Будьте очень внимательны, рассчитывая дозу. Если вы не уверены, как разделить дозу с помощью двух шприц-ручек, выберите и введите необходимую дозу новой шприц-ручкой. |  |
| 4. Введение дозы • Введите иглу под кожу так, как показал врач или медсестра. • Убедитесь, что вы видите счетчик дозы. Не закрывайте счетчик пальцами. |  |
| • Нажмите и удерживайте пусковую кнопку до тех пор, пока счетчик дозы не покажет 0. 0 должен выравниваться указателем дозы. Затем Вы можете услышать щелчок. |  |
| • Держите иглу в коже после того, как счетчик дозы вернется в 0 и считайте медленно до 6. Если иглу вытащить раньше, Вы можете увидеть поток раствора из кончика иглы. В этом случае правильная доза не будет введена. |  |
| • Выньте иглу из-под кожи. Если капля крови появилась в месте инъекции, легко прижмите это место, но не растирайте его. Вы можете увидеть каплю раствора на кончике иглы после введения. Это нормально и не влияет на объем введенной дозы. Всегда смотрите на указатель дозы, чтобы видеть, сколько миллиграммов раствора Вы ввели. Держите пусковую кнопку, пока указатель дозы не покажет 0. Как обнаружить заблокированную или поврежденную иглу? Если 0 не отображается на счетчике дозы после постоянного нажатия пусковой кнопки, можно использовать заблокированную или поврежденную иглу. В этом случае Вы не ввели нужное количество лекарственного средства, даже несмотря на то что счетчик дозы переместился от ранее установленной дозы. Как обращаться с заблокированной иглой? Снимите иглу как описано в пункте 5. «После инъекции» и повторите все шаги, начиная с Шага 1 «Подготовка шприц-ручки с новой иглой для использования» Никогда не прикасайтесь к счетчику дозы при введении. Это может прервать инъекцию. |  |
| 5. После инъекции Наденьте наружный колпачок на иглу, не касаясь иглы или наружной крышки иглы. |  |
| Как только игла накрывается, осторожно нажмите полностью на наружную крышку иглы. Осторожно выкрутите иглу и утилизируйте. |  |
| • Закрывайте крышкой шприц-ручку после каждого использования, чтобы защитить раствор от света. Всегда утилизируйте иглу после каждой инъекции, чтобы обеспечить удобство инъекции и предотвратить заблокирование иглы. Если игла заблокирована, вы не сможете ввести препарат. Когда ручка пуста, выбросьте ее без иглы по инструкции, полученным от врача, медсестры, фармацевта в соответствии с местными правилами. Никогда не пытайтесь надеть внутреннюю крышку иглы на иглу. Вы можете пораниться иглой. Всегда извлекайте иглу из ручки после каждой инъекции. Это может предотвратить заблокирование иглы, загрязнение, заражение, протекание раствора и неточную дозировку. |  |
| Важная информация Держите шприц-ручку и иглы в недоступном месте для других людей, особенно детей. Никогда не передавайте Вашу личную шприц-ручку или иглы другим людям. Ухаживающие лица должны быть очень осторожными при работе с использованными иглами, чтобы предотвратить травмирование иглой и перекрестное заражение. | |
| Уход за вашей шприц-ручкой Не оставляйте ручку в машине или в другом месте, где она может нагреться или слишком охладиться. Не вводить замороженный препарат. В случае введения ранее замороженного препарата Саксенда® Вы можете не получить ожидаемый лечебный эффект от этого препарата. Храните шприц-ручку от пыли, грязи и жидкостей. Не мойте, не замачивайте и не смазывайте шприц-ручку. При необходимости очистите ее с помощью ткани, смоченной мягким моющим средством. Предотвращайте падение и удары шприц-ручки о твердые поверхности. В случае падения или удара подсоедините новую иглу и проверьте подачу раствора перед вводом. Не пытайтесь наполнить шприц-ручку по окончании в ней лекарственного средства. Утилизируйте шприц-ручку по окончании раствора. Не пытайтесь отремонтировать автоинжектор или разобрать его. | |
Дети. Для детей в возрасте от 12 лет коррекция дозы не требуется. Безопасность и эффективность применения лекарственного средства Саксенда детям младше 12 лет не установлена (см. Фармакодинамика).
Противопоказания
повышенная чувствительность к действующему веществу или другим компонентам лекарственного средства.
Побочные эффекты
резюме профиля безопасности
Безопасность применение Саксенда® была оценена в 5 двойно слепых рандомизированных плацебо-контролируемых клинических исследованиях с участием 5813 взрослых пациентов с ожирением или избыточной массой тела с хотя бы одним сопутствующим заболеванием, связанным с избыточной массой тела. В целом наиболее частыми побочными реакциями были нарушения пищеварительной системы (67,9 %). (см. Описание отдельных побочных реакций).
Список побочных реакций
Ниже указаны побочные реакции, о которых сообщалось. Побочные реакции классифицированы по системам органов и частоте возникновения. Оценку частоты побочных реакций проводили по следующей шкале: очень часто (≥1/10), часто (от ≥1/100 до <1/10), нечасто (от ≥1/1000 до <1/100), редко (от ≥1/10000 до <1/1000), очень редко (<1/10000). В каждой группе побочные реакции приведены в порядке понижения их серьезности.
Со стороны иммунной системы: редко – анафилактические реакции.
Нарушения метаболизма и питания: часто – гипогликемия*; нечасто – обезвоживание.
Психические нарушения: часто – бессонница**.
Со стороны нервной системы: часто – головокружение, дисгевзия.
Со стороны сердечно-сосудистой системы: нечасто – тахикардия.
Со стороны пищеварительной системы: очень часто - тошнота, рвота, диарея, запор; часто - сухость во рту, диспепсия, гастрит, гастроэзофагеальная рефлюксная болезнь, боль в верхней части живота, метеоризм, отрыжка, вздутие живота; нечасто – панкреатит***, задержка стула желудка****.
Со стороны печени и желчных путей: часто желчнокаменная болезнь***; нечасто – холецистит***.
Со стороны кожи и подкожных тканей: нечасто – крапивница.
Со стороны почек и мочевыводящих путей: редко – ОПН, нарушение функции почек.
Общие расстройства и реакции в месте инъекции: часто – реакции в местах инъекций, астения, усталость; нечасто – недомогание.
Лабораторные исследования, часто – повышенный уровень липазы, повышенный уровень амилазы.
* Гипогликемия ( на основе сообщаемых симптомов не подтверждены измерениями уровня глюкозы в крови) произошла у пациентов, которые не имеют диабет 2 типа и, которые использовали Саксенда® в сочетании с диетой и физической активностью. Дополнительную информацию см. раздел Описание отдельных побочных реакций.
** Бессонница, в основном наблюдалась в течение первых 3 месяцев лечения.
*** См. ВЗАИМОДЕЙСТВИЯ.
**** Из контролируемой фазы 2, 3а и 3б клинических исследований.
Описание отдельных побочных реакций
Гипогликемия у пациентов, не страдающих сахарным диабетом 2 типа
В процессе клинических испытаний не было зафиксировано ни одного тяжелого случая возникновения гипогликемии (требующей посторонней помощи) у пациентов с избыточной массой тела или ожирением, которые не страдали сахарным диабетом 2 типа и применяли лекарственное средство Саксенда в сочетании с диетой и физической активностью. О возникновении симптомов гипогликемии сообщили 1,6% пациентов, получавших лекарственное средство Саксенда, и 1,1% пациентов, получавших плацебо. Однако эти случаи не были подтверждены измерением уровня глюкозы в крови. Большинство случаев легко переносились.
Гипогликемия у больных сахарным диабетом 2 типа
В клинических испытаниях, тяжелая гипогликемия было сообщено в 0,7% от избыточного веса или ожирения у пациентов с сахарным диабетом 2 типа , которые использовали Саксенда® в комбинации с диетой и физическими упражнениями и которые использовали сульфонилмочевины. Симптомы гипогликемии были также зарегистрированы у 43,6% пациентов, получавших Саксенда® и у 27,3% пациентов, получавших плацебо. Среди пациентов, не применявших сульфонилмочевину, было зафиксировано возникновение симптомов гипогликемии (определяются концентрацией глюкозы в плазме крови ≤3,9 ммоль/л) у 15,7% пациентов, получавших лекарственное средство Саксенда® и у 7,6% пациентов, получавших плацебо.
Гипогликемия у больных сахарным диабетом 2 типа, получающих инсулин
В процессе клинических исследований были сообщения о случаях тяжелой гипогликемии у 1,5% пациентов с избыточной массой тела или ожирением, страдавших сахарным диабетом 2 типа и применявших лираглутид по 3,0 мг/сут вместе с инсулином в сочетании с диетой и физической активностью. Среди пациентов было зафиксировано симптомы гипогликемии (определяются концентрацией глюкозы в плазме крови ≤3,9 ммоль/л) у 47,2% пациентов, получавших лираглутид, и у 51,8% пациентов, получавших плацебо. Среди пациентов, применявших сульфонилмочевину, были зафиксированы сообщения о возникновении симптомов гипогликемии у 60,9% пациентов, получавших лираглутид по 3,0 мг/сут, и у 60,0% пациентов, получавших плацебо.
Нарушения пищеварительной системы
Большинство случаев возникновения расстройств пищеварительной системы были легкой или умеренной тяжестью и не приводили к прекращению терапии. Обычно реакции возникали в течение первых недель лечения и снижались в течение нескольких дней или недель при продолжении лечения. У пациентов в возрасте от 65 лет при применении лекарственного средства Саксенда чаще наблюдаются нарушения со стороны пищеварительной системы. У пациентов с нарушением функции почек легкой или средней тяжести (клиренс креатинина ≥30 мл/мин) при применении лекарственного средства Саксенда могут чаще возникать нарушения со стороны пищеварительной системы. Острая почечная недостаточность Зафиксированы случаи возникновения ОПН у пациентов, применявших агонисты рецепторов ГПП-1. Большинство зарегистрированных случаев наблюдали у пациентов, страдающих тошнотой, рвотой и диареей, что и приводило к потере жидкости (см. ОСОБЫЕ УКАЗАНИЯ). Аллергические реакции Были сообщения о нескольких случаях возникновения анафилактических реакций, сопровождавшихся такими симптомами как гипотензия, повышенное сердцебиение, приступы удушья и отеки после применения лираглутида. Анафилактические реакции могут быть опасны для жизни. Поэтому, если есть подозрение на возникновение анафилактической реакции, следует прекратить применение лираглутида (см. ПРОТИВОПОКАЗАНИЯ). Реакции в месте инъекции Сообщалось о реакциях в месте введения лекарственного средства Саксенда. Эти реакции обычно были легкими, большинство из них исчезали в процессе последующего лечения. Тахикардия В процессе клинических исследований были сообщения о случаях тахикардии у 0,6% пациентов, получавших лекарственное средство Саксенда, и у 0,1% пациентов, получавших плацебо. Большинство случаев было легкой или умеренной степени тяжести. Случаи были единичными, большинство исчезали в процессе дальнейшего лечения. Дети В ходе клинического исследования с участием детей в возрасте от 12 лет с ожирением 125 пациентов принимали лекарственное средство Саксенда в течение 56 недель. В целом частота, тип и тяжесть нежелательных реакций у детей ≥ 12 лет с ожирением были сопоставимыми с взрослыми пациентами. По сравнению с взрослыми пациентами у детей от 12 лет вдвое чаще наблюдалась рвота. Процент пациентов, которые сообщили по крайней мере об одном эпизоде клинически значимой гипогликемии, был выше в группе применения лираглутида (1,6%), чем в группе плацебо (0,8%). Во время проведения исследования не зарегистрирован ни один эпизод тяжелой гипогликемии.
Особые указания
с целью улучшения наблюдения за биологическими лекарственными средствами название и номер серии вводимого препарата. должен быть четко указан.
Сердечная недостаточность
Нет терапевтического опыта лечения пациентов с застойной сердечной недостаточностью IV класса по классификации Нью-Йоркской ассоциации кардиологов (NYHA), поэтому лираглутид не рекомендуется применять этим пациентам.
Особые группы пациентов
Безопасность и эффективность применения лираглутида не установлены у пациентов:
– в возрасте ≥75 лет;
– применяющие другие лекарственные средства для коррекции массы тела;
– с вторичным ожирением, вызванным эндокринологическими расстройствами или расстройствами, связанными с питанием, или в результате применения лекарственных средств, которые могут вызвать увеличение массы тела;
– с тяжелыми нарушениями функции почек;
– с тяжелыми нарушениями функции печени.
Не используйте препарат Саксенда® эту группу пациентов (см. Дозировка). Поскольку исследования по применению лираглутида для коррекции массы тела у пациентов с легкими или умеренными нарушениями функции печени отсутствуют, его следует с осторожностью применять в этой группе пациентов (см. Фармакокинетика и Дозировка).
Опыт применения лираглутида больным с воспалительными заболеваниями кишечника и диабетическим гастропарезом ограничен. Применение лираглутида этим пациентам не рекомендовано, поскольку оно сопровождается временными побочными реакциями со стороны ЖКТ, в том числе тошнотой, рвотой и диареей.
Панкреатит
Случаи острого панкреатита наблюдались при применении аналогов рецептора ГПП-1.
Пациентов следует проинформировать о характерных симптомах острого панкреатита. При подозрении на панкреатит следует отменить лечение лираглутидом. Если подтверждается острый панкреатит, то повторное применение лираглутида не рекомендовано.
Желчнокаменная болезнь и холецистит
В клинических испытаниях при применении лираглутида для уменьшения массы тела процент возникновения желчнокаменной болезни и холецистита был выше по сравнению с пациентами, получавшими плацебо. Тот факт, что быстрая потеря массы тела может увеличить риск развития желчнокаменной болезни и, следовательно, холецистита, частично объясняет более высокую частоту возникновения данных заболеваний после применения лираглутида. Желчнокаменная болезнь и холецистит могут привести к госпитализации и холецистэктомии. Пациентов следует проинформировать о характерных симптомах желчнокаменной болезни и холецистита.
Заболевания щитовидной железы
В процессе клинических исследований сахарного диабета 2 типа отмечены побочные реакции со стороны щитовидной железы, такие как зоб, особенно у пациентов с уже имеющимися заболеваниями щитовидной железы. Поэтому лираглутид следует с осторожностью применять этим пациентам.
Частота сердечных сокращений
При клинических исследованиях лираглутида наблюдалось увеличение ЧСС (см. Фармакодинамика). ЧСС следует контролировать через равные промежутки времени. Пациенты должны быть проинформированы о симптомах увеличения частоты сердцебиения (повышенное сердцебиение или ощущение повышенного сердцебиения в покое). Пациентам, у которых наблюдается клинически устойчивое увеличение ЧСС в покое, лечение лираглутидом следует прекратить.
Обезвоживание
Симптомы обезвоживания, в том числе нарушения функции почек и ОПН, были зарегистрированы у пациентов, получающих агонист рецептора ГПП-1. Пациенты предписанных лираглутид должны быть информированы о возможности обезвоживания из-за нарушения пищеварения и необходимости принимать меры для обезвоживания.
Гипогликемия у больных сахарным диабетом 2 типа
У пациентов с сахарным диабетом 2 типа, получающих лираглутид в сочетании с инсулином и/или сульфонилмочевиной, может быть повышен риск гипогликемии. Риск возникновения гипогликемии может быть снижен за счет уменьшения дозы инсулина и/или сульфонилмочевины.
Дети. У детей ≥ 12 лет, получавших лечение лираглутидом, сообщалось об эпизодах клинически значимой гипогликемии. Пациентов следует проинформировать о характерных симптомах гипогликемии и соответствующих мерах.
Гипергликемия у больных сахарным диабетом 2 типа, получающих инсулин
Саксенда® не используется в качестве заменителя инсулина у диабетиков. Сообщалось о развитии диабетического кетоацидоза у пациентов, применявших инсулин, при быстром прекращении или уменьшении дозы в качестве заменителя инсулина (см. ПРИМЕНЕНИЕ).
Вспомогательные вещества
Саксенда содержит менее 1 ммоль натрия (23 мг), поэтому лекарственное средство можно считать несодержащим натрия.
Применение в период беременности и кормления грудью.
Беременность. Адекватные данные по применению лираглутида у беременных отсутствуют. Исследования на животных показали репродуктивную токсичность (см. Доклинические данные по безопасности). Потенциальный риск для людей неизвестен. Лираглутид не следует использовать во время беременности. Если пациентка хочет забеременеть или беременна, прием лираглутида необходимо отменить.
Период кормления грудью
Неизвестно, лираглутид экскретируется в грудное молоко человека. Исследования на животных показали, что в молоко попадает незначительное количество лираглутида и его близкородственных структурных метаболитов. Доклинические исследования выявили связанное с применением препарата уменьшение темпов роста новорожденных крысенок (см. Доклинические данные о безопасности применения). В связи с недостаточным опытом применения препарата в период кормления грудью не следует его применять в этот период.
Фертильность
Кроме незначительного уменьшения количества живых имплантированных эмбрионов, исследования на животных не выявили вредного воздействия препарата на репродуктивную способность (см. Доклинические данные о безопасности применения).
Способность влиять на скорость реакции при управлении транспортными средствами или другими механизмами. Саксенда® не влияет или оказывает незначительное влияние на способность управлять автомобилем и механизмами. Однако может возникать головокружение в течение первых 3 мес лечения лекарственным средством Саксенда®. Следует с осторожностью управлять автотранспортом или другими механизмами при возникновении головокружения.
Взаимодействия
In vitro лираглутид показал очень низкий потенциал воздействия на фармакокинетику других активных субстанций, обмен которых связан с цитохромом Р450, а также связывание с белками плазмы крови.
Лираглутид приводит к незначительной задержке стула желудка, что может повлиять на всасывание пероральных препаратов, применяемых одновременно. Исследования по взаимодействию не показали какого-либо клинически значимого замедления всасывания, поэтому коррекция дозы не требуется. Исследование взаимодействия проводили при применении лираглутида в дозе 1,8 мг. Влияние на скорость опорожнения желудка было эквивалентно лираглутиду в дозе 1,8 мг и 3,0 мг (парацетамол AUC0-300 мин). По крайней мере, один эпизод острой диареи был зарегистрирован у некоторых пациентов, получавших Саксенда®. Диарея может нарушать всасывание одновременно принимаемых пероральных лекарственных средств.
Варфарин и другие производные кумарина
Исследований лекарственного взаимодействия не проводили. Нельзя исключить клинически значимое взаимодействие с активной субстанцией, имеющей низкую растворимость или узкий терапевтический индекс, такой как варфарин. В начале лечения лираглутидом у пациентов, получающих варфарин или другие производные кумарина, рекомендуется чаще проводить контроль международного нормализованного соотношения (МНС).
Парацетамол
Лираглутид не изменял общую экспозицию парацетамола после однократной дозы 1000 мг. Cmax парацетамола снижалась на 31%, а время достижения максимальной концентрации (tmax ) увеличивалось до 15 мин. При одновременном применении парацетамола коррекция дозы не требуется.
Аторвастатин
Лираглутид не изменял общую экспозицию аторвастатина до клинически значимого уровня после его однократного введения в дозе 40 мг. В этой связи при одновременном применении с лираглутидом коррекция дозы аторвастатина не требуется. При одновременном введении с лираглутидом Cmax аторвастатина снижалась на 38%, а tmax увеличивался с 1 до 3 ч.
Гризеофульвин
Лираглутид не изменял общую экспозицию гризеофульвина после однократного его введения в дозе 500 мг. Cmax гризеофульвина увеличилась на 37%, а tmax не изменилась. Корректировка дозы при применении гризеофульвина и других низкорастворимых соединений с высокой проницаемостью не требуется.
Дигоксин
После однократного введения 1 мг дигоксина в сочетании с лираглутидом отмечено уменьшение значения AUC для дигоксина на 16%; Cmax уменьшился на 31%. Средний tmax дигоксина увеличивался с 1 до 1,5 ч. Исходя из данных результатов, коррекция дозы дигоксина не требуется.
Лизиноприл
После однократного введения 20 мг лизиноприла отмечено уменьшение AUC для лизиноприла на 15%, Cmax снижалось на 27%. Средний tmax лизиноприла увеличивался с 6 до 8 ч. Исходя из данных результатов, коррекция дозы лизиноприла не требуется.
Пероральные контрацептивы
При одновременном применении разовой дозы пероральных контрацептивов лираглутид снижал Cmax этинилэстрадиола или левоноргестрела на 12% и 13% соответственно, а tmax увеличивался на 1,5 ч. Это не оказало клинически значимого влияния на общую экспозицию этинилэстрадиола или левоноргестрела, что дает основание считать, что одновременный прием их с лираглутидом не повлияет на контрацептивный эффект этинилэстрадиола и левоноргестрела.
Дети. Исследования врачебного взаимодействия проводились только с участием взрослых пациентов.
Передозировка
в клинических исследованиях и сообщениях, поступивших после выведения лекарственного средства Саксенда на рынок, отмечены случаи превышения рекомендуемой поддерживающей дозы до 24 раз (72 мг). В целом пациенты сообщали о сильной тошноте, рвоте и диарее, но никто из пациентов не сообщал о тяжелой гипогликемии. Все больные поправились без осложнений. При передозировке следует проводить поддерживающее лечение в соответствии с имеющимися у пациента клиническими признаками и симптомами. Необходимо наблюдение за клиническими признаками на случай возникновения обезвоживания и гипогликемии.
Условия хранения
хранить в недоступном для детей месте. Хранить в холодильнике (2–8 °С) подальше от морозильной камеры. После первого применения хранить при температуре ниже 30 °С или в холодильнике (2-8 °С). Не замораживайтесь. Для предотвращения действия света храните шприц-ручку с закрытым колпачком.